

Abstract
Macrophages are first responders for the immune system. In this role, they have both effector functions for neutralizing pathogens and sentinel functions for alerting other immune cells of diverse pathologic threats, thereby initiating and coordinating a multipronged immune response. Macrophages are distributed throughout the body: they circulate in the blood, line the mucosal membranes, reside within organs, and survey the connective tissue. Several reviews have summarized their diverse roles in different physiological scenarios and in the initiation or amplification of different pathologies. In this review, we propose that both the effector and the sentinel functions of healthy macrophages rely on three hallmark properties: Response Specificity, Context Dependence, and Stimulus Memory. When these hallmark properties are diminished, the macrophage’s biological functions are impaired. That in turn results in increased risk for immune dysregulation, manifested by immune deficiency or auto-immunity. We review the evidence and the molecular mechanisms supporting these hallmark properties.
Keywords: innate immunity, macrophages, sentinel cells, stimulus specificity, polarization, epigenetic memory, NFκB, IRF, MAPK, Interferon, chromatin
Preface
Macrophages were first described in the late 19th century, when Ilya Mechnikov looked into his microscope and noted a striking observation: ‘white corpuscles’ moved to surround a small splinter embedded in a starfish larva (1). This first description of the cells now known as macrophages launched investigations of innate immunity.
Macrophages are first responders, capable of detecting tissue injury and pathogen threats. They respond to such threats with two broadly defined functions: 1) they have potent anti-pathogen effector functions as their name implies, and 2) they also have potent sentinel functions to call up and direct innate and adaptive immune cells. As effectors, macrophages phagocytose pathogens, upregulate antimicrobial peptides, or trigger cell death to limit intracellular pathogens (Figure 1a). As sentinels, macrophages initiate and coordinate local or systemic immune activation by secreting cytokines, chemokines and growth factors, and presenting antigen to adaptive immune cells (2).
Figure 1.

Functional hallmarks of macrophages. (a) Macrophages respond by performing a variety of functions. Categorization of these functions show that macrophages can perform as sentinel cells of the immune system or as immune effector cells. (b) Macrophage responses exhibit three hallmarks central to immunological function: Response Specificity, Context Dependence, and Stimulus Memory. Dashed and solid arrows represent deployment of specific functions to different degrees or speeds.
As first responders, macrophages are strategically placed. Almost every tissue in the body is populated with macrophages at a remarkably consistent density of ~500–1000/mm3 (3–5). Tissue resident alveolar macrophages, peritoneal macrophages, and Kupffer cells of the liver sense airborne pathogens or those from the digestive tract. In addition, bone-marrow-derived monocytes circulate the blood and readily extravasate into tissues upon sensing chemoattractants secreted by the very first responders. Upon sensing a pathogen-associated molecular pattern (PAMP) or damage-associated molecular pattern (DAMP) their differentiation into macrophages is accelerated, rapidly contributing effector and sentinel functions at the site of infection or injury.
Macrophages, despite having disparate ontogenies and a wide variety of physiological roles, share certain functional characteristics. These characteristics are less defined by developmental origin or their steady state (as seen in diverse epigenomic or transcriptomic profiles (6)), but rather by the manner in which they respond to immune threats. Here we propose three functional hallmarks that define healthy macrophages (Figure 1b): 1) Response Specificity – the capacity to mount threat-specific responses. 2) Context Dependence – the capacity to adapt immune threat-specific responses to the microenvironmental cytokine milieu. 3) Stimulus Memory – the capacity to record prior exposure to stimuli and adapt subsequent responses to immune threats.
Does the misregulation of these functional hallmarks contribute to risk for disease? The first hallmark of Response Specificity is a measure of the distribution overlap in sentinel responses to different ligands; single cell response distributions with abnormal spread may result in improper immune sequelae. Autoimmune diseases, where symptoms are often sporadic with unknown triggers, may involve the loss of healthy Response Specificity. Second, Context Dependence of macrophage responses are linked to diseases of aberrant cytokine conditioning. As opposed to the genetic changes that are often risk factors for autoimmune disease, infectious disease complications or inflammatory diseases like cancer are affected by microenvironment states such as aging and obesity, and may reflect poor regulation of context-dependent responses. Third, diseases of Memory may result when the primary stimulus improperly alters the epigenome, resulting in hyper- or hypo-inflammatory disorders upon a second encounter, rather than inducing innate immune memory that results in benefits against subsequent infectious agents. It is of great interest to delineate how the loss of the functional macrophage hallmarks may result in disease, prompting a focus on understanding of the molecular mechanisms that give rise to each functional hallmark.
The mechanisms underlying signaling and epigenome regulation of macrophage responses have been closely studied and reviewed (20–23). However, it has been less obvious how these molecularly-detailed biochemical and biophysical mechanisms give rise to the immunological functions of macrophages. Recent studies quantifying the stimulus-specificity of signaling dynamics (24), on feedforward and feedback mechanisms (25, 26), or on context- or exposure-dependent epigenomic changes (27, 28), have further elucidated these connections. Furthermore, new technology such as reliable live-cell microscopy, cost-effective scRNAseq and multi-dimensional flow cytometry, and algorithmic advances in analysis of the data, have allowed us to probe and measure these hallmarks accurately. In light of these advances, in this review, we place existing and recent mechanistic knowledge into the framework of these three functional hallmarks that characterize healthy macrophage identity.
1. Response Specificity
Macrophages express dozens of receptors and are capable of sensing hundreds of PAMPs, DAMPs, and cytokine ligands. Immune response genes are not expressed constitutively because they are in fact detrimental to the host. Hence, they must be deployed on an “only-as-needed” basis. Given that pathogens differ widely in their biology, different immune defenses are required to effectively counteract them. The “only-as-needed” rationale argues that healthy immune sentinel cell responses should be highly specific to the immune threat.
It is now appreciated that macrophages respond to ligands with stimulus-specific signaling profiles and stimulus-specific gene expression programs (9, 29, 30). Not only ligand, but also ligand dose, and ligand exposure dynamics and duration generate distinct responses (31–34). Yet, early studies of macrophages reported a common core response, emphasizing the common phagocytic and antigen-presenting functions of macrophages (35). Only the resulting adaptive immune responses were thought to provide specificity to the immune response. However, later studies revealed that macrophages, dendritic cells, and fibroblasts (36) produce gene expression programs that are in fact stimulus-specific (9, 37), and began to address the molecular mechanisms that allow for this Response Specificity.
Macrophages perform as individuals in their function as sentries; each has the capacity to sense molecular patterns and activate signaling pathways to generate a response, and the immune response relies on the response of the individual. Yet, individual macrophages are not identical, being subject to molecular stochasticity that affects gene expression (and hence the abundances of pathogen sensors and signaling proteins) and the cells’ spatial organization. Furthermore, macrophages within tissue are comprised of distinct cellular subsets (38), including subsets with distinct developmental origins (39). The resulting cell-to-cell heterogeneity may affect stimulus-response programs, and thus the Response Specificity ascribed to the population. Therefore, quantitative studies of Responses Specificity must involve measurements at single cell resolution.
1.1. Immune response signaling network
Response Specificity relies on molecular components and pathways that are activated stimulus-specifically. Given the large number of immune activating stimuli but a limited number of signaling factors, two principles have emerged that help to explain the stimulus-specific regulation of the molecular network: combinatorial and dynamic control of signaling pathways. These principles describe that stimulus-specific activation of immune response genes is dependent on 1) different stimuli activating different combinations of signaling regulators, and 2) different stimuli activating the same regulator but with different dynamic patterns of activity.
Specific responses are initiated by how cells sense ligands. Human macrophages sense PAMP and DAMP ligands with 10 Toll-like receptors (TLRs) (40), that reside in the plasma membrane and survey extra-cellular and endosomal environment, 22 cytosolic NOD-like receptors (NLRs) (41), 3 RIG-I-like-receptors (RLRs) (42), and DNA sensor cGAS (43, 44). Immune activating cytokines such as interferons, IL1, TNF and other TNF super family members are sensed by their cognate receptors in the plasma membrane. To elicit stimulus-specific responses the ligand-receptor interaction must be specific based on the specificity of the molecular interaction. Indeed, even low complexity PAMPs are distinguished by different TLR family members, and the biophysical basis for these this specificity has been studied (45). However, functional discrimination of ligands may also involve other mechanisms such as localization (46) or kinetic proof-reading, as for example in the case of RIG-I (47).
Despite the impressive number of pathogen sensors, the number of pathogens and potential PAMPs and DAMPs is even greater. Many receptors have been shown to mediate sensing of distinct ligands. TLR4, for example, the known endotoxin (LPS) sensor, also mediates sensing of DAMPs, such as HMGB1 (48), serum amyloid A (SAA) (49), and MRP8/MRP14 (50). The range of interactions may be expanded by soluble or membrane-bound co-factors such as CD14 (51). However, two ligands being recognized by the same receptor does not necessarily mean that they produce the same response. Ligand-receptor interactions are governed by specific kinetics of signaling adaptor recruitment, receptor internalization, and ligand degradation, which may produce not only stimulus-specific amplitudes of signaling but stimulus-specific signaling dynamics. Such dynamic coding mechanisms also allows for cells to mount dose-specific responses for the same stimulus via the same receptor.
Similar considerations govern how receptors activate downstream signaling pathways, which are mediated by a small number of primary signaling adaptors MyD88, TRIF, TRAF2/6, MAVS, STING, and ASC (52). In fact, all TLRs, except TLR3 utilize MyD88, as does the IL1 receptor. TRIF is engaged by TLR3 and TLR4. TRAFs are engaged by TNFR family members, ASC by NLRs, and STING by cGAS (53). However, specificity is possible because the relative strengths and dynamics of adaptor recruitment and signaling pathway activation may differ between receptors. For example, in response to lipopolysaccharide (LPS), TLR4 signals at the plasma membrane and initiates the oligomerization of the adapter Myd88, or can be internalized to signal through the endosome, where it interacts with a different adapter TRIF to initiate TRIF-dependent signaling. LPS dose-response specificity is controlled at the adapter level by the different oligomerization dynamics of Myd88 and TRIF (54).
Adapters and associated ubiquitin chains ultimately activate a limited set of kinase-transcription factor modules. While interferons activate the JAK-STAT pathways without a dedicated signaling adaptor, four primary immune response signaling pathways may be mapped onto these adaptors: IRF3 is activated by TRIF, MAVS and STING adaptors, MAPKp38 is strongly activated by MyD88, but NFκB and JNK/ERK pathways are activated by all adapters (in the case of ASC via the IL1b feedback loop) (53). Therefore, if Response Specificity solely relied on combinatorial coding only three patterns could be observed: NFκB + JNK/ERK (e.g. in response to TNF), NFκB + JNK/ERK + IRF3 (e.g. in response to poly(I:C)), NFκB + JNK/ERK + MAPKp38 (e.g. in response to Pam3CSK4), and NFκB + JNK/ERK + MAPKp38 + IRF3 (e.g. in response to LPS). However, stimulus-specific dynamic control of these pathways may allow for additional specificity (see next section).
Given that Response Specificity is not merely a function of biophysical interaction specificities that are genetically encoded in protein structures, but also of signaling dynamics in amplitude and time, it is subject to differences in the expression levels and localization of signal transducers. It is commonly appreciated that the PRR repertoire that macrophages express determines their responsiveness. The above discussion suggests that the Response Specificity of macrophages is diminished by cell-to-cell heterogeneity in the expression of PRRs, and other key signaling regulators (e.g. adaptors, kinases, etc), also. In response to LPS, the primary driver of the heterogeneity of NFκB responses was in fact found to be, not the TLR4 abundance, but the maturation time of the endosome, which determines the duration of TRIF signaling (54).
1.2. Immune response transcription factors
Response Specificity, the capacity to mount threat-specific responses, has been studied at the level of transcription factor activities, given the availability of quantitative assays. Biochemical studies performed in fibroblasts in the 2000’s elucidated both the temporal and combinatorial regulation of kinase-transcription factor activities mediating stimulus-specific mean responses (36, 55, 56) (Figure 2a). For example, NFκB activation was shown to be persistent in response to LPS, but oscillatory in response to TNF, mediated by negative feedback regulation from IκBα (24, 57). Similarly, JNK activation showed two distinguishable phases of activity (58). Combinatorially, while the kinase-TF modules IKK-NFκB and JNK-AP1 are ubiquitously, adapter-specific activation of TBK1-IRF3 by TRIF and MAPKp38 by Myd88 further contribute to Response Specificity of TF activity (9, 59, 60).
Figure 2.

Mechanisms and measurement approaches of Response Specificity. (a) Response Specificity relies on the ability of dozens of pathogen recognition receptors and cytokine receptors to recognize specific ligands. Ligand-receptor interactions activate specific signaling pathways with ligand-specific temporal and dose dynamics, which are recognized by gene regulatory mechanisms that decode the stimulus-specific combinations of temporally modulated transcription factor activities. Single cell heterogeneity in signaling network activation and transcriptional regulation all impact Response Specificity. (b) Single-cell measurements of signaling or epigenetic events that can be interrogated to quantify Response Specificity, (c) resulting in an understanding of ligand-response distributions in health versus disease. Abbreviations: ATAC-seq, assay for transposase-accessible chromatin with sequencing; FISH, fluorescence in situ hybridization; RNA-seq, RNA sequencing; PRR, pattern recognition receptor; TNFR, Tumor necrosis factor receptor; IFNAR, interferon alpha/beta receptor; AP1, activator protein 1; ATFs, Activating transcription factors; NFκB, nuclear factor kappa B; IRFs, interferon response factors; ISGF3, interferon stimulated gene factor 3; STATs, signal transducer and activator of transcription.
However, with the advent of single cell assays, the cell-to-cell heterogeneity of macrophage populations has come into focus. Studying Response Specificity must therefore not only address the distinction of mean responses with population level (bulk) assays, but about how well the distributions of responses across single cells may be distinguished. Not until the 2010’s was appropriate single cell technology and an analytical framework (based on information theory) developed to allow insight into the distinction of single-cell response distributions. Measuring NFκB activity levels at a single timepoint, TNF dose-response specificity was found to be low; while 12 different TNF doses spanning 4 orders of magnitude were measured cells were barely able to reliably distinguish the presence of absence of TNF (61). Subsequent studies showed that the temporal dynamics of signaling activity enabled by live-cell microscopy on fluorescently tagged proteins provides information for stimulus-distinction (24, 32, 62–64). Mechanisms of positive feedback from RelA were identified as one component enabling LPS dose-specificity (65). Furthermore, across a range of doses and an array of immune stimuli targeting both TLRs and cytokine receptors, six NFκB dynamical features, termed signaling codons, were shown to be key to maximally facilitating specificity to ligand dose and ligand identity (24). All six NFκB signaling codons are determined by precise modulation of IKK activity over time: the IκBα negative feedback loop serves to amplify small differences in IKK activity (low vs medium-low) by converting them into oscillatory vs non-oscillatory NFκB trajectories. However, which sources of molecular noise are the primary drivers of the heterogeneous deployment of each NFκB signaling codon remains unclear, and how such responses are modulated by microenvironmental or polarizing cytokines is still an active area of investigation.
Single-cell studies have also begun to quantify to what extent the combinatorial activation of signaling pathways is a contributor to Response Specificity. Simultaneous measurement of NFκB and MAPKp38 in single cell macrophages at a single timepoint revealed that dose-response curves for each pathway were distinct, with MAPKp38 being digitally activated above a ligand concentration threshold (32). Therefore the combination of NFκB and MAPK with differential dose response curves may have a larger overall dose-discrimination capacity than either pathway alone. Another study that measured single cell temporal dynamics for both NFκB and JNK indicated that the two pathways combined were biologically informative and reflective of different levels of threat from pathogenic vs non-pathogenic microbes (62). The development of kinase translocation reporters for MAPK ERK, p38, JNK activity may allow evaluation of the combinatorial and temporal control of these signal transducers in single cells (58). Less is known at the single cell level about the combinatorial and dynamic control and heterogeneity of IRF signaling.
1.3. Immune response gene expression
Downstream of signaling pathways, Response Specificity is evident and may be measured at the level of immune response gene expression programs. Bulk transcriptomic studies identified sets of genes controlled by stimulus-specific combinatorial activation of signaling pathway activity (9). These gene sets could be mapped to the immune response transcription factors, identifying sets regulated by single TFs NFκB, IRF, or AP1, but also those that required two pathways, such as a cytokine set whose transcriptional activation is mediated by NFκB and whose post-transcriptional regulation of mRNA half-life is mediated by MAPKp38. Ifnb1 and Ccl5 are other well-known examples of genes requiring the combinatorial activation of NFκB and IRF (66, 67).
Many previous studies relied on clustering algorithms of transcriptomic data to identify patterns of gene expression. These approaches readily identified a handful of distinct patterns when data from multiple ligands is available (9, 37), but closer inspection reveals many more patterns of expression, some of which may only be exhibited by a single gene (66, 68). Thus analytical approaches that have true single gene resolution are critical. For example, the duration of NFκB activity could be shown to be decoded gene-specifically using a mechanistic modeling approach with differential equations rather than a statistical evaluation that requires considerations of sets of genes; for each gene the contribution of duration decoding by mRNA half-life or chromatin mechanisms could be quantified (11). A similar mechanistic modeling approach has been developed to elucidate the combinatorial transcription factor control of gene expression at single gene resolution (60) but its implementation requires a very large amount of data from different conditions.
While prior gene expression studies at the population showed that transcriptional responses are ligand-specific and could elucidate regulatory mechanisms, the quantification Response Specificity requires single cell resolution data, as cell-to-cell heterogeneity affects this quantity. Interestingly, the cell-to-cell variability present in signaling pathway dynamics and activation levels may, in principle, be either buffered or amplified by the chromatin-associated or post-transcriptional regulatory mechanisms controlling the expression of each gene. Technological developments may be required to link signaling to gene expression in the same cell, such as advances in microfluidics, image analysis, smFISH or scRNAseq (Figure 2b) (69–71). In one study in the macrophage RAW246.7 cells, after measuring NFκB signaling dynamics in response to LPS and then profiling the transcriptome of each cell at the end point, it was found that the expression of some cytokine and feedback regulator genes were correlated to the cell’s NFκB dynamics (72). In addition, certain pairs of genes possibly controlled by the same enhancer elements maintained correlated expression levels across single cells. However, a key limitation of some single cell assays is the high degree of technical noise. Further work in profiling transcriptomic Response Specificity across multiple ligands and doses are needed to understand the extent to which signaling dynamics are decoded stimulus-specifically. As with studies of signaling dynamics in sentinel cells, both statistical analysis and mechanistic modeling may prove insightful in elucidating mechanisms and sources of biological noise in single cell gene regulation.
1.4. Cytokine loops
One product of the primary signaling response is the secretion of immune response cytokines that may then be sensed in an autocrine or paracrine manner. They may function as feedback and feedforward loops to contribute to Response Specificity.
The activation of the IRF pathway by PAMPs such as LPS and poly(I:C) induce the secretion of interferon-β (IFNβ), which acts in a feedforward loop to produce ISGF3 that reinforces the IRF3-driven gene expression programs in primary response cells, and produces an almost equivalent interferon-response program neighboring bystander cells (73). A single-cell study in dendritic cells responding to LPS showed that early paracrine secretion of IFNβ in just a handful of cells was important for antiviral gene expression in the population; at later timepoints, Ifnar and Stat1-dependent IFNβ paracrine signaling downregulated inflammatory genes not uniformly, but in a fraction of the cell population (74). This presence or absence of negative feedback from Type I interferons was shown to be biologically important for distinguishing Gram-negative versus Gram-positive bacteria in BMDM responses. Bacterial class-specific production of key cytokines such as CXCL1 and TNF was diminished in either IFNAR knockouts, or when IFNβ was exogenously supplied (25, 75).
Like interferons, TNF production also may further amplify or curtail Response Specificity. Single cell studies of NFκB signaling dynamics revealed that in the presence of TNFRII, a soluble TNF inhibitor, the responses to low dose CpG or LPS stimulation became less variable by reducing the oscillatory component from response trajectories (24). In another study, blocking TNF autocrine signaling decreased the heterogeneity of NFκB signaling profiles in response to LPS, suggesting that cell-to-cell variability of signaling was in part affected by the heterogeneity of cellular secretion of TNF (72).
In the case of NLR signaling, the secretion of IL1β and IL18 is in fact critical for the activation of NFκB and MAPK pathways (76, 77) – these are the pathways that allow for gene expression responses in both primary responders and bystanders. Thus, the production, secretion, and responses to soluble cytokines, and the single cell heterogeneity of these processes, may be a feature that can either expand or restrict sentinel ability to discriminate dose or identity of a pathogen or DAMP ligand.
1.5. Diseases of impaired Response Specificity
By proposing that Response Specificity is a property of healthy macrophage function, we suggest that impaired Response Specificity contributes to disease. Healthy Response Specificity may be characterized by particular response distributions to each immune threat, and both increases or decreases in the heterogeneity may result in disease (Figure 2c). As such, in these diseases, the behavior of outliers is critical. Rheumatoid arthritis, lupus, multiple sclerosis, and Sjogren’s Syndrome all have remitting and relapsing characteristics (78–80). The intermittent nature in the presentation of multiple autoimmune diseases hints that low probability outlier events may underlie its etiology.
Indeed, both aberrant TNF and IFNβ regulation have been implicated as opposing sides of different autoimmune diseases (81). Excessive IFNβ production from dendritic cells was postulated to be an initiator of the autoimmune disease systemic lupus erythematosus (SLE) (82). As IFNβ has both feedforward and negative feedback functions on neighboring cells, the improper production of IFNβ from even a subset of cells may have significant consequences on Response Specificity. A recent single-cell study more directly couched the autoimmune disease Sjogren’s Syndrome as involving loss of Response Specificity. Loss of the NFκB negative feedback regulator Nfkbia diminished the distinguishability of macrophage responses to TNF vs poly(I:C), interestingly through the increased expression of IRF target genes in a fraction of TNF-stimulated cells (24). TNF-induced IFNβ production through IRF1 has also been implicated in rheumatoid arthritis, which could be corrected through JAK inhibitor drugs (83). Thus, the misregulation of the TNF vs. IFNβ axes in autoimmune disease may provide clues into how to correct or control loss of Response Specificity (84).
2. Context Dependence
Macrophages populate all organ systems and are therefore exposed to different cytokine microenvironments that provide instructions for tailoring the function to the local physiological state. As a result, macrophage responses and functions have evolved to be highly adaptable. Interestingly, macrophages residing in the same organ system may have different developmental origins: a portion of the resident population are longer-lived derivatives of the yolk sac or fetal liver, while another portion are monocyte-derived and migrate to the tissue from the blood stream (85). However, their immunological functionality is a product of both ontogeny and cytokine cues of the local environment, such that all tissue-resident macrophages within an organ adopt similar functions (86). Polarizing cytokines in the microenvironment can further shift the response repertoire of macrophage to allow them to carry out more specialized roles. This tuning of function is important in normal physiology in the contexts of inflammation, injury, or repair.
We thus propose the second hallmark of healthy macrophage responses is Context Dependence. Context Dependence allows for the canalization of macrophage functions, in which both signaling networks and epigenetic states are tuned to the current microenvironment to promote specialized functions while the tuning cytokine is present (Figure 3a). Originally, for monocyte-derived macrophages, a simplified paradigm of Context Dependence existed within the framework of M1 and M2 macrophage polarization (87). M1 macrophages are canonically anti-microbial, producing pro-inflammatory cytokines, and upregulating their phagocytosis ability; M2 macrophages are canonically associated with repair, producing anti-inflammatory cytokines, or growth factors. While a much more complex topology of macrophages activation states is now known to exist, pro- and anti-inflammatory M1-IFNγ and M2-IL4 states generally represent opposing extremes of macrophage context-dependent functions (23, 88).
Figure 3.
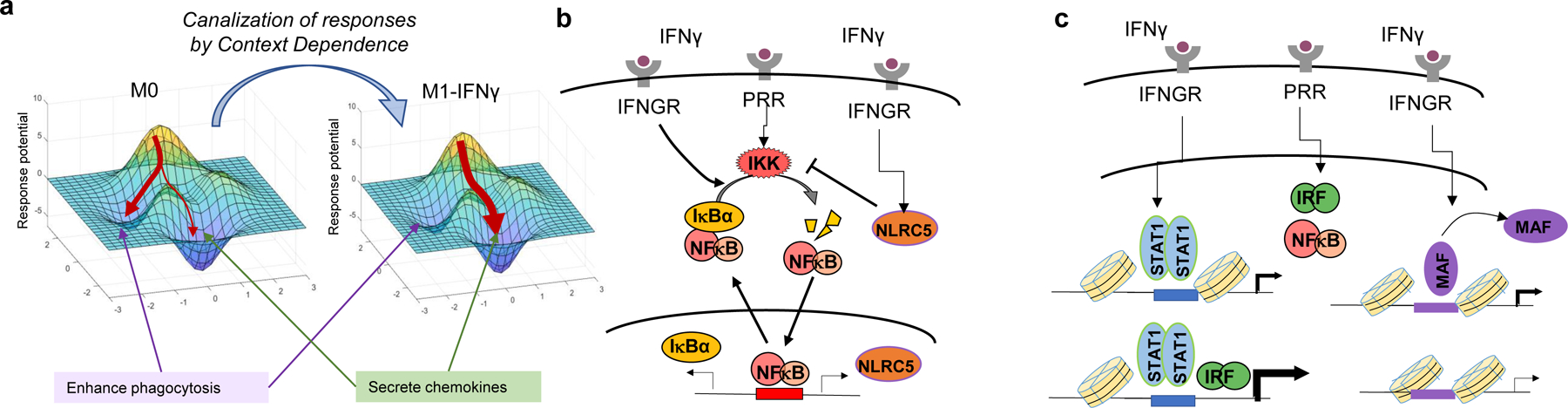
Context Dependence is mediated by microenvironmental signals that may lead to a canalization of the diverse stimulus-specific macrophage responses. (a) Cytokine context results in specialization of function by reversibly altering the epigenetic states of signaling and gene regulatory networks. Arrows pointing to regions of the response landscape represent possible responses given an inflammatory stimulus. (b) Positive and negative regulation of signaling feedback regulators by polarizing cytokines may generate context-dependent signaling profiles. (c) Epigenetic mechanisms that either hold open promoters or disassemble enhancers allow for gene-specific regulation of context-dependent responses in macrophages. Abbreviations: PRR, pattern recognition receptor; IFNGR, interferon gamma receptor.
2.1. Signaling mechanisms
What are molecular mechanisms that allow for Context Dependence? Both signaling crosstalk and epigenetic mechanisms allow macrophages to respond in relation to their microenvironment context. Generally, signaling networks that encode ligand-specific responses may be altered by polarizing cytokines that change the availability of signaling components (89). Such components may include receptors, adapters, transcription factors, feedback regulators, and even regulators of core machineries responsible for protein synthesis or decay.
Microenvironmental IFN can affect PAMP- or DAMP-induced signaling by altering expression levels of signaling pathway components: Type I interferon IFNα upregulated the expression of TLRs TLR3, TLR4, and TLR7 (90), and Type II interferon IFNγ upregulated transcription factor IRF1 that augments TLR-dependent IFN signaling (91). Modulation of feedback regulator activity is also critical: IFNβ increased NFκB activity by reducing the translation of IκBα, and in the late-phase, through increased IKK activity via expression of the viral RNA sensor RIG-I (92). IFNγ was shown to elevate NFκB activity through increased expression of proteasomal cap components that degrades the feedback inhibitors of NFκB, IκBα and IκBε (92, 93). Another cross-regulatory feedback protein has roles as a negative regulator of NFκB. NLRC5, a NOD-like receptor family member, is induced by IFNγ (94), and decreases NFκB activity via inhibition of IKKα/IKKβ phosphorylation. NLRC5 also negatively regulates type I interferon signaling at the receptor level via inhibition of RLR-mediated type I interferon responses (95) (Figure 3b).
Within the interferon pathway itself, polarizing cytokines induce negative regulators such as SOCS proteins that may also both enhance or suppress PAMP or DAMP-responses. Low dose IFNα increases baseline STAT2 and IRF9 expression without strongly activating the negative feedback regulators SOCS1, SOCS3, and USP18, thereby hypersensitizing cells to further interferon stimulation (26). On the other hand, higher levels of type I interferon may increase the expression of these negative regulators: recruitment of USP18 to the type I interferon receptor IFNAR desensitizes cells to any additional IFN-activating stimuli (96, 97), while increased SOCS1 expression suppresses responses to pathogens that activate interferon-pathway signaling like LPS (98). IFNγ was reported to upregulate the expression of SOCS3, which inhibits STAT3 activity and promote M1 macrophage activation (99). In contrast, IL4 was reported to upregulate SOCS1 but not SOSC3, thereby inhibiting STAT1 transcription factors and promoting the M2 macrophage phenotype (100). The strength of crosstalk is modulated by dose and duration of the polarizing cytokine cue, but may also be subject to other cell properties like cell-cycle phase (101).
2.2. Epigenetic mechanisms
Epigenetic mechanisms constitute a subsequent layer of control critical to context-dependent canalization of macrophage functions. Importantly, unlike signaling mechanisms, epigenetic control results in gene-specific rather than pathway-specific alterations. IFNγ-M1 and IL4-M2 polarization are associated with chromatin alterations at STAT1/IRF1 and STAT6 enriched genomic regions, respectively (102). Interestingly, macrophages exposed to multiple tuning cytokine signals simultaneously, such as the opposing polarization cytokines IFNγ and IL4, as might occur in tissue or bloodstream microenvironments, did not show extensive antagonism at the level of signaling pathway activation; instead cross-talk occurred more prominently at the gene regulatory level through gene-specific binding of STAT1 or STAT6 (103).
Interferon priming can potentiate some subsets of genes, while repressing others. In human macrophages, type II interferon IFNγ synergistically enhanced TLR-induced transcription at inflammatory genes TNF, IL6, and IL12B by recruiting STAT1 to enhancers and promoters to increase chromatin accessibility and prime genes without itself inducing transcription (91). Type I interferon IFNα/β also impacts chromatin. In cooperation with TNF, IFNα generated increased chromatin accessibility at specific sites to potentiate the pro-inflammatory effect of LPS (104). Monocytes stimulated with LPS from patients with systemic lupus erythematosus (SLE) displayed epigenomic similarity to those primed in vitro with IFNα and TNF, suggesting that IFNα exposure in vivo alters chromatin to contribute to inflammatory symptoms of SLE (104, 105). In addition to enhancing the expression of inflammatory genes, IFNγ was also found to repress M2-like genes by disassembling MAF and LDTFs PU.1 and C/EBPβ bound at select enhancers (28) (Figure 3c). At other genomic locations, IFNγ also suppressed enhancers associated with STAT3 (106). Notably, the genes at MAF enhancers were also repressed in macrophages from rheumatoid arthritis patients, suggesting that both potentiation and repression at the chromatin level by IFNγ priming may play roles in autoimmunity.
2.3. Specialized physiological functions via polarization
By affecting signaling and epigenetic mechanisms, contextual cytokines may alter macrophage functions to enhance protection of the host organism from pathogen threats or minimize collateral damage of inflammation. The presence of both type I and type II interferon in the microenvironment may prime macrophages for an LPS challenge, enhancing the expression of innate immune genes and cytokines (107). Polarization can also serve the function of resolving or more carefully regulating inflammatory processes. IFNγ priming was found to repress a portion of LPS-inducible genes, resulting in a reduction in the recruitment of neutrophils to the inflammatory site (108). These changes in the production of chemokines by macrophages can ultimately rewrite the script of systemic immune activation.
In addition to altering the level of expression of inflammatory gene programs, microenvironment context can also tune cell-to-cell heterogeneity, which is a determinant of Response Specificity. A single cell study measured ‘propagation of variance’ in gene expression within gene regulatory networks in human macrophages, and found that IFNγ+TNF, IL4, and IL10 cytokine environments each generated distinct changes in biochemical parameters within the signaling network, altering patterns of cellular heterogeneity (109). For example, IL10 signaling increased the phosphorylation and nuclear localization of ATF2, which in turn tuned the variability of ATF2 target gene expression. The adaptation of macrophage heterogeneity to cytokine microenvironments may be beneficial as a “bet-hedging” strategy that leverages an altered distribution in single cell responses to immune stimuli (109).
The microenvironment of different organ systems constitute different types of Context Dependence of macrophage function (110, 111). Tissue-specific environments selectively activate transcription factors that collaborate with PU.1 to establish distinct sets of enhancers and super-enhancer in the resident macrophage population (112, 113). This tissue-specific functional polarization of macrophages is reversible and held in place by transcription factors induced by the microenvironment. For the inflammatory potential of the peritoneum, retinoic acid (Vitamin A) from local tissues polarized peritoneal macrophages by inducing GATA6 (114). Interestingly, the expression of this peritoneal macrophage-specific gene was not required for peritoneal macrophage development and was decreased by depletion of Vitamin A signal, pointing to the role of GATA6 as a peritoneal polarization gene rather than a lineage-determining transcription factor. Thus, through constant surveillance of the contextual environment within the tissue, tissue-resident macrophages may be able to dynamically adjust their response potential (114).
Furthermore, the presence of polarizing cytokine contexts can also alter the macrophage’s metabolism. IFNβ has been shown to mediate metabolic reprogramming. For example, exposing BMDMs to live M. tuberculosis restrained glycolysis and mitochondrial stress, a phenotype recapitulated by IFNβ stimulation alone and abrogated in IFNAR knockout mice. This metabolic form of priming was hypothesized to affect macrophage responses to bacteria by decreasing macrophage energy metabolism during mycobacterial infection, though whether this phenomenon was beneficial or detrimental remains unclear (115). Thus, even in the absence of acute infection, the presence of type I interferons in the environmental milieu have marked effects on signaling nodes central to pathogen or cytokine responses (116).
2.4. Altered functions in disease contexts
While Context Dependence is a feature of healthy macrophage function, dysregulated microenvironments may also have detrimental effects on macrophage responses. Indeed, several chronic inflammatory diseases harbor aberrant cytokine microenvironments, which alter macrophage responses. Conditions common to human health include aging, which can involve an inflammatory component and has been termed inflammaging (117), or obesity, which is a disease of chronic metabolic inflammation (118, 119).
Both age and obesity have been linked to poorly regulated immune responses (120). For instance, a comparison of macrophages from old and young mice suggested that age impairs macrophage polarization, with significant decreases in M1 and M2 marker genes after exposure to polarizing ligands (121). Failure of macrophages to appropriately polarize may lead to less effective context-dependent immune responses. Similarly inflammation-associated aging may affect macrophage context-dependent responses through metabolic, signaling, and epigenetic mechanisms (122). This chronic low-grade inflammation has been linked to immunosenescence and is marked by increases in circulating pro-inflammatory cytokines such as TNF and IL6 (123–126). Furthermore, the Context Dependence of responses in aged macrophages is tissue specific. Within aging skeletal tissues, macrophages adopt a more M2-like phenotype (127), but the pro-inflammatory cytokine environment is hypothesized to produce age-associated pro-inflammatory M2-like macrophages (122). Further work may delineate the effect of age on the ligand responses of macrophages from different tissues and under a variety of polarization conditions.
Obesity is a second disease-associated inflammatory context that affects macrophage responses. Obesity causes inflammation of the adipose tissue, due to the release of IFNγ by natural killer cells (128). In lean mice, tissue-resident adipose macrophages retained an anti-inflammatory M2-like state. However, under a high fat diet, macrophages in the adipose tissue accumulate and adopt an M1-like pro-inflammatory phenotype marked by increased TNF and IL6 expression (129). Interestingly, macrophages in obese IFNγ knockout mice shifted towards an M2 phenotype, and organisms displayed improvements in insulin sensitivity (130). It remains to be seen by what mechanisms the polarized states of macrophages due to metabolic inflammation influences the effectiveness of their innate immune responses to immune threats.
3. Stimulus Memory
Stimulus Memory refers to a longer-term property of macrophages to record past exposures within its epigenetic state. The identity of differentiated cells is defined by the epigenomic enhancer landscape, which is held in place by the stable expression of a set of cell-type-specific transcription factors (131, 132). This category of transcription factors, termed lineage-determining transcription factors (LDTFs), are pioneer factors (133, 134) that have structural elements that enable them to bind to nucleosomal DNA and adjust the enhancer landscape during development (135). While the epigenetic landscape determines cell identity, epigenome plasticity allows differentiated cells to adapt their functions to environmental cues (Figure 4a).
Figure 4.
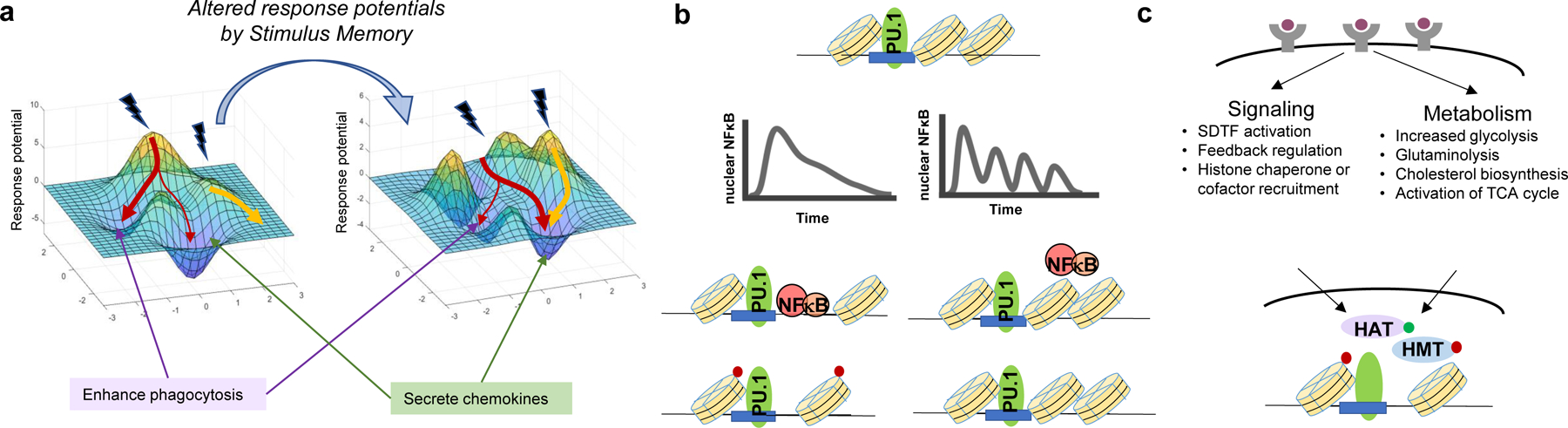
Stimulus Memory involves prior exposure altering epigenetic states of signaling and gene regulatory networks. (a) Stimulus Memory is mediated for example by changes to the chromatin enhancer landscape, altering response potential after the initial stimulus has subsided. Arrows pointing to regions of the response landscape represent possible responses given an inflammatory stimulus. (b) Stimulus-specific non-oscillatory activity of SDTFs opens chromatin in collaboration with cofactors and chromatin-remodeling enzymes. (c) Both signaling pathway activation and alterations to metabolic pathway activity are critical arms for generating innate immune memory. Abbreviations: HAT, histone acetyltransferase; HMT, histone methyltransferase; SDTF, signal-dependent transcription factor; TCA, tricarboxylic acid; NFκB, nuclear factor kappa B; PU.1, PU-box binding factor.
The third functional hallmark of macrophages responses, Stimulus Memory, concerns the malleability of the developmentally established epigenetic landscape, whereby signal-dependent transcription factors (SDTFs) are able to trigger the formation of new enhancers. When those stimulus-specific changes to the chromatin epigenome are not reversed when the SDTF activity ceases, epigenetic memory of the prior exposure event is formed. Stimulus Memory is thus distinguished from Context Dependence, in that that the inducing signal need not persist, and that signal is often generated by PAMPs and DAMPs rather than polarizing cytokines. Thus, Stimulus Memory stores marks of previous exposure to influence sentinel cell responses to future stimuli.
3.1. Transcription factors, nucleosome remodelers, metabolites
Several classes of molecules in the nucleus mediate Stimulus Memory: transcription factors, nucleosome remodelers, metabolites. For stimulus-induced epigenetic programming to occur, SDTFs, which bind to DNA, must be activated (136). SDTFs like AP1, NFκB and IRFs are activated stimulus-specifically by immune threats, but unlike LDTFs, they are not cell-type specific. Because the combinations and dynamics of SDTF activities are stimulus-specific, epigenetic memory may also be stimulus specific. Patterns of SDTF-DNA binding were shown to enable the stimulus-specific formation of de novo enhancers marked by H3K4me1 deposition, a covalent modification to the chromatin landscape (136). More recently, the mechanisms by which the temporal dynamics of SDTF activity control de novo enhancer formation have also been elucidated. In macrophages, non-oscillatory NFκB activity allowed the necessary continuous nuclear residence time for nucleosome eviction and eventual H3K4me1 deposition (27). Because cytokine and viral PAMPS induced oscillatory NFκB activity, while bacterial PAMPS induced non-oscillatory activity (24), the stimulus-specific dynamics of transcription factor activity determined the extent of epigenomic enhancer formation (27) (Figure 4b).
Because SDTFs like NFκB and IRF are not LDTFs, it seemed unlikely that they could impart long-term changes to the epigenome. However, biochemical and cryo-electron microscopy studies suggested that SDTFs like NFκB could bind to nucleosomal DNA and potentially displace histone H1 (137, 138). The spontaneous unwrapping and rewrapping ‘breathing’ of DNA around the histone core octamer suggests that even SDTFs could invade the nucleosomal DNA-histone octamer complex (139, 140), by preventing rewrapping of the SDTF-bound sections of DNA (141). However, the rates of spontaneous rewrapping were rapid enough that NFκB could only bind to its cognate motif when it was positioned at the edge of the nucleosome, and not its interior. Thus, while stimulus-specific epigenetic memory is mediated by the activation of SDTFs, only a specific nucleosomes may be targeted, and cooperative mechanisms from other proteins may be required (142).
LDTFs are distinct in each cell type, and their binding to chromatin regions contributes to establishing the cell type’s gene expression potential. In macrophages, PU.1 and C/EBPβ are the LDTFs that are known to promote nucleosome-free regions and establish macrophage identity (143). LDTFs, being pioneer factors, have the ability to directly perturb nucleosome structure (144) and thus cooperatively hold open chromatin regions for gene expression. LDTFs mark potential chromatin regions for the subsequent binding of non-pioneer factors and other nucleosome remodeling complexes, resulting in the deposition of histone modifications that produce epigenetic memory. In macrophages, the LDTF PU.1 colocalized with areas of SDTF binding and H3K4me1 deposition upon stimulation (136, 145). H3K4me1 marks active enhancers and persists even after H3K27ac and H3K4me3, a mark of active enhancers or promoters, is lost (146). Of note, the cooperative action of LDTFs results in cell-type specificity in which regions become epigenetically programmed upon SDTF activation (147, 148). Other histone chaperone proteins such as FACT (149), and ATP-dependent nucleosome remodelers such as SWI/SNF (human ortholog SMARCA/B) or RSC (human ortholog BAF) may catalyze unwrapping or nucleosome sliding (150, 151), thus allowing for SDTF binding (152).
Beyond chromatin modifying proteins, metabolites and alterations to metabolic flux are an integral arm of epigenetic memory (153). These mechanisms are driven by the reliance of many epigenetic modifications on metabolic processes, such as one-carbon metabolism for histone and DNA methylation (154) and generation of acetate pools from Acetyl-CoA for histone acetylation (155–157). Furthermore, mevalonate and cholesterol biosynthesis pathways are also downstream of Acetyl-CoA production, and influence the innate immune response through feedforward mechanisms at the receptor that activate PI3K signaling and mTOR (158). Increased activity of catabolic processes including glycolysis and glutaminolysis also mediate trained immunity: in human monocytes stimulated with β-glucan, glutaminolysis and cholesterol metabolism resulted in the accumulation of fumarate, which inhibited KDM5 histone demethylases to promote epigenetic reprogramming (159). Fumarate treatment of monocytes itself also mimicked β-glucan treatment by increasing both H3K4me3 and H3K27ac deposition. In human macrophages exposed to IL4, alpha-ketoglutarate, a TCA cycle intermediate, promoted demethylation of H3K27me3 in a manner dependent on Jmjd3, a histone demethylase (160). Therefore, rewiring of metabolic circuits is a key component for initiating and sustaining immune memory conveyed through histone modifications (161, 162) (Figure 4c).
3.2. Memory of prior infection
Stimulus Memory of past exposures serve the physiological purpose of changing future gene expression responses. Two main categories of innate immune memory, tolerance and trained immunity, are generated by different stimuli and alter responses in opposing directions (163). Tolerance was first described in mice surviving a lethal dose of endotoxin after having received a sublethal dose (164) . It was also observed in macrophages exposed to a primary stimulus of high concentrations of lipopolysaccharide (LPS), and after a washout of up to five days, stimulated again with a secondary stimulus (165). The resulting blunted second response was accompanied by nucleosome repositioning and histone H3 lysine methyltransferase G9a, which generated heterochromatin assembly and epigenetic silencing (166). Interestingly however, tolerance is dose-dependent: when high doses of LPS, P3CSK, and poly(I:C) are diluted 100–10000 fold, hyper-responsiveness rather than tolerance may result (163). Epigenetic changes resulting in tolerance are stimulus-specific, but it remains to be seen how much the responses to heterologous secondary stimulation are also altered stimulus-specifically.
Trained immunity, involving a hyper-response of key immune regulatory genes upon stimulation, is a key outcome of stimulus-specific epigenetic memory. In response to the fungal wall component β-glucan, or Candida albicans, monocytes responded to secondary stimulation with much higher production of key cytokines like TNF and IL6 (167, 168). Immune training was associated with increases in H3K4me3 and H3K4me1 (marking enhancers), even after loss of H3K27ac (marking active promoters), suggesting a stable epigenetic modification of enhancer regions help maintain trained immune memory. IFNγ secreted after initial challenge with C. neoformans was also shown to generate innate immune memory for up to 70 days, resulting in hyper responses of pro-inflammatory cytokines upon a secondary challenge (169). Furthermore, dendritic cells also show stimulus-specific trained immune memory. DCs treated with the fungal pathogen Cryptococcus neoformans, transplanted into naïve mice, and challenged again, showed increased interferon response gene expression, as well as increased production of C. neoformans cytokines. This apparent memory was inhibited by treatment with histone methylase inhibitors (170).
Though studies of the epigenetic plasticity and memory of innate immune responses have focused on immune cells like macrophages, there is emerging evidence that fibroblasts, stromal cells, and hematopoietic stem cells may also be pliable to stimulus-specific epigenetic programming (171). These cells have longer lifespans than circulating monocytes, and may thus be the optimal messengers to carrying memory of past exposures (172, 173). Other cell types with longer memory may include fibroblasts, as chromatin marks deposited after IFNβ stimulation led to faster and increased expression of interferon genes on a second stimulation (174). Epithelial stem cells were also shown to maintain memory of a primary response through sustained increase in chromatin accessibility at key inflammatory response genes, heightening responses to subsequent inflammatory stimuli (175).
3.3. Stimulus Memory via vaccination
Given its role in physiology, several attempts have been made to harness Stimulus Memory and the training of innate immunity through vaccination. The tuberculosis vaccine BCG (Bacillus Calmette-Guérin) is a well-known example (176), where vaccination with this attenuated bacterium provides broad protection against multiple bacterial and fungal organisms through hyper-response of key genes upon secondary stimulation (177). BCG-trained immunity not only affected monocytes via H3K27ac histone modifications (178), but also impacted the epigenetic landscape of hematopoietic stem cells (179, 180). BCG-trained HSCs led to epigenetically modified monocytes and macrophages that had alterations in H3K4me1, H3K4me3, and H3K27ac, and cleared tuberculosis infections more effectively than naïve macrophages (180). Trained immunity of progenitor cells like HSCs may explain the lasting effects of innate immune vaccination. Importantly, while programming the epigenetic landscape is specific to the stimulus, unlike vaccines targeted at adaptive immunity that aim to generate memory B-cells, innate immune vaccination by BCG provides heterologous effects and protects individuals from many other bacterial, viral, or fungal pathogen threats (181, 182).
Tolerance and immune training via treatment with LPS or BCG, respectively, has also been suggested as a potential avenue for the modulation of autoimmune diseases like systemic sclerosis (183), which is marked by fibrosis as a result of chronic but ‘sterile’ inflammation (184). Treatment of macrophages with low-dose LPS generated a tolerized phenotype that reduced inflammation-related fibrosis in a mouse model. On the other hand, BCG exposure generated a trained phenotype with increased production of pro-inflammatory cytokines, exacerbating the fibrotic process. LPS and BCG generated unique epigenomic changes, with gene-specific changes in chromatin marks, including H3K4me3 (183).
3.4. Diseases of dysregulated Stimulus Memory
Severe pathology can result from dysregulated immune memory. Sepsis, which involves hyperactivation of immune response as well as immune paralysis that prevents the clearance of bacteria in the bloodstream, affects millions of people yearly and nearly 1/3 of hospital deaths are caused by sepsis. Both tolerance and trained immunity play roles. Tolerance serves to eliminate hyper-response on secondary stimulation, but misapplied regulation of tolerance results in poor host defense to secondary exposures to bacterial stimuli. Interestingly, the metabolic output of TCA cycle decarboxylation, itaconate, promoted tolerance in human monocytes, while β-glucan inhibited IRG1, the enzyme that promotes itaconate synthesis, leading instead to an enhanced secondary immune response (185). The ability of specific stimuli to generate trained immunity and revert disease-causing tolerance could lead to additional methods to modulate the immune system during or after infection.
Another disease of dysregulated immune memory is hyper IgD syndrome (HIDS), an inborn error of metabolism where mevalonate kinase deficiency leads to accumulation of mevalonate (186). Monocytes and macrophages in these patients produce higher amounts of TNF, IL6, and IL1b, and anti-TNF and antii-IL1 therapies have only been partially effective (187). The metabolite mevalonate was shown to be critical in β-glucan- and oxLDL-induced trained immunity by driving the mTOR pathway, activating the TCA cycle, and generating acetyl-CoA needed for altered H3K27ac at inflammatory genes (188). The chronically trained immunity state of macrophages due to elevated mevalonate may be a cause of the ‘sterile’ inflammatory phenotype seen in these patients, such as febrile attacks, arthritis, and skin lesions (188). Importantly, administration of statins blocked the mevalonate-cholesterol synthesis pathway, attenuating trained immunity and reducing inflammatory attacks (189).
Outlook
The physiological roles of macrophages are many(190, 191), and they are precisely regulated. Here we have proposed that these varied physiological roles depend on a key set of three functional hallmarks. For each of these hallmarks, addressing outstanding questions will bring us closer to harnessing and controlling macrophage function, either for diagnostics or for the treatment of disease. We discuss a few open questions below and outline further potential lines of inquiry in the Future Issues box.
Might Response Specificity inform us about risk for inflammatory disease? Response Specificity, a property of macrophages that is affected by both cytokine context and immune memory, may prove a convenient metric for measuring health and disease states. In multiple immune diseases, noisy or ineffective recognition of an inflammatory threat lead to autoimmunity or faulty pathogen clearance. The functional health of the innate immune system, which is affected by context or prior exposures, could in the future be measured by perturbing monocytes isolated from peripheral blood, and profiling the resulting transcriptome or epigenome. The diagnosis or prognosis for a wide variety of diseases depends on immune system function, including autoimmune diseases, cancers, or neurodegenerative diseases, to name a few. It remains to be seen to what extent monocyte and macrophage Response Specificity is reflective of risk or disease stage of each of these.
Will understanding Context Dependence allow us to predictably tune macrophage responses with microenvironmental cues? A central difficulty in understanding Context Dependence has been to clarify the effect of different ligands or ligand combinations, their doses, their duration, or temporal order of exposure. In vitro studies have isolated the effects of particular combinations of contextual cytokines, often recapitulating Context Dependence responses that arise by exposure to real pathogens or from inflammatory diseases. Further work in uncovering the signaling and epigenetic mechanisms that channel macrophage responses, may in the future allow us to use specific adjuvants to manipulate favorable microenvironment conditions for cancer, atherosclerosis, or metabolic disorders.
Might we harness Stimulus Memory to strengthen innate immunity to improve human health? A recent study reported that four-weeks of aerobic exercise prior to surgery created a lasting phenotype of immune tolerance in Kupffer cells, improving ischemia-reperfusion injury outcomes (192). However, further study is needed to understand the physiological consequences of training innate immunity. For example, innate immune memory may play roles in post-COVID19 immunity or inflammatory sequelae. A recent study on convalescing COVID19 patients indicated altered monocyte subsets after COVID, with increased chromatin accessibility at inflammatory genes in patients covering from COVID19, suggestive of trained immunity. CD14+ and CD16+ monocytes from convalescing patients maintained epigenetic modifications and had increased IL6 and IL-1β production on subsequent stimulation with spike-nCoV pseudovirus (193). It remains to be determined whether this trained immunity results in an effect similar to vaccination, protecting the individual from subsequent infection, or whether the subsequent hyperinflammatory responses predisposes individuals to syndromes of long-COVID.
Summary Points.
The health of the immune system depends on the health of macrophage function, as they are the orchestrators of immune activation. Three functional hallmarks mark healthy macrophage responses: Response Specificity, Context Dependence, Stimulus Memory.
Response Specificity is the ability to single macrophage cells to selectively activate particular gene programs appropriate to the pathogen, and is evaluated by overlap of single cell ligand-response distributions.
Context Dependence refers to the canalization of macrophage responses relative to polarizing cytokines or signals from the microenvironment.
Stimulus Memory is the ability of macrophages to store specific marks of prior exposures stably within the epigenome, altering future responses.
Immunological diseases involving macrophage responses arise from combinatorial dysregulation of these functional properties. Understanding mechanisms that generate these functions will allow us to measure them for diagnosis or manipulate them for treatment.
Future Issues.
As regulators of Response Specificity, what mechanisms are the key sources of cell-to-cell heterogeneity and do they cause pathology?
What features of Response Specificity are predictive of innate immune health and which are impaired when immune health is compromised by so-called ‘pre-existing conditions’?
Context Dependence is regulated by polarizing cytokines, but what are the other microenvironmental components such as nutrients?
In understanding Context Dependence mechanisms operating on signaling and epigenetic networks may we be able to develop predictive interventions to improve macrophage function?
In encoding Stimulus Memory, what cofactors assist immune response transcription factors in evicting nucleosomes and establishing de novo enhancers?
Is Stimulus Memory sufficiently long and reliable such that trained immune precursor cells may produce trained differentiated macrophages that can provide health benefits?
Sidebar element.
In comparison to macrophages, other cell types can exhibit either effector or sentinel functions (7). Fibroblasts, which form part of the connective tissue, are tissue-resident sentinels, similarly express PAMP, DAMP, and cytokine receptors, and activate stimulus-specific immune response genes upon ligand challenge (8–11). Endothelial cells, placed in a prime position to respond to circulating endotoxins (12, 13), are also sentinels that produce inflammatory cytokines to mobilize other immune cells (14–16). However, these structural sentinel cells do not exhibit the strong effector functions of macrophages. Conversely, neutrophils and NK cells are not tissue-resident and do not serve in primary roles as stimulus-specific sentinels. However, they each share some of the effector functions of macrophages such as phagocytosis or release of anti-pathogen lytic enzymes, respectively (17–19).
Acknowledgments
We thank the Hoffmann lab for discussion. KMS is supported by the UCLA Medical Scientist Training Program (NIH NIGMS T32 GM008042). Research in the Hoffmann lab on described topics is funded by NIH R01AI127867, R01AI132835, R01AI127864, R01GM117134.
Terms and Definitions
- Sentinel cell
a cell that conveys messages about the environment to activate other cells
- PAMP
pathogen-associated molecular pattern; pathogen-derived molecules recognized by macrophage receptors
- DAMP
damage-associated molecular pattern; host-cell derived molecules indicative of damage like DNA, histones, ATP, and other nuclear or cytosolic proteins
- PRR
pattern recognition receptor; sensors that detect molecules related to pathogens (PAMPs) or cell damage (DAMPs)
- Polarization
the ability of macrophage to adopt specialized functions as an adaptation to contextual cytokines or other microenvironmental signals
- M1/M2 macrophages
two extremes of macrophage polarization, often generated by IFNγ or IL4 cytokine contexts, respectively
- Priming
microenvironment context sets up the epigenetic landscape for stronger induction upon PAMP, DAMP, or cytokine stimuli, without return to baseline
- SDTF
signal-dependent transcription factor; transcription factors activated upon an extracellular stress signal
- LDTF
lineage-determining transcription factor; transcription factors that establish cell type by remodeling the enhancer landscape
- Pioneer factor
a transcription factor that contains a DNA anchoring α helix that allows direct binding to nucleosomal DNA
- Tolerization
retainment of epigenetic histone modifications after a first exposure that suppresses the response to a second
- Trained immunity
retainment of epigenetic histone modifications after a first exposure that generates hyper-inflammatory response to a second
Footnotes
Disclosure Statement
The authors have no conflicts of interest to disclose.
References
- 1.Mechnikov I 1908. The Nobel Prize in Physiology or Medicine 1908 NobelPrize.org. www.nobelprize.org
- 2.Rivera A, Siracusa MC, Yap GS, Gause WC. 2016. Innate cell communication kick-starts pathogen-specific immunity. Nat Immunol 17(4):356–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sreejit G, Fleetwood AJ, Murphy AJ, Nagareddy PR. 2020. Origins and diversity of macrophages in health and disease. Clinical & Translational Immunology 9(12):e1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uderhardt S, Martins AJ, Tsang JS, Lämmermann T, Germain RN. 2019. Resident Macrophages Cloak Tissue Microlesions to Prevent Neutrophil-Driven Inflammatory Damage. Cell 177(3):541–555.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hume PS, Gibbings SL, Jakubzick CV, Tuder RM, Curran-Everett D, et al. 2020. Localization of Macrophages in the Human Lung via Design-based Stereology. Am J Respir Crit Care Med 201(10):1209–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Epelman S, Lavine KJ, Randolph GJ. 2014. Origin and Functions of Tissue Macrophages. Immunity 41(1):21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krausgruber T, Fortelny N, Fife-Gernedl V, Senekowitsch M, Schuster LC, et al. 2020. Structural cells are key regulators of organ-specific immune responses. Nature 583(7815):296–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bautista-Hernández LA, Gómez-Olivares JL, Buentello-Volante B, Bautista-de Lucio VM. 2017. Fibroblasts: the unknown sentinels eliciting immune responses against microorganisms. European Journal of Microbiology and Immunology 7(3):151–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng CS, Behar MS, Suryawanshi GW, Feldman KE, Spreafico R, Hoffmann A. 2017. Iterative Modeling Reveals Evidence of Sequential Transcriptional Control Mechanisms. Cell Systems 4(3):330–343.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davidson S, Coles M, Thomas T, Kollias G, Ludewig B, et al. 2021. Fibroblasts as immune regulators in infection, inflammation and cancer. Nature Reviews Immunology, pp. 1–14 [DOI] [PubMed] [Google Scholar]
- 11.Sen S, Cheng Z, Sheu KM, Chen YH, Hoffmann A. 2020. Gene Regulatory Strategies that Decode the Duration of NFκB Dynamics Contribute to LPS- versus TNF-Specific Gene Expression. Cell Syst 10(2):169–182.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Opitz B, Püschel A, Beermann W, Hocke AC, Förster S, et al. 2006. Listeria monocytogenes activated p38 MAPK and induced IL-8 secretion in a nucleotide-binding oligomerization domain 1-dependent manner in endothelial cells. J Immunol 176(1):484–90 [DOI] [PubMed] [Google Scholar]
- 13.Opitz B, Eitel J, Meixenberger K, Suttorp N. 2009. Role of Toll-like receptors, NOD-like receptors and RIG-I-like receptors in endothelial cells and systemic infections. Thromb Haemost 102(6):1103–9 [DOI] [PubMed] [Google Scholar]
- 14.Anand AR, Cucchiarini M, Terwilliger EF, Ganju RK. 2008. The tyrosine kinase Pyk2 mediates lipopolysaccharide-induced IL-8 expression in human endothelial cells. J Immunol 180(8):5636–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tissari J, Sirén J, Meri S, Julkunen I, Matikainen S. 2005. IFN-alpha enhances TLR3-mediated antiviral cytokine expression in human endothelial and epithelial cells by up-regulating TLR3 expression. J Immunol 174(7):4289–94 [DOI] [PubMed] [Google Scholar]
- 16.Andonegui G, Zhou H, Bullard D, Kelly MM, Mullaly SC, et al. 2009. Mice that exclusively express TLR4 on endothelial cells can efficiently clear a lethal systemic Gram-negative bacterial infection. J. Clin. Invest, p. JCI36411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galli SJ, Borregaard N, Wynn TA. 2011. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol 12(11):1035–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prame Kumar K, Nicholls AJ, Wong CHY. 2018. Partners in crime: neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res 371(3):551–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paul S, Lal G. 2017. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol 8: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akira S, Uematsu S, Takeuchi O. 2006. Pathogen Recognition and Innate Immunity. Cell 124(4):783–801 [DOI] [PubMed] [Google Scholar]
- 21.Beutler BA. 2009. TLRs and innate immunity. Blood 113(7):1399–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mogensen TH. 2009. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin Microbiol Rev 22(2):240–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, et al. 2014. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 41(1):14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adelaja A, Taylor B, Sheu KM, Liu Y, Luecke S, Hoffmann A. 2021. Six distinct NFκB signaling codons convey discrete information to distinguish stimuli and enable appropriate macrophage responses. Immunity 54(5):916–930.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gottschalk RA, Dorrington MG, Dutta B, Krauss KS, Martins AJ, et al. 2019. IFN-mediated negative feedback supports bacteria class-specific macrophage inflammatory responses. Elife 8: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kok F, Rosenblatt M, Teusel M, Nizharadze T, Gonçalves Magalhães V, et al. 2020. Disentangling molecular mechanisms regulating sensitization of interferon alpha signal transduction. Molecular Systems Biology 16(7):e8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng QJ, Ohta S, Sheu KM, Spreafico R, Adelaja A, et al. 2021. NF-κB dynamics determine the stimulus specificity of epigenomic reprogramming in macrophages. Science 372(6548):1349–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang K, Park SH, Chen J, Qiao Y, Giannopoulou E, et al. 2017. Interferon-γ Represses M2 Gene Expression in Human Macrophages by Disassembling Enhancers Bound by the Transcription Factor MAF. Immunity 47(2):235–250.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng Q, Behzadi F, Sen S, Ohta S, Spreafico R, et al. 2019. Sequential conditioning-stimulation reveals distinct gene- and stimulus-specific effects of Type I and II IFN on human macrophage functions. Scientific Reports 9(1):5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, et al. 2000. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between Toll-like receptors. Proceedings of the National Academy of Sciences 97(25):13766–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeFelice MM, Clark HR, Hughey JJ, Maayan I, Kudo T, et al. 2019. NF-κB signaling dynamics is controlled by a dose-sensing autoregulatory loop. Sci. Signal 12(579):eaau3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gottschalk RA, Martins AJ, Angermann BR, Dutta B, Ng CE, et al. 2016. Distinct NF-κB and MAPK Activation Thresholds Uncouple Steady-State Microbe Sensing from Anti-pathogen Inflammatory Responses. Cell Systems 2(6):378–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoffmann A, Levchenko A, Scott ML, Baltimore D. 2002. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science 298(5596):1241–45 [DOI] [PubMed] [Google Scholar]
- 34.Zhang Q, Gupta S, Schipper DL, Kowalczyk GJ, Mancini AE, et al. 2017. NF-κB Dynamics Discriminate between TNF Doses in Single Cells. Cell Systems 5(6):638–645.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nau GJ, Richmond JFL, Schlesinger A, Jennings EG, Lander ES, Young RA. 2002. Human macrophage activation programs induced by bacterial pathogens. Proceedings of the National Academy of Sciences 99(3):1503–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Werner SL, Barken D, Hoffmann A. 2005. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science 309(5742):1857–61 [DOI] [PubMed] [Google Scholar]
- 37.Amit I, Garber M, Chevrier N, Leite AP, Donner Y, et al. 2009. Unbiased Reconstruction of a Mammalian Transcriptional Network Mediating Pathogen Responses. Science 326(5950):257–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dehne N, Jung M, Mertens C, Mora J, Weigert A. 2016. Macrophage Heterogeneity During Inflammation. In Compendium of Inflammatory Diseases, ed Parnham MJ, pp. 865–74. Basel: Springer [Google Scholar]
- 39.Gordon S, Taylor PR. 2005. Monocyte and macrophage heterogeneity. Nat Rev Immunol 5(12):953–64 [DOI] [PubMed] [Google Scholar]
- 40.Kawai T, Akira S. 2011. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 34(5):637–50 [DOI] [PubMed] [Google Scholar]
- 41.Chen G, Shaw MH, Kim Y-G, Nuñez G. 2009. NOD-Like Receptors: Role in Innate Immunity and Inflammatory Disease. Annual Review of Pathology: Mechanisms of Disease 4(1):365–98 [DOI] [PubMed] [Google Scholar]
- 42.Rehwinkel J, Gack MU. 2020. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol 20(9):537–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ablasser A, Chen ZJ. 2019. cGAS in action: Expanding roles in immunity and inflammation. Science 363(6431):eaat8657. [DOI] [PubMed] [Google Scholar]
- 44.Takeuchi O, Akira S. 2010. Pattern Recognition Receptors and Inflammation. Cell 140(6):805–20 [DOI] [PubMed] [Google Scholar]
- 45.Botos I, Segal DM, Davies DR. 2011. The Structural Biology of Toll-like Receptors. Structure 19(4):447–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kawasaki T, Kawai T. 2014. Toll-Like Receptor Signaling Pathways. Front. Immunol 5: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Devarkar SC, Schweibenz B, Wang C, Marcotrigiano J, Patel SS. 2018. RIG-I Uses an ATPase-Powered Translocation-Throttling Mechanism for Kinetic Proofreading of RNAs and Oligomerization. Mol Cell 72(2):355–368.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, et al. 2010. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. PNAS 107(26):11942–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sandri S, Rodriguez D, Gomes E, Monteiro HP, Russo M, Campa A. 2008. Is serum amyloid A an endogenous TLR4 agonist? J Leukoc Biol 83(5):1174–80 [DOI] [PubMed] [Google Scholar]
- 50.Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, et al. 2007. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med 13(9):1042–49 [DOI] [PubMed] [Google Scholar]
- 51.Kim S, Kim SY, Pribis JP, Lotze M, Mollen KP, et al. 2013. Signaling of High Mobility Group Box 1 (HMGB1) through Toll-like Receptor 4 in Macrophages Requires CD14. Mol Med 19(1):88–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu S, Cai X, Wu J, Cong Q, Chen X, et al. 2015. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347(6227):aaa2630–aaa2630 [DOI] [PubMed] [Google Scholar]
- 53.Chen H, Jiang Z. 2013. The essential adaptors of innate immune signaling. Protein Cell 4(1):27–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng Z, Taylor B, Ourthiague DR, Hoffmann A. 2015. Distinct single-cell signaling characteristics are conferred by the MyD88 and TRIF pathways during TLR4 activation. Sci. Signal 8(385):ra69–ra69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Covert MW, Leung TH, Gaston JE, Baltimore D. 2005. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science 309(5742):1854–57 [DOI] [PubMed] [Google Scholar]
- 56.Shih VF, Kearns JD, Basak S, Savinova OV, Ghosh G, Hoffmann A. 2009. Kinetic control of negative feedback regulators of NF-kappaB/RelA determines their pathogen- and cytokine-receptor signaling specificity. Proc Natl Acad Sci U S A 106(24):9619–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoffmann A, Levchenko A, Scott ML, Baltimore D. 2002. The IκB-NF-κB Signaling Module: Temporal Control and Selective Gene Activation. Science 298(5596):1241–45 [DOI] [PubMed] [Google Scholar]
- 58.Regot S, Hughey JJ, Bajar BT, Carrasco S, Covert MW. 2014. High-sensitivity measurements of multiple kinase activities in live single cells. Cell 157(7):1724–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park JM, Greten FR, Wong A, Westrick RJ, Arthur JSC, et al. 2005. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis--CREB and NF-kappaB as key regulators. Immunity 23(3):319–29 [DOI] [PubMed] [Google Scholar]
- 60.Wang N, Lefaudeux D, Mazumder A, Li JJ, Hoffmann A. 2021. Identifying the combinatorial control of signal-dependent transcription factors. PLOS Computational Biology 17(6):e1009095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheong R, Rhee A, Wang CJ, Nemenman I, Levchenko A. 2011. Information transduction capacity of noisy biochemical signaling networks. science 334(6054):354–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lane K, Andres-Terre M, Kudo T, Monack DM, Covert MW. 2019. Escalating Threat Levels of Bacterial Infection Can Be Discriminated by Distinct MAPK and NF-κB Signaling Dynamics in Single Host Cells. Cell Systems 8(3):183–196.e4 [DOI] [PubMed] [Google Scholar]
- 63.Nelson RH, Nelson DE. 2018. Signal Distortion: How Intracellular Pathogens Alter Host Cell Fate by Modulating NF-κB Dynamics. Front Immunol 9:2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Selimkhanov J, Taylor B, Yao J, Pilko A, Albeck J, et al. 2014. Accurate information transmission through dynamic biochemical signaling networks. Science 346(6215):1370–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sung M-H, Li N, Lao Q, Gottschalk RA, Hager GL, Fraser IDC. 2014. Switching of the relative dominance between feedback mechanisms in lipopolysaccharide-induced NF-κB signaling. Sci Signal 7(308):ra6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tong AJ, Liu X, Thomas BJ, Lissner MM, Baker MR, et al. 2016. A Stringent Systems Approach Uncovers Gene-Specific Mechanisms Regulating Inflammation. Cell 165(1):165–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, et al. 2009. A Unifying Model for the Selective Regulation of Inducible Transcription by CpG Islands and Nucleosome Remodeling. Cell 138(1):114–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sheu KM, Luecke S, Hoffmann A. 2019. Stimulus-specificity in the responses of immune sentinel cells. Current Opinion in Systems Biology 18:53–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee RE, Walker SR, Savery K, Frank DA, Gaudet S. 2014. Fold change of nuclear NF-kappaB determines TNF-induced transcription in single cells. Mol Cell 53(6):867–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tay S, Hughey JJ, Lee TK, Lipniacki T, Quake SR, Covert MW. 2010. Single-cell NF-κB dynamics reveal digital activation and analogue information processing. Nature 466(7303):267–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Van Valen DA, Kudo T, Lane KM, Macklin DN, Quach NT, et al. 2016. Deep Learning Automates the Quantitative Analysis of Individual Cells in Live-Cell Imaging Experiments. PLoS Comput Biol 12(11):e1005177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lane K, Van Valen D, DeFelice MM, Macklin DN, Kudo T, et al. 2017. Measuring Signaling and RNA-Seq in the Same Cell Links Gene Expression to Dynamic Patterns of NF-κB Activation. Cell Systems 4(4):458–469.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ourthiague DR, Birnbaum H, Ortenlöf N, Vargas JD, Wollman R, Hoffmann A. 2015. Limited specificity of IRF3 and ISGF3 in the transcriptional innate-immune response to double-stranded RNA. Journal of Leukocyte Biology 98(1):119–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shalek AK, Satija R, Shuga J, Trombetta JJ, Gennert D, et al. 2014. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 510(7505):363–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peignier A, Parker D. 2021. Impact of Type I Interferons on Susceptibility to Bacterial Pathogens. Trends in Microbiology [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barker BR, Taxman DJ, Ting JP-Y. 2011. Cross-regulation between the IL-1β/IL-18 processing inflammasome and other inflammatory cytokines. Curr Opin Immunol 23(5):591–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Afonina IS, Müller C, Martin SJ, Beyaert R. 2015. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity 42(6):991–1004 [DOI] [PubMed] [Google Scholar]
- 78.Aletaha D, Smolen JS. 2018. Diagnosis and Management of Rheumatoid Arthritis: A Review. JAMA 320(13):1360–72 [DOI] [PubMed] [Google Scholar]
- 79.Buch MH, Eyre S, McGonagle D. 2021. Persistent inflammatory and non-inflammatory mechanisms in refractory rheumatoid arthritis. Nat Rev Rheumatol 17(1):17–33 [DOI] [PubMed] [Google Scholar]
- 80.Steinman L 2014. Immunology of Relapse and Remission in Multiple Sclerosis. Annual Review of Immunology 32(1):257–81 [DOI] [PubMed] [Google Scholar]
- 81.Banchereau J, Pascual V. 2006. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 25(3):383–92 [DOI] [PubMed] [Google Scholar]
- 82.Hall JC, Rosen A. 2010. Type I interferons: crucial participants in disease amplification in autoimmunity. Nat Rev Rheumatol 6(1):40–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bonelli M, Dalwigk K, Platzer A, Olmos Calvo I, Hayer S, et al. 2019. IRF1 is critical for the TNF-driven interferon response in rheumatoid fibroblast-like synoviocytes. Experimental & Molecular Medicine 51(7):1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vila-del Sol V, Punzón C, Fresno M. 2008. IFN-gamma-induced TNF-alpha expression is regulated by interferon regulatory factors 1 and 8 in mouse macrophages. J Immunol 181(7):4461–70 [DOI] [PubMed] [Google Scholar]
- 85.Sieweke MH, Allen JE. 2013. Beyond Stem Cells: Self-Renewal of Differentiated Macrophages. Science 342(6161):1242974–1242974 [DOI] [PubMed] [Google Scholar]
- 86.Blériot C, Chakarov S, Ginhoux F. 2020. Determinants of Resident Tissue Macrophage Identity and Function. Immunity 52(6):957–70 [DOI] [PubMed] [Google Scholar]
- 87.Martinez FO, Gordon S. 2014. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime reports 6: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Murray PJ. 2017. Macrophage Polarization. Annual Review of Physiology 79(1):541–66 [DOI] [PubMed] [Google Scholar]
- 89.Dorrington MG, Fraser IDC. 2019. NF-κB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front Immunol 10:705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sirén J, Pirhonen J, Julkunen I, Matikainen S. 2005. IFN-α Regulates TLR-Dependent Gene Expression of IFN-α, IFN-β, IL-28, and IL-29. The Journal of Immunology 174(4):1932–37 [DOI] [PubMed] [Google Scholar]
- 91.Qiao Y, Giannopoulou EG, Chan CH, Park S-H, Gong S, et al. 2013. Synergistic activation of inflammatory cytokine genes by interferon-γ-induced chromatin remodeling and toll-like receptor signaling. Immunity 39(3):454–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mitchell S, Mercado EL, Adelaja A, Ho JQ, Cheng QJ, et al. 2019. An NFκB Activity Calculator to Delineate Signaling Crosstalk: Type I and II Interferons Enhance NFκB via Distinct Mechanisms. Front Immunol 10:1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xu J, Zhou L, Ji L, Chen F, Fortmann K, et al. 2016. The REGγ-proteasome forms a regulatory circuit with IκBɛ and NFκB in experimental colitis. Nat Commun 7:10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kuenzel S, Till A, Winkler M, Häsler R, Lipinski S, et al. 2010. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J Immunol 184(4):1990–2000 [DOI] [PubMed] [Google Scholar]
- 95.Cui J, Zhu L, Xia X, Wang HY, Legras X, et al. 2010. NLRC5 Negatively Regulates the NF-κB and Type I Interferon Signaling Pathways. Cell 141(3):483–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arimoto K, Löchte S, Stoner SA, Burkart C, Zhang Y, et al. 2017. STAT2 is an essential adaptor in USP18-mediated suppression of type I interferon signaling. Nat Struct Mol Biol 24(3):279–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jiang Y, Hao N. 2021. Memorizing environmental signals through feedback and feedforward loops. Current Opinion in Cell Biology 69:96–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, et al. 2002. SOCS-1 Participates in Negative Regulation of LPS Responses. Immunity 17(5):677–87 [DOI] [PubMed] [Google Scholar]
- 99.Arnold CE, Whyte CS, Gordon P, Barker RN, Rees AJ, Wilson HM. 2014. A critical role for suppressor of cytokine signalling 3 in promoting M1 macrophage activation and function in vitro and in vivo. Immunology 141(1):96–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Whyte CS, Bishop ET, Rückerl D, Gaspar-Pereira S, Barker RN, et al. 2011. Suppressor of cytokine signaling (SOCS)1 is a key determinant of differential macrophage activation and function. J Leukoc Biol 90(5):845–54 [DOI] [PubMed] [Google Scholar]
- 101.Mudla A, Jiang Y, Arimoto K, Xu B, Rajesh A, et al. 2020. Cell-cycle-gated feedback control mediates desensitization to interferon stimulation. eLife 9:e58825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lawrence T, Natoli G. 2011. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nature Reviews Immunology 11(11):750–61 [DOI] [PubMed] [Google Scholar]
- 103.Piccolo V, Curina A, Genua M, Ghisletti S, Simonatto M, et al. 2017. Opposing macrophage polarization programs show extensive epigenomic and transcriptional cross-talk. Nature Immunology 18(5):530–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Park SH, Kang K, Giannopoulou E, Qiao Y, Kang K, et al. 2017. Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat Immunol 18(10):1104–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. 2001. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science 294(5546):1540–43 [DOI] [PubMed] [Google Scholar]
- 106.Kang K, Bachu M, Park SH, Kang K, Bae S, et al. 2019. IFN-γ selectively suppresses a subset of TLR4-activated genes and enhancers to potentiate macrophage activation. Nature Communications 10(1):3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Piaszyk-Borychowska A, Széles L, Csermely A, Chiang H-C, Wesoły J, et al. 2019. Signal Integration of IFN-I and IFN-II With TLR4 Involves Sequential Recruitment of STAT1-Complexes and NFκB to Enhance Pro-inflammatory Transcription. Front Immunol 10:1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hoeksema MA, Scicluna BP, Boshuizen MCS, Velden S van der, Neele AE, et al. 2015. IFN-γ Priming of Macrophages Represses a Part of the Inflammatory Program and Attenuates Neutrophil Recruitment. The Journal of Immunology 194(8):3909–16 [DOI] [PubMed] [Google Scholar]
- 109.Martins AJ, Narayanan M, Prüstel T, Fixsen B, Park K, et al. 2017. Environment Tunes Propagation of Cell-to-Cell Variation in the Human Macrophage Gene Network. Cell Syst 4(4):379–392.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hoeksema MA, Glass CK. 2019. Nature and nurture of tissue-specific macrophage phenotypes. Atherosclerosis 281:159–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mass E, Ballesteros I, Farlik M, Halbritter F, Günther P, et al. 2016. Specification of tissue-resident macrophages during organogenesis. Science [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, et al. 2014. Environment Drives Selection and Function of Enhancers Controlling Tissue-Specific Macrophage Identities. Cell 159(6):1327–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, et al. 2014. Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell 159(6):1312–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Okabe Y, Medzhitov R. 2014. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 157(4):832–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Olson GS, Murray TA, Jahn AN, Mai D, Diercks AH, et al. 2021. Type I interferon decreases macrophage energy metabolism during mycobacterial infection. Cell Reports 35(9): [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gough DJ, Messina NL, Clarke CJP, Johnstone RW, Levy DE. 2012. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity 36(2):166–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Franceschi C, Campisi J. 2014. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci 69 Suppl 1:S4–9 [DOI] [PubMed] [Google Scholar]
- 118.Dali-Youcef N, Mecili M, Ricci R, Andrès E. 2013. Metabolic inflammation: connecting obesity and insulin resistance. Ann Med 45(3):242–53 [DOI] [PubMed] [Google Scholar]
- 119.Tilg H, Zmora N, Adolph TE, Elinav E. 2020. The intestinal microbiota fuelling metabolic inflammation. Nat Rev Immunol 20(1):40–54 [DOI] [PubMed] [Google Scholar]
- 120.Philipose Z, Smati N, Wong CSJ, Aspey K, Mendall M. 2020. Obesity, old age, and frailty are the true risk factors for COVID-19 mortality and not chronic disease or ethnicity. medRxiv 2020.08.12.20156257 [Google Scholar]
- 121.Mahbub S, Deburghgraeve CR, Kovacs EJ. 2012. Advanced Age Impairs Macrophage Polarization. Journal of Interferon & Cytokine Research 32(1):18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.van Beek AA, Van den Bossche J, Mastroberardino PG, de Winther MPJ, Leenen PJM. 2019. Metabolic Alterations in Aging Macrophages: Ingredients for Inflammaging? Trends in Immunology 40(2):113–27 [DOI] [PubMed] [Google Scholar]
- 123.Franceschi C, Santoro A, Capri M. 2020. The complex relationship between Immunosenescence and Inflammaging: Special issue on the New Biomedical Perspectives. Semin Immunopathol 42(5):517–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Frasca D, Blomberg BB. 2016. Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology 17(1):7–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, et al. 2018. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front Immunol 8:1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Oishi Y, Manabe I. 2016. Macrophages in age-related chronic inflammatory diseases. npj Aging Mech Dis 2(1):1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cui C, Driscoll RK, Piao Y, Chia CW, Gorospe M, Ferrucci L. 2019. Skewed macrophage polarization in aging skeletal muscle. Aging Cell 18(6):e13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wensveen FM, Jelenčić V, Valentić S, Šestan M, Wensveen TT, et al. 2015. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol 16(4):376–85 [DOI] [PubMed] [Google Scholar]
- 129.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. 2003. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112(12):1796–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.O’Rourke RW, White AE, Metcalf MD, Winters BR, Diggs BS, et al. 2012. Systemic inflammation and insulin sensitivity in obese IFN-γ knockout mice. Metabolism 61(8):1152–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Allis CD, Jenuwein T. 2016. The molecular hallmarks of epigenetic control. Nature Reviews Genetics 17(8):487–500 [DOI] [PubMed] [Google Scholar]
- 132.Hayes JJ, Wolffe AP. 1992. The interaction of transcription factors with nucleosomal DNA. BioEssays 14(9):597–603 [DOI] [PubMed] [Google Scholar]
- 133.Heinz S, Romanoski CE, Benner C, Glass CK. 2015. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol 16(3):144–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, Zaret KS. 2015. Pioneer Transcription Factors Target Partial DNA Motifs on Nucleosomes to Initiate Reprogramming. Cell 161(3):555–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Garcia MF, Moore CD, Schulz KN, Alberto O, Donague G, et al. 2019. Structural Features of Transcription Factors Associating with Nucleosome Binding. Molecular Cell 0(0): [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, et al. 2013. Latent Enhancers Activated by Stimulation in Differentiated Cells. Cell 152(1–2):157–71 [DOI] [PubMed] [Google Scholar]
- 137.Angelov D, Lenouvel F, Hans F, Müller CW, Bouvet P, et al. 2004. The histone octamer is invisible when NF-kappaB binds to the nucleosome. J. Biol. Chem 279(41):42374–82 [DOI] [PubMed] [Google Scholar]
- 138.Lone IN, Shukla MS, Charles Richard JL, Peshev ZY, Dimitrov S, Angelov D. 2013. Binding of NF-κB to Nucleosomes: Effect of Translational Positioning, Nucleosome Remodeling and Linker Histone H1. PLoS Genetics 9(9):e1003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Li G, Widom J. 2004. Nucleosomes facilitate their own invasion. Nat Struct Mol Biol 11(8):763–69 [DOI] [PubMed] [Google Scholar]
- 140.Li G, Levitus M, Bustamante C, Widom J. 2005. Rapid spontaneous accessibility of nucleosomal DNA. Nature Structural & Molecular Biology 12(1):46–53 [DOI] [PubMed] [Google Scholar]
- 141.Zhu F, Farnung L, Kaasinen E, Sahu B, Yin Y, et al. 2018. The interaction landscape between transcription factors and the nucleosome. Nature 562(7725):76–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Comoglio F, Simonatto M, Polletti S, Liu X, Smale ST, et al. 2019. Dissection of acute stimulus-inducible nucleosome remodeling in mammalian cells. Genes Dev [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ghisletti S, Barozzi I, Mietton F, Polletti S, De Santa F, et al. 2010. Identification and Characterization of Enhancers Controlling the Inflammatory Gene Expression Program in Macrophages. Immunity 32(3):317–28 [DOI] [PubMed] [Google Scholar]
- 144.Zaret KS. 2020. Pioneer Transcription Factors Initiating Gene Network Changes. Annual Review of Genetics 54(1):367–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kaikkonen MU, Spann NJ, Heinz S, Romanoski CE, Allison KA, et al. 2013. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell 51(3):310–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Logie C, Stunnenberg HG. 2016. Epigenetic memory: A macrophage perspective. Semin Immunol 28(4):359–67 [DOI] [PubMed] [Google Scholar]
- 147.Barozzi I, Simonatto M, Bonifacio S, Yang L, Rohs R, et al. 2014. Coregulation of Transcription Factor Binding and Nucleosome Occupancy through DNA Features of Mammalian Enhancers. Molecular Cell 54(5):844–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhang DX, Glass CK. 2013. Towards an understanding of cell-specific functions of signal-dependent transcription factors. J Mol Endocrinol 51(3):T37–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Liu Y, Zhou K, Zhang N, Wei H, Tan YZ, et al. 2020. FACT caught in the act of manipulating the nucleosome. Nature 577(7790):426–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lorch Y, Zhang M, Kornberg RD. 2001. RSC Unravels the Nucleosome. Molecular Cell 7(1):89–95 [DOI] [PubMed] [Google Scholar]
- 151.Wagner FR, Dienemann C, Wang H, Stützer A, Tegunov D, et al. 2020. Structure of SWI/SNF chromatin remodeller RSC bound to a nucleosome. Nature 579(7799):448–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Brahma S, Henikoff S. 2020. Epigenome Regulation by Dynamic Nucleosome Unwrapping. Trends in Biochemical Sciences 45(1):13–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Penkov S, Mitroulis I, Hajishengallis G, Chavakis T. 2019. Immunometabolic Crosstalk: An Ancestral Principle of Trained Immunity? Trends Immunol 40(1):1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Serefidou M, Venkatasubramani AV, Imhof A. 2019. The Impact of One Carbon Metabolism on Histone Methylation. Front. Genet 10: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Baardman J, Licht I, de Winther MPJ, Van den Bossche J. 2015. Metabolic-epigenetic crosstalk in macrophage activation. Epigenomics 7(7):1155–64 [DOI] [PubMed] [Google Scholar]
- 156.O’Neill LAJ, Kishton RJ, Rathmell J. 2016. A guide to immunometabolism for immunologists. Nature Reviews Immunology 16(9):553–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Van den Bossche J, O’Neill LA, Menon D. 2017. Macrophage Immunometabolism: Where Are We (Going)? Trends in Immunology 38(6):395–406 [DOI] [PubMed] [Google Scholar]
- 158.Akula MK, Shi M, Jiang Z, Foster CE, Miao D, et al. 2016. Control of the innate immune response by the mevalonate pathway. Nat Immunol 17(8):922–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Arts RJW, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, et al. 2016. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab 24(6):807–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Liu P-S, Wang H, Li X, Chao T, Teav T, et al. 2017. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol 18(9):985–94 [DOI] [PubMed] [Google Scholar]
- 161.Arts RJW, Carvalho A, La Rocca C, Palma C, Rodrigues F, et al. 2016. Immunometabolic Pathways in BCG-Induced Trained Immunity. Cell Rep 17(10):2562–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cheng S-C, Quintin J, Cramer RA, Shepardson KM, Saeed S, et al. 2014. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345(6204):1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ifrim DC, Quintin J, Joosten LAB, Jacobs C, Jansen T, et al. 2014. Trained Immunity or Tolerance: Opposing Functional Programs Induced in Human Monocytes after Engagement of Various Pattern Recognition Receptors. Clin Vaccine Immunol 21(4):534–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Freudenberg MA, Galanos C. 1988. Induction of tolerance to lipopolysaccharide (LPS)-D-galactosamine lethality by pretreatment with LPS is mediated by macrophages. Infect Immun 56(5):1352–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Seeley JJ, Ghosh S. 2017. Molecular mechanisms of innate memory and tolerance to LPS. Journal of Leukocyte Biology 101(1):107–19 [DOI] [PubMed] [Google Scholar]
- 166.El Gazzar M, Liu T, Yoza BK, McCall CE. 2010. Dynamic and selective nucleosome repositioning during endotoxin tolerance. J Biol Chem 285(2):1259–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, et al. 2012. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 12(2):223–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Saeed S, Quintin J, Kerstens HHD, Rao NA, Aghajanirefah A, et al. 2014. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345(6204):1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wager CML, Hole CR, Campuzano A, Castro-Lopez N, Cai H, et al. 2018. IFN-γ immune priming of macrophages in vivo induces prolonged STAT1 binding and protection against Cryptococcus neoformans. PLOS Pathogens 14(10):e1007358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hole CR, Wager CML, Castro-Lopez N, Campuzano A, Cai H, et al. 2019. Induction of memory-like dendritic cell responses in vivo. Nat Commun 10(1):2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mitroulis I, Ruppova K, Wang B, Chen L-S, Grzybek M, et al. 2018. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 172(1–2):147–161.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Crowley T, Buckley CD, Clark AR. 2018. Stroma: the forgotten cells of innate immune memory. Clin Exp Immunol 193(1):24–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hamada A, Torre C, Drancourt M, Ghigo E. 2019. Trained Immunity Carried by Non-immune Cells. Front. Microbiol 9: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kamada R, Yang W, Zhang Y, Patel MC, Yang Y, et al. 2018. Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc Natl Acad Sci USA 115(39):E9162–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Naik S, Larsen SB, Gomez NC, Alaverdyan K, Sendoel A, et al. 2017. Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 550(7677):475–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Calmette A 1931. Preventive Vaccination Against Tuberculosis with BCG. Proc R Soc Med 24(11):1481–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Covián C, Fernández-Fierro A, Retamal-Díaz A, Díaz FE, Vasquez AE, et al. 2019. BCG-Induced Cross-Protection and Development of Trained Immunity: Implication for Vaccine Design. Front. Immunol 10: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Arts RJW, Moorlag SJCFM, Novakovic B, Li Y, Wang S-Y, et al. 2018. BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host & Microbe 23(1):89–100.e5 [DOI] [PubMed] [Google Scholar]
- 179.Cirovic B, de Bree LCJ, Groh L, Blok BA, Chan J, et al. 2020. BCG Vaccination in Humans Elicits Trained Immunity via the Hematopoietic Progenitor Compartment. Cell Host & Microbe 28(2):322–334.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, et al. 2018. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 172(1):176–190.e19 [DOI] [PubMed] [Google Scholar]
- 181.Chumakov K, Avidan MS, Benn CS, Bertozzi SM, Blatt L, et al. 2021. Old vaccines for new infections: Exploiting innate immunity to control COVID-19 and prevent future pandemics. PNAS 118(21): [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Pulendran B, Ahmed R. 2006. Translating Innate Immunity into Immunological Memory: Implications for Vaccine Development. Cell 124(4):849–63 [DOI] [PubMed] [Google Scholar]
- 183.Jeljeli M, Riccio LGC, Doridot L, Chêne C, Nicco C, et al. 2019. Trained immunity modulates inflammation-induced fibrosis. Nat Commun 10(1):5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Dowson C, Simpson N, Duffy L, O’Reilly S. 2017. Innate Immunity in Systemic Sclerosis. Curr Rheumatol Rep 19(1):2. [DOI] [PubMed] [Google Scholar]
- 185.Domínguez-Andrés J, Novakovic B, Li Y, Scicluna BP, Gresnigt MS, et al. 2019. The Itaconate Pathway Is a Central Regulatory Node Linking Innate Immune Tolerance and Trained Immunity. Cell Metab 29(1):211–220.e5 [DOI] [PubMed] [Google Scholar]
- 186.Drenth JPH, Cuisset L, Grateau G, Vasseur C, van de Velde-Visser SD, et al. 1999. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. Nat Genet 22(2):178–81 [DOI] [PubMed] [Google Scholar]
- 187.Mulders-Manders CM, Simon A. 2015. Hyper-IgD syndrome/mevalonate kinase deficiency: what is new? Semin Immunopathol 37(4):371–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden CDCC, et al. 2018. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 172(1–2):135–146.e9 [DOI] [PubMed] [Google Scholar]
- 189.Simon A, Drewe E, van der Meer JWM, Powell RJ, Kelley RI, et al. 2004. Simvastatin treatment for inflammatory attacks of the hyperimmunoglobulinemia D and periodic fever syndrome. Clin Pharmacol Ther 75(5):476–83 [DOI] [PubMed] [Google Scholar]
- 190.Murray PJ, Wynn TA. 2011. Protective and pathogenic functions of macrophage subsets. Nature Reviews Immunology 11(11):723–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Gordon S, Plüddemann A. 2017. Tissue macrophages: heterogeneity and functions. BMC Biology 15(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhang H, Chen T, Ren J, Xia Y, Onuma A, et al. 2021. Pre-operative exercise therapy triggers anti-inflammatory trained immunity of Kupffer cells through metabolic reprogramming. Nat Metab, pp. 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.You M, Chen L, Zhang D, Zhao P, Chen Z, et al. 2021. Single-cell epigenomic landscape of peripheral immune cells reveals establishment of trained immunity in individuals convalescing from COVID-19. Nat Cell Biol 23(6):620–30 [DOI] [PMC free article] [PubMed] [Google Scholar]