

Abstract
Suppressors of cytokine signaling (SOCS) provide negative regulation of inflammatory reaction. The role and precise cellular mechanisms of SOCS1 in control of endothelial dysfunction and barrier compromise associated with acute lung injury remain unexplored. Our results show that siRNA-mediated SOCS1 knockdown augmented lipopolysaccharide (LPS)-induced pulmonary endothelial cell (EC) permeability and enhanced inflammatory response. Consistent with in-vitro data, EC-specific SOCS1 knockout mice developed more severe lung vascular leak and accumulation of inflammatory cells in bronchoalveolar lavage fluid. SOCS1 overexpression exhibited protective effects against LPS-induced endothelial permeability and inflammation, which were dependent on microtubule (MT) integrity. Biochemical and image analysis of unstimulated EC showed SOCS1 association with the MT, while challenge with LPS or MT depolymerizing agent colchicine impaired this association. SOCS1 directly interacted with N2-domains of MT-associated proteins CLIP-170 and CLASP2. Furthermore, N-terminal region of SOCS1 was indispensable for these interactions and SOCS1-ΔN mutant lacking N-terminal 59 amino acids failed to rescue LPS-induced endothelial dysfunction. Depletion of endogenous CLIP-170 or CLASP2 abolished SOCS1 interaction with Toll-like receptor-4 and Janus kinase-2 leading to impairment of SOCS1 inhibitory effects on LPS-induced inflammation. Altogether, these findings suggest that endothelial barrier protective and anti-inflammatory effects of SOCS1 are critically dependent on its targeting to the MT.
Keywords: lung injury, vascular endothelium, endothelial permeability, inflammation, microtubules, cytoskeleton
Introduction
Suppressors of cytokine signaling (SOCS) family proteins are key regulators of cytokine signaling involved in immunity and inflammation (1, 2). Cytokines play an essential role in various cellular functions including cell survival, proliferation, differentiation, hematopoiesis, wound healing and immunity (3). Yet, it is imperative to tightly control the duration and intensity of cytokine-activated signaling to avoid detrimental pathophysiological effects. SOCS represent cytokine-inducible intracellular proteins that function as classical negative feedback loop to inhibit cytokine signaling pathways (4). SOCS family contain 8 members (SOCS1-SOCS7 and CIS, cytokine-inducible SH2-containing protein) that are characterized by a central SRC homology 2 (SH2) domain, a N-terminal domain of variable length and sequence, and a C-terminal conserved 40 amino acid module termed as SOCS box (5). The studies so far have proposed two models of SOCS inhibitory mechanisms of cytokine signaling: 1) the direct interaction of SH2 domains of SOCS proteins with activated cytokines receptors thereby inhibiting their activity; and 2) an interaction of SOCS box motif with elongin proteins that are a part of ubiquitin-mediated proteasomal degradation pathway (1, 4).
SOCS1 was simultaneously discovered by three groups using different approaches. One of them identified a protein harboring SH2-domain from an interleukin-6 (IL-6) unresponsive cDNA clone which was named SOCS1 (6). The other group discovered SOCS1 while investigating proteins that interact with catalytic domain of Janus Kinase 2 (JAK2) and named as JAK-binding domain (7). The third group identified this protein using specific monoclonal antibody against SH2 domain of signal transducers and activators of transcription 3 (STAT3) and named it “STAT-induced STAT-inhibitor” (8). Later, in vivo studies have revealed a critical physiological importance of SOCS1, since mice with SOCS1 knockout died within 3 weeks of age due to fatty degeneration of the liver, severe lymphopenia and macrophage infiltration of major organs such as pancreas, heart and lungs (9–11). These pathological consequences in mice lacking SOCS1 had been attributed to uncontrolled interferon (IFN) signaling, as IFN-γ administration in neonatal mice mimicked these complications and mice without SOCS1 and IFN-γ survived longer (12, 13).
SOCS1 is a potent negative regulator of the JAK-STAT pathway that is activated by various pro-inflammatory cytokines including interleukins and interferons (14, 15). JAK-mediated phosphorylation of cytokine receptors creates a docking site for SH2-containing signaling protein substrates. STATs represent one of the major substrates for JAK that mediate most of the cytokines signaling (16). SOCS1 directly interacts with JAK and inhibits its tyrosine kinase activity (17). The efficient inhibition of JAK by SOCS1 is facilitated by 12 amino acids located at immediate N-terminus of the SH2 domain of SOCS1 forming kinase inhibitory region (KIR) that act as a pseudo-substrate of JAK by blocking its substrate-binding cleft (17, 18). SOCS1 is also known to be activated by bacterial lipopolysaccharide (LPS) (19). SOCS1 deficient mice are hypersensitive to LPS, and macrophages isolated from these mice produce higher levels of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and IL-12 (19, 20). SOCS1 is suggested to exert its anti-inflammatory effects on LPS-induced pathologies by interfering with JAK-STAT and toll like receptor (TLR) signaling cascades (20, 21).
A number of inflammatory agents such as TNF-α and LPS cause endothelial barrier disruption that leads to an increase in endothelial permeability and inflammatory responses (22–24), a common pathological feature of multiple lung diseases such as edema, sepsis, acute lung injury (ALI), and acute respiratory distress syndrome (25, 26). A few studies have shown that downregulation of SOCS1 exacerbates lung inflammation (27, 28). Accordingly, exogenous expression of SOCS1 protects lung against hyperoxic injury by inhibiting IL-1β production (29). Similarly, SOCS1 overexpression has been shown to attenuate inflammation during smoke inhalation-induced ALI by inhibiting NALP3 inflammasome formation (30). In addition, overexpression of SOCS1 decreased bleomycin-induced lung inflammation and fibrosis in mice (31).
Endothelial cells (EC) form a semi-permeable barrier for fluids, macromolecules and circulating cells, and the maintenance of EC barrier integrity is especially critical in pathologic conditions such as infection, sepsis, chemical injury, trauma, etc. Endothelial permeability is directly controlled by rearrangements of actin cytoskeleton (32, 33). Prior studies have revealed a crucial role of microtubules (MT) dynamics in altering endothelial barrier function via crosstalk with actin cytoskeleton (34). Many MT plus-end binding proteins such as cytoplasmic linker protein -170 (CLIP-170) and cytoplasmic linker associated protein 2 (CLASP2) directly interact with endothelial adherens junction (AJ) and tight junction (TJ) proteins as well as actin that strengthens endothelial barrier (35). Involvement of SOCS1 in regulating EC responses to inflammatory agents remains to be elucidated. To date, one study has shown that TNF-α-induced apoptosis and inflammation are increased in EC from SOCS1 knockout (KO) mice due to the disassociation SOCS1-ASK1 complex, preventing the degradation of ASK1 (36). Thus, interrelation between SOCS1 and MT dynamics in EC and its impact on inflammatory EC barrier dysfunction remains unexplored.
In the present study, we examined the role of SOCS1 in attenuating LPS-induced EC permeability and inflammation in cultured human lung EC as well as in lungs from EC-specific SOCS1 KO mice. We also investigated the potential mechanisms of anti-inflammatory effects of SOCS1 on LPS-induced endothelial dysfunction and ALI. In addition, we also assessed the impact of MT dynamics and MT-associated proteins on SOCS1 function in EC.
Materials and Methods
Reagents.
LPS (E. coli 0111:B4) and antibodies to ICAM-1 (sc-8439), VCAM-1 (sc-8304), GFP (sc-9996), TLR4 (sc-10741), CLIP-170 (sc-25613) and Myc (sc-40) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). β-actin (A5441), and α-tubulin (T6199) antibodies were from Sigma (St. Louis, MO); phospho-NFκB antibody (3033) was from Cell Signaling (Beverly, MA) and CLASP2 antibody (PA5–13992) was from Invitrogen (Carlsbad, CA). EB1 antibody (610534) was obtained from BD Transduction Laboratories (San Diego, CA), and SOCS1 antibody (ab137384) was from Abcam (Cambridge, MA). Texas Red phalloidin and Alexa Flour 488-conjugated secondary antibodies were purchased form Molecular Probes (Eugene, OR). All other biochemical reagents were obtained from Sigma (St. Louis, MO) unless otherwise specified.
Cell culture and transfections.
Human pulmonary artery endothelial cells (HPAEC) and EGM-2 complete growth media were obtained from Lonza (Allendale, NJ). Cells were used at passages 5–8 and plasmid DNA transfections were done using Lipofectamine LTX with Plus reagent from Thermo Fisher Scientific (Waltham, MA) following manufacturer’s protocol. After 24 h of transfections, cells were subjected for various assays. DNA constructs of CLIP-170 (37) and CLASP2 (38) and plasmids encoding SOCS1 domains (17) have been previously described. To deplete endogenous SOCS1, CLIP-170, and CLASP2, pre-designed siRNAs were obtained from Dharmacon (Lafayette, CO) and transfections were done using RNAiMAX reagent from Thermo Fisher Scientific (Waltham, MA). After 72 h post-transfections, cells were either harvested for Western blot analysis or used for other assays.
Analysis of endothelial permeability.
The permeability across HPAEC monolayers was determined by utilizing two complementary techniques. First, transendothelial electrical resistance (TER) was monitored over indicated time periods under an electrical cell-substrate impedance sensing system ECIS Z (Applied Biophysics, Troy, NY) as described previously (39). All the experiments were carried out when the steady state resistance of cells grown on electrode array (8 wells, 10 microelectrodes/wells) reached to >1200 ohms. In the second assay to determine macromolecules permeability, express permeability testing (XPerT) assay was performed (40). Briefly, cells were grown on biotinylated gelatin-coated plates and at the end point of the experiments, FITC-avidin (25 μg/ml) was directly added to cells and incubated for 3 min. The unbound excess FITC was washed with PBS and coverslips were mounted on slides with DAPI-containing mounting solution. The images were captured in a Nikon Eclipse TE300 inverted microscope.
Co-immunoprecipitation and Western blotting.
At the end of agonist stimulation, cells were lysed on ice-cold TBS-NP40 lysis buffer (20 mM Tris pH 7.4, 150 mM NaCl, 1% NP40) supplemented with protease and phosphatase inhibitor cocktails (Roche, Indianapolis, IN). The lysates were clarified by centrifugation (13000xg, 5 min) and incubated with antibodies overnight at 4°C followed by incubation with Protein G agarose beads for 1 h. After washing 3 times with TBS-NP40 lysis buffer, the immunocomplexes were eluted with 2x Laemmli buffer by heating at 95°C for 5 min. For Western blotting, proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes followed by the incubation with primary antibodies at 4°C overnight. Then, after incubation with HRP-conjugated secondary antibodies at room temperature for 1 hour, membranes were developed using enhanced chemiluminescence substrate (Thermo Fisher, Waltham, MA). Equal protein loading was verified by reprobing the membranes with β-tubulin or β-actin antibodies. The relative intensities of the protein bands were quantified by scanning densitometry using Image Quant software (Molecular Dynamics, Sunnyvale, CA).
Pulldown assay.
To study the protein-protein interactions in vitro by pulldown assays, GST-tagged proteins were expressed in BL21 Escherichia coli strain and purified using glutathione resin (Clontech). Cells were lysed in buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 1% Triton X-100, 10% glycerol, and protease and phosphatase inhibitor cocktail. Clarified lysates were incubated with GST-fusion proteins for 2 h at 4°C and resin-bound proteins were eluted after washing 3 times with lysis buffer. Then, these samples were run on SDS-PAGE for Western blotting analysis.
MT fractionation.
MT fractionation was carried out to analyze MT-associated proteins as previously described (41). Briefly, cell lysis was performed in MT extraction buffer (100 mM PIPES pH 7.0, 1 mM MgCl2, 1mM EGTA, 0.5% NP-40) and lysates were clarified by centrifuging at 12000 rpm for 10 min. Proteins present in MT-enriched fractions (pellet) was dissolved in extraction buffer and run for Western blot analysis.
Immunofluorescence and image analysis.
For immunocytochemistry studies, cells were seeded on glass cover slips and after stimulation for desired periods, cells were subjected to fixation (3.7% formaldehyde for 10 min) followed by permeabilization (0.1% Triton X-100 in PBS for 30 min) and blocking (2% BSA in PBS for 30 min). The incubation with primary antibodies was performed for 1 hour at room temperature followed by staining with Alexa-488 or Alexa-544 conjugated secondary antibodies. To visualize actin filaments, cells were stained with Texas Red-conjugated phalloidin diluted in the blocking solution. The immunostained samples were visualized using a Nikon Eclipse TE300 inverted microscope connected to a Spot RT monochrome digital camera and image processor (Diagnostic Instruments, Sterling Heights, MI). The acquired images were processed in Adobe photoshop.
Proximity ligation assay.
The proximity ligation assay (PLA) was performed using a Duolink In Situ Red Starter Kit Mouse/Rabbit available from Millipore Sigma (Cat no. DUO92101) following manufacturer’s instructions. Briefly, after blocking, cells were incubated with rabbit polyclonal SOCS1 (Abcam, ab137384) and mouse monoclonal α-tubulin (Sigma, T6199) antibodies. After washing, incubation with mixture of secondary antibodies conjugated with oligonucleotides (anti-rabbit PLUS and anti-mouse MINUS PLA probes) was done and proximity ligation reaction was continued using Duolink In Situ Detection reagents. Red PLA signals were visualized, and images were captured in EVOS fluorescence microscope (ThermoFisher Scientific). Reaction without antibodies served as a negative control. PLA signals (red fluorescence) were analyzed using MetaVue 4.6 (Universal Imaging, Downington, PA). The values were statistically processed using Sigma Plot 7.1 (SPSS Science, Chicago, IL) software. For each experimental condition, at least 10 microscopic fields in each independent experiment were analyzed. To demonstrate co-localization of PLA signal with the MT, immunofluorescence staining of MT with β-tubulin antibodies was performed after the PLA assay.
Animal studies.
All animal care and treatment procedures were approved by the Institutional Animal Care & Use Committee of University of Chicago and University of Maryland. C57/BL6 mice were obtained from Jackson Laboratories (Bar Harbor, ME). EC-specific SOCS1 knockout mice were generated by cross-breeding of the endothelial VECad-Cre-ERT2 line (42) and SOCS1flox/flox mice (43) in the C57B6 background, as we previously described (44). Both sets of mice were anesthetized with an intraperitoneal injection of ketamine (75 mg/kg) and xylazine (7.5 mg/kg). Then, sterile saline solution or LPS (0.75 mg/kg body wt.) were given intratracheally. For the evaluation of lung injury parameters, BAL fluid was collected after intratracheal injection of 1 ml of sterile Hanks balanced salt buffer and total cells and protein content in BAL was measured as described previously (45). The vascular leak was analyzed by injecting Evans blue dye (30 mg/kg) into the external jugular vein 2 h before the end of the experiment as described earlier (45) and following perfusion excised lungs were imaged by a Kodak digital camera.
Statistical analysis.
Results are expressed as means ± SD of three to six independent experiments. Unpaired Student’s t-test was used to compare between stimulated groups from controls. For multiple groups comparison, we used one-way analysis of variance (ANOVA) followed by the post hoc Tukey test. P < 0.05 was considered statistically significant.
Results
Role of SOCS1 in modulation of LPS-induced endothelial permeability and inflammatory responses.
Two studies have reported the involvement of SOCS1 in attenuation of lung inflammation (29, 30), but the direct role of SOCS1 in modulating endothelial function remains unclear. Our results show that LPS-induced decrease in TER, an indication of EC barrier disruption, was markedly augmented in pulmonary EC with siRNA-mediated depletion of endogenous SOCS1 (Fig. 1A). Consistently, XPerT assay results demonstrated that knockdown of SOCS1 enhances LPS-induced EC permeability as evidenced by an increase in FITC fluorescence intensity (Fig. 1B). In the next set of experiments, we examined the anti-inflammatory effects of SOCS1 in EC by monitoring the levels of phosphorylated NF-κB as a measure of EC inflammatory activation. Silencing of SOCS1 resulted in elevation of phospho-NFκB levels following LPS challenge (Fig. 1C), suggesting an essential role of SOCS1 in repressing LPS-induced EC inflammation. The upregulation of EC adhesion molecules ICAM-1 and VCAM-1 is an essential step during transendothelial migration of neutrophils to initiate and propagate inflammation (46, 47). Our data showed a significant upregulation of LPS-induced ICAM-1 and VCAM-1 protein expression in SOCS1 siRNA-treated groups, as compared to non-transfected or control non-specific (NS) siRNA groups (Fig.1C). VE-cadherin, the major adherens junction protein, provides the framework for cell-cell contacts and is critical for controlling endothelial permeability (48). LPS caused a decrease in immunoreactivity of VE-cadherin, discontinuous cell-cell junctions, and formation of intercellular gaps. This effect was further escalated in SOCS1-depleted cells (Fig. 1D).
Figure 1. siRNA-mediated knockdown of SOCS1 exacerbates LPS-induced endothelial permeability and inflammation.
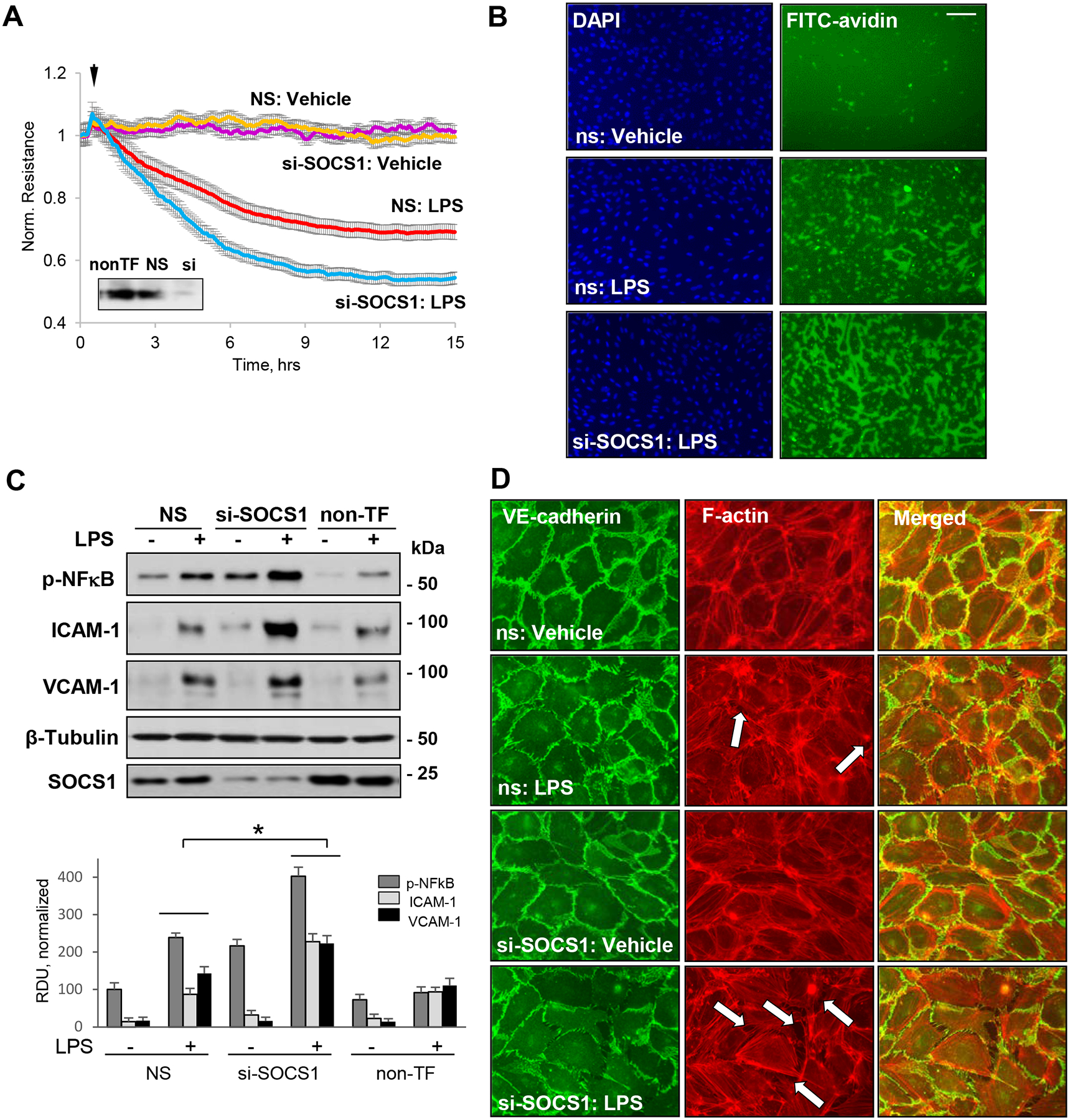
HPAEC transfected with control non-specific siRNA (NS) or siRNA targeting human SOCS1 (si-SOCS1) were stimulated with vehicle (Veh) or LPS (100 ng/ml). A – LPS-induced changes in transendothelial electrical resistance (TER) monitored for 15 h. siRNA-induced SOCS1 protein depletion was confirmed by western blot (inset). B – XPerT permeability assay. EC on coverslips coated with immobilized biotinylated gelatin were transfected with NS or SOCS1 siRNA and stimulated with LPS for 6 h followed by brief addition of FITC-avidin and washing step, as described in Methods. The coverslips were mounted on slides with DAPI-containing mounting solution and inspected by epifluorescence microscopy. Bar: 50 μm. C – Western blot analysis of phospho-NFκB, ICAM-1, and VCAM-1 after 6 h stimulation with LPS. Probing for β-tubulin was used as a loading control. Probing with SOCS1 antibody was performed to verify protein depletion. Bar graph depicts results of quantitative analysis of western blot data; n=6; *p<0.05. D – Immunostaining of control and LPS-treated (6 h) EC was performed with VE-cadherin antibody (green) and Texas Red phalloidin (red) to visualize F-actin. Intracellular gaps are marked by arrows. Bar: 20 μm.
EC-specific SOCS1 KO mice develop more severe ALI.
The role of SOCS1 in preserving endothelial function and reduction of inflammation was further evaluated in genetic models of conditional SOCS1 knockout. Endothelial-specific SOCS1 knockout mice were generated by cross-breeding of the endothelial VECad-Cre-ERT2 line (42) and SOCS1flox/flox mice (43) in the C57B6 background as described in Methods. Intratracheal challenge with LPS caused more prominent extravasation of Evans blue-labelled albumin into the lung parenchyma (Fig. 2A) in EC-SOCS1 KO groups compared to their littermate controls reflective of augmented vascular leak. Analysis of injury parameters bronchoalveolar lavage (BAL) fluid showed that, while BAL protein levels and cell count in SOCS1−/− mice were marginally elevated, they did not reach statistical significance. However, LPS challenge caused much higher elevation of cell counts and protein levels in BAL from EC-SOCS1 KO mice, as compared to their matching wild type controls (Fig. 2B).
Figure 2. EC-specific SOCS1 knockout mice are more susceptible to LPS-induced ALI.
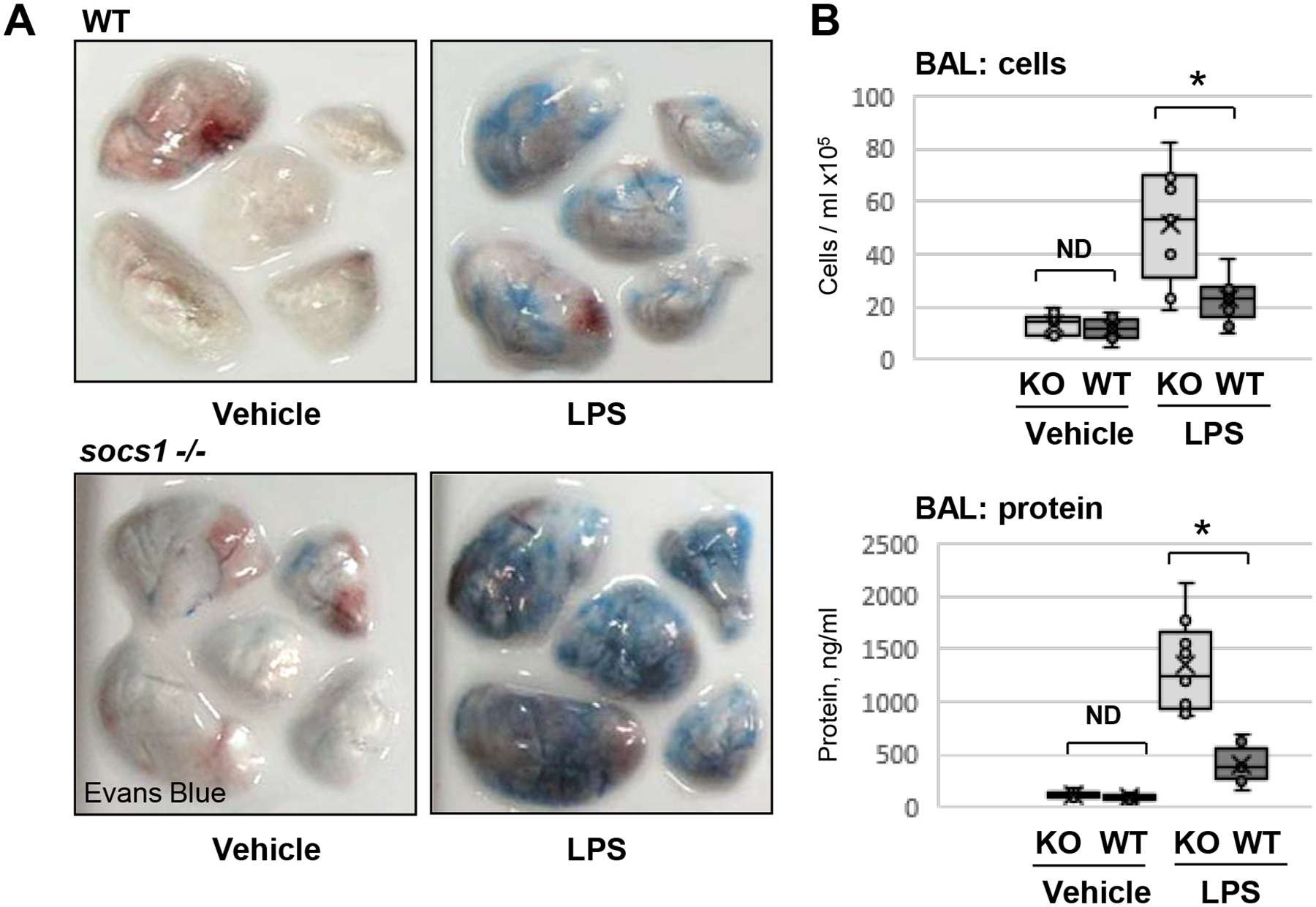
A – Wild type (WT) or EC-specific SOCS1 knockout (socs1 −/−) mice were injected with vehicle (saline) or LPS (intratracheally, 1 mg/kg) and Evans blue dye (30 mg/kg, i.v.) was injected 2 h before the end of the experiment. Lung vascular permeability was determined by visualized Evans blue-stained lungs. B – BAL was collected from control and LPS-exposed WT or socs1 −/− mice to analyze total cells (top) and protein content (bottom). Bar diagrams present individual data points, means (cross) and medians (line); N=6; *p<0.05.
SOCS1 overexpression attenuates LPS-induced endothelial barrier disruption and inflammation.
To further determine the role of SOCS1 in downregulation of agonist-induced EC inflammatory response, we investigated if overexpression of wild type SOCS1 had any impact on LPS-induced endothelial permeability and inflammation. TER analysis revealed that LPS-induced EC permeability was attenuated in cells overexpressing SOCS1 as compared to non-transfected groups (Fig. 3A). Endothelial barrier is formed and maintained by an interaction between various AJ and TJ proteins, while agonist-induced perturbation of this interaction increases endothelial permeability. Results of co-immunoprecipiation (co-IP) assays showed that ectopic expression of SOCS1 significantly attenuated LPS-induced decrease of VE-cadherin - ZO-1 interaction (Fig. 3B). These data suggest that SOCS1 activity is involved in the maintenance of endothelial barrier integrity. In consistence with the TER measurements data, LPS-induced increase in phospho-JAK2, phospho-NFκB, ICAM-1 and VCAM-1 protein levels was reduced in EC overexpressing SOCS1 (Fig. 3C). These results suggest that SOCS1 may act as rheostat controlling the magnitude of EC inflammatory response. We next analyzed the interaction of SOCS1 with key molecules of inflammatory signaling pathways in resting and agonist-stimulated EC. As expected, cAMP, a known barrier enhancing agent, enhanced the interaction of SOCS1 with JAK2, IRAK and NF-κB, while LPS caused a decrease of SOCS1 association with these proteins (Fig. 3D). TNF-α, an inflammatory cytokine, also reduced SOCS1-NFκB interaction but had no effect on interaction with other proteins tested in this study.
Figure 3. Forced expression of SOCS1 attenuates LPS-induced endothelial barrier disruption and inflammation.
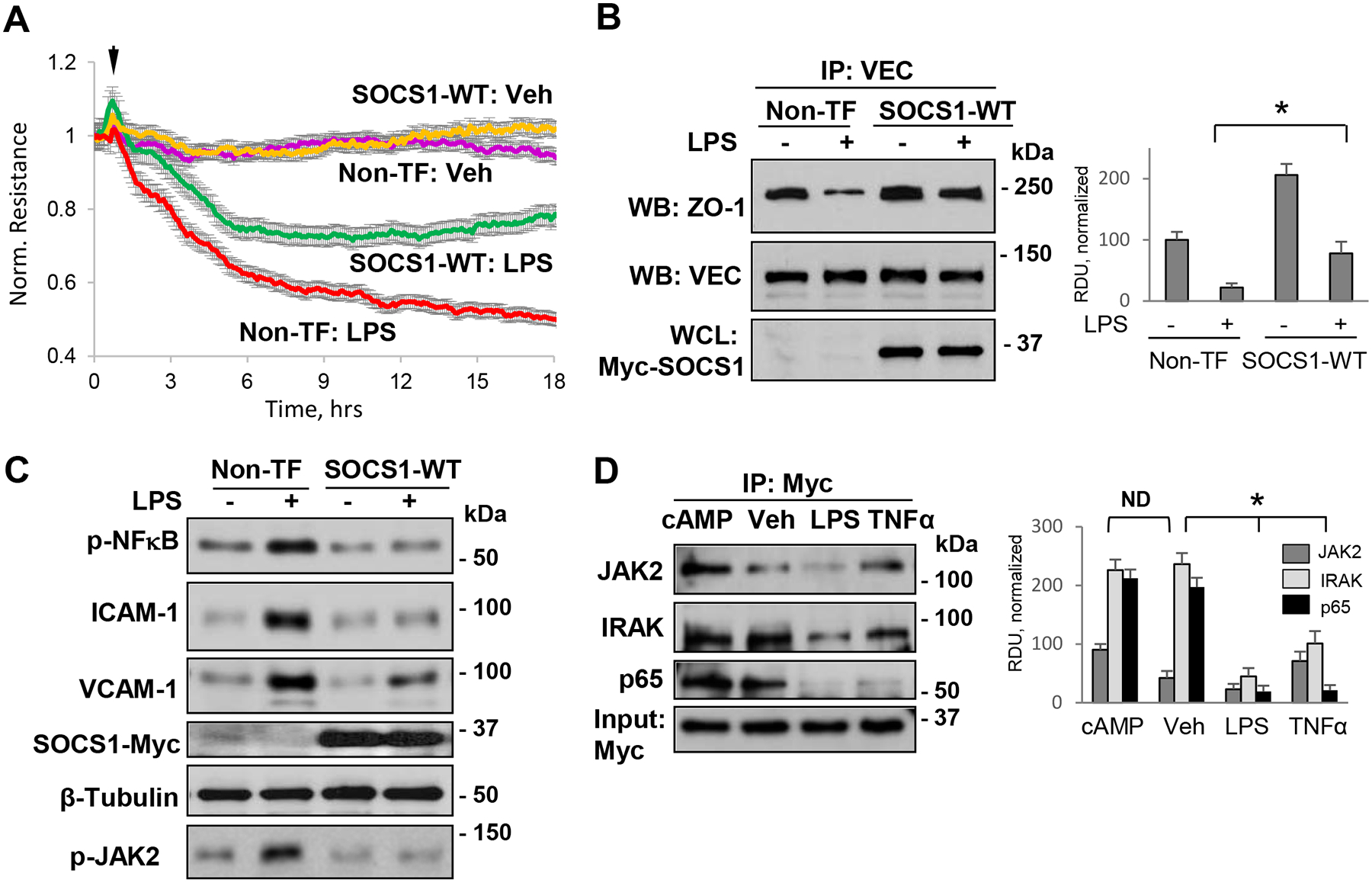
A – Cells were left non-transfected (Non-TF) or transfected with Myc-tagged wild type SOCS1 (SOCS1-WT) for 24 h, followed by stimulation with LPS and monitoring of TER over time. B – Non-transfected or Myc-SOCS1-WT-transfected cells were treated with LPS and cell lysates were analyzed in co-IP assays with VE-cadherin (VEC) antibody to evaluate VEC association with ZO-1. The blots were reprobed with VEC antibody to confirm uniform IP efficiency; SOCS1 overexpression was confirmed by probing whole cell lysates (WCL) with Myc antibody. Bar graph depicts results of quantitative analysis of Western blot data; n=5; *p<0.05. C– Non-transfected or SOCS1-WT transfected cells were exposed to LPS or vehicle for 6 h followed by analysis of phospho-JAK2 and phospho-NFκB reflecting their activation, VCAM-1, ICAM-1 protein levels in cell lysates. Probing with Myc was performed to verify SOCS1 overexpression, and β-tubulin served as a loading control. D – Cells overexpressing SOCS1-WT were stimulated with Br-cAMP (1 mM), LPS (100 ng/ml) or TNF-a (2 ng/ml) followed by co-IP analysis of SOCS1 interactions with JAK2, IRAK and NF-κB p65 using Myc antibody. Total cell lysates were probed with Myc antibody to confirm SOCS1 overexpression. Bar graphs depict results of quantitative analysis of Western blot data; n=5; *p<0.05, ND-no statistically significant difference.
SOCS1 is associated with microtubules.
Next studies focused on delineation of mechanism driving SOCS1 inflammation-inhibitory effects. Considering the emerging role of microtubules (MT) in maintaining endothelial barrier integrity and the link between MT destabilization and activated inflammatory signaling (49–51), we assessed an association between SOCS1 and the MT. Analysis of MT-enriched fractions by Western blot showed significant levels of SOCS1, which were comparable to known MT-associated proteins EB1 and CLASP2 (Fig. 4A). Association of SOCS1 with the MT was further evident when taxol, a MT stabilizing agent, increased SOCS1 protein levels in the MT fraction while colchicine, a MT disrupting agent, reduced SOCS1 levels in the MT fraction. Furthermore, co-immunostaining images showed the presence of endogenous SOCS1 along the MT network, but not with actin filaments (Fig. 4B). SOCS1 association with the MT was further confirmed by proximity ligation assay (PLA) (Fig. 4C). Panels with experimental data show that SOCS1-α-tubulin association (red) was decreased upon LPS challenge. MT staining with β-tubulin antibody (green) was performed after the PLA assay to demonstrate co-localization of PLA signal with the microtubules.
Figure 4. SOCS1 is associated with MT.
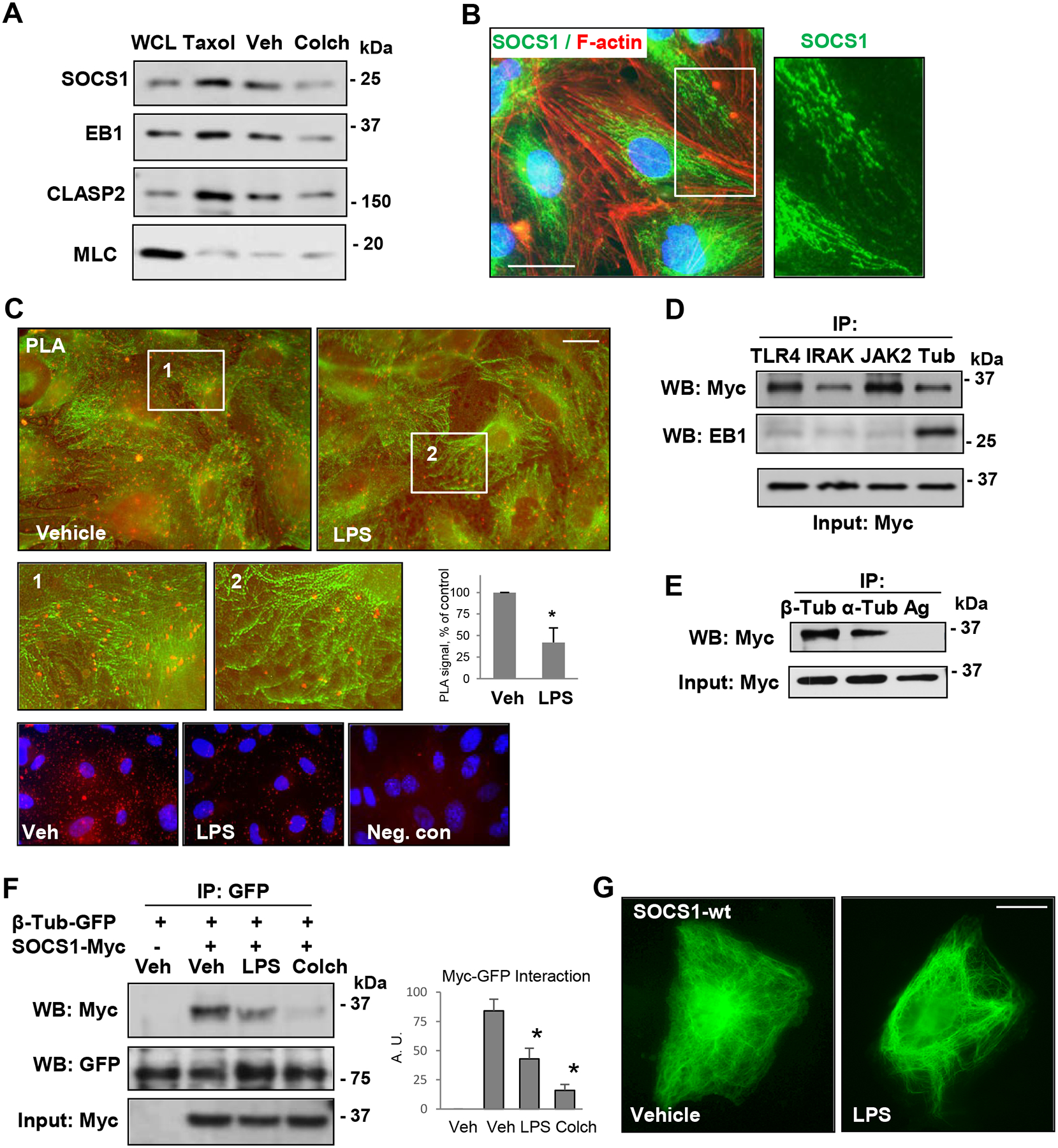
A – Cells were treated with MT polymerizing (Taxol, 0.5 μM, 30 min) and depolymerizing (Colchicine, 0.5 μM, 1 h) agents followed by MT fractionation. The first lane represents whole cell lysates (WCL). Probing with MLC antibody was used as a negative control. B – Immunofluorescence staining of unstimulated pulmonary EC with SOCS1 antibody (green) and Texas Red phalloidin (red). Higher magnification image (inset) depicts details of SOCS1 localization. Bar: 10 μm. C – Proximity ligation assay (PLA) in control and LPS-stimulated cells was performed using SOCS1 and α-tubulin antibodies followed by immunofluorescence staining with β-tubulin (green) antibody. PLA signal of SOCS1 co-localization with α-tubulin is reflected by red fluorescence. Bar: 10 μm. The insets represent higher magnification images; bar graph depicts quantification of PLA fluorescence signal; n=6; *p<0.05. Lower panel presents PLA signal (red) and nuclear counterstaining with DAPI (blue) in vehicle- and LPS-stimulated cells as well as a negative control (PLA reaction without primary SOCS1 antibody). D and E – Cells were transfected with Myc-SOCS1-WT, and co-IP assays were performed with indicated antibodies. SOCS1 interaction with proteins of interest was determined by probing with Myc antibody. Probing for EB1 was used as a negative control for SOCS1 interactions with TLR4, IRAK and JAK2 (D). IP with protein G agarose served as a negative control (E). Probing with Myc antibody was used as an input control for IP assays. F – EC co-expressing GFP-β-tubulin and Myc-SOCS1 were stimulated, with LPS (100 ng/ml, 6 h) or colchicine (0.5 μM, 1h) followed by co-IP with GFP antibody. The presence of SOCS1 in immunocomplexes was detected by western blot Myc antibody; membrane reprobing for GFP was used to verify uniform IP efficiency. Bar graph depicts the quantification of Western blot probed with Myc; n=5; *p<0.05. G – Cells transfected with Myc-SOCS1-WT plasmid were treated with vehicle or LPS followed by immunostaining with Myc antibody. Bar: 10 μm.
Ectopically expressed SOCS1 interacted with MT as strongly as EB1, and only SOCS1, but not EB1, showed interaction with inflammatory signaling proteins TLR4, IRAK1, and JAK2 (Fig. 4D). Likewise, exogenously expressed SOCS1 was detected in immunocomplexes pulled down by antibodies to α- and β-tubulins (Fig. 4E). A direct association of SOCS1 with the MT was further confirmed by co-immunoprecipitation analysis, where exogenously expressed β-tubulin and SOCS1 were detected in the same immunocomplex (Fig. 4F). The interaction between β-tubulin and SOCS1 was abolished in the presence of LPS and colchicine. Immunofluorescence staining of endothelial cells overexpressing Myc-tagged SOCS1 with Myc antibody showed MT-like pattern of SOCS1 localization, which changed to diffused staining after LPS stimulation (Fig. 4G). Altogether, these results provide a strong evidence of SOCS1 association with the MT, which becomes disrupted upon LPS challenge.
SOCS1-mediated control of endothelial barrier is MT-dependent.
We studied a functional significance of SOCS1 association with basal growing MT. To test it, we used a low dose of nocodazole to inhibit normal MT growth, but avoid global MT depolymerization, as we described previously (52). The results showed that preservation of endothelial barrier in LPS-challenged cells caused by ectopic expression of SOCS1 was impaired by nocodazole treatment (Fig. 5A). These data suggest a requirement of functional, non-disrupted MT network for the barrier protective function of SOCS1. A number of MT-associated proteins maintain MT stability, and some MT proteins have been also implicated in stabilizing EC junctions. We used co-immunoprecipitation assays to identify MT-associated proteins that could act as SOCS1 binding partners. The results showed that MT plus-end-tracking proteins CLIP-170 and CLASP2 interacted with SOCS1 (Fig. 5B). The association of SOCS1 with MT-binding proteins was also verified by overexpression approach. For this purpose, co-expression of GFP-tagged CLIP-170, CLASP2, EB1, and β-tubulin and Myc-tagged SOCS1 was followed by IP with GFP antibody and probing for Myc and by reverse co-IP assays. Both sets of co-IP assays showed that SOCS1 associated with CLIP-170, CLASP2, and tubulin, but not with EB1 (Fig. 5C, D). Complementary co-IP assays with SOCS1 antibody in EC with co-expression of Myc-SOCS1 and GFP-tagged EB1 or β -tubulin showed the presence of β-tubulin, but not EB1 in SOCS1 immunocomplexes (Fig. 5D, lower panels). More importantly, these interactions were impaired by cell pre-treatment with colchicine (Fig. 5E). These findings suggest that MT stability is essential for SOCS1 interaction with its binding partners.
Figure 5. SOCS1 function on EC depends on MT.
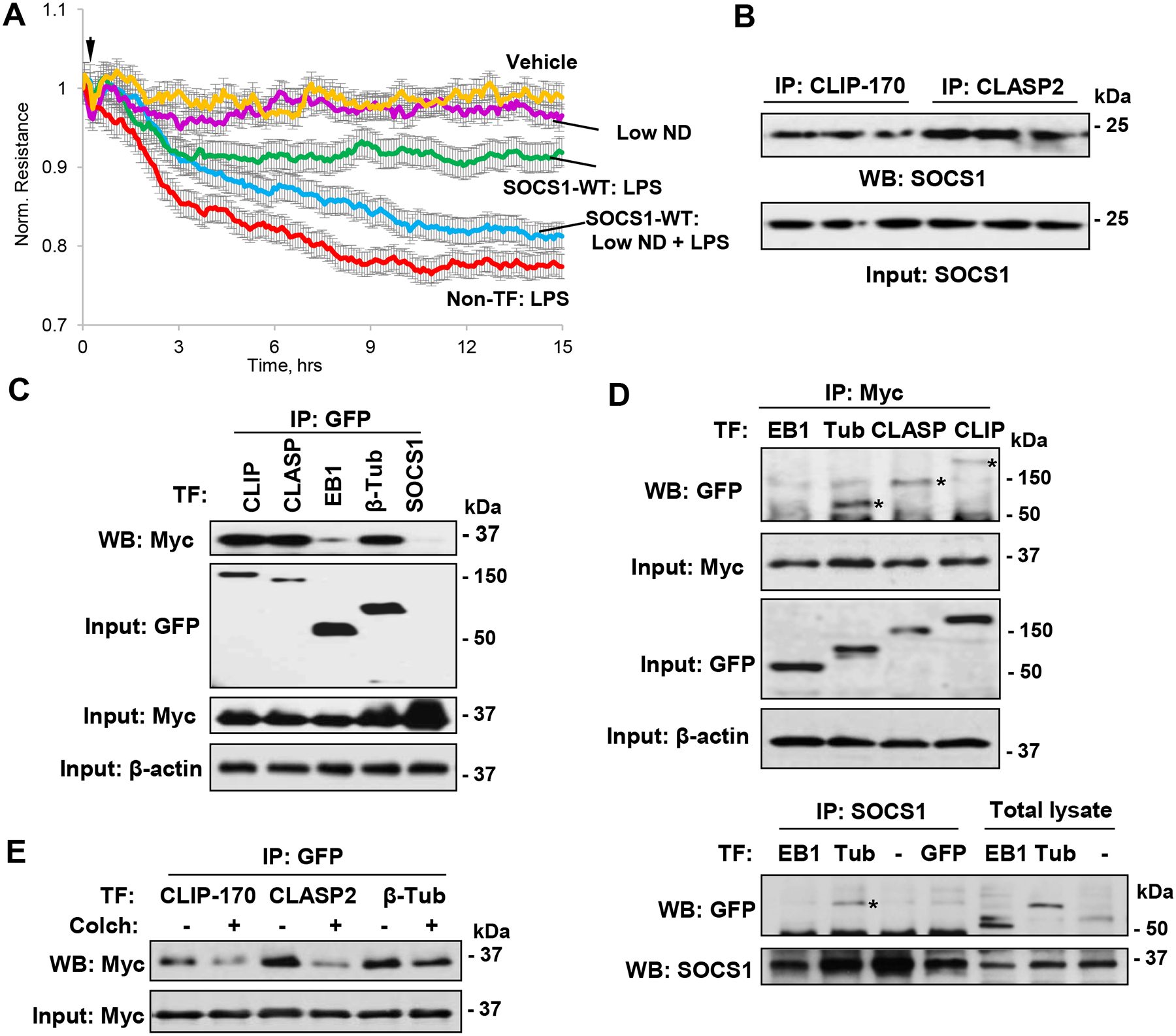
A – SOCS1-WT expressing EC were exposed to LPS in the presence or absence of low dose of nocodazole (0.05 nM) and EC permeability was determined by monitoring TER over indicated time period. B – Co-IP assays with CLIP-170 and CLASP2 antibodies, followed by immunoblotting with SOCS1 antibody. C – Pulmonary EC co-transfected with GFP-tagged CLIP-170, CLASP2, EB1 or β-tubulin and Myc-SOCS1 were tested in co-IP assay with GFP antibody followed by immunoblotting with Myc-antibody. Probing of total cell lysates with GFP and Myc antibodies was used to verify ectopic protein expression; membrane re-probing for β-actin was used to ensure equal sample loadings. D – Cells were co-transfected with GFP-tagged EB1, β-tubulin, CLASP2 or CLIP-170 and Myc-SOCS1 plasmids. Upper panels: Co-IP assay with Myc antibody. Western blot analysis of immunocomplexes with GFP antibody was performed to detect co-immunoprecipitated proteins (marked by asterisks). Probing of total lysates for GFP and Myc was used to verify successful overexpression, and β-actin was used as a loading control. Lower panels: Co-IP assay with SOCS1 antibody. E – Cells with co-expression of GFP-tagged CLIP-170, CLASP2 or β-tubulin and Myc-SOCS1 for 24 h were stimulated with colchicine (0.5 μM, 1 h), followed by co-IP with GFP and immunoblotting with Myc antibody. Whole cell lysates were probed for Myc to verify ectopic SOCS1 expression.
Characterization of binding domains involved in SOCS1 interaction with CLIP-170 and CLASP2.
By using a set of deletion mutants, we mapped the regions of SOCS1, CLIP-170, and CLASP2 that mediate their interactions. First, we examined intracellular localization patterns of Myc-tagged full length SOCS1 and its N-terminal and C-terminal deletion mutants. Immunostaining with Myc antibody revealed that wild type SOCS1 (WT, full length) shows MT-like localization pattern, and SOCS1 ΔC lacking C-terminal 40 aa, SOCS box region largely retains this property. In contrast, deletion of N-terminal 59 aa residues in SOCS1 ΔN mutant completely abolished SOCS1 ΔN colocalization with the MT (Fig. 6A). Next, wild type and ΔN SOCS1 constructs were co-transfected with GFP-tagged CLIP-170 or CLASP2 constructs and immunoprecipitated with GFP antibody. Immunoblotting with Myc showed the interaction of SOCS1 WT and ΔC, but not SOCS1 ΔN with both CLIP-170 and CLASP2 (Fig. 6B). These results suggested that N-terminal region of SOCS1 is indispensable for its interaction with MT-associated proteins. To further characterize the interacting domains of CLIP-170 and CLASP2 involved in binding with SOCS1, we performed pulldown assays with bacterially expressed and purified GST-fusion constructs containing various regions of CLIP-170 and CLASP2. SOCS1 from EC lysate showed a strong interaction with N2 domain of both CLIP-170 and CLASP2 (Fig. 6C). As a positive control to verify our pulldown assays, we also checked the binding of EB1 to immobilized GST beads fused to various regions of CLIP and CLASP. EB1 showed strong association with N2 domains of CLIP-170 and CLASP2 (Fig. 6D). The interactions of CLIP-170 and CLASP2 domains of varying lengths with SOCS1 were also evaluated after overexpression of SOCS1 in EC. Similarly to endogenous SOCS1, ectopically expressed Myc-tagged SOCS1 strongly interacted with N2 domains of both CLIP and CLASP2 (Fig. 6E). To determine the critical domain of SOCS1 essential for its binding to N2 domains of CLIP-170 and CLASP2, we performed a pulldown assay by overexpressing SOCS1 constructs that harbor different domains. The results showed that only WT and ΔC SOCS1 associated with N2 domains of CLIP-170 and CLASP2 (Fig. 6F). The lack of interaction between SOCS1 ΔN deletion mutant and CLIP-170/CLASP2 N2 domains again suggests that N-terminal region of SOCS1 is essential for its binding with these MT-associated proteins.
Figure 6. N-terminal of SOCS1 interacts with N2 domains of CLIP-170 and CLASP2.
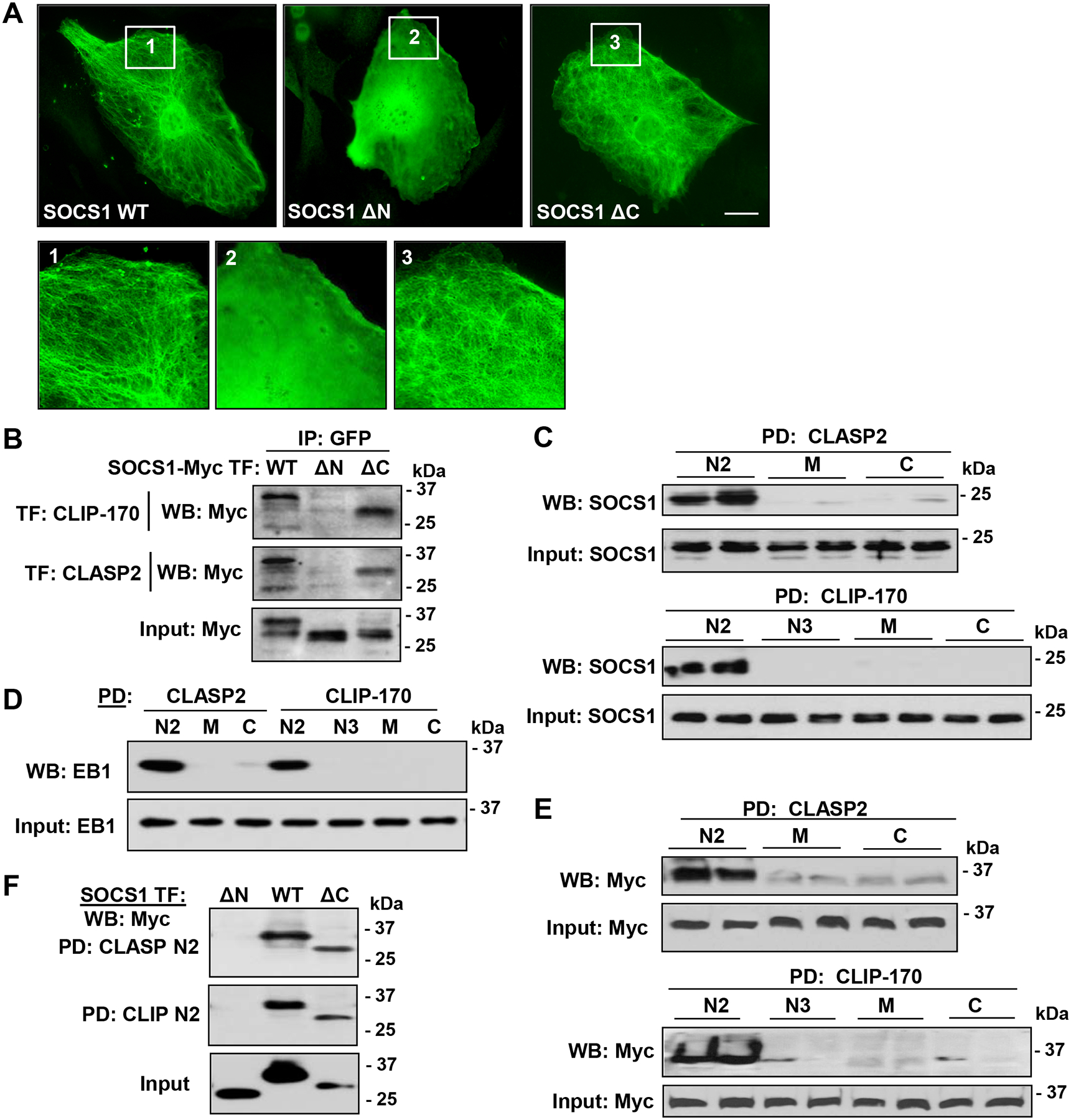
A – Cells were transfected with Myc-tagged SOCS-WT or its N-terminal (ΔN59, deletion of 59 aa at N-terminal region of SOCS1) or C-terminal (ΔC40, deletion of 40 aa of C-terminal region containing SOCS box) deletion mutant. Immunofluorescence staining with Myc antibody was performed to analyze SOCS1 intracellular localization. Higher magnification insets show details of SOCS1 localization. Bar: 5 μm. B – Cells were co-transfected with GFP-tagged CLIP-170 or CLASP2 and Myc-tagged wild type SOCS1 WT, ΔN59, and ΔC40. Co-IP assay was performed with GFP antibody; presence of SOCS1 mutants in immunocomplexes was detected by Myc antibody. Equal amounts of total cell lysates were used for Western blot to verify the overexpression of SOCS1 constructs. C and D – Pulldown assay was carried out with GST-fusion proteins containing N2, M, and C domains of CLASP2 or N2, N3, M, and C domains of CLIP-170. SOCS1 (C) and EB1 (D) association with immobilized CLIP-170 and CLASP2 domains was monitored by immunoblotting with SOCS1 antibody. E – Similar pulldown assays as in (C) were performed after overexpression of SOCS1-WT followed by probing with Myc antibody. Equal amounts of total cell lysates were analyzed by Western blot as input to verify successful plasmid DNA transfections. F – Cells were transfected with Myc-tagged SOCS1-WT, ΔN, or ΔC, followed by pulldown with GST-N2 domains of CLIP-170 and CLASP2. The proteins bound to immobilized GST beads were analyzed by Western blotting to detect Myc. The expression of SOCS1 was confirmed by immunoblotting of total cell lysates with Myc antibody.
N-terminal region of SOCS1 is essential for its barrier protective and anti-inflammatory functions.
Since our data provided evidence that N-terminal region of SOCS1 is required for its interaction with MT-binding proteins, we next examined whether this interaction has direct impact on SOCS1 function as a suppressor of inflammatory signaling. First, we investigated permeability changes in EC with ectopic expression of SOCS1 WT or SOCS1 ΔN after LPS stimulation. The results showed that the attenuating effects of SOCS1 on LPS-induced endothelial permeability were lost in EC with SOCS1 ΔN ectopic expression (Fig. 7A). We next tested the effects of overexpressed WT SOCS and its ΔN mutant on LPS-induced ICAM-1 protein expression as a measure of endothelial inflammation. Ectopic expression of SOCS1 WT markedly suppressed LPS-induced ICAM-1 upregulation, however EC expressing SOCS1-ΔN displayed similar induction of ICAM-1 expression in response to LPS as mock-transfected controls (Fig. 7B). Effects of ectopically expressed SOCS1 constructs on LPS-induced cytoskeletal remodeling were further analyzed by immunofluorescent staining of actin cytoskeleton. SOCS1 WT exhibited protective effects against LPS-induced F-actin reorganization and stress fibers formation, while such protection was lost in EC expressing SOCS1-ΔN (Fig. 7C). Taken together, these results strongly suggest the essential role for N-terminal region for barrier protective and anti-inflammatory activities of SOCS1 in pulmonary EC.
Figure 7. N-terminal region of SOCS1 is indispensable for its barrier protective and anti-inflammatory effects on EC.
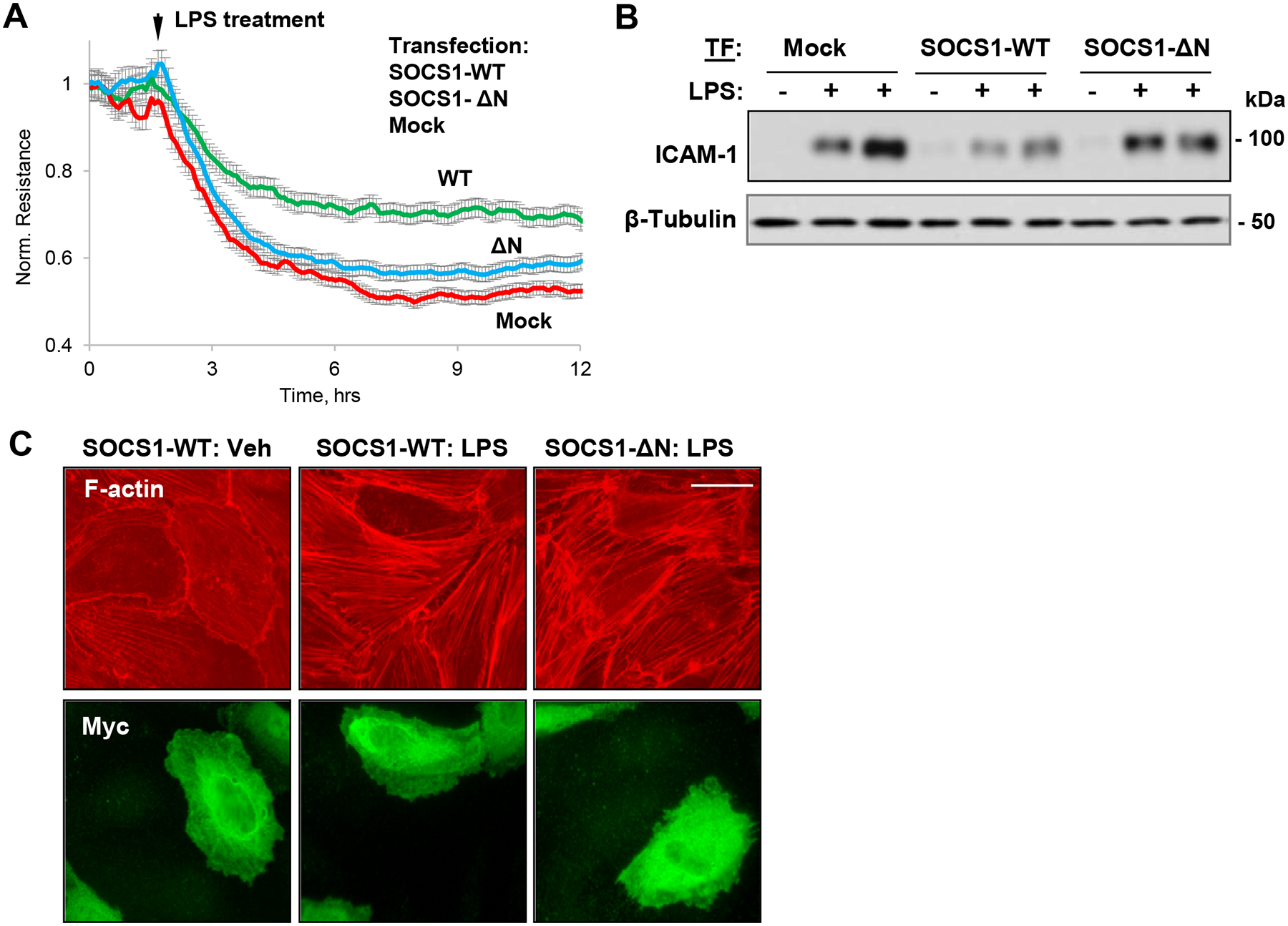
HPAEC expressing SOCS1-WT or its ΔN mutant were stimulated with LPS (100 ng/ml). A – Permeability changes were monitored by TER measurements over time. B – Western blot analysis of ICAM-1 protein levels in control and LPS-stimulated (6 h) cells; β-tubulin was used as a loading control. C – Cells were treated with LPS, and immunofluorescence staining was performed with Texas Red phalloidin to visualize actin cytoskeleton. Co-staining with Myc antibody was performed to visualize transfected cells. Bar: 10 μm.
Interaction of SOCS1 with MT-associated proteins is critical for its function.
To further evaluate the functional significance of SOCS1 interaction with MT-associated proteins CLIP-170 and CLASP2, we performed co-transfection experiments followed by co-immunoprecipitation assays. Specifically, we simultaneously transfected EC with siRNAs targeting CLIP-170 or CLASP2 and overexpressed SOCS1. Then, interaction of SOCS1 with two potential regulators of inflammatory signaling pathways was investigated. The association of SOCS1 with JAK2 was impaired by endogenous depletion of either CLIP-170 or CLASP2 proteins (Fig. 8A). Likewise, there was a significant decrease in the interaction between SOCS1 and TLR4 in EC transfected with CLIP-170- or CLASP2-specific siRNAs (Fig. 8A).
Figure 8. SOCS1 interactions with MT proteins is required for its anti-inflammatory function.
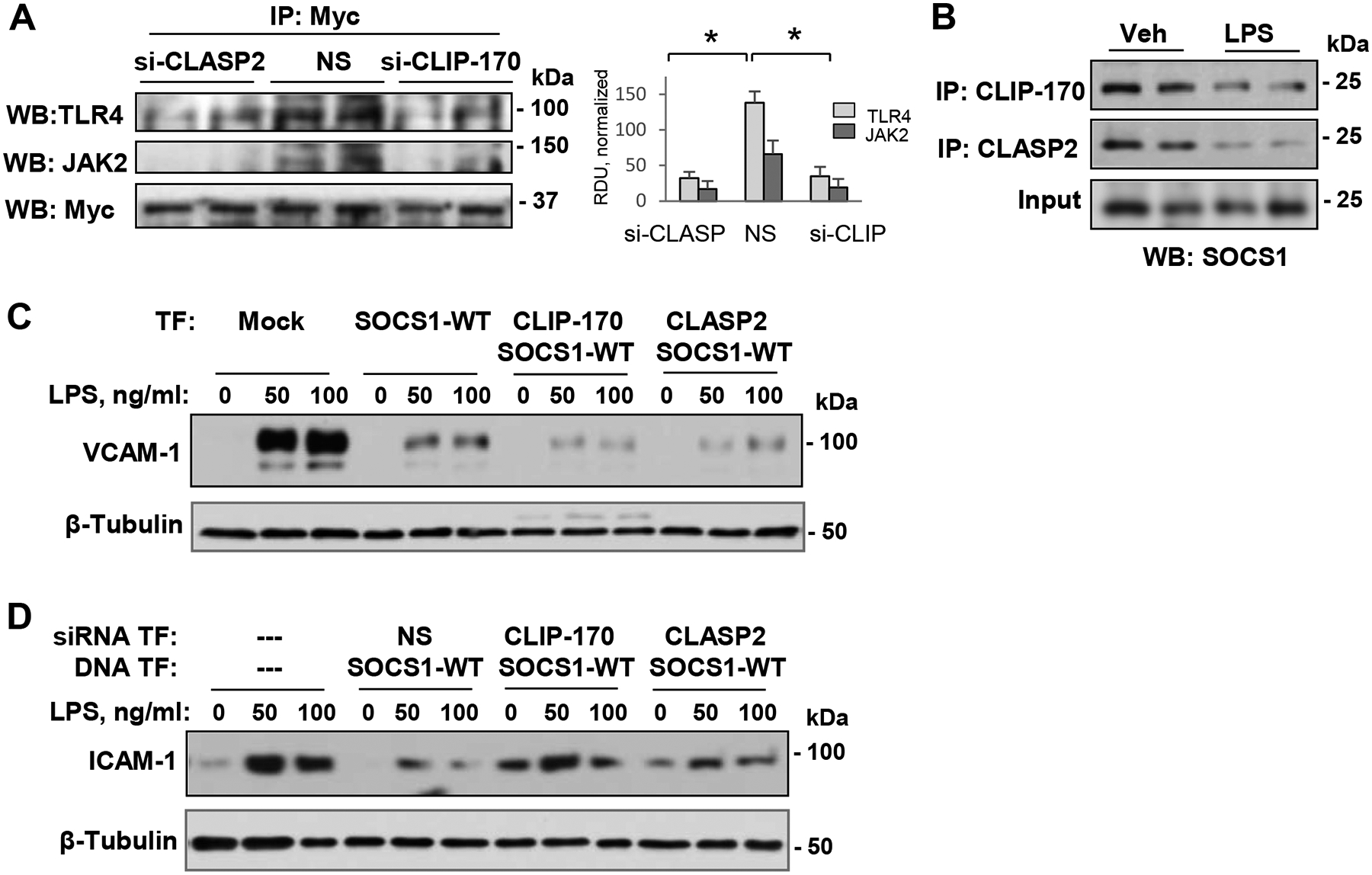
A – Pulmonary EC co-transfected with non-specific (NS) or gene-specific siRNAs targeting CLIP-170 or CLASP2 and Myc-SOCS1-WT plasmid DNA followed by co-IP with Myc antibody. Presence of TLR4 and JAK2 in SOCS1immunocomplexes was monitored by Western blot. Quantification of SOCS1 interaction with TLR4 and JAK2 is presented in a bar graph; n=5; p<0.05. B – Co-IP assays of LPS-stimulated EC using CLIP-170 or CLASP2 antibodies. Presence of SOCS1 in immunocomplexes was monitored by Western blot. Total lysates were probed with SOCS1 antibody to verify equal inputs for immunoprecipitation. C – VCAM-1 levels in control and LPS-stimulated (6 h) EC co-transfected with Myc-SOCS1-WT and GFP-tagged CLIP-170 or CLASP2 were detected by Western blot. β-Tubulin was used as a loading control. D – Cells were co-transfected with NS or gene-specific siRNAs targeting CLIP-170 and CLASP2 and SOCS1-WT plasmid DNA followed by LPS stimulation. Cell lysates were analyzed by Western blot to detect ICAM-1; β-tubulin served as a loading control.
Since our data so far indicated a critical importance of SOCS1-MT interactions in regulating SOCS1 function, we next determined whether this interaction could be disrupted in inflammatory settings. The data show that LPS caused a marked reduction in the SOCS1 interaction with CLIP-170 and CLASP2, which was evaluated by co-immunoprecipitation assays (Fig. 8B). An importance of SOCS1-CLIP/CLASP interaction in its anti-inflammatory effects was further evident when co-expression of WT SOCS1 with full length CLIP-170 or CLASP2 augmented the inhibition of LPS-induced increase in VCAM-1 expression (Fig. 8C). Consistently with these results, the knockdown of CLIP-170 or CLASP2 abolished the suppressive effect of SOCS1 overexpression on LPS-induced ICAM-1 induction (Fig. 8D). These results support the requirement for SOCS1 association with CLIP-170 and CLASP2 in order to fully display SOCS1 anti-inflammatory activities.
Discussion
This study provides a compelling evidence that SOCS1 plays a critical role in preserving endothelial function in inflammatory conditions. Furthermore, our data suggest that assembled MT reaching out the cell periphery are essential for optimal function of SOCS1 in the lung endothelium. Combined with its established anti-inflammatory effects and suppressing lung injury, our data reveal a novel mechanism of MT-dependent control of SOCS1 anti-inflammatory function.
The downregulation of SOCS1 expression has been previously linked to an increase in LPS or bleomycin-induced inflammation in mice lungs (28, 31). Our data show prominent vascular barrier protective and anti-inflammatory properties of SOCS1 in the LPS model of ALI, as illustrated by combination of knockdown, overexpression, and in vivo cell-specific knockout experiments. Our data strongly suggest that alteration in the expression or activity of SOCS1 may have a major impact on initiation and propagation of EC inflammation. These findings are further supported by reports that SOCS1 deficiency increases the susceptibility to inflammation and overexpression of SOCS1 ameliorates lung injury by suppressing inflammatory signaling (27–30, 36).
Mechanistically, our results suggest that barrier protective and anti-inflammatory functions of SOCS1 largely depend on MT dynamics. The immunostaining experiments showed SOCS1 colocalization with the MT network, and LPS stimulation disrupted this MT-like staining pattern (Fig. 4). A bona fide interaction of SOCS1 with the MT was further supported by PLA assays and was again markedly reduced by LPS challenge. The association of SOCS1 with the MT was also confirmed in functional assays with MT depolymerizing agent nocodazole. Even a low dose of nocodazole, which is only able to inhibit MT growth without global MT depolymerization (52), abolished barrier protective effects of SOCS1 overexpression on LPS-induced endothelial permeability. These results suggest that MT growth on its own may be important in proper targeting of SOCS1. These findings prompted us to evaluate MT plus-end binding proteins CLIP-170 and CLASP as interacting partners of SOCS1.
Destabilization of MT and breakdown of EC junctional protein complexes are commonly observed phenomenon during inflammatory endothelial dysfunction. Additionally, some MT-associated proteins such as stathmin and EB1 had been implicated in modulation of agonist-induced endothelial permeability (53, 54). More recently, we have shown an important role of CLASP2 in protecting against HKSA-induced endothelial barrier disruption via its interaction with p120-catenin (51). The results from this study demonstrate that SOCS1 interacts with CLASP2 and CLIP-170. We also determined the regions of CLIP-170 and CLASP2 involved in the interaction with SOCS1. Both endogenous and ectopically expressed SOCS1 showed strong association with N2 domains of CLIP and CLASP. Importantly, SOCS1-CLIP/CLASP interaction appears to be indispensable for SOCS1-mediated positive regulation of endothelial function. These conclusions are drawn from our data that SOCS1 interacted with both, CLASP2 and CLIP-170 (Fig. 6), and knockdown of either MT-end binding protein abolished both, SOCS1 association with TLR4-JAK2 inflammatory signaling complex (Fig. 8A) and SOCS1 inhibitory effect on ICAM1 expression (Fig. 8D). In turn, forced co-expression of either CLIP-170, or CLASP2 with SOCS1 augments anti-inflammatory activity of SOCS1 monitored by blocking LPS-induced ICAM1 induction (Fig. 8C). Based on these results we conclude that SOCS1 interaction with CLIP-170 and CLASP2 is interchangeable with respect to maximizing the SOCS1inflammation-inhibitory activity. This phenomenon may be explained by the notion that both, CLIP-170 and CLASP2 target SOCS1 to MT growing tips leading to optimal SOCS1 positioning at cell sub-membrane compartment for spatial alignment with TLR4/JAK2 signaling nexus.
At this point it is not clear whether SOCS1 delivery by the MT is the sole factor critical for SOCS1 function in endothelium, or complex interaction between MT and actin cytoskeleton and other signaling mechanisms are also involved in proper SOCS1 targeting and optimal activity. Nevertheless, our data show that silencing of CLIP-170 and CLASP2 impairs the interaction of SOCS1 with two major inflammatory signaling-related proteins TLR4 and JAK2 (Fig. 8A). LPS-stimulation of TLR4 mediates inflammatory responses by MyD88-dependent or independent pathways that also involve the downstream activation of the NF-κB pathway to generate inflammatory cytokines (55).
Our results indicate that SOCS1 association with MT proteins is required for SOCS1-dependent inhibition of JAK activity. It is also important to mention that, beyond its receptor‐proximal function in Jak/STAT signaling, SOCS1 operates as a ubiquitin ligase by virtue of its SOCS box participating in the formation of E3 ligase complexes, marking Jak2 and other protein targets (TEL‐Jak2 oncogene protein, Vav and insulin receptor substrates IRS1 and IRS2) mediating inflammatory signaling for proteasomal degradation (56, 57). Moreover, SOCS1 also mediates polyubiquitination and proteasomal degradation of nuclear p65, thus limiting prolonged p65 signaling and terminating the expression of NFκB inducible genes (58).
LPS-induced inflammatory response triggers generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) (59). The resulting oxidation and S-nitrosation modifications of cellular proteins may significantly affect their activity and function. Published studies show that S-nitrosation incapacitated SOCS1 binding to p65 and p65 targeting for degradation, leading to increased inflammation (60). On the other hand, our previous studies have demonstrated that LPS-induced disassembly of peripheral MT network is as result of LPS-induced ROS production and oxidative modifications of tubulin (49). This study shows that LPS-induced MT disassembly stimulated release of MT-associated CLIP-170/CLASP2 complexes. In turn, SOCS1 localization on MT and association with CLIP-170/CLASP2 was observed in non-stimulated but became impaired in LPS-stimulated cells. These results strongly suggest that in vascular EC SOCS1 binds to CLIP-170/CLASP2 complex associated with intact MT. Taken together, published findings and the data presented in this study suggest that LPS may impair anti-inflammatory activity of SOCS1 by a) partial inhibition of its ubiquitin ligase activity via S-nitrosation, and b) disturbed CLIP-170/CLASP2-assisted targeting of SOCS1 to the submembrane compartment caused by LPS-induced peripheral MT disassembly, which leads to decreased availability of SOCS1 at the subcortical compartment and impaired interactions with inflammatory proteins.
Our results of structure-function analysis show a vital role of N-terminal region of SOCS1 in its interaction with MT-associated proteins as well as its positive regulation of endothelial function. These data are consistent with the role of KIR located at N-terminal region of SOCS1 in inhibiting inflammatory signaling kinases such as JAK (17). C-terminal region of SOCS1 containing SOCS box motif is also reported to be important for its anti-inflammatory function (61). However, our results show that SOCS1 mutant lacking C-terminal region interacted normally with CLIP-170 and CLASP2, ruling out its role on SOCS1 regulation of endothelial function. This is consistent with the initial finding that deletion of up to 44 amino acid residues of C-terminal region of SOCS1 still retains the ability to inhibit STAT5 (17). On the contrary, SOCS1 N-terminal deletion mutant failed to associate with CLIP-170 or CLASP2 and more importantly, endothelial barrier protective and anti-inflammatory effects of SOCS1 were largely impaired with this mutation. These data strongly suggest that N-terminal region of SOCS1 is critical for its anti-inflammatory function on EC. One limitation of this study is that it is not clear whether only 12 amino acids comprising KIR at N-terminal domain of SOCS1 determine its regulation of EC or other adjacent regions are also involved, or whether this interaction is additionally controlled by protein phosphorylation events. Further studies are warranted to define the precise mechanism of SOCS1 interaction with MT-associated proteins and map the exact amino acid stretch within the N-terminal region of SOCS1 which mediates this interaction.
In summary, this study describes a novel modality in cytoskeletal regulation of SOCS1 anti-inflammatory activity and demonstrates a mechanism of SOCS1 targeting to TLR4 and Jak2 mediated by MT-CLIP170-CLASP2 module which downregulates LPS-induced inflammation and barrier dysfunction of the lung endothelium. This new principle of receptor targeting of anti-inflammatory molecules may be implied for development of future therapeutics for precise inhibition of inflammatory signaling complexes to improve the outcomes of ALI and other inflammatory syndromes.
Acknowledgements
This work was supported by the grants HL107920, HL130431 from the National Heart, Lung, and Blood Institute, and GM114171 from the National Institute of General Medical Sciences for A.A.B.; HL076259, HL087823 from the National Heart, Lung, and Blood Institute for K.G.B.; JSPS KAKENHI (S) JP17H06175, Challenging Research (P) JP18H05376, and AMED-CREST JP 20GM1110009 grants for A.Y.
Abbreviations
- AJ
adherens junction
- ALI
acute lung injury
- CLASP2
cytoplasmic linker associated protein 2
- CLIP-170
cytoplasmic linker protein -170
- EC
endothelial cells
- ECIS
Electric Cell-substrate Impedance Sensing
- HPAEC
human pulmonary artery endothelial cells
- JAK
Janus Kinase
- ICAM-1
intercellular adhesion molecule-1
- IL
interleukin
- IP
immunoprecipitation
- KO
knockout
- LPS
lipopolysaccharide
- MT
microtubules
- nsRNA
non-specific RNA
- PLA
proximity ligation assay
- SOCS
Suppressor of cytokine signaling
- STAT
signal transducers and activators of transcription
- TER
transendothelial electrical resistance
- TJ
tight junction
- TLR
toll like receptor
- TNF-α
tumor necrosis factor-α
- VE-cadherin
vascular endothelial cadherin
- VCAM-1
vascular cell adhesion molecule-1
- XPerT
express permeability testing
References
- 1.Alexander WS (2002) Suppressors of cytokine signalling (SOCS) in the immune system. Nat Rev Immunol 2, 410–416 [DOI] [PubMed] [Google Scholar]
- 2.Yoshimura A, Naka T, and Kubo M (2007) SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol 7, 454–465 [DOI] [PubMed] [Google Scholar]
- 3.Turner MD, Nedjai B, Hurst T, and Pennington DJ (2014) Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta 1843, 2563–2582 [DOI] [PubMed] [Google Scholar]
- 4.Kubo M, Hanada T, and Yoshimura A (2003) Suppressors of cytokine signaling and immunity. Nat Immunol 4, 1169–1176 [DOI] [PubMed] [Google Scholar]
- 5.Krebs DL, and Hilton DJ (2000) SOCS: physiological suppressors of cytokine signaling. J Cell Sci 113 (Pt 16), 2813–2819 [DOI] [PubMed] [Google Scholar]
- 6.Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, and Hilton DJ (1997) A family of cytokine-inducible inhibitors of signalling. Nature 387, 917–921 [DOI] [PubMed] [Google Scholar]
- 7.Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, Matsumoto A, Tanimura S, Ohtsubo M, Misawa H, Miyazaki T, Leonor N, Taniguchi T, Fujita T, Kanakura Y, Komiya S, and Yoshimura A (1997) A new protein containing an SH2 domain that inhibits JAK kinases. Nature 387, 921–924 [DOI] [PubMed] [Google Scholar]
- 8.Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, Nishimoto N, Kajita T, Taga T, Yoshizaki K, Akira S, and Kishimoto T (1997) Structure and function of a new STAT-induced STAT inhibitor. Nature 387, 924–929 [DOI] [PubMed] [Google Scholar]
- 9.Starr R, Metcalf D, Elefanty AG, Brysha M, Willson TA, Nicola NA, Hilton DJ, and Alexander WS (1998) Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc Natl Acad Sci U S A 95, 14395–14399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naka T, Matsumoto T, Narazaki M, Fujimoto M, Morita Y, Ohsawa Y, Saito H, Nagasawa T, Uchiyama Y, and Kishimoto T (1998) Accelerated apoptosis of lymphocytes by augmented induction of Bax in SSI-1 (STAT-induced STAT inhibitor-1) deficient mice. Proc Natl Acad Sci U S A 95, 15577–15582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marine JC, Topham DJ, McKay C, Wang D, Parganas E, Stravopodis D, Yoshimura A, and Ihle JN (1999) SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell 98, 609–616 [DOI] [PubMed] [Google Scholar]
- 12.Gresser I, Guy-Grand D, Maury C, and Maunoury MT (1981) Interferon induces peripheral lymphadenopathy in mice. J Immunol 127, 1569–1575 [PubMed] [Google Scholar]
- 13.Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, Kay TW, Nicola NA, Hertzog PJ, Metcalf D, and Hilton DJ (1999) SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell 98, 597–608 [DOI] [PubMed] [Google Scholar]
- 14.Schindler C, Levy DE, and Decker T (2007) JAK-STAT signaling: from interferons to cytokines. J Biol Chem 282, 20059–20063 [DOI] [PubMed] [Google Scholar]
- 15.Horvath CM (2004) The Jak-STAT pathway stimulated by interleukin 6. Sci STKE 2004, tr9. [DOI] [PubMed] [Google Scholar]
- 16.Ihle JN, and Kerr IM (1995) Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet 11, 69–74 [DOI] [PubMed] [Google Scholar]
- 17.Yasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, Ohtsuka S, Imaizumi T, Matsuda T, Ihle JN, and Yoshimura A (1999) The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. Embo J 18, 1309–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nicholson SE, Willson TA, Farley A, Starr R, Zhang JG, Baca M, Alexander WS, Metcalf D, Hilton DJ, and Nicola NA (1999) Mutational analyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction. EMBO J 18, 375–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, Yoshida H, Kubo M, and Yoshimura A (2002) SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity 17, 583–591 [DOI] [PubMed] [Google Scholar]
- 20.Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, Abe T, Seki E, Sato S, Takeuchi O, Takeda K, Akira S, Yamanishi K, Kawase I, Nakanishi K, and Kishimoto T (2002) SOCS-1 participates in negative regulation of LPS responses. Immunity 17, 677–687 [DOI] [PubMed] [Google Scholar]
- 21.Kimura A, Naka T, Muta T, Takeuchi O, Akira S, Kawase I, and Kishimoto T (2005) Suppressor of cytokine signaling-1 selectively inhibits LPS-induced IL-6 production by regulating JAK-STAT. Proc Natl Acad Sci U S A 102, 17089–17094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldblum SE, Hennig B, Jay M, Yoneda K, and McClain CJ (1989) Tumor necrosis factor alpha-induced pulmonary vascular endothelial injury. Infect Immun 57, 1218–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petrache I, Birukova A, Ramirez SI, Garcia JG, and Verin AD (2003) The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am J Respir Cell Mol Biol 28, 574–581 [DOI] [PubMed] [Google Scholar]
- 24.Birukova AA, Wu T, Tian Y, Meliton A, Sarich N, Tian X, Leff A, and Birukov KG (2013) Iloprost improves endothelial barrier function in lipopolysaccharide-induced lung injury. Eur Respir J 41, 165–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aird WC (2003) The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 101, 3765–3777 [DOI] [PubMed] [Google Scholar]
- 26.Oda M, Han JY, and Nakamura M (2000) Endothelial cell dysfunction in microvasculature: relevance to disease processes. Clin Hemorheol Microcirc 23, 199–211 [PubMed] [Google Scholar]
- 27.Chong MM, Metcalf D, Jamieson E, Alexander WS, and Kay TW (2005) Suppressor of cytokine signaling-1 in T cells and macrophages is critical for preventing lethal inflammation. Blood 106, 1668–1675 [DOI] [PubMed] [Google Scholar]
- 28.Wang W, Liu Z, Su J, Chen WS, Wang XW, Bai SX, Zhang JZ, and Yu SQ (2016) Macrophage micro-RNA-155 promotes lipopolysaccharide-induced acute lung injury in mice and rats. Am J Physiol Lung Cell Mol Physiol 311, L494–506 [DOI] [PubMed] [Google Scholar]
- 29.Galam L, Soundararajan R, Breitzig M, Rajan A, Yeruva RR, Czachor A, Harris F, Lockey RF, and Kolliputi N (2016) SOCS-1 rescues IL-1beta-mediated suppression of epithelial sodium channel in mouse lung epithelial cells via ASK-1. Oncotarget 7, 29081–29091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang L, Xu C, Chen X, Shi Q, Su W, and Zhao H (2018) SOCS-1 Suppresses Inflammation Through Inhibition of NALP3 Inflammasome Formation in Smoke Inhalation-Induced Acute Lung Injury. Inflammation [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakashima T, Yokoyama A, Onari Y, Shoda H, Haruta Y, Hattori N, Naka T, and Kohno N (2008) Suppressor of cytokine signaling 1 inhibits pulmonary inflammation and fibrosis. J Allergy Clin Immunol 121, 1269–1276 [DOI] [PubMed] [Google Scholar]
- 32.Kasa A, Csortos C, and Verin AD (2015) Cytoskeletal mechanisms regulating vascular endothelial barrier function in response to acute lung injury. Tissue Barriers 3, e974448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehta D, and Malik AB (2006) Signaling mechanisms regulating endothelial permeability. Physiol Rev 86, 279–367 [DOI] [PubMed] [Google Scholar]
- 34.Alieva IB, Zemskov EA, Smurova KM, Kaverina IN, and Verin AD (2013) The leading role of microtubules in endothelial barrier dysfunction: disassembly of peripheral microtubules leaves behind the cytoskeletal reorganization. J Cell Biochem 114, 2258–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alieva IB (2014) Role of microtubule cytoskeleton in regulation of endothelial barrier function. Biochemistry (Mosc) 79, 964–975 [DOI] [PubMed] [Google Scholar]
- 36.He Y, Zhang W, Zhang R, Zhang H, and Min W (2006) SOCS1 inhibits tumor necrosis factor-induced activation of ASK1-JNK inflammatory signaling by mediating ASK1 degradation. J Biol Chem 281, 5559–5566 [DOI] [PubMed] [Google Scholar]
- 37.Fukata M, Watanabe T, Noritake J, Nakagawa M, Yamaga M, Kuroda S, Matsuura Y, Iwamatsu A, Perez F, and Kaibuchi K (2002) Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell 109, 873–885 [DOI] [PubMed] [Google Scholar]
- 38.Watanabe T, Noritake J, Kakeno M, Matsui T, Harada T, Wang S, Itoh N, Sato K, Matsuzawa K, Iwamatsu A, Galjart N, and Kaibuchi K (2009) Phosphorylation of CLASP2 by GSK-3beta regulates its interaction with IQGAP1, EB1 and microtubules. J Cell Sci 122, 2969–2979 [DOI] [PubMed] [Google Scholar]
- 39.Birukova AA, Adyshev D, Gorshkov B, Bokoch GM, Birukov KG, and Verin AD (2006) GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol 290, L540–548 [DOI] [PubMed] [Google Scholar]
- 40.Dubrovskyi O, Birukova AA, and Birukov KG (2013) Measurement of local permeability at subcellular level in cell models of agonist- and ventilator-induced lung injury. Lab Invest 93, 254–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Birukova AA, Birukov KG, Gorshkov B, Liu F, Garcia JG, and Verin AD (2005) MAP kinases in lung endothelial permeability induced by microtubule disassembly. Am J Physiol Lung Cell Mol Physiol 289, L75–84 [DOI] [PubMed] [Google Scholar]
- 42.Monvoisin A, Alva JA, Hofmann JJ, Zovein AC, Lane TF, and Iruela-Arispe ML (2006) VE-cadherin-CreERT2 transgenic mouse: a model for inducible recombination in the endothelium. Dev Dyn 235, 3413–3422 [DOI] [PubMed] [Google Scholar]
- 43.Yoshida T, Ogata H, Kamio M, Joo A, Shiraishi H, Tokunaga Y, Sata M, Nagai H, and Yoshimura A (2004) SOCS1 is a suppressor of liver fibrosis and hepatitis-induced carcinogenesis. J Exp Med 199, 1701–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang X, Lin L, Woodling NS, Wang Q, Anacker C, Pan T, Merchant M, and Andreasson K (2011) Signaling via the prostaglandin E(2) receptor EP4 exerts neuronal and vascular protection in a mouse model of cerebral ischemia. The Journal of clinical investigation 121, 4362–4371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu P, Birukova AA, Xing J, Sammani S, Murley JS, Garcia JG, Grdina DJ, and Birukov KG (2009) Amifostine reduces lung vascular permeability via suppression of inflammatory signalling. Eur Respir J 33, 612–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frank PG, and Lisanti MP (2008) ICAM-1: role in inflammation and in the regulation of vascular permeability. Am J Physiol Heart Circ Physiol 295, H926–H927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sans M, Panes J, Ardite E, Elizalde JI, Arce Y, Elena M, Palacin A, Fernandez-Checa JC, Anderson DC, Lobb R, and Pique JM (1999) VCAM-1 and ICAM-1 mediate leukocyte-endothelial cell adhesion in rat experimental colitis. Gastroenterology 116, 874–883 [DOI] [PubMed] [Google Scholar]
- 48.Dejana E, Orsenigo F, and Lampugnani MG (2008) The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci 121, 2115–2122 [DOI] [PubMed] [Google Scholar]
- 49.Kratzer E, Tian Y, Sarich N, Wu T, Meliton A, Leff A, and Birukova AA (2012) Oxidative stress contributes to lung injury and barrier dysfunction via microtubule destabilization. Am J Respir Cell Mol Biol 47, 688–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tian X, Tian Y, Moldobaeva N, Sarich N, and Birukova AA (2014) Microtubule dynamics control HGF-induced lung endothelial barrier enhancement. PLoS One 9, e105912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karki P, Ke Y, Tian Y, Ohmura T, Sitikov A, Sarich N, Montgomery CP, and Birukova AA (2019) Staphylococcus aureus-induced endothelial permeability and inflammation are mediated by microtubule destabilization. J Biol Chem [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Higginbotham K, Tian Y, Gawlak G, Moldobaeva N, Shah A, and Birukova AA (2014) Hepatocyte growth factor triggers distinct mechanisms of Asef and Tiam1 activation to induce endothelial barrier enhancement. Cell Signal 26, 2306–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tian X, Tian Y, Sarich N, Wu T, and Birukova AA (2012) Novel role of stathmin in microtubule-dependent control of endothelial permeability. Faseb J 26, 3862–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tian Y, Tian X, Gawlak G, O’Donnell JJ 3rd, Sacks DB, and Birukova AA (2014) IQGAP1 regulates endothelial barrier function via EB1-cortactin cross talk. Mol Cell Biol 34, 3546–3558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu YC, Yeh WC, and Ohashi PS (2008) LPS/TLR4 signal transduction pathway. Cytokine 42, 145–151 [DOI] [PubMed] [Google Scholar]
- 56.Piessevaux J, Lavens D, Peelman F, and Tavernier J (2008) The many faces of the SOCS box. Cytokine Growth Factor Rev 19, 371–381 [DOI] [PubMed] [Google Scholar]
- 57.Linossi EM, and Nicholson SE (2012) The SOCS box-adapting proteins for ubiquitination and proteasomal degradation. IUBMB Life 64, 316–323 [DOI] [PubMed] [Google Scholar]
- 58.Strebovsky J, Walker P, Lang R, and Dalpke AH (2011) Suppressor of cytokine signaling 1 (SOCS1) limits NFkappaB signaling by decreasing p65 stability within the cell nucleus. FASEB J 25, 863–874 [DOI] [PubMed] [Google Scholar]
- 59.Mittal M, Siddiqui MR, Tran K, Reddy SP, and Malik AB (2014) Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 20, 1126–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baig MS, Zaichick SV, Mao M, de Abreu AL, Bakhshi FR, Hart PC, Saqib U, Deng J, Chatterjee S, Block ML, Vogel SM, Malik AB, Consolaro ME, Christman JW, Minshall RD, Gantner BN, and Bonini MG (2015) NOS1-derived nitric oxide promotes NF-kappaB transcriptional activity through inhibition of suppressor of cytokine signaling-1. J Exp Med 212, 1725–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang JG, Metcalf D, Rakar S, Asimakis M, Greenhalgh CJ, Willson TA, Starr R, Nicholson SE, Carter W, Alexander WS, Hilton DJ, and Nicola NA (2001) The SOCS box of suppressor of cytokine signaling-1 is important for inhibition of cytokine action in vivo. Proc Natl Acad Sci U S A 98, 13261–13265 [DOI] [PMC free article] [PubMed] [Google Scholar]