

Abstract
We calculate the forces required to package (or, equivalently, acting to eject) DNA into (from) a bacteriophage capsid, as a function of the loaded (ejected) length, under conditions for which the DNA is either self-repelling or self-attracting. Through computer simulation and analytical theory, we find the loading force to increase more than 10-fold (to tens of piconewtons) during the final third of the loading process; correspondingly, the internal pressure drops 10-fold to a few atmospheres (matching the osmotic pressure in the cell) upon ejection of just a small fraction of the phage genome. We also determine an evolution of the arrangement of packaged DNA from toroidal to spool-like structures.
The classic Hershey-Chase experiment (1) of almost 50 years ago is best known for confirming DNA as the genetic material. But it was significant also as the demonstration that a bacterial virus (phage) leaves its protein capsid outside the cell it infects. More explicitly, upon binding to its receptor protein in the outer membrane of the bacterial cell, the viral capsid is opened and its DNA is injected into the cytoplasm. Obviously, this transfer can only happen as a passive process if the DNA is sufficiently pressurized in the capsid. For the past several decades, a great deal of experimental work has been devoted to determining the arrangement of “packaged” DNA in phage capsids through techniques that include x-ray scattering (2, 3), Raman spectroscopy (4), chemical cross-linking (5, 6), and electron microscopy (7, 8, 9). Various competing models have been proposed in which the DNA molecule is organized in concentric rings as a “spool” (3), in parallel segments joined at sharp kinks (10), or as a folded toroid (11). The most recent electron microscopy results on bacteriophages T7 (8) and T4 (9) show concentric ring structures that lend support to a spool-like structural motif. The underlying theoretical problem is also formidable, because one is confronted with the statistical-mechanical challenge of accounting for how a semiflexible, highly charged chain can be confined in dimensions comparable to its persistence length ξ and yet hundreds of times smaller than its overall (contour) length L (12, 13). Although some estimates have been made of the pressure and elastic stress in a fully loaded capsid (12, 14), we are not aware of any attempt to treat the driving pressures during the course of ejection or the loading forces as a function of the extent of packaging (15).¶
In the present work, we connect the processes of loading DNA into, and ejecting it from, a phage capsid by calculating the energy of the chain as a function of the degree of loading or ejection. As already mentioned, the initial ejection is a passive process, being driven directly by the pressure difference inside and outside the capsid (16).‖ Indeed, we shall show that the energy decreases monotonically as successively shorter lengths of DNA are left confined in the capsid; this rate of decrease corresponds to a force driving the chain outside. By contrast, the loading of DNA into the virus must be an active process. Work is performed by the motor protein responsible for pushing the phage genome into the capsid volume whose size is small compared with the DNA length. As we demonstrate below, the force exerted by this motor takes on its maximum value at the end of the loading process, at which point the stored energy density (pressure) is sufficient to drive the ejection of the DNA when the capsid is eventually opened by binding to a new bacterial cell. It is significant that the loading process, both in vivo (17) and in vitro (18), can take place in the absence of polyvalent counterions or similar DNA-condensing agents. Under these conditions, the DNA is strongly self-repelling, making its packaging into the capsid still more difficult. In the presence of polyvalent counterions (e.g., spermidine, which is present in significant concentrations in most bacterial cells), the DNA is self-attracting, and yet we shall see that a comparable force is required to load it into the viral capsid.
We use complementary methods to investigate the evolution of structure and energetics along the loading coordinate. In our first approach, Brownian molecular dynamics computer simulations (19) of DNA loading into a viral capsid are performed by using a coarse-grained model, typical of polymer simulations, in which the double-stranded DNA chain is represented by a semirigid string of “beads.”** We treat the cases of self-attracting or self-repelling chains, corresponding to the presence or absence of polyvalent counterions, through either allowing full Lennard–Jones attractions between pairs of beads or by truncating the forces between them at the potential minimum to give a purely repulsive interaction. The capsid–particle interactions are described by a short-ranged repulsive potential that vanishes identically for particles within the radius RC of the capsid, but that increases steeply [as (R − RC)4 in our model] for particles at distances greater than RC from the capsid center. This potential is simply a convenient way to describe a hard interior wall, approximating the icosahedral capsid as a sphere and neglecting any specific interactions between the DNA and the capsid-wall interior, whose structural details (e.g., cationic charge distribution) are generally not known (20).‡‡ We also neglect small changes in capsid size that might accompany loading and ejection.
To simulate the loading of the chain into the capsid, each bead is first moved radially into the capsid at a fixed rate†† from an entrance position arbitrarily designated as the top of the capsid. All other beads—those already inside the capsid—are moved according to the forces they experience as a result of their interactions with other beads, the capsid wall, and the random forces exerted by the solvent. Once the bead being introduced has advanced one bond-length into the capsid, it is released from its constraint of constant loading rate, and the next bead in the chain takes its place to be loaded from the top of the capsid. In this way, previously loaded segments of the chain are able to respond to the gradual filling of the capsid as new beads are introduced. In our simulations, computational time constraints limit the total number of beads to somewhat over 1,000, corresponding to approximately one-sixth the length of the genome of a typical icosahedral bacteriophage. We scale the capsid size accordingly to obtain, upon full loading, a volume fraction approaching that of close packing.
Upon loading a length of only several capsid diameters of self-attracting chain, a donut-like toroidal structure (shown in Fig. 1A) forms spontaneously. As more and more chain enters the capsid, the donut-like structure expands parallel to its central axis into a spool-like structure with a nearly empty internal cylindrical core. During the final stages of loading, a portion of the chain ceases winding around the spool axis and, instead, fills in the core of the spool with strands parallel to this axis (as seen in Fig. 1B).
Figure 1.

Snapshots from Brownian dynamics simulations of self-attracting chain loading into spherical cavity. Only bonds are shown. The position of the wall potential is shown with short black dashes. (A) 199 beads, side view. (B) 1,120 beads, side view (Upper) and view down the spool axis (Lower). Interaction potentials correspond to: Lennard–Jones ɛ = 0.5 kBT; persistence length ξ = 32 σ; capsid radius RC = 6.25σ.
In the absence of attractions between nearby chain segments, a much more disordered structure is formed during loading. The transition between the ordered and disordered structures is kinetically inaccessible over the time-scale of the simulation (about 1 ms); i.e., ordered structures grown in the presence of attractions maintain much of their order when equilibrated in the absence of attractions. It is probable that the most stable structure for both attractive and repulsive cases has a high degree of local order at the high-packing fraction characteristic of the capsid in its final stages of loading. For this reason, the ordered structure formed during loading with attractions was used as a starting point for the force calculations both with and without attractions, as described below. (This situation also corresponds to the biological conditions during the phage infection cycle: polyvalent amines are available inside the bacterium for loading but will be exchanged for simple salt once the phage enters the surrounding medium.)
The average force resisting chain loading is shown in Fig. 2 with and without attractions. The resistive force on the self-attracting chain drops significantly upon progressive loading, as a condensed toroidal-spool structure forms within the capsid. It is here that the chain is able to benefit from its self-attraction. As the chain fills the capsid and the repulsive part of the bead–bead interaction becomes dominant, however, the force climbs steeply, reflecting the increasing curvature and packing strain of the condensate. Indeed, at this point, neighboring chain segments are up against each other's repulsive walls. In this regime, where the structure depends largely on repulsive interactions, the forces with and without attractions rise in parallel, offset by the difference in energy per unit length of chain between the two cases.
Figure 2.

Simulated mean force resisting loading from inside capsid with increasing degree of loading. Model potential is as for Fig. 1. ●, self-attracting chain (Lennard–Jones potential with cutoff of 2.5 σ); ○, purely repulsive chain (with truncated, shifted potential). Equilibrium mean force is determined by starting with a configuration from a loading simulation, fixing the position of the most recently entered bead at the capsid entrance, and averaging the force on that bead while the remainder of the chain is thermally equilibrated. Volume fraction is defined as though beads were hard spheres of radius σ inside a spherical cavity of radius RC.
The physics exposed by the above simulations are nicely illustrated by a simple phenomenological model, which has the further advantage that it can be straightforwardly calibrated to experiment. The goal again is to treat the forces and pressures associated with loading a long (L ≫ RC) semiflexible, self-attracting chain into a small capsid (with RC comparable to ξ). The condensates inside and outside the capsid are treated independently. In both cases, the condensate is assumed to occupy a volume with uniaxial symmetry, with chain circumferentially wound about a central axis. The cross-sectional profile of the condensate is not constrained to be a circle (as it is in a simple torus) but is allowed to assume an arbitrary shape to optimize the condensate's energy (21).§§
The elastic (bending) energy of the condensate per unit length is taken as the one-dimensional bending modulus κ times the square of the local curvature,¶¶ where (consistent with the assumption of circumferential winding about an axis) the curvature is the inverse distance R from the central axis:
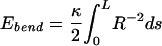 |
1 |
The cohesive energy per unit length of chain, ea, depends on how closely its strands are packed. The interhelical spacing d between locally parallel, hexagonally packed segments is related to the total volume occupied by a chain of length L by means of the following geometric relation:
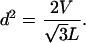 |
The function ea(d) was extracted from measurements on DNA condensed by the addition of polyvalent counterions and osmotically compressed to d-spacings less than the optimal value d0 of 28 Å (22, 23).‖‖ Then, the total cohesive energy Ebulk is the product of chain length and the cohesive energy per unit length, ea(d). A surface term is also included, proportional to the surface area A of the condensate:
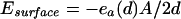 |
2 |
For an overall (fixed) chain length L, and for an arbitrary loaded length L* (0 < L* < L), we write the total energy of the DNA as
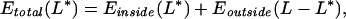 |
3 |
with each of the energies on the right-hand side given by
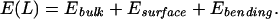 |
4 |
The optimal shape of the condensate outside the capsid, free of constraints, is found to be a torus with a circular profile. In contrast, the condensed chain inside the capsid, whose size and shape are limited by the constraint that it fits inside the spherical capsid of radius RC, evolves continuously from a torus into a spool-like shape in which the capsid is filled, except for a cylindrical core in the center. This progression, shown in Fig. 3, is in good agreement with that observed in the changing structure of the chain in our Brownian dynamics simulations of capsid loading.
Figure 3.

Cross-sectional profiles of condensates inside the capsid, optimized by continuum theory. One quadrant of a planar cross-section is shown, with the capsid boundary shown as a heavy black line; the uniaxial (toroid or spool) axis is the left-hand vertical line. Each profile is labeled with its fractional loaded length, L*/L. The relative lengths used in the calculations were chosen to represent bacteriophage λ: RC = 27.5 nm (0.55 ξ); L = 16.5 μm.
The minimum force required (of a molecular motor, for instance) to load the capsid is given by the derivative of the energy with respect to loaded length. This resistance to loading, plotted in Fig. 4, is a consequence both of the necessity to remove chain from the stable toroidal condensate outside the capsid and of the stresses imposed by the limited volume and the need for tight bending within the capsid. The force begins to increase dramatically at L* = 0.75 L to a maximum of 15 pN; this increase is accompanied by the onset of the compression of the condensate-to-interchain distances below its preferred value (d0) of 2.8 nm. This compression occurs as the elastic energy required to wind around the increasingly narrow inner core of the spool becomes prohibitive, and the spool is compressed against the capsid wall. The drop in interchain spacing at the later stages of loading also has been observed in experiments employing mutant bacteriophage DNA of variable length (3, 8).
Figure 4.

Total force resisting loading of capsid (lower curve) and interhelical spacing d (upper curve) as a function of loading fraction L*/L in the presence of polyvalent cation condensing agent, calculated from continuum theory.
Recent experiments by Bustamante and coworkers (24) have directly measured the force exerted by the motor protein that packages DNA in the particular instance of φ29 phage. Here the chain is self-repelling, because the studies are done in the absence of condensing agent. It is, therefore, of interest to consider whether the above results can be extrapolated to the purely repulsive case. At our calculated interhelical distance d of 2.5 nm in the fully loaded capsid, the experimentally measured osmotic pressures of DNA in the presence and absence of condensing agent are very similar (22). It is, perhaps, not surprising that the experimentally measured d-spacing in fully loaded bacteriophage is not changed by the addition or removal of condensing agent (3) because the balance between compression and bending is unaffected by the nature of the counterions at such high densities. The force against loading, on the other hand, will be greater in the purely repulsive case by roughly the difference in the work per unit length of DNA required to bring chains to this interhelical distance in the presence and absence of condensing agent. This difference will be sensitive to the ionic strength of the solution, but a very approximate estimate based on the experimental osmotic pressure data (22) suggests that removing condensing agent will increase the maximum loading force by roughly 20 pN to a value of about 35 pN, comparable to the measured internal force of ≈50 pN (24).
Fig. 5 shows the effect of outside osmotic pressure on the plot of energy vs. outside length. The solid curve is the energy from Eq. 1 (that is, the integral of the forces from Fig. 4), showing that, because the force resisting loading is always positive, the energy minimum is obtained for complete ejection, L* = 0. The dashed curve is a plot of the energy from Eq. 1 plus a term PosmVoutside. Here, Posm is the osmotic pressure outside the capsid because of, say, a fixed amount of a “stressing polymer” like polyethylene glycol in the solution external to the viral particle in in vitro studies. [In the in vivo case, Posm corresponds to the osmotic pressure inside the bacterial cell—believed to be on the order of a few atmospheres (T. Odijk, personal communication) because of high concentrations of cytoplasmic proteins.] Voutside is the volume of the ejected chain or, equivalently, of the chain that is yet-to-be loaded. Because Voutside is directly proportional to the length L-L*, through a factor of the cross-sectional area of the chain (of order d ), to a first approximation the PV term simply adds to Eq. 1 a straight line with slope Posm d
), to a first approximation the PV term simply adds to Eq. 1 a straight line with slope Posm d . The dashed curve in Fig. 5 shows the result for an osmotic pressure of 3 atm (1 atm = 101.3 kPa). Note that the ejection of DNA from the capsid is now only partial, i.e., the magnitude of the slope (and hence the force pushing the chain outside) vanishes when the outside length is only a small fraction of L (about 0.2 here). This predicted limitation of ejection at biological osmotic pressures suggests that the passive stage of phage DNA injection into bacteria should in general be incomplete; indeed, in some systems, this limitation has been established in detail, and translocation of the remaining DNA has been demonstrated to be transcription-dependent (16). The osmotic pressure required to suppress ejection completely is given by
. The dashed curve in Fig. 5 shows the result for an osmotic pressure of 3 atm (1 atm = 101.3 kPa). Note that the ejection of DNA from the capsid is now only partial, i.e., the magnitude of the slope (and hence the force pushing the chain outside) vanishes when the outside length is only a small fraction of L (about 0.2 here). This predicted limitation of ejection at biological osmotic pressures suggests that the passive stage of phage DNA injection into bacteria should in general be incomplete; indeed, in some systems, this limitation has been established in detail, and translocation of the remaining DNA has been demonstrated to be transcription-dependent (16). The osmotic pressure required to suppress ejection completely is given by
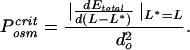 |
That is, P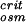 is determined by the maximum force in Fig. 4 divided by the cross-sectional area of the chain, and it corresponds to the total pressure inside the fully loaded viral capsid; this value is predicted by the present theory to be of the order of 30 atm in the presence of condensing agent.
is determined by the maximum force in Fig. 4 divided by the cross-sectional area of the chain, and it corresponds to the total pressure inside the fully loaded viral capsid; this value is predicted by the present theory to be of the order of 30 atm in the presence of condensing agent.
Figure 5.

Free energy E of ejection in the presence (dashed curve) and absence (solid curve) of 3 atm external osmotic pressure.
Acknowledgments
We heartily thank Drs. Françoise Livolant and Jean-Louis Sikorav for many extremely valuable discussions since the early stages of this work. For financial support, we acknowledge National Science Foundation Grant CHE99–88651 and American Chemical Society Grant PRF 36407-AC (to W.M.G.), Israel Science Foundation Excellence Center Grant 8013/00 (to A.B.S.), and U.S.–Israel Binational Science Foundation Grant 97–00205 (to A.B.S. and W.M.G.).
Footnotes
Beads are fed into the cavity at a rate of 3 × 10−5 σ/Δt, or at about 7500 μm/s. In actual packaging situations [see, for example, the in vitro measurements described in ref. 24] the loading rate decreases significantly with the force resisting loading. As explained later in this paper, however, we are careful to calculate our loading forces by stopping our loading at each of several different internal chain lengths and then averaging over long times the outward radial force on the last monomer.
Calculation details of our continuum theory results, reported in the present paper, are provided elsewhere [S.T., J.K., W.M.G., and A.-B.S., unpublished work], where we also treat more thoroughly the kinetics of loading/ejection and estimates of capsid pressures.
The bending modulus κ is set to 50 kBT nm, consistent with the experimentally determined persistence length for double-stranded DNA of ξ = 50 nm.
The dependence of d on osmotic pressure in the presence of condensing agent, as measured in the experiments of Rau and Parsegian (22) was fit to the form P = F0 {exp[−(d − d0)/c]−1}, with F0 = 0.12 kBT/nm3, c = 0.14 nm, and d0 = 2.8 nm. Integration of the pressure with respect to two-dimensional compression of the hexagonal lattice gives ea(d) to within a constant. The cohesive energy per unit length at d0 was chosen as −0.74 kBT/nm to give the known dimensions of toroidal condensates of DNA in solution (23).
Gabashvili and Grosberg (15) have treated several scenarios for the progressive kinetics of ejection, allowing for different sources of friction being dominant in the capsid and tail. Although they also make estimates of the ejection forces, our primary concern in the present work is to provide a more realistic and systematic theory of the thermodynamics underlying these forces and of the structures of packaged DNA in the capsid.
Later stages of the injection have been shown in many instances to be driven by bacterial-cell transcription of the leading portion of the translocated viral DNA; see, for example, ref. 16.
The “beads” are Lennard–Jones particles, linked to their nearest neighbors along the chain by harmonic stretching potentials centered at an interbead distance of σ, the Lennard–Jones diameter (corresponding approximately to the diameter of hydrated DNA, or about 2.5 nm). A harmonic bending potential, applied to the angle between neighboring bonds, dictates the intrinsic persistence length of the chain. The step size of our Brownian dynamics simulation, which corresponds to DΔt/kBT and where D is a diffusion constant, is 3 × 10−4 σ2/kBT. By the Stokes–Einstein relation for the diffusion constant of a sphere of diameter σ = 2.5 nm in a medium with the viscosity of water, this result translates to a time-step Δt of 10 ps. Hydrodynamic interactions between beads are neglected.
The first crystallographic structure determination of the capsid of an icosahedral double-stranded DNA bacteriophage (HK97), including information about the distribution of charge on the interior wall, was reported only recently (see ref. 20).
References
- 1.Hershey, A. D. & M. Chase, M. (1952) J. Gen. Physiol.36, 39–56. [DOI] [PMC free article] [PubMed]
- 2.North A C T, Rich A. Nature (London) 1961;191:1242–1245. doi: 10.1038/1911242a0. [DOI] [PubMed] [Google Scholar]
- 3.Earnshaw W C, Harrison C S. Nature (London) 1977;268:598–602. doi: 10.1038/268598a0. [DOI] [PubMed] [Google Scholar]
- 4.Aubrey K L, Casjens S R, Thomas G J., Jr Biochemistry. 1992;31:11835–11842. doi: 10.1021/bi00162a023. [DOI] [PubMed] [Google Scholar]
- 5.Widom J, Baldwin R L. J Mol Biol. 1983;171:419–437. doi: 10.1016/0022-2836(83)90038-4. [DOI] [PubMed] [Google Scholar]
- 6.Serwer P, Hayes S J, Watson R H. J Mol Biol. 1992;223:999–1101. doi: 10.1016/0022-2836(92)90258-l. [DOI] [PubMed] [Google Scholar]
- 7.Lepault J, Dubochet J, Baschong W, Kellenberger E. EMBO J. 1987;6:1507–1512. doi: 10.1002/j.1460-2075.1987.tb02393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cerritelli M E, Cheng N, Rosenberg A H, McPherson C E, Booy F P, Steven A C. Cell. 1997;91:271–280. doi: 10.1016/s0092-8674(00)80409-2. [DOI] [PubMed] [Google Scholar]
- 9.Olson N H, Gingery M, Eiserling F A, Baker T S. Virology. 2001;279:385–391. doi: 10.1006/viro.2000.0735. [DOI] [PubMed] [Google Scholar]
- 10.Black L W, Newcomb W W, Boring J W, Brown J C. Proc Natl Acad Sci USA. 1985;82:7960–7964. doi: 10.1073/pnas.82.23.7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hud N V. Biophys J. 1995;69:1355–1362. doi: 10.1016/S0006-3495(95)80002-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riemer S C, Bloomfield V A. Biopolymers. 1978;17:785–794. doi: 10.1002/bip.1978.360170317. [DOI] [PubMed] [Google Scholar]
- 13.Gabashvili I, Grosberg A, Yu, Kuznetsov D V, Mrevlishvili G M. Biophysics. 1992;36:782–789. [Google Scholar]
- 14.Odijk T. Biophys J. 1998;75:1223–1227. doi: 10.1016/S0006-3495(98)74041-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gabashvili I S, Grosberg A, Yu J Biomol Struct Dyn. 1992;9:911–920. doi: 10.1080/07391102.1992.10507966. [DOI] [PubMed] [Google Scholar]
- 16.Garcia L R, Molineux I J. J Bacteriol. 1996;178:6921–6929. doi: 10.1128/jb.178.23.6921-6929.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hafner E W, Tabor C W, Tabor H. J Biol Chem. 1979;254:12419–12426. [PubMed] [Google Scholar]
- 18.Rubinchik S, Parris W, Gold M. J Biol Chem. 1995;270:20059–20066. doi: 10.1074/jbc.270.34.20059. [DOI] [PubMed] [Google Scholar]
- 19.Ermak D L, McCammon J A. J Chem Phys. 1978;69:1352–1360. [Google Scholar]
- 20.Wikoff W R, Liljas L, Duda R L, Tsuruta H, Hendrix R W, Johnson J E. Science. 2000;289:2129–2133. doi: 10.1126/science.289.5487.2129. [DOI] [PubMed] [Google Scholar]
- 21.Ubbink J, Odijk T. Europhys Lett. 1996;33:353–358. [Google Scholar]
- 22.Rau D C, Parsegian V A. Biophys J. 1992;61:246–259. doi: 10.1016/S0006-3495(92)81831-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arscott P G, Li A Z, Bloomfield V A. Biopolymers. 1990;30:619–630. doi: 10.1002/bip.360300514. [DOI] [PubMed] [Google Scholar]
- 24.Smith D E, Tans S J, Smith S B, Grimes S, Anderson D L, Bustamante C. Nature (London) 2001;413:748–752. doi: 10.1038/35099581. [DOI] [PubMed] [Google Scholar]