

Abstract
The Diels-Alder cycloaddition is one of the most powerful approaches in organic synthesis and is often used in the synthesis of important pharmaceuticals. Yet, strictly controlling the stereoselectivity of the Diels-Alder reactions is challenging, and great efforts are needed to construct complex molecules with desired chirality via organocatalysis or transition metal strategies. Nature has evolved different types of enzymes to exquisitely control cyclization stereochemistry, however, most of the reported Diels-Alderases have been shown to only facilitate the energetically favorable diastereoselective cycloadditions. Here we report the discovery and characterization of CtdP, as a member of a new class of bifunctional oxidoreductase/Diels-Alderase, which was previously annotated as an NmrA-like transcriptional regulator. We demonstrate that CtdP catalyses the inherently unfavored cycloaddition to form the bicyclo[2,2,2]diazaoctane scaffold with a strict α-anti-selectivity. Guided by computational studies, we reveal a NADP+/NADPH-dependent redox mechanism for the CtdP-catalysed inverse electron demand Diels-Alder cycloaddition, which serves as the first example of a bifunctional Diels-Alderase that utilizes this mechanism.
Graphical Abstract
Introduction
The Diels-Alder (DA) reaction is one of the most fascinating synthetic approaches for building multiple C-C and C-heteroatom bonds to synthesize valuable pharmaceuticals with complex chiral centers1. The [4 + 2] DA reactions can be classified into two different types, normal electron demand Diels-Alder (NEDDA) and inverse electron demand Diels-Alder (IEDDA) reactions, based on the relative energies of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of the diene and the dienophile2 (Fig. 1a). In contrast to the NEDDA reaction, an electron-poor diene and an electron-rich dienophile are required for the IEDDA reaction, which is widely used to construct heterocycles in organic synthesis. However, fundamental challenges remain in controlling the stereoselectivity in the synthesis of complex chiral molecules by using traditional organocatalysts or transition metal catalysts1. Several classes of Diels-Alderases (DAases) have been characterized recently, including S-adenosyl-L-methionine-dependent methyltransferase (MT) like enzyme LepI3, flavindependent enzyme MaDA4, hydrolase TS5, β-barrel protein PyrI46, lipocalin-like protein PvhB7, short-chain dehydrogenases/reductases (SDRs) MalC and PhqE8, inverse electron demand Diels-Alderase IccD9, and unannotated protein EupfF10 (Extended Data Fig. 1a). While most of the DAases appear to have lost their ancestral functions and only facilitate the [4 + 2] DA reactions and stereocontrol in product formation, MalC and PhqE were demonstrated to simultaneously catalyse substrate reduction followed by an enantioselective DA reaction8. Meanwhile, most characterized DAases possess energetically favorable diastereoselectivity according to density functional theory (DFT) calculations11, and their biosynthetic substrates can undergo spontaneous DA cycloadditions to the corresponding racemic mixtures or diastereomers, including their enzymatic products.
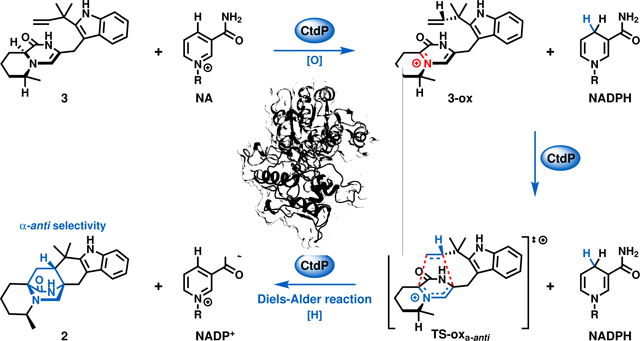
Fig. 1 |. Fungal bicyclo[2.2.2]diazaoctane alkaloids biosynthesis and functional characterization of CtdP.

a, Normal and inverse electron demand Diels-Alder reactions. EDG, electron-donating group; EWG, electron-withdrawing group; LUMO, lowest unoccupied molecular orbital; HOMO, highest occupied molecular orbital. b, The proposed azadiene intermediate and products with different configurations of the bicyclo[2.2.2]diazaoctane ring in the IMDA construction of PIAs. c, Chemical structures of natural PIAs with the α-syn or α-anti bicyclo[2.2.2]diazaoctane rings. d, The ctd gene cluster from P. citrinum ATCC 9849. e, LCMS analysis (extracted ion chromatogram, EIC) of P. citrinum WT and mutant ΔctdP. f, CtdP catalyses the conversion from 3 to 2, and spontaneous IMDA cycloadducts from 3. g, In vitro assays of 3 with CtdP. h, In vitro assays of 9 with CtdP and MalC, respectively. i, CtdP catalyses the conversion from 9 to 10, and MalC catalyses the conversion from 9 to (+)-11. The symbol * represents the compound identified by MS and UV spectra, and # represents the unknown compound.
Prenylated indole alkaloids (PIAs) containing the bicyclo[2.2.2]diazaoctane ring possess a wide range of biological activities, including anticancer stephacidins12, vasorelaxant malbrancheamides13, anthelmintic paraherquamides14, and insecticidal brevianamides15. The bicyclo[2.2.2]diazaoctane core arises from intramolecular [4+2] hetero-Diels-Alder (IMDA) cycloaddition after assembly by a nonribosomal peptide synthetase (NRPS), followed by a reverse prenyltransferase16. The reactive azadiene species has been acknowledged as the most plausible precursor for constructing the IMDA cycloaddition, which has not been defined as a NEDDA or an IEDDA mechanism16,17 (Fig. 1b). Theoretically, four products with different stereoisomeric bicyclo[2.2.2]diazaoctane rings could be furnished via IMDA reaction, including α-syn (α represents the lactam ring in α-position), β-syn (β represents the lactam ring in the β-position), α-anti, and β-anti (Fig. 1b). While several of these configurations are observed from different fungi, PIAs are commonly isolated as a single isomer, with respect to the bicyclic core16. Several monoketopiperazine PIAs, such as (+)-premalbrancheamide (11) and (+)-paraherquamides, were shown to be produced by the α-syn-selective DAases (MalC and PhqE)8, yet the biosynthetic origins of the energetically disfavored18 α-anti-selective PIAs, like the citrinadins19, citrinalin C20, and penioxamide A21, remained elusive (Fig. 1c and Extended Data Fig. 1b). In our previous investigation of the biosynthesis of 21R-citrinadin A (1)22, we identified an α-anti-bicyclo[2.2.2]diazaoctane intermediate, (+)-precitrinadin A (2), from Penicillium citrinum ATCC 9849 (Fig. 1c and Extended Data Fig. 1c). Since the wild-type (WT) P. citrinum strain was known to only produce the α-anti form of the PIAs, we hypothesized the existence of a dedicated DAase responsible for the strict diastereoselective and enantioselective cycloaddition for the assembly of the α-anti product 2, which has yet to be characterized in the citrinadin pathway.
Herein, we describe the discovery and characterization of an NmrA-like protein CtdP representing a new class of bifunctional oxidoreductase/DAase. Unlike the previously predicted function as a transcriptional regulator, CtdP catalyses the energetically demanding α-anti-selective cyclization of the bicyclo[2.2.2]diazaoctane ring. Guided by rigorous computational studies, we reveal an unexpected catalytic mechanism for CtdP, a NADP+/NADPH-dependent redox pathway that mediates the IEDDA cycloaddition with high stereoselectivity, which utilizes a biocatalytic LUMO lowering strategy via the active dienyl iminium to impart precise stereochemical control.
Results and Discussion
Probing the α-anti DAase in the ctd cluster.
To uncover the possible DAase in the ctd cluster (Fig. 1d), we first used the characterized SDR DAases, MalC and PhqE8, as bioinformatic probes to search for homologs that are likely responsible for the IMDA cycloaddition due to the similar bicyclo[2.2.2]diazaoctane core structures in premalbrancheamide/preparaherquamide and 2. We found that CtdN was the only homologous SDR in the ctd cluster, and showed high sequence identity with PhqE (50.57%) and MalC (45.77%) (Extended Data Fig. 2a and Supplementary Table 3). To investigate whether CtdN functions as an α-anti DAase, we obtained the ΔctdN mutant by using the split marker recombination approach23 and compared its metabolic profile to that in WT P. citrinum (Supplementary Fig. 3). Our results showed the production level of 1 was not abrogated by the deletion of ctdN (Extended Data Fig. 2b). Next, we synthesized its putative substrates 3 and 4 based on the analogous synthetic route for MalC substrates8 (Supplementary Fig. 1a, c) and the identification of pipecolate-tryptophan dipeptide precursors in the cnd pathway24 (Extended Data Fig. 1c). Compound 3 is relatively stable, and its structure was subsequently determined by 1D and 2D nuclear magnetic resonance (NMR) analyses (Supplementary Table 5). However, the zwitterionic 4 derived from the dienamine 3 using the previously reported synthetic approach was extremely unstable. The relative stabilities of 3 and 4 contrast significantly with the analogous compounds in the premalbrancheamide/preparaherquamide pathways, where the zwitterionic substrate was found to be more stable8. To verify the substrate and the function of CtdN in vitro, we overexpressed and purified the C-His-tagged form of the CtdN protein from Escherichia coli BL21(DE3) and performed in vitro assays. However, incubation of CtdN and 3 failed to show any desired α-anti IMDA products under both anaerobic and aerobic conditions compared with the no enzyme control, further indicating that the SDR enzyme CtdN might not act as an α-anti DAase in the ctd pathway (Extended Data Fig. 3a).
Investigating the function of the NmrA-like genes.
To identify other possible DAase candidates, further bioinformatic analysis shifted our focus to a group of NmrA-like family genes, including ctdO, ctdP, and ctdR, which are located adjacent to the NRPS gene ctdQ (Fig. 1d). The nitrogen metabolite repression protein NmrA was previously characterized as a transcriptional nitrogen metabolism repressor in Aspergillus nidulans25 and has a nicotinamide adenine dinucleotide (NAD)-binding motif and showed unexpected similarity with the structure of SDR family enzymes26. Several NmrA-like proteins in other organisms were identified as redox-sensing regulators, which can bind NAD(H) or NADP(H) cofactors but lack the function of catalysing dehydrogenation/hydrogenation reactions27,28.
To explore the potential functions of NmrA-like proteins in the citrinadin biosynthesis, deletions of each NmrA-like gene (ctdO, ctdP, and ctdR) were performed (Supplementary Fig. 3). Compared to WT P. citrinum, the LCMS metabolic profile of ΔctdO extract showed reduced production of citrinadins, while the ΔctdR mutant abolishes the production of 1 but not 2, and mainly accumulates a new metabolite R1 (m/z 464. 3 [M+H]+) that possesses the α-anti scaffold based on its NMR profiles (Extended Data Fig. 2c and Supplementary Table 10). Thus, CtdR is proposed to be involved in late-stage tailoring steps after the formation of 2. These in vivo results indicated that ctdO and ctdR are likely not responsible for the IMDA step in the citrinadin biosynthesis. Next, we determined that deletion of the ctdP abolished the production of both 1 and 2, while accumulating two new products, 5 (retention time, RT = 18.4 min, m/z 380.2 [M+H]+) and 6 (RT = 17.2 min, m/z 380.2 [M+H]+) (Fig. 1e, f). The UV and MS spectra of 5 and 6 suggested that they were possible hydroxy-substituted analogs of 2 (Supplementary Fig. 4). Through comprehensive NMR analysis, X-ray crystallographic data, together with electronic circular dichroism (ECD) spectra, 5 and 6 were demonstrated to be the oxidative IMDA products possessing β-syn and β-anti configurations, respectively (Extended Data Fig. 3b, d, CCDC 2127333 for 5). As the bioinformatic analysis of CtdP shows very low sequence similarity with MalC/PhqE8 (coverage/identity: ~30%/30%, Supplementary Table 4), our in vivo gene knockout study further indicated that this protein could function as a distinct DAase involved in the formation of an anti-bicyclo[2.2.2]diazaoctane intermediate en route to 1.
In vitro characterization of CtdP as an α-anti DAase.
To further validate our hypothesis, we overexpressed and purified all three NmrA-like proteins (CtdP, CtdR, and CtdO) as C-His-tagged proteins from E. coli BL21 (DE3). When 40 μM enzyme was incubated with 500 μM synthetic 3 and 1 mM NADP+, CtdP catalysed complete conversion of the substrate to the IMDA product 2 (Fig. 1g, traces ii, v), while no evident enzymatic product was detected after a 2 h reaction with CtdO or CtdR (Extended Data Fig. 3f). The kcat/KM value of CtdP with 3 and NADP+ was measured to be 27.9 ± 3.9 min−1 μM −1 (Supplementary Fig. 7a, b). By replacing the exogenous addition of NADP+ with NADPH or NAD+, the production of 2 was decreased significantly to lower levels, indicating the prebound NADP+ during CtdP purification (Fig. 1g, traces ii-iv, Extended Data Fig. 3f). Indeed, we quantified that 7.8 ± 2.6% NADP+ was detected in purified CtdP from E. coli (Supplementary Fig. 7c, d). Next, we added different concentrations of exogenous NADP+ in the reaction and observed the conversion of 2 increased from 10.4% to 63.9% with the increased amount of additional NADP+ from 0 to 1000 μM (Supplementary Fig. 7f). To demonstrate whether NADP+ is essential for the CtdP-catalysed IMDA cycloaddition, we removed the cofactor in CtdP protein by extensive dialysis29. In the absence of NADP+, the refolded CtdP could not catalyse the formation of 2, while it can restore the partial activity with the exogenous addition of NADP+ (Supplementary Fig. 7g). Thus, the cofactor NADP+ was deduced to be indispensable for CtdP catalysis. In the absence of CtdP catalysis, 3 could spontaneously transform to compounds 4–8 (Extended Data Fig. 3b, e), consistent with previous in vivo ctdP knockout study (Fig. 1e). To investigate whether the CtdP reaction requires oxygen for catalysis, we also performed enzymatic assays under rigorous anaerobic conditions (Extended Data Fig. 3a) and found oxygen was not needed in the production of 2 when 3 was used as the substrate. Thus, these assays unambiguously support that CtdP is responsible for converting 3 to the α-anti IMDA product 2 through an enantioselective and diastereoselective cycloaddition in the presence of NADP+ (Fig. 1f).
To further understand the exquisite stereoselectivity of CtdP, we overexpressed and purified the previously characterized DAase MalC and synthesized its cognate zwitterionic substrate 9 for comparative analyses8 (Supplementary Fig. 1b, e). Interestingly, when 9 was incubated with CtdP and NADPH, it was converted almost completely into a new product 10 (Fig. 1h, traces i, iii). Consistent with the stereochemistry of 2, 10 was elucidated as penicimutamide E30 with α-anti configuration confirmed by NMR and single-crystal X-ray diffraction analysis (CCDC 2127332, Fig. 1i). When NADPH was removed from the assay or replaced by NADP+, CtdP could not transform 9 to 10 (Fig. 1h, traces iv, vii), indicating that zwitterion 9 must be reduced by NADPH before initiation of the IMDA cycloaddition. The racemic syn-bicyclo[2.2.2]diazaoctane products of (±)-11 could be spontaneously generated in the presence of NADPH, consistent with previously reported assays8 (Fig. 1h, trace iii). Our biochemical data revealed that CtdP catalyses the α-anti-selective IMDA cyclization that converts the native substrate of dienamine 3 with cofactor NADP+ and also accepts zwitterionic 9 with the reductive rescue by NADPH cofactor. Thus, the indispensable cofactor NADP+ may play a role in oxidation in the CtdP assays that transforms the native substrate 3. To our best knowledge, CtdP represents a new class of NmrA-like DAase that catalyses anti-selective IMDA cyclization.
Crystal structure of CtdP and mutagenesis study.
To gain further insights into the catalytic mechanism of the NmrA-like DAase, the co-crystal structure of CtdP bound with the cofactor NADP+ and product 10 was determined at 2.5 Å resolution by selenomethionyl (SeMet) single-wavelength anomalous diffraction (SAD) phasing (Fig. 2a and Supplementary Table 13). The closest structural homolog to CtdP is HSCARG (24% sequence identity, PDB 2exx, 2.1 Å RMSD, 31.8 Z-score), which has been proposed to be a mammalian homolog of NmrA that acts as a negative regulator in cellular processes such as innate immunity and DNA damage repair in response to the cellular redox state28 (Extended Data Fig. 4 and Supplementary Table 14). Similar to HSCARG, CtdP contains an N-terminal NADP-binding domain and a C-terminal substrate-binding domain (Fig. 2b). The N-terminal domain contains the canonical Rossmann fold common to all NmrA-like and SDR family proteins, including the conserved GxxGxxG nucleotide-binding motif but lacking the YxxxK SDR catalytic motif26 (Extended Data Fig. 4).
Fig. 2 |. Crystal structure analysis of CtdP complex.

a, Overall structure of the CtdP asymmetric unit is shown in a cartoon model and colored by each chain. Ligands are represented by spheres with NADP+ in yellow and penicimutamide E (10) in salmon. b, Alignment of CtdP chains A and C are shown as a cartoon, with NADP-binding domain in grey and substrate-binding domain forest green (chain A) or pale yellow (chain C). NADP+ (yellow) and 10 (salmon) from chain A shown as sticks. The C-terminal tail (residues 329–346) is highlighted with bright orange. c, The simulated annealing omits maps (blue mesh, Fo–Fc, contoured at 3.0 σ) of the NADP+ and 10. d, Superimposed CtdP-NADP+−10 (forest green) and PhqE-NADP+-(+)-11 (orange, PDB 6NKK) complex structures aligned by the NADP-binding domain.
The structure was solved with four chains (A-D) in the asymmetric unit, all in complex with NADP+. Intriguingly, we found three chains (A, B, and D) in CtdP were bound with 10, while chain C was a ligand-free form (Fig. 2a). Chains A and B are nearly identical with an RMSD value of 0.290 Å (Extended Data Fig. 5) and strong electron densities are observed for both NADP+ and 10 (Fig. 2c). Superposition of chain A with chain C indicated remarkable conformational changes of the residues 162–174 (helices α12 to η3) and residues 260–273 (helices α8 to η2). The Cα atom of Pro268 (α12) in chain C moved 6.2 Å relative to chain A (Fig. 2b). Strikingly, a C-terminal tail (residues 329–346), which is disordered in chains C and D, is ordered in chains A and B, forming close contacts with product 10 (Fig. 2b). The C-terminal tail is proximal to the substrate and NADP+ binding sites, in which Ser340 forms a hydrogen bond (2.6 Å) with NADP+, Arg345 is in electrostatic interaction (3.0 Å) with Asp266, and Pro342 forms a hydrophobic interaction (3.8 Å) with the indole moiety (Extended Data Fig. 6a). To explore the function of the C-terminal tail in CtdP, we constructed four truncated CtdP mutants (CtdPΔ330–367, CtdPΔ335–367, CtdPΔ342–367, and CtdPΔ348–367). In vitro assays with substrate 3 indicated that the truncated CtdP proteins Δ330–367, Δ335–367, Δ342–367, and Δ348–367 retained 55.2 ± 0.7%, 46.5 ± 2.9%, 61.5 ± 2.3%, and 91.5 ± 4.0 % activity respectively. While chain D is complex with NADP+ and 10, the C-terminal tail is disordered, and a symmetry mate occupies the same space as the ordered tail. The electron density of 10 in chain D is significantly weaker than that in chains A and B, which could be a result of the disordered C-terminal tail. Our data suggested that the C-terminal tail may interact with the substrate-binding domain to promote product binding but was not observed to play a significant catalytic role in CtdP.
To further investigate the structural characteristics of CtdP, the common Rossmann fold of the NmrA-like CtdP and the SDR DAase PhqE were superimposed, which revealed significant differences, particularly in the orientation of the bound products (Fig. 2d), suggesting that the substrate-binding domain is the key determinant of stereoselective catalysis. Product 10 is positioned over the nicotinamide group of NADP+ with a relatively close distance (4 Å) of C16 to C4 in the pyridine ring of NADP+ (Fig. 3a), indicating a typical π-π stacking interaction. The relative positioning of the product and NADP+ is also similarly present in the previous PhqE co-crystal structure8. However, superimposing the common bicyclo[2.2.2]diazaoctane rings of syn-product (+)-11 of PhqE with anti-product 10 in the CtdP active site revealed a major steric clash with bulky residues Trp160, Phe170, and Phe174, indicating these residues could sterically block the syn conformation from forming (Fig. 3b). At the interface between the cofactor NADP+ and product 10 in CtdP, Asn164 forms a hydrogen bond with the nicotinamide group of NADP+ (2.8 Å), and Tyr161 interacts with the phosphate oxygen atoms of the bound NADP+ (3.0 Å and 3.5 Å, Fig. 3a). Compared with WT CtdP, N164A and Y161A mutations caused more than 90% loss of enzymatic activity when using 3 as the substrate (Fig. 3c).
Fig. 3 |. CtdP active site view and mutagenesis study.

a, Active site of CtdP. The CtdP residues are shown in blue, while NADP+ and 10 are shown in yellow and salmon, respectively. b, Superposition of the common bicyclo[2.2.2]diazaoctane ring in (+)-11 (green) with that in 10 (salmon) in CtdP-NADP+−10 complex. c, Analysis of the formation of 10 with CtdP mutants relative to wild-type (WT) production. Data represents as mean ± s.d. from triplicate independent experiments (n = 3). The relative ratio of product 2 formation in the mutant compared to the WT CtdP is based on the peak area of UV at 280 nm.
The substrate-binding domain of CtdP contains a hydrophobic binding pocket (Extended Data Fig. 6b) surrounding product 10. Trp160, Phe170, Phe174, Phe277, and Pro342 form close contacts with the aromatic ring of 10 (Fig. 3a). Mutants W160A, F170A, F277A, F174A, and P342A retained 4.9 ± 0.3%, 1.7 ± 0.4%, 1.5 ± 0.1%, 90.7 ± 0.9%, and 95.5 ± 3.0% activity, respectively (Fig. 3c). These adjacent hydrophobic residues Trp160, Phe170, and Phe277, together with the NADP+ cofactor, likely play a critical role in the substrate binding and conformation preorganization through π-π stacking and hydrophobic interactions (Fig. 3a). Four residues Tyr280, Leu134, Val133, and Gln118, are proximal to the proline ring or bicyclo[2.2.2]diazaoctane ring of the bound product 10. Mutants Y280A and L134A completely abolished the enzyme activity with 3 while variants of Q118A and V133A retained 34.9 ± 1.4% and 80.2 ± 1.6% enzymatic activity, respectively. To explore whether the Ar-OH group of Tyr280 is critical to its function, we tested the Y280F mutant with 3, which only retained 18.7 ± 0.5% enzymatic activity. Therefore, Tyr280 may assist the formation of the reactive diene, and Leu134 likely facilitates orienting the corresponding substrate dienophile. Additionally, polar residues Ser273 and Tyr269 near the indole nitrogen of 10 are not critical for catalysis since variants S273A and Y269A still retained 89.2 ± 2.1% and 83.7 ± 3.8% enzymatic activity, respectively (Fig. 3c).
Probing the redox catalytic mechanism of CtdP.
To elucidate the catalytic mechanism of CtdP, we performed DFT computations to investigate two mechanistic pathways for transforming native substrate 3 into product 2 (Fig. 4a). In the traditional tautomerization pathway, 3 undergoes two tautomerizations to form reactive diene 3-taut, which subsequently undergoes a DA reaction to form 2-taut and finally tautomerizes to product 2. However, the two tautomerizations needed to form the reactive diene are energetically costly: reactive diene 3-taut is higher in free energy than native substrate 3 by 21.4 kcal mol−1, and this in turn causes the DA reaction to form 2-taut to have a high overall free energy barrier of 44.6 kcal mol−1. Even with the benefit of an enzyme catalyst (CtdP), such a high free energy barrier is unlikely to be overcome, and we therefore reasoned that the NADP+ cofactor mediates redox chemistry enabled by its proximity to the substrate.
Fig. 4 |. Proposed mechanism of CtdP-catalysed α-anti-selective IMDA reaction.

a, Comparison of the energetics of a tautomerization pathway (left pathway in purple) and a redox-mediated pathway (right pathway in blue) using the NADP+ cofactor present in the CtdP active site. b, MS spectra of 2 and 2H-2 produced in CtdP and (S)-[4-2H] NADPH assay. c, Monitoring the two dihedral angles ϕdiene and ϕdienophile in substrate 3-ox that controls the stereochemistry of the IMDA adduct during three 1.2-microsecond MD simulations. Each point corresponds to an MD snapshot where both DA forming bonds are 4.0 Å or less; to the right are the α-syn and α-anti snapshots with the shortest DA bond lengths. d, The proposed mechanistic pathway of CtdP-catalysed α-anti-selective IMDA reaction. Free energies are in kcal mol−1 and bond lengths are in Å.
In this redox-mediated pathway, native substrate 3 is first oxidized by NADP+ via a hydride transfer reaction to form reactive diene 3-ox, which subsequently undergoes a DA reaction to form 2-ox. Finally, the NADPH generated from substrate oxidation reduces 2-ox to product 2 via a second hydride transfer reaction to regenerate NADP+. Substrate oxidation by NADP+ (modeled computationally by 1-methylnicotinamide) is much less energetically costly than substrate tautomerization: the reactive dienyl iminium species 3-ox is only higher in free energy than native substrate 3 by 7.1 kcal mol−1, and this allows the DA reaction to form 2-ox with a much lower free energy barrier of 29.6 kcal mol−1. Further orbital calculations indicated that a major role of NADP+ is to lower the LUMOdiene energy of 3-ox (–2.14 eV) in comparison to the LUMOdiene energy of 3-taut (–0.10 eV), thus enabling an IEDDA reaction in 3-ox between a low-lying cationic diene LUMO and the dienophile HOMO. In contrast, 3-taut would react via a NEDDA reaction between the diene HOMO and the dienophile LUMO, as these orbitals are closer to each other in energy (6.50 eV gap) than the diene LUMO and the dienophile HOMO (8.68 eV gap). Thus, DFT computations provide strong evidence that the α-anti-selective IMDA cycloaddition catalysed by CtdP occurs through a redox-mediated pathway in which NADP+ oxidation lowers the LUMOdiene and enables an IEDDA reaction.
Further evidence supporting the redox-mediated pathway is derived from exchanging NADP+ with NADPH, which fails CtdP to catalyse the DA reaction (Fig. 1g, trace iii). To further verify the role of a reduction step by NADPH during the CtdP redox-mediated pathway, the enzymatic assay was performed in the presence of (S)-[4-2H] NADPH31–34. A one Da increase at m/z 365.3 [M’ + H]+ was observed in the DA adduct 2H-2, indicating that the deuterium was transferred to 2H-2 from (S)-[4-2H] NADPH (Fig. 4b and Extended Data Fig. 7a). In accord with the computational results, this observation demonstrates that reduction by NADPH occurs during the CtdP catalysed DA reaction, converting intermediate adduct 2-ox to the final product 2. Taken together, our computational data and isotopic labeling results reveal a unique redox mechanism for CtdP, in which direct involvement of NADP+ lowers the free energy barrier. This requires that the NADP+ cofactor should be in an ideal position to oxidize substrate 3 to the active dienyl iminium intermediate 3-ox, thus lowering the energy of the LUMOdiene and enabling an accelerated IEDDA reaction35. To the best of our knowledge, CtdP is the first natural biocatalyst that employs a redox-mediated LUMOdiene-lowering strategy for catalysing a DA reaction.
Computational studies on the mechanism of α-anti selectivity.
To begin understanding why CtdP exclusively forms α-anti IMDA cycloadduct 2, we compared the free energies of the four possible diastereomeric transition states, α-syn (TS-oxα-syn), α-anti (TS-oxα-anti), β-syn (TS-oxβ-syn), and β-anti (TS-oxβ-anti), in the redox-mediated pathway. In the absence of CtdP, the α-anti transition state has the highest free energy (ΔG‡ = 29.6 kcal mol−1) and is not inherently favored over the other diastereomers (Extended Data Fig. 7b). The transition state with the lowest free energy is the β-syn transition state (ΔG‡ = 27.3 kcal mol−1), likely owing to slightly smaller steric interactions of non-reacting methyl groups and consistent with the experimental finding that the major oxidized DA cycloadduct 5 formed both in the ΔctdP mutant and non-enzymatic reactions of 3 have β-syn stereochemistry (Supplementary Fig. 8). Therefore, quantum mechanical computations demonstrate that the exclusive stereoselective formation of α-anti-IMDA cycloadduct 2 by CtdP must be catalyst-controlled, and not mediated by the inherent properties of the native substrate 3.
To obtain a more detailed understanding of how CtdP controls the stereoselectivity and facilitates the cycloaddition reaction, we turned to classical molecular dynamics (MD) simulations (Fig. 4c). The immediate precursor 3-ox of the DA reaction was docked into the CtdP active site with the NADP+/NADPH cofactor simulated in the form present just before the DA reaction (as NADPH). During three long-time 1.2-microsecond MD simulations, we monitored the distances between the atoms of 3-ox that form the two new sigma bonds as well as two dihedral angles ϕdiene and ϕdienophile that control the stereochemistry of the IMDA adduct. By examining snapshots taken every 2.4 nanoseconds from all three MD simulations, we identified a total of 510 snapshots where both DA forming bonds had distances less than 4.0 Å. Of these, 125 snapshots had dihedral angles consistent with the formation of α-syn cycloadduct, 385 snapshots were oriented to form α-anti cycloadduct, and no snapshots were oriented to form β-syn or β-anti cycloadducts (Fig. 4c). Thus, the MD simulations reveal that the CtdP active site binds oxidized substrate 3-ox in an orientation that favors the formation of α-anti-selective IMDA cycloadduct 2 by more than a 3 to 1 ratio, in reasonable agreement with the experimental finding that only the α-anti IMDA cycloadduct is formed in the CtdP assays.
In addition to revealing how CtdP controls the α-anti stereoselectivity of the IMDA cycloaddition, MD simulations also identified a second major catalytic role of the enzyme: maintaining the substrate carbon C16 near the NADP+ cofactor so it is poised for oxidation. Although the MD simulations are performed after the initial hydride transfer from 3 to the NADP+ cofactor, the oxidized carbon in 3-ox remains within 4 Å of the hydride throughout the 1.2-microsecond simulation time in all three MD replicas (Extended Data Fig. 8a). Therefore, the MD simulations revealed that CtdP plays a major role in enabling the oxidation step that makes IMDA cycloaddition possible. MD simulations also revealed a persistent hydrogen bond between the amide N–H of 3-ox (H-bond donor) and the ribose 2’-OH of NADPH (H-bond acceptor) that prevents the substrate from flipping into cycloadducts with other conformations. This hydrogen bond is maintained at a length of approximately 3 Å throughout two of the three MD replicas (Extended Data Fig. 8b). Moreover, the MD snapshot with the shortest DA bond lengths in which substrate 3-ox has an α-anti conformation revealed a key π-π stacking interaction between the positively charged substrate and the nicotinamide group of NADPH, as well as possible polar interaction with residue Tyr280 (Extended Data Fig. 8c). Concurrently, the nonpolar residues Trp160, Phe170, and Phe277 identified through mutagenesis as essential to the activity of CtdP may facilitate preorganization of the substrate by creating a substrate-binding pocket that best accommodates the α-anti conformation. This binding mode is likely enhanced by the π-π stacking interaction identified between substrate 3/3-ox and NADP+/NADPH. Based on these data, we proposed a detailed picture of the unique redox mechanism of CtdP catalysis (Fig. 4d). Driven by the close shape complementarity of the substrate-binding pocket and cofactor NADP+, substrate 3 is preorganized into an α-anti conformation and oxidized by NADP+ to generate intermediate 3-ox with an active iminium diene, which is likely stabilized by the 2’-OH of NADPH and residue Tyr280. This active dienyl iminium 3-ox then undergoes a highly stereoselective IEDDA cycloaddition to form iminium adduct 2-ox, and a final reductive rescue by NADPH generates α-anti-cycloadduct 2.
Just as CtdP uses NADP+ to convert native dienamine substrate 3 to α-anti-cycloadduct 2 via a NADP+/NADPH-dependent redox pathway, it is also able to convert zwitterionic substrate 9 to α-anti-cycloadduct 10 via an NADPH-dependent reduction rescue. We sought to understand why the native substrate for CtdP is the reduced dienamine form 3 while the native substrate for MalC is the oxidized zwitterionic form 9. This difference in substrate oxidation states was surprising given that the only other difference between the two native substrates is that the 5-membered pyrrolidine ring in the MalC substrate has been replaced by a 6-membered methylpiperidine ring in the CtdP substrate. However, DFT computations revealed that CtdP native substrate 3 (6-membered methylpiperidine ring) is over 1000 times more energetically demanding to oxidize than the corresponding substrate bearing a 5-membered pyrrolidine ring (Extended Data Fig. 7c). Thus, DFT computations clarified why the CtdP native substrate, present in reduced dienamine form 3, requires NADP+ for initial oxidation.
Finally, to further explore the anti-selective DAases, sequence similarity network (SSN) analysis of amino acids sequences of CtdP homologs revealed a clear boundary of NmrA-like proteins (Extended Data Fig. 9). Moreover, most of the CtdP homologs that appear in the clusters remain uncharacterized, further genome mining approaches will aid to explore more fascinating biocatalysts.
Conclusions
DAases involved in natural product biosynthesis had remained elusive for decades, recent efforts have revealed the remarkable functional and structural diversity within this non-traditional enzyme family. Unlike traditional enzyme families that are related by their sequence and structures, DAases are derived from a wide range of progenitor proteins with orthogonal catalytic functions discovering new types of DAases is particularly challenging11. In this work, we have characterized the function, structure, and mechanism of CtdP, an NmrA-like DAase that is responsible for the formation of α-anti-bicyclo[2.2.2]diazaoctane scaffold of 21R-citrinadin A (1). This represents the first NmrA-like DAase, while this family of proteins has typically been characterized as transcriptional regulators26 or redox sensors27. We propose a redox mechanism in that CtdP utilizes NADP+ to oxidize substrate 3, producing the electron-deficient diene required for the favored IEDDA cyclization that yields α-anti-cycloadduct 2-ox, and is subsequently reduced by NADPH to generate 2. CtdP is distinct in its ability to catalyse the formation of the electron-deficient diene for cycloaddition, which involves a LUMO lowering strategy. By contrast, MalC/PhqE syn-DAases also produce the reactive diene, but they catalyse its reductive formation that is required for NEDDA reaction8. Furthermore, enzymes that catalyse IEDDA cycloadditions are exceedingly rare, with only two other known examples, LepI3, and IccD9. Finally, cocrystallization of CtdP and MD simulations revealed that NADP+ and bulky residues Trp160, Phe170, and Phe277 position the substrate for conversion into the α-anti configuration, leading to CtdP’s exquisite stereoselectivity.
In conclusion, CtdP represents a novel addition to the expanding family of natural DAases. Based on the biosynthetic elucidation of both α-anti- and α-syn- bicyclo[2.2.2]diazaoctane core formation in the ctd and mal/phq pathways, we propose that CtdP and other NmrA-like DAases have played a critical role in the evolution and expansion of structural diversity in PIA natural products. The mechanistic insights gained from this study highlight the precise stereocontrol that nature employs in the assembly of complex metabolites, and also reveal new opportunities for utilizing DAases as biocatalytic tools.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgments, peer review information; details of author contributions and competing interests; and statements of data and code availability are available.
Methods
Strains and materials.
The fungus Penicillium citrinum ATCC 9849 was purchased from the American Type Culture Collection (ATCC, https://www.atcc.org/). Escherichia coli TOP10 was used as the host for plasmid propagation and E. coli BL21(DE3) (Novagen) was used for protein expression. Primers and functional genes malC and BmGDH were synthesized by Integrated DNA Technologies, Inc. (Coralville, USA). DNA sequencing was tested at GENEWIZ, Inc. (South Plainfield, USA). Kits for plasmid preparation and DNA extraction were purchased from Vazyme Biotech Co., Ltd (Nanjing, CN). Optical pure 21R-citrinadin A (1) and (+)-precitrinadin A (2) were respectively purified from the extract of P. citrinum ATCC 9849 and its ΔctdU mutant as previously described22. Cofactors NADP+, NAD+, NADPH, NADH, and FAD, as well as the NADP/NADPH quantitation kit, were purchased from Millipore Sigma (Burlington, USA).
Gene inactivation in P. citrinum ATCC 9849.
For targeted gene inactivation, split-marker recombination25 was performed in P. citrinum ATCC 9849 (Supplementary Fig. 3). Two specific homologous regions (~1.5 kb) were designed and amplified from P. citrinum ATCC 9849 genome, and the glufosinate (bar) resistant marker was amplified from plasmid pBARKS1. By using Gibson assembly, the DNA fragments were further assembled into a pUC57 vector. PCR amplification and gel purification were subsequently performed to obtain 10 μg of each DNA fragment for homologous recombination. The protoplasts of P. citrinum ATCC 9849 were obtained after 4 hours of digestion of the conidia (shake at 100 RPM at 30 °C) by Yatalase (2 mg/mL) and lysing enzyme (3 mg/mL), and further centrifuged and resuspended to the concentration of 108-109. After that, the prepared DNA fragments (each 10 μg) were mixed and incubated with the fresh protoplast for 50 min at 4 °C. Then the polyethylene glycol (PEG) mediated recombinations were performed on GMM (10 g/L glucose, 7 mM salts without nitrate, 10 mM ammonium tartrate, 0.1% trace elements, 16 g/L agar, pH = 6.5) plates containing 10% glufosinate and 1.2 M sorbitol. Colonies possessing correct genotype were verified by PCR reaction with the primers listed in Supplementary Table 2. The PCR results were illustrated in Supplementary Fig. 3.
Compounds purification and spectroscopic analyses.
The mutants of P. citrinum were cultured on a YES medium (150 g/L sucrose, 20 g/L yeast extract, 0.5 g/L magnesium sulfate) at 28 °C for 5 days. The fungal cells were extracted with ethyl acetate and ultrasonic-assisted three times. The EtOAc extracts were subsequently evaporated to dryness and further separated by silica chromatography to obtain the fractions containing the target compounds. After combination and concentration, the targeted fractions were further purified on Sephadex LH-20 chromatography (40–70 μm; GE Healthcare Life Science, USA). The obtained subfractions were finally purified by a preparative Agilent 1260 Infinity II LC System (Agilent Technologies, Inc., Santa Clara, USA) equipped with a semi-preparative column (Ultimate XB-C18, 10 × 250 mm, 5 μm, Welch, China). A linear gradient program of 30%−70% acetonitrile (v/v) with 0.01% triethylamine in H2O (v/v) over 30 min at a 4 mL/min flow rate was used for compounds purification. The target compounds were collected and dried for further structural identification. NMR spectra were measured on a Bruker NEO 600 MHz High-Performance Digital NMR (BrukerBiospin, Sweden) equipped with Bruker NMR TOPSPIN 3.6.2 software. NMR samples dissolved in deuterated solvents (CDCl3, CD3CN, DMSO-d6, Cambridge Isotope Laboratories). Electronic circular dichroism (ECD) spectra were recorded on a Jasco-810 spectropolarimeter (Jasco Inc., Easton, MD). High-resolution mass spectrometry (HRMS) was tested on an Agilent 1290 Infinity/6230 TOF LCMS system using electrospray ionization (ESI) in positive mode.
Proteins overexpression and purification.
The target genes were amplified and cloned into a modified pETDuet-1 vector with a C-terminal 6×histidine tag. After sequencing verification, the plasmids were transformed into E. coli BL21(DE3) for protein expression. E.coli cells were cultured in 1 L LB broth containing 100 μg/mL ampicillin at 37 °C until the OD600 value reached 0.5, and then protein expression was induced with 0.24 mM IPTG for 14 h at 16 °C. All purification steps were conducted at 4 °C. The cells were harvested by centrifugation (4000 g) for 20 min, and then resuspended in 30 mL lysis buffer (50 mM Tris-HCl, 300 mM NaCl, 5 mM imidazole, 1.0 mM TCEP, pH 8.0), and lysed by sonication. Subsequently, high-speed centrifugation (12,000 g, 30 min) was applied to obtain the lysate soluble fraction. The soluble fraction was added to 0.5 mL of Ni-NTA resin (QIAGEN) for protein binding (1 h), and then the mixture was loaded into a gravity-flow column. Proteins were washed with washing buffer (50 mM Tris-HCl, 300 mM NaCl, 20 mM imidazole, 1.0 mM TCEP, pH 8.0) and eluted with elution buffer (50 mM Tris-HCl, 300 mM NaCl, 300 mM imidazole, 1.0 mM TCEP, pH 8.0). The purified proteins were finally exchanged with the exchange buffer (50 mM Tris-HCl, 300 mM NaCl, 10% glycerol, 1.0 mM TCEP, pH 8.0) and then concentrated. The obtained proteins were used for in vitro assays and stored at −80 °C.
Dialysis of refolded CtdP preparations.
The purified C-His-tagged CtdP protein was diluted with denaturing buffer (8 M urea, 50 mM Tris-HCl, and 300 mM NaCl, pH 8.0). 1 mL solution containing CtdP was transferred into a dialysis bag and incubated in denaturing buffer (4 L) for 24 h dialysis. To remove residual urea and refold the protein, the denaturing buffer was subsequently exchanged with refolding buffer (0.5 M L-arginine, 50 mM Tris-HCl, and 300 mM NaCl, pH 8.0) for 12 h incubation. Finally, to remove the L-arginine, a further buffer exchange step was performed in the reaction buffer (50 mM Tris-HCl and 300 mM NaCl, pH 8.0) and incubation for 5 h. All the exchange steps were performed at 4 °C.
Chemical synthesis of compounds 3 and 9.
Substrate 3 was synthesized following the steps as shown in Supplementary Fig. 1a (synthetic steps from S1 to S7 were conducted by WuXi Apptec Co., Ltd., CN). The NMR data of 3 is shown in Supplementary Table 5. Substrate 9 was synthesized following reference8. The synthetic steps from S1 to 9 are shown in Supplementary Fig. 1b. The NMR and MS spectra of 9 are identical to those of the zwitterion11. The NMR spectra of 9 are shown in Supplementary Fig. 13.
In vitro activity assays.
The standard enzyme assay containing 500 μM substrate, 1 mM NADP+ and 40 μM enzyme in 50 μL reaction buffer (50 mM Tris-HCl, pH 7.0) was performed at 28 °C for 2 hours. The reactions were quenched with 50 μL LCMS grade methanol and centrifuged to remove solid material. The samples were analysed on an Agilent 6120B Single Quadrupole LC-MS using an Agilent Poroshell 120 EC-C18 column (3.0 × 150 mm) with the following time program: 5% acetonitrile over 5 minutes, 5%−60% acetonitrile over 25 minutes, 95% acetonitrile for 5 minutes, 95%−5% acetonitrile over 1 minute, and 5% acetonitrile for 4 minutes. 0.1% of formic acid was added to H2O. The flow rate was 0.5 mL/min and the reactions were monitored at 280 nm. LCMS data was collected on Agilent Openlab CDS ChemStation Edition software, version C.01.08 [210].
Anaerobic assays.
Anaerobic assays were performed in a Whitley A35 anaerobic workstation (Don Whitley Scientific Limited, West Yorkshire, UK) at 30 °C. The O2 concentration inside the chamber is monitored within 0~0.01%. The reaction buffer and other used components were degassed with nitrogen mixture (5% H2, 10% CO2) before transfer to the chamber, and placed in the chamber over 1 h gas exchange before usage in the reaction. The 50 μL reactions were quenched with 50 μL LCMS grade methanol in the anaerobic workstation. After centrifugation of each sample, the supernatant was analysed on LCMS. All experiments were repeated three times independently with similar results.
(S)-[4-2H] NADPH assays.
(S)-[4-2H] NADPH was generated by the incubation of glucose dehydrogenase BmGDH, NADP+, and [1-2H] D-glucose. The recombinant protein BmGDH was expressed in E. coli and purified before reactions31. 40 μM CtdP, 2 mM NADP+, 500 μM substrate 3, 4 mM [1-2H] D-glucose (Cambridge Isotope Laboratories) and 100 μM BmGDH were added in total 100 μL Tris-HCl buffer (50 mM, pH 7.0) for reaction. Controls were set as minus CtdP and minus [1-2H] D-glucose, respectively. Then the solutions were incubated at 30 °C for 2 h. Finally, the reactions were quenched with 100 μL LCMS grade methanol. After centrifugation of each sample, the supernatant was analysed on LCMS.
Mutagenesis of CtdP.
Primers for ctdP mutagenesis were ordered from IDT. After PCR amplification and gel purification, the mutated DNA fragments were cloned into modified pETDuet-1 vectors using Gibson assembly. The mutant plasmids were verified by DNA sequencing and transformed into E. coli BL21(DE3) for protein expression. E. coli cells were cultured in 0.5 L LB medium to an optical density (OD600) value of 0.5. Protein expression was induced with 0.24 mM IPTG for 13–16 h at 16 °C. The proteins were purified using the general purification method and subsequently involved in the enzymatic assays. The conversion and the relative activities of CtdP mutants were measured by the relative ratio of product 2 formation in the mutant compared to the wild-type. The concentration of product 2 was estimated by its standard carve that was generated from peak areas at 280 nm (UV) by LCMS. The data is shown in Supplementary Fig. 7a. The error bars represent the standard deviation (s.d.) of three independent replicates.
Single-crystal X-ray diffraction analyses of small molecules 5 and 10.
Colorless needles of compounds 5 and 10 were obtained from CH3CN solution by slow evaporation at room temperature, respectively. Data collection was performed on a Rigaku Oxford Diffraction XtalLAB Synergy-S using Cu Kα radiation at Rigaku Corp. The structures were solved with the SHELXT structure solution program using Intrinsic Phasing36 and refined with the SHELXL refinement package using Least Squares minimization37, performing on Olex238. The final refined crystallographic data of 5 and 10 have been deposited at the Cambridge Crystallographic Data Centre with deposition numbers CCDC 2127333 (5) and CCDC 2127332 (10). The crystallographic data and final refinement of 5 and 10 are presented in Supplementary Tables 11 and 12, respectively.
Overexpression and Purification of SeMet CtdP for Crystallization.
CtdP was subcloned from previously constructed plasmids into a modified pET28 vector with a TEV-cleavable, N-terminal 8×histidine-tag. Once validated, the plasmid was transformed into E. coli BL21(DE3) for protein overexpression. These transformed cells were then grown in 1 L of M9 minimal media and 50 g/mL of kanamycin at 37 °C with shaking at 200 RPM to OD600 = ~1.0. Cultures were subsequently supplemented with an amino acid solution (100 mg each of lysine, threonine, and phenylalanine, 50 mg each of leucine, isoleucine, and valine) including selenomethionine (SeMet, 50 mg) and induced with 1 mM IPTG, chilled to 18 °C with shaking at 200 RPM, and incubated for 18–20 h. Cells were harvested by centrifugation and stored at −80 °C.
For purification of SeMet CtdP, 10 g of cells were resuspended in lysis buffer (0.5 mg/mL lysozyme, 10% w/v glycerol, 50 mM NaCl, 20 mM imidazole, 50 mM HEPES pH 7.6, 0.1 mM NADP+, 0.2 mM TCEP). The resuspended solution was then subjected to sonication followed by high-speed centrifugation (60,000 g, 30 min) to obtain a clarified lysate. The clarified lysate was then applied to a Ni-NTA Histrap column and washed with 7 columns volumes of Ni-NTA buffer (25 mM potassium phosphate buffer pH 7.8, 300 mM NaCl, 20 mM imidazole, 10% w/v glycerol, 0.2 mM TCEP, 0.1 mM NADP+). SeMet CtdP was eluted using a solution of 250 mM imidazole pH 8.0, 0.2 mM TCEP, 0.1 mM NADP+, and 10% w/v glycerol. Fractions containing SeMet CtdP were pooled and combined with His-tagged tobacco etch virus (TEV) protease in a 1 : 30 (TEV : CtdP) ratio and dialyzed overnight at 4 °C into 10 mM HEPES pH 7.6, 50 mM NaCl, 0.2 mM TCEP, and 0.1 mM NADP+. The dialyzed protein solution was subsequently passed through a Ni-NTA Histrap column to remove the remaining His-tagged SeMet CtdP and TEV to obtain exclusively cleaved SeMet CtdP. Finally, the cleaved SeMet CtdP was applied to a GE PD-10 desalting column using storage buffer (10 mM HEPES pH 7.6, 50 mM NaCl, 0.2 mM TCEP, and 0.1 mM NADP+). Purified protein was drop-frozen into liquid nitrogen and stored at −80 °C.
Crystallization and structure determination of CtdP complex.
For crystallization of SeMet CtdP, a solution of CtdP (13 mg/mL), compound 9 (1 mM), and NADPH (2 mM) was made in storage buffer. The solution was incubated at room temperature for 1 h to allow the production of compound 10. The protein solution was mixed 1:1 v/v with precipitant solution (0.1 M BisTris pH 6.75, 25% PEG monomethyl ether 2000, 0.1 M CaCl2). Crystals formed after 24–48 h, harvested into a cryo-protectant solution (10% ethylene glycol, 25% PEG monomethyl ether 2000, 0.1 M CaCl2, 0.1 M BisTris pH 6.75, 1 mM NADP+, 0.5% DMSO) and flash-frozen in liquid nitrogen. Diffraction data were collected at beamline 23-ID-B at the Advanced Photon Source (APS) using an X-ray wavelength of 0.979 Å (360° of data, 100 K, 0.2° image width). Data were processed with XDS39 and the structure of SeMet crystals was solved by SAD phasing with Autosol40. Coot41 was used for model building and refinement was conducted using PHENIX.refine42. Cycles of manual building in Coot and refinement in PHENIX.refine were used to produce a final model. The structure of CtdP crystals was solved as a tetramer (chains A, B, C, D) in the asymmetric unit with NADP+ bound in each chain and compound 10 in three chains (chains A, B, D). MolProbity43 was used to validate the structures. Figures were generated using PyMol (https://pymol.org/2/).
Density functional theory calculations.
Conformational searches were performed using the Schrödinger MacroModel44 software package to identify the lowest energy conformers for quantum mechanical calculations. All quantum mechanical calculations were performed using the Gaussian 16 software package45. Structures were optimized at the M06–2X46/6–31+G(d) level of theory using the CPCM47–49 solvent model for water, which was chosen to mimic the role of the enzyme active site in stabilizing charges; frequency calculations were used to confirm the presence of local minima (no imaginary frequencies) and transition states (one imaginary frequency). Thermochemistry corrections to obtain enthalpies and free energies were computed at 298 K and 1 M, with Truhlar corrections50 applied to all frequencies below 100 cm−1 using the GoodVibes51 software package. To obtain more accurate energetics, single-point energy calculations were performed on the optimized structures at the M06–2X46/6–311++G(2d,2p) level of theory using the CPCM47−49solvent model for water. Orbital energies were also obtained at this level of theory. Visualizations of all computed structures were created using the CYLview2052 software package.
Molecular dynamics simulations.
Molecular dynamics simulations were prepared and equilibrated using the GPU code (pmemd)53 of the AMBER 16 package54. Parameters for the substrate 3-ox and the NADPH cofactor were generated within the antechamber module using the general AMBER force field (gaff)55, with partial charges set to fit the electrostatic potential generated at the HF/6–31(d) level by the RESP model56. The partial charges were calculated according to the Merz–Singh–Kollman scheme57,58 using the Gaussian 16 package45. Each protein was immersed in a pre-equilibrated cubic box with a 10 Å buffer of TIP3P59 water molecules using the leap module, resulting in the addition of around 19,000 solvent molecules. The systems were neutralized by the addition of explicit counter ions (Na+ and Cl−). All subsequent calculations were done using the widely tested Stony Brook modification of the Amber14 force field (ff14sb)60. Water molecules were treated with the SHAKE algorithm such that the angle between the hydrogen atoms was kept fixed. For the heating and equilibration steps, long-range electrostatic effects were modeled using the particle-mesh-Ewald method61. An 8 Å cutoff was applied to Lennard–Jones and electrostatic interactions. First, a geometry optimization was performed on each system to minimize the positions of solvent molecules and ions while imposing positional restraints on the protein backbone and ligands using a harmonic potential with a force constant of 2 kcal mol−1·Å−2. Second, each system was gently and continuously heated over 1 ns from 0 K to 300 K under constant-volume and periodic-boundary conditions. Harmonic restraints of 2 kcal mol−1 were applied to the protein backbone and ligands, and the Andersen equilibration scheme was used to control and equalize the temperature. The time step was kept at 1 fs during the heating stages, allowing potential inhomogeneities to self-adjust. Third, each system was then equilibrated for a total of 4 ns at a constant pressure of 1 atm with a Berendson barostat with a 2 fs time step; harmonic restraints of 2 kcal mol−1 were applied for the first 2 ns and harmonic restraints of 0.5 kcal mol−1 was applied for the second 2 ns to the protein backbone and ligands. Finally, production trajectories without harmonic restraints were run on the Anton 2 supercomputer62 for 1200 ns with a 2.5 fs time step at 300 K and 1 atm using the default NPT integrator and the default u-series treatment of electrostatic interactions. Three production trajectories were obtained for the CtdP enzyme with the substrate 3-ox and the NADPH cofactor present in the active site.
Extended Data
Extended Fig 1-.

Representations of different classes of DielsAlderases and the proposed biosynthetic pathway of 21Rcitrinadin A.
Extended Fig 2-.

Comparison analysis of the biosynthetic gene clusters of PIAs and in vivo characterization of NmrA-like proteins CtdO, CtdR, and CtdP.
Extended Fig 3-.

In vitro assays of 3 in anaerobic and aerobic conditions.
Extended Fig 4-.

Sequence alignment of CtdP and homologous NmrA-like proteins.
Extended Fig 5-.

Superposition of the different chains in CtdP.
Extended Fig 6-.

C-terminal tail and electrostatic surface structures of CtdP.
Extended Fig 7-.

Density functional theory calculations for tautomerization and redox mechanisms.
Extended Fig 8-.

MD simulations of substrate 3-ox in CtdP active site
Extended Fig 9-.

Sequence similarity network analysis of NmrAlike protein CtdP.
Supplementary Material
Acknowledgments
We thank Prof. László Kürti and Young-Do Kwon for their assistance with HRMS experiments, Prof. Jeffrey D. Hartgerink for sharing the instruments for electronic circular dichroism measurements, and Prof. Caroline Ajo-Franklin and Siliang Li for sharing the anaerobic workstation, and Prof. Janet Smith for providing generous resources for protein crystallization, APS beamline access, and software for structure determination; and finally, 23-IDB@GM/CA beamline staff. This work was supported by the National Institute of Health (NIH) grant (R35GM138207) and the Robert A. Welch Foundation (C-1952) to [X.G], the NIH grant (R35GM11810), and the Hans W. Vahlteich Professorship to [D.H.S], and the NIH grant (AI141481) to [K.N.H]. J.N.S. acknowledges the support of the National Institute of General Medical Sciences (NIGMS) of the NIH under an F32 individual postdoctoral fellowship (F32GM122218). S.R. acknowledges support from the Michigan Chemistry-Biology Interface Training Program, funded by the NIGMS, NIH T32 Training Grant (5T32GM132046). Computational resources for DFT computations were provided by the UCLA Institute for Digital Research and Education (IDRE) and by the San Diego Supercomputing Center (SDSC) through XSEDE (ACI-1548562). Microsecond molecular dynamics simulations were performed using Anton 2 computer time provided by the Pittsburgh Supercomputing Center (PSC) through Grant R01GM116961 from NIH. The Anton 2 machine at PSC was generously made available by D.E. Shaw Research.
Footnotes
Additional information
Supplementary Information is available for this paper at www.---.
Competing interests
The authors declare no competing interests.
Data availability
Data supporting the findings of this work are available within the Article, Extended Data, Source Data and the Supplementary files. Data supporting the current study are also available from the corresponding author upon request. The genome sequences of P. citrinum ATCC 9849 can be accessible from GeneBank database (BCKA00000000.1). Coordinates and associated structure factors of CtdP have been deposited in the Protein Data Bank (PDB) database (PDB code: 7UF8). The crystallographic data of small molecules have been deposited at the CCDC with 2127333 for 5 and 2127332 for 10. Energies and molecular coordinates of calculated structures are provided in the Supplementary Information file.
References
- 1.Nicolaou KC, Snyder SA, Montagnon T. & Vassilikogiannakis G. The Diels-Alder reaction in total synthesis. Angew. Chem. Int. Ed 41, 1668–1698 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Houk KN, Liu F, Yang Z. & Seeman JI Evolution of the Diels–Alder reaction mechanism since the 1930s: Woodward, Houk with Woodward, and the influence of computational chemistry on understanding cycloadditions. Angew. Chem. Int. Ed 60, 12660–12681 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Ohashi M. et al. SAM-dependent enzyme-catalysed pericyclic reactions in natural product biosynthesis. Nature 549, 502–506 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao L. et al. FAD-dependent enzyme-catalysed intermolecular [4+2] cycloaddition in natural product biosynthesis. Nat. Chem 12, 620–628 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Caputi L. et al. Missing enzymes in the biosynthesis of the anticancer drug vinblastine in Madagascar periwinkle. Science 360, 1235–1239 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Tian Z. et al. An enzymatic [4+2] cyclization cascade creates the pentacyclic core of pyrroindomycins. Nat. Chem. Biol 11, 259–265 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Tan D. et al. Genome-mined Diels–Alderase catalyzes formation of the cis-octahydrodecalins of varicidin A and B. J. Am. Chem. Soc 141, 769–773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dan Q. et al. Fungal indole alkaloid biogenesis through evolution of a bifunctional reductase/Diels-Alderase. Nat. Chem 11, 972–980 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Z. et al. Enzyme-catalyzed inverse-electron demand Diels–Alder reaction in the biosynthesis of antifungal ilicicolin H. J. Am. Chem. Soc 141, 5659–5663 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Q. et al. Enzymatic intermolecular hetero-Diels–Alder reaction in the biosynthesis of tropolonic sesquiterpenes. J. Am. Chem. Soc 141, 14052–14056 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jamieson CS, Ohashi M, Liu F, Tang Y. & Houk KN The expanding world of biosynthetic pericyclases: cooperation of experiment and theory for discovery. Nat. Prod. Rep 36, 698–713 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qian-Cutrone J. et al. Stephacidin A and B: two structurally novel, selective inhibitors of the testosterone-dependent prostate LNCaP cells. J. Am. Chem. Soc 124, 14556–14557 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Madariaga-Mazón A, Hernández-Abreu O, Estrada-Soto S. & Mata R. Insights on the vasorelaxant mode of action of malbrancheamide. J. Pharm. Pharmacol 67, 551–558 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Zinser EW et al. Anthelmintic paraherquamides are cholinergic antagonists in gastrointestinal nematodes and mammals. J. Vet. Pharmacol. Ther 25, 241–250 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Paterson RRM, Simmonds MSJ & Blaney WM Mycopesticidal effects of characterized extracts of Penicillium isolates and purified secondary metabolites (including mycotoxins) on Drosophila melanogaster and Spodoptora littoralis. J. Invertebr. Pathol 50, 124–133 (1987). [Google Scholar]
- 16.Klas KR et al. Structural and stereochemical diversity in prenylated indole alkaloids containing the bicyclo[2.2.2]diazaoctane ring system from marine and terrestrial fungi. Nat. Prod. Rep 35, 532–558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fraley AE & Sherman DH Enzyme evolution in fungal indole alkaloid biosynthesis. FEBS J. 287, 1381–1402 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Domingo LR, Zaragozá RJ & Williams RM Studies on the biosynthesis of paraherquamide A and VM99955. A theoretical study of intramolecular Diels−Alder cycloaddition. J. Org. Chem 68, 2895–2902 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Tsuda M. et al. Citrinadin A, a novel pentacyclic alkaloid from marine-derived fungus Penicillium citrinum. Org. Lett 6, 3087–3089 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Mercado-Marin EV et al. Total synthesis and isolation of citrinalin and cyclopiamine congeners. Nature 509, 318–324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang P, Li X-M, Liu H, Li X. & Wang B-G Two new alkaloids from Penicillium oxalicum EN-201, an endophytic fungus derived from the marine mangrove plant Rhizophora stylosa. Phytochem. Lett 13, 160–164 (2015). [Google Scholar]
- 22.Liu Z. et al. Structural basis of the stereoselective formation of the spirooxindole ring in the biosynthesis of citrinadins. Nat. Commun 12, 4158 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goswami RS Targeted gene replacement in fungi using a split-marker approach. Methods Mol. Biol 835, 255–269 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Chen M, Liu C-T & Tang Y. Discovery and biocatalytic application of a PLP-dependent amino acid γ-substitution enzyme that catalyzes C–C bond formation. J. Am. Chem. Soc 142, 10506–10515 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andrianopoulos A, Kourambas S, Sharp JA, Davis MA & Hynes MJ Characterization of the Aspergillus nidulans nmrA gene involved in nitrogen metabolite repression. J. Bacteriol 180, 1973–1977 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stammers DK et al. The structure of the negative transcriptional regulator NmrA reveals a structural superfamily which includes the short-chain dehydrogenase/reductases. EMBO J. 20, 6619–6626 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zang W. & Zheng X. Structure and functions of cellular redox sensor HSCARG/NMRAL1, a linkage among redox status, innate immunity, DNA damage response, and cancer. Free Radic. Biol. Med 160, 768–774 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng X. et al. Restructuring of the dinucleotide-binding fold in an NADP(H) sensor protein. Proc. Natl. Acad. Sci. U. S. A 104, 8809–8814 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trottmann F. et al. Pathogenic bacteria remodel central metabolic enzyme to build a cyclopropanol warhead. Nat. Chem 14, 884–890 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu CJ, Li CW, Gao H, Huang XJ & Cui CB Penicimutamides D–E: two new prenylated indole alkaloids from a mutant of the marine-derived Penicillium purpurogenum G59. RSC Adv. 7, 24718–24722 (2017). [Google Scholar]
- 31.Makino Y, Negoro S, Urabe I. & Okada H. Stability-increasing mutants of glucose dehydrogenase from Bacillus megaterium IWG3. J. Biol. Chem 264, 6381–6385 (1989). [PubMed] [Google Scholar]
- 32.Zhang G. et al. Mechanistic insights into polycycle formation by reductive cyclization in ikarugamycin biosynthesis. Angew. Chem. Int. Ed 53, 4840–4844 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Wen W-H et al. Reductive inactivation of the hemiaminal pharmacophore for resistance against tetrahydroisoquinoline antibiotics. Nat. Commun 12, 7085 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Z, Chen L, Liu L, Su X. & Rabinowitz JD Chemical basis for deuterium labeling of fat and NADPH. J. Am. Chem. Soc 139, 14368–14371 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laina-Martín V, Fernández-Salas JA & Alemán J. Organocatalytic strategies for the development of the enantioselective inverse-electron-demand hetero-Diels-Alder reaction. Chem. Eur. J 27, 12509–12520 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheldrick GM SHELXT-integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr 71, 3–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheldrick GM Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Cryst. Struct. Commun 71, 3–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JA & Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr 42, 339–341 (2009). [Google Scholar]
- 39.Kabsch W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Terwilliger TC et al. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. Sect. D Biol. Crystallogr 65, 582–601 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emsley P, Lohkamp B, Scott WG & Cowtan K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schrödinger Release 2017–2: MacroModel, version 11.2.014. (Schrödinger, 2017). [Google Scholar]
- 45.Frisch MJ et al. Gaussian 16 Revision C.01. (Gaussian, 2016). [Google Scholar]
- 46.Zhao Y. & Truhlar DG The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc 120, 215–241 (2008). [Google Scholar]
- 47.Miertuš S, Scrocco E. & Tomasi J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys 55, 117–129 (1981). [Google Scholar]
- 48.Miertus S. & Tomasi J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys 65, 239–245 (1982). [Google Scholar]
- 49.Pascual-ahuir JL, Silla E. & Tuñon I. GEPOL: an improved description of molecular surfaces. III. A new algorithm for the computation of a solvent-excluding surface. J. Comput. Chem 15, 1127–1138 (1994). [Google Scholar]
- 50.Ribeiro RF, Marenich AV, Cramer CJ & Truhlar DG Use of solution-phase vibrational frequencies in continuum models for the free energy of solvation. J. Phys. Chem 115, 14556–14562 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Luchini G, Alegre-Requena J, Funes-Ardoiz I. & Paton R. GoodVibes: automated thermochemistry for heterogeneous computational chemistry data [version 1; peer review: 2 approved with reservations]. F1000Research 9 (2020). [Google Scholar]
- 52.Legault CY Legault CY CYLview20. (Université de Sherbrooke, 2020). [Google Scholar]
- 53.Salomon-Ferrer R, Götz AW, Poole D, Le Grand S. & Walker RC Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput 9, 3878–3888 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Case DA et al. AMBER 2016. (University of California, San Francisco, 2016). [Google Scholar]
- 55.Wang J, Wolf RM, Caldwell JW, Kollman PA & Case DA Development and testing of a general amber force field. J. Comput. Chem 25, 1157–1174 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Bayly CI, Cieplak P, Cornell W. & Kollman PA A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Chem. Phys 97, 10269–10280 (1993). [Google Scholar]
- 57.Besler BH, Merz KM Jr. & Kollman PA Atomic charges derived from semiempirical methods. J. Comput. Chem 11, 431–439 (1990). [Google Scholar]
- 58.Singh UC & Kollman PA An approach to computing electrostatic charges for molecules. J. Comput. Chem 5, 129–145 (1984). [Google Scholar]
- 59.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW & Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 79, 926–935 (1983). [Google Scholar]
- 60.Maier JA et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput 11, 3696–3713 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Darden T, York D. & Pedersen L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys 98, 10089–10092 (1993). [Google Scholar]
- 62.Shaw DE et al. Anton 2: raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer. in SC ‘14: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis 41–53 (2014). [Google Scholar]
- 63.Li H. et al. Asperversiamides, linearly fused prenylated indole alkaloids from the marine-derived fungus Aspergillus versicolor. J. Org. Chem 83, 8483–8492 (2018). [DOI] [PubMed] [Google Scholar]
- 64.Kagiyama I. et al. Taichunamides: prenylated indole alkaloids from Aspergillus taichungensis (IBT 19404). Angew. Chem. Int. Ed 55, 1128–1132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fraley AE et al. Molecular basis for spirocycle formation in the paraherquamide biosynthetic pathway. J. Am. Chem. Soc 142, 2244–2252 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fraley AE et al. Flavin-dependent monooxygenases NotI and NotI’ mediate spiro-oxindole formation in biosynthesis of the notoamides. ChemBioChem 21, 2449–2454 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sievers F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 7, 539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robert X. & Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gerlt JA et al. Enzyme function initiative-enzyme similarity tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta 1854, 1019–1037 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shannon P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the findings of this work are available within the Article, Extended Data, Source Data and the Supplementary files. Data supporting the current study are also available from the corresponding author upon request. The genome sequences of P. citrinum ATCC 9849 can be accessible from GeneBank database (BCKA00000000.1). Coordinates and associated structure factors of CtdP have been deposited in the Protein Data Bank (PDB) database (PDB code: 7UF8). The crystallographic data of small molecules have been deposited at the CCDC with 2127333 for 5 and 2127332 for 10. Energies and molecular coordinates of calculated structures are provided in the Supplementary Information file.