

Abstract
Targeted protein degradation (TPD) is emerging as a strategy to overcome the limitations of traditional small-molecule inhibitors. Proteolysis-targeting chimera (PROTAC) technology can be used to target proteins by hijacking the ubiquitin-proteasome system. Conceptually, PROTAC aims to target the “undruggable” majority of proteins in the human proteome. Through constant exploration and optimization of PROTACs and the exploitation of other TPD strategies over two decades, TPD has expanded from theoretical studies to clinical strategies, with practical applications in oncological, immunological, and other diseases. In this review, we introduce the mechanisms, features, and molecular targets of orthodox PROTACs and summarize the PROTAC drugs under study as cancer therapeutics in clinical trials. We also discuss PROTAC derivatives and other TPD strategies, such as lysosome-targeting chimeras, autophagy-targeting chimeras, and molecular glue strategies. Collectively, the studies summarized herein support the full potential of TPD in the biomedical industry.
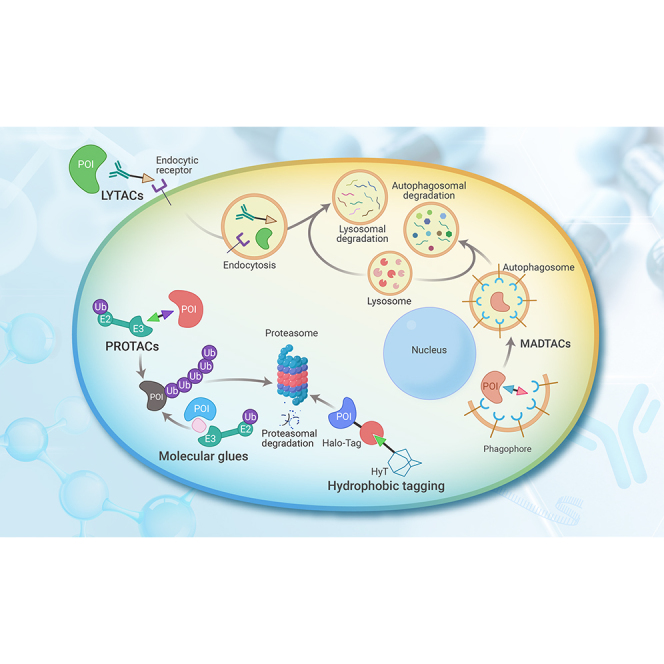
Graphical abstract
Public summary
-
•
Targeted protein degradation provides new opportunities for cancer therapy.
-
•
Proteolysis-targeting chimeras (PROTACs) can target “undruggable” proteins.
-
•
PROTAC drugs have entered the clinic and have the potential to become “best in class”.
Introduction
Proteins play essential roles in processes that control the health and survival of cells. Accordingly, any abnormality in a protein can affect its constitution, functionality, homeostasis, and/or other properties, leading to various diseases.1 Studies have shown that proteins comprise the most important class of drug targets.2 In the past 20 years, efforts to inhibit or disrupt the biological functions of target proteins have led to several targeted cancer therapies, such as antibodies and small-molecule inhibitors (SMIs).3,4 Antibodies have been widely used in cancer treatment because of their biological specificity.5 However, antibodies have a high molecular weight and low permeativity and, thus, primarily target membrane-associated proteins, which has limited their clinical application.6,7 According to the US Food and Drug Administration (FDA) database, 52 SMIs targeting protein kinases have been identified via large-scale screening and optimization efforts. These SMIs inhibit the biological activity of their target proteins by binding to the active sites of the proteins.8,9 Compared with antibodies, SMIs have much lower molecular weights and are more suitable for targeting intracellular, membrane-associated, and extracellular proteins. However, SMI therapy faces a significant disadvantage: long-term administration of these drugs increases the risk of developing resistance via mutations in the protein targets.10
Approximately 16,000 proteins with no targeted active sites have been identified, accounting for 85% of the human proteome. These proteins, which include pseudokinases, transcription factors, and scaffolding proteins or adaptors, have long been known as undruggable targets.11,12,13,14 Gene knockdown techniques, such as RNA interference and the CRISPR-Cas9 system, have been used to expand the range of druggable targets. However, the nucleic acid-based molecules used in these approaches face significant challenges, such as instability, poor cell permeativity, and inefficiency due to enzyme-catalyzed hydrolysis.15,16,17 To overcome these problems, attention has increasingly been directed toward novel strategies that exploit the physiological protein degradation machinery, including the ubiquitin-proteasome system (UPS)-mediated proteolysis-targeting chimera (PROTAC),18,19 autophagy-lysosome pathway-mediated autophagy-targeting chimera (AUTAC),20 and endosome-lysosome pathway-mediated lysosome-targeting chimera (LYTAC);21 molecular glue; and other PROTAC derivatives.22,23,24
In this review, we discuss the development and derivatives of PROTACs. In addition, we review the drug-discovery applications of PROTACs and other targeted protein degradation (TPD) strategies in academia and industry.
The development of PROTACs
The ubiquitin-proteasome system
In eukaryotic cells, the UPS is one of the primary pathways through which intracellular proteins are degraded to maintain cell homeostasis.25,26 The UPS consists of ubiquitin (Ub), enzymes, the proteasome, and specified substrates that play crucial roles in various cellular metabolic processes, such as the regulation of cell signaling and transcription and protein turnover.27,28 Ubiquitin, a highly evolutionarily conserved protein, is a 76-amino-acid polypeptide that is used to mark target proteins for proteolysis in the UPS. Protein ubiquitination is carried out via a reversibly ubiquitinating enzymatic cascade comprising the E1 ubiquitin-activating enzyme (E1), E2 ubiquitin-conjugating enzyme (E2), and ubiquitin (E3) ligase. E1 activates ubiquitin for conjugation and transfers it to an E2. Through catalysis by E3 ligase, ubiquitin is transferred directly from E2 to a substrate protein via the ε-amino group on a lysine residue or the N terminus. Ubiquitin comprises seven lysine residues and an N-terminal methionine residue, which allow further ubiquitination to form a polyubiquitin chain via E3 ligase.29 A polyubiquitinated protein can be recognized by the 26S proteasome, transported to the 20S proteasome, and hydrolyzed into oligopeptides via various enzymes, which are eventually released from the proteasome (Figure 1A).30,31
Figure 1.

Background information on PROTACs
The mechanism of action of PROTACs
PROTACs have attracted considerable attention owing to their great potential for use in cancer treatment.32 In this strategy, the PROTAC, a heterobifunctional molecule, contains a “warhead” specific for a protein of interest (POI) and a ligand for an E3 ligase, which are joined by an intermediate linker. The PROTAC can simultaneously engage an E3 ubiquitin ligase and a POI in an event-driven manner (Figure 1B). This spatial proximity allows for the ubiquitination and subsequent proteasomal degradation of the POI. Rather than being directly degraded, PROTAC molecules are recycled to target other proteins.33,34 The PROTAC system has opened up a new and promising drug-development pathway, leading to remarkable achievements, such as the reversal of drug resistance,35,36 targeting of “undruggable” proteins,37,38 and enhancement of drug selectivity and specificity.39,40
Structural analysis of PROTACs
The formation of a stable, high-affinity ternary complex (POI-PROTAC-E3 ligase) is a key step in the mechanism of action (MOA) of PROTAC. Ciulli and colleagues were the first to solve the crystal structure of a ternary complex comprising BRD4BD2 (BRD4, a member of the bromo- and extraterminal [BET] family proteins; BRD4BD2, the second bromodomain [BD] of BRD4), MZ1, and VCB (VHL [von Hippel-Lindau protein], ElonginC, and ElonginB) at a 2.7-Å resolution (PDB: 5T35).41 MZ1, which consists of the BET inhibitor JQ1, VHL ligand VH032, and a polyethylene glycol (PEG) linker, is bound within a bowl-shaped interface formed by extensive protein-protein interactions (PPIs) between BRD4BD2 and VHL.42 The bowl-shaped interface is formed mainly from hydrophobic and electrostatic contacts between proteins and ligands and PPIs, including (1) the formation of a WPF shelf (W374, P375, F376) and an extended PWPF (P71, W374, P375, F376) shelf via the interaction of BRD4BD2 with VHL, (2) the interaction between the helical turn of BRD4BD2 and the hydrophobic side chains of VHL, (3) the embedding of JQ1 in the acetyl-lysine binding pocket of BRD4BD2 and binding of VHL032 to the hydroxyproline binding site of VHL, and (4) van der Waals interactions and a hydrogen bond due to an interaction with the PEG linker. With this system, stabilized and selective target degradation is attributed to the extensive burial of the ternary complex surface area (from 1,933 to 2,621 Å2) and the formation of new PPIs (Figure 1C).41
A few other examples of the X-ray crystal structures of ternary complexes are available. Fischer and colleagues solved multiple X-ray structures of BRD4-PROTAC-CRL4CRBN (CRBN, cereblon) complexes and showed that plasticity results in several distinct low-energy binding conformations selectively bound by ligands (PDB: 5FQD).43 The ternary complex crystal structures of ACBI1, a potent and cooperative PROTAC of SMARCA2/4 and PBRM1, have also been solved by the Ciulli team, who optimized the structures rationally using biophysical data (PDB: 6HAY).44
Characteristics of PROTACs
Crews and Deshaie’s group pioneered the concept of PROTACs in 2001 with PROTAC-1. PROTAC-1 was shown to simultaneously bind methionine aminopeptidase-2 (MetAP2) and the E3 ligase Skp1-Cullin-F boxβ-TrCP (SCFβ-TrCP) to induce degradation of MetAP2 in a crude cell extract.45 However, peptide-based PROTACs have poor chemical stability and cellular permeativity, which limits their clinical applications.46 Small-molecule-based PROTACs take advantage of small molecules to engage E3 ligases, thus advancing the development of PROTAC technology. Some small-molecule ligands of common E3 ligases, including mouse double minute 2 homolog (MDM2), inhibitors of apoptosis (IAPs), VHL, and CRBN, have been reported.47,48,49,50
Given its unique MOA, a PROTAC does not need to bind within the target of a functionally bioactive site to achieve degradation of the POI. PROTACs have been verified as a feasible option for targeting proteins with scaffolding functions51,52 or mutations,53,54 as well as those that are overexpressed,55,56 aggregated, or present in different isoforms,57,58,59,60 and thus can overcome the limitation of traditional SMIs, which are not capable of effective and selective employment. Most orthodox PROTACs are used in the field of oncology (Table S1), and this use is described and discussed in detail in later sections. In addition to PROTACs that target various cancer-related proteins, PROTACs relevant to hematology, immunology, neurology, and antiviral treatment have been the focus of active research in academia and industry,37,61,62,63 but are not introduced in this review.
Targeting various molecules using orthodox PROTACs
Targeting receptor tyrosine kinases (RTKs)
RTKs, a subgroup of tyrosine kinases, are embedded in cell membranes and mediate intercellular communication. Dysregulation of RTKs has been correlated with many diseases, especially cancer. Chromosomal rearrangements, gain-of-function mutations, overexpression, and genomic amplification are the main factors contributing to the oncogenic activation of RTKs and leading to resistance to pharmaceutical inhibitors.64 To date, most PROTAC warheads have been inhibitors that act as ligands to recruit POIs. Viable PROTAC strategies to degrade RTKs, such as anaplastic lymphoma kinase (ALK), epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor 2 (VEGFR-2), and tropomyosin receptor kinase (TRK) family members, have been validated (Figure 2; Table S1). Alectinib-based degrader 17 was shown to exhibit potent ALK-binding affinity and antiproliferative activity in an ALK-dependent cell line, but not in exclusively ALK fusion-negative cells.65 The ability of compounds 2/10 to induce EGFR degradation, with respect to the half-maximal degradation concentration (DC50) values of 45.2 and 34.8 nM, and induce apoptosis in a cell line lacking EGFR exon 19 has been validated.66 Binding of a lapatinib-based PROTAC-1 to a VHL ligand effectively induces the degradation of wild-type and FLAG-labeled exon-20-mutated EGFR.67 A series of selective EGFRL858R/T790M degraders exclusively target mutated EGFR, with nanomolar DC50 values, but do not degrade wild-type EGFR.67,68,69 PROTACs based on the antiangiogenesis agents S7 and ABT-869, especially PROTAC-2 and PROTAC-5, have been shown to induce a specific decrease in VEGFR-2 levels and exhibit considerable antiproliferative activity against human umbilical vein endothelial cells.70 Compounds 5 (CG416) and 6 (CG428) induce the selective degradation of the TPM3-TrkA fusion protein.71 CRBN-based degrader 4 was shown to induce TrkC degradation in breast cancer cells, with an estimated DC50 value of 0.1–1.0 μM.72 PROTACs that target other RTKs are listed in Table S1.
Figure 2.

PROTACs of different POIs
Targeting non-receptor tyrosine kinases (nRTKs)
Another subgroup of tyrosine kinases, nRTKs, includes cytosolic enzymes involved in signal transduction in activated T and B lymphocytes, the dysregulation of which can lead to hematological malignancies, such as lymphomas, leukemias, and myelomas. Point mutations resulting in oncogene formation and chromosomal translocation caused by gene fusion can cause the aberrant activation of nRTKs.73 VHL-, CRBN-, or IAP-based PROTACs can degrade various nRTKs, including the BCR-ABL fusion gene, aurora kinase A (AURKA), B cell lymphoma 2 (BCL-2) and BCL-extra large (BCL-xL), induced myeloid leukemia cell differentiation protein (MCL-1), Bruton’s tyrosine kinase (BTK), focal adhesion kinase (FAK), Janus kinase (JAK), and Src homology 2 (SH2) domain-containing phosphatase 2 (SHP2), and these PROTACs have been shown to induce cytotoxicity in multiple types of hematological tumor cells (Figure 2; Table S1). Ponatinib- and dasatinib-based PROTACs P19P and P22D have been developed to degrade the mutant protein ABLT315I and thus overcome resistance to dasatinib and asciminib.74 The thalidomide-based degraders JB170 and JB301 induce highly specific AURKA degradation at a nanomolar scale, causing profound toxicity in MV4-11 cells.75,76 The PROTAC DT2216, which is currently being studied in a clinical trial, links the relative inhibitor ABT263, which binds BCL-2 and BCL-xL, to a VHL ligand. DT2216 has been shown to specifically target BCL-xL and exhibit increased antitumor activity and reduced platelet toxicity compared with ABT263, the BCL-xL inhibitor.77,78 The reversible non-covalent PROTAC NC-1 has been reported to be a potent degrader that induces the depletion of wild-type and some mutated forms of BTK in chronic lymphocytic leukemia (CLL) cells from patients in vitro.79 Compared with NC-1, the reversible covalent PROTAC RC-3 displays enhanced selectivity toward BTK.80 The defactinib-based PROTAC-3 effectively degraded FAK and suppressed its activity (autophosphorylation of Y397) in the PC-3 cell line. PROTAC-3 has been shown to reduce the invasiveness of MDA-MB-231 cells by ∼65%, whereas no obvious effect was observed with defactinib treatment.81 The inhibitors pyrimidine 1 and quinoxaline 2 have been chosen as warheads that selectively bind JAKs.82 The PROTACs JP-1 to JP-6, which bear IAP ligands, have been validated as efficient inducers of JAK1 and JAK2 degradation.83 VHL-based SHP2-D26 mediates the depletion of SHP2, leading to a decrease in the level of the downstream protein pERK, with half-maximal inhibitory concentration (IC50) values of 0.66 μM and 9.9 nM in the KYSE-520 and MV4-11 cell lines, respectively.82 PROTACs that target other nRTKs are listed in Table S1.
Targeting serine/threonine protein kinases (STKs)
STKs are characterized by their ability to phosphorylate serine or threonine residues, and they play roles in cell proliferation, differentiation, programmed cell death, and embryonic development. For example, BRAFV600E, an oncogenic mutant of BRAF that monomerically transmits a signal in the absence of activated RAS, is selectively degraded by SJF-0628, a vemurafenib-based PROTAC that does not affect the wild-type form or other RAF family members, such as ARAF and CRAF.84 PROTACs that target cyclin-dependent kinase 12 (CDK12), such as BSJ-4-116 and PP-C8, have been reported to degrade functional mutants of CDK12.85,86 However, two CDK12 mutations have exhibited resistance to BSJ-4-116, demonstrating a potential mechanism by which tumor cells can evade TPD.85 Compound 9, a conjugate of interleukin-1 receptor-associated kinase 4 (IRAK4) inhibitor 1, which binds to IRAK4, and pomalidomide, which binds to CRBN, induces substantial degradation of IRAK4 in the ABC-diffuse large B cell lymphoma (DLBCL) cell lines OCI-LY10 and TMD8.87 PROTAC_RIPK2 was shown to degrade receptor-interacting serine/threonine protein kinase 2 (RIPK2) in a highly specific manner, and at nanomolar concentrations, it leads to a decrease in the level of downstream mitogen-activated protein kinase (MAPK) in a VHL-dependent manner.88 Degrader 23 (MS423), which is composed of PD0325901 and a VHL ligand, inhibits mitogen-activated protein kinase kinases 1/2 (MEK1/2) kinase activity in vitro and degrades target proteins in colorectal cancer (CRC) and melanoma cell lines, leading to decreases in the levels of p-MEK and p-ERK and inhibition of the proliferation of CRC and melanoma cell lines.89 PROTACs SJFα and SJFδ have been shown to degrade the p38 MAPK family isoforms p38α and p38δ, respectively.90 SJFδ has been shown to target both wild-type and mutant p38δK220E/T221E. SGK3-PROTAC1 mediates the selective degradation of serum/glucocorticoid regulated kinase family member 3 (SGK3), but not SGK1/2, at a DC50 of <100 nM.91 Combination therapy with PI3K inhibitors and SGK3-PROTAC1 at a micromolar concentration has been shown to outperform the effects of a conventional PI3K inhibitor (GDC0941) in the CAMA-1 breast cancer cell line (Figure 2).91 PROTACs that target other STKs are listed in Table S1.
Targeting proteins in transcriptional regulation
Transcription factors (TFs) work alone or with other proteins in complexes to regulate gene transcription and thus contribute to cell-fate determination. Historically, TFs have been classified as undruggable targets due to their smooth surface structure and lack of a pocket resulting from protein-DNA interactions or PPIs.92 Nuclear hormone receptors, such as estrogen receptor (ER), androgen receptor (AR), retinoic acid receptor (RAR), and cellular retinoic acid-binding protein (CRABP), have been reported as targets of PROTACs.47,56,93,94 SD-36, a conjugate of the signal transducer and activator of transcription 3 (STAT3) inhibitor SI-109 and a CRBN ligand, is the first reported selective degrader of STAT3 protein. It has been shown to achieve complete and continuous tumor suppression in a Molm-16 mouse xenograft model at well-tolerated dose schedules.95 In addition to targeting TFs, PROTECs are designed to target epigenetic-related proteins, such as BET family members (BRD2/3/4/7/9), P300/CBP-associated factor (PCAF), histone deacetylases (HDACs), and Polycomb repressive complex 2 (PRC2), as dysregulation of these proteins and, subsequently, the epigenome promotes cancer onset and progression (Table S1).96 For example, SMIs (JQ1 and I-BET726) and typical PROTACs (MZ1, ARV-825, and ARV-771) have been shown to target BET family members and inhibit downstream oncogene expression.41,57,58,97,98,99 The PROTAC GSK983 mediates the degradation of PCAF and its homologous protein general control non-derepressible 5 (GCN5) in a concentration-dependent manner in both macrophages and monocyte-derived dendritic cells, leading to a reduction in inflammatory cytokine production.100 UNC6852, which selectively degrades PRC2, has been shown to reduce the levels of EZH2, EED, and SUZ12, leading to a decrease in the level of trimethylation of lysine 27 on histone H3 (H3K27me3). UNC6852 inhibits the proliferation of DB and Pfeiffer cells (DLBCL-related cell lines bearing mutated EZH2) (Figure 2).101 PROTACs that target other proteins involved in transcriptional regulation are listed in Table S1.
Clinical proof of concept for orthodox PROTACs
AR-PROTACs
Bavdegalutamide (ARV-110; Arvinas) is a first-in-class PROTAC that has been shown to degrade wild-type AR and several forms of mutant AR (T878A, T878S, H875Y, F877L, and M895V), leading to a prostate-specific antigen level decline ≥50% (PSA50) rate in 46% of patients. Patients receiving ARV-110 treatment have achieved tumor reductions, suggesting that this PROTAC may be useful as a form of precision medicine for metastatic castration-resistant prostate cancer (mCRPC). However, bavdegalutamide cannot penetrate the blood-brain barrier (BBB). Furthermore, it does not degrade ARL702H, a form of AR containing a point mutation that is present in 3%–10% of patients with mCRPC, or AR-V7, a splice variant that lacks the ligand binding domain of AR.102 AC0176 (Accutar Biotech) potently degrades both wild-type AR and the prevalent AR mutants (e.g., L702H, T878A, H875Y, W742, and C247) associated with drug resistance to current AR-targeted therapies. CC-94676 (AR-LDD; Bristol Myers Squibb) is another AR-PROTAC with preclinical activity similar to that of ARV-110 in terms of its favorable pharmacokinetic properties, ability to effectively degrade AR, and ability to induce continuous tumor regression in a VCaP mouse model (sources: public data from the companies) (Table 1).
Table 1.
Ongoing clinical trials of bifunctional degraders (PROTACs)
| Clinical trial NCT no. | Highest phase | Degrader | ROA | POI | E3 ligase | Indications | Sponsor |
|---|---|---|---|---|---|---|---|
| NCT03888612 | II | ARV-110 | oral | AR | CRBN | PC | Arvinas |
| NCT05067140 | I | ARV-766 | oral | AR | undisclosed | PC | Arvinas |
| NCT05241613 | I | AC0176 | oral | AR | undisclosed | PC | Accutar Biotech |
| NCT04428788 | I | CC-94676 | oral | AR | CRBN | PC | Bristol Myers Squibb |
| NCT05252364 | I | HP518 | oral | AR | undisclosed | PC | Hinova |
| NCT05428449 | I | GT20029 | topical | AR | undisclosed | acne vulgaris, androgenetic alopecia | Kintor |
| NCT04072952 | II | ARV-471 | oral | ER | CRBN | BC | Arvinas/Pfizer |
| NCT05080842 | I | AC0682 | oral | ER | CRBN | BC | Accutar Biotech |
| NCT05487170 | II | RNK05047 | i.v. | BRD4 | chaperone | DLBCL | Ranok |
| IND-e | CFT8634 | oral | BRD9 | CRBN | SS, SMARCB1-null solid tumors | C4 Therapeutics | |
| NCT04965753 | I | FHD-609 | i.v. | BRD9 | undisclosed | SS | Foghorn Therapeutics |
| NCT04772885 | I | KT-474 | oral | IRAK4 | undisclosed | multiple immunoinflammatory diseases: HS, AD, RA, others | Kymera/Sanofi |
| NCT05233033 | I | KT-413 | i.v. | IRAK4 | CRBN | MYD88-mutant DLBCL | Kymera |
| NCT04830137 | I | NX-2127 | oral | BTK | CRBN | B cell malignancies | Nurix Therapeutics |
| NCT05131022 | I | NX-5948 | oral | BTK | CRBN | B cell malignancies and autoimmune diseases | Nurix Therapeutics |
| NCT04861779 | I | HSK29116 | oral | BTK | undisclosed | B cell malignancies | HAISCO |
| NCT05006716 | I | BGB-16673 | oral | BTK | undisclosed | B cell malignancies | BeiGene |
| NCT05225584 | I | KT-333 | i.v. | STAT3 | undisclosed | liquid and solid tumors | Kymera |
| NCT04886622 | I | DT2216 | i.v. | BCL-xL | VHL | liquid and solid tumors | Dialectic Therapeutics |
| IND-e | CFT8919 | oral | EGFRL858R | CRBN | NSCLC | C4 Therapeutics | |
| CTR20222742 | II | CG001419 | oral | TRK | CRBN | cancer and other indications | Cullgen |
| IND-e | CFT1946 | oral | BRAFV600X | undisclosed | melanoma, CRC, NSCLC | C4 Therapeutics | |
| IND-e | KT-253 | undisclosed | MDM2 | undisclosed | liquid and solid tumors | Kymera |
AD, atopic dermatitis; AR, androgen receptor; BC, breast cancer; BCL-xL, B cell lymphoma-extra large; BRD9, bromodomain-containing protein 9; BTK, Bruton’s tyrosine kinase; CRBN, cereblon; CRC, colorectal cancer; DLBCL, diffuse large B cell lymphoma; EGFR, epidermal growth factor receptor; ER, estrogen receptor; HS, hidradenitis suppurativa; IND-e, in IND-enabling preclinical studies; IRAK4, interleukin-1 receptor-associated kinase 4; i.v., intravenous; MDM2, mouse double minute 2 homolog; NSCLC, non-small-cell lung cancer; PC, prostate cancer; PROTAC, proteolysis-targeting chimera; RA, rheumatoid arthritis; ROA, route of administration; SS, synovial sarcoma; STAT3, signal transducer and activator of transcription 3; TRK, tropomyosin receptor kinase; VHL, von Hippel-Lindau.
ER-PROTACs
ARV-471 (Arvinas) is believed to be the only trial of an ER-targeting therapy requiring prior CDK4/6 treatment for all patients. ARV-471 is well tolerated at doses ranging from 30 to 700 mg, but the dose-limiting toxicity (DLT) and maximal tolerated dose (MTD) have not yet been determined. Recently, ARV-471 was shown to yield a clinical benefit rate of 38% in evaluable patients and it has continued to show a favorable tolerability profile in its phase II expansion trial.103 AC0682 (Accutar Biotech) has been shown to potently and selectively degrade both wild-type and mutant forms of ERα. AC0682 has favorable pharmacological properties, can penetrate the BBB, and may offer a new form of breast cancer treatment with an MOA distinct from that of fulvestrant (sources: public data from the companies) (Table 1).
BRD9-PROTACs
CFT8634 (C4 Therapeutics) is a highly selective BRD9-PROTAC currently being investigated in a phase I clinical trial. It has been shown to specifically inhibit the growth of SMARCB1-perturbed (SMARCB1, SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1) cells and to be robustly effective in a clinically relevant patient-derived synovial sarcoma (SS) xenograft model.104 FHD-609 (Foghorn Therapeutics) is an intravenous BRD9-PROTAC under investigation to determine the primary (DLT and adverse events) and secondary outcome measures (pharmacokinetics [PK], objective response rate, duration of response, time to response, progression-free survival, and overall survival) (sources: public data from the companies) (Table 1).
IRAK4-PROTACs
KT-474 (Kymera/Sanofi) selectively degrades IRAK4. It has been shown to reduce IRAK4 expression levels in peripheral blood mononuclear cells (PBMCs) and block cytokine induction at considerably lower doses than those required for toll-like receptor (TLR) agonists. At day 14, KT-474 shows consistent degradation of 92% at the lowest dose level (25 mg) and 96%–98% at the two highest dose levels (100 and 200 mg). KT-474, delivered in multiple doses, has been shown to be well tolerated in blood and skin for at least 14–21 days. KT-413 (Kymera) is a CRBN-based IRAK4 degrader that is being investigated in a phase I clinical trial to determine its safety, PK/pharmacodynamics (PD), and preliminary efficacy in DLBCL and to explore its target knockdown and downstream effects in PBMCs and tumors (sources: public data from the companies) (Table 1).
Other PROTACs
KT-333 (Kymera) degrades STAT3 and is expected to receive investigational new drug (IND) clearance for evaluation of its metrics in liquid (e.g., peripheral T cell lymphoma, cutaneous T cell lymphoma, large granular lymphocytic leukemia) and solid tumors. KT-253 (Kymera) is an MDM2 degrader that stabilizes p53, as wild-type p53 exists in approximately 50% of tumor cells. KT-253 shows antitumor activity at picomolar concentrations, with a potency >200-fold greater than that of clinically active MDM2 SMIs. It achieves its effects by blocking the feedback loop, which upregulates MDM2 production and drives tumor cells to undergo rapid apoptosis. CFT1946 (C4 Therapeutics) is an on-mechanism, CRBN-based PROTAC that selectively degrades BRAFV600X; it was shown to potently inhibit MAPK signaling and promote tumor regression in a BRAFV600E A375 xenograft mouse model. CFT1946 also acts against BRAFV600E/NRASQ61K, a model of clinical resistance to BRAF inhibitors, and has exhibited potential efficacy as a TPD-based therapy in non-V600X-mutated BRAF-driven cancers. CFT7455 (C4 Therapeutics) is a potent small molecule that degrades IKZF1/3. It has been shown to exhibit enhanced catalytic activity, resulting in a >1,000-fold improvement in potency compared with pomalidomide, and has been shown to be efficacious in a model of systemic multiple myeloma. NX-2127 (Nurix Therapeutics) is a heterobifunctional, orally administered, CRBN-based BTK degrader that has been shown to catalyze the neo-substrate degradation of IKZF1/3 in DLBCL cell lines, including cells harboring the ibrutinib-resistance mutation BTKC481S, and in the cynomolgus monkey.105 NX-5948 (Nurix Therapeutics) degrades BTK and has been shown to penetrate the BBB in preclinical models to degrade BTK in both microglia and CNS-resident lymphoma cells and to exert antilymphoma activity in a primary model of CNS lymphoma. CFT8919 (C4 Therapeutics) specifically induces EGFRL858R degradation at low (nanomolar) concentrations without affecting wild-type EGFR, and its activity is equivalent to that of EGFR inhibitors. CF8919 has been shown to have antitumor activity in multiple tumor models, including NCI-H1975 EGFRL858R/T790M xenograft, BaF3 allograft, and H1975-LUC (EGFRL858R/T790M) brain metastasis models (sources: public data from the companies) (Table 1).
Major concerns of PROTAC
The “hook effect”
A PROTAC forms a cooperative ternary complex to degrade its target. At concentrations above a certain threshold, however, PROTACs tend to form ineffective binary complexes (e.g., PROTAC-POI or PROTAC-E3 ligase) (Figure 3). The competition between these binary and active ternary complexes (e.g., POI-PROTAC-E3 ligase) is described as the “hook effect” and it reduces the degradation efficiency of the PROTAC.90,106 The hook effect reduces the potency of the PROTAC, and it typically occurs at micromolar concentrations and is difficult to evade.83,99,107 Unfortunately, in vivo data on the hook effect are scarce, and the MTDs identified from preclinical profiles in animal studies indicate that the PROTAC concentration threshold may be too high to enable adequate degradation of POIs.108,109 To confirm the most appropriate doses, PROTACs should be tested over a wide concentration gradient in vitro and in vivo. Intriguingly, the study of ligand-bound structures can promote the design of next-generation PROTACs. By modifying some components, PROTACs can be used to form new ligand-induced PPIs, yielding PROTAC catalytic activity via a principle similar to enzyme-substrate convergence.110 AT1, a structure-designed compound, was developed by updating the BET degrader MZ1 and it possesses a more stable ternary complex (POI-PROTAC-E3 ligase) and a higher hook effect threshold.41
Figure 3.

The advantages and challenges of PROTACs
Low tissue/cell permeativity
PROTACs are iterative (Figure 3) and, unlike SMIs, degrade POIs in an event-driven, rather than occupancy-driven, manner. Most known orthodox PROTACs are beyond Lipinski’s rule of five, which helps distinguish the druggability of molecular determinants with undesirably high molecular weights (1,000–2,000 Da). PROTACs have high molecular weights, and the resulting low permeativity and solubility contribute to their lack of ideal cellular uptake and target degradation, as well as a high rate of active transporter-mediated efflux (Figure 3).111 The highly polar surface of a PROTAC limits its ability to cross physiological barriers and cell membranes. In clinical settings, CRBN is strongly preferred over other E3 ligases due to its lower molecular weight, fewer hydrogen bond donors, and smaller number of rotatable bonds, even though VHL-based PROTACs are more likely than other PROTACs to form functional ternary structures.112 Various properties of PROTAC-delivery agents (e.g., prodrugs, nanoparticles, and protein- or nucleic acid-based formulations), such as their ability to improve solubility, intracellular accumulation, and site-specific distribution and minimal side effects, have been summarized.113 The problem of cell permeativity has been addressed by attaching cell-permeative peptides (e.g., poly-D-arginine sequences) to E3 ligands.46 Furthermore, an in-cell click-formed proteolysis-targeting chimera (CLIPTAC) contains two individual precursors—one each is bound to POI and E3 ligase—that form a heterobifunctional molecule intracellularly via a bio-orthogonal “click” reaction (Figure 4). Upon entering cells, a tetrazine-tagged thalidomide derivative (Tz-thalidomide) and TCO-tagged inhibitor of POI can self-assemble to recruit CRBN to the POI, thus triggering its ubiquitination and degradation.114 Compared with a high-molecular-weight compound, it is easier for two compounds with lower molecular weights to enter a cell. In addition to the direct modification of PROTACs, nanoparticles can be used to improve the action of PROTACs. The encapsulation of PROTACs with different modified nanoparticles was shown to increase tumor permeability via the enhanced permeability and retention (EPR) effect controlled by delivering high concentrations of PROTACs. The improved cell permeability relies on the MOA of nanoparticles via endocytosis with reduced PROTAC metabolism.115
Figure 4.

Derivates from orthodox PROTACs
Inefficiency of PROTAC design
Traditionally, PROTAC design has required extensive experiments and trials, and the generation of linkers has played a crucial role in the physiochemical properties and degradation activities of PROTACs.55,116 As the development of POI warheads and E3 is fundamentally identical to that of other small molecules, de novo linker design methods, such as a graph-based deep generator (DeLinker) and a language model (SyntaLinker), are emphasized and exploited to fill the gap in linker generation.117,118 However, the open-source database of PROTACs is small (∼2,300 PROTACs),119 and thus, linkers designed using these de novo methods are not practical for developing druggable PROTACs. Hence, it is difficult to train a model capable of designing PROTACs with ideal properties and activity both in vitro and in vivo. PROTAC-RL, a novel deep generative model for rational PROTAC design, combines an augmented transformer architecture with memory-assisted reinforcement learning (RL). As a proof of concept, 5,000 PROTACs were generated to target BRD4 during a 49-day discovery period, and one exclusive candidate was validated to have antiproliferative activity in vivo.120 The biotech company Differentiated Therapeutics built Auto/dx, a unique platform that integrates proprietary protein interaction dynamics, AI, and synthetic biology methodologies for molecular simulation. The company announced the completion of a $5 million seed financing round. The combination of drug discovery and cutting-edge computational tools is likely to be essential for advancing the development of TPD strategies (Figure 3).
Expansion of the E3 ligase landscape
Peptide-based E3 ligands are used to recruit β-TrCP and VHL, but their use is restricted due to poor cell permeativity. Although more than 600 human-genome-encoded E3 ligases have been identified, the lack of specific high-affinity ligands has limited the scope of application of PROTAC technology.121 Further exploration of E3 ligases is needed to address the following challenges: (1) in tumor cells, drug resistance inactivates degraders; (2) resistance to components of immunomodulatory imide drugs (IMiDs), such as pomalidomide, lenalidomide, and thalidomide, cause genomic alterations that affect neo-substrates (IKZF1/IKZF3)122; and (3) there is a lack of effective E3 ligases known to be exclusive to tumor cells. Beyond the ligases initially used in PROTAC technology (CRBN, VHL, MDM2, and IAP), other E3 ligases may be useful in PROTAC development and other TPD strategies. These include damage-specific DNA binding protein 1 (DDB1)-CUL4-associated factor 16 (DCAF16), DCAF15, ring-finger protein 4 (RNF4), RNF114, Kelch-like ECH-associated protein 1 (KEAP1), fem-1 homolog B (FEM1B), and the aryl hydrocarbon receptor (AhR) (Table 2).
Table 2.
List of E3 ligases and related POIs
| E3 ligase | Complex | POI | Reference |
|---|---|---|---|
| CRBN | CUL4-RBX1-DDB1-CRBN | POIs involved in cancers, immune diseases, neology, and HCV | de Wispelaere et al.,62 Bassi et al.,100 Sun et al.,123 Silva et al.124 |
| VHL | CUL2-RBX1-ElonginB-ElonginC-VHL | KRAS, EGFR, BCR-ABL, etc. | Bond et al.,53 Zhao et al.,68 Khan et al.,77 Sun et al.,123 |
| MDM2 | BTK, PARP1 | Sun et al.,125 Zhao et al.126 | |
| IAP | RAR | Itoh et al.47 | |
| DCAF16 | CUL4-RBX1-DDB1-DCAF16 | BRD4, FKBP12 | Zhang et al.127 |
| DCAF15 | CUL4-RBX1-DDB1-DCAF15 | RBM39, BRD4 | Li et al.,128 Han et al.,129 Bussiere et al.130 |
| RNF4 | BRD4 | Ward et al.131 | |
| RNF114 | BRD4 | Spradlin et al.132 | |
| KEAP1 | KEAP1-CUL3 | BRD4 | Tong et al.133 |
| FEM1B | BRD4 | Henning et al.134 | |
| AhR | CUL4-RBX1-DDB1-AhR | CRABP1 | Ohoka et al.135 |
On-target off-tumor toxicity
On-target toxicity commonly results from interactions of the drug with its intended target. In the context of this review, a PROTAC degrades its POI in both tumor and normal cells, meaning that the therapeutic and toxic targets are the same (Figure 3). Accordingly, prolonged TPD eliminates entire proteins, including enzymatic and scaffolding subunits. If the POI is essential for normal cellular functions, TPD may cause intolerance and significant adverse effects.81,95,136 For example, the phase II trial of ARV-110 revealed that treatment-related adverse events (TRAEs) such as nausea, fatigue, vomiting, weight loss, and anemia are common in treated patients. Hence, the future development of PROTACs is challenged by the need to exclusively target tumor cells or the tumor microenvironment to avoid on-target toxicity.
PROTACs with additional agents that target specific cell membrane receptors (e.g., nucleolin [NCL], c-type lectin-like molecule-1 [CLL1], human EGFR 2 [HER2], and folate receptor α [FORL1]) have been introduced to improve off-tumor toxicity and further expand the practical applicability of PROTAC technology. For example, AS1411 is a 26-base, guanine-rich, single-stranded DNA aptamer that binds to NCL with high affinity and specificity. NCL is typically overexpressed on the plasma membrane of tumor cells relative to normal cells. As AS1411 can be internalized into cells by NCL-dependent micropinocytosis, it provides both suitable tumor targeting and antitumor activity. Aptamer-PROTAC conjugates (APCs) that use AS1411 as the aptamer have been designed to target tumors specifically. The BET degrader APR has been shown to degrade BRD2/3/4 and exhibit effective tumor-targeting and antitumor activity.137 Folate has a strong affinity for FOLR1, which is highly expressed in many tumor cells. Hence, the development of PROTACs conjugated with folate represents a strategy for delivering PROTACs into specific tumor cells for on-target degradation by targeting FOLR1. The degradation of POIs (e.g., BRD, MEK, ALK, and IKZF1/3) via folate-PROTACs or molecular glues has been validated.138,139 Given the outstanding successes of antibody-drug conjugates (ADCs) in the field of anticancer therapeutics,140 antibody-PROTAC conjugates have also been brought forward as a concept for achieving tissue-specific degradation. For example, PROTACs that target the cell membrane proteins CLL1 and HER2 have been used to degrade BRD4 in a specific target-positive manner.22,141,142,143
Off-target toxicity
Drugs that target non-POI proteins, such as receptors or enzymes, may have severe downstream effects.37,144 Theoretically, PROTACs are more selective than traditional SMIs because of the strictness and ingenuity of forming a viable ternary complex that enables polyubiquitination and degradation via the UPS. Compared with the occupancy-driven MOA of SMIs, PROTACs frequently induce new PPIs that alter the selectivity of the parent compounds.145 For instance, foretinib, a multikinase inhibitor that targets more than 130 kinases, has been used to generate PROTACs with different E3 ligands (VHL and CRBN). The formation of bifunctional degraders reduces the target binding selectivity of VHL- and CRBN-PROTACs to 52 and 62 kinases, respectively, and the stricter condition of degradation selectivity reduces the numbers of targets to only 9 and 14 kinases, respectively.146 Many similar reductions in the number of targeted kinases have been reported, suggesting that the selective degradation of POIs is not dependent solely on target ligand binding selectivity or affinity.147,148,149,150 In addition, the match between the E3 ligase and the POI is one of the most crucial factors in the formation of ternary complexes with preferred PROTACs and optimal E3 ligases. Degradation selectivity is largely associated with E3 ligase preference. Pairings of different E3 ligases with a given POI can alter the degradation selectivity and efficiency, as demonstrated by the CDK4/6 selectivity of a VHL-based palbociclib degrader151 and the weak HDAC6 selectivity of an IAP-based degrader.152 There is one exception regarding unexpected neo-substrates for IMiDs, such as IKZF1/3 for CRBN. It remains uncertain whether E3 ligases can recruit nonspecific substrates in an IMiD-like manner.153 NX-2127, a CRBN-based BTK degrader from Nurix Therapeutics, has been shown to catalyze the neo-substrate degradation of IKZF1/3 in DLBCL cell lines (Figure 3).
PROTAC derivates
BacPROTACs
BacPROTACs, which are small-molecule degraders, hijack bacterial ClpC:ClpP (ClpCP) proteases to extend inducible protein-targeted degradation techniques that interfere with microbial infection. Phosphorylated arginine residue (pArg)-containing BacPROTACs consist of a POI ligand, chemical linker, and pArg bound to the ClpCNTD domain to induce its reassembly and activation. BacPROTAC targets ClpCNTD and activates ClpC, transforming the resting unfoldase into its functional state. The successful use of a BacPROTAC for degradation in mycobacteria has provided new ideas for the development of antimicrobial compounds (Figure 4A).154
Antibody-based PROTACs (AbTACs)
AbTACs are recombinant bispecific antibodies used to tether cell-surface E3 ligases to transmembrane proteins to induce target degradation in vitro and in vivo. AC-1, a fully recombinant bispecific IgG, was developed to target programmed death-ligand 1 (PD-L1), a membrane protein not targeted by orthodox PROTACs. AC-1 recruits ring-finger protein 43 (RNF43), an E3 ligase on the cell surface, to degrade PD-L1 via internalization and lysosome-mediated degradation (Figure 4B).22 The abilities of multiple Wnt-responsive ligases (e.g., RNF43 and zinc- and ring-finger 3) to induce cancer-specific degradation in an “on-demand” manner have been validated,155 and these findings have partially addressed the issues of poor permeativity and difficulties in targeting membrane-associated proteins.
Ribonuclease-targeting chimeras (RIBOTACs)
Since various TPD strategies have been shown to successfully degrade target proteins, decaying RNA by related nucleases is considered a prospective mimicry. More than 80% of the human genome is transcribed into RNA, of which less than 2% comprises mRNAs that are translated into proteins. Non-coding RNAs can regulate gene expression and, thus, are potential drug targets. RIBOTACs consist of an RNA binder, a small molecule (2′-5′-linked tetra-adenylate) that recruits and activates a local latent ribonuclease (RNase L), and a linker that binds these two components. RIBOTACs thus recruit monomeric RNase L and dimerize it to form an active structure (Figure 4C). The first RIBOTAC molecule was shown to selectively cleave the precursor of microRNA 96 (miR-96), and the subsequent silencing of miR-96 derepressed a proapoptotic TF, leading to the selective apoptosis of breast cancer cells.156 The compound TGP-210-RL has been shown to selectively degrade pre-miR-210 and thus block the formation of miR-210, which is essential for cancer cell survival in a hypoxic microenvironment.157 DNA-encoded libraries have been screened to identify new binders that can recruit RNase L. The RTK inhibitor dovitinib and a binder have been incorporated into the design of a next-generation RIBOTAC intended to target the miRNA-21 precursor.158 The potential use of RIBOTACs for selective RNA degradation has broadened the prospect of targeting vulnerabilities in RNA-based diseases.
Phosphatase-recruitment chimeras (PHORCs)
Disease onset is frequently caused by the dysregulation of signal transduction, especially the hyperphosphorylation, of oncoproteins. PHORCs have been designed to reduce the phosphorylation of target proteins via proximity to a relevant phosphatase. DDO3711, the first PHORC, comprises an activator of protein phosphatase 5 (PP5; a serine/threonine phosphatase), an SMI of apoptosis signal-regulated kinase 1 (ASK1), and a chemical linker. Overexpression of ASK1 has been linked to the progression of multiple cancers. PP5 is characterized by its explicit reversal of ASK1 Thr838 autophosphorylation (p-ASK1T838); however, PP5 is autoinhibited in some cancer types, leading to an excessive level of p-ASK1T838. Activation of PP5 and colocalization of PP5 with its substrate ASK1 are expected to accelerate the specific dephosphorylation of p-ASK1T838 (Figure 4D). DDO3711 has shown desirable antiproliferative activity (IC50 0.5 μM) against cancer cells, whereas the individual components (ASK1, SMI, and PP5 activator) have shown no effects in cancer cells.159
Oligonucleotide-based PROTACs
Some oligonucleotide-based PROTACs, including RNA-PROTACs, programmable oligonucleotide PROTACs (O’PROTACs), and TF-targeting chimeras (TRAFTACs), have been developed according to the characteristic abilities of RNA-binding proteins (RBPs) and TFs to bind RNA and dsDNA consensus sequences, respectively (Figure 4E). Such constructs have been demonstrated to degrade Lin-28 homolog A (LIN28A) and the TFs, including ETS TF (ERG), lymphoid enhancer-binding factor 1 (LEF1), NF-κB, and brachyury.24,160,161,162,163,164 The PROTAC ZL216 comprises AS1411 as the warhead and a VHL ligand to enable the selective internalization and degradation of NCL, and it has been shown to exert antiproliferative activity against breast cancer cells in vitro and in vivo.165
Phospho-dependent PROTACs (phosphoPROTACs)
Phosphorylation, a post-translational modification, is a crucial step in the activation of proteins, especially kinases, and their downstream signaling pathways. PhosphoPROTACs represent an update of orthodox PROTACs, wherein PROTAC-mediated protein degradation is coupled with the activation state of a particular signaling pathway.30 RTKs are activated by autophosphorylation, and their downstream signaling cascades are amplified by recruiting and phosphorylating molecules with phosphotyrosine-binding (PTB) and SH2 domains. A phosphoPROTAC contains a warhead with a common RTK phosphorylation sequence that can be phosphorylated by activated RTKs, and an E3 ligand is a residue of HIF-1α binding to VHL. Poly-D-Arg is linked to the E3 ligand to improve cell permeativity. Under specific situations involving RTK activation, the controllable phosphoPROTAC can be phosphorylated, which is followed by the recruitment of target proteins with PTB and SH2 domains. These target proteins are ubiquitinated and consequently degraded via the VHL-mediated ubiquitin-proteasome pathway. The phosphoPROTACs TrkAPPFRS2α and ErbB2PPPI3K (FRS2α and PI3K are the downstream proteins of TrkA and ErbB2, respectively) have been shown to inactivate the TrkA- and ErbB2/ErbB3-regulated signaling pathways, respectively, in breast cancer cell lines (Figure 4E).166
Light-controllable PROTACs
Light-controllable PROTACs can be divided into two main groups, namely photoswitchable PROTACs and photocaged PROTACs, which are characterized by reversible or irreversible light control, respectively, at a high spatiotemporal resolution. Photoswitchable PROTACs induce reversible TPD via an optically controlled moiety on the linker or E3 ligand. Exposure to light at a specific wavelength induces an “inactive-active” transition in the PROTAC that enables the formation of a stable ternary complex (Figure 4E).167,168,169 The incorporation of azobenzene photoswitches into PROTACs has led to a series of BET and FKBP12 degraders whose precise ability to regulate activation and inactivation is regulated under two conditions: blue-violet light (380–440 nM) and darkness.169
Photocaged PROTACs are characterized by the binding of a photolabile blocking group to the E3 ligand, which blocks the interaction between the PROTAC and the E3 ligase. Upon light stimulation, the PROTAC is released from the photocaging group, enabling the active conformation of the ternary complex (Figure 4E).170,171,172,173 The degraders dBET1 and dALK, along with a photolabile caging group (nitroveratryloxy-carbonyl group), have been conjugated to the E3 ligand pomalidomide, and photolysis has been shown to be induced by ultraviolet A irradiation. The substrates of pomalidomide, dBET, and dALK (including IKZF1/3, BRD2/3/4, and ALK) were shown to be degraded in a light-controllable manner.173
Dual/trivalent PROTACs
Inspired by dual-targeting drugs,174 especially bispecific antibodies, Li and colleagues first presented the concept of a dual PROTAC that could degrade two distinct POIs (EGFR and PARP).175 The EGFR inhibitor gefitinib, the PARP inhibitor olaparib, and a CRBN/VHL ligand were linked with a star-type core linker of trifunctional amino acids. The dual PROTACs DP-C-1 and DP-V-4 have demonstrated potent ability to degrade both EGFR and PARP in a dose- and time-dependent manner. However, DP-V-4 was shown to possess a weaker antiproliferative activity (IC50 19.92 ± 1.08 μM) than its parent inhibitors in H1299 cells, potentially because its high molecular weight led to low solubility and poor cell permeativity.175
To address incomplete target degradation and the hook effect due to unproductive high concentrations of ternary complexes, a trivalent PROTAC SIM1 was developed to degrade the BET domain family members BRD2, BRD3, and BRD4. A trivalent PROTAC comprises bivalent POI ligands and a moiety for recruiting E3 ligase, tethered by a branched linker. As the most potent trivalent PROTAC, SIM1 can degrade target proteins over a wide range of concentrations (10 pM–30 μM), thus eliminating the hook effect.176 This avidity- and cooperativity-enhanced ternary complex outperforms the BET degraders MZ1 and MT1 by forming a 1:1:1 complex with VHL and the tandem bromodomain BD1–BD2 with conformational change. Moreover, SIM1 has been shown to degrade its targets rapidly and at a lower concentration than that reported for the BET degraders ARV-771 and MZ1, and it has been shown to have a higher residence time rate and longer half-life in vivo.176 In principle, trivalent PROTACs use the same MOA as the above-mentioned dual PROTACs (Figure 4E).
Chaperone-mediated protein degradation (CHAMP)
CHAMP, another important PROTAC derivative, directly hijacks the chaperone molecule HSP90, instead of E3 ligase, to achieve target degradation. The compound BRD4-CHAMP, which comprises a BRD4:CHAMP:HSP90 ternary complex, has been shown to selectively degrade BRD4 and exert antitumor activity in vitro and in vivo.177 First, the CHAMP compound RNK05047 (CHAMP-1) is being explored in a clinical trial as a potential DLBCL treatment (Figure 4E).
Homo-PROTACs
Homo-PROTACs have been designed and validated to mediate the self-destruction of E3 ligases, such as VHL, MDM2, and CRBN (Figure 4E).178,179,180 For example, CM11, a homo-PROTAC, has been shown to induce the selective depletion of a VHL isoform (pVHL30) at a nanomolar concentration. However, this exclusive and highly isoform-selective degradation of pVHL30 was unexpected because CM11 does not differentiate between pVHL19 and pVHL30 in ternary complexes.180
BioPROTACs
A bioPROTAC was exploited at an early stage after the first PROTAC debut. This hybrid molecule comprises a small-molecule warhead and a phosphopeptide. BioPROTAC activity against some clinically relevant cancer targets, such as HER2, MYC, and KRAS, has been validated in human cancer cell lines and mouse xenograft tumor models.181,182,183,184 However, bioPROTACs cannot be administered orally due to the high molecular weights of their peptide components, and a suitable drug-delivery system needs to be chosen.
Other TPD therapies
Macroautophagy degradation-targeting chimeras (MADTACs) AUTACs/ATTECs/AUTOTACs
Macroautophagy is another degradation pathway in cells, wherein lysosomes engulf and degrade cytoplasmic substrates.185 Many studies of TPD technologies that engage lysosomal pathways through different mechanisms have been summarized.186 Because many intracellular proteins are not targeted by the UPS for degradation, AUTACs have been designed to degrade proteins and dysfunctional mitochondria via autophagy.187 AUTACs consist of three parts: a warhead that specifically targets the proteins to be degraded, a tag for selective autophagic degradation, and a linker connecting the other two parts. In addition to their target proteins, AUTACs can degrade disease-related debris, such as fragmented mitochondria. Compared with earlier PROTACs, AUTACs can potentially degrade a broader range of disease-related organelles and intracellular pathogens.188 Once macroautophagy is initiated, the area of cytoplasm containing the cargo to be degraded is elongated and isolated.189 The cytoplasmic membrane subsequently closes around the contents and matures to form an autophagosome, which then fuses with a lysosome to degrade the cargo.190 S-Guanylation (cysteine conjugation of 8-nitro-cGMP) is a post-translational modification important for K63 ubiquitination that is used to clear intracellular bacterial pathogens.191 S-guanylate may be an ideal tag for substrate targeting. The pairing of a degradation tag (e.g., guanine derivatives such as FBnG) and a ligand for the target can be characterized by the induction of K63-polyubiquitination to mediate substrate S-guanylation for selective autophagy. AUTAC platforms have been proven effective against MetAP2, FKBP12, BRD4, and mitochondria.
ATTECs have been shown to provide a more direct approach, interacting with both the mutant huntingtin protein (mHTT) with an expanded polyglutamine (polyQ) tract, which causes Huntington disease, and the autophagosome protein microtube-associated protein 1A/1B light chain 3 (LC3), but not with wild-type HTT.192 Using small-molecule-microarray-based screening technology, compounds that can conjugate LC3 and mHTT have been exploited to degrade mHTT rather than wild-type HTT, with high specificity. As these compounds recognize polyQ tracts in their targets, they might be useful for degrading other disease-related polyQ-containing proteins.193 In addition to polyQ as the LC3 ligand, GW5074 and ispinesib have been used to design ATTECs that have been verified to effectively degrade the oncoproteins BRD4 and nicotinamide phosphoribosyltransferase (NAMPT).194,195 Furthermore, non-proteinaceous targets, such as lipid droplets (LDs), cannot be degraded using orthodox PROTAC technology but are potential targets for ATTECs because they are known to be degraded via autophagy. ATTEC degraders are able to clear LDs almost completely and rescue LD-related phenotypes in vitro and in vivo, thus expanding the scope of TPD platform applications.196
AUTOTAC is generated by connecting a POI ligand to a p62-binding autophagy-targeting ligand via an intermediate linker. AUTOTACs recruit POIs in tandem with the binding ZZ domain of the otherwise dormant autophagy receptor p62/Sequestosome-1/SQSTM1.197 This interaction stimulates self-polymerization in complex with the cargo and the macroautophagy induction cascade in a p62-dependent manner. AUTOTACs have been designed and verified to mediate the degradation of various oncoproteins (e.g., ERβ, AR, and MetAP2) and degradation-resistant misfolded protein aggregates associated with neurodegeneration in vitro and in vivo.197
Lysosome-targeting chimeras
Although the first AbTAC was able to target the cell-surface protein PD-L1 with a ligandable intracellular domain,22 most extracellular and membrane-associated proteins still cannot be targeted by PROTACs. Approximately 40% of the proteins in the human proteome are secreted or membrane proteins, and this proportion is even higher in the pancreas, salivary gland, and liver.198 Therefore, LYTACs have been designed to target extracellular and membrane-associated proteins, using conjugates that bind to both a cell-surface lysosome-targeting receptor (LTR) and the extracellular domain of the target protein.21 A LYTAC is a heterobifunctional molecule comprising a warhead that binds to the protein targeted for degradation and a tail containing chemically synthesized glycopeptide ligands that can hijack the cation-independent mannose-6-phosphate receptor (CI-M6PR), a prototypical LTR. When the POI is linked to CI-M6PR by M6Pn-LYTAC molecules, TPD is triggered, and the POI is transported to the lysosome and degraded. The ability of M6Pn-LYTACs to effectively degrade clusters of targets, such as the extracellular protein apolipoprotein E4 (APOE4) and membrane proteins (e.g., EGFR, PD-L1, and CD71), has been validated, thus expanding the scope of the TPD platform.21
Drawing on experiences from the first generation of systemically non-specific LYTACs, another tissue-specific LTR has been chosen to develop a localized-targeting LYTAC. N-acetylgalactosamine (tri-GalNAc), a ligand of the liver-specific LTR asialoglycoprotein receptor (ASGPR) that can be internalized via clathrin-mediated endocytosis, was selected as the LYTAC warhead.199 Antibody-based GalNAc-LYTACs have been shown to successfully degrade membrane-associated proteins (EGFR, HER2, and integrins) in the hepatocellular carcinoma cell line HepG2.200,201 In addition, molecular degraders of extracellular proteins through ASGPR (MoDE-As) have been generated as the first non-proteinogenic synthetic tri-GalNAc conjugates, and their ability to effectively degrade extracellular proteins in lysosomes has been validated both in vitro and in vivo.202
Taken together, these studies have shown the potential use and feasibility of the LYTAC platform for lysosomal degradation of extracellular and membrane-associated proteins; accordingly, LYTAC is a powerful solution to the drawbacks of PROTACs.
Hydrophobic tagging
In hydrophobic tagging (HyT) technology, bifunctional small molecules bind to a bacterial dehalogenase (the HaloTag protein) and present a hydrophobic group on the surface of the POI. Two possible MOAs of HyT-mediated degradation have been proposed as follows. First, the hydrophobic tag destabilizes the POI irreversibly, enabling the recruitment of endogenous chaperones and the shuttling of the POI to the proteasome for degradation. Second, chaperones recognize the hydrophobic tag directly and mediate proteasomal degradation of the tagged protein.203,204 Hydrophobic tags can be divided into three types: typical adamantane,23,205 tert-butyl carbamate-protected arginine,206,207 and carborane.208 HyT molecules, which target POIs such as HER3, Tau, and PDEδ, have validated degradation efficacy and antiproliferation activity in human cancer cell lines and mouse xenograft tumor models.61,205,209,210 Selective ER degraders (SERDs; e.g., fulvestrant, RU 58668) for the treatment of estrogen-responsive cancers and the selective AR degrader (SARD) (e.g., SARD279) used to treat prostate cancer have similar functions: a hydrophobic chain is exposed on the surface of a POI to mimic a misfolded protein state and, thus, induce degradation.211,212
Molecular glues
Molecular glues are commonly used to induce proteolysis of a target and have an MOA similar to that of PROTACs. Unlike PROTACs, however, a molecular glue contacts and is optimized for both the target protein and the E3 ligase via insertion into a naturally occurring PPI interface. Molecular glues have the obvious advantages of a low molecular weight and good druggability. Due to a limited understanding of the controlling factors, however, molecular glue design remains challenging, and few successful cases have been reported.
Most molecular glues induce protein degradation via E3 ligases, including iMIDs (thalidomide, lenalidomide, and pomalidomide) via CRBN binding,153,213,214,215,216,217 aryl sulfonamide via engagement with DCAF15,218 and other small molecules that adhere to the adaptor proteins DDB1,219,220,221 SIAH1,222 UBR7,223 or SCFβ-TrCP.223 Molecular glues for TPD, such as the neo-substrates (IKZF1/3 and CK1α) of iMIDs and POIs (RBM39,129,224,225 BCL6,222 and CDK220) for some SMIs, were discovered serendipitously. Other molecular glues have been shown to induce TPD via an autophagy-mediated degradation pathway, wherein LC3 is hijacked to degrade mHTT via the compounds AN1, AN2, 10O5, and 8F20 derived from microarray-based high-throughput screening. The mechanisms of these autophagic degraders remain to be clarified.193
Clinical trials of the IKZF1/3 degraders iberdomide (CC-220),226,227 CC-92480, CC-99282, and CFT7455 for hematologic malignancies, including multiple myeloma, acute myeloid leukemia, chronic myeloid leukemia, and non-Hodgkin’s lymphoma, are beginning. CC-90009 is a CRBN-based GSPT1 degrader with antileukemic activity and is currently in a dose-escalation phase and combination trial. DKY709, a degrader of IKZF2, is being used as a monotherapy and combination therapy with PDR001, a ligand-blocking IgG4 monoclonal antibody specific for PD-1. GBD-9 combines PROTAC and molecular glue strategies to yield a dual-mechanism inhibitor that effectively degrades BTK and GSPT1 and inhibits cancer cell growth by recruiting the E3 ligase CRBN (Table 3).228
Table 3.
Ongoing clinical trials of bifunctional degraders (molecular glues)
| Clinical trial NCT no. | Highest phase | Degrader | POI | E3 ligase | Indications | Sponsor |
|---|---|---|---|---|---|---|
| NCT02773030 | II | CC-220 | IKZF1/3 | CRBN | MM | Bristol Myers Squibb |
| NCT03989414 | II | CC-92480 | IKZF1/3 | CRBN | MM | Bristol Myers Squibb |
| NCT02848001/NCT04336982 | II | CC-90009 | GSPT1 | CRBN | AML | Bristol Myers Squibb |
| NCT01421524 | I | CC-122 | IKZF1/3 | CRBN | MM | Bristol Myers Squibb |
| NCT05144334 | I | BTX-1188 | IKZF1/3 | CRBN | AML and solid tumor | Biotheryx |
| NCT04434196/NCT03930953 | I | CC-99282 | IKZF1/3 | CRBN | CML and NHL | Bristol Myers Squibb |
| NCT04756726 | I | CFT7455 | IKZF1/3 | CRBN | MM and lymphoma | C4 Therapeutics |
| NCT04283097 | I | KPG-818 | IKZF1/3 | CRBN | Hematological malignancies and SLE | Kangpu |
| NCT03569280 | I | KPG-121 | IKZF1/3 | CRBN | CRPC | Kangpu |
| IND-e | ICP-490 | IKZF1/3 | CRBN | MM and NHL | InnoCare | |
| NCT03891953 | I | DKY709 | IKZF2 | CRBN | NSCLC | Novartis |
AML, acute myeloid leukemia; CML, chronic myelogenous leukemia; MM, multiple myeloma; NHL, non-Hodgkin’s lymphoma; NSCLC, non-small-cell lung cancer; SLE, systemic lupus erythematosus.
Conclusions
Since the debut of PROTAC technology in 2001, TPD has expanded through two decades of development. The clinical proof of concept of PROTACs has been completed, and a series of TPD spin-offs have emerged consecutively, allowing the potential applications of this technology to be continuously tapped for biomedicine. The entry of the first PROTAC drug into the clinic has validated the potential of this technology to become the best in class. The characteristics of clinical practicability and an orally bioactive medication have promoted the transition of PROTACs from academia to industry. However, the on-target/off-tumor toxicity, potentially off-target effects, and other challenges associated with PROTACs will continue to hinder future developmental efforts. The development and application of an expanded toolbox of E3 ligases for PROTACs should be another focal point. Some tissue-specific E3 ligases are expected to serve as passive targeting carriers.110,229 In this review, we comprehensively list various target proteins of PROTACs, including kinases and transcription regulators, and provide verification of the effectiveness of these treatments for cancers, autoimmune diseases, and other pathologies. In the clinic, PROTAC drugs mainly target the AR, ER BRD9, and BTK proteins, and progress in the treatment of mCRPC, ER+/HER− breast cancer, SS, and liquid tumors has been gratifying. PROTAC derivatives have expanded the spectrum of POIs of various forms that can be inactivated. Other TPD strategies, such as AUTACs, ATTECs, and LYTACs, have expanded the platforms for degrading target proteins to the autophagy and lysosomal degradation pathways, which can target proteins in different locations, including cytosolic, intramembrane, and extracellular proteins.
Acknowledgments
This review is supported by the National Natural Science Foundation of China (82172386 and 81922081 to C.L.), the Department of Education of Guangdong Province (2021KTSCX104 to C.L.), the 2020 Guangdong Provincial Science and Technology Innovation Strategy Special Fund (Guangdong-Hong Kong-Macau Joint Lab) (2020B1212030006 to A.L.), the Guangdong Basic and Applied Basic Research Foundation (2022A1515012164 to C.L.), and the Science, Technology and Innovation Commission of Shenzhen (JCYJ20210324104201005 to C.L.).
Author contributions
C.L. and A.L. supervised and revised the manuscript. J.L. and X.C. wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Declaration of interests
The authors declare no competing interests.
Published Online: March 15, 2023
Footnotes
It can be found online at https://doi.org/10.1016/j.xinn.2023.100413.
Contributor Information
Aiping Lu, Email: aipinglu@hkbu.edu.hk.
Chao Liang, Email: liangc@sustech.edu.cn.
Lead contact website
The lead contact website is at https://faculty.sustech.edu.cn/liangc/.
Supplemental information
References
- 1.Bastola P., Oien D.B., Cooley M., Chien J. Emerging cancer therapeutic targets in protein homeostasis. AAPS J. 2018;20:94. doi: 10.1208/s12248-018-0254-1. [DOI] [PubMed] [Google Scholar]
- 2.Miklos G.L.G., Maleszka R. Protein functions and biological contexts. Proteomics. 2001;1:169–178. doi: 10.1002/1615-9861(200102)1:2<169::AID-PROT169>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 3.Santos R., Ursu O., Gaulton A., et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017;16:19–34. doi: 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Powers E.T., Morimoto R.I., Dillin A., et al. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 5.Chau C.H., Steeg P.S., Figg W.D. Antibody–drug conjugates for cancer. Lancet. 2019;394:793–804. doi: 10.1016/S0140-6736(19)31774-X. [DOI] [PubMed] [Google Scholar]
- 6.Wolska-Washer A., Robak T. Safety and tolerability of antibody-drug conjugates in cancer. Drug Saf. 2019;42:295–314. doi: 10.1007/s40264-018-0775-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coats S., Williams M., Kebble B., et al. Antibody–drug conjugates: future directions in clinical and translational strategies to improve the therapeutic index. Clin. Cancer Res. 2019;25:5441–5448. doi: 10.1158/1078-0432.CCR-19-0272. [DOI] [PubMed] [Google Scholar]
- 8.Roskoski R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019;144:19–50. doi: 10.1016/j.phrs.2019.03.006. [DOI] [PubMed] [Google Scholar]
- 9.Roskoski R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: a 2020 update. Pharmacol. Res. 2020;152:104609. doi: 10.1016/j.phrs.2019.104609. [DOI] [PubMed] [Google Scholar]
- 10.Ohashi K., Maruvka Y.E., Michor F., Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J. Clin. Oncol. 2013;31:1070–1080. doi: 10.1200/JCO.2012.43.3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferguson F.M., Gray N.S. Kinase inhibitors: the road ahead. Nat. Rev. Drug Discov. 2018;17:353–377. doi: 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- 12.Fischer P.M. Approved and experimental small-molecule oncology kinase inhibitor drugs: a mid-2016 overview. Med. Res. Rev. 2017;37:314–367. doi: 10.1002/med.21409. [DOI] [PubMed] [Google Scholar]
- 13.Harmsen S., Kok R.J. Kinase inhibitor conjugates. Curr. Pharm. Des. 2012;18:2891–2900. doi: 10.2174/138161212800672778. [DOI] [PubMed] [Google Scholar]
- 14.Finan C., Gaulton A., Kruger F.A., et al. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 2017;9:eaag1166. doi: 10.1126/scitranslmed.aag1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fellmann C., Gowen B.G., Lin P.-C., et al. Cornerstones of CRISPR–Cas in drug discovery and therapy. Nat. Rev. Drug Discov. 2017;16:89–100. doi: 10.1038/nrd.2016.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh R., Tabrizi S.J. Gene suppression approaches to neurodegeneration. Alzheimer's Res. Ther. 2017;9:82. doi: 10.1186/s13195-017-0307-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wittrup A., Lieberman J. Knocking down disease: a progress report on siRNA therapeutics. Nat. Rev. Genet. 2015;16:543–552. doi: 10.1038/nrg3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burslem G.M., Crews C.M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell. 2020;181:102–114. doi: 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y., Jiang X., Feng F., et al. Degradation of proteins by PROTACs and other strategies. Acta Pharm. Sin. B. 2020;10:207–238. doi: 10.1016/j.apsb.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi D., Arimoto H. Targeting selective autophagy by AUTAC degraders. Autophagy. 2020;16:765–766. doi: 10.1080/15548627.2020.1718362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banik S.M., Pedram K., Wisnovsky S., et al. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature. 2020;584:291–297. doi: 10.1038/s41586-020-2545-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cotton A.D., Nguyen D.P., Gramespacher J.A., et al. Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J. Am. Chem. Soc. 2021;143:593–598. doi: 10.1021/jacs.0c10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neklesa T.K., Tae H.S., Schneekloth A.R., et al. Small-molecule hydrophobic tagging–induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 2011;7:538–543. doi: 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samarasinghe K.T.G., Jaime-Figueroa S., Burgess M., et al. Targeted degradation of transcription factors by TRAFTACs: TRAnscription Factor TArgeting Chimeras. Cell Chem. Biol. 2021;28:648–661.e5. doi: 10.1016/j.chembiol.2021.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y.-J., Wu H., Shen X.-Z. The ubiquitin–proteasome system and its potential application in hepatocellular carcinoma therapy. Cancer Lett. 2016;379:245–252. doi: 10.1016/j.canlet.2015.06.023. [DOI] [PubMed] [Google Scholar]
- 26.Nandi D., Tahiliani P., Kumar A., Chandu D. The ubiquitin-proteasome system. J. Biosci. 2006;31:137–155. doi: 10.1007/BF02705243. [DOI] [PubMed] [Google Scholar]
- 27.Dikic I. Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 2017;86:193–224. doi: 10.1146/annurev-biochem-061516-044908. [DOI] [PubMed] [Google Scholar]
- 28.Hershko A., Ciechanover A. The ubiquitin system. Annu. Rev. Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 29.Pohl C., Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. 2019;366:818–822. doi: 10.1126/science.aax3769. [DOI] [PubMed] [Google Scholar]
- 30.Gu S., Cui D., Chen X., et al. PROTACs: an emerging targeting technique for protein degradation in drug discovery. Bioessays. 2018;40:1700247. doi: 10.1002/bies.201700247. [DOI] [PubMed] [Google Scholar]
- 31.Livneh I., Cohen-Kaplan V., Cohen-Rosenzweig C., et al. The life cycle of the 26S proteasome: from birth, through regulation and function, and onto its death. Cell Res. 2016;26:869–885. doi: 10.1038/cr.2016.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong J., Cheng X.-D., Zhang W.-D., Qin J.-J. Recent update on development of small-molecule STAT3 inhibitors for cancer therapy: from phosphorylation inhibition to protein degradation. J. Med. Chem. 2021;64:8884–8915. doi: 10.1021/acs.jmedchem.1c00629. [DOI] [PubMed] [Google Scholar]
- 33.Khan S., He Y., Zhang X., et al. Proteolysis targeting chimeras (PROTACs) as emerging anticancer therapeutics. Oncogene. 2020;39:4909–4924. doi: 10.1038/s41388-020-1336-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X., Song Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020;13:50. doi: 10.1186/s13045-020-00885-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kregel S., Wang C., Han X., et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia. 2020;22:111–119. doi: 10.1016/j.neo.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun Y., Ding N., Song Y., et al. Degradation of Bruton’s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia. 2019;33:2105–2110. doi: 10.1038/s41375-019-0440-x. [DOI] [PubMed] [Google Scholar]
- 37.Jiang B., Wang E.S., Donovan K.A., et al. Development of dual and selective degraders of cyclin-dependent kinases 4 and 6. Angew. Chem. Int. Ed. Engl. 2019;58:6321–6326. doi: 10.1002/anie.201901336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Z., Pinch B.J., Olson C.M., et al. Development and characterization of a Wee1 kinase degrader. Cell Chem. Biol. 2020;27:57–65.e9. doi: 10.1016/j.chembiol.2019.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao H., Wu Y., Sun Y., et al. Design, synthesis, and evaluation of highly potent FAK-targeting PROTACs. ACS Med. Chem. Lett. 2020;11:1855–1862. doi: 10.1021/acsmedchemlett.9b00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Popow J., Arnhof H., Bader G., et al. Highly selective PTK2 proteolysis targeting chimeras to probe focal adhesion kinase scaffolding functions. J. Med. Chem. 2019;62:2508–2520. doi: 10.1021/acs.jmedchem.8b01826. [DOI] [PubMed] [Google Scholar]
- 41.Gadd M.S., Testa A., Lucas X., et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017;13:514–521. doi: 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li K., Crews C.M. PROTACs: past, present and future. Chem. Soc. Rev. 2022;51:5214–5236. doi: 10.1039/d2cs00193d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nowak R.P., DeAngelo S.L., Buckley D., et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol. 2018;14:706–714. doi: 10.1038/s41589-018-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farnaby W., Koegl M., Roy M.J., et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 2019;15:672–680. doi: 10.1038/s41589-019-0294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakamoto K.M., Kim K.B., Kumagai A., et al. Protacs: chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schneekloth J.S., Jr., Fonseca F.N., Koldobskiy M., et al. Chemical genetic control of protein levels: selective in vivo targeted degradation. J. Am. Chem. Soc. 2004;126:3748–3754. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- 47.Itoh Y., Ishikawa M., Naito M., Hashimoto Y. Protein knockdown using methyl bestatin-ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J. Am. Chem. Soc. 2010;132:5820–5826. doi: 10.1021/ja100691p. [DOI] [PubMed] [Google Scholar]
- 48.Schneekloth A.R., Pucheault M., Tae H.S., Crews C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008;18:5904–5908. doi: 10.1016/j.bmcl.2008.07.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Testa A., Lucas X., Castro G.V., et al. 3-Fluoro-4-hydroxyprolines: synthesis, conformational analysis, and stereoselective recognition by the VHL E3 ubiquitin ligase for targeted protein degradation. J. Am. Chem. Soc. 2018;140:9299–9313. doi: 10.1021/jacs.8b05807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winter Georg E., Buckley Dennis L., Paulk J., et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao H., Zheng C., Du J., et al. FAK-targeting PROTAC as a chemical tool for the investigation of non-enzymatic FAK function in mice. Protein Cell. 2020;11:534–539. doi: 10.1007/s13238-020-00732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen Y., Gao G., Yu X., et al. Discovery of first-in-class protein arginine methyltransferase 5 (PRMT5) degraders. J. Med. Chem. 2020;63:9977–9989. doi: 10.1021/acs.jmedchem.0c01111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bond M.J., Chu L., Nalawansha D.A., et al. Targeted degradation of oncogenic KRASG12C by VHL-recruiting PROTACs. ACS Cent. Sci. 2020;6:1367–1375. doi: 10.1021/acscentsci.0c00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zeng M., Xiong Y., Safaee N., et al. Exploring targeted degradation strategy for oncogenic KRAS G12C. Cell Chem. Biol. 2020;27:19–31.e6. doi: 10.1016/j.chembiol.2019.12.006. [DOI] [PubMed] [Google Scholar]
- 55.Han X., Wang C., Qin C., et al. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J. Med. Chem. 2019;62:941–964. doi: 10.1021/acs.jmedchem.8b01631. [DOI] [PubMed] [Google Scholar]
- 56.Lee G.T., Nagaya N., Desantis J., et al. Effects of MTX-23, a novel PROTAC of androgen receptor splice variant-7 and androgen receptor, on CRPC resistant to second-line antiandrogen therapy. Mol. Cancer Ther. 2021;20:490–499. doi: 10.1158/1535-7163.MCT-20-0417. [DOI] [PubMed] [Google Scholar]
- 57.Lu J., Qian Y., Altieri M., et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2015;22:755–763. doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zengerle M., Chan K.-H., Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS. Chem. Biol. 2015;10:1770–1777. doi: 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cao F., de Weerd S., Chen D., et al. Induced protein degradation of histone deacetylases 3 (HDAC3) by proteolysis targeting chimera (PROTAC) Eur. J. Med. Chem. 2020;208:112800. doi: 10.1016/j.ejmech.2020.112800. [DOI] [PubMed] [Google Scholar]
- 60.Yang K., Wu H., Zhang Z., et al. Development of selective histone deacetylase 6 (HDAC6) degraders recruiting von Hippel–Lindau (VHL) E3 ubiquitin ligase. ACS. Med. Chem. Lett. 2020;11:575–581. doi: 10.1021/acsmedchemlett.0c00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao N., Chu T.-T., Li Q.-Q., et al. Hydrophobic tagging-mediated degradation of Alzheimer's disease related Tau. RSC Adv. 2017;7:40362–40366. [Google Scholar]
- 62.de Wispelaere M., Du G., Donovan K.A., et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat. Commun. 2019;10:3468. doi: 10.1038/s41467-019-11429-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao Y., Shu Y., Lin J., et al. Discovery of novel BTK PROTACs for B-Cell lymphomas. Eur. J. Med. Chem. 2021;225:113820. doi: 10.1016/j.ejmech.2021.113820. [DOI] [PubMed] [Google Scholar]
- 64.Du Z., Lovly C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer. 2018;17:58. doi: 10.1186/s12943-018-0782-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xie S., Sun Y., Liu Y., et al. Development of alectinib-based PROTACs as novel potent degraders of anaplastic lymphoma kinase (ALK) J. Med. Chem. 2021;64:9120–9140. doi: 10.1021/acs.jmedchem.1c00270. [DOI] [PubMed] [Google Scholar]
- 66.Zhang H., Zhao H.-Y., Xi X.-X., et al. Discovery of potent epidermal growth factor receptor (EGFR) degraders by proteolysis targeting chimera (PROTAC) Eur. J. Med. Chem. 2020;189:112061. doi: 10.1016/j.ejmech.2020.112061. [DOI] [PubMed] [Google Scholar]
- 67.Burslem G.M., Smith B.E., Lai A.C., et al. The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chem. Biol. 2018;25:67–77.e3. doi: 10.1016/j.chembiol.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao H.-Y., Yang X.-Y., Lei H., et al. Discovery of potent small molecule PROTACs targeting mutant EGFR. Eur. J. Med. Chem. 2020;208:112781. doi: 10.1016/j.ejmech.2020.112781. [DOI] [PubMed] [Google Scholar]
- 69.Zhao H.-Y., Wang H.-P., Mao Y.-Z., et al. Discovery of potent PROTACs targeting EGFR mutants through the optimization of covalent EGFR ligands. J. Med. Chem. 2022;65:4709–4726. doi: 10.1021/acs.jmedchem.1c01827. [DOI] [PubMed] [Google Scholar]
- 70.Shan Y., Si R., Wang J., et al. Discovery of novel anti-angiogenesis agents. Part 11: development of PROTACs based on active molecules with potency of promoting vascular normalization. Eur. J. Med. Chem. 2020;205:112654. doi: 10.1016/j.ejmech.2020.112654. [DOI] [PubMed] [Google Scholar]
- 71.Chen L., Chen Y., Zhang C., et al. Discovery of first-in-class potent and selective tropomyosin receptor kinase degraders. J. Med. Chem. 2020;63:14562–14575. doi: 10.1021/acs.jmedchem.0c01342. [DOI] [PubMed] [Google Scholar]
- 72.Zhao B., Burgess K. TrkC-targeted kinase inhibitors and PROTACs. Mol. Pharm. 2019;16:4313–4318. doi: 10.1021/acs.molpharmaceut.9b00673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Siveen K.S., Prabhu K.S., Achkar I.W., et al. Role of non receptor tyrosine kinases in hematological malignances and its targeting by natural products. Mol. Cancer. 2018;17:31. doi: 10.1186/s12943-018-0788-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang Y., Gao H., Sun X., et al. Global PROTAC toolbox for degrading BCR–ABL overcomes drug-resistant mutants and adverse effects. J. Med. Chem. 2020;63:8567–8583. doi: 10.1021/acs.jmedchem.0c00967. [DOI] [PubMed] [Google Scholar]
- 75.Adhikari B., Bozilovic J., Diebold M., et al. PROTAC-mediated degradation reveals a non-catalytic function of AURORA-A kinase. Nat. Chem. Biol. 2020;16:1179–1188. doi: 10.1038/s41589-020-00652-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bozilovic J., Eing L., Berger B.-T., et al. Novel, highly potent PROTACs targeting AURORA-A kinase. Curr. Res. Chem. Biol. 2022;2:100032. [Google Scholar]
- 77.Khan S., Zhang X., Lv D., et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019;25:1938–1947. doi: 10.1038/s41591-019-0668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pal P., Thummuri D., Lv D., et al. Discovery of a novel BCL-XL PROTAC degrader with enhanced BCL-2 inhibition. J. Med. Chem. 2021;64:14230–14246. doi: 10.1021/acs.jmedchem.1c00517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shorer Arbel Y., Katz B.-Z., Gabizon R., et al. Proteolysis targeting chimeras for BTK efficiently inhibit B-cell receptor signaling and can overcome ibrutinib resistance in CLL cells. Front. Oncol. 2021;11:646971. doi: 10.3389/fonc.2021.646971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gabizon R., Shraga A., Gehrtz P., et al. Efficient targeted degradation via reversible and irreversible covalent PROTACs. J. Am. Chem. Soc. 2020;142:11734–11742. doi: 10.1021/jacs.9b13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cromm P.M., Samarasinghe K.T.G., Hines J., Crews C.M. Addressing kinase-independent functions of Fak via PROTAC-mediated degradation. J. Am. Chem. Soc. 2018;140:17019–17026. doi: 10.1021/jacs.8b08008. [DOI] [PubMed] [Google Scholar]
- 82.Wang M., Lu J., Wang M., et al. Discovery of SHP2-D26 as a first, potent, and effective PROTAC degrader of SHP2 protein. J. Med. Chem. 2020;63:7510–7528. doi: 10.1021/acs.jmedchem.0c00471. [DOI] [PubMed] [Google Scholar]
- 83.Shah R.R., Redmond J.M., Mihut A., et al. Hi-JAK-ing the ubiquitin system: the design and physicochemical optimisation of JAK PROTACs. Bioorg. Med. Chem. 2020;28:115326. doi: 10.1016/j.bmc.2020.115326. [DOI] [PubMed] [Google Scholar]
- 84.Alabi S., Jaime-Figueroa S., Yao Z., et al. Mutant-selective degradation by BRAF-targeting PROTACs. Nat. Commun. 2021;12:920. doi: 10.1038/s41467-021-21159-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jiang B., Gao Y., Che J., et al. Discovery and resistance mechanism of a selective CDK12 degrader. Nat. Chem. Biol. 2021;17:675–683. doi: 10.1038/s41589-021-00765-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Niu T., Li K., Jiang L., et al. Noncovalent CDK12/13 dual inhibitors-based PROTACs degrade CDK12-Cyclin K complex and induce synthetic lethality with PARP inhibitor. Eur. J. Med. Chem. 2022;228:114012. doi: 10.1016/j.ejmech.2021.114012. [DOI] [PubMed] [Google Scholar]
- 87.Chen Y., Ning Y., Bai G., et al. Design, synthesis, and biological evaluation of IRAK4-targeting PROTACs. ACS Med. Chem. Lett. 2021;12:82–87. doi: 10.1021/acsmedchemlett.0c00474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bondeson D.P., Mares A., Smith I.E.D., et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015;11:611–617. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wei J., Hu J., Wang L., et al. Discovery of a first-in-class mitogen-activated protein kinase kinase 1/2 degrader. J. Med. Chem. 2019;62:10897–10911. doi: 10.1021/acs.jmedchem.9b01528. [DOI] [PubMed] [Google Scholar]
- 90.Smith B.E., Wang S.L., Jaime-Figueroa S., et al. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019;10:131. doi: 10.1038/s41467-018-08027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tovell H., Testa A., Zhou H., et al. Design and characterization of SGK3-PROTAC1, an isoform specific SGK3 kinase PROTAC degrader. ACS Chem. Biol. 2019;14:2024–2034. doi: 10.1021/acschembio.9b00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bushweller J.H. Targeting transcription factors in cancer — from undruggable to reality. Nat. Rev. Cancer. 2019;19:611–624. doi: 10.1038/s41568-019-0196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Han X., Zhao L., Xiang W., et al. Discovery of highly potent and efficient PROTAC degraders of androgen receptor (AR) by employing weak binding affinity VHL E3 ligase ligands. J. Med. Chem. 2019;62:11218–11231. doi: 10.1021/acs.jmedchem.9b01393. [DOI] [PubMed] [Google Scholar]
- 94.Itoh Y., Kitaguchi R., Ishikawa M., et al. Design, synthesis and biological evaluation of nuclear receptor-degradation inducers. Bioorg. Med. Chem. 2011;19:6768–6778. doi: 10.1016/j.bmc.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 95.Zhou H., Bai L., Xu R., et al. Structure-based discovery of SD-36 as a potent, selective, and efficacious PROTAC degrader of STAT3 protein. J. Med. Chem. 2019;62:11280–11300. doi: 10.1021/acs.jmedchem.9b01530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hogg S.J., Beavis P.A., Dawson M.A., Johnstone R.W. Targeting the epigenetic regulation of antitumour immunity. Nat. Rev. Drug Discov. 2020;19:776–800. doi: 10.1038/s41573-020-0077-5. [DOI] [PubMed] [Google Scholar]
- 97.Roy M.J., Winkler S., Hughes S.J., et al. SPR-measured dissociation kinetics of PROTAC ternary complexes influence target degradation rate. ACS Chem. Biol. 2019;14:361–368. doi: 10.1021/acschembio.9b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Raina K., Lu J., Qian Y., et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA. 2016;113:7124–7129. doi: 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chan K.-H., Zengerle M., Testa A., Ciulli A. Impact of target warhead and linkage vector on inducing protein degradation: comparison of bromodomain and extra-Terminal (BET) degraders derived from triazolodiazepine (JQ1) and tetrahydroquinoline (I-BET726) BET inhibitor scaffolds. J. Med. Chem. 2018;61:504–513. doi: 10.1021/acs.jmedchem.6b01912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bassi Z.I., Fillmore M.C., Miah A.H., et al. Modulating PCAF/GCN5 immune cell function through a PROTAC approach. ACS Chem. Biol. 2018;13:2862–2867. doi: 10.1021/acschembio.8b00705. [DOI] [PubMed] [Google Scholar]
- 101.Potjewyd F., Turner A.-M.W., Beri J., et al. Degradation of polycomb repressive complex 2 with an EED-targeted bivalent chemical degrader. Cell Chem. Biol. 2020;27:47–56.e15. doi: 10.1016/j.chembiol.2019.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gao X., Burris H.A., Iii, Vuky J., et al. Phase 1/2 study of ARV-110, an androgen receptor (AR) PROTAC degrader, in metastatic castration-resistant prostate cancer (mCRPC) J. Clin. Oncol. 2022;40:17. [Google Scholar]
- 103.Flanagan J.J., Qian Y., Gough S.M., et al. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer. Cancer Res. 2019;79 P5-04-18-P05-04-18. [Google Scholar]
- 104.Jackson K.L., Agafonov R.V., Carlson M.W., et al. Abstract ND09: the discovery and characterization of CFT8634: a potent and selective degrader of BRD9 for the treatment of SMARCB1-perturbed cancers. Cancer Res. 2022;82:ND09. [Google Scholar]
- 105.Mato A.R., Wierda W.G., Ai W.Z., et al. NX-2127-001, a first-in-human trial of NX-2127, a Bruton's tyrosine kinase-targeted protein degrader, in patients with relapsed or refractory chronic lymphocytic leukemia and B-cell malignancies. Blood. 2022;140:2329–2332. [Google Scholar]
- 106.Cecchini C., Pannilunghi S., Tardy S., Scapozza L. From conception to development: investigating PROTACs features for improved cell permeability and successful protein degradation. Front. Chem. 2021;9:672267. doi: 10.3389/fchem.2021.672267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Anderson N.A., Cryan J., Ahmed A., et al. Selective CDK6 degradation mediated by cereblon, VHL, and novel IAP-recruiting PROTACs. Bioorg. Med. Chem. Lett. 2020;30:127106. doi: 10.1016/j.bmcl.2020.127106. [DOI] [PubMed] [Google Scholar]
- 108.Chamberlain P.P., Hamann L.G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019;15:937–944. doi: 10.1038/s41589-019-0362-y. [DOI] [PubMed] [Google Scholar]
- 109.Edmondson S.D., Yang B., Fallan C. Proteolysis targeting chimeras (PROTACs) in ‘beyond rule-of-five’ chemical space: recent progress and future challenges. Bioorg. Med. Chem. Lett. 2019;29:1555–1564. doi: 10.1016/j.bmcl.2019.04.030. [DOI] [PubMed] [Google Scholar]
- 110.Békés M., Langley D.R., Crews C.M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 2022;21:181–200. doi: 10.1038/s41573-021-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li X., Pu W., Zheng Q., et al. Proteolysis-targeting chimeras (PROTACs) in cancer therapy. Mol. Cancer. 2022;21:99. doi: 10.1186/s12943-021-01434-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Garber K. The PROTAC gold rush. Nat. Biotechnol. 2022;40:12–16. doi: 10.1038/s41587-021-01173-2. [DOI] [PubMed] [Google Scholar]
- 113.Chen Y., Tandon I., Heelan W., et al. Proteolysis-targeting chimera (PROTAC) delivery system: advancing protein degraders towards clinical translation. Chem. Soc. Rev. 2022;51:5330–5350. doi: 10.1039/d1cs00762a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lebraud H., Wright D.J., Johnson C.N., Heightman T.D. Protein degradation by in-cell self-assembly of proteolysis targeting chimeras. ACS Cent. Sci. 2016;2:927–934. doi: 10.1021/acscentsci.6b00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Juan A., Del Mar Noblejas-López M., Arenas-Moreira M., et al. Options to improve the action of PROTACs in cancer: development of controlled delivery nanoparticles. Front. Cell Dev. Biol. 2021;9:805336. doi: 10.3389/fcell.2021.805336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zoppi V., Hughes S.J., Maniaci C., et al. Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von Hippel–Lindau (VHL) based dual degrader probe of BRD9 and BRD7. J. Med. Chem. 2019;62:699–726. doi: 10.1021/acs.jmedchem.8b01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Imrie F., Bradley A.R., van der Schaar M., Deane C.M. Deep generative models for 3D linker design. J. Chem. Inf. Model. 2020;60:1983–1995. doi: 10.1021/acs.jcim.9b01120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yang Y., Zheng S., Su S., et al. SyntaLinker: automatic fragment linking with deep conditional transformer neural networks. Chem. Sci. 2020;11:8312–8322. doi: 10.1039/d0sc03126g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Weng G., Shen C., Cao D., et al. PROTAC-DB: an online database of PROTACs. Nucleic Acids Res. 2021;49:D1381–D1387. doi: 10.1093/nar/gkaa807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zheng S., Tan Y., Wang Z., et al. Accelerated rational PROTAC design via deep learning and molecular simulations. Nat. Mach. Intell. 2022;4:739–748. [Google Scholar]
- 121.Cao C., He M., Wang L., et al. Chemistries of bifunctional PROTAC degraders. Chem. Soc. Rev. 2022;51:7066–7114. doi: 10.1039/d2cs00220e. [DOI] [PubMed] [Google Scholar]
- 122.Gooding S., Ansari-Pour N., Towfic F., et al. Multiple cereblon genetic changes are associated with acquired resistance to lenalidomide or pomalidomide in multiple myeloma. Blood. 2021;137:232–237. doi: 10.1182/blood.2020007081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sun X., Gao H., Yang Y., et al. PROTACs: great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019;4:64. doi: 10.1038/s41392-019-0101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Silva M.C., Ferguson F.M., Cai Q., et al. Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife. 2019;8:e45457. doi: 10.7554/eLife.45457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sun Y., Zhao X., Ding N., et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018;28:779–781. doi: 10.1038/s41422-018-0055-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhao Q., Lan T., Su S., Rao Y. Induction of apoptosis in MDA-MB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chem. Commun. (Camb.) 2019;55:369–372. doi: 10.1039/c8cc07813k. [DOI] [PubMed] [Google Scholar]
- 127.Zhang X., Crowley V.M., Wucherpfennig T.G., et al. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol. 2019;15:737–746. doi: 10.1038/s41589-019-0279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li L., Mi D., Pei H., et al. In vivo target protein degradation induced by PROTACs based on E3 ligase DCAF15. Signal Transduct. Target. Ther. 2020;5:129. doi: 10.1038/s41392-020-00245-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Han T., Goralski M., Gaskill N., et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science. 2017;356:eaal3755. doi: 10.1126/science.aal3755. [DOI] [PubMed] [Google Scholar]
- 130.Bussiere D.E., Xie L., Srinivas H., et al. Structural basis of indisulam-mediated RBM39 recruitment to DCAF15 E3 ligase complex. Nat. Chem. Biol. 2020;16:15–23. doi: 10.1038/s41589-019-0411-6. [DOI] [PubMed] [Google Scholar]
- 131.Ward C.C., Kleinman J.I., Brittain S.M., et al. Covalent ligand screening uncovers a RNF4 E3 ligase recruiter for targeted protein degradation applications. ACS Chem. Biol. 2019;14:2430–2440. doi: 10.1021/acschembio.8b01083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Spradlin J.N., Hu X., Ward C.C., et al. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Biol. 2019;15:747–755. doi: 10.1038/s41589-019-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tong B., Luo M., Xie Y., et al. Bardoxolone conjugation enables targeted protein degradation of BRD4. Sci. Rep. 2020;10:15543. doi: 10.1038/s41598-020-72491-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Henning N.J., Manford A.G., Spradlin J.N., et al. Discovery of a covalent FEM1B recruiter for targeted protein degradation applications. J. Am. Chem. Soc. 2022;144:701–708. doi: 10.1021/jacs.1c03980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ohoka N., Tsuji G., Shoda T., et al. Development of small molecule chimeras that recruit AhR E3 ligase to target proteins. ACS Chem. Biol. 2019;14:2822–2832. doi: 10.1021/acschembio.9b00704. [DOI] [PubMed] [Google Scholar]
- 136.Nunes J., McGonagle G.A., Eden J., et al. Targeting IRAK4 for degradation with PROTACs. ACS Med. Chem. Lett. 2019;10:1081–1085. doi: 10.1021/acsmedchemlett.9b00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.He S., Gao F., Ma J., et al. Aptamer-PROTAC conjugates (APCs) for tumor-specific targeting in breast cancer. Angew. Chem. Int. Ed. Engl. 2021;60:23299–23305. doi: 10.1002/anie.202107347. [DOI] [PubMed] [Google Scholar]
- 138.Chen H., Liu J., Kaniskan H.Ü., et al. Folate-guided protein degradation by immunomodulatory imide drug-based molecular glues and proteolysis targeting chimeras. J. Med. Chem. 2021;64:12273–12285. doi: 10.1021/acs.jmedchem.1c00901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liu J., Chen H., Liu Y., et al. Cancer selective target degradation by folate-caged PROTACs. J. Am. Chem. Soc. 2021;143:7380–7387. doi: 10.1021/jacs.1c00451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lambert J.M., Berkenblit A. Antibody–drug conjugates for cancer treatment. Annu. Rev. Med. 2018;69:191–207. doi: 10.1146/annurev-med-061516-121357. [DOI] [PubMed] [Google Scholar]
- 141.Dragovich P.S., Pillow T.H., Blake R.A., et al. Antibody-mediated delivery of chimeric BRD4 degraders. Part 1: exploration of antibody linker, payload loading, and payload molecular properties. J. Med. Chem. 2021;64:2534–2575. doi: 10.1021/acs.jmedchem.0c01845. [DOI] [PubMed] [Google Scholar]
- 142.Dragovich P.S., Pillow T.H., Blake R.A., et al. Antibody-mediated delivery of chimeric BRD4 degraders. Part 2: improvement of in vitro antiproliferation activity and in vivo antitumor efficacy. J. Med. Chem. 2021;64:2576–2607. doi: 10.1021/acs.jmedchem.0c01846. [DOI] [PubMed] [Google Scholar]
- 143.Pillow T.H., Adhikari P., Blake R.A., et al. Antibody conjugation of a chimeric BET degrader enables in vivo activity. ChemMedChem. 2020;15:17–25. doi: 10.1002/cmdc.201900497. [DOI] [PubMed] [Google Scholar]
- 144.Hsu J.H.-R., Rasmusson T., Robinson J., et al. EED-targeted PROTACs degrade EED, EZH2, and SUZ12 in the PRC2 complex. Cell Chem. Biol. 2020;27:41–46.e17. doi: 10.1016/j.chembiol.2019.11.004. [DOI] [PubMed] [Google Scholar]
- 145.Guenette R.G., Yang S.W., Min J., et al. Target and tissue selectivity of PROTAC degraders. Chem. Soc. Rev. 2022;51:5740–5756. doi: 10.1039/d2cs00200k. [DOI] [PubMed] [Google Scholar]
- 146.Bondeson D.P., Smith B.E., Burslem G.M., et al. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 2018;25:78–87.e5. doi: 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cruite J.T., Dann G.P., Che J., et al. Cereblon covalent modulation through structure-based design of histidine targeting chemical probes. RSC Chem. Biol. 2022;3:1105–1110. doi: 10.1039/d2cb00078d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Huang H.-T., Dobrovolsky D., Paulk J., et al. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem. Biol. 2018;25:88–99.e6. doi: 10.1016/j.chembiol.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lai A.C., Toure M., Hellerschmied D., et al. Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew. Chem. Int. Ed. Engl. 2016;55:807–810. doi: 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang L., Shao X., Zhong T., et al. Discovery of a first-in-class CDK2 selective degrader for AML differentiation therapy. Nat. Chem. Biol. 2021;17:567–575. doi: 10.1038/s41589-021-00742-5. [DOI] [PubMed] [Google Scholar]
- 151.Steinebach C., Ng Y.L.D., Sosič I., et al. Systematic exploration of different E3 ubiquitin ligases: an approach towards potent and selective CDK6 degraders. Chem. Sci. 2020;11:3474–3486. doi: 10.1039/d0sc00167h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Xiong Y., Donovan K.A., Eleuteri N.A., et al. Chemo-proteomics exploration of HDAC degradability by small molecule degraders. Cell Chem. Biol. 2021;28:1514–1527.e4. doi: 10.1016/j.chembiol.2021.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Krönke J., Udeshi Namrata D., Narla A., et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301–305. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Morreale F.E., Kleine S., Leodolter J., et al. BacPROTACs mediate targeted protein degradation in bacteria. Cell. 2022;185:2338–2353.e18. doi: 10.1016/j.cell.2022.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Marei H., Tsai W.-T.K., Kee Y.-S., et al. Antibody targeting of E3 ubiquitin ligases for receptor degradation. Nature. 2022;610:182–189. doi: 10.1038/s41586-022-05235-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Costales M.G., Matsumoto Y., Velagapudi S.P., Disney M.D. Small molecule targeted recruitment of a nuclease to RNA. J. Am. Chem. Soc. 2018;140:6741–6744. doi: 10.1021/jacs.8b01233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Costales M.G., Suresh B., Vishnu K., Disney M.D. Targeted degradation of a hypoxia-associated non-coding RNA enhances the selectivity of a small molecule interacting with RNA. Cell Chem. Biol. 2019;26:1180–1186.e5. doi: 10.1016/j.chembiol.2019.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Meyer S.M., Tanaka T., Zanon P.R.A., et al. DNA-encoded library screening to inform design of a ribonuclease targeting chimera (RiboTAC) J. Am. Chem. Soc. 2022;144:21096–21102. doi: 10.1021/jacs.2c07217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhang Q., Wu X., Zhang H., et al. Protein phosphatase 5-recruiting chimeras for accelerating apoptosis-signal-regulated kinase 1 dephosphorylation with antiproliferative activity. J. Am. Chem. Soc. 2023;145:1118–1128. doi: 10.1021/jacs.2c10759. [DOI] [PubMed] [Google Scholar]
- 160.Ghidini A., Cléry A., Halloy F., et al. RNA-PROTACs: degraders of RNA-binding proteins. Angew. Chem. Int. Ed. Engl. 2021;60:3163–3169. doi: 10.1002/anie.202012330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Shao J., Yan Y., Ding D., et al. Destruction of DNA-binding proteins by programmable oligonucleotide PROTAC (O'PROTAC): effective targeting of LEF1 and ERG. Adv. Sci. 2021;8:e2102555. doi: 10.1002/advs.202102555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Liu J., Chen H., Kaniskan H.Ü., et al. TF-PROTACs enable targeted degradation of transcription factors. J. Am. Chem. Soc. 2021;143:8902–8910. doi: 10.1021/jacs.1c03852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Samarasinghe K.T.G., An E., Genuth M.A., et al. OligoTRAFTACs: a generalizable method for transcription factor degradation. RSC Chem. Biol. 2022;3:1144–1153. doi: 10.1039/d2cb00138a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pei H., Guo W., Peng Y., et al. Targeting key proteins involved in transcriptional regulation for cancer therapy: current strategies and future prospective. Med. Res. Rev. 2022;42:1607–1660. doi: 10.1002/med.21886. [DOI] [PubMed] [Google Scholar]
- 165.Zhang L., Li L., Wang X., et al. Development of a novel PROTAC using the nucleic acid aptamer as a targeting ligand for tumor selective degradation of nucleolin. Mol. Ther. Nucleic Acids. 2022;30:66–79. doi: 10.1016/j.omtn.2022.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hines J., Gough J.D., Corson T.W., Crews C.M. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc. Natl. Acad. Sci. USA. 2013;110:8942–8947. doi: 10.1073/pnas.1217206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jin Y.-H., Lu M.-C., Wang Y., et al. Azo-PROTAC: novel light-controlled small-molecule tool for protein knockdown. J. Med. Chem. 2020;63:4644–4654. doi: 10.1021/acs.jmedchem.9b02058. [DOI] [PubMed] [Google Scholar]
- 168.Pfaff P., Samarasinghe K.T.G., Crews C.M., Carreira E.M. Reversible spatiotemporal control of induced protein degradation by bistable photoPROTACs. ACS Cent. Sci. 2019;5:1682–1690. doi: 10.1021/acscentsci.9b00713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Reynders M., Matsuura B.S., Bérouti M., et al. PHOTACs enable optical control of protein degradation. Sci. Adv. 2020;6:eaay5064. doi: 10.1126/sciadv.aay5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Xue G., Wang K., Zhou D., et al. Light-induced protein degradation with photocaged PROTACs. J. Am. Chem. Soc. 2019;141:18370–18374. doi: 10.1021/jacs.9b06422. [DOI] [PubMed] [Google Scholar]
- 171.Kounde C.S., Shchepinova M.M., Saunders C.N., et al. A caged E3 ligase ligand for PROTAC-mediated protein degradation with light. Chem. Commun. 2020;56:5532–5535. doi: 10.1039/d0cc00523a. [DOI] [PubMed] [Google Scholar]
- 172.Naro Y., Darrah K., Deiters A. Optical control of small molecule-induced protein degradation. J. Am. Chem. Soc. 2020;142:2193–2197. doi: 10.1021/jacs.9b12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Liu J., Chen H., Ma L., et al. Light-induced control of protein destruction by opto-PROTAC. Sci. Adv. 2020;6:eaay5154. doi: 10.1126/sciadv.aay5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Anighoro A., Bajorath J., Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J. Med. Chem. 2014;57:7874–7887. doi: 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- 175.Zheng M., Huo J., Gu X., et al. Rational design and synthesis of novel dual PROTACs for simultaneous degradation of EGFR and PARP. J. Med. Chem. 2021;64:7839–7852. doi: 10.1021/acs.jmedchem.1c00649. [DOI] [PubMed] [Google Scholar]
- 176.Imaide S., Riching K.M., Makukhin N., et al. Trivalent PROTACs enhance protein degradation via combined avidity and cooperativity. Nat. Chem. Biol. 2021;17:1157–1167. doi: 10.1038/s41589-021-00878-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Foley K.P., Ye L., Wang M., et al. Abstract 971: chaperone-mediated protein degradation (CHAMP): a novel technology for tumor-targeted protein degradation. Cancer Res. 2021;81:971. [Google Scholar]
- 178.He S., Ma J., Fang Y., et al. Homo-PROTAC mediated suicide of MDM2 to treat non-small cell lung cancer. Acta Pharm. Sin. B. 2021;11:1617–1628. doi: 10.1016/j.apsb.2020.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Steinebach C., Lindner S., Udeshi N.D., et al. Homo-PROTACs for the chemical knockdown of cereblon. ACS Chem. Biol. 2018;13:2771–2782. doi: 10.1021/acschembio.8b00693. [DOI] [PubMed] [Google Scholar]
- 180.Maniaci C., Hughes S.J., Testa A., et al. Homo-PROTACs: bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat. Commun. 2017;8:830. doi: 10.1038/s41467-017-00954-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Li X., Shen L., Zhang J., et al. Degradation of HER2 by Cbl-based chimeric ubiquitin ligases. Cancer Res. 2007;67:8716–8724. doi: 10.1158/0008-5472.CAN-06-3731. [DOI] [PubMed] [Google Scholar]
- 182.Hatakeyama S., Watanabe M., Fujii Y., Nakayama K.I. Targeted destruction of c-Myc by an engineered ubiquitin ligase suppresses cell transformation and tumor formation. Cancer Res. 2005;65:7874–7879. doi: 10.1158/0008-5472.CAN-05-1581. [DOI] [PubMed] [Google Scholar]
- 183.Pan T., Zhang Y., Zhou N., et al. A recombinant chimeric protein specifically induces mutant KRAS degradation and potently inhibits pancreatic tumor growth. Oncotarget. 2016;7:44299–44309. doi: 10.18632/oncotarget.9996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ma Y., Gu Y., Zhang Q., et al. Targeted degradation of KRAS by an engineered ubiquitin ligase suppresses pancreatic cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2013;12:286–294. doi: 10.1158/1535-7163.MCT-12-0650. [DOI] [PubMed] [Google Scholar]
- 185.Khandia R., Dadar M., Munjal A., et al. A comprehensive review of autophagy and its various roles in infectious, non-infectious, and lifestyle diseases: current knowledge and prospects for disease prevention, novel drug design, and therapy. Cells. 2019;8:674. doi: 10.3390/cells8070674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ding Y., Xing D., Fei Y., Lu B. Emerging degrader technologies engaging lysosomal pathways. Chem. Soc. Rev. 2022;51:8832–8876. doi: 10.1039/d2cs00624c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Takahashi D., Moriyama J., Nakamura T., et al. AUTACs: cargo-specific degraders using selective autophagy. Mol. Cell. 2019;76:797–810.e10. doi: 10.1016/j.molcel.2019.09.009. [DOI] [PubMed] [Google Scholar]
- 188.Nedelsky N.B., Todd P.K., Taylor J.P. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim. Biophys. Acta. 2008;1782:691–699. doi: 10.1016/j.bbadis.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mari M., Griffith J., Rieter E., et al. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol. 2010;190:1005–1022. doi: 10.1083/jcb.200912089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Glick D., Barth S., Macleod K.F. Autophagy: cellular and molecular mechanisms. J. Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ito C., Saito Y., Nozawa T., et al. Endogenous nitrated nucleotide is a key mediator of autophagy and innate defense against bacteria. Mol. Cell. 2013;52:794–804. doi: 10.1016/j.molcel.2013.10.024. [DOI] [PubMed] [Google Scholar]
- 192.Li Z., Zhu C., Ding Y., et al. ATTEC: a potential new approach to target proteinopathies. Autophagy. 2020;16:185–187. doi: 10.1080/15548627.2019.1688556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Li Z., Wang C., Wang Z., et al. Allele-selective lowering of mutant HTT protein by HTT–LC3 linker compounds. Nature. 2019;575:203–209. doi: 10.1038/s41586-019-1722-1. [DOI] [PubMed] [Google Scholar]
- 194.Pei J., Pan X., Wang A., et al. Developing potent LC3-targeting AUTAC tools for protein degradation with selective autophagy. Chem. Commun. (Camb.) 2021;57:13194–13197. doi: 10.1039/d1cc04661f. [DOI] [PubMed] [Google Scholar]
- 195.Dong G., Wu Y., Cheng J., et al. Ispinesib as an effective warhead for the design of autophagosome-tethering chimeras: discovery of potent degraders of nicotinamide phosphoribosyltransferase (NAMPT) J. Med. Chem. 2022;65:7619–7628. doi: 10.1021/acs.jmedchem.1c02001. [DOI] [PubMed] [Google Scholar]
- 196.Fu Y., Chen N., Wang Z., et al. Degradation of lipid droplets by chimeric autophagy-tethering compounds. Cell Res. 2021;31:965–979. doi: 10.1038/s41422-021-00532-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ji C.H., Kim H.Y., Lee M.J., et al. The AUTOTAC chemical biology platform for targeted protein degradation via the autophagy-lysosome system. Nat. Commun. 2022;13:904. doi: 10.1038/s41467-022-28520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Uhlén M., Fagerberg L., Hallström B.M., et al. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 199.Spiess M. The asialoglycoprotein receptor: a model for endocytic transport receptors. Biochemistry. 1990;29:10009–10018. doi: 10.1021/bi00495a001. [DOI] [PubMed] [Google Scholar]
- 200.Ahn G., Banik S.M., Miller C.L., et al. LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat. Chem. Biol. 2021;17:937–946. doi: 10.1038/s41589-021-00770-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhou Y., Teng P., Montgomery N.T., et al. Development of triantennary N-acetylgalactosamine conjugates as degraders for extracellular proteins. ACS Cent. Sci. 2021;7:499–506. doi: 10.1021/acscentsci.1c00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Caianiello D.F., Zhang M., Ray J.D., et al. Bifunctional small molecules that mediate the degradation of extracellular proteins. Nat. Chem. Biol. 2021;17:947–953. doi: 10.1038/s41589-021-00851-1. [DOI] [PubMed] [Google Scholar]
- 203.Lai A.C., Crews C.M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017;16:101–114. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Neklesa T.K., Crews C.M. Greasy tags for protein removal. Nature. 2012;487:308–309. doi: 10.1038/487308a. [DOI] [PubMed] [Google Scholar]
- 205.Tae H.S., Sundberg T.B., Neklesa T.K., et al. Identification of tydrophobic tags for the degradation of stabilized proteins. Chembiochem. 2012;13:538–541. doi: 10.1002/cbic.201100793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Long M.J.C., Gollapalli D.R., Hedstrom L. Inhibitor mediated protein degradation. Chem. Biol. 2012;19:629–637. doi: 10.1016/j.chembiol.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Shi Y., Long M.J.C., Rosenberg M.M., et al. Boc3Arg-linked ligands induce degradation by localizing target proteins to the 20S proteasome. ACS Chem. Biol. 2016;11:3328–3337. doi: 10.1021/acschembio.6b00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Asawa Y., Nishida K., Kawai K., et al. Carborane as an alternative efficient hydrophobic tag for protein degradation. Bioconjug. Chem. 2021;32:2377–2385. doi: 10.1021/acs.bioconjchem.1c00431. [DOI] [PubMed] [Google Scholar]
- 209.Xie T., Lim S.M., Westover K.D., et al. Pharmacological targeting of the pseudokinase Her3. Nat. Chem. Biol. 2014;10:1006–1012. doi: 10.1038/nchembio.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Guo M., He S., Cheng J., et al. Hydrophobic tagging-induced degradation of PDEδ in colon cancer cells. ACS Med. Chem. Lett. 2022;13:298–303. doi: 10.1021/acsmedchemlett.1c00670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.McDonnell D.P., Wardell S.E., Norris J.D. Oral selective estrogen receptor downregulators (SERDs), a breakthrough endocrine therapy for breast cancer. J. Med. Chem. 2015;58:4883–4887. doi: 10.1021/acs.jmedchem.5b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Xiang W., Wang S. Therapeutic strategies to target the androgen receptor. J. Med. Chem. 2022;65:8772–8797. doi: 10.1021/acs.jmedchem.2c00716. [DOI] [PubMed] [Google Scholar]
- 213.Fischer E.S., Böhm K., Lydeard J.R., et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512:49–53. doi: 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Lopez-Girona A., Mendy D., Ito T., et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012;26:2326–2335. doi: 10.1038/leu.2012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Sievers Q.L., Petzold G., Bunker R.D., et al. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science. 2018;362:eaat0572. doi: 10.1126/science.aat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Petzold G., Fischer E.S., Thomä N.H. Structural basis of lenalidomide-induced CK1α degradation by the CRL4CRBN ubiquitin ligase. Nature. 2016;532:127–130. doi: 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- 217.Gemechu Y., Millrine D., Hashimoto S., et al. Humanized cereblon mice revealed two distinct therapeutic pathways of immunomodulatory drugs. Proc. Natl. Acad. Sci. USA. 2018;115:11802–11807. doi: 10.1073/pnas.1814446115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kim S.A., Jo S.-H., Cho J.H., et al. Aryl sulfonamides induce degradation of aryl hydrocarbon receptor nuclear translocator through CRL4DCAF15 E3 ligase. Mol. Cells. 2020;43:935–944. doi: 10.14348/molcells.2020.0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Mayor-Ruiz C., Bauer S., Brand M., et al. Rational discovery of molecular glue degraders via scalable chemical profiling. Nat. Chem. Biol. 2020;16:1199–1207. doi: 10.1038/s41589-020-0594-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Słabicki M., Kozicka Z., Petzold G., et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature. 2020;585:293–297. doi: 10.1038/s41586-020-2374-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Lv L., Chen P., Cao L., et al. Discovery of a molecular glue promoting CDK12-DDB1 interaction to trigger cyclin K degradation. Elife. 2020;9:e59994. doi: 10.7554/eLife.59994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Słabicki M., Yoon H., Koeppel J., et al. Small-molecule-induced polymerization triggers degradation of BCL6. Nature. 2020;588:164–168. doi: 10.1038/s41586-020-2925-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Simonetta K.R., Taygerly J., Boyle K., et al. Prospective discovery of small molecule enhancers of an E3 ligase-substrate interaction. Nat. Commun. 2019;10:1402. doi: 10.1038/s41467-019-09358-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Uehara T., Minoshima Y., Sagane K., et al. Selective degradation of splicing factor CAPERα by anticancer sulfonamides. Nat. Chem. Biol. 2017;13:675–680. doi: 10.1038/nchembio.2363. [DOI] [PubMed] [Google Scholar]
- 225.Faust T.B., Yoon H., Nowak R.P., et al. Structural complementarity facilitates E7820-mediated degradation of RBM39 by DCAF15. Nat. Chem. Biol. 2020;16:7–14. doi: 10.1038/s41589-019-0378-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Matyskiela M.E., Zhang W., Man H.-W., et al. A cereblon modulator (CC-220) with improved degradation of Ikaros and Aiolos. J. Med. Chem. 2018;61:535–542. doi: 10.1021/acs.jmedchem.6b01921. [DOI] [PubMed] [Google Scholar]
- 227.Bjorklund C.C., Kang J., Amatangelo M., et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia. 2020;34:1197–1201. doi: 10.1038/s41375-019-0620-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Yang Z., Sun Y., Ni Z., et al. Merging PROTAC and molecular glue for degrading BTK and GSPT1 proteins concurrently. Cell Res. 2021;31:1315–1318. doi: 10.1038/s41422-021-00533-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Schapira M., Calabrese M.F., Bullock A.N., Crews C.M. Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discov. 2019;18:949–963. doi: 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.