

Summary
Many double-stranded DNA viruses, including tailed bacteriophages (phages) and herpesviruses, use the HK97-fold in their major capsid protein to make the capsomers of the icosahedral viral capsid. Following the genome packaging at near-crystalline densities, the capsid is subjected to a major expansion and stabilization step that allows it to withstand environmental stresses and internal high pressure. Several different mechanisms for stabilizing the capsid have been structurally characterized, but how these mechanisms have evolved is still not understood. Using cryo-EM structure determination of ten capsids, structural comparisons, phylogenetic analyses, and Alphafold predictions, we have constructed a detailed structural dendrogram describing the evolution of capsid structural stability within the actinobacteriophages. We show that the actinobacteriophage major capsid proteins can be classified into 15 groups based upon their HK97-fold.
eTOC blurb
The HK97-fold is widespread and found in protein shells used by bacteria and some viruses. Understanding the evolutionary links between such divergent proteins is challenging. Podgorski et al. used the major capsid protein HK97-fold of bacteriophages (viruses that infect bacteria) to build a structural dendrogram to analyze the evolutionary relationships.
Graphical Abstract
Introduction
The HK97-fold (Figure 1 A) is ubiquitous in the biosphere and has been identified in viruses that infect the three domains of life1,2,3, as well as encapsulins4: protein shells used by bacteria for gene transfer and reaction confinement5. It is found across the Caudovirales order (the double-stranded DNA tailed phages), which is one of the largest groups of viruses in the biosphere and plays major roles in bacterial evolution and in carbon/nitrogen/phosphorus cycling6. Actinobacteriophages (bacteriophages infecting actinobacterial hosts, such as Mycobacteria, Streptomyces and Rhodococcus) have been intensively studied with over 20,000 individual isolates, the vast majority of which are dsDNA phages. These phages are the central focus of integrated research-education programs7,8, have provided tools for Mycobacterium genetics9, and show promise as therapies for drug-resistant Mycobacterium infections10,11.
Figure 1. The HK97 bacteriophage: the prototypical HK97-fold.

A) A single major capsid protein subunit from the HK97 bacteriophage showing the conserved domains of the HK97-fold: A-domain (teal), E-loop (purple), G-loop (orange), P-domain (magenta), and N-arm (green). The two amino acids (Lysine169 and Asparagine356) of the isopeptide bond are shown, as is Glu363, which catalyzes the bond formation. B) Model of the mature HK97 bacteriophage (1OHG) on the left and on the right a ribbon diagram highlighting the concatenated rings that are formed by the isopeptide bond. Coloring is the same as in A. Scalebar = 10 nm. C) A zoomed-in image of B showing the local 3-fold axis where the isopeptide bond is formed and where many stabilizing interactions in other bacteriophage capsids have been described.
Bacteriophages are known to use at least four different folds in their major capsid protein12. To date, all the structurally characterized tailed bacteriophages use the HK97-fold13 (Figure 1A) as the foundational block to build the capsid (Figure 1B). The HK97-fold has several conserved domains13,14. They include the A domain (Figure 1A, teal) that forms the central core of the hexamer and pentamer capsomers of the capsid; the P-domain (Figure 1A, magenta) where the long spine helix is located; as well as the E-loop (Figure 1A, purple) and N-arm (Figure 1A, green). The E-loop and N-arm make long-range contacts with other major capsid proteins and play important roles in capsid stability15. The viral major capsid protein that uses the HK97-fold is assembled into an icosahedral shell consisting of eleven pentamers and different numbers of hexamers of the major capsid protein depending on the size and shape of the capsid. The icosahedral capsid is described by a T-number that defines the number of major capsid proteins in the icosahedral shell - equal to the T-number multiplied by 6016. Within the dsDNA tailed phages, one pentamer is replaced by the portal complex to which the tail is bound and through which the DNA is packaged and then released.
The stability of the mature capsid is a key factor in the evolutionary success of phages17. The capsid needs to withstand various environmental conditions, and the pressure of the packed DNA genome18,19,20. The local 3-fold axes for each capsomer, where the P-domains of the pentamer and hexamer capsomers intersect, are thought to be important for capsid stability17 (Figure 1B/C highlights one such 3-fold axis). The tailed phages use several different mechanisms to stabilize the capsid local 3-fold axes. The most common is a minor capsid protein, or ‘cement’/’decoration’, that binds to the local 3-fold axis and makes several contacts with the surrounding major capsid subunits21,22. Others use catenated rings, with either non-covalent23 or covalent bonding24 mechanisms, that connect the major capsid proteins around the local 3-fold axis15. The major capsid protein of the HK97 phage (in which the HK97-fold was first characterized) uses a covalent isopeptide bond to cross-link a conserved asparagine (Asn356) in the P-domain of one major capsid protein with a conserved lysine (Lys169) in an adjacent E-loop of a different major capsid protein24 (Figure 1B/C). The cross-linking is catalyzed by a nearby glutamic acid (Glu363) on a third major capsid protein subunit and aided by three other amino acids that form a hydrophobic pocket25. The cross-linking of all the major capsid proteins results in the catenated rings or “protein chainmail” that stabilizes the capsid around the local 3-fold axes (Figure 1C). Other phages have been characterized, for example, P2226, Sf627, epsilon1528, phageL29, T530, T731, and phiRSA132, that rely solely on intracapsomeric interactions and do not use cement or non-covalent/covalent bonding mechanisms. These different mechanisms of capsid stabilization make the HK97-fold highly adaptable and able to survive a wide variety of environments, from soil to hot springs and allows for the formation of structurally very diverse capsids that range in size from relatively small 50 nm diameter capsids33,34,35 to hundreds of nanometers in diameter “giant” capsids36,37.
High-resolution structures of over 25 tailed phage capsids 22,23,24,33,35,38,39,40,28,41,42,26,43,44,45,32,46,47,48,27,49,50,30,31,51,52, viruses that infect archea2, and the human pathogenic Herpesvirus3 show that the HK97-fold is well conserved, even among viral capsids sharing little or no amino acid sequence similarity and using several different capsid stability mechanisms. However, these structures are from viruses that infect diverse hosts across all three domains of life and are so divergent from one another that only limited conclusions can be made about their evolution. We, therefore, carried out a systematic investigation of closely related phages infecting actinobacterial hosts to understand how capsid stability mechanisms are conserved and how they may have evolved. We built a structural dendrogram of actinobacteriophage major capsid protein HK97-fold structures and showed that they can be classified into 15 structural groups. We obtained ten capsid structures solved to resolutions between 2.2–4 Ångstroms that revealed that eight of them exhibit major capsid proteins that are linked by a covalent cross-linking (isopeptide bond) between subunits that was first described in the HK97 phage. However, three of the closely related phages do not exhibit such an isopeptide bond as demonstrated by both our cryo-EM maps and the lack of the required residue. This work raises questions about the importance of previously described capsid stabilization mechanisms.
Results
The actinobacteriophages have 42 major capsid protein phamilies
There are currently (August 2022) over 4000 sequenced and annotated actinobacteriophages, which can be grouped into over 139 clusters and sub-clusters. Clustering is based on shared gene content between phage genomes, such that a phage is included in a cluster if it shares at least 35% of its genes with any member of that cluster (e.g. Cluster A, Cluster B, etc). Therefore, phages within a cluster are generally more globally similar to one another than to phages in other clusters. Some clusters can be similarly divided into sub-clusters (e.g. Subcluster A1, Subcluster A2, etc). Additionally, there are 66 “Singletons” (August 2022), those phages that have a genome that does not fit into an existing cluster. These cluster/subcluster/singleton groupings do not reflect firm biological distinctions, as phage genomes are pervasively architecturally mosaic, and phage populations likely span a continuum of diversity53.
The shared gene content comparison used for clustering is done at the protein level after genes have been translated and their products sorted into protein “phamilies” using Phamerator54 and a pipeline built on MMseqs2 (Gauthier and Hatfull, manuscript in preparation)55. Proteins within a phamily typically have a minimum pairwise 20% amino acid identity54. Amino acid sequence analysis of approx. 3200 major capsid proteins shows that there are 42 major capsid protein phamilies within the actinobacteriophages database (July 2021).
The F1 sub-cluster contains three major capsid protein phamilies
The majority of the 139 phage clusters use only one of the 42 major capsid protein phamiles, however, because of the mosaic nature of phage genomes, some cluster/subcluster groups (A, BC, CZ, DN, and F) include multiple major capsid protein phamilies. Subcluster F1 has the most with three different major capsid protein phamilies. Previous structural studies with the Escherichia coli CUS-3 and Salmonella P22 phages have shown that major capsid proteins with minimal amino acid sequence identity (less than 15%) can result in almost identical capsid morphologies and HK97-folds26,27. We, therefore, started the systematic investigation of the actinobacteriophages with the F1 major capsid proteins to address whether the three major capsid protein phamilies in the F1 subcluster are the same HK97-fold with highly diverged amino acid sequences, or whether they are three distinct folds.
Cryo-electron microscopy (see Table S1 for collection parameters, analysis, and final resolutions). was used to determine a sub 3 Å map (Figure 2A) for a representative phage from each of the F1 subcluster major capsid protein phamilies (Table 1). The capsids all use the T=9 icosahedral architecture, with 540 copies of the major capsid protein, and are of similar size (740 Å diameter) and internal volumes (approx. 3×107 Å3), which is expected since they package double-stranded DNA genomes of very similar length (Table S2).
Figure 2. Structures of the F1 bacteriophages.

A) Surface representations of cryo-EM maps of capsids from the three representative major capsid protein phamilies of the F1 subcluster: Che8, Bobi, and Ogopogo. Each phamily number is displayed below the bacteriophage name. The capsids have been colored by the radial distance from the center of the capsid (the color key is shown on the right-hand side). Scalebar = 10 nm. B) Models of the HK97-fold of each representative bacteriophage with select areas highlighted and labeled. The lysines and aspartic acid/asparagine involved in the isopeptide bond of Bobi and Ogopogo are also labeled (Che8 does not have an isopeptide bond). A zoomed-in and rotated image of the A3-loop and G-loop of Ogopogo is shown bottom right. Throughout the paper, the cryo-EM derived HK97-folds shown are of equivalent positions in the capsid and are the hexamer subunit adjacent to the pentamer subunit in the asymmetric unit.
Table 1.
Representative F1 subcluster bacteriophages. Note: phamily numbers are subject to change but are accurate at the time of writing (August 2022).
| Phage name | Phamily/pham number | Major capsid protein gene number |
|---|---|---|
| Che8 | 4611 | 6 |
| Bobi | 15199 | 7 |
| Ogopogo | 57445 | 8 |
A comparison of the three experimentally determined HK97-folds, which have ~15% amino acid sequence identity, showed that they are very similar to one another (Figure 2), with Root Mean-Square Deviation (RMSD) of atomic position values <1.35 Å. While each fold is structurally similar to the original HK97-fold of the HK97 virus24, some key differences exist. Che8 lacks the G-loop that is found near the C-terminal end of the spine helix in the HK97 phage major capsid protein (Figure 1A), therefore, Che8 has a continuous spine helix (Figure 2B). Che8 also lacks the “protein chainmail” of the HK97 phage; the cryo-EM map revealed that there was no density to suggest isopeptide bond formation, nor were there any amino acids in the correct location to potentially form an isopeptide bond. Bobi and Ogopogo both have a G-loop, although they are extended when compared to the original HK97 fold. Ogopogo has an A-loop that extends over the G-loop and makes important stabilizing contacts with the G-loop and spine helix (Figure 2B, bottom right). The A-loop is in the same position as the A-loop of phage T7 where it was first characterized31. Bobi has an additional loop between the A and P domains, in a similar position to the A-pocket described in phage T731, which Ogopogo and Che8 both lack. Bobi and Ogopogo have an isopeptide bond, with clear density in the cryoEM map showing the covalent link between a lysine in the E-loop and either aspartic acid or asparagine in the P-domain (Figure 2B) of the adjacent major capsid protein: this demonstrates that they form the characteristic “protein chainmail” like the original HK97 fold. Ogopogo and Bobi both have an extended P-loop within the P-domain and three of these are found in close contact around the local 3-fold axis of the capsid. This structural comparison of the three F1 major capsid protein phamilies suggested that Che8 may be more divergent from Bobi and Ogopogo.
For simplicity, from this point on the major capsid protein phams will be called by the corresponding representative phage used in Figure 2; the Che8-like phages (pham 4631); the Ogopogo-like phages (pham 57445) and the Bobi-like phages (pham 15199).
The three major capsid protein phamilies of the F1 subcluster constitute two structural groups of major capsid protein in the actinobacteriophages
To confirm the structural observations, we next put the three F1 major capsid protein phamilies into the broader context of the major capsid proteins from the actinobacteriophages database since focusing on just the three F1 phamilies would likely not reveal much insight into their level of evolutionary relationship due to their relatively high structural similarity. We, therefore, created a structural dendrogram of all the major capsid protein phamilies annotated in the actinobacteriophages (Figure 3). Previously, structural comparison of distantly related, yet conserved, protein folds has been used successfully to imply evolutionary links between viral capsid proteins; for example, with the PRD1 and other double jelly-roll viral capsid proteins56,12,57, as well as showing a link between the dsDNA tailed phages and Herpes virus3.
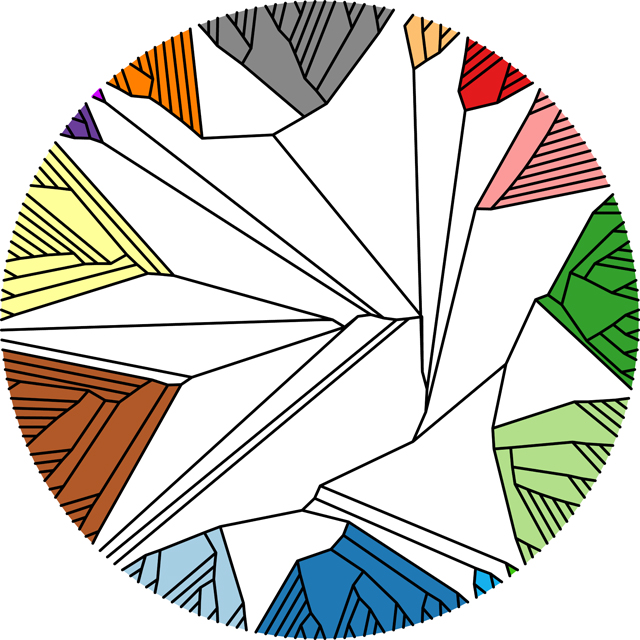
Figure 3. Structural dendrogram of the actinobacteriophages.

A) 201 Alphafold-predicted representative major capsid protein HK97-folds, representing 99% of the actinobacteriophages, were clustered based upon their structural similarity using the Homologous Structure Finder algorithm. Each cluster is designated with a Group number, and the percentages in parenthesis after the group number are the number of actinobacteriophages (from the 4000+ in phagesdb.org) that are found within each structural group. The map is colored to highlight each structural group. B) A zoomed-in image of the structural dendrogram highlighting Structural Groups 1 and 2 that contain the three F1 major capsid protein phamilies. Representative bacteriophage names are shown and are colored by cluster. Within Groups 1 and 2 are subgroups of proteins from the same phamily, as indicated.
To create the structural dendrogram we used Alphafold58 to predict the three-dimensional HK97-folds of the major capsid proteins. Folding every major capsid protein in the actinobacteriophage database (over 3000 entries when the analysis was carried out in July 2021) was not feasible from a computational standpoint. Therefore, we selected a representative major capsid protein from each cluster (139 total clusters at the time of analysis), as well as for every Singleton (62 at the time of analysis), as each Singleton could represent a future cluster distinct from the extant groups. For those clusters (A, BN, CZ, DN, and F) with more than one major capsid protein phamily, we folded a representative of each major capsid protein phamily from that cluster. In total, the structure of 201 major capsid proteins were predicted using Alphafold and represent the 42 annotated major capsid protein phamilies of the actinobacteriophages. These clusters and Singletons span the different morphologies of capsids, including Sipho-,Myo, and Podoviridae, as well as various length elongated prolate capsids. We validated a subset of the Alphafold predictions with cryo-EM derived structures (Figure S1), revealing excellent agreement for most of the HK97-fold (RMSD values between 0.8 – 1 Ångstrom).
These 201 major capsid protein predictions were then used to create a major capsid protein structural dendrogram of the actinobacteriophages using the Homologous Structure Finder algorithm56 (Figure 3A). The algorithm compares the three-dimensional Alphafold predictions and classifies the major capsid proteins on their structural similarity. The sophisticated classification methodology allows for the creation of a structural dendrogram whereby common structural elements between the major capsid proteins are identified and a common structural ancestor can be inferred. It has been used successfully for other protein fold lineages56,59. The major advantage of this methodology is for detecting similarities in protein folds even when no similarities remain in the amino acid sequences. The structural map of the actinobacteriophages revealed that the 42 major capsid protein phamilies can be classified into 15 structural groups (Figure 3A) that are likely to be evolutionarily related. Beyond the 15 structural groups are several “structural Singletons”. The structural dendrogram supports the initial structural comparison, in that the Che8-like phamily, sorted into Group 1 (Figure S2), is more diverged from the Ogopogo and Bobi-like phamilies found in Group 2 (Figure 3B/ Figure S3).
The Bobi-like (15199) phamily can form differently-sized capsids
We next investigated how the HK97-fold and capsid stability mechanisms are conserved within closely related phages. We chose to concentrate on the Group 2 phages since they use the protein “chainmail” found in HK97. Alignment of all the Alphafold-predicted major capsid proteins from Group 2 shows lysine and aspartic acid/asparagine at the expected positions in the E-loop and P-domain in the majority of the Group 2 phages, apart from those in the Zuko-like (9942) sub-group and a subset of the Cluster K phages in the Bobi-like (15199) sub-group.
It was surprising to identify some of the K subcluster lacking the lysine residue needed for the isopeptide bond since all the other Bobi-like (15199) phages were predicted to use it. Removal of the isopeptide bond in HK97 by mutating the E-loop lysine to tyrosine results in non-viable phage particles indicating that the isopeptide bond is required for infectious capsids to be made60. The Bobi-like (15199) phages, therefore, provided an opportunity to characterize phage structures with and without the isopeptide bond and investigate how/and if the capsid compensates for its absence. We carried out cryo-EM on five other members of the Bobi-like (15199) phage capsids to yield maps of < 4 Å resolution (Table S1). We found that some of the Bobi-like (15199) phages formed T=9 capsids while others formed T=7 capsids (Figure 4). In each of the six phages (including Bobi), only the major capsid protein was identified in the cryo-EM map; no minor capsid proteins were observed. Bridgette of Cluster FA, however, did have a decoration protein (Gp7, Figure S4) that bound as dimers around the 5-fold axis of the pentamer, reminiscent of the phi29 phage spike proteins33.
Figure 4. Cryo-EM maps of representative bacteriophages from the Bobi-like (15199 phamily) of major capsid proteins.

Muddy, Bridgette, and Oxtober96 all form T=7 capsids. Ziko, Bobi, and Adephagia all form T=9 capsids. Bridgette has a decoration protein that assembles as dimers around the pentamer. The scale bar is 10 nm. Maps are radially colored. Muddy, Bridgette, and Oxtober use one radial color scheme, and Ziko, Bobi, and Adephagia another. For clarity, names are colored by cluster using the same colors as in Figure 3. The bacteriophage cluster name is shown in brackets after the bacteriophage name. The Bobi map is reproduced here from Figure 2 for comparison with the other five cryo-EM maps. Scalebar = 100 Å.
The HK97-folds of the Bobi-like phamily (Pham 15199) are highly conserved, but exhibit structural diversity in the loop regions
Comparison of the HK97-folds of the representative Bobi-like (pham 15199) phages showed very high similarity between them (Figure S5), with the highest RMSD value of 1.2 Å (Table S3). The protein fold is highly conserved and near identical, especially in the secondary structure alpha helices and beta sheets and can be overlaid without much deviation along the protein fold. It is within five loop regions that the most structural diversity is observed (Figure 5). We have designated these loop regions A1-A4 because of their position in the A domain, as well as the P-loop found in the P-domain. The A3 and A4 loops were first described in the HK97-fold of the phage T7 major capsid protein and named the A-loop (A3 loop) and A-pocket (A4 loop)31. We have renamed them here because of the extra loops we have identified. All the structurally characterized Bobi-like (pham 15199) phages have both A3 and A4 loops. However the interactions each loop makes are not conserved. Within Bobi, Oxtober96, and Adephagia, the two loops are of similar length and make intramolecular interactions but do not interact with one another. The other three phages, Bridgette, Muddy, and Ziko, all have increasingly long A3 and A4 loops, with those of Ziko being the longest (Figure 5B) and make intermolecular interactions with an adjacent major capsid protein (Figure 6A). Only a single tryptophan (W224 in Bobi) is conserved in the A3 loop across the Bobi-like (15199) major capsid proteins. It forms a pi-pi interaction with a conserved phenylalanine (Phe127 in Bobi) that presumably stabilizes the A3 loop (Figure S6). No residues are conserved in the A4 loop and the A4 loop is not universal amongst the Group 2 phages (Figure S3): for example, the Ogopogo-like (57445 phamily) and Bxb1like (15229 phamily) phages. The A1 and A2 loops are found at the top of the A domain (Figure 5D), with the A2 loop inserting into the center of the hexamer or pentamer capsomere. Oxtober96, Ziko, and Muddy all have elongated A2 loops. A comparison of the A1 and A2 loops in the context of the capsid (Figure S7) shows that there is a conserved salt bridge between an Arginine in the A2 loop of one major capsid protein and a Glutamic acid in the A1 loop of an adjacent major capsid protein (Figure 6B). However, in Bridgette, the salt bridge is between the two A2 loops (Figure S7). The elongated A2 loops do not appear to result in a consistent increase in intermolecular or intramolecular interactions between the major capsid proteins (Table S4) and no other amino acids are conserved.
Figure 5. The variable loops of the Bobi-like (15199) major capsid proteins.

A) Major capsid protein subunits for each phage with all the variable loops highlighted. B, C, and D show zoomed-in images of the same major capsid protein as in A.
Figure 6. Intermolecular interactions between Bobi-like (15199) major capsid proteins.

A) Interactions between the A3 and A4 loops of one major capsid protein subunit with an adjacent subunit for phages Bridgette, Muddy, and Ziko. B) The interaction between the A1 and A2 loops of one major capsid protein subunit and an adjacent major capsid protein in Adephagia (top) and a zoomed-in viewpoint of the same structure with the intermolecular interactions between sidechains shown and the conserved salt bridge highlighted with a black box (bottom). The Arginine/Glutamic acid salt bridge between the A2 loop (R198) and the A1 loop (E162) is shown. C) The P-loops of three major capsid protein subunits in Adephagia are colored in three different shades of magenta. The black triangle shows the center of the local 3-fold axis.
The P-loop (Figure 5C) makes contact with other P loops around the local 3-fold axis, creating small “turrets” that stick outward from the capsid (Figure 6C). The P-loops make several hydrogen bonds and salt bridges (Table S5) between adjacent P-loops that are likely to play a role in the stabilization of the local 3-fold axis. Adephagia has one of the longest P-loops and makes the most salt bridges and hydrogen bonds between the P loops out of all six Bobi-like phages.
The G-loop structure is well conserved across all of the Bobi-like (15199) phages and is a clear structural marker for this phamily of phage major capsid proteins despite only having a single conserved glycine and proline across the Bobi-like (15199) major capsid proteins (Figure S3).
Covalent crosslink residues are not found in all of the Bobi-like major capsid proteins
Alignment of the Bobi-like (15199) phamily major capsid protein amino acid sequences revealed that a subset (48%) of the Cluster K phages had the lysine substituted by isoleucine (Figure 7A). Within the subset of Cluster K phages, there are nearby lysine residues that we hypothesized could act as the lysine for the isopeptide bond. Depending on the position of this misplaced lysine, these Cluster K phages can be divided into two groups. We have termed one group the Adephagia-like phages, and the other the Cain-like phages (Figure 7A).
Figure 7. The isopeptide bond.

A) Alignment of Bobi and the three main groups of the K cluster. B) Bobi isopeptide bond is shown with the cryo-EM map shown in light blue. C) Adephagia is shown that lacks the isopeptide bond.
A consequence of the isopeptide bond is that all the major capsid proteins are covalently linked to one another, forming large complexes. Previous SDS-PAGE analysis of the capsid of the HK97, where the isopeptide bond was first demonstrated (Figure 1), showed that the major capsid protein was unable to enter the gel due to its extensive crosslinking and large size61. We, therefore, ran Ziko (with isopeptide bond) and Adephagia (no isopeptide bond) capsids on SDS-PAGE (Figure S8). Within the gel, no major capsid protein band was observed for Ziko at the expected molecular weight of 34 kDa but instead, there were dark areas at the top of the gel reminiscent of the HK97 SDS-PAGE analysis. Conversely, a large band consistent with the major capsid protein of Adephagia was detected on the gel at approximately 32 kDa (predicted size of Adephagia major capsid protein is 32.7 kDa), showing that Adephagia does not have the isopeptide bond.
Analysis of the six cryo-EM maps from the Bobi-like (pham 15199) phages (Figure 4), as well as the map of the Cluster K Cain phage (Figure S9A) confirmed that all the phages, except Adephagia and Cain, had clear density for the isopeptide bond between the lysine in the E-loop and an aspartic acid in the P-domain of the adjacent major capsid protein (Figure 7B). The lysine residues within Adephagia and Cain that we hypothesized could form the isopeptide bond were therefore found not to be involved in covalent bond formation (Figure 7C and Figure S9B).
Structural groups do not have a conserved hydrogen bond network
The lack of an isopeptide bond in Adephagia and the other Cluster K phages raised the question as to how they compensate for the absence of the covalent isopeptide bond around the local 3-fold axis for capsid stability. Removal of the isopeptide bond in HK97 results in unviable capsids, suggesting it is critical for the survival of virion. With no minor capsid protein or other accessory protein to compensate for the loss of the isopeptide bond, we examined the hydrogen bonds and salt bridges around the local 3fold axis, hypothesizing that there would be an increased number of such interactions around the local 3-fold of Adephagia when compared to Bobi, its closest relative in the Bobi-like (15199) pham. MAFFT alignment and phylogenetic analysis of the major capsid proteins showed that the Cluster K phages (Figure S10) are most closely related to the Cluster F phages with approx. 70% amino acid sequence identity. We observed an increase in both salt bridges and hydrogen bonds between the P-loops, with Adephagia having three times the salt bridges and double the number of hydrogen bonds when compared to Bobi (Table S5). This equates to an increase of 240 kJ/mol free energy between Adephagia and Bobi at the site of the P-loops, although this must be contrasted with a loss of 1068 kJ/mol in free energy because of the removal of the isopeptide bonds. However, despite the increase in interactions at the P-loop, which is located at the center of the local 3-fold axis, there was no increase in the number of hydrogen bonds and salt bridges between Adephagia and Bobi around the wider local 3-fold axis that takes into account all nine interacting major capsid proteins (Table S5 and Figure S11). We next expanded the analysis to the other members of the Bobi-like (15199) pham, which revealed that all the capsids had similar numbers of hydrogen bonds and salt bridges around the local 3-fold axis but only a handful of these were structurally conserved, with different distribution patterns of the interactions for each phage (Table S5 and Figure S11). We, therefore, conclude that extra stabilization around the local 3-fold axis is not required to compensate for the missing isopeptide bond.
We next hypothesized that the handful of conserved amino acids found in the Bobi-like (15199) phages that were involved in capsid stability would be conserved across all of Structural Group 2; these phages all had very similar major capsid protein HK97-folds and we predicted they would use similar mechanisms to maintain the stability of the capsid around the local 3-fold. However, analysis of the amino acid sequences of the major capsid proteins of Structural Group 2 revealed almost no conserved amino acid sequence identity. A single conserved aspartic acid (D122 in Bobi) is found in all of Structural Group 2, located at the top of the spine helix (C-terminal end). In Bobi (and the other Bobi-like phages that we structurally characterized) this aspartic acid makes a hydrogen bond with the N-arm of the same major capsid protein chain.
Due to the lack of amino acid sequence identity, we turned to the models we had derived from the cryo-EM maps of the phages. We examined the structurally conserved interactions across Structural Group 2, once again hypothesizing that the most critical of these interactions would be conserved. We overlaid the local 3-fold axis of all six of the Bobi-like (pham 15199) phages and Ogopogo (pham 57445). To better represent more members of Structural Group 2 in this analysis we also included Cozz, a close relative of Ogopogo and also part of the 57445 pham (Figure S10). Cozz was subjected to cryoEM analysis and a sub-3 Å resolution map was obtained that allowed for de novo model building. This structural comparison showed that there were no salt bridges conserved between the different phages in Structural Group 2, nor was there a conserved hydrogen bond network (Figure S11). Each phage used a different pattern of bond formation between the major capsid proteins for intermolecular stability. We, therefore, conclude that what unifies a Structural Group is the capsid configuration and that the phages have evolved different bond networks to achieve the same final product.
Discussion
Capsid stability
Tailed phages have been found in a wide range of environmental habitats, ranging from relatively benign soil to hot springs and stomach acid32. In addition to these harsh external environments, the capsid is also under stress from the inside: the predicted pressure that the packaged dsDNA exerts on the inside of the capsid has been estimated at 30 atmospheres62. The phage capsid must be stable enough to survive these two main stresses. Many stabilization mechanisms have been characterized at the local three-fold axis of the capsid, implying they play an important role. These typically include many inter-capsomer interactions, for example, the interaction of the Pdomains around the local three-fold axis of the capsid; the N-arm reaching across to make contacts with adjacent major capsid proteins, and the E-loop interacting with the P-domain. Only HK97 has had its isopeptide bond structurally characterized, although phages have been shown to use the isopeptide bond biochemically. Since then, each structurally characterized phage has been found to lack the isopeptide bond but uses alternate mechanisms to compensate for the lack of this bond to stabilize the local three-fold axis. These include extra domains in the HK97-fold, for example, the I domains found in P2226 and T450, that have been shown to play a role in capsid stability63. Additional capsid proteins have also been characterized that are thought to play a role in stability. This includes minor capsid proteins/cement proteins found in several tailed phages and which form trimers/dimers throughout the capsid between the hexamers and pentamers. Other proteins, called decoration or ancillary proteins, have also been characterized that may be involved in stability, although in many cases they are not vital for capsid viability (for example, the soc protein of T464 and the decoration protein of phage L29). Finally, more diverse mechanisms have been characterized such as the lasso-like interactions in the E-loop observed in two phages isolated in hot springs43,44.
However, some phages, for example, T731 and the recently structurally characterized phage phiRSA132, show that some phages rely solely on the electrostatic and hydrophobic interactions between the major capsid proteins32. Here, we have described a similar lack of stabilizing mechanisms in the T=9 actinobacteriophage Che8, which is an even more simplified example of the HK97-fold than phiRSA1; Che8 lacks the isopeptide bond and any other previously characterized capsid stabilizing interactions, instead relying on only a handful of protein: protein interactions between the major capsid proteins that are found in every HK97-fold major capsid protein. The Che8 major capsid protein also lacks the G-loop, which has been shown to play an important role in capsid assembly65. Furthermore, it lacks any potential loop that could compensate for the G-loop, demonstrating that the roles of the G-loop in the HK97 capsid are not required across all other HK97-folds. Additionally, Che8 has no minor capsid proteins, decoration proteins, or I-loops/other extended loops or embellishments that may contribute to capsid stability. This suggests that the core HK97-fold is all that is needed for capsid stabilization and that Che8 is likely to be more similar to the earliest HK97fold. This is further supported by the Structural Group 9 phages that all have relatively small genomes (< 30 kbp) and are predicted to form T=4 or smaller capsids (unpublished data). All of these phages lack a G-loop and are similar in structure to the Che8 HK97-fold with a long spine helix. This leads us to speculate that the earliest common ancestor to these phages lacked the G-loop. It also suggests different assembly mechanisms between the different HK97-folds since the G-loop in HK97 was shown to play an important role in assembly and mutations in the G-loop led to the formation of aberrant particles65.
The isopeptide bond is a covalent bond between two neighboring major capsid protein subunits and is critical to the viability of HK97 virions. Here, we have structurally characterized other tailed phages that also use the isopeptide bond in their capsid. The Bobi-like (pham 15199) phages all use the same isopeptide bond as in HK97, although they substitute asparagine for aspartate in the P-domain to create the bond. The use of an aspartic acid to form the isopeptide bond has not been observed in the tailed phages before but has been characterized in bacterial proteins66. Also, the mechanism by which the isopeptide is formed may be subtly different. The catalytic glutamic acid residue is still present in the Bobi-like phages, but they lack two of the residues known to form the hydrophobic pocket that is important for the catalysis of the bond25. There are no obvious analogs in the Bobi-like phages for those two residues, and these phages may create the hydrophobic pocket through other means. However, not all of the Bobi-like phages use the isopeptide bond; a small subset of the Cluster K phages, which we term the Adephagia and Cain-like phages, do not use the isopeptide bond and the lysine in the E-loop is substituted with isoleucine. This resulting residue chemistry prevents the formation of an isopeptide bond. Phylogenetic analysis of the Bobi-like phages (Figure S10) suggests that the Cluster K phages diverged from within the Bobi-like phamily. Although this is speculative, it does support the model that the Adephagia-like and Cainlike phages had the isopeptide bond at some point before it was lost, as opposed to being an intermediate between non-isopeptide bond phages that then evolved to have the isopeptide bond. This is further supported by the other Cluster K phages having the correct lysine for isopeptide formation and presumably forming that isopeptide bond. We were unable to identify any unique increase in inter-capsid interactions in the Adephagia- and Cain-like phages that would compensate for the loss of the isopeptide bond. This suggests that, at least in the Bobi-like phages, the isopeptide bond is not critical to the viability of the phage capsid and that compensatory mechanisms, for example, minor capsid proteins, are not needed. This raises the question as to the role of the isopeptide bond, and why some phages do not require the extra stabilization it affords. A potential explanation is that Cain- and Adephagia-like phages package less dsDNA, exerting less internal pressure on the capsid than those that use the isopeptide bond. However, this correlation cannot yet be made as the amount of dsDNA packaged has not been measured, although we observe that both phages have cos-type genome ends that typically means that the DNA packaged is the same as the genome length.
Capsid size
The tailed phages make protein shells of variable sizes. The smallest to date are the T=4 capsids of P6835 and the T=3 prolate phi2933. The majority are predicted to be T=7, although many “jumbo” phages have been characterized with very large T numbers37. How capsid size is controlled is still an open question. However, many major capsid protein mutants that change the size of the final capsid have been identified in the model phages P22 and T4. The major capsid protein mutants in P22, where the capsid protein is referred to as the coat protein, all result in the wild-type T=7 capsid with the ability of also creating smaller T=4 capsids or aberrant particles67. Within the prolate phage T4, the mutants result in “giant” capsids that have lost the ability to regulate the length of the prolate caps and form very long prolate heads68. The work on Staphylococcus aureus infecting phages and the mechanisms that this bacterium uses to co-opt the phage capsids for the use of the bacteria all result in smaller capsids48,39. Here we have identified several closely related phages that use related major capsid proteins from that same protein phamily, but make either T=7 or T=9 capsids. There are no obvious differences in structure or amino acid conservation between these T=9 and T=7 phage capsids (from phamily 15199) that explains the difference in size. The T=7 capsids use a different phamily of scaffold proteins than the T=9. supporting the role of the different scaffolding proteins as the main mechanism of capsid size determination.
However, further work is needed to characterize the mechanisms by which these phages assemble. The actinobacteriophages are a rich resource for these types of studies. For example, the structural Group 1 phages contain both Che8 (a T=9 capsid) and Myrna, a T=16 capsid that uses minor capsid proteins69. Further study of the Group 1 phages could provide important insights into how minor capsid proteins are first incorporated into the capsid and how larger capsids evolve.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Simon White (simon.white@uconn.edu). Requests for the bacteriophages and bacterial strains should be directed to Graham Hatfull. Questions about Homologous Structure Finder should be directed to Janne Ravantti.
Materials availability
This study did not generate new unique reagents.
Data and code availability
All the models have been deposited in PDB. The cryo-EM maps have been deposited in EMDB. The raw cryo-EM micrographs have been deposited in EMPIAR. The raw cryo-EM micrographs for Muddy have not been deposited in EMPIAR due to on-going research. All data are publicly available as of the data of publication. The accession numbers are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Adephagia | Actinobacteriophage database (University of Pittsburgh) | Accession number: JF704105 |
| Bobi | Actinobacteriophage database (University of Pittsburgh) | Accession number: KF114874 |
| Bridgette | Actinobacteriophage database (University of Pittsburgh) | Accession number: MH834603 |
| Cain | Actinobacteriophage database (University of Pittsburgh) | Accession number: MF324913 |
| Che8 | Actinobacteriophage database (University of Pittsburgh) | Accession number: AY129330 |
| Cozz | Actinobacteriophage database (University of Pittsburgh) | Accession number: KU998239 |
| Muddy | Actinobacteriophage database (University of Pittsburgh) | Accession number: KF024728 |
| Ogopogo | Actinobacteriophage database (University of Pittsburgh) | Accession number: MG925354 |
| Oxtober96 | Actinobacteriophage database (University of Pittsburgh) | Accession number: MT024864 |
| Ziko | Actinobacteriophage database (University of Pittsburgh) | Accession number: MK919478 |
| Arthrobacter globiformis B-2979 | Actinobacteriophage database (University of Pittsburgh) | NCBI:txid1077972 |
| Gordonia terrae 3612 | Actinobacteriophage database (University of Pittsburgh) | NCBI:txid2055 |
| Microbacterium foliorum NRRL B-24224 | Actinobacteriophage database (University of Pittsburgh) | NCBI:txid104336 |
| Mycobacterium smegmatis mc 2 155 | Actinobacteriophage database (University of Pittsburgh) | NCBI:txid246196 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Middlebrook 7H9 Broth Base | Sigma-Aldrich | M0178–500G |
| Glycerol | Fisher Scientific | BP229–1 |
| Sodium chloride | Fisher Scientific | S271–10 |
| Dextrose (D-Glucose) Anhydrous | Fisher Scientific | D16–500 |
| Albumin, Bovine, Cohn Fraction V 98% | Fisher Scientific | AAJ6573122 |
| Tween-80 | Fisher Scientific | BP338–500 |
| Calcium Chloride | Sigma-Aldrich | C1016–500G |
| Agar | Fisher Scientific | BP1423–500 |
| LB Broth, Lennox | Fisher Scientific | BP1427–500 |
| Yeast Extract | Sigma-Aldrich | Y1625–1KG |
| Peptone | Fisher Scientific | BP1420 500 |
| Tris Base | Millipore Sigma | 648311–1KG |
| Magnesium Sulfate Anhydrous | Fisher Scientific | M65–500 |
| Cesium Chloride | Fisher Scientific | BP1591–1 |
| Ethane (research grade) | Airgas | ET R35 |
|
| ||
| Deposited data | ||
|
| ||
| Adephagia | This paper | PDB: 8EC2 |
| Adephagia | This paper | EMD-28012 |
| Adephagia | This paper | EMPIAR-11200 |
| Bobi | This paper | PDB: 8EC8 |
| Bobi | This paper | EMD-28015 |
| Bobi | This paper | EMPIAR-11201 |
| Bridgette | This paper | PDB: 8ECI |
| Bridgette | This paper | EMD-28016 |
| Bridgette | This paper | EMPIAR-11209 |
| Cain | This paper | PDB: 8ECJ |
| Cain | This paper | EMD-28017 |
| Cain | This paper | EMPIAR-11205 |
| Che8 | This paper | PDB: 8E16 |
| Che8 | This paper | EMD-27824 |
| Che8 | This paper | EMPIAR-11190 |
| Cozz | This paper | PDB: 8ECK |
| Cozz | This paper | EMD-28018 |
| Cozz | This paper | EMPIAR-11206 |
| Muddy | This paper | PDB: 8EDU |
| Muddy | This paper | EMD-28039 |
| Ogopogo | This paper | PDB: 8ECN |
| Ogopogo | This paper | EMD-28020 |
| Ogopogo | This paper | EMPIAR-11207 |
| Oxtober96 | This paper | PDB: 8ECO |
| Oxtober96 | This paper | EMD-28021 |
| Oxtober96 | This paper | EMPIAR-11208 |
| Ziko | This paper | PDB: 8EB4 |
| Ziko | This paper | EMD-27992 |
| Ziko | This paper | EMPIAR-11195 |
|
| ||
| Software and algorithms | ||
|
| ||
| Relion v3.1.1 | Zivanov et al.70 | https://github.com/3dem/relion |
| Alphafold v2.0 | Jumper et al.58 | https://github.com/deepmind/alphafold |
| ChimeraX 1.3 | Goddard et al.71 | https://www.cgl.ucsf.edu/chimerax/ |
| Coot v0.9.2 | Emsley et al.72 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix v1.19.2–4158 | Liebschner et al.73 | https://phenix-online.org/ |
| Isolde v1.3 | Croll74 | https://isolde.cimr.cam.ac.uk/ |
| MAFFT v7.453 | Katoh and Standley75 | https://mafft.cbrc.jp/alignment/software/ |
| IQTree v1.6.6 | Minh et al.76 | http://www.iqtree.org/ |
| FigTree v1.4.4 | Rambaut77 | https://github.com/rambaut/figtree/releases |
| Homologous Structure Finder | Ravantti et al.56 | N/A |
This paper does not report original code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mycobacterium was grown on Luria agar plates and all other bacteria were grown on PyCa plates. Incubation temperatures can be found in Table S2.
METHOD DETAILS
Production and purification of Phages for Cryo-Electron Microscopy
Phages were produced as previously described69. Twenty webbed plates were made for each phage with their host (Table S2) in top agar on Luria agar plates (for Mycobacterium) or PYCa top agar and PYCa agar plates (for all other bacteria) and incubated overnight at the temperatures shown in Table S2. Phages were extracted from the webbed plates using 5 mL of Phage Buffer (10 mM Tris-HCl pH 7.5, 10 mM MgSO4, 68 mM NaCl, 1 mM CaCl2) and incubated overnight at room temperature to allow diffusion of the phages into the Phage Buffer. The lysate was aspirated from the plates and centrifuged at 12,000× g for 15 min at 4 °C to remove cell debris. Phage particles were then pelleted using an SW41Ti swinging bucket rotor (Beckman Coulter, Brea, CA) at 30,000 rpm for 3 hours using 12.5 mL open-top poly clear tubes (Seton Scientific, Petaluma, CA). The phage particles in the pellet were then resuspended in 7 mL of Phage Buffer by gentle rocking overnight at 4 °C. The new phage lysate was subjected to isopycnic centrifugation with the addition of 5.25 g of CsCl to the 7 mL of phage lysate. The CsCl/phage solutions were centrifuged at 40,000 rpm in an S50-ST swinging bucket rotor (Thermofisher Scientific, Waltham, MA) for 16 h and the phage particle band (that appeared roughly halfway down the tube) was removed via side puncture with a syringe and needle. Phage particles were then dialyzed three times against Phage Buffer to remove CsCl. To do this the ~1 mL of purified phages was placed into a Tube-O-Dialyzer Micro (G-Biosciences, St Louis, MO) with a 50 kDa molecular weight cut-off. The phages were then concentrated a final time by pelleting them at 75000 rpm in an S120-AT2 fixed angle rotor (Beckman Coulter, Brea, CA). The phage particles were then resuspended in 20 μL of Phage Buffer with gentle pipetting.
Preparation of Cryo-Electron Microscopy Grids
Five microliters of concentrated phage particles (approximately 10 mg/mL) were added to Au-flat 2/2 (2 μm hole, 2 μm space) cryo-electron microscopy grid (Protochips, Morrisville, NC, USA) using a Vitrobot Mk IV (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Grids were blotted for 5 s with a force of 5 (a setting on the Vitrobot) before being plunged into liquid ethane. For Muddy phage, three microliters of concentrated phage particles were added to a freshly glow-discharged Quantifoil R2/1 grid (Quantifoil Micro Tools GmbH, Großlöbicha, Germany) and plunge-frozen with a Vitrobot Mk IV into a 50:50 mixture of liquid ethane:propane78.
Cryo-Electron Microscopy
Data were collected on a 300 keV Titan Krios (Thermo Fisher Scientific, Waltham, Massachusetts, USA) at the Pacific Northwest Center for Cryo-EM with either a K3 or Falcon 3 direct electron detector (Gatan, Pleasanton, CA, USA). The data for Muddy was collected on a 300 keV Titan Krios 3Gi at the University of Pittsburgh with a Falcon 3 direct electron detector (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Table S1 provides the collection parameters for each phage.
Cryo-Electron Microscopy Data Analysis
Relion 3.1.170 was used for phage capsid reconstructions using the standard workflow. CTF Refinement was performed using the default settings. Bayesian polishing was not performed since it made little improvement on resolution (approx. 0.1 Å for Bobi when attempted) for the computational time. Ewald sphere correction was carried out for each particle using the relion_image_handler command that is included with Relion. The mask_diameter value used in the Ewald sphere correction is reported in Table S1.
De novo model building
The amino acid sequences of the major capsid proteins were folded with Alphafold58 version 2.0 using the default settings on a local workstation. The highest-ranked prediction model was fitted into the cryo-EM map using ChimeraX71 version 1.3 and the “Fit in Map” command. Coot72 version 0.9.2 was then used to manually fit the model into the density using the “Stepped sphere refine active chain” provided by the python script developed by Oliver Clarke79. Any remaining protein backbone that was incorrectly placed was then manually moved into the correct density. All maps were of sufficient quality for side chains to be easily recognizable. The real-space refinement tool of Phenix73 version 1.19.2–4158 was used with default settings to refine the model and Coot was then used to manually fix the majority of the issues identified through Phenix.
The final step was to use the ChimeraX plugin, Isolde74 version 1.3, to refine the major capsid protein model. The whole model simulation was used with a temperature of 20°K. All other parameters were default. After the first model was completed, the asymmetric unit of the capsid was created using a similar workflow with a final Isolde refinement of the entire asymmetric unit.
Phylogenetic analysis of the major capsid protein amino acid sequences
Amino acid sequences of the three major capsid protein phams (named as of July 2021: 4631, 15199, 57445) were downloaded from PhagesDB80 and merged into a single multifasta file. A phamily54 is defined as a group of related proteins and although built with k-mer-based methods, proteins within a phamily typically have a minimum pairwise 20% amino acid identity. The divergent nature of these protein sequences required an alignment algorithm that could permit a large number of gaps in our multiple sequence alignment. To that end, we aligned the major capsid proteins using MAFFT (v7.453)75 with the following parameters: globalpair, unalignlevel 0.8, leavegappyregion, and maxiterate 1000.
A maximum likelihood phylogeny was created from the multiple sequence alignment using IQTree (v1.6.6)76 with the following parameters: ModelFinder Plus81 (-m MFP), and 100 non-parametric bootstraps (-bb 100). The model finder chose an LG model with empirical frequencies and five rate categories (LG+F+R5) as the most likely model based on the Bayesian information criterion. The resulting phylogeny was visualized in Figtree (v1.4.4)77. Nodes were collapsed only when the collapsed node contained a single pham from a single phage subcluster.
Alphafold of major capsid proteins
To create the structural dendrogram, we used Alphafold to predict the three-dimensional protein fold of a representative major capsid protein from each cluster (139 total clusters), as well as every Singleton (62) major capsid protein. All protein sequences were obtained from the actinobacteriophage database (PhagesDB)80 and Phamerator54 in July 2021. For the few clusters (A, BN, CZ, DN, and F) that have more than one major capsid protein phamily, we folded a representative of each major capsid protein phamily from that cluster. There are forty-two annotated major capsid protein phamilies in the actinobacteriophages, spread across the 201 clusters and Singletons. In total, five clusters (DK, DS, EK, EM, and FC) and eight Singletons have no annotated major capsid protein and were therefore excluded from this analysis. The 18 total excluded phages account for less than 0.5% of the total number of annotated actinobacteriophages, so their exclusion is unlikely to skew the results. Cluster BO, which contains two phages, was also excluded from this analysis since they do not use the HK97-fold in their major capsid protein and are part of the Tectiviridae family of viruses. In total, 201 major capsid proteins were predicted with the default Alphafold settings and the major capsid protein amino acid sequence as input. The model with the highest confidence was used in the structural map. Approximately 35% of the Alphafold predicted major capsid proteins from the actinobacteriophages had an N-terminal extension that was similar in size to the HK97 phage delta domain (needed for the assembly of the empty capsid into which the viral DNA genome is packaged82). Some of the predicted delta domains in the actinobacteriophage major capsid proteins were almost as large (300 amino acids) as the major capsid protein and it is not possible to predict the cleavage site between the delta domain and the post-cleavage N-arm. We therefore manually truncated all the Alphafold predicted major capsid proteins to remove the N-terminal arm and the delta domain, if present, to make sure that we did not introduce bias from the N-arm and delta domains into the structural comparison. The N-arm was truncated to approximately where the N-arm crosses behind the spine helix of the major capsid protein. The fasta files and PDB files of the predicted fulllength and truncated major capsid proteins can be found in Supplemental Information.
Creation of a structural dendrogram using Homologous Structure Finder
In this study, we applied automatic structure alignment and the structure-based classification method Homologous Structure Finder (HSF)56, which allows comprehensive comparisons of proteins, not only within a protein family (such as RNA-dependent RNA polymerase)83 but also between protein families and superfamilies, significantly extending the depth of sequence-based phylogenies56. HSF identifies the equivalent residues for a pair of protein structures by comparing a set of amino acid properties (e.g., physiochemical properties of amino acids, local geometry, backbone direction, local alignment, and Cα distances)56. The two protein structures that are the most similar based on the properties are merged into a common structural core which then represents the pair in the later iterations. Next, the structure or a core from a previous iteration, best matching to an existing core or to a single structure not in any core yet, is merged either to a core or to another structure. The iterations are continued until all the protein structures are part of a clustering and a single structural core is identified for all the proteins in the data set. The equivalent residues in the structural core can be considered homologous, similar to high-scoring columns of multiple sequence alignment.
Pairwise comparison of the properties of the residues in the homologous positions of the common structural core between the original structures results in a pairwise distance matrix, which can be then used for constructing a structure-based distance tree56. The distances in such structure-based distance trees do not necessarily scale with respect to time, as changes in protein structure may not be continuous. However, the clustering of proteins in the structure-based distance tree constructed using HSF has been shown to follow the sequence-based classification of proteins into protein families, even when the common core contains less than 40 residues83. Thus, structure-based analysis is appropriate for a rough estimation of evolutionary events and relationships between protein families when the proteins share little or no detectable sequence similarity, and the accuracy of estimation of the evolutionary events increases as the sequence similarity increases.
Quantification and statistical analysis
Cryo-EM data collection and refinement statistics are shown in Table S1 and Table S6.
Supplementary Material
Highlights.
Structural classification of HK97-fold capsid proteins from actinobacteriophages
Fifteen structural groups of major capsid proteins
CryoEM of one structural group shows use of isopeptide bond to stabilize the capsid
Acknowledgments
We thank Dr. Gabrielle Valles for a helpful review of the paper. We acknowledge the hard work and dedication of all those involved (those at the University of Pittsburgh and HHMI) in the creation and continued support of the SEA-PHAGES program. Specifically, we thank the following students from the SEA-PHAGES program for the isolation of each phage:
Bridgette: Kira Zack and others at the University of Pittsburgh, PHIRE Program
Muddy: Lilli Hoist and others at the University of Kwazulu-Natal, PHIRE Program
Oxtober96: Lijia Xin and others are the University of Connecticut, SEA-PHAGES Program
Ziko: Anna Bondonese and others are at the University of Pittsburgh, SEA-PHAGES Program
Bobi: Margaret Korty and Stephanie Maas and others are Purdue University, SEA-PHAGES Program
Adephagia: Jordan L. Mosier and others at the University of North Texas, SEA-PHAGES Program
Cain: Thomas Cast and Kara Gallo and others at Gonzaga University, SEA-PHAGES Program
Che8: V. Kumar and others at the Albert Einstein College of Medicine
Ogopogo: Kaylee Nicholson and others at the University of California, Santa Cruz, SEA-PHAGES Program
Cozz: Matthew Montgomery and others at the University of Pittsburgh, PHIRE Program
We also want to thank the myriad of students and faculty who have contributed to the 201 phages we included in our bioinformatic analyses.
A portion of this research was supported by NIH grant U24GM129547 and performed at the PNCC at OHSU and accessed through EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research.
This work was supported by National Institutes of Health grants GM131729 and Howard Hughes Medical Institute grants GT12053 (to GFH). The University of Pittsburgh Titan Krios microscope and Falcon 3 camera were supported by the Office of the Director, National Institutes of Health, under award numbers S10 OD025009 and S10 OD019995, respectively (JFC).
We also thank the following scientists at PNCC for the data collection: Theo Humphreys, Omar Davulcu, Nancy Meyer, and Rose Marie Haynes.
Footnotes
Declaration of Interests: G.F.H. is a compensated consultant for Tessera and for Janssen Inc. The remaining authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Helgstrand C.et al. The Refined Structure of a Protein Catenane: The HK97 Bacteriophage Capsid at 3.44Å Resolution. Journal of Molecular Biology 334, 885–899 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Pietilä MK et al. Structure of the archaeal head-tailed virus HSTV-1 completes the HK97 fold story. Proc Natl Acad Sci U S A 110, 10604–10609 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dai X.& Zhou ZH Structure of the herpes simplex virus 1 capsid with associated tegument protein complexes. Science 360, eaao7298 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sutter M.et al. Structural basis of enzyme encapsulation into a bacterial nanocompartment. Nat. Struct. Mol. Biol. 15, 939–947 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Nichols RJ., Cassidy-Amstutz C., Chaijarasphong T. & Savage DF. Encapsulins: molecular biology of the shell. Critical Reviews in Biochemistry and Molecular Biology 52, 583–594 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Suttle CA Marine viruses — major players in the global ecosystem. Nat Rev Microbiol 5, 801–812 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Jordan TC et al. A Broadly Implementable Research Course in Phage Discovery and Genomics for First-Year Undergraduate Students. mBio 5, e01051–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teaching Scientific Inquiry. https://www.science.org/doi/10.1126/science.1136796. [Google Scholar]
- 9.Hatfull GF Actinobacteriophages: Genomics, Dynamics, and Applications. Annual Review of Virology 7, 37–61 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dedrick RM et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med 25, 730–733 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diacon AH et al. Mycobacteriophages to Treat Tuberculosis: Dream or Delusion? Respiration 101, 1–15 (2022). [DOI] [PubMed] [Google Scholar]
- 12.Krupovic M.& Koonin EV Multiple origins of viral capsid proteins from cellular ancestors. PNAS 114, E2401–E2410 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duda RL & Teschke CM The amazing HK97 fold: versatile results of modest differences. Current Opinion in Virology 36, 9–16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suhanovsky MM & Teschke CM Nature’s favorite building block: Deciphering folding and capsid assembly of proteins with the HK97-fold. Virology 479–480, 487–497 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou ZH & Chiou J.Protein chainmail variants in dsDNA viruses. AIMS Biophys 2, 200–218 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caspar DL & Klug A.Physical principles in the construction of regular viruses. Cold Spring Harb. Symp. Quant. Biol. 27, 1–24 (1962). [DOI] [PubMed] [Google Scholar]
- 17.Gertsman I., Fu C-Y., Huang R., Komives EA. & Johnson JE. Critical Salt Bridges Guide Capsid Assembly, Stability, and Maturation Behavior in Bacteriophage HK97. Mol Cell Proteomics 9, 1752–1763 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evilevitch A.et al. Effects of Salt Concentrations and Bending Energy on the Extent of Ejection of Phage Genomes. Biophysical Journal 94, 1110–1120 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.São-José C, de Frutos M, Raspaud E, Santos MA & Tavares P.Pressure Built by DNA Packing Inside Virions: Enough to Drive DNA Ejection in Vitro, Largely Insufficient for Delivery into the Bacterial Cytoplasm. Journal of Molecular Biology 374, 346–355 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Kindt J, Tzlil S, Ben-Shaul A.& Gelbart WM DNA packaging and ejection forces in bacteriophage. PNAS 98, 13671–13674 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lander GC et al. Bacteriophage lambda stabilization by auxiliary protein gpD: timing, location, and mechanism of attachment determined by cryoEM. Structure 16, 1399–1406 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang C, Zeng J.& Wang J.Structural basis of bacteriophage lambda capsid maturation. Structure (2022) doi: 10.1016/j.str.2021.12.009. [DOI] [PubMed] [Google Scholar]
- 23.Zhang X.et al. A new topology of the HK97-like fold revealed in Bordetella bacteriophage by cryoEM at 3.5 Å resolution. eLife 2, e01299 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wikoff WR et al. Topologically Linked Protein Rings in the Bacteriophage HK97 Capsid. Science 289, 2129–2133 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Tso D., Peebles CL., Maurer JB., Duda RL. & Hendrix RW. On the catalytic mechanism of bacteriophage HK97 capsid crosslinking. Virology 506, 84–91 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hryc CF et al. Accurate model annotation of a near-atomic resolution cryo-EM map. PNAS 114, 3103–3108 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao H.et al. Structure of a headful DNA-packaging bacterial virus at 2.9 Å resolution by electron cryo-microscopy. PNAS 114, 3601–3606 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baker ML et al. Validated near-atomic resolution structure of bacteriophage epsilon15 derived from cryo-EM and modeling. PNAS 110, 12301–12306 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newcomer RL et al. The phage L capsid decoration protein has a novel OB-fold and an unusual capsid binding strategy. eLife 8, e45345 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huet A, Duda RL, Boulanger P.& Conway JF Capsid expansion of bacteriophage T5 revealed by high resolution cryoelectron microscopy. PNAS 116, 21037–21046 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo F.et al. Capsid expansion mechanism of bacteriophage T7 revealed by multistate atomic models derived from cryo-EM reconstructions. PNAS 111, E4606–E4614 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Effantin G, Fujiwara A, Kawasaki T, Yamada T.& Schoehn G.High Resolution Structure of the Mature Capsid of Ralstonia solanacearum Bacteriophage ϕRSA1 by Cryo-Electron Microscopy. Int J Mol Sci 22, 11053 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu J, Wang D, Gui M.& Xiang Y.Structural assembly of the tailed bacteriophage ϕ29. Nat Commun 10, 2366 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aksyuk AA. et al. Structural investigations of a Podoviridae streptococcus phage C1, implications for the mechanism of viral entry. Proc Natl Acad Sci U S A 109, 14001–14006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hrebík D.et al. Structure and genome ejection mechanism of Staphylococcus aureus phage P68. Science Advances 5, eaaw7414 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalez B.et al. Phage G structure at 6.1 Å resolution, condensed DNA, and host identity revision to a Lysinibacillus. J Mol Biol 432, 4139–4153 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hua J.et al. Capsids and Genomes of Jumbo-Sized Bacteriophages Reveal the Evolutionary Reach of the HK97 Fold. mBio 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nováček J.et al. Structure and genome release of Twort-like Myoviridae phage with a double-layered baseplate. Proc Natl Acad Sci USA 113, 9351–9356 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dearborn AD et al. Competing scaffolding proteins determine capsid size during mobilization of Staphylococcus aureus pathogenicity islands. eLife 6, e30822 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cui N.et al. Capsid Structure of Anabaena Cyanophage A-1(L). Journal of Virology 95, e01356–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamiya R.et al. Acid-stable capsid structure of Helicobacter pylori bacteriophage KHP30 by single-particle cryoelectron microscopy. Structure (2021) doi: 10.1016/j.str.2021.09.001. [DOI] [PubMed] [Google Scholar]
- 42.Jin H.et al. Capsid Structure of a Freshwater Cyanophage Siphoviridae Mic1. Structure 27, 1508–1516.e3 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Bayfield OW et al. Cryo-EM structure and in vitro DNA packaging of a thermophilic virus with supersized T=7 capsids. PNAS 116, 3556–3561 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stone NP., Demo G., Agnello E. & Kelch BA. Principles for enhancing virus capsid capacity and stability from a thermophilic virus capsid structure. Nat Commun 10, 4471 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson MC et al. Structure, proteome and genome of Sinorhizobium meliloti phage ΦM5: A virus with LUZ24-like morphology and a highly mosaic genome. Journal of Structural Biology 200, 343–359 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Liu X.et al. Structural changes in a marine podovirus associated with release of its genome into Prochlorococcus. Nat Struct Mol Biol 17, 830–836 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bárdy P.et al. Structure and mechanism of DNA delivery of a gene transfer agent. Nat Commun 11, 3034 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hawkins NC, Kizziah JL, Penadés JR & Dokland T.Shape shifter: redirection of prolate phage capsid assembly by staphylococcal pathogenicity islands. Nat Commun 12, 6408 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gipson P.et al. Protruding knob-like proteins violate local symmetries in an icosahedral marine virus. Nat Commun 5, 4278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Z.et al. Cryo-EM structure of the bacteriophage T4 isometric head at 3.3-Å resolution and its relevance to the assembly of icosahedral viruses. PNAS 114, E8184–E8193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Z.et al. Structure of the Marine Siphovirus TW1: Evolution of Capsid-Stabilizing Proteins and Tail Spikes. Structure 26, 238–248.e3 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Hardy JM et al. The architecture and stabilisation of flagellotropic tailed bacteriophages. Nat Commun 11, 3748 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pope WH & Hatfull GF Adding pieces to the puzzle: New insights into bacteriophage diversity from integrated research-education programs. Bacteriophage 5, e1084073 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cresaw SG. et al. Phamerator: a bioinformatic tool for comparative bacteriophage genomics. BMC Bioinformatics 12, 395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Steinegger M.& Söding J.MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol 35, 1026–1028 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Ravantti J, Bamford D.& Stuart DI Automatic comparison and classification of protein structures. J Struct Biol 183, 47–56 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Ravantti JJ, Martinez-Castillo A.& Abrescia NGA Superimposition of Viral Protein Structures: A Means to Decipher the Phylogenies of Viruses. Viruses 12, 1146 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jumper J.et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mönttinen HAM, Ravantti JJ & Poranen MM Structural comparison strengthens the higher-order classification of proteases related to chymotrypsin. PLOS ONE 14, e0216659 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ross PD et al. Crosslinking renders bacteriophage HK97 capsid maturation irreversible and effects an essential stabilization. EMBO J 24, 1352–1363 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Duda RL, Martincic K, Xie Z.& Hendrix RW Bacteriophage HK97 head assembly. FEMS Microbiol Rev 17, 41–46 (1995). [DOI] [PubMed] [Google Scholar]
- 62.Molineux IJ & Panja D.Popping the cork: mechanisms of phage genome ejection. Nat Rev Microbiol 11, 194–204 (2013). [DOI] [PubMed] [Google Scholar]
- 63.D’Lima NG. & Teschke CM. A Molecular Staple: D-Loops in the I Domain of Bacteriophage P22 Coat Protein Make Important Intercapsomer Contacts Required for Procapsid Assembly. Journal of Virology (2015) doi: 10.1128/JVI.01629-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steven AC, Greenstone HL, Booy FP, Black LW & Ross PD Conformational changes of a viral capsid protein: Thermodynamic rationale for proteolytic regulation of bacteriophage T4 capsid expansion, co-operativity, and super-stabilization by soc binding. Journal of Molecular Biology 228, 870–884 (1992). [DOI] [PubMed] [Google Scholar]
- 65.Tso D, Hendrix RW & Duda RL Transient contacts on the exterior of the HK97 procapsid that are essential for capsid assembly. J Mol Biol 426, 2112–2129 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hagan RM et al. NMR Spectroscopic and Theoretical Analysis of a Spontaneously Formed Lys-Asp Isopeptide Bond. Angew Chem Int Ed Engl 49, 8421–8425 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Suhanovsky MM & Teschke CM Bacteriophage P22 capsid size determination: Roles for the coat protein telokin-like domain and the scaffolding protein amino-terminus. Virology 417, 418–429 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Earnshaw WC, King J, Harrison SC & Eiserling FA The structural organization of DNA packaged within the heads of T4 wild-type, isometric and giant bacteriophages. Cell 14, 559–568 (1978). [DOI] [PubMed] [Google Scholar]
- 69.Podgorski J.et al. Structures of Three Actinobacteriophage Capsids: Roles of Symmetry and Accessory Proteins. Viruses 12, 294 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zivanov J.et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goddard TD et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Emsley P, Lohkamp B, Scott WG & Cowtan K.Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liebschner D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Cryst D 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Croll TI ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Cryst D 74, 519–530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Katoh K.& Standley DM MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Minh BQ et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Molecular Biology and Evolution 37, 1530–1534 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rambaut A.FigTree v1. 4. (2012). [Google Scholar]
- 78.Tivol WF, Briegel A.& Jensen GJ An Improved Cryogen for Plunge Freezing. Microsc Microanal 14, 375–379 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Clarke O.Coot-trimmings. [Google Scholar]
- 80.Russell DA & Hatfull GF PhagesDB: the actinobacteriophage database. Bioinformatics 33, 784–786 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A.& Jermiin LS ModelFinder: fast model selection for accurate phylogenetic estimates. Nature Methods 14, 587–589 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oh B, Moyer CL, Hendrix RW & Duda RL The delta domain of the HK97 major capsid protein is essential for assembly. Virology 456–457, 171–178 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mönttinen HAM, Ravantti JJ & Poranen MM Common Structural Core of Three- Dozen Residues Reveals Intersuperfamily Relationships. Molecular Biology and Evolution 33, 1697–1710 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the models have been deposited in PDB. The cryo-EM maps have been deposited in EMDB. The raw cryo-EM micrographs have been deposited in EMPIAR. The raw cryo-EM micrographs for Muddy have not been deposited in EMPIAR due to on-going research. All data are publicly available as of the data of publication. The accession numbers are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Adephagia | Actinobacteriophage database (University of Pittsburgh) | Accession number: JF704105 |
| Bobi | Actinobacteriophage database (University of Pittsburgh) | Accession number: KF114874 |
| Bridgette | Actinobacteriophage database (University of Pittsburgh) | Accession number: MH834603 |
| Cain | Actinobacteriophage database (University of Pittsburgh) | Accession number: MF324913 |
| Che8 | Actinobacteriophage database (University of Pittsburgh) | Accession number: AY129330 |
| Cozz | Actinobacteriophage database (University of Pittsburgh) | Accession number: KU998239 |
| Muddy | Actinobacteriophage database (University of Pittsburgh) | Accession number: KF024728 |
| Ogopogo | Actinobacteriophage database (University of Pittsburgh) | Accession number: MG925354 |
| Oxtober96 | Actinobacteriophage database (University of Pittsburgh) | Accession number: MT024864 |
| Ziko | Actinobacteriophage database (University of Pittsburgh) | Accession number: MK919478 |
| Arthrobacter globiformis B-2979 | Actinobacteriophage database (University of Pittsburgh) | NCBI:txid1077972 |
| Gordonia terrae 3612 | Actinobacteriophage database (University of Pittsburgh) | NCBI:txid2055 |
| Microbacterium foliorum NRRL B-24224 | Actinobacteriophage database (University of Pittsburgh) | NCBI:txid104336 |
| Mycobacterium smegmatis mc 2 155 | Actinobacteriophage database (University of Pittsburgh) | NCBI:txid246196 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Middlebrook 7H9 Broth Base | Sigma-Aldrich | M0178–500G |
| Glycerol | Fisher Scientific | BP229–1 |
| Sodium chloride | Fisher Scientific | S271–10 |
| Dextrose (D-Glucose) Anhydrous | Fisher Scientific | D16–500 |
| Albumin, Bovine, Cohn Fraction V 98% | Fisher Scientific | AAJ6573122 |
| Tween-80 | Fisher Scientific | BP338–500 |
| Calcium Chloride | Sigma-Aldrich | C1016–500G |
| Agar | Fisher Scientific | BP1423–500 |
| LB Broth, Lennox | Fisher Scientific | BP1427–500 |
| Yeast Extract | Sigma-Aldrich | Y1625–1KG |
| Peptone | Fisher Scientific | BP1420 500 |
| Tris Base | Millipore Sigma | 648311–1KG |
| Magnesium Sulfate Anhydrous | Fisher Scientific | M65–500 |
| Cesium Chloride | Fisher Scientific | BP1591–1 |
| Ethane (research grade) | Airgas | ET R35 |
|
| ||
| Deposited data | ||
|
| ||
| Adephagia | This paper | PDB: 8EC2 |
| Adephagia | This paper | EMD-28012 |
| Adephagia | This paper | EMPIAR-11200 |
| Bobi | This paper | PDB: 8EC8 |
| Bobi | This paper | EMD-28015 |
| Bobi | This paper | EMPIAR-11201 |
| Bridgette | This paper | PDB: 8ECI |
| Bridgette | This paper | EMD-28016 |
| Bridgette | This paper | EMPIAR-11209 |
| Cain | This paper | PDB: 8ECJ |
| Cain | This paper | EMD-28017 |
| Cain | This paper | EMPIAR-11205 |
| Che8 | This paper | PDB: 8E16 |
| Che8 | This paper | EMD-27824 |
| Che8 | This paper | EMPIAR-11190 |
| Cozz | This paper | PDB: 8ECK |
| Cozz | This paper | EMD-28018 |
| Cozz | This paper | EMPIAR-11206 |
| Muddy | This paper | PDB: 8EDU |
| Muddy | This paper | EMD-28039 |
| Ogopogo | This paper | PDB: 8ECN |
| Ogopogo | This paper | EMD-28020 |
| Ogopogo | This paper | EMPIAR-11207 |
| Oxtober96 | This paper | PDB: 8ECO |
| Oxtober96 | This paper | EMD-28021 |
| Oxtober96 | This paper | EMPIAR-11208 |
| Ziko | This paper | PDB: 8EB4 |
| Ziko | This paper | EMD-27992 |
| Ziko | This paper | EMPIAR-11195 |
|
| ||
| Software and algorithms | ||
|
| ||
| Relion v3.1.1 | Zivanov et al.70 | https://github.com/3dem/relion |
| Alphafold v2.0 | Jumper et al.58 | https://github.com/deepmind/alphafold |
| ChimeraX 1.3 | Goddard et al.71 | https://www.cgl.ucsf.edu/chimerax/ |
| Coot v0.9.2 | Emsley et al.72 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix v1.19.2–4158 | Liebschner et al.73 | https://phenix-online.org/ |
| Isolde v1.3 | Croll74 | https://isolde.cimr.cam.ac.uk/ |
| MAFFT v7.453 | Katoh and Standley75 | https://mafft.cbrc.jp/alignment/software/ |
| IQTree v1.6.6 | Minh et al.76 | http://www.iqtree.org/ |
| FigTree v1.4.4 | Rambaut77 | https://github.com/rambaut/figtree/releases |
| Homologous Structure Finder | Ravantti et al.56 | N/A |
This paper does not report original code.