

SUMMARY
Dynamic changes in protein-protein interaction (PPI) networks underlie all physiological cellular functions and drive devastating human diseases. Profiling PPI networks can, therefore, provide critical insight into disease mechanisms and identify new drug targets. Kinases are regulatory nodes in many PPI networks, yet facile methods to systematically study kinase interactome dynamics are lacking. We describe kinobead competition and correlation analysis (kiCCA), a quantitative mass spectrometry-based chemoproteomic method for rapid and highly multiplexed profiling of endogenous kinase interactomes. Using kiCCA, we identified 1,154 PPIs of 238 kinases across 18 diverse cancer lines, quantifying context-dependent kinase interactome changes linked to cancer type, plasticity, and signaling states, thereby assembling an extensive knowledgebase for cell signaling research. We discovered drug target candidates, including an endocytic adapter-associated kinase (AAK1) complex that promotes cancer cell epithelial-mesenchymal plasticity and drug resistance. Our data demonstrate the importance of kinase interactome dynamics for cellular signaling in health and disease.
INTRODUCTION
Proteins form dynamic protein-protein interaction (PPI) networks that cooperatively carry out their biological functions. PPI network topology is regulated at the level of protein abundance and through signaling events like protein post-translational modifications (PTMs), affecting network composition and connectivity.1,2 Numerous human diseases alter signaling pathways and protein homeostasis, leading to the rewiring of PPI networks and disease progression.3–6 The 538 protein kinases in the human kinome are central players in cell signaling, embedded within large PPI networks that respond to physiological and pathological cues.7–10 Greater understanding of how the kinome integrates with cellular PPI networks can provide insights into physiological processes, identify disease mechanisms, and discover specific kinase complexes that are unique drivers of disease; such complexes may serve as valuable, novel drug targets and biomarkers.11
Large-scale maps of PPI networks have been generated with mass spectrometry (MS)-based approaches like affinity purification (AP)-MS12,13 and proximity labeling-MS methods like BioID14,15 and APEX.16–18 However, AP-MS and proximity labeling-MS lack the necessary throughput and multiplexing capabilities to globally profile PPI network dynamics across many samples and conditions. Furthermore, these methods typically require the expression of epitope-tagged proteins, which can interfere with native cell signaling and is difficult to implement in primary cells and tissues. Size-exclusion chromatography-MS19 and protein crosslinking-MS20 allow multiplexed detection of native protein complexes, but currently lack the required sensitivity to detect low-abundance PPIs such as kinase-dependent signaling and transcription factor complexes.17 Sensitive and high-throughput methods are urgently needed to map native kinase PPIs and their dynamic interactomes.
To address this need, we developed kinobead competition and correlation analysis (kiCCA), a MS-based chemoproteomic method for rapid and highly multiplexed profiling of native kinase PPIs in cell and tissue lysates. kiCCA uses a panel of multi-targeted kinase probes to compete kinases and their interaction partners from immobilized kinase inhibitor beads (kinobeads or multiplexed inhibitor beads, MIBs)21–25 to identify kinase PPIs. We used kiCCA to systematically map kinase interactomes in 18 diverse cancer cell lines and to interrogate PPI changes in the context of cancer type, plasticity, and signaling states; this revealed that PPI network topologies are highly dynamic and context dependent. Through our profiling efforts, we identified and quantified 1,154 high-confidence PPIs between 238 kinases and 684 non-kinase proteins, which we compiled into an extensive and easily accessible kinase interactome knowledgebase (Table S3 and https://quantbiology.org/kiCCA).
Using this knowledgebase, we discovered that kinase PPIs can describe kinase functional states and their integration into signaling pathways. We found that cancer cells showing epithelial-mesenchymal plasticity (EMP) and increased therapy resistance26–28 drastically rewired their kinase PPI networks. Specifically, we found that EMP altered endocytic and vesicle trafficking pathways that are controlled by an adapter-associated kinase 1 (AAK1) interaction network. RNAi-mediated knockdown of AAK1 complex components affected EMP marker expression and greatly sensitized cancer cells to targeted therapy, highlighting that kiCCA can provide actionable leads for drug target discovery. Collectively, our kiCCA method and kinase interactome knowledgebase are invaluable tools for studying kinase PPI networks and can help understand how signaling events influence disease states.
RESULTS
kiCCA, a method for rapid and highly multiplexed profiling of native kinase interactomes
We and others have shown that kinobeads enrich kinases along with their interactors,21,25,29–31 yet assigning iterators to specific kinase complexes remained challenging. We demonstrated that selective, ATP-competitive kinase inhibitors (KIs) can be used to identify specific kinase interactors by monitoring the co-competition of kinases and non-kinase proteins (Figure 1A).32,33 We reasoned that if one selective KI displaces a protein complex from the kinobeads, then broadly selective KIs will compete for multiple kinase complexes in a single experiment. By correlating abundances of kinases and their interaction partners across a panel of competition experiments using multiple broad-selectivity KIs with orthogonal kinase binding affinities, hereafter referred to as kinase interactome probes (KIPs), our kiCCA approach would simultaneously identify hundreds of kinobead-bound kinase complexes (Figure 1B).
Figure 1. Overview of kinobead competition and correlation analysis (kiCCA), a chemoproteomic approach for rapid, sensitive, and highly multiplexed kinase protein-protein interaction (PPI) profiling.
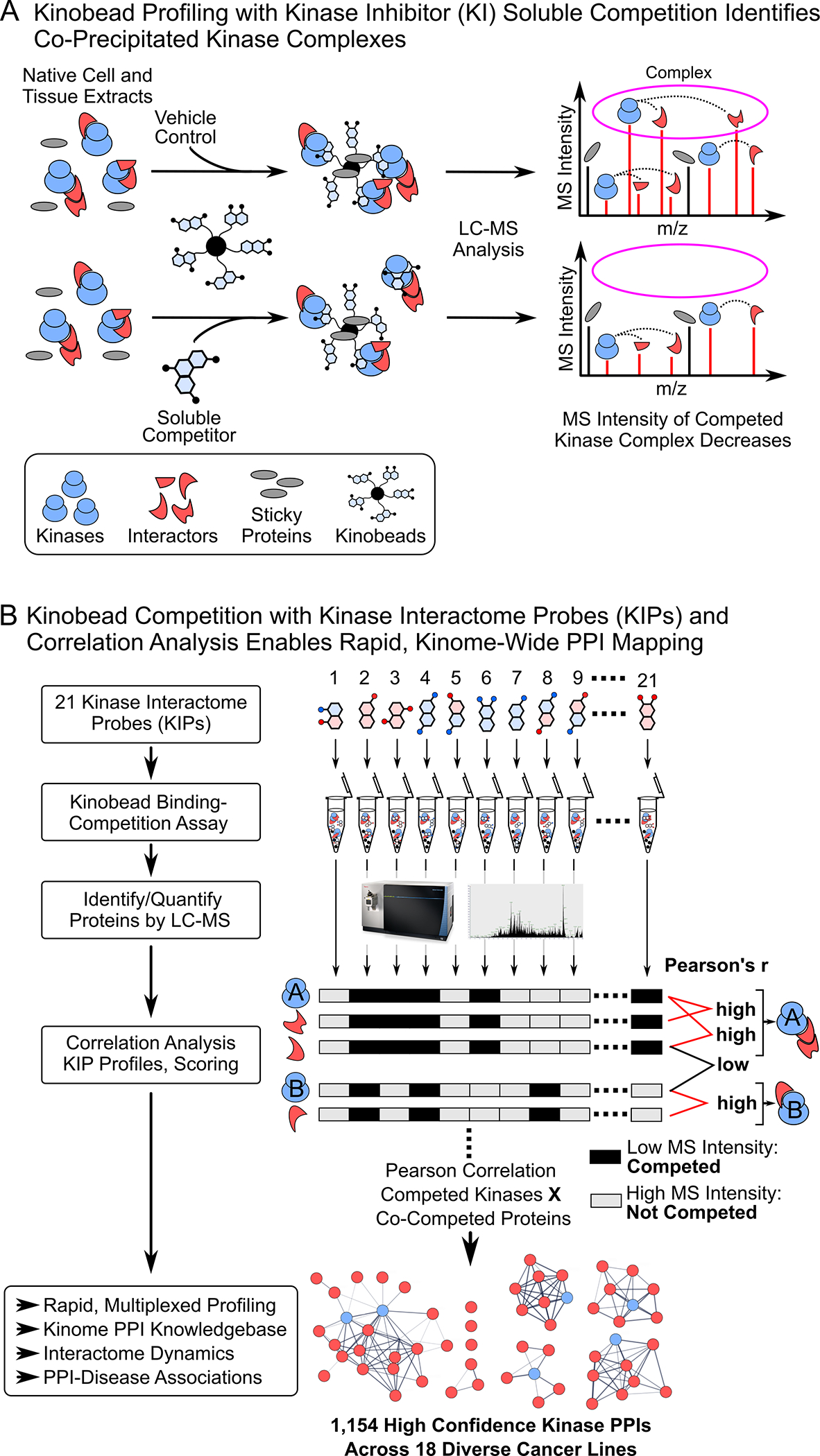
(A) Kinome profiling with kinobead/LC-MS and kinase inhibitor (KI) soluble competition identifies co-precipitating kinase complexes.
(B) Workflow of our kiCCA analysis using 21 kinase interactome probes (KIPs) to identify multiple kinase complexes in the same experiment.
To identify suitable KIPs, we collated kinobead competition data (i.e., MS intensity ratios) from a study profiling targets of 243 KIs22 and applied pairwise Pearson correlation of MS intensity ratios for all KIs, followed by unsupervised hierarchical clustering of the resulting matrix of r-values (Table S1); this identified 12 distinct groups of KIs with orthogonal kinome binding profiles. On average, clusters 1–5, 8–10, and 12 contained more broadly selective inhibitors compared to clusters 6, 7, and 11. Consequently, we chose one to five KIs from clusters 1–5, 8–10, and 12 to obtain 21 KIPs with broad kinome coverage (Figure 2A and Table S1).
Figure 2. Selecting kinase interactome probes (KIPs), proof-of-concept in HeLa lysate, and kiCCA profiling of a diverse 18-cancer line panel.
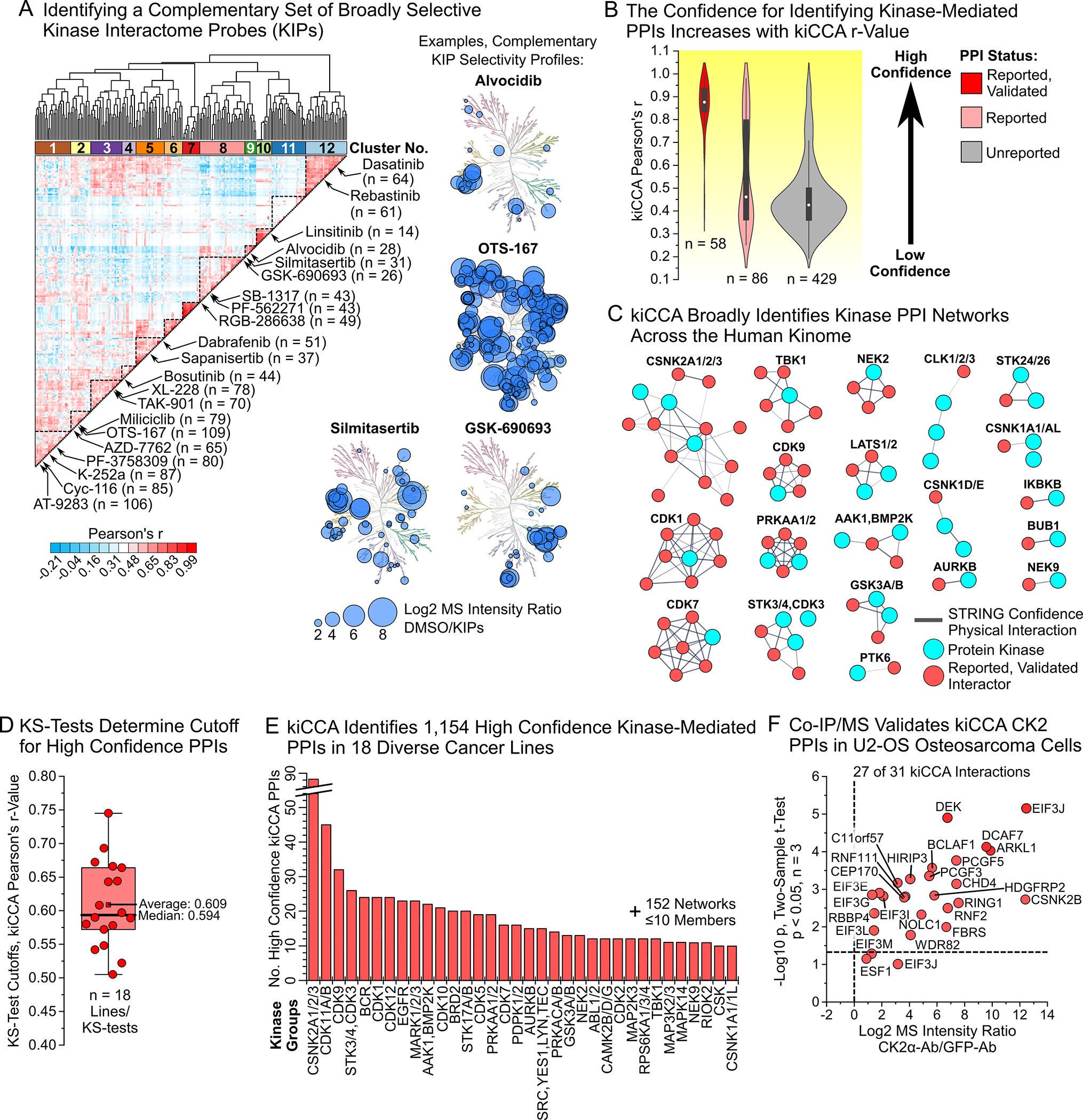
(A) Unsupervised hierarchical clustering of kinase inhibitors (KIs) by their kinome selectivity profiles identifies 21 complementary KIPs.
(B) kiCCA accurately identified previously reported PPIs in HeLa cells with high, positive Pearson’s r-values. Violin plot of r-value distributions for previously reported and independently validated (left), reported but unvalidated (center), and unreported kinase PPIs (right).
(C) kiCCA identified kinase PPI networks across the human kinome. Shown are previously reported, independently validated PPI networks of 37 kinase groups identified using kiCCA in unstimulated HeLa cell lysates. Network models created with STRING 11.5.38
(D) Kolmogorov-Smirnov (KS) tests of kiCCA data from each of the 18 cancer lines showed that r > 0.6 identified most previously reported PPIs.
(E) kiCCA in 18 diverse cancer cell lines identified 1,154 high confidence kinase interactions between 684 proteins and 238 kinase groups.
(F) Co-immunoprecipitation/MS (Co-IP/MS) experiments with casein kinase 2 catalytic and regulatory subunits (CK2α and β) antibodies validated a CK2 PPI network identified by kiCCA in U2-OS cells (two sample t-test, p < 0.05, n = 3), demonstrating kiCCA’s high accuracy.
To validate that our KIPs compete most expressed kinases, we evaluated their kinome selectivity, each at a single high concentration in HeLa cell lysate (10–50 μM, STAR Methods) using our kinobead/LC-MS workflow and label-free quantification.23,32 Of the 232 kinases quantified, 199 (86%) were efficiently competed by at least one KIP (log2 MS Intensity ratio >0.75, two sample t-test p < 0.1, n = 2) and kinase binding profiles of individual KIPs were highly dissimilar (average Pearson’s r-value = 0.27, Figures S1A, S1B and Table S2). Because highly homologous kinases like the adenosine monophosphate-activated kinases AMPK1 and 2 (PRKAA1 and 2) showed very similar KIP binding profiles (Pearson’s r ≥ 0.9), we combined these kinases into 54 groups of two to four members, with the other 239 kinase groups comprising one member each (293 kinase groups total, Figure S1C, Table S2, STAR Methods). Our results show that by binding and competing kinases of all major families, our 21 KIPs can broadly identify kinase PPIs in kiCCA.
kiCCA accurately and broadly identifies native kinase PPIs
Seeing that our KIPs competed not only kinases but also non-kinase proteins in HeLa cell lysate, we next sought to identify specific kinase PPIs in our HeLa data using correlation analysis (Figure 1B, STAR Methods). We correlated the MS intensity values of all 199 competed kinases and 573 co-competed non-kinase proteins across the 21 KIP panel and the DMSO control experiment and called the kinase group showing the highest Pearson’s r-value for each non-kinase protein its most likely interactor. Matching the resulting list of PPIs with the BioGRID protein interaction database v.4.4.20034, the BioPlex 3.0 dataset13, and the kinome-centric AP-MS dataset from Buljan et al.12 showed that 144 of 573 kiCCA interactions had been previously reported, with independent validation for 58 of these. Validated PPIs had highly positive Pearson’s r-values (median r = 0.88, Figure 2B and Table S3), demonstrating that kiCCA can identify known kinase PPIs. Previously reported but unvalidated kinase PPIs showed a bimodal distribution of r-values, with many kiCCA interactions showing high r-values (n = 38, r > 0.5). In contrast, unreported kiCCA interactions generally had low r-values (median r = 0.43) although a tail of kiCCA interactions with highly positive r-values suggested that novel kinase PPIs were identified (Figure 2B). Importantly, previously reported, and validated interactions were distributed among 37 distinct kinase groups from all kinome sub-families, showing that kiCCA achieves broad and unbiased coverage of kinase PPIs (Figures 2C and S1D, Table S3).
To explore kiCCA’s ability to identify kinase interactomes across different model systems, we analyzed 17 additional cell lines representing distinct types of cancers, including carcinoma (10 HCC lines), neuroblastoma (SK-N-SH and SH-SY5Y), glioblastoma (A172), osteosarcoma (U2-OS), as well as myeloid malignancies such as chronic myeloid leukemia (K562), mantle cell lymphoma (JeKo1), and T-cell leukemia (Jurkat, Tables S2 and S3). Across all 18 lines, our KIPs competed 357 kinases (66% of the human kinome) and 4136 non-kinase proteins. kiCCA identified 10,791 interactions of these proteins with 294 kinase groups, of which 1783 PPIs were previously reported.12,13,34 The kiCCA data from our full cell line panel recapitulated our observations in HeLa cell lysates (Figure S1E). Comparing the r-value distributions of reported and unreported interactions from each of the 18 cell lines using Kolmogorov–Smirnov (KS)-tests (Figure S2), we found that most reported PPIs were identified above a median Pearson’s r-value ≥ 0.594, and that hundreds of unreported PPIs had r-values surpassing this threshold (Figure S1E, Tables S2 and S3). We concluded that an r-value of > 0.6 identifies kinase PPIs, both previously reported and unreported, with high confidence.
Applying our r > 0.6 selection rule to our kiCCA dataset of 18 diverse cancer lines, we mapped 1,154 high confidence kinase PPIs, of which 692 interactions (60%) were not previously reported. Each kinase group interacted on average with six proteins, with the casein kinase 2 group (CK2 or CSNK2A1, 2, and 3) forming the largest interaction network of 88 members (Figure 2E and Table S3). For proteins interacting with several kinases, we introduced a compound score, hereafter referred to as the kiCCA score, to identify its most likely kinase interactor. This kiCCA score considers both the kiCCA r-value and the frequency of identification across our 18-cancer line panel (STAR Methods) and can be used to annotate kinase PPIs in kinobead/LC-MS profiling data. For example, while 14–3-3 binding protein YWHAZ can interact with multiple kinase groups, including the MAP3K2/3 (kiCCA score = 0.21) and the PDPK1/2 group (kiCCA score = 0.08), the higher kiCCA score suggested that the MAP3K2/3-YWHAZ PPI is the more likely interaction (Table S3).
To experimentally validate the accuracy of kiCCA, we performed co-immunoprecipitation/MS (Co-IP/MS) experiments in the U2-OS osteosarcoma line using antibodies specific to the catalytic CK2α subunit and the regulatory CK2β subunit, as well as GFP (control). kiCCA identified 31 CK2 interactions in U2-OS cells, and our antibody-based Co-IP/MS study confirmed 24 (83%) of these interactions (Figures 2F, S3A, and Table S3). Collectively, these results highlight the high accuracy of kiCCA and its potential to identify cancer cell-type specific kinase PPIs.
kiCCA quantifies kinase interactome changes linked to cancer cell plasticity
Cancer cells dramatically rewire signaling pathways in response to drugs and changing conditions in the tumor microenvironment, resulting in cellular plasticity that promotes therapy escape and metastasis.24,26,28 To explore if kiCCA can quantify differences in kinase interactomes associated with pathway rewiring and plasticity, we compared the abundances of high confidence kiCCA PPIs between related cell lines with distinct phenotypic marker gene expression (Figure S3B). From our 18-cell line panel, we chose the neuroblastoma line SK-N-SH and its subclone SH-SY5Y as models of cancer cell plasticity. Previous reports showed that the parental SK-N-SH line has mesenchymal stem cell-like characteristics, whereas SH-SY5Y exhibits a noradrenergic neuronal phenotype, suggesting the noradrenergic-mesenchymal plasticity (NMP) of these lines (Figure 3A).35,36 Our own analysis of mRNA expression data from the Cancer Cell Line Encyclopedia (CCLE)37 confirmed high expression of mesenchymal and low expression of noradrenergic neuronal markers in SK-N-SH compared to SH-SY5Y cells (Figure S3B and C). Furthermore, our differential expression analysis (DEA, STAR Methods) of kinases confirmed that SK-N-SH cells express a kinome profile typical for mesenchymal-like cancer cells, including high expression of the receptor tyrosine kinase UFO (AXL) and the transforming growth factor β receptor TGFBR2,31 whereas SH-SY5Y cells show high expression of neuronal-specific kinases like the anaplastic lymphoma kinase ALK and the β-adrenergic receptor kinase ADRBK2 (Figure 3A and Table S4, Student’s t-test, Benjamini-Hochberg (BH)-FDR < 0.05, n = 22). Together, these results suggested that neuroblastoma line NMP is associated with wide-spread changes in kinase expression and a rewiring of kinome-dependent signaling networks.
Figure 3. kiCCA quantifies kinome interactome changes caused by cancer cell plasticity and acute signaling events.
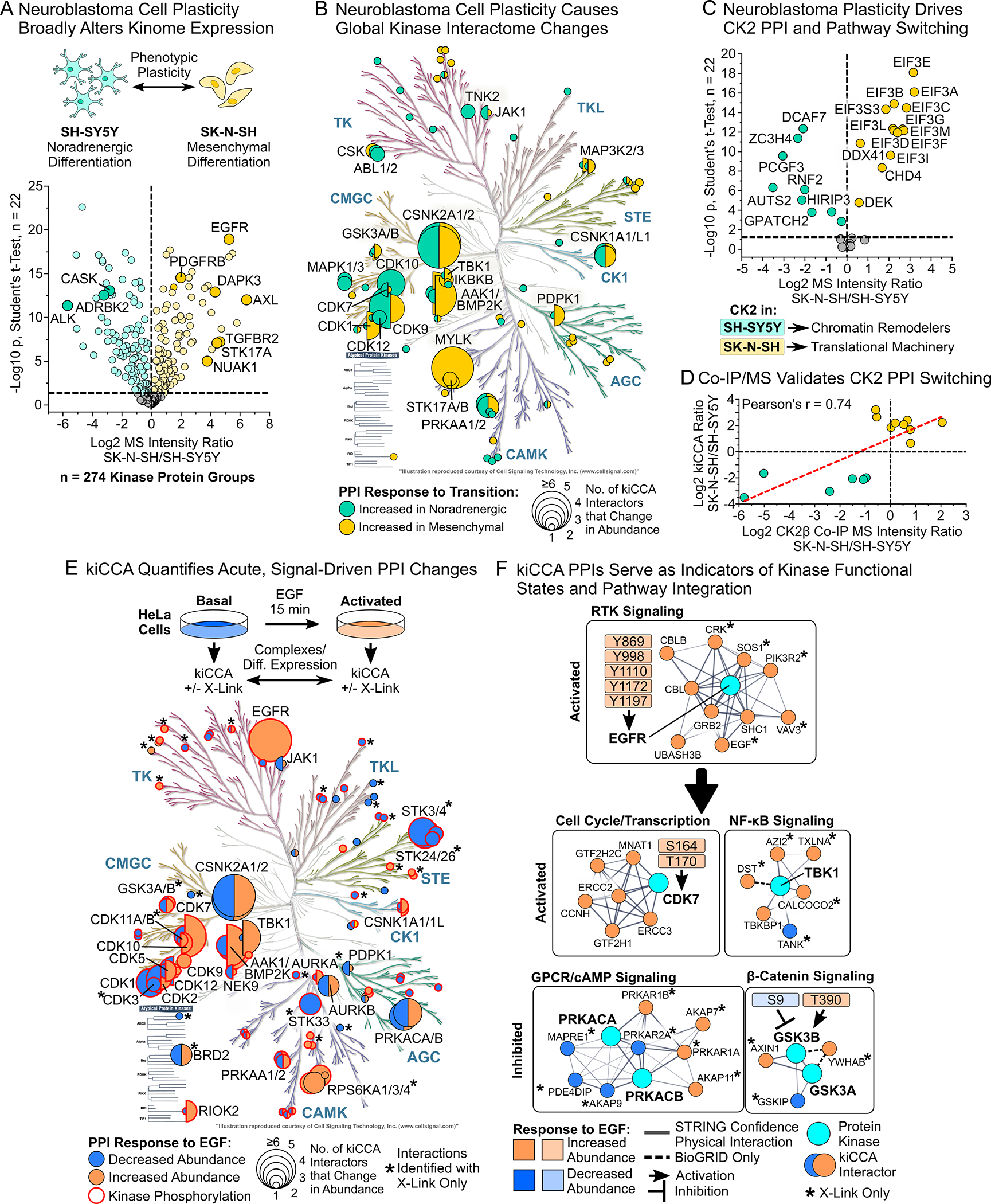
(A) Differential expression analysis (DEA) of kiCCA data revealed that the neuroblastoma lines SK-N-SH and SH-SY5Y have kinome profiles indicative of mesenchymal-noradrenergic plasticity (NMP, Student’s t-test, Benjamini-Hochberg (BH)-FDR < 0.05, n = 22). Kinases marking the noradrenergic and mesenchymal phenotype are highlighted.
(B) Kinases with altered PPI abundance between the SK-N-SH and SH-SY5Y neuroblastoma lines (n = 44 kinase groups, DEA statistics see (A)). Circle size scales with the number of kinase-binding partners that change in abundance.
(C) DEA of high confidence kiCCA interactions reveals that 20 members of a CK2 interaction network showed an altered abundance between neuroblastoma lines (statistics see (A)). Pathway enrichment analysis with STRING 11.538 showed that differentially abundant CK2 interactors in either cell line participated in distinct signaling pathways.
(D) Co-IP/MS experiments in the SK-N-SH and SH-SY5Y neuroblastoma lines using specific CK2α/β antibodies confirmed altered abundance of CK2 interaction partners as determined by kiCCA.
(E) Overview of kiCCA of EGF-stimulated HeLa cells and annotated kinases with altered PPI abundance upon EGF treatment (n = 63 kinase groups, for DEA statistics see (A)). Kinases with co-regulated changes in phosphorylation and PPIs are highlighted in red. Kinases marked with an asterisk (*) have PPI changes only detected with protein crosslinking.
(F) kiCCA of EGF-stimulated HeLa cells revealed that abundance changes of PPIs correlated with changes in kinase functional states, connecting the EGFR to several non-canonical EGFR-signaling pathways. Network models were created using STRING 11.5.38
See also Figure S4.
To clarify if kinome rewiring was accompanied by kinase interactome changes, we next subjected high confidence kiCCA PPIs in the two neuroblastoma lines to DEA. We discovered that NMP affected interactions between 44 kinase groups and 90 proteins (Figures 3B and S4), indicating that neuroblastoma cell plasticity caused widespread changes in PPI network topology. The largest rearrangements affected CK2, myosin light-chain kinase (MYLK), and cyclin-dependent kinase (CDK) networks (Table S4). Unlike alterations in kinase abundance, changes in interactor abundance only weakly correlated with changes in mRNA expression between the two lines (CCLE data, R2 = 0.18, Figure S5A), suggesting that changes in kinase PPI networks were caused by post-translational events. For instance, 20 CK2 interactors significantly differed in abundance, half of them showing higher expression in SK-N-SH than SH-SY5Y cells, and vice versa, suggesting that CK2 integrated with distinct pathways in a NMP-dependent manner (Figure 3C). Pathway enrichment analysis using STRING 11.538 showed that CK2 preferentially integrated into Polycomb repressive complex (PRC1)-like chromatin remodeling complexes in noradrenergic SH-SY5Y cells, while binding more to components of the eukaryotic translation initiation factor 3 (eIF-3) complex in SK-N-SH cells. Notably, the PRC1-like complex also contained the autism susceptibility candidate AUTS2 that together with CK2 has been shown to alleviate transcriptional repression and promote expression of neuronal genes, suggesting that CK2 promotes the neuroblastoma cell NMP program.39 To validate that kiCCA accurately quantified CK2 interactome rewiring and pathway integration, we used CK2α- and CK2β-targeted antibodies and Co-IP/MS to quantify abundance changes in CK2 interaction partners between SK-N-SH and SH-SY5Y cells (Figure 3D). This analysis showed that 15 CK2 interactions significantly differed in abundance in both kiCCA and Co-IP/MS, including the NMP-associated CK2-AUTS2 interaction, and that MS intensity ratios between the two analyses were tightly correlated (Pearson’s r = 0.74). Together, our results showed that kiCCA accurately quantifies plasticity-associated changes in kinase interactomes and their involvement in specific cellular pathways, and that CK2 can switch roles between regulating chromatin structure and translation in neuroblastoma cell NMP.
kiCCA quantifies dynamic changes in kinase interactomes induced by acute signaling events
We next asked if kiCCA can also quantify rapid and transient changes in kinase PPI networks caused by acute signaling events like growth factor stimulation. We stimulated HeLa cells with 50 ng/mL EGF for 15 min, analyzed cell lysates with kiCCA, and identified 163 high confidence kinase PPIs (Table S3). To learn which of these PPIs were EGF-responsive, we performed DEA of our kiCCA data from unstimulated and EGF-stimulated HeLa cells, identifying 57 high-confidence interactions of 33 kinase groups that significantly changed in abundance (Student’s t-test, BH-FDR < 0.05, n = 22, Figure 3E and S5B). Members of the EGFR interactome including the signaling adapters GRB2 and SHC1, and the E3 ubiquitin ligases CBL and CBLB, increased in abundance up to ~110-fold (Figures 3E, 3F, and S5B, Table S4). These results suggested that kiCCA can map acute changes in kinase PPI networks, yet the overall number of EGF-sensitive PPIs appeared low given that EGFR signaling can affect the phosphorylation states of at least 120 other kinases.32,40
Reasoning that kiCCA, like other AP-MS approaches, may miss transient and low affinity PPIs, we repeated the experiment using formaldehyde-mediated protein crosslinking to stabilize kinase signaling complexes (STAR Methods). kiCCA with protein crosslinking identified 127 EGF-responsive high confidence PPIs involving 32 additional kinase groups, more than doubling our coverage of EGF-responsive kinase interactions (Figures 3E and S5B). These PPIs included additional EGFR interactions, e.g., with the GTP-activating proteins SOS1 and VAV3, and the phosphatidylinositol 3-kinase regulatory subunit PIK3R2, as well as PPIs of several other kinases in the EGFR pathway, including mitogen-activated protein kinases (MAPKs) and the ribosomal S6 kinases (Figures 3E, 3F, and S5B, Table S3). These results indicated that crosslinking greatly expands kiCCA’s ability to detect transient and weak kinase PPIs. To see if kinase interactome changes were indeed tied to phosphorylation changes triggered by EGFR signaling, we compared kinases with PPI abundance changes to kinases that we previously found differentially phosphorylated upon EGF treatment.32 This revealed co-regulation of PPI and phosphorylation changes in 44 of 65 kinase groups (Figure 3E, hypergeometric test, p = 1.8E-6), confirming that PPI rewiring was signal-dependent and validating kiCCA’s accuracy for quantifying acute kinase interactome changes.
kiCCA PPIs are indicators of kinase functional states and pathway integration
We next asked if changes in kinase interactomes could indicate kinases in distinct functional states. Kinase phosphorylation sites with known regulatory roles41 were associated with kiCCA PPIs known to affect kinase function; for example, tyrosine phosphorylation of the EGFR recruited its known signaling partners to activate ERK and PI3K-AKT signaling, T-loop S164 and T170 phosphorylation and activation of CDK7 correlated with recruited components of the CDK activating kinase complex, and activating T390 phosphorylation of GSK3B coincided with binding of AXIN1 and the β-catenin destruction complex; in contrast, inhibitory phosphorylation of S9 on GSK3B was accompanied by binding of its inhibitor GSKIP (Figure 3F); this confirms that kiCCA PPIs can serve as proxies for phosphorylation events that affect kinase functional states. We also observed changes in PPIs for kinases that are not regulated by phosphorylation, but rather by binding to second messengers or regulatory subunits, such as cAMP-activated protein kinase A (PKA) that bound more of its regulatory subunit following EGF treatment, indicating inactivation. PKA also switched interactions between the A-kinase anchoring proteins AKAP9 and AKAP7 and AKAP11, indicating cellular re-localization. Similarly, the NF-κB activating kinase B (TBK1) dissociated from the signaling scaffold TANK, marking inactivation (Figure 3F). kiCCA PPIs are, therefore, also proxies for phosphorylation-independent signaling events affecting kinase activity and subcellular localization. Searching the 1,154 high confidence kiCCA PPIs from our interactome knowledgebase in the BioGRID34 and UniProt42 databases and associated primary literature, we identified 140 interactions of 85 diverse kinase groups that can affect kinase activation state and/or cellular localization; we hereafter refer to these as functional marker PPIs (fmPPIs) (Figure S6A, Table S3).
Next, to explore if kiCCA PPIs can also indicate the activity of specific cellular pathways, we studied the pathway membership of high confidence PPIs in our EGF-activated HeLa dataset. Analyzing EGF-sensitive kinase PPIs with STRING pathway enrichment analysis,38 we found, in addition to the expected EGFR signaling complex, many kinase interactors participating in cellular processes and pathways distinct from canonical EGFR signaling, including G-protein coupled receptor (GPCR)/cAMP signaling, WNT/β-catenin signaling, NF-κB signaling, the cell cycle, and autophagy (Figure 3F, Table S4). Encouraged by these results, we mapped high confidence kiCCA interactors from our knowledgebase to 32 distinct, disease relevant signaling pathways and cellular processes using gene ontology – biological process (GOBP) terms (Figure S6B and Table S3); hypothesizing that an interaction of a kinase with a specific non-kinase pathway member integrates the kinase into the corresponding pathway, we mapped 492 (72%) of kiCCA interactors, and the 169 kinase groups that interact with them, to at least one pathway or process term. Each of the 32 GO terms comprised on average 35 non-kinase interactors, representing an extensive resource for kinase pathway integration (Figure S6B and Table S3). Collectively, using fmPPIs and GOBP term mapping, our kiCCA knowledgebase determined kinase functional states and integrated kinases into cellular pathways through specific PPIs.
kiCCA identifies kinase functional states and pathways underlying hepatocellular carcinoma therapy resistance
Hepatocellular carcinoma (HCC) is the most common form of primary liver cancer and the second most deadly malignancy worldwide.43 HCCs are highly resistant to therapy which is, in part, caused by cancer cell EMP.31,44 We previously showed that ~50% HCC CCLE cell lines are resistant to kinase inhibitors of clinically relevant HCC targets such as cell cycle kinases, PI3K/mTOR, and FGFRs, and that resistance was tightly linked to EMP.45 To demonstrate the utility of kiCCA in identifying disease mechanisms and drug targets, and to understand how HCC cell EMP rewires the kinome to promote therapy resistance, we compared kiCCA data from the archetypical epithelial-like and drug sensitive HuH-7 line to the mesenchymal-like and drug resistant SNU761 line (Figures 4A, S3B and Table S3). Applying DEA to the kinases quantified in the two HCC lines, we discovered that 170 kinases significantly differed in abundance (Student’s t-test, BH-FDR < 0.05, n = 22), and that the changes in the kinome showed a profile typical for HCC cell epithelial-mesenchymal transition (EMT), including ~1,200-fold increased abundance of AXL in SNU761 cells and increased expression of fibroblast growth factor receptor (FGFR) isoforms 2, 3, and 4, and various polarity and cell cycle-related kinases in HuH-7 cells (Figure 4A). DEA of the 227 high confidence kiCCA PPIs identified in the two HCC lines showed that 127 PPIs significantly differed in abundance. Like our analysis of neuroblastoma lines (Figure 3B and S5A), abundance changes of kiCCA interactors correlated poorly with CCLE mRNA expression differences while kinase abundance changes were better correlated (Figure S6C), suggesting that EMP-associated kinome rewiring was driven by post-translational events. Mapped fmPPIs in the mesenchymal-like SNU761 line indicated elevated activation of the survival-promoting tyrosine kinases SRC, EGFR, and ACK1 (TNK2), the WNT pathway kinases GSK3A and B, and the NF-κB activating kinase CHUK. Surprisingly, SNU761 cells also showed elevated activation of numerous proliferation suppressor kinases, including the Hippo kinases STK3 and 4 (MST1 and 2) and STK38 (NDR1), and the liver kinase B1 (LKB1 or STK11, Figure 4B). These results suggest that mesenchymal-like HCC cells evade therapies by simultaneously activating kinases that promote survival along with kinases that blunt proliferation and the cell cycle, thereby evading drugs that preferentially kill rapidly proliferating cells.
Figure 4. kiCCA analysis of hepatocellular carcinoma (HCC) lines identifies changes in kinase functional states and pathways correlating with plasticity and drug resistance.
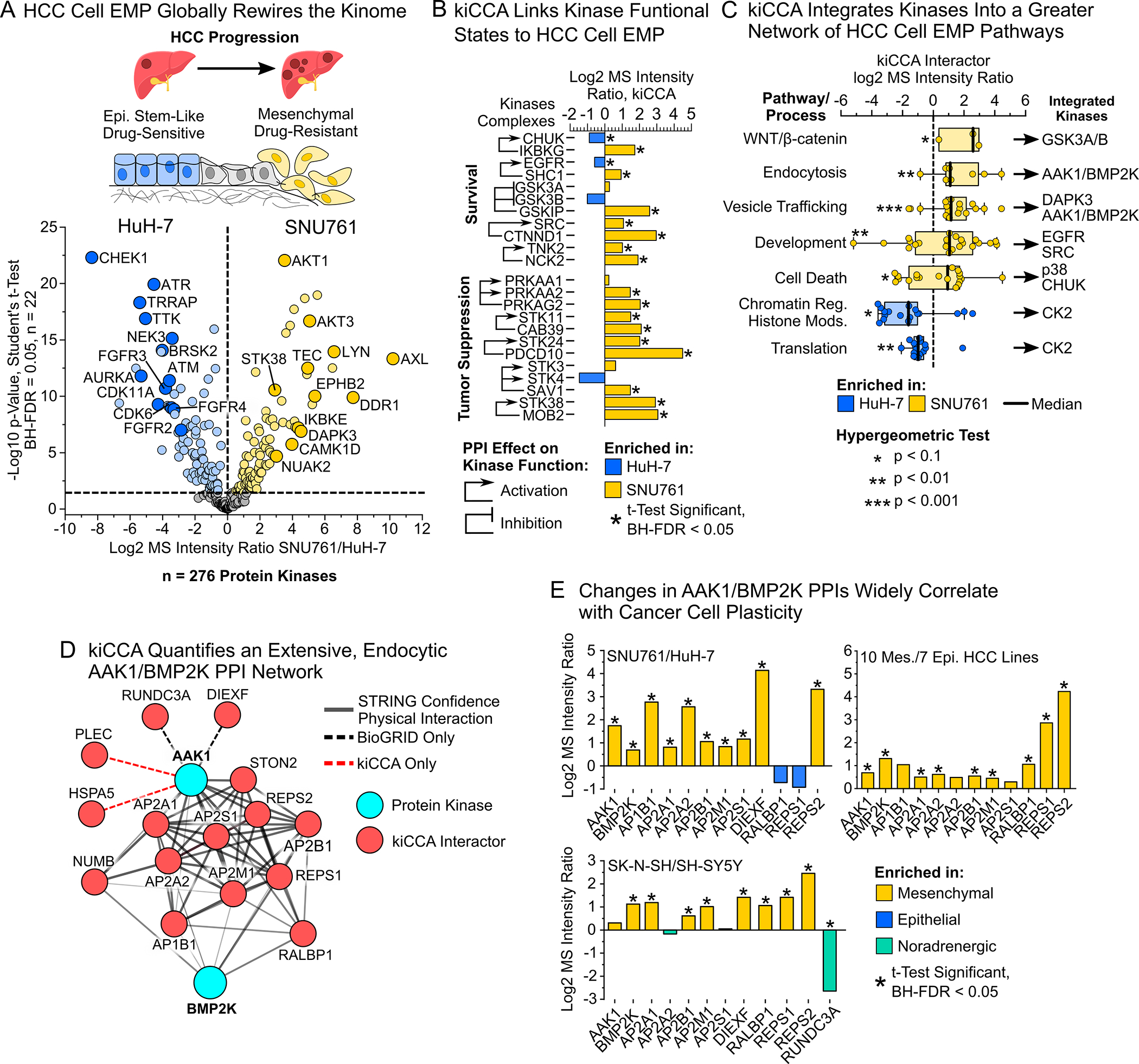
(A) Epithelial-mesenchymal plasticity (EMP) promotes HCC progression, metastatic spread, and therapy resistance. kiCCA DEA data revealed extensive kinome expression changes between epithelial-like HuH-7 cells and mesenchymal-like SNU761 cells in HCC cell EMP (Student’s t-test, BH-FDR < 0.05, n = 22)
(B) kiCCA and DEA analysis of the HCC cell EMP models SNU761 and HuH-7 mapped 127 kinase PPIs that significantly differed in abundance. Cross-referencing our curated set of fmPPIs revealed that kinases involved in cell survival and proliferation suppression are broadly activated in the mesenchymal-like SNU761 line (for DEA statistics see (A))
(C) Gene ontology-biological process (GOBP) term analysis of kiCCA interactors identifies cellular pathways and processes associated with HCC cell EMP, integrating specific kinases into EMP pathways through their PPIs. GOBP term enrichment was determined by hypergeometric test (p < 0.1).
(D) kiCCA quantifies an extensive AAK1/BMP2K interaction network involved in endocytosis and vesicle trafficking across the 18-cancer cell line panel (Created using STRING 11.538).
(E) Members of the AAK1/BMP2K PPI network were broadly associated with cancer cell plasticity in the SNU761 and HuH-7 HCC lines and SK-N-SH and SH-SY5Y neuroblastoma lines (for DEA statistics see (A)), and in the larger 17-member HCC line panel (two sample t-test mesenchymal vs epithelial, BH-FDR < 0.05).45
Next, to systematically understand how EMP affects kinase-mediated pathways, we analyzed GOBP terms for kiCCA PPIs differing in abundance between the HuH7 and SNU761 lines (Figure S6D). Translation and chromatin remodeling pathways related to cell proliferation were enriched in HuH-7 cells, with mainly CK2 integrating into these pathways. In contrast, SNU761 cells were enriched in endocytosis and vesicle trafficking pathways involving the adapter-associated kinase 1 (AAK1) and the BMP-2-inducible protein kinase (BMP2K), as well as developmental pathways like GSK3A and B inactivation-driven canonical WNT/β-catenin signaling, and EGFR and SRC signaling (Figures 4C and S6D). To validate these findings, we applied gene set enrichment analysis (GSEA) with GOBP terms46,47 (STAR Methods) to our kiCCA data from the HuH7 and SNU761 lines, and our published kinobead/LC-MS profiling data from seven epithelial-like and ten mesenchymal-like HCC lines (Figure S6E and Table S4). We confirmed that endocytosis, vesicle trafficking, survival, and EMP-related pathways were highly enriched in mesenchymal-like HCC cells. Components of the endocytosis and vesicle trafficking machinery have been previously linked to HCC progression,48,49 and to cancer cell EMP in pancreatic ductal adenocarcinoma cells,50–52 but whether aberrant endocytosis and vesicle trafficking promotes HCC cell EMP remains unclear. Hypothesizing that dysregulated AAK1/BMP2K interaction networks can promote cancer cell plasticity, we examined abundance changes of AAK1/BMP2K PPIs (Figure 4D) in our kiCCA data from HuH-7 and SNU761 HCC models, SK-N-SH and SH-SY5Y models, and our 17 HCC cell line panel.45 Abundance of the Ral GTPase effectors and endocytic adapters RalBP1-associated Eps domain-containing protein 1 and 2 (REPS1 and 2) were consistently increased in mesenchymal-like cells (up to 16-fold, Figure 4E) and the scaffolding protein and known REPS1 and 2 interactor RalA-binding protein 1 (RALBP1, Figure 4D) also showed increased abundance in mesenchymal-like cells, albeit with lower ratios (Figure 4E), suggesting that the AAK1/BMP2K interaction network may be important for acquiring plasticity.
An endocytic AAK1 interaction network promotes HCC cell EMP and therapy resistance
We next sought to clarify if the AAK1/BMP2K PPI network can drive HCC cell EMP and therapy resistance or merely acts as a bystander. AAK1 has been shown to function in clathrin-mediated endocytosis53 and canonical Notch and Wnt signaling that can both contribute to cancer cell EMP,54,55 whereas the function of BMP2K is poorly understood. REPS1 and 2 regulate receptor tyrosine kinase endocytosis and recycling downstream of Ras-related proteins RALA and B,56 cell migration, and NF-κB pathway activation.57,58 Likewise, RALBP1 acts in RALA- and B-mediated receptor endocytosis and serves as a GTPase activating protein (GAP) for CDC42 and RAC.59,60 First, to see if RALBP1, REPS1 and 2 interact with AAK1 or BMP2K, we performed a kinobead soluble competition experiment in FOCUS cell lysate using the selective AAK1 inhibitor LP-935509 (Figure 5A, Table S4),61 revealing that REPS1 and 2, RALBP1, and the four subunits of the adapter protein 2 (AP-2) complex were selectively competed along with AAK1; this was concordant with our kiCCA results from CYC116 competition in FOCUS cell lysates (Figure S7A) and strongly suggested that REPS1 and 2, and RALBP1 interacted with AAK1 rather than BMP2K. We further validated these results by Co-IP/MS using antibodies specific to AAK1, RALBP1, and REPS1 in FOCUS cell lysate (Figure S7B and Table S4), establishing AAK1 as the central kinase of the PPI network.
Figure 5. An AAK1 interaction network promotes EMP and therapy resistance in HCC and exhibits characteristics of a proliferation suppressor.
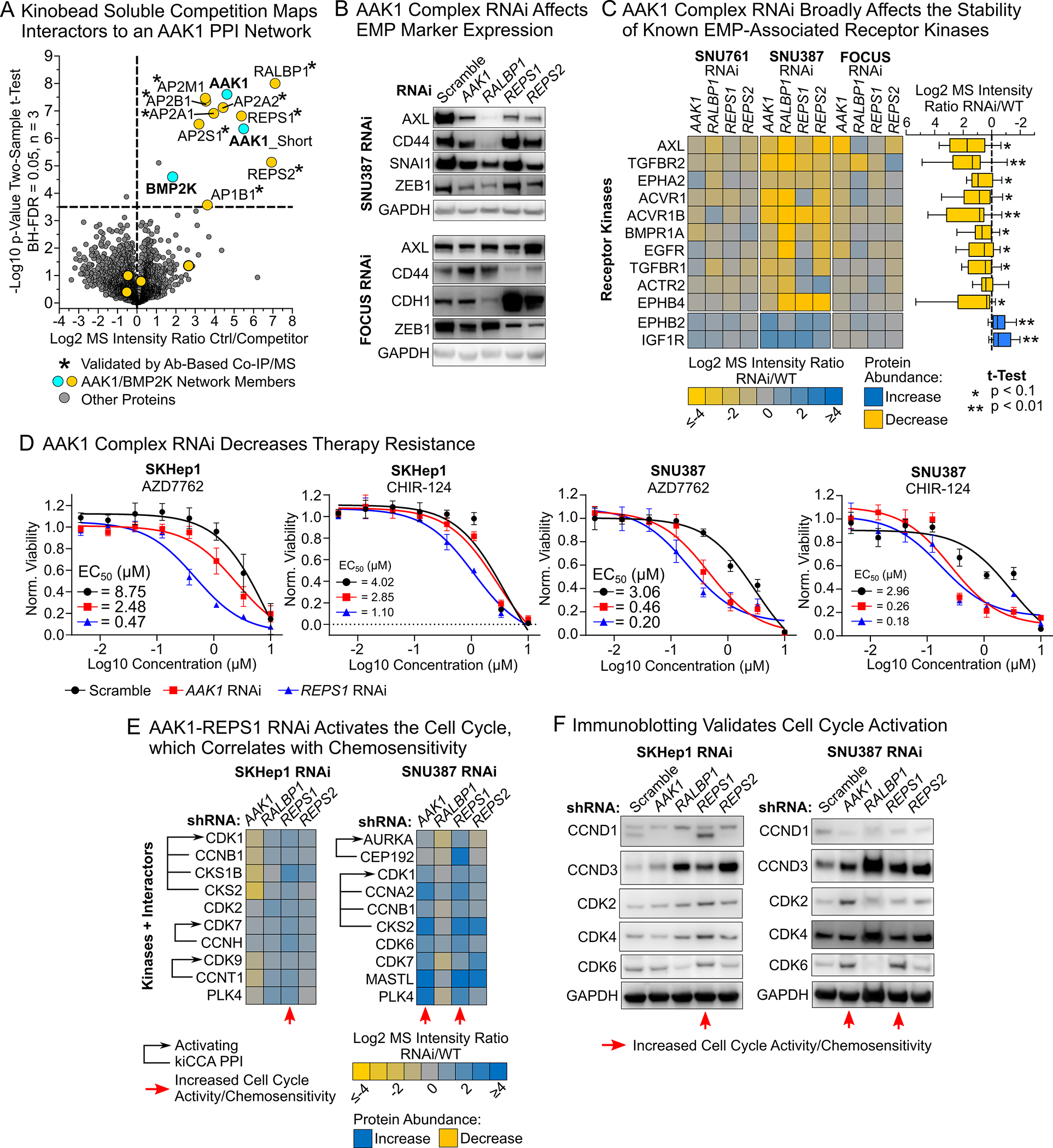
(A) Kinobead/LC-MS soluble competition experiments using 1 μM of the selective AAK1 inhibitor LB-935509 show that the PPI network is centered on AAK1, not BMP2K.
(B) Immunoblotting of EMP markers in SNU387 and FOCUS AAK1 network RNAi lines indicated alterations in EMP state.
(C) Heatmap showing EMP-associated receptor kinases that change in abundance in response to AAK1 PPI network RNAi in mesenchymal-like HCC cells. Kinase abundance differences were determined by kinobead/LC-MS profiling.
(D) Drug screen results demonstrating that AAK1 network RNAi lines are up to 18-fold more sensitive to the cell cycle checkpoint kinase (CHEK1) inhibitors AZD-7762 and CHIR-124.
(E) Kinobead/LC-MS profiling of SKHep1 and SNU387 lines shows that AAK1 and REPS1 RNAi causes upregulation of cell cycle-related kinases and their activating PPIs specifically in lines with increased sensitivity to CHEK1 inhibitors.
(F) Immunoblotting confirmed activation of the cell cycle specifically in AAK1 and REPS1 RNAi lines, suggesting that they act as proliferation suppressors in mesenchymal-like cells.
To test if AAK1 network components promote HCC cell EMP and drug resistance, we stably expressed shRNAs targeting AAK1, RALBP1, REPS1 and REPS2 or a scrambled sequence in the mesenchymal-like FOCUS, SKHep1, SNU761, and SNU387 cell lines (STAR Methods). By qPCR and immunoblotting, we consistently achieved near-complete knockdown of RALBP1, REPS1 and REPS2, and reduced AAK1 expression by 2- to 4-fold (Figure S8A, and S8B). Immunoblot analysis of different EMP markers in our RNAi lines showed reduced expression of the central EMP transcription factor ZEB1 in FOCUS, SNU387, and SNU761 cells, and revealed that RNAi also affected the expression of AXL, CD44, E-cadherin CDH1, and the transcriptional repressor Snail (SNAI1), albeit in a cell line-dependent manner (Figure 5B and S9); this suggested that the AAK1 complex can indeed promote EMP. Hypothesizing that the AAK1 network’s function in EMP may be linked to its role in kinase receptor endocytosis and recycling, we profiled the kinome of RNAi lines using kinobead/LC-MS (Table S4). In addition to validating successful knockdown of AAK1 PPI network components (Figure S10A), this revealed that the known EMP driver kinases AXL and TGFBR2, as well as the EGFR, the ephrin receptor A2 (EPHA2), and five additional members of the TGFβ receptor superfamily were consistently downregulated across three of the four HCC lines tested (Figure 5C, Table S4). Applying GSEA to our kinobead profiling data confirmed that AAK1 complex RNAi causes the downregulation of endocytosis-related pathways, HCC cell EMP related pathways like cell adhesion and WNT signaling, and survival signaling through protein kinase B and the mammalian target of rapamycin (mTOR) (Figure S10B and Table S4). In contrast, pathways related to insulin signaling, chromatin remodeling, and the cell cycle increased in response to RNAi, confirming transition to a more epithelial, hepatocyte-like state. Together, these results suggested that the AAK1-RALBP1-REPS1/2 network can promote HCC cell EMP and drug resistance by stabilizing specific kinase receptors that drive developmental and survival signaling.
To learn if the AAK1 complex could serve as a target to sensitize HCC cells to drug treatment, we conducted a screen for cell viability using ten kinase-targeted drugs and doxorubicin in scramble control versus AAK1 complex RNAi lines (Figure 5D and Table S4). RNAi lines, particularly SNU387 and SKHep1, showed up to 18-fold down-shifts in EC50s for inhibitors targeting the cell cycle checkpoint kinases CHEK1 (AZD7762 and CHIR-124), inhibitors that we found previously to be highly effective in killing rapidly proliferating, epithelial-like HCC cells compared to mesenchymal-like cells.45 To identify pathways mediating the RNAi-dependent increase in drug efficacy, we revisited our kinome profiling data of the most chemosensitized lines, SKHep1 and SNU387 (Figure 5E and Table S4). Cross-referencing fmPPIs in our interactome knowledgebase revealed that these cell lines activated cell cycle-related kinases and their signaling complexes specifically in response to AAK1 and REPS1 RNAi, which coincided with the greatest sensitization to CHEK1 inhibitors (Figure 5E). This also agreed with our GSEA of knockdown lines, as pathways related to cell cycle progression and DNA repair were upregulated with RNAi of the AAK1 network (Figure S10B and Table S4). Likewise, immunoblot analysis validated that AAK1 and REPS1 RNAi increased the expression of cell cycle-driving proteins (Figure 5F and Figure S11). These results indicated (1) that AAK1 and REPS1 act as proliferation suppressors in mesenchymal-like HCC cells,62 and (2) that AAK1 and REPS1 RNAi-mediated activation of proliferation exposes a vulnerability of drug resistant HCC cells that can be targeted with cell cycle checkpoint kinase inhibitors.
Systematic mapping of kinase PPIs in kinome profiling data from clinical tissue specimens
Mapping disease-associated interactome changes in pre-clinical animal models and clinical tissue specimens is a critical milestone in translating PPI network information into biomarkers and drug targets. Studies using kinobead/LC-MS profiling to characterize kinome aberrations in pre-clinical and clinical tissue specimens have transformed our understanding of disease mechanisms (Figure 6A);24,45,63–65 these studies contain valuable kinase interactome data that could greatly expand our understanding of in vivo disease biology, yet computational approaches to accurately interpret this information are lacking.
Figure 6. Applying our kiCCA interactome knowledgebase to map kinase PPIs in kinome-centric chemoproteomic datasets from clinical and pre-clinical tissues.
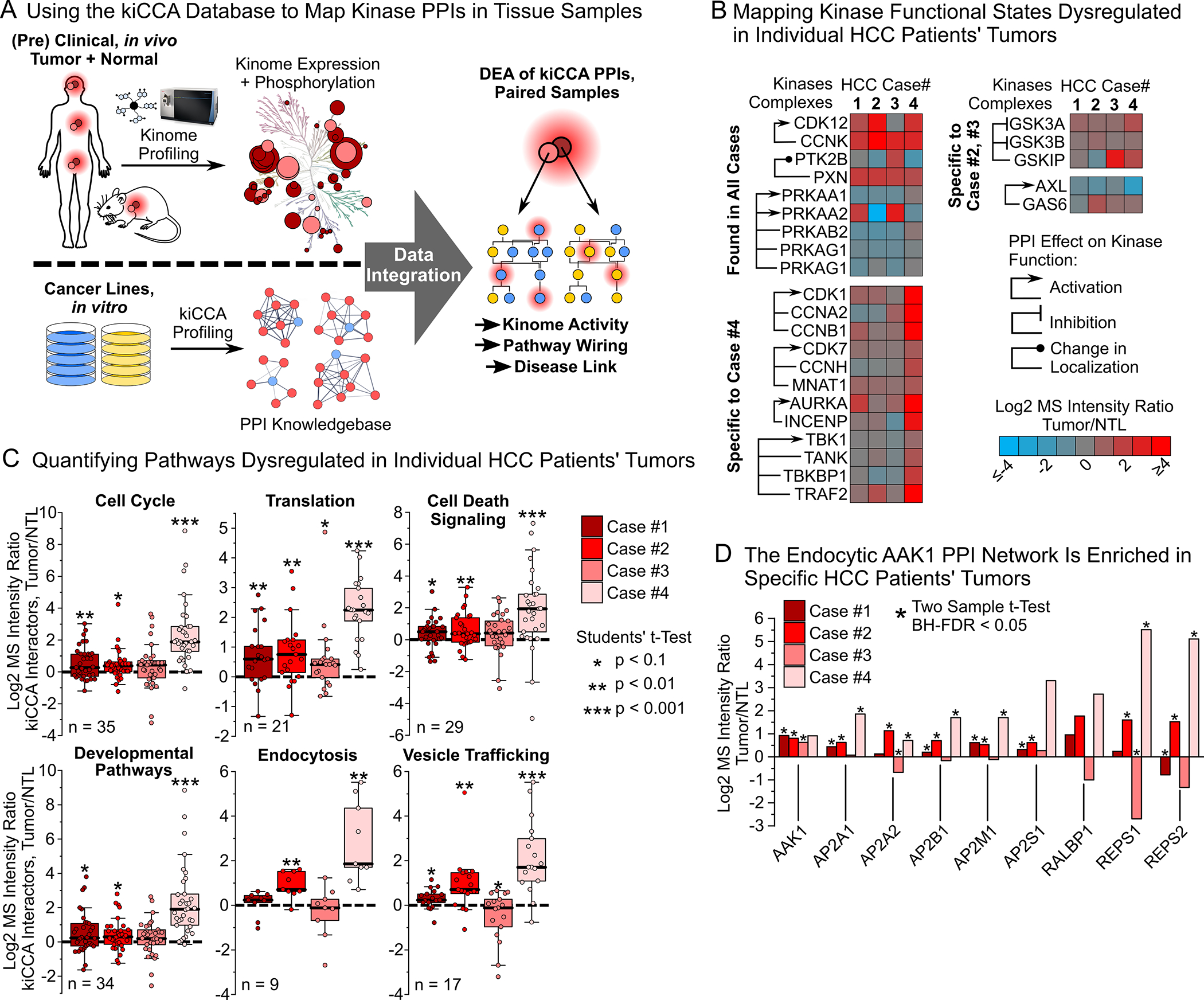
(A) Workflow for interpreting in vivo kinome profiling data from clinical specimens using our kiCCA knowledgebase.
(B) Integrating our kiCCA knowledgebase with kinobead profiling data from four, paired HCC patients’ tumor and non-tumor liver (NTL) samples revealed differential abundance of kinase PPIs, and thus aberrations in kinase functional states in vivo (two sample t-test, BH-FDR = 0.05, n = 5 or 6).
(C) Pathway mapping of kinase PPIs altered between HCC patients’ tumors and paired NTL tissues revealed dysregulation of cellular pathways (for statistics, see (B)). Each datapoint is the log2 MS intensity tumor/NTL ratio of a kiCCA interactor with significantly different abundance in at least one HCC case; all interactions were then compared across all HCC cases.
(D) DEA of the AAK1-mediated PPI network between HCC tumor and NTL tissues revealed frequent upregulation in tumors, suggesting important roles in HCC progression and drug resistance in vivo (for statistics, see (B)).
See also Table S3
To explore if our kiCCA interactome data can identify kinome PPIs in kinobead profiling data from clinical tissue specimens, we mapped high-confidence interactions in our knowledgebase to our previously published kinobead profiling dataset of four paired, clinical HCC and non-tumor liver (NTL) tissue samples (Figure 6A)45 and identified the interactions between 133 kinase groups and 275 non-kinase proteins (Table S4). Because the kiCCA knowledgebase aggregates kinome PPI information across a range of biological contexts and some proteins may have been observed to interact with more than one kinase, we applied our kiCCA score that considers both the kiCCA r-value and the frequency of identification across our 18-cancer line panel (STAR Methods), to determine the 275 most likely kinase PPIs; 199 of these interactions integrated 75 kinase groups into specific signaling pathways, and 39 fmPPIs specified the functional states of 24 kinase groups (Figure 6B and 6C and Table S4).
DEA between tumors and paired NTL tissues showed that 189 of these PPIs showed altered abundance, including several functional PPIs affecting kinases with important roles in HCC progression. For example, we found that the activating CDK12-CCNK interaction,66 and the paxillin (PXN)-PTK2B interaction that directs kinase activity to focal adhesions were upregulated in all four tumor samples compared to NTL,67 whereas the activating interactions of AMPK (PRKAA1 and 2) with its regulatory subunits were consistently downregulated in all tumors.68 This provides evidence for the broad relevance of CDK12 activity and focal adhesion signaling in HCC progression, and AMPK’s tumor suppressor function in HCC. We also identified functional PPIs specific to individual HCC cases; for instance, activating PPIs of the cell cycle kinases CDK1 and 7, and aurora kinase B (AURKB), and the NF-κB kinase TBK1 were enriched in tumor #4, inhibitory interactions of the WNT/β-catenin kinases GSK3A and B were enriched in tumor #3, and the activating interaction of the EMP and survival kinase AXL and GAS6 was specifically enriched in tumor #2 (Figure 6B). These results show that our interactome knowledgebase and kiCCA scoring can identify kinase activities in individual patient’s tumors, highlighting kiCCA’s utility for precision oncology.
To uncover in vivo mechanisms controlling HCC progression, we analyzed the pathways associated with differentially expressed kinase PPIs. This confirmed activation of the cell cycle in tumor #4, and upregulation of protein translation, developmental pathways, and apoptosis in most tumor samples compared to NTL tissue (Figure 6C), consistent with a cycle of proliferation and apoptosis in hepatocytes within the inflamed, cirrhotic liver that underlies most HCC cases.43 Strikingly, our pathway analysis revealed that endocytosis and vesicle transport pathways were highly active in tumors #2 and #4 compared to NTL, the same pathways that we found to be activated in mesenchymal-like and therapy resistant HCC lines. Specifically, REPS1 and REPS2 were significantly increased in abundance in two of four HCC tissues compared to NTL (Figure 6D). These results indicate that dysregulation of an endocytic AAK1 network may contribute to HCC EMP and therapy resistance in vivo, and that AAK1 and its interaction partners may serve as novel drug target candidates with high translational potential.
DISCUSSION
We introduced kiCCA, a chemoproteomic approach that exploits kinase inhibitor polypharmacology for highly multiplexed interactome mapping of the kinome. kiCCA allows the high-throughput profiling of various cell states and model systems using native cell and tissue lysates, entirely avoiding the use of antibodies and the expression of genetically tagged bait proteins. We demonstrated that kiCCA is a powerful approach for cell signaling and cancer research as it: (1) broadly quantifies kinase interactome changes associated with cancer types, cellular phenotypes like neuroblastoma cell NMP and HCC cell EMP; and (2) prioritizes kinase complexes for mechanistic studies and drug target discovery, as demonstrated by our identification of an AAK1 complex promoting HCC cell EMP and therapy resistance. We also showed that kiCCA combined with protein crosslinking captured the rapid, kinome-wide rearrangement of transient and low affinity signaling complexes caused by acute hormone stimuli, highlighting that kiCCA presents a unique tool for cell signaling research. We applied our kinase interactome knowledgebase from 18 diverse cancer lines to identify kinase signaling complexes in kinobead profiling data of clinical tumor samples to gain novel insights into kinase-dependent PPI aberrations in patients’ tumors. While our kinome PPI knowledgebase provides the broadest range of kinome PPIs mapped to date, future kiCCA experiments will increase the diversity of cell and tissue types while additional bespoke kinome PPI databases can be developed from specific cell lines or tissues to match a particular biological context. Our general approach shows the value of PPI network information in studying dysregulated signaling in disease and provides a roadmap for obtaining similar kinobead-based PPI networks for clinical proteomics.
Collectively, we curated an extensive reference set of PPIs that can determine kinase functional states and kinase–pathway integration, thereby allowing us to interpret the biological significance of changes in kinase interactomes. Kinome PPI abundance information derived from biochemical enrichment by kinobeads or other kinome-centric chemoproteomic tools 69 provide quantitative proteomics data that is distinct and complementary to phosphoproteomics data. We expect that the combination of both kinome PPI and phosphorylation site data will provide a more specific and sensitive measurement of kinome activity that would be important for comparing signaling activity between different biological states.
We demonstrated that kiCCA interactome data can identify disease mechanisms and drug targets by characterizing an AAK1-mediated PPI network that links endocytosis and vesicle trafficking pathways to HCC cell EMP and therapy resistance. Knockdown of AAK1 and its interaction partners REPS1 and REPS2, and RALBP1 reduced HCC cell EMP marker expression and decreased drug resistance, showing that kiCCA can identify kinase target candidates as well as non-kinase target candidates like REPS1 whose RNAi is better tolerated than AAK1 RNAi, and strongly sensitizes mesenchymal-like cancer cells to targeted therapy. Therefore, inhibiting the AAK1-REPS1 interaction may be a promising strategy to minimize drug cytotoxicity and maximize cancer therapy responses.8
The limitations of our kiCCA approach are the same as for any other AP-MS-based method, for instance, difficulties identifying weak and transient PPIs, and the lack of sub-cellular spatial resolution.17 Here, we demonstrated that kiCCA with protein crosslinking can increase the coverage of weak and transient PPIs. We speculate that subcellular fractionation, e.g., into cytosolic, membrane, and nuclear fractions, followed by kiCCA could further resolve the localization of kinase signaling complexes and reduce sample complexity to increase the number of identifiable signaling and transcription factor complexes. The throughput of kiCCA can be further increased iteratively by using smaller sets of KIPs and isobaric TMT labeling for analysis of entire interactomes in single LC-MS runs.
In summary, we presented a unique and powerful approach for studying kinase interactome dynamics in virtually any model system that can be broadly implemented in biomedical labs. We collated our kinase interactome data into a knowledgebase that is easily accessible through our supplementary tables (Tables S3) and an interactive Shiny web application (https://quantbiology.org/kiCCA), serving as an important resource for cancer and cell signaling researchers.
Limitations of the Study
kiCCA enables high-throughput kinome interactomics, however, it currently cannot identify kinase-kinase interactions or determine multiple kinase interactions of a protein in the same sample, therefore missing potentially important signaling events. Future iterations of kiCCA will integrate the kinase binding affinities of KIPs with kinobead competition-binding profiles to distinguish direct probe binding from kinase-kinase co-competition, and will utilize improved computational algorithms like network propagation methods70 to identify multiple kinase interactors of co-precipitating proteins. kiCCA with protein crosslinking identified numerous weak and transient PPIs, yet kiCCA still requires cell lysis, which dilutes cellular contents ~100-fold, and causes the dissociation of protein complexes. Future iterations of kiCCA will utilize soluble kinome affinity probes to capture kinase interactomes in situ. Studying how dysregulated AAK1 PPI network promoted HCC cell EMP and therapy resistance, we showed that RNAi of network components affected the stability of multiple EMP-associated receptor kinases, however, if this is caused by altered transcription or recycling and degradation of receptors, and if the effect can be exploited for pharmacological intervention will have to be clarified in future studies. Finally, the application of the kiCCA knowledgebase in a new tissue or disease model may not be accurate if there is a new cell/tissue-specific set of kinome PPIs that are not previously captured in the knowledgebase; in such situations, it may be necessary to generate a new model-specific kiCCA database.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Shao-En Ong, Department of Pharmacology, University of Washington, Seattle, WA 98195, USA, shaoen@u.washington.edu
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
MS .raw files, MaxQuant output files, and kiCCA correlation matrices generated by this study have been uploaded to the MassIVE repository of the University of San Diego under the acquisition number: MSV000088067. Original western blot images have been deposited at Mendeley Data and are publicly available as of the date of publication. A Shiny app for real time interrogation of the kinase interactomics data generated in this study can be accessed at https://quantbiology.org/kiCCA.
This study did not generate new code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and tissue culture conditions
C3A, SNU398, Hep3B2.1–7, U2-OS, SK-N-SH, SH-SY5Y, JeKo-1, HeLa, Jurkat, A-172, K562, and SNU449 cell lines were purchased from the American Type Culture Collection (ATCC). SNU761 and SNU886 were purchased from the Korean Cell Line Bank (KCLB). JHH6 and HuH-7 cells were purchased from the JRCB Cell Bank. FOCUS WT cells were obtained from the Laboratory of J. Wands, Brown University 72. FOCUS AXL RNAi cells were obtained from Dr. Taranjit Gujral of the Fred Hutchinson Cancer Research Institute, Seattle. All cells were grown at 37°C under 5% CO2, 95% ambient atmosphere. Fifteen cryo-frozen cell stocks were generated from the original vial from the cell bank or from the collaborator’s lab (passage 3). Experiments were performed with cells at <10 passages from the original vial. All cell media used were supplemented with 100x penicillin-streptomycin-glutamine (Thermo Fisher Scientific, Waltham, MA). FOCUS and HuH-7 cells were grown in Dulbecco’s minimum essential medium (DMEM) supplemented with 10% FBS (VWR Life Science, Seradigm). C3A, SNU398, Hep3B2.1–7, U2-OS, SK-N-SH, SH-SY5Y, JeKo-1, HeLa, Jurkat, A-172, K562, and SNU449 lines were grown in the ATCC-recommended medium. JHH6 cells were grown in William’s E medium, and SNU761 and SNU886 lines in RPMI 1640 medium all supplemented with 10% FBS. Cells were harvested when reaching 90% confluency or a density of 1×106 cells/ml.
METHOD DETAILS
RNAi knockdown experiments
Three shRNA sequences each targeting AAK1, RALBP1, REPS1, and REPS2 mRNAs were obtained from The RNAi Consortium (TRC) of the Broad Institute web portal (https://www.broadinstitute.org/rnai-consortium/rnai-consortium-shrna-library, ID Numbers: TRCN0000001943, TRCN0000199939, TRCN0000082348, TRCN0000053363, TRCN0000423162, TRCN0000436095, TRCN0000423057, TRCN0000428939, TRCN0000056210, TRCN0000305689, TRCN0000047918, TRCN0000047920) and cloned into the lentiviral pLKO.1 vector (Plasmid#10878, Dr. David Root’s Lab, Addgene, Watertown, MA) as previously described.78 Lentiviral particles were produced from individual pLKO.1 vectors, the pMD2.G plasmid (envelope, plasmid #12259, Didier Trono, Addgene), and the pCMVR8.74 plasmid (packaging, plasmid #22036, Didier Trono, Addgene) according to the manufacturer’s instructions (Addgene). Virus particle-containing cell culture supernatants were sterile filtered over 0.22 μM PES syringe filters (Millex-GP, Sigma Millipore, Burlington, MA), mixed 1:1 with fresh growth medium, 8 μg/mL final polybrene was added and the mixture added to target cells (70–80% confluency). Cells were incubated for 24h, the medium exchanged, and stable cell lines selected using puromycin (FOCUS: 4 μg/mL; SNU387 and SKHep1: 6 μg/mL; SNU761: 8 μg/mL) for 7–14 days. Puromycin-resistant cells were maintained in growth medium containing half of the selection concentration of puromycin. Target knockdown was validated using qPCR, immunoblotting, and kinobead/LC-MS profiling, and the stable cell lines with the highest knockdown among the three shRNAs used for each target were chosen to perform all downstream experiments.
Immunoblot analysis and antibodies
Antibodies used for immunoblotting were anti-E-cadherin (24E10, Cell Signaling Technology, CST, Cat # 3195), anti-AXL (C89E7, CST, Cat # 8661), anti-Snail (C15D3, CST, Cat # 3879), anti-ZEB1 (E2G6Y, CST, Cat # 70512), anti-CD44 (E7K2Y, CST, Cat # 37259), anti-GAPDH HRP conjugate (D16H11, CST, Cat # 8884), anti-AAK1 (E8M3P, CST, Cat # 61527), anti-RALBP1 (D87H8, CST, Cat # 5739), anti-REPS1 (D6F4, CST, Cat # 6404), anti-CDK4 (D9G3E, CST Cat # 12790), anti-CDK6 (DCS83, CST, Cat # 3136), anti-CDK2 (78B2, CST, Cat # 2546), anti-Cyclin D1 (92G2, CST, Cat # 2978), and anti-Cyclin D3 (DCS22, CST, Cat # 2936). Cell lysis and immunoblotting experiments were performed using standard procedures. Briefly, cells were rinsed twice with ice-cold phosphate buffered saline (PBS), lysed in modified RIPA buffer V1 (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40 (v/v), 0.25% Na-deoxycholate (w/v), 1 mM EDTA, 10 mM NaF, 5% glycerol (v/v), pH 7.8) supplemented with HALT protease inhibitor (100x, Thermo Fisher Scientific, Waltham, MA), and lysates clarified by centrifugation at 21,000 rcf for 20 minutes at 4°C. Protein concentration was quantified using the Piece 660 nm Protein Assay Reagent (Pierce, Rockford, IL). Lysates were mixed with NuPAGE LDS Sample Buffer (4X, Thermo Fisher Scientific) containing 50 mM DTT and heated for 5 min at 95°C. 20 μg of protein was separated on Bolt 4–12% Bis-Tris Protein Gels (Thermo Fisher Scientific) and electro-transferred onto nitrocellulose membranes. The buffer used for blocking and antibody incubation was 5% BSA in TBS-T (50 mM NaCl, 150 mM Tris-HCl, 1% Tween-20, pH = 7.8). Membranes were incubated with goat anti-rabbit HRP conjugate, and bands visualized using the Clarity Western ECL Substrate (Bio-Rad, Hercules, CA) and the Fluor Chem E imaging system (Protein Simple, San Jose, CA).
Quantitative Real-Time PCR (qPCR) Analysis of mRNA Expression
shRNA-mediated knockdown was validated by quantifying the target’s mRNA expression levels using quantitative real-time PCR (qPCR). Briefly, cells were cultured on 35 mm dishes until reaching 80–90% confluency and total mRNA was isolated using the TRIzol reagent according to manufacturer’s instructions (Thermo Fisher Scientific). mRNA quality was controlled by running 1% agarose gels and assessing the presence of sharp, clear 28S and 18S rRNA bands. 0.5 μg of total RNA was used to generate first-strand cDNA using the Protoscript II First Strand cDNA Synthesis Kit (New England Biolabs, Ipswich, MA). The resulting cDNA was subjected to qPCR using human gene-specific primers for AAK1, RALBP1, REPS1, and REPS2, and two housekeeping genes, i.e., PSMB2 and RAB7A. The qPCR reaction was performed using QuantStudio 5 Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific) using the following program:
| Step | Temp (°C) | Time (mm.ss) | Cycles |
|---|---|---|---|
| Hold (Enzyme Active) | 50, 95 | 02:00, 10:00 | 1 |
| PCR (Denature, Anneal, Extend) | 95, 50, 60 | 00:15, 00:15, 01:00 | 50 |
| Dissociation/Melting Curve | 95, 60, 95 | 00:15, 01:00, 00:15 | 5 |
The mRNA levels of each gene were normalized relative to the mean levels of the two housekeeping genes and compared with the data obtained from cell lines carrying a stably incorporated scramble shRNA using the 2-ΔΔCt method. According to this method, the normalized level of a mRNA, X, is determined using Equation (1):
| (1) |
where Ct is the threshold cycle (the number of the cycle at which an increase in reporter fluorescence above a baseline signal is detected), GOI refers to the gene of interest, and CTL refers to a control housekeeping gene. This method assumes that Ct is inversely proportional to the initial concentration of mRNA and that the amount of product doubles with every cycle.
Inhibitor treatment of RNAi and scramble lines for the growth inhibition assay
1800 cells/well were seeded onto white flat bottom half area 96-well plates (Greiner Bio-One, Kremsmünster, AT) in 50 μl of growth medium and allowed to attach in an incubator for 24 h. Then the drugs in DMSO and/or DMSO vehicle controls as 11X solutions in growth medium were added to a total volume of 55 μl and 0.1% DMSO final. The cells were grown in an incubator for another 72 h. Then, 55 μl of CellTiter-Glo 2.0 (Promega, Madison, WI) reagent/well were added according to the manufacturer’s instructions and luminescence was quantified with a SpectraMax 190 plate reader (Molecular Devices, San Jose, CA). The drugs AZD7762 (CHEK1 inhibitor, Selleckchem, Houston, TX), CHIR-124 (CHEK1 inhibitor, ApexBio, Houston, TX), Selumetinib (MEK1/2 inhibitor, Selleckchem), Dasatinib (SRC inhibitor, Selleckchem), Lenvatinib (FGFR inhibitor, Selleckchem), Sorafenib and Regorafenib (BRAF inhibitors, Selleckchem), Cabozantinib (AXL and MET inhibitor, Selleckchem), and Doxorubicin (Cytotoxic/TopII inhibitor, Selleckchem) were applied at 8 different concentrations between 10 μM and 4.6 nM (3-fold dilution steps). The drugs Dinaciclib (CDK inhibitor, Selleckchem) and Volasertib (PLK1/BRD2 inhibitor, Selleckchem) were applied at 8 different concentrations between 1 μM and 0.46 nM (3-fold dilution steps). Experiments were performed in four biological replicates. Growth inhibition curves were fitted using the GraphPad Prism software package (V5.0a) with a least-squares nonlinear regression model for curve fitting (One site - Fit logIC50 function).
Preparation of optimized kinobead mixture
The seven used kinobead affinity reagents were either synthesized in house or custom synthesized through Bellen Chemistry (Beijing, China), and kinobeads were prepared as previously described 23,32,79. For optimal coverage of the human kinome an optimized mixture of the seven kinobead reagents was prepared as previously described.32 Briefly, 1 ml of reagent 1, 0.5 ml of reagents 2, 3 and 7, respectively, and 0.25 ml of reagents 4, 5 and 6, respectively, were mixed to yield 3.25 ml of the complete kinobead mixture. All reagents were a 50% slurry in 20% aq. ethanol.
Kinase affinity enrichment, KI competition and on-bead digestion
Kinase affinity enrichment, KI competition, and on-bead digestion was performed as previously described.23,30,71 Briefly, to 150 μL of cell lysate (5 mg protein per mL) in modified RIPA buffer V1 (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40 (v/v), 0.25% Na-deoxycholate (w/v), 1 mM EDTA, 10 mM NaF, 5% glycerol (v/v), pH 7.8) containing HALT protease inhibitor cocktail (100x, Thermo Fisher Scientific, Waltham, MA) and phosphatase inhibitor cocktail II and III (100x, Sigma-Aldrich, St Louis, MO) 1.5 μL DMSO (vehicle control) or the corresponding inhibitor solution in DMSO (competition) were added to a final concentration 1% DMSO. The lysate was vortexed at intermediate speed intermittently every 5 min for 20 min while being kept on ice. Meanwhile, 40 μl of a 50% slurry of the in-house-made, optimized kinobead mixture in 20% aq. ethanol were prepared for each pulldown experiment. The beads were washed twice with 400 μl modified RIPA buffer and lysates containing DMSO, or inhibitor were added. The mixture was incubated on a tube rotator for 3h at 4°C and then the beads were pelleted rapidly at 2000 Xg on a benchtop centrifuge (5s). After removal of the supernatant, the beads were rapidly washed twice with 400 μl of ice-cold mod. RIPA buffer and three times with 400 μl ice-cold tris-buffered saline (TBS, 50 mM tris, 150 mM NaCl, pH 7.8) to remove detergents. 100 μl of freshly prepared denaturing buffer (8M urea, 100 mM Tris, pH 8.5) containing 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP*HCl) and 10 mM chloroacetamide (CAM), were added and the slurry agitated on a thermomixer at 37°C and 1400 rpm for 30 min. The mixture was diluted 2-fold with 100 mM triethylamine bicarbonate (TEAB), the pH adjusted to 8–9 by addition 1 N aq. NaOH; 2 μg LysC were added, and the mixture agitated on a thermomixer at 1400 rpm at 37°C for 2 h. Then, the mixture was diluted another 2-fold with 100 mM TEAB, 2 μg MS-grade trypsin (Thermo Fisher Scientific, Waltham, MA) were added, and the mixture agitated on a thermomixer at 1400 rpm at 37°C overnight. Then, 6 μL of formic acid (FA) were added (1.5% FA final) to adjust to pH 3 and peptides were extracted and desalted using C18 StageTips according to the published protocol.80 For kinobead/LC-MS profiling of RNAi cell lines the same protocol was applied except that the lysates were not preincubated with DMSO or inhibitor. The following kinase inhibitors were used as KIPs for competition experiments at the given final concentrations: GSK-690693 (10 μM, MedChemExpress, MCE, Monmouth Junction, NJ), Miliciclib (10 μM, MCE), Rebastinib (10 μM, MCE), AT9283 (10 μM, MCE), TAK-901 (10 μM, MCE), RGB-286638 (10 μM, MCE), Flavopiridol*HCl (10 μM, MCE), PF-562271 besylate (10 μM, MCE), Dabrafenib mesylate (10 μM, MCE), OTSSP167*HCl (10 μM, MCE), CYC-116 (10 μM, MCE), Silmitasertib (10 μM, MCE), SB1317 (10 μM, MCE), XL228 (10 μM, MCE), Sapanisertib (10 μM, MCE), PF-3758309 (10 μM, ApexBio), Staurosporine (replacing the structurally closely related K-252a, 1 μM, LC Labs, Woburn, MA), AZD-7762 (10 μM Selleckchem), Bosutinib (10 μM, Selleckchem), Dasatinib (10 μM, Selleckchem), LP-935509 (1 μM, MCE), and Linsitinib (10 μM, ApexBio).
Kinobead/LC-MS competition with the 21 KIPs and formaldehyde-mediated protein crosslinking
Kinobead competition with formaldehyde-mediated protein crosslinking was performed as described in ‘Kinase affinity enrichment, KI competition, and on-bead digestion’ above with the following modifications. To 150 μL of cell lysate (5 mg protein per mL) in modified RIPA buffer V2 (50 mM HEPES, 150 mM NaCl, 1% NP-40 (v/v), 0.25% Na-deoxycholate (w/v), 1 mM EDTA, 10 mM NaF, 5% glycerol (v/v), pH 7.8) containing HALT protease inhibitor cocktail (100x, Thermo Fisher Scientific, Waltham, MA) and phosphatase inhibitor cocktail II and III (100x, Sigma-Aldrich, St Louis, MO) 1.5 μL DMSO (vehicle control) or the corresponding inhibitor solution in DMSO (competition) were added to a final concentration 1% DMSO. The lysate was vortexed at intermediate speed intermittently every 5 min for 20 min while being kept on ice. The mixture was added to the kinobeads and incubated on a tube rotator for 3h at 4°C and then 4 μL of 37 wt% aq. Formaldehyde solution was added (1% concentration final). The mixture was incubated on a tube rotator for an additional 30 min at 4°C and then the beads were pelleted rapidly at 2000 rpm on a benchtop centrifuge (5s). After removal of the supernatant, the beads were rapidly washed twice with 400 μl of ice-cold mod. RIPA buffer V2 and three times with 400 μl ice-cold HEPES-buffered saline (HBS, 50 mM HEPES, 150 mM NaCl, pH 7.8) to remove detergents. 100 μl of the denaturing buffer (6M Gdn*HCl, 100 mM Tris-HCl, pH 8.5) containing 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP*HCl) and 10 mM chloroacetamide (CAM), were added and the slurry agitated on a thermomixer at 70°C and 1400 rpm for 30 min to reverse crosslinking. The mixture was then subjected to the same digestion protocol and downstream handling as described above “Kinase affinity enrichment, KI competition, and on-bead digestion”.
Co-immunoprecipitation/MS (Co-IP/MS) analyses of AAK1 and CK2 PPI networks
200 μl of the corresponding cell lysate in modified RIPA buffer V1 (5 mg/mL protein) containing protease and phosphatase inhibitors (see ‘Kinase affinity enrichment, KI competition and on-bead digestion’) were incubated with antibodies against AAK1, RALBP1, or REPS1 (FOCUS cell lysate, see ‘Immunoblot analysis and antibodies’), CK2α or CK2β (U2-OS cell lysate, Novus Biologicals, CK2α Ab: polyclonal, # NB100–378, CK2β Ab: polyclonal, #NBP1–06515) or GFP (control, clone D5.1, CST, Cat # 2956) at the manufacturer’s recommended concentrations, respectively, and agitated overnight on a tube rotator at 4°C. The next day, Pierce Protein A Agarose (Thermo Fisher Scientific) was washed twice with ice-cold modified RIPA buffer V1 and 40 μL aliquots of a 50% bead slurry were added to each lysate/antibody mixture. The slurry was agitated for 3h on a tube rotator at 4°C, the supernatant was aspirated, and then the beads were washed twice with ice-cold modified RIPA buffer V1 and three times with TBS. Proteins were reduced, alkylated, and eluted by adding 100 μL denaturing buffer (8M urea, 5mM TCEP, 10mM CAM, 100 mM Tris pH 7.8) to beads and agitating them on a thermomixer at 1,400 rpm for 30 min at 37°C. The supernatant containing the protein was transferred to a new tube and diluted two-fold with 100 mM TEAB, and the pH was adjusted to 8.5 using 1N NaOH solution. 2 μg of Lys-C (Wako Chemicals) were added, and samples agitated on a thermo mixer at 1,400 rpm for 2 h at 37°C. Then, samples were diluted another two-fold with 100 mM TEAB and 2 μg of MS-grade trypsin (Thermo Fisher Scientific) were added. The mixture was agitated on a thermomixer at 1,400 rpm overnight at 37°C. The resulting peptide solution was acidified with formic acid to achieve pH <3 (1.5% FA final) and desalted using C18 StageTips according to the published protocol.80 Co-IP/MS experiments were performed in three biological replicates per antibody.
nanoLC-MS/MS analyses
LC-MS analyses were performed as described previously with the following minor modifications.23,32 Peptide samples were separated on an EASY-nLC 1200 System (Thermo Fisher Scientific) using 20 cm long fused silica capillary columns (100 μm ID, laser pulled in-house with Sutter P-2000, Novato CA) packed with 3 μm 120 Å reversed phase C18 beads (Dr. Maisch, Ammerbuch, DE). The LC gradient was 120 min long with 5–35% B at 300 nL/min. LC solvent A was 0.1% (v/v) aq. acetic acid and LC solvent B was 20% 0.1% (v/v) acetic acid, 80% acetonitrile. MS data was collected with a Thermo Fisher Scientific Orbitrap Fusion Lumos. Data-dependent analysis was applied using Top15 selection with CID fragmentation.
Computation of MS raw files
Data .raw files were analyzed by MaxQuant/Andromeda73 version 1.5.2.8 using protein, peptide and site FDRs of 0.01 and a score minimum of 40 for modified peptides, 0 for unmodified peptides; delta score minimum of 17 for modified peptides, 0 for unmodified peptides. MS/MS spectra were searched against the UniProt human database (updated July 22nd, 2015). MaxQuant search parameters: Variable modifications included Oxidation (M) and Phospho (S/T/Y). Carbamidomethyl (C) was a fixed modification. Max. missed cleavages was 2, enzyme was Trypsin/P and max. charge was 7. The MaxQuant “match between runs” feature was enabled. The initial search tolerance for FTMS scans was 20 ppm and 0.5 Da for ITMS MS/MS scans.
QUANTIFICATION AND STATISTICAL ANALYSIS
MaxQuant output data processing
MaxQuant output files were processed, statistically analyzed and clustered using the Perseus software package v1.5.6.0.74 Human gene ontology (GO) terms (GOBP, GOCC and GOMF) were loaded from the ‘mainAnnot.homo_sapiens.txt’ file downloaded on 02.03.2020. Expression columns (protein and phosphopeptide intensities) were log2 transformed and normalized by subtracting the median log2 expression value from each expression value of the corresponding data column. Potential contaminants, reverse hits and proteins only identified by site were removed. Reproducibility between LC-MS/MS experiments was analyzed by column correlation (Pearson’s r) and replicates with a variation of r > 0.25 compared to the mean r-values of all replicates of the same experiment (cell line or knockdown experiment) were considered outliers and excluded from the analyses. Data imputation was performed in Perseus using a modeled distribution of MS intensity values downshifted by 1.8 and having a width of 0.2.
Kinobead competition correlation analysis (kiCCA)
For each cell line and condition tested, 21 KIP competition experiments and one DMSO control experiment were performed in biological duplicate, resulting in 44 kinobead pulldown and LC-MS experiments per condition/cell line. We called a kinase or non-kinase protein competed if it showed a log2 MS intensity ratio of ≥ 0.75 and passed a two-sample t-test p < 0.1 with at least one of the 21 KIs used, i.e., comparing the two DMSO control experiments and two corresponding KI competition experiments. We then correlated MS intensity values of all competed kinases and co-competed non-kinase proteins using Pearson moment correlation (n = 44). Then, kinases that showed similar competition behavior were combined into groups (see “Determining kinase groups with similar KIP binding profiles” and Table S2), and the maximum r-value of members retained for that group. Next, we removed kinase groups that showed very large numbers of PPIs with high positive r-values, which was caused by systemic shifts in MS intensity for certain non-kinase proteins between biological replicates. These kinases show a systematic upshift in PPI r-values that can be recognized by an unusually high median and 3rd quartile (Q3) value for the kiCCA r-value distribution. Accordingly, we calculated the Q3 for each kinase group, determined high, positive outliers of Q3 values using box plots (outlier = 1.5*IQR), and removed kinase groups that appeared as outliers from downstream analysis. Finally, the kinase group which showed the highest r-value for each non-kinase protein was determined, representing the most probably kinase group–protein interaction among all possible interaction for each cell line or condition.
Differential Expression Analysis (DEA)
To identify differentially expressed proteomic features between cell lines, tissues, and treatment conditions, we applied either a two-sample t-test or a student’s t-test against the null hypothesis, applying Benjamini-Hochberg correction for multiple hypothesis testing (FDR < 0.05, discovery mode in kinobead profiling and kiCCA data), or we applied a simple p < 0.05 (validation mode in kinobead and Co-IP/MS data). Briefly, for two sample t-testing of protein MS intensity differences in kinobead profiling data, we calculated the differences of the mean log2 MS intensities in each population (n = 5 or 6, log2 MS intensity ratio) and tested for significant differences between the two populations. For Student’s t-testing of differential protein expression between the kiCCA datasets of each cell line or condition, we first calculated paired mean differences of log2 MS intensities for each kinome interactome probe (KIP) or DMSO control (n = 22), and then tested the population of ratio values against the null hypothesis, the mean log2 differences across the probe panel representing protein expression changes.
Determining kinase groups with similar KIP binding profiles
Like kiCCA correlation analysis, kinase MS Intensity values were correlated with one another for each cell line and condition tested to identify kinases with very similar KIP binding profiles. Thus, kinases that show an r-value > 0.9 in at least 7 of 21 tested cell line were combined into kinase groups, defining that the interactors of kinases in these groups cannot be distinguished using kiCCA. Examples are PRKAA1 and 2, AAK1 and BMPK, as well as STK24 and STK26 (see Table S2, Tab ‘Kinase Groups’)
Plotting STRING Interaction Networks
PPI network models were plotted using the STRING web application version 11.5 with the following settings: Edges were scaled with confidence, and only ‘physical subnetwork’ interactions were considered, i.e., only considering text mining, experiments, and databases.38
KS-test analysis and Receiver Operating Characteristic (ROC) plots
Combined PPIs from BioGrid (v.4.4.200, July 25 2021)34, BioPlex13, and Buljan et. al. 202012 were used to populate the ‘known’ PPIs in our dataset and used as the binary classifier. The kiCCA Pearson’s r-value was used as the discrimination variable. KS plots were generated with the ‘ROCit’ package in R. ROC p-values were determined with the ‘verification’ package in R.
Kinome dendrograms
Kinome dendrograms were prepared using the KinMap web application (http://kinhub.org/kinmap/).77
Mapping high confidence kiCCA PPIs that determine kinase functional states
High confidence kinase interaction partners identified by kiCCA were searched in the BioGRID34 and UniProt42 databases. PubMed IDs were retrieved for publications that previously described the effect of a PPI on kinase function, i.e., activation, inhibition, or a change in cellular localization. Kinase PPIs for which there was evidence from a closely related kinase only, for example the Src-family kinases SRC and FYN, were marked with ‘By similarity’ (see Table S3).
Mapping high confidence kiCCA interactors to 32, disease relevant GOBP terms
The search strings presented in Table S2 were used to annotate pathway membership of the 684 high confidence kiCCA interactors in gene ontology-biological process (GOBP) terms. Proteins were annotated using Perseus and the ‘mainAnnot.homo_sapiens.txt’ file downloaded on 02.03.2020.
Calculating the kiCCA score
To determine the rank order of most likely kinase interactors of non-kinase proteins that can interact with several kinases in our kinase interactome knowledgebase, we introduced a kiCCA score for each kinase PPI that considers both the mean kiCCA r-value and the number of times the interaction was identified in the 21 cancer cell lines and cell states. The kiCCA score can be calculated according to the formula (2)
| ()2 |
Where the mean kiCCA Pearson’s r-value across all samples and n is the number of cell lines and states tested (number of samples, in our case 21).
GSEA Analysis
For gene set enrichment analysis (GSEA), we used the ssGSEA2.0 script in R together with the Gene Ontology: Biological Process (GOBP) gene set of the MSigDB database (‘c5.bp.v7.0.symbols’) according to the published protocol with the following minor modifications.46 To rank gene names, we calculated a compound score using the two sample t-test of Student’s t-test log2 MS intensity ratio multiplied by the -log10 p-value. The parameters used for GSEA were: sample.norm.type = “none”, weight = 1, statistic = “area.under.RES”, output.score.type = “NES”, nperm = 1e3, min.overlap = 10, correl.type = “z.score”, par = T, spare.cores = 1, export.signat.gct = T, extended.output = T. To display GSEA results in the heatmap Figure S10B, we calculated an adjusted normalized enrichment score (NES), by multiplying the NES with the -log10 FDR.
ADDITIONAL RESOURCES
An interactive web application of the kinome interactome data in Supplemental Table 3 is available: https://quantbiology.org/kiCCA
Supplementary Material
Table S4. Results from kiCCA DEA, kinobead/LC-MS profiling of RNAi lines and HCC tissues, and GSEA; Supplement to Figures 3–6, S4–7, and S10. MaxQuant protein groups output data from kiCCA analysis of HuH7, SNU761, SK-N-SH, SH-SY5Y, and EGF-stimulated HeLa cells, as well as kinobead/LC-MS profiling data from AAK1 complex RNAi knockdown lines and AAK1 kinobead competition experiments. Drug Screening results in AAK1 network RNAi lines. All GSEA results.
Table S1. Determining an optimal set of kinase interactome probe (KIP); Supplement to Figures 2 and S1. Kinobead/LC-MS competition data from Klaeger et al., Science, 2017 and correlation clustering analysis results.
Table S2. Supporting data for KIP competition, kinase groups, and plasticity markers. Supplement to Figures 2–4 and S3. MaxQuant protein groups output data obtained for KIP competition in unstimulated HeLa Cells, list of kiCCA kinase groups, and list of cancer cell plasticity markers used for unsupervised clustering.
Table S3. kiCCA interactome knowledgebase, functional marker PPIs, and pathway associations; Supplement to Figures 3–6, and S6. Summary spreadsheet of all kiCCA interactions across the 18-cancer cell line panel to serve as reference knowledgebase, list of curated fmPPIs with literature references indicating functional consequences of PPIs, and list of GOBP pathway annotations for all identified high-confidence kiCCA interactors.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Snail (C15D3) | Cell Signaling Technology | Cat# 3879, RRID: AB_2255011 |
| E-Cadherin (24E10) Rabbit mAb | Cell Signaling Technology | Cat# 3195, RRID: AB_2291471 |
| ZEB1 (E2G6Y) | Cell Signaling Technology | Cat# 70512 |
| CD44 (E7K2Y) | Cell Signaling Technology | Cat# 37259, RRID: AB_2750879 |
| GAPDH (D16H11, HRP conjugate) | Cell Signaling Technology | Cat# 8884, RRID: AB_11129865 |
| AAK1 (E8M3P) | Cell Signaling Technology | Cat# 61527 |
| RALBP1 (D87H8) | Cell Signaling Technology | Cat# 5739, RRID: AB_10697484 |
| Axl (C89E7) Rabbit mAb | Cell Signaling Technology | Cat# 8661, RRID: AB_11217435 |
| REPS1 (D6F4) | Cell Signaling Technology | Cat# 6404, RRID: AB_11220228 |
| CDK4 (D9G3E) | Cell Signaling Technology | Cat# 12790, RRID: AB_2631166 |
| CDK6 (DCS83) | Cell Signaling Technology | Cat# 3136, RRID: AB_2229289 |
| CDK2 (78B2) | Cell Signaling Technology | Cat# 2546, RRID: AB_2276129 |
| Cyclin D1 (92G2) | Cell Signaling Technology | Cat# 2978, RRID: AB_2259616 |
| Cyclin D3 (DCS22) | Cell Signaling Technology | Cat# 2936, RRID: AB_2070801 |
| GFP (D5.1) | Cell Signaling Technology | Cat# 2956, RRID: AB_1196615 |
| CK2α (polyclonal) | Novus Biologicals | Cat# NB100-378, RRID: AB_2230121 |
| CK2β (polyclonal) | Novus Biologicals | Cat# NBP1-06515, RRID: AB_1582302 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Kinobead affinity capture reagents | Laboratory of Dustin J. Maly, University of Washington71 | Compounds #1, 2, 3, 4, 5, 6 and 7 |
| AZD7762 | Selleckchem | Cat#: S1532, CAS#: 860352-01-8 |
| CHIR-124 | APExBIO | Cat#: A8394, CAS#: 405168-58-3 |
| GSK-690693 | MedChemExpress | Cat#: HY-10249, CAS#: 937174-76-0 |
| Milciclib | MedChemExpress | Cat#: HY-10424, CAS#: 802539-81-7 |
| Rebastinib | MedChemExpress | Cat#: HY-13024, CAS#: 1020172-07-9 |
| AT9283 | MedChemExpress | Cat#: HY-50514, CAS#: 896466-04-9 |
| TAK-901 | MedChemExpress | Cat#: HY-12201, CAS#: 934541-31-8 |
| RGB-286638 | MedChemExpress | Cat#: HY-15504, CAS#: 784210-87-3 |
| Flavopiridol*HCl | MedChemExpress | Cat#: HY-10006, CAS#: 131740-09-5 |
| PF-562271 besylate | MedChemExpress | Cat#: HY-10458, CAS#: 939791-38-5 |
| Dabrafenib mesylate | MedChemExpress | Cat#: HY-14660A, CAS#: 1195768-06-9 |
| OTSSP167*HCl | MedChemExpress | Cat#: HY-15512A, CAS#: 1431698-10-0 |
| CYC-116 | MedChemExpress | Cat#: HY-10558, CAS#: 693228-63-6 |
| Silmitasertib | MedChemExpress | Cat#: HY-50855, CAS#: 1009820-21-6 |
| SB1317 | MedChemExpress | Cat#: HY-15166, CAS#: 937270-47-8 |
| XL228 | MedChemExpress | Cat#: HY-15749, CAS#: 898280-07-4 |
| Sapanisertib | MedChemExpress | Cat#: HY-13328, CAS#: 1224844-38-5 |
| PF-3758309 | APExBIO | Cat#: A3716, CAS#: 898044-15-0 |
| Staurosporine | LC-Labs | Cat#: S-9300, CAS#: 62996-74-1 |
| Bosutinib | Selleckchem | Cat#: S1014, CAS#: 380843-75-4 |
| Dasatinib | Selleckchem | Cat#: S1021, CAS#: 302962-49-8 |
| Linsitinib | APExBIO | Cat#: A8334, CAS#: 867160-71-2 |
| LP-935509 | MedChemExpress | Cat#: HY-117626, CAS#: 1454555-29-3 |
| Selumetinib (AZD6244) | Selleckchem | Cat#: S1008, CAS#: 606143-52-6 |
| Lenvatinib | Selleckchem | Cat#: S1164, CAS#: 417716-92-8 |
| Sorafenib | Selleckchem | Cat#: S7397, CAS#: 284461-73-0 |
| Regorafenib | Selleckchem | Cat#: S1178, CAS#: 755037-03-7 |
| Cabozantinib | Selleckchem | Cat#: S1119, CAS#: 849217-68-1 |
| Dinaciclib | Selleckchem | Cat#: S2768, CAS#: 779353-01-4 |
| Volasertib (BI 6727) | Selleckchem | Cat#: S2235, CAS#: 755038-65-4 |
| Doxorubicin | Selleckchem | Cat#: E2516, CAS#: 23214-92-8 |
| Seradigm Fetal Bovine Serum (FBS) | VWR Life Science | Cat#: 97068-085 |
| Lysyl Endopeptidase, Mass Spectrometry Grade (Lys-C) | Wako | Cat#: 125-05061 |
| Pierce Trypsin Protease, MS Grade | Thermo Fisher Scientific | Cat#: 90058 |
| HALT Protease Inhibitor | Thermo Fisher Scientific | Cat#: 78430 |
| NuPAGE LDS Sample Buffer | Thermo Fisher Scientific | Cat#: NP0007 |
| Bolt 4-12% Bis-Tris Protein Gels | Thermo Fisher Scientific | Cat#: NW04120BOX and NW04127BOX |
| TRIzol Reagent | Thermo Fisher Scientific | Cat#: 15596026 |
| Phosphatase Inhibitor Cocktail 2 | Sigma-Aldrich | Cat#: P5726-5ML |
| Phosphatase Inhibitor Cocktail 3 | Sigma-Aldrich | Cat#: P0044-5ML |
| Pierce Protein A Agarose | Thermo Fisher Scientific | Cat#: 20333 |
| ReproSil-Pur 120 C18-AQ, 3 µm | Dr. Maisch | Cat#: r13.aq. |
| Critical Commercial Assays | ||
| Pierce 660 nm Protein Assay Reagent | Thermo Fisher Scientific | Cat#: 22660 |
| RNeasy Mini Kit | Quiagen | Cat#: 74104 |
| CellTiter-Glo 2.0 Assay | Promega | Cat#: G9241 |
| Clarity Western ECL Substrate | Bio-Rad | Cat#: 1705060 |
| Protoscript II First Strand cDNA Synthesis Kit | New England Biolabs | Cat#: E6560L |
| Deposited Data | ||
| All MS raw files and MaxQuant output files | MassIVE Repository of the University of California, San Diego | Acquisition#: MSV000088067 |
| Experimental Models: Cell Lines | ||
| Human: SNU449, <10 Passages | ATCC | Cat#: CRL-2234 |
| Human: HuH-7, <10 Passages | JRCB Cell Bank | Cat#: JCRB0403 |
| Human: FOCUS AXL RNAi, <10 Passages | Laboratory of Taranjit Gujral, Fred Hutchinson Cancer Center, Seattle, WA31 | N/A |
| Human: FOCUS, <10 Passages | Laboratory of J. Wands, Brown University72 | N/A |
| Human: C3A, <10 Passages | ATCC | Cat#: CRL-10741 |
| Human: SNU398, <10 Passages | ATCC | Cat#: CRL-2233 |
| Human: Hep3B2.1-7, <10 Passages | ATCC | Cat#: HB-8064 |
| Human: A-172, <10 Passages | ATCC | Cat#: CRL-1620 |
| Human: SNU761, <10 Passages | Korean Cell Line Bank (KCLB) | Cat#: 00761 |
| Human: SNU886, <10 Passages | Korean Cell Line Bank (KCLB) | Cat#: 00886 |
| Human: HeLa, <10 Passages | ATCC | Cat#: CRM-CCL-2 |
| Human: JHH6, <10 Passages | JRCB Cell Bank | Cat#: JCRB1030 |
| Human: SH-SY5Y, <10 Passages | ATCC | Cat#: CRL-2266 |
| Human: U-2 OS, <10 Passages | ATCC | Cat#: HTB-96 |
| Human: SK-N-SH, <10 Passages | ATCC | Cat#: HTB-11 |
| Human: JeKo-1, <10 Passages | ATCC | Cat#: CRL-3006 |
| Human: Jurkat, <10 Passages | ATCC | Cat#: PTS-TIB-152 |
| Human: K562, <10 Passages | ATCC | Cat#: CCL-243 |
| Human: SKHep1, <10 Passages | ATCC | Cat#: HTB-52 |
| Human: SNU387, <10 Passages | ATCC | Cat#: CRL-2237 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| AAK1 shRNA#1 | The RNAi Consortium (TRC) | TRCN0000001943 |
| AAK1 shRNA#2 | The RNAi Consortium (TRC) | Cat#: TRCN0000199939 |
| AAK1 shRNA#3 | The RNAi Consortium (TRC) | Cat#: TRCN0000082348 |
| REPS1 shRNA#1 | The RNAi Consortium (TRC) | Cat#: TRCN0000053363 |
| REPS1 shRNA#2 | The RNAi Consortium (TRC) | Cat#: TRCN0000423162 |
| REPS1 shRNA#3 | The RNAi Consortium (TRC) | Cat#: TRCN0000436095 |
| REP2 shRNA#1 | The RNAi Consortium (TRC) | Cat#: TRCN0000423057 |
| REP2 shRNA#2 | The RNAi Consortium (TRC) | Cat#: TRCN0000428939 |
| REP2 shRNA#3 | The RNAi Consortium (TRC) | Cat#: TRCN0000056210 |
| RALBP1 shRNA#1 | The RNAi Consortium (TRC) | Cat#: TRCN0000305689 |
| RALBP1 shRNA#2 | The RNAi Consortium (TRC) | Cat#: TRCN0000047918 |
| RALBP1 shRNA#3 | The RNAi Consortium (TRC) | Cat#: TRCN0000047920 |
| Recombinant DNA | ||
| Lentiviral pLKO.1 Vector Plasmid | Addgene | Cat#: 10878 |
| pMD2.G Lentiviral Envelope Plasmid | Addgene | Cat#: 12259 |
| pCMVR8.74 Lentiviral Packaging Plasmid | Addgene | Cat#: 22036 |
| Software and Algorithms | ||
| MaxQuant/Andromeda, v1.5.2.8 | https://www.biochem.mpg.de/5111795/maxquant | Cox, J. et al.73 |
| Perseus, v1.5.6.0 | https://www.biochem.mpg.de/5111810/perseus | Tayanova, S. et al.74 |
| R-package, gplots v3.0.1, gplots::heatmap.2 | https://www.rdocumentation.org/packages/gplots/versions/3.0.1 | N/A |
| R-package, ssGSEA2.0 | https://github.com/broadinstitute/ssGSEA2.0 | Krug, K. et al.46 |
| GraphPad Prism, V7 | https://www.graphpad.com/ | N/A |
| STRING 11.5, PPI Networks and Enrichment Analysis | https://string-db.org/ | Szklarczyk, D. et al.38 |
| BioGRID | https://thebiogrid.org/ | Chatr-Aryamontri, A. et al.75 |
| PhosphoSite Plus | https://www.phosphosite.org/homeAction | Hornbeck, P.V. et al.41 |
| BioVenn | http://www.biovenn.nl/ | Hulsen, T. et al.76 |
| KinMap | http://kinhub.org/kinmap/ | Eid, S. et al.77 |
| Other | ||
| Fluor Chem E Imager | ProteinSimple/Bio-Techne Brand | N/A |
| QuantStudio 5 Real-Time PCR System | Applied Biosystems, Thermo Fisher Scientific | Cat#: A34322 |
| SpectraMax 190 Multimode Plate Reader | Molecular Devices | N/A |
| P-2000 Laser Puller | Sutter | N/A |
| EASY-nLC 1200 System | Thermo Fisher Scientific | Cat#: LC140 |
| Orbitrap Fusion Lumos Tribrid Mass Spectrometer | Thermo Fisher Scientific | Cat#: IQLAAEGAAPFADBMBHQ |
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health issued under the award numbers R01GM129090 (S-E.O.), R03TR003308 (M.G.), and R01GM086858 (D.J.M). This work used an EASY-nLC1200 UHPLC and Thermo Scientific Orbitrap Fusion Lumos Tribrid mass spectrometer purchased with funding from a National Institutes of Health SIG grant S10OD021502 (S-E.O.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We gratefully acknowledge Dr. Taran Gujral from the Fred Hutch Cancer Center for providing the FOCUS AXL RNAi cell line, and Dr. David Root from the Broad Institute of MIT and Harvard and Dr. Didier Trono from the École Polytechnique Fédérale de Lausanne for providing the lentiviral vectors through Addgene.
Footnotes
DECLARATION OF INTERESTS
The authors declare that there are no competing financial interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. We support inclusive, diverse, and equitable conduct of research.
REFERENCES
- 1.Vidal M, Cusick ME, and Barabasi AL (2011). Interactome networks and human disease. Cell 144, 986–998. 10.1016/j.cell.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nussinov R (2013). The spatial structure of cell signaling systems. Phys Biol 10, 045004. 10.1088/1478-3975/10/4/045004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng F, Zhao J, Wang Y, Lu W, Liu Z, Zhou Y, Martin WR, Wang R, Huang J, Hao T, et al. (2021). Comprehensive characterization of protein-protein interactions perturbed by disease mutations. Nat Genet 53, 342–353. 10.1038/s41588-020-00774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. 10.1038/nature22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor IW, and Wrana JL (2012). Protein interaction networks in medicine and disease. Proteomics 12, 1706–1716. 10.1002/pmic.201100594. [DOI] [PubMed] [Google Scholar]
- 6.Duan G, and Walther D (2015). The roles of post-translational modifications in the context of protein interaction networks. PLoS Comput Biol 11, e1004049. 10.1371/journal.pcbi.1004049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manning G, Whyte DB, Martinez R, Hunter T, and Sudarsanam S (2002). The protein kinase complement of the human genome. Science 298, 1912–1934. 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson FM, and Gray NS (2018). Kinase inhibitors: the road ahead. Nat Rev Drug Discov 17, 353–377. 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- 9.Fleuren ED, Zhang L, Wu J, and Daly RJ (2016). The kinome ‘at large’ in cancer. Nat Rev Cancer 16, 83–98. 10.1038/nrc.2015.18. [DOI] [PubMed] [Google Scholar]
- 10.Moret N, Liu C, Gyori BM, Bachman JA, Steppi A, Hug C, Taujale R, Huang L-C, Berginski ME, Gomez SM, et al. (2021). A resource for exploring the understudied human kinome for research and therapeutic opportunities. bioRxiv, 2020.2004.2002.022277. 10.1101/2020.04.02.022277. [DOI] [Google Scholar]
- 11.Lu H, Zhou Q, He J, Jiang Z, Peng C, Tong R, and Shi J (2020). Recent advances in the development of protein-protein interactions modulators: mechanisms and clinical trials. Signal Transduct Target Ther 5, 213. 10.1038/s41392-020-00315-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buljan M, Ciuffa R, van Drogen A, Vichalkovski A, Mehnert M, Rosenberger G, Lee S, Varjosalo M, Pernas LE, Spegg V, et al. (2020). Kinase Interaction Network Expands Functional and Disease Roles of Human Kinases. Mol Cell 79, 504–520 e509. 10.1016/j.molcel.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huttlin EL, Bruckner RJ, Navarrete-Perea J, Cannon JR, Baltier K, Gebreab F, Gygi MP, Thornock A, Zarraga G, Tam S, et al. (2021). Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 184, 3022–3040 e3028. 10.1016/j.cell.2021.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roux KJ, Kim DI, Raida M, and Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196, 801–810. 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goos H, Kinnunen M, Salokas K, Tan Z, Liu X, Yadav L, Zhang Q, Wei GH, and Varjosalo M (2022). Human transcription factor protein interaction networks. Nat Commun 13, 766. 10.1038/s41467-022-28341-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, and Ting AY (2013). Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339, 1328–1331. 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards AL, Eckhardt M, and Krogan NJ (2021). Mass spectrometry-based protein-protein interaction networks for the study of human diseases. Mol Syst Biol 17, e8792. 10.15252/msb.20188792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qin W, Cho KF, Cavanagh PE, and Ting AY (2021). Deciphering molecular interactions by proximity labeling. Nat Methods 18, 133–143. 10.1038/s41592-020-01010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kristensen AR, Gsponer J, and Foster LJ (2012). A high-throughput approach for measuring temporal changes in the interactome. Nat Methods 9, 907–909. 10.1038/nmeth.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Reilly FJ, and Rappsilber J (2018). Cross-linking mass spectrometry: methods and applications in structural, molecular and systems biology. Nat Struct Mol Biol 25, 1000–1008. 10.1038/s41594-018-0147-0. [DOI] [PubMed] [Google Scholar]
- 21.Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, et al. (2007). Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol 25, 1035–1044. 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 22.Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig PA, Reinecke M, Ruprecht B, Petzoldt S, Meng C, et al. (2017). The target landscape of clinical kinase drugs. Science 358. 10.1126/science.aan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golkowski M, Vidadala RS, Lombard CK, Suh HW, Maly DJ, and Ong SE (2017). Kinobead and Single-Shot LC-MS Profiling Identifies Selective PKD Inhibitors. Journal of proteome research 16, 1216–1227. 10.1021/acs.jproteome.6b00817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, et al. (2012). Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 149, 307–321. 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daub H, Olsen JV, Bairlein M, Gnad F, Oppermann FS, Korner R, Greff Z, Keri G, Stemmann O, and Mann M (2008). Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol Cell 31, 438–448. 10.1016/j.molcel.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Shibue T, and Weinberg RA (2017). EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 14, 611–629. 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lamouille S, Xu J, and Derynck R (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 15, 178–196. 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boumahdi S, and de Sauvage FJ (2020). The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov 19, 39–56. 10.1038/s41573-019-0044-1. [DOI] [PubMed] [Google Scholar]
- 29.Sharma K, Kumar C, Keri G, Breitkopf SB, Oppermann FS, and Daub H (2010). Quantitative analysis of kinase-proximal signaling in lipopolysaccharide-induced innate immune response. Journal of proteome research 9, 2539–2549. 10.1021/pr901192p. [DOI] [PubMed] [Google Scholar]
- 30.Golkowski M, Perera GK, Vidadala VN, Ojo KK, Van Voorhis WC, Maly DJ, and Ong SE (2018). Kinome chemoproteomics characterization of pyrrolo[3,4-c]pyrazoles as potent and selective inhibitors of glycogen synthase kinase 3. Mol Omics 14, 26–36. 10.1039/c7mo00006e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Golkowski M, Lau HT, Chan M, Kenerson H, Vidadala VN, Shoemaker A, Maly DJ, Yeung RS, Gujral TS, and Ong SE (2020). Pharmacoproteomics Identifies Kinase Pathways that Drive the Epithelial-Mesenchymal Transition and Drug Resistance in Hepatocellular Carcinoma. Cell systems. 10.1016/j.cels.2020.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Golkowski M, Vidadala VN, Lau HT, Shoemaker A, Shimizu-Albergine M, Beavo J, Maly DJ, and Ong SE (2020). Kinobead/LC-MS Phosphokinome Profiling Enables Rapid Analyses of Kinase-Dependent Cell Signaling Networks. J Proteome Res 19, 1235–1247. 10.1021/acs.jproteome.9b00742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ong SE, Schenone M, Margolin AA, Li X, Do K, Doud MK, Mani DR, Kuai L, Wang X, Wood JL, et al. (2009). Identifying the proteins to which small-molecule probes and drugs bind in cells. Proc Natl Acad Sci U S A 106, 4617–4622. 10.1073/pnas.0900191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oughtred R, Stark C, Breitkreutz BJ, Rust J, Boucher L, Chang C, Kolas N, O’Donnell L, Leung G, McAdam R, et al. (2019). The BioGRID interaction database: 2019 update. Nucleic Acids Res 47, D529–D541. 10.1093/nar/gky1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ross RA, Spengler BA, and Biedler JL (1983). Coordinate morphological and biochemical interconversion of human neuroblastoma cells. J Natl Cancer Inst 71, 741–747. [PubMed] [Google Scholar]
- 36.Gautier M, Thirant C, Delattre O, and Janoueix-Lerosey I (2021). Plasticity in Neuroblastoma Cell Identity Defines a Noradrenergic-to-Mesenchymal Transition (NMT). Cancers (Basel) 13. 10.3390/cancers13122904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, et al. (2021). The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res 49, D605–D612. 10.1093/nar/gkaa1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao Z, Lee P, Stafford JM, von Schimmelmann M, Schaefer A, and Reinberg D (2014). An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 516, 349–354. 10.1038/nature13921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, and Mann M (2006). Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648. 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 41.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43, D512–520. 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.UniProt C (2021). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49, D480–D489. 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Villanueva A (2019). Hepatocellular Carcinoma. N Engl J Med 380, 1450–1462. 10.1056/NEJMra1713263. [DOI] [PubMed] [Google Scholar]
- 44.Giannelli G, Koudelkova P, Dituri F, and Mikulits W (2016). Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol 65, 798–808. 10.1016/j.jhep.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 45.Golkowski M, Lau HT, Chan M, Kenerson H, Vidadala VN, Shoemaker A, Maly DJ, Yeung RS, Gujral TS, and Ong SE (2020). Pharmacoproteomics Identifies Kinase Pathways that Drive the Epithelial-Mesenchymal Transition and Drug Resistance in Hepatocellular Carcinoma. Cell Syst 11, 196–207 e197. 10.1016/j.cels.2020.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krug K, Mertins P, Zhang B, Hornbeck P, Raju R, Ahmad R, Szucs M, Mundt F, Forestier D, Jane-Valbuena J, et al. (2019). A Curated Resource for Phosphosite-specific Signature Analysis. Mol Cell Proteomics 18, 576–593. 10.1074/mcp.TIR118.000943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He H, Dai F, Yu L, She X, Zhao Y, Jiang J, Chen X, and Zhao S (2002). Identification and characterization of nine novel human small GTPases showing variable expressions in liver cancer tissues. Gene Expr 10, 231–242. 10.3727/000000002783992406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang W, Jia WD, Hu B, and Pan YY (2017). RAB10 overexpression promotes tumor growth and indicates poor prognosis of hepatocellular carcinoma. Oncotarget 8, 26434–26447. 10.18632/oncotarget.15507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mellman I, and Yarden Y (2013). Endocytosis and cancer. Cold Spring Harb Perspect Biol 5, a016949. 10.1101/cshperspect.a016949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corallino S, Malabarba MG, Zobel M, Di Fiore PP, and Scita G (2015). Epithelial-to-Mesenchymal Plasticity Harnesses Endocytic Circuitries. Front Oncol 5, 45. 10.3389/fonc.2015.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aiello NM, Maddipati R, Norgard RJ, Balli D, Li J, Yuan S, Yamazoe T, Black T, Sahmoud A, Furth EE, et al. (2018). EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev Cell 45, 681–695 e684. 10.1016/j.devcel.2018.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conner SD, and Schmid SL (2002). Identification of an adaptor-associated kinase, AAK1, as a regulator of clathrin-mediated endocytosis. J Cell Biol 156, 921–929. 10.1083/jcb.200108123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Agajanian MJ, Walker MP, Axtman AD, Ruela-de-Sousa RR, Serafin DS, Rabinowitz AD, Graham DM, Ryan MB, Tamir T, Nakamichi Y, et al. (2019). WNT Activates the AAK1 Kinase to Promote Clathrin-Mediated Endocytosis of LRP6 and Establish a Negative Feedback Loop. Cell Rep 26, 79–93 e78. 10.1016/j.celrep.2018.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gupta-Rossi N, Ortica S, Meas-Yedid V, Heuss S, Moretti J, Olivo-Marin JC, and Israel A (2011). The adaptor-associated kinase 1, AAK1, is a positive regulator of the Notch pathway. J Biol Chem 286, 18720–18730. 10.1074/jbc.M110.190769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakashima S, Morinaka K, Koyama S, Ikeda M, Kishida M, Okawa K, Iwamatsu A, Kishida S, and Kikuchi A (1999). Small G protein Ral and its downstream molecules regulate endocytosis of EGF and insulin receptors. EMBO J 18, 3629–3642. 10.1093/emboj/18.13.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Penninkhof F, Grootegoed JA, and Blok LJ (2004). Identification of REPS2 as a putative modulator of NF-kappaB activity in prostate cancer cells. Oncogene 23, 5607–5615. 10.1038/sj.onc.1207750. [DOI] [PubMed] [Google Scholar]
- 58.Oshiro T, Koyama S, Sugiyama S, Kondo A, Onodera Y, Asahara T, Sabe H, and Kikuchi A (2002). Interaction of POB1, a downstream molecule of small G protein Ral, with PAG2, a paxillin-binding protein, is involved in cell migration. J Biol Chem 277, 38618–38626. 10.1074/jbc.M203453200. [DOI] [PubMed] [Google Scholar]
- 59.Jullien-Flores V, Dorseuil O, Romero F, Letourneur F, Saragosti S, Berger R, Tavitian A, Gacon G, and Camonis JH (1995). Bridging Ral GTPase to Rho pathways. RLIP76, a Ral effector with CDC42/Rac GTPase-activating protein activity. J Biol Chem 270, 22473–22477. 10.1074/jbc.270.38.22473. [DOI] [PubMed] [Google Scholar]
- 60.Jullien-Flores V, Mahe Y, Mirey G, Leprince C, Meunier-Bisceuil B, Sorkin A, and Camonis JH (2000). RLIP76, an effector of the GTPase Ral, interacts with the AP2 complex: involvement of the Ral pathway in receptor endocytosis. J Cell Sci 113 (Pt 16), 2837–2844. [DOI] [PubMed] [Google Scholar]
- 61.Kostich W, Hamman BD, Li YW, Naidu S, Dandapani K, Feng J, Easton A, Bourin C, Baker K, Allen J, et al. (2016). Inhibition of AAK1 Kinase as a Novel Therapeutic Approach to Treat Neuropathic Pain. J Pharmacol Exp Ther 358, 371–386. 10.1124/jpet.116.235333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lenoir WF, Morgado M, DeWeirdt PC, McLaughlin M, Griffith AL, Sangree AK, Feeley MN, Esmaeili Anvar N, Kim E, Bertolet LL, et al. (2021). Discovery of putative tumor suppressors from CRISPR screens reveals rewired lipid metabolism in acute myeloid leukemia cells. Nat Commun 12, 6506. 10.1038/s41467-021-26867-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dustin D, Gu G, Beyer AR, Herzog SK, Edwards DG, Lin H, Gonzalez TL, Grimm SL, Coarfa C, Chan DW, et al. (2021). RON signalling promotes therapeutic resistance in ESR1 mutant breast cancer. Br J Cancer 124, 191–206. 10.1038/s41416-020-01174-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kurimchak AM, Shelton C, Duncan KE, Johnson KJ, Brown J, O’Brien S, Gabbasov R, Fink LS, Li Y, Lounsbury N, et al. (2016). Resistance to BET Bromodomain Inhibitors Is Mediated by Kinome Reprogramming in Ovarian Cancer. Cell Rep 16, 1273–1286. 10.1016/j.celrep.2016.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mundt F, Rajput S, Li S, Ruggles KV, Mooradian AD, Mertins P, Gillette MA, Krug K, Guo Z, Hoog J, et al. (2018). Mass Spectrometry-Based Proteomics Reveals Potential Roles of NEK9 and MAP2K4 in Resistance to PI3K Inhibition in Triple-Negative Breast Cancers. Cancer Res 78, 2732–2746. 10.1158/0008-5472.CAN-17-1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang C, Wang H, Lieftink C, du Chatinier A, Gao D, Jin G, Jin H, Beijersbergen RL, Qin W, and Bernards R (2020). CDK12 inhibition mediates DNA damage and is synergistic with sorafenib treatment in hepatocellular carcinoma. Gut 69, 727–736. 10.1136/gutjnl-2019-318506. [DOI] [PubMed] [Google Scholar]
- 67.Yam JW, Tse EY, and Ng IO (2009). Role and significance of focal adhesion proteins in hepatocellular carcinoma. J Gastroenterol Hepatol 24, 520–530. 10.1111/j.1440-1746.2009.05813.x. [DOI] [PubMed] [Google Scholar]
- 68.Jiang X, Tan HY, Teng S, Chan YT, Wang D, and Wang N (2019). The Role of AMP-Activated Protein Kinase as a Potential Target of Treatment of Hepatocellular Carcinoma. Cancers (Basel) 11. 10.3390/cancers11050647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, et al. (2011). In situ kinase profiling reveals functionally relevant properties of native kinases. Chem Biol 18, 699–710. 10.1016/j.chembiol.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bello T, Chan M, Golkowski M, Xue AG, Khasnavis N, Ceribelli M, Ong SE, Thomas CJ, and Gujral TS (2021). KiRNet: Kinase-centered network propagation of pharmacological screen results. Cell Rep Methods 1. 10.1016/j.crmeth.2021.100007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Golkowski M, Maly DJ, and Ong SE (2017). Proteomic Profiling of Protein Kinase Inhibitor Targets by Mass Spectrometry. Methods Mol Biol 1636, 105–117. 10.1007/978-1-4939-7154-1_8. [DOI] [PubMed] [Google Scholar]
- 72.He L, Isselbacher KJ, Wands JR, Goodman HM, Shih C, and Quaroni A (1984). Establishment and characterization of a new human hepatocellular carcinoma cell line. In Vitro 20, 493–504. [DOI] [PubMed] [Google Scholar]
- 73.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, and Mann M (2011). Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10, 1794–1805. 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 74.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13, 731–740. 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 75.Chatr-Aryamontri A, Oughtred R, Boucher L, Rust J, Chang C, Kolas NK, O’Donnell L, Oster S, Theesfeld C, Sellam A, et al. (2017). The BioGRID interaction database: 2017 update. Nucleic Acids Res 45, D369–D379. 10.1093/nar/gkw1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hulsen T, de Vlieg J, and Alkema W (2008). BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9, 488. 10.1186/1471-2164-9-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eid S, Turk S, Volkamer A, Rippmann F, and Fulle S (2017). KinMap: a web-based tool for interactive navigation through human kinome data. BMC Bioinformatics 18, 16. 10.1186/s12859-016-1433-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298. 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 79.Golkowski M, Brigham JL, Perera GK, Romano GE, Maly DJ, and Ong SE (2014). Rapid profiling of protein kinase inhibitors by quantitative proteomics. MedChemComm 5, 363–369. 10.1039/C3MD00315A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature protocols 2, 1896–1906. 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S4. Results from kiCCA DEA, kinobead/LC-MS profiling of RNAi lines and HCC tissues, and GSEA; Supplement to Figures 3–6, S4–7, and S10. MaxQuant protein groups output data from kiCCA analysis of HuH7, SNU761, SK-N-SH, SH-SY5Y, and EGF-stimulated HeLa cells, as well as kinobead/LC-MS profiling data from AAK1 complex RNAi knockdown lines and AAK1 kinobead competition experiments. Drug Screening results in AAK1 network RNAi lines. All GSEA results.
Table S1. Determining an optimal set of kinase interactome probe (KIP); Supplement to Figures 2 and S1. Kinobead/LC-MS competition data from Klaeger et al., Science, 2017 and correlation clustering analysis results.
Table S2. Supporting data for KIP competition, kinase groups, and plasticity markers. Supplement to Figures 2–4 and S3. MaxQuant protein groups output data obtained for KIP competition in unstimulated HeLa Cells, list of kiCCA kinase groups, and list of cancer cell plasticity markers used for unsupervised clustering.
Table S3. kiCCA interactome knowledgebase, functional marker PPIs, and pathway associations; Supplement to Figures 3–6, and S6. Summary spreadsheet of all kiCCA interactions across the 18-cancer cell line panel to serve as reference knowledgebase, list of curated fmPPIs with literature references indicating functional consequences of PPIs, and list of GOBP pathway annotations for all identified high-confidence kiCCA interactors.
Data Availability Statement
MS .raw files, MaxQuant output files, and kiCCA correlation matrices generated by this study have been uploaded to the MassIVE repository of the University of San Diego under the acquisition number: MSV000088067. Original western blot images have been deposited at Mendeley Data and are publicly available as of the date of publication. A Shiny app for real time interrogation of the kinase interactomics data generated in this study can be accessed at https://quantbiology.org/kiCCA.
This study did not generate new code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.