

Abstract
Aberrant splicing is typically attributed to splice‐factor (SF) mutation and contributes to malignancies including acute myeloid leukemia (AML). Here, we discovered a mutation‐independent means to extensively reprogram alternative splicing (AS). We showed that the dysregulated expression of eukaryotic translation initiation factor eIF4E elevated selective splice‐factor production, thereby impacting multiple spliceosome complexes, including factors mutated in AML such as SF3B1 and U2AF1. These changes generated a splicing landscape that predominantly supported altered splice‐site selection for ~800 transcripts in cell lines and ~4,600 transcripts in specimens from high‐eIF4E AML patients otherwise harboring no known SF mutations. Nuclear RNA immunoprecipitations, export assays, polysome analyses, and mutational studies together revealed that eIF4E primarily increased SF production via its nuclear RNA export activity. By contrast, eIF4E dysregulation did not induce known SF mutations or alter spliceosome number. eIF4E interacted with the spliceosome and some pre‐mRNAs, suggesting its direct involvement in specific splicing events. eIF4E induced simultaneous effects on numerous SF proteins, resulting in a much larger range of splicing alterations than in the case of mutation or dysregulation of individual SFs and providing a novel paradigm for splicing control and dysregulation.
Keywords: acute myeloid leukemia, eIF4E, splicing
Subject Categories: Chromatin, Transcription & Genomics; Translation & Protein Quality
Elevated expression of eIF4E leads to widescale changes in alternative splicing in the absence of splice‐factor mutations.

Introduction
Studies into the genomics, epigenetics, and transcriptomes of cancer have yielded important insights into its pathogenesis. However, proteomic studies revealed that the transcriptome does not always predict the proteome (de Sousa Abreu et al, 2009). This disconnect is due, in part, to post‐transcriptional regulation, e.g., splicing, nuclear RNA export, and translation. Dysregulation of these events can elevate the production and/or alter the structure and function of proteins involved in all facets of malignancy (Mars et al, 2021; Borden, 2022). Altered splicing is well known to produce a variety of biological impacts on the cells. Splicing is the removal of introns and joining of flanking exons in pre‐messenger RNAs (pre‐mRNA) and some noncoding RNAs (Wahl et al, 2009). Most splicing is catalyzed by the major spliceosome, an intricate assembly of > 150 proteins and 5 uridine‐rich small nuclear UsnRNAs (U1, U2, U4, U5, and U6 snRNAs) (Wahl et al, 2009). This machine recognizes elements in the 5′ splice‐site (5′SS), 3′SS, and the branch site to catalyze the excision of targeted introns. Alternative splicing (AS) generates greater diversity in the proteome by producing multiple mRNAs from the same pre‐mRNA (Urbanski et al, 2018). About 95% of multi‐exonic genes undergo AS (Pan et al, 2008). AS events include the altered selection of the 5′SS or 3′SS, skipped exons (SE), inclusion of mutually exclusive exons (MXE), or intron retention (IR) (Pan et al, 2008). AS products can have opposing functions to their constitutive counterparts, lead to transcript and protein mislocalization, generate highly labile transcripts that are rapidly degraded causing protein loss, or other effects (Effenberger et al, 2017; Urbanski et al, 2018).
Dysregulation of splicing contributes to hematologic malignancies, solid tumors, and genetic diseases (Saez et al, 2017; Urbanski et al, 2018; Taylor & Lee, 2019). In AML, ~30% of expressed genes are aberrantly spliced compared with CD34+ cells from healthy individuals (Adamia et al, 2014; Dvinge & Bradley, 2015). The best‐characterized modality for driving aberrant splicing in AML involves mutations of splice factors (SF). The most frequently mutated SFs in hematological malignancies are SF3B1, SRSF2, and U2AF1 in myelodysplastic syndromes (MDS), and these mutations are associated with progression to AML, with ~5–10% of AML patients harboring these (Saez et al, 2017; Urbanski et al, 2018; Taylor & Lee, 2019; Visconte et al, 2019). Intriguingly, these mutations do not disrupt the splicing of all transcripts but rather have targeted effects. For instance, SF3B1 mutations lead to 83 altered splicing events in AML (Hershberger et al, 2021). Furthermore, in AML, aberrant splicing is much more widespread than the frequency of SF mutations suggesting that there are additional means to modify splicing (Rivera et al, 2021). For example, two components of the spliceosome (PRPF6 and SF3B1) are elevated in secondary AML specimens without SF mutations, relative to healthy volunteers (Crews et al, 2016). In a diverse set of solid tumors, SRSF1, SRSF2, and U2AF2 levels are upregulated (Urbanski et al, 2018). In these malignancies, the underlying mechanisms for SF elevation are not understood. Identification of modalities by which splicing becomes dysregulated is critical since it will enable us to unravel the molecular basis for reprogramming splicing in the absence of SF mutations, which in turn can provide new directions for therapeutic targeting.
The eukaryotic translation initiation factor eIF4E is dysregulated in hematological malignancies including AML (Topisirovic et al, 2003; Assouline et al, 2009, 2015). eIF4E can be found in the nucleus and in the cytoplasm where it plays distinct roles in RNA metabolism, which contribute to AML and other malignancies (Cohen et al, 2001; Topisirovic et al, 2003, 2009a; Culjkovic et al, 2005, 2006; Culjkovic‐Kraljacic et al, 2012, 2020a; Volpon et al, 2017; Davis et al, 2019). Consistently, high‐eIF4E AML patient specimens are characterized by both elevation of eIF4E and its nuclear enrichment (Topisirovic et al, 2003; Assouline et al, 2009, 2015; Osborne & Borden, 2015) (Appendix Fig S1A and B). This eIF4E phenotype can be targeted in AML patients with ribavirin leading to clinical benefit (Assouline et al, 2009, 2015). Indeed, the nuclear distribution of eIF4E is observed prior to ribavirin treatment in high‐eIF4E AML patients, but then during clinical responses including complete remissions eIF4E is found mainly in the cytoplasm with its return to the nucleus an indicator of clinical relapse (Assouline et al, 2009, 2015). Molecular studies revealed that eIF4E entry into the nucleus was mediated by Importin 8 (Volpon et al, 2016). Interestingly, Importin 8 binds the cap‐binding site of eIF4E preventing its association with RNAs and thus selectively imports RNA‐free eIF4E (Volpon et al, 2016, 2017). Ribavirin, or excess cap analogs, disrupt this eIF4E‐Importin 8 interaction preventing nuclear entry of eIF4E and thus provides a molecular basis for the mainly cytoplasmic staining of eIF4E during clinical responses (Volpon et al, 2016, 2017). Upon chemical deactivation of ribavirin, eIF4E once again binds Importin 8, which allows nuclear entry and thus re‐engagement of dysregulated nuclear activities of eIF4E associated with disease progression (Zahreddine et al, 2014; Volpon et al, 2016, 2017).
At the molecular level, eIF4E binds the m7G cap (referred to as the “cap”), on the 5′ end of RNAs, an activity conserved between nuclear and cytoplasmic compartments. In this way, eIF4E interacts with the cap to mediate nuclear RNA export, and translation (Borden & Volpon, 2020). While these RNAs are capped, other signals within the RNAs, which are referred to as USER codes, provide selectivity (Keene & Tenenbaum, 2002; Blackinton & Keene, 2014). For example, to be a nuclear RNA export target, RNAs must not only be capped but also contained an element known as an eIF4E sensitivity element (4ESE) within their 3′UTRs. This element recruits the export machinery forming an active export complex with eIF4E‐LRPPRC‐4ESE RNA and CRM1 (Culjkovic et al, 2005, 2006; Topisirovic et al, 2009a; Volpon et al, 2017). For translation selectivity, RNAs must contain complex, structured 5′UTRs in addition to their cap (Gingras et al, 1999). In these cases, eIF4E does not alter transcript levels. Thus, eIF4E can amplify transcriptional signals for a subset of RNAs containing appropriate USER codes by promoting their nuclear export and/or enhancing their translation efficiency (Culjkovic‐Kraljacic & Borden, 2018; Borden, 2020).
More recently, eIF4E has been implicated in nuclear RNA maturation including m7G capping, as well as cleavage and polyadenylation (CPA) of selected RNAs (Davis et al, 2019; Culjkovic‐Kraljacic et al, 2020a; Osborne et al, 2022). Thus, eIF4E plays broader roles in mRNA maturation, and eIF4E can be considered a cap chaperone (Borden, 2016; Mars et al, 2021). Similar to its selection of nuclear RNA export targets, selectivity arises due to USER codes in target RNAs (Davis et al, 2019; Culjkovic‐Kraljacic et al, 2020a). For the case of both m7G capping and CPA, studies revealed a pattern whereby eIF4E: (i) elevates the levels of the CPA and capping machinery through increased export of the transcripts encoding these factors; and (ii) eIF4E physically associated with selected components of the capping and CPA machinery (Davis et al, 2019; Culjkovic‐Kraljacic et al, 2020a; Osborne et al, 2022). Given these observations, we reasoned that eIF4E similarly impacts the splicing of selected RNAs. Here, we demonstrate that in the nucleus eIF4E bound to capped RNAs that encode SFs stimulated their RNA export and resultant protein production without altering their transcription or RNA stability. Additionally, nuclear eIF4E was physically interacted with components of the spliceosome and specific pre‐mRNAs indicative of a direct role in some splicing events. Consistent with these observations, eIF4E drives wide‐ranging changes to the splicing of a broad array of RNAs in multiple cellular contexts. In all, this work uncovers novel roles for eIF4E beyond amplifying the protein‐coding capacity of transcripts, to include molding their physical nature. It also provides a novel paradigm by which to reprogram splicing in the absence of SF mutation. In this way, our studies provide a unique opportunity to dissect mechanisms driving dysregulated splicing independent of these mutations. In turn, this could substantially expand the use of splicing inhibitors beyond patients harboring SF mutations.
Results
eIF4E overexpression impacts protein levels of splice factors through its RNA export function
As a first step to investigate the possible role of eIF4E in splicing, we inspected a previously collected endogenous nuclear eIF4E RIP‐seq dataset derived from an aggressive B‐cell lymphoma LY1 cell line where nuclear eIF4E was associated with ~3,000 transcripts (Culjkovic‐Kraljacic et al, 2016). Here, our analysis revealed that eIF4E was associated with ~100 RNAs, which encoded factors involved in splicing regulation of which 53 RNAs encoded components of the major spliceosome including SF3B1 and U2AF1, factors frequently mutated in AML. Interestingly, STRING analysis revealed a significant enrichment of all the major spliceosome complexes with FDRs ranging from 0.0016 to 2.19 × 10−49 depending on the splicing group (Fig 1A). These analyses prompted us to examine whether endogenous eIF4E could modulate splice‐factor production and thus splicing.
Figure 1. eIF4E modifies the SF landscape via its nuclear export activity.
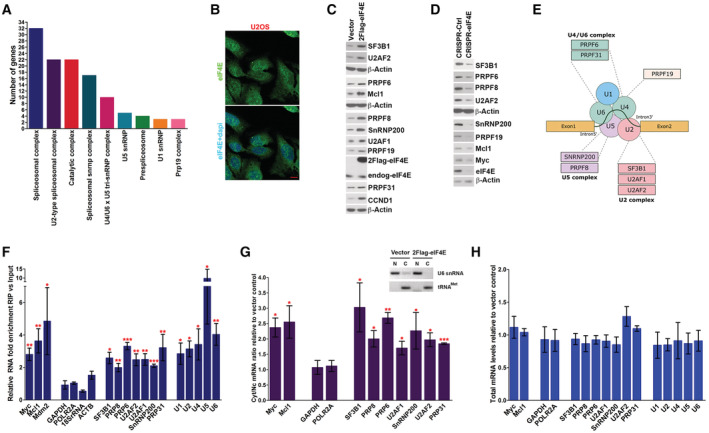
- STRING analysis of nuclear eIF4E RIPs from LY1 cells indicates that eIF4E is physically associated with RNAs encoding components of each of the major snRNPs. Number of RNAs in each group is shown. All enrichments were statistically significant with false discovery rates (FDR) ranging from 0.0016 to 2.16 × 10−49.
- Localization of eIF4E in U2OS cells. Confocal micrographs of cells stained with anti‐eIF4E antibodies to detect endogenous eIF4E and DAPI as a nuclear marker. Single (eIF4E) and overlaid (eIF4E + DAPI) channels are shown. Micrographs are single sections through the plane of the cells with 63× magnification. Scale bar, 10 μm.
- Western blot (WB) analysis of splicing factors for Vector and 2FLAG‐eIF4E U2OS cell lines. Myc, Mcl1, and cyclin D1 (CCND1) served as positive controls and β‐actin as a loading control. Both 2FLAG‐eIF4E (2Flag‐eIF4E) and endogenous eIF4E (endog‐eIF4E) are shown. Each β‐actin blot corresponds to the above western blots. Experiments were carried out at least three independent times, and one representative experiment is shown. These are quantified in concert with the S53A mutant in Fig 2A.
- WB analysis of splicing factors as a function of eIF4E reduction using CRISPR‐eIF4E and CRISPR‐Ctrl U2OS cell lines. Myc, Mcl1, and CCND1 served as positive controls and β‐actin a loading control. Each β‐actin blot corresponds to the western blots immediately above. Experiments were carried out at least three independent times, and one representative experiment is shown.
- eIF4E is positioned to influence multiple facets of spliceosome activity. Schematic representation summarizing the splicing factors targeted by eIF4E. Proteins are grouped based on their activity and/or association with a specific complex of the major spliceosome. eIF4E also physically interacts with all UsnRNAs shown (panel F).
- The enrichment of RNAs in RIPs of endogenous eIF4E versus input RNAs from the nuclear fractions of Vector control U2OS cells monitored by RT–qPCR. Data were normalized to input samples and presented as a fold change. The mean, standard deviation, and P‐values were derived from five independent experiments (each carried out in triplicate). MYC and MCL1 are established eIF4E nuclear targets and served as positive controls, while ACTB, GAPDH, POLR2A, and 18S rRNA were negative controls.
- RNA export assays for 2FLAG‐eIF4E and Vector control U2OS cell lines. Levels of the transcript were measured in nuclear and cytoplasmic compartments by RT–qPCR with fractionation controls shown in the inset. Data were normalized to Vector control and shown as a fold change. ACTB was used as a normalizer. The mean, standard deviation, and P‐values were derived from three independent replicates, i.e., from three different cell lines for each condition, with each carried out in triplicate. Inset. Semi‐qPCR for U6 snRNA and tRNAMet as controls for the nuclear and cytoplasmic fractions, respectively, corresponding to the export assay shown; N = nuclear fraction, C = cytoplasmic fraction. Representative fractionation control set (out of three biological replicates) is shown.
- Total RNA levels monitored by RT–qPCR corresponding to mRNA export assays shown in (G). Data were normalized to Vector control to calculate fold change. The mean and standard deviation, where no P‐values were < 0.05 (using Student t‐test), were derived from three biological experiments, each carried out in technical triplicate.
Data information: Student t‐test, *P < 0.05, **P < 0.01, ***P < 0.001.
To interrogate the potential eIF4E‐splicing axis, we prioritized studies using U2OS cells for several reasons. First, unlike the lymphoma cells used above, U2OS cells have endogenous eIF4E levels, which are similar to healthy volunteer cells enabling us to measure the impact of eIF4E overexpression (Appendix Fig S1A). Second, we observed endogenous and overexpressed eIF4E in the nucleus, consistent with a potential role in splicing, as well as in the cytoplasm (Culjkovic et al, 2005; Topisirovic et al, 2009a) (Fig 1B–H and Appendix Fig S1B). Third, eIF4E plays well‐established cap‐dependent roles in capping, CPA, nuclear RNA export, and translation in these cells (Culjkovic et al, 2005, 2006; Topisirovic et al, 2009a; Culjkovic‐Kraljacic et al, 2012, 2020a; Volpon et al, 2017; Davis et al, 2019) suggestive that splicing could also be impacted. Finally, a conservation of these interactions between lymphoma and U2OS cells would suggest that this is a general function of eIF4E and not a phenomenon restricted to lymphoma. With this in mind, we generated 3 stable 2FLAG‐eIF4E or Vector control cell lines as described (Culjkovic‐Kraljacic et al, 2020a). RNA sequence data revealed that neither U2OS cells nor 2FLAG‐eIF4E nor Vector stable cell lines harbored SF mutations frequently associated with cancer, e.g., SF3B1, U2AF1, or SRSF2 (Data ref: Culjkovic‐Kraljacic et al, 2020b). Additionally, the extent of eIF4E elevation in these cells was physiologically relevant; 2FLAG‐eIF4E U2OS cells had an average 2.7‐fold elevation in eIF4E protein levels relative to Vector controls over the three clones (Fig 1C with quantification in Fig 2A). By comparison, AML specimens had up to ~8‐fold elevation of eIF4E protein levels relative to healthy tissues (Assouline et al, 2009, 2015).
Figure 2. Analysis of the impacts of S53A eIF4E mutant on its capacity to alter the SF landscape and the role of translation for this activity.
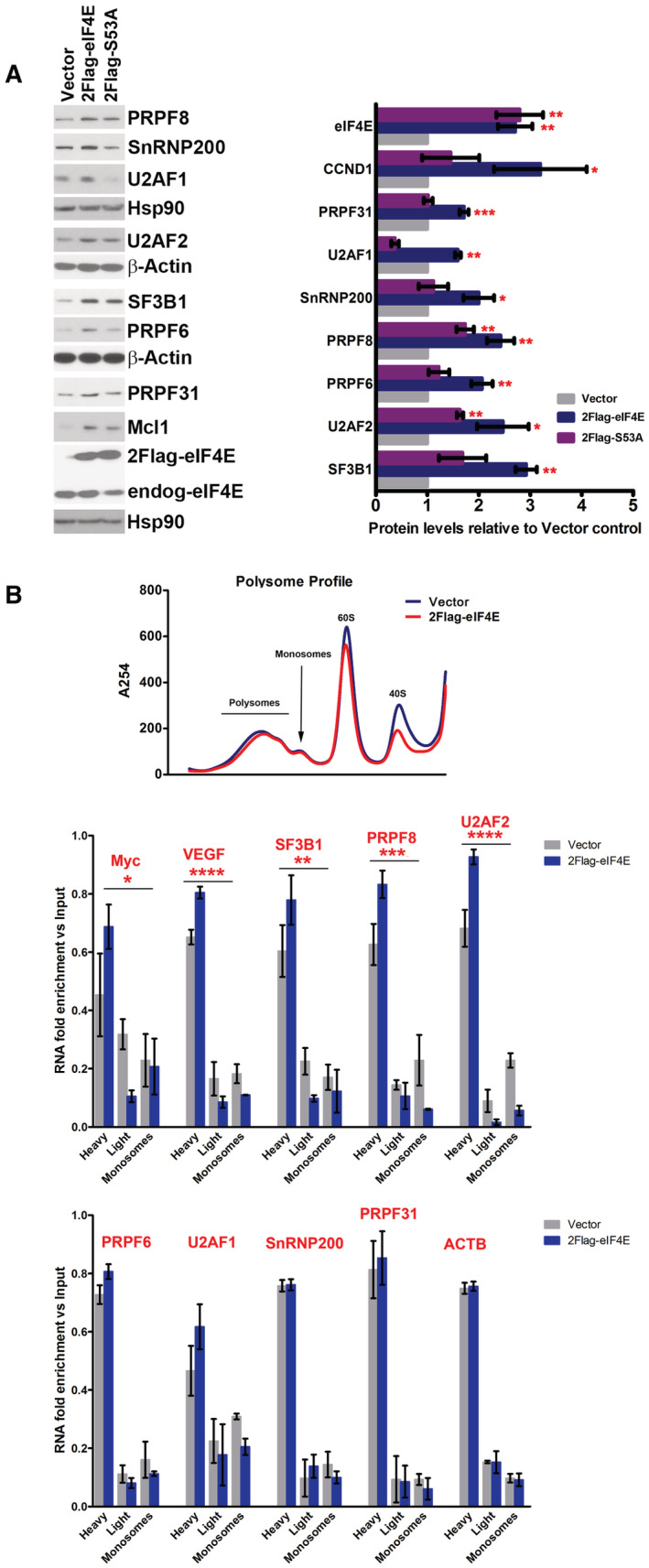
- WB analysis of the impacts of S53A 2FLAG‐eIF4E (2Flag‐S53A) relative to WT 2FLAG‐eIF4E (2Flag‐eIF4E) on SF production. CCND1 served as positive controls and β‐actin or HSP90 as loading controls. Both 2FLAG‐eIF4E (2Flag‐eIF4E) and endogenous eIF4E (endog‐eIF4E) are shown. Each β‐actin or HSP90 blot corresponds to the above western blots. S53A eIF4E protein levels are indistinguishable from eIF4E. Experiments were carried out using three separate clones for eIF4E, S53A, and Vector controls, and one representative experiment is shown. (Right) Western blots were quantified by FIJI, and intensities normalized to Vector control and plotted using PRISM. The mean, standard deviation, and P‐values are shown. Student t‐test: *P < 0.05, **P < 0.01, ***P < 0.001.
- Polysomal loading analysis was measured to assess the relevance of eIF4E's capacity to increase translation efficiency on SF protein production. Top panel shows the polysomal profile (at 254 nm) demonstrating that 2FLAG‐eIF4E overexpression did not alter the profile relative to Vector controls, consistent with previous studies (Culjkovic‐Kraljacic et al, 2016, 2020a). Polysomes were isolated by size exclusion chromatography (SEC) and thus the heaviest polysomes elute first and monosomes last. (Middle and Bottom panels) RNAs were monitored on heavy, light, or monosome polysomes using RT–qPCR and presented as a fraction of the given RNA. Known translation targets of eIF4E, MYC, and VEGF were shifted to higher polysomes in eIF4E overexpressing cells and SF‐encoding RNAs with altered polysomes (middle) or those that were unchanged by eIF4E including the negative control ACTB are in the bottom panel (Culjkovic‐Kraljacic et al, 2016, 2020a). Experiments were carried out in biological duplicates using different clones, each carried out in technical triplicate. Means are shown and P‐values calculated using ANOVA (PRISM).
Data information: ANOVA test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We first set out to ascertain if eIF4E modulated levels of SFs. To assess these, we prepared total cell lysates and monitored protein levels by western blot. SF3B1, U2AF1, U2AF2, PRPF6, PRPF8, PRPF19, PRPF31, SNRNP200 protein levels were elevated by ~2‐ to 3‐fold in 2FLAG‐eIF4E cells relative to Vector controls (Fig 1C). Cyclin D1 and Mcl1 served as positive controls while β‐actin provides a negative control as it is not an eIF4E target (Culjkovic et al, 2006; Topisirovic et al, 2009a; Culjkovic‐Kraljacic et al, 2012, 2020a; Davis et al, 2019) (Fig 1C). We next examined whether depletion of endogenous eIF4E reduced levels of these SFs using CRISPR‐eIF4E or CRISPR‐Ctrl U2OS cell lines as described (Culjkovic‐Kraljacic et al, 2020a). CRISPR‐Ctrl cell lines were generated using guide RNAs to Galaxidae coral Azami‐green transcripts (Culjkovic‐Kraljacic et al, 2020a). CRISPR‐eIF4E cell lines were heterozygous for eIF4E as its complete deletion was lethal (Culjkovic‐Kraljacic et al, 2020a). As expected, CRISPR‐eIF4E cells had reduced levels of PRPF6, PRPF8, PRPF19, SF3B1, SNRNP200, and U2AF2 proteins relative to CRISPR‐Ctrl cells (Fig 1D). These findings indicated that endogenous eIF4E impacted the production of the splicing machinery, and thus, eIF4E's effects on SFs were not limited to circumstances characterized by eIF4E elevation. In all, we demonstrate that SF production can be driven in an eIF4E‐dependent manner. SFs targeted by eIF4E are found in each of the major spliceosome complexes (Fig 1A and E) and included SFs that are often mutated in AML and other cancers, e.g., SF3B1 and U2AF1 (de Necochea‐Campion et al, 2016; Visconte et al, 2019; Rivera et al, 2021). In all, we demonstrate that eIF4E overexpression is sufficient to elevate the production of a wide array of SFs including components of the spliceosome. Thus, eIF4E is positioned to influence several splicing steps.
Next, we investigated the mechanism underpinning these effects on SF protein production. Given the fact that mRNAs encoding factors involved in capping and CPA were eIF4E‐dependent RNA export targets (Davis et al, 2019; Culjkovic‐Kraljacic et al, 2020a) and that SF‐coding mRNAs were associated with nuclear eIF4E in B‐cell lymphoma (Fig 1A), we reasoned that eIF4E was positioned to modulate levels of SF proteins by driving nuclear export of their corresponding RNAs thereby increasing their cytoplasmic availability to the translation machinery (Davis et al, 2019; Culjkovic‐Kraljacic et al, 2020a). As a first step to assess this possibility, we carried out eIF4E RIPs from nuclear lysates to ascertain if eIF4E is bound to SF‐encoding RNAs in the U2OS cells. Nuclear lysates were crosslinked with formaldehyde to prevent reassortment during the IP and RNAs were monitored by real‐time quantitative PCR (RT–qPCR). We observed that endogenous nuclear eIF4E RIPs with SF3B1, SNRNP200, PRPF6, PRPF8, PRPF31, U2AF1, U2AF2, which were enriched by ~2‐ to 4‐fold versus nuclear input (Fig 1F). Negative control RNAs ACTB, GAPDH, POLR2A RNAs, and 18S rRNA were not enriched in the RIPs relative to input while positive controls MCL1, MDM2, and MYC were present consistent with previous studies (Culjkovic et al, 2005, 2006; Davis et al, 2019; Culjkovic‐Kraljacic et al, 2020a). Given this physical interaction, we examined whether these SF‐encoding RNAs were targets of eIF4E‐dependent nuclear export. For this purpose, we monitored RNA levels in nuclear and cytoplasmic fractions by RT–qPCR in 2FLAG‐eIF4E cells relative to Vector controls, using the three different cell lines as above. As a proof‐of‐principle, we monitored SF3B1, U2AF1, U2AF2, SNRNP200, PRPF6, PRPF8, and PRPF31 all of which had ~2‐fold increase in cytoplasmic/nuclear ratios upon eIF4E overexpression indicating their nuclear export was elevated by eIF4E overexpression (Fig 1G). The positive controls MYC and MCL1 were elevated while the negative controls GAPDH and POLR2A were unchanged as expected (normalized to ACTB). Fraction quality was assessed by semiquantitative PCR monitoring U6 snRNA and tRNAMet for controls for the nuclear and cytoplasmic fractions, respectively (Fig 1G, inset) (Culjkovic‐Kraljacic et al, 2012). In all, these findings indicate that eIF4E enhanced the nuclear export of SF‐encoding RNAs, which provides a mechanism for eIF4E‐mediated elevation of SF protein production.
In addition to driving SF production via nuclear RNA export, it is possible that eIF4E could also indirectly drive transcription and/or alter transcript stability of SF‐encoding RNAs as a parallel means to drive their protein production. However, we noted that typical of eIF4E targets in the literature, eIF4E overexpression did not modulate total levels of any of the RNAs examined by RT–qPCR (Fig 1H). Moreover, our previously collected RNA‐Seq data comparing 2FLAG‐eIF4E and Vector control U2OS cells revealed only ~400 transcripts with significant changes (Culjkovic‐Kraljacic et al, 2020a; Data ref: Culjkovic‐Kraljacic et al, 2020b). Further inspection of this data revealed there were no alterations to the levels of SF‐encoding RNA. Thus, eIF4E does not impact on steady‐state transcription or RNA stability of these SF‐encoding transcripts.
To dissect the relative contribution of eIF4E‐dependent RNA export relative to its translation function on SF protein production, we used a separation‐of‐function mutant. The eIF4E S53A mutant is fully structured, binds the m7G cap, and promotes eIF4E‐dependent translation and wild‐type eIF4E; however, it is unable to promote eIF4E‐dependent nuclear RNA export (Kaufman et al, 1993; Zhang et al, 1995; Culjkovic‐Kraljacic et al, 2012). Importantly, this mutant no longer oncogenically transforms cells (Lazaris‐Karatzas et al, 1990; Culjkovic‐Kraljacic et al, 2012). We compared SF protein production in 2FLAG‐eIF4E, S53A mutant, and Vector controls. We monitored 3 stable clones for each cell line and note that wild‐type and S53A eIF4E are expressed at similar levels (Fig 2A). As expected for control RNAs, which are established eIF4E‐RNA export targets such as CCND1 (Culjkovic‐Kraljacic et al, 2012), the S53A mutant could not stimulate the production of cyclin D1 protein yielding levels similar to Vector controls, while levels were elevated in wild‐type eIF4E‐overexpressing cells as anticipated. Analysis of a subset of SFs revealed a similar pattern for SNRNP200, PRPF6, PRPF31, and U2AF1 whereby the levels of SFs were roughly equivalent between S53A and Vector controls indicating that the RNA export function was the primary means by which to enhance levels of these proteins. For some SFs such as PRPF8 and U2AF2, there was an intermediary impact of S53A mutant, elevating protein relative to Vector cells but not to the same extent as wild‐type eIF4E. This predicts that for PRPF8 and U2AF2 RNAs eIF4E impacted both the nuclear export and translation efficiency of these. The negative controls β‐actin and HSP90 were unchanged by mutant or wild‐type protein, as expected. In all, we observed that the S53A mutant did not promote protein expression to the same extent as wild‐type eIF4E for tested SFs confirming a nuclear export role for eIF4E in SF protein elevation.
To delve deeper into the possibility of co‐regulation of some SFs at both the RNA export and translation levels, we used polysome loading to directly measure translation efficiency, which is defined as the number of ribosomes loaded per transcript (Fig 2B). Polysome fractions were isolated using size exclusion chromatography according to (Yoshikawa et al, 2018, 2021) and thus the heaviest polysomes elute in the first fractions and monosomes last (Fig 2B). As expected, overall polysome profiles from 2FLAG‐eIF4E and Vector cells were indistinguishable (Fig 2B). Transcripts found in the heavier fractions have more ribosomes per transcript and thus are more efficiently translated than those found in the lighter polysome fractions. We compared SF‐encoding RNA content in monosomes, light and heavy polysome fractions in 2FLAG‐eIF4E and Vector U2OS cell lines using RT–qPCR (Fig 2B). RNAs that were impacted at the translation levels were expected to have an increase in the heavy polysome fraction with concomitant decreases in the light polysome and monosome fractions. As noted above, transcript levels were unchanged for measured targets upon eIF4E overexpression (Fig 1H). We found ACTB mRNAs unaltered by eIF4E expression as expected while positive controls MYC and VEGF were shifted to heavier polysomes as expected. We found no change in polysome loading for PRP31, U2AF1, SNRNP200, or PRPF6 transcripts. However, PRPF8, SF3B1, and U2AF2 mRNAs were characterized by an eIF4E‐dependent shift to heavy polysomes and reduction in light polysome and monosome fractions (Fig 2B, middle panel). Consistently with the protein levels observed with the S53A mutant, we observed that most eIF4E targets here were nuclear export targets; however, a few were sensitive to eIF4E at both the export and translation level making them much more sensitive to eIF4E levels.
Together the above studies demonstrated that eIF4E physically interacted with and promoted the nuclear export of many SF‐encoding RNAs providing a mechanism for the eIF4E‐mediated elevation of these factors. Further, eIF4E did not modulate the steady‐state transcription or stability of these transcripts. The studies with the S53A mutant strongly support a major role for the nuclear RNA export function of eIF4E in driving SF protein production, but for some SFs, there is also a translation contribution, making these latter RNAs even more sensitive to eIF4E dysregulation.
eIF4E drives the production of SFs in AML cell lines and primary AML specimens
Given the impact of eIF4E on the production of SFs in U2OS cells, we investigated whether eIF4E drove their production in a high‐eIF4E cancer opting to study high‐eIF4E AML given eIF4E has been targeted with ribavirin in early phase clinical trials leading to objective responses including remissions (Assouline et al, 2009, 2015). In these high‐eIF4E patients, eIF4E is typically elevated ~3‐ to 8‐fold and predominantly localized to the nucleus and characterized by elevated eIF4E‐dependent nuclear RNA export (Topisirovic et al, 2003; Assouline et al, 2009, 2015) (e.g., Appendix Fig S1A and B). Further supporting this choice of indication, AML is often associated with dysregulated splicing, which is typically attributed to mutation in specific SFs (Adamia et al, 2014; Crews et al, 2016; de Necochea‐Campion et al, 2016; Visconte et al, 2019; Rivera et al, 2021). We used two experimental systems to explore the impact of eIF4E on SFs in AML: the NOMO‐1 AML cell line, and primary specimens from AML patients. Given the close link between dysregulated splicing and mutations in U2AF, SF3B1, or SRSF2 in AML (Adamia et al, 2014; de Necochea‐Campion et al, 2016; Visconte et al, 2019; Rivera et al, 2021), we inspected our RNA‐Seq data to ensure that NOMO‐1 (Appendix Fig S1C) and the primary AML specimens (Data ref: Leucegene, 2015) examined did not harbor these mutations. In NOMO‐1 cells, eIF4E levels are very similar to those in healthy volunteers or normal‐eIF4E AML specimens (Appendix Fig S1A and B); thus, these cells provided a unique opportunity to engineer high‐eIF4E AML cells. To genetically dissect the role of eIF4E in an AML context and to compare to our U2OS system to ascertain if these eIF4E mechanisms are conserved in diverse cell types, eIF4E or Vector NOMO‐1 cell lines were produced as stable pools. NOMO‐1 eIF4E cells produced eIF4E protein levels and localization similar to high‐eIF4E AML specimens (Fig 3A, Appendix Fig S1A and B). We noted that eIF4E overexpression in NOMO‐1 cells did not lead to SF mutations commonly found in AML using targeted RNA‐sequencing (Appendix Fig S1C). We observed elevation of PRPF6, PRPF8, PRP31, SF3B1, SNRNP200, U2AF1, and U2AF2 proteins in eIF4E NOMO‐1 relative to Vector NOMO‐1 cells (Fig 3A). Further, positive controls Myc and cyclin D1 were elevated while the negative control β‐actin was unchanged. To assess the impact of eIF4E inhibition, we monitored the effect of the eIF4E inhibitor ribavirin, which has been used to target eIF4E in early‐phase clinical trials (Assouline et al, 2009, 2015; Dunn et al, 2017; Kosaka et al, 2017). We observed reduced SF levels upon eIF4E inhibition with clinically achievable doses of ribavirin in both conditions relative to vehicle‐treated controls consistent with ribavirin targeting eIF4E (Fig 3B). Thus, overexpression of eIF4E in AML cells led to changes in the splicing machinery similar to those observed in U2OS cell lines suggesting that eIF4E can dysregulate SF production in multiple contexts. Further, its effects on SF production can be reversed by a clinically used eIF4E inhibitor (Fig 3B) with similar results to the genetic reduction observed by CRISPR‐4E in U2OS cells (Fig 1D).
Figure 3. eIF4E reprograms the SF landscape in AML.
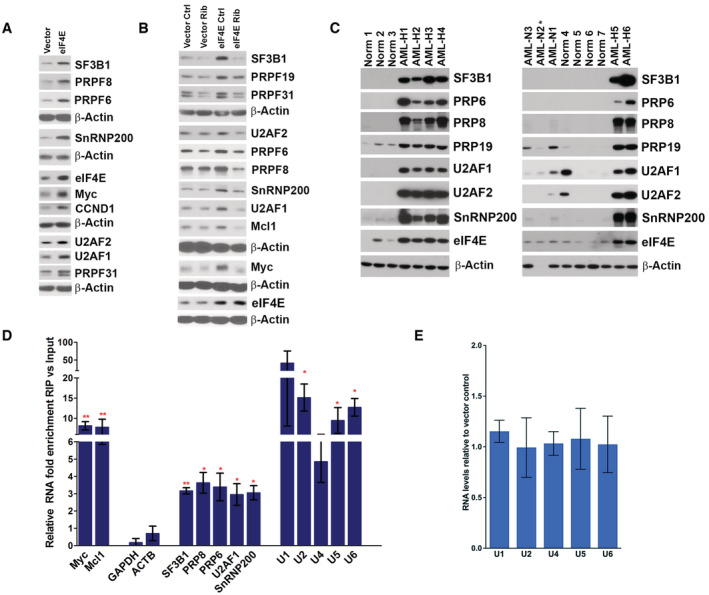
- WB analysis of splicing factors for Vector and eIF4E overexpressing NOMO‐1 cell lines. Myc and cyclin D1 (CCND1) served as positive controls and β‐actin as a loading control. Each β‐actin blot corresponds to the above western blots. Experiments were carried out at least three independent times, and one representative experiment is shown.
- Western blot analysis of untreated (Ctrl) and Ribavirin treated (Rib) Vector and eIF4E NOMO‐1 cell lines. Myc and Mcl1 served as positive controls and β‐actin as a loading control. Each β‐actin blot corresponds to the above WBs. Experiments were carried out at least three independent times, and one representative experiment is shown. Ribavirin dose was 10 μM, which is clinically achievable (Assouline et al, 2009, 2015).
- WB analysis of Splicing Factor levels in primary AML samples with high (AML‐H) or normal (AML‐N) eIF4E levels, as well as bone marrow mononuclear cells from healthy volunteers (Norm for normal). Numbers refer to different individuals. β‐actin was used as a loading control. (* = degraded sample).
- The enrichment of mRNAs and UsnRNAs in RIPs of endogenous eIF4E versus input RNAs from the nuclear fractions of NOMO‐1 cells monitored by RT–qPCR. Data were normalized to input samples and presented as a fold change. The mean, standard deviation, and P‐values (using Student t‐test) were derived from three independent experiments (each carried out in triplicate). MYC and MCL1 are known eIF4E nuclear targets and served as positive controls, while ACTB and GAPDH were used as negative controls. Student t‐test: *P < 0.05, **P < 0.01.
- Total levels of UsnRNA in NOMO‐1 Vector and eIF4E cells monitored by RT–qPCR. Data were normalized to Vector control and shown as a fold change. The mean and standard deviation, where no P‐values were P < 0.05 (using Student t‐test), were derived from three biological replicates each carried out in triplicate for all panels.
Next, we examined whether eIF4E status correlated with levels of SFs in primary AML specimens. We isolated de‐identified AML patient blasts using FACS (Assouline et al, 2009, 2015; Kraljacic et al, 2011; Zahreddine et al, 2017) and compared these with bone marrow mononuclear cells or CD34+ cells from healthy volunteers. Analysis of extracted proteins revealed that high‐eIF4E AML specimens had elevated levels of PRPF6, PRPF8, PRPF19, SF3B1, U2AF1, U2AF2, and SNRNP200 relative to healthy volunteers or normal‐eIF4E AMLs (Fig 3C). There was 1/7 healthy volunteer who had elevated U2AF1, for unknown reasons. Longer exposures of the western blots revealed the presence of these factors in the healthy volunteer specimens and normal‐eIF4E AML patient specimens. Indeed, this highlights the extreme elevation of these SFs in high‐eIF4E AML. Thus, the elevation of these SFs was not a general feature of AML patient specimens but correlated with eIF4E levels consistent with our studies in U2OS and NOMO‐1 cells. While eIF4E is correlated with elevated levels of these factors, it is likely that there are other mechanisms that can also drive the elevation of SFs, this would be an interesting avenue of future study.
eIF4E physically interacts with the splicing machinery
Our above studies demonstrate that eIF4E modulated SF production in several contexts and further this is driven mainly by the RNA export activity of eIF4E. Previous studies showed that eIF4E interacted with RNA processing machinery involved in capping, CPA, nuclear export, and translation (Kapp & Lorsch, 2004; Culjkovic et al, 2006; Topisirovic et al, 2009a; Volpon et al, 2017; Davis et al, 2019; Culjkovic‐Kraljacic et al, 2020a). Thus, we examined whether eIF4E similarly interacted with SFs; providing an additional means by which eIF4E could impact splicing. Nuclear lysates were crosslinked with formaldehyde prior to RIPs to prevent reassortment during processing. We observed that endogenous eIF4E immunoprecipitated with U1, U2, U4, U5, and U6 snRNAs relative to inputs with a ~3 to 10‐fold enrichment in nuclear lysates from U2OS cells (Fig 1F), as well as in NOMO‐1 cells (Fig 3D). Endogenous eIF4E did not associate with negative controls (e.g., GAPDH, ACTB, etc) indicating these RIPs were specific (Figs 1F and 3D). Additionally, levels of the UsnRNAs were not altered upon eIF4E overexpression in U2OS or NOMO‐1 cells suggesting that eIF4E did not lead to the generation of more spliceosomes (Figs 1H and 3E). Given UsnRNAs play both structural and catalytic roles in the spliceosome (Wahl et al, 2009), we investigated whether eIF4E is physically associated with protein components of the spliceosome. We found that endogenous eIF4E also immunoprecipitated with protein components of each of the major spliceosome complexes including PRPF6, PRPF8, PRPF19, PRPF31, SF3B1, U2AF1, U2AF2, and SNRNP200 in U2OS (Fig 4A) and NOMO‐1 cells (Fig 4B). Moreover, eIF4E did not IP with the negative control β‐actin. Notably, these proteins that physically interacted with eIF4E also had their production driven by eIF4E (Figs 1C and D, and 3A–C). Thus, as we observed for eIF4E‐dependent capping and CPA, eIF4E promoted the production of SFs presumably to increase its capacity to physically interact with their protein forms. In all, eIF4E binds to several SFs and UsnRNAs of the major spliceosome and elevated SF protein levels in both AML and U2OS cell contexts. Thus, eIF4E is positioned to modulate splicing in different cellular contexts, suggesting this could be a broadly applicable property of eIF4E.
Figure 4. eIF4E physically interacts with the splicing machinery in an RNA and cap‐dependent manner.
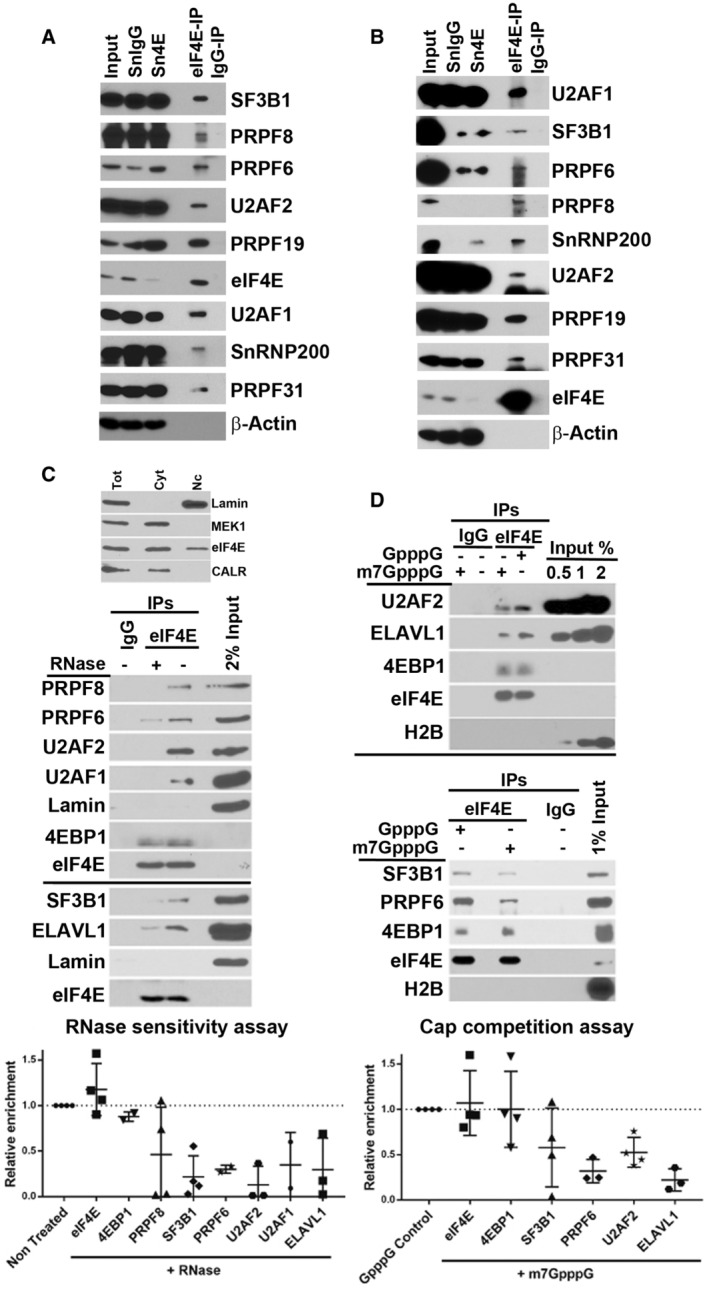
- Endogenous eIF4E co‐immunoprecipitated with SFs in the nuclear fractions of U2OS cells. Immunoprecipitations (IP) were carried out using U2OS nuclear lysates and anti‐eIF4E antibody (eIF4E‐IP) or appropriate IgG control (IgG‐IP), carried out at least three independent times, and one representative experiment is shown. IP samples along with Input (2%) and supernatants (Sn) after IPs were analyzed by WB using antibodies as indicated.
- eIF4E co‐immunoprecipitated with SFs in the nuclear fractions of NOMO‐1 cells. IPs were carried out using eIF4E overexpressing NOMO‐1 nuclear lysates and anti‐eIF4E antibody (eIF4E‐IP) or appropriate IgG control (IgG‐IP). IP samples were analyzed by WB as above and carried out in three biological replicates.
- eIF4E interaction with splicing factors depends on the presence of RNAs in U2OS cells. The eIF4E‐IP was divided into two and treated with buffer or RNase. After elution, 2% of the nuclear extract and the IPs were resolved on SDS–PAGE and analyzed by WB with indicated antibodies. Representative gels of three biological replicates are shown (middle panel) and intensities measured using FIJI are shown in scatterplot (bottom panel) generated in PRISM with means depicted as horizontal lines and standard deviation as vertical lines. Each point represents an independent experiment. Fraction purity was assessed with the indicated antibodies (C top panel).
- eIF4E interaction with SF is cap‐dependent. eIF4E‐IPs were competed with m7GpppG and the negative control GpppG treatment in U2OS cells. Representative gels of three biological replicates are shown (top and middle panel), and quantification as in panel (C) (bottom panel) is shown.
Data information: (C, D) The number of independent experiments for each target is shown, which varies from n = 3 to 4 replicates. Error bars represent standard deviations (vertical line).
The interaction of eIF4E with the splicing machinery is RNA and m7G cap‐sensitive
We sought to determine whether RNA played a role in the interactions between eIF4E and SFs. To this end, we carried out endogenous eIF4E RIPs followed by RNAse treatment. RNAse A and T1 were used in combination to efficiently degrade both duplex and loop structures, respectively (Edy et al, 1976). Immunoprecipitations were carried out as described above with the exception that due to the RNAse step, there was no formaldehyde crosslinking of nuclear lysates from U2OS cells; we note that even in the absence of crosslinking, the same interactions were observed in the untreated controls between SFs and eIF4E (Fig 4A vs. C). We observed that SF3B1, U2AF1, U2AF2, PRPF6, and PRPF8 were all present in untreated controls and that this interaction was reduced by ~5‐fold with RNAse treatment (Fig 4C). Similarly, a factor involved in specific splicing events, the AU‐rich RNA‐binding protein ELAVL1/HuR (Abdelmohsen & Gorospe, 2010) bound to eIF4E and was reduced by ~5‐fold by RNAse. Importantly, the capacity of eIF4E to immunoprecipitate with itself was unaffected by RNAse treatment. Moreover, the interaction with the eIF4E‐binding protein 1 (4EBP1) was not impacted by RNAse treatment consistent with the direct protein–protein interactions that characterizes the eIF4E‐4EBP1 interaction (Osborne & Borden, 2015). There were no SFs found in the IgG negative controls (Fig 4C). Fractionation controls indicated that Lamin as a nuclear marker was enriched in these nuclear lysates while MEK1 a cytoplasmic marker was absent (Fig 4C), and moreover, eIF4E did not immunoprecipitate with Lamin in either treated or untreated samples serving a negative control for the IP (Fig 4C). Thus, eIF4E‐SF interactions required intact RNAs.
Conscious of the fact that RNAse treatment targets both substrate m7G‐capped pre‐mRNAs and UsnRNAs, we examined the impact of treatment with an m7G cap analog, which would specifically disrupt m7G‐capped mRNA interactions and not UsnRNA‐mediated interactions. We conducted eIF4E RIPs as a function of m7GpppG or the negative control GpppG and monitored SF association by western blot (Fig 4D). As for the RNAse treatments, there was no formaldehyde crosslinking of nuclear lysates and eIF4E immunoprecipitated itself equally well in m7GpppG‐ or GpppG‐treated samples (Fig 4D). Further, the eIF4E‐4EBP1 interaction was not influenced by m7GpppG or GpppG as expected nor did eIF4E‐IP with the negative control H2B. We observed that the interactions between eIF4E and PRPF6, U2AF3, and SF3B1 were reduced ~2‐5‐fold by m7GpppG treatment relative to GpppG controls (Fig 4D). Moreover, HuR/ELAVL1‐eIF4E complexes were also depleted by ~5‐fold for m7GpppG relative to GpppG (Fig 4D). There were no SFs found in the IgG controls. Given that eIF4E was associated with the majority of RNAs via the m7G cap interaction, these studies suggested that eIF4E and SF interactions are mediated and/or stabilized by substrate m7G‐capped RNAs. These observations strongly suggest that eIF4E recruited selected m7G‐capped RNAs to the spliceosome and/or stabilized their interaction once there.
eIF4E overexpression alone is sufficient to alter splicing programs
Given these findings, we monitored the impact of eIF4E on splicing. RNAs from total cell lysates were isolated from the three 2FLAG‐eIF4E and Vector U2OS cell lines and subjected to RNA‐Sequencing. Differences in splicing profiles were quantified using replicate Multivariate Analysis of Transcript Splicing (rMATS) (Shen et al, 2012). rMATS calculates the “inclusion level differences” for splicing events such as exon skipping (ES), inclusion of mutually exclusive exons (MXE), intron retention (IR), and alternative splice‐site usage. For example, it calculates the extent an exon is included, i.e., not skipped, in Vector versus 2FLAG‐eIF4E cells. In this case, positive values indicate an exon is more included in Vector and negative values that this exon is more included in eIF4E‐overexpressing cells. Here, a value of +1 for SE events indicated that the relevant exon is 100% included in Vector cells and 0% in 2FLAG‐eIF4E cells. Many of the same events (129; Dataset EV1) were also observed using EBSeq, which maps exons, but in contrast to rMATS, EBSeq does not provide splice‐site event information. Thus, we focused on rMATS. Finally, as stated above, we note that eIF4E overexpression did not induce mutation in SF3B1, U2AF1, or SF3B1, as observed by our RNA‐Sequencing data (Data ref: Culjkovic‐Kraljacic et al, 2020b). We note that RNAs were ribo‐depleted to remove ribosomal RNA. Ribodepletion has the advantage over polyA selection that RNAs with short polyA tails or with no polyA tails can be captured in contrast to polyA selection strategies. Given eIF4E has impacts on the production of CPA factors (Davis et al, 2019) and impacts on APA for hundreds of transcripts (MG and KLBB, our unpublished observation), we deemed this a necessary consideration.
Using rMATS, we observed that ~890 splicing events for ~760 transcripts were altered (FDR‐adjusted P‐value < 0.15; inclusion differences of > 0.05 or < −0.05), or using more stringent cutoffs (P‐value < 0.1, inclusion level differences of > 0.1 or < −0.1), we observed 555 splicing events for 493 transcripts (Fig 5B, Dataset EV2). In the RNA‐Seq experiments, a total of 5,738 annotated transcripts were detected with > 10 TPM indicating that ~15% of annotated transcripts were differentially spliced in an eIF4E‐dependent manner. SE was the most frequent event (~55%) followed by MXE events (~30%) (Dataset EV2; Fig 5A). More rarely, Intron Retention (IR) and altered 3′/5′ splice‐site usage were impacted. Most transcripts affected were coding RNAs, but some noncoding RNAs were also impacted (Dataset EV2). Hierarchical clustering analyses based on the inclusion levels across replicates revealed that all types of splicing events (e.g., SE, MXE, IR) were segregated solely based on eIF4E levels (Fig 5B, Appendix Fig S2A). Positive and negative inclusion level differences of splice events were distributed roughly equally (45 vs. 50%, respectively) suggesting splicing reprogramming of selected RNAs. In all, these findings support a role for eIF4E as a mediator of alternative splicing (AS) for a subgroup of transcripts; importantly, our studies indicate that eIF4E did not elicit global changes to splicing when overexpressed in U2OS cell lines. Also, eIF4E overexpression did not lead to a global reprogramming of the transcriptome, with only 402 differentially expressed genes in Vector versus 2FLAG‐eIF4E U2OS cells (Culjkovic‐Kraljacic et al, 2020a; Data ref: Culjkovic‐Kraljacic et al, 2020b). Additionally, only ~2% of the differentially expressed transcripts (13 targets) undergo AS upon eIF4E overexpression, suggesting that eIF4E‐related AS is not generally correlated with RNA levels in these cells (Appendix Fig S2B, Dataset EV3).
Figure 5. eIF4E overexpression drives widescale reprogramming of splicing.
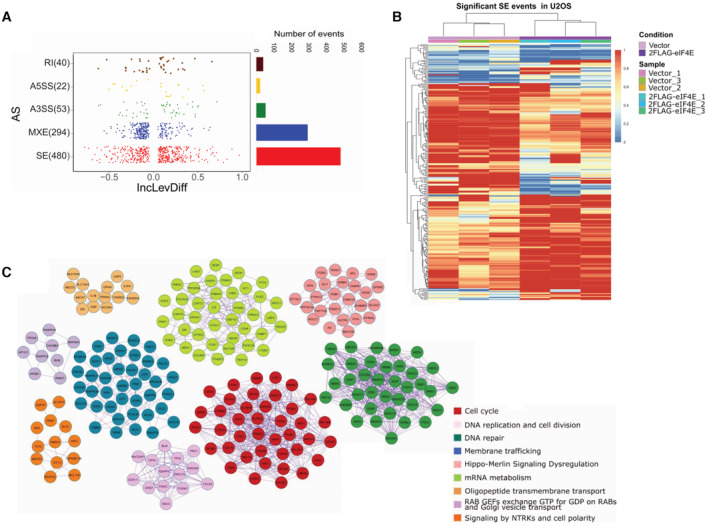
- Left. Dot plot showing the distribution of Inclusion Level Differences (IncLevDiff) values (IncLev (2FLAG‐eIF4E) – IncLev (Vector)) for each splicing category. Right. Histogram showing the number of events for each splicing category.
- Splicing heatmap showing the values of IncLev for all the Skipping exon (SE) events between Vector (n = 3, biological replicates) and 2FLAG‐eIF4E (n = 3, biological replicates) U2OS cells. Other events are shown in Appendix Fig S3.
- Protein interaction networks identified for alternatively spliced genes. Pathway and process enrichment analysis was applied for each MCODE component, and the best‐scoring term by P‐value is shown.
To understand the biological impact of eIF4E‐dependent alterations to splicing, we carried out pathway and process enrichment analyses. Enrichment of GO terms for all significant events indicated that eIF4E influenced pathways that could support its oncogenic phenotype (Dataset EV4). Top GO categories include cell cycle, membrane trafficking, DNA repair, and microtubule cytoskeletal organization. Protein–protein interaction enrichment analyses are consistent with the GO terms with the top hits being cell cycle, RNA metabolism, and membrane trafficking (Fig 5C, Dataset EV5). The enrichment of RNA metabolism is particularly interesting and suggestive of a positive feedback loop resulting from eIF4E dysregulation and is consistent with its known multiple functions.
eIF4E levels are correlated with differential splicing programs in AML
We next examined the relevance of eIF4E‐dependent splicing in AML (Mehterov et al, 2021; Rivera et al, 2021) and because targeting eIF4E with ribavirin in AML provided clinical benefits including remissions in a subset of AML patients in early stage clinical trials (Assouline et al, 2009, 2015). As a first step, we analyzed RNA‐Seq data from 437 de‐identified AML patient specimens and 17 specimens of normal CD34+ cells derived from human cord blood (www.leucegene.ca) (Fig 6A–C). Dividing the entire Leucegene cohort into two groups (above and below median RNA expression of eIF4E), we observed a substantial and significant reduction in survival for the high‐eIF4E group (Appendix Fig S3A). For further analysis, we pre‐screened specimens by inspection of the RNA‐Seq data to ensure they did not harbor the major mutations reported in AML, i.e., those in U2AF1, SRSF2, or SF3B1. We used the normal CD34+ cells to benchmark eIF4E levels to categorize AML specimens into high‐ or normal‐eIF4E groups (Fig 6A and B). For rMATS analysis, we selected 10 AML specimens with the highest eIF4E levels and 11 with normal‐eIF4E levels, i.e., overlapping with or below levels in normal CD34+ cells (Fig 6A–C). Corresponding clinical data revealed that the 10 high‐eIF4E AML patients had drastically reduced overall survival (median survival 136 days) when compared to the 11 patients with lowest eIF4E levels in AML (1,396 days, Fig 6C).
Figure 6. eIF4E‐dependent alternative splicing in AML.
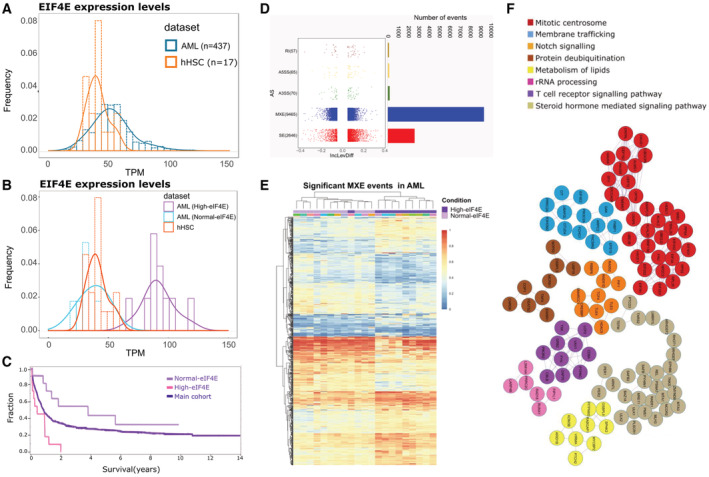
- Graph showing the distribution of eIF4E‐RNA levels in AML patients' specimens (blue, AML) in comparison with normal human CD34+ cells derived from cord blood (orange, hHSC). Data are derived from the Leucegene cohort (437 patients).
- High‐eIF4E (purple) and normal‐eIF4E (blue) classification of AML patients' specimens used for our analysis. eIF4E levels in healthy volunteer CD34+ cells are shown for comparison. These specimens were selected based on both their eIF4E levels and the absence of common SF mutations as observed from the Leucegene RNA‐Seq dataset (Dataset EV21).
- Survival analyses for High‐eIF4E patients and Normal‐eIF4E patients for which specimens were studied (Dataset EV21). The rest of the cohort is shown for comparison. The log‐rank test was performed and shows a significant difference in survival between the High‐eIF4E group (10 specimens) and the rest of the cohort (432 specimens), as well as between High‐eIF4E and Normal‐eIF4E groups (11 specimens) (P‐value < 0.05).
- Left. Dot plot showing the distribution of IncLevDiff values (IncLev (High‐eIF4E) – IncLev (Normal‐eIF4E)) for each splicing category. Right. Histogram showing the number of events for each splicing category.
- Splicing heatmap showing the values of IncLev for all the splicing events between Normal‐eIF4E and High‐eIF4E AML samples.
- MCODE networks identified for MXE targets between Normal‐eIF4E and High‐eIF4E AML patients' samples. Pathway and process enrichment analysis was applied for each MCODE component and the best‐scoring term by P‐value.
We compared the splicing profiles of these high‐eIF4E and normal‐eIF4E specimens using rMATS. We observed differences in ~1,600 splicing events impacting ~1,500 RNAs (FDR‐adjusted P‐value < 0.1, absolute value of inclusion level differences of > 0.1) and if the threshold is lowered, ~12,000 events for ~4,600 transcripts (absolute inclusion value difference of > 0.05 and FDR‐adjusted P‐value < 0.15; Fig 6D, Dataset EV6). In terms of transcript levels, ~14,000 genes had an average TPM value > 10. Thus alternative splicing of ~15–30% of detected transcripts corresponds with eIF4E levels, depending on the cutoffs used. Of these events, ~75% arose due to MXE, ~20% SE, and the remainder was attributable to retained introns or altered 3′/5′ splice‐site usage (Fig 6D). As for U2OS, high‐eIF4E levels correlated with a roughly equal number of increased and decreased inclusion levels across AS splicing events. The maximum magnitude of the average inclusion level differences across patients was ~0.4 (corresponding to ~40% change in population) versus ~0.8 (~80%) for U2OS cells (Figs 5A vs. 6D). The reduction in the magnitude of these effects in primary specimens likely arises due to heterogeneity among patients' transcriptomes (Hershberger et al, 2021; Rivera et al, 2021). As in U2OS cells, we did not generally observe any correlation between AS reprogramming and changes in RNA levels, since only ~7% of the alternatively spliced targets were differentially expressed (Appendix Fig S3C, Datasets EV7 and EV8).
To establish whether eIF4E levels were the factor most significantly influencing splicing, we conducted an unsupervised hierarchical clustering analysis. There was a very tight correlation between eIF4E status in AML patients and MXE events. Specifically, 20/21 AML specimens clustered based on eIF4E levels when monitoring MXE events (Fig 6E). Similar analysis of RI events revealed groups clustered on eIF4E levels with the exception of two high‐eIF4E patients that clustered with normal‐eIF4E patients (Appendix Fig S3B). Finally, analyses of SE events indicated two distinct clusters of high‐eIF4E AML patients, one cluster was more similar to normal‐eIF4E patients (Appendix Fig S3B). Interestingly, we note that unlike the overexpression of eIF4E in U2OS cells where only five SFs were differentially spliced (Dataset EV2), eIF4E elevation in AML patients correlates with splicing reprogramming of a wide array of spliceosome components (~20 targets), including PRPF3, PRPF8, PRPF4B, PRPF39, PRPF31, PRPF6, PRPF40A, SF1, SRSF10, SF3B3, SF3B2, SRSF5, and U2AF1 (Dataset EV6). Thus, eIF4E appears to have a broader impact on the splicing of SFs in the context of AML. Similar to U2OS cells, we observed that the top GO enrichment terms for eIF4E‐dependent MXE targets were highly similar to those in the U2OS datasets: cell cycle, DNA repair, membrane trafficking, and RNA metabolism (Dataset EV9). Protein interaction networks were enriched in mitotic centrosomes, membrane trafficking, and notch signaling (Fig 6F, Dataset EV10).
We reasoned that the identification of splicing events conserved across disparate cell types would reveal pan‐cancer core networks that underpin, at least in part, eIF4E's oncogenic effects (Figs 5 vs. 6). A comparison of all eIF4E‐dependent splicing targets in U2OS and AML datasets evinces a core set of ~450 common RNA targets (Fig 7A, Dataset EV11; the absolute value of inclusion level difference > 0.05; FDR‐adjusted P‐value < 0.15). Indeed, 60% of targets identified in the U2OS cells were in common with the AML targets. In terms of transcript expression, AML specimens and U2OS cells had 4,705 transcripts in common (> 10 TPM). About 10% of these transcripts were also eIF4E‐dependent splicing targets. Note that core targets were not necessarily characterized by the same splice event in each cell type. GO term analysis of RNA targets revealed that the RNAs enriched between U2OS and AML were a general reflection of those identified in the analysis of either cell type alone. For example, analyses identified cell cycle, DNA repair, membrane trafficking, chromatin modification, and RNA metabolism for GO terms (Dataset EV12) and DNA repair, adaptive immune system, MYC activation, and others for protein–protein interaction networks (Fig 7B, Dataset EV13).
Figure 7. Identification and validation of core splicing networks and physical interactions with selected pre‐mRNAs AS targets.
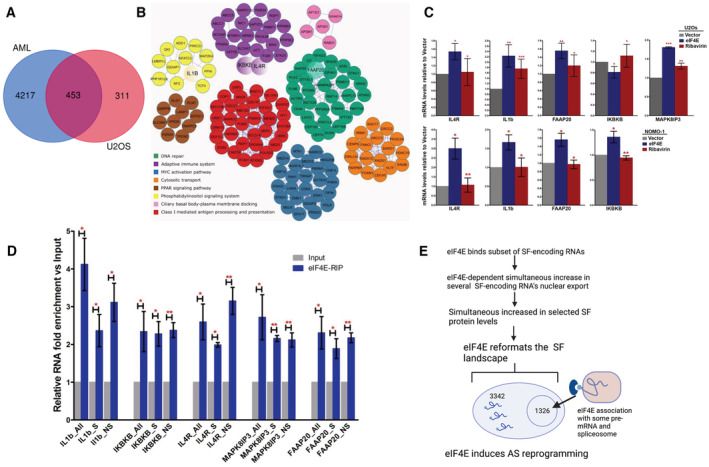
- Venn diagram showing the common targets affected by eIF4E‐dependent alternative splicing in high‐eIF4E AML patients' cells and 2FLAG‐eIF4E U2OS cells highlighting a core network.
- Protein interaction networks for alternative splicing targets common between high‐eIF4E AML patients' samples and 2FLAG‐eIF4E U2OS cells. Pathway and process enrichment analysis has been applied for each MCODE component and the best‐scoring term by P‐value. Common targets selected for subsequent validation by RT–qPCR are highlighted as larger circles (IkBkB, IL4R, FAAP20, IL1B).
- Validation of splicing targets identified by rMATs in U2OS (upper panel) and NOMO‐1 (lower panel) cell lines. RT–qPCR analysis using specific primers for each splicing event normalized to the corresponding total levels of that given transcript (using primers specific to common regions, see Dataset EV22). Data were normalized to Vector control to calculate fold change. The mean and standard deviation, as well as P‐values, were derived from three biological replicates. P‐values were calculated between eIF4E overexpressing and Vector cells (when the asterisk is over eIF4E), and eIF4E cells treated with Ribavirin versus eIF4E overexpressing untreated cells (for the asterisk over Ribavirin).
- eIF4E associates with both pre‐mRNA splicing substrates and their spliced products. The enrichment of mRNAs for splicing targets in eIF4E‐RIPs versus input mRNAs from the nuclear fractions of 2FLAG‐eIF4E U2OS cells monitored by RT–qPCR using primers for the specific splice event (S for “spliced”), pre‐mRNA specific primers to the relevant intron or intron‐exon boundary (NS for “unspliced”), and primers specific to common regions present in all spliced and not‐spliced mRNAs (All). Data were normalized to input samples and presented as a fold change. The mean, standard deviation, and P‐values (Student t‐test) were derived from three independent experiments (each carried out in triplicate).
- Model summarizing the mechanisms by which eIF4E can modulate alternative splicing. Blue semicircle represents eIF4E, the light blue circle the cap, and RNA as a line with USER codes as loops. Figure produced in Biorender.
Data information: Student t‐test, *P < 0.05, **P < 0.01, ***P < 0.001.
Validation of eIF4E‐induced alternative splicing events derived from rMATS analysis using genetic and pharmacological means
Next, we validated several of these core splicing targets (Fig 7C). We selected factors of biological interest to eIF4E's oncogenic activities. Using RT–qPCR, we validated splicing events identified from the rMATS analysis for the following targets as a function of eIF4E overexpression: IL1B, IL4R, IkBkB, MAPK8IP3, and FAAP20 in U2OS cells and IL1B, IL4R, IkBkB, and FAAP20 in NOMO‐1 cells. As expected, we validated the splicing as found in rMATS but with one exception. Interestingly, the event that we validated in U2OS cells for IkBkB was identified in the AML patients (skipping exon 2) rather than the one predicted by rMATS in the U2OS cells (skipping exon 21) confirming the known limitations of rMATS despite its high efficacy and reproducibility when compared to other event‐based AS analysis programs (Mehmood et al, 2020). Next, we examined the impact of eIF4E inhibition with clinically relevant concentrations of ribavirin (Assouline et al, 2009, 2015). We observed in both cell lines that ribavirin treatment reverted the splicing events in eIF4E overexpressing cells, i.e., eIF4E‐dependent splicing events were reversed back to those observed in Vector cells after treatment (Fig 7C). To establish the relevance of SFs regulated by eIF4E to these splice events, we employed RNAi methods to knockdown PRPF8, U2AF2, and SF3B1 in eIF4E‐FLAG U2OS cells and then compared these with Vector controls. Unfortunately, the single knockdown of any one of these factors led to the elevation of the other two factors suggestive of cellular compensation. Further, their reduction was associated with massive cell death even under conditions of partial knockdown. Intriguingly, reduction in PRPF8 also decreased eIF4E protein levels. Due to these issues, we were unable to employ this strategy to ascertain which SF components were the most relevant to eIF4E‐dependent AS. In all, we validated rMATS‐predicted eIF4E‐dependent AS targets and showed that these splicing effects were reversed by the addition of eIF4E inhibitor ribavirin.
eIF4E binds intron‐containing RNAs
Given eIF4E modulated splicing of specific RNAs and physically interacted with spliceosome components in a cap‐sensitive manner, we examined whether eIF4E was associated with both pre‐mRNA splicing substrates and their spliced products. In this way, we investigated whether eIF4E is recruited to the pre‐mRNA prior to (or along with the spliceosome) or if these eIF4E‐transcript associations were specific to the spliced products. To address this, we carried out nuclear eIF4E RIPs using formaldehyde crosslinking and analyzed RNA content using primers specific to the specific splice event with product‐specific primers spanning exon‐exon junctions or substrate‐specific primers to the relevant intron (or intron‐exon boundary) using RT–qPCR. Results were compared with levels of RNAs obtained with primers to both substrate and product RNAs, which act as normalization controls. We observe that eIF4E RIPs with transcripts that have not yet undergone the targeted splice event (“unspliced”), as well as those that have undergone that splice event (“spliced”) for IL1B, IL4R, IKBKB, FAAP20, and MAPK8IP3 (Fig 7D). For example, IL1B showed more retained intron in Vector cells, and its splicing is enhanced by eIF4E elevation (Fig 7C, Dataset EV2). In this case, eIF4E RIPs with both intron‐containing IL1B and spliced IL1B as observed using exon‐intron and exon‐exon primers, respectively (Fig 7D). Given eIF4E is physically associated with the spliceosome (Fig 4) and pre‐mRNA (Fig 7D), eIF4E could play a role in the recruitment of some substrate RNAs. These findings implicate eIF4E in a direct role in splicing for at least some splicing targets. Moreover, these were the first studies to show that eIF4E binds to any pre‐mRNAs, which has important implications for eIF4E function.
A subset of eIF4E AS targets physically interact with nuclear eIF4E
To globally estimate the extent to which eIF4E physical association with AS splice targets, we compared the RNAs identified as physically associated with nuclear eIF4E in the RIP‐Seq dataset to the RNAs identified as eIF4E‐dependent AS targets across AML patient data from rMATs. LY1 and AML share ~9,000 transcripts (with > 10 TPM) as observed from the RNA‐Seq data and are both derived from hematological malignancy serving as a rationale for this comparison. Interestingly, 1,326/~2,800 transcripts in the eIF4E RIP were also identified in the ~4,600 eIF4E‐dependent splicing targets in AML. This constitutes ~50% of the RNAs in the nuclear eIF4E RIP and 1,326/4,600, ~30% of AS targets. These include RNAs for which we validated for both eIF4E interaction and eIF4E‐dependent splicing, e.g., IkBkB and MAPK8IP3 (Fig 7C and D). These data indicate that there are two classes of AS targets, those that physically interact with nuclear eIF4E and those that do not. Those RNAs that bind to eIF4E in nuclear RIPs but are not eIF4E‐dependent AS targets are presumably targets of other eIF4E nuclear functions, e.g., nuclear export, capping, or polyadenylation (Volpon et al, 2017; Davis et al, 2019; Culjkovic‐Kraljacic et al, 2020a and references therein).
Characterization of eIF4E‐dependent AS targets
A key aspect to our understanding of eIF4E‐dependent AS is to elucidate the principles of target RNA selection. As a first step, we examined features related to the basic structure of target RNAs with regard to the splice events identified in the rMATS analysis. We monitored exon and intron length, GC content of introns, exon position within the RNA, number of exons within the transcript as a measure of transcript complexity and splice‐site usage, each as a function of the splicing event, as well as of positive or negative inclusion level differences (Appendix Figs S4 and S5). We selected the most significant splice targets to increase the likelihood of identifying commonalities and thus interrogated both AML and U2OS datasets using the threshold of > 0.1 for the absolute value of inclusion level differences and FDR‐adjusted P‐value < 0.1. We observed that longer introns were associated with eIF4E‐dependent MXE and SE splicing events. Introns were up to ~9,000–15,000 bp in average length for eIF4E‐dependent events in U2OS cells and ~6,000–9,500 bp in AML specimens relative to average intron length for all introns, which was ~4,000–5,000 bp (Dataset EV14). We noted that AS targets of eIF4E contained overall more exons than nontarget transcripts (P < 0.001) for all categories of AS in both U2OS and AML, which suggested that more complex transcripts and eIF4E‐sensitive (Appendix Figs S4F and S5F). We observed a trend toward higher GC content for high‐eIF4E AML specimens for A3′SS, A5′SS, and SE events (Appendix Fig S5E) and for A5′SS and MXE events in 2FLAG‐eIF4E relative to Vector controls (Appendix Fig S4E). We did not observe significant alterations in splice‐site usage in either cell context (Appendix Figs S4D and S5D); consistent with the low number of these events identified by rMATS (Datasets EV2 and EV6). We noted no significant eIF4E‐dependent differences for exon length or position (Appendix Figs S4A and B, and S5A and B). In all, exons involved in eIF4E‐dependent splicing events tend to be bracketed by longer than average introns, which likely contain relevant sequence features or secondary structure elements that act as USER codes for eIF4E‐dependent splicing.
PRP8, U2AF2, and ARE‐binding proteins are identified potential regulators of eIF4E‐dependent AS target RNAs
To further dissect sequence elements that impact RNA sensitivity and to determine whether these could be contained within the large introns we identified above, we employed the Atlas of UTR Regulatory Activity (AURA2) database regulatory enrichment tool to identify RBP‐binding sites within eIF4E‐AS targets focusing on the core transcripts shared between U2OS and AML groups. We searched for known motifs of RNA‐binding proteins and other regulatory cis‐elements within 3′ or 5′ UTR regions in the eIF4E‐AS core target RNAs. Strikingly, about 60% of the core AS target RNAs contained binding sites for ELAVL1, AGO, eIF4A3, TIA, and IGF2BP, which play roles in alternative splicing (Dataset EV15). Also, binding sites for PRPF8 and U2AF2 factors, identified here as both bound to eIF4E and regulated by eIF4E, were found in > 20% of core eIF4E‐dependent AS target transcripts. Indeed, 60 RNA‐binding proteins (RBPs) were predicted to co‐regulate > 20% of core eIF4E‐AS target RNAs and the top‐scoring terms associated with these RBPs involved in RNA splicing and processing of capped, intron‐containing pre‐mRNAs (Appendix Fig S6). RNAs contained interaction motifs for several factors indicating that transcripts could be regulated by multiple factors simultaneously. We note that this analysis did not change if RNAs were grouped into those AS targets that physically interacted with eIF4E and those that did not. Taken together, this suggests that a plethora of co‐factors exists to act in eIF4E‐AS and that these could be regulated combinatorically as anticipated from the RNA regulon model.
Remarkably, many of the RBPs that target > 60% of AS‐eIF4E target transcripts recognize AU‐ or U‐rich elements including ELAVL1 (also known as HuR), TIA1, TIAL1, AGO (Dataset EV15). These factors were also implicated in alternative splicing and play oncogenic roles consistent with an interplay with eIF4E (Izquierdo et al, 2005; Zhu et al, 2006, 2008; Izquierdo, 2008, 2010; Batsche & Ameyar‐Zazoua, 2015). Of these, ELAVL1 is the best‐characterized ARE‐binding factor (Mukherjee et al, 2011; Simone & Keene, 2013; Bakheet et al, 2018), and furthermore, ELAVL1 regulates the RNA stability of eIF4E (Topisirovic et al, 2009b) suggesting a feedback pathway. Above, we demonstrated above that endogenous ELAVL1 physically interacted with endogenous eIF4E in the nucleus in an RNAse and cap‐sensitive manner (Fig 4). This provides a proof‐of‐principle for mRNA‐dependent interactions of eIF4E with ARE‐binding proteins.
To ascertain whether these ARE regions were found within the introns, we examined the enrichment of these sequences within eIF4E‐dependent AS transcripts and determined the location of these elements within the transcripts using the ARE‐containing mRNA database (ARED, Dataset EV16). ARED analysis demonstrated that ~80% of the core AS‐eIF4E targets RNAs contain AU‐rich elements (Dataset EV16). Cluster 1‐ and 2‐type AREs constitute ~60% of elements in eIF4E‐AS targets. About 94% of identified AREs were found in introns (Bakheet et al, 2018). Inspection of targeted introns of our validated targets, IL4R, MAPK8IP3, FAAP20, and IKBKB revealed these contained AREs, which is consistent with interactions between eIF4E and ELAVL1 (Fig 4, Dataset EV16). In all, our studies suggest that the presence of AU‐rich elements within introns contributes to the selectivity of eIF4E‐dependent AS target RNAs. This will be further dissected in future.
Discussion
In this study, we report a novel paradigm for reprogramming splicing with far‐reaching impacts, e.g., ~15% of transcripts in U2OS cells and ~15–30% of transcripts in AML patient specimens. Here, eIF4E simultaneously elevates the production of multiple SFs thereby generating an altered splicing environment to support its widescale effects on splice‐site selection as manifested by altered SE and MXE profiles. In more limited cases, eIF4E is also positioned to modify splicing through physical association with selected pre‐mRNAs and the splicing machinery. Modulation of eIF4E did not induce well‐known mutations in SFs, nor did it alter SF‐encoding transcript levels. Thus, classical genomic and transcriptomic approaches would be blind to eIF4E‐dependent splicing and likely explain why this function has, until now, gone unobserved. Moreover, the dysregulation of eIF4E‐dependent splicing in primary high‐eIF4E AML patient specimens highlights the biomedical relevance of this new pathway.
Our mechanistic dissection revealed the biochemical basis for the capacity of eIF4E to elicit widescale reprogramming of SF protein production (Fig 7E). These studies revealed that nuclear eIF4E was physically associated with SF‐encoding RNAs and drove their nuclear export. Enhanced nuclear export of these transcripts in turn increased their availability to the translation machinery and elevated SF protein levels. Consistently, our separation‐of‐function S53A mutant demonstrated that loss of nuclear export activity severely impaired eIF4E's capacity to promote SF protein production (Fig 2). Direct measurement of translation efficiency showed that eIF4E had a two‐tier impact on a subset of SF‐encoding RNAs whereby it promoted not only their nuclear export but also enhanced translation efficiency making these exquisitely sensitive to eIF4E. We also note that eIF4E modulated the splicing of 5 SFs in U2OS (Dataset EV2) and ~20 SFs in AML (Dataset EV6) suggesting that splicing reprogramming also contributes to the modified SF landscape, which can be investigated in future. Importantly, eIF4E did not alter SF‐encoding RNA levels and thus did not influence the steady‐state transcription or stability of these transcripts. In all, eIF4E reformats the spliceosome landscape in multiple cellular contexts underpinning its ability to reprogram splicing.
The impact of eIF4E on splicing was selective rather than global. In this way, the effects of eIF4E on splicing were not a simple product of elevated SF levels increasing the capacity to produce the same splicing outcomes. Indeed, eIF4E did not alter levels of UsnRNAs themselves, suggesting that eIF4E did not alter the number of spliceosomes. Rather, the ability of eIF4E to modulate selected SF levels suggests that it modifies the composition and subsequent activity of the splicing machinery to favor certain splicing outcomes. Interestingly, eIF4E‐sensitive SFs include U2 snRNP components SF3B1 and U2AF1, which function in splice‐site selection. Indeed, these SFs can be present in sub‐stoichiometric quantities relative to the enzymatic machinery of the spliceosome (Wahl et al, 2009), and thus, their modulation by eIF4E likely provides a means to shift splice‐site preferences.
While all splice events modulated by eIF4E occur in the context of reprogrammed SF protein production and thus are influenced by this new splicing environment; eIF4E is additionally positioned to influence splicing through physical interactions with some pre‐mRNAs and the spliceosome (Fig 7E). Indeed, our analysis suggested that up to 30% of eIF4E‐dependent AS targets were physically associated with nuclear eIF4E, and we experimentally determined that eIF4E binds both unspliced and spliced forms of selected AS target RNAs. Moreover, we found that eIF4E bound U1, U2, U4, U5, and U6 snRNPs and related SFs (Figs 1 and 3). These interactions were sensitive to RNAse and m7G cap analog addition suggesting these were stabilized by capped mRNAs (Fig 4). Consistently, previous work in Drosophila found RNA‐dependent physical interactions between eIF4E and components of the U2 snRNP, and that eIF4E modulated splicing of the SXL transcript supporting our findings here (Graham et al, 2011). It is important to note that the spliceosome does not exist as a preformed complex, but rather, UsnRNPs must assemble on each splice site and undergo substantial remodeling during splicing (Wahl et al, 2009). Yet, we observed that eIF4E binds to representative factors of all 5 major UsnRNPs (Figs 1E and 4). These interactions could arise due to nonmutually exclusive reasons: (i) eIF4E‐m7G‐RNA complexes represent a snapshot of a population of the same species of transcript at different stages of splicing, (ii) eIF4E interacts with the precatalytic spliceosome (complex B), the only complex to contain all five UsnRNPs, and/or (iii) eIF4E‐RNA complexes are occupied by snRNPs at the eIF4E‐dependent targeted AS splice site, but U1 and U2 snRNPs subsequently mark splice sites on unrelated intron‐exon boundaries for other splicing events which other groups suggest can enhance splicing assembly for upcoming splice events (Braun et al, 2018). Further studies will be required to dissect these possibilities. Finally, in terms of AS RNA target‐eIF4E associations, many eIF4E‐dependent targeted splicing events were characterized by long introns. Interestingly, splicing of long introns is often considered to be slow and to occur post‐transcriptionally at splicing speckles (Boutz et al, 2015). Consistent with this hypothesis, older studies suggested that eIF4E may co‐localize with Sc35, a marker of these speckles (Dostie et al, 2000), and in this way, eIF4E could play a role in splicing at speckles. This is an interesting future direction of study.
The above studies position eIF4E as the second cap‐binding protein to be involved in splicing acting in much the same manner as the cap‐binding complex CBC does for bulk pre‐mRNA during splicing (Izaurralde et al, 1994; Lewis et al, 1996; Gornemann et al, 2005; Rambout & Maquat, 2020). Interestingly, CBC is linked to splicing of the first intron of target transcripts (Lewis et al, 1996), a preference we did not observe for eIF4E‐sensitive transcripts (Appendix Figs S4 and S5). We note that despite many years of study the exact biochemical role that CBC plays in splicing is still not fully understood, even though its requirement for many splicing events is clear (Rambout & Maquat, 2020). Given eIF4E acts as a cap chaperone (Borden, 2016; Mars et al, 2021), it could escort newly spliced capped RNAs to other processing steps, to the export machinery, and/or possibly is exchanged for the CBC. eIF4E‐CBC cap‐chaperone switching was observed in studies of eIF4E‐dependent nuclear RNA export (Topisirovic et al, 2009a). Here, cellular RNAs containing specific USER codes, e.g., 50‐nucleotide 4ESE element, are enriched in nuclear eIF4E RIPs and favor the eIF4E‐dependent export pathway in high‐eIF4E conditions; while in normal‐eIF4E conditions, these transcripts are more equally distributed between eIF4E and CBC and the resultant export pathways used (Culjkovic et al, 2005; Topisirovic et al, 2009a). USER codes may play a role in the selection of CBC or eIF4E but have not yet been identified but likely lie within the long introns and A‐/U‐rich regions that characterize eIF4E‐AS targets. For the ~70% of AS‐target RNAs that did not physically associate with nuclear eIF4E but are eIF4E‐dependent splice targets, these are presumably chaperoned by the CBC.
Analysis of AS targets indicates that eIF4E sensitivity is not based a priori on sensitivity to eIF4E regulation at other levels of RNA processing. For instance, CCND1, MCL1, and MYC are found in eIF4E nuclear RIPs and are targets of eIF4E‐dependent capping and nuclear export but were not observed as eIF4E‐dependent splicing targets according to the rMATS analysis (Datasets EV2 and EV6) (Culjkovic et al, 2005, 2006; Culjkovic‐Kraljacic et al, 2016, 2020a). A comparison of datasets reveals that six targets of eIF4E‐dependent capping (Culjkovic‐Kraljacic et al, 2020a) are also AS targets (MDM2, AGRN, TUBB6, CATSPER1, CTNNB1, and ERCC2; Appendix Fig S7B, Dataset EV19) indicating that co‐regulation of capping and splicing can occur. Thus, we posit that there are USER codes within the RNA targets that sensitize them to eIF4E‐dependent splicing and not universally to all eIF4E‐related processes.
eIF4E‐sensitive AS targets were characterized by a greater number of exons and involved longer introns than nontarget transcripts. They were also enriched in binding sites for AU‐rich binding proteins including ELAVL1 and for SFs including those elevated by eIF4E, e.g., PRPF8. In this way, eIF4E could be involved in a positive feedback loop regarding its AS activity. Moreover, we found that eIF4E is physically associated with both ELAVL1 and PRPF8 proteins in an RNAse manner. This would support a model where eIF4E and ELAVL1 are recruited to A/U‐rich elements in AS targets to facilitate splicing, this will be examined in future. Importantly, these sequence features were not altered whether we included all eIF4E‐AS target RNAs in the analysis or separated transcripts based on whether they had a physical interaction with eIF4E. Thus, further studies are required to characterize splicing USER codes that recruit or exclude appropriate factors to favor eIF4E‐dependent AS and to predict the identity of the subset of AS target RNAs that are physically escorted through splicing via eIF4E.
Many of the SFs elevated by eIF4E were shown in other studies to be mutated in AML, e.g., SF3B1, U2AF1, and SRSF2. Interestingly, these mutations do not occur simultaneously in the same AML cell likely because due to synthetic lethality (Visconte et al, 2019). However, we observed that eIF4E coopts these factors by simultaneously driving their production and in some cases through their physical interactions with eIF4E. Indeed, SF elevation and mutation are likely to lead to different phenotypes. Given the pervasive effects of eIF4E, it is extremely unlikely that its splicing signature would be attributable to the elevation of and/or interaction with any single SF. Consistent with this notion, in secondary AML (sAML) specimens without SF mutations, PRPF6 and SF3B1 protein levels were elevated relative to healthy volunteer specimens (Crews et al, 2016). Only 363/4,670 of eIF4E‐AS targets (363/963 for secondary AML targets) were in common between high‐eIF4E AML and sAML specimens (Appendix Fig S7C, Dataset EV17). Thus, the elevation of PRPF6 and SF3B1 alone was not sufficient to recapitulate the much broader eIF4E‐dependent splicing profile. Moreover, splicing patterns arising from eIF4E overexpression were not simply a recapitulation of those observed in AML cells with SF mutations. For instance, a comparison of splicing events arising in high‐eIF4E AML cells relative to AML cells with SF3B1 mutation (SF3B1K700 and other SF3B1 mutations were also included; Hershberger et al, 2021) showed only a modest overlap (47/4670) of eIF4E targets but included a substantial fraction of SF3B1 targets (47/83 of SF3B1) (Appendix Fig S7C, Dataset EV20). Recent findings indicate that Myc hyperactivation stimulates splicing factor SF3A3 translation through eIF machinery, leading to a metabolic reprogramming that amplifies the oncogenic potential of Myc (Ciesla et al, 2021). A comparison of splicing targets arising due to Myc dysregulation with eIF4E targets reveals a common 146/764 of targets for eIF4E and 146/1,442 of Myc targets (Appendix Fig S7D, Dataset EV18; Phillips et al, 2020). In all, these analyses suggest that eIF4E‐dependent reprogramming of splicing is not readily recapitulated by any single downstream target of eIF4E highlighting the extensive reprogramming. These studies with eIF4E provide a possible mechanism for the simultaneous dysregulation of multiple splicing factors, which has been reported in some solid tumors. For example, PRPF8, SRSF1, SRSF2, and U2AF2 protein levels are upregulated, rather than mutated, in some solid tumors (Urbanski et al, 2018), and thus, eIF4E could provide a mechanism for their elevation, a hypothesis that could be tested in future. Consistent with the findings presented here, others showed that modulation of the components of the splicing machinery such as SF3B1 does not elicit global effects on splicing but rather targets specific pre‐mRNAs (Papasaikas et al, 2015).
In all, the collective alterations to the SF content in combination in some cases with eIF4E's interaction with substrate RNAs and splicing machinery likely combinatorially drive the broad‐ranging altered splicing patterns we observed in U2OS and AML cells. eIF4E‐dependent AS targets act in processes that could underpin, at least in part, the oncogenic activities of eIF4E, and likely represent an eIF4E‐dependent splicing RNA regulon. Indeed, the eIF4E S53A mutant has lost both its oncogenic capacity (Lazaris‐Karatzas et al, 1990; Culjkovic‐Kraljacic et al, 2012), and its ability to reprogram SF protein production (Fig 2) strongly supporting a link between splicing and oncogenic capacity. Moreover, we show for the first time that eIF4E binds to pre‐mRNA and mature transcripts (Fig 7D) substantially broadening its potential cap‐chaperone roles in the cell. This study reveals a novel mechanism for dysregulated splicing and showcases that eIF4E can both amplify and rewrite the mRNA message.
Materials and Methods
Plasmids, antibodies, and reagents
pcDNA‐2Flag‐eIF4E wild‐type, S53A mutant and vector, as well as MSCV‐pgk‐GFP‐eIF4E and control constructs were previously described (Topisirovic et al, 2003; Culjkovic et al, 2006; Culjkovic‐Kraljacic et al, 2012, 2016). Antibodies for immunoblotting: Mouse monoclonal anti‐eIF4E (BD Biosciences), mouse monoclonal anti‐β‐actin (Sigma‐Aldrich), rabbit polyclonal anti‐Mcl‐1 (S‐19) (Santa Cruz), mouse monoclonal anti‐HSP90α/β (F‐8) (Santa Cruz), rabbit polyclonal anti‐Myc (ab32072 Abcam), rabbit polyclonal antiCyclinD1 (ab134175 Abcam), rabbit polyclonal SF3B1 (Cell Signaling), rabbit polyclonal anti‐PRP8 (Bethyl), rabbit polyclonal anti‐PRP6 (Bethyl), rabbit polyclonal anti‐SnRNP200 (Bethyl), rabbit polyclonal anti‐U2AF1 (Bethyl), rabbit polyclonal anti‐PRP31 (Bethyl), mouse monoclonal anti‐PRP19 (Santa Cruz), and mouse monoclonal anti‐U2AF2 (Santa Cruz).
Cell culture
U2OS cells were obtained from ATCC and maintained at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) (ThermoFisher Scientific) supplemented with 10% fetal bovine serum (FBS) (ThermoFisher Scientific) and 1% penicillin–streptomycin (ThermoFisher Scientific). NOMO‐1 cells (obtained from DSMZ) were maintained at 37°C and 5% CO2 in Roswell Park Memorial Institute (RPMI) 1640 medium (ThermoFisher Scientific) supplemented with 10% FBS and 1% penicillin–streptomycin. Vector and 2FLAG‐eIF4E wild‐type U2OS cell lines were generated as described previously (Culjkovic et al, 2006; Culjkovic‐Kraljacic et al, 2016), and maintained as U2OS cells with the addition of G‐418 (1 mg/ml, Wisent Bioproducts). MSCV‐pgk‐GFP‐eIF4E wild‐type was used for retroviral transduction of NOMO‐1 cells (Topisirovic et al, 2003). Internal ribosomal entry site (IRES) is placed between GFP and eIF4E, and thus, eIF4E is not produced as a fusion protein. Transduced cells were isolated using FACSAria cell sorter (BD Biosciences). Cell lines were authenticated using STR profiling (Wyndham Forensic Group). Cultured cells were routinely checked to ensure that there was no mycoplasma contamination by PCR (Sung et al, 2006). U2OS cells were treated with 20 micromolar ribavirin (Kemprotec, UK) for 72 h, and NOMO‐1 cells were treated with 10 micromolar ribavirin for 72 h.
Primary specimens
De‐identified patient specimens for analysis of splicing factor levels were obtained from the Banque de Cellules Leucémiques du Québec with institutional ethics approval. BCLQ obtained written informed consent in accordance with the Declaration of Helsinki. Samples for WB in Fig 3C and in Appendix Fig S1B were obtained from AML patients prior to treatment with ribavirin combination in the clinical trial (ClinicalTrials.gov NCT02073838) and received institutional review board and Health Canada approval. Blasts were isolated using flow cytometry as described (Assouline et al, 2009). Specimens from healthy volunteers were obtained from ATCC. Samples used for rMATS analysis of Leucegene.ca are listed in Dataset EV21.
Western blot analysis
Western blot analysis was performed as described previously (Zahreddine et al, 2017). Blots were blocked in 5% milk in TBS–Tween 20. Primary antibodies were diluted in 5% milk. Quantification of blots was carried out using FIJI and plotted in PRISM.
Immunofluorescence and laser‐scanning confocal microscopy
U2OS cells were grown on 4 well glass slides (Millicell EZ SLIDE 4 well glass, Millipore Sigma PEZGS0416). After washing three times in 1× PBS (pH 7.4), cells were fixed in prechilled Methanol at −20°C for 10 min, and air dried at room temperature for 30 min. NOMO‐1 cells were harvested and washed three times in 1×PBS in Eppendorf tubes, with centrifugation of 5 min at 300 x g. Washed pellets were resuspended in 1×PBS at 50,000 cells/10 μl, and 20 μl cell suspensions was spotted on glass slides. After drying for 30 min at room temperature, NOMO‐1 cells were fixed in prechilled methanol at −20°C for 10 min, and air dried at room temperature for 30 min. After drying, slides were blocked for 1 h in Blocking solution (10% FBS and 0.1% Tween 20 in PBS) and incubated with eIF4E‐FITC (BD Biosciences) directly conjugated antibody diluted in Blocking solution (1:50) overnight at 4°C. After washing three times in PBS, cells were mounted in antifade mounting medium with DAPI (Vector Laboratories). The cells were washed four times with PBS and mounted in mounting media with DAPI (Vector Laboratories, H‐2000). Analysis was carried out using a laser‐scanning confocal microscope (LSM700 META; Carl Zeiss, Inc.), with excitation at 405 and 488 nm, 63× oil objective and numerical aperture of 1.4. Channels were detected separately, with no cross‐talk observed. Confocal micrographs represent single sections through the plane of the cell. Images were obtained from ZEN software (Carl Zeiss, Inc.) and displayed using Adobe Photoshop CS6 (Adobe).
Cellular fractionation and RNA export assay
About 5 × 107 U2OS cells were collected and washed twice in ice‐cold PBS (300 × g for 3–5 min) and then resuspended with slow pipetting in 0.5 ml of lysis buffer B (10 mM Tris (pH 8.4), 140 mM NaCl, 1.5 mM MgCl2, 0.25% Nonidet P‐40, 1 mM DTT, 100 U/ml RNase inhibitors). The lysate was centrifuged at 1,000 × g for 3 min at 4°C, and the supernatant (cytoplasmic fraction) was transferred into a fresh microtube. The pellet (nuclear fraction) was resuspended in 1 Volume of lysis buffer B and transferred to a round‐bottomed polypropylene tube, and 1/10 volume of detergent stock (3.3% sodium deoxycholate, 6.6% Tween 40 in DEPC H2O) was added with slow vortexing (to prevent the nuclei from clumping) and incubated on ice for 5 min. The suspension was transferred to a microtube and centrifuged at 1,000 × g for 3 min at 4°C. Supernatant (postnuclear fraction) was added to the cytoplasmic fraction. RNA was extracted from the different fractions by adding TRIzol reagent (ThermoFisher Scientific) and isolated using Direct‐zol RNA Mini‐prep Kit (Zymo Research).
Polysome fractionation was carried out using size exclusion chromatography as described (Yoshikawa et al, 2018, 2021). Briefly, cells grown in two 15 cm plates grown as described above (70–80% confluency) were treated with 50 μg/ml cycloheximide for 15 min under 37°C and 5% CO2, washed twice with ice‐cold PBS containing 50 μg/ml cycloheximide, scraped in 600 μl of ice‐cold polysome extraction buffer (20 mM Hepes‐NaOH (pH 7.4), 130 mM NaCl, 10 mM MgCl2, 1% CHAPS, 0.2 mg/ml heparin, 2.5 mM DTT, 50 μg/ml cycloheximide, 20 U RNase inhibitor, cOmplete EDTA‐free Protease inhibitor) and lysed for 30 min on ice. Lysates were centrifuged at 16,000 × g for 10 min at 4°C and subsequently filtered through 0.45 μm Ultrafree‐MC HV centrifugal filter units by 12,000 × g for 5 min. Protein concentration in lysates was quantified by BCA protein assays. Separation of polysomes was done using Agilent Bio SEC‐5 1,000 Å column equilibrated with two column volumes (CV) of filtered SEC buffer (20 mM Hepes‐NaOH (pH 7.4), 60 mM NaCl, 10 mM MgCl2, 0.3% CHAPS, 0.2 mg/ml heparin, 2.5 mM DTT) (all column conditioning and separation were at room temperature). After monitoring the column condition by injecting standards, ~500 μg cell lysates were injected into a column. The chromatogram was monitored by measuring UV absorbance at 215, 260, and 280 nm. The flow rate was 0.8 ml/min and fractions were collected. RNAs from each fraction were extracted either by TRIzol LS reagent or Direct‐zol RNA Micro‐prep Kit.
Co‐immunoprecipitation and RIP
Nuclei isolated using the Cellular Fractionation protocol were rinsed 2× with 1×PBS and fixed with 1% PFA for 10 min at RT with rotation, quenched 5 min with 0.15 M Glycine (RT with rotation), then washed three times with 1×PBS and lysed in 0.5 ml NT‐2 buffer by three times 6 s bursts (with 30 s pause between each burst) using microtip at 25% power (Sonic Dismembrator Model 500, Fisher, Max Output 400W). NT‐2 buffer: 150 mM NaCl, 50 mM Tris–HCl (pH 7.4), 2.5 mM MgCl2, 0.05% Nonidet P‐40, 8 supplemented with 1 mM DTT, 1× protease inhibitors without EDTA, 200 U/ml RNaseOut. Nuclear lysates were centrifuged at 10,000 × g for 10 min, and supernatants were transferred into fresh tubes. After adjusting the concentration to be no more than 1 mg/ml, nuclear extracts were precleared with 50 μl protein G conjugated superparamagnetic beads (Dynabeads Protein G, ThermoFisher Scientific) for 30 min at 4°C. Precleared lysates (1 mg) were incubated with 10 μg of anti‐eIF4E antibody (RN001P, MBL) or 10 μg of appropriate IgG as a control, and 0.5 mg/ml yeast tRNA (Sigma‐Aldrich), overnight at 4°C with rotation. After ON incubation, 50 μl of Dynabeads were added and incubated for an additional 3 h at 4°C with rotation. Beads were washed once with NT‐2 buffer supplemented with 1 mg/ml heparin (Sigma‐Aldrich) for 5 min at 4°C with rotation and an additional six times with NT‐2 buffer with 300 mM NaCl. After washing, beads were resuspended in 2×Laemmli Buffer with β‐mercaptoethanol and incubated for 5 min at 98°C. Co‐immunoprecipitated proteins were resolved on SDS–PAGE and visualized by Western blotting. To isolate RNAs from immunoprecipitated reactions, beads were resuspended in Elution Buffer (100 mM Tris–HCl (pH 6.8), 4% (w/v) SDS, 20% (v/v) glycerol, 12% (v/v) β‐mercaptoethanol), and incubated for 5 min at 98°C. RNA was isolated using TRIzol reagent (ThermoFisher) and Direct‐zol RNA Micro‐prep Kit (Zymo Research).
eIF4E immunoprecipitation as a function of RNase treatment and m7G cap competition
Cells were collected and fractionated as described previously (Culjkovic‐Kraljacic & Borden, 2022). After fractionation, nuclei have been lysed by sonication (20% power, 3×6s with Sonic Dismembrator Model 500, Fisher, Max Output 400W) in 0.5 ml per 4 × 107 cells of NT‐2 buffer supplemented with 1× protease inhibitors. Lysates have been cleared by centrifugation at 10,000 × g 10 min at 4°C and transferred into a clean tube, and the concentration determined by BCA assay before being adjusted to 1 mg/ml. Nuclear Lysate has been cleared with 30 μl of Dynabeads G per mg of extract at 4°C for 40 min. For 1 mg IP, 33 μl of Dynabeads G (Invitrogen) were preincubated with 10 μg of anti‐eIF4E (rabbit, MBL) or rabbit IgGs at RT for 20 min. After five washes, beads are resuspended with precleared lysate and incubated with rotation at 4°C o/n. IPs have been washed five times with 1 ml of NT‐2 buffer. IPs have been resuspended in 1× beads volume of TE (10 mM Tris–pH 8, 1 mM EDTA) +0.5 ng/μl RNaseA (Qiagen 158922) and 0.05 U/μl of RNaseT1 (Sigma R1003) and incubate at RT for 15 min with agitation, supernatant has been kept in a new tube. IPs have been rinsed with 1× beads volume of TE, and supernatant has been pooled with the eluate. IPs have been washed once with NT‐2 buffer, and beads have been resuspended in 2× LB and incubated at 95°C for 10 min. Samples have been resolved by SDS–PAGE and visualized by Western Blot.
For cap elution, nuclear lysates were obtained as above and precleared with 30 μl of Dynabeads G per mg of extract at 4°C for 40 min and then treated with 100 or 200 micromolar or GpppG (NEB S1407S) or m7GpppG (NEBS 1404S) with rotation. For 1 mg IP, 33 μl of Dynabeads G (Invitrogen) were preincubated with 10 μg of anti‐eIF4E (rabbit, MBL) or anti‐eIF4E (mouse, SantaCruz) or IgGs (mouse or rabbit as appropriate, Millipore) at RT for 20 min. After five washes, beads were resuspended with the treated precleared lysate and incubated with rotation at 4°C o/n. IPs were washed one time with NT‐2 buffer supplemented with 1 mg/ml Heparin and five times with 1 ml of NT‐2 buffer. Proteins were analyzed by western blot.
Reverse transcription and quantitative PCR
RNA samples were reversed transcribed using SuperScript VILO cDNA synthesis kit (for RIP experiments) (ThermoFisher Scientific) or MMLV reverse transcription kit and oligo‐dT or Random hexamers (ThermoFisher Scientific) for mRNA or UsnRNA analyses, respectively. qPCR analyses were performed using SensiFastSybr Lo‐Rox Mix (Bioline, MA, USA, Cat# BIO‐94020) in Applied Biosystems Viia7 or QuantStudio7 thermal cyclers using the relative standard curve method (Applied Biosystems User Bulletin #2). All the primers are listed in Dataset EV22, and all conditions were described previously (Culjkovic‐Kraljacic et al, 2016).
Analysis of alternative splicing
Alternative splicing events were detected in RNA‐seq data and the probability that the differences in isoform abundance were evaluated statistically with rMATS version 4.1.1. Sequenced fragments were treated as nonstranded to allow a uniform treatment of both stranded and nonstranded samples, and variable read lengths were allowed in the analysis. JC and JCEC quantifications have been combined for the complete analysis. Events with a false discovery rate below 0.1 or 0.15 and an absolute inclusion level higher than 0.1 or 0.05 were considered significant and carried further in the analysis. Maxent was used to evaluate the strength of significant splice‐site events. MAXENT ranges considered for the analysis were: Strong: MAXENT score ≥ 7, Intermediate: MAXENT score ≥ 3 and < 7, Weak: MAXENT score < 3.
Data analysis and visualization
DESeq2 version 1.30.1 (Love et al, 2014) was used to normalize gene read counts (nonstranded) for the 21 AML samples. Significant differentially expressed genes (DEGs) are typically those with padj lower than 0.05 and an absolute fold change > 2. Data visualization, graphics, and plots were made using R package ggplot2 and related packages digest, glue, grDevices, grid, gTable, isoband, MASS, mgcv, rlang, scales, stats, tibble, withr, and dplyr. “pheatmap” package was applied to construct heat maps and hierarchical clustering analyses. Process and pathway enrichment analyses were performed using METASCAPE (Zhou et al, 2019). Briefly, for each gene list, pathway and process enrichment analysis have been carried out with the following ontology sources: KEGG Pathway, GO Biological Processes, Reactome Gene Sets, Canonical Pathways, WikiPathways, and PANTHER Pathway. All genes in the genome have been used as the enrichment background. Terms with a P‐value < 0.01, a minimum count of 3, and an enrichment factor > 1.5 (the enrichment factor is the ratio between the observed counts and the counts expected by chance) are collected and grouped into clusters based on their membership similarities. More specifically, P‐values are calculated based on the accumulative hypergeometric distribution, and q‐values are calculated using the Benjamini–Hochberg procedure to account for multiple testing. Protein–protein interaction analysis was completed with METASCAPE (Zhou et al, 2019) and CYTOSCAPE (Shannon et al, 2003). For each target gene list, protein–protein interaction enrichment analysis has been carried out with the following databases: STRING, BioGrid, OmniPath, and InWeb_IM. Only physical interactions in STRING (physical score > 0.132) and BioGrid are used. The resultant network contains the subset of proteins that form physical interactions with at least one other member in the list. If the network contains more than five proteins, the Molecular Complex Detection (MCODE) algorithm has been applied to identify densely connected network components. Pathway and process enrichment analysis has been applied to each MCODE component independently, and the best‐scoring term by P‐value has been retained as the functional description of the corresponding component. Venn diagrams were constructed using Bioinformatics and evolutionary genomics webtool (http://bioinformatics.psb.ugent.be/webtools/Venn/). Figures and cartoons were edited and laid out using the open‐source Vector graphics editor Inkscape (https://inkscape.org/).
Author contributions
Mehdi Ghram: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Gavin Morris: Conceptualization; investigation. Biljana Culjkovic‐Kraljacic: Conceptualization; resources; formal analysis; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Jean‐Clément Mars: Formal analysis; validation; investigation; visualization; methodology; writing – review and editing. Patrick Gendron: Data curation; investigation; visualization. Lucy Skrabanek: Investigation. Maria Victoria Revuelta: Investigation. Leandro Cerchietti: Investigation. Monica L Guzman: Investigation. Katherine L B Borden: Conceptualization; supervision; investigation; writing – original draft; writing – review and editing.
Disclosure and competing interests statement
MG, GM, BCK, JCM, PG, LS, MVR, LC, MLG, and KLBB have no disclosure or competing interests. Of note, BCK and KLBB hold patents related to ribavirin use in AML; however, they have received no royalties or other benefit from these: Combination therapy using ribavirin as elF4E inhibitor, Inventors: Katherine Borden, Hiba Zahreddine, Biljana Culjkovic‐Kraljacic (targeting inducible drug glucuronidation). US10342817B2; translation dysfunction‐based therapeutic inventors: Gordon Jamieson, Katherine Borden, Biljana Culjkovic, Alex Kentsis: US8497292B2. KLBB has held Leukemia and Lymphoma Society and NIH funds listed above to study AML. MGL has research funding and consulting from BridgeMedicines, Equity SeqRX, LLC, and holds other AML funding from the NIH (R21CA245454 and R01CA234478).
Supporting information
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Dataset EV10
Dataset EV11
Dataset EV12
Dataset EV13
Dataset EV14
Dataset EV15
Dataset EV16
Dataset EV17
Dataset EV18
Dataset EV19
Dataset EV20
Dataset EV21
Dataset EV22
Acknowledgements
We are grateful for assistance from the Genomics Platform at IRIC. Primary human specimens were obtained from the Banque de cellules leucémiques du Québec (BCLQ), supported by the Cancer Research Network of the Fonds de Recherche du Québec—Santé (FRQS). KLBB acknowledges funding from Leukemia and Lymphoma Society United States and Canada (TRP R6513‐20), the Canadian Institutes for Health Research (PJT 159785), and was supported by the National Cancer Institutes of Health under Award numbers RO1CA098571 and RO1CA080728, the content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. KLBB holds a Canada Research Chair in Molecular Biology of the Cell Nucleus (950‐231848), and MG holds Cole Foundation and Baumgartner fellowships and JC a Bourse postdoctorale Power Corporation du Canada de la relève scientifique.
The EMBO Journal (2023) 42: e110496
Data availability
The datasets and computer code used in this study are available in the following databases: RNA‐Seq data: Gene Expression Omnibus GSE158728 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158728); RNA‐Seq data: Gene Expression Omnibus GSE67040 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE67040); Modeling computer scripts: rMATS (https://rnaseq‐mats.sourceforge.net/rmats4.1.1/).
References
- Abdelmohsen K, Gorospe M (2010) Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip Rev RNA 1: 214–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamia S, Haibe‐Kains B, Pilarski PM, Bar‐Natan M, Pevzner S, Avet‐Loiseau H, Lode L, Verselis S, Fox EA, Burke J et al (2014) A genome‐wide aberrant RNA splicing in patients with acute myeloid leukemia identifies novel potential disease markers and therapeutic targets. Clin Cancer Res 20: 1135–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assouline S, Culjkovic B, Cocolakis E, Rousseau C, Beslu N, Amri A, Caplan S, Leber B, Roy DC, Miller WH Jr et al (2009) Molecular targeting of the oncogene eIF4E in acute myeloid leukemia (AML): a proof‐of‐principle clinical trial with ribavirin. Blood 114: 257–260 [DOI] [PubMed] [Google Scholar]
- Assouline S, Culjkovic‐Kraljacic B, Bergeron J, Caplan S, Cocolakis E, Lambert C, Lau CJ, Zahreddine HA, Miller WH Jr, Borden KL (2015) A phase I trial of ribavirin and low‐dose cytarabine for the treatment of relapsed and refractory acute myeloid leukemia with elevated eIF4E. Haematologica 100: e7–e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakheet T, Hitti E, Al‐Saif M, Moghrabi WN, Khabar KSA (2018) The AU‐rich element landscape across human transcriptome reveals a large proportion in introns and regulation by ELAVL1/HuR. Biochim Biophys Acta Gene Regul Mech 1861: 167–177 [DOI] [PubMed] [Google Scholar]
- Batsche E, Ameyar‐Zazoua M (2015) The influence of Argonaute proteins on alternative RNA splicing. Wiley Interdiscip Rev RNA 6: 141–156 [DOI] [PubMed] [Google Scholar]
- Blackinton JG, Keene JD (2014) Post‐transcriptional RNA regulons affecting cell cycle and proliferation. Semin Cell Dev Biol 34: 44–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden KL (2016) The eukaryotic translation initiation factor eIF4E wears a “cap” for many occasions. Translation (Austin) 4: e1220899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden KLB (2020) The nuclear pore complex and mRNA export in cancer. Cancers (Basel) 13: 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden KLB (2022) Cancer cells hijack RNA processing to rewrite the message. Biochem Soc Trans 50: 1447–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden KLB, Volpon L (2020) The diversity, plasticity, and adaptability of cap‐dependent translation initiation and the associated machinery. RNA Biol 17: 1239–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutz PL, Bhutkar A, Sharp PA (2015) Detained introns are a novel, widespread class of post‐transcriptionally spliced introns. Genes Dev 29: 63–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun JE, Friedman LJ, Gelles J, Moore MJ (2018) Synergistic assembly of human pre‐spliceosomes across introns and exons. Elife 7: e37751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciesla M, Ngoc PCT, Cordero E, Martinez AS, Morsing M, Muthukumar S, Beneventi G, Madej M, Munita R, Jonsson T et al (2021) Oncogenic translation directs spliceosome dynamics revealing an integral role for SF3A3 in breast cancer. Mol Cell 81: 1453–1468 [DOI] [PubMed] [Google Scholar]
- Cohen N, Sharma M, Kentsis A, Perez JM, Strudwick S, Borden KL (2001) PML RING suppresses oncogenic transformation by reducing the affinity of eIF4E for mRNA. EMBO J 20: 4547–4559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews LA, Balaian L, Delos Santos NP, Leu HS, Court AC, Lazzari E, Sadarangani A, Zipeto MA, La Clair JJ, Villa R et al (2016) RNA splicing modulation selectively impairs leukemia stem cell maintenance in secondary human AML. Cell Stem Cell 19: 599–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culjkovic B, Topisirovic I, Skrabanek L, Ruiz‐Gutierrez M, Borden KL (2005) eIF4E promotes nuclear export of cyclin D1 mRNAs via an element in the 3′UTR. J Cell Biol 169: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culjkovic B, Topisirovic I, Skrabanek L, Ruiz‐Gutierrez M, Borden KL (2006) eIF4E is a central node of an RNA regulon that governs cellular proliferation. J Cell Biol 175: 415–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culjkovic‐Kraljacic B, Borden KLB (2018) The impact of post‐transcriptional control: better living through RNA regulons. Front Genet 9: 512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culjkovic‐Kraljacic B, Borden KLB (2022) Subcellular fractionation suitable for studies of RNA and protein trafficking. Methods Mol Biol 2502: 91–104 [DOI] [PubMed] [Google Scholar]
- Culjkovic‐Kraljacic B, Baguet A, Volpon L, Amri A, Borden KLB (2012) The oncogene eIF4E reprograms the nuclear pore complext to promote mRNA export and oncogenic transformation. Cell Rep 2: 207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culjkovic‐Kraljacic B, Fernando TM, Marullo R, Calvo‐Vidal N, Verma A, Yang S, Tabbo F, Gaudiano M, Zahreddine H, Goldstein RL et al (2016) Combinatorial targeting of nuclear export and translation of RNA inhibits aggressive B‐cell lymphomas. Blood 127: 858–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culjkovic‐Kraljacic B, Skrabanek L, Revuelta MV, Gasiorek J, Cowling VH, Cerchietti L, Borden KLB (2020a) The eukaryotic translation initiation factor eIF4E elevates steady‐state m7G capping of coding and noncoding transcripts. Proc Natl Acad Sci USA 117: 26773–26783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culjkovic‐Kraljacic B, Skrabanek LA, Revuelta M, Gasiorek J, Cowling VH, Cerchietti L, Borden KL (2020b) Gene Expression Omnibus GSE158728 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158728). [DATASET] [DOI] [PMC free article] [PubMed]
- Davis MR, Delaleau M, Borden KLB (2019) Nuclear eIF4E stimulates 3′‐end cleavage of target RNAs. Cell Rep 27: 1397–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Necochea‐Campion R, Shouse GP, Zhou Q, Mirshahidi S, Chen CS (2016) Aberrant splicing and drug resistance in AML. J Hematol Oncol 9: 85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sousa Abreu R, Penalva LO, Marcotte EM, Vogel C (2009) Global signatures of protein and mRNA expression levels. Mol Biosyst 5: 1512–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostie J, Lejbkowicz F, Sonenberg N (2000) Nuclear eukaryotic initiation factor 4E (eIF4E) colocalizes with splicing factors in speckles. J Cell Biol 148: 239–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn LA, Fury MG, Sherman EJ, Ho AA, Katabi N, Haque SS, Pfister DG (2017) Phase I study of induction chemotherapy with afatinib, ribavirin, and weekly carboplatin and paclitaxel for stage IVA/IVB human papillomavirus‐associated oropharyngeal squamous cell cancer. Head Neck 40: 233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Bradley RK (2015) Widespread intron retention diversifies most cancer transcriptomes. Genome Med 7: 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edy VG, Szekely M, Loviny T, Dreyer C (1976) Action of nucleases on double‐stranded RNA. Eur J Biochem 61: 563–572 [DOI] [PubMed] [Google Scholar]
- Effenberger KA, Urabe VK, Jurica MS (2017) Modulating splicing with small molecular inhibitors of the spliceosome. Wiley Interdiscip Rev RNA 8: e1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N (1999) eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem 68: 913–963 [DOI] [PubMed] [Google Scholar]
- Gornemann J, Kotovic KM, Hujer K, Neugebauer KM (2005) Cotranscriptional spliceosome assembly occurs in a stepwise fashion and requires the cap binding complex. Mol Cell 19: 53–63 [DOI] [PubMed] [Google Scholar]
- Graham PL, Yanowitz JL, Penn JK, Deshpande G, Schedl P (2011) The translation initiation factor eIF4E regulates the sex‐specific expression of the master switch gene Sxl in Drosophila melanogaster. PLoS Genet 7: e1002185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger CE, Moyer DC, Adema V, Kerr CM, Walter W, Hutter S, Meggendorfer M, Baer C, Kern W, Nadarajah N et al (2021) Complex landscape of alternative splicing in myeloid neoplasms. Leukemia 35: 1108–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izaurralde E, Lewis J, McGuigan C, Jankowska M, Darzynkiewicz E, Mattaj IW (1994) A nuclear cap binding protein complex involved in pre‐mRNA splicing. Cell 78: 657–668 [DOI] [PubMed] [Google Scholar]
- Izquierdo JM (2008) Hu antigen R (HuR) functions as an alternative pre‐mRNA splicing regulator of Fas apoptosis‐promoting receptor on exon definition. J Biol Chem 283: 19077–19084 [DOI] [PubMed] [Google Scholar]
- Izquierdo JM (2010) Heterogeneous ribonucleoprotein C displays a repressor activity mediated by T‐cell intracellular antigen‐1‐related/like protein to modulate Fas exon 6 splicing through a mechanism involving Hu antigen R. Nucleic Acids Res 38: 8001–8014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo JM, Majos N, Bonnal S, Martinez C, Castelo R, Guigo R, Bilbao D, Valcarcel J (2005) Regulation of Fas alternative splicing by antagonistic effects of TIA‐1 and PTB on exon definition. Mol Cell 19: 475–484 [DOI] [PubMed] [Google Scholar]
- Kapp LD, Lorsch JR (2004) The molecular mechanics of eukaryotic translation. Annu Rev Biochem 73: 657–704 [DOI] [PubMed] [Google Scholar]
- Kaufman RJ, Murtha‐Riel P, Pittman DD, Davies MV (1993) Characterization of wild‐type and Ser53 mutant eukaryotic initiation factor 4E overexpression in mammalian cells. J Biol Chem 268: 11902–11909 [PubMed] [Google Scholar]
- Keene JD, Tenenbaum SA (2002) Eukaryotic mRNPs may represent posttranscriptional operons. Mol Cell 9: 1161–1167 [DOI] [PubMed] [Google Scholar]
- Kosaka T, Maeda T, Shinojima T, Nagata H, Ryuchi R, Oya M (2017) A clinical study to evaluate the efficacy and safety of docetaxal with ribavirin in patients with progressive castration resistant prostate cancer who have previously received docetaxol alone. J Clin Oncol 35: e14010 [Google Scholar]
- Kraljacic BC, Arguello M, Amri A, Cormack G, Borden K (2011) Inhibition of eIF4E with ribavirin cooperates with common chemotherapies in primary acute myeloid leukemia specimens. Leukemia 25: 1197–1200 [DOI] [PubMed] [Google Scholar]
- Lazaris‐Karatzas A, Montine KS, Sonenberg N (1990) Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature 345: 544–547 [DOI] [PubMed] [Google Scholar]
- Leucegene (2015) AML Sequencing Gene Expression Omnibus GSE67040 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE67040). [DATASET]
- Lewis JD, Izaurralde E, Jarmolowski A, McGuigan C, Mattaj IW (1996) A nuclear cap‐binding complex facilitates association of U1 snRNP with the cap‐proximal 5′ splice site. Genes Dev 10: 1683–1698 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mars JC, Ghram M, Culjkovic‐Kraljacic B, Borden KLB (2021) The cap‐binding complex CBC and the eukaryotic translation factor eIF4E: co‐conspirators in cap‐dependent RNA maturation and translation. Cancers (Basel) 13: 6185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehmood A, Laiho A, Venalainen MS, McGlinchey AJ, Wang N, Elo LL (2020) Systematic evaluation of differential splicing tools for RNA‐seq studies. Brief Bioinform 21: 2052–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehterov N, Kazakova M, Sbirkov Y, Vladimirov B, Belev N, Yaneva G, Todorova K, Hayrabedyan S, Sarafian V (2021) Alternative RNA splicing‐the trojan horse of cancer cells in chemotherapy. Genes (Basel) 12: 1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M Jr, Tuschl T, Ohler U, Keene JD (2011) Integrative regulatory mapping indicates that the RNA‐binding protein HuR couples pre‐mRNA processing and mRNA stability. Mol Cell 43: 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne MJ, Borden KL (2015) The eukaryotic translation initiation factor eIF4E in the nucleus: taking the road less traveled. Immunol Rev 263: 210–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne MJ, Volpon L, Memarpoor‐Yazdi M, Pillay S, Thambipillai A, Czarnota S, Culjkovic‐Kraljacic B, Trahan C, Oeffinger M, Cowling VH et al (2022) Identification and characterization of the interaction between the Methyl‐7‐guanosine cap maturation enzyme RNMT and the cap‐binding protein eIF4E. J Mol Biol 434: 167451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high‐throughput sequencing. Nat Genet 40: 1413–1415 [DOI] [PubMed] [Google Scholar]
- Papasaikas P, Tejedor JR, Vigevani L, Valcarcel J (2015) Functional splicing network reveals extensive regulatory potential of the core spliceosomal machinery. Mol Cell 57: 7–22 [DOI] [PubMed] [Google Scholar]
- Phillips JW, Pan Y, Tsai BL, Xie Z, Demirdjian L, Xiao W, Yang HT, Zhang Y, Lin CH, Cheng D et al (2020) Pathway‐guided analysis identifies Myc‐dependent alternative pre‐mRNA splicing in aggressive prostate cancers. Proc Natl Acad Sci USA 117: 5269–5279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambout X, Maquat LE (2020) The nuclear cap‐binding complex as choreographer of gene transcription and pre‐mRNA processing. Genes Dev 34: 1113–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera OD, Mallory MJ, Quesnel‐Vallieres M, Chatrikhi R, Schultz DC, Carroll M, Barash Y, Cherry S, Lynch KW (2021) Alternative splicing redefines landscape of commonly mutated genes in acute myeloid leukemia. Proc Natl Acad Sci USA 118: e2014967118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez B, Walter MJ, Graubert TA (2017) Splicing factor gene mutations in hematologic malignancies. Blood 129: 1260–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Huang J, Dittmar KA, Lu ZX, Zhou Q, Carstens RP, Xing Y (2012) MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA‐seq data. Nucleic Acids Res 40: e61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone LE, Keene JD (2013) Mechanisms coordinating ELAV/Hu mRNA regulons. Curr Opin Genet Dev 23: 35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung H, Kang SH, Bae YJ, Hong JT, Chung YB, Lee CK, Song S (2006) PCR‐based detection of mycoplasma species. J Microbiol 44: 42–49 [PubMed] [Google Scholar]
- Taylor J, Lee SC (2019) Mutations in spliceosome genes and therapeutic opportunities in myeloid malignancies. Genes Chromosomes Cancer 58: 889–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topisirovic I, Guzman ML, McConnell MJ, Licht JD, Culjkovic B, Neering SJ, Jordan CT, Borden KL (2003) Aberrant eukaryotic translation initiation factor 4E‐dependent mRNA transport impedes hematopoietic differentiation and contributes to leukemogenesis. Mol Cell Biol 23: 8992–9002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topisirovic I, Siddiqui N, Lapointe VL, Trost M, Thibault P, Bangeranye C, Pinol‐Roma S, Borden KL (2009a) Molecular dissection of the eukaryotic initiation factor 4E (eIF4E) export‐competent RNP. EMBO J 28: 1087–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topisirovic I, Siddiqui N, Orolicki S, Skrabanek LA, Tremblay M, Hoang T, Borden KL (2009b) Stability of eukaryotic translation initiation factor 4E mRNA is regulated by HuR, and this activity is dysregulated in cancer. Mol Cell Biol 29: 1152–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanski LM, Leclair N, Anczukow O (2018) Alternative‐splicing defects in cancer: splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip Rev RNA 9: e1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visconte V, Nakashima MO, Rogers HJ (2019) Mutations in splicing factor genes in myeloid malignancies: significance and impact on clinical features. Cancers (Basel) 11: 1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpon L, Culjkovic‐Kraljacic B, Osborne MJ, Ramteke A, Sun Q, Niesman A, Chook YM, Borden KL (2016) Importin 8 mediates m7G cap‐sensitive nuclear import of the eukaryotic translation initiation factor eIF4E. Proc Natl Acad Sci USA 113: 5263–5268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpon L, Culjkovic‐Kraljacic B, Sohn HS, Blanchet‐Cohen A, Osborne MJ, Borden KLB (2017) A biochemical framework for eIF4E‐dependent mRNA export and nuclear recycling of the export machinery. RNA 23: 927–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Luhrmann R (2009) The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718 [DOI] [PubMed] [Google Scholar]
- Yoshikawa H, Larance M, Harney DJ, Sundaramoorthy R, Ly T, Owen‐Hughes T, Lamond AI (2018) Efficient analysis of mammalian polysomes in cells and tissues using Ribo Mega‐SEC. Elife 7: e36530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa H, Sundaramoorthy R, Mariyappa D, Jiang H, Lamond AI (2021) Efficient and rapid analysis of polysomes and ribosomal subunits in cells and tissues using Ribo Mega‐SEC. Bio Protoc 11: e4106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahreddine HA, Culjkovic‐Kraljacic B, Assouline S, Gendron P, Romeo AA, Morris SJ, Cormack G, Jaquith JB, Cerchietti L, Cocolakis E et al (2014) The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature 511: 90–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahreddine HA, Culjkovic‐Kraljacic B, Emond A, Pettersson F, Midura R, Lauer M, Del Rincon S, Cali V, Assouline S, Miller WH et al (2017) The eukaryotic translation initiation factor eIF4E harnesses hyaluronan production to drive its malignant activity. Elife 6: e29830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Klein HL, Schneider RJ (1995) Role of Ser‐53 phosphorylation in the activity of human translation initiation factor eIF‐4E in mammalian and yeast cells. Gene 163: 283–288 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK (2019) Metascape provides a biologist‐oriented resource for the analysis of systems‐level datasets. Nat Commun 10: 1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Hasman RA, Barron VA, Luo G, Lou H (2006) A nuclear function of Hu proteins as neuron‐specific alternative RNA processing regulators. Mol Biol Cell 17: 5105–5114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Hinman MN, Hasman RA, Mehta P, Lou H (2008) Regulation of neuron‐specific alternative splicing of neurofibromatosis type 1 pre‐mRNA. Mol Cell Biol 28: 1240–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]