

SUMMARY
The choroid plexus (ChP) is the blood-cerebrospinal fluid (CSF) barrier and the primary source of CSF. Acquired hydrocephalus, caused by brain infection or hemorrhage, lacks drug treatments due to obscure pathobiology. Our integrated, multi-omic interrogation of post-infectious (PIH) and post-hemorrhagic hydrocephalus (PHH) models revealed lipopolysaccharide and blood-breakdown products trigger highly similar TLR4-dependent immune responses at the ChP-CSF interface. The resulting CSF “cytokine storm”, elicited from peripherally-derived and border-associated ChP macrophages, causes increased CSF production from ChP epithelial cells via phospho-activation of the TNF receptor-associated kinase SPAK, which serves as a regulatory scaffold of a multi-ion transporter protein complex. Genetic or pharmacological immunomodulation prevents PIH and PHH by antagonizing SPAK-dependent CSF hypersecretion. These results reveal the ChP as a dynamic, cellularly heterogeneous tissue with highly regulated immune-secretory capacity, expand our understanding of ChP immune-epithelial cell crosstalk, and reframe PIH and PHH as related neuroimmune disorders vulnerable to small molecule pharmacotherapy.
Graphical Abstract
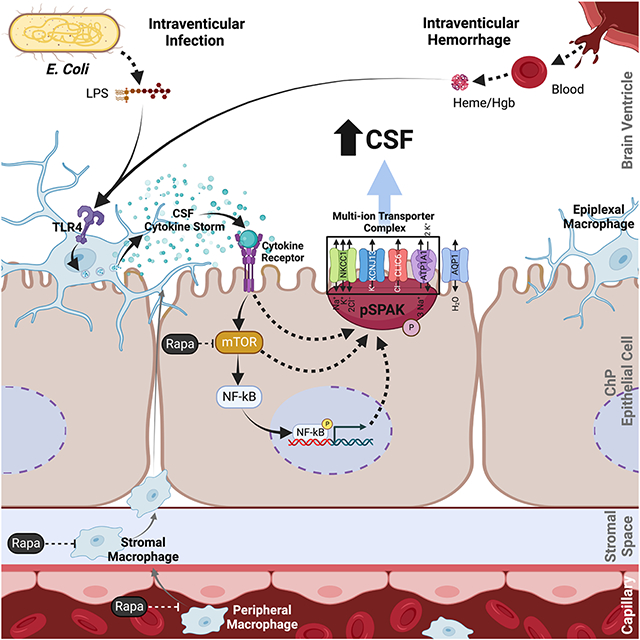
IN BRIEF
Infectious and hemorrhagic hydrocephalus converge on highly similar immune and secretory responses at the choroid plexus that drive pathological cerebrospinal fluid secretion, thus opening up immunomodulation as a potential non-surgical intervention.
INTRODUCTION
The choroid plexus (ChP) projects into each of the four cerebrospinal fluid (CSF)-filled ventricles and comprises an epithelial cell sheet populated by immune and mesenchymal cell types1,2. The ChP is the body’s secretory epithelium par excellence, producing a half-liter of CSF per day via the concerted action of its multiple ion and water transport molecules3. The ChP is also the blood-CSF barrier, gating circulating immune cell entry into the CSF in the setting of infection or tissue damage4-8. The ChP’s location at the nexus of the CSF, brain parenchyma, and systemic circulation allows it to sense perturbations and transduce danger signals into homeostatic responses. The cellular and molecular mechanisms that coordinate the immune and secretory functions of the ChP are poorly understood.
Hydrocephalus is characterized by the expansion of the cerebral ventricles (ventriculomegaly). Post-infectious hydrocephalus (PIH) and post-hemorrhagic hydrocephalus (PHH), the most common forms of hydrocephalus, are treated by neurosurgical CSF diversion with or without ChP cauterization to decrease CSF production9-11. These life-saving operations have significant long-term morbidity and failure rates and are unavailable in impoverished areas10,12,13. ChP cauterization may also disrupt normal ChP functions important for brain development and immune function14. Drug treatments for hydrocephalus remain unavailable14. An improved understanding of ChP biology could identify therapeutic targets for hydrocephalus and other CSF disorders and neuroimmune diseases4,8,14-17.
PIH and PHH are commonly attributed to intraventricular CSF accumulation secondary to decreased CSF reabsorption due to obstruction of intraventricular CSF flow and/or blockage of the extraventricular arachnoid granulations18. In contrast, the role of ChP’s immune-secretory functions to the pathogenesis of hydrocephalus remains under-investigated. Interestingly, human PIH and PHH exhibit similar CSF profiles of immune cells and cytokines19,20, and CSF neutrophilic pleocytosis can predict the development of PHH21. These observations suggest an important yet uncharacterized role of ChP and CSF space inflammation in the pathogenesis of acquired hydrocephalus.
Systemic epithelia respond to pro-inflammatory stimuli by increasing rates of fluid transport22 to clear organisms or tissue debris23-25. However, inappropriately triggered or maladaptively sustained epithelial inflammation can dysregulate ion transport homeostasis. Examples include chemical, autoimmune, and infectious forms of pleuritis, colitis, and pancreatitis25-28. Intraventricular hemorrhage (IVH) is known to cause inflammation-induced ChP CSF hypersecretion in PHH models 29-33. Nonetheless, the mechanism(s) by which the ChP contributes to PHH and PIH remain obscure, hindering the development of non-surgical treatment strategies.
Lipopolysaccharide (LPS)-expressing bacteria commonly cause PIH34-36. LPS is the canonical pathogen-associated molecular pattern (PAMP) for toll-like receptor-4 (TLR4)37,38. PHH-derived hemoglobin and possibly other blood products are TLR4-stimulating damage-associated molecular patterns (DAMPs)39,40. We hypothesized PIH and PHH have convergent pathophysiology that dysregulates ChP immune-secretory function. To test this, we created rat models of PIH and PHH and conducted a multi-omics investigation of these animals to dissect the physiologic, cellular, molecular pathology of PIH and PHH. Our results suggest that PIH and PHH are related neuroinflammatory disorders amenable to systemic immunomodulation.
RESULTS
E. coli post-infectious hydrocephalus models exhibit ChP-mediated CSF hypersecretion
Escherichia coli (E. coli) CSF infection is a common cause of post-infectious hydrocephalus (PIH) in resource-limited countries34-36. PIH is characterized by CSF space inflammation and the acute development of ventriculomegaly10. To investigate mechanisms of CSF infection on ChP function in PIH, we administered infectious material into the brain by intracerebroventricular (ICV) delivery. Wild-type E. coli (E. coli+LPS) or E. coli genetically-engineered to lack LPS in the outer membrane (E. coli−LPS, see Methods), were infused in sterile artificial CSF (aCSF) into the lateral ventricles of 8-week-old Wistar rats for 72h via stereotactically-placed infusion pumps (Fig. 1A). Control animals received ICV infusions of sterile aCSF. We found the lateral ventricular volume of animals 72h after infusion of E. coli+LPS was increased ~3-fold relative to control rats (Fig. 1, B and C). The ventriculomegaly in animals subjected to 72h ICV infusion of LPS alone (10 ng/μL in aCSF) resembled E. coli+LPS-treated animals (Fig. 1, B and C) and persisted when evaluated two weeks after treatment (fig. S1A). These findings suggest ICV-delivered LPS can model the acute development and persistence of ventriculomegaly characteristic of human PIH.
Fig. 1. Models of E. coli post-infectious hydrocephalus exhibit ChP-mediated CSF hypersecretion.

(A) Illustration of two models of post-infectious hydrocephalus (PIH). Insertion of an infusion pump in the lateral ventricle (LV) for E. coli+LPS, E. coli−LPS, LPS, or artificial CSF (aCSF, Ctl) administration over 72h. Left, illustration of the subcutaneous pump/catheter and intraventricular catheter placement. Right, schematic demonstrating pump/catheter placement and resulting ventricular changes. CC, corpus callosum, AC, anterior commissure. (B) Representative immunohistochemical images showing lateral ventricles 72h after infusion of control, E. coli+LPS, E. coli−LPS, or LPS (DAPI, blue). (C) Quantification of lateral ventricular size (% brain volume) after 72h infusion of aCSF, E. coli+LPS, E. coli−LPS, or LPS. Volume was calculated from sequential slices through the entire lateral ventricular system (n=5-7 animals per condition; see Methods). (D) Ventricular system infusion of Evans blue dye (injected into the LV of Ctl and LPS-treated animals), demonstrating flow through cerebral aqueduct (CA) and fourth ventricle (4V). (E) Body weight-normalized CSF secretion rates (μL/min/kg) in control (aCSF), E. coli+LPS (24h, 48h, or 72h), E. coli−LPS, LPS (72 h), or LPS + bumetanide (72 h)-treated animals (n=3-6 animals per condition). (F) Representative immunohistochemical images of LVs in Ctl vs LPS-treated Tlr4+/+ and Tlr4−/− rats (DAPI, blue). (G) Quantitation of LV size (% brain volume) in Ctl and LPS-treated Tlr4+/+ and Tlr4−/− rats (n=5-6 animals per condition). (H) Body weight-normalized CSF secretion rates (μL/min/kg) of Tlr4+/+ Ctl, Tlr4+/+ LPS-treated, Tlr4−/− Ctl, and Tlr4−/− LPS-treated rats (n=4-6 animals per condition). 2.5x mag, scale bars 0.25m. Error bars, mean ± sem; each symbol represents one animal. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant, by one-way ANOVA.
The pathways that regulate CSF homeostasis are poorly understood. However, acute ventriculomegaly in PIH could arise from impaired CSF efflux (e.g., due to obstruction of the cerebral aqueduct causing reduced bulk flow or reduced CSF egress via glymphatic and meningeal lymphatic pathways) and/or augmented CSF volume flux via an increase in ChP-dependent CSF production41-43. To examine CSF outflow in our PIH model, we injected the CSF tracer Evans Blue into the lateral ventricles of PIH rats after 72h of LPS infusion and found that dye distribution was unimpeded through the cerebral aqueduct and 4th ventricle, suggestive of aqueductal patency and preservation of bulk flow CSF (Fig. 1D). In vivo magnetic resonance imaging (MRI) of control and LPS-treated animals confirmed increased lateral ventricle volume in LPS-treated animals with normal intracranial volume (fig. S1, B-F), as well as cerebral aqueduct patency (fig. S1, G-H).
To examine glymphatic function in our PIH model, we performed in vivo dynamic contrast-enhanced, T1-weighted MRI after CSF infusion of gadoteric acid (see Methods)44-46. The MRI data were analyzed using computational fluid dynamics analysis based on regularized optimal mass theory which derives metrics describing the transport of the Gd-tagged contrast tracer in the brain47. Quantification of glymphatic v-flux (mm3) and glymphatic mean tissue speed (mm/s) data revealed no significant differences in brain-wide glymphatic transport between LPS-treated and control animals (fig. S1, I-K). Notably, this nomenclature refers to ‘glymphatic’ transport because modeling included both diffusion and advection drivers for solute transport47. However, other perivascular transport models including the ‘mixing model’ and ‘intra-mural periarterial drainage’ (IPAD) have been proposed and will be considered in future studies48-50. While these results suggest PIH is associated with the development of acute, communicating ventriculomegaly, as is seen in many human patients51,52, they do not exclude the possibility of alterations in CSF dynamics along other egress pathways48.
We next directly measured the effect of LPS on the rate of CSF secretion using a validated method in live animals (Fig. 1E)30,53,54. In this method, mineral oil blocks CSF exit from the third ventricle at the level of the Sylvian aqueduct, preventing contributions to measured CSF secretion from CSF reabsorption pathways distal to this block (fig. S1L). Therefore, this technique measures bona fide lateral ventricle CSF production by the ChP30,53,54. LPS-treated PIH rats exhibited a >2.5-fold increase in CSF secretion relative to controls (Fig. 1E). A similar increase in CSF secretion was observed in E. coli+LPS hydrocephalic rats but not E. coli−LPS control rats (Fig. 1E). ICV infusion of aCSF in naive rats at a rate approximating the difference in CSF secretion rates between that of control rats and LPS-induced or E. coli+LPS hydrocephalic rats recapitulated the ventriculomegaly observed in both conditions. ICV-delivered bumetanide, an inhibitor of ChP-mediated CSF secretion30,55,56, reduced LPS-induced CSF secretion by >70% (Fig. 1E). LPS-induced CSF hypersecretion and ventriculomegaly were attenuated in Tlr4 knockout rats (Tlr4−/−)57 (Fig. 1, F-H). These results suggest acute ventriculomegaly in PIH models results from an LPS-triggered, TLR4-dependent increase in bumetanide-sensitive CSF secretion from the ChP.
Post-infectious hydrocephalus models are associated with robust ChP-CSF interface inflammation
To gain molecular insight into the observed ChP-mediated acute CSF hypersecretory response in our models of PIH, we generated transcriptomic and proteomic datasets of micro-dissected lateral ventricle ChP from LPS-induced hydrocephalic and control rats using bulk RNA-sequencing and quantitative mass spectroscopy (Fig. 2A). Differential expression analyses of RNA transcripts (Fig. 2B, genes) and polypeptides (Fig. 2C, proteins) revealed significant differences between LPS-treated and control ChP. Wiki pathway, Gene Ontology (GO), and Mouse Gene Atlas analyses of the top 100 differentially-expressed genes and proteins in PIH versus control animals revealed enrichment of terms related to innate immunity and inflammation, such as “positive regulation of myeloid leukocyte mediated immunity” and “microglia pathogen phagocytosis pathway” (Fig. 2, D and E, and Table S1).
Figure 2. Post-infectious hydrocephalus is associated with robust ChP-CSF interface inflammation.

(A) Schematic of integrated multi-omic analysis. (B-C) Heatmaps of the most highly differentially expressed ChP genes (B) and proteins (C) from LPS-treated and control rats (n=3-5 animals per condition). (D) Gene Ontology (GO) and (E) pathway analysis of differentially expressed genes (DEGs). (F) UMAP clustering of ChP immune cells and (G, left) gene expression module heatmap for individual cell types using a scRNAseq brain macrophage atlas58. (G, upper right) Hypergeometric enrichment analysis of LPS-induced DEGs/DEPs in gene co-expression modules (G, lower right); Module 11 gene enrichment demonstrating dendritic cell (DC) and monocyte expression. (H) Module 11 GO biological process analysis (see also Methods, Table S1*). (I) CSF cytokine/chemokine expression (pg/mL) in Ctl and LPS-treated Tlr4+/+ and Tlr4−/− rats (n=3 animals per condition). (J) Representative IHC of Ctl and LPS-treated Tlr4+/+ and Tlr4−/− rat ChP. Iba1+ (green)/CD68+ (red) cells located apically (epiplexus macrophages, white arrowheads) and within stroma/capillaries (white arrows); activated macrophages (pink arrowheads). Apical membrane tNkcc1 (white); nuclei (DAPI, blue); scale bars, 25μm. (K) Representative merged (left) and magnified merged and single-channel IHC (right) of Ki-67 (green), ED1 (blue), Iba1 (red), and DAPI (white) in Ctl and LPS-treated Tlr4+/+ and Tlr4−/− rat ChP; senescent (arrows) and activated (arrowheads) Iba1+ cells. Scale bars, 50μm; insets at right, 2x enlarged. (L-N) Quantitation of (K) showing DAPI-normalized % Iba1+ (L), ED1+ (M), and Ki67+ cells (N) (n=5-6 animals per condition). (O) Scatterplots and (P) quantitation of FACS-isolated CD45+ (left) and CD3+ ChP cells (right) (n=4-6 animals per condition). Error bars, mean ± sem; each symbol represents one animal. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant, by one-way ANOVA.
We next integrated our bulk multi-omics ChP data with published single-cell and single-nucleus RNA-sequencing (sc- and snRNAseq, respectively) of the ChP and brain macrophages2,58. We constructed 21 modules of co-expressed genes from a published scRNAseq dataset of rodent ChP58 (Fig. 2F) and found that PIH-associated transcripts converged within a single module associated with monocytes that included dendritic cells (cDC1s, cDC2s, and migDCs) (Module 11, Fig. 2G)59. The most upregulated transcripts and proteins in PIH converged in another immune cell-specific co-expression module (Module 17, Fig. 2G)59. Genes in Modules 11 and 17 were both significantly enriched in pathways related to the recruitment of circulating myeloid/lymphoid cells and chemokine/interferon receptor signaling, as well as ChP secretory activity including chloride ion binding and ligand-gated ion channel activity (Fig. 2H, Table S1).
Multiple cytokines responsible for inflammatory cell signaling and modulation, and multiple chemokines responsible for the recruitment of circulating myeloid and lymphoid cells, were significantly increased in the CSF of LPS-treated animals compared to controls (Fig. 2I and fig. S1M). Among these, the most highly up-regulated (>30-fold increase, p=0.0017) was C–C motif chemokine ligand 2 (Ccl2). Levels of Ccl5 and C-X-C motif ligand 5 (Cxcl5), tumor necrosis factor-alpha (Tnfα), Il-1ß, Il-6, and other interferons and interleukins were also significantly increased. These findings indicate that CSF of PIH animals exhibits a pro-inflammatory state absent from normocephalic (non-hydrocephalic) control animals and similarly treated Tlr4−/− rats (Fig. 2I).
LPS-treated animals demonstrated accumulation and proliferation of Iba1+ macrophages at the apical, CSF-facing ChP surface and ChP stromal compartment between the basolateral membrane and endothelium (Fig. 2, J-N). Iba1+ cells likely include immune cells recruited from the periphery4,58 and CNS border-associated macrophages (BAMs), consisting of epiplexal (Kolmer) and stromal subtypes. In contrast to their quiescent appearance in controls58, BAMs in PIH rats exhibited amoeboid-like shape, increased circularity (Fig. 2, J and K)60, and increased CD68/ED1-positivity (Fig. 2, K and M), all indicative of increased phagocytic activity61. ChP-associated Iba1+ and CD68/ED1+ cells were reduced in similarly treated Tlr4−/− rats (Fig. 2, K-N). In comparison to E. coli+LPS-treated animals, the ChP of E. coli−LPS rats exhibited a milder inflammatory reaction (fig. S2, A-C). Like human patients with PIH62-65, E. coli+LPS-treated animals, and to a lesser extent E. coli−LPS animals, also exhibited immune cell activation in the meninges lining the CSF spaces and cortex (fig. S2).
Fluorescence-activated cell sorting (FACS) of micro-dissected, dissociated lateral ventricle ChP labeled with an antibody against CD45, a glycoprotein that marks immune cells of peripheral origin66,67, revealed higher CD45+ cell numbers in the ChP of PIH animals than of controls (Fig. 2, O and P, left panels). CD3+ T-cell number was also increased (Fig. 2, O and P, right panels). LPS-mediated increases in ChP-associated CD45+ peripheral immune cells and CD3+ T cells were attenuated in Tlr4−/− animals (Fig. 2, O and P). These data suggest PIH is associated with TLR4-dependent accumulation and activation of peripherally-derived immune cells and resident BAMs at the ChP that elicit a robust innate immune response characterized by a “CSF cytokine storm.” This is consistent with the ChP-CSF-brain interface inflammation in patients with PIH and other forms of acquired hydrocephalus65,68-70.
SPAK kinase links pro-inflammatory CSF cytokine signaling to ChP-mediated CSF secretion
Analysis of ChP snRNAseq results2 (fig. S3, A-E) revealed high baseline Tlr4 expression in ChP-associated macrophages and myeloid lineage immune cells, but scant expression in ChP epithelial cells (fig. S3B). Expression profiles of immune-related genes in adult ChP epithelial cells further revealed 3 unique subclusters (Table S2), all with low-level Tlr4 expression. In contrast, even under basal conditions, ChP epithelial cells exhibited significant levels of expression for multiple receptors of pro-inflammatory cytokines upregulated in CSF of PIH animals (Fig. 2I) and humans20, including TNFα, IL1ß, IL6, CCL2, and IFNγ. We hypothesized CSF cytokines elaborated by activated immune cells at the ChP stimulate their receptors on ChP epithelial cells to promote CSF secretion. Consistent with this hypothesis, quantitative real-time PCR of micro-dissected ChP epithelial cells showed higher expression of Ccr2, Relt, and Il1r1 in LPS-treated animals than in controls (fig. S3F). The Tnfα receptor Relt was also significantly up-regulated at the apical membrane in PIH rats, but not in similarly-treated Tlr4−/− rats (fig. S3, G and H).
The bumetanide-sensitivity of the acute CSF hypersecretory response in PIH implicates the NKCC1 cotransporter in disease pathogenesis. NKCC1 phosphorylation at Thr203, Thr207, and Thr212 by SPAK kinase is required for NKCC1 activity71. SPAK phosphorylation at Ser373 by WNK1 kinase is required for SPAK kinase activity72. SPAK transduces extracellular stress signals, including NF-κB-dependent cytokines, into downstream signaling events73. RELT binds SPAK via its C-terminal domain74 and TNFα and IL-1β stimulate SPAK kinase activity in a TLR4- and NF-κB-dependent manner to increase intestinal epithelial transport25,73. We hypothesized SPAK transduces TLR4-dependent pro-inflammatory CSF signals into a ChP epithelial CSF hypersecretory response by increasing NKCC1 phosphorylation.
Consistent with SPAK functioning downstream of TLR4-mediated immune cell activation in a non-cell-autonomous manner, expression of Spak and Nkcc1 in ChP epithelial cells far exceeded that in other ChP cell types (fig. S3, I and J). The phosphorylated, activated species of SPAK (pSpak) and NKCC1 (pNkcc1)71,72,75,76 were up-regulated at the ChP apical membrane in PIH rats but not in similarly treated Tlr4−/− rats (fig. S3, K-M). While Spak knockout had no effect on LPS-induced increases in ChP-associated immune cell infiltration and activation or Relt expression (fig. S4, A-B)77, it prevented the up-regulation of pSpak-pNkcc1 in ChP epithelial cells (fig. S3, K-M) and attenuated LPS-induced increases in CSF secretion and ventriculomegaly (fig. S3, N and O). These data suggest SPAK mediates molecular crosstalk between TLR4-dependent ChP immune cell activation and NKCC1-dependent CSF secretion by ChP epithelial cells via cytokine receptor engagement.
SPAK is a regulatory scaffold of a multi-ion transporter complex at the ChP apical membrane
CSF secretion requires the coordinated function of multiple ion and water transport proteins (Fig. 3A)3,56. NKCC1 is the canonical SPAK target; however, SPAK regulates multiple ion transport proteins26. To investigate whether other ion transporters are involved in the SPAK-dependent CSF hypersecretory response, we immunoprecipitated SPAK from micro-dissected ChP of adult Spak−/− and Spak+/+ rats, and adult pigs (a model with increased ChP volume per animal) and analyzed the composition of immunoprecipitates using LC-MS-MS (Fig. 3B)30. Spak was the most abundant protein in the purified immune complexes from wild-type pig and rat ChP but absent from Spak−/− ChP. Nkcc1, the Na+/K+ ATPase α-1 subunit Atp1a1, the Cl− channel regulatory protein Clic6, the K+ channel Kcnj13, and Wnk1 were among the most significantly enriched Spak-bound proteins in immunoprecipitates from wild-type pig and rat ChP (Fig. 3C). These proteins were absent from Spak−/− ChP immunoprecipitates. Pulldown experiments validated these findings (Fig. 3D).
Figure 3. SPAK kinase is a regulatory scaffold for an apically-localized multi-ion transporter complex in ChP epithelial cells.

(A) Schematic illustrating the rat ventricular system and ChP, highlighting important ion transporters and channels in the apical and basolateral membranes of the ChP epithelium. BAM, border-associated macrophage; CSF, cerebrospinal fluid; RBC, red blood cell. (B, left) Illustration of SPAK immunoprecipitation and LC-MS/MS analysis of SPAK-interacting proteins detected in the micro-dissected ChP of Spak+/+ and Spak−/− rats, and wildtype pig (see Methods). (B, right) Western blot of SPAK, illustrating removal of gel lanes (area of the dashed box) corresponding to SPAK/SPAK-associated proteins for analysis with LC-MS/MS. (C) Identification of 24 highly SPAK-bound polypeptides, shared between Spak+/+ rats and wild-type pig ChP, and absent from immunoprecipitates from lysates of Spak−/− rat ChP. (D) Immunoblot of SPAK (upper blot; beta-actin, loading control) and (lower blots) co-immunoprecipitations of SPAK with LC-MS/MS-identified SPAK-bound proteins WNK1, NKCC1, ATP1A1 (*non-specific band), and KCNJ13 (SDS-resistant tetramer shown) in Spak+/+ and Spak−/− rat ChP. (E) Representative IHC co-staining of pSPAK (red) with ATP1A1 (green, top row), KCNJ13 (green, second row), CLIC6 (green, third row), and AQP1 (green, last row) in ChP of WT, Spak−/−, and Tlr4−/− animals +/− LPS treatment (n=6 ChP per genotype, 3 animals per condition). Insets show magnification; apical membrane (Ap), basolateral membrane (Bl) of the choroid plexus. 40x, Scale bars 25 μm.
snRNAseq analysis of ChP epithelial cells showed high expression of Wnk1, Stk39 (Spak), Slc12a2 (Nkcc1), Atp1a1, Kcnj13, and Clic6 in each ChP epithelial cell subcluster at levels significantly higher than other ChP cell types (fig. S4C). Spak co-localized with Atp1a1, Kcnj13, and Clic6 at the ChP apical membrane (Fig. 3E). PIH rats had increased Tlr4- and Spak-dependent co-localization of all three apical transporters, as well as of water channel Aqp1, with pSpak at the ChP apical membrane (Fig. 3E). These findings suggest SPAK is both a transducer of immune signals and regulator of ion transporters that are integral to PIH pathophysiology in ChP epithelial cells.
Post-infectious and -hemorrhagic hydrocephalus share common ChP immune-secretory mechanisms
Intraventricular organisms in PIH and blood products in PHH are similarly associated with CSF immune cell infiltration and cytokine production in patients 19,78,79. Bacteria-derived LPS and autologous blood products (e.g., heme) are well-known TLR4 pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), respectively80,81. We hypothesized convergent pathophysiology affecting ChP immune-secretory capacity drives the development of acute ventriculomegaly in both PIH and PHH.
To test this, we generated transcriptomic and proteomic datasets of micro-dissected lateral ventricle ChP from a validated rat model of intraventricular hemorrhage (IVH)-induced PHH30 and control rats. The PHH model resembles the PIH model, except that blood from the tail vein is harvested and injected directly into the lateral ventricles of the same animal (see Methods). Comparison of the PHH datasets with those from age- and sex-matched PIH rats revealed a striking overlap of the most differentially-expressed genes and proteins (Fig. 4, A-D and Table S3). Highly enriched pathways were related to macrophage-mediated innate immunity, toll-like receptor signaling, cytokine receptor transduction, and phosphoinositide 3-kinase signaling (Fig. 4, D-F and Table S3).
Figure 4. Post-hemorrhagic and -infectious hydrocephalus models exhibit highly similar ChP immune-secretory pathophysiology.

(A) Volcano plot and heatmaps of the most highly significant (sig) (B) DEGs and (C) DEPs compared to control from bulk RNAseq analyses of ChP from Ctl, IVH, and LPS-treated animals (n=3-5 animals per condition). (D) Venn diagram and GO biological process analysis of genes upregulated in IVH versus LPS conditions, and those common to both conditions. Outlined box at the bottom shows the top 20 DEGs common to IVH and LPS. (E) Hypergeometric enrichment analysis of gene co-expression modules for DEG/DEPs between LPS and IVH, and shared (LPS/IVH) DEG/DEPs (dashed box highlights shared enrichment in Module 17). (F) Module 17 pathway analysis. (G) Representative IHC of Iba1 (red), Ki67 (green), and ED1 (blue) cells in ChP of Ctl and IVH-treated rats (n=5 animals per condition) (ChP border denoted by dashed white outline; EP, ependyma). Left panel inset, ChP magnification; right panel inset, magnification Iba1+ cell (white arrows). Scale bars, 50μm. (H) Quantitative IHC in (G) showing % (normalized to DAPI) of ED1+, Ki67+, and Iba1+ cells (n=5 animals per condition). (I, J) FACS quantification of (I) CD45+ and (J) CD3+ cells in ChP from IVH-treated Tlr4+/+ and Tlr4−/− rats (n=4-6 animals per condition). (K) Quantitation of lateral ventricular size (% brain volume) and (L) body weight-normalized CSF secretion in WT, Tlr4−/−, and Spak−/− IVH-treated (48h) rats (n=3-6 animals per condition). (M) Representative ChP IHC of pNKCC1 (green) and pSPAK (red) expression in Ctl and IVH-treated Spak+/+ and Spak−/− animals. Scale bars, 25 μm. Quantitation of (N) pNKCC1 (Interval Density/cell #) and (R) pSPAK IHC in (O) (n=6 ChP, 3 animals per condition). Error bars, mean ±sem; each symbol represents one animal. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant; unpaired t-test (K) or one-way ANOVA (L-O, Q, R). [D-G, abbreviations: pos reg = positive regulation, neg reg = negative regulation, pw = pathway, act = activity, a/o = acting on, FA = focal adhesion, Mϕ = macrophage].
Integrated analysis of the top differentially expressed genes and proteins in PHH with scRNAseq data showed convergence to the same immune cell-specific co-expression module identified in PIH (Module 17, Fig. 4, E and F, and Table S3). Like PIH rats, PHH rats exhibited increased Iba1+, ED1+, and Ki67+ ChP-associated macrophages (Fig. 4, G and H) and Tlr4-dependent increases in CD45+ and CD3+ T-cells at the ChP (Fig. 4, I and J). PHH also rats resembled PIH rats in their >2-fold increased Tlr4- and Spak-dependent ventriculomegaly (Fig. 4K); >2-fold increased CSF hypersecretion (Fig. 4L); and up-regulated pSpak-pNkcc1 apical membrane ChP expression (Fig. 4, M-O). These results show PIH and PHH share a pathophysiological signature characterized by TLR4-mediated ChP immune cell signaling and SPAK-dependent ChP transepithelial ion transport.
scRNAseq uncovers molecular crosstalk between peripherally-derived and resident immune cells and ChP epithelial cells
Given the similarities of the ChP immune response to LPS and IVH, we performed single-cell RNA sequencing (scRNAseq) on microsurgically-dissected ChP (Fig. 5A) from control, PIH, and PHH animals (Fig. 5). We then compared the CD45+ and CD45− cell expression profiles (Fig. 5, B and I) using markers (Fig. 5C and fig. S5A) defined in the literature2,58. The integrated CD45+ dataset consisted of 1427 control, 3331 PHH, and 5529 PIH ChP immune cells. The integrated CD45− dataset consisted of 2036 control, 718 PHH, and 1603 PIH ChP non-immune cells. Clustering of CD45+ immune cells in control ChP resembled findings previously reported58.
Figure 5. scRNAseq uncovers crosstalk between peripheral and resident immune cells and epithelial cells at the ChP.

(A) Schematic illustrating the scRNA experimental design and downstream analyses. (B) UMAP clustering of ChP CD45+ cells across control, IVH-treated and LPS-treated conditions, colored by cell type. (C) DotPlot showing gene expression signatures of CD45+ cell clusters, with dot size representing the percentage of cells expressing the gene and the color representing average expression within a cluster. (D) Heatmap of the top differentially expressed genes (DEGs) comparing ChP CD45+ cell expression profiles from Ctl, IVH, and LPS-treated animals. (E) Volcano plots depicting differences of cluster abundance in IVH-treated and LPS-treated ChP CD45+ cells compared to control plotting fold change (log10) against p-value (−log10) based on beta-binomial regression. The red horizontal dashed line indicates the significance threshold. (F) UMAP of ChP myeloid cell clusters, including peripheral blood macrophages, CP stromal macrophages and CP epiplexus macrophages, colored by cell type. (G) Heatmap of the top DEGs for myeloid subclusters. (H) UMAP of ChP epiplexus macrophage subclusters, colored by group, showing transformed and proliferating epiplexus macrophages in control, IVH, and LPS conditions. (I) UMAP clustering of ChP CD45− cells across IVH-treated, LPS-treated, and control conditions, colored by cell type. (J) Heatmap of top DEGs comparing ChP CD45− epithelial cell expression profiles from Ctl, IVH, and LPS-treated animals. (K) UMAP of ChP epithelial subclusters, colored by group.
Comparing CD45+ immune cells in the ChP from PIH and PHH animals demonstrated significantly different immune cell profiles compared to control. However, the inflammatory profiles of the ChP from PIH and PHH animals closely resembled each other (Fig. 5B and D), with similar increases in macrophage/monocytes and neutrophils (Fig. 5B and E). Heatmap analysis of the most differentially expressed genes in the CD45+ dataset showed highly significant overlap for PIH and PHH compared to control (Fig. 5D). Interestingly, plasmacytoid DCs (pDCs) and CD4+ T cells were particularly increased in PIH (Fig 5B and E).
The myeloid cell population was further separated into three main subclusters, in agreement with recently defined markers58, which included stromal BAMs (CP_BAM; C1qahiCsf1rhiCd14lo), ChP-epiplexus cells (CP_Epi; Sall1+), and peripherally-derived blood monocytes (Pbm, Cd14hiC1qalo) (Fig. 5, F and G). The Pbm sub-cluster was increased in the ChP from PIH and PHH compared to control, reflecting the infiltration of peripheral macrophages (Fig. 5, F and G)58. As top markers of the Pbm-2 subcluster included interferon-stimulated genes (such as Irf7 Isg15, Isg20, Ifit2, Ifit3 and Ifitm3), Pbm recruitment is likely a response to upregulated interferon signaling.
The significant increase in Pbm, neutrophils, and pDCs in the ChP of PIH and PHH animals prompted pathway analyses of the top markers for these cell populations (fig. S5, B-J). Pbm pathway analysis revealed enrichment of pathways related to macrophage function and chemokine/interferon signaling, macrophage and neutrophil chemotaxis and migration, and regulation of dendritic differentiation (fig. S5B). Neutrophil pathway analysis showed enrichment of interferon/chemokine signaling and PI3K-Akt-mTOR signaling pathways (fig. S5D). pDC pathway analysis also identified significant enrichment of TLR signaling, chemokine signaling, and PI3K-Akt-mTOR signaling pathways (fig. S5, E and F).
The ChP epiplexus macrophage (Kolmer cells) cell population transformed similarly in PIH and PHH ChP compared to controls (Fig 5F). When further subclustered, both PIH and PHH exhibited two new populations of ChP-associated Kolmer cells (Fig 5H, blue and green). Pathway analysis revealed enrichment of DNA replication and mitotic division processes in sub-cluster 1 (fig. S5G), suggesting a subpopulation of replicating Kolmer cells. Pathway analysis for top markers of Kolmer cell sub-cluster 2 showed enrichment of LPS-mediated signaling, TLR signaling, and chemokine receptor binding, among others (fig. S5H).
Clustering of the ChP CD45− non-immune cell populations demonstrated findings like those previously reported (Fig 5I)2). Heatmap analysis of the most differentially expressed genes in the CD45− dataset again revealed substantial overlap for PIH and PHH ChP compared to control ChP (Fig. 5J). Subclustering of the epithelial cell (CD45−) population uncovered a new ChP epithelial subcluster in PIH and PHH (Fig. 5K; subcluster 2), which expressed Cxcl17, CxcI9, Ccl5, II18bp, and Irf7 (Table S4, fig. S6). Inflammatory and immune signaling pathways, particularly type I interferon signaling, were enriched in this subcluster in both PIH and PHH (fig. S5, I and J).
To compare ligand-receptor interactions in control, PIH, and PHH animals, we merged the CD45+ and CD45− datasets and applied CellChat82, a scRNAseq tool used for inferring intercellular communication networks based on the expression of known ligand-receptor pairs in different cell clusters (Fig. 6A). Our analysis identified 6 ligand-receptor pathways present in both PIH and PHH ChP that were absent in the control ChP (Fig 6B). Among these, the Osteopontin (Spp1) ligand-receptor pathway showed very similar ligand-receptor interaction profiles for both PIH and PHH ChP (Fig 6C). In both groups, the most significant ligand-receptor interactions originated from ChP epiplexus macrophages to ChP resident macrophages, monocytes, T cells, B cells, and DCs (Fig 6C). CellChat analysis also identified the ligand-receptor pairs Spp1-Cd44 and Spp1-(Itga4_Itgb1) as the most significant signaling contributing to the Spp1 communication pathway (Fig 6D). These interactions are known to activate the PI3K-Akt-mTOR signaling pathway83,84.
Figure 6. scRNAseq CellChat analysis reveals ligand-receptor pairs enabling ChP immune-epithelial cell communication.

(A) Schematic illustrating CellChat downstream analyses. (B) Identification of 6 ligand-receptor pathways shared between IVH-treated and LPS-treated rats and absent in control. (C) Circle plot depicting ligand-receptor interactions of the SPP1 pathway in IVH-treated and LPS-treated ChP. Edge width represents communication probability. (D) Bubble plot showing significant ligand-receptor interactions for the SPP1 pathway in IVH-treated and LPS-treated ChP (no significant interactions were found for control). X-axis shows the cell groups associated with the interactions. Y-axis shows the ligand-receptor pairs. Dot color reflects communication probabilities and dot size represents computed p-values. Empty space means zero communication probability. p-values reflect a one-sided permutation test. (E) Upregulated ligand-receptor pairs for IVH-treated and LPS-treated ChP compared to control, showing significant overlap of upregulated pathways in IVH and LPS, with increased signal in SEMA, CD45, MHC-I, FN1, CXCL, CCL, COMPLEMENT pathway ligand-receptor pairs for both conditions. Dot color reflects communication probabilities and dot size represents computed p-values (from a one-sided permutation test). (F) Significant signaling pathways ranked based on differences in the overall information flow within the inferred networks between epithelial and CD45+ cells, comparing LPS (left) and IVH conditions (right) to control. The overall information flow of a signaling network is calculated by summarizing all communication probabilities in that network. Rows with high blue-to-red ratio indicate higher ligand-receptor pathway activity for IVH or LPS conditions. (G) Circle plot comparing CXCL pathway ligand-receptor interactions among control, IVH-treated, and LPS-treated ChP epithelial and CD45+ cell groups. Edge width represents communication probability. (H) Comparison of control, IVH, and LPS bubble plots showing significant ligand-receptor interactions between epithelial and CD45+ cells for the CXCL pathway. X-axis shows the cell groups associated with the interactions. Y axis shows the ligand-receptor pairs. Dot color reflects communication probabilities, and dot size represents computed p-values (from a one-sided permutation test). Empty space means zero communication probability.
We next examined ligand-receptor signaling pathways present in all three conditions but independently upregulated in PIH or PHH ChP compared to the control ChP. Cell-cell communications were very similar for PIH and PHH, showing significant upregulation in inflammatory pathways including MHC-I, CCL, CXCL, and FN1 (Fig 6E). We then compared the ligand-receptor signaling pathways of epithelial and immune cells in PIH and PHH ChP to control ChP. We found inflammatory and cell adhesion pathways such as chemokine (CXCL), Galectin, and Periostin to be activated in both PIH and PHH ChP between epithelial and immune clusters (Fig 6F). The significant CXCL interactions between ChP epithelial and immune cells were mainly sent from epithelial cells to T cells and to dendritic cells for both PIH and PHH compared to control (Fig 6G). Cell-cell communication analysis revealed that ligand-receptor pairs Cxcl11-Cxcr3 and Cxcl9-Cxcr3 contributed most significantly to the CXCL epithelial-immune cell communication pathway (Fig 6H) in both PIH and PHH, suggesting that ChP epithelial cells play active roles during the inflammatory response.
Repurposed systemic immunomodulators treat hydrocephalus by antagonizing ChP-mediated CSF hypersecretion
The importance of highly similar ChP immune-secretory mechanisms to the development of acute ventriculomegaly in PIH and PHH suggested the possibility of shared therapeutic strategies. No clinically approved drugs target TLR4 or SPAK, but multiple agents are in clinical trials or in preclinical development42,85,86. We therefore mined our ChP -omics data for pathways enriched in both PIH and PHH models that are targeted by clinically approved drugs. Among top-ranked hits, the PI3K-Akt-mTOR-signaling pathway (WikiPathways WP2841; z-score 12.13) (Fig. 2E) was an appealing target because of its role in both immune cells and SPAK activation87-89. Indeed, the top differentially expressed genes and proteins common to PIH and PHH were enriched in multiple phosphatidylinositol pathways, the most significant of which was 1-phosphatidylinositol-3-kinase regulator activity (GO: 0046935; z-score 141.43) (Fig. 4, D and F)90. Integration of differentially expressed genes with published scRNAseq data revealed enrichment of the Focal Adhesion-PI3K-Akt-mTOR-signaling pathway (WP2841) in co-expression module 11 (z-score 4.16) (Table S1). Consistent with these findings, phosphorylation of S6 ribosomal protein (pS6), indicative of mTOR pathway activation, was significantly increased in both ED-1+ ChP immune cells and ChP epithelial cells in both PIH and PHH rats, but not in similarly treated Tlr4−/− rats (Fig. 7A). snRNAseq analysis revealed high Mtor and Nfkb expression in ChP epithelial cells and hematopoietic cells (Fig. 7B and fig. S7, A-B). These results suggest PI3K/Akt/mTOR signaling is a therapeutic target in PHH and PIH.
Figure 7. Repurposed systemic immunomodulators treat hydrocephalus by antagonizing ChP-mediated CSF hypersecretion.

(A) Representative IHC of pS6 (green) and ED1 (red) in ChP-associated immune cells (arrows) and epithelial cells (arrowheads) in WT and Tlr4−/− animals +/− LPS, and WT LPS and IVH conditions +/− Rapamycin (Rapa) (n=5). Scale bars, 30μm. DAPI, blue. (B) snRNAseq analysis (left panel) and violin plot (right) demonstrating Mtor in ChP epithelial (Ep) cells and ChP-associated hematopoietic cells (Epithelial, Avg Log2FC=7.88e-2, p=1.23e-17). (C) Representative IHC of Iba1 (red), Ki67 (red), and ED1 (blue) expression in the ChP of WT rats treated with LPS or IVH, in the presence of Rapa. (D-F) Quantitative IHC of the ChP immune response in (C) in WT LPS and IVH-treated animals +/−Rapa. Graphs represent DAPI-normalized % of cells identified as (D) Iba1+, (E) Ki67+, (F) ED1+ in Ctl, LPS, LPS+Rapa, IVH, and IVH+Rapa-treated animals (n=4-6). (G) Quantitation of FACS-isolated CD45+ and CD3+ cells in Ctl, LPS, LPS+Rapa, IVH, IVH+Rapa-treated rats (n=5-6). (H) Weight-normalized CSF secretion and (I) lateral ventricular size (% brain volume) in Ctl, LPS, LPS+Rapa, IVH, and IVH+Rapa-treated animals (n=4-6). Representative IHC of (J) pNKCC1 and (K) pSPAK expression in WT Ctl, LPS +/−Rapa, and IVH +/−Rapa. (L) Quantitation (Interval Density/cell #) of pNkcc1 and pSPAK IHC in (J) and (K), respectively (n=6 ChP, 3 animals). Error bars, mean ± sem; each symbol represents one animal. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant; one-way ANOVA.
We tested the hypothesis that systemic treatment with rapamycin, an immunosuppressive mTOR inhibitor, would attenuate the development of acute PIH and PHH by antagonizing inflammation-dependent CSF hypersecretion from ChP epithelial cells. In both PIH and PHH rats, intraperitoneal rapamycin (6 mg/kg administered 2-3h prior to initiating LPS infusion or 2-3h pre-IVH) decreased up-regulated pS6 immunostaining in ChP immune cells and epithelial cells (Fig. 7A), reduced numbers of ChP-associated Iba1+, ED1+, and Ki67+ cells (Fig. 7, C-F), and attenuated the LPS- and IVH-induced influx of CD45+ cells (Fig. 7G). Rapamycin also attenuated both LPS- and IVH-induced increases in CSF secretion (Fig. 5H), ventriculomegaly (Fig. 7I and fig. S7, C-E), and pSPAK-pNKCC1 abundance at the ChP apical membrane (Fig. 7, J-L). Strikingly, delay of rapamycin administration until 3 h after initiating LPS infusion had similar effects, including attenuation of LPS-mediated ventriculomegaly (fig. S7, C-E) and a decrease in ChP-associated Iba1+ and ED1+ cells (fig. S7, F-G). These results suggest that repurposed systemic immunomodulators can ameliorate acute PIH and PHH by antagonizing ChP epithelial cell CSF hypersecretion.
DISCUSSION
Results from multi-modal analysis of CSF dynamics in E. coli models of PIH suggest that acute ventriculomegaly results primarily from an LPS-induced increase in ChP CSF secretion. These data may revise or augment existing paradigms emphasizing obstruction of intraventricular CSF flow. The relevance of these findings is supported by observations in patients that: (i) PIH can develop within hours of CNS infection91 and can precede radiographic evidence of aqueductal obstruction92; (ii) destruction of the ChP (with or without an endoscopic third ventriculostomy [ETV]), can effectively treat PIH93 and some other forms of hydrocephalus; and (iii) increased CSF secretion can cause hydrocephalus in humans with ChP hyperplasia in the setting of ChP tumors94-96 and certain genetic syndromes97-100. Although multiple models of CSF clearance have been proposed (e.g., via vascular basement membranes, the intramural periarterial drainage pathway, perineuronal routes, and skull base lymphatics), their role in hydrocephalus, particularly in chronic hydrocephalus when inflammation-dependent scarring from tissue damage could impact resorptive mechanisms101,102, will be important topics of future investigation.
While we demonstrated that LPS, likely E. coli’s most immunogenic virulence factor, is necessary and sufficient to cause CSF hypersecretion and ventriculomegaly in our PIH model, the full immune response to E. coli at the ChP-CSF interface likely includes components besides LPS, such as flagellar H-antigens and capsular K-antigens103,104. Indeed, animals treated with LPS-deficient E. coli−LPS still exhibit ChP-CSF interface inflammation, albeit less severe than with E. coli+LPS, and exhibit a trend towards ventricular enlargement 105,106. Moreover, the immune response to E. coli and to other infectious agents impacts the meninges and parenchyma105,106. Future studies will explore the involvement of other portals of immune cell entry into the brain and whether PAMPs from other PIH-causing bacteria (e.g., the TLR5 agonist flagellin107) trigger ChP CSF hypersecretion.
Our data supports BAMs on the apical ChP membrane as the “first responders” to microorganisms in the CSF108-111. ChP macrophages express TLRs, which bind PAMPs to activate innate immune responses111,112. Our integrated -omics analysis showed dependence of the acute ChP hypersecretory response in PIH on LPS-induced, TLR4-mediated recruitment of both peripheral blood monocytes and accumulation, proliferation, and activation of resident stromal BAMs (C1qahiCsf1rhiCd14lo) and ChP-epiplexus macrophages (CP_Epi, Sall1+). A similar TLR4-dependent immune cell response was triggered in PHH by intraventricular hemorrhage-derived DAMPs such as heme40,80,113,114. Notably, ChP macrophage depletion attenuates ventriculomegaly following ICV-injection of the proinflammatory blood product peroxiredoxin-2115. These data show the remarkable similarity of the innate immune cell responses to CNS infection and to hemorrhage, and highlight the ChP as a dynamic, cellular heterogeneous tissue with highly regulated immune-secretory capacity.
We found acute PIH and PHH are both characterized by a CSF “cytokine storm” analogous to sepsis-associated elevations of blood circulating cytokines in the settings of systemic infection or widespread tissue damage116,117. These observations correspond to cytokine and immune cell responses in human CSF stimulated by intraventricular infection and hemorrhage19,78,79. CSF cytokines likely engage their cognate receptors on ChP epithelial cells to modulate epithelial cell function. Among up-regulated cytokines, Ccl2 and its ChP epithelial cell receptor Ccr2 recruits macrophages to the ChP-CSF interface in response to maternal immune activation 4. The ChP's ability to detect infection or tissue damage and initiate a counter-response may also be an Achille’s heel that renders the brain susceptible to attack by circulating immune cells in multiple sclerosis and other diseases8,118,119.
SPAK kinase links ChP immune cell signaling and epithelial function via its interactions with upstream cytokine receptors and downstream ion transport proteins73. The TNF-α receptor RELT binds SPAK to regulate the innate immune system74 and increase epithelial transport in experimental inflammatory bowel disease73,120,121. SPAK is a master regulator of the SLC12A family cation-Cl− cotransporters such as NKCC1122. SPAK phospho-activation in the ChP, triggered by LPS or IVH, leads to increased ChP CSF production in part by phosphorylating and thereby stimulating NKCC1. These data accord with the importance of bumetanide-sensitive (NKCC1-mediated) ChP transport for CSF production in humans123, dogs55, rats30, and mice124, and are consistent with the finding that patients with loss-of-function NKCC1 mutations exhibit decreased epithelial secretion and “slit-ventricle” brain morphology indicative of decreased intraventricular CSF volumes125.
NKCC1 operates close to its equilibrium electrochemical potential in the ChP as in other epithelia. This suggests the capacity for regulated, bidirectional net ion and fluid transport across the ChP75,124 and might account for reported differences in the vector of NKCC1-mediated net ion transport (inward vs. outward), and in net CSF secretion and/or CSF clearance in various developmental126 and/or disease contexts30. We showed Spak binds not only Nkcc1122,127, but also other ion transport proteins implicated in CSF secretion including Atp1a1. We predict that SPAK functions as a regulatory scaffold that senses extracellular and/or intracellular signals (e.g., changes in CSF ionic composition), and then transduces these signals into phosphorylation events to coordinate the activities of an ensemble of associated ion transport proteins.
Drugs targeting TLR4 or SPAK have not been clinically approved42,128,129. Our -omics data converged upon PI3K-Akt-mTOR signaling as important for ChP immune-secretory mechanisms in both PIH and PHH, consistent with this pathway’s contributions to immune cell and epithelial cell homeostasis130, and to TLR4131,132 and SPAK signaling88,133. “Rapalog” mTOR inhibitors are approved for systemic treatment of allograft rejection and for adjunct treatment of subependymal cell astrocytomas134,135. The immunosuppressive effects of mTOR inhibition likely prevail over other growth-mediated pathways in rapamycin's therapeutic effect on our acquired hydrocephalus models. Other potential beneficial effects of rapamycin may include its autophagy-inducing effects in brain microglia136 and inhibition of hypoxia-inducible factor 1a137. The clinical utility of rapamycin will be better assessed by testing later time points of administration. Repurposed drugs modulating ChP immune-secretory function might be a viable non-surgical treatment strategy for multiple forms of acquired hydrocephalus and for other neuroimmune brain disorders associated with ChP dysfunction.
Limitations of the Study
Limitations of our study include our reliance on the rat as a model organism, chosen for its amenability to in vivo measurements of CSF secretion and for its CSF volume higher than that of mouse. Future development of non-invasive, imaging-based assays to measure ChP-mediated CSF production, and of ChP-specific gene targeting approaches should overcome present limitations of our direct surgical CSF secretion assay and of current indirect methods to interrogate in vivo transporter activity. Additional limitations include the lack of correlative scRNAseq analysis of CSF from PIH and PHH patients, and lack of clinical data testing efficacy of rapamycin and other immunomodulators as treatments for hydrocephalus. The latter could be studied in patients with high-grade aneurysm rupture complicated by intraventricular hemorrhage and PHH, all currently managed with indwelling intraventricular drains and intermittent imaging.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Requests for further information should be directed to and will be fulfilled by the Lead Contact, Kristopher Kahle (kahle.kristopher@mgh.harvard.edu).
Materials Availability
Mouse lines generated in this study have been deposited to the Knockout Mouse Project (KOMP), Charles River, albino wister rats, strain code 003.
Data and Code Availability
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE139 partner repository with the dataset identifier PXD030678. Any FACS or microscopy data, confocal, or other, reported in this paper will be shared by the lead contact upon request. All materials and associated protocols can be found within the methods section of this paper, and any relevant papers are also cited for further reference. In addition to the methods provided here, for additional explanations of the IVH protocol or direct CSF measurements, please see published works32,53,138, or additional information can be addressed via contact with the corresponding article of this manuscript.
This paper does not report original code. All previously published algorithms are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| AQP1, rabbit recombinant monoclonal | Abcam | Ab168387 |
| ATP1A, rabbit polyclonal | Alomone | ANP-001 |
| CD68, rabbit monoclonal | CST | 97778 |
| CLIC6, mouse monoclonal | Santa Cruz | 365303 |
| ED1/CD68, mouse monoclonal | Millipore | MAB1435 |
| IBA1, goat polyclonal | Thermo | PA5-18039 |
| KCNJ13, mouse monoclonal | Santa Cruz | 398810 |
| Ki67, rabbit monoclonal | CST | 9129 |
| pNKCC1, rabbit polyclonal | Millipore | ABS 1004 |
| pSPAK, rabbit polyclonal | Millipore | 07-2273 |
| RELT, rabbit polyclonal | Novus | NBP2-56851 |
| NKCC1, rabbit monoclonal | CST | 85403S |
| Anti-WNK1 | University of Dundee | S079B |
| Anti-SPAK | University of Dundee | S365D |
| Anti-NKCC1 | University of Dundee | S022D |
| Anti-ATP1A1 | DSHB | a5 |
| Anti-HRP | Pierce | 21130 |
| Anti-beta actin | Abcam | Ab8227 |
| CD16-32 | Biolegend | Cat#: 101302 |
| CD45-PE/cy7 | Biolegend | Cat#: 202207 |
| CD3-647 | Biolegend | Cat#: 201408 |
| Alexa Fluor 488 | Invitrogen | Cat#: A32723 |
| Alexa Fluor 555 | Invitrogen | Cat#: A32816 |
| Alexa Fluor 647 | Invitrogen | Cat#: A32849 |
| Bacterial and virus strains | ||
| E. Coli+LPS (BD21) | Lucigen | E. cloni EXPRESS BL21(DE3) |
| E. Coli−LPS (ClearColi) | Lucigen | CLEARCOLI BL21 |
| Biological samples | ||
| N/A | ||
| Chemicals, peptides, and recombinant proteins | ||
| Bumetanide | Sigma-Aldrich | CAS#: 28395-03-1 |
| aCSF | Harvard Apparatus | Item#: 59-7316 |
| Rapamycin | Cayman | Item#: 13346 |
| Critical commercial assays | ||
| N/A | ||
| Deposited data | ||
| Bulk RNA seq | GEO | GSE192946 |
| Mass spectrometry proteomics data | PRIDE | PXD030678 |
| ScRNA seq | GEO | GSE218143 |
| Experimental models: Cell lines | ||
| N/A | ||
| Experimental models: Organisms/strains | ||
| Wistar rats, albino | Charles River | Strain code 003 |
| Oligonucleotides | ||
| N/A | ||
| Recombinant DNA | ||
| N/A | ||
| Software and algorithms | ||
| Seurat | Hao et al.151 | https://satijalab.org/seurat/ |
| Monocle3 | Cao et al.152 | https://cole-trapnelllab.github.io/monocle3/ |
| Cellchat | Jin et al.82 | https://github.com/sqjin/CellChat |
| Other | ||
| N/A | ||
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Rodent Model
Animal experiments were performed under a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of Yale University and in accordance with the guidelines and regulations in the NIH Guide for the Care and Use of Laboratory Animals. Male and Female Wistar rats (Charles River, Wilmington, MA, USA) of wild type and Stk39−/− 77 and Tlr4−/− 57 genotypes, aged 8 weeks (200–350g) and bred in accordance with an IACUC-approved protocol at the Yale University School of Medicine, New Haven, CT were anesthetized (90 mg/kg body weight ketamine plus 7.5 mg/kg body weight xylazine, i.p.) and allowed to breathe room air spontaneously. Body temperature was maintained at 37 ± 1 °C (Harvard Apparatus, Holliston, MA) throughout the course of the experiments. This study randomly chose animals for either control or experimental conditions and the researchers were not blinded. Female rats were excluded from this study to avoid introducing sex as an additional variable. Animals were determined to be healthy by the veterinary staff, were immunocompetent, no involved in previous procedures (other than those directly related to this project and described below in the Methods Details section), were drug and test naïve (other than those directly related to this project and described below in the Methods Detials section), and were kept under standard, IACUC approved husbandry and housing conditions at Yale.
Porcine Model
All porcine studies were performed in collaboration with the Dardik laboratory at Yale University and in compliance with federal guidelines and protocols approved by the Yale University Institutional Animal Care and Use Committee. Yorkshire male pigs, with mean age of 3.4 months, were delivered to our facility 7-10 days prior to any procedures, kept under standard and IACUC approved husbandry and housing conditions at Yale, and were determined to be healthy after examination by veterinary staff. Animals had not undergone any previous procedures and were drug and test naïve.
METHOD DETAILS
Model of post-hemorrhagic hydrocephalus
IVH was modeled using our previously published methods30, and detailed here. In an anesthetized animal, the hair on the dorsal side of the skull was initially shaved using clippers. A 2.5-cm incision was made in the proximal tail (beginning 2.5 cm below the rectum) using a scalpel (Bard-Parker, #10 Carbon Steel Surgical Blade) and the tail artery was then aseptically cannulated using a flexible catheter (PE-20) pre-loaded with heparinized saline. The rat was then mounted in a stereotactic apparatus (Stoelting Co., Wood Dale, IL), a 2.5-cm midline scalp incision was made using a new scalpel blade to expose the aponeurosis, and the skin was retracted using Barraquer retractors (Harvard Apparatus). The aponeurosis was then cut via a midline incision and carefully retracted using the same scalpel. The surface of the skull was cleaned using 0.9% Sodium Chloride (Hospira, Lake Forest, IL) and dried using 16 ply 4x4 sterile gauze sponges (Covidien, Mansfield, MA). Any bleeding from the skull was burned using a cautery pen (Bovie, Clearwater, FL). After the skull was dry, a 1 mm burr hole was made using a high-speed drill over the right lateral ventricle (coordinates, x= −0.8, y= −1.7 mm from bregma) and the skull was subsequently cleaned and allowed to dry using the same method as indicated above. Approximately 150 μL of blood was then drawn from the tail artery catheter into a 1 mL syringe initially filled with 500 μL of heparinized saline and was subsequently discarded. An additional 50 μL of blood was then drawn into a new, sterile 1 mL syringe and immediately transferred into a 500 μL syringe (Hamilton, Reno, NV), which was then mounted to the stereotactic frame. Under stereotactic guidance, 50 μL of freshly collected autologous blood, free from anticoagulants, was infused into the right lateral ventricle (coordinates, x= −0.8, y= −1.7, z= −4.5 mm from bregma), over the course of 5 minutes, and the 26-gauge needle was held in place for an additional 20 minutes to prevent blood backflow from the the burr hole upon needle removal. The incisions were then sutured with a simple uninterrupted stich using 4-0 2.0 Metric DemeGUT absorbable surgical suture (DemeTECH, Miami Lakes, FL). Intraventricular sterile aCSF (Harvard Apparatus) infused in the same manner served as the IVH control condition.
Model of post-infectious hydrocephalus
As in the model of post-hemorrhagic hydrocephalus outlined above, an anesthetized rat with shaved dorsal skull was mounted into a stereotactic apparatus (Stoelting Co.), and a 1-inch midline scalp incision was made with a scalpel blade to expose the fascia on the dorsal side of the skull. The aponeurosis was then incised midline and carefully retracted with the same scalpel blade. The surface of the skull was then cleaned using 0.9% Sodium Chloride, dried with 16 ply 4x4 sterile gauze sponges, and any skull bleeding cauterized by cautery pen. A high-speed drill was used to make a 1 mm burr hole in the dry skull over the right lateral ventricle (coordinates, x= −0.8, y= −1.7 mm from bregma), and the skull subsequently cleaned and allowed to dry as above. After the skull was sufficiently dry, a brain infusion cannula with depth adjusted to 4.5 mm using 1 spacer (Alzet, Cupertino, CA; brain infusion and cannula kit, #2) was mounted with veterinary surgical cyanoacrylate adhesive (Covetrus, Dublin, OH) into the right lateral ventricle (x= −0.8, y= −1.7 relative to bregma). A small horizonal incision was then made just above the spinotrapezius muscle overlying the neck and upper back. An osmotic pump (Alzet, 1003D, rate: 1mL/hr), filled 12h earlier with lipopolysaccharide (LPS; Enzo, Serotype O55:B5) diluted in aCSF (10ng/mL) or E. coli (Lucigen) diluted in aCSF (see below), was then placed subcutaneously in the caudal portion of the dorsum and connected to the cannula attached to the skull using PE tubing provided in the Alzet brain infusion kit. The incisions were then sutured via a simple uninterrupted stich using 4-0 2.0 Metric DemeGUT absorbable surgical suture. Control animals were treated identically, but with aCSF loaded into the osmotic pump.
ICV-infusion of E. coli
Wild-type E. coli and ClearColi (E. coli lacking functional surface LPS; Lucigen BD21; https://www.nature.com/articles/nmeth.f.367.pdf?origin=ppub) were cultured originally on standard LB-agar plates. A single colony from each strain was used for inoculation in 10 mLs of standard LB broth and incubated overnight at 37 degrees Celsius. To get a specific number of cells per milliliter, 10 mLs of LB was then inoculated with 100 μl of the overnight culture and grown at 37 degrees Celsius for 3 hours to achieve bacterial growth at log phase. The cells were harvested by centrifugation at 2000RPM at 4 degrees Celsius for 10 minutes. The pellet was collected and resuspended in 50ml of PBS. 1 μl of the 50ml was then added to 10ml of LB. 10 μl of the primary dilution was added to 300 μl of LB and plated on a 10cm LB-agar plate. The plates were incubated at 37 degrees Celsius overnight. The number of bacterial colonies were counted, and the colonies/ml determined. This was then used to determine the volume of the 50 mL stock bacteria to be used for brain infusion. The 50ml stock bacteria was stored in 1xPBS with 10% glycerol at −80 degrees Celsius. E. coli cells were diluted into sterile aCSF and delivered ICV via osmotic pump (Alzet 1003D, rate 1μl/hr) as detailed above for our model of PIH.
Measurement of CSF production rates
Rates of CSF production were measured using our published method30,53, and as described here. Anesthetized rats were mounted in a stereotactic apparatus and a 1.3 mm burr hole was made over the left lateral ventricle (coordinates, x= −0.8, y= +1.7 relative to bregma). The mounted head was rotated on the ear-bars 90°, nose-down, and the suboccipital muscles were dissected to the cisterna magna to expose the atlanto-occipital ligament. The ligament was punctured, and a 23-gauge flexible catheter (PE-20) was advanced 5 mm through the foramen of Magendie to the 4th ventricle. Sterile, molecular grade mineral oil (100 μL; Sigma Aldrich, St. Louis, MO) was infused into the 4th ventricle to occlude the aqueduct of Sylvius, thereby creating a closed system of CSF circulation. With the rat in the same position, a glass capillary tube (cat # CV8010-300; borosilicate; OD, 1 mm; ID, 0.8 mm; length, 30 cm; VitroCom, Mountain Lakes, NJ) was advanced through the burr hole into the right lateral ventricle (4.5 mm beyond entry). The volume (V) of CSF that had formed at a given timepoint was calculated as: V (mm3) = π · r2 · d, where r is the radius of the capillary tube and d is the distance CSF traveled within the capillary. Pictures were taken every 5 min over the course of 30 min. The rate of CSF formation (μL/min) could be calculated from the slope of the volume–time relationship.
Ventricular volume analysis
Ventricular volume analysis was performed as we previously published30, and as described here. Rats were transcardially perfused with 40 mL of ice-cold normal TBS (pH=7.60) followed by 40 mL of ice-cold 10% neutral buffered formalin (NBF; EMD Millipore Corporation). Brains were harvested and kept in 10% NBF overnight, then transferred to a 30% sucrose solution for cryoprotection. The brains were then removed, placed in Optimal Cutting Temperature Compound (OCT; Tissue-Tek, Sakura) for 1hr at room temperature and frozen in dry ice. The mounted brains were kept at −80°C until transfer to the cryostat, then left 1hr at −20°C. Brains were then serially sectioned in 25 μm sections). To prevent distortion from cryosectioning and slide mounting, we took high resolution pictures of serial coronal sections (200 μm apart, 14 levels) while the brain was mounted in the cryostat, using uniform parameters of camera positioning, magnification, and external lighting. Imaging software Gimp-2.10 was used to obtain a pixel count of the lateral ventricular and whole brain area of each section. A pixel to mm conversion factor was generated using the standard distance 22 mm sized block. Pixels were converted to area in mm2, summed over 14 levels and multiplied by the distance between levels (0.2 mm) to calculate ventricular and brain volume. The ventricular volume was divided by the brain volume to correct for varying brain size among animals.
ICV bumetanide delivery
As previously described30,140, at the time of CSF collection prior to rotating the rat's head to a vertical orientation, a 28-gauge cannula from an Alzet brain infusion kit (#2; Durect) with a single spacer to adjust the depth to 4.5 mm, was stereotactically placed with a single spacer to adjust the depth to 4.5 mm into the burr hole over the left lateral ventricle (coordinates, x= −0.8, y= +1.7 relative to bregma) and secured to the skull with cyanoacrylate adhesive. The cannula was connected via a preloaded PE-20 catheter to a 1-ml syringe containing bumetanide (2.7 mM, pH 9; Sigma-Aldrich) diluted in aCSF (Harvard Apparatus). The syringe was loaded into a syringe infusion apparatus (Pump elite 11, Harvard Apparatus) and maintained at 37 °C. To determine the rate of CSF formation during intraventricular drug infusion, the 'actual infusion rate' of the drug (1.93 μl/min) was subtracted from the 'measured outflow rate' to obtain the operational 'calculated rate of CSF formation'. To assess the effect of a drug, the baseline rate of CSF formation was determined during spontaneous CSF formation (no drug infusion), and the calculated rate of formation was then determined after infusion of bumetanide.
Systemic rapamycin administration
Rapamycin (6mg/kg in normal saline, IP, Cayman) was administered IP 2-3hr prior to the start of IVH and LPS surgery. At 48h for IVH-treated rats and 72h for LPS-treated rats, rats were then either (i) euthanized and transcardially perfused with TBS (pH=7.60) and 10% NBF and processed for ventriculomegaly or immunohistochemistry analysis as described above, or (ii) anesthetized for surgery to allow measurement of the rate of CSF secretion as described above.
ICV injection of Evans Blue Dye
Based on our previous methods to determine communication between the cerebral ventricles53, Evans Blue Dye (Sigma Aldrich; 0.5% in sterile aCSF) was loaded into a 1mL syringe (BD Syringe, Franklin Lakes, NJ) and connected to a cannula (Alzet; brain infusion and cannula kit, #2) using PE tubing. Anesthetized rats were mounted in a stereotactic apparatus and a 1.0 mm burr hole was made over the right lateral ventricle (coordinates, x= −0.8, y= −1.7 relative to bregma). The cannula was placed into the right lateral ventricle and dye was infused at a nominal rate of 2μL/min for 5 minutes with a syringe pump (Harvard Apparatus; Elite 11). Following infusion, the cannula remained in the ventricle for 30 minutes to prevent back flow and assure adequate circulation of the dye within the CSF spaces. Animals were then transcardially perfused with ice cold TBS (pH = 7.60), brains were harvested and immediately cut sagittally along the interhemispheric line. Images were taken to show the level of patency of the cerebral aqueduct.
CSF cytokine collection
48h after surgery for the PHH model or 72h following surgery for the PIH model, anesthetized rats were mounted in a stereotactic apparatus. The rat’s head was rotated on the ear-bars 90°, nose-down, and the suboccipital muscles were dissected to the cisterna magna to expose the atlanto-occipital ligament. The ligament was punctured using a 27G PrecisionGlide needle (BD, Franklin Lakes, NJ) attached to a 1mL syringe (BD, Franklin Lakes, NJ) and inserted into the cisterna magna. CSF (~100mL) was slowly withdrawn into a syringe and immediately transferred to a 1.5 mL Eppendorf tube on dry ice. Rats were immediately euthanized following CSF collection. Samples were stored at −80°C until shipment on dry ice to Eve Technologies (Calgary, CA) for analysis using their Rat Cytokine Array / Chemokine Array 27 Plex (RD27).
Rodent choroid plexus epithelium harvesting
Immediately following transcardial perfusion with ice cold TBS (pH = 7.60), the brain was isolated and placed in an ice-cold saline bath. Brains were cut sagittally along the interhemispheric line to expose the lateral ventricles, and choroid plexuses were rapidly but carefully dissected from both lateral ventricles using sharp forceps under magnification. Each brain yielded ~3 mg of choroid plexus tissue total, which was collected into a 1.5 mL tube, snap frozen in liquid nitrogen, and stored at −80°C until use.
Immunohistochemistry
Rats were transcardially perfused with ice-cold TBS followed by 10% NBF. Brains were dissected, kept in formalin overnight and dehydrated before paraffin embedding. 5-micron paraffin sections were rehydrated (briefly, slides were incubated in Xylene x10 min (x2), 100% ethanol x5 min (x2), then 95% ethanol x5 min (x2), then 70% ethanol (x1), then washed briefly in diH2O) prior to antigen-retrieval at 95°C for 40 min using a citrate-based buffer (Retrieve-all system 1, Biolegend). After three TBST washes, the sections were incubated in blocking solution (5% donkey serum solution / TBST) for 1h at room temperature. The primary antibodies (diluted 1:200 in blocking solution) were incubated overnight at 4°C. After three TBST washes, secondary antibodies diluted 1:500 (Alexa Fluor 488, Alexa Fluor 555, Alexa Fluor 647; Invitrogen, Molecular probes) and DAPI (5ug/ml, Biolegend) was added to the sections and incubated for 1 hour at room temperature and washed in TBST before mounting in Prolong Gold Antifade reagent (Invitrogen) and analysis by epifluorescence microscopy (Nikon Eclipse 90i, Nikon Instruments). The following antibodies were used: AQP1 (Abcam, Rabbit recombinant monocolonal, ab168387), ATP1A1 (Alomone, rabbit polyclonal, ANP-001), CD68 (CST, rabbit monoclonal, 97778), CLIC6 (Santa Cruz, mouse monoclonal, 365303), ED1/CD68 (Millipore, mouse monoclonal, MAB1435), IBA1 (Thermo, goat polyclonal, PA5-18039), KCNJ13 (Santa Cruz, mouse monoclonal 398810), Ki67 (CST, rabbit monoclonal 9129), pNKCC1 (Millipore, rabbit polyclonal, ABS 1004), pSPAK (Millipore, rabbit polyclonal, 07-2273), RELT (Novus, rabbit polyclonal, NBP2-56851), NKCC1 (CST, rabbit monoclonal, 85403S). Colocalization analysis was performed using the Fiji Coloc 2 plugin141.
Tissue dissociation and flow cytometry
Animals were anesthetized with Ketamine/Xylazine and transcardially perfused with ice cold TBS before brain removal. Choroid plexi from bilateral lateral ventricles were microdissected in ice cold HBSS and digested at 37°C for 20 min with Collagenases-I (10 U/mL) and -IV (400 U/mL) and DNaseI (30 U/mL) (this protocol was adapted from Van Hove et al58). The cell suspension was blocked with CD16-CD32 antibodies and stained 30 min at 4°C with anti-rat CD45-PE/cy7, CD3-647 or corresponding isotype antibodies (Biolegend) and DAPI- co-stained to monitor cell viability. Cells were subsequently washed in FACS staining buffer (0.5% BSA, 2mM EDTAin PBS without Calcium/Magnesium) and centrifuged at 350 rcf for 5 minutes. For FACS, cells were resuspended in 300ul of FACS buffer and sorted on a BD ARIA instrument, with gating and analysis by FlowJo software.
Antibodies for western blots
Primary antibodies used were: anti-WNK1 (University of Dundee, S079B), anti-SPAK (University of Dundee, S365D), anti-NKCC1 total antibody (University of Dundee, S022D), anti-Na(+)/K(+) ATPase alpha-1 (ATP1A1) antibody (DSHB, a5), anti-beta Actin antibody (Abcam, ab8227). Horseradish peroxidase-coupled (HRP) secondary antibodies used for immunoblotting were from Pierce. IgG used in control immunoprecipitation experiments was affinity-purified from pre-immune serum using Protein G-Sepharose.
Buffers for Western blots
Buffer A contained 50 mM Tris/HCl, pH 7.5 and 0.1 mM EGTA. Lysis buffer was 50 mM Tris/HCl, pH 7.5, 1 mM EGTA, 1 mM EDTA, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1% (w/v) NP40, 0.27 M sucrose, 0.1% (v/v) 2-mercaptoethanol, and protease inhibitors (complete protease inhibitor cocktail tablets, Roche, 1 tablet per 50 mL). TBS-Tween buffer (TTBS) was Tris/HCl, pH 7.5, 0.15 M NaCl and 0.2% (v/v) Tween-20. SDS sample buffer was 1X NuPAGE™ LDS Sample Buffer (NP0007, Invitrogen™), containing 1% (v/v) 2-mercaptoethanol. Protein concentrations were determined following centrifugation of the lysate at 16,000 x g at 4°C for 20 minutes using the using a Pierce™ Coomassie (Bradford) Protein Assay Kit (23200, Thermo Scientific) with bovine serum albumin as the standard.
Immunoblot and immunoprecipitation analyses
Rat choroid plexus lysates were subjected to immunoblot and immunoprecipitation as previously described30,142, and is detailed here. Rat choroid plexus lysates (20 μg) were boiled at 75°C in sample buffer for 10 minutes, resolved by 7.5% sodium dodecyl sulfate polyacrylamide-gel electrophoresis and electrotransferred onto a polyvinylidene difluoride membrane. Membranes were incubated for 30 min with TBST (Tris-buffered saline, 0.05% Tween-20) containing 5% (w/v) skim milk. Blots were then washed three times with TBST and incubated for 1 h at room temperature with secondary HRP-conjugated antibodies diluted 5000-fold in 5% (w/v) skim milk in TBST. After repeating the washing steps, signals were detected with enhanced chemiluminescence reagent. Immunoblots were developed using ChemiDoc™ Imaging Systems (Bio-Rad, Feldkir). Figures were generated using Photoshop/Illustrator (Adobe).
SPAK was immunoprecipitated from rat choroid plexus lysates clarified by centrifugation at 16,000 x g at 4°C for 20 minutes, using SPAK antibody coupled to protein G–Sepharose30,142 at a ratio of 1 mg antibody per 1 mL beads. Clarified cell lysate (2 mg) was incubated 2 h at 4°C with 15 μg of antibody conjugated to 15 μL of protein-G–Sepharose with gentle agitation. Beads were washed three times with 1 mL lysis buffer containing 0.15 M NaCl and twice with 1 mL buffer A. Bound proteins were eluted with 1X LDS sample buffer.
qPCR analysis
Microdissected tissues were snap frozen and stored at −80°C until total RNA was extracted using RNeasy Micro plus kit (Qiagen, 74034) and reverse transcribed (Biorad, 170-8890). Gene expression analysis was determined by quantitative reverse-transcription PCR (Bimake, B21202) using a CFX96 PCR machine (Bio-Rad). The qPCR primer sequences are provided in Table S5. The data were expressed using the comparative threshold cycle (ΔCT) method (GOI, Gene Of Interest vs. Gapdh), and the mRNA ratios were given by 2−ΔCT compared to Housekeeping genes. Gapdh expression was also compared to another Housekeeping Gene (Hprt1), as an additional control.
Bulk RNAseq
Full length high quality total cell RNA isolated by Qiagen RNAeasy micro kit (Quigen, https://www.qiagen.com/us/shop/pcr/rneasy-micro-kit/). ChP cells were homogenized in 350ul RLT buffer with beta-mercaptoethanol in Tissuelyzer for 10 mins and proceed according to the instructions in RNAeasy Micro RNA kit and RNAs was eluted in 18ul dH2O. 16ul of elutant was recovered and 1.2ul was used for the QC. Libraries were run on an Illumina HiSeq 2500 at the Yale Center for Genome Analysis. Expression level for each gene was estimated using Python program HTSeq143. Specifically, htseq-count function of HTSeq accumulated the number of aligned reads that falls under the exons of the gene. The R/Bioconductor package “DESeq2” was used to identify differentially expressed genes between cell populations derived from control, IVH-treated and LPS-treated rats144. Genes with a log2-fold change expression greater than 3 and a q-value less than 0.01 were considered significant. The R ComplexHeatmap package was used to visualize the heatmap expression of highly variable genes145. Volcano plots were generated using the R package EnhancedVolcano.
scRNAseq sample preparation
Animals were anesthetized with Ketamine/Xylazine and transcardially perfused with ice cold TBS before brain removal. Choroid plexi from bilateral lateral ventricles were microdissected in ice cold HBSS with 30uM Actinomycin D (ActD, Sigma, cat#A1410 ) and digested at 37°C for 20 min with Collagenases-I and -IV (10 U/mL, 400 U/mL final, Worthington, Cat# LS004194, and LS004184) and DNase I (30 U/mL, Roche, cat# 04716728001) and ActD (3uM) (this protocol was adapted from Van Hove et al58). For Flow sorting, the cell suspension was blocked with CD16-CD32 antibodies (1:100, biolegend, cat#101302) and stained 30 min at 4°C with anti-rat CD45-PE/cy7 (1:100, biolegend, cat#202207) to and DAPI (50ug/mL). Cells were subsequently washed in FACS staining buffer (0.5% BSA, 2mM EDTA in PBS without Calcium/Magnesium) and centrifuged at 350 rcf for 5 minutes. Cells were resuspended in 300ul of FACS buffer and sorted by CD45 positivity on a BD ARIA instrument. CD45 positive and CD45 negative cells were collected in 0.4% BSA/PBS and 3uM ActD and sent for 10X 3prime library preparation and sequencing at Yale Center for Genome Analysis.
Anesthesia and CM catheter placement
The rats were anesthetized with ketamine-xylazine (90 mg/kg bodyweight ketamine plus 7.5 mg/kg bodyweight xylazine, i.p.) and after loss righting reflex they breathed an Air:O2 mix (1:1) spontaneously via a nose cone. Anesthesia was maintained with KX administered every 30 min via an IP catheter. When surgical plane anesthesia was assured by toe pinch before placing the rats in a stereotaxic frame. The dura above the cisterna magna (CM) was exposed and a small 5-mm copper tube (0.32 mm o.d., Nippon Tockushukan, MFG. CO., LTD, Tokyo, Japan) attached to a PE 10 microcatheter was positioned into the CSF compartment and secured in place using cyanoacrylate glue. After surgery, the rats were transferred to the MRI animal bed equipped with a heating waterbed and nose cone.
Magnetic resonance imaging (MRI)
All MRI acquisitions were performed on a Bruker 9.4T/16 magnet (Bruker BioSpin, Billerica MA) interfaced to an Avance III console controlled by Paravision 6.0 software. We used a 30-mm ID planar surface radiofrequency (RF) coil (Bruker BioSpin, Billerica, MA, USA) which was placed under the head of the rats for RF signal reception and a custom-made volume transmit RF coil designed with an internal diameter of 50 mm was used as RF signal transmitter. While in the MRI instrument, the rats were in supine position and breathed an Air:O2 mix (1:1) spontaneously via a nose cone. Heart rate (HR) via oxygen saturation monitoring, respiratory rate and body temperature were continuously monitored (SA Instruments, Stony Brook, NY, USA). The body temperature was kept within ~ 37.0°C using a heated waterbed and oxygen saturation was kept ≥ 97% during the MRI experiments. Brain morphometry: A single flip angle spoiled gradient echo (SPGR) sequence was used to acquire 3D proton density weighted (PDW) MRIs: (repetition time (TR) = 50ms, echo time (TE) = 4ms, flip angle (FA) = 7°, Average = 2, field of view (FOV) = 30x30x15mm, spatial resolution = 0.234x0.234x0.234mm, scan time ~ 14min. Dynamic contrast enhanced MRI: We acquired pre-contrast (baseline) T1-weighted images and post-contrast T1-weighted imaging of the whole brain. The 3D T1 weighted scans were acquired using a single flip angle spoiled gradient echo (SPGR) sequence: TR=15ms, TE=4ms, FA=15°, Average = 2, FOV = 32x30x30mm, the spatial resolution= 0.302x0.300x0.300mm, acquisition time/scan = 5mins). After three baseline T1-weighted scans, 30 μL of 1:10 gadoteric acid (Gd-DOTA, DOTAREM, Guerbert LLC, Carol Stream IL) diluted in sterile water was infused at a rate of 1.5 μL/min into CSF through the CM catheter using an infusion pump for a total of 20 min. Post-contrast T1-weighted scans continued for a total 160 min.
Porcine choroid plexus epithelium harvesting
All porcine studies were performed in collaboration with the Dardik laboratory at Yale University and in compliance with federal guidelines and protocols approved by the Yale University Institutional Animal Care and Use Committee. Male Yorkshire pigs of ~48 kg and aged 3.4 months were euthanized via lethal injection of Euthasol (0.22 ml/kg)54. Euthanized pigs were placed prone on the operating table for brain dissection. A semicircular scalp incision was made around the vertex of the cranium, the skin was retracted, and the periosteum removed. Using an oscillating bone saw (Dremel Multi-max; Illinois, USA) a frontal craniectomy was performed to expose the underlying dura and cerebral hemispheres. The dura was excised at the midline to gain access to the superior sagittal sinus. The cerebral hemispheres were separated using surgical forceps and the lateral and third ventricles were exposed. The choroid plexus, identified as a highly vascular intraventricular structure, was carefully removed via forceps and placed into sterile saline on ice for molecular analysis.
Mass-spectrometry (MS) proteomics
Frozen pellets of ChP cells were lysed by sonication in RIPA buffer plus 1× PhosSTOP phosphatase inhibitor (Roche) and 1× cOmplete protease inhibitor cocktail (Roche). Protein (200 μg) for each sample underwent reduction, alkylation, and chloroform-methanol precipitation. Samples were resuspended in 100 mM TEAB (pH 8.5) and digested with trypsin (Promega, 1:50 w/w) overnight at 37°C. Digested samples were acidified and desalted on a C18 Macro Spin Column (The Nest Group). Peptides were eluted in 80% acetonitrile/0.1% trifluoroacetic acid (TFA), then dried by a SpeedVac. The dried peptide pellet from each sample was then TMT labeled according to manufacturer’s protocol (Thermo Fisher Scientific) using a TMT10Plex Isobaric Labeling Reagent Kit (Cat#:90110). A label efficiency test was performed on 1/50th of each sample prior to equally mixing the 10 channels, then samples were dried prior to fractionation. Pooled TMT labeled peptides were then dissolved in buffer A and injected onto a Waters ACQUITY UPLC (BEH) C18 column (Waters XBridge BEH130 C18 3.5 μm 4.6 × 250 mm) at a flow rate of 0.4 mL/min. An orthogonal high pH reverse phase separation was carried out using a Waters H-class UPLC system with Buffers (A:95% water/5% acetonitrile (ACN) with 10 mM ammonium acetate, pH 10; B: 5% water/95% acetonitrile (ACN) with 10 mM ammonium acetate, pH 10) were used to separate the peptides with a 60 min gradient 15% – 55% with buffer B. Total 60 fractions were collected. The fractions were then pooled into 8 fractions, dried and reconstituted in 0.1% formic acid.
Mass spectrometry data were obtained on an Orbitrap Fusion Tribrid Mass Spectrometer (ThermoFisher Scientific, San Jose, CA, USA) coupled to a nanoACQUITY UPLC system (Waters Corporation, Milford, MA, USA) in the Keck MS & Proteomics Resource at Yale University as described in Lee et. al146, and detailed here. Individual pooled fractions were loaded into a trapping column (nanoACQUITY UPLC Symmetry C18 Trap column, 180 μm × 20 mm and separated with a C18 column (nanoACQUITY column Peptide BEH C18, 75 μm × 250 mm) at a flow rate of 5 μL/min. Peptides were eluted with a gradient from 6% to 20% mobile phase B over 120 min and then to 40% mobile phase B for another 45 min at a flow rate of 300 nL/min at a flowrate of 300 nL/min with buffers (A: 0.1% formic acid in water and B: 0.1% formic acid in acetonitrile). The data were acquired with data-dependent mode with multinotch synchronous precursor selection (SPS)-MS3 scanning for TMT tags as described previously147. The ions above with an intensity threshold of 5000 was selected for collision-induced dissociation (CID)-MS fragmentation. The top 10 fragment ions for each peptide MS2 were notched out and co-fragmented with higher-energy collision dissociation (HCD) to produce MS3 scans and were analyzed in the Orbitrap at a resolution of 60,000.
The proteomics raw data files were processed and quantified by MaxQuant (https://maxquant.net/maxquant/) version 1.6.1.0 with Andromeda search engine using the Rat UniProt FASTA database (http://coxdocs.org/doku.php?id=maxquant:andromeda:start). Default value of parameters were used in Maxquant unless explicitly stated. Briefly, false-discovery rate (FDR) was set 1% for proteins and peptides respectively and match between runs was enabled with a default time window of 0.7 min. Search results (TMT reporter ion intensities) were exported as text files. The differentially expressed proteins were identified using the Perseus framework (https://maxquant.net/perseus/). Proteins with a log2-fold change expression greater than 1 and a q-value less than 0.05 were considered significant. The R ComplexHeatmap package was used to visualize the heatmap expression of highly variable genes145.
SPAK Immunoprecipitation for LC-MS/MS
ChP cells were lysed in a lysis buffer containing protease and phosphatase inhibitor by incubating 10 min on ice and then centrifuged at 18.000 g for 5 min at 4°C. Supernatants containing the proteins were first precleared with Protein G beads for 30 min at 4°C and then were split in half and transferred into two clean 1.5 mL Eppendorf tubes. One half of the supernatant was incubated with Protein G beads only (IgG) as control for non-specific binding; the other half of the supernatant was incubated with Protein G beads + anti-SPAK 1°Ab. Incubation with gentle agitation was conducted at 4°C for overnight. Supernatant was removed and beads were washed 4 times with RIPA containing 150mM NaCl. Bound proteins were then eluted twice with 2M urea (for in-solution trypsin digestion) or SDS sample buffer for 5 minutes (In-gel trypsin digestion). The elutions of SDS samples buffer were then loaded onto a mini SDS PAGE gel (4-12%, Bio-Rad Laboratories, Hercules, CA). Gel bands/plugs were excised and processed for LCMS/MS analysis148. Gel bands/plugs were first cut into small pieces and subjected to the following washes with agitation: 50% (v/v) acetonitrile (5 min), 50% (v/v) acetonitrile/10 mM NH4HCO3 (30 min). Gel pieces were dried with a SpeedVac, resuspended in 30 μL of 10 mM NH4HCO3/0.2 μg digestion grade trypsin (Promega), and incubated for 16 h at 37 °C. Peptides were then acidified with 0.1% (v/v) trifluoroacetic acid (TFA) prior to LC MS/MS analysis on an Orbitrap Fusion mass spectrometer coupled to a nanoACQUITY UPLC149 located within the Keck MS & Proteomics Resource at Yale.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis
Statistical analysis of CSF secretion, CSF cytokine analysis, ventriculomegaly, and IHC quantification used Prism (GraphPad, San Diego, CA) for one-way ANOVA analysis with Tukey’s multiple comparisons, unpaired t-test, or paired t-test. Transcriptomic and proteomic data was analyzed using RStudio (RStudio Inc., Boston, MA). Non-parametric Wilcoxon rank sum test was used to determine statistically significant gene markers in scRNAseq datasets. Numerical data in text and figures are presented as mean ± standard error of the mean (SE). Rates (μL/min) were calculated as the slope of the volume-time relationship, based on data collected over 20 min or more. Rates were determined for individual animals and averaged across individuals. Sample size calculations were based on two previous studies that used the same model of IVH 30 and method for measuring rates of CSF production 30,53. The statistical details of each experiment can be found in the figures and corresponding figure legends (including the specific statistical test used, value of n and what each n represents, and the dispersion and precision measures used), except for the scRNAseq datasets. Details for the specific analysis used for the scRNAseq datasets are detailed in the Method Details of the STAR Methods section. Any additional information can be addressed via contact with the corresponding article of this manuscript.
snRNA-seq rat ChP data analysis
As described previously2, the preprocessing and clustering analysis for snRNA mouse choroid plexus cell dataset was completed using Seurat150. For each nucleus, the number of genes for which there was at least one mapped read, was quantified, and then all nuclei with fewer than 500 detected genes were excluded. Furthermore, all nuclei with more than 10% of total UMI’s mapped to mitochondrial genes were excluded. Genes detected in fewer than 10 nuclei were excluded. Expression values Ei,j for gene i in nucleus j were calculated by dividing UMI counts for gene i by the sum of the UMI counts in nucleus j, to normalize for differences in coverage, and then multiplying by 10,000 to create TPM-like values (TP10K), and finally computing log2(TP10K + 1).
Selection of variable genes was performed by fitting a logistic regression model to the cellular detection fraction (often referred to as α), using the total number of UMIs per cell/nucleus as a predictor, as in Montoro et al., 2018151. Outliers from this curve are genes expressed in a lower fraction of cells/nuclei than would be expected, given the total number of UMIs mapping to that gene; that is, likely cell-type or state-specific genes. A threshold of deviance between < −0.15 and < −0.3 was used.
The expression matrix was restricted to the subsets of variable genes and high-quality cells/nuclei noted above, and then centered and scaled values before inputting them into principal component analysis (PCA), implemented using ‘RunPCA’ in Seurat which runs the irlba function. For the snRNA-seq data, which included mixed sexes, Y chromosome and X-inactivation genes (Xist, Tsix) were removed from the variable genes prior to running PCA. The cell embeddings were either the singular vectors themselves or the singular vectors multiplied with the singular values depending on the cell type. After PCA, significant principal components were identified using the elbow-method, when looking at the distribution of singular values. Scores from only those significant principal components were used as the input to further analysis. To embed cells in a graph structure, first a KNN graph was constructed based on the euclidean distance in PCA space, and the edge weights were refined between any two cells based on the shared overlap in their local neighborhoods (Jaccard similarity). This step was performed using the FindNeighbors (reduction=‘pca’, dims=1:50) command by taking the first 50 principal components as input. To cluster the cells in an unsupervised fashion, the Louvain algorithm (default) was applied to iteratively group cells together, with the goal of optimizing the standard modularity function. The FindClusters(resolution=0.5) function was implemented for this procedure. For visualization purposes, the dimensionality of the datasets was further reduced to 2D embeddings using UMAP on the significant PCs via RunUMAP() functions of the Seurat package in R152. Non-parameteric Wilcoxon rank sum test was used to identify differentially expressed markers across all clusters by running FindAllMarkers(dataset, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25). VlnPlot(), which shows expression probability distributions across clusters, and FeaturePlot(), which visualizes feature expression on a UMAP plot) were used to visualize key genes differentially expressed for individual clusters. Adult epithelial cells were isolated and further sub-clustered in an unsupervised fashion using the Louvain algorithm.
Nucleus doublets were identified using the R package DoubletFinder (version DoubletFinder_2.0.2153. Briefly, artificial doublets are generated from the data, and for each real data point the fraction of artificial doublets of the nearest neighbors in a latent space is computed. If this fraction is above a certain cutoff, the data point is a doublet. Doublets were calculated for each sample separately after initial data filtering by genes detected and mitochondrial gene expression as per instructions. For each sample, an optimal pK (defines the PC neighborhood size) was chosen by computing a parameter sweep (paramSweep_v3 function), and a pK was chosen that maximized the BCmvn (mean-variance-normalized bimodality coefficient). Doublets were then calculated using the function doubletFinder_v3() with parameters; PCs = 1:30, pN = 0.25, nEXP = 15% of cells in the sample, and removed from downstream analysis.
PIH and PHH rat ChP scRNAseq datasets
The preprocessing and clustering analysis for scRNA rat choroid plexus CD45+ and CD45− cell datasets for was completed using Seurat 150. Cells with low numbers of genes (200), low numbers of UMIs (500) or high numbers of UMIs (>10000) and mitochondrial gene percentage greater than 10% were removed. The normalization and initial feature selection for each of the six datasets (control, IVH-treated, LPS-treated conditions for CD45+ and CD45− cells) were completed individually. The filtered matrices were normalized using ‘LogNormalize’ methods in Seurat using a size factor of 10,000 molecules for each cell. 2,000 most variable genes were identified by variance stabilizing transformation for each dataset by implementing the FindVariableFeatures function in Seurat v4 (selection.method = “vst”). The experimental conditions were integrated using Seurat’s integration pipeline for CD45+ and CD45− cell datasets. For integration, 2,000 shared highly variable genes were identified using Seurat’s ‘SelectIntegrationFeatures()’ function. Integration anchors were identified based on these genes using canonical correlation analysis using the ‘FindIntegrationAnchors()’ function. The data were then integrated using ‘IntegrateData()’ and scaled again using ‘ScaleData()’. Overall, integrated CD45+ dataset consisted of 1427 control, 3331 IVH-treated and 5529 LPS-treated cells whereas integrated CD45− dataset consisted of 2036 control, 718 IVH-treated and 1603 LPS-treated cells.
Principal component analysis (PCA) and uniform manifold approximation and projection (UMAP) dimension reduction with 50 principal components were performed. A KNN graph was constructed based on the euclidean distance in PCA space to embed cells in a graph structure, and the edge weights were refined between any two cells based on the shared overlap in their local neighborhoods (Jaccard similarity). This step was performed using the FindNeighbors (reduction=‘pca’, dims=1:50) command by taking the first 50 principal components as input. To cluster the cells in an unsupervised fashion, the Louvain algorithm (default) was applied to iteratively group cells together, with the goal of optimizing the standard modularity function. The FindClusters(resolution=0.4) function was implemented for this procedure. For visualization purposes, the dimensionality of the datasets was further reduced to 2D embeddings using UMAP on the significant PCs via RunUMAP() functions of the Seurat package in R152. Non-parameteric Wilcoxon rank sum test was used to identify differentially expressed markers across all clusters by running FindAllMarkers(dataset, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25). Further sub-clustering of certain cell populations imcluding myeloid cells, T-cells, epithelial cells were conducted in an unsupervised fashion using the Louvain algorithm. The R ComplexHeatmap package was used to visualize the heatmap expression of highly variable genes145. VlnPlot, FeaturePlot and DotPlot functions were used to visualize the gene expression profiles across clusters and conditions
To infer cell–cell interactions based on the expression of known ligand–receptor pairs in different cell types, CellChat was applied to our experimental conditions after merging CD45+ and CD45− for each condition. The official workflow and databases were implemented. Briefly, the normalized counts were loaded into CellChat after which the preprocessing functions identifyOverExpressedGenes, identifyOverExpressedInteractions and projectData with standard parameters set were applied to our datasets. The main analyses were conducted using the functions computeCommunProb, computeCommunProbPathway and aggregateNet with fixed randomization seeds.
ScRNA-seq mouse ChP immune cell data analysis
As described previously 2, the preprocessing and clustering analysis for ScRNA mouse choroid plexus immune cell dataset was completed using Seurat 150. Outlier cells were first identified based on three metrics (library size, number of expressed genes and mitochondrial proportion); cells were tagged as outliers when they were four median absolute deviations distant from the median value of each metric across all cells. Secondly, a principal component analysis plot was generated based on the following metrics:‘pct_counts_in_top_100_features’,‘total_features_by_counts’,‘pct_counts_feature_control’,‘total_featur es_by_counts_feature_control’, ‘log10_total_counts_endogenous’ and ‘log10_total_counts_feature_control’. Outlier cells in this principal component analysis plot were identified using the R package mvoutlier. Low-abundance genes were removed using the ‘calcAverage’ function and the proposed workflow. The raw counts were normalized and log2 transformed by first calculating ‘size factors’ that represented the extent to which counts should be scaled in each library. Highly variable genes were detected using the proposed workflow of the scran R package and by applying false discovery rate ≤0.05 and var.out$bio ≥0.01 as cutoffs. Highly variable genes were subsequently used for unsupervised dimensionality reduction techniques and principal component analysis.
To embed cells in a graph structure, first a KNN graph was constructed based on the euclidean distance in PCA space, and the edge weights were refined between any two cells based on the shared overlap in their local neighborhoods (Jaccard similarity). This step was performed using the FindNeighbors (reduction=‘pca’, dims=1:50) command by taking the first 50 principal components as input. To cluster the cells in an unsupervised fashion, the Louvain algorithm (default) was applied to iteratively group cells together, with the goal of optimizing the standard modularity function. The FindClusters (resolution=0.5) function was implemented for this procedure. For visualization purposes, the dimensionality of the datasets was further reduced to 2D embeddings using UMAP on the significant PCs via RunUMAP() functions of the Seurat package in R152. Non-parameteric Wilcoxon rank sum test was used to identify differentially expressed markers across all clusters by running FindAllMarkers (dataset, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25). VlnPlot(), which shows expression probability distributions across clusters, and FeaturePlot(), which visualizes feature expression on a UMAP plot) were used to visualize key genes differentially expressed for individual clusters.
Induction of immediate early genes (IEGs) was observed in some of the Choroid Plexus cells in which two clearly separated macrophage clusters differed only in expression of IEGs and heat shock proteins were observed. These differentially expressed genes closely resembled those identified in a subset of muscle satellite cells and shown to be dissociation-induced genes. Single-cell isolation using either a standard or ActD modified protocol was compared to confirm that the IEGs induced in our clusters were also dissociation-induced. This allowed us to identify 224 genes that were upregulated in standard versus ActD samples in Choroid Plexus Border Associated Macrophages, comprising the dissociation-induced gene list. To correct for dissociation-induced gene expression, which may mask biologically relevant results, this gene list was used to detect cells that were ‘dissociation-affected’ by plotting expression of all these genes over all cells. The corresponding histogram revealed a clear cutoff that split the cells between ‘dissociation-affected’ and unaffected cells. This information was added to the metadata attribute of the Seurat object, and this unwanted source of variation was subsequently removed using the ‘Regress out’ function of the Seurat R package.
Gene co-expression network creation
The gene co-expression modules for the preprocessed and clustered choroid plexus dataset were constructed by using Monocle3’s differential expression analysis pipeline154. Monocle3’s graph_test (neighbor_graph="knn") function was used to identify genes that vary between groups of cells in UMAP space. This function employs a statistic from spatial autocorrelation analysis called Moran’s I, which Cao et al. showed to be effective in finding genes that vary in single-cell RNA-seq datasets154. Genes with FDR-adjusted p-values of less than 0.05 were used to construct gene co-expression networks via UMAP and community analyses. 21 gene modules were created using this pipeline.
Hypergeometric enrichment test analysis
Module gene lists were obtained via Monocle3 as described above. Hypergeometric enrichment analysis was conducted using the R package hypeR155. Syndromic and non-syndromic gene lists for both high confidence and probable genes were used to find modules enriched with Craniosynostosis-related genes. Modules with FDR adjusted p-values less than 0.05 were considered significant.
Cluster abundance analysis
In order to test the difference in cell counts for IVH-treated and LPS-treated ChP compared to control, we used beta-binomial generalized linear model in package aod::betabin for both CD45+ and CD45− clustering. In our generalized linear model, we set the count of the cell type of interest and the total count of cells of each experimental condition to be the response variable and the experimental condition of the cells to be the independent variable. We tested for Pearson’s correlation between the frequency of each cell cluster. We adjusted the p value threshold using Bonferroni correction (0.05/32 = 0.00156 for CD45+ dataset and 0.05/20 = 0.0025 for CD45− dataset).
Gene Ontology enrichment analysis
The gene and protein lists for differentially expressed genes and proteins for IVH and LPS conditions from the RNAseq and proteomics experiments, as well as the modules significantly enriched with these genes/proteins, were further studied for gene ontology, pathway and upstream transcription factor enrichment using the Enrichr R package156. The Enrichr contains a diverse and up-to-date collection of over 100 gene set libraries available for analysis and download. The databases for these analyses include gene ontologies (biological processes, cellular components, and molecular functions), biological pathways (Wiki pathways Human and Mouse) and upstream transcription factors (TRANSFAC and JASPAR). Adjusted p-value of less than 0.05 was considered significant and combined Z-score was used for ranking.
MRI data analysis
Morphometry of CSF and brain parenchyma (grey matter + white matter) volumes for each rat was performed as previously described (ref). Briefly, The 3D PDW images were corrected for intensity inhomogeneity using the N4 bias field correction algorithm157, and then segmented into grey matter (GM), white matter (WM) and CSF brain compartments to calculate their brain parenchymal (GM+WM) and CSF volumes. All the spatial segmentations were performed on SPM12 (http://www.fil.ion.ucl.ac.uk/spm) software package platform, using our previously custom-made tissue probability maps158. The tissue probability maps generated by segmentation of the individual rat brains were thresholded at 0.5, yielding GM, WM, and CSF binary masks in native space. Spatially normalized average CSF compartment volume maps of the sham controls (N=5) and LPS treated (N=6) rats were subsequently created. In addition to SPM analysis, for each rat manual segmentation of the lateral ventricle volume was performed on the PWD and T1-weighted baseline MRIs using PMOD (PMOD, version 3.908, PMOD Technologies LLC, Zürich, Switzerland). Analysis of DCE-MRI data included correction for head motion, followed by intensity normalization, smoothing, and then voxel-wise percent signal change to baseline was calculated using SPM12 (https://www.fil.ion.ucl.ac.uk/spm/)159. The ‘percent signal change from baseline images’ were then used for optimal mass transport (rOMT) analysis160. From each rat’s whole brain DCE-MRI data series the rOMT analysis produced whole brain binary pathlines endowed with solute speed (representing the local speed and spatial distribution of the pathline networks) from which glymphatic flux and mean tissue solute speed was derived.47
Supplementary Material
Supplementary Fig. 1. LPS mediates acute and chronic ventriculomegaly, with preserved patency of the cerebral aqueduct and glymphatic transport, and increases cytokine/chemokine expression, Related to Figure 1. (A) Quantification of lateral ventricular volume at 72 hours and 14 days after an initial 72h infusion of aCSF (control) or LPS. Volume calculated using sequential slices through entire lateral ventricular system (n=5-6 animals per condition). (B-H) Spatially normalized population averaged CSF probability maps overlaid on proton density weighted anatomical MRI brain images of live, anesthetized animals treated with aCSF (control) (B, C) or LPS (D, E). CSF filled ventricles are represented as high intensity signal areas. Top panels (B, D) show MRI images (axial plane view) at the level of the lateral ventricles obtained from voxel wise morphometric analysis of the CSF compartment and bottom panels (C, E) show the corresponding 3D volume reconstructions of the lateral ventricles. (F) Morphometric quantification of the lateral ventricular volume (% brain volume) and total intracranial volume (mm3) for control (sham) and LPS treated PIH animals. (G, H) CSF contrast enhanced T1-weighted MRI images (sagittal plane view) of live, anesthetized animals demonstrating patency of the cerebral aqueduct (Aq) in control (G) and PIH animals (H). (I, J) Average glymphatic flux with superimposed color-coded solute speed maps (μm/s) for control (I) and LPS-treated PIH animals (J). Representative speed maps demonstrating glymphatic transport in sagittal (top panels) and axial (bottom panels) MRI view planes. (K) Quantification of glymphatic transport metrics including glymphatic v-flux (mm3) and glymphatic mean tissue speed (mm/s). (L) Schematic illustrating in vivo CSF secretion measurements. Mineral oil is administered into the cerebral aqueduct (aqueduct) through a catheter (yellow line and arrow) inserted into the aqueduct, which blocks flow of cerebrospinal fluid (CSF). A glass capillary tube is then placed through a burr hole in the skull into the lateral ventricle (blue line and arrow). The distance of CSF flow through the capillary tube is measured using a ruler (black) and rate is calculated over time. (M) CSF cytokine/chemokine expression (from Fig. 2) concentrations (from Fig. 2, pg/mL) of Il-1b, Il-6, Il-10, Ifnγ, Il-17a, Il1-8, Ccl2, Cxcl1, Vegf, Cxcl5, Tnfα, Ccl5, IL-10, Mip-1α, and Il-5 in Ctl and 72h LPS-treated Tlr4+/+ and Tlr4−/− rats. For all experimental conditions, n=4-6 animals per condition. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant, by one-way ANOVA (A) and unpaired t-test with Welch correction (K)
Supplementary Fig. 2. Choroid plexus, meningeal, and cortical immune reaction in PIH animals treated with E. coli +LPS, Related to Fig. 2. (A) Illustration of a representative coronal brain slice (left panel) indicating location of the ChP (black box) imaged for analysis. Representative IHC (right panel) of Iba1+ (red) and ED1+ (blue) positive cells in Ctl and E. coli +/− LPS rat ChP in single-channel IHC (Dapi, white), as well as merged images. (B-C) Quantification of (A) showing DAPI-normalized % ED1+ (B) and Iba1+ (C) cells. (D) Illustration of a representative coronal brain slice (left panel) indicating location of the meninges (black box) imaged for analysis. Representative IHC of Iba1+ (red) and ED1+ (blue) positive cells in Ctl and E. coli +/− LPS rat meninges in single-channel IHC (Dapi, white), as well as merged images. (E-F) Quantification of (D) showing DAPI-normalized % Iba+ (E) and ED1+ (F) cells. (G) Illustration of a representative coronal brain slice (left panel) indicating location of the cortex (black box) imaged for analysis. Representative IHC of Iba1+ (red) and ED1+ (blue) positive cells in Ctl and E. coli +/− LPS rat meninges in single-channel IHC (Dapi, white), as well as merged images. (H-I) Quantification of (G) showing DAPI-normalized % Iba+ (H) and ED1+ (H) cells. Scale bars, 20μm; n=4-6 animals per condition. Error bars, mean ± sem; each symbol represents one animal. *p<0.05, **p<0.01, ****p<0.0001, by one-way ANOVA.
Supplementary Fig. 3. SPAK kinase transduces pro-inflammatory CSF signals in ChP epithelial cells to stimulate NKCC1-dependent CSF secretion, Related to Fig. 2. (A) UMAP of single nucleus transcript profiles of adult ChP-associated cells2, colored by cell type. (B) Tlr4 expression in Chp-associated cells (Avg Log2FC = 1.0; p=5.14e-214). (C) UMAP of ChP epithelial cell subclusters, colored by group and by expression of (D) Tlr4, (E, left) interleukin-1 receptor accessory protein (Il1rap) (Avg Log2FC=0.96, p<10e-300), and (E, right) interleukin 6 receptor (Il6ra) (Avg Log2FC=0.23, p=1.87e-211). (F) Quantitative PCR (qPCR) delta-delta Ct (ddCT) of cytokines Il1b, Tnfa, Mcp1, Ifng, and their receptors Il1r1, Relt, Ccr2, IfngrI1 (n=5 animals per condition; see Table S5*). (G) Representative IHC of ChP TNF receptor RELT (red) and Iba1 (white) in Tlr4+/+ and Tlr4−/− Ctl and LPS-treated animals (inset, 4x magnification). (H) Quantitation of (G) (normalized intensity density (IntDen)/cell #) (n=6 ChP per genotype, 3 animals per condition). (I) UMAP of cell ontology (left, as in (A) and violin plots (right) showing expression of Stk39 (Avg Log2FC=1.1, p<10e-300; Epithelial, Avg Log2FC=4.46, p=4.22e-233) and (J) Slc12a2 (Avg Log2FC=1.3, p<10e-300; Epithelial, Avg Log2FC=2.70, p=8.15e-215). (K) Representative IHC of phospho-activated NKCC1 (pNKCC1; green, top row) and phospho-activated SPAK (pSPAK; red, bottom row) in Ctl and LPS-treated WT, Tlr4−/− and Spak−/− rats. Quantitation (Interval Density/cell #) of (L) pNKCC1 and (M) pSPAK from (K) (n=6 ChP per genotype, 3 animals per condition). (N) Body weight-normalized CSF secretion rate (μL/min/kg) and (O) lateral ventricular size (% brain volume) in Spak+/+ Ctl, Spak+/+ LPS, Spak−/− Ctl, and Spak−/− LPS rats (n=5-6 animals per condition). Scale bars, 25 μm. Error bars, mean ± sem; each symbol represents one animal. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant; (F) Unpaired t-test; (H, L-O) one-way ANOVA.
Supplementary Fig. 4. Relt expression in Spak KO rats and ChP expression of Atp1a, Kcnj13, Clic6, and Aqp1, Related to Fig. 2&3. (A) Representative IHC of Relt (green) and Iba1 (white) in Spak−/− Ctl and LPS-treated animals (DAPI, blue) and (B) Quantitation of Relt expression (normalized intensity density (IntDen)/cell #) of SPAK−/− Ctl and LPS (n=6 ChP, 3 animals for all conditions). *p<0.05. Error bars show mean +/− sem, and each symbol represents average data obtained from an individual animal. (C) Expression analysis of SPAK-interacting proteins in the ChP. UMAP of cell ontology (as in fig. S3A, left panels) and violin plots (right panels) of cellular expression of (A) Atp1a1, (B) Kcnj13, (C) Clic6, and (D) Aqp1 in epithelial and hematopoietic cells.
Supplementary Fig. 5. Pathway and gene ontology (GO) analysis of the top markers for peripherally derived macrophages (Pbm), neutrophils, and plasmacytoid dendritic cells (pDC), Related to Fig. 5. A) Heatmap of top markers for CD45+ dataset clusters. (B) Pbm1 GO Biological Processes, (C) Pbm2 GO Biological Processes, (D) Neutrophils GO Wiki Pathways, (E) pDC GO Biological Processes, (F) pDC GO Wiki Pathways, (G) Epiplexus Macrophages Subcluster 1 GO Biological Processes, (H) Epiplexus Macrophages Subcluster 2 GO Biological Processes, (I) IVH vs control epithelial cells Wiki Pathways, (J) LPS vs control epithelial Wiki Pathways.
Supplementary Fig. 6. Choroid plexus epithelial and myeloid cells demonstrate expression of key genes in the mTOR/Irf7 pathway, Related to Fig. 5. (A) Feature plots showing expression of key genes in mTOR/Irf7 pathway in myeloid clusters (B) Feature plots showing expression of key genes in mTOR/Irf7 pathway in epithelial clusters.
Supplementary Fig. 7. ChP expression of Mtor and Nfkb and treatment of PIH and PHH animals with Rapamycin to prevent ventriculomegaly and the ChP immune response, Related to Fig. 7. (A-B) snRNAseq analysis of Mtor and Nfkb in the ChP. (A) UMAP of Mtor expression across ChP epithelial cell clusters. (B) UMAP of cell ontology (as in fig. S3A) (left panel), ChP epithelial cell clusters (middle panel), and violin plot (right panel) demonstrating Nfkb expression. (C) Representative immunohistochemical images of the lateral ventricles (LV) in control, LPS, IVH, LPS +/− Rapamycin, and IVH +/− Rapamycin. Rapamycin (RAPA) indicates animals treated with rapamycin prior to LPS administration, RAPA 3h indicated animal treated with rapamycin after LPS administration. Dashed white line indicating border of lateral ventricle. CC, corpus callosum; ChP, choroid plexus, mag 2.5x (D) Quantification of lateral ventricle volume (% brain volume) in control, LPS, IVH, LPS +/− Rapamycin, and IVH +/− Rapamycin (E) Lateral ventricular size (% brain volume) in Ctl, LPS, and LPS + Rapamycin 3h after initial LPS infusion (Rapa 3h post) treated animals. (F) Representative IHC of Iba1 (red) and ED1 (blue) expression in the ChP of rats treated with LPS alone or LPS + Rapa 3h post. DAPI, white. Scale bars 20 μm. (G) Quantitative IHC of the ChP immune response in (F) in control, LPS, and LPS + Rapa 3h post treated animals. Graphs represent DAPI-normalized % of cells identified as Iba1+ or ED1+ in Ctl, LPS, LPS + Rapa 3h post. N=3-6 animals per condition. For all graphs, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant, one-way ANOVA. Error bars show mean +/− sem, and each symbol represents average data obtained from an individual animal.
Table S1. Differentially expressed genes (DEG) and differentially expressed proteins (DEP) in PIH animals, Related to Fig. 2
Table S3. Bulk RNAseq and proteomic analysis of DEGs and DEPs shared between PIH and PHH, Related to Fig. 4
LIFE SCIENCE TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Snail | Cell Signaling Technology | Cat#3879S; RRID: AB_2255011 |
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 |
| Rabbit polyclonal anti-BMAL1 | This paper | N/A |
| Bacterial and virus strains | ||
| pAAV-hSyn-DIO-hM3D(Gq)-mCherry | Krashes et al.1 | Addgene AAV5; 44361-AAV5 |
| AAV5-EF1a-DIO-hChR2(H134R)-EYFP | Hope Center Viral Vectors Core | N/A |
| Cowpox virus Brighton Red | BEI Resources | NR-88 |
| Zika-SMGC-1, GENBANK: KX266255 | Isolated from patient (Wang et al.2) | N/A |
| Staphylococcus aureus | ATCC | ATCC 29213 |
| Streptococcus pyogenes: M1 serotype strain: strain SF370; M1 GAS | ATCC | ATCC 700294 |
| Biological samples | ||
| Healthy adult BA9 brain tissue | University of Maryland Brain & Tissue Bank; http://medschool.umaryland.edu/btbank/ | Cat#UMB1455 |
| Human hippocampal brain blocks | New York Brain Bank | http://nybb.hs.columbia.edu/ |
| Patient-derived xenografts (PDX) | Children's Oncology Group Cell Culture and Xenograft Repository | http://cogcell.org/ |
| Chemicals, peptides, and recombinant proteins | ||
| MK-2206 AKT inhibitor | Selleck Chemicals | S1078; CAS: 1032350-13-2 |
| SB-505124 | Sigma-Aldrich | S4696; CAS: 694433-59-5 (free base) |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124-87-8 |
| Human TGF-β | R&D | 240-B; GenPept: P01137 |
| Activated S6K1 | Millipore | Cat#14-486 |
| GST-BMAL1 | Novus | Cat#H00000406-P01 |
| Critical commercial assays | ||
| EasyTag EXPRESS 35S Protein Labeling Kit | PerkinElmer | NEG772014MC |
| CaspaseGlo 3/7 | Promega | G8090 |
| TruSeq ChIP Sample Prep Kit | Illumina | IP-202-1012 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE63473 |
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Nanog STILT inference | This paper; Mendeley Data | http://dx.doi.org/10.17632/wx6s4mj7s8.2 |
| Affinity-based mass spectrometry performed with 57 genes | This paper; Mendeley Data | Table S8; http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Experimental models: Cell lines | ||
| Hamster: CHO cells | ATCC | CRL-11268 |
| D. melanogaster: Cell line S2: S2-DRSC | Laboratory of Norbert Perrimon | FlyBase: FBtc0000181 |
| Human: Passage 40 H9 ES cells | MSKCC stem cell core facility | N/A |
| Human: HUES 8 hESC line (NIH approval number NIHhESC-09-0021) | HSCI iPS Core | hES Cell Line: HUES-8 |
| Experimental models: Organisms/strains | ||
| C. elegans: Strain BC4011: srl-1(s2500) II; dpy-18(e364) III; unc-46(e177)rol-3(s1040) V. | Caenorhabditis Genetics Center | WB Strain: BC4011; WormBase: WBVar00241916 |
| D. melanogaster: RNAi of Sxl: y[1] sc[*] v[1]; P{TRiP.HMS00609}attP2 | Bloomington Drosophila Stock Center | BDSC:34393; FlyBase: FBtp0064874 |
| S. cerevisiae: Strain background: W303 | ATCC | ATTC: 208353 |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J | The Jackson Laboratory | JAX: 006494 |
| Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J | The Jackson Laboratory | RRID: IMSR_JAX:008471 |
| Zebrafish: Tg(Shha:GFP)t10: t10Tg | Neumann and Nuesslein-Volhard3 | ZFIN: ZDB-GENO-060207-1 |
| Arabidopsis: 35S::PIF4-YFP, BZR1-CFP | Wang et al.4 | N/A |
| Arabidopsis: JYB1021.2: pS24(AT5G58010)::cS24:GFP(-G):NOS #1 | NASC | NASC ID: N70450 |
| Oligonucleotides | ||
| siRNA targeting sequence: PIP5K I alpha #1: ACACAGUACUCAGUUGAUA | This paper | N/A |
| Primers for XX, see Table SX | This paper | N/A |
| Primer: GFP/YFP/CFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC | This paper | N/A |
| Morpholino: MO-pax2a GGTCTGCTTTGCAGTGAATATCCAT | Gene Tools | ZFIN: ZDB-MRPHLNO-061106-5 |
| ACTB (hs01060665_g1) | Life Technologies | Cat#4331182 |
| RNA sequence: hnRNPA1_ligand: UAGGGACUUAGGGUUCUCUCUAGGGACUUAG GGUUCUCUCUAGGGA | This paper | N/A |
| Recombinant DNA | ||
| pLVX-Tight-Puro (TetOn) | Clonetech | Cat#632162 |
| Plasmid: GFP-Nito | This paper | N/A |
| cDNA GH111110 | Drosophila Genomics Resource Center | DGRC:5666; FlyBase:FBcl0130415 |
| AAV2/1-hsyn-GCaMP6-WPRE | Chen et al.5 | N/A |
| Mouse raptor: pLKO mouse shRNA 1 raptor | Thoreen et al.6 | Addgene Plasmid #21339 |
| Software and algorithms | ||
| ImageJ | Schneider et al.7 | https://imagej.nih.gov/ij/ |
| Bowtie2 | Langmead and Salzberg8 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al.9 | http://samtools.sourceforge.net/ |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al.10 | https://github.com/ChristophRau/wMICA |
| ICS algorithm | This paper; Mendeley Data | http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Other | ||
| Sequence data, analyses, and resources related to the ultra-deep sequencing of the AML31 tumor, relapse, and matched normal | This paper | http://aml31.genome.wustl.edu |
| Resource website for the AML31 publication | This paper | https://github.com/chrisamiller/aml31SuppSite |
PHYSICAL SCIENCE TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| QD605 streptavidin conjugated quantum dot | Thermo Fisher Scientific | Cat#Q10101MP |
| Platinum black | Sigma-Aldrich | Cat#205915 |
| Sodium formate BioUltra, ≥99.0% (NT) | Sigma-Aldrich | Cat#71359 |
| Chloramphenicol | Sigma-Aldrich | Cat#C0378 |
| Carbon dioxide (13C, 99%) (<2% 18O) | Cambridge Isotope Laboratories | CLM-185-5 |
| Poly(vinylidene fluoride-co-hexafluoropropylene) | Sigma-Aldrich | 427179 |
| PTFE Hydrophilic Membrane Filters, 0.22 μm, 90 mm | Scientificfilters.com/TischScientific | SF13842 |
| Critical commercial assays | ||
| Folic Acid (FA) ELISA kit | Alpha Diagnostic International | Cat# 0365-0B9 |
| TMT10plex Isobaric Label Reagent Set | Thermo Fisher | A37725 |
| Surface Plasmon Resonance CM5 kit | GE Healthcare | Cat#29104988 |
| NanoBRET Target Engagement K-5 kit | Promega | Cat#N2500 |
| Deposited data | ||
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Structure of compound 5 | This paper; Cambridge Crystallographic Data Center | CCDC: 2016466 |
| Code for constraints-based modeling and analysis of autotrophic E. coli | This paper | https://gitlab.com/elad.noor/sloppy/tree/master/rubisco |
| Software and algorithms | ||
| Gaussian09 | Frish et al.1 | https://gaussian.com |
| Python version 2.7 | Python Software Foundation | https://www.python.org |
| ChemDraw Professional 18.0 | PerkinElmer | https://www.perkinelmer.com/category/chemdraw |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al.2 | https://github.com/ChristophRau/wMICA |
| Other | ||
| DASGIP MX4/4 Gas Mixing Module for 4 Vessels with a Mass Flow Controller | Eppendorf | Cat#76DGMX44 |
| Agilent 1200 series HPLC | Agilent Technologies | https://www.agilent.com/en/products/liquid-chromatography |
| PHI Quantera II XPS | ULVAC-PHI, Inc. | https://www.ulvac-phi.com/en/products/xps/phi-quantera-ii/ |
HIGHLIGHTS.
Integrated multi-omics study of models of infectious and hemorrhagic hydrocephalus
Blood or bacteria in CSF elicit highly similar ChP immune and secretory responses
Crosstalk between ChP immune and epithelial cells drives pathologic CSF secretion
Immunomodulators treat PIH/PHH by antagonizing a SPAK-regulated ChP transportome
ACKNOWLEDGMENTS
K.T.K. is supported by grants from the NIH (1R01NS109358-01; 1R01NS111029-01A1), Rudi Schulte Organization, Hydrocephalus Association, Simons Foundation, and March of Dimes. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Inclusion and Diversity:
We support the inclusive, diverse, and equitable conduct of research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest:
The authors declare no competing interests.
Data and materials availability:
All materials and associated protocols can be found within the methods section of this paper, or the papers referenced. For additional explanations of the IVH protocol please see published works 32 and 138. For a detailed protocol on the direct methods of CSF measurement, please see53. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE139 partner repository with the dataset identifier PXD030678.
References
- 1.Cui J, Xu H, and Lehtinen MK (2021). Macrophages on the margin: choroid plexus immune responses. Trends Neurosci 44, 864–875. 10.1016/j.tins.2021.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dani N, Herbst RH, McCabe C, Green GS, Kaiser K, Head JP, Cui J, Shipley FB, Jang A, Dionne D, et al. (2021). A cellular and spatial map of the choroid plexus across brain ventricles and ages. Cell 184, 3056–3074 e3021. 10.1016/j.cell.2021.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Damkier HH, Brown PD, and Praetorius J (2013). Cerebrospinal fluid secretion by the choroid plexus. Physiol Rev 93, 1847–1892. 10.1152/physrev.00004.2013. [DOI] [PubMed] [Google Scholar]
- 4.Cui J, Shipley FB, Shannon ML, Alturkistani O, Dani N, Webb MD, Sugden AU, Andermann ML, and Lehtinen MK (2020). Inflammation of the Embryonic Choroid Plexus Barrier following Maternal Immune Activation. Dev Cell 55, 617–628 e616. 10.1016/j.devcel.2020.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engelhardt B, Vajkoczy P, and Weller RO (2017). The movers and shapers in immune privilege of the CNS. Nat Immunol 18, 123–131. 10.1038/ni.3666. [DOI] [PubMed] [Google Scholar]
- 6.Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, and Sallusto F (2009). C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol 10, 514–523. 10.1038/ni.1716. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz M, and Baruch K (2014). The resolution of neuroinflammation in neurodegeneration: leukocyte recruitment via the choroid plexus. EMBO J 33, 7–22. 10.1002/embj.201386609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodriguez-Lorenzo S, Konings J, van der Pol S, Kamermans A, Amor S, van Horssen J, Witte ME, Kooij G, and de Vries HE (2020). Inflammation of the choroid plexus in progressive multiple sclerosis: accumulation of granulocytes and T cells. Acta Neuropathol Commun 8, 9. 10.1186/s40478-020-0885-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coulter IC, Dewan MC, Tailor J, Ibrahim GM, and Kulkarni AV (2021). Endoscopic third ventriculostomy and choroid plexus cauterization (ETV/CPC) for hydrocephalus of infancy: a technical review. Childs Nerv Syst 37, 3509–3519. 10.1007/s00381-021-05209-5. [DOI] [PubMed] [Google Scholar]
- 10.Kahle KT, Kulkarni AV, Limbrick DD Jr., and Warf BC (2016). Hydrocephalus in children. Lancet 387, 788–799. 10.1016/S0140-6736(15)60694-8. [DOI] [PubMed] [Google Scholar]
- 11.Warf BC (2005). Comparison of endoscopic third ventriculostomy alone and combined with choroid plexus cauterization in infants younger than 1 year of age: a prospective study in 550 African children. J Neurosurg 103, 475–481. 10.3171/ped.2005.103.6.0475. [DOI] [PubMed] [Google Scholar]
- 12.Anderson IA, Saukila LF, Robins JMW, Akhunbay-Fudge CY, Goodden JR, Tyagi AK, Phillips N, and Chumas PD (2018). Factors associated with 30-day ventriculoperitoneal shunt failure in pediatric and adult patients. J Neurosurg 130, 145–153. 10.3171/2017.8.JNS17399. [DOI] [PubMed] [Google Scholar]
- 13.Kulkarni AV, Riva-Cambrin J, Butler J, Browd SR, Drake JM, Holubkov R, Kestle JR, Limbrick DD, Simon TD, Tamber MS, et al. (2013). Outcomes of CSF shunting in children: comparison of Hydrocephalus Clinical Research Network cohort with historical controls: clinical article. J Neurosurg Pediatr 12, 334–338. 10.3171/2013.7.PEDS12637. [DOI] [PubMed] [Google Scholar]
- 14.Karimy JK, Reeves BC, Damisah E, Duy PQ, Antwi P, David W, Wang K, Schiff SJ, Limbrick DD Jr., Alper SL, et al. (2020). Inflammation in acquired hydrocephalus: pathogenic mechanisms and therapeutic targets. Nat Rev Neurol 16, 285–296. 10.1038/s41582-020-0321-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fleischer V, Gonzalez-Escamilla G, Ciolac D, Albrecht P, Kury P, Gruchot J, Dietrich M, Hecker C, Muntefering T, Bock S, et al. (2021). Translational value of choroid plexus imaging for tracking neuroinflammation in mice and humans. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2025000118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shipley FB, Dani N, Xu H, Deister C, Cui J, Head JP, Sadegh C, Fame RM, Shannon ML, Flores VI, et al. (2020). Tracking Calcium Dynamics and Immune Surveillance at the Choroid Plexus Blood-Cerebrospinal Fluid Interface. Neuron 108, 623–639 e610. 10.1016/j.neuron.2020.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muller J, Sinnecker T, Wendebourg MJ, Schlager R, Kuhle J, Schadelin S, Benkert P, Derfuss T, Cattin P, Jud C, et al. (2022). Neurol Neuroimmunol Neuroinflamm 9. 10.1212/NXI.0000000000001147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Symss NP, and Oi S (2013). J Neurosurg Pediatr 11, 170–177. 10.3171/2012.3.PEDS0934. [DOI] [PubMed] [Google Scholar]
- 19.Habiyaremye G, Morales DM, Morgan CD, McAllister JP, CreveCoeur TS, Han RH, Gabir M, Baksh B, Mercer D, and Limbrick DD Jr. (2017). Fluids Barriers CNS 14, 35. 10.1186/s12987-017-0083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lepennetier G, Hracsko Z, Unger M, Van Griensven M, Grummel V, Krumbholz M, Berthele A, Hemmer B, and Kowarik MC (2019). J Neuroinflammation 16, 219. 10.1186/s12974-019-1601-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cuoco JA, Guilliams EL, Klein BJ, Benko MJ, Darden JA, Olasunkanmi AL, Witcher MR, Rogers CM, Marvin EA, Patel BM, and Entwistle JJ (2021). World Neurosurg 156, e338–e344. 10.1016/j.wneu.2021.09.059. [DOI] [PubMed] [Google Scholar]
- 22.Berkes J, Viswanathan VK, Savkovic SD, and Hecht G (2003). Gut 52, 439–451. 10.1136/gut.52.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doyle WJ, Skoner DP, Hayden F, Buchman CA, Seroky JT, and Fireman P (1994). Ann Otol Rhinol Laryngol 103, 59–69. 10.1177/000348949410300111. [DOI] [PubMed] [Google Scholar]
- 24.Wilson R, Alton E, Rutman A, Higgins P, Al Nakib W, Geddes DM, Tyrrell DA, and Cole PJ (1987). Eur J Respir Dis 70, 272–279. [PubMed] [Google Scholar]
- 25.Yan Y, Dalmasso G, Nguyen HT, Obertone TS, Charrier-Hisamuddin L, Sitaraman SV, and Merlin D (2008). Am J Pathol 173, 1013–1028. 10.2353/ajpath.2008.080339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shekarabi M, Zhang J, Khanna AR, Ellison DH, Delpire E, and Kahle KT (2017). Cell Metab 25, 285–299. 10.1016/j.cmet.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 27.Yan Y, Nguyen H, Dalmasso G, Sitaraman SV, and Merlin D (2007). Biochim Biophys Acta 1769, 106–116. 10.1016/j.bbaexp.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weidenfeld S, and Kuebler WM (2017). Front Immunol 8, 393. 10.3389/fimmu.2017.00393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gram M, Sveinsdottir S, Cinthio M, Sveinsdottir K, Hansson SR, Morgelin M, Akerstrom B, and Ley D (2014). J Neuroinflammation 11, 200. 10.1186/s12974-014-0200-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karimy JK, Zhang J, Kurland DB, Theriault BC, Duran D, Stokum JA, Furey CG, Zhou X, Mansuri MS, Montejo J, et al. (2017). Nat Med 23, 997–1003. 10.1038/nm.4361. [DOI] [PubMed] [Google Scholar]
- 31.Purohit D, Finkel DA, Malfa A, Liao Y, Ivanova L, Kleinman GM, Hu F, Shah S, Thompson C, Joseph E, et al. (2021). Front Cell Neurosci 15, 633185. 10.3389/fncel.2021.633185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simard PF, Tosun C, Melnichenko L, Ivanova S, Gerzanich V, and Simard JM (2011). Transl Stroke Res 2, 227–231. 10.1007/s12975-011-0070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan X, Chen J, Keep RF, Xi G, and Hua Y (2020). Stroke 51, 1578–1586. 10.1161/STROKEAHA.119.028672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guillen-Pinto D, Malaga-Espinoza B, Ye-Tay J, Rospigliosi-Lopez ML, Montenegro-Rivera A, Rivas M, Stiglich ML, Villasante-Valera S, Lizama-Olaya O, Tori A, et al. (2020). Rev Peru Med Exp Salud Publica 37, 210–219. 10.17843/rpmesp.2020.372.4772. [DOI] [PubMed] [Google Scholar]
- 35.Huo L, Fan Y, Jiang C, Gao J, Yin M, Wang H, Yang F, and Cao Q (2019). J Child Neurol 34, 11–16. 10.1177/0883073818799155. [DOI] [PubMed] [Google Scholar]
- 36.Li L, Padhi A, Ranjeva SL, Donaldson SC, Warf BC, Mugamba J, Johnson D, Opio Z, Jayarao B, Kapur V, et al. (2011). J Neurosurg Pediatr 7, 73–87. 10.3171/2010.9.PEDS10162. [DOI] [PubMed] [Google Scholar]
- 37.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, and Akira S (1999). J Immunol 162, 3749–3752. [PubMed] [Google Scholar]
- 38.Park BS, and Lee JO (2013). Exp Mol Med 45, e66. 10.1038/emm.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janciauskiene S, Vijayan V, and Immenschuh S (2020). Front Immunol 11, 1964. 10.3389/fimmu.2020.01964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kwon MS, Woo SK, Kurland DB, Yoon SH, Palmer AF, Banerjee U, Iqbal S, Ivanova S, Gerzanich V, and Simard JM (2015). Int J Mol Sci 16, 5028–5046. 10.3390/ijms16035028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shemie S, Jay V, Rutka J, and Armstrong D (1997). Ann Emerg Med 29, 524–528. 10.1016/s0196-0644(97)70227-0. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J, Bhuiyan MIH, Zhang T, Karimy JK, Wu Z, Fiesler VM, Zhang J, Huang H, Hasan MN, Skrzypiec AE, et al. (2020). Nat Commun 11, 78. 10.1038/s41467-019-13851-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarica C, Tanrikulu B, Sahin Y, Dagcinar A, Baltacioglu F, and Bayri Y (2018). Pediatr Neurosurg 53, 247–253. 10.1159/000488458. [DOI] [PubMed] [Google Scholar]
- 44.Xue Y, Liu X, Koundal S, Constantinou S, Dai F, Santambrogio L, Lee H, and Benveniste H (2020). Sci Rep 10, 14592. 10.1038/s41598-020-71582-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xue Y, Gursky Z, Monte B, Koundal S, Liu X, Lee H, Michurina TV, Mellanson KA, Zhao L, Nemajerova A, et al. (2022). Fluids Barriers CNS 19, 20. 10.1186/s12987-022-00319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thalen S, Maanja M, Sigfridsson A, Maret E, Sorensson P, and Ugander M (2019). J Cardiovasc Magn Reson 21, 71. 10.1186/s12968-019-0580-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen X, Liu X, Koundal S, Elkin R, Zhu X, Monte B, Xu F, Dai F, Pedram M, Lee H, et al. (2022). Nat Aging 2, 214–223. 10.1038/s43587-022-00181-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao L, Tannenbaum A, Bakker E, and Benveniste H (2022). Physiology (Bethesda) 37, 0. 10.1152/physiol.00015.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Proulx ST (2021). Cell Mol Life Sci 78, 2429–2457. 10.1007/s00018-020-03706-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hladky SB, and Barrand MA (2022). Fluids Barriers CNS 19, 9. 10.1186/s12987-021-00282-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Doctor BA, Newman N, Minich NM, Taylor HG, Fanaroff AA, and Hack M (2001). Clin Pediatr (Phila) 40, 473–480. 10.1177/000992280104000901. [DOI] [PubMed] [Google Scholar]
- 52.Deopujari CE, Padayachy L, Azmi A, Figaji A, and Samantray SK (2018). Childs Nerv Syst 34, 1905–1914. 10.1007/s00381-018-3901-z. [DOI] [PubMed] [Google Scholar]
- 53.Karimy JK, Kahle KT, Kurland DB, Yu E, Gerzanich V, and Simard JM (2015). J Neurosci Methods 241, 78–84. 10.1016/j.jneumeth.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu G, Mestre H, Sweeney AM, Sun Q, Weikop P, Du T, and Nedergaard M (2020). Cell Rep 33, 108524. 10.1016/j.celrep.2020.108524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Javaheri S, and Wagner KR (1993). In vivo evidence for NaCl cotransport in the central nervous system. J Clin Invest 92, 2257–2261. 10.1172/JCI116829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karimy JK, Duran D, Hu JK, Gavankar C, Gaillard JR, Bayri Y, Rice H, DiLuna ML, Gerzanich V, Marc Simard J, and Kahle KT (2016). Neurosurg Focus 41, E10. 10.3171/2016.8.FOCUS16278. [DOI] [PubMed] [Google Scholar]
- 57.Ferguson C, McKay M, Harris RA, and Homanics GE (2013). Alcohol 47, 595–599. 10.1016/j.alcohol.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, Vandamme N, De Schepper S, Van Isterdael G, Scott CL, et al. (2019). Nat Neurosci 22, 1021–1035. 10.1038/s41593-019-0393-4. [DOI] [PubMed] [Google Scholar]
- 59.Karman J, Chu HH, Co DO, Seroogy CM, Sandor M, and Fabry Z (2006). J Immunol 177, 7750–7760. 10.4049/jimmunol.177.11.7750. [DOI] [PubMed] [Google Scholar]
- 60.McWhorter FY, Wang T, Nguyen P, Chung T, and Liu WF (2013). Proc Natl Acad Sci U S A 110, 17253–17258. 10.1073/pnas.1308887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chistiakov DA, Killingsworth MC, Myasoedova VA, Orekhov AN, and Bobryshev YV (2017). Lab Invest 97, 4–13. 10.1038/labinvest.2016.116. [DOI] [PubMed] [Google Scholar]
- 62.Dodge PR, and Swartz MN (1965). Ii. Special Neurologic Problems, Postmeningitic Complications and Clinicopathological Correlations. N Engl J Med 272, 1003–1010 CONCL. 10.1056/NEJM196505132721906. [DOI] [PubMed] [Google Scholar]
- 63.Vergouwen MD, Schut ES, Troost D, and van de Beek D (2010). Neurocrit Care 13, 217–227. 10.1007/s12028-010-9387-5. [DOI] [PubMed] [Google Scholar]
- 64.Dickson LG, and Yassin MW (1969). J Egypt Public Health Assoc 44, 349–355. [PubMed] [Google Scholar]
- 65.Engelen-Lee JY, Brouwer MC, Aronica E, and van de Beek D (2016). Acta Neuropathol Commun 4, 26. 10.1186/s40478-016-0297-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakano A, Harada T, Morikawa S, and Kato Y (1990). Acta Pathol Jpn 40, 107–115. 10.1111/j.1440-1827.1990.tb01549.x. [DOI] [PubMed] [Google Scholar]
- 67.Rangaraju S, Raza SA, Li NX, Betarbet R, Dammer EB, Duong D, Lah JJ, Seyfried NT, and Levey AI (2018). Front Immunol 9, 405. 10.3389/fimmu.2018.00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sharma S, Goyal MK, Sharma K, Modi M, Sharma M, Khandelwal N, Prabhakar S, Sharma N, R S, Gairolla J, et al. (2017). J Neurol Sci 379, 131–136. 10.1016/j.jns.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 69.Thwaites GE, Macmullen-Price J, Tran TH, Pham PM, Nguyen TD, Simmons CP, White NJ, Tran TH, Summers D, and Farrar JJ (2007). Lancet Neurol 6, 230–236. 10.1016/S1474-4422(07)70034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lolansen SD, Rostgaard N, Oernbo EK, Juhler M, Simonsen AH, and MacAulay N (2021). Dis Markers 2021, 8834822. 10.1155/2021/8834822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vitari AC, Thastrup J, Rafiqi FH, Deak M, Morrice NA, Karlsson HK, and Alessi DR (2006). Biochem J 397, 223–231. 10.1042/BJ20060220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vitari AC, Deak M, Morrice NA, and Alessi DR (2005). Biochem J 391, 17–24. 10.1042/BJ20051180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yan Y, and Merlin D (2008). World J Gastroenterol 14, 6115–6121. 10.3748/wjg.14.6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Polek TC, Talpaz M, and Spivak-Kroizman T (2006). Biochem Biophys Res Commun 343, 125–134. 10.1016/j.bbrc.2006.02.125. [DOI] [PubMed] [Google Scholar]
- 75.Delpire E, and Gagnon KB (2008). Biochem J 409, 321–331. 10.1042/BJ20071324. [DOI] [PubMed] [Google Scholar]
- 76.Richardson C, and Alessi DR (2008). J Cell Sci 121, 3293–3304. 10.1242/jcs.029223. [DOI] [PubMed] [Google Scholar]
- 77.Dwinell MR, Lazar J, and Geurts AM (2011). Mamm Genome 22, 466–475. 10.1007/s00335-011-9346-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Savman K, Blennow M, Hagberg H, Tarkowski E, Thoresen M, and Whitelaw A (2002). Acta Paediatr 91, 1357–1363. 10.1111/j.1651-2227.2002.tb02834.x. [DOI] [PubMed] [Google Scholar]
- 79.Schmitz T, Heep A, Groenendaal F, Huseman D, Kie S, Bartmann P, Obladen M, and Felderhoff-Muser U (2007). 10.1203/pdr.0b013e31805341f1. [DOI] [PubMed] [Google Scholar]
- 80.Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, Oliveira MF, Oliveira PL, Graca-Souza AV, and Bozza MT (2007). J Biol Chem 282, 20221–20229. 10.1074/jbc.M610737200. [DOI] [PubMed] [Google Scholar]
- 81.Molteni M, Gemma S, and Rossetti C (2016). Mediators Inflamm 2016, 6978936. 10.1155/2016/6978936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, Myung P, Plikus MV, and Nie Q (2021). Nat Commun 12, 1088. 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim J, Erikson DW, Burghardt RC, Spencer TE, Wu G, Bayless KJ, Johnson GA, and Bazer FW (2010). Matrix Biol 29, 369–382. 10.1016/j.matbio.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 84.Bazer FW, Song G, Kim J, Erikson DW, Johnson GA, Burghardt RC, Gao H, Carey Satterfield M, Spencer TE, and Wu G (2012). Mol Cell Endocrinol 354, 22–33. 10.1016/j.mce.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 85.Anwar MA, Shah M, Kim J, and Choi S (2019). Med Res Rev 39, 1053–1090. 10.1002/med.21553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Josiah SS, Meor Azlan NF, and Zhang J (2021). Int J Mol Sci 22. 10.3390/ijms22031232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nishida H, Sohara E, Nomura N, Chiga M, Alessi DR, Rai T, Sasaki S, and Uchida S (2012). Hypertension 60, 981–990. 10.1161/HYPERTENSIONAHA.112.201509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sorensen MV, Saha B, Jensen IS, Wu P, Ayasse N, Gleason CE, Svendsen SL, Wang WH, and Pearce D (2019). JCI Insight 5. 10.1172/jci.insight.126910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weichhart T, Hengstschlager M, and Linke M (2015). Nat Rev Immunol 15, 599–614. 10.1038/nri3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu F, Na L, Li Y, and Chen L (2020). Cell Biosci 10, 54. 10.1186/s13578-020-00416-0. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 91.Robert SM, Reeves BC, Marlier A, Duy PQ, DeSpenza T, Kundishora A, Kiziltug E, Singh A, Allington G, Alper SL, and Kahle KT (2021). Childs Nerv Syst 37, 3341–3353. 10.1007/s00381-021-05255-z. [DOI] [PubMed] [Google Scholar]
- 92.Garoon RB, Foroozan R, and Vaphiades MS (2017). Surv Ophthalmol 62, 383–386. 10.1016/j.survophthal.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 93.Stone SS, and Warf BC (2014). J Neurosurg Pediatr 14, 439–446. 10.3171/2014.7.PEDS14152. [DOI] [PubMed] [Google Scholar]
- 94.Buxton N, and Punt J (1997). Pediatr Neurosurg 27, 108–111. 10.1159/000121236. [DOI] [PubMed] [Google Scholar]
- 95.Eisenberg HM, McComb JG, and Lorenzo AV (1974). J Neurosurg 40, 381–385. 10.3171/jns.1974.40.3.0381. [DOI] [PubMed] [Google Scholar]
- 96.Milhorat TH, Hammock MK, Davis DA, and Fenstermacher JD (1976). I. Proof of cerebrospinal fluid overproduction. Childs Brain 2, 273–289. [PubMed] [Google Scholar]
- 97.Anei R, Hayashi Y, Hiroshima S, Mitsui N, Orimoto R, Uemori G, Saito M, Sato M, Wada H, Hododuka A, and Kamada K (2011). Neurol Med Chir (Tokyo) 51, 437–441. 10.2176/nmc.51.437. [DOI] [PubMed] [Google Scholar]
- 98.Cataltepe O, Liptzin D, Jolley L, and Smith TW (2010). J Neurosurg Pediatr 5, 518–522. 10.3171/2009.12.PEDS0960. [DOI] [PubMed] [Google Scholar]
- 99.Furey C, Antwi P, Duran D, Timberlake AT, Nelson-Williams C, Matouk CC, DiLuna ML, Gunel M, and Kahle KT (2018). Cold Spring Harb Mol Case Stud 4. 10.1101/mcs.a003145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jin SC, Dong W, Kundishora AJ, Panchagnula S, Moreno-De-Luca A, Furey CG, Allocco AA, Walker RL, Nelson-Williams C, Smith H, et al. (2020). Nat Med 26, 1754–1765. 10.1038/s41591-020-1090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Erickson MA, Hartvigson PE, Morofuji Y, Owen JB, Butterfield DA, and Banks WA (2012). J Neuroinflammation 9, 150. 10.1186/1742-2094-9-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Manouchehrian O, Ramos M, Bachiller S, Lundgaard I, and Deierborg T (2021). J Neuroinflammation 18, 34. 10.1186/s12974-021-02082-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fratamico PM, DebRoy C, Liu Y, Needleman DS, Baranzoni GM, and Feng P (2016). Front Microbiol 7, 644. 10.3389/fmicb.2016.00644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kaczmarek A, Budzynska A, and Gospodarek E (2014). Folia Microbiol (Praha) 59, 419–422. 10.1007/s12223-014-0315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hu X, Deng Q, Ma L, Li Q, Chen Y, Liao Y, Zhou F, Zhang C, Shao L, Feng J, et al. (2020). Cell Res 30, 229–243. 10.1038/s41422-020-0287-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Papadopoulos Z, Herz J, and Kipnis J (2020). J Immunol 204, 286–293. 10.4049/jimmunol.1900838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hajam IA, Dar PA, Shahnawaz I, Jaume JC, and Lee JH (2017). Exp Mol Med 49, e373. 10.1038/emm.2017.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Engelhardt B, Wolburg-Buchholz K, and Wolburg H (2001). Microsc Res Tech 52, 112–129. . [DOI] [PubMed] [Google Scholar]
- 109.Ghersi-Egea JF, Strazielle N, Catala M, Silva-Vargas V, Doetsch F, and Engelhardt B (2018). Acta Neuropathol 135, 337–361. 10.1007/s00401-018-1807-1. [DOI] [PubMed] [Google Scholar]
- 110.Gu C, Hao X, Li J, Hua Y, Keep RF, and Xi G (2019). J Cereb Blood Flow Metab 39, 1936–1948. 10.1177/0271678X19836117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stridh L, Ek CJ, Wang X, Nilsson H, and Mallard C (2013). Transl Stroke Res 4, 220–227. 10.1007/s12975-012-0248-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shimada A, and Hasegawa-Ishii S (2021). Toxicol Rep 8, 520–528. 10.1016/j.toxrep.2021.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Akira S, and Takeda K (2004). Nat Rev Immunol 4, 499–511. 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 114.Laird MD, Shields JS, Sukumari-Ramesh S, Kimbler DE, Fessler RD, Shakir B, Youssef P, Yanasak N, Vender JR, and Dhandapani KM (2014). Glia 62, 26–38. 10.1002/glia.22581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen T, Tan X, Xia F, Hua Y, Keep RF, and Xi G (2021). Biomolecules 11. 10.3390/biom11050654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Merle NS, Paule R, Leon J, Daugan M, Robe-Rybkine T, Poillerat V, Torset C, Fremeaux-Bacchi V, Dimitrov JD, and Roumenina LT (2019). Proc Natl Acad Sci U S A 116, 6280–6285. 10.1073/pnas.1814797116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nader E, Romana M, and Connes P (2020). Front Immunol 11, 454. 10.3389/fimmu.2020.00454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Haas J, Rudolph H, Costa L, Faller S, Libicher S, Wurthwein C, Jarius S, Ishikawa H, Stump-Guthier C, Tenenbaum T, et al. (2020). Front Immunol 11, 618544. 10.3389/fimmu.2020.618544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim H, Lim YM, Kim G, Lee EJ, Lee JH, Kim HW, and Kim KK (2020). J Neurol Sci 415, 116904. 10.1016/j.jns.2020.116904. [DOI] [PubMed] [Google Scholar]
- 120.Yan Y, Dalmasso G, Nguyen HT, Obertone TS, Sitaraman SV, and Merlin D (2009). PLoS One 4, e5049. 10.1371/journal.pone.0005049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yan Y, Laroui H, Ingersoll SA, Ayyadurai S, Charania M, Yang S, Dalmasso G, Obertone TS, Nguyen H, Sitaraman SV, and Merlin D (2011). J Immunol 187, 1496–1505. 10.4049/jimmunol.1002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Alessi DR, Zhang J, Khanna A, Hochdorfer T, Shang Y, and Kahle KT (2014). Sci Signal 7, re3. 10.1126/scisignal.2005365. [DOI] [PubMed] [Google Scholar]
- 123.Chaplin ER, Goldstein GW, Myerberg DZ, Hunt JV, and Tooley WH (1980). Pediatrics 65, 901–909. [PubMed] [Google Scholar]
- 124.Steffensen AB, Oernbo EK, Stoica A, Gerkau NJ, Barbuskaite D, Tritsaris K, Rose CR, and MacAulay N (2018). Nat Commun 9, 2167. 10.1038/s41467-018-04677-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Stodberg T, Magnusson M, Lesko N, Wredenberg A, Martin Munoz D, Stranneheim H, and Wedell A (2020). Neurol Genet 6, e478. 10.1212/NXG.0000000000000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Xu H, Fame RM, Sadegh C, Sutin J, Naranjo C, Della S, Cui J, Shipley FB, Vernon A, Gao F, et al. (2021). Nat Commun 12, 447. 10.1038/s41467-020-20666-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Thastrup JO, Rafiqi FH, Vitari AC, Pozo-Guisado E, Deak M, Mehellou Y, and Alessi DR (2012). Biochem J 441, 325–337. 10.1042/BJ20111879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Karimy JK, Reeves BC, and Kahle KT (2020). Expert Opin Ther Targets 24, 525–533. 10.1080/14728222.2020.1752182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Matsunaga N, Tsuchimori N, Matsumoto T, and Ii M (2011). Mol Pharmacol 79, 34–41. 10.1124/mol.110.068064. [DOI] [PubMed] [Google Scholar]
- 130.Kotani T, Setiawan J, Konno T, Ihara N, Okamoto S, Saito Y, Murata Y, Noda T, and Matozaki T (2020). Sci Rep 10, 13810. 10.1038/s41598-020-70655-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhou M, Xu W, Wang J, Yan J, Shi Y, Zhang C, Ge W, Wu J, Du P, and Chen Y (2018). EBioMedicine 35, 345–360. 10.1016/j.ebiom.2018.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yu M, Kang X, Xue H, and Yin H (2011). Acta Biochim Biophys Sin (Shanghai) 43, 940–947. 10.1093/abbs/gmr093. [DOI] [PubMed] [Google Scholar]
- 133.Sengupta S, Lorente-Rodriguez A, Earnest S, Stippec S, Guo X, Trudgian DC, Mirzaei H, and Cobb MH (2013). Proc Natl Acad Sci U S A 110, 18826–18831. 10.1073/pnas.1318676110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T, and Franz DN (2010). N Engl J Med 363, 1801–1811. 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 135.Li J, Kim SG, and Blenis J (2014). Cell Metab 19, 373–379. 10.1016/j.cmet.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ye X, Zhu M, Che X, Wang H, Liang XJ, Wu C, Xue X, and Yang J (2020). J Neuroinflammation 17, 18. 10.1186/s12974-019-1644-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mengke NS, Hu B, Han QP, Deng YY, Fang M, Xie D, Li A, and Zeng HK (2016). Mol Med Rep 14, 4957–4966. 10.3892/mmr.2016.5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lodhia KR, Shakui P, and Keep RF (2006). Acta Neurochir Suppl 96, 207–211. 10.1007/3-211-30714-1_45. [DOI] [PubMed] [Google Scholar]
- 139.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). Nucleic Acids Res 47, D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Garate I, Garcia-Bueno B, Madrigal JL, Caso JR, Alou L, Gomez-Lus ML, and Leza JC (2014). J Neuroinflammation 11, 8. 10.1186/1742-2094-11-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Nat Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhang J, Gao G, Begum G, Wang J, Khanna AR, Shmukler BE, Daubner GM, de Los Heros P, Davies P, Varghese J, et al. (2016). Sci Rep 6, 35986. 10.1038/srep35986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Anders S, Pyl PT, and Huber W (2015). Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Love MI, Huber W, and Anders S (2014). Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gu Z, Eils R, and Schlesner M (2016). Bioinformatics 32, 2847–2849. 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 146.Lee AM, Mansuri MS, Wilson RS, Lam TT, Nairn AC, and Picciotto MR (2021). Front Mol Neurosci 14, 657064. 10.3389/fnmol.2021.657064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). Anal Chem 86, 7150–7158. 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Miller MB, Wilson RS, Lam TT, Nairn AC, and Picciotto MR (2018). Proteomes 6. 10.3390/proteomes6040042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Janostiak R, Rauniyar N, Lam TT, Ou J, Zhu LJ, Green MR, and Wajapeyee N (2017). Cell Rep 21, 2829–2841. 10.1016/j.celrep.2017.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Cell 184, 3573–3587 e3529. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, Yuan F, Chen S, Leung HM, Villoria J, et al. (2018). Nature 560, 319–324. 10.1038/s41586-018-0393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Nat Biotechnol. 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 153.McGinnis CS, Murrow LM, and Gartner ZJ (2019). Cell Syst 8, 329–337 e324. 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. (2019). Nature 566, 496–502. 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Federico A, and Monti S (2020). Bioinformatics 36, 1307–1308. 10.1093/bioinformatics/btz700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Nucleic Acids Res 44, W90–97. 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tustison NJ, Avants BB, Cook PA, Zheng Y, Egan A, Yushkevich PA, and Gee JC (2010). IEEE Trans Med Imaging 29, 1310–1320. 10.1109/TMI.2010.2046908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Koundal S, Liu X, Sanggaard S, Mortensen K, Wardlaw J, Nedergaard M, Benveniste H, and Lee H (2019). Neuroscience 404, 14–26. 10.1016/j.neuroscience.2019.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Smith NM, Ford JN, Haghdel A, Glodzik L, Li Y, D'Angelo D, RoyChoudhury A, Wang X, Blennow K, de Leon MJ, and Ivanidze J (2022). Front Aging Neurosci 14, 849932. 10.3389/fnagi.2022.849932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chen X, Liu X, Koundal S, Elkin R, Zhu X, Monte B, Xu F, Dai F, Pedram M, Lee H, et al. (2022). Nature Aging 2, 214–223. 10.1038/s43587-022-00181-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. LPS mediates acute and chronic ventriculomegaly, with preserved patency of the cerebral aqueduct and glymphatic transport, and increases cytokine/chemokine expression, Related to Figure 1. (A) Quantification of lateral ventricular volume at 72 hours and 14 days after an initial 72h infusion of aCSF (control) or LPS. Volume calculated using sequential slices through entire lateral ventricular system (n=5-6 animals per condition). (B-H) Spatially normalized population averaged CSF probability maps overlaid on proton density weighted anatomical MRI brain images of live, anesthetized animals treated with aCSF (control) (B, C) or LPS (D, E). CSF filled ventricles are represented as high intensity signal areas. Top panels (B, D) show MRI images (axial plane view) at the level of the lateral ventricles obtained from voxel wise morphometric analysis of the CSF compartment and bottom panels (C, E) show the corresponding 3D volume reconstructions of the lateral ventricles. (F) Morphometric quantification of the lateral ventricular volume (% brain volume) and total intracranial volume (mm3) for control (sham) and LPS treated PIH animals. (G, H) CSF contrast enhanced T1-weighted MRI images (sagittal plane view) of live, anesthetized animals demonstrating patency of the cerebral aqueduct (Aq) in control (G) and PIH animals (H). (I, J) Average glymphatic flux with superimposed color-coded solute speed maps (μm/s) for control (I) and LPS-treated PIH animals (J). Representative speed maps demonstrating glymphatic transport in sagittal (top panels) and axial (bottom panels) MRI view planes. (K) Quantification of glymphatic transport metrics including glymphatic v-flux (mm3) and glymphatic mean tissue speed (mm/s). (L) Schematic illustrating in vivo CSF secretion measurements. Mineral oil is administered into the cerebral aqueduct (aqueduct) through a catheter (yellow line and arrow) inserted into the aqueduct, which blocks flow of cerebrospinal fluid (CSF). A glass capillary tube is then placed through a burr hole in the skull into the lateral ventricle (blue line and arrow). The distance of CSF flow through the capillary tube is measured using a ruler (black) and rate is calculated over time. (M) CSF cytokine/chemokine expression (from Fig. 2) concentrations (from Fig. 2, pg/mL) of Il-1b, Il-6, Il-10, Ifnγ, Il-17a, Il1-8, Ccl2, Cxcl1, Vegf, Cxcl5, Tnfα, Ccl5, IL-10, Mip-1α, and Il-5 in Ctl and 72h LPS-treated Tlr4+/+ and Tlr4−/− rats. For all experimental conditions, n=4-6 animals per condition. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant, by one-way ANOVA (A) and unpaired t-test with Welch correction (K)
Supplementary Fig. 2. Choroid plexus, meningeal, and cortical immune reaction in PIH animals treated with E. coli +LPS, Related to Fig. 2. (A) Illustration of a representative coronal brain slice (left panel) indicating location of the ChP (black box) imaged for analysis. Representative IHC (right panel) of Iba1+ (red) and ED1+ (blue) positive cells in Ctl and E. coli +/− LPS rat ChP in single-channel IHC (Dapi, white), as well as merged images. (B-C) Quantification of (A) showing DAPI-normalized % ED1+ (B) and Iba1+ (C) cells. (D) Illustration of a representative coronal brain slice (left panel) indicating location of the meninges (black box) imaged for analysis. Representative IHC of Iba1+ (red) and ED1+ (blue) positive cells in Ctl and E. coli +/− LPS rat meninges in single-channel IHC (Dapi, white), as well as merged images. (E-F) Quantification of (D) showing DAPI-normalized % Iba+ (E) and ED1+ (F) cells. (G) Illustration of a representative coronal brain slice (left panel) indicating location of the cortex (black box) imaged for analysis. Representative IHC of Iba1+ (red) and ED1+ (blue) positive cells in Ctl and E. coli +/− LPS rat meninges in single-channel IHC (Dapi, white), as well as merged images. (H-I) Quantification of (G) showing DAPI-normalized % Iba+ (H) and ED1+ (H) cells. Scale bars, 20μm; n=4-6 animals per condition. Error bars, mean ± sem; each symbol represents one animal. *p<0.05, **p<0.01, ****p<0.0001, by one-way ANOVA.
Supplementary Fig. 3. SPAK kinase transduces pro-inflammatory CSF signals in ChP epithelial cells to stimulate NKCC1-dependent CSF secretion, Related to Fig. 2. (A) UMAP of single nucleus transcript profiles of adult ChP-associated cells2, colored by cell type. (B) Tlr4 expression in Chp-associated cells (Avg Log2FC = 1.0; p=5.14e-214). (C) UMAP of ChP epithelial cell subclusters, colored by group and by expression of (D) Tlr4, (E, left) interleukin-1 receptor accessory protein (Il1rap) (Avg Log2FC=0.96, p<10e-300), and (E, right) interleukin 6 receptor (Il6ra) (Avg Log2FC=0.23, p=1.87e-211). (F) Quantitative PCR (qPCR) delta-delta Ct (ddCT) of cytokines Il1b, Tnfa, Mcp1, Ifng, and their receptors Il1r1, Relt, Ccr2, IfngrI1 (n=5 animals per condition; see Table S5*). (G) Representative IHC of ChP TNF receptor RELT (red) and Iba1 (white) in Tlr4+/+ and Tlr4−/− Ctl and LPS-treated animals (inset, 4x magnification). (H) Quantitation of (G) (normalized intensity density (IntDen)/cell #) (n=6 ChP per genotype, 3 animals per condition). (I) UMAP of cell ontology (left, as in (A) and violin plots (right) showing expression of Stk39 (Avg Log2FC=1.1, p<10e-300; Epithelial, Avg Log2FC=4.46, p=4.22e-233) and (J) Slc12a2 (Avg Log2FC=1.3, p<10e-300; Epithelial, Avg Log2FC=2.70, p=8.15e-215). (K) Representative IHC of phospho-activated NKCC1 (pNKCC1; green, top row) and phospho-activated SPAK (pSPAK; red, bottom row) in Ctl and LPS-treated WT, Tlr4−/− and Spak−/− rats. Quantitation (Interval Density/cell #) of (L) pNKCC1 and (M) pSPAK from (K) (n=6 ChP per genotype, 3 animals per condition). (N) Body weight-normalized CSF secretion rate (μL/min/kg) and (O) lateral ventricular size (% brain volume) in Spak+/+ Ctl, Spak+/+ LPS, Spak−/− Ctl, and Spak−/− LPS rats (n=5-6 animals per condition). Scale bars, 25 μm. Error bars, mean ± sem; each symbol represents one animal. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant; (F) Unpaired t-test; (H, L-O) one-way ANOVA.
Supplementary Fig. 4. Relt expression in Spak KO rats and ChP expression of Atp1a, Kcnj13, Clic6, and Aqp1, Related to Fig. 2&3. (A) Representative IHC of Relt (green) and Iba1 (white) in Spak−/− Ctl and LPS-treated animals (DAPI, blue) and (B) Quantitation of Relt expression (normalized intensity density (IntDen)/cell #) of SPAK−/− Ctl and LPS (n=6 ChP, 3 animals for all conditions). *p<0.05. Error bars show mean +/− sem, and each symbol represents average data obtained from an individual animal. (C) Expression analysis of SPAK-interacting proteins in the ChP. UMAP of cell ontology (as in fig. S3A, left panels) and violin plots (right panels) of cellular expression of (A) Atp1a1, (B) Kcnj13, (C) Clic6, and (D) Aqp1 in epithelial and hematopoietic cells.
Supplementary Fig. 5. Pathway and gene ontology (GO) analysis of the top markers for peripherally derived macrophages (Pbm), neutrophils, and plasmacytoid dendritic cells (pDC), Related to Fig. 5. A) Heatmap of top markers for CD45+ dataset clusters. (B) Pbm1 GO Biological Processes, (C) Pbm2 GO Biological Processes, (D) Neutrophils GO Wiki Pathways, (E) pDC GO Biological Processes, (F) pDC GO Wiki Pathways, (G) Epiplexus Macrophages Subcluster 1 GO Biological Processes, (H) Epiplexus Macrophages Subcluster 2 GO Biological Processes, (I) IVH vs control epithelial cells Wiki Pathways, (J) LPS vs control epithelial Wiki Pathways.
Supplementary Fig. 6. Choroid plexus epithelial and myeloid cells demonstrate expression of key genes in the mTOR/Irf7 pathway, Related to Fig. 5. (A) Feature plots showing expression of key genes in mTOR/Irf7 pathway in myeloid clusters (B) Feature plots showing expression of key genes in mTOR/Irf7 pathway in epithelial clusters.
Supplementary Fig. 7. ChP expression of Mtor and Nfkb and treatment of PIH and PHH animals with Rapamycin to prevent ventriculomegaly and the ChP immune response, Related to Fig. 7. (A-B) snRNAseq analysis of Mtor and Nfkb in the ChP. (A) UMAP of Mtor expression across ChP epithelial cell clusters. (B) UMAP of cell ontology (as in fig. S3A) (left panel), ChP epithelial cell clusters (middle panel), and violin plot (right panel) demonstrating Nfkb expression. (C) Representative immunohistochemical images of the lateral ventricles (LV) in control, LPS, IVH, LPS +/− Rapamycin, and IVH +/− Rapamycin. Rapamycin (RAPA) indicates animals treated with rapamycin prior to LPS administration, RAPA 3h indicated animal treated with rapamycin after LPS administration. Dashed white line indicating border of lateral ventricle. CC, corpus callosum; ChP, choroid plexus, mag 2.5x (D) Quantification of lateral ventricle volume (% brain volume) in control, LPS, IVH, LPS +/− Rapamycin, and IVH +/− Rapamycin (E) Lateral ventricular size (% brain volume) in Ctl, LPS, and LPS + Rapamycin 3h after initial LPS infusion (Rapa 3h post) treated animals. (F) Representative IHC of Iba1 (red) and ED1 (blue) expression in the ChP of rats treated with LPS alone or LPS + Rapa 3h post. DAPI, white. Scale bars 20 μm. (G) Quantitative IHC of the ChP immune response in (F) in control, LPS, and LPS + Rapa 3h post treated animals. Graphs represent DAPI-normalized % of cells identified as Iba1+ or ED1+ in Ctl, LPS, LPS + Rapa 3h post. N=3-6 animals per condition. For all graphs, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns = not significant, one-way ANOVA. Error bars show mean +/− sem, and each symbol represents average data obtained from an individual animal.
Table S1. Differentially expressed genes (DEG) and differentially expressed proteins (DEP) in PIH animals, Related to Fig. 2
Table S3. Bulk RNAseq and proteomic analysis of DEGs and DEPs shared between PIH and PHH, Related to Fig. 4
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE139 partner repository with the dataset identifier PXD030678. Any FACS or microscopy data, confocal, or other, reported in this paper will be shared by the lead contact upon request. All materials and associated protocols can be found within the methods section of this paper, and any relevant papers are also cited for further reference. In addition to the methods provided here, for additional explanations of the IVH protocol or direct CSF measurements, please see published works32,53,138, or additional information can be addressed via contact with the corresponding article of this manuscript.
This paper does not report original code. All previously published algorithms are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| AQP1, rabbit recombinant monoclonal | Abcam | Ab168387 |
| ATP1A, rabbit polyclonal | Alomone | ANP-001 |
| CD68, rabbit monoclonal | CST | 97778 |
| CLIC6, mouse monoclonal | Santa Cruz | 365303 |
| ED1/CD68, mouse monoclonal | Millipore | MAB1435 |
| IBA1, goat polyclonal | Thermo | PA5-18039 |
| KCNJ13, mouse monoclonal | Santa Cruz | 398810 |
| Ki67, rabbit monoclonal | CST | 9129 |
| pNKCC1, rabbit polyclonal | Millipore | ABS 1004 |
| pSPAK, rabbit polyclonal | Millipore | 07-2273 |
| RELT, rabbit polyclonal | Novus | NBP2-56851 |
| NKCC1, rabbit monoclonal | CST | 85403S |
| Anti-WNK1 | University of Dundee | S079B |
| Anti-SPAK | University of Dundee | S365D |
| Anti-NKCC1 | University of Dundee | S022D |
| Anti-ATP1A1 | DSHB | a5 |
| Anti-HRP | Pierce | 21130 |
| Anti-beta actin | Abcam | Ab8227 |
| CD16-32 | Biolegend | Cat#: 101302 |
| CD45-PE/cy7 | Biolegend | Cat#: 202207 |
| CD3-647 | Biolegend | Cat#: 201408 |
| Alexa Fluor 488 | Invitrogen | Cat#: A32723 |
| Alexa Fluor 555 | Invitrogen | Cat#: A32816 |
| Alexa Fluor 647 | Invitrogen | Cat#: A32849 |
| Bacterial and virus strains | ||
| E. Coli+LPS (BD21) | Lucigen | E. cloni EXPRESS BL21(DE3) |
| E. Coli−LPS (ClearColi) | Lucigen | CLEARCOLI BL21 |
| Biological samples | ||
| N/A | ||
| Chemicals, peptides, and recombinant proteins | ||
| Bumetanide | Sigma-Aldrich | CAS#: 28395-03-1 |
| aCSF | Harvard Apparatus | Item#: 59-7316 |
| Rapamycin | Cayman | Item#: 13346 |
| Critical commercial assays | ||
| N/A | ||
| Deposited data | ||
| Bulk RNA seq | GEO | GSE192946 |
| Mass spectrometry proteomics data | PRIDE | PXD030678 |
| ScRNA seq | GEO | GSE218143 |
| Experimental models: Cell lines | ||
| N/A | ||
| Experimental models: Organisms/strains | ||
| Wistar rats, albino | Charles River | Strain code 003 |
| Oligonucleotides | ||
| N/A | ||
| Recombinant DNA | ||
| N/A | ||
| Software and algorithms | ||
| Seurat | Hao et al.151 | https://satijalab.org/seurat/ |
| Monocle3 | Cao et al.152 | https://cole-trapnelllab.github.io/monocle3/ |
| Cellchat | Jin et al.82 | https://github.com/sqjin/CellChat |
| Other | ||
| N/A | ||