

SUMMARY
Growing evidence suggests prevalence of transcriptional condensates on chromatin, yet their mechanisms of formation and functional significance in disease remain unclear. In human cancer, a series of mutations in the histone acetylation reader ENL create gain-of-function mutants with increased transcriptional activation ability. Here we show that these mutations, clustered in ENL’s structured acetyl-reading YEATS domain, trigger aberrant condensates at native genomic targets through multivalent homotypic and heterotypic interactions. Mechanistically, mutation-induced structural changes in the YEATS domain, ENL’s two disordered regions of opposing charges, and the incorporation of extrinsic elongation factors are all required for ENL condensate formation. Extensive mutagenesis establishes condensate formation as a driver of oncogenic gene activation. Furthermore, expression of ENL mutants beyond the endogenous level leads to non-functional condensates. Our findings provide new mechanistic and functional insights into cancer-associated condensates and support condensate dysregulation as an oncogenic mechanism.
eTOC Blurb
Song et al show that ENL cancer mutations trigger aberrant condensate formation at genomic targets through multivalent homotypic and heterotypic interactions and in a dose-dependent manner. Perturbing these interactions or overexpressing ENL mutant disrupts condensate formation and/or properties, leading to defects in oncogenic gene activation.
Graphical Abstract
INTRODUCTION
Precise control of gene expression is fundamental for normal development and tissue homeostasis; accordingly, dysregulation of this process is a driver of many diseases, notably cancer (Bradner et al., 2017). Cancer genome sequencing studies have revealed genetic abnormalities that target diverse gene regulatory proteins including transcription factors (TF), chromatin regulators, and histone proteins (Garraway and Lander, 2013). While these genetic alterations have been found to drive oncogenic gene expression, their precise mechanisms of action remain largely elusive.
Recent studies revealed that some gene regulatory proteins form dynamic, locally concentrated assemblies on chromatin (Cho et al., 2018; Chong et al., 2018; Cisse et al., 2013; Mir et al., 2018; Sabari et al., 2018). These assemblies, often described as condensates or hubs, form through multivalent and non-stoichiometric interactions among their constituents and could have biophysical properties and regulatory mechanisms distinct from stable protein complexes driven by ‘lock and key’ type of high-affinity interactions. Here, we use ‘condensates’ to refer to such assemblies, making no assumption regarding the process by which they form or their biophysical properties (Mittag and Pappu, 2022). Transcriptional condensates at certain genomic loci are proposed to form through liquid-liquid phase separation (LLPS) (Boija et al., 2018; Guo et al., 2019; Hnisz et al., 2017; Lu et al., 2018; Sabari et al., 2018), a process underlying the formation of diverse membrane-less organelles (Alberti et al., 2019; Banani et al., 2017; Shin and Brangwynne, 2017). It has been shown that RNA Polymerase II (Pol II) and several TFs and co-activators contain intrinsically disordered regions (IDRs) that are prone to phase separation in vitro, and in cells these proteins often form nuclear puncta in an IDR-dependent manner (Boehning et al., 2018; Cai et al., 2019; Chong et al., 2018; Kwon et al., 2013; Lu et al., 2018; Sabari et al., 2018). While IDRs are widely studied contributors to condensate formation, they rarely exist in isolation and are typically connected to structured domains, whose role in condensate regulation is underexplored (Martin et al., 2021).
While the discovery of transcriptional condensates offers a framework for how transcriptional machinery is organized, their functional significance remains largely unclear. In some cases, deleting or heavily mutating the IDRs of some TFs and co-activators compromises their ability to phase separate in vitro and to activate transcription in cells, although derivatives with fewer total mutations and specific phase separation defects are needed to establish the causal relationship (Hahn, 2018; Mir et al., 2019). Recent studies have implicated dysregulation of transcriptional condensates in disease (Boija et al., 2021; Cai et al., 2021). Oncogenic fusion proteins EWS-FLI1 and NUP98-HOXA9 involve fusing an IDR to a TF DNA binding domain, and their oncogenic capabilities are associated with the ability to form condensates (Ahn et al., 2021; Boulay et al., 2017; Chandra et al., 2021; Chong et al., 2018; Terlecki-Zaniewicz et al., 2021). In addition, disease-associated repeat expansions in the IDRs of several TFs (e.g., HOXD13) alter their capacity to co-condense with components of transcriptional machinery (Basu et al., 2020). While these findings suggest a link between condensate formation and transcription, a better understanding of how condensates form at native genomic targets would inform specific perturbation strategies to assess their function.
This study explores a condensate-driven oncogenic function of the chromatin reader Eleven-Nineteen-Leukemia (ENL) (encoded by MLLT1). ENL binds to acetylated histones via its YEATS domain, thereby recruiting associated complexes (e.g., Super Elongation Complex, or SEC) to promote Pol II elongation (Erb et al., 2017; Wan et al., 2017). ENL has been well implicated in leukemia, where it is frequently fused with the mixed lineage leukemia protein (MLL1, or KMT2A) through chromosomal translocation (Krivtsov and Armstrong, 2007; Winters and Bernt, 2017). Wildtype (WT) ENL is also required to support oncogenic gene expression programs necessary for acute myeloid leukemia (AML) maintenance (Erb et al., 2017; Liu et al., 2022; Wan et al., 2017). More recently, hotspot mutations in the ENL YEATS domain have been found in patients with Wilms tumor and AML (T1-T8, Figure 1A) (Gadd et al., 2015, 2017; Hetzner et al., 2018; Wan et al., 2019). We previously showed that these mutations increase ENL occupancy at cancer-related genes, leading to aberrant gene activation and impaired kidney differentiation in cellular models. Compared to WT, ENL mutants have a stronger tendency to form droplet-like condensates upon overexpression, leading to our proposal that ENL mutations drive oncogenesis through a gain of condensation propensity (Wan et al., 2019). However, it remained unclear 1) whether ENL mutants can form condensates at native genomic targets under physiologically relevant conditions, 2) and if so, how mutations in a structured domain induce such a property, and 3) whether the condensate formation underlies ENL mutation-induced oncogenic gene activation.
Figure 1. Near endogenous levels of ENL mutants form submicron-sized condensates at native genomic targets.

(A) Bottom, schematic of ENL protein structure. The mutated regions are in purple. IDR, intrinsically disordered region. AHD, ANC1 homologue domain. Top, protein sequences for ENL WT and mutant (T1-T8).
(B-D) Representative images (B) and quantification in HEK293 cells transfected with indicated mCherry-ENL constructs. B, all cells shown had similar mCherry-ENL intensities. C, plot showing puncta formation and mCherry intensities. Boxes highlight the top 25% highest mCherry-ENL levels among cells without detectable puncta. D, fraction of in-puncta fluorescence intensity as a function of mean nuclear intensity. Each dot indicates one cell (n at the range of 49~78).
(E) Representative images and quantification of in vitro droplet formation by indicated ENL(FL) constructs (375 nM).
(F) Western blotting showing near endogenous levels of WT and mutant Flag-ENL transgenes in HEK293 cells. V, empty vector.
(G-I) Flag IF staining (G) and quantification (H and I) in HEK293 cells. H, percentage of nuclei with and without Flag-ENL puncta. I, the number of puncta in each nucleus. Center lines indicate median and box limits are set to the 25th and 75th percentiles. H and I: cell number at the range of 32~49.
(J) IF staining of Flag-ENL (green) and H3K27ac (magenta) in HEK293 cells. The enrichment of H3K27ac at ENL puncta (magenta line) and random sites (gray line) was quantified. T1, n = 28 cells; T1(Y78A), n = 26 cells.
(K) Representative images (K) and quantification (L) showing co-localization of Halo-ENL-T1 puncta with HOXA11 nascent RNA FISH foci (n = 20 cells).
(M and N) ChIP-qPCR analysis of Flag-ENL occupancy at the HOXA locus (M) and mRNA expression (normalized to GAPDH) of HOXA genes (N) in HEK293 cells expressing endogenous levels of indicated Flag-ENL transgenes.
H and L: Chi-square test. E, I, M, N: two-tailed unpaired Student’s t-test. *** P < 0.001. E, M, N
Data represent Mean ± S.D.
See also Figures S1 and S2.
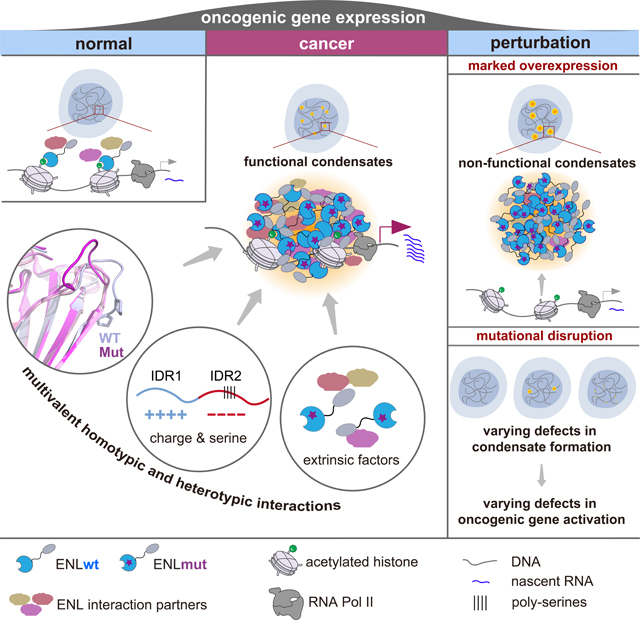
Integrating structural, molecular, and quantitative imaging studies, we show that oncogenic ENL mutants expressed at the endogenous level form aberrant condensates at target genes through a network of multivalent homotypic and heterotypic interactions. Extensive mutagenesis establishes condensate formation as a driver of oncogenic gene activation. We also show that overexpressing ENL mutant beyond the endogenous level leads to non-functional condensates. Our results provide new mechanistic and functional insights into cancer-associated condensates and support condensate dysregulation as a key oncogenic mechanism.
RESULTS
A series of hotspot mutations in ENL enhance its intrinsic propensity to form condensates
A series of ENL mutations found in cancer are somatic, heterozygous, and tightly clustered within the structured acetyl-binding YEATS domain (Figure 1A). Expression of mutant (T1/2/3) but not WT ENL in the human embryonic kidney cell line HEK293 induces Wilms tumor-associated gene expression (Wan et al., 2019), making this cell line a suitable model for investigating the basic gene regulatory mechanisms of these ENL mutations.
To assess puncta formation ability, we transiently transfected mCherry-ENL variants into HEK293 cells. Cell-by-cell analysis revealed that ENL mutants form nuclear puncta at a much lower concentration threshold than WT (Figures 1B and 1C). The overall degree of puncta formation by each ENL mutant positively correlates with its expression level (Figure 1D). We next probed the internal dynamics of puncta formed by overexpressed ENL mutants. Fluorescence recovery after photobleaching (FRAP) showed ~100% recovery for ENL mutants within 80 seconds (Figures S1A and S1B), and time-lapse imaging showed rapid fusion of these puncta (Figure S1C). In vitro, full-length (FL) ENL mutant proteins exhibited an increased ability to form droplets (Figure 1E). Together, these results suggest that these mutations commonly enhance intrinsic propensity of ENL to form condensates.
Of note, the large droplet-like condensates formed by overexpressed ENL mutants occurred at regions with low DNA density and did not co-localize with H3K27ac, a histone mark recognized by ENL (Figures S1D and S1E) (Erb et al., 2017; Wan et al., 2017). Such chromatin exclusion behavior has been noted for condensates formed under similar expression conditions (Nott et al., 2016; Shin et al., 2018), which calls for caution in linking such condensates to biological function. Thus, we asked whether endogenous levels of ENL mutants can form condensates on chromatin.
Near endogenous levels of ENL mutants form submicron-sized condensates at native genomic targets
We expressed Flag-tagged ENL WT or mutant transgenes at near endogenous levels using doxycycline-mediated induction in HEK293 cells (Figure 1F). We observed submicron-sized nuclear puncta formed by mutant but not WT Flag-ENL (Figures 1G–1I). Unlike puncta formed upon overexpression, these ENL mutant puncta localized at H3K27ac-marked chromatin (Figures 1J and S1F) and recovered slower after photobleaching (Figures S1G and S1H). Next, we asked whether ENL mutants gain the ability to form puncta because of increased binding to histone acetylation. We measured the binding affinity of purified WT and mutant YEATS domains with an H3K27ac peptide and found that mutant YEATS domains exhibited varying degrees of decrease in H3K27ac binding when compared to WT (Figures S1I and S1J). Introducing a point mutation (Y78A) known to disrupt acetyl-binding activity did not abolish puncta formation in ENL T1 or T2, but rather the puncta no longer localized to H3K27ac-marked chromatin (Figures 1J and S1F). These results indicate that while the acetyl-binding activity is not the driver of ENL mutant puncta formation, it is essential for puncta localization to chromatin.
Next, we asked whether ENL mutant puncta localize at specific genomic loci. We focused on HOXA genes, whose expression is most upregulated by ENL mutants in Wilms tumors (Gadd et al., 2015, 2017; Wan et al., 2019). We expressed Halo-ENL-T1 or T2 at near endogenous levels in HEK293 cells and performed Halo staining with concurrent nascent RNA fluorescence in situ hybridization (FISH). We found that ~100% of cells harbored one or more HOXA11 but not GAPDH FISH foci that overlapped with ENL mutant puncta (Figures 1K and 1L, S1K and S1L). The presence of the mutant puncta at the HOXA locus was accompanied by increased ENL chromatin occupancy and expression of HOXA genes compared to WT (Figure 1M and 1N). Furthermore, similar results were observed when we expressed ENL mutants in the Wilms tumor cell line WiT49 (Figures S2A–S2H). Thus, near endogenous levels of ENL mutants form aberrant condensates on chromatin that coincide with increased ENL occupancy and target gene expression.
Overexpressed ENL mutants form large, non-chromatin-associated condensates that fail to activate transcription
Given that the degree of condensate formation by ENL mutants is positively correlated with expression levels (Figures 1B–1D), we explored how increasing levels of ENL mutant impact target gene expression. We first stably expressed Halo-ENL-T1 at near endogenous levels in HEK293 cells followed by transient transfection with mEGFP-ENL-T1, resulting in a cell population with heterogenous expression of mEGFP-ENL-T1 (Figure 2A). Over 80% of cells with non-detectable or low levels of mEGFP-ENL-T1 contained HOXA11 FISH foci that co-localized with Halo-ENL-T1 puncta (Figures 2B–2D). As mEGFP-ENL-T1 level increased, larger condensates consisting of both mEGFP-ENL-T1 and Halo-ENL-T1 appeared, accompanied by a loss of Halo-ENL-T1 puncta at the HOXA locus. Furthermore, the percentage of cells with detectable HOXA11 FISH foci was reduced upon mEGFP-ENL-T1 overexpression (Figures 2E and 2F), indicating a decrease in HOXA11 expression. Consistently, Halo-ENL-T1-induced activation of HOXA genes gradually decreased as mEGFP-ENL-T1 level increased (Figures 2G and 2H). These results indicate that condensate properties and function highly depend on the expression level of ENL mutant and that marked overexpression could result in non-functional condensates (Figure 2I).
Figure 2. Overexpressed ENL mutants form large, non-chromatin-associated condensates that fail to activate transcription.

(A) Schematic for transient transfection of mEGFP-ENL-T1 into HEK293 cells that express Halo-ENL-T1 at near endogenous levels and the following experimental design.
(B) Representative images indicating the co-localization of Halo-ENL-T1 puncta with HOXA11 RNA FISH foci in HEK293 cells in the absence (top) and presence (bottom) of overexpressed mEGFP-ENL-T1.
(C and D) Percentage of cells (n = 20) containing HOXA11 (C) or GAPDH (D) RNA FISH foci overlapping with Halo-ENL-T1 puncta.
(E and F) Percentage of cells with or without detectable HOXA11 (E, n = 37 cells) or GAPDH (F, n = 64 cells) RNA FISH foci.
(G) Western blotting showing the protein levels of transiently expressed mEGFP-ENL-T1 and endogenous ENL in sorted cell populations. Increasing mEGFP-ENL-T1 levels are depicted by wedge.
(H) mRNA expression (normalized to GAPDH) of HOXA genes in sorted HEK293 cell populations.
(I) Schematic showing the formation of functional and non-functional condensates in cells depending on the expression level of oncogenic ENL mutants.
C-F: Chi-square test. H: Data presents Mean ± S.D., two-tailed unpaired Student’s t-test. *** P < 0.001. n.s., no significance.
Insertion and deletion mutations induce consensus structural changes in the ENL YEATS domain
Next, we investigated the molecular basis underlying ENL mutant condensate formation, hypothesizing that the eight YEATS domain mutations might induce key structural changes that confer this ability. We determined the crystal structures of two insertion (T1 and T4) and two deletion (T2 and T3) mutant YEATS domains in complex with an H3K27ac peptide at resolutions of 1.9, 2.3, 2.9, and 2.4 Å, respectively (Figures 3A and 3B). Mutant YEATS domains adopt an eight-stranded β-sandwich fold with the H3K27ac peptide snugly attached to an acidic surface at the top and the flat acetylamide group of K27ac sandwiched by the aromatic residues F59 and Y78 (Figures 3C and 3D), an overall conformation similar to that of the WT YEATS:H3K27ac complex (PDB 5J9S) (Wan et al., 2017), indicating that oncogenic mutations do not significantly alter the β-sheet central core and acetyl-binding pocket of the YEATS domain.
Figure 3. Insertion and deletion mutations induce consensus structural changes in the ENL YEATS domain.

(A) Top, schematic comparing loop L8 and β8 protein sequences in indicated ENL YEATS domains. Key residues are colored in purple. β8 strand is highlighted in cyan box. Right, changes in the number of residues in loop L8 and β8 in each mutant compared to WT.
(B) Overall structures of H3K27ac-bound ENL T1 (blue), T2 (magenta), T3 (cyan), and T4 (salmon) YEATS domain superimposed with WT (gray) YEATS:H3K27ac complex (PDB: 5J9S). H3K27ac is depicted as stick, and differences in loop L8 and β8 region are boxed. H3K27ac peptide in WT, T2, T3, T4 structures is in yellow; H3K27ac peptide in T1 structure is in blue.
(C) Details of H3K27ac-binding pocket of T1 YEATS domain.
(D) Comparison of key residues in WT, T1, T2, T3, and T4 YEATS domains that are involved in H3K27ac binding showing nearly identical conformations.
(E-H) Detailed analysis of local conformational changes between WT and mutant YEATS domains. Left, enlarged view of alignment of loop L8 and β8 region of WT and indicated mutant YEATS domains. Right, key residues forming extra hydrogen bonds and H3 peptides are highlighted.
(I) Close-up view of loop L8 in ENL WT (left and middle, PDB: 5J9S) and T3 (right) YEATS structures. Loop L8: green in WT, yellow in T3. P112P113 (PP) bulge is highlighted. Hydrogen bonds are shown as red dashed lines, water molecules as red spheres, and key residues as sticks.
(J) Crystal packing analysis of T3 YEATS structure revealing a YEATS-YEATS interaction mode that is mediated by β8-β8 association. Key residues and hydrogen bonds are shown in the red box.
(K) Quantification of in vitro droplet formation with purified ENL WT and T1 YEATS domains (60 μM and 240 μM).
(L) Schematic showing the use of LacI-LacO assay to test YEATS domain association in cells. (M and N) Representative images and quantification showing mCherry-ENL(YD) enrichment at the LacO array. M, yellow squares indicate the LacO array. N, enrichment of mCherry above 1 suggests YEATS domain self-association. n = 8, 14, 9, 36, 42, 41 cells from left to right.
K and N: Data represent Mean ± S.D., two-tailed unpaired Student’s t-test; *** P < 0.001. See also Figure S3 and Table 1.
However, we found substantial alterations in the β8 strand and loop L8, regions in which all eight mutations occur. The WT β8 strand starts after residue P113, and in T1 and T4, due to the insertion of three amino acids, H116 aligns with WT P113, and thus β8 strand in T1/T4 begins with H116 followed by L117 and the inserted residues (Figures 3E and 3F). Therefore, the insertion increases the β8 length in T1/T4 by one residue (+1) when compared to WT. On the other hand, residues V114 and N115 in T1/T4 reside in the loop L8, rendering L8 longer than that of WT by two residues (+2). The longer L8 in T1/T4 becomes more open and folds away from the β-sheet core (Figures 3E and 3F). We previously reported the role of loop L8 in AF9 (ENL paralog) in binding histone H3 residues at the N-terminus of the acetyl-lysine (Li et al., 2014). Structural overlay of T1/T4:H3K27ac and AF9:H3K9ac complexes (PDB: 4TMP) revealed that the longer loop L8 in T1/T4 could exert steric clash with the H3 N-terminal region (Figure S3A). In support, molecular dynamics simulations revealed that T1 loop L8 exhibits more contacts with the N-terminal region of H3 than WT (Figure S3B). The predicted steric clash is consistent with the observations that T1 YEATS bind to the H3(17–28) K27ac peptide weaker than the H3(24–27) K27ac peptide harboring a shorter N-terminal region, whereas WT YEATS shows comparable binding to both peptides (Figures S3C and S3D). Additionally, a hydrogen bond formed by β1 strand’s L19 residue and loop L8’s N115 residue in T1/T4 was absent in WT YEATS and may help stabilize the local loop L8 conformation in mutants (Figures 3E and 3F). We modelled T7 and T8 YEATS structures based on that of T1/T4 and observed similarly extended β8 strand and loop L8 (Figure S3E), suggesting that these structural changes are induced by all insertion mutations.
In T2 and T3, the substitution of residues NPP to a single leucine (L111 in T2) or lysine (K111 in T3) also resulted in β8 extension (Figures 3G and 3H). Prolines contain a rigid ring structure and often function as secondary structure breakers (Imai and Mitaku, 2005). Upon P112P113 removal, the resultant adjacent residues in T2 (N111L112) and T3 (K111V112) contribute to an extended (+1) β8 formation. Unlike the insertion mutants, loop L8 in T2/T3 was shortened by 3 residues (−3) when compared with WT YEATS, leading to a more rigid loop that is packed closer to the β-sheet core. The shorter loop L8 in T2/T3 exhibits no hindrance to H3 N-terminal end when aligned with the AF9:H3K9ac structure (Figure S3F), consistent with the minimal effect of deletion mutations on histone acetylation binding (Figures S1J, S3C, S3D). Compared with WT YEATS, there is an extra hydrogen bond formed by β1 strand’s L19 and loop L8’s G110 in T2/T3, which may help stabilize the local loop L8 conformation in T2/T3 (Figures 3G and 3H). Notably, deletion of the consecutive prolines is a common feature of all deletion mutants (Figure 1A), and structural modeling of T5 and T6 YEATS domains revealed changes consistent with T2/T3 (Figure S3G).
Collectively, our structural analyses reveal shared and distinct structural changes in the ENL YEATS domain caused by insertion and deletion mutations. Among them, β8 extension is a key consensus structural change. Moreover, in WT YEATS, the consecutive proline is stabilized by multiple hydrogen bonds and acts as a blocker bulge at one end of β8 (Figure 3I), and such a “PP-bulge” is shifted (insertion) or removed (deletion) away in oncogenic ENL mutants, which likely facilitates YEATS-mediated interactions that may benefit from the β8 extension. In fact, as exemplified by crystal packing analysis of T3 structure, the extended β8 mediates “YEATS-YEATS” association in the crystalline state that may resemble a crowded environment under LLPS (Figures 3J and S3H), and such an interaction mode is incompatible with the existence of “PP-bulge” due to steric clashes (Figure S3I).
To test the effect of oncogenic mutations on YEATS domain self-association, we performed analytical ultracentrifugation with purified WT and T1 YEATS proteins and found that they exist predominantly as monomers, indicating that the T1 mutation does not promote stable dimerization/oligomerization of the YEATS domains in vitro (Figure S3J). However, the T1 mutation enhanced YEATS domain’s ability to form droplets at higher concentrations (Figure 3K), suggesting a role of oncogenic mutations in promoting transient and weak YEATS-YEATS interactions under crowding conditions. To measure YEATS domain self-association in cells, we co-expressed EYFP-labeled ENL YEATS fused with LacI (EYFP-YD-LacI) and mCherry-labeled ENL YEATS (mCherry-YD) into U2OS cells that contain a synthetic Lac operator (LacO) array in the genome, and then we measured the enrichment of mCherry-YD at the LacO array (Figure 3L) (Chong et al., 2018; Janicki et al., 2004). The LacO array recruited EYFP-YD-LacI molecules through LacI-LacO interactions, forming bright foci in the nucleus (Figure 3M). We observed an increased enrichment of T1 and T2 mCherry-YD at the LacO array when compared with WT (Figure 3N). Together, our data reveal structural changes induced by ENL mutations and suggest their potential role in promoting weak homotypic interactions of YEATS domains, although we do not rule out that these structural changes could also engage in other modes of molecular interactions.
Reverting ENL mutation-induced structural changes abolishes condensate formation and function
We next wanted to test the importance of oncogenic mutation-induced β8 extension in ENL condensate formation. In T1 YEATS, the β8 strand starts with residue H116 and is longer than WT β8 by one residue (+1). In WT YEATS, the corresponding position is a proline (P113), which is located at the juncture of loop L8 and the β8 strand (Figure 3E). As prolines typically function as secondary structure breakers, we predicted that mutating H116 in T1 to a proline (H116P) would shorten T1 β8 to the same length as WT β8 (Figure 4A). The crystal structure of T1(H116P) YEATS in complex with an H3K27ac peptide (1.8 Å) revealed that it adopted an overall conformation similar to that of T1 (Figure 4B). As predicted, β8 in T1(H116P) starts with L117, making it the same length as WT β8. Due to a shortened β8, the adjacent loop L8 in T1(H116P) is longer than T1 and WT by one (+1) and three (+3) residues, respectively (Figures 4C and 4D). H116P substitution abolished T1-induced increase in YEATS self-association in vitro and in cells (Figures S4A–S4C) without significant impact on its acetyl-binding affinity or protein stability (Figure S4D and S4E). Furthermore, H116P substitution impaired ENL T1’s ability to form droplets in vitro and in cells (Figures S4F–S4I). Consistently, T1-induced puncta formation and gain-of-function on chromatin was abolished by H116P substitution (Figures 4E–4I, S4J and S4K).
Figure 4. Reverting ENL mutation-induced structural changes abolishes condensate formation and function.

(A) Top, schematic comparing loop L8 and β8 protein sequences in indicated ENL YEATS domains. Key residues are colored in purple. β8 strand is highlighted in cyan box. Right, changes in the number of residues in loop L8 and β8 in each mutant when compared with WT.
(B) Overall structures of H3K27ac-bound T1 (blue) and T1(H116P) (green) YEATS domain superimposed with WT (gray) YEATS:H3K27ac complex. H3K27ac is depicted as stick and differences in loop L8 and β8 region are boxed.
(C and D) Detailed analysis of sequences (left) and local structural changes (right) between T1 and T1(H116P) (C) or WT and T1(H116P) (D).
(E-G, J-L, O-Q) Flag IF staining (E, J, O) and quantification in HEK293 cells expressing near endogenous levels of indicated Flag-ENL. F, K, P: percentage of nuclei with and without Flag-ENL puncta. G, L, Q: number of Flag-ENL puncta in each nucleus. Center lines indicate median and box limits are set to the 25th and 75th percentiles. Each dot indicates one cell (n at the range of 34~48).
(H, I, M, N, R, S) ChIP-qPCR analysis of Flag-ENL occupancy at the HOXA locus (H, M, R) and mRNA expression (normalized to GAPDH) of HOXA genes (I, N, S) in HEK293 cells. Data presents Mean ± S.D..
F, K, P: Chi-square test; G-I, L-N, Q-S: Two-tailed unpaired Student’s t-test; *** P < 0.001. See also Figure S4 and Table 1.
The “PP” deletion in T2 and T3 allows the residue at position 111, which resides in loop L8 in WT, to become part of the β8 strand. Structural modeling predicted that substituting residue 111 to a proline would abolish β8 extension induced by T2 and T3 mutations (Figure S4L). We found that N111P substitution reduced T2 YEATS self-association in the LacO assay (Figures S4M and S4N) and compromised ENL T2’s ability to form droplets in vitro and in cells (Figures S4F, S4G, S4O and S4P). At near endogenous levels, ENL T2(N111P) exhibited reduced puncta formation, chromatin occupancy, and target gene expression (Figures 4J–4N, S4Q, and S4R) when compared with ENL T2. Similar defects were observed with T3(K111P) (Figures 4O–4S and S4S–S4V). Collectively, our results reveal a critical role of mutation-induced structural changes in ENL condensate formation and function.
Multiple regions of ENL mutants are required for condensate formation and function
In addition to its N-terminal YEATS domain, ENL also contains a central IDR as well as a C-terminal ANC1 homology domain (AHD) which mediates interactions with other complexes. To investigate the contribution of these regions to condensate formation (Figure 5A), we transiently expressed mCherry-ENL deletion variants into HEK293 cells and found that deletion of the YD, IDR, or AHD in ENL T1 abolished its condensate formation. IDR, AHD, or IDR-AHD also failed to form condensates upon overexpression (Figures 5B–5D). We next compared the incorporation of mCherry-ENL deletion variants into condensates formed by EGFP-ENL-T1 (Figure S5A). Deleting YD, IDR, or AHD individually in mCherry-ENL-T1 decreased its incorporation into EGFP-ENL-T1 condensates (Figures S5B and S5C). Moreover, the presence of the T1 mutation in different mCherry-ENL backbones all resulted in increased incorporation into EGFP-ENL-T1 condensates when compared with the WT counterparts (Figures S5B and S5D). To test the requirement of individual domains in ENL mutants’ function, we expressed different deletion variants at near endogenous levels (Figures 5A, S5E and S5F) and found that all deletions compromised T1-induced condensate formation, increased chromatin occupancy, and gene activation (Figures 5E–5I). Largely similar results were observed for ENL T2 (Figures 5J–5N and S5G–S5J). Although T2(ΔIDR) retained some ability to form nuclear puncta, the puncta formed were fewer in number (Figure 5L), smaller in size (Figure S5I), and had a lower partition ratio (Figure S5J) compared with those formed by T2 (FL). Together, our data show that multivalent interactions contributed by multiple regions in oncogenic ENL mutants are collectively required for condensate formation and function.
Figure 5. Multiple regions of ENL mutants are required for condensate formation and function.

(A) Schematic showing different ENL variants and their ability to form puncta under overexpression (O/E) and form puncta and exhibit gain-of-function (GOF) on chromatin at endogenous levels.
(B-D) Representative images (B) and fraction of in-puncta fluorescence intensity quantification (C and D) of HEK293 cells transiently transfected with indicated mCherry-ENL variants. B, all cells shown had similar mCherry-ENL intensities. C and D, each dot indicates one cell (n > 20).
(E and J) Flag IF staining in HEK293 cells expressing endogenous levels of indicated Flag-ENL. J, the yellow arrows show smaller nuclear puncta formed by T2(ΔIDR2).
(F, G, K, L) Percentage of nuclei with and without Flag-ENL puncta (F and K) and the number of Flag-ENL puncta in each nucleus (G and L) in HEK293 cells. Each dot indicates one cell (n at the range of 30~60). Center lines indicate median and box limits are set to the 25th and 75th percentiles. (H, I, M, N) ChIP-qPCR analysis of Flag-ENL occupancy at HOXA locus (H and M) and mRNA expression (normalized to GAPDH) of HOXA genes (I and N) in HEK293 cells with endogenous levels of indicated Flag-ENL. Data shown are Mean ± S.D..
F and K, Chi-square test. G-I, L-N: Two-tailed unpaired Student’s t-test. *** P < 0.001, ** P < 0.01, *P < 0.05.
See also Figure S5.
Two oppositely charged IDRs play distinct yet complementary roles in the initiation and growth of ENL mutant condensates
We next investigated the molecular mechanism underlying the condensate formation defect upon ENL IDR deletion. The ENL IDR has non-uniform charge distribution, in which the first segment (IDR1) is highly positively charged and the second segment (IDR2) is highly negatively charged (Figures 6A and 6B). Moreover, polar amino acids, most notably serine, are enriched in IDR1 and IDR2 (Figure 6C). We hypothesized that these sequence features could play a regulatory role in condensate formation. To test this, we compared condensate formation, chromatin occupancy, and target gene activation in cells expressing ENL T1 with a series of alterations in IDR1 or IDR2.
Figure 6. Two oppositely charged IDRs play distinct yet complementary roles in the initiation and growth of ENL mutant condensates.

(A) Sequence features of human ENL protein. Top, values > 0.5 in IUPred prediction indicate regions of protein disorder. Middle, blue and red colors in NCPR (net charge per residue) indicate positively or negatively charged residues, respectively. Bottom, sequence conservation of 79 placental mammal ENL orthologs.
(B) Net charge of each residue was analyzed across 79 placental mammal orthologous ENL sequences and binned for ENL (FL), IDR1, or IDR2.
(C) Heatmap showing the amino acid composition of ENL IDRs. The percentage of each residue in IDR1(201–343), IDR2 (344–450), or IDR (201–450) was shown in the table (right).
(D and K) Schematic showing different ENL variants and their ability to form puncta under overexpression (O/E) and form puncta and exhibit gain-of-function on chromatin at endogenous levels.
(E and L) Schematic showing the impacts of disrupting IDR1 (E) or IDR2 (L) on condensate formation and target gene activation.
(F-H) Flag IF staining (F) and quantification (G and H) in HEK293 cells expressing near endogenous levels of indicated Flag-ENL. G, percentage of nuclei with and without Flag-ENL puncta. H, the number of Flag-ENL puncta in each nucleus. Each dot indicates one cell (n at the range of 37~87). Center lines indicate median values and box limits are set to the 25th and 75th percentiles.
(I, J, P, Q) ChIP-qPCR analysis of Flag-ENL occupancy at the HOXA locus (I and P) and mRNA expression (normalized to GAPDH) of HOXA genes (J and Q) in HEK293 cells with near endogenous levels of indicated Flag-ENL. Data shown are Mean ± S.D..
(M-O) Flag IF staining (M) and quantification (N and O) in HEK293 cells expressing near endogenous levels of indicated Flag-ENL. M, the yellow arrow indicates smaller nuclear puncta formed by IDR2 mutants. N, puncta size (n at the range of 109~436). O, the ratio of in-puncta/out-of-puncta intensity (n at the range of 18~60). Center lines indicate median and box limits are set to the 25th and 75th percentiles.
G: Chi-square test. H-J, N-Q: Two-tailed unpaired Student’s t-test. *** P < 0.001, ** P < 0.01, * P < 0.05. n.s., no significance.
See also Figures S6 and S7.
We first focused on the IDR1 (Figures 6D and 6E). Deletion of IDR1 in ENL T1 decreased its ability to form droplet-like puncta upon overexpression (Figures S6A and S6B). Although T1(ΔIDR1) retained some ability to form nuclear puncta when expressed at near endogenous levels, the puncta number was decreased (Figures 6F–6H, S6C and S6D). These puncta, however, are similar in size and partition ratio to those of T1(FL) (Figure S6E and S6F), suggesting that IDR1 primarily contributes to the initiation of ENL mutant condensates. Consequently, T1-induced increase in chromatin occupancy and gene activation were compromised upon IDR1 deletion (Figures 6I and 6J). Despite low sequence conservation in ENL IDR1, its charge property is well conserved (Figures 6A and 6B). Thus, we speculated that the positive charge density rather than the motif encoded in the primary amino acid sequence confers the role of IDR1. We scrambled the IDR1 sequence and found that scrambled variants (IDR1_Scr1/2) behave indistinguishably from ENL T1 in condensate formation, chromatin occupancy, and gene activation (Figures 6F–6J and S6A–S6F). We also generated a mutant in which a subset of serines, the most over-represented amino acid in IDR1, were mutated to alanines (IDR1_S/A21) (Figures 6C and 6D). Like the scrambled mutants, the serine mutant retained T1-induced condensate formation and function (Figures 6F–6J and S6G–S6L). To directly test the importance of the positive charge density, we generated a mutant in which 21 out of 29 lysine residues in IDR1 (excluding lysines within the NLS in IDR1) were substituted with charge-neutral glutamine (K/Q21) (Figure 6D). Unlike the scrambled and serine mutants, K/Q21 mutation phenocopied the effects of IDR1 deletion (Figures 6F–6J and S6A–S6F). These results indicate that the IDR1 contributes to the initiation of ENL mutant condensates mainly through its positive charge density.
To investigate the mechanism by which the IDR2 contributes to ENL mutant condensate formation, we tested a series of IDR2 variants in ENL T1 (Figure 6K and 6L). First, deletion of IDR2 compromised ENL T1’s ability to form droplet-like puncta upon transient transfection (Figures S7A and S7B). While near endogenous levels of T1(ΔIDR2) formed a similar number of puncta as T1 (FL), these puncta were smaller in size and had a lower partition ratio (Figures 6M–6O and S7C–S7F), suggesting that IDR2 regulates the growth of ENL mutant condensates (Figure 6L). While IDR2 deletion only resulted in a slight decrease in ENL T1’s chromatin occupancy (Figure 6P), it induced an evident decrease in target gene expression (Figure 6Q), indicating that changes in local ENL concentration within the puncta can impact transcriptional output beyond regulating the amount of chromatin bound ENL molecules. To assess the role of negative charges in IDR2, we generated two mutants: IDR2_ED/A29, in which all negatively charged residues (17 glutamic acid and 12 aspartic acid) were substituted with alanines, and IDR2_E/Q17, in which all glutamic acid residues were substituted with charge-neutral glutamines. These two alterations recapitulated IDR2 deletion with respect to condensate formation, chromatin occupancy, and gene activation (Figures 6M–6Q and S7A–S7F). IDR2 is highly enriched in serine residues and contains a conserved polyserine (polyS) region. To test the importance of serine, we generated two mutants: one in which all 33 serine residues were substituted with alanines (S/A33) and another where the polyS region was deleted (Figure 6K). These IDR2 mutants had a weaker ability to form droplet-like puncta (Figures S7G and S7H). When expressed at near endogenous levels (Figures S7D and S7I), the serine mutants phenocopied IDR2 deletion in their defects in condensate formation and function (Figures 6M–6Q, Figures S7E and S7F). These data suggest that negative charge density and serine-rich sequences in IDR2 regulate the local protein concentration within ENL condensates. Collectively, our results show that two oppositely charged IDRs in ENL mutants play distinct yet complementary roles in the initiation and growth of condensates to drive target gene activation.
Interactions with extrinsic factors are critical for ENL condensate formation and function
We hypothesized that ENL mutant condensates enrich a high local concentration of co-factors. Indeed, imaging analysis showed that several well-known ENL-associated proteins, including AFF4 (SEC subunit), CDK9 and Cyclin T1 (P-TEFb subunits), and DOT1L, were significantly enriched in ENL mutant condensates (Figure 7A). Previously, we showed that ENL mutant expression led to increased Pol II S2 phosphorylation (Pol II S2P) at key target genes, indicating enhanced elongation activity (Figure 7B) (Wan et al., 2019). To ask if condensate formation is required for mutation-induced transcription elongation, we compared Pol II S2P occupancy at HOXA genes in cells expressing ENL T1 variants with varying condensate-forming abilities. H116P substitution or mutational disruption of IDR1/IDR2 reduced T1-induced Pol II S2P at HOXA genes, and the degree of reduction correlated with the defect in condensate formation (Figure 7B). Hence, ENL mutant condensates represent “pathogenic elongation condensates” that harbor aberrantly high elongation activity.
Figure 7. Interactions with extrinsic factors are critical for ENL mutant condensate formation and function.

(A) IF imaging (left) and quantification (right) showing enrichment of endogenous Halo-AFF4, Halo-CDK9, Cyclin T1, and DOT1L in Flag-ENL-T1 puncta over random sites. n = 185, 377, 62, 325 Flag-ENL-T1 puncta/random sites for AFF4, CDK9, Cyclin T1, DOT1L, respectively. (B) ChIP-qPCR comparing Pol II S2P occupancy at the HOXA locus in HEK293 cells expressing near endogenous levels of indicated Flag-ENL. Data shown are Mean ± S.D..
(C) Schematic showing different ENL variants and their ability to form puncta under overexpression (O/E) and form puncta and exhibit gain-of-function (GOF) on chromatin at endogenous levels.
(D) Immunoprecipitation followed by western blotting showing interactions between indicated Flag-ENL variants and known ENL interaction partners.
(E-I) Flag IF staining (E) and quantification (F-I) in HEK293 cells expressing near endogenous levels of indicated Flag-ENL. E, yellow arrows indicate smaller puncta formed by T1(M1) and T1(M2). F, percentage of nuclei with and without Flag-ENL puncta; G, the number of Flag-ENL puncta in each nucleus. F and G: cell number at the range of 40~55. Center lines indicate median and box limits are set to the 25th and 75th percentiles. H, puncta size (n at the range of 42~273). I, the in puncta/out-of-puncta intensity ratio (n at the range of 20~28).
(J and K) ChIP-qPCR analysis of Flag-ENL occupancy at the HOXA locus (J) and mRNA expression analysis (normalized to GAPDH) of HOXA genes (K) in HEK293 cells with near endogenous levels of indicated Flag-ENL. Data shown are Mean ± S.D..
F: Chi-square test; B, G-K: two-tailed unpaired Student’s t-test; *** P < 0.001, ** P < 0.01. n.s., no significance.
See also Figure S7.
To ask whether interactions of ENL mutants with partners contribute to condensate formation, we generated three ENL T1 variants in which key residues predicted to mediate ENL interaction with SEC/P-TEFb and DOT1L were mutated (Figures 7C and S7J) (Leach et al., 2013). Co-immunoprecipitation confirmed that each of the three AHD mutations (M1/M2/M3), as well as AHD deletion, disrupted interactions of ENL T1 with SEC/P-TEFb (AFF4, ELL2, CDK9) and DOT1L (Figure 7D). Unlike AHD deletion (Figures 5B–5D, 7C), AHD mutations had little impact on ENL-T1’s ability to form condensates under overexpression conditions in cells (Figures S7K and S7L) or in vitro (Figure S7M), suggesting that AHD could contribute to ENL condensation independent of its role in interacting with other proteins and that these interactions are not essential for condensate formation when ENL mutant proteins are abundant. And yet, at near endogenous levels (Figures S7N and S7O), T1 AHD mutants exhibited substantial defects in puncta formation, chromatin occupancy, and target gene activation when compared with ENL T1 (Figures 7E–7K). These data reveal that heterotypic interactions with extrinsic factors are important for oncogenic ENL mutants to form condensates on chromatin particularly under physiologically relevant expression conditions.
DISCUSSION
The functional significance of transcriptional condensates in normal and diseased states remains largely elusive, partially due to poor understanding of how condensates form at native genomic targets and hence lack of strategies to specifically modulate their formation for functional interrogation. The high specificity and gain-of-function nature of oncogenic ENL mutations make them a powerful model for addressing these outstanding questions. We show that a series of ENL mutants form submicron-sized condensates at select genomic targets under physiologically relevant conditions. Moreover, our extensive mutagenesis study strongly supports a functional requirement of these condensates for ENL mutation-driven oncogenic gene activation. Thus, our work offers crucial experimental evidence linking condensate formation and transcriptional output and helps establish condensate dysregulation as a mechanism underlying pathogenic gene expression in cancer and potentially other diseases.
Our study reveals that the properties and function of condensates are highly dependent on the expression level of oncogenic ENL mutants. At endogenous levels, ENL mutants form submicron-sized condensates on chromatin, which exhibit partial recovery in FRAP experiments and correlate with hyper-activation of target genes. Upon overexpression, ENL mutants form larger and highly dynamic droplet-like condensates away from chromatin, possibly through LLPS upon crossing the critical saturation concentration. These large condensates prevent the formation of small condensates at target genes and fail to activate transcription (Figure 2I). Although the biophysical mechanism governing such a transition remains unclear (A and Weber, 2019), a recent study described a prewetting transition of pioneer transcription factors on DNA (Morin et al., 2022), a principle that might help explain the differences between ENL mutant condensates formed under different expression conditions. We also show that an ability to form condensates under overexpression conditions does not guarantee an ability to do so in an endogenous context and that interactions with extrinsic factors play a particularly important role in condensate formation by endogenous level of ENL mutants (Figure 7C). This is consistent with the notion that heterotypic interactions dominate endogenous condensates, and the formation and properties of heterotypic condensates can be modulated by their components (Choi et al., 2019; Mittag and Pappu, 2022; Riback et al., 2019; Wei et al., 2020). We speculate that the local chromatin environment, such as histone modifications, nucleic acids, and chromatin-associated proteins, could all play a role in regulating the formation and properties of ENL mutant condensates at specific genomic loci. Our observation that increasing expression of ENL mutant beyond endogenous levels can suppress its gene activation activity is reminiscent of a recent study which showed that ectopically expressed EWS IDR can repress EWS-FLI driven transcription (Chong et al., 2022). These results together indicate that finely controlled levels of multivalent interactions are needed for the formation of functional condensates, underscoring a critical need for the field to investigate condensate formation and function in physiologically relevant contexts. These results also reveal a dosesensitive function of oncogenic proteins which could open new therapeutic avenues.
While IDRs have emerged in recent years as key regulators of biomolecular condensates (Chong and Mir, 2020; Uversky, 2017), the roles of structured protein domains in condensate regulation remain less clear. ENL oncogenic mutations represent a prominent example in which disease-causing mutations in a structured protein domain can trigger condensate formation to aberrantly augment the protein’s biological function. During the course of our study, Ni et al reported Apo structures for two ENL YEATS mutants (T1 and T3) (Ni et al., 2021) and speculated changes in the loop L8 as a key mechanism underlying ENL mutant function. However, structural changes in the loop L8 are largely opposite between insertion and deletion mutants, unlikely explaining the highly similar function across oncogenic ENL mutants. Instead, our structural studies reveal β8 extension and PP-bulge elimination as key consensus structural changes induced by both insertion and deletion ENL mutations. We propose that without the PP-bulge-induced restriction, the resultant β8 extension in ENL oncogenic mutants could enable extra homotypic and possibly heterotypic interactions to increase its multivalence for the benefit of condensate formation. Most importantly, reverting β-strand extension by single point mutagenesis is sufficient to suppress ENL mutant condensate formation and gene activation. These results present a pioneering example in which mutation-induced structural changes can trigger prominent changes in condensation behaviors and offer a model for understanding the role of structural domains in condensate regulation. In addition to the YEATS domain, we show that two ENL IDRs of opposing charges play distinct yet complementary roles in regulating the initiation and growth of ENL condensates, raising the question as to whether other condensate-forming proteins may also evolve to use distinct sequence features to regulate different condensate properties. Detailed investigations into the aforementioned interactions will further illuminate how ENL condensates form and function and inform strategies to perturb these condensates.
To date, the involvement of condensates during transcriptional elongation has remained elusive. The cyclin T1 subunit in P-TEFb has been shown to use its condensation capacity to target Pol II for hyperphosphorylation and efficient elongation (Lu et al., 2018), and yet phosphorylation of Pol II has been shown to disrupt condensate formation (Boehning et al., 2018; Kwon et al., 2013). Furthermore, it has been proposed that low levels of RNA at the early stage of transcription stimulate condensate formation, while high levels of RNA resulting from productive elongation favor condensate dissolution (Henninger et al., 2021). We show that ENL mutant condensates enrich an aberrantly high number of elongation factors and exhibit increased elongation activity, and such an activity is dependent on condensate formation. Thus, ENL mutant condensates represent one of the first examples of “pathogenic elongation condensates”, which likely harbor properties and regulatory mechanisms distinct from those of normal elongation machinery. Future work into ENL mutant condensates and potentially other disease-associated transcriptional condensates would bring fundamental insights into gene regulation and dysregulation that could potentially be exploited for therapeutic interventions.
Limitations of the study
While we reveal that oncogenic mutation-induced structural changes in the ENL YEATS domain promotes weak YEATS-YEATS interactions, they may also engage in other modes of molecular interactions (e.g., heterotypic interactions with other factors) to promote condensate formation, a hypothesis that awaits further investigations. Our study lacks details on the biophysical process by which ENL mutant condensates form, how they nucleate at specific genomic loci, and how the formation of condensates influence the molecular dynamics of their constituents to alter transcription. Future studies employing single-molecule imaging and live imaging of transcription will address these outstanding questions. In addition, our discovery of concentration-dependent formation of functional and non-functional condensates begs the questions as to how this transition occurs at the molecular and biophysical levels. Finally, while the formation of condensates clearly underlies ENL mutants’ gene activation activity in the cellular system that we tested, how such a property drives tumorigenesis in clinically relevant in vivo models remains to be determined.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Liling Wan (Liling.Wan@Pennmedicine.upenn.edu).
Materials availability
All materials generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
Original western blot and microscopy images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-FLAG | Sigma-Aldrich | Cat# F1804-1MG |
| Rabbit anti-Histone H3 (acetyl K27) | Abcam | Cat# Ab4729 |
| Mouse anti-Cyclin T1 | Santa Cruz | Cat# sc-271348 |
| Rabbit anti-DOT1L (D1W4Z) | Cell Signaling Technology | Cat# 77087s |
| Rabbit anti-ENL | EMD Millipore | Cat# ABE2596-100UG |
| Mouse anti-β-Actin Antibody (AC-15) | Novus Biologicals | Cat# NB600-501 |
| Mouse anti-AFF4 | Santa Cruz | Cat# sc-390130 |
| Mouse anti-ELL2 | Santa Cruz | Cat# sc-376611 |
| Rabbit anti-Pol II S2P | Abcam | Cat# Ab5095 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074s |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7076s |
| goat anti-Rabbit IgG (H+L) Alexa Fluor® 488 | Invitrogen | Cat# A32732 |
| goat anti-Mouse IgG (H+L) Alexa Fluor® 488 | Invitrogen | Cat# A32723 |
| goat anti-Rabbit IgG (H+L) Alexa Fluor® 568 | Invitrogen | Cat# A11011 |
| goat anti-Mouse IgG (H+L) Alexa Fluor® 568 | Invitrogen | Cat# A11031 |
| Bacterial and virus strains | ||
| NEB® 5-alpha Competent E. coli (High Efficiency) | NEB | Cat# C2987I |
| NEB® Stable Competent E. coli (High Efficiency) | NEB | Cat# C3040I |
| Escherichia coli (E. coli) strain BL21 (DE3) | Novagen | Cat# 69450 |
| Chemicals, peptides, and recombinant proteins | ||
| EMEM | Thermo Fisher Scientific |
Cat# MT10009CV |
| DMEM/F12 | Sigma | Cat# D8062 |
| DMEM | Thermo Fisher Scientific |
Cat# MT10013CV |
| low-glucose DMEM | Thermo Fisher Scientific |
Cat# 10567014 |
| OPTI-MEM | Gibco | Cat# 31985062 |
| Penicillin-streptomycin | Thermo Fisher Scientific |
Cat# 15-140-122 |
| FBS | HyClone | Cat# SH30910.03 |
| Lipofectamine 3000 | Invitrogen | Cat# L3000008 |
| Polyethylene Glycol 8000 (PEG) (1kg) | Fisher Scientific | Cat# BP233-1 |
| PEI MAX | Fisher Scientific | Cat# NC1038561 |
| Polybrene | EMD Millipore | Cat# TR-1003-G |
| Doxycycline | Sigma-Aldrich | Cat# D9891-1G |
| SYBR Green PCR Master Mix | Fisher Scientific | Cat# A25778 |
| Anti-Flag M2-conjugated agarose beads | Sigma-Aldrich | Cat# A2220-1ML |
| Ribonucleoside vanadyl complex (RVC) | Sigma-Aldrich | Cat# R3380-5ML |
| DPBS | Mediatech | Cat# MT21030CM |
| Janelia Fluor® 549 (JF549) | Laboratory of Mustafa A. Mir | N/A |
| Paraformaldehyde (PFA) | Sigma-Aldrich | Cat# 158127-500G |
| Triton X-100 | Fisher Scientific | Cat# BP151500 |
| Tween-20 | Fisher Scientific | Cat# BP337500 |
| Stellaris® FISH Probes, Human GAPDH_intron with Quasar® 670 Dye | Biosearch Technologies |
Cat# ISMF-2151-5 |
| Stellaris 5 × RNA FISH Wash buffer A | Biosearch Technologies |
Cat# SMF-WA1-60 |
| Stellaris RNA FISH hybridization buffer | Biosearch Technologies |
Cat# SMF-HB1-10 |
| Stellaris RNA FISH Wash Buffer B | Biosearch Technologies |
Cat# SMF-WB1-20 |
| Deionized Formamide | VWR | Cat# EM4650 |
| Fluoroshield™ with DAPI, histology mounting medium | Sigma-Aldrich | Cat# F6057-20ML |
| Goat serum | Cell Signaling Technology | Cat# 5425s |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat# A7906-10G |
| 16% Paraformaldehyde Aqueous Solution, EM Grade | Electron microscope scientific | Cat# 15710 |
| Tris-HCl | Fisher Scientific | Cat# BP1531 |
| NaDOC | Sigma-Aldrich | Cat# 35915 |
| N-Lauroylsarcosine sodium salt | Sigma-Aldrich | Cat# L9150-50G |
| DTT | Sigma-Aldrich | Cat# D9163-5G |
| cOmplete™ EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# 11836170001 |
| Protein A Dynabeads | Fisher Scientific | Cat# 10-002-D |
| EDTA | Sigma-Aldrich | Cat# E5134-500G |
| SDS | Thermo Fisher Scientific |
Cat# BP8200500 |
| Lithium chloride (LiCl) | Sigma-Aldrich | Cat# 213233-500G |
| NaCl | Fisher Scientific | Cat# BP358-1 |
| Glycerol | Fisher Scientific | Cat# BP2291 |
| 2-Mercaptoethanol | Thermo Fisher Scientific |
Cat# 21985023 |
| Glycine | Fisher Scientific | Cat# BP3815 |
| Tris Base | Fisher Scientific | Cat# BP152-1 |
| Pierce ECL Western Blotting Substrate | Pierce Endogen | Cat# 32106 |
| Blotting-Grade Blocker | Bio-Rad | Cat# 1706404 |
| MgCl | Sigma-Aldrich | Cat# M0250-500G |
| NP-40 | BioVision | Cat# 2111-100 |
| Protein K | Sigma-Aldrich | Cat# 03115879001 |
| RNase A | Fisher Scientific | Cat# 9001-99-4 |
| RNase inhibitor | Thermo Fisher Scientific |
Cat# N8080119 |
| LB Broth | Thermo Fisher Scientific |
Cat# BP1426-2 |
| Ampicillin | Fisher Scientific | Cat# BP176025 |
| Kanamycin | Fisher Scientific | Cat# BP906-5 |
| HEPES-Na | Sigma-Aldrich | Cat# H7006 |
| Polyethylene Glycol 4000 | Thermo Fisher Scientific |
Cat# A1615130 |
| sodium citrate | Sigma-Aldrich | Cat# 1613859 |
| 2-propanol | Sigma-Aldrich | Cat# I9516 |
| sodium formate | Sigma-Aldrich | Cat# 247596 |
| sodium acetate trihydrate | Sigma-Aldrich | Cat# 236500 |
| PowerUp SYBR Green Master Mix | Applied Biosystems Inc. |
Cat# A25742 |
| Alexa 488 Fluor™ 488 NHS | Invitrogen | Cat# A20000 |
| Ni IDA Beads 6FF | Smart-Lifesciences | Cat# SA00501L |
| Strep-4FF beads | Smart-Lifesciences | Cat# SA053100 |
| Alexa Fluor™ 488 | Thermo Fisher Scientific |
Cat# A30005 |
| Alexa Fluor™ 568 | Thermo Fisher Scientific |
Cat# A20003 |
| Isopropyl β-D-thiogalactoside | Sigma-Aldrich | Cat# 10724815001 |
| imidazole | Sigma-Aldrich | Cat# 12399 |
| phenylmethylsulfonyl fluoride (PMSF) | VWR Chemicals | Cat# 329-98-6 |
| Desthiobiotin | IBA Life Sciences | Cat# 2-1000-002 |
| SYPRO Orange stain | Thermo Fisher Scientific |
Cat# S6650 |
| H317-28K27ac | This paper | SciLight Biotechnology, LLC |
| H324-27K27ac | This paper | SciLight Biotechnology, LLC |
| ENL wildtype YEATS protein with his-tag, residues 1148 | This paper | Accession number: NP_005925.2 |
| ENL T1 mutant YEATS protein with his-tag, insNHL, residues 1–151 | This paper | N/A |
| ENL T2 mutant YEATS protein with his-tag, PPV-> L, residues 1–146 | This paper | N/A |
| ENL T3 mutant YEATS protein with his-tag, NPP-> K, residues 1–146 | This paper | N/A |
| ENL T4 mutant YEATS protein with his-tag, insHLR, residues 1–151 | This paper | N/A |
| Critical commercial assays | ||
| RNeasy kit | Qiagen | Cat# 74106 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher | Cat# 4368814 |
| Miniprep kit | Qiagen | Cat# 27106 |
| Gel extraction kit | Qiagen | Cat# 28704 |
| PCR purification kit | Qiagen | Cat# 28106 |
| HisTrap HP 5mL column | GE Healthcare | Cat# 17524705 |
| Superdex 75 10/300 GL | GE healthcare | Cat# 10146535 |
| Deposited data | ||
| ENL T1 YEATS domain in complex with H3K27ac | This paper | PDB ID: 7X8B |
| ENL T2 YEATS domain in complex with H3K27ac | This paper | PDB ID: 7X88 |
| ENL T3 YEATS domain in complex with H3K27ac | This paper | PDB ID: 7E74 |
| ENL T4 YEATS domain in complex with H3K27ac | This paper | PDB ID: 7X8F |
| ENL T1(H116P) YEATS domain in complex with H3K27ac | This paper | PDB ID: 7X8G |
| Mendeley Database | This paper | DOI: 10.17632/bfrkxbhkw p.1 |
| Experimental models: Cell lines | ||
| HEK293 | ATCC | Cat# CRL-1573 |
| HEK293T | ATCC | Cat# CRL-3216 |
| U2OS | Laboratory of Shasha Chong | N/A |
| WiT49 | Laboratory of Herman Yeger Lab | N/A |
| Oligonucleotides | ||
| Oligonucleotides are listed in Table S1 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: mCherry-hsENL-WT-NLS | Wan et al., 2019 | N/A |
| Plasmid: mCherry-hsENL-T1-NLS | Wan et al., 2019 | N/A |
| Plasmid: mCherry-hsENL-T1 (H116P)-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T2-NLS | Wan et al., 2019 | N/A |
| Plasmid: mCherry-hsENL-T3-NLS | Wan et al., 2019 | N/A |
| Plasmid: mCherry-hsENL-T4-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T5-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T6-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T7-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T8-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T2 (N111P)-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T3 (K111P)-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-WT-YD-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T1-YD-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-WT-ΔIDR-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T1-ΔIDR-NLS | Wan et al., 2019 | N/A |
| Plasmid: mCherry-hsENL-WT-ΔAHD-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T1-ΔAHD-NLS | Wan et al., 2019 | N/A |
| Plasmid: mCherry-hsENL-ΔYD-NLS | Wan et al., 2019 | N/A |
| Plasmid: mCherry-hsENL-IDR-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-AHD-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-ΔIDR1-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-ΔIDR2-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-IDR1_Scr1-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-IDR1_Scr2-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-IDR1_K/Q21-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-IDR1_S/A21-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-IDR2_ED/A29-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-IDR2_E/Q17-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-IDR2_S/A33-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-ΔpolyS-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T1-M1-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T1-M2-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T1-M3-NLS | This paper | N/A |
| Plasmid: mCherry-Vector | Wan et al., 2019 | N/A |
| Plasmid: EYFP-hsENL-WT-YD-LacI | This paper | N/A |
| Plasmid: EYFP-hsENL-T1-YD-LacI | This paper | N/A |
| Plasmid: EYFP-hsENL-T2-YD-LacI | This paper | N/A |
| Plasmid: EYFP-hsENL-T1 (H116P)-YD-LacI | This paper | N/A |
| Plasmid: EYFP-hsENL-T2 (N111P)-YD-LacI | This paper | N/A |
| Plasmid: mEGFP-hsENL-T1-NLS | This paper | N/A |
| Plasmid: EGFP-hsENL-T1-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-WT-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T2-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T3-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T4-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T5-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T6-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T7-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T8-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-Vector | Wan et al., 2019 | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1 (H116P)-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T2 (N111P)-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T3 (K111P)-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-WT-YD-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-YDNLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T2-YDNLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-WT-ΔIDR-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-ΔIDR-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T2-ΔIDR-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-WT-ΔAHD-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-ΔAHD-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T2-ΔAHD-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-ΔYD-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-ΔIDR1-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-ΔIDR2-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-IDR1_Scr1-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-IDR1_Scr2-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-IDR1_K/Q21-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-IDR2_ED/A29-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-IDR2_E/Q17-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-IDR1_S/A21-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-IDR2_S/A33-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-ΔpolyS-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-M1-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-M2-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-M3-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-WT-M1-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-WT-M2-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-WT-M3-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-Halo-hsENL-T1-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-Halo-hsENL-T2-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-Halo-hsENL-T1(Y78A)-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-Halo-hsENL-T2(Y78A)-NLS | This paper | N/A |
| Plasmid: pSpCas9(BB)-2A–GFP (PX458) | Addgene | Cat# 48138 |
| Plasmid: pSpCas9(BB)-2A–sgAFF4-GFP | This paper | N/A |
| Plasmid: pSpCas9(BB)-2A–sgCDK9-GFP | This paper | N/A |
| pET28b-ENL wildtype and mutant YEATS, residues 1148 | This paper | N/A |
| pET28b-ENL YEATS Y78A, residues 1–148 | This paper | N/A |
| pET28b-full length ENL with N-terminal Strep II tag and C-terminal His-tag, residues 1–559 | This paper | N/A |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism | GraphPad | https://www.graphpad.com/ |
| Snapgene | Snapgene | https://www.snapgene.com/snapgene-viewer/ |
| Biorender | Biorender | https://biorender.com/ |
| HKL2000 | Otwinowski and Minor, 1997 | https://www.hkl-xray.com/hkl-2000 |
| MOLREP | Vagin and Teplyakov, 2010 | http://www.ccp4.ac.uk/ |
| COOT | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PHENIX | Adams et al., 2010 | https://www.phenix-online.org |
| PYMOL | Molecular Graphics System, Version 1.8 Schrödinger |
https://pymol.org/2/ |
| CFX Manager | BioRad | N/A |
| Origin 7.0 | OriginLab | N/A |
| CCP4 | Collaborative Computational Project, Number 4, 1994 |
http://www.ccp4.ac.uk/ |
| Imaris 9.3.1 | Oxford Instruments | N/A |
| NIS-Elements AR | Nikon | N/A |
| Astra 6 software program | Wyatt Technology | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
In this study, HEK293 (ATCC® CRL-1573) and HEK293T (ATCC® CRL-3216) were obtained from ATCC. HEK293 cells were cultured in EMEM with 10% FBS and 100 U/mL penicillin-streptomycin. HEK293T cells were cultured in DMEM supplemented with 10% FBS and 100 U/mL penicillin-streptomycin. U2OS cell line (human female osteosarcoma cells) containing a LacO array (~40,000 LacO elements) in the genome was a kind gift from Shasha Chong Lab (Caltech), and U2OS cells were maintained in low-glucose DMEM with 10% FBS and 100 U/mL penicillin-streptomycin. WiT49 cells were cultured in DMEM/F12 (Sigma, D8062) with 10% FBS and 100 U/mL penicillin-streptomycin. Cells were cultured in a humidified incubator at 37°C with 5% CO2. All cell lines were mycoplasma-negative and were tested for authentication.
METHOD DETAILS
Cell transient transfection
HEK293 (180K/well) and U2OS (200K/well) cells were seeded in 24-well plate containing cover glasses. 12 hours later, cells were transfected with the target fluorescent protein construct using lipofectamine 3000 (Invitrogen) transfection reagent in accordance with the manufacturer’s instruction and grown for 18 hours.
Lentivirus generation, concentration, and transduction
HEK293T cells were used for lentiviral packaging, where HEK293T (450K/dish) cells were plated in 10 cm dish, and 24 hours later, cells were transfected using polyethylenimine “Max” (PEI MAX) reagent (Fisher Scientific) (DNA (μg): PEI (μL) = 1: 4) according to manufacturer’s instruction. Virus concentration and transduction were performed as described previously (Wan et al., 2019). In brief, medium containing virus was collected 2 and 3 days after transfection and concentrated using 5 × PEG8000 solution. Cells were incubated with concentrated viral supernatant in the presence of 10 μg/mL polybrene. Infected HEK293 cells were selected 48 hours post-transduction. To achieve near endogenous levels of ENL transgenes, HEK293 and WiT49 cells stably expressing lenti-teton-3xFlag-ENL or lenti-teton-3xFlag-Halo-ENL were treated with low concentrations of doxycycline (4 to 20 ng/ml) for 48 hours.
Live cell confocal imaging
Cells were grown on (No. 1.5, 14 mm diameter) 35 mm dishes (MatTek, P35G-1.5–14-C). For fluorescence recovery after photobleaching (FRAP) assay and puncta fusion imaging in the transient transfection system, mCherry-ENL constructs were transfected into HEK293 cells. For FRAP assay performed at near endogenous levels, cells expressing Halo-ENL constructs were cultured in regular growth medium containing 150 nM JF549 for 15 min. Cells were quickly rinsed with PBS twice and then incubated in regular growth medium for at least 30 min in incubator at 37°C with 5% CO2.
Live cell imaging experiments were performed on a LSM880 confocal microscope (Zeiss) which is equipped with an incubation chamber maintaining 37°C and 5% CO2. Images were acquired with 561 nm laser and 63 × 4 oil DIC objective under control of the Zeiss Zen software.
For FRAP experiment, frames (mCherry-ENL: 500~1000, Halo-ENL: 800~1000) were acquired at one frame per 0.2 sec. To measure the baseline fluorescence of the bleach spot and the whole nucleus, the first 5 frames were acquired before bleach pulse. Photobleaching was performed under 561 nm laser (80% laser power with dwell time adjusted to 0.64 μs for mCherry-ENL images; 100% laser power with dwell time adjusted to 0.63 μs for Halo-ENL images). A circular spot (radius of mCherry-ENL image, ~0.7 μm; radius of Halo-ENL image, ~0.6 μm) was chosen in a region with homogenous fluorescence and at least 1 μm from nuclear or nucleolar boundaries. To track puncta of ENL mutants in live cells, HEK293 cells expressing mCherry-ENL-T1 were imaged for over 5 min under 561 nm laser with a frame interval time of 0.2 sec.
RNA fluorescence in situ hybridization (FISH) and imaging
HEK293 or WiT49 cells stably expressing plenti-teton-3xFlag-Halo-ENL-T1/T2 were treated with 20 ng/ml doxycycline for 48 hours to obtain close-to-endogenous levels of Halo-ENL-T1/T2. Cells expressing Halo-ENL were cultured in regular growth medium containing 150 nM JF549 for 15 min. Cells were quickly rinsed with PBS twice and then incubated in regular growth medium for at least 30 min in incubator at 37°C with 5% CO2.
To make an RNase-free environment for RNA-FISH experiment, 1% ribonucleoside vanadyl complex (RVC) was added to all the solutions to prevent RNA degradation. JF549 labeled cells were quickly rinsed with PBS once and then fixed with 4% PFA in PBS for 10 min at room temperature (RT). Cells were quickly washed once with PBS and then washed with PBS containing 0.05% Triton X-100 three times, 5 min/time. Fixed cells were permeabilized in PBS containing 2.5% Triton X-100 for 3 min. Permeabilized cells were quickly rinsed with PBS once and incubated with 70% ethanol for at least 1 hour at 4°C. Ethanol was aspirated off and cells were incubated with Wash Buffer A (containing 20% Stellaris 5 × RNA FISH Wash buffer A (Biosearch Technologies, SMF-WA1–60), 10% Deionized Formamide, and 70% nuclease-free water) for 5 min at RT. Cells were hybridized with hybridization solution (containing 90% stellaris RNA FISH hybridization buffer (Biosearch Technologies, SMF-HB1–10), 10% Deionized Formamide, 12.5 μM Stellaris RNA FISH probes which were designed to hybridize introns of target genes). Cells with hybridization solution were incubated overnight at 37°C. Cells were then washed with Wash Buffer A for 30 min at 37 °C and nuclei were stained with 5 ng/ml 4’,6-diamidino-2-phenylindole (DAPI) in Wash buffer A for 30 min at 37°C. Cells were washed with Wash Buffer B (Biosearch Technologies, SMF-WB1–20) for 5 min at RT. Cover glass was mounted with mounting medium. RNA FISH probes were labeled with Quasar® 670 dye. Images were captured on a widefield Leica microscope with 63 × oil objective and illuminated with a mercury lamp and standard filters for DAPI, Cy2, and Cy5.
Immunofluorescence staining and confocal microscopy
Cells (100K/well) were seeded on 24-well plate containing cover glasses. After 24 hours, cells were treated with 4 ng/ml doxycycline for another 48 hours as describe in the previous section. Cells were fixed with 4% PFA in PBS for 15 min, washed with PBS (three times, 5 min/time), and then permeabilized with PBS containing 0.1% Triton-X 100 for 10 min, and washed with PBS again (three times, 5 min/time). Subsequently, cells were blocked in PBS with 10% goat serum for 30 min and incubated overnight with primary antibodies. After three washes in PBS (5 min/time), cells were incubated with secondary antibodies for 1 hour in the dark. Cells were washed with PBS (5 min/time, three times) and mounted with DAPI-containing mounting medium. The primary antibody: Flag (Sigma-Aldrich, F1804–1MG) diluted 1:300, H3K27ac (Abcam, ab4729) diluted 1:1000, Cyclin T1 (Santa Cruz, sc-271348) diluted 1:50, DOT1L (Cell Signaling Technology, 77087s) diluted 1:50 in PBS with 1% BSA and 0.1% Tween-20. The secondary antibody: goat anti-Rabbit IgG (H+L) Alexa Fluor® 488 (Invitrogen, A32732), goat anti-Mouse IgG (H+L) Alexa Fluor® 488(Invitrogen, A32723), goat anti-Rabbit IgG (H+L) Alexa Fluor® 568(Invitrogen, A11011), and goat anti-Mouse IgG (H+L) Alexa Fluor® 568 (Invitrogen, A11031) was diluted 1:250 in PBS with 1% BSA and 0.1% Tween-20.
Images were captured on an inverted laser scanning confocal microscope (Zeiss, LSM880) with 63 × oil DIC objective under control of the Zeiss Zen software. Z stacks were acquired with 63 × oil DIC objective, a pinhole size of 1 airy unit, and a slice interval of 0.25 μm. For confocal fluorescence imaging, 488 nm, 514 nm, and 561 nm lasers were used to excite fluorescence of EGFP/mEGFP, EYFP-ENL-LacI, and mCherry-ENL respectively. Before acquiring fluorescence images, we carefully set the laser intensity and microscope detectors to make sure that no pixel in the images was saturated. For simultaneous 2-color imaging, we used proper emission filters and ensured no bleed-through between the two channels by imaging fixed cell samples that contain either fluorophore under the same 2-color imaging settings.
RT-qPCR analysis
Total RNA was isolated using the RNeasy kit (Qiagen) and reverse transcribed with the high-capacity cDNA reverse transcription kit (Applied Biosystems) in accordance with the manufacturer’s instructions. Quantitative PCR was performed using the SYBR Green PCR Master Mix (Fisher Scientific, A25778) with the ViiA 7 Real-time PCR System (Thermo Fisher). qPCR primers used are provided in Table S1.
ChIP-qPCR analysis
In general, cells (20~30 million cells/sample) were collected, washed, and cross-linked with freshly made 1% formaldehyde for 10 min at room temperature (Flag-ChIP) or 37°C (Pol IIS2P-ChIP), and incubated with 125 mM glycine for 5 min to stop cross-linking. Subsequently, fixed cells were resuspended and sonicated in RIPA 0.3 buffer (0.1% SDS for Flag-ChIP and 0.2% SDS for Pol IIS2P-ChIP, 1% Triton X-100, 10 mM Tris-HCl (pH 7.4), 1 mM EDTA (pH 8.0), 0.1% NaDOC, 0.3 M NaCl, 0.25% sarkosyl, 1mM DTT, and protease inhibitors) using a Covaris Ultrasonicator. For one sample, 6 μg Flag-M2 antibody (Sigma-Aldrich) or 8 μg Pol IIS2P antibody (Abcam) was pre-incubated with 75 μl Protein A Dynabeads at 4°C for at least 4 hours. After three washes of the Dynabeads in PBS containing 0.01% tween-20, the sonicated samples were added and incubated overnight at 4°C. The immunoprecipitates were washed twice with low salt wash buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, and 0.1% SDS), twice with high salt wash buffer (50 mM Tris pH 8.0, 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, and 0.1% SDS), twice with LiCl wash buffer (50 mM Tris pH 8.0, 150 mM LiCl, 1 mM EDTA, 1% NP-40, and 0.5% Na-Deoxycholate, 0.1% SDS), and once with TE buffer (1 mM EDTA, 10 mM Tris-HCl pH8.0) plus 50 mM NaCl. Bound DNA was eluted using 200 μl ChIP elution buffer (1% SDS, 50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 200 mM NaCl), then reverse crosslinked, treated with protein K (40 μg per sample) at 55°C and RNase A (1 μg per sample) at 37°C, and then purified using PCR purification kit (Qiagen, 28106) in a final volume of 50 μl. 10 μl ChIP DNA was diluted 100-fold using nuclease-free water for quantitative real-time PCR using ViiA 7 Real-time PCR System (Thermo Fisher) and the Power SYBR Green PCR Master Mix (Fisher scientific, A25778).
Western blot
Cells were collected, washed once with PBS, and heated in freshly made 1 × SDS loading buffer (5 × SDS loading buffer: 0.25M Tris-HCl (PH6.8), 0.5M DTT, 10% SDS, 50% glycerol, 0.25% BPB, 1% β-mercaptoethanol (β-ME) distilled in H2O) at 95°C for 20 min. Lysates were separated on 10% SDS-PAGE gels in 1 × running buffer (2.5mM Tris Base, 19.2 mM Glycine, 0.01% SDS in H2O, pH 8.3). Proteins were then transferred onto a PVDF membrane (Millipore) in 1 × transferring buffer (1.2 mM Tris Base, 9.6 mM Glycine in H2O, pH 8.3). After transfer, the membrane was blocked with 4% blocking buffer (Bio-Rad) for at least 30 min at RT with shaking. The membrane was incubated with the primary antibody in blocking buffer overnight at 4 °C with shaking. The next day, after three washes (10 min per wash) with PBST (PBS with 0.05% Tween-20), the membrane was incubated with the secondary antibody in blocking buffer for 1 hour at RT with shaking and subsequently washed three times in PBST (10 min per wash). Finally, the membrane was illuminated using Pierce ECL Western Blotting Substrate (Pierce Endogen) according to the manufacturer’s instructions. The primary antibodies: ENL (EMD Millipore, ABE2596–100UG) diluted 1:1000, Flag M2 (Sigma-Aldrich, F1804–1MG) diluted 1:2000, β-actin (Novus Biologicals, NB600–501) diluted 1:2000, AFF4 (Santa Cruz, sc-390130) diluted 1:1000, CDK9 (Santa Cruz, sc-13130) diluted 1:1000, ELL2 (Santa Cruz, sc-376611), and DOT1L (Cell Signaling Technology, 77087s) diluted 1:500 in blocking buffer. The secondary antibodies: Anti-mouse IgG, HRP-linked Antibody and Anti-mouse IgG, and HRP-linked Antibody diluted 1:2000 in blocking buffer.
Co-immunoprecipitation (Co-IP)
Cells were collected, washed once with ice-cold PBS, and lysed in IP lysis buffer (15mM Tris-HCl pH 7.4, 1mM EDTA, 150mM NaCl, 1mM MgCl2, 10% glycerol, 0.1% NP-40 in H2O, 1:100 protease inhibitor (1 table in 0.5 ml H2O), 1 mM DTT, 0.5 mM PMSF) for 20 min on ice. Anti-Flag M2-conjugated agarose beads (Sigma-Aldrich) were pre-washed once with IP lysis buffer for 5min. The lysates were incubated with anti-Flag M2-conjugated agarose beads overnight at 4 °C. The next day, the beads were washed 3 times with IP lysis buffer (5 min per wash) with rotating. The bound proteins were eluted in 2 × SDS buffer and analyzed by western blot.
Generation of Halo Tag CRISPR knock-in cell line
Single-guide RNA (sgRNA) targeting ± 60 bp around the stop codon of AFF4 or CDK9 gene was designed using the web-based CRISPR design tool (https://benchling.com/). The criteria to select sgRNA is that the sgRNA spans the stop codon and with a high on-target score. DNA oligonucleotides with restriction sites, BbsI, were ordered from Genewiz. pSpCas9(BB)-2A-GFP (PX458) vector was digested using BbsI enzyme and ligated with annealed genomic RNA (gRNA) fragments to generate pSpCas9(BB)-2A-sgAFF4-GFP and pSpCas9(BB)-2A-sgCDK9-GFP constructs. The AFF4-HaloTag-KI and CDK9-HaloTag-KI donors were generated by Genewiz. Specifically, the homology repair fragment spanning the AFF4 or CDK9 codon (500 bp on each side) and containing the Halo protein-coding gene and linker sequence was synthesized, which was cloned into pUC-GW-Amp construct. Next, we co-transfected pSpCas9(BB)-2A-sgRNA-GFP and HaloTag-KI donors into HEK293 cells using lipofectamine 3000 transfection reagent. The GFP-high cells were sorted 48 h after transfection using FACS. The sorted cells were cultured and expanded for another week. To select Halo-positive cells, cells were stained with 200 nM JF549 Halo dye for 15min, washed three times with PBS, and cultured in regular medium for at least 30 min in the incubator. The Halo-positive cells were sorted out using FACS, which were used to generate stable cell lines.
In vitro droplet formation assay
In vitro droplet formation assay was performed in droplet formation buffer: 150 mM NaCl, 5% glycerol, 2 mM β-ME, and 25 mM HEPES-Na (pH 7.4 for full-length protein, pH 7.0 for YEATS domain). Proteins were fluorescently labeled for microscopy using Microscale Protein Labeling kits from Thermo Scientific: Alexa Fluor™ 488 (A30005) and Alexa Fluor™ 568 (A20003). Labeled proteins were added to unlabeled proteins at a 1:10 molar ratio. Droplets were assembled in 384 low-binding multi-well 0.17-mm microscopy plates (384-well microscopy plates) (In Vitro Scientific) and sealed with optically clear adhesive film. After quick centrifugation, droplets were imaged under Olympus FV1200 using 100 × oil objectives. The results were processed by Imaris 9.3.1 (Oxford Instrument).
Protein purification, crystallization, data collection, and structure determination
Recombinant ENL YEATS (residues 1–148, accession number: NP_005925.2) was cloned into pET28b vector and expressed with N-terminal 6 × His tag in E. coli strain BL21(DE3) (Novagen) and induced overnight by 0.4 mM isopropyl β-D-thiogalactoside at 16°C in LB medium. Overnight-induced cells were collected and suspended in lysis buffer: 500 mM NaCl, 25 mM Tris, pH 7.5, 20 mM imidazole, 2 mM β-ME, and 1 mM PMSF. Then the cells were lysed using an Emulsiflex C3 (Avestin) high-pressure homogenizer. After centrifugation, the supernatant was applied to a HisTrap (GE Healthcare) nickel column, and proteins were eluted with a linear imidazole gradient from 20 mM to 500 mM. The resultant proteins were treated with thrombin (Sigma) digestion overnight for His-tag removal. The tag-free ENL YEATS proteins were further purified and polished over a Superdex 75 10/300 (GE Healthcare) gel filtration column using AKTA Purifier 10 systems (GE Healthcare). The monomer peak under elution buffer (500 mM NaCl, 25 mM Tris, pH 7.5, and 2 mM β-ME) was pooled, concentrated to about 8 mg/ml, and stored at −80°C. ENL YEATS mutants were generated using overlapping PCR or QuikChange (Stratagene) methods and verified by sequencing. Recombinant mutant YEATS domains of human ENL (1–148) were expressed and purified with essentially the same method as WT ENL YEATS.
For recombinant full-length ENL WT (residues 1–559, accession number: NP_005925.2) and mutants, their respective cDNA were inserted into the modified pET28b expression vector which includes an N-terminal 2 × strep-tag II and a C-terminal 6 × His tag in the frame. The protein was expressed in the E. coli BL21(DE3) (Novagen) expression system induced with 0.4 mM IPTG for 16 hours at 16°C. Cells were harvested and re-suspended in lysis buffer (25 mM Tris-HCl pH 7.5, 500 mM NaCl, 5% glycerol, 50 mM imidazole, 1 mM PMSF). After lysis using cell disrupter and centrifugation, the recombinant protein was homogenized with HisTrap column and washed with lysis buffer and high salt buffer (25 mM Tris-HCl pH 7.5, 1 M NaCl, 5% glycerol). ENL proteins were eluted using lysis buffer with 500 mM Imidazole, and the elution was diluted with strep buffer (25 mM HEPES-Na pH 7.4, 500 mM NaCl, 5% glycerol, 1 mM EDTA, 2 mM β-ME) to decrease the imidazole concentration. The diluted elution was incubated with strep-4FF beads (Smart-Lifesciences) and washed with strep buffer. Then ENL proteins on the beads were eluted using strep buffer with 2.5 mM desthiobiotin (IBA Life Sciences).
Crystallization was performed by the sitting-drop vapor diffusion method under 18°C by mixing equal volumes (1–2 μl) of protein and reservoir solution. For complex crystallization, ENL YEATS proteins (0.2 mM) were first incubated with a histone H3(24–27)K27ac peptide (SciLight Biotechnology) at a molar ratio of 1: 5 for approximately 2 hours. The complex samples were then concentrated to ~8 mg/ml before crystallization. Crystals of ENL T1/T4/T1(H116P) YEATS-H3(24–27)K27ac complex were obtained under 20% (w/v) PEG4000, 0.1 M sodium citrate pH 5.6, and 20% (v/v) 2-propanol. Crystals of ENL T2 YEATS-H3(24–27)K27ac complex were obtained under 20% (w/v) PEG3350, 0.1 M sodium citrate/citric acid pH 4.0, 0.2 M sodium citrate tribasic. Crystals of ENL T3 YEATS-H3(24–27)K27ac complex were obtained under 2 M sodium formate, 0.1 M sodium acetate trihydrate pH 4.6.
Crystals were briefly soaked in a cryoprotectant comprising reservoir solution supplemented with 20% glycerol and were flash-frozen in liquid nitrogen for data collection at 100 K. Complex datasets were collected at beamline BL17U1/18U1/19U1 at the Shanghai Synchrotron Radiation Facility. All data were indexed, integrated, and merged using the HKL2000 software package (Otwinowski and Minor, 1997). The complex structures were solved by molecular replacement using MOLREP (Vagin and Teplyakov, 2010). All structures were refined using PHENIX (Adams et al., 2010), with iterative manual model-building using COOT (Emsley and Cowtan, 2004). Model geometry was analyzed with PROCHECK (Laskowski et al., 1996). Detailed structural refinement statistics are presented in Table 1. All structural figures were created using PYMOL (https://pymol.org/2/).
Table 1.
Data collection and refinement statistics
| ENL T1 YEATS-H3K27ac (PDB code: 7X8B) | ENL T2 YEATS-H3K27ac (PDB code: 7X88) | ENL T3 YEATS-H3K27ac (PDB code: 7E74) | ENL T4 YEATS-H3K27ac (PDB code: 7X8F) | ENL T1(H116P) YEATS-H3K27ac (PDB code: 7X8G) | |
|---|---|---|---|---|---|
|
| |||||
| Data collection | |||||
| Space group | C2 | P3121 | P212121 | C2 | C2C2 |
| Cell dimensions | |||||
| a, b, c (Å) | 156.7, 48.7, | 52.1, 52.1, | 82.8, 91.3, | 156.6, 48.6, | 155.1, 48.5, |
| 47.7 | 136.4 | 165.8 | 47.3 | 47.8 | |
| a, β, γ(°) | 90, 101, 90 | 90,90,120 | 90,90,90 | 90, 101, 90 | 90, 101, 90 |
| Unique reflections | 15861(1466) | 10730(994) | 28461(2770) | 13193(1305) | 27089(2697) |
| Resolution Å) | 30.00–2.30 | 50.00–2.25 | 50.00–2.91 | 40.00–2.45 | 30.00–1.90 |
| (2.34–2.30) | (2.29–2.25) | (2.96–2.91) | (2.49–2.45) | (1.93–1.90) | |
| R sym | 0.091(0.333) | 0.132(0.735) | 0.239(0.955) | 0.149(0.931) | 0.124(0.423) |
| I/σ(I) | 37.2(13.4) | 32.7(3.0) | 8.3(2.0) | 11.4(1.3) | 17.7(5.0) |
| Completeness (%) | 98.7(92.4) | 99.1(96.8) | 99.5(98.0) | 99.8(99.0) | 99.1(99.0) |
| CC1/2 | 0.992(0.932) | 0.999(0.960) | 0.984(0.863) | 0.987(0.571) | 0.975(0.924) |
| Redundancy | 5.1(5.5) | 15.2(7.7) | 13.1(13.4) | 6.4(5.0) | 6.5(6.4) |
| Refinement | |||||
| Resolution (Å) | 17.29–2.23 | 45.16–2.25 | 44.02–2.91 | 38.40–2.45 | 25.79–1.90 |
| No. reflections | 15858 | 10676 | 28455 | 13189 | 27077 |
| Rwork/Rfree | 18.8/22.6 | 21.3/24.4 | 20.4/24.4 | 20.2/25.2 | 19.7/21.6 |
| No. atoms | |||||
| Protein | 2467 | 1193 | 5697 | 2541 | 2470 |
| Ligand | / | 13 | / | 1 | / |
| Solvent | 122 | 16 | 7 | 42 | 250 |
| B factors | |||||
| Protein |
31.5 |
50.9 |
45.9 |
52.8 |
30.0 |
| Ligand | / | 80.7 | / | 51.1 | / |
| Solvent | 32.8 | 49.9 | 33.1 | 47.8 | 36.0 |
| R.m.s. deviations | |||||
| Bond lengths (Å) | 0.009 | 0.015 | 0.012 | 0.011 | 0.00.009 |
| Bond angles (°) | 0.97 | 1.54 | 1.26 | 1.32 | 1.24 |
| Ramchandran | |||||
| Statistics(%) | |||||
| Favored | 99.3 | 97.1 | 97.2 | 97.6 | 99.0 |
| Allowed | 0.0 | 2.9 | 1.9 | 2.1 | 1.01.0 |
| Outliers | 0.7 | 0.0 | 0.9 | 0.3 | 0.00.0 |
Values in parentheses are for highest-resolution shell.
Isothermal titration calorimetry (ITC) assay
All ITC titrations were performed at 25°C using MicroCal iTC200 system (Malvern). Both synthetic histone H3 peptides (H3(17–28)K27ac) and H3 peptides (H3(24–27)K27ac) (SciLight Biotechnology, LLC) and the recombinant ENL YEATS proteins were extensively dialyzed against ITC buffer (25 mM Tris, pH 7.5, 500 mM NaCl, and 5 % glycerol). All samples were centrifuged at 13,000 rpm at 25°C for ~10–15 minutes before titration. ITC cells were rinsed with buffer and then protein solution at test concentration was added to the cell. Contents were pipetted up/down several times to mix with any trace buffer in the cell. A small volume of protein solution was removed from the cell for concentration re-check using NanoDrop. Each ITC titration consisted of 17 successive injections with 0.5 μl for the first and 2.4 μl for the rest. The intervals between injections were 150 s, and the stirring speed was 750 rpm. Peptides were titrated into proteins in all experiments. Usually, H3 peptides at 2.0–2.2 mM were titrated into proteins at 0.15 mM. The resultant ITC curves were processed using the Origin (v.7.0) software (OriginLab) in accordance with the “One Set of Sites” fitting model. First injection data is always excluded from the analysis.
Thermal shift assay (TSA)
The fluorescence-based TSA was performed with a CFX96TM real-time PCR instrument (Bio-Rad). A typical TSA assay consists of ~3 mg/mL ENL YEATS proteins and 5 × Sypro Orange (Invitrogen) in a total volume of 20 μl. The assay buffer contains 500 mM NaCl and 25 mM Tris (pH 7.5). During the assay, all samples were heated from 25°C to 95°C with an increased rate of 0.5°C per min. Protein denaturation was monitored by the increased fluorescence signal of Sypro Orange, which captures exposed hydrophobic residues during thermal unfolding. The recorded curves were analyzed by the software CFX-Manager (Bio-Rad). The temperature corresponding to the inflection point was defined as melting temperature (Tm).
Molecular dynamics simulations
Molecular dynamics simulations were performed with the Desmond package in the 2019–3 release of Schrödinger Maestro. Protein structures of WT and T1 ENL YEATS (PDB: 5J9S, 7X8B, respectively) were imported. Structure pre-processing in Maestro was executed to add hydrogens and remove redundant water molecules. Positioning of the histone H3 K27ac peptide (18–27) was based on the poses of histone H3 K27ac peptide (24–27) in the imported crystallography structures. The solvent environment was then set up with SPC model and OPLS3e force field. A 100-ns simulation with 100-ps intervals was then run with default condition (300 K, 1.0 bar). Molecular dynamics simulation reports were generated by running the Simulation Interaction Diagram in Maestro.
Protein disordered analysis and net charge per residue (NCPR) analysis
The intrinsically disordered region (IDR) in ENL was predicted using the publicly available IUPred3 which allows identification of IDR using ANCHOR2. An amino acid in a protein was considered disordered if the IUPred score was > 0.5. NCPR in human ENL(IDR) was calculated using publicly available CIDER v1.7 (5-residue sliding window) which allows for the calculation of many different parameters associated with disordered protein sequences (Holehouse et al., 2017).
Sequence conservation analysis
Sequence conservation was calculated by computing the sequence alignment. 79 placental mammal orthologous ENL/MLLT1 sequences were downloaded from Ensembl database (https://useast.ensembl.org/index.html), and conservation was determined using the analysis for conservation in Jalview (Waterhouse et al., 2009). The net charge per residue is calculated using the local-CIDER analysis package (Holehouse et al., 2017).
QUANTIFICATION AND STATISTICAL ANALYSIS
Image analysis
To measure the fraction of in-puncta fluorescence intensity of mCherry-ENL in individual cells, we first created a mask that covered all the puncta in the single-cell image using the “threshold” tool of ImageJ and integrated the fluorescence intensity of all the pixels within the foci mask. Next, we created another mask that covered the entire cell nucleus and integrated the fluorescence intensity of all the pixels within the nuclear mask. Finally, we calculated the total in-puncta to total in-nucleus intensity ratio as a measure of the mCherry-ENL in-puncta intensity fraction. To measure the mean nuclear fluorescence intensity of mCherry-ENL or Flag-ENL in individual cells, we included all the pixels within the cell nucleus in the single-cell image. For saturation concentration, we identified it as the 25% highest levels of mCherry-ENL expression among cells without detectable ENL puncta. To measure Flag-ENL puncta size, if the image was a Z stack image, we first performed Z-projection for the image stack of a single cell using the maximum intensity algorithm in ImageJ software. Then we created a mask that covered all the puncta in the single-cell image using the “threshold” tool of ImageJ and integrated the fluorescence intensity of all the pixels within the puncta mask. Then we measured the Feret diameter using the function of “analyzed particles”. To quantify the ratio of in-puncta/out-of-puncta intensity in Flag-ENL images, we first performed Z-projection for the image stack of a single cell using the maximum intensity algorithm in ImageJ software, created a mask that covered all the puncta in the single-cell image using the “threshold” tool of ImageJ and integrated the area size (A1) and intensity of all the pixels within the puncta mask (I1), then created a mask that includes puncta and the area immediately outside the periphery location with an interval ranging from 0.23 to 0.45 μm and extracted the area size (A2) and intensity of all the pixels (I2). Finally, we calculated the ratio using the formula: I1(A2-A1)/(I2A2-I1A1). To quantify the enrichment of mCherry-ENL at LacO arrays bound by EYFP-ENL-LacI or at EGFP-ENL-T1 puncta, we manually located the LacO array or EYFP-ENL-T1 condensate center in the cell nucleus and plotted the radial profiles surrounding this center pixel in the mCherry-channel. The EYFP or EGFP intensity radial profile allowed us to locate the concentration peak and periphery of the LacO-associated hub or EGFP-ENL-T1 condensates. Next, from the mCherry intensity radial profile, we extracted the intensity at the peak location (Ipeak) and averaged two intensity values immediately outside the periphery location with an interval ranging from 0.23 to 0.45 μm (Iperiphery). Finally, we calculated the peak to periphery intensity ratio (Ipeak/Iperiphery) as a measure of mCherry-ENL enrichment at the LacO array/EGFP-ENL-T1 condensate.
FRAP quantification
FRAP analysis for mCherry-ENL puncta formed under transient transfection condition was performed as described previously (Wan et al., 2019). Briefly, to correct for xy-drift of the bleached spot due to living cell movement during movie acquisition, we used a published ImageJ plugin “Template Matching and Slice Alignment” (https://sites.google.com/site/qingzongtseng/template-matching-ij plugin#description). Then we used three steps of normalization to quantify the FRAP signal in the bleach spot. First, we normalized the mean intensity of the bleach spot and the whole nucleus at time t to the respective pre-bleaching baseline intensity, i.e., Ifrap(t)/Ifrap-pre and Iwhole(t)/Iwhole-pre. Secondly, we normalized the relative bleach spot intensity to the relative nuclear intensity by
| (1) |
Then we calculated the bleach depth (ΔIfrap), i.e., the difference between the double-normalized FRAP intensity before and at the first frame (t1) after bleach pulse by
| (2) |
Finally, we normalized the bleach depth to 100%, and the triple-normalized FRAP intensity became
| (3) |
To analyze FRAP for puncta formed by endogenous levels of Halo-ENL, we adapted a published pipeline (Mir et al., 2018). Briefly, to measure the mean value of the background at time t (Ibackground), a dark spot located outside the nucleus was selected. Then a 600 nm diameter circle was identified from the metadata for the bleached spot and used to calculate the mean intensity at time t (Ifrap(t)). To correct for background, the mean of the dark spot was subtracted from the corresponding bleach spot value for each frame, i.e., Ifrap(t)-Ibackground. To correct for the reduction in total nuclear fluorescence, the data was normalized by dividing the mean of the bleach spot value by its mean value for the five pre-bleach frames ((Ifrap(t)-Ibackground)/Ifrap-pre). Similar to the analysis for the mCherry-ENL puncta, we calculated the bleach depth and normalized the bleach depth to 100% for all curves. Between 29~35 recovery curves were analyzed in each condition.
These curves were then averaged, and the mean recovery curve was used in Figure S1H. Code is available at https://gitlab.com/mir-lab/frap-code-for-song-2022. The pipeline performs the interpolation and includes quality control functions to obtain feedback from the user to correct for any drift.
Radial Enrichment Analysis
The radial distribution was determined by computing the mean intensity of the pixels at a given Euclidean distance from the maximum Z-projection of each Flag-ENL puncta or random point. This distribution was fit to a Gaussian function of the form , where d is the lateral distance from the puncta centroid and F is the full-width at half-maximum of the Gaussian function. Each profile was then normalized to a baseline of 0 to quantify enrichment above the local background.
To quantify the enrichment distribution of each condition, individual radial profiles of the Flag-ENL puncta and random point signals were measured and fit as above, and the enrichment was measured as the difference between the signal at the puncta centroid or at a random point to the baseline of the radial profile fit. Welch’s t-test was used to determine p-values.
Supplementary Material
Table S1. Oligonucleotides, Related to STAR Methods
Highlights.
Distinct cancer mutations induce consensus structural changes that trigger condensation
Oppositely charged IDRs in ENL and extrinsic factors contribute to condensate formation
Condensates formed by endogenous level of ENL mutant drive oncogenic gene activation
Marked overexpression of ENL mutant can lead to non-functional condensates
ACKNOWLEDGMENTS
We thank members of the Wan laboratory for scientific input throughout the study; B. Black, G.A. Blobel, J. Shorter, and R. Marmorstein for critical reading of the manuscript; K. Budinich and S. Yuan for manuscript editing; the CDB Microscopy Core at the University of Pennsylvania and the China National Center for Protein Sciences Beijing for facility support; the staff members at beamlines BL02U/17U1/18U1 of the Shanghai Synchrotron Radiation Facility and S. Fan at Tsinghua Center for Structural Biology for their assistance in data collection. The research was supported by funding from the University of Pennsylvania (L.W.), NIH (1DP2HG012443 and R00CA226399 to L.W., DP2OD030635-01 to M.M., T32GM008216 to K.M., T32GM132039 to A.P.B.), Pew-Stewart Scholars Program (L.W.), National Natural Science Foundation of China (31725014 and 92153302 to H.L, 31871283 and 31922016 to Y.L.), National Key R&D Program of China (2021YFA1300103 and 2020YFA0803303 to H.L.), China Association for Science and Technology for the Young Elite Scientists Sponsorship Program (YESS20170075 to Y.L.), Margaret Q. Landenberger Foundation (M.M.).
Footnotes
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
DECLARATION OF INTERESTS
L.W. serves as a consultant for Bridge Medicines and Panorama Medicine. Other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Weber A (2019). Evidence for and against Liquid-Liquid Phase Separation in the Nucleus. Non-Coding Rna 5, 50. 10.3390/ncrna5040050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D Biological Crystallogr 66, 213–221. 10.1107/s0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn JH, Davis ES, Daugird TA, Zhao S, Quiroga IY, Uryu H, Li J, Storey AJ, Tsai Y-H, Keeley DP, et al. (2021). Phase separation drives aberrant chromatin looping and cancer development. Nature 595, 591–595. 10.1038/s41586-021-03662-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S, Gladfelter A, and Mittag T (2019). Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 176, 419–434. 10.1016/j.cell.2018.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA, and Rosen MK (2017). Biomolecular condensates: organizers of cellular biochemistry. Nature Reviews. Molecular Cell Biology 18, 285–298. 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Mackowiak SD, Niskanen H, Knezevic D, Asimi V, Grosswendt S, Geertsema H, Ali S, Jerković I, Ewers H, et al. (2020). Unblending of Transcriptional Condensates in Human Repeat Expansion Disease. Cell 181, 1062–1079.e30. 10.1016/j.cell.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehning M, Dugast-Darzacq C, Rankovic M, Hansen AS, Yu T, Marie-Nelly H, McSwiggen DT, Kokic G, Dailey GM, Cramer P, et al. (2018). RNA polymerase II clustering through carboxy-terminal domain phase separation. Nat Struct Mol Biol 25, 833–840. 10.1038/s41594-018-0112-y. [DOI] [PubMed] [Google Scholar]
- Boija A., Klein IA., Sabari BR., Dall’Agnese A., Coffey EL., Zamudio AV., Li CH., Shrinivas K., Manteiga JC., Hannett NM., et al. (2018). Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 175, 1842–1855.e16. 10.1016/j.cell.2018.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boija A, Klein IA, and Young RA (2021). Biomolecular condensates and cancer. Cancer Cell 39, 174–192. 10.1016/j.ccell.2020.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay G, Sandoval GJ, Riggi N, Iyer S, Buisson R, Naigles B, Awad ME, Rengarajan S, Volorio A, McBride MJ, et al. (2017). Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 171, 163–178.e19. 10.1016/j.cell.2017.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JE, Hnisz D, and Young RA (2017). Transcriptional Addiction in Cancer. Cell 168, 629–643. 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Feliciano D, Dong P, Flores E, Gruebele M, Porat-Shliom N, Sukenik S, Liu Z, and Lippincott-Schwartz J (2019). Phase separation of YAP reorganizes genome topology for long-term YAP target gene expression. Nat Cell Biol 21, 1578–1589. 10.1038/s41556-019-0433-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Liu Z, and Lippincott-Schwartz J (2021). Biomolecular Condensates and Their Links to Cancer Progression. Trends Biochem Sci 46, 535–549. 10.1016/j.tibs.2021.01.002. [DOI] [PubMed] [Google Scholar]
- Chandra B, Michmerhuizen NL, Shirnekhi HK, Tripathi S, Pioso BJ, Baggett DW, Mitrea DM, Iacobucci I, White MR, Chen J, et al. (2021). Phase Separation mediates NUP98 Fusion Oncoprotein Leukemic Transformation. Cancer Discov 12, 1152–1169. 10.1158/2159-8290.cd-21-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W-K, Spille J-H, Hecht M, Lee C, Li C, Grube V, and Cisse II (2018). Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415. 10.1126/science.aar4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J-M, Dar F, and Pappu RV (2019). LASSI: A lattice model for simulating phase transitions of multivalent proteins. Plos Comput Biol 15, e1007028. 10.1371/journal.pcbi.1007028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S, and Mir M (2020). Towards Decoding the Sequence-Based Grammar Governing the Functions of Intrinsically Disordered Protein Regions. J Mol Biol 433, 166724. 10.1016/j.jmb.2020.11.023. [DOI] [PubMed] [Google Scholar]
- Chong S., Dugast-Darzacq C., Liu Z., Dong P., Dailey GM., Cattoglio C., Heckert A., Banala S., Lavis L., Darzacq X., et al. (2018). Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361, eaar2555–39. 10.1126/science.aar2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S, Graham TGW, Dugast-Darzacq C, Dailey GM, Darzacq X, and Tjian R (2022). Tuning levels of low-complexity domain interactions to modulate endogenous oncogenic transcription. Mol Cell 82, 2084–2097.e5. 10.1016/j.molcel.2022.04.007. [DOI] [PubMed] [Google Scholar]
- Cisse II, Izeddin I, Causse SZ, Boudarene L, Senecal A, Muresan L, Dugast-Darzacq C, Hajj B, Dahan M, and Darzacq X (2013). Real-Time Dynamics of RNA Polymerase II Clustering in Live Human Cells. Science 341, 664–667. 10.1126/science.1239053. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D Biological Crystallogr 60, 2126–2132. 10.1107/s0907444904019158. [DOI] [PubMed] [Google Scholar]
- Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo H-S, Souza A, Roberts JM, Dastjerdi S, Buckley DL, et al. (2017). Transcription control by the ENL YEATS domain in acute leukaemia. Nature 543, 270–274. 10.1038/nature21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd S, Arold ST, Radhakrishnan A, Gerhard DS, Jennings L, Huff V, Auvil JMG, Davidsen TM, Dome JS, Meerzaman D, et al. (2015). MLLT1 YEATS domain mutations in clinically distinctive Favourable Histology Wilms tumours. Nature Communications 6, 1–10. 10.1038/ncomms10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd S, Huff V, Walz AL, Ooms AHAG, Armstrong AE, Gerhard DS, Smith MA, Auvil JMG, Meerzaman D, Chen Q-R, et al. (2017). A Children’s Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nature Genetics 49, 1487–1494. 10.1038/ng.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, and Lander ES (2013). Lessons from the Cancer Genome. Cell 153, 17–37. 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Guo YE, Manteiga JC, Henninger JE, Sabari BR, Dall’Agnese A, Hannett NM, Spille J-H, Afeyan LK, Zamudio AV, Shrinivas K, et al. (2019). Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572, 543–548. 10.1038/s41586-019-1464-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn S (2018). Phase Separation, Protein Disorder, and Enhancer Function. Cell 175, 1723–1725. 10.1016/j.cell.2018.11.034. [DOI] [PubMed] [Google Scholar]
- Henninger JE, Oksuz O, Shrinivas K, Sagi I, LeRoy G, Zheng MM, Andrews JO, Zamudio AV, Lazaris C, Hannett NM, et al. (2021). RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 184, 207–225.e24. 10.1016/j.cell.2020.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetzner K., Garcia-Cuellar M-P., Büttner C., and Slany RK. (2018). The interaction of ENL with PAF1 mitigates polycomb silencing and facilitates murine leukemogenesis. Blood 131, 662–673. 10.1182/blood-2017-11-815035. [DOI] [PubMed] [Google Scholar]
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK, and Sharp PA (2017). A Phase Separation Model for Transcriptional Control. Cell 169, 13–23. 10.1016/j.cell.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holehouse AS, Das RK, Ahad JN, Richardson MOG, and Pappu RV (2017). CIDER: Resources to Analyze Sequence-Ensemble Relationships of Intrinsically Disordered Proteins. Biophys J 112, 16–21. 10.1016/j.bpj.2016.11.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K, and Mitaku S (2005). Mechanisms of secondary structure breakers in soluble proteins. Biophysics 1, 55–65. 10.2142/biophysics.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tal Y, Bertrand E, Singer RH, et al. (2004). From Silencing to Gene Expression Real-Time Analysis in Single Cells. Cell 116, 683–698. 10.1016/s0092-8674(04)00171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, and Armstrong SA (2007). MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 7, 823–833. 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- Kwon I, Kato M, Xiang S, Wu L, Theodoropoulos P, Mirzaei H, Han T, Xie S, Corden JL, and McKnight SL (2013). Phosphorylation-Regulated Binding of RNA Polymerase II to Fibrous Polymers of Low-Complexity Domains. Cell 155, 1049–1060. 10.1016/j.cell.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, and Thornton JM (1996). AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol Nmr 8, 477–486. 10.1007/bf00228148. [DOI] [PubMed] [Google Scholar]
- Leach BI, Kuntimaddi A, Schmidt CR, Cierpicki T, Johnson SA, and Bushweller JH (2013). Leukemia fusion target AF9 is an intrinsically disordered transcriptional regulator that recruits multiple partners via coupled folding and binding. Structure (London, England : 1993) 21, 176–183. 10.1016/j.str.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wen H, Xi Y, Tanaka K, Wang H, Peng D, Ren Y, Jin Q, Dent SYR, Li W, et al. (2014). AF9 YEATS Domain Links Histone Acetylation to DOT1L-Mediated H3K79 Methylation. Cell 159, 558–571. 10.1016/j.cell.2014.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Li Q, Alikarami F, Barrett DR, Mahdavi L, Li H, Tang S, Khan TA, Michino M, Hill C, et al. (2022). Small-molecule inhibition of the acyl-lysine reader ENL as a strategy against acute myeloid leukemia. Cancer Discov 10.1158/2159-8290.cd-21-1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H., Yu D., Hansen AS., Ganguly S., Liu R., Heckert A., Darzacq X., and Zhou Q. (2018). Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 558, 318–323. 10.1038/s41586-018-0174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EW, Thomasen FE, Milkovic NM, Cuneo MJ, Grace CR, Nourse A, Lindorff-Larsen K, and Mittag T (2021). Interplay of folded domains and the disordered low-complexity domain in mediating hnRNPA1 phase separation. Nucleic Acids Res 49, 2931–2945. 10.1093/nar/gkab063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir M, Stadler MR, Ortiz SA, Hannon CE, Harrison MM, Darzacq X, and Eisen MB (2018). Dynamic multifactor hubs interact transiently with sites of active transcription in Drosophila embryos. Elife 7, e40497. 10.7554/elife.40497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir M, Bickmore W, Furlong EEM, and Narlikar G (2019). Chromatin topology, condensates and gene regulation: shifting paradigms or just a phase? Development 146, dev182766. 10.1242/dev.182766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittag T, and Pappu RV (2022). A conceptual framework for understanding phase separation and addressing open questions and challenges. Mol Cell 82, 2201–2214. 10.1016/j.molcel.2022.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin JA, Wittmann S, Choubey S, Klosin A, Golfier S, Hyman AA, Jülicher F, and Grill SW (2022). Sequence-dependent surface condensation of a pioneer transcription factor on DNA. Nat Phys 18, 271–276. 10.1038/s41567-021-01462-2. [DOI] [Google Scholar]
- Ni X, Londregan AT, Owen DR, Knapp S, and Chaikuad A (2021). Structure and Inhibitor Binding Characterization of Oncogenic MLLT1 Mutants. Acs Chem Biol 16, 571–578. 10.1021/acschembio.0c00960. [DOI] [PubMed] [Google Scholar]
- Nott TJ, Craggs TD, and Baldwin AJ (2016). Membraneless organelles can melt nucleic acid duplexes and act as biomolecular filters. Nat Chem 8, 569–575. 10.1038/nchem.2519. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326. 10.1016/s0076-6879(97)76066-x. [DOI] [PubMed] [Google Scholar]
- Riback JA, Zhu L, Ferrolino MC, Tolbert M, Mitrea DM, Sanders DW, Wei M-T, Kriwacki RW, and Brangwynne CP (2019). Composition dependent phase separation underlies directional flux through the nucleolus. Biorxiv 809210. 10.1101/809210. [DOI] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958. 10.1126/science.aar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y, and Brangwynne CP (2017). Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382–13. 10.1126/science.aaf4382. [DOI] [PubMed] [Google Scholar]
- Shin Y, Chang Y-C, Lee DSW, Berry J, Sanders DW, Ronceray P, Wingreen NS, Haataja M, and Brangwynne CP (2018). Liquid Nuclear Condensates Mechanically Sense and Restructure the Genome. Cell 175, 1481–1491.e13. 10.1016/j.cell.2018.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terlecki-Zaniewicz S., Humer T., Eder T., Schmoellerl J., Heyes E., Manhart G., Kuchynka N., Parapatics K., Liberante FG., Müller AC., et al. (2021). Biomolecular condensation of NUP98 fusion proteins drives leukemogenic gene expression. Nat Struct Mol Biol 28, 190–201. 10.1038/s41594-020-00550-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN (2017). Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr Opin Struc Biol 44, 18–30. 10.1016/j.sbi.2016.10.015. [DOI] [PubMed] [Google Scholar]
- Vagin A, and Teplyakov A (2010). Molecular replacement with MOLREP. Acta Crystallogr Sect D Biological Crystallogr 66, 22–25. 10.1107/s0907444909042589. [DOI] [PubMed] [Google Scholar]
- Wan L, Wen H, Li Y, Lyu J, Xi Y, Hoshii T, Joseph JK, Wang X, Loh Y-HE, Erb MA, et al. (2017). ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 543, 265–269. 10.1038/nature21687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Chong S, Xuan F, Liang A, Cui X, Gates L, Carroll TS, Li Y, Feng L, Chen G, et al. (2019). Impaired cell fate through gain-of-function mutations in a chromatin reader. Nature 577, 121–126. 10.1038/s41586-019-1842-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, and Barton GJ (2009). Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M-T, Chang Y-C, Shimobayashi SF, Shin Y, Strom AR, and Brangwynne CP (2020). Nucleated transcriptional condensates amplify gene expression. Nat Cell Biol 22, 1187– 1196. 10.1038/s41556-020-00578-6. [DOI] [PubMed] [Google Scholar]
- Winters AC, and Bernt KM (2017). MLL-Rearranged Leukemias—An Update on Science and Clinical Approaches. Frontiers in Pediatrics 5, 691–21. 10.3389/fped.2017.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Oligonucleotides, Related to STAR Methods
Data Availability Statement
Original western blot and microscopy images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-FLAG | Sigma-Aldrich | Cat# F1804-1MG |
| Rabbit anti-Histone H3 (acetyl K27) | Abcam | Cat# Ab4729 |
| Mouse anti-Cyclin T1 | Santa Cruz | Cat# sc-271348 |
| Rabbit anti-DOT1L (D1W4Z) | Cell Signaling Technology | Cat# 77087s |
| Rabbit anti-ENL | EMD Millipore | Cat# ABE2596-100UG |
| Mouse anti-β-Actin Antibody (AC-15) | Novus Biologicals | Cat# NB600-501 |
| Mouse anti-AFF4 | Santa Cruz | Cat# sc-390130 |
| Mouse anti-ELL2 | Santa Cruz | Cat# sc-376611 |
| Rabbit anti-Pol II S2P | Abcam | Cat# Ab5095 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074s |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7076s |
| goat anti-Rabbit IgG (H+L) Alexa Fluor® 488 | Invitrogen | Cat# A32732 |
| goat anti-Mouse IgG (H+L) Alexa Fluor® 488 | Invitrogen | Cat# A32723 |
| goat anti-Rabbit IgG (H+L) Alexa Fluor® 568 | Invitrogen | Cat# A11011 |
| goat anti-Mouse IgG (H+L) Alexa Fluor® 568 | Invitrogen | Cat# A11031 |
| Bacterial and virus strains | ||
| NEB® 5-alpha Competent E. coli (High Efficiency) | NEB | Cat# C2987I |
| NEB® Stable Competent E. coli (High Efficiency) | NEB | Cat# C3040I |
| Escherichia coli (E. coli) strain BL21 (DE3) | Novagen | Cat# 69450 |
| Chemicals, peptides, and recombinant proteins | ||
| EMEM | Thermo Fisher Scientific |
Cat# MT10009CV |
| DMEM/F12 | Sigma | Cat# D8062 |
| DMEM | Thermo Fisher Scientific |
Cat# MT10013CV |
| low-glucose DMEM | Thermo Fisher Scientific |
Cat# 10567014 |
| OPTI-MEM | Gibco | Cat# 31985062 |
| Penicillin-streptomycin | Thermo Fisher Scientific |
Cat# 15-140-122 |
| FBS | HyClone | Cat# SH30910.03 |
| Lipofectamine 3000 | Invitrogen | Cat# L3000008 |
| Polyethylene Glycol 8000 (PEG) (1kg) | Fisher Scientific | Cat# BP233-1 |
| PEI MAX | Fisher Scientific | Cat# NC1038561 |
| Polybrene | EMD Millipore | Cat# TR-1003-G |
| Doxycycline | Sigma-Aldrich | Cat# D9891-1G |
| SYBR Green PCR Master Mix | Fisher Scientific | Cat# A25778 |
| Anti-Flag M2-conjugated agarose beads | Sigma-Aldrich | Cat# A2220-1ML |
| Ribonucleoside vanadyl complex (RVC) | Sigma-Aldrich | Cat# R3380-5ML |
| DPBS | Mediatech | Cat# MT21030CM |
| Janelia Fluor® 549 (JF549) | Laboratory of Mustafa A. Mir | N/A |
| Paraformaldehyde (PFA) | Sigma-Aldrich | Cat# 158127-500G |
| Triton X-100 | Fisher Scientific | Cat# BP151500 |
| Tween-20 | Fisher Scientific | Cat# BP337500 |
| Stellaris® FISH Probes, Human GAPDH_intron with Quasar® 670 Dye | Biosearch Technologies |
Cat# ISMF-2151-5 |
| Stellaris 5 × RNA FISH Wash buffer A | Biosearch Technologies |
Cat# SMF-WA1-60 |
| Stellaris RNA FISH hybridization buffer | Biosearch Technologies |
Cat# SMF-HB1-10 |
| Stellaris RNA FISH Wash Buffer B | Biosearch Technologies |
Cat# SMF-WB1-20 |
| Deionized Formamide | VWR | Cat# EM4650 |
| Fluoroshield™ with DAPI, histology mounting medium | Sigma-Aldrich | Cat# F6057-20ML |
| Goat serum | Cell Signaling Technology | Cat# 5425s |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat# A7906-10G |
| 16% Paraformaldehyde Aqueous Solution, EM Grade | Electron microscope scientific | Cat# 15710 |
| Tris-HCl | Fisher Scientific | Cat# BP1531 |
| NaDOC | Sigma-Aldrich | Cat# 35915 |
| N-Lauroylsarcosine sodium salt | Sigma-Aldrich | Cat# L9150-50G |
| DTT | Sigma-Aldrich | Cat# D9163-5G |
| cOmplete™ EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# 11836170001 |
| Protein A Dynabeads | Fisher Scientific | Cat# 10-002-D |
| EDTA | Sigma-Aldrich | Cat# E5134-500G |
| SDS | Thermo Fisher Scientific |
Cat# BP8200500 |
| Lithium chloride (LiCl) | Sigma-Aldrich | Cat# 213233-500G |
| NaCl | Fisher Scientific | Cat# BP358-1 |
| Glycerol | Fisher Scientific | Cat# BP2291 |
| 2-Mercaptoethanol | Thermo Fisher Scientific |
Cat# 21985023 |
| Glycine | Fisher Scientific | Cat# BP3815 |
| Tris Base | Fisher Scientific | Cat# BP152-1 |
| Pierce ECL Western Blotting Substrate | Pierce Endogen | Cat# 32106 |
| Blotting-Grade Blocker | Bio-Rad | Cat# 1706404 |
| MgCl | Sigma-Aldrich | Cat# M0250-500G |
| NP-40 | BioVision | Cat# 2111-100 |
| Protein K | Sigma-Aldrich | Cat# 03115879001 |
| RNase A | Fisher Scientific | Cat# 9001-99-4 |
| RNase inhibitor | Thermo Fisher Scientific |
Cat# N8080119 |
| LB Broth | Thermo Fisher Scientific |
Cat# BP1426-2 |
| Ampicillin | Fisher Scientific | Cat# BP176025 |
| Kanamycin | Fisher Scientific | Cat# BP906-5 |
| HEPES-Na | Sigma-Aldrich | Cat# H7006 |
| Polyethylene Glycol 4000 | Thermo Fisher Scientific |
Cat# A1615130 |
| sodium citrate | Sigma-Aldrich | Cat# 1613859 |
| 2-propanol | Sigma-Aldrich | Cat# I9516 |
| sodium formate | Sigma-Aldrich | Cat# 247596 |
| sodium acetate trihydrate | Sigma-Aldrich | Cat# 236500 |
| PowerUp SYBR Green Master Mix | Applied Biosystems Inc. |
Cat# A25742 |
| Alexa 488 Fluor™ 488 NHS | Invitrogen | Cat# A20000 |
| Ni IDA Beads 6FF | Smart-Lifesciences | Cat# SA00501L |
| Strep-4FF beads | Smart-Lifesciences | Cat# SA053100 |
| Alexa Fluor™ 488 | Thermo Fisher Scientific |
Cat# A30005 |
| Alexa Fluor™ 568 | Thermo Fisher Scientific |
Cat# A20003 |
| Isopropyl β-D-thiogalactoside | Sigma-Aldrich | Cat# 10724815001 |
| imidazole | Sigma-Aldrich | Cat# 12399 |
| phenylmethylsulfonyl fluoride (PMSF) | VWR Chemicals | Cat# 329-98-6 |
| Desthiobiotin | IBA Life Sciences | Cat# 2-1000-002 |
| SYPRO Orange stain | Thermo Fisher Scientific |
Cat# S6650 |
| H317-28K27ac | This paper | SciLight Biotechnology, LLC |
| H324-27K27ac | This paper | SciLight Biotechnology, LLC |
| ENL wildtype YEATS protein with his-tag, residues 1148 | This paper | Accession number: NP_005925.2 |
| ENL T1 mutant YEATS protein with his-tag, insNHL, residues 1–151 | This paper | N/A |
| ENL T2 mutant YEATS protein with his-tag, PPV-> L, residues 1–146 | This paper | N/A |
| ENL T3 mutant YEATS protein with his-tag, NPP-> K, residues 1–146 | This paper | N/A |
| ENL T4 mutant YEATS protein with his-tag, insHLR, residues 1–151 | This paper | N/A |
| Critical commercial assays | ||
| RNeasy kit | Qiagen | Cat# 74106 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher | Cat# 4368814 |
| Miniprep kit | Qiagen | Cat# 27106 |
| Gel extraction kit | Qiagen | Cat# 28704 |
| PCR purification kit | Qiagen | Cat# 28106 |
| HisTrap HP 5mL column | GE Healthcare | Cat# 17524705 |
| Superdex 75 10/300 GL | GE healthcare | Cat# 10146535 |
| Deposited data | ||
| ENL T1 YEATS domain in complex with H3K27ac | This paper | PDB ID: 7X8B |
| ENL T2 YEATS domain in complex with H3K27ac | This paper | PDB ID: 7X88 |
| ENL T3 YEATS domain in complex with H3K27ac | This paper | PDB ID: 7E74 |
| ENL T4 YEATS domain in complex with H3K27ac | This paper | PDB ID: 7X8F |
| ENL T1(H116P) YEATS domain in complex with H3K27ac | This paper | PDB ID: 7X8G |
| Mendeley Database | This paper | DOI: 10.17632/bfrkxbhkw p.1 |
| Experimental models: Cell lines | ||
| HEK293 | ATCC | Cat# CRL-1573 |
| HEK293T | ATCC | Cat# CRL-3216 |
| U2OS | Laboratory of Shasha Chong | N/A |
| WiT49 | Laboratory of Herman Yeger Lab | N/A |
| Oligonucleotides | ||
| Oligonucleotides are listed in Table S1 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: mCherry-hsENL-WT-NLS | Wan et al., 2019 | N/A |
| Plasmid: mCherry-hsENL-T1-NLS | Wan et al., 2019 | N/A |
| Plasmid: mCherry-hsENL-T1 (H116P)-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T2-NLS | Wan et al., 2019 | N/A |
| Plasmid: mCherry-hsENL-T3-NLS | Wan et al., 2019 | N/A |
| Plasmid: mCherry-hsENL-T4-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T5-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T6-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T7-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T8-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T2 (N111P)-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T3 (K111P)-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-WT-YD-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T1-YD-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-WT-ΔIDR-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T1-ΔIDR-NLS | Wan et al., 2019 | N/A |
| Plasmid: mCherry-hsENL-WT-ΔAHD-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T1-ΔAHD-NLS | Wan et al., 2019 | N/A |
| Plasmid: mCherry-hsENL-ΔYD-NLS | Wan et al., 2019 | N/A |
| Plasmid: mCherry-hsENL-IDR-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-AHD-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-ΔIDR1-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-ΔIDR2-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-IDR1_Scr1-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-IDR1_Scr2-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-IDR1_K/Q21-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-IDR1_S/A21-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-IDR2_ED/A29-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-IDR2_E/Q17-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-IDR2_S/A33-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-ΔpolyS-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T1-M1-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T1-M2-NLS | This paper | N/A |
| Plasmid: mCherry-hsENL-T1-M3-NLS | This paper | N/A |
| Plasmid: mCherry-Vector | Wan et al., 2019 | N/A |
| Plasmid: EYFP-hsENL-WT-YD-LacI | This paper | N/A |
| Plasmid: EYFP-hsENL-T1-YD-LacI | This paper | N/A |
| Plasmid: EYFP-hsENL-T2-YD-LacI | This paper | N/A |
| Plasmid: EYFP-hsENL-T1 (H116P)-YD-LacI | This paper | N/A |
| Plasmid: EYFP-hsENL-T2 (N111P)-YD-LacI | This paper | N/A |
| Plasmid: mEGFP-hsENL-T1-NLS | This paper | N/A |
| Plasmid: EGFP-hsENL-T1-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-WT-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T2-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T3-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T4-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T5-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T6-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T7-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T8-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-Vector | Wan et al., 2019 | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1 (H116P)-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T2 (N111P)-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T3 (K111P)-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-WT-YD-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-YDNLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T2-YDNLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-WT-ΔIDR-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-ΔIDR-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T2-ΔIDR-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-WT-ΔAHD-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-ΔAHD-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T2-ΔAHD-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-ΔYD-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-ΔIDR1-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-ΔIDR2-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-IDR1_Scr1-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-IDR1_Scr2-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-IDR1_K/Q21-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-IDR2_ED/A29-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-IDR2_E/Q17-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-IDR1_S/A21-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-IDR2_S/A33-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-ΔpolyS-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-M1-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-M2-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-T1-M3-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-WT-M1-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-WT-M2-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-HA-hsENL-WT-M3-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-Halo-hsENL-T1-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-Halo-hsENL-T2-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-Halo-hsENL-T1(Y78A)-NLS | This paper | N/A |
| Plasmid: plenti-teton-miniCMV-3xflag-Halo-hsENL-T2(Y78A)-NLS | This paper | N/A |
| Plasmid: pSpCas9(BB)-2A–GFP (PX458) | Addgene | Cat# 48138 |
| Plasmid: pSpCas9(BB)-2A–sgAFF4-GFP | This paper | N/A |
| Plasmid: pSpCas9(BB)-2A–sgCDK9-GFP | This paper | N/A |
| pET28b-ENL wildtype and mutant YEATS, residues 1148 | This paper | N/A |
| pET28b-ENL YEATS Y78A, residues 1–148 | This paper | N/A |
| pET28b-full length ENL with N-terminal Strep II tag and C-terminal His-tag, residues 1–559 | This paper | N/A |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism | GraphPad | https://www.graphpad.com/ |
| Snapgene | Snapgene | https://www.snapgene.com/snapgene-viewer/ |
| Biorender | Biorender | https://biorender.com/ |
| HKL2000 | Otwinowski and Minor, 1997 | https://www.hkl-xray.com/hkl-2000 |
| MOLREP | Vagin and Teplyakov, 2010 | http://www.ccp4.ac.uk/ |
| COOT | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PHENIX | Adams et al., 2010 | https://www.phenix-online.org |
| PYMOL | Molecular Graphics System, Version 1.8 Schrödinger |
https://pymol.org/2/ |
| CFX Manager | BioRad | N/A |
| Origin 7.0 | OriginLab | N/A |
| CCP4 | Collaborative Computational Project, Number 4, 1994 |
http://www.ccp4.ac.uk/ |
| Imaris 9.3.1 | Oxford Instruments | N/A |
| NIS-Elements AR | Nikon | N/A |
| Astra 6 software program | Wyatt Technology | N/A |