

Abstract
Background
Molecular characterization of late-onset Alzheimer’s disease (LOAD), the leading cause of age-related dementia, has revealed transcripts, proteins, and pathway alterations associated with disease. Assessing these postmortem signatures of LOAD in experimental model systems can further elucidate their relevance to disease origins and progression. Model organisms engineered with human genetic factors further link these signatures to disease-associated variants, especially when studies are designed to leverage homology across species. Here we assess differential gene splicing patterns in aging mouse models carrying humanized APOE4 and/or the Trem2*R47H variant on a C57BL/6J background. We performed a differential expression of gene (DEG) and differential splicing analyses on whole brain transcriptomes at multiple ages. To better understand the difference between differentially expressed and differentially spliced genes, we evaluated enrichment of KEGG pathways and cell-type specific gene signatures of the adult brain from each alteration type. To determine LOAD relevance, we compared differential splicing results from mouse models with multiple human AD splicing studies.
Results
We found that differentially expressed genes in Trem2*R47H mice were significantly enriched in multiple AD-related pathways, including immune response, osteoclast differentiation, and metabolism, whereas differentially spliced genes were enriched for neuronal related functions, including GABAergic synapse and glutamatergic synapse. These results were reinforced by the enrichment of microglial genes in DEGs and neuronal genes in differentially spliced genes in Trem2*R47H mice. We observed significant overlap between differentially spliced genes in Trem2*R47H mice and brains from human AD subjects. These effects were absent in APOE4 mice and suppressed in APOE4.Trem2*R47H double mutant mice relative to Trem2*R47H mice.
Conclusions
The cross-species observation that alternative splicing observed in LOAD are present in Trem2*R47H mouse models suggests a novel link between this candidate risk gene and molecular signatures of LOAD in neurons and demonstrates how deep molecular analysis of new genetic models links molecular disease outcomes to a human candidate gene.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-023-09280-x.
Keywords: Genetics, Alzheimer’s disease, Mouse models, Genomics, Splicing, Transcriptomics
Background
Late-onset Alzheimer’s disease (LOAD) is the leading cause of dementia but has uncertain etiology. Recent characterization of molecular changes associated with AD pathology from postmortem brain studies have identified abundant alterations in transcripts, proteins, and processes, including differential gene splicing. In a recent study, deep proteomic analysis of brain tissue identified alternations in abundance of RNA binding proteins, suggesting a potential role for aberrant RNA splicing in AD pathogenesis [1]. Transcriptome-wide analysis of aging prefrontal cortex of human subjects identified genetic variants and trans acting splicing factors involved in dysregulation of mRNA splicing affecting brain transcriptomes in Alzheimer’s disease [2]. In a second study, proteomic profiles of Alzheimer’s disease brains identified an increased aggregation of insoluble U1 snRNP, a small nuclear RNA (snRNA) component of the spliceosomal complex, suggesting that the core splicing machinery may be altered in Alzheimer’s disease [3]. With this growing list of disrupted splicing events in AD brains, it provides a great opportunity to investigate the factors that contribute to mis-splicing events, understand how they lead to disease and conceive ways to correct pathological splicing through targeted therapeutics, and use splice variations as robust biomarkers.
Animal models provide the experimental platforms to study disease-relevant molecular changes in vivo, including alternative splicing, and enable translation through pre-clinical testing [4, 5]. Mouse models have been developed to understand and treat splicing defects associated with many diseases including AD and tauopathies [6–8]. In a previous study, neuron-specific alternative splicing of amyloid precursor protein (APP) was identified in brain tissue of hAPP transgenic mice, suggesting a role of splicing defects of hAPP in the development of AD-type brain changes [9]. However, these studies have largely focused on alternative splicing of selected candidate genes, and investigations of global splicing events in LOAD mouse models are scarce. Using transcriptome-wide analyses of new mouse models, we recently found upregulation of the spliceosome pathway in an aging mouse model carrying the AD risk-associated R47H point mutation in the Trem2 gene [10]. This work motivated us to further investigate alternative splicing events in these mouse models and identify possible functional consequences that represent AD pathogenesis.
Trem2 (triggering receptor expressed on myeloid cells 2) is a receptor protein expressed in dendritic cells, macrophages, and microglia either on the plasma membrane, intracellularly, or as a soluble factor [11, 12]. Viewed primarily as an immune responsive factor, when expressed on the plasma membrane Trem2 binds to phospholipids, apolipoproteins, and DNA to activate pathways related to cytokine and chemokine production, motility, survival, and phagocytosis [13]. Downstream signaling leads to alterations in microglia activation states and neuroinflammation [14]. In the brain, amyloid beta has also been shown to bind to Trem2 on homeostatic microglia responsible for sensing and clearing the neural environment [15]. In the context of the central nervous system, mutations to Trem2 are associated with serious human disease. The R47H mutation in Trem2 is one of the strongest genetic risk factors of late-onset Alzheimer’s disease (LOAD) [16–19]. However, following CRISPR-mediated insertion of the R47H point mutation into humanized-Trem2 mice, we noticed a novel, cryptic splice acceptor site in Exon 2 that is not observed in humans. The resultant splice variant leads to a pronounced decrease in expression. Since, we have generated additional, humanized Trem2*R47H alleles that produce normal levels of transcript in the brains of mouse hosts. Characterization and comparison of these strains is ongoing which will provide insight into the consequences of this human disease risk factor on mouse health. It is well described in humans that increased inflammation follows with increasing age [20] and is associated with development of systemic and neurological pathologies. Thus, it is possible that a reduction in Trem2 expression is protective against AD progression via reduced neuroinflammation. However, microglia sensing, pruning, and clearing of apoptotic neurons is also highly dependent on Trem2 receptors and justifies more attention in the developmental context of AD [21].
Alternative splicing is an important post-transcriptional regulatory mechanism through which multiple mRNAs are produced from a single pre-mRNA molecule. Alternative splicing is a major contributor to cellular diversity in both normal tissues and disease. Alternative splicing affects over 95% of mammalian genes [22, 23], contributing significantly to the functional diversity of proteins expressed in tissues [24]. Tissue specific alternative splicing events tend to be associated with specific functions of the tissue. For instance, brain-specific alternative splicing events are often associated with neural-specific functions [24]. Alternative splicing is abundant in human nervous system tissues and contributes to phenotypic differences within and between individuals [25]. The mechanisms underlying altered splicing involve either disruption of cis-acting elements within the affected gene and/or trans-acting factors (e.g., RNA-binding proteins) that are required for normal splicing or splicing regulation such as the heterogeneous ribonucleoproteins (hnRNPs) [4, 26]. Splicing defects can cause disease, modify the severity of disease phenotypes, or correspond to disease susceptibility [4, 26]. There are many diseases that result from disrupted alternative splicing, such as amyotrophic lateral sclerosis (ALS), autism, and cancer [4, 27]. Disruptions in mRNA splicing are also associated with age-related disorders, such as frontotemporal lobar dementia (FTD) [28], Parkinson’s disease [29] and Alzheimer’s disease [9, 30].
Here, by applying multiple analytic methods, we investigated genome-wide splicing events in new LOAD mouse models created by knocking-in human APOE4 sequence and the R47H mutation in Trem2. We annotated the differentially spliced genes using functional and cell type enrichment analyses. Moreover, we compared the splicing events in the LOAD mouse models with splicing events reported in human AD studies [1–3]. We identified a number of differential splicing events in mouse models that mimic results from human AD studies, including differential isoform usage in the heterogeneous ribonucleoprotein M (hnRNPM). In order to assess the effects of isoform switch in hnRNPM, we investigated the binding sites of disrupted RNA-binding proteins on other differentially spliced genes in the LOAD mice. Overall, we identified numerous splicing alterations in our Trem2.R47H mouse model that were also observed in human AD cohorts, suggesting that the mechanisms of AD risk involve splice alterations in neurons.
Results
Differential gene expression and functional analyses of the brain transcriptomes identified age-dependent molecular changes associated with AD pathologies in the APOE4, Trem2*R47H and APOE4.Trem2*R47H mice on a C57BL/6J background [10]. Notably, in 12-month-old Trem2*R47H male mice, significantly increased expression was observed for genes associated with the spliceosome pathway, suggesting the possibility of differential splicing events in this mouse model [10]. This effect was not observed in double-mutant APOE4.Trem2*R47H mice. These findings motivated us to further investigate alternative splicing events in the same panel of mouse models that might correspond to AD pathogenesis in humans. Here we systematically investigate the splicing events in mouse models (Fig. 1; Supplementary Fig. 1).
Fig. 1.
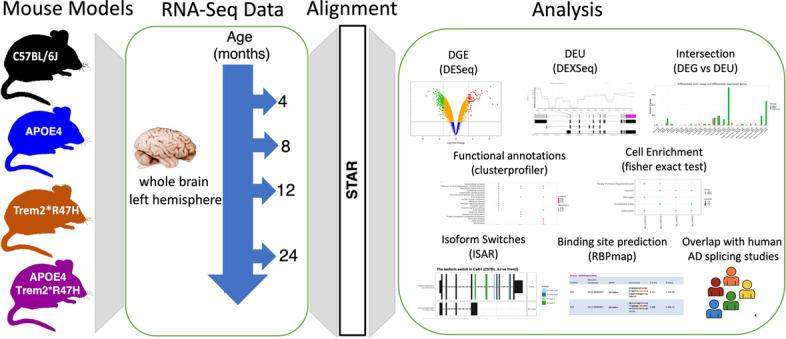
Schematic overview of the analysis. Bulk RNA-Seq data from mouse whole brain hemispheres were processed by our parallelized RNA-Seq analysis pipeline. We performed differential gene expression analysis, differential exon usage analysis, and differential isoform usage analysis. After performing differential analysis, we used the software clusterProfiler to perform functional enrichment analysis followed by cell type enrichment analysis. The RBPmap webserver was used to predict Binding sites of RNA-binding proteins on differentially spliced genes. In addition, we performed hypergeometric tests to identify enrichment of differentially spliced gene set in human Alzheimer’s cases
Differentially spliced genes were enriched in neuronal functions associated with AD
First, we applied DEXSeq [31] on processed transcriptomic data to identify genes with differential exon usages (DEU) to infer differential splicing events in the LOAD mouse models compared to age and sex-matched B6 controls. The greatest number of DEU genes were observed in Trem2*R47H mice at 12 months of age, which agrees with differential expression patterns and corresponds to the age at which spliceosome gene abundances were altered [10]. In this strain, we identified a total of 50 genes with significant DEU (padj < 0.1) at 4 months, 71 genes with significant DEU at 8 months (padj < 0.1), 480 genes with significant DEU (padj < 0.1) at 12 months, and 50 genes with significant DEU (padj < 0.1) at 24 months (Table 1; Supplementary Table 1). Functional analysis of these DEU genes returned multiple AD associated biological processes (Supplementary Table 2). Interestingly, DEU genes in 12-month-old Trem2*R47H male mice were enriched for neuron-specific process such as ‘GABAergic synapse’ and ‘glutamatergic synapse’ (Fig. 2; Supplementary Table 2), suggesting perturbation in microglial gene Trem2 leads to differential splicing in genes expressed in neurons.
Table 1.
Differentially exon usages genes in mouse models used in this study. Number of genes with differential exon usage in each mouse model compared to age and sex-matched B6 control mice. Number in parentheses shows the overlap between genes with DEU and differentially expressed genes (DEGs)
| MOUSE MODELS | AGE (in months) | Genes with differential exon usage (DEU) | |
|---|---|---|---|
| MALE | FEMALE | ||
| APOE4 | 4 | 40 (0) | 22 (0) |
| 8 | 69 (3) | 49 (1) | |
| 12 | 4 (1) | 6 (1) | |
| 24 | 12 (0) | 12 (0) | |
| Trem2*R47H | 4 | 9 (0) | 41 (0) |
| 8 | 35 (0) | 36 (0) | |
| 12 | 207 (8) | 273 (9) | |
| 24 | 23 (0) | 27 (2) | |
| APOE4.Trem2*R47H | 4 | 77 (0) | 11 (0) |
| 8 | 46 (0) | 13(0) | |
| 12 | 48 (1) | 56 (1) | |
| 24 | 20 (0) | 31 (2) | |
Fig. 2.

Functional Analysis of Differentially Exon Usages (DEU) genes. Significantly enriched KEGG pathways (p < 0.05) in the set of DEU genes across all mouse models using enrichKEGG function build under clusterprofiler R package (mo. indicates months)
Relative to Trem2*R47H brains, DEU was lower in both APOE4 and APOE4.Trem2*R47H mice. In humanized APOE4 mice, we identified a total of 62 genes with significant DEU (padj < 0.1) at 4 months, 118 genes with significant DEU (padj < 0.1) at 8 months, 10 genes with significant DEU (padj < 0.1) at 12 months, and 24 genes with significant DEU (padj < 0.1) at 24 months (Table 1; Supplementary Table 1). Functional analysis showed enrichment for DEU genes involved in ‘synaptic vesicle cycle’, and ‘mTOR signaling’ related pathways in 24-month-old APOE4 female mice (Fig. 2; Supplementary Table 2). In APOE4.Trem2*R47H mice, we observed a total of 89 genes with significant DEU (padj < 0.1) at 4 months, 58 genes with significant DEU (padj < 0.1) at 8 months, 104 genes with significant DEU (padj < 0.1) at 12 months, and 51 genes with significant DEU (padj < 0.1) in 24-month-old mice (Table 1; Supplementary Table 1). Functional analysis showed enrichment for DEU genes involved in ‘endocytosis’, and ‘protein processing in ER’ related pathways in 8 months old APOE4.Trem2*R47H mice, respectively (Fig. 2; Supplementary Table 2). The reduction of DEU in APOE4.Trem2*R47H mice relative to Trem2*R47H mice recapitulates the DEG patterns previously observed [10] and further supports a suppression of mid-life Trem2*R47H mutation effects by the presence of humanized APOE4.
Next, we performed a comparative analysis between differentially spliced genes and differentially expressed genes across all mouse models. We did not observe any significant overlap between sets of DEU genes and differentially expressed genes (DEGs) for any mouse models (Fig. 3A; Supplementary Table 3). Furthermore, in 12 months old Trem2*R47H male mice, differentially expressed genes were enriched for RNA splicing related functions [10], whereas DEU genes were enriched for neuronal related functions (Fig. 3B; Supplementary Table 2). To better understand the difference between DEGs and DEU genes, we evaluated enrichment of cell-type specific genes [32] in these gene sets for each mouse model. We identified that DEU genes in Trem2*R47H and APOE4.Trem2*R47H male mice were enriched in neuronal signatures, while DEGs were enriched in the microglial signatures (Fig. 3C). In APOE4 male mice, DEU genes were enriched in the neuronal signatures, while DEGs were enriched for the astrocytes and the myelinating oligodendrocytes signatures. DEU genes in the APOE4 female mice were enriched for microglial cell type signatures, while DEGs were enriched in the endothelial cells (Fig. 3C). Overall, we identified more alternatively spliced genes in 12-month-old Trem2*R47H mice compared to other LOAD mouse models, which were further enriched for neuronal associated functions.
Fig. 3.

Differential splicing analysis using DEXSeq. A Number of genes with differential exon usage (DEU) and differential expressed genes (DEG) in each mouse model. Common infers gene overlap between DEG and DEU genes. B Significantly enriched KEGG pathways in the set of differential exon usage (DEU) and differentially expressed genes (DEG) in the 12 months old Trem2*R47H mice (p < 0.05). C Cell type enrichment analysis in the gene set with differential exon usage (DEU) and differentially expressed genes (DEG) in each mouse model using Fisher exact test. Significant cell type enrichment (p < 0.1) are shown by solid dots
Identification of alternative splicing events in Trem2 knockout model suggest possible role of Trem2 gene in disrupting AD through alternative splicing mechanism
The Trem2*R47H mouse model used in this study exhibits both expression of the R47H mutation and expression levels reduced by approximately 50% [10]. To investigate whether differential splicing events are due to altered expression of Trem2 gene or specific to R47H mutation in Trem2 gene, we performed differential splicing analysis using DEXSeq on processed transcriptomic data from cortex and hippocampus brain regions of Trem2 Knockout (KO) and respective WT mice from a recent study [33]. Since this study assessed only male mice, we compared all results from Trem2 KO mice with Trem2.R47H male mice.
Interestingly, we identified more DEU genes in Trem2 KO mice compared to Trem2*R47H mice, even at younger ages (Table 2; Supplementary Table 4). Next, we computed the intersection between DEU genes in Trem2 KO mice and 12-month-old Trem2.R47H mice and found significant gene overlaps (p < 0.05, hypergeometric test). Combined across both tissues, 56 DEU genes were common between 4-month-old Trem2 KO and 12-month-old Trem2.R47H, and 55 DEU genes were common between 8-month-old Trem2 KO and 12-month-old Trem2.R47H (Table 2). We did not observe a significant overlap between DEU genes in 4-month-old Trem2*R47H mice and Trem2 KO mice (Table 2), suggesting perturbations in splicing start earlier in mice with Trem2 deletions compared to mice carrying the R47H mutation in Trem2.
Table 2.
Differentially spliced genes in Trem2 KO mice. Number of genes showing differential exon usage in Trem2 KO male mice compared to respective B6 control mice in Cortex and hippocampus brain regions. Overlap between DEU genes in Trem2 KO male mice and Trem2*R47H male mice at 4, 8, 12, and 24 months. Significance of overlaps were computed using hypergeometric test
| Brain Tissue | Age (in months) | DEU Genes in Trem2.KO mice | Overlapped with DEU genes in Trem2.R47H mice | |||
|---|---|---|---|---|---|---|
| 12 months Trem2.R47H | 4 mo. Trem2.R47H | 8 mo. Trem2.R47H | 24 mo. Trem2.R47H | |||
| Cortex | 4 | 734 | 38 (p = 4.29E-13) | 1 (p = 0.36) | 5 (p = 0.02) | 2 (p = 0.28) |
| 8 | 874 | 34 (p = 1.51E-08) | 2 (p = 0.09) | 3 (p = 0.30) | 1 (p = 0.72) | |
| Hippocampus | 4 | 782 | 26 (p = 1.50E-05) | 1 (p = 0.37) | 4 (p = 0.09) | 4 (p = 0.02) |
| 8 | 723 | 30 (p = 4.29E-13) | 2 (p = 0.07) | 2 (p = 0.48) | 1 (p = 0.65) | |
Similar to Trem2.R47H mice, we did not observe any significant overlap between DEGs and DEU genes in Trem2 KO mice (Supplementary Fig. 2A). Functional analyses revealed enrichment of ‘synaptic vesicle cycle’ and ‘protein processing in ER’ pathways in DEU genes in hippocampus brain region of 8 months old Trem2 KO mice (Supplementary Fig. 2B; Supplementary Table 2). Further, DEGs are mostly enriched for microglial and astrocytes signatures, while DEU genes are enriched for neuronal and oligodendrocytes and astrocytes cell signatures in Trem2 KO mice (Supplementary Fig. 2C). All together, these results suggest that differential splicing is due to reduced expression of Trem2 gene possibly combined with a loss-of-function from the Trem2.R47H variant, as we observed similar pattern of splicing events (enriched in neuronal signatures) in Trem2 KO mice models as Trem2.R47H.
Differential spliced genes in human AD cases significantly overlapped with differentially spliced genes in the Trem2.R47H mice
We next compared our findings with results from multiple studies of alternative splicing events in human Alzheimer’s disease populations [1–3]. In these studies, different approaches were employed to assess aberrant mRNA splicing events in distinct human AD cohorts. We computed overlaps between gene sets identified as differentially spliced in LOAD mouse models and differentially spliced genes reported in human AD cases [1–3].
In the first study, Towfique et al. identified 84 genes with alternative splicing events related to AD by measuring differential intron usages in human AD cases compared to controls [2]. We identified 67 genes out of the 84 with mouse orthologs (Methods). Cell enrichment analysis identified that these 67 genes were significantly enriched for newly formed oligodendrocytes cell signatures (p < 0.1) (Supplementary Fig. 3). Next, we found that 12 out of the 67 genes were identified as differentially spliced (genes with DEU) across all our mouse models, representing a significant overlap (hypergeometric p = 0.004). Notably, 8 of these 12 genes (ENO2, LUC7I3, KIF21A, MAP4K4, PPP3CA, MACF1, NDRG4, and PRRC2C) were differentially spliced in 12-month-old Trem2*R47H mice, which was highly significant (hypergeometric p = 2.78 × 10–6) (Table 3). We did not observe any significant overlap with any other mouse models.
Table 3.
Summary of overlap between differentially spliced genes identified in multiple human AD splicing studies and mouse models. Number of differentially spliced genes identified in splicing analyses of human AD cohort in multiple studies. Significant overlap with differentially exon usage genes in mouse models were computed using hypergeometric test (p < 0.05). Control-AsymAD represents comparison between asymptomatic AD cases and controls. AsymAD-AD represents comparison between AD cases and asymptomatic AD cases. Control-AD represents comparison between AD cases and controls
| Differentially Spliced Genes |
Overlap with 12 mo. Trem2 male mice (207 DEU) |
Overlap across all mouse models (1167 DEU) |
|
|---|---|---|---|
| ROSMAP | 67 | 8 (p = 2.78E-06) | 12 (p = 0.004) |
|
TMT Proteomic Study (Control-AsymAD) |
155 | 6 (p = 0.017) | 27 (p = 0.001) |
| TMT Proteomic Study (AsymAD-AD) | 291 | 10 (p = 0.005) | 34 (p = 0.10) |
|
TMT Proteomic Study (Control-AD) |
237 | 5 (p = 0.20) | 26 (p = 0.18) |
| Emory/Kentucky | 536 | 13 (p = 0.03) | 55 (p = 0.01) |
In the second study, Johnson et al. investigated RNA splicing events by predicting alternative exon-exon junction splicing events across all subjects (symptomatic AD (n = 20), asymptomatic AD (n = 14), and control (n = 13) cases) [1]. For comparison, we extracted mouse orthologs for 237 proteins identified with differential alternative exon-exon junction in human symptomatic AD cases compared to controls (Control-AD); 155 proteins with differential alternative exon-exon junction in human asymptomatic AD cases compared to controls (Control-AsymAD); and 291 proteins with differential alternative exon-exon junction in human symptomatic AD cases compared to asymptomatic AD cases (AsymAD-AD) (Table 3). Cell enrichment analysis identified that these genes were enriched in oligodendrocytes (p < 0.05) (Supplementary Fig. 3). We found that 10 DEU genes (ENO2, KIF21A, ATL3, GLS, STXBP1, PPP3CA, MACF1, MAP1B, DPP6, and SMS) in 12-month-old Trem2*R47H mice overlapped with differentially spliced genes identified in human AD cases compared to AsymAD cases (hypergeometric p = 0.005) (Table 3). Similarly, 5 genes (EFTUD2, DNM1L, STXBP1, DPP6, and SMS) with DEU in 12-month-old Trem2*R47H mice overlapped (hypergeometric p = 0.2) with differentially spliced genes in human AD cases compared to controls; and 6 genes (EFTUD2, KIF21A, DNM1L, MACF1, IST1, and PPP1R9B) with DEU in 12 months old Trem2*R47H mice overlapped (hypergeometric p = 0.02) with differential spliced gene sets in human asymptomatic AD cases compared to controls (Table 3). We identified more DEU genes in 12-month-old Trem2*R47H mice overlapped with human AsymAD-AD group compared to Control-AsymAD group, which could be due to the fact that more differential splicing events were identified in human symptomatic AD cases compared to asymptomatic AD cases. Two genes EFTUD2 and DNM1L were differentially spliced in both asymptomatic and symptomatic AD cases compared to controls, while two genes DPP6 and SMS were differentially spliced in symptomatic AD cases compared to both asymptomatic AD cases and controls.
In the third study, Bai et al. investigated proteomic profiles of Alzheimer’s disease brains and identified accumulation of 36 proteins including insoluble U1-70 K and other small nuclear ribonucleoprotein spliceosome components, suggesting a possible loss of nuclear spliceosome activity in Alzheimer’s disease [3]. In this study 631 genes were identified with differential splicing deficiency scores in AD cases. We identified 536 genes out of the 631 with mouse orthologs (Methods). Further, cell enrichment analysis identified enrichment of neuronal cell signatures in these 536 genes with differential splicing efficiency in human AD cases (p < 0.05) (Supplementary Fig. 3). A total of 55 genes out of 536 genes with differential splicing efficiency in human AD cases were also identified as DEU genes across all mouse models (p = 0.01) (Table 3). Thirteen of these 55 genes overlapped with DEU genes in 12-month-old Trem2*R47H male (p = 0.03) and 14 genes with Trem2*R47H female mice (p = 0.07) (Table 3). Next, 4 of these 55 genes (GPR107, EFTUD2, MAN1C1, and GRSF1) overlapped (p = 0.03) with DEU genes in 8 months old Trem2*R47H male mice. We did not observe any significant overlap with any other mouse models.
In summary, two genes (ENO2 and PPP3CA) that were identified as DEU in 12-month-old Trem2*R47H male mice were identified as differentially spliced in all three human studies [1–3], while STXBP1 and EFTUD2 were identified as differentially spliced in Johnson et al. and Bai et al. [1, 3]. Moreover, two genes (KIF21A and MACF1) that overlapped among 12-month-old Trem2*R47H male mice, human Control-AsymAD, and AsymAD-AD group were also identified as differentially spliced in prefrontal cortex of AD cases by Towfique et al. [2]. MACF1 is a F-actin binding protein, and suggested to play role in AD pathogenesis as it is critical for neuron migration, neurite formation, synaptic function, and hippocampus-dependent learning and memory [34]. KIF21A is a family member of KIF21B and has similar amino acid composition. Kif21A is expressed throughout neurons, while KIF21B is mainly expressed in dendrites [35, 36]. Both KIF21A and KIF21B are linked to several human diseases. Enhanced KIF21B expression has been associated with severe AD pathology [35], while KIF21A is linked to congenital fibrosis of the extraocular muscles type 1 (CFEOM1), a disease characterized by absence of motor neurons of the midbrain [37]. Together, this suggests that aberrant splicing in KIF21A may disrupt neuronal functions associated with AD.
Differential isoform usage analyses identified isoform switch in RNA binding proteins associated with splicing mechanism
We identified multiple genes with differential exon usages, specifically in 12-month-old Trem2*R47H mice, using DEXSeq [31]. This approach pinpoints the location of the difference by focusing on specific exons, but it is also informative to determine the resulting transcripts with altered abundances. Therefore, we have implemented IsoformSwitchAnalyzeR (ISAR) to identify isoform switches (reduced abundance of one isoform and increased abundance of another isoform of a given gene) and predict potential functional consequences of the identified isoform switches such as loss/gain of protein domains, signal peptides, and intrinsically disordered regions [38].
We identified many genes with differential isoform switches across LOAD mouse models (Table 4). We observed the greatest number of differential isoform switches in 12-month-old Trem2*R47H male (256 isoform switches across 215 genes) and 24-month-old Trem2*R47H female mice (267 isoform switches across 218 genes) (Table 4; Supplementary Table 5). Hereafter, we refer to genes with at least one differential isoform switch as genes with differential transcript usages (DTU). In order to identify if genes with differential isoform usage are with differential used exons, we computed overlap between DTU and DEU genes and found only 6 genes (Ppp1r9b, Celf4, Fars2,Gabra1, Safb, and Pgbd5) common between both gene sets for 12-month-old Trem2*R47H male mice, and 4 genes (Morf4l2, Mcoln1, Atp2a2, and Usp18) common between both gene sets for 12-month-old Trem2*R47H female mice (Table 5). For other mouse models either none or only one gene were common between DTU and DEU genes. We identified few genes with differential isoform usages that also has differentially used exons, suggesting either changes in single exons may be compensated by changes in other exons for a given isoform, or isoform usage might be driven by sub-significant changes in multiple exons. Hence, we further investigated genes with differential isoform switches in order to identify more signal associated with alternative splicing that might be missed by DEU analyses.
Table 4.
Differentially isoform usages genes identified in mouse models using IsoformSwitchAnalyzerR (ISAR). Number of genes showing differential isoform (or transcript) usage in each mouse model compared to age and sex-matched B6 control mice
| Mouse Models | Age | Sex | All Isoform with differential usage | ||
|---|---|---|---|---|---|
| nrIsoforms | nrSwitches | nrGenes | |||
| APOE4 | 4 | Male | 66 | 81 | 58 |
| 4 | Female | 77 | 88 | 63 | |
| 8 | Male | 54 | 66 | 49 | |
| 8 | Female | 39 | 51 | 35 | |
| 12 | Male | 62 | 86 | 57 | |
| 12 | Female | 45 | 65 | 40 | |
| 24 | Male | 28 | 34 | 24 | |
| 24 | Female | 36 | 39 | 27 | |
| Trem2*R47H | 4 | Male | 54 | 60 | 47 |
| 4 | Female | 36 | 41 | 31 | |
| 8 | Male | 54 | 65 | 45 | |
| 8 | Female | 56 | 61 | 50 | |
| 12 | Male | 273 | 256 | 215 | |
| 12 | Female | 94 | 91 | 77 | |
| 24 | Male | 21 | 24 | 17 | |
| 24 | Female | 276 | 267 | 218 | |
| APOE4.Trem2*R47H | 4 | Male | 88 | 106 | 77 |
| 4 | Female | 39 | 46 | 31 | |
| 8 | Male | 62 | 72 | 55 | |
| 8 | Female | 46 | 50 | 39 | |
| 12 | Male | 41 | 49 | 34 | |
| 12 | Female | 78 | 99 | 72 | |
| 24 | Male | 18 | 23 | 15 | |
| 24 | Female | 41 | 44 | 34 | |
Table 5.
Overlap between DEGs, DTU, and DEU genes in Trem2*R47H mice models. Number of genes showing differential expression (DEGs), differential exon usage (DEUs), and differential isoform usage (DTUs) in Trem2*R47H mice at 12 and 24 months compared to age and sex-matched B6 control mice and genes overlapped between three groups
|
Differentially expressed genes (DEGs) |
Differentially transcript usage genes (gDTU) |
Differentially exon usage genes (gDEU) |
Overlap between DEGs and gDTU | Overlap between gDTU and gDEU | |
|---|---|---|---|---|---|
| 12-month-old Trem2 male mice | 206 | 215 | 207 | 7 | 6 |
| 12-month-old Trem2 female mice | 285 | 77 | 274 | 3 | 4 |
| 24-month-old Trem2 male mice | 144 | 17 | 24 | 0 | 0 |
| 24-month-old Trem2 female mice | 748 | 218 | 28 | 27 | 0 |
First, we computed the intersection between DTU genes and differentially expressed genes (DEGs) across all mouse models. As with DEU genes, we did not observe any significant overlap between DTU genes and DEGs for any mouse model (Fig. 4A; Supplementary Table 3). Further, cell enrichment analysis determined that DTU genes in 12-month-old Trem2*R47H mice were not significantly enriched for neuronal signatures like DEU genes (Fig. 4B). However, functional analyses identified enrichment of splicing and neuronal related biological process in DTU genes in 12-month-old Trem2*R47H male mice (Fig. 4C; Supplementary Table 6), while DTU genes in 12-month-old Trem2*R47H female mice were enriched for protein folding and catabolic associated processes (Supplementary Table 6).
Fig. 4.

Differential splicing analysis using IsoformSwitchAnalyzerR (ISAR). A Number of genes with differential transcript usage (DTU) and differential expressed genes in each mouse model. Common infers gene overlap between DEG and DTU genes. B Significantly enriched biological process in the set of differential transcript usage (DTU) in the 12 months old Trem2*R47H mice (p < 0.05). C Cell type enrichments in the gene set with differential exon usage (DEU) and differentially expressed genes (DEG) in each mouse model using Fisher exact test. Significant cell type enrichment (p < 0.1) are shown by solid dots
To compare with differentially spliced genes reported in human AD cohorts [1–3], we found that DTU genes in 12-month-old Trem2*R47H male mice showed significant overlap with differentially spliced genes reported in Johnson et al. and Bai et al. [1, 3]. A total of 7 genes out of 215 DTU genes in 12-month-old Trem2*R47H male mice overlapped (hypergeometric p = 0.004) with differentially spliced genes in human asymptomatic AD cases compared to controls (PPP1R9B, TMED4, HYOU1, NT5DC1, PFN2, CCDC91, and PSMD2), symptomatic AD cases compared to controls (EIF5B, HNRNPM, CDK5, NPEPPS, KIF3A, MRPS7, and CANX), and symptomatic AD cases compared to asymptomatic AD cases (YWHAE, SPTBN1, CDK5, MGLL, NT5DC1, PFN2, and MAGI2) [1] (Table 6). Further, 15 genes (SUB1, TCF25, NDUFV1, R3HDM2, CALM2, CALB1, PUM2, PFN2, ACTN2, ARHGEF3, SLMAP, CELF4, MED28, FARS2, and PSMD2) were found common (hypergeometric p = 0.003) between DTU genes in 12-month-old Trem2*R47H male mice and differentially spliced genes reported in Bai et al. [3] (Table 6). On the other hand, we found only one gene (Clu) common between DTU genes in 12-month-old Trem2*R47H male mice and differentially spliced genes reported in Towfique et al. [2] (Table 6).
Table 6.
Summary of differentially spliced genes identified in multiple human AD splicing studies and overlap with mouse models. Number of differentially spliced genes identified in splicing analyses of human AD cohort in multiple studies and overlap with differentially spliced genes in mouse models were identified using differential isoform usage approach (IsoformSwitchAnalyzerR). Control-AsymAD represents comparison between asymptomatic AD cases and controls. AsymAD-AD represents comparison between AD cases and asymptomatic AD cases. Control-AD represents comparison between AD cases and controls
| Differentially Spliced Genes |
Overlap with 12 mo. Trem2 male mice (215 DTU) |
Overlap across all mouse models (1410 DTU) |
|
|---|---|---|---|
| ROSMAP | 67 | 1 (p = 0.58) | 4 (p = 0.83) |
|
TMT Proteomic Study (Control-AsymAD) |
155 | 7 (p = 0.004) | 16 (p = 0.23) |
| TMT Proteomic Study (Asym-AD) | 291 | 7 (p = 0.08) | 16 (p = 0.97) |
|
TMT Proteomic Study (Control-AD) |
237 | 7 (p = 0.03) | 17 (p = 0.79) |
| Emory/Kentucky | 536 | 15 (p = 0.003) | 44 (p = 0.6) |
Next, we investigated DTU genes enriched for splicing associated functions (Fig. 4C; Supplementary Table 2) and identified five of these genes (Hnrnpm, Celf5, Snrpb2, Bcl2l2, and Rbm39) with RNA binding domains; i.e. these genes encode for RNA-binding proteins (RBPs). Interestingly, the HNRNPM protein was also found to be differentially spliced in human AD cases compared to controls as reported in Johnson et al. [1]. In 12-month-old Trem2*R47H male mice, we did not observe a significant increase (p > 0.05) in overall expression of the Hnrnpm gene, but ISAR identified significantly increased usage of the non-coding isoform of Hnrnpm gene (isoform that do not contain RNA recognition motif (RRM)) and decreased usage of primary isoforms with RRM in Hnrnpm gene (Supplementary Fig. 4). RRM is the most abundant RNA binding domain [39] and is required for mRNA processing and splicing, suggesting a possible loss of nuclear spliceosome activity of HNRNPM protein due to differential transcript usage.
Differentially spliced genes are predicted to have multiple binding sites of disrupted RNA binding proteins
Disruption of RNA binding proteins can potentially affect the splicing of their targets. To test this, we first investigated the binding sites of RNA binding protein RBM25 (encoded by significantly differentially expressed Rbm25 gene in 12-month-old Trem2*R47H male mice that are associated with the spliceosome pathway) on differentially spliced genes identified by our approaches and reported in at-least one human AD splicing study. RBM25 is U1 small nuclear ribonucleoprotein with BAD domains and has been suggested to have increased aggregation in Alzheimer’s disease [40]. We identified binding sites of RBM25 protein on differentially used exons of multiple DEU genes such as PPP1R9B, CELF4, FARS2, MACF1, ENO2, STXBP1 (Supplementary Table 7), and on multiple exonic regions of all DTU genes (Supplementary Table 8). Identification of binding sites of RBM25 in these differentially spliced genes suggests that significant increased expression of RBM25 protein in 12-month-old Trem2*R47H male mice may lead to the differential splicing of genes identified in our study. We could not investigate binding sites of other differentially expressed genes (Acin1, Rp9, Snrnp27 and Prpf38b) enriched for spliceosomes pathways in 12-month-old Trem2*R47H male mice, as they were not present in the experimentally verified RBPmap database.
Further, ISAR identified the differential splicing of another type of RNA binding protein HNRNPM. RNA abundances of hnRNPs are altered in many types of cancer and neurodegenerative diseases, such as spinal muscular atrophy (SMA), amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), and frontotemporal lobe dementia (FTLD) [39]. hnRNP M proteins are an abundant group of hnRNPs that have been shown to bind avidly to poly(G) and poly(U) RNA homopolymers [41]. hnRNP M family members can induce exon skipping and promote exon inclusion, suggesting that the proteins may broadly contribute to the fidelity of splice site recognition and alternative splicing regulation [42]. Neurons are non-dividing cells and therefore need a tight regulation of mRNA homeostasis, so they are highly vulnerable to dysfunction of RNA-binding proteins (RBPs), including the hnRNPs [39]. Therefore, we hypothesized that RNA splicing factors, such as hnRNPM might be disrupted in Trem2*R47H mice due to increased usage of non-coding isoforms, which further results in splicing defects in other neuronal cell genes regulated by hnRNP M proteins.
To test this hypothesis, we investigated binding sites of differentially spliced RNA-binding proteins HNRNPM and CELF5 on differentially spliced genes (genes with differentially used exons and/or differentially transcript usages) in Trem2*R47H, as these two regulatory proteins were present in the RBPmap webserver [43]. For this study, we primarily focused on genes that are either identified as differentially spliced using both DEXSeq and ISAR tools and reported in at-least one human AD splicing studies (Ppp1r9b, Celf4, and Fars2) or reported in multiple human AD splicing studies and exhibit either differentially used exons or differential transcript usages (MACF1, PPP3CA, ENO2, KIF21A, EFTUD2, STXBP1, PFN2, and PSMD2) (Table 7). We assess the binding sites of the HNRNPM protein on all exons (input region included 10 bp before exon starts and 10 bp exon end position) of these selected genes.
Table 7.
Mouse genes identified either in both approaches/multiple human AD splicing studies: Genes that are either identified as differentially spliced using both DEXSeq and ISAR tools and further reported to be differentially spliced in at-least one human AD splicing study or reported to be differentially spliced in multiple human AD splicing studies and also identified by any one of these tools in 12 months old Trem2*R47H male mice
| Splicing Analysis Tools | Human AD splicing Studies | |||||
|---|---|---|---|---|---|---|
| SYMBOL | ENSEMBL | DEXSeq | ISAR | ROSMAP | TMT-Proteomics | Emory/Kentucky |
| Ppp1r9b | ENSMUSG00000038976 | Y | Y | - | Y | - |
| Celf4 | ENSMUSG00000024268 | Y | Y | - | - | Y |
| Fars2 | ENSMUSG00000021420 | Y | Y | - | - | Y |
| Macf1 | ENSMUSG00000028649 | Y | N | Y | Y | - |
| Ppp3ca | ENSMUSG00000028161 | Y | N | Y | Y | Y |
| Eno2 | ENSMUSG00000004267 | Y | N | Y | Y | Y |
| Stxbp1 | ENSMUSG00000026797 | Y | N | - | Y | Y |
| Kif21a | ENSMUSG00000022629 | Y | N | Y | Y | - |
| Eiftud2 | ENSMUSG00000020929 | Y | N | - | Y | Y |
| Pfn2 | ENSMUSG00000027805 | N | Y | - | Y | Y |
| Psmd2 | ENSMUSG00000006998 | N | Y | - | Y | Y |
First, we examined the binding sites of HNRNPM on differentially used exons of DEU genes from the list (PPP1R9B, CELF4, FARS2, MACF1, PPP3CA, ENO2, KIF21A, EFTUD2, and STXBP1) and found multiple binding sites on differentially used exon in Ppp1r9b and Fars2 gene located at 3’end of gene (Supplementary Table 7). No binding sites of the HNRNPM protein were found in differentially used exon of rest of the selected genes. Next, we assess the binding sites on all the exons of DTU genes (PPP1R9B, CELF4, FARS2, PFN2, and PSMD2) from the list, and identified binding sites of HNRNPM protein on multiple exons of these DTU genes (Supplementary Table 8). We extended our analysis on all other DTU and DEU genes that were also reported differentially spliced in human AD studies and found binding sites of HNRNPM protein on all DTU genes except the Clu gene, and 10 other DEU genes (Supplementary Tables 7 and 8). Similarly, we investigated binding site of CELF5 and identified binding sites on multiple differentially spliced genes (Supplementary Tables 7 and 8). All together, these results suggest that disruption of RNA binding proteins may lead to differential splicing of other target genes.
Discussion and conclusions
In this study, we investigated alternative gene splicing events in aging brain transcriptomes of LOAD mouse models in both sexes using state-of-the-art analytic methods. Notably, we observed more alternative gene splicing events in middle-aged Trem2.R47H mice that were further enriched for AD associated neuronal functions. Similar splicing alterations were observed in Trem2 knockout mice, implying that these events are not uniquely caused by the R47H mutation and at least partially driven by the reduced Trem2 transcript expression in our Trem2*R47H mouse model [44]. However, splicing differences appeared at a younger age in the full knockout suggesting a more severe effect in mice lacking Trem2. Interestingly, we did not observe a significant overlap between differential expressed genes and differential spliced genes. We further determined that differentially expressed and differentially spliced genes represent distinct biological processes associated with AD, suggesting that splicing analysis identified novel associations and uncovered functional diversity that had been missed by conventional approaches evaluating gene expression only.
Further assessment identified enrichment for neuronal cell signatures in the differentially spliced genes, while DEGs were enriched for microglial cell signatures. Since Trem2 is expressed in microglia and not neurons, it is not immediately clear how its perturbation leads to differential splicing in neuronal genes. In our prior characterization of these models, we did not observe any significant changes in neuron or microglial cell abundances [10]. Bidirectional communication between neurons and glial cells is required for microglia housekeeping functions and preservation of neuronal homeostasis [45, 46]. Neurons express many receptors that are activated by microglia released molecules, enabling microglial control of neurotransmission [45]. For instance, growth factors secreted by microglia (NGF, BDNF) are required by neurons for proper functioning and may not be secreted at sufficient levels. Altered expression of Trem2 gene may perturb expression of signaling ligands secreted by microglia and thus lead to aberrant splicing in neuronal genes. Expression of cytokines and growth factors secreted by microglia involved in modulation of neuronal functions did not exhibit significant transcriptomic changes in the middle-aged Trem2.R47H mice. However, we observed significant reduced level of BDNF in 4 months old Trem2 KO mice in cortex tissue, as well as reduced expression levels of other cytokines and growth factors (although not significantly). This further suggests that the effect of aberrant splicing is stronger and occurs at a younger age with absence of Trem2 gene. Finally, our analysis of whole brain hemispheres precludes resolution of region-specific effects. Deeper investigation is required to properly unravel the involvement of microglia in functional synaptic regulation and understand the physiological role of microglia-neuron interactions.
There are numerous bioinformatics methods for detecting differential splicing events using different approaches. In our study, to infer differential splicing events we adopted two approaches (exon-centric and isoform-centric) [38, 47] that use same DEXSeq method [47]. Still, we did not observe significant overlap between number of genes that had been identified differentially spliced by both approaches. This reflects that different methods capture different signals and/or are biased to certain features, such as exon-centric DEXSeq is expected to be biased against small exons while isoform usage based methods can be biased against detection of differential splicing genes with many isoforms [48]. Hence, it is important to use consensus results from different approaches to infer potential differential splicing events and/or compare with human studies based on different methods [1–3]. Therefore, we have focused on differentially spliced genes that were either identified by both approaches and/or identified as differentially spliced in human AD studies [1–3]. Overall, we identified significant overlap between differentially spliced genes identified in 12-month-old Trem2.R47H mice and multiple human AD studies. A number of genes, including Kif21a,Macf1, Eno2, Ppp3ca, Stxbp1, and Eftud2, that were reported in more than one human AD study were also found differentially spliced in 12-month-old Trem2.R47H male mice. These genes represent potential candidate for further studies of disease-related changes linked to aberrant splicing.
One important finding was identification of differential isoform usages in RNA binding proteins, specifically in Hnrnpm gene. Family members of hnRNPs has been previously associated with Alzheimer’s disease [39] and, in particular, the gene HNRNPM was identified as differentially spliced in symptomatic AD cases compared to controls [1]. We observed decreased use of the primary isoform of the hnrnpm gene that contains an RNA recognition motif required for proper mRNA processing and splicing, suggesting that reduced usage of primary isoform of hnrnpm gene may cause aberrant splicing of target genes that may trigger AD associated pathogenesis resulting from TREM2 mutations or similar disease etiologies. We identified multiple binding sites of hnRNPM in alternatively spliced genes in the Trem2.R47H mouse, including gene markers of AD. Notably, the hnRNPM target Ppp1r9b was identified differentially spliced using both analytical methods (ISAR and DEXSeq), and has also been reported in human AD [1]. Ppp1r9b encodes a scaffold protein (known as neurabin-2 and spinophilin) that interacts with actin-filament and protein phosphatase 1a. The encoded protein acts in the nervous system to regulate spine morphology and density, synaptic plasticity, and neuronal migration [49, 50], and a reduction in its abundance was associated with increased plaque burden in a mouse model [51]. The PPP1R9B gene was also reported as a potential biomarker for AD in neuronal cultures exposed to Aβ [52]. Together, these findings suggest that hnRNPM-mediated changes Ppp1r9b encoded protein levels may disrupt synaptic transmission associated with AD pathology.
In summary, a global splicing analysis in mouse models of LOAD implicated the possible role of the Trem2 gene in disrupting AD associated neuronal signaling process through an alternative splicing mechanism. Further studies focused on alternative splicing in specific cell types may reveal a microglial-mediated mechanism affecting alterative splicing in aging neurons. In this study, we provided a foundation for such a multi-cellular hypothesis relevant to late-onset Alzheimer’s disease, highlighting the importance of splicing analyses in approaches to evaluating gene expression levels in model systems.
Methods
Mouse gene expression data and data processing
All mouse data (raw fastq files and count matrices) analyzed in this study were obtained from the characterization of recently created mouse models based on late-onset AD genetics. We have provided information and Synapse ID for each fast file associated with each sample and count matrices used in this study in Supplementary Table 9. Data and experimental details are available on the AD Knowledge Portal [53] (www.synapse.org/#!Synapse:syn17095980). Briefly, RNA-Seq data were obtained from whole left hemisphere brain samples from APOE4 KI mouse, carrying a humanized version of the prominent APOEε4 genetic risk factor for LOAD, and the Trem2*R47H mouse, carrying the R47H allele of Trem2 gene. In addition, a mouse model expressing both human APOEε4 and the Trem2*R47H mutation (referred to as LOAD1) [10] was used to compare the transcriptional changes in mice carrying both variants to mice carrying only a single risk allele and B6 controls (Table 8). In all cases, mice were homozygous for the engineered allele(s). Transcriptomes from left brain hemispheres were collected at 4, 8, 12, and 24 months of age from both sexes, with six replicates of each group. RNA-Seq data were processed by a parallelized and automatic bioinformatics workflow (Supplementary Fig. 1). Reads were quality trimmed and filtered using the Trimmomatic tool (v0.33) [54]. Reads passing quality filtering were mapped to the mouse genome (GRCm38.p6) augmented by human APOE sequence using the RNA-Seq aligner STAR (v2.5.3) [55]. Gene expression was quantified in two ways, to enable multiple analytical methods: transcript per million (TPM) using RSEM (v1.2.31) [56], and raw counts using HTSeq-count (v0.8.0) [57]. Differential expression in mouse models was assessed using the R Bioconductor package DESeq2 (v1.16.1) [47] and genes with the Benjamini–Hochberg adjusted p-values < 0.05 were considered as significantly differentially expressed genes.
Table 8.
Study population. Whole brain left hemispheres were collected at 4, 8, 12, and 24 months of age from both sexes from mouse models based on late-onset AD genetics and C57BL/6J control mice
| Mouse Models | 4 months | 8 months | 12 months | 24 months | ||||
|---|---|---|---|---|---|---|---|---|
| Male | Female | Male | Female | Male | Female | Male | Female | |
| C57BL/6J | 12 | 12 | 6 | 6 | 6 | 6 | 7 | 6 |
| APOE4 KI | 13 | 12 | 6 | 6 | 4 | 5 | 5 | 6 |
| TREM2*R47H | 12 | 12 | 6 | 6 | 6 | 6 | 6 | 3 |
| APOE4.TREM2*R47H | 10 | 12 | 5 | 6 | 8 | 5 | 7 | 6 |
Differential splicing analysis
Differential splicing analyses were performed using two bioinformatics tools: DEXSeq (v1.40.0), a Bioconductor package which measures differential exon usage (DEU) as a surrogate to infer differential splicing events in RNA-Seq data [31]; and IsoformSwitchAnalyzerR (ISAR) (v1.20.0), which measures differential isoform usage to identify isoform switches and predict the resulting functional consequences [38]. In DEXSeq analysis, exons with FDR < 0.1 were considered significantly differentially used. ISAR measures isoform usage via isoform fraction (IF) values, which quantify the fraction of the parent gene expression originating from a specific isoform (calculated as isoform_exp/gene_exp). Difference in isoform usage is quantified as the difference in isoform fraction (dIF). ISAR implemented DEXSeq based tests as the default way to identify isoform switches [38]. In ISAR analysis, the cutoff for change in absolute dIF was set to 0.05 to be considered potential isoform switch and switches with FDR < 0.05 were considered significant. We chose different threshold for ISAR analysis due to greater sensitivity than to DEXSeq analysis.
Functional enrichment analysis
Functional annotations and enrichment analyses were performed using the R Bioconductor package clusterProfiler [58], with Gene Ontology terms and KEGG pathways [59–61] enrichment analyses by functions enrichGO and enrichKEGG, respectively. The function compareCluster was used to compare enriched functional categories of each gene module. The significance threshold for all enrichment analyses was set to 0.05 using Benjamini–Hochberg adjusted p-values.
Cell type enrichment analysis
Different brain cell types (neurons, endothelial cell, astrocytes, microglia, oligodendrocyte precursor cells, newly formed oligodendrocytes, and myelinating oligodendrocytes) specific gene signatures were selected from an RNA-Sequencing database [32]. Fisher exact test was used to test enrichment of cell type signatures in each input gene set. Resulting p-values were corrected for multiple tests using the Benjamini–Hochberg method and the significance threshold was set to 0.1.
Predicting binding sites of RNA-binding proteins
Binding sites of RNA-binding proteins on differentially spliced genes were predicted using RBPmap webtool [43]. We selected RNA binding proteins from RBPmap’s database of experimentally defined human/mouse motifs. Within the scope of this study, we explicitly looked for RNA-binding proteins that were either identified as differentially expressed or differentially spliced through our analyses. We used RBPmap webserver at default settings for mouse genome (GRCm38/mm10 assembly).
Trem2 Knockout (KO) mice transcriptomic data
RNA-Seq data (fastq files) of brain tissue samples for Trem2 KO and respective control mice was obtained from the Gene Expression Omnibus data repository (accession number: GSE104381). Details on brain sample collection, tissue and RNA preparation, and sequencing can be found in previously published work on this data [33]. Further, we have re-processed RNA-Seq data using our RNA-Seq processing pipeline (as described above).
Human splicing studies in AD cohort
Differentially spliced genes in the human brain associated with AD were obtained from three distinct studies. In the first study, dorsolateral prefrontal cortex (DLPFC) of 450 subjects from the Religious order Study (ROS) and the Memory and Aging Project (MAP) was deeply sequenced followed by identification of alternative splicing events related to AD by measuring differential intron usages [2]. In the second study, authors have used a new proteogenomic approach (TMT-LysC) to investigate RNA splicing events by predicting alternative exon-exon junction splicing events across symptomatic AD (n = 20), asymptomatic AD (n = 14), and control (n = 13) cases [1]. In the third study, authors have performed deep RNA-Sequencing of frontal cortex RNAs using sample groups from Emory university (four controls and five AD cases); and the university of Kentucky (three controls and AD cases) followed by identification of genes associated with RNA processing and splicing abnormalities in AD cases [3]. Genomic information on orthologous genes was obtained via the latest ENSEMBL build for human genome version GRCh38. All orthologous relationships between human and mouse were downloaded via BioMart [62]. Next, we performed hypergeometric test to compute significant overlaps (p < 0.05) between differentially spliced genes identified in each human study and each mouse model.
Supplementary Information
Additional file 1: Supplementary Fig. 1. Bioinformatics workflow with each step of the splicing analysis.
Additional file 2: Supplementary Text 1. Documentation of each tool and command used for each step mentioned in the bioinformatics workflow.
Additional file 3: Supplementary Fig. 2. Differential splicing analysis in Trem2.KO mouse models. (A) Number of differentially expressed genes (DEGs) and genes with differential exon usage (DEU) in Trem2.KO mice at 4 and 8 months of age in cortex and hippocampus brain tissues. (B) Enrichment of KEGG pathways (p < 0.05) in DEU genes in Trem2 KO mice. (C) Cell type enrichment analysis in the gene set with differential exon usage (DEU) and differentially expressed genes (DEG) in each mouse model using Fisher exact test. Significant cell type enrichment are shown by solid dots (p < 0.1).
Additional file 4: Supplementary Fig. 3. Cell type enrichment in differentially spliced genes in human AD cohorts. Cell type enrichment analysis in the differentially spliced genes in human AD populations were computed using Fisher exact test. Solid circles represent significant enrichment (p < 0.1) of cell type.
Additional file 5: Supplementary Fig. 4. ISAR identified differential isoform usage in Hnrnpm, which encode for RNA binding protein (RBP) HNRNPM. Significant increased usage ( FDR < 0.05 was observed for non-coding isoform in 12 months old Trem2*R47H male mice compared to B6 mice.
Additional file 6: Supplementary Table 1. Differentially exon usage (DEU) genes identified in mouse models. The attached table depicts the differentially expressed usage genes in LOAD mouse strains compared to C7BL/6 J mice using DEXSeq tool.
Additional file 7: Supplementary Table 2. Enriched KEGG pathways in DEU genes in each mouse model. The attached table depicts the KEGG pathway annotations for the differentially exon usage genes in LOAD mouse strains and Trem2 KO mice compared to C7BL/6 J mice.
Additional file 8: Supplementary Table 3. Overlap between differentially expressed genes and differentially spliced in each mouse model. The attached tables depicts overlap between the differentially exon usage genes (DEUs) and differentially expressed genes (DEGs), and differentially isoform usage genes (DTUs) and differentially expressed genes (DEGs) in LOAD mouse strains compared to C7BL/6 J mice.
Additional file 9: Supplementary Table 4. Differentially exon usage genes in Trem2 KO mice. The attached table depicts the differentially expressed usage genes in Trem2 KO mice compared to C7BL/6 J mice using DEXSeq tool.
Additional file 10: Supplementary Table 5. Differentially isoform usage (DTU) genes identified in each mouse model. The attached table depicts the differentially expressed usage genes in LOAD mouse strains compared to C7BL/6 J mice using IsoformSwitchAnalyzeR (ISAR) tool.
Additional file 11: Supplementary Table 6. Enrichment of Gene ontology biological processes analyses in differentially isoform usage (DTU) genes. The attached table depicts the gene 2 ontology enrichment for the differentially isoform usage genes in Trem2*R47H male and female mice compared to age and sex-matched C7BL/6 J mice.
Additional file 12: Supplementary Table 7. List of differentially DEU genes with binding sites of RNA binding proteins. The attached file contains the differentially exon usage genes in Trem2*R47H male mice with binding sites for RNA-binding proteins.
Additional file 13: Supplementary Table 8. List of DTU genes with binding sites of RNA binding proteins. The attached file contains the differentially isoform usage genes in Trem2*R47H male mice with binding sites for RNA-binding proteins.
Additional file 14: Supplementary Table 9. Information about each sample and unique Synapse ID for each sample for each associated raw FASTQ files and raw gene count matrices used in this study.
Acknowledgements
We thank the many institutions and their staff that provided support for this study and who were involved in this collaboration.
Abbreviations
- AD
Alzheimer’s disease
- AsymAD
Asymptomatic AD
- AMP-AD
Accelerating Medicines Partnership—Alzheimer's Disease
- B6
C57BL/6J
- DEG
Differentially expressed genes
- DEU
Differentially exon usage
- DTU
Differential transcript usage
- LOAD
Late-onset Alzheimer’s disease
- RBP
RNA binding protein
- ROS/MAP
Religious Orders Study and Memory and Aging Project
- RRM
RNA recognition motif
Authors’ contributions
RSP curated the data, investigated and performed the bioinformatics analyses. KPK, GRH and MS performed mouse experiments. GWC and GRH supervised and designed the project. RSP and GWC wrote the manuscript. All authors edited and approved the final manuscript.
Funding
This work was supported by the National Institute of Health grant U54AG054345.
Availability of data and materials
The B6.APOE4.Trem2*R47H (LOAD1) data sets are available via the AD Knowledge Portal (https://adknowledgeportal.org). The AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the Accelerating Medicines Partnership (AMP-AD) Target Discovery Program and other National Institute on Aging (NIA)-supported programs to enable open-science practices and accelerate translational learning. The data, analyses and tools are shared early in the research cycle without a publication embargo on secondary use. Data is available for general research use according to the following requirements for data access and data attribution (https://adknowledgeportal.org/DataAccess/Instructions). The results published here are in whole or in part based on data obtained from the AD Knowledge Portal. Study data were provided by the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago. Study data were provided through the Accelerating Medicine Partnership for AD (U01AG046161 and U01AG061357) based on samples provided by the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago. Data collection was supported through funding by NIA grants P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36836, U01AG32984, U01AG46152, the Illinois Department of Public Health, and the Translational Genomics Research Institute.
For access to content described in this manuscript see: https://doi.org/10.7303/syn23631984. Detailed documentation reporting each tool (software name and version) and commands used to analyze the mouse data are provided in Supplementary Text 1.
Declarations
Ethics approval and consent to participate
No human subjects or data was used in this study. All mouse data were reported previously with animal care standards [10].
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Johnson ECB, Dammer EB, Duong DM, Yin L, Thambisetty M, Troncoso JC, et al. Deep proteomic network analysis of Alzheimer’s disease brain reveals alterations in RNA binding proteins and RNA splicing associated with disease. Mol Neurodegener. 2018;13(1):52. doi: 10.1186/s13024-018-0282-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raj T, Li YI, Wong G, Humphrey J, Wang M, Ramdhani S, et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer's disease susceptibility. Nat Genet. 2018;50(11):1584–1592. doi: 10.1038/s41588-018-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bai B, Hales CM, Chen P-C, Gozal Y, Dammer EB, Fritz JJ, et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc Natl Acad Sci. 2013;110(41):16562. doi: 10.1073/pnas.1310249110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montes M, Sanford BL, Comiskey DF, Chandler DS. RNA Splicing and Disease: Animal Models to Therapies. Trends Genet. 2019;35(1):68–87. doi: 10.1016/j.tig.2018.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oblak AL, Forner S, Territo PR, Sasner M, Carter GW, Howell GR, et al. Model organism development and evaluation for late-onset Alzheimer's disease: MODEL-AD. Alzheimer's & dementia (New York, N Y) 2020;6(1):e12110-e. doi: 10.1002/trc2.12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Avale ME, Rodríguez-Martín T, Gallo J-M. Trans-splicing correction of tau isoform imbalance in a mouse model of tau mis-splicing. Hum Mol Genet. 2013;22(13):2603–2611. doi: 10.1093/hmg/ddt108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hinrich AJ, Jodelka FM, Chang JL, Brutman D, Bruno AM, Briggs CA, et al. Therapeutic correction of ApoER2 splicing in Alzheimer's disease mice using antisense oligonucleotides. EMBO Mol Med. 2016;8(4):328–345. doi: 10.15252/emmm.201505846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wobst HJ, Denk F, Oliver PL, Livieratos A, Taylor TN, Knudsen MH, et al. Increased 4R tau expression and behavioural changes in a novel MAPT-N296H genomic mouse model of tauopathy. Sci Rep. 2017;7:43198. doi: 10.1038/srep43198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rockenstein EM, McConlogue L, Tan H, Power M, Masliah E, Mucke L. Levels and alternative splicing of amyloid & #x3b2; protein precursor (APP) transcripts in brains of APP transgenic mice and humans with alzheimer's disease (*) J Biol Chem. 1995;270(47):28257–28267. doi: 10.1074/jbc.270.47.28257. [DOI] [PubMed] [Google Scholar]
- 10.Kotredes KP, Oblak AL, Pandey RS, Lin PB, Garceau D, Williams HM, et al. Uncovering disease mechanisms in a novel mouse model expressing humanized APOEε4 and Trem2*R47H. Front Aging Neurosci. 2021;13:735524. [DOI] [PMC free article] [PubMed]
- 11.Boutajangout A, Wisniewski T. The innate immune system in Alzheimer's disease. Int J Cell Biol. 2013;2013:576383. doi: 10.1155/2013/576383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daws MR, Lanier LL, Seaman WE, Ryan JC. Cloning and characterization of a novel mouse myeloid DAP12-associated receptor family. Eur J Immunol. 2001;31(3):783–791. doi: 10.1002/1521-4141(200103)31:3<783::AID-IMMU783>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 13.Zhao Y, Wu X, Li X, Jiang LL, Gui X, Liu Y, et al. TREM2 is a receptor for beta-amyloid that mediates microglial function. Neuron. 2018;97(5):1023–31 e7. doi: 10.1016/j.neuron.2018.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C, Zhao B, Lin C, Gong Z, An X. TREM2 inhibits inflammatory responses in mouse microglia by suppressing the PI3K/NF-kappaB signaling. Cell Biol Int. 2019;43(4):360–372. doi: 10.1002/cbin.10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. 2016;91(2):328–340. doi: 10.1016/j.neuron.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 16.Cheng-Hathaway PJ, Reed-Geaghan EG, Jay TR, Casali BT, Bemiller SM, Puntambekar SS, et al. The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer's disease. Mol Neurodegener. 2018;13(1):29. doi: 10.1186/s13024-018-0262-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368(2):107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pottier C, Wallon D, Rousseau S, Rovelet-Lecrux A, Richard AC, Rollin-Sillaire A, et al. TREM2 R47H variant as a risk factor for early-onset Alzheimer’s disease. J Alzheimers Dis. 2013;35(1):45–49. doi: 10.3233/JAD-122311. [DOI] [PubMed] [Google Scholar]
- 20.Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4–9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 21.Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014;6(243):243ra86. doi: 10.1126/scitranslmed.3009093. [DOI] [PubMed] [Google Scholar]
- 22.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40(12):1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 24.Kornblihtt AR, Schor IE, Alló M, Dujardin G, Petrillo E, Muñoz MJ. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol. 2013;14(3):153–165. doi: 10.1038/nrm3525. [DOI] [PubMed] [Google Scholar]
- 25.Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science. 2012;338(6114):1587. doi: 10.1126/science.1230612. [DOI] [PubMed] [Google Scholar]
- 26.Wang G-S, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8(10):749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 27.Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016;17(1):19–32. doi: 10.1038/nrg.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 29.Trabzuni D, Wray S, Vandrovcova J, Ramasamy A, Walker R, Smith C, et al. MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies. Hum Mol Genet. 2012;21(18):4094–4103. doi: 10.1093/hmg/dds238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Rev. 2000;33(1):95–130. doi: 10.1016/S0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 31.Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22(10):2008–2017. doi: 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carbajosa G, Malki K, Lawless N, Wang H, Ryder JW, Wozniak E, et al. Loss of Trem2 in microglia leads to widespread disruption of cell coexpression networks in mouse brain. Neurobiol Aging. 2018;69:151–166. doi: 10.1016/j.neurobiolaging.2018.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Qi Y, Zhou X, Zhang G, Fu C. Alteration of scaffold: possible role of MACF1 in Alzheimer’s disease pathogenesis. Med Hypotheses. 2019;130:109259. doi: 10.1016/j.mehy.2019.109259. [DOI] [PubMed] [Google Scholar]
- 35.Kreft KL, van Meurs M, Wierenga-Wolf AF, Melief M-J, van Strien ME, Hol EM, et al. Abundant kif21b is associated with accelerated progression in neurodegenerative diseases. Acta Neuropathol Commun. 2014;2:144. doi: 10.1186/s40478-014-0144-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bianchi S, van Riel WE, Kraatz SHW, Olieric N, Frey D, Katrukha EA, et al. Structural basis for misregulation of kinesin KIF21A autoinhibition by CFEOM1 disease mutations. Sci Rep. 2016;6(1):30668. doi: 10.1038/srep30668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamada K, Andrews C, Chan W-M, McKeown CA, Magli A, de Berardinis T, et al. Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1) Nat Genet. 2003;35(4):318–321. doi: 10.1038/ng1261. [DOI] [PubMed] [Google Scholar]
- 38.Vitting-Seerup K, Sandelin A. IsoformSwitchAnalyzeR: analysis of changes in genome-wide patterns of alternative splicing and its functional consequences. Bioinformatics. 2019;35(21):4469–4471. doi: 10.1093/bioinformatics/btz247. [DOI] [PubMed] [Google Scholar]
- 39.Geuens T, Bouhy D, Timmerman V. The hnRNP family: insights into their role in health and disease. Hum Genet. 2016;135(8):851–867. doi: 10.1007/s00439-016-1683-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bishof I, Dammer EB, Duong DM, Kundinger SR, Gearing M, Lah JJ, et al. RNA-binding proteins with basic-acidic dipeptide (BAD) domains self-assemble and aggregate in Alzheimer’s disease. J Biol Chem. 2018;293(28):11047–11066. doi: 10.1074/jbc.RA118.001747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Datar KV, Dreyfuss G, Swanson MS. The human hnRNP M proteins: identification of a methionine/arginine-rich repeat motif in ribonucleoproteins. Nucleic Acids Res. 1993;21(3):439–446. doi: 10.1093/nar/21.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hovhannisyan RH, Carstens RP. Heterogeneous ribonucleoprotein M is a splicing regulatory protein that can enhance or silence splicing of alternatively spliced exons *. J Biol Chem. 2007;282(50):36265–36274. doi: 10.1074/jbc.M704188200. [DOI] [PubMed] [Google Scholar]
- 43.Paz I, Kosti I, Ares M, Jr., Cline M, Mandel-Gutfreund Y. RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014;42(Web Server issue):W361–W7. doi: 10.1093/nar/gku406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiang X, Piers TM, Wefers B, Zhu K, Mallach A, Brunner B, et al. The Trem2 R47H Alzheimer's risk variant impairs splicing and reduces Trem2 mRNA and protein in mice but not in humans. Mol Neurodegen. 2018;13(1):49. doi: 10.1186/s13024-018-0280-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marinelli S, Basilico B, Marrone MC, Ragozzino D. Microglia-neuron crosstalk: Signaling mechanism and control of synaptic transmission. Semin Cell Dev Biol. 2019;94:138–151. doi: 10.1016/j.semcdb.2019.05.017. [DOI] [PubMed] [Google Scholar]
- 46.Szepesi Z, Manouchehrian O, Bachiller S, Deierborg T. Bidirectional microglia-neuron communication in health and disease. Front Cell Neurosci. 2018;12:323. doi: 10.3389/fncel.2018.00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hooper JE. A survey of software for genome-wide discovery of differential splicing in RNA-Seq data. Hum Genomics. 2014;8(1):3. doi: 10.1186/1479-7364-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferrer I, Verdugo-Sivianes EM, Castilla MA, Melendez R, Marin JJ, Muñoz-Galvan S, et al. Loss of the tumor suppressor spinophilin (PPP1R9B) increases the cancer stem cell population in breast tumors. Oncogene. 2016;35(21):2777–2788. doi: 10.1038/onc.2015.341. [DOI] [PubMed] [Google Scholar]
- 50.Hsieh-Wilson LC, Allen PB, Watanabe T, Nairn AC, Greengard P. Characterization of the neuronal targeting protein spinophilin and its interactions with protein phosphatase-1. Biochemistry. 1999;38(14):4365–4373. doi: 10.1021/bi982900m. [DOI] [PubMed] [Google Scholar]
- 51.Palavicini JP, Wang H, Bianchi E, Xu S, Rao JS, Kang DE, et al. RanBP9 aggravates synaptic damage in the mouse brain and is inversely correlated to spinophilin levels in Alzheimer’s brain synaptosomes. Cell Death Dis. 2013;4(6):e667-e. doi: 10.1038/cddis.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Henriques AG, Müller T, Oliveira JM, Cova M, da Cruz e Silva CB, da Cruz e Silva OAB. Altered protein phosphorylation as a resource for potential AD biomarkers. Sci Rep. 2016;6(1):30319. doi: 10.1038/srep30319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Greenwood AK, Montgomery KS, Kauer N, Woo KH, Leanza ZJ, Poehlman WL, et al. The AD knowledge portal: a repository for multi-omic data on Alzheimer’s disease and aging. Curr Protoc Hum Genet. 2020;108(1):e105. doi: 10.1002/cphg.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2012;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12(1):323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28(11):1947–1951. doi: 10.1002/pro.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51(D1):D587–D592. doi: 10.1093/nar/gkac963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21(16):3439–3440. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplementary Fig. 1. Bioinformatics workflow with each step of the splicing analysis.
Additional file 2: Supplementary Text 1. Documentation of each tool and command used for each step mentioned in the bioinformatics workflow.
Additional file 3: Supplementary Fig. 2. Differential splicing analysis in Trem2.KO mouse models. (A) Number of differentially expressed genes (DEGs) and genes with differential exon usage (DEU) in Trem2.KO mice at 4 and 8 months of age in cortex and hippocampus brain tissues. (B) Enrichment of KEGG pathways (p < 0.05) in DEU genes in Trem2 KO mice. (C) Cell type enrichment analysis in the gene set with differential exon usage (DEU) and differentially expressed genes (DEG) in each mouse model using Fisher exact test. Significant cell type enrichment are shown by solid dots (p < 0.1).
Additional file 4: Supplementary Fig. 3. Cell type enrichment in differentially spliced genes in human AD cohorts. Cell type enrichment analysis in the differentially spliced genes in human AD populations were computed using Fisher exact test. Solid circles represent significant enrichment (p < 0.1) of cell type.
Additional file 5: Supplementary Fig. 4. ISAR identified differential isoform usage in Hnrnpm, which encode for RNA binding protein (RBP) HNRNPM. Significant increased usage ( FDR < 0.05 was observed for non-coding isoform in 12 months old Trem2*R47H male mice compared to B6 mice.
Additional file 6: Supplementary Table 1. Differentially exon usage (DEU) genes identified in mouse models. The attached table depicts the differentially expressed usage genes in LOAD mouse strains compared to C7BL/6 J mice using DEXSeq tool.
Additional file 7: Supplementary Table 2. Enriched KEGG pathways in DEU genes in each mouse model. The attached table depicts the KEGG pathway annotations for the differentially exon usage genes in LOAD mouse strains and Trem2 KO mice compared to C7BL/6 J mice.
Additional file 8: Supplementary Table 3. Overlap between differentially expressed genes and differentially spliced in each mouse model. The attached tables depicts overlap between the differentially exon usage genes (DEUs) and differentially expressed genes (DEGs), and differentially isoform usage genes (DTUs) and differentially expressed genes (DEGs) in LOAD mouse strains compared to C7BL/6 J mice.
Additional file 9: Supplementary Table 4. Differentially exon usage genes in Trem2 KO mice. The attached table depicts the differentially expressed usage genes in Trem2 KO mice compared to C7BL/6 J mice using DEXSeq tool.
Additional file 10: Supplementary Table 5. Differentially isoform usage (DTU) genes identified in each mouse model. The attached table depicts the differentially expressed usage genes in LOAD mouse strains compared to C7BL/6 J mice using IsoformSwitchAnalyzeR (ISAR) tool.
Additional file 11: Supplementary Table 6. Enrichment of Gene ontology biological processes analyses in differentially isoform usage (DTU) genes. The attached table depicts the gene 2 ontology enrichment for the differentially isoform usage genes in Trem2*R47H male and female mice compared to age and sex-matched C7BL/6 J mice.
Additional file 12: Supplementary Table 7. List of differentially DEU genes with binding sites of RNA binding proteins. The attached file contains the differentially exon usage genes in Trem2*R47H male mice with binding sites for RNA-binding proteins.
Additional file 13: Supplementary Table 8. List of DTU genes with binding sites of RNA binding proteins. The attached file contains the differentially isoform usage genes in Trem2*R47H male mice with binding sites for RNA-binding proteins.
Additional file 14: Supplementary Table 9. Information about each sample and unique Synapse ID for each sample for each associated raw FASTQ files and raw gene count matrices used in this study.
Data Availability Statement
The B6.APOE4.Trem2*R47H (LOAD1) data sets are available via the AD Knowledge Portal (https://adknowledgeportal.org). The AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the Accelerating Medicines Partnership (AMP-AD) Target Discovery Program and other National Institute on Aging (NIA)-supported programs to enable open-science practices and accelerate translational learning. The data, analyses and tools are shared early in the research cycle without a publication embargo on secondary use. Data is available for general research use according to the following requirements for data access and data attribution (https://adknowledgeportal.org/DataAccess/Instructions). The results published here are in whole or in part based on data obtained from the AD Knowledge Portal. Study data were provided by the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago. Study data were provided through the Accelerating Medicine Partnership for AD (U01AG046161 and U01AG061357) based on samples provided by the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago. Data collection was supported through funding by NIA grants P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36836, U01AG32984, U01AG46152, the Illinois Department of Public Health, and the Translational Genomics Research Institute.
For access to content described in this manuscript see: https://doi.org/10.7303/syn23631984. Detailed documentation reporting each tool (software name and version) and commands used to analyze the mouse data are provided in Supplementary Text 1.