

Abstract
Background
Crohn’s disease (CD) is highly heterogenous and may be complicated by stricturing behavior. Personalized prediction of stricturing will inform management. We aimed to create a stricturing risk stratification model using genomic/clinical data.
Methods
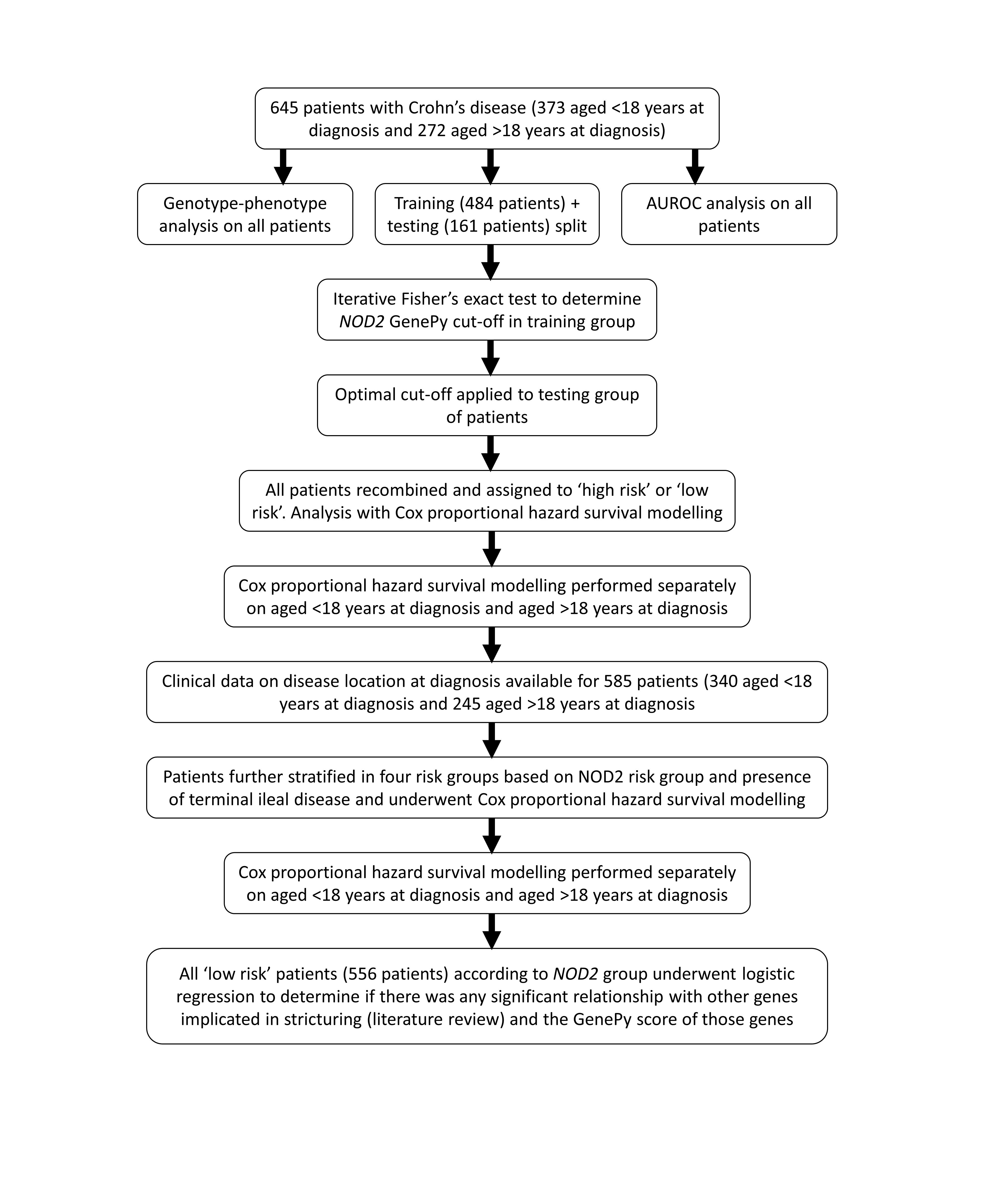
Exome sequencing was performed on CD patients, and phenotype data retrieved. Biallelic variants in NOD2 were identified. NOD2 was converted into a per-patient deleteriousness metric (“GenePy”). Using training data, patients were stratified into risk groups for fibrotic stricturing using NOD2. Findings were validated in a testing data set. Models were modified to include disease location at diagnosis. Cox proportional hazards assessed performance.
Results
Six hundred forty-five patients were included (373 children and 272 adults); 48 patients fulfilled criteria for monogenic NOD2-related disease (7.4%), 24 of whom had strictures. NOD2 GenePy scores stratified patients in training data into 2 risk groups. Within testing data, 30 of 161 patients (18.6%) were classified as high-risk based on the NOD2 biomarker, with stricturing in 17 of 30 (56.7%). In the low-risk group, 28 of 131 (21.4%) had stricturing behavior. Cox proportional hazards using the NOD2 risk groups demonstrated a hazard ratio (HR) of 2.092 (P = 2.4 × 10-5), between risk groups. Limiting analysis to patients diagnosed aged < 18-years improved performance (HR-3.164, P = 1 × 10-6). Models were modified to include disease location, such as terminal ileal (TI) disease or not. Inclusion of NOD2 risk groups added significant additional utility to prediction models. High-risk group pediatric patients presenting with TI disease had a HR of 4.89 (P = 2.3 × 10-5) compared with the low-risk group patients without TI disease.
Conclusions
A NOD2 genomic biomarker predicts stricturing risk, with prognostic power improved in pediatric-onset CD. Implementation into a clinical setting can help personalize management.
Keywords: NOD2, Crohn’s disease, stricturing, personalized, prediction
Key messages.
What is already known?
NOD2 is highly implicated in Crohn’s disease and has been linked to a stricturing phenotype.
What is new here?
By using NOD2 as a genomic biomarker, we are able to predict high-risk stricturing patients; disease location data also further improved prediction.
In those diagnosed at younger than 18 years of age, the high-risk group had a 5x increased risk of stricturing compared with low-risk patients.
How can this study help patient care?
Routine utilisation of NOD2 as a genomic biomarker may allow risk stratification of Crohn’s disease patients at diagnosis.
Personalizing management based stricturing risk may be possible.
Stratified randomized trials of high-risk patients will be important.
Introduction
Crohn’s disease is a chronic, relapsing, and remitting condition characterized by inflammatory change throughout the gastrointestinal tract, commonly seen in the terminal ileum. Prediction of disease severity and behavior is extremely challenging at the point of diagnosis. Differentiating between patients who will develop inflammatory, penetrating, and stricturing phenotypes could potentially enable targeted therapy to impact disease course.1 The interplay between genetic risk and environmental exposure leads to disease pathogenesis, something which appears increasingly likely to be specific to a patient or family.2 Specific disease traits and responses to therapy have been linked to genetic defects, gene expression modules, or microbiome profiles.3–5 Previous attempts to translate molecular data to predict clinical outcomes have produced promising results, although no testing has routinely entered clinical practice to date.6,7
NOD2 is the best characterized risk gene for development of Crohn’s disease, coding for a vital intracellular microbial pattern recognition and response protein, triggering downstream innate immune response.8,9 Recent data have pointed towards a potential monogenic role for NOD2 in a subset of Crohn’s disease patients who appear to be at high risk of developing a stricturing disease phenotype.10,11 Despite this, NOD2 is largely viewed as a risk gene without a clear clinical role for routine genotyping.12 Previous studies have largely focused on the 3 most common risk variants within NOD2 (R702W, G908R, and 1007fs) and have failed to account for the role of a rarer variation, epistasis, or accumulation of multiple variants with modest deleteriousness.13 Data from our group utilizing GenePy, a contemporary in silico mutational burden tool across the whole gene, has demonstrated additional NOD2 variation playing a role in Crohn’s disease phenotype.11 Accounting for cumulative burden of pathogenic variation within genes is likely to have a discovery uplift when considering non-Mendelian complex disease.14 The role of pathogenic variation throughout NOD2 as a single-gene contributor to adult onset disease is also poorly elucidated. Although NOD2 is the strongest genetic signal for Crohn’s disease and stricturing disease, additional genetic risk loci have been identified for fibrostenotic disease, including genes within the NOD2-signaling pathway such as ATG16L1.15 It increasingly appears that the role of NOD2 in Crohn’s disease is not yet fully understood.
Utilizing genomic biomarkers—measurable genomic characteristics that predict a specific clinical outcome or response to treatment—is an exciting avenue of personalized medicine. This study aimed to develop and optimize NOD2 as a genomic biomarker capable of stratifying Crohn’s disease patients into high- and low-risk groups for development of fibrotic stricturing disease, providing a tool for translation into clinical practice. Additionally, we aimed to characterize NOD2 genotypes in both pediatric and adult patients and determine the prevalence of deleterious variants across the age spectrum.
Methods
Recruitment
Patients were included from the Wessex regional pediatric inflammatory bowel disease (IBD) service at Southampton Children’s Hospital and the adult IBD service at University Hospital Southampton. Patients were recruited from 2010 to the present. All patients within the cohort had a confirmed histological diagnosis of either Crohn’s disease, ulcerative colitis, or IBD-unclassified, in line with the Porto criteria or British Society of Gastroenterology guidelines.16 Patients with Crohn’s disease were extracted for this analysis. There are no exclusion criteria, if a patient has a confirmed diagnosis of IBD and is able to give informed consent.
Longitudinal Data Collection
Endoscopy, small bowel magnetic resonance imaging (MRI), abdominal ultrasound, and computed tomography (CT) abdomen scan reports were retrieved from the electronic patient records (EHRs) of the University Hospital Southampton. These records, including clinic letters, imaging reports, and endoscopy reports, were searched for stricturing keywords (fibrosis, fibrotic, stricture, stricturing, narrowing, narrowed, pre-stenotic dilatation, stenotic, reduced diameter) to reduce the number of reports requiring clinical curation. Records without key words were recorded as a nonstricturing phenotype, and the remaining were manually checked by 2 clinicians (J.J.A. and M.K.) to assign patients as having stricturing or nonstricturing phenotypes. Where there was uncertainty, a further clinician (R.M.B.) was consulted to give a final classification. As this study was focused on fibrotic or predominately fibrotic disease, strictures that resolved without surgery or dilatation were presumed to be purely inflammatory; and these patients were assigned as having a nonstricturing phenotype. As described previously, we used the strict definition of fibrotic stricturing as “histologically proven, or narrowing demonstrable on 2 consecutive MRIs, with prestenotic dilatation” to define a specific disease phenotype.11 Date of stricturing was recorded, and time from diagnosis to stricture was calculated. Duration of follow-up was calculated for all patients. During the follow-up period (diagnosis to most recent clinical contact), all patients were assessed for occurrence of fibrotic strictures if they had narrowing initially assigned as inflammatory and grouped accordingly. Presence of terminal ileal disease at diagnosis was retrieved from endoscopy and imaging reports.
Whole Exome Sequencing Data Processing
DNA was extracted from blood samples collected in ethylenediaminetetraacetic using the salting-out method, or from saliva, as previously described.17 An estimated 20 µg of DNA was used for whole exome sequencing.
Raw fastq sequencing data from patients in the cohort were processed using our in-house pipeline in line with the Genome Analysis Tool Kit 4 (GATK 4) best practice (https://github.com/UoS-HGIG/WES_2022_QC_pipeline).18 Alignment was performed against the human reference genome (GRCh38 assembly with decoy human leucocyte antigen [HLA] regions) using BWA-mem (version 0.7.15).19 Joint variant calling of all samples in the cohort was restricted to the 150 bp padded union of the Agilent SureSelect All Exon V5 and V6 capture kits.
VerifyBamID was utilized to check the presence of DNA contamination across the cohort.20 We applied our in-house fingerprint panel to confirm sample identity and provenance.21 In addition, following the GATK built-in Variant Quality Score Recalibration, data were assessed for sequencing depth, genotyping quality, and variant allele frequency (AF).
Variant called format (VCF) file annotation was performed using Ensembl-VEP (v.103),22 using default databases, deleteriousness scores databases (dbnsfp35c, CADD v.1.6),23 dbSNP147, and the human genetic mutation database (HGMD Pro 2021).24 Variant allele frequencies were sourced through the genome aggregation exome database (gnomAD),25 v2.1.1. We referred to the canonical NOD2 transcript ENST00000300589 (GENCODE) for the annotation of coding variants unless otherwise specified.
Lollipop plot of NOD2 coding variants was generated using the MutationMapper tool of cBioPortal,26 and variants were mapped to the Pfam domains of NOD2 (Figure 1) to visualize the distribution of variants with respect to deleteriousness metrics (CADD v.1.6) and allele frequency (gnomAD) within our cohort.
Figure 1.

A, Lollipop plots showing homozygous variants in NOD2 identified in our cohort. B. All variants identified in the cohort and (C) variants with CADD scores >15 (deleterious variants) within the cohort. C, An area of 171 amino acids in the NOD domain are free from variants predicted to be deleterious. In all figures, variants are shown by position within NOD2 (x axis) and frequency (y axis).
Application of GenePy
GenePy provides a per gene, per individual single metric of deleteriousness, facilitating genes to be incorporated into downstream risk stratification modeling. Whole exome sequencing data were transformed into GenePy scores for patient stratification (https://github.com/UoS-HGIG/GenePy-1.4).14 Firstly, the joint called aggregated cohort VCF underwent recommended quality control filtration steps,27 such that only good quality biallelic variants were retained in a VCF for annotation as described previously. The GenePy score algorithm was applied to exonic variants, with a CADD Phred score >15 (as per developer guidance for determining deleterious variants), and GenePy scores were retrieved for analysis.23
Monogenic NOD2 Disease
To stratify the risk of stricturing in individuals based on NOD2 genotype, all patients were screened for biallelic NOD2 variants. Variants were initially annotated with functional evidence from the literature, as previously described.11 Variants that were functionally demonstrated to impact NOD2 function, including reduced/absent protein function, impact downstream signaling, nonsense mediated decay or deletions, were included in line with American College of Medical Genetics guidelines (ACMG) for “pathogenic” or “likely pathogenic” variants.28 Patients who were homozygous, or had 2 or more heterozygous variants, were denoted “NOD2-related disease.”
We determined the number of patients with putative deleterious NOD2 variation but without functional evidence to meet ACMG criteria to be in silico NOD2-related disease. These patients were homozygous or had 2 or more heterozygous variants, where the variants met in silico criteria for deleteriousness-allele frequency (gnomAD) <0.05 and a CADD-PHRED score of >15.23
We have previously demonstrated that all potential NOD2 compound heterozygous variants in pediatric-onset patients had confirmed variant segregation and were biallelic.23 In this study, it was not possible to perform segregation analysis in patients with adult-onset disease due to lack of parental DNA.
Incidence of stricturing disease in patients was retrieved, and we assessed enrichment for a stricturing vs nonstricturing phenotype in each group through a Chi-squared test. Data were visualized using a dumbbell plot. A summary of the methodology can be seen in Supplementary Figure 1.
Stricturing Disease Prediction Modeling
Receiver operator curve analysis
To determine the stricturing disease classification ability of NOD2 mutation burden, we performed an area under receiver operator curve (AUROC) analysis (SPSS, IBM v27). NOD2 GenePy score was the test variable. The analyses were performed on all patients, and then separately on the subgroup of patients diagnosed <18 years of age.
Group Optimization: Training and Testing Data Sets
Patients were split into training and testing (validation) sets for risk-group determination utilizing the caTools R package (training proportion = 0.75). Utilising Cutoff Finder,29 a biomarker optimization software, an iterative Fisher exact test was used to determine the optimal number of risk groups and NOD2 GenePy score boundaries. All groups and boundaries were initially determined on the training data, with assessment of model performance on the testing data using a 2 test.
Survival Analysis and Model Performance
Following confirmation of valid group boundaries, all data were combined to determine model performance metrics. To account for variable follow-up duration between patients, survival analysis was performed using a Cox proportional hazards (CPH) model to give final model performance metrics. Survival analysis was performed on all patients. Additionally, analysis was performed on patients diagnosed younger than 18 years of age to determine if prediction was improved in patients with a presumed higher heritable component to their disease. All statistical analyses were performed in SPSS (IBM v27).
Inclusion of Disease Location Data
NOD2 variants are known to predispose to terminal ileal (TI) inflammation. To assess the independent role of NOD2 in the prediction of stricturing disease, we determined whether adding the presence or absence of TI inflammation as a variable to the risk stratification model improved or negated the predictive ability of NOD2 risk groups. Previously determined NOD2 risk groups were further stratified into the following groups: group 1, low-risk NOD2 group and no TI disease; group 2, low-risk NOD2 group and TI disease; group 3, high-risk NOD2 group and no TI disease; and group 4, high-risk NOD2 group and TI disease. Survival modeling was performed on these groups, including separate analysis for pediatric-onset patients.
Non-NOD2 Genetic Determinants of Stricturing Disease
We hypothesized that in patients who developed strictures in the absence of significant NOD2 variation, an alternative genetic driver may be identified to further stratify these individuals. In patients stratified to a low-risk group based on NOD2 as a biomarker, we performed a logistic regression with the GenePy scores (calculated for each gene as described previously) of ATG16L1 and 15 additional genes identified through literature review to impact stricturing disease risk (ie, CX3CR1, FUT2, IL12B, IL23R, JAK2, MAGI1, MMP3, TGFB1, SLC22A4, ICAM1, SELP, SELL, IL10, TNFSF15, and WWOX), as independent variables, and stricturing disease status as the dependant variable.
Patient and Public Involvement
Patients and families were involved in the design and conduct of this research. Patient priorities for research have determined priority analyses and dissemination.
Ethics
This study has University of Southampton category A ‘ERGO’ 2 ethics approval (30630) and research ethics committee approval from Southampton and South West Hampshire Research Ethics Committee (09/H0504/125).
Results
Six hundred forty-five patients with a confirmed diagnosis of Crohn’s disease were included. Of these, 373 were diagnosed younger than 18 years of age, and 272 were diagnosed as adults.
Within the cohort, we identified 112 distinct variants within NOD2. Of these variants, 15 had functional evidence impacting protein function or downstream signaling, identified through review of the literature (Supplementary Data 1). There were 11 NOD2 variants with functional evidence and a further 32 variants fulfilling in silico criteria for deleteriousness (CADD >15 and AF <0.05). Characteristics of these variants are summarized in Supplementary Data 1.
Variant location within the NOD2 gene were assessed and visualized (Figure 1). Pathogenic, likely pathogenic, and in silico deleterious variants were present throughout the gene, apart from a 171 amino acid region (positions 441-612, GENCODE transcript ENST00000300589) within the nucleotide binding domain (NOD) in which no deleterious variants were found. Variants were observed in both caspase recruitment domains and throughout the remaining NOD and leucine-rich repeat domains.
Monogenic NOD2-Related Disease
In adult-onset patients, we were unable to segregate variants due to lack of parental DNA; however, all potential compound heterozygote NOD2 pediatric-onset patients were previously confirmed to be biallelic.11 We treated all potential compound heterozygote variants in the cohort as presumed compound heterozygote.
ACMG Pathogenic or Likely Pathogenic Criteria for NOD2-Related Disease
To stratify patient risk of structuring, we considered NOD2 as an autosomal recessive cause of Crohn’s disease. We identified patients who fulfilled ACMG criteria for harboring causative variants (pathogenic or likely pathogenic). Across the entire cohort, 48 patients (7.4%) fulfilled ACMG criteria for NOD2-related disease, including 19 patients who were homozygote and 29 patients who were (presumed) compound heterozygote for a variant with functional evidence. Table 1.
Table 1.
Number of patients with harboring NOD2 variants fulfilling American College of Medical Genetics criteria for Likely Pathogenic or Pathogenic, and variants predicted to be deleterious but with no functional evidence. For functional evidence refer to Supplementary Data 1. Prevalence of stricturing disease within each group is reported.
| All Patients (N = 645) | Patients Diagnosed Younger than 18 Years Only (n = 373) | Patients Diagnosed 18 Years or Older Only (n = 272) | ||||
|---|---|---|---|---|---|---|
| NOD2-Related Diseasea (number with stricturing phenotype, %) | In silico NOD2-Related Diseaseb (number with stricturing phenotype, %) | NOD2-Related Diseasea (number with stricturing phenotype, %) | In silico NOD2-Related Diseaseb (number with stricturing phenotype, %) | NOD2-Related Diseasea (number with stricturing phenotype, %) | In silico NOD2-Related Diseaseb (number with stricturing phenotype, %) | |
| Homozygote | 19 (9 patients, 47.4%) | 0 (0 patients) | 13 (5 patients, 38.5%) | 0 (0 patients) | 6 (4 patients, 66.7%) | 0 (0 patients) |
| Presumed compound heterozygote | 29 (15 patients, 51.7%) | 15c (5 patients, 33.3%) | 17 (9 patients, 52.9%) | 10c (3 patients, 30%) | 12 (6 patients, 50%) | 5c (2 patients, 40%) |
| 9 d (5 patients, 55.6%) | 5 d (3 patients, 60%) | 4 d (2 patients, 50%) | ||||
| Total patients | 48 (24 patients, 50%) | 24 (10 patients, 41.7%) | 30 (14 patients, 46.7%) | 15 (6 patients, 40%) | 18 (10 patients, 55.6%) | 9 (4 patients, 44.4%) |
a Two or more variants that functionally impact NOD2 function, including reduced/absent protein function, impact downstream signaling, nonsense mediated decay or deletions, in line with American College of Medical Genetics guidelines.
b Either, one variant that functionally impacts NOD2 function including reduced/absent protein function, impact downstream signaling, nonsense-mediated decay or deletions, in line with American College of Medical Genetics guidelines AND one variant had a minor allele frequency (MAF) (gnomAD_AF) <0.05 and a CADD-PHRED 1.6 score of >15, OR 2 variants had a MAF (gnomAD_AF) <0.05 and a CADD-PHRED 1.6 score of >15.
c Patients with one variant that functionally impacts on NOD2 function, including reduced/absent protein function, impact downstream signaling, nonsense mediated decay or deletions, in line with American College of Medical Genetics guidelines AND one variant with a MAF (gnomAD_AF) <0.05 and a CADD-PHRED 1.6 score of >15.
d Patients with 2 variants of a MAF (gnomAD_AF) <0.05 and a CADD-PHRED 1.6 score of >15.
We stratified patients by age at diagnosis. Of those younger than 18 years of age at diagnosis, 30 of 373 patients (8%) had NOD2-related disease compared with 18 of 272 patients (6.6%) diagnosed when older than 18 years of age (P = .5).
In Silico NOD2-Related Disease
We characterized patients with deleterious variation within NOD2 but insufficient functional evidence to fulfil ACMG pathogenic or likely pathogenic criteria. No patients were homozygous for deleterious in silico NOD2 variants. We identified 24 patients (3.7%) with either 1 variant with published evidence for functional impact NOD2 function and 1 variant assessed to have in silico evidence of potential functional impact (an AF [gnomAD] <0.05 and a CADD PHRED score of >15) or 2 variants with an AF (gnomAD) <0.05 and a CADD PHRED score of >15 (Table 1).
In patients aged younger than 18 at diagnosis, 15 of 373 patients (4%) had in silico NOD2-related disease compared with 9 of 272 patients (3.3%) diagnosed 18 years of age and older (P = .6).
Stricturing Phenotype-Genotype Assessment
We assessed the relationship between NOD2-genotype and stricturing phenotype (Table 1). In patients fulfilling ACMG pathogenic or likely pathogenic criteria, 24 of 48 patients (50%) had strictures compared with 156 of 597 patients (26.1%) not fulfilling these criteria (P = .0004). When considering the in silico NOD2-genotype group, 10 of 24 patients (41.7%) had strictures.
Combining the 2 groups (NOD2-related disease and in silico NOD2-related disease) demonstrated 34 of 70 (48.6%) patients had developed stricturing disease compared with 147 of 575 (25.5%) patients not fulfilling either criterion for a deleterious NOD2-genotype (P = .00005).
NOD2 as a Genomic Biomarker for Stricturing Phenotype
We assessed the ability of NOD2 GenePy score to classify all patients by stricturing outcome using an AUROC analysis. For all patients (N = 645), NOD2 showed modest power to discriminate stricturing disease behavior (AUROC, 0.586; P = .001; Supplementary Figure 2A). Performance improved when considering only patients diagnosed younger than 18 years of age (n = 373; AUROC, 0.654; P = .000024; Supplementary Figure 2B).
To better utilize NOD2 as a genomic biomarker in a clinical setting, we stratified patients into high- and low-risk groups for stricturing disease using an easily automatable bioinformatic process.
Group Number and Cutoff Optimization
To determine optimal risk groups, patients were split into training (484 patients) and testing (161 patients) sets. The training and testing data sets were balanced according to the number of stricturing patients (the minority class). We employed an iterative Fisher exact test within the training set to determine the number of risk groups and GenePy score cutoff values.
This analysis identified 2 risk groups derived from the training data (Table 2). The absolute GenePy cutoff values were then applied to the testing set of patients, where ≥1.078 indicated high risk and <1.078 indicated low risk for stricturing disease. Within the testing set of patients, the high-risk group demonstrated a 56.7% stricturing rate compared with 21.4% stricturing risk in the low-risk group (P = .0001).
Table 2.
Training and testing data for identification of predictive risk groups for stricturing disease using summed deleteriousness across the whole gene (GenePy score). Performance of the cutoffs derived from training data is shown in the test data.
| Groupsa | NOD2 CADD >15 Variants (training set = 484) | NOD2 CADD >15 Variants (testing set = 161) | ||||
|---|---|---|---|---|---|---|
| Absolute Cut Off Value of GenePy Scorea | Number of Patients in Group (%) | Number of Stricturing Patients in Group | Absolute Cutoff Values | Number of Patients in Group (%) | Number of Stricturing Patients in Group | |
| Group 1 (high risk) | ≥1.078 | 59 (12.2%) | 27 (45.7%) | ≥1.078 | 30 (18.6%) | 17 (56.7%) |
| Group 2 (low risk) | <1.078 | 425 (87.8%) | 108 (25.4%) | <1.078 | 131 (81.4%) | 28 (21.4%) |
aGroups determined by Fisher exact test.
Survival Analysis
To assess model performance, all patients were combined for survival modeling. We employed a CPH model to account for variable follow-up duration.
Considering all patients, the risk groups demonstrated ability to stratify patients by stricturing risk, based only on NOD2 genomic data, Figure 2A. Patients in the high-risk group (n = 89), as determined by NOD2, had higher rates of stricturing at all timepoints from diagnosis, with 44 patients stricturing over this time (49.4%), β = 2.092, P = .000024. At maximal follow-up, over 80% of high-risk group patients had stricturing disease compared with less than 60% of low-risk group patients.
Figure 2.

A, Cox proportional hazard survival plot for all patients. Patients are divided into high risk of stricturing (group 1) and low risk of stricturing (group 2). High risk compared with low risk, hazard ratio of 2.092. B, Restriction of survival analysis to patients diagnosed younger than 18 years of age. High risk compared with low risk, hazard ratio of 3.164.
Paediatric-Onset Patients
We hypothesized that genetic determinants of disease would be more prominent in patients with younger age of onset. Survival modeling using only patients diagnosed < 18 years (n = 373) was performed. Analysis demonstrated improved performance, with 27 of 57 (47.4%) patients in the high-risk group having stricturing disease compared with only 53 of 315 (16.8%) of patients in the low-risk group, β = 3.164, P = .000001. Figure 2B. At maximal follow-up, nearly 80% of high-risk patients had strictures compared with an estimated 35% of low-risk patients.
Refinement of Prediction Using Disease Location Data
Disease location data, at the point of diagnosis, were available for 585 patients including 340 pediatric-onset individuals. Patients were split into those with TI disease and those without. As expected, presence of TI disease at diagnosis was associated with stricturing phenotype, odds ratio 2.5, P = .00018.
To determine the impact of NOD2-risk group combined with disease location, we performed a CPH survival model. Patients were stratified into combined NOD2 and disease location risk groups (group 1-4). Considering patients diagnosed as adults and children, the addition of NOD2-risk group, derived from whole exome sequencing data to disease location, resulted in a significant increase in predictive ability. When compared with group 1 (low-risk NOD2 and no TI disease), the hazard ratio (HR) increased from 1.66 (P = .028) for group 2 (low-risk NOD2 and TI disease) to 3.19 (P = .00001) for group 4 (TI disease plus NOD2 high-risk group; Figure 3A). Only a small number of patients were in group 3, which included the high risk-NOD2 group with no TI disease involvement (n = 7).
Figure 3.

A, Cox proportional hazard survival plot for all patients. Patients are divided into 4 groups. When comparing the lowest risk patients in group 1 to both group 2 and group 4, there are significant increases in the risk of developing stricturing disease. B, Restriction of survival analysis to patients diagnosed younger than 18 years of age. The highest risk of stricturing disease occurred in group 4, with a hazard ratio of 4.89 (P = .000023) compared with group 1.
We performed the same analysis for patients diagnosed younger than 18 years of age, given the previous data indicating a stronger predictive value of NOD2 in younger patients. There was further improvement in predictive ability. There was no significant difference between group 1 and 2 (HR, 1.67; P = .146). However, when comparing group 1 with group 4, the HR was 4.89 (P = .000023; Figure 3B). Again, only a very small number of patients were in group 3 (ie, high risk-NOD2 group) and had no TI disease involvement (n = 6).
Identification of Additional Genomic Factors Implicated in Stricturing Disease
We attempted to determine whether patients who did not harbor a high burden of NOD2 variants but still had stricturing disease had an alternative genetic driver of this disease behavior. Patients defined as high-risk of stricturing according to GenePy NOD2 biomarker stratification were excluded from further analysis, leaving 556 patients defined as low-risk for stricturing according to NOD2. Of these patients, 136 (24.5%) still developed stricturing disease.
All 556 patients were included in a logistic regression model, which did not reveal any significant relationships between the GenePy score of any gene previously implicated in development of stricturing phenotype by literature review (Supplementary Table 1). Despite this, ATG16L1 approached statistical significance for a positive association with stricturing phenotype (β = 3.434; P = .064).
Discussion
These data demonstrate the potential utility of NOD2 as a genomic biomarker for the prediction of stricturing phenotype in patients with Crohn’s disease. We were able to stratify patients into highly significant high- and low-risk groups for development of stricturing disease. This could be a clinically useful tool that complements clinician decision-making for individual patient management. When combined with disease location data, we are able to refine predictive ability whilst demonstrating the additional utility of the NOD2 biomarker. Although NOD2 is a well-established risk locus for Crohn’s disease, a patient’s distinct genetic variation in this gene is not currently utilized in the clinical setting. These data also add additional weight to the hypothesis that for some patients there is an autosomal recessive inheritance of NOD2 variants, leading to disease.10,11
Recent functional work has pointed to a mechanism by which impaired NOD2 function may lead directly to fibrotic disease.30 Our data provide a bridge between this elegant functional work and a clinically applicable tool that can be used to stratify patients at diagnosis by the risk of stricturing disease. Additionally, contemporary data have pointed towards the importance of genetic variation within the wider NOD-signaling pathway, with direct impact on transcription levels in patients with pediatric-onset inflammatory bowel disease.31 There is the possibility that in some patients, a stricturing disease phenotypes will be associated with rare genetic defects across the NOD-signaling pathway, and further refinement of predictive models may be possible with future integration. Data from genome wide association study have pointed towards stricturing disease in relation to NOD2 purely being a function of its predisposition to trigger terminal ileal disease.32 However, these analyses fail to account for any rare variation and are limited to association with phenotype derived from single nucleotide polymorphism data. Newer evidence from whole exome sequencing would appear to suggest that NOD2 leads to stricturing disease, regardless of disease location at diagnosis.10,11 Furthermore, NOD2 genomic data alone still predict stricturing disease occurrence at the point of diagnosis, regardless of any clinical features at diagnosis. It appears increasingly likely that the full role of NOD2 in the development of Crohn’s disease is not yet understood.
Construction of predictive models for complicated Crohn’s disease have proven challenging. Kugathasan and colleagues previously detailed a joint model for stricturing and penetrating complications, performing with borderline significance (specificity 63%, sensitivity 66%).7 Interestingly, an NOD2 genotype (analysis of the 3 common variants only—rs2066844, rs2066845, and rs2066847) was not a significant predictor in the competing-risk model, whereas CBir1 seropositivity and an extracellular matrix gene expression signature were positively associated with a B2 disease phenotype. Our data point to the importance of analysis of deleterious variation across the whole NOD2 gene, rather than limiting this to specific, more frequent variants. Additional predictive models for Crohn’s disease have taken alternative approaches, including T-cell specific transcription and microbiome signatures.5,33 Although yielding impressive results in a research setting, these data have not yet moved into routine practice. Furthermore, the prediction of long-term disease activity is heavily restricted by the lack of a longitudinal disease activity metric.
Long-range sequencing of NOD2 presents an opportunity to refine any predictive model, providing additional data on regulatory, promotor, and intronic regions. It is possible that some of the missing predictive power for stricturing disease will be accounted for within these NOD2 noncoding regions. Refinement of our predictive model was possible by limiting to patients diagnosed younger 18 years of age. Previous data have pointed to higher heritability in patients diagnosed as children.34 However, the long-term phenotype of Crohn’s disease does not appear to significantly differ between adults and children.35 This points to additional, yet unknown, factors in the development of stricturing disease, with NOD2 variation being the most common in early-onset disease.
Translating genomic data into clinical practice within IBD has huge potential. The importance of integration of genomic data with longitudinal and well-phenotyped data sets, such as our own, has recently been highlighted specifically in relation to preventing tissue remodeling and fibrosis.36 To date, the use of next generation sequencing has yielded significant advances in the diagnosis and management of patients with monogenic forms of IBD; however, these only account for a very small number of the total number of patients.37 Our data point to the importance of inclusion of clinical data to aid refinement of genomic prediction tools. Utilizing these technologies for wider patient benefit has lagged significantly behind. Limited pharmacogenomic testing is now starting to emerge, with the opportunity for routine screening of TPMT,38NUDT15,39 and HLA-DQA1*0540 to prevent thiopurine toxicity, myelosuppression, and formation of anti-tumour necrosis factor antibodies, respectively. Our model requires either targeted NOD2 sequencing or whole exome or whole genome sequencing, which would incur an additional cost. However, preventing complications, or treating them early, is likely to have a net saving over the lifetime of a patient. We envisage that our prediction model could enable routine monitoring through regular small bowel imaging, alongside the potential for preemptive monoclonal therapy and randomized control trials of established and new therapies, based on genomic risk stratification.
To better utilize genomic data in clinical practice, there is likely to be an increased need to employ powerful machine-learning algorithms to make sense of genomic and big clinical data.41 These tools have already been widely employed for autoimmune diseases, including IBD; however, the results are variable.42,43 Recent studies seem to indicate a shift in applications from diagnostics to prediction of outcomes. There is an accompanying increase in the number of studies being published, but the clinical translation of these data, including from complex algorithms such as neural networks, is currently limited.41
This study has several key strengths. The genomic data are high quality with stringent quality control processing. Longitudinal phenotyping data is extracted and processed in a standardized fashion from a single institution’s electronic health record. The integration of data follows a novel methodology, revealing new predictive power of genomic data. We acknowledge limitations in this work. There was a necessity for retrospective phenotyping of patients, although this was performed in a systematic way with structured clinician validation. Due to the strict definition of fibrotic stricturing disease, it is possible that some patients may be misassigned to a nonstricturing category if they were early in their disease course. It appears likely that with improved follow-up duration, the performance of the model in predicting tructuring disease would improve. In patients diagnosed as adults, we were unable to perform segregation analysis to determine phase of variants or analyze noncoding variants—both areas that would be bolstered by long-range sequencing. We accept that although patients in the high-risk NOD2 group are at up to a 3-fold increased risk of developing fibrostenotic stricturing disease, this group only accounts for 25% of all stricturing cases. We hypothesize that additional factors including genetic and environmental variables are contributing to the development of strictures in the remaining patients, which we are currently unable to integrate into the model to improve its performance. Future research must focus on identification of risk and protective factors that can be used to further refine these models, including extended sequencing of the NOD2 region to determine the impact of noncoding sequences. Whether NOD2 variants predispose to fibrostenotic stricturing or whether variants predispose to ileal inflammation, leading to strictures, has been raised as a potential confounder of utilizing NOD2 as a predictive tool.44 Data present here and previously published from our group indicate that NOD2 is significantly associated with stricturing disease, even when accounting for presence (or absence) of ileal inflammation.11 Utilization of NOD2 as a predictive biomarker has the additional advantage that it is not limited by aging or by technical difficulties at endoscopy to identify disease location. Combining genomic and clinical predictors appears to be a highly useful strategy for future modeling.
Conclusions
NOD2 is a powerful predictive tool for stratification of patients by their risk of stricturing disease. Further refinement of the model through addition of disease location improved performance. Future improvement may be possible with the addition of long-range phased sequencing data. The next steps are construction of predictive tools for additional complications and diseases behaviors. Translation of these methods into a clinical application and accessible tool is an important step towards personalized medicine in IBD.
Supplementary Material
{kind=link}
Contributor Information
James J Ashton, Department of Human Genetics and Genomic Medicine, University of Southampton, Southampton, UK; Department of Paediatric Gastroenterology, Southampton Children’s Hospital, Southampton, UK.
Guo Cheng, Department of Human Genetics and Genomic Medicine, University of Southampton, Southampton, UK; NIHR Southampton Biomedical Research Centre, University Hospital Southampton, Southampton, UK.
Imogen S Stafford, Department of Human Genetics and Genomic Medicine, University of Southampton, Southampton, UK; Institute for Life Sciences, University of Southampton, Southampton, UK.
Melina Kellermann, Department of Human Genetics and Genomic Medicine, University of Southampton, Southampton, UK.
Eleanor G Seaby, Department of Human Genetics and Genomic Medicine, University of Southampton, Southampton, UK.
J R Fraser Cummings,, Department of Gastroenterology, University Hospital Southampton, Southampton, UK.
Tracy A F Coelho, Department of Paediatric Gastroenterology, Southampton Children’s Hospital, Southampton, UK.
Akshay Batra, Department of Paediatric Gastroenterology, Southampton Children’s Hospital, Southampton, UK.
Nadeem A Afzal, Department of Paediatric Gastroenterology, Southampton Children’s Hospital, Southampton, UK.
R Mark Beattie, Department of Paediatric Gastroenterology, Southampton Children’s Hospital, Southampton, UK.
Sarah Ennis, Department of Human Genetics and Genomic Medicine, University of Southampton, Southampton, UK.
Author Contributions
J.J.A., R.M.B., and S.E. conceived the study. Analyses were performed by J.J.A., I.S.S., and G.C. under the guidance of S.E. J.J.A. wrote the manuscript with help from all authors. All authors approved the final manuscript prior to submission.
Funding
J.J.A. was funded during this work by an Action Medical Research training fellowship. J.J.A. is currently funded by an NIHR clinical lectureship and an ESPR post-doctoral grant. This study is supported by the National Institute for Health Research (NIHR) Southampton Biomedical Centre. The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.
Conflicts of Interest
F.C has served as consultant, advisory board member, or speaker for AbbVie, Amgen, Celltrion, Falk, Ferring, Gilead, Janssen, MSD, Napp Pharmaceuticals, Pfizer, Pharmacosmos, Sandoz, Biogen, Samsung, Tillotts and Takeda. He has received research funding from Biogen, Amgen, Hospira/Pfizer, Celltrion, Takeda, Janssen, GSK and AstraZeneca. No other authors declare any conflicts of interest.
Data Availability
Whole exome sequencing (WES) data will be available through collaborative agreement. Due to consent signed by participants, WES data cannot be deposited within a public repository.
References
- 1. Ashton JJ, Mossotto E, Ennis S, Beattie RM.. Personalising medicine in inflammatory bowel disease—current and future perspectives. Translational Pediatrics 2019;8:56. Accessed June 27, 2019. http://www.ncbi.nlm.nih.gov/pubmed/30881899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Graham DB, Xavier RJ.. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature 2020;578:527–539. Accessed March 1, 2020. http://www.nature.com/articles/s41586-020-2025-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Verstockt B, Smith KG, Lee JC.. Genome-wide association studies in Crohn’s disease: past, present and future. Clin Transl Immunology 2018;7:e1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Biasci D, Lee JC, Noor NM, et al. . A blood-based prognostic biomarker in IBD. Gut 2019;68:1386–1395. doi: 10.1136/gutjnl-2019-318343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Douglas GM, Hansen R, Jones CMA, et al. . Multi-omics differentially classify disease state and treatment outcome in pediatric Crohn’s disease. Microbiome 2018;6:13. Accessed February 6, 2018. https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-018-0398-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Verstockt B, Verstockt S, Dehairs J, et al. . Low TREM1 expression in whole blood predicts anti-TNF response in inflammatory bowel disease. EBioMedicine 2019;40:733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kugathasan S, Denson LA, Walters TD, et al. . Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: a multicentre inception cohort study. The Lancet 2017;389:1710–1718. Accessed August 9, 2018. http://www.ncbi.nlm.nih.gov/pubmed/28259484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ogura Y, Bonen DK, Inohara N, et al. . A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001;411:603–606. Accessed June 11, 2019. http://www.ncbi.nlm.nih.gov/pubmed/11385577 [DOI] [PubMed] [Google Scholar]
- 9. Caruso R, Warner N, Inohara N, Núñez G.. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity 2014;41:898–908. Accessed January 29, 2019. http://www.ncbi.nlm.nih.gov/pubmed/25526305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Horowitz JE, Warner N, Staples J, et al. . Mutation spectrum of NOD2 reveals recessive inheritance as a main driver of Early Onset Crohn’s Disease. Sci Rep. 2021;11. Accessed November 16, 2021. https://pubmed.ncbi.nlm.nih.gov/33692434/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ashton JJ, Mossotto E, Stafford IS, et al. . Genetic sequencing of paediatric patients identifies mutations in monogenic inflammatory bowel disease genes that translate to distinct clinical phenotypes. Clinical and Translational Gastroenterology 2020;11:e00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Uhlig HH, Charbit-Henrion F, Kotlarz D, et al. . Clinical Genomics for the Diagnosis of Monogenic Forms of Inflammatory Bowel Disease: A Position Paper From the Paediatric IBD Porto Group of European Society of Paediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2021;72:456–473. Accessed November 16, 2021. https://journals.lww.com/jpgn/Fulltext/2021/03000/Clinical_Genomics_for_the_Diagnosis_of_Monogenic.25.aspx [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abreu MT, Taylor KD, Lin Y-C, et al. . Mutations in NOD2 are associated with fibrostenosing disease in patients with Crohn’s disease. Gastroenterology 2002;123:679–688. [DOI] [PubMed] [Google Scholar]
- 14. Mossotto E, Ashton JJ, O’Gorman L, et al. . GenePy: a score for estimating gene pathogenicity in individuals using next-generation sequencing data. BMC Bioinf. 2019;20:254. Accessed May 24, 2019. https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2877-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Verstockt B, Cleynen I.. Genetic influences on the development of fibrosis in Crohn’s DISEASE. Frontiers in Medicine 2016;3:24. Accessed April 15, 2019. http://journal.frontiersin.org/Article/10.3389/fmed.2016.00024/abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Levine A, Koletzko S, Turner D, et al. . ESPGHAN revised porto criteria for the diagnosis of inflammatory bowel disease in children and adolescents. J Pediatr Gastroenterol Nutr. 2014;58:795–806. [DOI] [PubMed] [Google Scholar]
- 17. Miller SA, Dykes DD, Polesky HF.. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van der Auwera GA, Carneiro MO, Hartl C, et al. . From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Current Protocols in Bioinformatics 2013;43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Genomics 2013;1303:3997. [Google Scholar]
- 20. Jun G, Flickinger M, Hetrick KN, et al. . Detecting and Estimating Contamination of Human DNA Samples in Sequencing and Array-Based Genotype Data. The American Journal of Human Genetics 2012;91:839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pengelly RJ, Gibson J, Andreoletti G, et al. . A SNP profiling panel for sample tracking in whole-exome sequencing studies. Genome Med. 2013;5:1–7. Accessed January 27, 2022. https://genomemedicine.biomedcentral.com/articles/10.1186/gm492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karczewski KJ, Francioli LC, Tiao G, et al. . The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:7809. 2020;581:434–443. Accessed January 7, 2022. https://www.nature.com/articles/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M.. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894. Accessed July 26, 2021. https://academic.oup.com/nar/article/47/D1/D886/5146191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stenson PD, Mort M, Ball E, et al. . The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136:665–677. Accessed January 22, 2019. http://www.ncbi.nlm.nih.gov/pubmed/28349240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lek M, Karczewski KJ, Minikel E, et al. . Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gao J, Aksoy BA, Dogrusoz U, et al. . Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6. Accessed January 18, 2022. https://pubmed.ncbi.nlm.nih.gov/23550210/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carson AR, Smith EN, Matsui H, et al. . Effective filtering strategies to improve data quality from population-based whole exome sequencing studies. BMC Bioinf. 2014;15:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Richards S, Aziz N, Bale S, et al. . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. http://www.ncbi.nlm.nih.gov/pubmed/25741868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Budczies J, Klauschen F, Sinn B, et al. . Cutoff finder: a comprehensive and straightforward web application enabling rapid biomarker cutoff optimization. PLoS One. 2012;7:e51862. Accessed November 17, 2021. https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0051862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nayar S, Morrison JK, Giri M, et al. . A myeloid–stromal niche and gp130 rescue in NOD2-driven Crohn’s disease. Nature 2021;1:9. Accessed April 8, 2021. http://www.nature.com/articles/s41586-021-03484-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ashton JJ, Boukas K, Stafford IS, et al. . Deleterious genetic variation across the NOD signaling pathway is associated with reduced NFKB signaling transcription and upregulation of alternative inflammatory transcripts in pediatric inflammatory bowel disease. Inflamm Bowel Dis. 2022;1:11. 10.1093/ibd/izab318/6492639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cleynen I, Boucher G, Jostins L, et al. . Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. The Lancet 2016;387:156–167. Accessed September 28, 2018. https://www.sciencedirect.com/science/article/pii/S0140673615004651#fig2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee JC, Lyons PA, McKinney EF, et al. . Gene expression profiling of CD8+ T cells predicts prognosis in patients with Crohn disease and ulcerative colitis. J Clin Investig. 2011;121:4170–4179. Accessed August 9, 2018. http://www.ncbi.nlm.nih.gov/pubmed/21946256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Khor B, Gardet A, Xavier RJ, et al. . Genetics and pathogenesis of inflammatory bowel disease. Nature 2011;474:307–317. http://www.ncbi.nlm.nih.gov/pubmed/21677747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jakobsen C, Bartek J, Wewer V, et al. . Differences in phenotype and disease course in adult and pediatric inflammatory bowel disease--a population-based study. Aliment Pharmacol Ther. 2011;34:1217–1224. Accessed July 11, 2019. http://www.ncbi.nlm.nih.gov/pubmed/21981762 [DOI] [PubMed] [Google Scholar]
- 36. Lamb CA, Saifuddin A, Powell N, et al. . The future of precision medicine to predict outcomes and control tissue remodeling in inflammatory bowel disease. Gastroenterology 2022;0. Accessed January 19, 2022. http://www.gastrojournal.org/article/S0016508521040695/fulltext [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bolton C, Smillie CS, Pandey S, et al. . An Integrated Taxonomy for Monogenic Inflammatory Bowel Disease. Gastroenterology 2021;162(3):859–876. doi: 10.1053/j.gastro.2021.11.014. Epub 2021 Nov 13. [DOI] [PubMed] [Google Scholar]
- 38. Coelho T, Andreoletti G, Ashton JJ, et al. . Genes implicated in thiopurine-induced toxicity: Comparing TPMT enzyme activity with clinical phenotype and exome data in a pediatric IBD cohort. Sci Rep. 2016;6:34658. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5050412/pdf/srep34658.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Walker GJ, Harrison JW, Heap GA, et al. . Association of genetic variants in NUDT15 with thiopurine-induced myelosuppression in patients with inflammatory bowel disease. JAMA 2019;321:753–761. Accessed January 18, 2022. https://pubmed.ncbi.nlm.nih.gov/30806694/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sazonovs A, Kennedy NA, Moutsianas L, et al. . HLA-DQA1*05 carriage associated with development of anti-drug antibodies to infliximab and adalimumab in patients with Crohn’s disease. Gastroenterology 2020;158:189–199. Accessed January 18, 2022. https://pubmed.ncbi.nlm.nih.gov/31600487/ [DOI] [PubMed] [Google Scholar]
- 41. Brooks-Warburton J, Ashton J, Dhar A, et al. . Artificial intelligence and inflammatory bowel disease: practicalities and future prospects. Frontline Gastroenterology. 2021;0. flgastro-2021-102003. Accessed December 14, 2021. https://fg.bmj.com/content/early/2021/12/09/flgastro-2021-102003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stafford IS. A systematic review of the applications of artificial intelligence and machine learning in autoimmune diseases. NPJ Digital Medicine 2020;3:30. doi: 10.1038/s41746-020-0229-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stafford IS, Gosink MM, Mossotto E, et al. . A systematic review of artificial intelligence and machine learning applications to inflammatory bowel disease, with practical guidelines for interpretation. Inflamm Bowel Dis. 2022;izac115. doi: 10.1093/ibd/izac115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yoo JH, Holubar S, Rieder F.. Fibrostenotic strictures in Crohn’s disease. Intestinal Research 2020;18:379. Accessed January 19, 2022. http://pmc/articles/PMC7609387/ [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole exome sequencing (WES) data will be available through collaborative agreement. Due to consent signed by participants, WES data cannot be deposited within a public repository.