

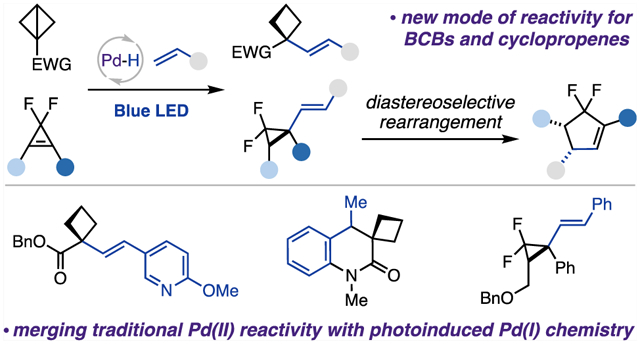
Abstract
We report the first palladium hydride enabled hydroalkenylation of strained molecules. This new mild protocol proceeds via a regio- and chemoselective hydropalladation step, followed by a photoinduced radical alkyl Heck reaction. This methodology represents a new reactivity mode for strained molecules and opens new avenues for photoinduced palladium catalysis. The reaction is compatible with a wide range of functional groups and can be applied to complex structures, delivering a diverse array of highly valuable and modifiable alkenylated cyclobutanes and cyclopropanes. A hydroalkenylation/diastereoselective rearrangement cascade toward a cyclopentene scaffold has also been demonstrated.
Graphical Abstract
1. INTRODUCTION
In recent years, 3- and 4-membered strained carbocycles have attracted significant attention in synthesis1 and bioconjugation.2 They serve as unusual bioisosteres3 in the development of pharmaceuticals,4 owing to their unique chemical and physical properties. One appealing and atom economical approach toward these molecules is from precursors with even higher energies, thus harnessing a thermodynamically favored strain-release process5 (Scheme 1A). Among these, bicyclo[1.1.0]butanes (BCBs) A and cyclopropenes B display remarkable ring strain,6 which enable them to undergo transformations that are challenging or unfeasible for non-strained ring systems. A particularly attractive strategy toward incorporation of small ring systems in organic molecules is a C−C bond formation between strained carbocycles and alkenes. Along this line, Gryko and co-workers disclosed an elegant Co-catalyzed addition of BCBs to a wide range of Michael acceptors. This transformation proceeds via the generation of alkyl radical intermediates, which upon Giese-type addition deliver disubstituted cyclobutanes C (Scheme 1B).5i In addition, the groups of Glorius, Brown, and Procter independently reported efficient [2 + 2] cycloaddition of BCBs with alkenes enabled by thioxanthone (TXT)-photosensitized energy transfer5j,k or SmI2-catalysis,5l thus delivering bicyclo[2.1.1]hexanes D. Due to the inherent nature of these catalytic systems, however, all these transformations offer access to reduced products C or D, and thus a valuable and modifiable alkene moiety is sacrificed. Accordingly, the development of a new protocol that would preserve synthetically important olefin functionality is highly desirable. Likewise, it would be extremely appealing to develop the analogous alkenylation method for a cyclopropene core, which is another ideal candidate for strain-release transformations.1b,7
Scheme 1. Reactivity of Strained Molecules with Alkenesa.
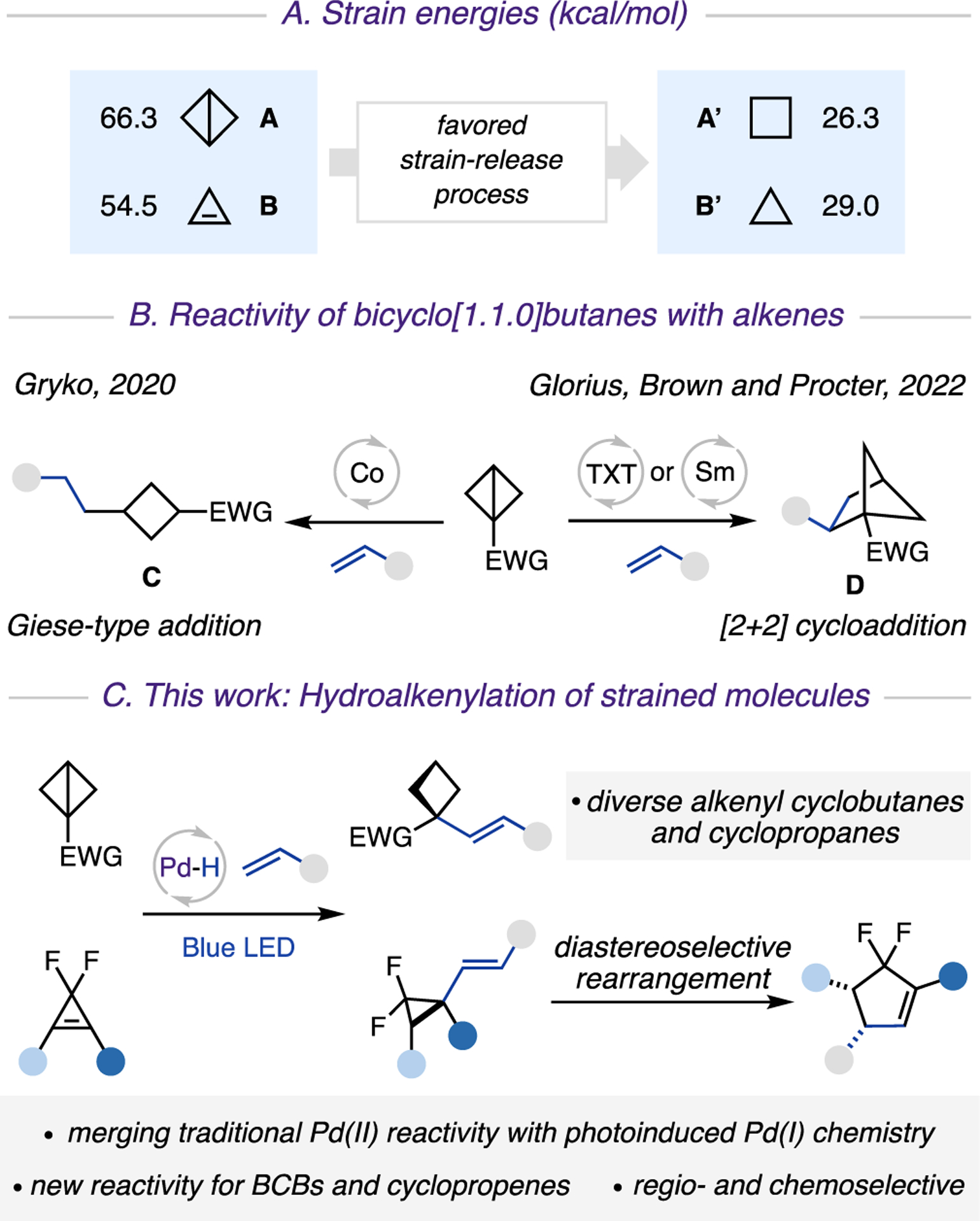
a(A) Strained energies (kcal/mol). (B) The reactivity of bicyclo[1.1.0] butanes with alkenes. (C) This work: hydroalkenylation of strained molecules.
Herein, we report the first palladium hydride enabled hydroalkenylation approach of strained molecules, BCBs and cyclopropenes, with vinyl arenes and heteroarenes, which proceeds via a sequential regio- and chemoselective hydro-palladation of strained C−C bonds, followed by a photo-induced generation of hybrid palladium C(sp3)-centered radicals, and a Heck-type coupling reaction (Scheme 1C). Furthermore, this report also outlines our preliminary findings on diastereoselective rearrangement of vinyl cyclopropanes into cyclopentenes.
2. REACTION DESIGN
To achieve alkenylation of small rings, we sought a palladium catalysis known for its facile oxidative end-game.8 Recently, photoinduced palladium catalysis has become an emerging field of study.9 We and others have established mild and efficient generation of carbon-centered radicals from (pseudo) halides or redox-active esters via single electron transfer (SET) from photoexcited Pd0 catalysts. Apparently, such an activation mode is not applicable to BCBs and cyclopropenes. Hence, we searched for generation of radicals from strained molecules via an alternative Pd-catalyzed strategy. In our recently developed method for a photoinduced Pd-catalyzed Heck reaction of diazo compounds,10 we suggested that the generation of alkyl radicals may proceed through a denitrogenative reaction between palladium hydride (PdH) and a diazo compound (Scheme 2A). Accordingly, we hypothesized that the desired cycloalkyl radicals could be accessed from highly reactive strained molecules with the aid of putative PdH species.
Scheme 2. Viability of Photoinduced Palladium Hydride Catalytic Systema.
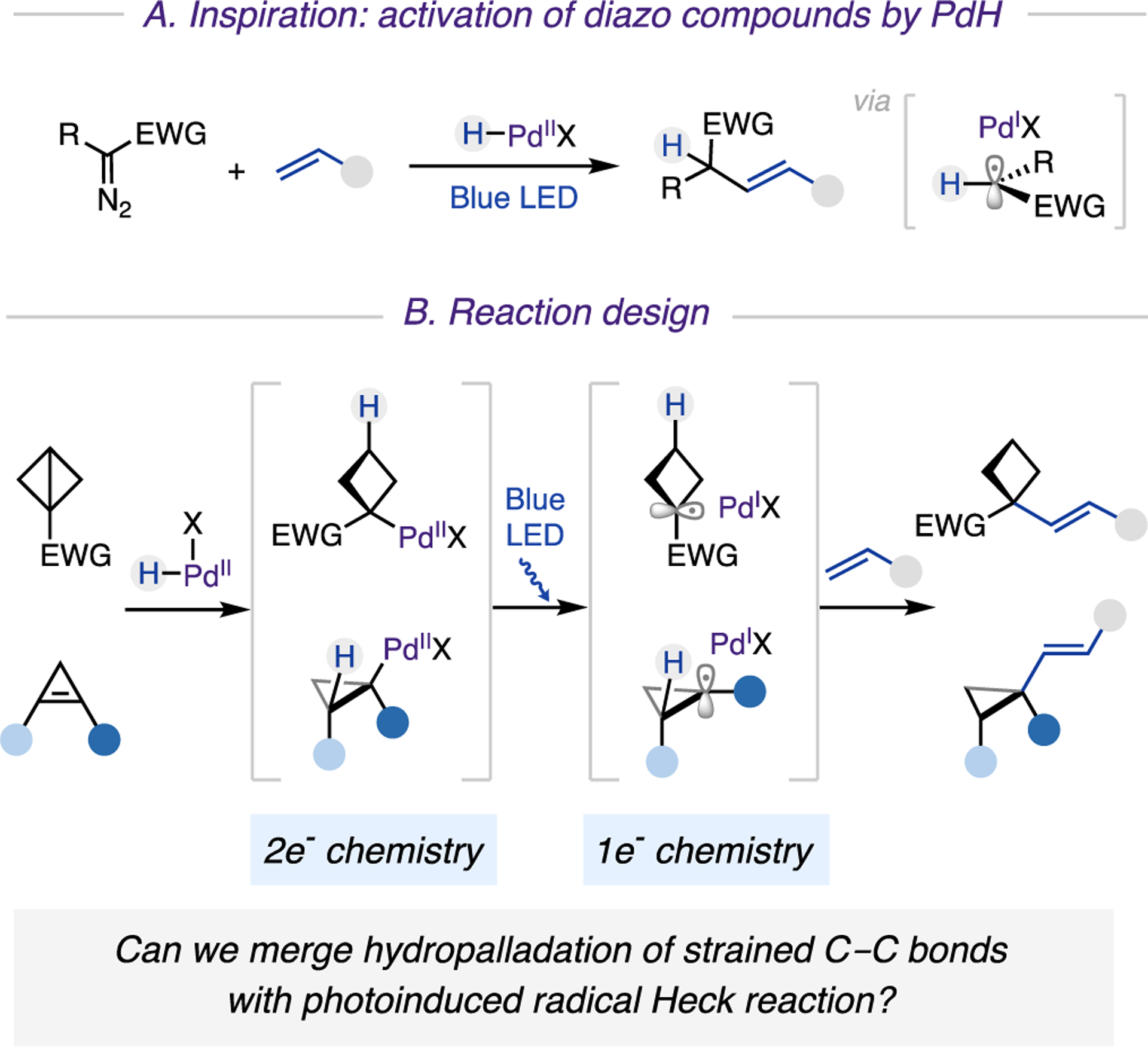
a(A) Inspiration: activation of diazo compounds with PdH. (B) Reaction design.
We envisioned achieving hydroalkenylation of strained molecules with alkenes via the design plan depicted in Scheme 2B, which combines traditional Pd(II) reactivity with photo-induced Pd(I) chemistry. Since two types of (pseudo) alkene substrates would be involved in the reaction, they must be differentiated in order to achieve the desired coupling reaction. As a design principle, we recognized that a π-like central C−C bond of BCBs or a strained double bond of cyclopropenes should be more reactive toward hydropalladation with PdH compared to that of its nonstrained counterpart. Therefore, the in situ generated PdH species is expected to add onto strained molecules in a chemoselective manner. The resulting alkyl Pd(II) complex, upon visible-light-induced homolysis of the Pd−C bond, would then generate hybrid Pd(I) cyclobutyl or cyclopropyl radical species. The capture of these key intermediates with an alkene and a successive facile β-H elimination would lead to the formation of aimed vinylcyclobutane and vinylcyclopropane products. If successful, this approach would represent a new reaction profile of the photoinduced Pd 0/I/II manifold, by introducing a hydro-palladation process and encompassing a new class of substrates. Moreover, the ability to directly couple strained molecules with easily accessible alkenes would facilitate a practical synthesis of small rings with valuable alkenyl functionality.11
3. RESULTS AND DISCUSSION
3.1. Reaction Optimization.
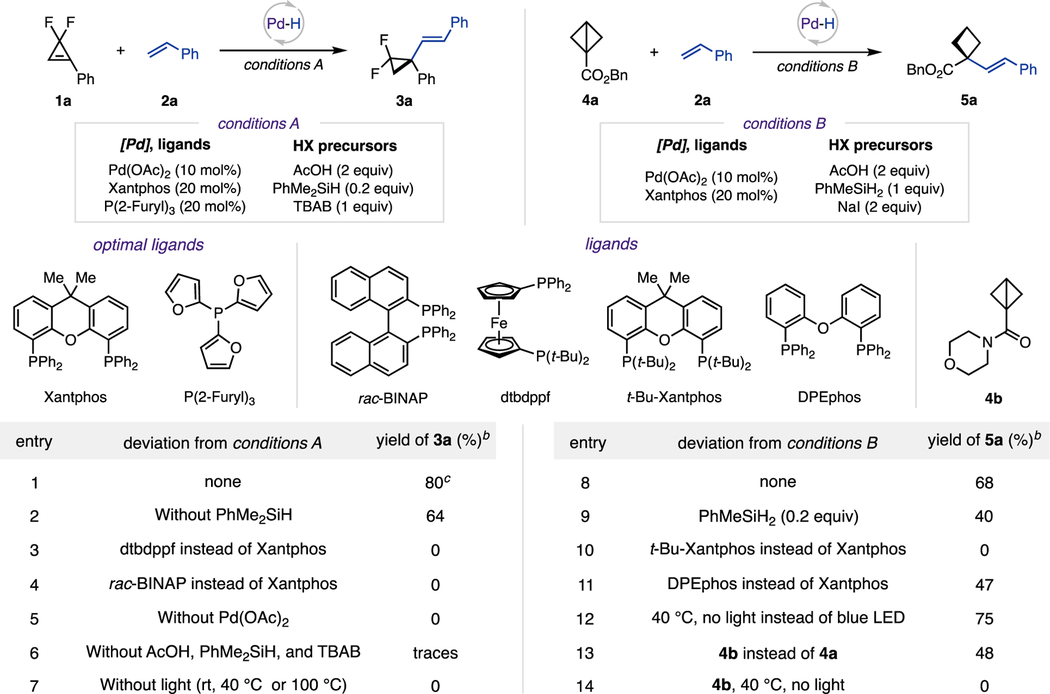
We commenced our studies with examining a model reaction between gem-difluorocyclopropene12 1a and styrene 2a under standard visible light/Pd conditions (Table 1). Consistently with our previous studies,10,13 the combination of Pd(OAc)2 and bidentate Xantphos proved to be the most efficient catalytic system. An extensive evaluation14 of monodentate additive ligand10,13e,15 indicated P(2-Furyl)3 as a superior ligand. Evidently, we recognized the need for additives to promote the in situ generation of PdH species. Therefore, in contrast to the previously developed basic conditions for photoinduced Heck reactions,10,13,16 we introduced an acidic environment, where acetic acid was identified as the key beneficial hydrogen donor, operating with dimethylphenylsilane as a hydride codonor. It was also found that employment of tetrabutylammonium bromide (TBAB), an exogenous halide counterion source, was crucial for further improvement of the reaction efficiency.17 Gratifyingly, under these conditions, the desired hydro-alkenylation product 3a was formed in 80% yield (condition A, entry 1). Reaction was less efficient in the absence of hydrosilane (entry 2). Reactions with other bidentate phosphines failed to provide any product (entries 3 and 4). Control experiments demonstrated that both Pd catalyst and HX precursors were essential for this transformation (entries 5, 6). Likewise, thermal reactions under dark conditions did not lead to any product (entry 7).
Table 1.
Reaction Optimization and Control Experimentsa
|
0.1 mmol scale; 1a:2a = 1:2. 4a:2a = 1:2. Conditions A: Pd(OAc)2 (10 mol %), Xantphos (20 mol %), P(2-Furyl)3 (20 mol %), acetic acid (2 equiv), PhMe2SiH (0.2 equiv), TBAB (1 equiv), 1,4-dioxane (0.15 M), blue LED (40 W, 427 nm), 16 h. Conditions B: Pd(OAc)2 (10 mol %), Xantphos (20 mol %), acetic acid (2 equiv), PhMeSiH2 (1 equiv), NaI (2 equiv), DCE (0.15 M), blue LED (40 W, 427 nm), 16 h.
Yields were determined by 1H NMR spectroscopy using CH2Br2 as an internal standard.
0.15 mmol scale, isolated yields.
Motivated by the successful employment of cyclopropenes in the hydroalkenylation reaction, we then turned our attention to BCBs. After re-evaluation of initial reaction parameters,14 we found modified conditions, which allowed hydroalkenylation reaction of BCB 4a with styrene 2a to be efficiently performed (condition B, entry 8). Employment of substochio-metric amounts of hydrosilane hampered the reaction performance (entry 9). Similarly to the reactions with cyclopropenes, employment of other bidentate phosphine ligands resulted in diminished yields (entries 10, 11). Lastly, control experiments revealed that certain BCB substrates, such as strained ester 4a, engaged in the reaction in the absence of light13b (entry 12), whereas amide 4b was completely unreactive under such conditions (entries 13, 14).
3.2. Substrate Scope.
With the optimized conditions in hand, the scope of strained molecules in reactions with styrene and derivatives was examined first (Table 2). It was found that gem-difluorocyclopropenes possessing different aryl substituents (3b and 3c) are all capable partners. Moreover, 1,2-disubstituted gem-difluorocyclopropenes proved to be viable substrates, delivering hydroalkenylation products 3d−3g with good diastereoselectivity. Notably, 3d was obtained with excellent regiocontrol.18 Likewise, various BCBs, including ester 4a, amide 4b, and nitrile 4c, were competent radical precursors. This reaction can also be performed in an intramolecular fashion. Thus, 6-exo-trig cyclization of 4d furnished an interesting spiro-cyclobutyl benzolactam 5g in reasonable yield.
Table 2.
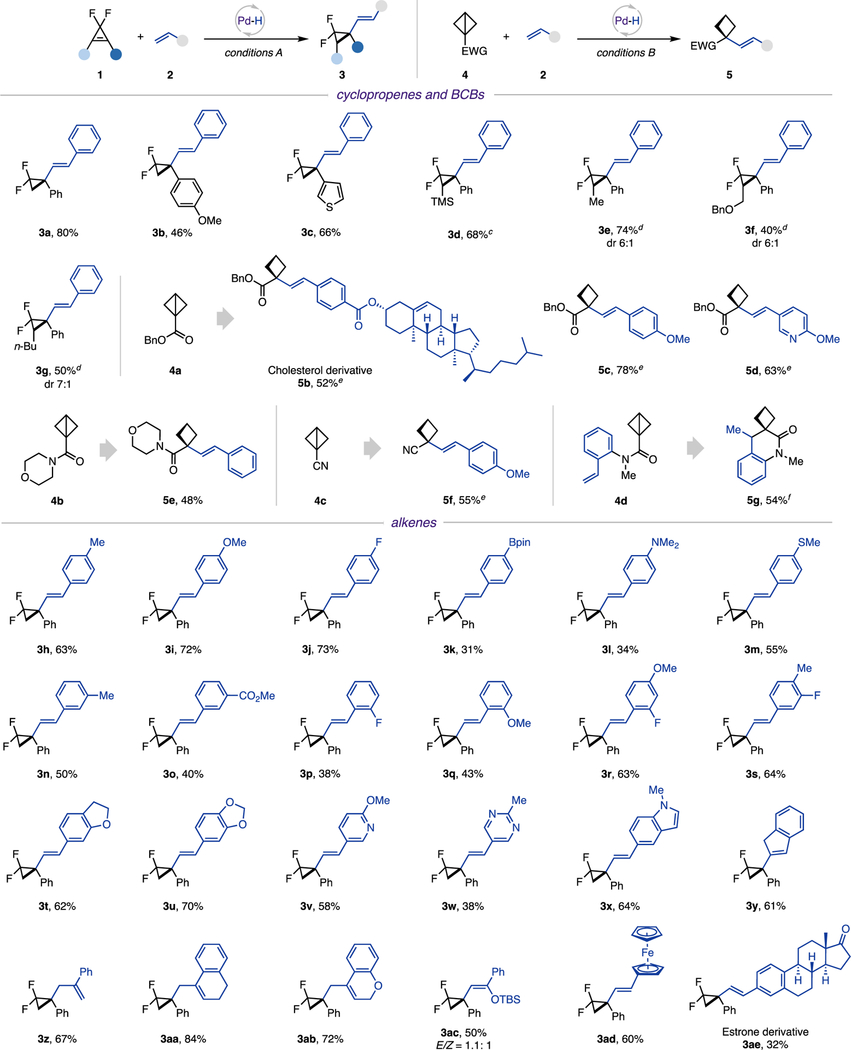
|
0.15 mmol scale, 1:2 = 1:2, 4:2 = 1:2, isolated yields.
Diastereomeric ratio (dr) and E/Z ratio was determined by 1H NMR analysis of crude reaction mixtures, using CH2Br2 as an internal standard.
Single diastereomer.
Xantphos Pd G3 (10 mol %) was used as a catalyst, and 1 equiv of PhMe2SiH was used. Xantphos Pd G3: [(4,5-bis(diphenylphosphino)-9,9-dimethylxanthene)-2-(2′-amino-1,1′-biphenyl)]palladium(II) methane-sulfonate.
Reaction was performed at 40 °C without light.
Styrene was not added.
Next, the generality of alkenes was evaluated. Various para-substituted styrenes, including methyl, methoxy, fluoro, boronic ester, amine, and thioether groups, reacted smoothly to provide the corresponding products 3h−3m in moderate to good yields. Analogously, meta- and ortho-substituted styrenes furnished vinyl difluorocyclopropane 3n−3q in reasonable yields. Disubstituted substrates also showed good reactivity (3r and 3s). Hydroalkenylation reaction with vinyl heteroarenes proceeded uneventfully, producing targeted vinyl difluorocyclopropanes possessing dihydrobenzofuran (3t), benzodioxole (3u), pyridine (3v), pyrimidine (3w), and indole (3x) rings. Notably, indene (3y), as well as α-substituted alkenes (3z−3ab), reacted well in this hydroalkenylation process. In addition, TBS-enol ether was proven to be a capable coupling partner, affording cyclopropyl silyl enol ether 3ac. Finally, the reaction of 1a with a vinyl derivative of ferrocene and estrone generated product 3ad and 3ae, highlighting the applicability of this photoinduced hydroalkenylation protocol in a complex setting. While all reactions were run until full conversion of substrates, in some cases, formation of notable amounts of the reduced cyclopropane side products was observed.
Interestingly, during the investigation of the cyclopropene scope, we discovered a hydroalkenylation/diastereoselective rearrangement cascade toward the difluorocyclopentene scaffold (Table 3). Preliminary study of the scope of this transformation indicated that gem-difluorocyclopropenes possessing two phenyl groups at C1, 2 (1b) or phenyl and thioether substituent (1c) underwent hydroalkenylation reaction with alkenes 2, followed by a facile ring-expansion cycloisomerization to produce cyclopentenes 6. Using this strategy, tricyclic products 6c and 6e were smoothly obtained from indene. Notably, this rearrangement is highly diastereoselective, producing cis-substituted cyclopentenes exclusively.
Table 3.
Hydroalkenylation/Diastereoselective Rearrangement Cascadea
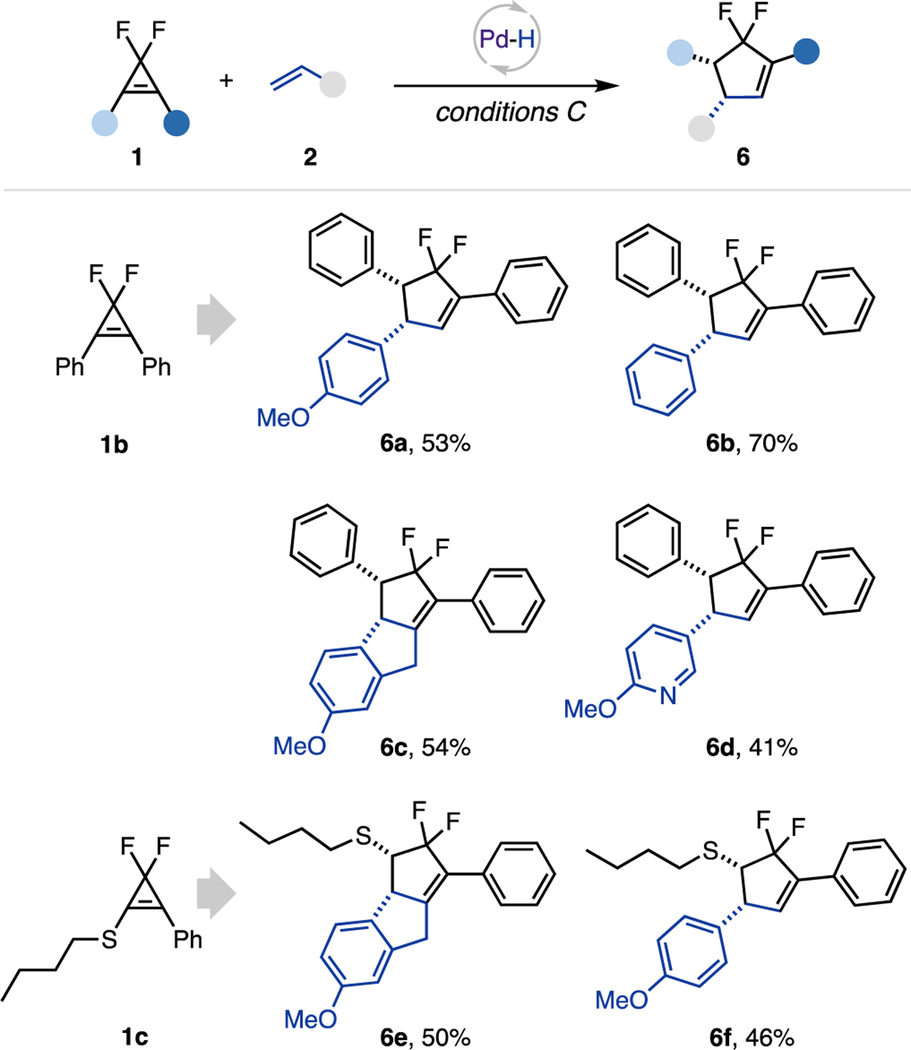
|
0.15 mmol scale, 1:2 = 1:2, isolated yields. Conditions C: Xantphos Pd G3 (10 mol %), Xantphos (20 mol %), acetic acid (4 equiv), Et3SiH (2 equiv), TBAB (1 equiv), 1,4-dioxane/toluene (0.15 M), blue LED (40 W, 427 nm), 16 h.
3.3. Scalability and Diverse Transformations.
The developed hydroalkenylation platform proved to be easily scalable. Thus, reaction on a 1 mmol scale was performed without any additional optimization, furnishing product 3a in 73% yield (Scheme 3A). In addition, in a nonoptimized experiment, it was found that this reaction can also be applied to hydrazones13g to deliver cyclopropyl-containing hydrazone 3af in reasonable yield (Scheme 3B).
Scheme 3. Scalability and Diverse Transformationsa.
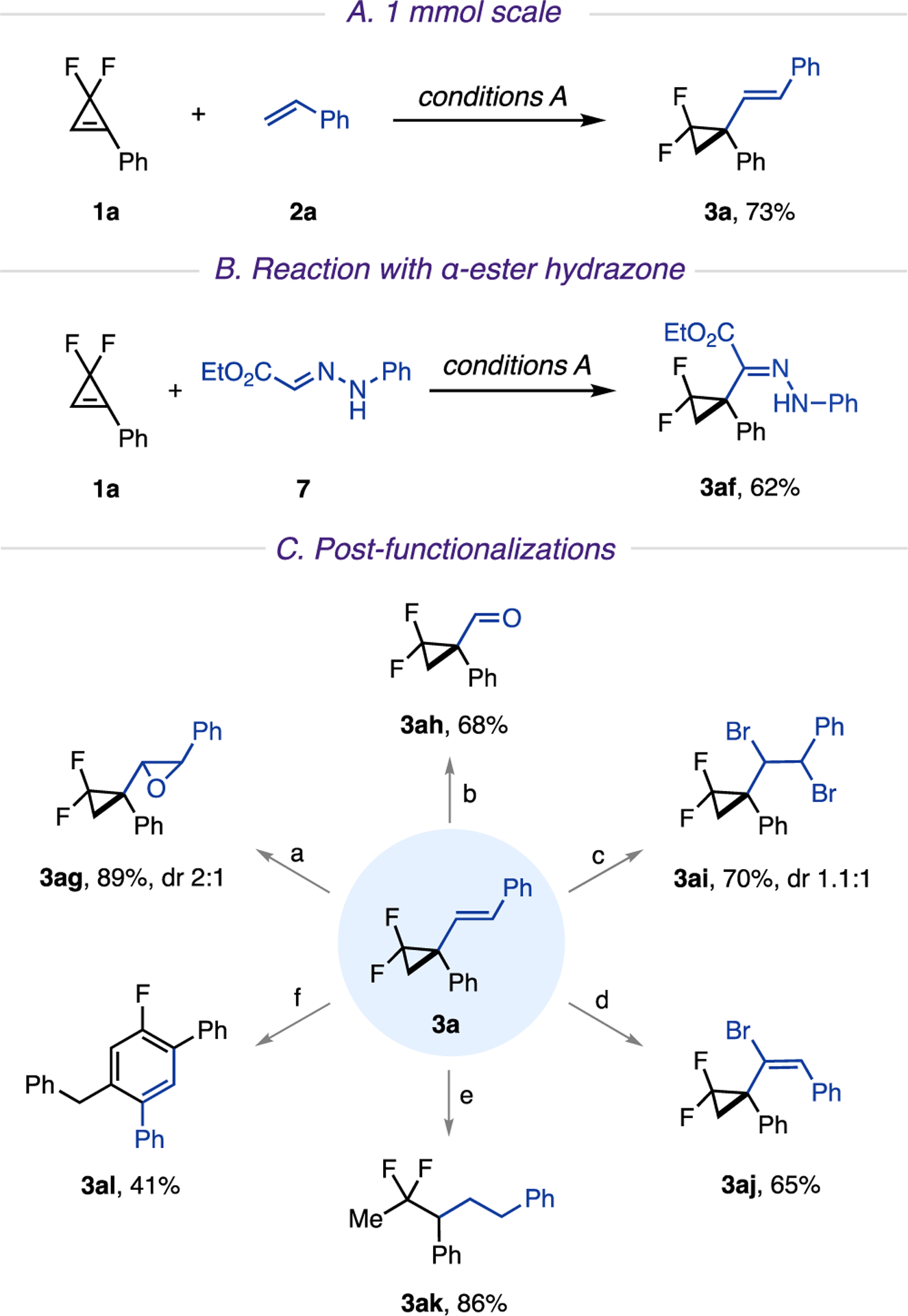
a(A) 1 mmol scale. (B) Reaction with α-ester hydrazone. (C) Postfunctionalizations. Reaction conditions: (a) m-CPBA (1.5 equiv), DCM (0.1 M), rt, 24 h; (b) 4-nitrobenzonitrile (1.5 equiv), MeCN (0.1 M), rt, 390 nm LED, 16 h; (c) LiBr (2 equiv), NaIO4 (0.5 equiv), H2SO4 (0.3 equiv), MeCN (0.05 M), rt, 24 h; (d) Br2 (1.2 equiv), DCM (0.05 M), 0 °C, 2 h, then KOH (2 equiv), THF/MeOH (1/1), 80 °C, 3 h. (e) Pd/C (10 mol %), H2 (1 atm), EtOAc (0.1 M), rt, 2 h; (f) phenylacetylene (2 equiv), Pd(TFA)2 (10 mol %), P(t-Bu)3·HBF4 (12 mol %), Cs2CO3 (2 equiv), THF (0.2 M), 60 °C, 18 h. m-CPBA: meta-chloroperoxybenzoic acid. Pd(TFA)2: palladium-(II) trifluoroacetate.
Synthetic usefulness of the obtained alkenylated gem-difluorocyclopropanes was highlighted by the following transformations (Scheme 3C). Epoxidation and photoinduced oxidative cleavage19 of the alkene proceeded smoothly, furnishing epoxide 3ag and aldehyde 3ah, respectively. Dibromination20 of the olefin provided product 3ai in good yield with two new functionalizable reaction sites. Additionally, a semi-one-pot dibromination/dehydrobromination of 3a delivered alkenyl bromide 3aj. Expectedly, hydrogenation of an alkene together with regioselective hydrogenolysis21 of the distal C2−C3 bond of cyclopropane ring produced difluoropentane 3ak. Finally, alkenylated gem-difluorocyclopropane 3a was successfully employed in the Pd-catalyzed C−C bond activation/F elimination process,22 followed by alkynylation with phenylacetylene and cycloisomerization, to deliver aryl fluoride 3al in reasonable yield.
3.4. Mechanistic Investigations and Proposed Mechanism.
Naturally, we were eager to elucidate the mechanism of this novel two-component coupling reaction (Scheme 4). The involvement of a key PdH species was unambiguously supported by the following experiments. First, the reaction of Pd(OAc)2, phosphine ligands, and additives was monitored by 1H NMR (Scheme 4A). The appearance of a new resonance signal in the high-field region (−11.48 ppm) was detected in 1H NMR spectra, which was attributed to the newly formed palladium hydride complex.17 Upon addition of BCB 4a and styrene 2a to this reaction mixture, the expected hydro-alkenylation product 5a was produced in 43% yield. Next, upon addition of the independently synthesized palladium(II) hydride complex,17c HPdCl(PPh3)2, to cyclopropene 1a and styrene 2a, the hydroalkenylation reaction proceeded smoothly without any exogenous additives (Scheme 4B).
Scheme 4. Mechanistic Studiesa.
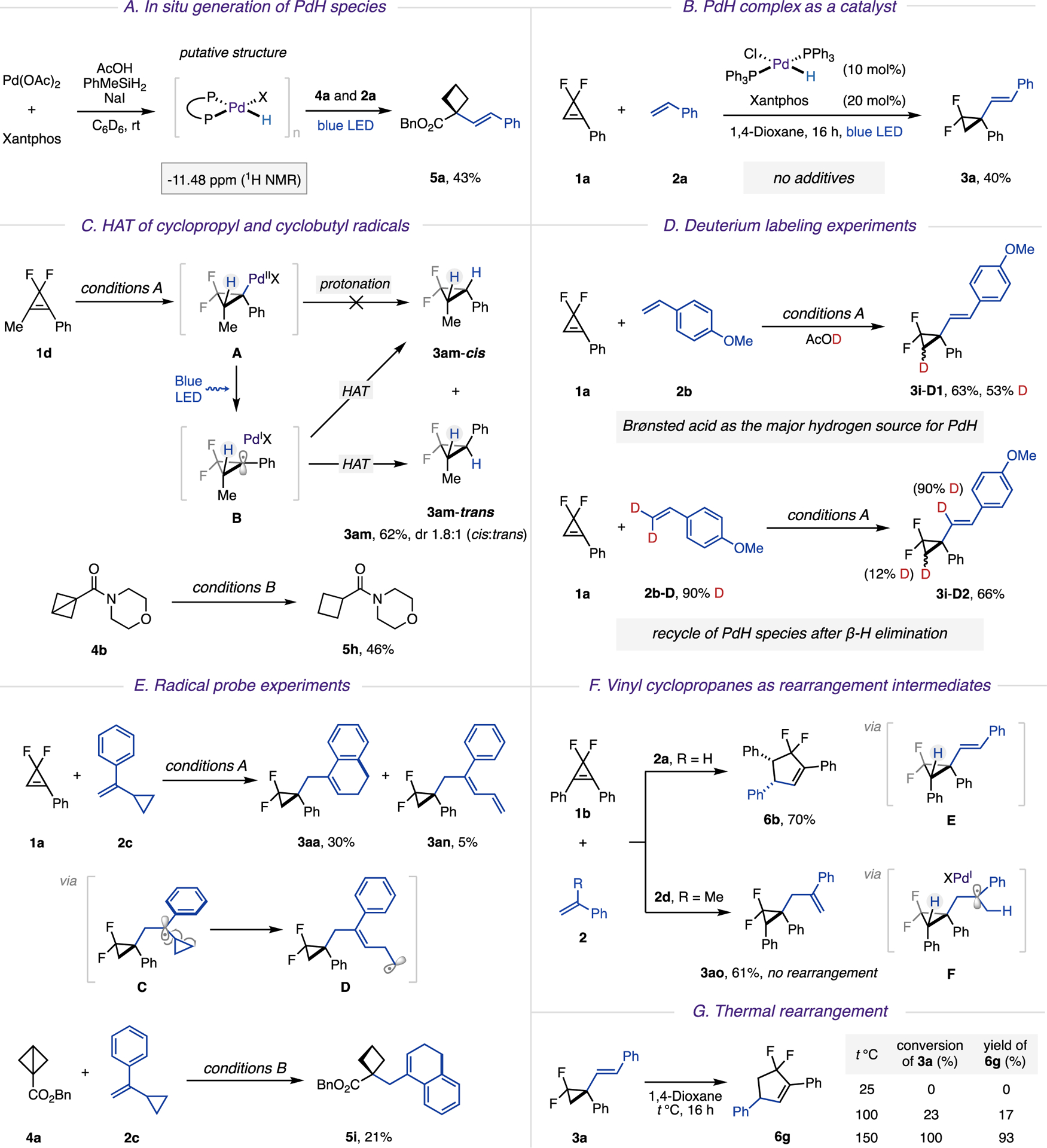
a(A) In situ generation of PdH species. (B) PdH complex as a catalyst. (C) HAT of cyclopropyl and cyclobutyl radicals. (D) Deuterium labeling experiments. (E) Radical probe experiments. (F) Vinyl cyclopropanes as rearrangement intermediates. (G) Thermal rearrangement
To validate the radical nature of this transformation, we examined the reaction of cyclopropene 1d in the absence of styrene under otherwise identical catalytic conditions (Scheme 4C). It was expected that upon hydropalladation of 1d (A) and a subsequent photoinduced homolysis, the hybrid palladium cyclopropyl radical B could be produced, which would be capable of intermolecular hydrogen atom transfer (HAT) from either face. Indeed, a diastereomeric mixture of reduced products cyclopropane 3am-cis and 3am-trans (62%, cis:trans = 1.8:1) was formed, which provided additional support for the radical pathway for this transformation. It should be noted that no conversion of the starting material was observed in the absence of light, thus indicating that a direct protonation of alkyl palladium intermediate A, derived from syn hydro-palladation, is unlikely. Analogously, when BCB 4b was tested under alkene-free conditions, the reduced product, cyclobutane 5h, was obtained.
Then, we performed a series of deuterium labeling experiments to reveal the H-source in this reaction (Scheme 4D). Thus, AcOD was used in the experiment of 1a with 2b. The D-incorporation of product 3i-D1 at the β position to the vinyl group, clearly suggested the Brønsted acid as the major hydrogen source for the formation of PdH species. Furthermore, when deuterium-labeled alkene 2b-D was subjected to the reaction, hydroalkenylation product 3i-D2 was obtained with minor deuterium incorporation at the cyclopropyl ring, which indicated the recycle of PdH species after the β-H elimination step.
The employment of radical probes and radical traps further confirmed the radical nature of this transformation. Hence, reaction of gem-difluorocyclopropene 1a with 2c, possessing a cyclopropyl substituent, underwent ring-opening of methylenecyclopropyl radical C into a homoallylic radical D. Its subsequent cyclization at the aryl ring produced bicyclic product 3aa, whereas competing β-hydrogen loss delivered linear diene 3an (Scheme 4E). Likewise, a radical probe experiment of BCB 4a delivered bicyclic product 5i. It was also shown that reactions of both substrates 1a and 4a were inhibited in the presence of radical traps, such as (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO).14
Lastly, a pathway of a hydroalkenylation/diastereoselective rearrangement cascade was investigated. As mentioned above, the reaction of 1,2-diphenyl cyclopropene 1b with styrene 2a delivered cyclopentene 6b (Scheme 4F). Thermal ring expansion of difluorinated alkenyl cyclopropanes containing an electron-withdrawing group is documented.23 Accordingly, we presumed that the cascade reaction is likely to proceed via intermediacy of vinyl cyclopropane E. To validate this assumption, we subjected the same substrate 1b to the reaction with α-methylstyrene 2d. It was anticipated that hybrid Pd(I) tertiary alkyl radical F would undergo β-H elimination from a less substituted site, thus delivering an allyl cyclopropane product not capable of cycloisomerization. Indeed, the experiment indicated formation of allyl cyclo-propane 3ao, as a single reaction product. Additional evidence was obtained by resubjecting the isolated, β-nonsubstituted vinyl cyclopropane 3a to thermal reactions (Scheme 4G). It was found that elevated temperatures (150 °C) were required to initiate its ring expansion, whereas β-phenyl-substituted akenyl difluorocyclopropane E, likely due to lower activation barriers,23 underwent spontaneous rearrangement at room temperature.
Based on the above mechanistic studies, the following mechanism for photoinduced PdH-catalyzed hydroalkenylation of strained molecules is proposed (Scheme 5). Upon an oxidative addition of Pd(0) into HX precursor, the catalytically active H−Pd(II)−X species (X = Br− or I−) is formed. A following regio- and chemoselective migratory insertion of PdH into the double bond of cyclopropenes or π-like central C−C bond of BCBs provides alkyl−Pd(II)−X complex A or A′. A subsequent homolysis of the Pd−C bond under light irradiation generates the key hybrid Pd(I) cyclobutyl or cyclopropyl radical species B or B′. Addition of the latter at the alkene produces a benzylic radical intermediate C or C′. Finally, β-H-elimination delivers hydroalkenylation product 3 or 5, while the resulting H−Pd(II)−X complex returns to the catalytic cycle.
Scheme 5.
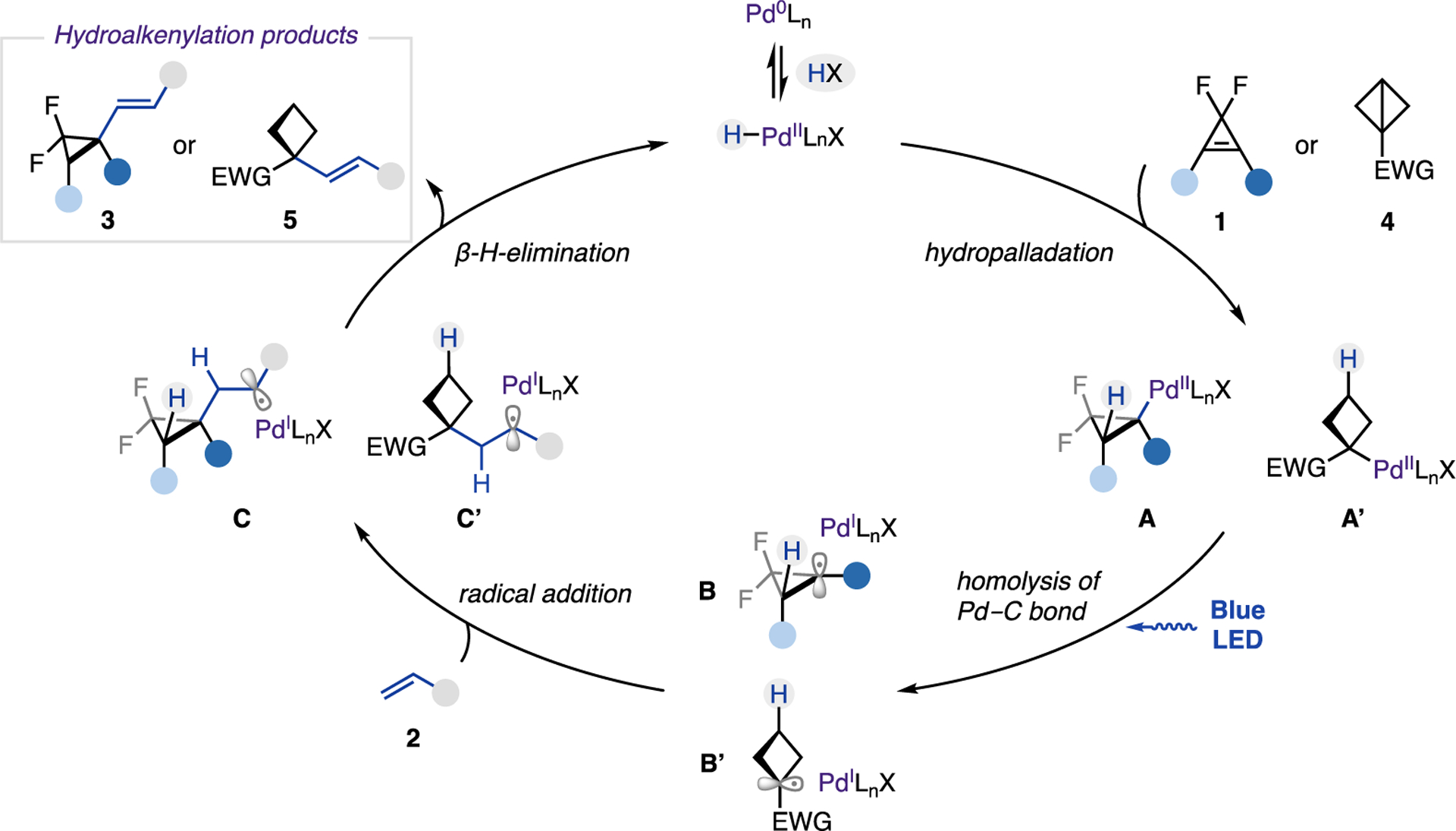
Proposed Mechanism
4. CONCLUSION
In summary, we developed the first light-induced Pd-catalyzed hydroalkenylation reaction of strained molecules, which allows for expedient synthesis of alkenylated cyclobutanes and cyclopropanes. Notably, this transformation highlights the merger of a traditional two-electron hydropalladation process and photoinduced hybrid Pd-radical chemistry, as intermediacy of both PdH and radical species were confirmed by mechanistic studies. This transformation demonstrates broad functional group tolerance and is amenable to late-stage functionalization of complex molecules. It is anticipated that this mild method would find broad applications in synthesis and would inspire development of new transformations.
Supplementary Material
ACKNOWLEDGMENTS
We thank the National Institutes of Health (GM120281), National Science Foundation (CHE-1955663), and Welch Foundation (Chair, AT-0041) for financial support.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c09045.
Additional experimental details, materials, methods, and characterization data for all new compounds. (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.2c09045
The authors declare no competing financial interest.
Contributor Information
Ziyan Zhang, Department of Chemistry and Biochemistry, The University of Texas at Dallas, Richardson, Texas 75080-3021, United States.
Vladimir Gevorgyan, Department of Chemistry and Biochemistry, The University of Texas at Dallas, Richardson, Texas 75080-3021, United States.
REFERENCES
- (1).(a) de Meijere A; Kozhushkov SI; Schill H Three-Membered-Ring-Based Molecular Architectures. Chem. Rev 2006, 106, 4926–4996. [DOI] [PubMed] [Google Scholar]; (b) Souillart L; Cramer N Catalytic C−C Bond Activations via Oxidative Addition to Transition Metals. Chem. Rev 2015, 115, 9410–9464. [DOI] [PubMed] [Google Scholar]; (c) Walczak MAA; Krainz T; Wipf P Ring-Strain-Enabled Reaction Discovery: New Heterocycles from Bicyclo[1.1.0]butanes. Acc. Chem. Res 2015, 48, 1149–1158. [DOI] [PubMed] [Google Scholar]; (d) Kanazawa J; Uchiyama M Recent Advances in the Synthetic Chemistry of Bicyclo[1.1.1]pentane. Synlett 2019, 30, 1–11. [Google Scholar]; (e) Turkowska J; Durka J; Gryko D Strain Release − an Old Tool for New Transformations. Chem. Commun 2020, 56, 5718–5734. [DOI] [PubMed] [Google Scholar]
- (2).(a) Zhang P; Zhuang R; Wang X; Liu H; Li J; Su X; Chen X; Zhang X Highly Efficient and Stable Strain-Release Radio-iodination for Thiol Chemoselective Bioconjugation. Bioconjugate Chem. 2018, 29, 467–472. [DOI] [PubMed] [Google Scholar]; (b) Tokunaga K; Sato M; Kuwata K; Miura C; Fuchida H; Matsunaga N; Koyanagi S; Ohdo S; Shindo N; Ojida A Bicyclobutane Carboxylic Amide as a Cysteine-Directed Strained Electrophile for Selective Targeting of Proteins. J. Am. Chem. Soc 2020, 142, 18522–18531. [DOI] [PubMed] [Google Scholar]
- (3).(a) Meanwell NA Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem 2011, 54, 2529–2591. [DOI] [PubMed] [Google Scholar]; (b) Yang Y; Tsien J; Hughes JME; Peters BK; Merchant RR; Qin T An Intramolecular Coupling Approach to Alkyl Bioisosteres for the Synthesis of Multisubstituted Bicycloalkyl Boronates. Nat. Chem 2021, 13, 950–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Boström J; Brown DG; Young RJ; Keserü GM Expanding the Medicinal Chemistry Synthetic Toolbox. Nat. Rev. Drug Discovery 2018, 17, 709–727. [DOI] [PubMed] [Google Scholar]; (b) Bauer MR; Di Fruscia P; Lucas SCC; Michaelides IN; Nelson JE; Storer RI; Whitehurst BC Put a Ring on it: Application of Small Aliphatic Rings in Medicinal Chemistry. RSC Med. Chem 2021, 12, 448–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Walczak MAA; Wipf P Rhodium(I)-Catalyzed Cycloisomerizations of Bicyclobutanes. J. Am. Chem. Soc 2008, 130, 6924–6925. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Panish R; Chintala SR; Boruta DT; Fang Y; Taylor MT; Fox JM Enantioselective Synthesis of Cyclobutanes via Sequential Rh-catalyzed Bicyclobutanation/Cu-catalyzed Homo-conjugate Addition. J. Am. Chem. Soc 2013, 135, 9283–9286. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gianatassio R; Lopchuk JM; Wang J; Pan C-M; Malins LR; Prieto L; Brandt TA; Collins MR; Gallego GM; Sach NW; Spangler JE; Zhu H; Zhu J; Baran PS Strain-Release Amination. Science 2016, 351, 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lopchuk JM; Fjelbye K; Kawamata Y; Malins LR; Pan C-M; Gianatassio R; Wang J; Prieto L; Bradow J; Brandt TA; Collins MR; Elleraas J; Ewanicki J; Farrell W; Fadeyi OO; Gallego GM; Mousseau JJ; Oliver R; Sach NW; Smith JK; Spangler JE; Zhu H; Zhu J; Baran PS Strain- Release Heteroatom Functionalization: Development, Scope, and Stereospecificity. J. Am. Chem. Soc 2017, 139, 3209–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Fawcett A; Biberger T; Aggarwal VK Carbopalladation of C−C σ-Bonds Enabled by Strained Boronate Complexes. Nat. Chem 2019, 11, 117–122. [DOI] [PubMed] [Google Scholar]; (f) Silvi M; Aggarwal VK Radical Addition to Strained σ-Bonds Enables the Stereocontrolled Synthesis of Cyclobutyl Boronic Esters. J. Am. Chem. Soc 2019, 141, 9511–9515. [DOI] [PubMed] [Google Scholar]; (g) Fawcett A; Murtaza A; Gregson CHU; Aggarwal VK Strain-Release-Driven Homologation of Boronic Esters: Application to the Modular Synthesis of Azetidines. J. Am. Chem. Soc 2019, 141, 4573–4578. [DOI] [PubMed] [Google Scholar]; (h) Ernouf G; Chirkin E; Rhyman L; Ramasami P; Cintrat J-C Photochemical Strain-Release- Driven Cyclobutylation of C(sp3)-Centered Radicals. Angew. Chem., Int. Ed 2020, 59, 2618–2622. [DOI] [PubMed] [Google Scholar]; (i) Ociepa M; Wierzba AJ; Turkowska J; Gryko D Polarity-Reversal Strategy for the Functionalization of Electrophilic Strained Molecules via Light-Driven Cobalt Catalysis. J. Am. Chem. Soc 2020, 142, 5355–5361. [DOI] [PubMed] [Google Scholar]; (j) Guo R; Chang Y-C; Herter L; Salome C; Braley SE; Fessard TC; Brown MK Strain-Release [2π + 2σ] Cycloadditions for the Synthesis of Bicyclo[2.1.1]hexanes Initiated by Energy Transfer. J. Am. Chem. Soc 2022, 144, 7988–7994. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Kleinmans R; Pinkert T; Dutta S; Paulisch TO; Keum H; Daniliuc CG; Glorius F Intermolecular [2π+2σ]-Photocycloaddition Enabled by Triplet Energy Transfer. Nature 2022, 605, 477–482. [DOI] [PubMed] [Google Scholar]; (l) Agasti S; Beltran F; Pye E; Kaltsoyannis N; Crisenza G; Procter D A Catalytic Alkene Insertion Approach to Bicyclo[2.1.1]hexane Bioisosteres. ChemRxiv 2022, DOI: 10.26434/chemrxiv-2022-v93kv, (accessed 2022-03-23). [DOI] [PubMed] [Google Scholar]
- (6).(a) Wiberg KB The Concept of Strain in Organic Chemistry. Angew. Chem., Int. Ed. Engl 1986, 25, 312–322. [Google Scholar]; (b) Khoury PR; Goddard JD; Tam W Ring Strain Energies: Substituted Rings, Norbornanes, Norbornenes and Norbornadienes. Tetrahedron 2004, 60, 8103–8112. [Google Scholar]; (c) Bach RD; Dmitrenko O Strain Energy of Small Ring Hydrocarbons. Influence of C−H Bond Dissociation Energies. J. Am. Chem. Soc 2004, 126, 4444–4452. [DOI] [PubMed] [Google Scholar]
- (7).(a) Rubin M; Rubina M; Gevorgyan V Recent Advances in Cyclopropene Chemistry. Synthesis 2006, 2006, 1221–1245. [Google Scholar]; (b) Rubin M; Rubina M; Gevorgyan V Transition Metal Chemistry of Cyclopropenes and Cyclopropanes. Chem. Rev 2007, 107, 3117–3179. [DOI] [PubMed] [Google Scholar]; (c) Vicente R Recent Progresses towards the Strengthening of Cyclopropene Chemistry. Synthesis 2016, 48, 2343–2360. [Google Scholar]; (d) Fumagalli G; Stanton S; Bower JF Recent Methodologies That Exploit C−C Single-Bond Cleavage of Strained Ring Systems by Transition Metal Complexes. Chem. Rev 2017, 117, 9404–9432. [DOI] [PubMed] [Google Scholar]; (e) Li P; Zhang X; Shi M Recent Developments in Cyclopropene Chemistry. Chem. Commun 2020, 56, 5457–5471. [DOI] [PubMed] [Google Scholar]; (f) Vicente R C−C Bond Cleavages of Cyclopropenes: Operating for Selective Ring-Opening Reactions. Chem. Rev 2021, 121, 162–226. [DOI] [PubMed] [Google Scholar]; (g) Dian L; Marek I Asymmetric Preparation of Polysubstituted Cyclopropanes Based on Direct Functionalization of Achiral Three-Membered Carbocycles. Chem. Rev 2018, 118, 8415–8434. [DOI] [PubMed] [Google Scholar]
- (8).Bräse S; Meijere AD Cross-Coupling of Organyl Halides with Alkenes: the Heck Reaction. Metal-Catalyzed Cross-Coupling Reactions 2004, 217–315. [Google Scholar]
- (9).(a) Chuentragool P; Kurandina D; Gevorgyan V Catalysis with Palladium Complexes Photoexcited by Visible Light. Angew. Chem., Int. Ed 2019, 58, 11586–11598. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kurandina D; Chuentragool P; Gevorgyan V Transition-Metal-Catalyzed Alkyl Heck-Type Reactions. Synthesis 2019, 51, 985–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kancherla R; Muralirajan K; Sagadevan A; Rueping M Visible Light-Induced Excited-State Transition-Metal Catalysis. Trends Chem. 2019, 1, 510–523. [Google Scholar]; (d) Cheng W-M; Shang R Transition Metal-Catalyzed Organic Reactions under Visible Light: Recent Developments and Future Perspectives. ACS Catal. 2020, 10, 9170–9196. [Google Scholar]; (e) Cheung KPS; Sarkar S; Gevorgyan V Visible Light-Induced Transition Metal Catalysis. Chem. Rev 2022, 122, 1543–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhang Z; Kvasovs N; Dubrovina A; Gevorgyan V Visible Light Induced Brønsted Acid Assisted Pd-Catalyzed Alkyl Heck Reaction of Diazo Compounds and N-Tosylhydrazones. Angew. Chem., Int. Ed 2022, 61, No. e202110924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).For examples of transition-metal-catalyzed approaches to access vinyl cyclopropanes, see:; (a) Müller DS; Werner V; Akyol S; Schmalz H-G; Marek I Tandem Hydroalumination/Cu-Catalyzed Asymmetric Vinyl Metalation as a New Access to Enantioenriched Vinylcyclopropane Derivatives. Org. Lett 2017, 19, 3970–3973. [DOI] [PubMed] [Google Scholar]; (b) Bruffaerts J; Pierrot D; Marek I Efficient and Stereodivergent Synthesis of Unsaturated Acyclic Fragments Bearing Contiguous Stereogenic Elements. Nat. Chem 2018, 10, 1164–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang H; Huang W; Wang T; Meng F Cobalt-Catalyzed Diastereo- and Enantioselective Hydroalkenylation of Cyclopropenes with Alkenylboronic Acids. Angew. Chem., Int. Ed 2019, 58, 11049–11053. [DOI] [PubMed] [Google Scholar]; (d) Cohen A; Chagneau J; Marek I Stereoselective Preparation of Distant Stereocenters (1,5) within Acyclic Molecules. ACS Catal. 2020, 10, 7154–7161. [Google Scholar]; (e) Ritchie NFC; Zahara AJ; Wilkerson-Hill SM Divergent Reactivity of α,α-Disubstituted Alkenyl Hydrazones: Bench Stable Cyclopropylcarbinyl Equivalents. J. Am. Chem. Soc 2022, 144, 2101–2106. [DOI] [PubMed] [Google Scholar]; (f) Jiang Z-T; Chen Z; Zeng Y; Shi J-L; Xia Y Enantioselective Formation of All- Carbon Quaternary Stereocenters in gem-Difluorinated Cyclopropanes via Rhodium-Catalyzed Stereoablative Kinetic Resolution. Org. Lett 2022, 24, 6176–6181. [DOI] [PubMed] [Google Scholar]
- (12).For synthesis of gem-difluorinated cyclopropanes, see:; (a) Wang F; Luo T; Hu J; Wang Y; Krishnan HS; Jog PV; Ganesh SK; Prakash GKS; Olah GA Synthesis of gem-Difluorinated Cyclopropanes and Cyclopropenes: Trifluoromethyl-trimethylsilane as a Difluorocarbene Source. Angew. Chem., Int. Ed 2011, 50, 7153–7157. [DOI] [PubMed] [Google Scholar]; (b) Li L; Wang F; Ni C; Hu J Synthesis of gem-Difluorocyclopropa (e)nes and O-, S-, N-, and P- Difluoromethylated Compounds with TMSCF2Br. Angew. Chem., Int. Ed 2013, 52, 12390–12394. [DOI] [PubMed] [Google Scholar]; For significance to incorporate fluoroalkyl groups into drug molecules, see:; (c) Müller K; Faeh C; Diederich F Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]; (d) Hagmann WK The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem 2008, 51, 4359–4369. [DOI] [PubMed] [Google Scholar]; (e) Purser S; Moore PR; Swallow S; Gouverneur V Fluorine in Medicinal Chemistry. Chem. Soc. Rev 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]; (f) Meanwell NA Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- (13).(a) Kurandina D; Parasram M; Gevorgyan V Visible Light-Induced Room-Temperature Heck Reaction of Functionalized Alkyl Halides with Vinyl Arenes/Heteroarenes. Angew. Chem., Int. Ed 2017, 56, 14212–14216. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kurandina D; Rivas M; Radzhabov M; Gevorgyan V Heck Reaction of Electronically Diverse Tertiary Alkyl Halides. Org. Lett 2018, 20, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chuentragool P; Yadagiri D; Morita T; Sarkar S; Parasram M; Wang Y; Gevorgyan V Aliphatic Radical Relay Heck Reaction at Unactivated C(sp3)-H Sites of Alcohols. Angew. Chem., Int. Ed 2019, 58, 1794–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cheung KPS; Kurandina D; Yata T; Gevorgyan V Photoinduced Palladium- Catalyzed Carbofunctionalization of Conjugated Dienes Proceeding via Radical-Polar Crossover Scenario: 1,2-Aminoalkylation and Beyond. J. Am. Chem. Soc 2020, 142, 9932–9937. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kvasovs N; Iziumchenko V; Palchykov V; Gevorgyan V Visible Light-Induced Pd-Catalyzed Alkyl- Heck Reaction of Oximes. ACS Catal. 2021, 11, 3749–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Jia X; Zhang Z; Gevorgyan V Three-Component Visible-Light-Induced Palladium- Catalyzed 1,2-Alkyl Carbamoylation/Cyanation of Alkenes. ACS Catal. 2021, 11, 13217–13222. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Kvasovs N; Gevorgyan V Accessing Illusive E Isomers of α-Ester Hydrazones via Visible-Light-Induced Pd-Catalyzed Heck-Type Alkylation. Org. Lett 2022, 24, 4176–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).See Supporting Information for details.
- (15).(a) Cheng W-M; Shang R; Fu Y Irradiation-induced Palladium-catalyzed Decarboxylative Desaturation Enabled by a Dual Ligand System. Nat. Commun 2018, 9, 5215. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhao B; Shang R; Wang GZ; Wang S; Chen H; Fu Y Palladium-Catalyzed Dual Ligand-Enabled Alkylation of Silyl Enol Ether and Enamide under Irradiation: Scope, Mechanism, and Theoretical Elucidation of Hybrid Alkyl Pd(I)-Radical Species. ACS Catal. 2020, 10, 1334–1343. [Google Scholar]
- (16).(a) Wang G-Z; Shang R; Cheng W-M; Fu Y Irradiation-Induced Heck Reaction of Unactivated Alkyl Halides at Room Temperature. J. Am. Chem. Soc 2017, 139, 18307–18312. [DOI] [PubMed] [Google Scholar]; (b) Wang G-Z; Shang R; Fu Y Irradiation-Induced Palladium-Catalyzed Decarboxylative Heck Reaction of Aliphatic N-(Acyloxy)phthalimides at Room Temperature. Org. Lett 2018, 20, 888–891. [DOI] [PubMed] [Google Scholar]; (c) Koy M; Sandfort F; Tlahuext-Aca A; Quach L; Daniliuc CG; Glorius F Palladium-Catalyzed Decarboxylative Heck-Type Coupling of Activated Aliphatic Carboxylic Acids Enabled by Visible Light. Chem.—Eur. J 2018, 24, 4552–4555. [DOI] [PubMed] [Google Scholar]; (d) Koy M; Bellotti P; Katzenburg F; Daniliuc CG; Glorius F Synthesis of All-Carbon Quaternary Centers by Palladium-Catalyzed Olefin Dicarbofunctionalization. Angew. Chem., Int. Ed 2020, 59, 2375–2379. [DOI] [PubMed] [Google Scholar]; (e) Lee GS; Kim D; Hong SH Pd-catalyzed Formal Mizoroki−Heck Coupling of Unactivated Alkyl Chlorides. Nat. Commun 2021, 12, 991. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Yao W; Zhao G; Wu Y; Zhou L; Mukherjee U; Liu P; Ngai M-Y Excited-State Palladium-Catalyzed Radical Migratory Mizoroki−Heck Reaction Enables C2-Alkenylation of Carbohydrates. J. Am. Chem. Soc 2022, 144, 3353–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).(a) Kudo K; Hidai M; Murayama T; Uchida Y A New Route to Hydrido-palladium Complexes: Oxidative Addition Reactions of Hydrogen Chloride to Palladium(0) Complexes. J. Chem. Soc. D 1970, 1701b−1702. [Google Scholar]; (b) Grushin VV Hydrido Complexes of Palladium. Chem. Rev 1996, 96, 2011–2034. [DOI] [PubMed] [Google Scholar]; (c) Hills ID; Fu GC Elucidating Reactivity Differences in Palladium-Catalyzed Coupling Processes: The Chemistry of Palladium Hydrides. J. Am. Chem. Soc 2004, 126, 13178–13179. [DOI] [PubMed] [Google Scholar]; (d) Li H; Dong K; Neumann H; Beller M Palladium-Catalyzed Hydroamidocarbonylation of Olefins to Imides. Angew. Chem., Int. Ed 2015, 54, 10239–10243. [DOI] [PubMed] [Google Scholar]
- (18).For regiochemistry of hydropalladation, see, for example:; (a) Trost BM; Sorum MT; Chan C; Rühter G Palladium-Catalyzed Additions of Terminal Alkynes to Acceptor Alkynes. J. Am. Chem. Soc 1997, 119, 698–708. [Google Scholar]; (b) Shimamoto T; Chimori M; Sogawa H; Yamamoto K Cationic Palladium-Catalyzed Hydro-silylative Cross-Coupling of Alkynes with Alkenes. J. Am. Chem. Soc 2005, 127, 16410–16411. [DOI] [PubMed] [Google Scholar]; (c) Jahier C; Zatolochnaya OV; Zvyagintsev NV; Ananikov VP; Gevorgyan V General and Selective Head-to-Head Dimerization of Terminal Alkynes Proceeding via Hydropalladation Pathway. Org. Lett 2012, 14, 2846–2849. [DOI] [PubMed] [Google Scholar]; (d) Zatolochnaya OV; Gordeev EG; Jahier C; Ananikov VP; Gevorgyan V Carboxylate Switch between Hydro- and Carbopalladation Pathways in Regiodivergent Dimerization of Alkynes. Chem.—Eur. J 2014, 20, 9578–9588. [DOI] [PubMed] [Google Scholar]; (e) Pradhan TR; Kim HW; Park JK Regiodivergent Synthesis of 1,3- and 1,4-Enynes through Kinetically Favored Hydropalladation and Ligand-Enforced Carbopalladation. Angew. Chem., Int. Ed 2018, 57, 9930–9935. [DOI] [PubMed] [Google Scholar]
- (19).(a) Wise DE; Gogarnoiu ES; Duke AD; Paolillo JM; Vacala TL; Hussain WA; Parasram M Photoinduced Oxygen Transfer Using Nitroarenes for the Anaerobic Cleavage of Alkenes. J. Am. Chem. Soc 2022, 144, 15437–15442. [DOI] [PubMed] [Google Scholar]; (b) Ruffoni A; Hampton C; Simonetti M; Leonori D Photoexcited Nitroarenes for the Oxidative Cleavage of Alkenes. Nature 2022, 610, 81. [DOI] [PubMed] [Google Scholar]
- (20).Karabal PU; Chouthaiwale PV; Shaikh TM; Suryavanshi G; Sudalai A NaIO4/LiBr-Mediated Aziridination of Olefins Using Chloramine-T. Tetrahedron Lett. 2010, 51, 6460–6462. [Google Scholar]
- (21).Isogai K; Nishizawa N; Saito T; Sakai J.-i. Catalytic Hydrogenolysis of 1,1-Difluoro-2-phenyl- and 1,1-Difluoro-3-methyl-2- phenylcyclopropane. Bull. Chem. Soc. Jpn 1983, 56, 1555–1556. [Google Scholar]
- (22).Ahmed E-AMA; Suliman AMY; Gong T-J; Fu Y Access to Divergent Fluorinated Enynes and Arenes via Palladium-Catalyzed Ring-Opening Alkynylation of gem-Difluorinated Cyclopropanes. Org. Lett 2020, 22, 1414–1419. [DOI] [PubMed] [Google Scholar]
- (23).Orr D; Percy JM; Harrison ZA A Computational Triage Approach to the Synthesis of Novel Difluorocyclopentenes and Fluorinated Cycloheptadienes Using Thermal Rearrangements. Chem. Sci 2016, 7, 6369–6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.