

Abstract
Forkhead box O (FOXO) transcription factors (TFs) are a subclass of the larger family of forkhead TFs. Mammalians express four members FOXO1, FOXO3, FOXO4, and FOXO6. The interest in FOXO function stems mostly from their observed role in determining lifespan, where in model organisms, increased FOXO activity results in extended lifespan. FOXOs act as downstream of several signaling pathway and are extensively regulated through post‐translational modifications. The transcriptional program activated by FOXOs in various cell types, organisms, and under various conditions has been described and has shed some light on what the critical transcriptional targets are in mediating FOXO function. At the cellular level, these studies have revealed a role for FOXOs in cell metabolism, cellular redox, cell proliferation, DNA repair, autophagy, and many more. The general picture that emerges hereof is that FOXOs act to preserve equilibrium, and they are important for cellular homeostasis. Here, we will first briefly summarize the general knowledge of FOXO regulation and possible functions. We will use genomic stability to illustrate how FOXOs ensure homeostasis. Genomic stability is critical for maintaining genetic integrity, and therefore preventing disease. However, genomic mutations need to occur during lifetime to enable evolution, yet their accumulation is believed to be causative to aging. Therefore, the role of FOXO in genomic stability may underlie its role in lifespan and aging. Finally, we will come up with questions on some of the unknowns in FOXO function, the answer(s) to which we believe will further our understanding of FOXO function and ultimately may help to understand lifespan and its consequences.
Keywords: aging, DNA damage, signal transduction, transcription
Changes in the extracellular and intracellular environment cause ‘cellular stress’ that is potentially detrimental and can force an equilibrium shift from healthy to diseased and eventually organismal death. Forkhead box O (FOXO) transcription factors are regulators of the equilibrium and ensure optimal cellular function and lifespan. There are four FOXO isoforms, and hence, the question addressed here: Do FOXOs act to preserve the equilibrium as a family or in an isoform‐specific manner?

Abbreviations
- CR3
conserved region 3
- DBD
DNA‐binding domain (also known as Forkhead domain)
- DDR
DNA damage response
- FOXO
forkhead box O
- HSC
hematopoietic stem cell
- IDR
intrinsically disordered regions
- LLPS
liquid–liquid phase separation
- NLS
nuclear localization sequence
- PI3K
phosphoinositide 3′‐kinase
- PKB/AKT
protein kinase B/c‐AKT
- PTMs
post‐translational modifications
- ROS
reactive oxygen species
- SBS
single‐base substitutions
- TFs
transcription factors
- TLS
trans lesion synthesis
Introduction
The large family of forkhead box (FOX) transcription factors (TFs) consists of 45–50 members depending on species and whether or not counting gene duplications, and this family is subdivided into 19 classes labeled A to S. The forkhead box family members are defined through their evolutionarily conserved ‘forkhead’ or ‘winged‐helix’ DNA‐binding domain (DBD) [1]. The mammalian FOXO family contains four TFs, namely FOXO1, FOXO3, FOXO4, and FOXO6. They all bind to the consensus DNA sequence 5′‐TTGTTTAC‐3′ [2] and share a common structural layout with intrinsically disordered regions (IDR) of various length N‐ and C‐terminally flanking the structured DNA‐binding domain. FOXOs are the mammalian orthologues of the Caenorhabditis elegans DAF‐16 gene. Genetic studies in C. elegans showed that DAF‐16 mediates the longevity phenotype of reduced DAF‐2 signaling [3]. DAF‐2 is the orthologue of the mammalian insulin receptor, and subsequently, it was shown that FOXOs are also regulated by insulin signaling through phosphoinositide 3′‐kinase (PI3K)‐mediated protein kinase B/c‐AKT (PKB/AKT) signaling [4].
The timely process of aging, the deterioration of an organism that accompanies its lifespan and ultimately results in death, determines longevity and is affected by many (cellular) processes. Among many theories describing the nature of process(es) driving aging, the prominent one is the concept of cellular damage and dysfunction due to reactive oxygen species (ROS) as a driving force of aging [5]. ROS react with all cellular constituents, DNA, RNA, protein, lipid, etc., and this can result in modifications that may be coined damage when leading to loss of function. Alternatively, ROS‐induced oxidation or modification may regulate function and thereby partake in signaling in a manner similar to, for example, phosphorylation [6]. In agreement, with its effect on longevity and the suggested role of ROS, FOXOs have been implicated in regulating cellular redox, thereby reducing the potential damage induced by ROS. Furthermore, FOXOs are themselves in turn regulated by cellular redox and FOXOs are involved in maintaining cellular homeostasis by regulating processes that deal with cellular damage, including DNA repair and proteostasis (autophagy) [7].
Here, we will summarize some aspects of FOXO regulation and FOXO function, to provide background to discuss some of the outstanding questions. The answers to these questions may aid in understanding the complex phenotype of aging, age‐related diseases, and the role FOXOs play herein.
Signaling pathways regulate FOXO activity through post‐translational modifications
FOXO activity is under the control of several cellular signaling pathways through post‐translational modifications (PTMs). Genetic studies on lifespan in C. elegans implicated phosphoinositide 3′ kinase (PI3K) (DAF‐18 in C. elegans) to regulate DAF‐16 activity [8]. Protein kinase B/c‐AKT (PKB/AKT) acts as a major downstream signaling component of PI3K, and consequently, it was shown that FOXOs are regulated by PKB/AKT‐mediated phosphorylation at three evolutionary conserved sites ([9, 10, 11] and see Fig. 1). In keeping with genetic data, PKB/AKT‐mediated phosphorylation inhibits FOXO activity and this is accomplished through PKB/AKT phosphorylation‐dependent 14‐3‐3 binding and translocation of FOXOs from the nucleus to the cytosol. In the absence of PKB/AKT phosphorylation, FOXO accumulates in the nucleus, which facilitates interaction with gene regulatory regions to induce expression of target genes. Nuclear localization per se is likely not sufficient to drive full transcriptional activity and opposite PI3K/PKB/AKT, and other signaling pathways can positively regulate FOXO. Most importantly, from the perspective of aging, a disbalance in cellular redox, for example, through increased formation of ROS activates FOXOs through stress‐kinase (JNK, p38, MST1)‐mediated phosphorylation. Also, other types of cellular stress, for example, DNA damage (CDK1, CDK2, CDK4/6), energy stress (AMPK), either directly regulate FOXOs, or regulate in conjunction with indirect changes in cellular redox evoked by these types of cellular stress. For more detailed reviews, see, for example Refs [7, 12, 13].
Fig. 1.
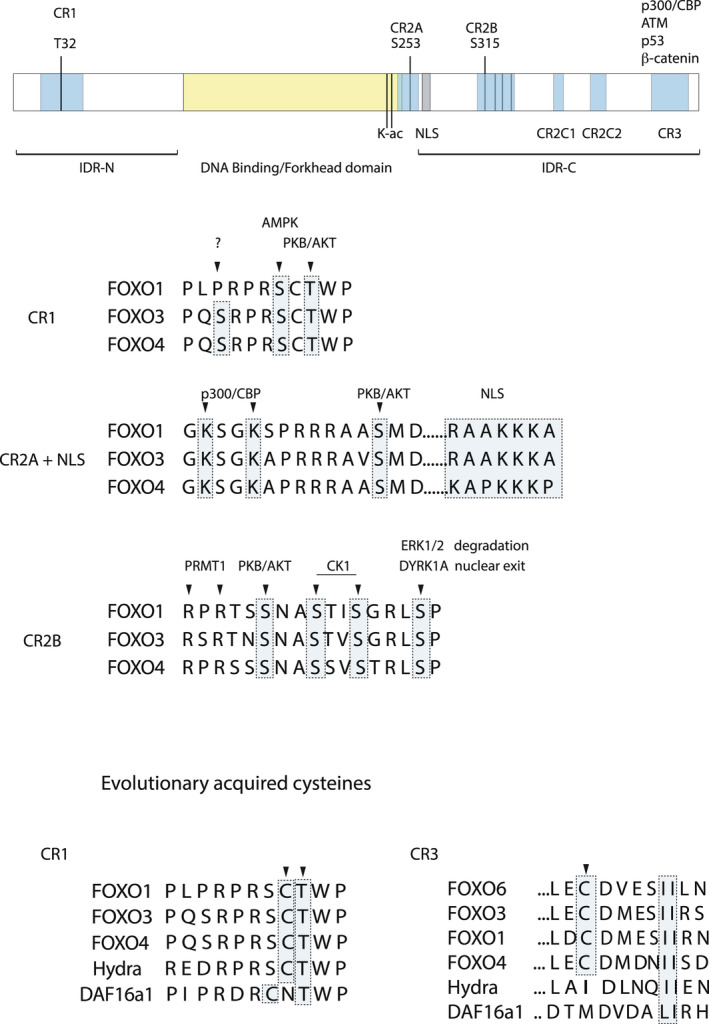
A cartoon representation of FOXO structure. Upper panel: schematic overview of the location of the various conserved FOXO regions. T32, S253, and S315 are the respective PKB/AKT phosphorylation sites numbering from human FOXO3. The forkhead domain or DNA‐binding domain is flanked by two intrinsically disordered domains N terminally (IDR‐N) and C terminally (IDR‐C). Sequences of regulatory elements that are discussed in terms of differential post‐translational modifications are provided. In CR1 relative to the PKB/AKT site, the position of AMPK phosphorylation sites conserved in the FOXOs is indicated as well as the serine residues in FOXO3 and FOXO4 that are listed in phosphosite.org but for which the kinase is unknown. For CR2A and NLS, this is similarly indicated. For CR2B, the homology is indicated and thereby the likelihood of similar regulation by upstream kinases, but evidence is variable as discussed in the text. In the lower panel, the cysteines present in CR1 and CR3 are indicated, and for the cysteine in CR3, it has been shown to be involved in p300/CBP binding. This illustrates positive evolutionary selection for cofactor recruitment through cysteine‐disulfide‐dependent binding (see for discussion Putker et al. [172]).
Beside phosphorylation, a number of additional PTMs have been described to regulate FOXO activity and function, which include ubiquitination, acetylation, lysine/arginine methylation, and O‐GlcNAcylation [14].
O‐GlcNAcylation
O‐GlcNAcylation is a reversible PTM that consists of the attachment of N‐acetylglucosamine (GlcNAc) to serine or threonine residues of proteins [15]. The enzyme O‐GlcNAc transferase (OGT) catalyzes the addition of GlcNAc, using the substrate UDP‐GlcNAc. This is produced by the hexosamine biosynthetic pathway, and a small fraction of the glucose entering the cell is used for the synthesis of UDP‐GlcNAc. Therefore, O‐GlcNAcylation regulates protein functions according to glucose availability. O‐GlcNAcylation has been shown for FOXOs (FOXO1 [16, 17], FOXO3 [18] and FOXO4 [19]). However, the serine/threonine residues modified by O‐GlcNAcylation remain to be determined [20]. Irrespective, this modification does not appear to compete with phosphorylation by PKB/AKT of FOXOs [16].
Methylation
Methylation can occur at lysine and/or arginine residues through the action of multiple enzymes. Lysine methylation mediated by G9a has been described for FOXO1 [21] whereas a different methyltransferase, Set9, has been described for FOXO3 [22, 23]. The identified lysines, K273 FOXO1 and K270/K271 FOXO3, are located within the conserved nuclear localization sequence (NLS), and as such, it is likely that also FOXO4 will be methylated at homologous residues (Fig. 1). The consequence of methylation is not entirely clear. G9a‐mediated methylation would increase binding of the E3 ubiquitin ligase Skp2 and consequently enhance degradation [22], whereas for FOXO3 inhibition of DNA binding or enhanced transcriptional activity, combined with increased degradation [23] has been reported.
Arginine methylation was described for FOXO1 [24] and mediated by PRMT1. The residues identified were all located within the consensus sequence for PKB/AKT phosphorylation (RXRXXS/T), and consequently, PRMT1‐mediated arginine methylation was shown to activate FOXOs by inhibiting subsequent PKB/AKT‐mediated FOXO regulation. Other arginine methyltransferases may also impinge on FOXOs, and for FOXO1, PRMT6‐mediated methylation has been shown in the absence of PRMT1 [25].
Detection of methylation usually relies on the quality of commercial site‐specific antibodies, the quality and specificity of which is not necessarily sufficient to draw conclusions. Recently, we have been involved in developing a novel NMR‐based approach to detect arginine methylation in vivo and this may possibly enable studying level and dynamics of arginine methylation more accurate in the future [26].
Ubiquitination
A number of E3 ubiquitin ligases for the various FOXO members have been identified. Mdm2 can mediate polyubiquitination and subsequent FOXO protein degradation [27, 28] but also induce mono‐ubiquitination and FOXO activation [28]. This differential regulation likely depends on additional signaling, and ERK‐mediated phosphorylation has been shown to steer toward polyubiquitination [28], whereas low redox stress favors mono‐ubiquitination and consequent FOXO activation [29]. Other proposed E3 ligases are Skp2 [30], the binding of which is shown to depend on PKB/AKT phosphorylation and Itch [31]. USP7 is reported to deubiquitinate mono‐ubiquitinated FOXO [32], and as such, regulation by ubiquitination of FOXOs shows strong resemblance to p53 and probably represents an important signaling connection to simultaneously coregulate FOXO and p53 function.
Acetylation
Phosphosite.org (https://www.phosphosite.org/) lists a variety of reported FOXO acetylation sites, some of which are confirmed by mass‐spectrometry and some of which are identified on the basis of mutagenesis. Acetylation of two lysines within the C‐terminal end of the DNA‐binding domain and preceding the NLS is conserved for all FOXOs (K245 and K248 in FOXO1). Also, there is acetylation described of a lysine near the CR3 domain for all FOXOs, but the sequence surrounding the identified lysine shows little conservation. All other reported acetylated lysines appear isoform‐specific. Acetylation of the lysines near the NLS has been shown to weaken DNA binding [33] and consequently enhances PKB/AKT‐mediated phosphorylation. The consequence of acetylation of other residues remains mostly unclear. This is in part due to the different effects described for FOXO deacetylases. With respect to aging, the role of the sirtuin family of deacetylases has been extensively studied. Initially, (oxidative) stress‐induced binding between FOXO and SIRT1 was described to repress FOXO function [34, 35] or to selectively enhance transcription of FOXO‐regulated genes involved in (oxidative) stress regulation and cell cycle regulation [36] or to activate gene transcription [37]. Following the described interaction with SIRT1, FOXOs have been reported to interact with SIRT2 [38], SIRT3 [39], and SIRT6 (mice [40] and C. elegans [41]). In addition, as another layer of interaction SIRT1 regulates expression of SIRT6 together with FOXO [42].
In addition to the apparent extensive interaction between FOXOs and the sirtuin family, other classes of deacetylases also interact with FOXOs. During fasting, AMPK mediates phosphorylation and translocation of class IIa HDACs (HDAC 4/5) to the nucleus, where these recruit HDAC3 resulting in FOXO deacetylation and activation [43]. A similar scheme applies for KDM5 that binds FOXO to recruit HDAC4 [44].
Understanding intrinsically disordered regions, the example of FOXO
In order to regulate transcription, TFs need to accommodate the recruitment of a large variety of regulatory proteins and protein complexes to the genome. For many TFs, this recruitment proceeds through binding of accessory proteins to so called IDR within the TFs. IDRs do not adopt a defined three‐dimensional structure and in contrast to structured domains, such as the DNA‐binding domain, allow for more flexibility, faster interaction dynamics and the ability to provide binding interfaces for a larger set of interactors. The N‐ and C‐terminal region of FOXOs flanking the DBD are IDRs (Fig. 1). Within the IDR, there are small regions that are conserved between FOXO members and these conserved regions have propensity to fold [45, 46, 47]. These regions encompass besides the forkhead domain the 3 PKB/AKT phosphorylation sites (CR1, CR2A and CR2B), the NLS and nuclear export signal the latter being part of the CR2B, two lysine rich domains (CR2C1 and CR2C2) involved in the histone acetyl transferases CBP/p300 binding [45] and finally the transactivation domain at the end of the C‐terminal domain (CR3) which is implicated in binding various coregulators (Fig. 1 for schematic representations and for sequence comparison between FOXO isoforms, see Fig. 2).
Fig. 2.

Protein sequence comparison of FOXO isofroms. Sequence comparison for all conserved regulatory elements (except the Forkhead domain) as found in human FOXOs and for which functional evidence is present. FOXO6 does not harbor clear sequence homology to CR2B, CR2C1, and CR2C2. For sequence alignment, we used the clustalw program (website: https://www.genome.jp/tools‐bin/clustalw). (*) positions that have a single and fully conserved residue; (:) conservation between groups of strongly similar properties, with a score greater than .5 on the PAM 250 matrix; (.) conservation between groups of weakly similar properties with a score less than or equal to .5 on the PAM 250 matrix.
Binding of the IDRs of TFs to the structured regions present in co‐activators requires tight regulation by PTMs. Within the conserved regions, there is by and large consensus as to how these PTMS are established, but some questions remain to be resolved. For example, AMPK regulation of CR1 has been described for FOXO3 [48], but not for, for example, FOXO4 whereas the residue within CR1 is conserved (Fig. 1).
A region within an IDR involved in binding, usually displays low binding affinity toward a certain cofactor and binding to a structured region, therefore requires multiple discrete small low‐affinity regions within the IDR, as to generate a high‐affinity synergistic binding (reviewed in Ref. [49]). Interestingly, FOXOs are regulated by redox signaling and this proceeds through cysteine oxidation. This can result in cysteine‐dependent disulfide binding between FOXO and its interacting protein. We have described this type of covalent interaction for CBP/p300 and transportin‐1 [50]. Importantly, in this way, cysteine‐dependent disulfide interaction represents an alternative mode for IDRs to engage in high‐affinity binding.
FOXO‐binding cofactors
A variety of FOXO interactors has been published, and these fall into two categories, first those that put PTMs on FOXO and the second the interactors that coregulate transcriptional output. The interaction of FOXO with the acetyl transferases p300/CBP results in acetylation of both FOXO itself and histones. At present, it is unclear whether this entails specific histone acetylation (e.g., H3K27 acetylation) events or that this results in a general increase in histone acetylation. In general, the latter contributes to activation of gene transcription whereas the consequence(s) of FOXO acetylation itself are less clear (see section above). Surprisingly, little is known to date as to how other chromatin‐remodeling complexes, besides p300/CBP, interact with FOXOs to regulate gene transcription. DAF‐16 interacts with the SWI/SNF chromatin‐remodeling complex [51], and a RNAi screen for epigenetic modifiers in C. elegans identified four genes to be involved in lifespan regulation, and of these UTX‐1, a H3K27me demethylase was validated to act with DAF‐16 [52].
Several examples of FOXO regulating gene expression in conjunction with other TFs are known. In many cases, this is due to the presence of a DNA consensus sequence for a certain TF adjacent to a FOXO consensus sequence, but some of these interactions appear more extensive. Firstly, SMADs are transcriptional regulators downstream of TGF‐β signaling and genetic interaction between SMADs and FOXO has been described in C. elegans [53] and later also in mammalian cells [54, 55]. TGF‐β‐induced immediate early gene expression in part depends on both FOXO and Smad function, and FOXOs were shown to be essential for the induction of 11 of these genes [55]. The reverse, namely the relevance of SMAD for the induction of FOXO‐dependent genes, has not been studied as far as we know.
The E2F class of TFs also shows extensive interaction with FOXO. E2F1 regulates FOXO1/3 expression [56]. This E2F1 transcriptional‐dependent increase in FOXO expression generates a feed‐forward loop as FOXO binds E2F1 and activates a subset of E2F1 genes involved in apoptosis [57]. The reverse is also observed, and binding of E2F is shown to inhibit FOXO [58].
Besides extensive similarities in their mode of regulation through PTMs, p53 and FOXO also physically interact. Structural determinants of FOXO p53 binding have been determined [46], and this involves the forkhead and CR3 domains of FOXO. Nutrient‐sensitive physical interaction was observed between FOXO3a and p53 in the induction of SIRT1 expression [59]. Recently, disruption of the p53‐FOXO4 interaction through treatment of cells with a so called FOXO4‐DRI peptide was shown to induce apoptosis of senescent cells and thereby improving healthy aging [60].
In senescent cells, FOXO4 and p53 colocalize in PML bodies and it has been shown that the PML tumor suppressor prevents cancer by inactivating PKB/AKT inside the nucleus [61]. PML specifically recruits the PKB/AKT phosphatase PP2a as well as phosphorylated PKB/AKT into PML nuclear bodies. Consequently, Pml‐null cells show impaired PP2a phosphatase activity toward PKB/AKT and thus accumulate active nuclear PKB/AKT. How this precisely relates to induction and maintenance of senescence through FOXO remains to be investigated.
PI3K signaling intertwines with WNT/TCF/β‐catenin signaling at various levels, and we have shown redox‐sensitive interaction between FOXOs and β‐catenin [62]. Consistent with this observation, genome wide profiling of β‐catenin chromatin binding in the absence of TCF/LEF shows β‐catenin overlaps with FOXO binding [63]. Binding of β‐catenin to FOXO inversely correlates with Wnt/TCF‐mediated transcription, and this shift of β‐catenin from TCF to FOXO has been shown to play a role in various biological processes such as colon cancer metastasis [64], osteoblast differentiation [65], liver metabolism [66], and kidney fibrosis [67].
Furthermore, WNT is shown to regulate FOXO1 nuclear exclusion resulting in FOXO1 inhibition in mammary stem cells through PKB/AKT [68] and β‐catenin regulation by PI3K/PKB/AKT signaling possibly through GSK3 has been shown in many studies, but remains debated [69].
Finally, FOXOs do not only bind and regulate proteins involved in transcriptional regulation but also have been shown to bind ATM to regulate the DNA damage response (DDR) [70] and ATG7 to regulate autophagy [71].
FOXOs and the genomic equilibrium
Maintaining genome stability, or preventing genomic instability, is essential for an organism to enable faithful genome transmission toward offspring. On the other hand, for selective pressure driving evolution to function, the occurrence of genomic mutations is inevitable. To meet these seemingly contradictive demands, cells have intricate and complicated ways to deal with DNA damage and to maintain genome stability. Recently, the notion that DNA damage is the driving force of aging has been reinforced and given the evolutionary conserved impact of FOXOs on aging, we discuss here specifically the role of FOXOs in prevention of DNA damage and response to DNA damage.
It is estimated that approximately 70 000 DNA‐lesion events take place in each human cell daily, (reviewed in Ref. [72]). Both endogenous genotoxic stress and exogenous genotoxic stress induce DNA base modifications that will result in DNA damage and ultimately genomic instability if not properly repaired. At present, 50–60 different base modifications have been described [73]. These modifications form important precursors for future mutations and/or can cause aberrant gene expression directly. For this reason, several DNA modifications have been unequivocally linked to cancer [74]. FOXOs have been shown to control cellular redox, and therefore, we will focus on DNA base modifications and consequent genome instability due to ROS.
Reactive oxygen species are oxygen containing derivatives comprised of highly unstable oxygen‐free radicals. They can be divided into free radicals and nonradicals. Free radicals contain an unpaired electron, and this unpaired electron makes free radicals highly reactive and short lived. Besides ROS, there are also reactive nitrogen species (reviewed in Ref. [75]). Superoxide () is the precursor of most intracellular ROS and is formed through the single electron reduction of oxygen (O2) [76]. Superoxide dismutases (SOD) can convert two superoxide molecules into the nonradical hydrogen peroxide (H2O2) [77]. Subsequently, H2O2 through the Fenton reaction, which requires free Fe2+, produces a hydroxyl radical (HO·), which is a highly reactive radical that can damage protein, lipids, and DNA [78]. Alternatively, H2O2 can be reduced to H2O by glutathione peroxidases (GPX), peroxiredoxins (PRX), and catalase.
The most common form of oxidative DNA damage is the formation of 8‐oxo‐7,8‐dihydroguanine (8‐oxo‐dG and its tautomer 8‐OH‐dG). Oxidation of nucleotides can also occur in the free nucleotide pool, after which 8‐oxo‐dG can be incorporated into the genomic DNA [79]. The other nucleotide bases can also be oxidized but guanine has the lowest oxidation potential and is thereby most susceptible to ROS [80]. Where guanine normally pairs with a cytosine, 8‐oxoG frequently mis‐pairs during replication with an adenine resulting in a C>A/G>T transversion [80]. 8‐oxo‐dG is excised by 8‐oxoguanine glycosylase 1 (OGG1), which is part of the base excision repair [81]. This creates a so called apurinic site (AP site). The AP‐nuclease 1 (APE‐1) processes this into a single‐stranded break and further repair is followed. ~ 15 000–30 000 AP and a few thousand 8‐oxoG sites per cell are estimated to be present at steady state in the DNA [82]. 8‐oxo‐dG mispairing with an adenine results after replication in a C>A/G>T transversion [82]. The MutY homolog (MUTYH) recognizes and excises the adenine opposite to an 8‐oxoG [82], to prevent mispairing. MUTYH will in this way also prevent these transversions when 8‐oxo‐dG is incorporated from the oxidized nucleotide pool.
In case repair fails, ROS can ultimately induce single‐ and double‐stranded breaks, deletions, base modifications, chromosomal rearrangements, and hypo‐ and hypermethylation. Specific signatures, of mutations, which represent patterns of all the six single‐base substitutions (SBS), namely C>A, C>G, C>T, T>A, T>C, and T>G), can be attributed to various DNA damaging agents, and Alexandrov et al. [83, 84] have categorized these signatures, where signature SBS18 (predominantly C>A), SBS36 (predominantly C>A), SBS17a (predominantly T>C), and SBS17b (predominantly T>G) are attributed to the action of ROS.
Reactive oxygen species can be generated at several cellular locations as a consequence of normal cellular function. Oxidative metabolism in mitochondria produces ROS, but also NADPH oxidases localized at the plasma membrane produce ROS during growth factor signaling. ROS are produced by peroxisomes (as by‐product of lipid beta‐oxidation) and even in the nucleus certain enzymes produce ROS as enzymatic product (histone and DNA re‐modeling enzymes). These nuclear enzymes belong to the enzyme class of dioxygenases that use O2 in catalysis, for example, the histone demethylase LSD1 uses O2 and vitC/Fe2+ to catalyze histone lysine demethylation, and this produces H2O2 and local oxidation of DNA [85]. However, it remains to be established whether ROS generation at these different cellular locations will have similar impact in generating oxidative DNA lesions.
FOXOs have been shown to regulate/reduce the level of ROS in various ways, and the underlying assumption thereof yet not formally proven is that this will lower the number of DNA lesions that require repair. However, FOXOs have also been shown to regulate the DDR at various levels as to ensure that proper repair is facilitated. If all fails, FOXOs have also been shown to regulate cell fate (senescence, apoptosis) that accompanies excessive unrepairable DNA damage, in order to minimize detrimental consequences of mutational overload. Interestingly, when comparing cellular and organismal response to DNA damage, the consequence of DNA damage as well as how to deal with DNA damage appears different and consequently also the involvement of FOXO. At the cellular level, both induction of apoptosis and senescence by FOXO after DNA damage will stop cell proliferation and consequent establishment of DNA mutations. At the organismal level, in case of C. elegans development, it has been shown that persistent DNA damage, as occurring, for example, during aging, leads to a transient developmental arrest. In contrast to the DAF‐16‐dependent developmental arrest induced by food starvation (dauer formation), now DAF‐16 combined with the GATA factor EGL‐27 is responsible for driving development when DNA damage persists [86, 87]. It will be interesting to study how this DAF‐16/FOXO‐dependent developmental phenotype relates to other organisms or to the process of aging in humans, especially, since all cells except germ cells in adult C. elegans are postmitotic, and thus can accumulate DNA damage yet not DNA mutations, as opposed to cells in, for example, adult humans.
Regulation of cellular redox by FOXOs
The verb ‘Dosis facit venenum’ especially accounts for ROS. In cells, there exists a tightly controlled balance between ROS production and clearance [88]. This provides an equilibrium at which ROS can function in normal cellular processes such as cell division and signal transduction. Excessive production of ROS or defective clearance will result in a disbalance that is usually referred to as oxidative stress. This stress can be counterbalanced by reducing ROS production or increasing antioxidant systems. If oxidative stress cannot be handled properly by cells, it will result in damage to cellular constituents, including DNA, as described above. In case of excessive damage that cannot be repaired, cells can respond in different ways, cell cycle arrest either transient or permanent (senescence), or cell death (apoptosis, ferroptosis). The cellular redox balance can differ between cells, and, for example, cancer cells in general produce higher levels of ROS that is, however, counterbalanced by upregulation of certain antioxidant systems. Thus, also cancer cells maintain a redox equilibrium albeit at higher redox potential.
FOXOs are regulated by changes in cellular redox in several ways (reviewed in Ref. [7]). Most importantly, ROS‐induced activation of JNK and/or p38 stress kinases results in phosphorylation of FOXOs although the phospho‐acceptor sites appear differentially positioned within the N‐ and C‐terminal domains of the various FOXO isoforms [89, 90]. FOXOs also employ cysteine oxidation and formation of cysteine‐dependent disulfide bridges to bind cofactors such as CBP/p300 and nuclear importers (TNPO1, IPO7 and IPO8), and this combined results in activation of FOXO‐dependent transcription (reviewed in Ref. [50]).
FOXOs regulate expression of a variety of antioxidant genes. MnSOD/SOD2 was the first to be reported both in C. elegans [91] and mammalian cells [92]. In addition to MnSOD, regulation of a number of other genes involved in redox control has been described. These include catalase, glutathione peroxidase 1 (GPX1), SOD1, peroxiredoxin III, and Jafrac1, a Drosophila ortholog of human peroxiredoxin II and sestrins (reviewed in Ref. [7]). FOXO also regulates isocitrate dehydrogenases 1 (IDH1) expression, thereby increasing the cytoplasmic levels of NADPH [93]. Notably, cytoplasmic NADPH can reduce GSSG (glutathione disulfide, oxidized) to GSH (glutathione, reduced), which is involved in ROS detoxification. In agreement with this, mice engineered with triple deletion of FOXO1, FOXO3, and FOXO4 display increased ROS level and decreased expression of certain ROS detoxification proteins, accompanied with hematopoietic stem cell (HSC) depletion. Furthermore, treatment with antioxidant agents rescues to some extend the depletion of HSCs after FOXO deletion [94].
Regulating metabolism and mitochondria
Mitochondria are a major intracellular site of ROS production. FOXOs regulate mitochondrial function in several ways. During hypoxia, a number of nuclear‐encoded genes related to mitochondrial function are repressed by FOXO3 in a c‐Myc‐dependent manner, leading to reduction of mitochondrial respiratory activity and ROS production from mitochondria [95, 96]. This observation is corroborated by the observation that in HSC, that reside in a hypoxic niche, display elevated ROS level [97]. We recently showed that FOXOs affect the mitochondrial fission fusion cycle to reduce mitochondrial number and activity during intestinal differentiation [98]. SIRT3 is localized in mitochondria and interacts with FOXO3; consequently, it is suggested that FOXO3 also resides in mitochondria [99, 100]. To reside in mitochondria, proteins are imported through the TOMM20 complex and cleaved at consensus sequences [101]. These are not clearly present in FOXO3, but a AMPK/ERK‐dependent mechanism of cleavage and import has been described [102].
FOXOs regulate general autophagy and also mitophagy which removes dysfunctional mitochondria, for example, as a consequence of excessive ROS leakage [103]. Notably, FOXOs transcriptionally regulate expression of PTEN‐induced putative kinase protein 1 (PINK1), a kinase which plays a critical role in mitophagy [104].
Taken together, by regulating mitochondrial metabolism, FOXOs represent an important first line of defense against endogenous metabolism that can cause DNA damage.
FOXOs mediate DNA damage response
When prevention fails and DNA damage occurs, cells respond to this damage by mounting a DDR [105]. This response entails damage sensing followed by recruitment of repair proteins and activation of cellular processes, such as cell cycle control, that enable timely repair. FOXOs are implicated at various steps within this response.
The prime mediators of DNA damage sensing are ATM, ATR, and DNA‐PK, kinases that belong to the family of PI3K‐related kinases, but are protein kinases rather than lipid kinases [106].
ATM responds to DNA damage as well as oxidative stress (reviewed in Ref. [107]). In DNA damage, sensing ATM is activated by binding to the MRN complex and single‐stranded DNA, whereas ROS can induce ATM dimerization through cysteine‐dependent disulfide linkage [107]. ATM mutants have been constructed that specifically respond to either DNA damage or ROS, but in many experimental settings, it will be hard to disentangle these as DNA damage oftentimes comes along with changes in cellular redox and vice versa. FOXOs bind to ATM, and it has been shown that this binding is necessary for ATM activation [70], probably through FOXO‐dependent recruitment of TIP60 [108]. The latter mediates ATM acetylation and activation [109]. Intriguingly, it was initially shown that Tip60 is recruited by ATM through binding to the FATC domain of ATM [110]. It is unknown at present how the mechanism proposed for FOXO‐dependent ATM activation and Tip60 recruitment intertwines with that of the ROS‐induced dimerization. To add an additional layer to the ATM‐FOXO interaction, FOXO has also been shown to regulate ATM expression [111]. As for FOXO, ATM does not only sense cellular redox changes. ATM‐dependent regulation of G6PD activity regulates the flux through the pentose phosphate pathway and consequently the levels of NADPH, required for ROS detoxification [112]. FOXO also links to DNA‐PK and ATR. We have shown binding of FOXO to Ku70 [113], which in complex with Ku80 and other proteins acts to recruit DNA‐PK to DNA damage sites to regulate repair by nonhomologous end‐joining [114]. Interestingly, DNA‐PK regulates PKB/AKT activity by phosphorylating ser473 [115], and thus, FOXO and DNA‐PK may constitute a feedback loop, whereby FOXO aids DNA‐PK in resolving DNA damage, and this is then terminated by DNA‐PK‐induced PKB/AKT‐mediated FOXO inhibition. Recently, we showed a role for FOXO in resolving replication stress in G2/M and this is dependent on ATR [116]. Whether this relates to the binding of FOXOs to FANCD2 [117, 118] remains to be determined. Similarly, stalled replication forks are resolved through trans lesion synthesis (TLS) and a role for FOXO1 on TLS has been shown through binding replication protein A (RPA1) that coats single‐stranded DNA and acts as a scaffold for TLS [119]. As for ATM, ATR expression is also controlled by FOXO [120]. Besides direct interaction with regulators of the DDR, FOXOs also induce transcription of genes involved in DNA repair. The first example being GADD45a [121], how GADD45a acts in DNA repair, is not fully clear, but it has been implicated in removal of DNA methylation [122].
Regulation of cell cycle progression is important to allow for timely DNA damage repair, and FOXOs are controlled to regulate cell cycle progression at various phases. FOXO regulates G1 by transcriptional control of p27 [123] and in some conditions p21 [124]. Recently, we showed that FOXOs by binding to E2F regulate Emi expression and thereby APC/C control of G2 [116]. Furthermore, S phase entry has also been shown to be partially controlled by FOXO [125]. Thus, FOXO activation can arrest cells at different cell cycle phases and this is likely regulated by the DNA damage that may occur at/during these different phases.
DNA damage inhibits CDK activity and CDK1 and CDK2 phosphorylate FOXOs [76], and this was shown to inhibit FOXO1 activity, via cytoplasmic retention. Consequently, following DNA damage this negative regulation is alleviated resulting in FOXO1 nuclear relocalization and FOXO‐mediated arrest.
A proteomic survey of ATM substrates listed FOXO1 as ATM substrate [126], and all FOXOs harbor numerous SQ sequences that represent potential ATM/ATR phosphorylation sites. The consequence(s) hereof has not yet been studied.
If all fails, cells either remain permanently arrested (senescence for recent review [127]) or will die mostly through apoptosis (reviewed in Ref. [128]). Both senescence and cell death controlled by FOXO are nonreversible, which represent a diversion in terms of equilibrium. However, from an organismal point of view, these processes are essential for survival. For further discussion on senescence and apoptosis, we refer to aforementioned reviews.
An outstanding question: Isoform specificity
The freshwater polyp Hydra expresses one FOXO allele [129], whereas in mammalians, four FOXO isoforms are expressed. This raises the question whether in higher eukaryotes, the different FOXO isoforms mediate the same function, yet their role is determined by context, or that FOXO function has diverged between these isoforms.
In mice, genetic deletion of individual Foxo genes shows that loss of Foxo1 is embryonic lethal due to incomplete vascular development [130], loss of Foxo3 affects in an age‐dependent manner female sterility [130, 131], and loss of Foxo4 results in no detectable phenotype [130]. However, conditional deletion of all three Foxo isoforms results in a cancer‐prone phenotype that includes age‐progressive thymic cancers and hemangiomas, indicating that Foxo1, Foxo3, and Foxo4 are redundant tumor suppressor genes specifically involved in endothelial growth suppression [132].
These results may suggest distinct functions for the FOXO isoforms, but do not exclude the possibilities that functions are similar yet tissue‐specific expression of isoforms, or differential timely expression, or even relative level of isoforms within one cell underlies these phenotypes.
Also, C. elegans expresses multiple DAF‐16 isoforms albeit from a single genetic locus. Although not entirely clear, two isoforms (daf‐16a and daf‐16d/f) appear most relevant in determining the longevity phenotype. Bansal et al. [133] provide clear evidence that next to upstream (PI3K/ PKB/AKT) regulation there appears to be also a gene dosage effect. This implies first that transcriptional regulators of DAF‐16 expression possibly affect longevity in a DAF‐16‐dependent manner. Second, protein level also impacts on the cellular location whereby low daf‐16a expression results in mostly nuclear and high daf‐16a expression mostly in cytosolic location. This expression‐dependent cellular location will affect regulation by upstream signaling and thus may underlie apparent isoform‐specific regulation by upstream signaling.
Also, in mice there may be a dosage‐dependent effect in FOXO function. Whereas, in contrast to FOXO1 null mice, FOXO3 and FOXO4 null mice did not show a hemangioma phenotype, deletion of FOXO3 and FOXO4 in a FOXO1 null background did, however, increase severity of the hemangioma phenotype. This suggests that FOXO1 is the relevant and most important isoform in maintaining endothelial function. However, considering the gene dosage example in C. elegans, it may also reflect relative expression level rather than an isoform‐specific function.
Taken together, the possibility of dosage effects combined with dosage‐dependent either cellular location or signaling differences warrants caution in interpreting mammalian data using overexpression and/or more or less incomplete shRNA‐mediated knockdown to indicate isoform‐specific function.
In mammalians, it has also been suggested that FOXOs have a nontranscriptional function. Examples of this nontranscriptional function of FOXOs are as follows: the interaction of FOXO3 with ATM [70] regulating the DDR and FOXO1 with ATG7 [71] regulating autophagy. The interaction interface with ATM on FOXO3 has been partially mapped [134], and this entails the CR3 domain and Forkhead domain of FOXO3. These domains are actually conserved between FOXO isoforms, and indeed, ATM also binds to other FOXO isoforms (B. M. T. Burgering, unpublished data). In addition, FOXOs also transcriptionally regulate gene expression of genes involved in DNA repair, including ATM and ATR [111, 120] and genes involved autophagy [135]. Although an attractive possibility, it remains to be determined how relevant these nontranscriptional functions are compared to transcriptional regulation of the same process and whether these may underlie isoform specificity.
There are additional confounding issues in determining isoform specificity. First, FOXO3 has been shown to regulate FOXO1 expression [136], and consequently, a FOXO3 knockdown will concomitantly reduce expression of FOXO1. Second, FOXOs induce complex feedback signaling, the best described example being mTORC2‐mediated PI3K/AKT activation and consequent inhibition of FOXO activity [137, 138]. Thus, ectopic expression of an, for example, constitutive active FOXO3 mutant will reduce activity of FOXO1 and FOXO4, suggesting that formally the observed phenotype could be due to the other isoforms rather than FOXO3. Third, as for many TFs, it is unclear whether or not FOXOs act both as transcriptional activator and repressor. Evidence indicates that FOXOs act predominantly as transcriptional activator [139, 140, 141], so gene repression is in most cases a secondary/indirect event and will be likely dependent not only on the nature of the transcriptional repressor regulated by FOXOs but also on the level of induction. This confounds interpretation of downregulated gene signatures after FOXO activation. In addition, and at least as shown for FOXO3 [139, 140], FOXOs appear to regulate transcription by and large through enhancers. We have shown for FOXO3 that the basal state of an enhancer is a strong determinant for the ability of FOXO3 to activate an enhancer and the transcription of a nearby gene. Consequently, the pre‐existing enhancer landscape and the genomic 3‐D architecture will strongly affect the ability of FOXO3 to induce gene transcription. The genomic 3‐D structure also likely underlies the observed cell type‐specific gene signatures induced by FOXO3 [140]. Whether the importance of enhancers also accounts for the other FOXOs remains to be determined. Irrespective, given the above considerations, FOXO isoform‐specific function remains largely not understood.
During evolution, gene duplication allows one allele to retain its original function whereas the other allele may acquire other functions due to mutational gains or losses. Comparison of the 4 FOXO alleles suggests that FOXO1/4 and FOXO3/6 resulted from gene duplications ([142] and see Fig. 3A). However, when comparing protein sequence (see Fig. 2), it is clear that except for FOXO6 all relevant functional elements described thus far have been retained during evolution. This may implicate that these elements are essential to maintain FOXO function and that as such FOXO1, FOXO3, and FOXO4 have not been evolutionary selected for novel acquired functions (see for more extensive discussion on this issue [143]). Thus, the main function of FOXOs has likely remained evolutionary conserved within the FOXO members and apparently depends on these conserved regulatory features. We interpret this as FOXOs mediating a family response that given the context is conveyed by either one or more family members.
Fig. 3.
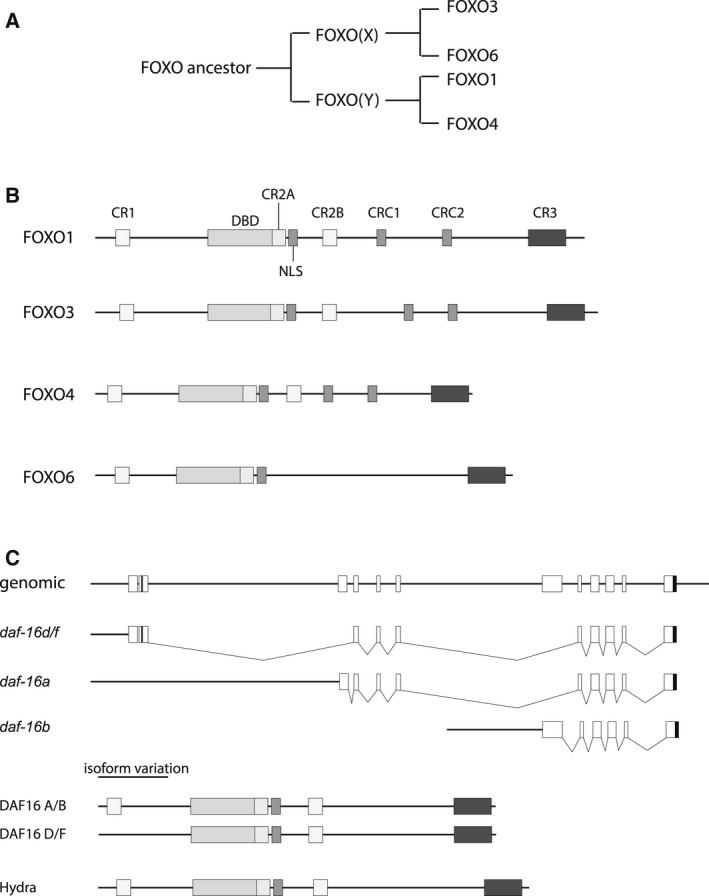
Comparison of FOXO isoforms. (A) Evolutionary tree for FOXO duplications. (B) Presence of the various conserved elements within the various FOXO isoforms. (C) Cartoon representation of the DAF‐16 genomic locus (for further discussion see Ref. [160]) and the splicing events leading to the DAF‐16 isoforms a, b, and d/f. The resulting protein sequences and the location of the conserved elements are indicated and compared to FOXO from Hydra vulgaris.
So, we think it is at present not yet determined whether there are clear isoform‐specific functions and this raises the questions whether there will be even an isoform‐specific function? and what function actually could be isoform specific? If based on evolutionary biology, the increase in FOXO members did not associate with discrete functional divergence, what is then actually shared between isoforms, and how do unique and/or shared regulation determine phenotype. These questions remain relevant, if not for understanding aging.
FOXOs and aging, all about FOXO3?
The freshwater polyp Hydra has an apparent infinite lifespan and expresses only one FOXO isoform [129] (see also Fig. 3C). Longevity is phenotypically linked to FOXO activity in other organisms, and longevity or the aging process that is inevitably associated with lifespan could be considered the overarching FOXO‐dependent phenotype. In humans, genome wide association studies have shown that FOXO3 is reproducibly associated with human longevity [144]. This could suggest that FOXO1 and FOXO4 have diverged from this ancestral function and that it is retained by FOXO3. This being the case, the question becomes first what is the function that is critical in affecting lifespan that is then specific to FOXO3 and second what additional functions are acquired by FOXO1 and/or FOXO4.
The remarkable immortality of Hydra is due to the asexual mode of reproduction through budding. In essence, this therefore represents an endless ongoing tissue renewal and thus requires stem cells with continuous self‐renewal capacity [145]. FOXO regulates proliferation of Hydra interstitial stem cells, whereby overexpression in interstitial stem cells increases stem cell proliferation and in addition induces expression of stem cell genes in differentiated progeny [145]. The latter may illustrate a case of transdifferentiation or cellular plasticity by which differentiated cells can fall back into a stem cell fate. In contrast, reduced FOXO expression slows down proliferation and increases differentiation. Thus, these results comply with the model that lifespan is a function of stem cell (proliferation) and this involves FOXO. These compelling data, however, also raise questions with regard to mammalians.
First, the control of proliferation appears in contrast to what is observed in mammalian cells. FOXO wild‐type overexpression has little effect on cell proliferation, but FOXO activation, through expression of PKB/AKT site mutants, inhibits proliferation, through induction of cell cycle inhibitors such as p27. Reduced FOXO expression has little to no effect on proliferation. However, this has been studied mostly in the context of cancer cell lines and their ability to differentiate is compromised. The colon and intestine of mammalians are in several ways comparable to Hydra, the epithelial lining constantly needs to regenerate, and this is also driven by a continuous cycling stem cell, the LGR5‐positive columnar‐based crypt (CBC) cell [146]. These stem cells express both FOXO1 and FOXO3, and, in agreement with Hydra, knockdown of FOXO expression in stem cells present within intestinal organoids also slows down stem cell proliferation and enhances differentiation [98]. In contrast to Hydra, the effect of overexpression of wild‐type FOXO is not known but expression of a mutant that is no longer inhibited by PI3K/PKB/AKT signaling, stops cell cycle progression, and results in the collapse of organoid growth as stem cells can no longer contribute to organoid growth (M. Ludikhuize, B. M. T. Burgering & M. Rodriguez‐Colman, unpublished data).
Whereas the LGR5‐positive intestinal stem cell represents a class of highly proliferative mammalian adult stem cells, muscle and hematopoietic stem cells are examples of quiescent stem cells. Importantly, FOXOs are similarly implicated in quiescent stem cell control. This is best studied for the hematopoietic stem cell compartment (HSC). Genetic ablation of FOXO1/3/4 and to a large extend ablation of FOXO3 only impairs HSC function. Mechanistically, the importance of FOXOs in maintaining HSC function is related to their role in redox control and cell proliferation [94].
In skeletal muscle, satellite cells represent quiescent stem cells (reviewed in Ref. [147]). In response to muscle fiber injury, satellite cells are activated, proliferated, and differentiated into multinucleated myofibers or self‐renew. Upon completion of self‐renewal, a subset of satellite cells returns to quiescence. Self‐renewal of satellite cells during muscle regeneration requires FOXO3 [148], and it was recently shown that FOXOs are essential to maintain the quiescent state until old age [149]. Furthermore, FOXO3 may also induce cell death of satellite cells under pathological conditions [150].
In contrast to skeletal muscle, heart muscle appears to lack stem cells [151], although this remains a controversial subject. Consequently, a role for FOXOs remains unknown, although there may be a role for FOXO4 in recovery after myocardial infarct [152].
Finally, also in brain, FOXO3 plays a role in maintaining the neuronal stem cell pool [153]. Thus, in terms of stem cell control and in agreement with role of stem cells in aging, FOXO3 appears the dominant FOXO, however, in some organs, for example, FOXO1 in the intestine. FOXOs may act redundant. Importantly, in the context of cancer as an age‐related disease, FOXOs are also required for maintaining cancer stem cells [154, 155]. Again, most research focus here lies on FOXO3 but this does not exclude yet the role of other isoforms in cancer stem cell function. Finally, although the regenerative potential of stem cells strongly affects aging in humans, the role of FOXOs in stem cells unlikely represents the full impact of FOXOs on lifespan and aging. C. elegans has no regenerative potential and harbors no equivalent of human adult stem cells, yet the notion of genetic impact on aging and the role of DAF‐16/FOXO has been discovered there in C. elegans.
In Hydra, loss of FOXO also resulted in reduced expression of antimicrobial genes (reviewed in Ref. [156]) and this was shown to affect the ability of Hydra to cope with foreign microbes and to re‐establish its specific microbe symbiosis after stem cell transfer. This implies that next to, or combined with, the control of stem cell function, a role for FOXO in the control of innate immunity represents an ancestral function. Functional immunity declines with age, and interventions preventing infectious disease, for example, hygiene, have a major impact on lifespan. With respect to innate immunity, FOXOs appear to play vary diverse roles, including regulating expression of antimicrobial peptides [157]. In addition to this protective function, the example of Hydra shows that FOXOs are also involved in establishing the symbiosis between microbes and their host organism. Given the proposed role of the gut microbiome in many human age‐related diseases, it will be of interest to explore the relation between gut microbiome function and FOXOs.
Finally, Hydra appears to lack any signs of cellular senescence, and cellular senescence has been put forward as a major driver of aging (reviewed in Ref. [158]) and selective removal of senescent cells improves healthy aging [60, 159]. Whether and how different isoforms affect induction and/or maintenance of senescence is yet to be studied in detail. Recently, it was shown that FOXO4‐DRI a peptide based on the interaction interface of FOXO4 with p53 clears senescent cells and improves healthy aging, suggesting FOXO4 to be an important player [60].
From an evolutionary perspective on aging, the control of stem cell function, regulation of innate immunity, and the induction of senescence are the primary phenotypes linked to FOXO function. In humans and mice, these functions appear mostly mediated by FOXO3 with possibly a role for FOXO1 and FOXO4 in specific settings. It therefore appears that FOXO3 has retained most of these ancestral functions and that FOXO1 and FOXO4 can be redundant and may have acquired additional roles that may or may not be relevant for aging.
Interestingly, from this perspective is that FOXO3 and FOXO6 represent one arm and FOXO1 and FOXO4 the other arm of evolutionary development ([142] and see Fig. 3A). Whereas in the FOXO3/6 branch there is apparent divergence between FOXO6 and FOXO3 as illustrated by the loss of the PKB/AKT phosphorylation site (CR2B) sequence and the CR2B and CR2C leucine‐rich domains (Fig. 2), this has not occurred within the FOXO1/4 branch. In addition, whereas FOXOs act as reciprocal transcriptional activators, FOXO6 appears to repress FOXO3 transcription. Thus, from an evolutionary point of view, FOXO6 appears the only member that is functionally drifting out of the family.
The family function of FOXOs is also observed in C. elegans aging. Different DAF‐16 isoforms are produced through differential splicing from one single DAF‐16 gene locus (Fig. 3B). Although debated, it appears that both DAF‐16a and DAF‐16d/f are important in mediating the DAF‐2‐dependent longevity phenotype. Given the context, either of the one isoforms appears more relevant, but overall, also longevity in C. elegans appears a family trait mediated by the combined action of DAF‐16a and DAF‐16d/f. Interestingly, a major difference between DAF‐16a and DAF‐16d/f is the absence of the first AKT/PKB site in DAF‐16d/f (Fig. 3C). This would result at least in a partial lack of response to DAF‐2 signaling mediated by PI3K/PKB/AKT, yet gene dosage effects appear to compensate for this partial loss in DAF‐2 regulation [160], thus reinforcing the notion that gene dosage also in other organisms might be relevant in establishing phenotypic response.
What is the difference between isoforms in specific gene regulation?
Assuming a difference between FOXO3 on the one hand and FOXO1 and FOXO4 on the other hand, the question becomes how FOXO isoform‐specific transcriptional programs are established. All isoforms bind to the same DNA consensus, so isoform‐specific binding to gene regulatory regions will not be based on the presence/absence of the DNA consensus motif. Chromatin context could direct the differential binding of FOXOs. For transcription to be proceeded, chromatin needs to be in an open conformation, and the opening of closed chromatin is established by so called pioneer factors. FOXA, like FOXO, is a member of the larger forkhead family and has been shown to act as a pioneer factor in establishing chromatin opening at regulatory regions to enable subsequent recruitment of transcription regulators (reviewed in Ref. [161]). In vitro experiments indicate FOXO1 binding can disrupt histone‐DNA contacts, resulting in chromatin opening [162, 163]. This has not been described for the other FOXO members so potentially there could be a differing ability between FOXOs to regulate transcription in chromatin dense versus open areas. Also, recruitment of the chromatin‐remodeling complex SWI/SNF to DAF‐16/FOXO bound C. elegans genomic locations has been taken to suggest that FOXO may act as a pioneer factor [51]. However, genome wide analysis of FOXO3 binding shows preference for binding to open chromatin, suggesting FOXO3 in general not to act as a pioneer factor [139, 140]. Clearly, more detailed knowledge as to how chromatin context regulates FOXO role in transcription regulation and whether this context determines an isoform‐specific activation of gene programs that mediate specific phenotypic outcome is needed.
A novel kid on the block, phase separation
In addition to mediating binding to structured domains, IDRs can also engage in weak interactions that are a driving force for liquid–liquid phase separation (LLPS) [164]. Phase separation is a physicochemical process by which molecules separate into a dense phase and a dilute phase. A growing body of literature indicates that during active transcription RNA polymerase II, TFs and co‐activators are clustered in hubs or transcription factories [165] that display characteristics of LLPS [166, 167]. Phase‐separated biomolecular condensates have been observed throughout the cell with prominent examples being stress granules, P‐bodies, and nucleoli. However, only recently it has been shown that components of the transcriptional co‐activator mediator and Pol II form condensates at sites of active transcription and rely on their IDRs to do so [168].
The presence of IDRs in the N‐ and C‐terminal domains of FOXOs may indicate that also for FOXOs their ability to partake (or not) in the formation of transcriptionally active biocondensates will be relevant for understanding function. As the primary amino acid sequence of FOXO isoforms differs mostly within their IDR region, the ability to enter and/or form biocondensates provides and interesting territory to explore with respect to understanding isoform specificity.
Interaction specifics and upstream regulation
Recruitment of specific co‐acting TFs or gene regulatory complexes by FOXOs could also provide a mechanism for differential gene expression. A well‐studied example is the connection between FOXO and SMADs. Convergence of TGF‐β and insulin/PI3K signaling in aging has been described for C. elegans [53] and to a lesser extent in Hydra as well [169]. However, SMADs can bind to all FOXO1, FOXO3, and FOXO4 [54, 55]. Also, for other co‐acting TFs, there is no apparent isoform‐specific connection. Most have been identified on the basis of DNA consensus sequence elements in the vicinity of FOXO‐binding elements so in principle these can be corecruited also by all FOXO isoforms. Binding to other regulators such as p53 and β‐catenin is mediated by conserved sequence elements, so these bind to all FOXOs albeit with possibly differing affinity.
Isoform‐specific function may not be an intrinsic characteristic but could be established by differential upstream regulation. However, at present there is a plethora of regulatory inputs described, and to address this possibility, a concerted effort needs to be made as literature is to diffuse to reach understanding here.
For example, JNK and p38 phosphorylate all FOXO isoforms yet the positions differ and sequence context mostly shows little conservation. This may suggest that this results in different regulation, however, as mentioned phosphorylation is most abundant in the IDR. Therefore, one can argue that opposed to phosphorylation of structured domains, position is less relevant, and it is only the mere fact of phosphorylation that is relevant.
Furthermore, also when analyzing phosphorylation in conserved regions within the FOXO IDR, it is hard to reach unambiguous conclusions. For example (Fig. 1), the third PKB/AKT phosphorylation site has been shown to act as a gate‐keeper for subsequent casein kinase I phosphorylation [170]. A negatively charged interface is further generated by DYRK1‐mediated phosphorylation as shown for FOXO1 and FOXO4 ([171] and B. M. T. Burgering, unpublished data). However, despite its sequence conservation, MAPK/ERK has been suggested to phosphorylate this DYRK1 site within FOXO3 [28]. Although overall the consequence (negative regulation) is similar, the detailed described modification mechanism was not. These issues need to be resolved in a structured manner before we can draw conclusions concerning the role of upstream regulation in defining isoform‐specific roles.
Conclusion
Erwin Schrödinger in his collection of lectures bundled in ‘what is life’ pointed to the importance and consequences of the equilibrium as determined by the second law of thermodynamics, for understanding life, also in a metaphysical sense. FOXOs, or at least FOXO3, are tightly linked with lifespan, and thereby in this frame of reasoning, FOXOs are essential in maintaining the equilibrium. Full understanding of FOXOs as an example how in biology, systems deal with this conundrum of the equilibrium that supports life, will help not only in understanding the molecular underpinning of age‐associated diseases, but maybe lifespan and how to deal with it.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
TG and BMTB discussed content, figure outlay, and cowrote the manuscript.
Acknowledgements
We wish to thank all members of the Burgering lab for support and discussion. The work in the Burgering lab is financially supported by ‘the Oncode Institute’, Dutch Cancer Foundation (KWF) and the Netherlands Organization for Science (NWO). TG was financially supported through the Chinese Scholarship Council (grant no. 201604910624).
Contributor Information
Tianshu Gui, Email: t.gui-2@umcutrecht.nl.
Boudewijn M. T. Burgering, Email: b.m.t.burgering@umcutrecht.nl.
References
- 1. Kaestner KH, Knochel W & Martinez DE (2000) Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev 14, 142–146. [PubMed] [Google Scholar]
- 2. Furuyama T, Nakazawa T, Nakano I & Mori N (2000) Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF‐16 homologues. Biochem J 349, 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lin K, Dorman JB, Rodan A & Kenyon C (1997) daf‐16: an HNF‐3/forkhead family member that can function to double the life‐span of Caenorhabditis elegans . Science 278, 1319–1322. [DOI] [PubMed] [Google Scholar]
- 4. Burgering BM (2008) A brief introduction to FOXOlogy. Oncogene 27, 2258–2262. [DOI] [PubMed] [Google Scholar]
- 5. Beckman KB & Ames BN (1998) The free radical theory of aging matures. Physiol Rev 78, 547–581. [DOI] [PubMed] [Google Scholar]
- 6. Sies H & Jones DP (2020) Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol 21, 363–383. [DOI] [PubMed] [Google Scholar]
- 7. Eijkelenboom A & Burgering BM (2013) FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol 14, 83–97. [DOI] [PubMed] [Google Scholar]
- 8. Dorman JB, Albinder B, Shroyer T & Kenyon C (1995) The age‐1 and daf‐2 genes function in a common pathway to control the lifespan of Caenorhabditis elegans . Genetics 141, 1399–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J & Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868. [DOI] [PubMed] [Google Scholar]
- 10. Kops GJ, de Ruiter ND, De Vries‐Smits AM, Powell DR, Bos JL & Burgering BM (1999) Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature 398, 630–634. [DOI] [PubMed] [Google Scholar]
- 11. Rena G, Guo S, Cichy SC, Unterman TG & Cohen P (1999) Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem 274, 17179–17183. [DOI] [PubMed] [Google Scholar]
- 12. Calissi G, Lam EW & Link W (2021) Therapeutic strategies targeting FOXO transcription factors. Nat Rev Drug Discov 20, 21–38. [DOI] [PubMed] [Google Scholar]
- 13. Carter ME & Brunet A (2007) FOXO transcription factors. Curr Biol 17, R113–R114. [DOI] [PubMed] [Google Scholar]
- 14. Brown AK & Webb AE (2018) Regulation of FOXO factors in mammalian cells. Curr Top Dev Biol 127, 165–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hart GW, Housley MP & Slawson C (2007) Cycling of O‐linked beta‐N‐acetylglucosamine on nucleocytoplasmic proteins. Nature 446, 1017–1022. [DOI] [PubMed] [Google Scholar]
- 16. Kuo M, Zilberfarb V, Gangneux N, Christeff N & Issad T (2008) O‐glycosylation of FoxO1 increases its transcriptional activity towards the glucose 6‐phosphatase gene. FEBS Lett 582, 829–834. [DOI] [PubMed] [Google Scholar]
- 17. Fardini Y, Masson E, Boudah O, Ben Jouira R, Cosson C, Pierre‐Eugene C, Kuo MS & Issad T (2014) O‐GlcNAcylation of FoxO1 in pancreatic beta cells promotes Akt inhibition through an IGFBP1‐mediated autocrine mechanism. FASEB J 28, 1010–1021. [DOI] [PubMed] [Google Scholar]
- 18. Shin H, Cha HJ, Na K, Lee MJ, Cho JY, Kim CY, Kim EK, Kang CM, Kim H & Paik YK (2018) O‐GlcNAcylation of the tumor suppressor FOXO3 triggers aberrant cancer cell growth. Cancer Res 78, 1214–1224. [DOI] [PubMed] [Google Scholar]
- 19. Ho SR, Wang K, Whisenhunt TR, Huang P, Zhu X, Kudlow JE & Paterson AJ (2010) O‐GlcNAcylation enhances FOXO4 transcriptional regulation in response to stress. FEBS Lett 584, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fardini Y, Perez‐Cervera Y, Camoin L, Pagesy P, Lefebvre T & Issad T (2015) Regulatory O‐GlcNAcylation sites on FoxO1 are yet to be identified. Biochem Biophys Res Commun 462, 151–158. [DOI] [PubMed] [Google Scholar]
- 21. Chae YC, Kim JY, Park JW, Kim KB, Oh H, Lee KH & Seo SB (2019) FOXO1 degradation via G9a‐mediated methylation promotes cell proliferation in colon cancer. Nucleic Acids Res 47, 1692–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xie Q, Hao Y, Tao L, Peng S, Rao C, Chen H, You H, Dong MQ & Yuan Z (2012) Lysine methylation of FOXO3 regulates oxidative stress‐induced neuronal cell death. EMBO Rep 13, 371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Calnan DR, Webb AE, White JL, Stowe TR, Goswami T, Shi X, Espejo A, Bedford MT, Gozani O, Gygi SP et al. (2012) Methylation by Set9 modulates FoxO3 stability and transcriptional activity. Aging (Albany NY) 4, 462–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamagata K, Daitoku H, Takahashi Y, Namiki K, Hisatake K, Kako K, Mukai H, Kasuya Y & Fukamizu A (2008) Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol Cell 32, 221–231. [DOI] [PubMed] [Google Scholar]
- 25. Choi S, Jeong HJ, Kim H, Choi D, Cho SC, Seong JK, Koo SH & Kang JS (2019) Skeletal muscle‐specific Prmt1 deletion causes muscle atrophy via deregulation of the PRMT6‐FOXO3 axis. Autophagy 15, 1069–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang F, Kerbl‐Knapp J, Rodriguez Colman MJ, Meinitzer A, Macher T, Vujić N, Fasching S, Jany‐Luig E, Korbelius M, Kuentzel KB et al. (2021) Global analysis of protein arginine methylation. Cell Rep Methods 1, 100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fu W, Ma Q, Chen L, Li P, Zhang M, Ramamoorthy S, Nawaz Z, Shimojima T, Wang H, Yang Y et al. (2009) MDM2 acts downstream of p53 as an E3 ligase to promote FOXO ubiquitination and degradation. J Biol Chem 284, 13987–14000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, Lang JY, Lai CC, Chang CJ, Huang WC et al. (2008) ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2‐mediated degradation. Nat Cell Biol 10, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brenkman AB, de Keizer PL, van den Broek NJ, Jochemsen AG & Burgering BM (2008) Mdm2 induces mono‐ubiquitination of FOXO4. PLoS One 3, e2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang H, Regan KM, Wang F, Wang D, Smith DI, van Deursen JM & Tindall DJ (2005) Skp2 inhibits FOXO1 in tumor suppression through ubiquitin‐mediated degradation. Proc Natl Acad Sci USA 102, 1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xiao N, Eto D, Elly C, Peng G, Crotty S & Liu YC (2014) The E3 ubiquitin ligase Itch is required for the differentiation of follicular helper T cells. Nat Immunol 15, 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van der Horst A, de Vries‐Smits AM, Brenkman AB, van Triest MH, van den Broek N, Colland F, Maurice MM & Burgering BM (2006) FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol 8, 1064–1073. [DOI] [PubMed] [Google Scholar]
- 33. Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K & Fukamizu A (2005) Acetylation of Foxo1 alters its DNA‐binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci USA 102, 11278–11283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M & Guarente L (2004) Mammalian SIRT1 represses forkhead transcription factors. Cell 116, 551–563. [DOI] [PubMed] [Google Scholar]
- 35. Yang Y, Hou H, Haller EM, Nicosia SV & Bai W (2005) Suppression of FOXO1 activity by FHL2 through SIRT1‐mediated deacetylation. EMBO J 24, 1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY et al. (2004) Stress‐dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015. [DOI] [PubMed] [Google Scholar]
- 37. van der Horst A, Tertoolen LG, de Vries‐Smits LM, Frye RA, Medema RH & Burgering BM (2004) FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). J Biol Chem 279, 28873–28879. [DOI] [PubMed] [Google Scholar]
- 38. Jing E, Gesta S & Kahn CR (2007) SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab 6, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A & Gupta MP (2009) Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a‐dependent antioxidant defense mechanisms in mice. J Clin Invest 119, 2758–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tao R, Xiong X, DePinho RA, Deng CX & Dong XC (2013) FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)‐cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. J Biol Chem 288, 29252–29259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chiang WC, Tishkoff DX, Yang B, Wilson‐Grady J, Yu X, Mazer T, Eckersdorff M, Gygi SP, Lombard DB & Hsu AL (2012) C. elegans SIRT6/7 homolog SIR‐2.4 promotes DAF‐16 relocalization and function during stress. PLoS Genet 8, e1002948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A, Vazquez‐Ortiz G, Jeong WI, Park O, Ki SH et al. (2010) Hepatic‐specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab 12, 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud PD, Yu RT, Alvarez JG, Downes M, Evans RM, Montminy M & Shaw RJ (2011) Class IIa histone deacetylases are hormone‐activated regulators of FOXO and mammalian glucose homeostasis. Cell 145, 607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu X, Greer C & Secombe J (2014) KDM5 interacts with Foxo to modulate cellular levels of oxidative stress. PLoS Genet 10, e1004676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang F, Marshall CB, Yamamoto K, Li GY, Gasmi‐Seabrook GM, Okada H, Mak TW & Ikura M (2012) Structures of KIX domain of CBP in complex with two FOXO3a transactivation domains reveal promiscuity and plasticity in coactivator recruitment. Proc Natl Acad Sci USA 109, 6078–6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang F, Marshall CB, Yamamoto K, Li GY, Plevin MJ, You H, Mak TW & Ikura M (2008) Biochemical and structural characterization of an intramolecular interaction in FOXO3a and its binding with p53. J Mol Biol 384, 590–603. [DOI] [PubMed] [Google Scholar]
- 47. Bourgeois B, Gui T, Hoogeboom D, Hocking HG, Richter G, Spreitzer E, Viertler M, Richter K, Madl T & Burgering BMT (2021) Multiple regulatory intrinsically disordered motifs control FOXO4 transcription factor binding and function. Cell Rep 36, 109446. [DOI] [PubMed] [Google Scholar]
- 48. Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP & Brunet A (2007) The energy sensor AMP‐activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem 282, 30107–30119. [DOI] [PubMed] [Google Scholar]
- 49. Wright PE & Dyson HJ (2015) Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol 16, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Putker M, Vos HR & Dansen TB (2014) Intermolecular disulfide‐dependent redox signalling. Biochem Soc Trans 42, 971–978. [DOI] [PubMed] [Google Scholar]
- 51. Riedel CG, Dowen RH, Lourenco GF, Kirienko NV, Heimbucher T, West JA, Bowman SK, Kingston RE, Dillin A, Asara JM et al. (2013) DAF‐16 employs the chromatin remodeller SWI/SNF to promote stress resistance and longevity. Nat Cell Biol 15, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Maures TJ, Greer EL, Hauswirth AG & Brunet A (2011) The H3K27 demethylase UTX‐1 regulates C. elegans lifespan in a germline‐independent, insulin‐dependent manner. Aging Cell 10, 980–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA & Ruvkun G (1997) The Fork head transcription factor DAF‐16 transduces insulin‐like metabolic and longevity signals in C. elegans . Nature 389, 994–999. [DOI] [PubMed] [Google Scholar]
- 54. Seoane J, Le HV, Shen L, Anderson SA & Massague J (2004) Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 117, 211–223. [DOI] [PubMed] [Google Scholar]
- 55. Gomis RR, Alarcon C, He W, Wang Q, Seoane J, Lash A & Massague J (2006) A FoxO‐Smad synexpression group in human keratinocytes. Proc Natl Acad Sci USA 103, 12747–12752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nowak K, Killmer K, Gessner C & Lutz W (2007) E2F–1 regulates expression of FOXO1 and FOXO3a. Biochim Biophys Acta 1769, 244–252. [DOI] [PubMed] [Google Scholar]
- 57. Shats I, Gatza ML, Liu B, Angus SP, You L & Nevins JR (2013) FOXO transcription factors control E2F1 transcriptional specificity and apoptotic function. Cancer Res 73, 6056–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xie Q, Peng S, Tao L, Ruan H, Yang Y, Li TM, Adams U, Meng S, Bi X, Dong MQ et al. (2014) E2F transcription factor 1 regulates cellular and organismal senescence by inhibiting Forkhead box O transcription factors. J Biol Chem 289, 34205–34213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nemoto S, Fergusson MM & Finkel T (2004) Nutrient availability regulates SIRT1 through a forkhead‐dependent pathway. Science 306, 2105–2108. [DOI] [PubMed] [Google Scholar]
- 60. Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA et al. (2017) Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169, 132–147.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon‐Cardo C & Pandolfi PP (2006) Identification of a tumour suppressor network opposing nuclear Akt function. Nature 441, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Essers MA, de Vries‐Smits LM, Barker N, Polderman PE, Burgering BM & Korswagen HC (2005) Functional interaction between beta‐catenin and FOXO in oxidative stress signaling. Science 308, 1181–1184. [DOI] [PubMed] [Google Scholar]
- 63. Doumpas N, Lampart F, Robinson MD, Lentini A, Nestor CE, Cantu C & Basler K (2019) TCF/LEF dependent and independent transcriptional regulation of Wnt/beta‐catenin target genes. EMBO J 38, e98873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tenbaum SP, Ordonez‐Moran P, Puig I, Chicote I, Arques O, Landolfi S, Fernandez Y, Herance JR, Gispert JD, Mendizabal L et al. (2012) beta‐catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat Med 18, 892–901. [DOI] [PubMed] [Google Scholar]
- 65. Almeida M, Han L, Martin‐Millan M, O'Brien CA & Manolagas SC (2007) Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta‐catenin from T cell factor‐ to forkhead box O‐mediated transcription. J Biol Chem 282, 27298–27305. [DOI] [PubMed] [Google Scholar]
- 66. Liu H, Fergusson MM, Wu JJ, Rovira II, Liu J, Gavrilova O, Lu T, Bao J, Han D, Sack MN et al. (2011) Wnt signaling regulates hepatic metabolism. Sci Signal 4, ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rao P, Pang M, Qiao X, Yu H, Wang H, Yang Y, Ren X, Hu M, Chen T, Cao Q et al. (2019) Promotion of beta‐catenin/Foxo1 signaling ameliorates renal interstitial fibrosis. Lab Invest 99, 1689–1701. [DOI] [PubMed] [Google Scholar]
- 68. Sreekumar A, Toneff MJ, Toh E, Roarty K, Creighton CJ, Belka GK, Lee DK, Xu J, Chodosh LA, Richards JS et al. (2017) WNT‐mediated regulation of FOXO1 constitutes a critical axis maintaining pubertal mammary stem cell homeostasis. Dev Cell 43, 436–448.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ng SS, Mahmoudi T, Danenberg E, Bejaoui I, de Lau W, Korswagen HC, Schutte M & Clevers H (2009) Phosphatidylinositol 3‐kinase signaling does not activate the wnt cascade. J Biol Chem 284, 35308–35313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tsai W‐B, Chung YM, Takahashi Y, Xu Z & Hu MCT (2008) Functional interaction between FOXO3a and ATM regulates DNA damage response. Nat Cell Biol 10, 460–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L & Zhu WG (2010) Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol 12, 665–675. [DOI] [PubMed] [Google Scholar]
- 72. Lindahl T & Barnes DE (2000) Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol 65, 127–133. [DOI] [PubMed] [Google Scholar]
- 73. Sood AJ, Viner C & Hoffman MM (2019) DNAmod: the DNA modification database. J Cheminform 11, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Olinski R, Zastawny T, Budzbon J, Skokowski J, Zegarski W & Dizdaroglu M (1992) DNA base modifications in chromatin of human cancerous tissues. FEBS Lett 309, 193–198. [DOI] [PubMed] [Google Scholar]
- 75. Zarkovic N (2020) Roles and functions of ROS and RNS in cellular physiology and pathology. Cells 9, 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sena LA & Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48, 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J Physiol 552, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Winterbourn CC (1995) Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett 82–83, 969–974. [DOI] [PubMed] [Google Scholar]
- 79. Rosen JE, Prahalad AK & Williams GM (1996) 8‐Oxodeoxyguanosine formation in the DNA of cultured cells after exposure to H2O2 alone or with UVB or UVA irradiation. Photochem Photobiol 64, 117–122. [DOI] [PubMed] [Google Scholar]
- 80. Pluskota‐Karwatka D (2008) Modifications of nucleosides by endogenous mutagens‐DNA adducts arising from cellular processes. Bioorg Chem 36, 198–213. [DOI] [PubMed] [Google Scholar]
- 81. Boiteux S, Gellon L & Guibourt N (2002) Repair of 8‐oxoguanine in Saccharomyces cerevisiae: interplay of DNA repair and replication mechanisms. Free Radic Biol Med 32, 1244–1253. [DOI] [PubMed] [Google Scholar]
- 82. Poetsch AR (2020) The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput Struct Biotechnol J 18, 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN et al. (2020) The repertoire of mutational signatures in human cancer. Nature 578, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Alexandrov LB, Nik‐Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen‐Dale AL et al. (2013) Signatures of mutational processes in human cancer. Nature 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Perillo B, Ombra MN, Bertoni A, Cuozzo C, Sacchetti S, Sasso A, Chiariotti L, Malorni A, Abbondanza C & Avvedimento EV (2008) DNA oxidation as triggered by H3K9me2 demethylation drives estrogen‐induced gene expression. Science 319, 202–206. [DOI] [PubMed] [Google Scholar]
- 86. Bianco JN & Schumacher B (2018) MPK‐1/ERK pathway regulates DNA damage response during development through DAF‐16/FOXO. Nucleic Acids Res 46, 6129–6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mueller MM, Castells‐Roca L, Babu V, Ermolaeva MA, Muller RU, Frommolt P, Williams AB, Greiss S, Schneider JI, Benzing T et al. (2014) DAF‐16/FOXO and EGL‐27/GATA promote developmental growth in response to persistent somatic DNA damage. Nat Cell Biol 16, 1168–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kong H & Chandel NS (2018) Regulation of redox balance in cancer and T cells. J Biol Chem 293, 7499–7507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Essers MA, Weijzen S, de Vries‐Smits AM, Saarloos I, de Ruiter ND, Bos JL & Burgering BM (2004) FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J 23, 4802–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ho KK, McGuire VA, Koo CY, Muir KW, de Olano N, Maifoshie E, Kelly DJ, McGovern UB, Monteiro LJ, Gomes AR et al. (2012) Phosphorylation of FOXO3a on Ser‐7 by p38 promotes its nuclear localization in response to doxorubicin. J Biol Chem 287, 1545–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Honda Y & Honda S (1999) The daf‐2 gene network for longevity regulates oxidative stress resistance and Mn‐superoxide dismutase gene expression in Caenorhabditis elegans . FASEB J 13, 1385–1393. [PubMed] [Google Scholar]
- 92. Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH & Burgering BM (2002) Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419, 316–321. [DOI] [PubMed] [Google Scholar]
- 93. Charitou P, Rodriguez‐Colman M, Gerrits J, van Triest M, Groot Koerkamp M, Hornsveld M, Holstege F, Verhoeven‐Duif NM & Burgering BM (2015) FOXOs support the metabolic requirements of normal and tumor cells by promoting IDH1 expression. EMBO Rep 16, 456–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo‐Kallanian S, Williams IR & Sears C (2007) FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128, 325–339. [DOI] [PubMed] [Google Scholar]
- 95. Ferber EC, Peck B, Delpuech O, Bell GP, East P & Schulze A (2012) FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ 19, 968–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Jensen KS, Binderup T, Jensen KT, Therkelsen I, Borup R, Nilsson E, Multhaupt H, Bouchard C, Quistorff B, Kjær A et al. (2011) FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J 30, 4554–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Rimmelé P, Liang R, Bigarella CL, Kocabas F, Xie J, Serasinghe MN, Chipuk J, Sadek H, Zhang CC & Ghaffari S (2015) Mitochondrial metabolism in hematopoietic stem cells requires functional FOXO3. EMBO Rep 16, 1164–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ludikhuize MC, Meerlo M, Gallego MP, Xanthakis D, Burgaya Julia M, Nguyen NTB, Brombacher EC, Liv N, Maurice MM, Paik JH et al. (2020) Mitochondria define intestinal stem cell differentiation downstream of a FOXO/notch axis. Cell Metab 32, 889–900.e7. [DOI] [PubMed] [Google Scholar]
- 99. Jacobs KM, Pennington JD, Bisht KS, Aykin‐Burns N, Kim H‐S, Mishra M, Sun L, Nguyen P, Ahn B‐H, Leclerc J et al. (2008) SIRT3 interacts with the daf‐16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int J Biol Sci 4, 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Tseng AH, Shieh S‐S & Wang DL (2013) SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic Biol Med 63, 222–234. [DOI] [PubMed] [Google Scholar]
- 101. Pfanner N, Warscheid B & Wiedemann N (2019) Mitochondrial proteins: from biogenesis to functional networks. Nat Rev Mol Cell Biol 20, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Celestini V, Tezil T, Russo L, Fasano C, Sanese P, Forte G, Peserico A, Signorile ML, Longo G & De Rasmo D (2018) Uncoupling FoxO3A mitochondrial and nuclear functions in cancer cells undergoing metabolic stress and chemotherapy. Cell Death Dis 9, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lee J, Giordano S & Zhang J (2012) Autophagy, mitochondria and oxidative stress: cross‐talk and redox signalling. Biochem J 441, 523–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mei Y, Zhang Y, Yamamoto K, Xie W, Mak TW & You H (2009) FOXO3a‐dependent regulation of Pink1 (Park6) mediates survival signaling in response to cytokine deprivation. Proc Natl Acad Sci USA 106, 5153–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Jeggo PA, Pearl LH & Carr AM (2016) DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer 16, 35–42. [DOI] [PubMed] [Google Scholar]
- 106. Lovejoy CA & Cortez D (2009) Common mechanisms of PIKK regulation. DNA Repair (Amst) 8, 1004–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lee JH & Paull TT (2020) Mitochondria at the crossroads of ATM‐mediated stress signaling and regulation of reactive oxygen species. Redox Biol 32, 101511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Adamowicz M, Vermezovic J & d'Adda di Fagagna F (2016) NOTCH1 inhibits activation of ATM by impairing the formation of an ATM‐FOXO3a‐KAT5/Tip60 complex. Cell Rep 16, 2068–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Sun Y, Xu Y, Roy K & Price BD (2007) DNA damage‐induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol Cell Biol 27, 8502–8509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sun Y, Jiang X, Chen S, Fernandes N & Price BD (2005) A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci USA 102, 13182–13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yalcin S, Zhang X, Luciano JP, Mungamuri SK, Marinkovic D, Vercherat C, Sarkar A, Grisotto M, Taneja R & Ghaffari S (2008) Foxo3 is essential for the regulation of ataxia telangiectasia mutated and oxidative stress‐mediated homeostasis of hematopoietic stem cells. J Biol Chem 283, 25692–25705. [DOI] [PubMed] [Google Scholar]
- 112. Cosentino C, Grieco D & Costanzo V (2011) ATM activates the pentose phosphate pathway promoting anti‐oxidant defence and DNA repair. EMBO J 30, 546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Brenkman AB, van den Broek NJ, de Keizer PL, van Gent DC & Burgering BM (2010) The DNA damage repair protein Ku70 interacts with FOXO4 to coordinate a conserved cellular stress response. FASEB J 24, 4271–4280. [DOI] [PubMed] [Google Scholar]
- 114. Collis SJ, DeWeese TL, Jeggo PA & Parker AR (2005) The life and death of DNA‐PK. Oncogene 24, 949–961. [DOI] [PubMed] [Google Scholar]
- 115. Park J, Feng J, Li Y, Hammarsten O, Brazil DP & Hemmings BA (2009) DNA‐dependent protein kinase‐mediated phosphorylation of protein kinase B requires a specific recognition sequence in the C‐terminal hydrophobic motif. J Biol Chem 284, 6169–6174. [DOI] [PubMed] [Google Scholar]
- 116. Hornsveld M, Feringa FM, Krenning L, van den Berg J, Smits LMM, Nguyen NBT, Rodriguez‐Colman MJ, Dansen TB, Medema RH & Burgering BMT (2021) A FOXO‐dependent replication checkpoint restricts proliferation of damaged cells. Cell Rep 34, 108675. [DOI] [PubMed] [Google Scholar]
- 117. Li J, Du W, Maynard S, Andreassen PR & Pang Q (2010) Oxidative stress‐specific interaction between FANCD2 and FOXO3a. Blood 115, 1545–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Li X, Li J, Wilson A, Sipple J, Schick J & Pang Q (2015) Fancd2 is required for nuclear retention of Foxo3a in hematopoietic stem cell maintenance. J Biol Chem 290, 2715–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Daitoku H, Kaneko Y, Yoshimochi K, Matsumoto K, Araoi S, Sakamaki JI, Takahashi Y & Fukamizu A (2016) Non‐transcriptional function of FOXO1/DAF‐16 contributes to translesion DNA synthesis. Mol Cell Biol 36, 2755–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Dall'Acqua A, Sonego M, Pellizzari I, Pellarin I, Canzonieri V, D'Andrea S, Benevol S, Sorio R, Giorda G, Califano D et al. (2017) CDK6 protects epithelial ovarian cancer from platinum‐induced death via FOXO3 regulation. EMBO Mol Med 9, 1415–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ Jr, DiStefano PS, Chiang LW & Greenberg ME (2002) DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 296, 530–534. [DOI] [PubMed] [Google Scholar]
- 122. Niehrs C & Schafer A (2012) Active DNA demethylation by Gadd45 and DNA repair. Trends Cell Biol 22, 220–227. [DOI] [PubMed] [Google Scholar]
- 123. Medema RH, Kops GJ, Bos JL & Burgering BM (2000) AFX‐like Forkhead transcription factors mediate cell‐cycle regulation by Ras and PKB through p27kip1. Nature 404, 782–787. [DOI] [PubMed] [Google Scholar]
- 124. de Keizer PL, Packer LM, Szypowska AA, Riedl‐Polderman PE, van den Broek NJ, de Bruin A, Dansen TB, Marais R, Brenkman AB & Burgering BM (2010) Activation of forkhead box O transcription factors by oncogenic BRAF promotes p21cip1‐dependent senescence. Cancer Res 70, 8526–8536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Tudzarova S, Trotter MW, Wollenschlaeger A, Mulvey C, Godovac‐Zimmermann J, Williams GH & Stoeber K (2010) Molecular architecture of the DNA replication origin activation checkpoint. EMBO J 29, 3381–3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y et al. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166. [DOI] [PubMed] [Google Scholar]
- 127. Bourgeois B & Madl T (2018) Regulation of cellular senescence via the FOXO4‐p53 axis. FEBS Lett 592, 2083–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Zhang X, Tang N, Hadden TJ & Rishi AK (2011) Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta 1813, 1978–1986. [DOI] [PubMed] [Google Scholar]
- 129. Bridge D, Theofiles AG, Holler RL, Marcinkevicius E, Steele RE & Martinez DE (2010) FoxO and stress responses in the cnidarian Hydra vulgaris . PLoS One 5, e11686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Hosaka T, Biggs WH 3rd, Tieu D, Boyer AD, Varki NM, Cavenee WK & Arden KC (2004) Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci USA 101, 2975–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Castrillon DH, Miao L, Kollipara R, Horner JW & DePinho RA (2003) Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science 301, 215–218. [DOI] [PubMed] [Google Scholar]
- 132. Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR et al. (2007) FoxOs are lineage‐restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 128, 309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Bansal A, Kwon ES, Conte D Jr, Liu H, Gilchrist MJ, MacNeil LT & Tissenbaum HA (2014) Transcriptional regulation of Caenorhabditis elegans FOXO/DAF‐16 modulates lifespan. Longev Healthspan 3, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Wang F, Marshall CB & Ikura M (2015) Forkhead followed by disordered tail: the intrinsically disordered regions of FOXO3a. Intrinsically Disord Proteins 3, e1056906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. van der Vos KE, Eliasson P, Proikas‐Cezanne T, Vervoort SJ, van Boxtel R, Putker M, van Zutphen IJ, Mauthe M, Zellmer S, Pals C et al. (2012) Modulation of glutamine metabolism by the PI(3)K‐PKB‐FOXO network regulates autophagy. Nat Cell Biol 14, 829–837. [DOI] [PubMed] [Google Scholar]
- 136. Franz F, Weidinger C, Krause K, Gimm O, Dralle H & Fuhrer D (2016) The transcriptional regulation of FOXO genes in thyrocytes. Horm Metab Res 48, 601–606. [DOI] [PubMed] [Google Scholar]
- 137. Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ & Sabatini DM (2006) Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt‐FOXO and PKCalpha, but not S6K1. Dev Cell 11, 859–871. [DOI] [PubMed] [Google Scholar]
- 138. Chen CC, Jeon SM, Bhaskar PT, Nogueira V, Sundararajan D, Tonic I, Park Y & Hay N (2010) FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor. Dev Cell 18, 592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Eijkelenboom A, Mokry M, de Wit E, Smits LM, Polderman PE, van Triest MH, van Boxtel R, Schulze A, de Laat W, Cuppen E et al. (2013) Genome‐wide analysis of FOXO3 mediated transcription regulation through RNA polymerase II profiling. Mol Syst Biol 9, 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Eijkelenboom A, Mokry M, Smits LM, Nieuwenhuis EE & Burgering BM (2013) FOXO3 selectively amplifies enhancer activity to establish target gene regulation. Cell Rep 5, 1664–1678. [DOI] [PubMed] [Google Scholar]
- 141. Webb AE, Pollina EA, Vierbuchen T, Urban N, Ucar D, Leeman DS, Martynoga B, Sewak M, Rando TA, Guillemot F et al. (2013) FOXO3 shares common targets with ASCL1 genome‐wide and inhibits ASCL1‐dependent neurogenesis. Cell Rep 4, 477–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Wang M, Zhang X, Zhao H, Wang Q & Pan Y (2009) FoxO gene family evolution in vertebrates. BMC Evol Biol 9, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Schmitt‐Ney M (2020) The FOXO's advantages of being a family: considerations on function and evolution. Cells 9, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Morris BJ, Willcox DC, Donlon TA & Willcox BJ (2015) FOXO3: a major gene for human longevity – a mini‐review. Gerontology 61, 515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Boehm AM, Khalturin K, Anton‐Erxleben F, Hemmrich G, Klostermeier UC, Lopez‐Quintero JA, Oberg HH, Puchert M, Rosenstiel P, Wittlieb J et al. (2012) FoxO is a critical regulator of stem cell maintenance in immortal Hydra . Proc Natl Acad Sci USA 109, 19697–19702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Sato T & Clevers H (2013) Growing self‐organizing mini‐guts from a single intestinal stem cell: mechanism and applications. Science 340, 1190–1194. [DOI] [PubMed] [Google Scholar]
- 147. Cheung TH & Rando TA (2013) Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol 14, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Gopinath SD, Webb AE, Brunet A & Rando TA (2014) FOXO3 promotes quiescence in adult muscle stem cells during the process of self‐renewal. Stem Cell Reports 2, 414–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Garcia‐Prat L, Perdiguero E, Alonso‐Martin S, Dell'Orso S, Ravichandran S, Brooks SR, Juan AH, Campanario S, Jiang K, Hong X et al. (2020) FoxO maintains a genuine muscle stem‐cell quiescent state until geriatric age. Nat Cell Biol 22, 1307–1318. [DOI] [PubMed] [Google Scholar]
- 150. Dentice M, Ambrosio R, Damiano V, Sibilio A, Luongo C, Guardiola O, Yennek S, Zordan P, Minchiotti G, Colao A et al. (2014) Intracellular inactivation of thyroid hormone is a survival mechanism for muscle stem cell proliferation and lineage progression. Cell Metab 20, 1038–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Kretzschmar K, Post Y, Bannier‐Helaouet M, Mattiotti A, Drost J, Basak O, Li VSW, van den Born M, Gunst QD, Versteeg D et al. (2018) Profiling proliferative cells and their progeny in damaged murine hearts. Proc Natl Acad Sci USA 115, E12245–E12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Zhu M, Goetsch SC, Wang Z, Luo R, Hill JA, Schneider J, Morris SM Jr & Liu ZP (2015) FoxO4 promotes early inflammatory response upon myocardial infarction via endothelial Arg1. Circ Res 117, 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Renault VM, Rafalski VA, Morgan AA, Salih DA, Brett JO, Webb AE, Villeda SA, Thekkat PU, Guillerey C, Denko NC et al. (2009) FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell 5, 527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, Nakao S, Motoyama N & Hirao A (2010) TGF‐beta‐FOXO signalling maintains leukaemia‐initiating cells in chronic myeloid leukaemia. Nature 463, 676–680. [DOI] [PubMed] [Google Scholar]
- 155. Sykes SM, Lane SW, Bullinger L, Kalaitzidis D, Yusuf R, Saez B, Ferraro F, Mercier F, Singh H, Brumme KM et al. (2011) AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell 146, 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Mortzfeld BM & Bosch TC (2017) Eco‐Aging: stem cells and microbes are controlled by aging antagonist FoxO. Curr Opin Microbiol 38, 181–187. [DOI] [PubMed] [Google Scholar]
- 157. Becker T, Loch G, Beyer M, Zinke I, Aschenbrenner AC, Carrera P, Inhester T, Schultze JL & Hoch M (2010) FOXO‐dependent regulation of innate immune homeostasis. Nature 463, 369–373. [DOI] [PubMed] [Google Scholar]
- 158. Campisi J (2005) Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120, 513–522. [DOI] [PubMed] [Google Scholar]
- 159. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A et al. (2016) Naturally occurring p16(Ink4a)‐positive cells shorten healthy lifespan. Nature 530, 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Kwon ES, Narasimhan SD, Yen K & Tissenbaum HA (2010) A new DAF‐16 isoform regulates longevity. Nature 466, 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Lalmansingh AS, Karmakar S, Jin Y & Nagaich AK (2012) Multiple modes of chromatin remodeling by Forkhead box proteins. Biochim Biophys Acta 1819, 707–715. [DOI] [PubMed] [Google Scholar]
- 162. Hatta M & Cirillo LA (2007) Chromatin opening and stable perturbation of core histone:DNA contacts by FoxO1. J Biol Chem 282, 35583–35593. [DOI] [PubMed] [Google Scholar]
- 163. Hatta M, Liu F & Cirillo LA (2009) Acetylation curtails nucleosome binding, not stable nucleosome remodeling, by FoxO1. Biochem Biophys Res Commun 379, 1005–1008. [DOI] [PubMed] [Google Scholar]
- 164. Shin Y & Brangwynne CP (2017) Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382. [DOI] [PubMed] [Google Scholar]
- 165. Iborra FJ, Pombo A, Jackson DA & Cook PR (1996) Active RNA polymerases are localized within discrete transcription “factories’ in human nuclei. J Cell Sci 109 (Pt 6), 1427–1436. [DOI] [PubMed] [Google Scholar]
- 166. Sabari BR, Dall'Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC et al. (2018) Coactivator condensation at super‐enhancers links phase separation and gene control. Science 361, eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Hnisz D, Shrinivas K, Young RA, Chakraborty AK & Sharp PA (2017) A phase separation model for transcriptional control. Cell 169, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Boija A, Klein IA, Sabari BR, Dall'Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM et al. (2018) Transcription factors activate genes through the phase‐separation capacity of their activation domains. Cell 175, 1842–1855.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Mortzfeld BM, Taubenheim J, Klimovich AV, Fraune S, Rosenstiel P & Bosch TCG (2019) Temperature and insulin signaling regulate body size in Hydra by the Wnt and TGF‐beta pathways. Nat Commun 10, 3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Rena G, Woods YL, Prescott AR, Peggie M, Unterman TG, Williams MR & Cohen P (2002) Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. EMBO J 21, 2263–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Woods YL, Rena G, Morrice N, Barthel A, Becker W, Guo S, Unterman TG & Cohen P (2001) The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem J 355, 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Putker M, Vos HR, van Dorenmalen K, de Ruiter H, Duran AG, Snel B, Burgering BM, Vermeulen M & Dansen TB (2015) Evolutionary acquisition of cysteines determines FOXO paralog‐specific redox signaling. Antioxid Redox Signal 22, 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]