

Abstract
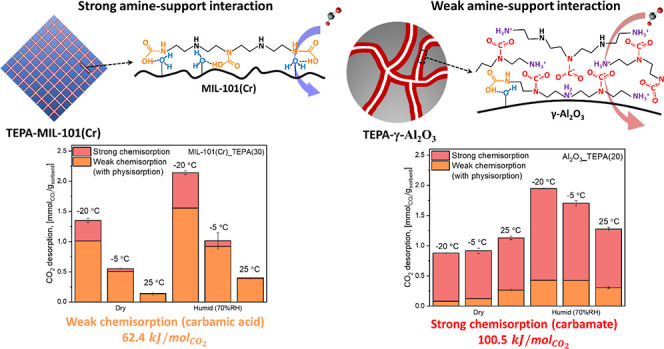
A variety of amine-impregnated porous solid sorbents for direct air capture (DAC) of CO2 have been developed, yet the effect of amine-solid support interactions on the CO2 adsorption behavior is still poorly understood. When tetraethylenepentamine (TEPA) is impregnated on two different supports, commercial γ-Al2O3 and MIL-101(Cr), they show different trends in CO2 sorption when the temperature (−20 to 25 °C) and humidity (0–70% RH) of the simulated air stream are varied. In situ IR spectroscopy is used to probe the mechanism of CO2 sorption on the two supported amine materials, with weak chemisorption (formation of carbamic acid) being the dominant pathway over MIL-101(Cr)-supported TEPA and strong chemisorption (formation of carbamate) occurring over γ-Al2O3-supported TEPA. Formation of both carbamic acid and carbamate species is enhanced over the supported TEPA materials under humid conditions, with the most significant enhancement observed at −20 °C. However, while equilibrium H2O sorption is high at cold temperatures (e.g., −20 °C), the effect of humidity on a practical cyclic DAC process is expected to be minimal due to slow H2O uptake kinetics. This work suggests that the CO2 capture mechanisms of impregnated amines can be controlled by adjusting the degree of amine-solid support interaction and that H2O adsorption behavior is strongly affected by the properties of the support materials. Thus, proper selection of solid support materials for amine impregnation will be important for achieving optimized DAC performance under varied deployment conditions, such as cold (e.g., −20 °C) or ambient temperature (e.g., 25 °C) operations.
Introduction
Fast decarbonization of the energy sector requires the deployment of avoided emission technologies like carbon capture and sequestration from point sources as well as the deployment of negative emission technologies (NETs) such as direct air capture (DAC) coupled with carbon storage.1 DAC has several advantages over other NETs, such as bioenergy with carbon capture and storage,2 due to its lower requirement for land and water utilization.3 However, DAC must overcome challenges associated with high capital costs and moderate energy costs.
DAC has been intensively studied in recent years, and a variety of adsorbent materials have been developed to directly capture CO2 from the air. Among them, amine-functionalized porous materials are attractive adsorbents for DAC and are widely studied due to their promising performance under humid conditions, lower amine volatilization rates compared to amine solvents, and the strong affinity of amine groups for CO2.4−6 However, due to the high CO2 binding energy of amines, amine-based adsorbent materials require higher energy for CO2 desorption compared to physisorbents such as zeolites and MOFs. While the amine-containing solid adsorbents are tolerant to moisture in the air, it is generally known that physisorbents are not effective adsorbents for CO2 capture in the presence of moisture due to competitive adsorption between CO2 and H2O, which yields high sorbent regeneration costs. Thus, to utilize physisorbents for DAC, a two-stage process for the dehydration of air will likely be required.7
The behavior of solid-supported amines can be impacted by the structure and composition of the support materials because the porous structure of the support impacts amine dispersion and thus the access of CO2 to the amine sites.5 Moreover, differences in surface chemistries in support materials can result in different binding motifs between the amine and the support, which also has the potential to influence the CO2 capture performance. A variety of porous support materials have been used for amine impregnation, including silica,8−40 γ-alumina,41−43 MOFs,44−46 zeolite,47 and mixed metal oxides.48,49 Despite this, the effects of amine-solid support interactions on CO2 adsorption behavior are still poorly understood. Furthermore, most prior DAC studies are limited to adsorption temperatures above indoor ambient room temperature (>20 °C). Only our previous study on amine-impregnated MIL-101(Cr) reported detailed DAC behavior under sub-ambient temperature conditions (from −20 to 25 °C), including both dry35 and humid gases.44 Since there will likely be no optimal sorbent covering all conditions, including day/night, summer/winter, and rainy/dry conditions, designing and operating a DAC process will be complex. A high-performing adsorbent will, therefore, be a sorbent that operates well under all conditions, rather than one that is optimized for a narrow set of conditions. To this end, investigating the DAC performance of sorbent materials under varied conditions is crucial for the practical deployment of DAC technologies.50
As noted earlier, the adsorption mechanism (e.g., physisorption, the formation of carbamic acid, the formation of ammonium carbamate ion pairs, and the formation of ammonium bicarbonate) may vary depending on the degree of interaction between the impregnated amines and support. For example, specific molecular interactions between amines and MOFs like Mg2(dobpdc) lead to unique CO2 adsorption mechanisms51−57 compared to more ill-defined systems based on MOFs and PEI.58−61 We hypothesize that the CO2 adsorption pathways for impregnated amines within different support materials and/or pore structures may differ depending on adsorption temperature and humidities. Thus, to design optimized adsorbents for DAC processes operating in an ever-changing environment (outdoor air),50 an in-depth understanding of the roles of support materials, temperature, humidity on CO2 adsorption kinetics, and thermodynamics is required.
Here, to systematically probe the effect of support materials on the DAC performance of impregnated amines [tetraethylenepentamine (TEPA)], amine-based solid sorbents were synthesized with two different porous solid supports, MIL-101(Cr) and γ-Al2O3. TEPA was used for amine impregnation because this chemical mainly consists of primary and secondary amines, which have a higher potential for CO2 adsorption (e.g., heat of CO2 adsorption = −80 to −110 kJ/molCO2) compared to tertiary amines (−20 kJ/molCO2),62,63 as well as shorter amine chains that likely result in enhanced CO2 kinetics relative to the branched poly(ethylenimine) typically employed, especially under cold temperature conditions. Commercial γ-Al2O3 was selected for comparison with MIL-101(Cr) supports since they are already well-investigated support materials for amine impregnation and currently offer a more accessible pathway for scale up and deployment than MOFs. The materials’ CO2 adsorption behavior under both dry and humid (70% RH) 400 ppm CO2 conditions over a range of adsorption temperatures (−20 to 25 °C) was probed by CO2/H2O temperature-programed desorption (TPD) experiments and in situ FT-IR spectroscopy. We demonstrate that the properties of the porous solid support materials, such as pore size, pore volume, surface area, and support composition, have a significant effect on the CO2 adsorption pathways of the impregnated amines. The results presented here suggest that proper porous support materials should be considered for amine impregnation to operate an optimized DAC process, depending on the sub-ambient to ambient temperature conditions and humidities anticipated.
Experimental Section
Material Synthesis
Synthesis of MIL-101(Cr) powders was conducted based on the literature.46,64 The detailed synthesis process is described in our previous work.44 The synthesized MOF powders were dried under a high vacuum (about 10 mTorr) at 150 °C overnight for further analysis and amine impregnation. The MIL-101(Cr) and γ-Al2O3 powders were physically impregnated with 30 and 20 wt % TEPA loadings, respectively, to achieve a similar extent of pore filling (about 25%). The detailed procedure of amine impregnation is described in the Supporting Information.
Characterization
Nitrogen Physisorption
A surface area and porosity (SAP) system (autosorb iQ/Quantachrome) was used for N2 physisorption experiments at 77 K. About 100 mg of MIL-101(Cr) and γ-Al2O3 were activated at 150 and 110 °C, respectively, under a vacuum for 15 h before the measurement. For the TEPA-impregnated MIL-101(Cr) and γ-Al2O3 powder sorbents, the activation was conducted at 60 °C under a vacuum for 3 h. The BET surface area was estimated using the N2 physisorption data in the P/P0 range of 0.05–0.2. Pore volumes of the materials were determined based on the N2 physisorption at a partial pressure of 0.995 and normalized per gram of support (MIL-101(Cr) or γ-Al2O3) in the powder sorbents.
CO2 Breakthrough Experiments under Dry and Humid Conditions with 400 ppm CO2
A custom-built fixed bed reactor described in the literature44 was used to perform dry and humid 400 ppm CO2 breakthrough experiments with TEPA-impregnated MIL-101(Cr) and γ-Al2O3 under a wide range of temperatures (−20, −5, and 25 °C) and humidities (0–70% RH). About 50 mg (wet basis) of powder sorbents were loaded inside the 5 cm fixed bed (1/4″ stainless steel tube with 4 mm ID) using glass wool on both ends of the column. A more detailed procedure is described in Supporting Information. In this study, small amounts of powder sorbents (<50 mg) were tested inside a 1/4″ stainless steel fixed bed column under an extremely diluted CO2 gas stream (400 ppm CO2/N2). The bed was always immersed in liquid (50% water/50% ethylene glycol) continuously circulating inside the bath of chiller (Julabo CD-600F) for efficient removal of the heat generated during the 400 ppm CO2 breakthrough. To confirm an isothermal condition of the fixed bed system, the convective heat transfer coefficient, h, needed to keep the temperature of the powder sorbents less than 1 °C above the flowing simulated air was estimated. For example, the required h was estimated to be 2.36 × 10–3 W/m2/K for 0.01 W of the heat generation rate during 400 ppm CO2 adsorption with the TEPA-impregnated γ-Al2O3 in the fixed bed system (45 mg of the sorbent, 94 m2/gsorbent, −ΔH = 100 kJ/molCO2, and 2.64 × 10–4 mmol/g/s adsorption rate). The estimated value was much lower than the h of natural convection in the air (2.5–25 W/m2/K, produced by density differences of air),65 indicating that convection cooling by flowing adsorption gas is enough to maintain an isothermal condition due to the slow heat generation rate of CO2 adsorption under an extremely diluted CO2 gas stream (400 ppm CO2/N2). Thus, it was assumed that the temperature change of the fixed bed column is negligible during the 400 ppm CO2 adsorption.
Measurement of Energy for Dry/Wet CO2 and H2O Desorption
The interaction strength between CO2 and the amine-impregnated MIL-101(Cr) and γ-Al2O3 powder sorbents are expected to be different under varied dry and humid adsorption temperature conditions. Thus, it is crucial to quantify the binding energy of dry and humid CO2 (also H2O) to estimate an energy requirement for sorbent regeneration. In this study, TPD experiments were performed with varied heating rates to directly evaluate the energy required for desorption of CO2 (dry and humid) and H2O from amine-impregnated MIL-101(Cr) and γ-Al2O3 powder sorbents.66 Detailed procedures are described in Supporting Information, including a definition of the desorption energy used in this work.
In Situ Diffuse Reflectance Infrared Fourier Transform Spectroscopy
In situ FT-IR spectroscopy was performed on a Nicolet iS10 IR spectrometer with a low-temperature diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) cell (CHC-CHA-4, Harrick Scientific Products Inc.). For the dry and wet 400 ppm CO2 adsorption studies, a sample holder inside the DRIFTS cell chamber was filled with about 50 mg of sorbent powders. The sample was then activated at 60 °C under 40 sccm N2 flow for 3 h. After the sample temperature cooled to the adsorption temperature (−20 °C or 25 °C), the inlet gas flow was switched to dry or wet (70% RH) 400 ppm CO2/N2 (40 sccm). During the adsorption, spectra were collected with 64 scans at a resolution of 4 cm–1 every 5 min. The humidity for the wet gas was precisely controlled by a wet gas generator (WETSYS/SETARAM).
Results and Discussion
Characterization of Amine (TEPA)-Impregnated MIL-101(Cr) and γ-Al2O3
To investigate the effect of support material structure and composition on the CO2 capture behavior of impregnated amines under a similar extent of pore filling (about 25%), MIL-101(Cr) and γ-Al2O3 porous supports were physically impregnated with TEPA to 30 and 20 wt % loadings, respectively. Key physical properties, including pore volumes, pore sizes, and BET surface areas of the bare support and amine-loaded sorbent materials were determined from N2 physisorption isotherms (Figure S1), and the results are summarized in Table 1. The particle size of two support materials is also included. The mean particle size of γ-Al2O3 was estimated based on SEM images (Figure S2). The properties of the MIL-101(Cr) materials are comparable to the prior literature.44,46 MIL-101(Cr) has a smaller average pore size (2.4 nm) than γ-Al2O3 (16.1 nm), with a much higher pore volume and BET surface area, suggesting that impregnated amines may interact with MIL-101(Cr) more significantly than γ-Al2O3 at similar loadings. Table 1 shows the extent of pore filling of sorbents determined based on the N2 physisorption isotherms of the bare support materials and amine-impregnated powder sorbents (Figure S1). The theoretical extent of pore filling was also calculated with an assumption that all TEPA was impregnated inside the pores of the support materials. The 30 wt % TEPA-impregnated MIL-101(Cr) [MIL-101(Cr)_TEPA(30)] and 20 wt % TEPA-impregnated γ-Al2O3 [Al2O3_TEPA(20)] show almost identical extents of pore filling (25.4–27.6%). Close values between the measured and theoretical pore filling indicate that TEPA was effectively impregnated inside the pores of both MIL-101(Cr) and γ-Al2O3.
Table 1. Physical Properties of Support Materials and Adsorbents.
| support materials | MIL-101(Cr) | γ-Al2O3 |
|---|---|---|
| particle size, [μm] | ∼0.544 | 6.9a |
| pore volume, [cc/g] | 1.84 | 0.95 |
| pore size (mode), [nm] | 2.4 | 16.1 |
| BET surface area, [m2/g] | 3270 | 153 |
| amine (TEPA) loadings, [wt %] | 30 | 20 |
| powder sorbents | MIL-101(Cr)_TEPA(30) | Al2O3_TEPA(20) |
| pore filling, [ %] | 25.4 (by N2 physisorption) | 27.6 (by N2 physisorption) |
| 23.3 (by calculation) | 26.5 (by calculation) |
Mean particle size was determined based on SEM images (Figure S2).
Humid and Dry CO2 Adsorption Behavior of Amine (TEPA)-Impregnated MIL-101(Cr) and γ-Al2O3 at Ambient and Cold Temperatures
To study the effects of temperature and humidity on the CO2 adsorption behavior of impregnated amines (TEPA) within different porous support materials, dry and humid (70% RH) 400 ppm CO2 breakthrough experiments were conducted with MIL-101(Cr)_TEPA(30) and Al2O3_TEPA(20) powder sorbents at −20, −5, and 25 °C. Figure S3 shows the dry and humid (70% RH) CO2 breakthrough curves of the powder sorbents under different temperature conditions. Pseudo-equilibrium 400 ppm CO2 adsorption capacities of MIL-101(Cr)_TEPA(30) and Al2O3_TEPA(20) were determined based on the breakthrough curves, and the results are shown in Figure 1a,b, respectively, with amine efficiencies.
Figure 1.

Pseudo-equilibrium 400 ppm CO2 adsorption capacities and amine efficiencies of (a) 30 wt % TEPA-impregnated MIL-101(Cr) and (b) 20 wt % TEPA-impregnated γ-Al2O3 powder adsorbents under dry and humid (70% RH) conditions at −20, −5, and 25 °C. Both materials have similar pore filling with TEPA.
The 400 ppm CO2 uptake of MIL-101(Cr)_TEPA(30) under dry conditions was significantly enhanced with decreasing temperature, from 0.11 mmol/g (25 °C) to 1.11 mmol/g (−20 °C) because the overall CO2 capture capacities are dominantly affected by thermodynamics rather than mass transfer due to the low degree of pore filling (25.4%, see Table 1).44 On the other hand, TEPA in γ-Al2O3 showed a different trend. The 400 ppm CO2 uptake of Al2O3_TEPA(20) (1.10 mmol/g) at 25 °C gradually reduced with decreasing temperature down to −20 °C (0.81 mmol/g) even with a much larger pore size (16.1 nm > 2.4 nm) and similar extent of pore filling compared to MIL-101(Cr)_TEPA(30) (see Table 1). Interestingly, at −20 °C, the MIL-101(Cr)_TEPA(30) showed higher 400 ppm CO2 uptake (1.11 mmol/g) with lower amine efficiency (0.14) than Al2O3_TEPA(20) (0.81 mmol/g with 0.15 amine efficiency), suggesting that a larger amount of CO2 physisorption probably occurred in the CO2 capture by the MIL-101(Cr)_TEPA(30) due to the smaller pore size of MIL-101(Cr) than γ-Al2O3 (2.4 nm < 16.1 nm).
Under humid conditions, the overall DAC performances of the two powder sorbents were enhanced. Similar observations for amine sorbents in the presence of humidity have been attributed to more carbamate/carbamic acid species forming due to the freeing of amines from support interactions17,67,68 or the formation of bicarbonate under humid conditions.69 The form(s) of captured CO2 under dry and humid conditions are further discussed below with insight from in situ FT-IR experiments. Moreover, MIL-101(Cr)_TEPA(30) was shown to be more significantly affected by the presence of humidity than Al2O3_TEPA(20). These different CO2 capture behaviors of the MIL-101(Cr)_TEPA(30) and Al2O3_TEPA(20) under dry and humid conditions strongly suggest that the local environment of TEPA within each support is different. To further probe the impact of the support materials (or pore structures) on the CO2 adsorption mechanism(s) of the impregnated amine (TEPA), CO2/H2O temperature-programed desorption (TPD) and in situ FT-IR experiments were conducted.
Representative CO2/H2O TPD experimental results are shown in Figure S4. The desorbed CO2 and H2O during the TPD experiments were quantified as a function of time (Figure S5). The total amounts of desorbed H2O from the MIL-101(Cr)_TEPA(30) and Al2O3_TEPA(20) materials after humid (70% RH) 400 ppm CO2 breakthroughs are plotted in Figure 2a as a function of adsorption temperature. Since the humid CO2 capture experiments were conducted at constant relative humidity (RH) conditions, 70% RH, under varied adsorption temperatures from −20 to 25 °C, the absolute humidity range was very wide (870 ppm–2.2% by volume). As a result, we observed that the H2O uptake by MIL-101(Cr)_TEPA(30) did not consistently change with adsorption temperature conditions. While the H2O uptake slightly increased from 25 °C (32.7 mmol/g) to −5 °C (35.2 mmol/g), it decreased at −20 °C (28.7 mmol/g), likely due to the extremely low absolute humidity (870 ppm) at this temperature. Overall, these uptake values are consistent with liquid-like conditions in the pores of the MIL-101(Cr). In the case of Al2O3_TEPA(20), the H2O sorption continuously decreased with decreasing adsorption temperature, from 25 °C (13.5 mmol/g) to −20 °C (8.3 mmol/g).
Figure 2.
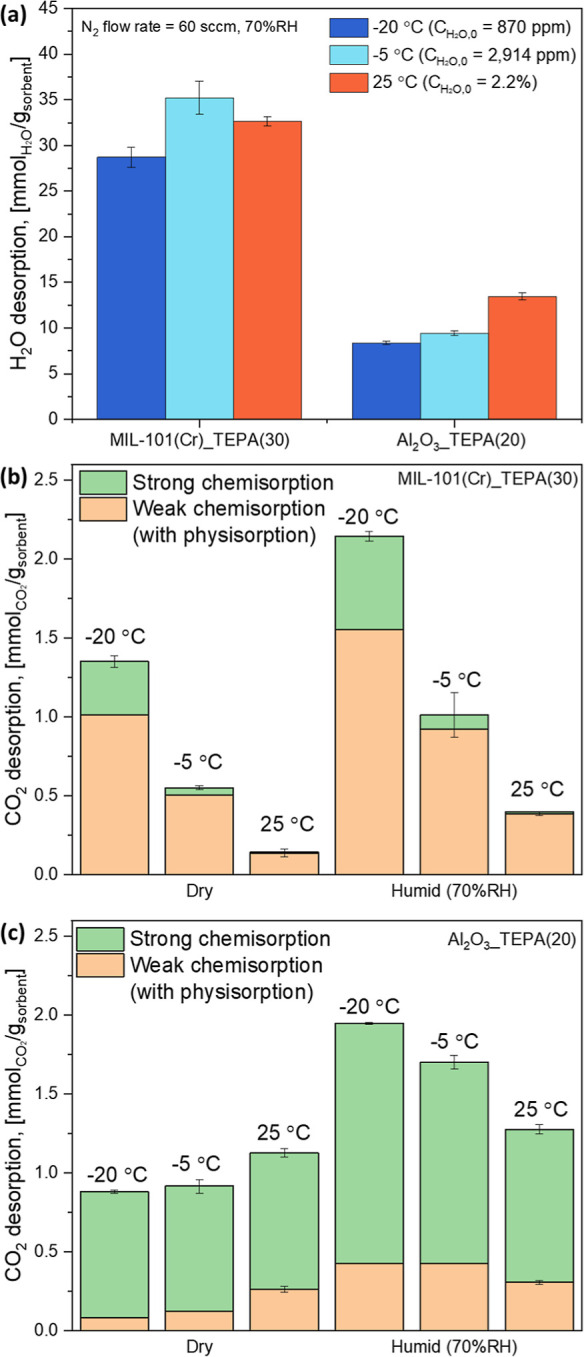
(a) Amount of desorbed H2O from TEPA-impregnated MIL-101(Cr) and γ-Al2O3 powder adsorbents during the CO2/H2O TPD. Amount of desorbed dry/humid CO2 from (b) 30 wt % TEPA-impregnated MIL-101(Cr) and (c) 20 wt % TEPA-impregnated γ-Al2O3 powder adsorbents during the CO2/H2O TPD. Strong chemisorption: CO2 desorption above 25 °C; weak chemisorption: CO2 desorption under 25 °C.
Interestingly, the MIL-101(Cr)_TEPA(30) showed significantly higher H2O uptake than the Al2O3_TEPA(20) under all temperature conditions. This is likely due to the much smaller pore size of MIL-101(Cr) (2.4 nm) than γ-Al2O3 (16.1 nm) (Table 1), yielding higher capillary condensation at lower partial pressures of water, as shown in Figure S6. It was reported that the process of capillary condensation of MIL-101(Cr) begins at 35% RH at 25 °C,70 while γ-Al2O3 begins at 65% RH at 22–25 °C,71,72 which is consistent with measured water vapor adsorption isotherm in this study (Figure S6). When compared to the humid 400 ppm CO2 adsorption results shown in Figure 1a,b, one can see that the H2O uptake is more significant during the humid DAC process at all temperature conditions. Thus, assuming similar ΔHads(H20) for these materials, more energy will be required for the regeneration of MIL-101(Cr)_TEPA(30) than for Al2O3_TEPA(20) for both ambient and cold humid DAC processes due to the significant H2O uptake. However, it should be noted that the H2O uptake kinetics are extremely slow at cold temperature conditions due to the low absolute humidity and the effect of temperature on the overall kinetics (Figure S7). Thus, if water sorption is kinetically limited, regeneration energies can be significantly reduced.
In this study, weak chemisorption (including physisorption) is defined as CO2 desorption below 25 °C and strong chemisorption is defined as CO2 desorption above 25 °C, as discussed in Supporting Information (Figure S8). The amounts of weak and strong chemisorption were quantified based on Figure S5 and the results are plotted in Figure 2b,c for the MIL-101(Cr)_TEPA(30) and Al2O3_TEPA(20), respectively. The figures clearly show that the impregnated TEPA within MIL-101(Cr) and γ-Al2O3 exhibit different CO2-amine interactions. Inside smaller pores (2.4 nm) with higher remaining pore volume (1.84 cc/g), as in MIL-101(Cr), TEPA showed weak chemisorption dominant behaviors.44 Inside a larger pore (16.1 nm) support with lower pore volume (1.03 cc/g), as in γ-Al2O3, strong chemisorption was the dominant CO2 capture mechanism of TEPA. Both weak and strong chemisorption was enhanced under humid conditions.
Effect of Support Materials on the CO2 Adsorption Mechanism(s) of Impregnated Amines (TEPA) under Dry Conditions
To identify the adsorbed CO2 species associated with weak and strong chemisorption, in situ FT-IR experiments were conducted at 25 and −20 °C under dry 400 ppm CO2 environments (Figure 3). It should be noted that these spectra were obtained after subtracting the activated sorbent spectra from the IR spectra after a given time of exposure to dry 400 ppm CO2 to observe contributions more clearly from CO2 adsorption. The relevant peak assignments are summarized in Table S1 with supporting references.
Figure 3.

In situ FT-IR spectra of (a/b) 20 wt % TEPA-impregnated γ-Al2O3 and (c/d) 30 wt % TEPA-impregnated MIL-101(Cr) powder adsorbents as a function of adsorption time at (a/c) 25 °C and (b/d) −20 °C with the activated sample as the background. Orange: carbamic acid, red: carbamate ion, purple: ammonium ion. Adsorption conditions: gas, 400 ppm CO2/N2; flow rate, 40 sccm; relative humidity, dry; activation, 60 °C under 40 sccm N2 for 2–3 h.
Results shown in Figure 3a,c indicate that the dominant adsorbed CO2 species on the Al2O3_TEPA(20) and MIL-101(Cr)_TEPA(30) at 25 °C are different. The TEPA-impregnated MIL-101(Cr) appears to dominantly form carbamic acid with a small amount of ammonium carbamate ion pairs, while the TEPA-impregnated γ-Al2O3 adsorbents only or primarily form ammonium carbamate ion pairs at the same conditions. It is known that carbamate ion species (strong chemisorption) have higher binding energies than carbamic acid species (weak chemisorption),73,74 and the form of adsorbed CO2 here correlates well with the ease of desorption, as determined by the TPD experiments.
The measured FT-IR spectra of Al2O3_TEPA(20) and MIL-101(Cr)_TEPA(30) under dry 400 ppm CO2 flow at −20 °C (sub-ambient) are shown in Figure 3b,d, respectively. It appears that the same adsorbed CO2 species formed at 25 °C were also formed at −20 °C on each sorbent, while there are differences in the CO2 adsorption kinetics and the total amount of adsorbed CO2 species at equilibrium under the different temperature conditions. Obvious increases in the intensity of adsorbed CO2 in both adsorbents were observed as a function of time at 25 °C, whereas the intensities relatively slowly increased at −20 °C, indicating reduced CO2 adsorption kinetics at cold temperature conditions, as also observed in breakthrough experiments described above. The overall intensity of adsorbed CO2 on Al2O3_TEPA(20) at equilibrium slightly decreased from 25 to −20 °C, suggesting that the formation of ammonium carbamate ion pairs may be suppressed at sub-ambient temperature conditions. Given that carbamate formation requires cooperative interaction of two amine sites, lower amine chain mobility at low temperatures may impede carbamate formation. In the case of carbamic acid formation on the MIL-101(Cr)_TEPA(30) powder sorbents, sharper and higher equilibrium intensities were observed at −20 °C, even with reduced CO2 adsorption kinetics, indicating the enhanced formation of carbamic acid. These in situ FT-IR results are consistent with the CO2 desorption data shown in Figure 2b,c. The effects of temperature on the formation of carbamic acid and ammonium carbamate ion pairs will be further discussed below.
Also noteworthy is that an apparent physisorbed CO2 peak at 2335 cm–1 was observed on the MIL-101(Cr)_TEPA(30) at −20 °C, while the Al2O3_TEPA(20) did not show any distinct CO2 physisorption peak, as depicted in Figure S9. Thus, it can be inferred that the higher pseudo-equilibrium 400 ppm CO2 uptake of MIL-101(Cr)_TEPA(30) vs Al2O3_TEPA(20) with lower amine efficiency at −20 °C (Figure 1) is due to a relatively larger amount of CO2 physisorption over MIL-101(Cr)_TEPA(30). We infer that TEPA impregnation inside the small pores of MIL-101(Cr) (2.4 nm) does two things: (i) it leaves relatively isolated primary amines leading to carbamic acid formation, and (ii) it leaves behind suitable micropore space for physisorption of CO2.
The dry CO2 TPD profiles of MIL-101(Cr)_TEPA(30) and Al2O3_TEPA(20) shown in Figure 4a clearly show the difference in CO2 desorption trends (weak and strong chemisorption). A desorption peak of MIL-101(Cr)_TEPA(30) was shown at a relatively low temperature between 30 and 35 °C for the 25 °C adsorption temperature, and it was shifted to 2.2 °C when decreasing the adsorption temperature down to −20 °C, indicating that the weak chemisorption became more significant at colder adsorption temperatures.44 The in situ FTIR results shown in Figure 3 strongly indicate that the weak chemisorption is related to the formation of carbamic acid. In contrast, a desorption peak of Al2O3_TEPA(20) was confirmed at a much higher temperature (53 °C), consistent with the strong chemisorption mechanism associated with formation of ammonium carbamate ion pairs (Figure 3).
Figure 4.
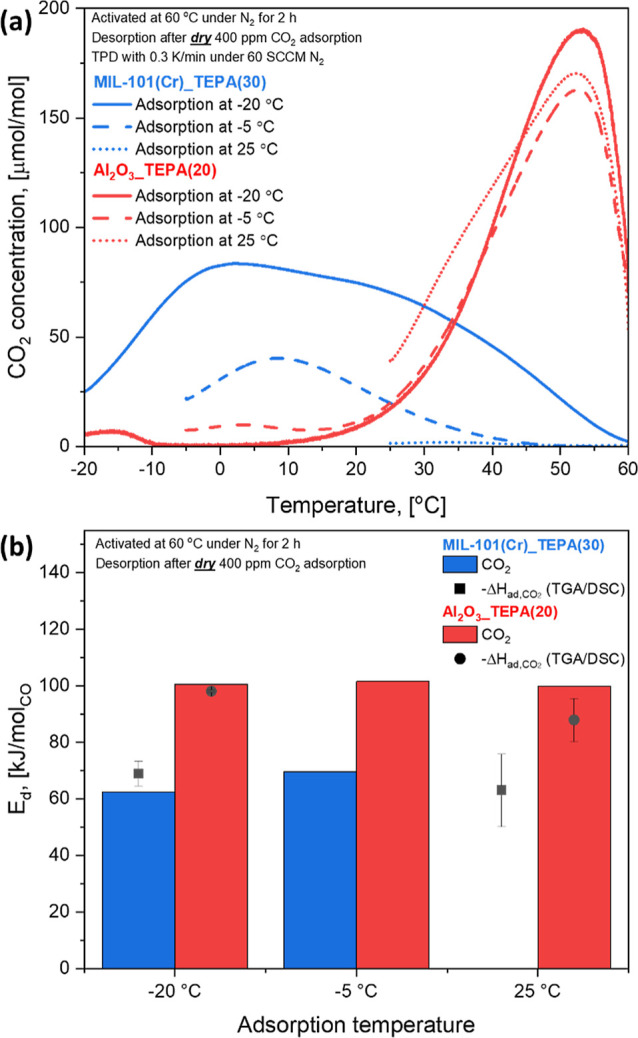
(a) CO2 TPD profiles and (b) energy of CO2 desorption of 30 wt % TEPA-impregnated MIL-101(Cr) and 20 wt % TEPA-impregnated γ-Al2O3 powder adsorbents for dry 400 ppm adsorption at −20, −5, and 25 °C. ΔHad,CO2: measured heat of dry 400 ppm CO2 adsorption by TGA/DSC.
Interestingly, the desorption peak position and the amount of desorbed CO2 (peak area) of the Al2O3_TEPA(20) adsorbent were not significantly changed when decreasing the adsorption temperature, indicating that the formation of ammonium carbamate (a strongly exothermic reaction) is not significantly affected by the adsorption temperature, although lower temperatures enthalpically favor the adsorption process. On the other hand, the formation of carbamic acid (a less exothermic reaction) was enhanced at cold temperatures (Figure 4a). These different trends are probably due to the effects of temperature on amine mobility, in which amine mobility dramatically decreases with decreasing temperature conditions.75 The formation of ammonium carbamate ion pairs occurs via a reaction between two amine groups (primary or secondary) and CO2 with a 2:1 molar ratio, while the reaction ratio is 1:1 for the formation of carbamic acid. We hypothesize that the formation of the ammonium carbamate ion pairs may be more strongly impacted by the decrease in amine mobility at cold temperatures than the formation of carbamic acid due to the higher amine to CO2 reaction ratio (2 > 1). Thus, the expected large thermodynamic enhancement of the more exothermic carbamate ion pair formation is mitigated to some degree by arrested amine chain mobility. In contrast, chain motion is less important for carbamic acid formation, requiring only a single adsorption site, and thus, thermodynamic effects are more prominent. Thus, the MIL-101(Cr)_TEPA(30) and Al2O3_TEPA(20) show different DAC behavior at ambient and sub-ambient temperature conditions (Figure 1).
The CO2 TPD results shown in Figure 4a indicate that the interaction strength between CO2 and the impregnated TEPA is different depending on the support materials (MIL-101(Cr) and γ-Al2O3) and adsorption temperature conditions. To quantify the binding energy of dry CO2 onto the adsorbent materials, CO2 TPD experiments were performed with varied heating rates and the energy of CO2 desorption, Ed, was calculated based on the method described in the Supporting Information. The procedure is described in more detail in Figures S10 and S11. The heat of CO2 adsorption is an indicator of the strength of the interaction between the CO2 molecules and amine sites on sorbents, and it is released as heat during the exothermic CO2 adsorption process. The total energy of CO2 desorption, as defined in this work, is the energy required to overcome the thermodynamic barrier for desorption (ΔHads) + the energy needed to overcome any kinetic barrier to desorption (EAdes). Sensible heats are typically small under these conditions and are therefore often neglected.
The
results are plotted in Figure 4b for MIL-101(Cr)_TEPA(30) and Al2O3_TEPA(20), as a function of adsorption temperature. The Ed of MIL-101(Cr)_TEPA(30) for 25 °C adsorption
was not determined because most of captured CO2 was desorbed
during the N2 purging period at 25 °C (Figure S5e). The heat of dry 400 ppm CO2 adsorption, as measured in situ by TGA/DSC, is
also plotted. Since the energy required for the desorption process
(Ed) is comparable to the heat released
during the adsorption process (ΔHads), the kinetic barrier for desorption (e.g., activation energy, EAdes) is very small or negligible, and thus, the desorption
energy determined by the TPD method approximates the heat of adsorption.76 The Ed of MIL-101(Cr)_TEPA(30)
was slightly reduced (69.6 kJ/molCO2 to 62.4
kJ/molCO2) with decreasing the adsorption temperature
from −5 to −20 °C because of enhanced weak chemisorption
(or formation of carbamic acid) at cold temperature conditions (Figure 4a). In contrast,
the CO2 desorption peak of the Al2O3_TEPA(20) was not shifted under different adsorption temperature
conditions (Figure 4a), resulting in a similar Ed (99.9–101.5
kJ/molCO2), consistent with strong chemisorption
(or formation of ammonium carbamate pairs) at all adsorption temperature
conditions, assuming ideal mixing (i.e., 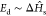 , the sorption enthalpy). These results
clearly show that the impregnated TEPA has different CO2 capture mechanisms (carbamic acid vs carbamate) within different
support materials, and the ammonium carbamate pair (100.5 kJ/molCO2) has stronger binding energy than carbamic acid
(62.4 kJ/molCO2).74,77,78 The regeneration of MIL-101(Cr)_TEPA(30) and Al2O3_TEPA(20) saturated with dry 400 ppm CO2 at −20 °C was monitored with in situ FTIR, and the results shown in Figure S12 support the assertion that carbamate species are stronger chemisorption
products than carbamic acid.
, the sorption enthalpy). These results
clearly show that the impregnated TEPA has different CO2 capture mechanisms (carbamic acid vs carbamate) within different
support materials, and the ammonium carbamate pair (100.5 kJ/molCO2) has stronger binding energy than carbamic acid
(62.4 kJ/molCO2).74,77,78 The regeneration of MIL-101(Cr)_TEPA(30) and Al2O3_TEPA(20) saturated with dry 400 ppm CO2 at −20 °C was monitored with in situ FTIR, and the results shown in Figure S12 support the assertion that carbamate species are stronger chemisorption
products than carbamic acid.
CO2 Adsorption Via Carbamic Acid Versus Ammonium Carbamate Ion Pair Formation
The abovementioned results are consistent with the previous work of Bacsik and Hedin (2011), who previously elucidated the formation of carbamate species vs carbamic acid species over amine-based sorbents.79,80 Their in situ FT-IR study revealed that the relatively low amine-loaded silica (AMS-6) adsorbents captured CO2 by forming both carbamic acid and ammonium carbamate pairs, while the high amine-loaded silica primarily formed ammonium carbamate ion pairs during CO2 adsorption (Figure S13a). Another work by Didas et al. (2014) showed similar results (Figure S13b).81 Yoo et al. (2015)73 controlled the formation of carbamic acid and ammonium carbamate pairs when amines react with CO2 by functionalizing a mesoporous silica SBA-15 with single (MONO) and triaminosilanes (TRI), as shown in Figure S14. Since the length of the MONO molecules was estimated to be sufficient for the interaction between primary amines (−NH2) and silanols (−OH) on the surface of SBA-15 but not for interaction with other amines,82,83 the MONO grafted adsorbents captured CO2 via interaction between primary amines and surface silanols, forming carbamic acid species stabilized by surface silanols with low isosteric heats of adsorption (∼−46 kJ/mol).73 On the other hand, the TRI grafted adsorbents captured CO2 via intramolecular amine–amine interactions within a single chain, forming ammonium carbamate ion pairs that have higher heats of adsorption of ∼−73 to −78 kJ/mol than the MONO adsorbents. It can be inferred from this prior study that the formation of carbamic acid is probably dominant when the interaction between impregnated amines and support materials is strong. These studies indicate that enough surface silanols are required (strong amine-support material interactions) to form carbamic acid stabilized by surface silanol groups, while ammonium carbamate pairs are the dominant species of adsorbed CO2 when amine–amine interactions are strong via amine clustering.
In the same manner, one possible cause for the different CO2 adsorption mechanisms of impregnated TEPA within the two support materials studied here, MIL-101(Cr) and γ-Al2O3, could be the result of different extents of interaction between the impregnated amines and the support materials. As shown in Table 1, MIL-101(Cr) has a much higher BET surface area to pore volume ratio of 1.78 nm–1 than γ-Al2O3 (0.16 nm–1), indicating that an equivalent amount of impregnated amines (filling a similar pore volume percentage) may more strongly interact with MIL-101(Cr) than γ-Al2O3. This is consistent with the observation that the dominant CO2 adsorbed species on MIL-101(Cr)_TEPA(30) adsorbents were carbamic acids, while the Al2O3_TEPA(20) primarily formed ammonium carbamate pairs during dry 400 ppm CO2 adsorption, as shown in Figure 3.
The empirical formula of MIL-101(Cr) is Cr3(O)OH(bdc)3(H2O), where bdc is benzene-1,4-dicarboxylate. The octahedral trinuclear Cr(III)3O building units of MIL-101(Cr) are saturated with one hydroxyl group and two water molecules. MIL-101(Cr) is generally activated at 150 °C under a high vacuum to remove the water molecules, creating potential Lewis acid sites.84−86 Since the MIL-101(Cr)_TEPA(30) was activated at 60 °C under 1 atm N2 flow, the open metal sites of MIL-101(Cr) saturated with water during the amine impregnation process with MeOH solution might be mostly occupied with water molecules even after the activation process due to the mild conditions used. The theoretical maximum amount of hydroxyl groups and water molecules on the surface of MIL-101(Cr) is 4.2 mmol OH and H2O/g MOFs. Thus, as shown in Figure S15a, the formed carbamic acid on the MIL-101(Cr)_TEPA(30) adsorbents might be stabilized by water molecules or hydroxyl groups occupying the open metal sites of MIL-101(Cr).
The Al2O3_TEPA(20) may also contain hydroxyl groups on the surface of alumina because it was confirmed that traces of water are generated from γ-Al2O3 during heating, even after activation at 1000 °C.87 Hydroxylation of the alumina surface occurs by the dissociative adsorption of water to Al3+, where Lewis acid sites and residing protons on lattice oxygens form surface hydroxyl groups.88 The reported heat of water adsorption on γ-Al2O3 at 25 °C under low water coverage (0.4 H2O/nm2) is −158.8 kJ/mol,71 indicating that the activated Al2O3_TEPA(20) at 60 °C under N2 flow certainly contains surface hydroxyl groups. Lagauche et al. also reported that the coverage of surface OH groups on the activated γ-Al2O3 at 100 °C for 4 h is about 11.7 OH/nm2 or 4.4 mmol OH/g alumina,89 which is comparable to the theoretical maximum of MIL-101(Cr). Although γ-Al2O3 has a similar amount of surface hydroxyl groups to MIL-101(Cr), the degree of interaction between impregnated amine groups and hydroxyl groups on the surface of alumina will be much lower than in the case of MIL-101(Cr) due to the much lower BET surface area to pore volume ratio of γ-Al2O3 (0.16 nm–1) than that of MIL-101(Cr) (1.78 nm–1), as discussed above. Thus, it can be inferred that ammonium carbamate pairs are dominantly formed via amine–amine interactions during the CO2 adsorption by Al2O3_TEPA(20), probably due to the lower degree of interaction between impregnated TEPA and γ-Al2O3 (Figure S15c).
The amine-impregnated MIL-101(Cr) powder sorbents also adsorbed 400 ppm CO2 via the formation of ammonium carbamate pairs when amine loading was high enough for significant amine clustering to occur (50 wt % TEPA). As shown in Figure S16, the CO2 TPD44 and in situ FT-IR studies revealed that both strong (ammonium carbamate) and weak chemisorption (carbamic acid) are involved in the CO2 adsorption by high amine loaded MIL-101(Cr) adsorbents (Figure S15b), while the low amine loaded adsorbents only showed weak chemisorption (carbamic acid) dominant behavior at −20 °C (Figure S15a). It appears that the CO2 capture mechanisms of amine-impregnated porous solid sorbents, including the formation of carbamic acid (weak chemisorption) or ammonium carbamate (strong chemisorption), can be controlled by adjusting the extent of pore filling and interactions between amine and the surface of support materials. Thus, the selection of support materials for amine impregnation could be an important factor in determining the DAC performance of the amine-impregnated solid adsorbent materials.
Effect of Moisture on CO2 Adsorption Behavior of Amine (TEPA) within Different Support Materials (MIL-101(Cr) and γ-Al2O3)
The dry/humid 400 ppm CO2 breakthrough experiment results described in Figure 1 clearly show that moisture has a positive effect on the DAC performance of the TEPA-impregnated MIL-101(Cr) and γ-Al2O3 powder sorbents at both ambient (25 °C) and sub-ambient (−5 and −20 °C) temperatures. Similar results were also reported in our previous work with the amine-impregnated MIL-101(Cr).44 To gain in-depth understanding of the effects of humidity, the CO2/H2O TPD profiles after humid (70% RH) CO2 adsorption are compared with the results of dry CO2 adsorption at different adsorption temperatures (25, −5, and −20 °C) in Figure S17. At the same time, the measured in situ FT-IR spectra (1750–1250 cm–1) during the humid 400 ppm CO2 adsorption (70% RH) at 25 and −20 °C are shown in Figure S18 to support the discussion of the effects of humidity. Figure S19 shows overall in situ FT-IR spectra (3750–1250 cm–1) under the same conditions. After introducing humid 400 ppm CO2 into the DRIFTS cell, a dramatic peak increase was observed at 3750 cm–1, which is attributed to H2O adsorption.74,90,91 Much faster H2O uptake was observed at 25 °C, while the uptake kinetics at −20 °C were extremely slow, as discussed in Figure S7. In situ FT-IR desorption experiments were also conducted after humid 400 ppm CO2 adsorption at 25 °C to investigate the thermal decomposition behavior of carbamic acid and ammonium carbamate ion pairs formed under humid environments (Figure S20).
The CO2/H2O TPD and in situ FT-IR results show that the formation of carbamic acid and ammonium carbamate ion pairs on MIL-101(Cr)_TEPA(30) and Al2O3_TEPA(20), respectively, was enhanced upon humid 400 ppm CO2 adsorption. The enhancement in CO2 adsorption under humid conditions became particularly more significant at −20 °C, indicating that the effect of humidity on the DAC performance becomes more significant with decreasing adsorption temperature. The enhanced carbamic acid formation under humid conditions may be because more water-stabilized carbamic acid was formed (via hydrogen bonding) with adsorbed water molecules.74 More ammonium carbamate ion pairs also can be formed with water vapor adsorption due to water’s ability to free amine sites for amine–amine or amine-support interactions.17,80,92,93 More detailed discussion on the effect of moisture is described in Supporting Information.
Interestingly, a very small peak for the symmetric νCOO– vibration of bicarbonate anions was observed at 1358 cm–1 during humid 400 ppm CO2 adsorption with the Al2O3_TEPA(20) at 25 and −20 °C, while clear evidence for the formation of bicarbonate under humid conditions was not observed from the MIL-101(Cr)_TEPA(30) (Figure S18). This is probably because ammonium bicarbonate is formed from a paired ammonium carbamate precursor.74,81,94 As shown in Figures 3 and S18, since the formation of ammonium carbamate ion pairs is the dominant CO2 capture mechanism of Al2O3_TEPA(20), the formation of bicarbonate within Al2O3_TEPA(20) may be more favorable under humid conditions when compared to the MIL-101(Cr)_TEPA(30). However, it has been reported that observation of bicarbonate formation on solid-supported amines under humid CO2 capture conditions is challenging with in situ FT-IR conditions due either to the small amounts of bicarbonate formed or due to extinction coefficients that make resolution difficult.95,96 Thus, the formation of bicarbonate under humid conditions should be further studied with 13C solid-state NMR in the future.96−100
The CO2/H2O TPD profiles of MIL-101(Cr)_TEPA(30) and Al2O3_TEPA(20) are shown in Figure 5a,b, respectively, for different humid CO2 adsorption temperature conditions (−20, −5, and 25 °C). The amount of desorbed CO2 from the two powder sorbents dramatically increased with decreasing adsorption temperatures from 25 to −20 °C, indicating that the effect of moisture on 400 ppm CO2 adsorption became more significant. The binding energy of CO2 on the powder sorbents may be affected by H2O adsorption. Thus, quantifying the binding energy of CO2 and H2O under humid conditions is necessary to estimate the energy required for sorbent regeneration. Generally, the isosteric heat of adsorption is calculated from multiple isotherms measured at different temperature conditions. However, measuring the adsorption isotherm of CO2 under humid conditions (co-adsorption of CO2 and H2O) is challenging. Thus, the energies of CO2 and H2O desorption, Ed, for humid CO2 adsorption, were determined based on the TPD method described in Supporting Information. Figures S21 and S22 show the CO2/H2O TPD profiles of the MIL-101(Cr)_TEPA(30) and Al2O3_TEPA(20), respectively, with varied heating rates for humid CO2 adsorption.
Figure 5.

(a/b) CO2/H2O TPD profiles and (c/d) energy of CO2 and H2O desorption of (a/c) 30 wt % TEPA-impregnated MIL-101(Cr) and (b/d) 20 wt % TEPA-impregnated γ-Al2O3 powder adsorbents for humid (70% RH) 400 ppm adsorption at −20, −5, and 25 °C.
The determined Eds of CO2 and H2O for humid CO2 adsorption are plotted in Figure 5c for MIL-101(Cr)_TEPA(30) and (d) for Al2O3_TEPA(20) along with the results of dry CO2 desorption (Figure 4c,d) as a function of adsorption temperature conditions. It should be noted that Tms for some conditions could not be identified in the TPD profiles due to significant CO2 or H2O desorption during the N2 purging step, and thus, we were not able to determine Ed for those conditions. The determined desorption energy of water for Al2O3_TEPA(20) was 46.4 kJ/molH2O, which is comparable to the heat of vaporization of water in the range of 0–40 °C and 43.4–45.1 kJ/molH2O, supporting the condensation-like sorption of water by Al2O3_TEPA(20). A previous study directly measured the heat of adsorption of γ-Al2O3 via microcalorimetry of water adsorption, and it was −44 kJ/molH2O at a relative water saturation pressure of 0.7 at 25 °C.71 In the case of MIL-101(Cr)_TEPA(30), the measured desorption energy of water (47.6–59.4 kJ/molH2O) was slightly higher than that of Al2O3_TEPA(20). The reported isosteric heat of water adsorption on MIL-101(Cr) is from −53 to −44 kJ/molH2O.70 The higher heat of water desorption on the MIL-101(Cr)_TEPA(30) than Al2O3_TEPA(20) might be linked to the smaller pore size of the MOF, as shown in Table 1. Thus, the MIL-101(Cr)_TEPA(30) showed much higher H2O uptake than Al2O3_TEPA(20) under all adsorption temperature conditions (Figure 2a).
The calculated energy required for desorption of dry and humid CO2, shown in Figure 5c,d, indicates that the binding energy of CO2 increases under humid conditions, and the effect of moisture on CO2 adsorption is more significant with decreasing adsorption temperature. The difference between wet and dry CO2 desorption energies increased with decreasing adsorption temperatures from 25 to −20 °C. At −20 °C, the desorption energy of CO2 for Al2O3_TEPA(20) increased from 100.5 kJ/molCO2 (dry) to 121.1 kJ/molCO2 under humid conditions. The MIL-101(Cr)_TEPA(30) also showed similar behavior. It can be inferred from the CO2 TPD profiles in Figure S17e,f that the increased CO2 desorption energy for humid CO2 adsorption could be mostly attributed to enhanced strong chemisorption (desorption above 25 °C), which is related to the formation of carbamate species. The ammonium carbamate ion pairs formed under humid conditions may have higher binding energies than the carbamate species formed under dry conditions.101,102 Cyclic diamine 2-(aminomethyl)piperidine (2-ampd) functionalized Mg2(dobpdc) MOFs also show similar trends. While the DFT-calculated heat of CO2 adsorption was −70 kJ/mol for the formation of ammonium carbamate chains under dry conditions, the calculated humid CO2 adsorption energy was −88 kJ/mol in the case of the co-adsorption of 1 CO2 and 1 H2O per diamine, indicating that H2O enhances the CO2 binding energy for the ammonium carbamate chain formation.103
The results in Figure 5c,d initially suggest that much higher desorption energies could be required for operation of DAC process under humid sub-ambient conditions (e.g., −20 °C) than ambient conditions (e.g., 25 °C) due to the significant increase in the binding energy of chemisorbed CO2 under humid conditions at −20 °C. However, it should be noted that all the data reported in this study are pseudo-equilibrium values. As discussed in Figure S7, the two powder sorbents have extremely slow H2O uptake kinetics when compared to CO2 uptake kinetics, especially at sub-ambient temperature conditions (e.g., −20 °C), due to an extremely low absolute humidity level and the effect of cold temperatures on kinetics of chemical processes. Thus, in realistic sub-ambient DAC operation, the H2O adsorption working capacity could be very low, depending on the adsorption time and the effect of adsorbed H2O on the 400 ppm CO2 adsorption may not be significant due to the kinetically limited H2O uptake at sub-ambient temperature conditions (e.g., −20 °C).
To support the abovementioned argument, H2O uptake profiles of the MIL-101(Cr)_TEPA(30) and Al2O3_TEPA(20) were obtained by integrating the H2O breakthrough curves in Figure S7 and are plotted in Figure S23 as a function of time. The dashed lines in the figures depict H2O adsorption working capacity at the time required to reach pseudo-equilibrium states (95% of breakthrough) of humid 400 ppm CO2 adsorption during the breakthrough experiments in Figure S3. Figure S23 indicates that if the DAC process is operated until the pseudo-equilibrium states at each adsorption temperature, the H2O adsorption working capacity could dramatically decrease with decreasing temperatures due to significant kinetic limitation of H2O uptake at colder temperatures. The H2O adsorption working capacities of MIL-101(Cr)_TEPA(30) and Al2O3_TEPA(20) were decreased from 27.0 and 13.5 mmol/g at 25 °C to 7.7 and 4.1 mmol/g at −20 °C, respectively. The H2O uptakes of both sorbent materials were reduced by 3.3–3.5 times when decreasing the adsorption temperature from 25 to −20 °C, indicating that significant energy saving for water desorption may be achieved under sub-ambient DAC operation.
The CO2/H2O uptakes and total energies for CO2 and H2O desorption of the two sorbent materials are compared in Table 2 to estimate the energy requirements for sorbent regeneration. These energies are broken down into the energy for desorption (ΔHads + EAdes) and the sensible heat for heating the sample from the adsorption to the desorption temperature. The energies of CO2 and H2O desorption were calculated based on the measured CO2 and H2O uptakes (Figure 2) and the Ed for CO2/H2O desorption determined by the TPD method (Figures 4 and 5). Under dry conditions, the MIL-101(Cr)_TEPA(30) sorbents are better sorbent materials for sub-ambient temperature DAC operation (e.g., −20 °C) due to their higher CO2 uptakes and lower regeneration energies than Al2O3_TEPA(20). For dry ambient temperature operations (e.g., 25 °C), the Al2O3_TEPA(20) appears to be an advantaged sorbent because of its higher CO2 uptake under such conditions. When the sorbent materials are fully saturated with water, their DAC performance is significantly enhanced. However, the sorbent regeneration energy requirement dramatically increases at the same time due to significant amounts of H2O adsorption. In particular, DAC operation with MIL-101(Cr)_TEPA(30) is very energy-intensive if water is completely removed in every cycle because of its high H2O uptake. However, as discussed above, the sorbent materials are never fully saturated with H2O, especially at sub-ambient temperature conditions (e.g., −20 °C) due to kinetically limited H2O adsorption. When the estimated H2O working capacity (Figure S23) is considered under kinetic conditions in the calculation, both sorbent materials show much lower regeneration energy requirements for DAC operation at −20 than at 25 °C, indicating the advantage of sub-ambient DAC operation.
Table 2. DAC Performance and Energies for CO2/H2O Desorption of MIL-101(Cr)_TEPA(30) and Al2O3_TEPA(20) Sorbent Materials (Desorption at 60 °C Under N2).
| MIL-101(Cr)_TEPA(30) |
Al2O3_TEPA(20) |
|||
|---|---|---|---|---|
| adsorption temperature, [°C] | –20 | 25 | –20 | 25 |
| Dry | ||||
| qCO2, [mol/kg] | 1.11 | 0.11 | 0.81 | 1.10 |
| qH2O, [mol/kg] | 0.00 | 0.00 | 0.00 | 0.00 |
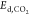 , [kJ/mol] , [kJ/mol] |
62.4 | 62.4 | 100.5 | 99.8 |
 , [kJ/mol] , [kJ/mol] |
53.5 | 46.4 | ||
| energy for CO2 desorption, [GJ/ton CO2] | 1.42 | 1.42 | 2.28 | 2.27 |
| energy for H2O desorption, [GJ/ton CO2] | 0.00 | 0.00 | 0.00 | 0.00 |
| sensible heat for sorbent, [GJ/ton CO2] | 1.46 | 6.45 | 1.92 | 0.62 |
| Kinetically Limited H2O Adsorption | ||||
| qCO2, [mol/kg] | 1.87 | 0.38 | 1.98 | 1.33 |
| qH2O, [mol/kg] | 7.70 | 27.00 | 4.10 | 13.46 |
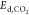 , [kJ/mol] , [kJ/mol] |
91.7 | 62.4 | 121.1 | 104 |
 , [kJ/mol] , [kJ/mol] |
53.5 | 46.4 | ||
| energy for CO2 desorption, [GJ/ton CO2] | 2.08 | 1.42 | 2.75 | 2.36 |
| energy for H2O desorption, [GJ/ton CO2] | 5.01 | 86.4 | 2.18 | 10.7 |
| sensible heat for sorbent, [GJ/ton CO2] | 0.87 | 1.87 | 0.78 | 0.51 |
| Full H2O Adsorption | ||||
| qCO2, [mol/kg] | 1.87 | 0.38 | 1.98 | 1.33 |
| qH2O, [mol/kg] | 28.7 | 32.7 | 8.3 | 13.5 |
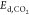 , [kJ/mol] , [kJ/mol] |
91.7 | 62.4 | 121.1 | 104 |
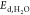 , [kJ/mol] , [kJ/mol] |
53.5 | 46.4 | ||
| energy for CO2 desorption, [GJ/ton CO2] | 2.08 | 1.42 | 2.75 | 2.36 |
| energy for H2O desorption, [GJ/ton CO2] | 18.7 | 104.6 | 4.44 | 10.7 |
| sensible heat for sorbent, [GJ/ton CO2] | 0.87 | 1.87 | 0.78 | 0.51 |
For DAC operations, Table 2 indicates that the Al2O3_TEPA(20) will be better sorbents than MIL-101(Cr)_TEPA(30) due to its lower H2O uptake. However, it should be noted that the MIL-101(Cr) has a weak chemisorption dominant mechanism, and thus, a small temperature swing can be applied. The DAC performance and energies of CO2/H2O desorption for small temperature swing operation (adsorption at −20 °C and regeneration at 25 °C) are summarized in Table S2. Due to the weak chemisorption dominant mechanism, MIL-101(Cr)_TEPA(30) shows a much higher CO2 working capacity, 1.55 mmol/g, than Al2O3_TEPA(20), 0.43 mmol/g, for the small temperature swing operation, resulting in lower sorbent regeneration energy.
Experimental results of this study demonstrate that adjusting interactions between amine and the surface of the support materials can control the CO2 capture mechanisms of amine-impregnated sorbent materials, varying from the formation of carbamic acid (weak chemisorption) to ammonium carbamate (strong chemisorption). DAC performances, including CO2 uptake and energy requirement for sorbent regeneration, of these two different mechanisms were strongly affected by adsorption temperatures and humidity. These condition-dependent results demonstrate the complexity of designing and operating a DAC process, as there will likely be no optimal sorbent under all conditions in most locations (day/night; summer/winter; rainy/dry conditions), and a material that operates with acceptable performance under all or most of the conditions likely to be encountered is required.50
Conclusions
TEPA (tetraethylenepentamine) was physically impregnated into two mesoporous solid support materials, MIL-101(Cr) and γ-Al2O3, to explore the effect of support materials on CO2 adsorption mechanisms of a model-impregnated amine. These supports have different BET surface area to pore volume ratios and surface chemistry, and the DAC behavior of the amine-impregnated powder sorbents was investigated under a wide range of temperatures (−20 to 25 °C) and humidities (0–70% RH) conditions.
Analysis of 400 ppm CO2 and H2O adsorption/desorption data on comparably filled MIL-101(Cr) and γ-Al2O3 provided a means to infer if impregnated amines behave differently within different pore structures when capturing CO2. The CO2/H2O TPD and in situ FT-IR results revealed that the impregnated TEPA within MIL-101(Cr) captures CO2 primarily by forming carbamic acid (via weak chemisorption, 62.4 kJ/molCO2), while the TEPA within γ-Al2O3 forms ammonium carbamate ion pairs (via strong chemisorption, 100.5 kJ/molCO2) under 400 ppm CO2 conditions. Since MIL-101(Cr) has a much higher BET surface area to the pore volume ratio of 1.78 nm–1 vs γ-Al2O3 (0.16 nm–1), impregnated amines may more strongly interact with MIL-101(Cr) than γ-Al2O3. Thus, the carbamic acid within the TEPA-impregnated MIL-101(Cr) adsorbents might be stabilized by surface water molecules or hydroxyl groups. In contrast, ammonium carbamate pairs were primarily formed in the TEPA-impregnated γ-Al2O3 via amine–amine interactions (as opposed to amine-surface interactions, like in the MIL-101(Cr) samples) during the CO2 adsorption, probably due to the weaker interaction between impregnated amines and γ-Al2O3.
Interestingly, we found that the formation of carbamic acid in the MIL-101(Cr)_TEPA(30) is enhanced at cold temperatures, showing better DAC performance at sub-ambient (e.g., −20 and −5 °C) over ambient (e.g., 25 °C) conditions. Conversely, the formation of ammonium carbamate (a strongly exothermic reaction) on the Al2O3_TEPA(20) is not significantly affected by the adsorption temperature. We speculate that the different trends are because amine mobility dramatically decreases with decreasing temperature conditions. Although the formation of both carbamate and carbamic acid is affected by decreased amine mobility at cold temperatures, it appears that the formation of the ammonium carbamate ion pairs is more strongly impacted by the decrease in amine mobility at cold temperatures than the formation of carbamic acid, due to the higher amine to CO2 reaction stoichiometry (2 vs 1). Thus, the thermodynamically favorable reaction at lower temperatures may be suppressed by the decreased amine mobility at cold temperatures (kinetically limited) in the alumina-supported case, where carbamate ion pairs are mainly formed.
Under humid conditions, the formation of ammonium carbamate ion pairs on the Al2O3_TEPA(20) and carbamic acid species on the MIL-101(Cr)_TEPA(30) was enhanced when compared to dry conditions. The enhancement in DAC performance under humid conditions was the most distinct at −20 °C, indicating that the effect of humidity on the DAC performance of the two adsorbents becomes more significant with decreasing adsorption temperatures. The difference between wet and dry CO2 desorption energies also increased with decreasing adsorption temperatures from 25 to −20 °C. Additionally, it appears that the two adsorbent materials have different H2O adsorption behaviors. The MIL-101(Cr)_TEPA(30) showed much higher H2O uptake (28.8–35.2 mmol/g) than the Al2O3_TEPA(20) (8.3–13.5 mmol/g) under all temperature conditions, likely due to the much smaller pore size. However, it is expected that the effect of humidity on the 400 ppm CO2 adsorption may not be significant, especially at sub-ambient temperature conditions (e.g., −20 °C) due to the kinetically limited H2O uptake. The estimated H2O adsorption working capacities of two sorbent materials were reduced by 3.3–3.5× via decreasing the adsorption temperature from 25 to −20 °C. These results suggest that significant energy savings associated with H2O and CO2 desorption energies could be achieved by operating a DAC process at sub-ambient temperatures (e.g., −20 °C).
There are several limitations to our study. First, although the analysis of different amine-impregnated porous support materials clearly showed that the support materials could affect the CO2 adsorption mechanism of impregnated amines, more experiments with a variety of support materials will be required to develop an in-depth understanding of the full range of amine-surface effects. Second, while we observed a very small stretch for bicarbonate anions in the FT-IR spectra of the Al2O3_TEPA(20) upon humid 400 ppm CO2 adsorption, it is not clear and definitive evidence for the formation of (or absence of) significant bicarbonate under humid conditions. As discussed in this article, observation of bicarbonate formation on solid-supported amines under humid CO2 capture conditions is challenging with in situ FT-IR. Finally, this study evaluated powder materials, and any scalable, low pressure drop contactor design will need to incorporate such materials into alternate forms, such as fibers, monoliths, or other structures, which may result in deviations in amine-surface interactions from those found in powder studies.104−107
While only two different porous support materials were investigated in this study, the results clearly demonstrate that the CO2 capture mechanisms of impregnated amines could be controlled to vary from the formation of carbamic acid (weak chemisorption) to ammonium carbamate (strong chemisorption) by adjusting interactions between the amine and the surface of the support materials. The results also show that H2O adsorption behavior is strongly affected by the properties of solid supports. Thus, to achieve optimized DAC performance depending on the range of sub-ambient and ambient temperature operations, proper support materials should be considered for amine impregnation.
Acknowledgments
The authors thank the Georgia Tech Direct Air Capture Center, DirACC, for fruitful discussions. This research was supported by the National Energy Technology Laboratory of the U.S. Department of Energy, under award no. DE-FE-FE0031952, and Zero Carbon Partners, LLC.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c12707.
Experimental section, characterization data (N2 physisorption and pore size distribution), CO2 adsorption behavior (dry and humid 400 ppm CO2 breakthrough and CO2/H2O-TPD data) of TEPA-impregnated MIL-101(Cr) and γ-alumina powder sorbents, and in situ FT-IR spectra of TEPA-impregnated MIL-101(Cr) and γ-alumina powder sorbents during CO2 adsorption (PDF)
The authors declare the following competing financial interest(s): C.W.J. has a financial interest in Global Thermostat LLC, which seeks to commercialize CO2 capture from air. This work is not affiliated with Global Thermostat, LLC. C.W.J. has a conflict-of-interest management plan in place at Georgia Tech.
Supplementary Material
References
- Ozkan M. Direct air capture of CO2: A response to meet the global climate targets. MRS Energy Sustain. 2021, 8, 51–56. 10.1557/s43581-021-00005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes H. E.; Lively R. P.; Realff M. J. Defining Targets for Adsorbent Material Performance to Enable Viable BECCS Processes. JACS Au. 2021, 1, 795–806. 10.1021/jacsau.0c00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrman J.; McJeon H.; Patel P.; Doney S. C.; Shobe W. M.; Clarens A. F. Food-energy-water implications of negative emissions technologies in a +1.5 oC future. Nat. Clim. Change 2020, 10, 920–927. 10.1038/s41558-020-0876-z. [DOI] [Google Scholar]
- Cherevotan A.; Raj J.; Peter S. C. An overview of porous silica immobilized amines for direct air CO2 capture. J. Mater. Chem. A 2021, 9, 27271–27303. 10.1039/d1ta05961k. [DOI] [Google Scholar]
- Zhu X. C.; Xie W. W.; Wu J. Y.; Miao Y. H.; Xiang C. J.; Chen C. P.; Ge B. Y.; Gan Z. Z.; Yang F.; Zhang M.; O’Hare D.; Li J.; Ge T. S.; Wang R. Z. Recent advances in direct air capture by adsorption. Chem. Soc. Rev. 2022, 51, 6574–6651. 10.1039/d1cs00970b. [DOI] [PubMed] [Google Scholar]
- Ozkan M.; Akhavi A. A.; Coley W. C.; Shang R. X.; Ma Y. Progress in carbon dioxide capture materials for deep decarbonization. Chem 2022, 8, 141–173. 10.1016/j.chempr.2021.12.013. [DOI] [Google Scholar]
- Song M.; Rim G.; Kong F.; Priyadarshini P.; Rosu C.; Lively R. P.; Jones C. W. Cold-Temperature Capture of Carbon Dioxide with Water Coproduction from Air Using Commercial Zeolites. Ind. Eng. Chem. Res. 2022, 61, 13624–13634. 10.1021/acs.iecr.2c02041. [DOI] [Google Scholar]
- Sayari A.; Liu Q.; Mishra P. Enhanced Adsorption Efficiency through Materials Design for Direct Air Capture over Supported Polyethylenimine. ChemSusChem 2016, 9, 2796–2803. 10.1002/cssc.201600834. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Wang X. X.; Liu S. M.; He L. L.; Song C. S.; Jiang X.; Blach T. P. Discovering Inherent Characteristics of Polyethylenimine-Functionalized Porous Materials for CO2 Capture. ACS Appl. Mater. Interfaces 2019, 11, 36515–36524. 10.1021/acsami.9b08496. [DOI] [PubMed] [Google Scholar]
- Wang D. X.; Wang X. X.; Ma X. L.; Fillerup E.; Song C. S. Three-dimensional molecular basket sorbents for CO2 capture: Effects of pore structure of supports and loading level of polyethylenimine. Catal. Today 2014, 233, 100–107. 10.1016/j.cattod.2014.01.038. [DOI] [Google Scholar]
- Lou F. J.; Zhang A. F.; Zhang G. H.; Ren L. M.; Guo X. W.; Song C. S. Enhanced kinetics for CO2 sorption in amine-functionalized mesoporous silica nanosphere with inverted cone-shaped pore structure. Appl. Energy 2020, 264, 114637. 10.1016/j.apenergy.2020.114637. [DOI] [Google Scholar]
- Wang X. X.; Song C. S. Temperature-programmed desorption of CO2 from polyethylenimine-loaded SBA-15 as molecular basket sorbents. Catal. Today 2012, 194, 44–52. 10.1016/j.cattod.2012.08.008. [DOI] [Google Scholar]
- Zhang Z. H.; Ma X. L.; Wang D. X.; Song C. S.; Wang Y. G. Development of silica-gel-supported polyethylenimine sorbents for CO2 capture from flue gas. AIChE J. 2012, 58, 2495–2502. 10.1002/aic.12771. [DOI] [Google Scholar]
- Wang X. X.; Ma X. L.; Schwartz V.; Clark J. C.; Overbury S. H.; Zhao S. Q.; Xu X. C.; Song C. S. A solid molecular basket sorbent for CO2 capture from gas streams with low CO2 concentration under ambient conditions. Phys. Chem. Chem. Phys. 2012, 14, 1485–1492. 10.1039/c1cp23366a. [DOI] [PubMed] [Google Scholar]
- Wang D. X.; Wang X. X.; Song C. S. Comparative Study of Molecular Basket Sorbents Consisting of Polyallylamine and Polyethylenimine Functionalized SBA-15 for CO2 Capture from Flue Gas. ChemPhysChem 2017, 18, 3163–3173. 10.1002/cphc.201700828. [DOI] [PubMed] [Google Scholar]
- Wang X. X.; Ma X. L.; Song C. S.; Locke D. R.; Siefert S.; Winans R. E.; Möllmer J.; Lange M.; Möller A.; Gläser R. Molecular basket sorbents polyethylenimine-SBA-15 for CO2 capture from flue gas: Characterization and sorption properties. Microporous Mesoporous Mater. 2013, 169, 103–111. 10.1016/j.micromeso.2012.09.023. [DOI] [Google Scholar]
- Wang X. X.; Schwartz V.; Clark J. C.; Ma X. L.; Overbury S. H.; Xu X. C.; Song C. S. Infrared Study of CO2 Sorption over ″Molecular Basket″ Sorbent Consisting of Polyethylenimine-Modified Mesoporous Molecular Sieve. J. Phys. Chem. C 2009, 113, 7260–7268. 10.1021/jp809946y. [DOI] [Google Scholar]
- Xu X. C.; Song C. S.; Miller B. G.; Scaroni A. W. Influence of moisture on CO2 separation from gas mixture by a nanoporous adsorbent based on polyethylenimine-modified molecular sieve MCM-41. Ind. Eng. Chem. Res. 2005, 44, 8113–8119. 10.1021/ie050382n. [DOI] [Google Scholar]
- Xu X. C.; Song C. S.; Andresen J. M.; Miller B. G.; Scaroni A. W. Preparation and characterization of novel CO2 ″molecular basket″ adsorbents based on polymer-modified mesoporous molecular sieve MCM-41. Microporous Mesoporous Mater. 2003, 62, 29–45. 10.1016/s1387-1811(03)00388-3. [DOI] [Google Scholar]
- Xu X. C.; Song C. S.; Andresen J. M.; Miller B. G.; Scaroni A. W. Novel polyethylenimine-modified mesoporous molecular sieve of MCM-41 type as high-capacity adsorbent for CO2 capture. Energy Fuels 2002, 16, 1463–1469. 10.1021/ef020058u. [DOI] [Google Scholar]
- Fayaz M.; Sayari A. Long-Term Effect of Steam Exposure on CO2 Capture Performance of Amine-Grafted Silica. ACS Appl. Mater. Interfaces 2017, 9, 43747–43754. 10.1021/acsami.7b15463. [DOI] [PubMed] [Google Scholar]
- Lashaki M. J.; Ziaei-Azad H.; Sayari A. Insights into the Hydrothermal Stability of Triamine-Functionalized SBA-15 Silica for CO2 Adsorption. ChemSusChem 2017, 10, 4037–4045. 10.1002/cssc.201701439. [DOI] [PubMed] [Google Scholar]
- Lashaki M. J.; Sayari A. CO2 capture using triamine- grafted SBA-15: The impact of the support pore structure. Chem. Eng. J. 2018, 334, 1260–1269. 10.1016/j.cej.2017.10.103. [DOI] [Google Scholar]
- Liu F. Q.; Wang L.; Huang Z. G.; Li C. Q.; Li W.; Li R. X.; Li W. H. Amine-Tethered Adsorbents Based on Three-Dimensional Macroporous Silica for CO2 Capture from Simulated Flue Gas and Air. ACS Appl. Mater. Interfaces 2014, 6, 4371–4381. 10.1021/am500089g. [DOI] [PubMed] [Google Scholar]
- Belmabkhout Y.; Serna-Guerrero R.; Sayari A. Amine-bearing Mesoporous Silica for CO2 Removal from Dry and Humid Air. Chem. Eng. Sci. 2010, 65, 3695–3698. 10.1016/j.ces.2010.02.044. [DOI] [Google Scholar]
- Zerze H.; Tipirneni A.; McHugh A. J. Reusable poly(allylamine)-based solid materials for carbon dioxide capture under continuous flow of ambient air. Sep. Sci. Technol. 2017, 52, 2513–2522. 10.1080/01496395.2017.1345943. [DOI] [Google Scholar]
- Meng Y.; Jiang J. G.; Aihemaiti A.; Ju T. Y.; Gao Y. C.; Liu J. W.; Han S. Y. Feasibility of CO2 Capture from O2-Containing Flue Gas Using a Poly(ethylenimine)-Functionalized Sorbent: Oxidative Stability in Long-Term Operation. ACS Appl. Mater. Interfaces 2019, 11, 33781–33791. 10.1021/acsami.9b08048. [DOI] [PubMed] [Google Scholar]
- Min K.; Choi W.; Kim C.; Choi M. Oxidation-stable amine-containing adsorbents for carbon dioxide capture. Nat. Commun. 2018, 9, 726. 10.1038/s41467-018-03123-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y.; Ju T. Y.; Han S. Y.; Gao Y. C.; Liu J. W.; Jiang J. G. Exploring the stability on exposure to acid impurities of polyethyleneimine-functionalized silica for post-combustion CO2 capture. Chem. Eng. J. 2021, 421, 127754. 10.1016/j.cej.2020.127754. [DOI] [Google Scholar]
- Kim J. Y.; Woo J. M.; Jo S. H.; Lee S. Y.; Moon J. H.; Kim H.; Yi C. K.; Lee H.; Snape C. E.; Stevens L.; Sun C. G.; Liu H.; Liu J. J.; Park Y. C. Continuous testing of silica-PEI adsorbents in a lab.-scale twin bubbling fluidized-bed system. Int. J. Greenh. Gas Control 2019, 82, 184–191. 10.1016/j.ijggc.2019.01.007. [DOI] [Google Scholar]
- Al-Absi A. A.; Mohamedali M.; Domin A.; Benneker A. M.; Mahinpey N. Development of in situ polymerized amines into mesoporous silica for direct air CO2 capture. Chem. Eng. J. 2022, 447, 137465. 10.1016/j.cej.2022.137465. [DOI] [Google Scholar]
- Rosu C.; Narayanan P.; Leisen J. E.; Jones C. W. Sequential polymer infusion into solid substrates (SPISS): Impact of processing on sorbent CO2 adsorption properties. Sep. Purif. Technol. 2022, 292, 121042. 10.1016/j.seppur.2022.121042. [DOI] [Google Scholar]
- Min Y. J.; Ganesan A.; Realff M. J.; Jones C. W. Direct Air Capture of CO2 Using Poly(ethyleneimine)-Functionalized Expanded Poly(tetrafluoroethylene)/Silica Composite Structured Sorbents. ACS Appl. Mater. Interfaces 2022, 14, 40992–41002. 10.1021/acsami.2c11143. [DOI] [PubMed] [Google Scholar]
- Wijesiri R. P.; Knowles G. P.; Yeasmin H.; Hoadley A. F. A.; Chaffee A. L. CO2 Capture from Air Using Pelletized Polyethylenimine Impregnated MCF Silica. Ind. Eng. Chem. Res. 2019, 58, 3293–3303. 10.1021/acs.iecr.8b04973. [DOI] [Google Scholar]
- Miao Y. H.; He Z. J.; Zhu X. C.; Izikowitz D.; Li J. Operating temperatures affect direct air capture of CO2 in polyamine-loaded mesoporous silica. Chem. Eng. J. 2021, 426, 131875. 10.1016/j.cej.2021.131875. [DOI] [Google Scholar]
- Kumar D. R.; Rosu C.; Sujan A. R.; Sakwa-Novak M. A.; Ping E. W.; Jones C. W. Alkyl-Aryl Amine-Rich Molecules for CO2 Removal via Direct Air Capture. ACS Sustain. Chem. Eng. 2020, 8, 10971–10982. 10.1021/acssuschemeng.0c03706. [DOI] [Google Scholar]
- Kumar R.; Bandyopadhyay M.; Pandey M.; Tsunoji N. Amine-impregnated Nanoarchitectonics of Mesoporous Silica for Capturing Dry and Humid 400 ppm Carbon Dioxide: A Comparative Study. Microporous Mesoporous Mater. 2022, 338, 111956. 10.1016/j.micromeso.2022.111956. [DOI] [Google Scholar]
- Sujan A. R.; Kumar D. R.; Sakwa-Novak M.; Ping E. W.; Hu B.; Park S. J.; Jones C. W. Poly(glycidyl amine)-Loaded SBA-15 Sorbents for CO2 Capture from Dilute and Ultradilute Gas Mixtures. ACS Appl. Polym. Mater. 2019, 1, 3137–3147. 10.1021/acsapm.9b00788. [DOI] [Google Scholar]
- Pang S. H.; Lee L. C.; Sakwa-Novak M. A.; Lively R. P.; Jones C. W. Design of Aminopolymer Structure to Enhance Performance and Stability of CO2 Sorbents: Poly(propylenimine) vs Poly(ethylenimine). J. Am. Chem. Soc. 2017, 139, 3627–3630. 10.1021/jacs.7b00235. [DOI] [PubMed] [Google Scholar]
- Bai F. T.; Liu X.; Sani S.; Liu Y. M.; Guo W.; Sun C. G. Amine Functionalized Mesocellular Silica Foam as Highly Efficient Sorbents for CO2 Capture. Sep. Purif. Technol. 2022, 299, 121539. 10.1016/j.seppur.2022.121539. [DOI] [Google Scholar]
- Chaikittisilp W.; Kim H. J.; Jones C. W. Mesoporous Alumina-Supported Amines as Potential Steam-Stable Adsorbents for Capturing CO2 from Simulated Flue Gas and Ambient Air. Energy Fuels 2011, 25, 5528–5537. 10.1021/ef201224v. [DOI] [Google Scholar]
- Sakwa-Novak M. A.; Yoo C. J.; Tan S.; Rashidi F.; Jones C. W. Poly(ethylenimine)-Functionalized Monolithic Alumina Honeycomb Adsorbents for CO2 Capture from Air. ChemSusChem 2016, 9, 1859–1868. 10.1002/cssc.201600404. [DOI] [PubMed] [Google Scholar]
- Sakwa-Novak M. A.; Jones C. W. Steam Induced Structural Changes of a Poly(ethylenimine) Impregnated gamma-Alumina Sorbent for CO2 Extraction from Ambient Air. ACS Appl. Mater. Interfaces 2014, 6, 9245–9255. 10.1021/am501500q. [DOI] [PubMed] [Google Scholar]
- Rim G.; Kong F. H.; Song M. Y.; Rosu C.; Priyadarshini P.; Lively R. P.; Jones C. W. Sub-Ambient Temperature Direct Air Capture of CO2 using Amine-Impregnated MIL-101(Cr) Enables Ambient Temperature CO2 Recovery. JACS Au. 2022, 2, 380–393. 10.1021/jacsau.1c00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M. Z.; Wu H.; Lv L.; Meng H.; Yun J.; Jin J. S.; Mi J. G. A Highly Efficient and Stable Composite of Polyacrylate and Metal-Organic Framework Prepared by Interface Engineering for Direct Air Capture. ACS Appl. Mater. Interfaces 2021, 13, 21775–21785. 10.1021/acsami.1c03661. [DOI] [PubMed] [Google Scholar]
- Darunte L. A.; Oetomo A. D.; Walton K. S.; Sholl D. S.; Jones C. W. Direct Air Capture of CO2 Using Amine Functionalized MIL-101(Cr). ACS Sustain. Chem. Eng. 2016, 4, 5761–5768. 10.1021/acssuschemeng.6b01692. [DOI] [Google Scholar]
- Fisher J. C.; Tanthana J.; Chuang S. S. C. Oxide-Supported Tetraethylenepentamine for CO2 Capture. Environ. Prog. Sustain. Energy 2009, 28, 589–598. 10.1002/ep.10363. [DOI] [Google Scholar]
- Zhu X. C.; Ge T. S.; Yang F.; Lyu M.; Chen C. P.; O’Hare D.; Wang R. Z. Efficient CO2 capture from ambient air with amine-functionalized Mg-Al mixed metal oxides. J. Mater. Chem. A 2020, 8, 16421–16428. 10.1039/d0ta05079b. [DOI] [Google Scholar]
- Zhao M.; Xiao J. W.; Gao W. L.; Wang Q. Defect-rich Mg-Al MMOs supported TEPA with enhanced charge transfer for highly efficient and stable direct air capture. J. Energy Chem. 2022, 68, 401–410. 10.1016/j.jechem.2021.12.031. [DOI] [Google Scholar]
- Kong F.; Rim G.; Song M. G.; Rosu C.; Priyadarshini P.; Lively R. P.; Realff M. J.; Jones C. W. Research Needs Targeting Direct Air Capture of Carbon Dioxide: Material & Process Performance Characteristics Under Realistic Environmental Conditions. Korean J. Chem. Eng. 2022, 39, 1–19. 10.1007/s11814-021-0976-0. [DOI] [Google Scholar]
- Dinakar B.; Forse A. C.; Jiang H. Z. H.; Zhu Z. T.; Lee J. H.; Kim E. J.; Parker S. T.; Pollak C. J.; Siegelman R. L.; Milner P. J.; Reimer J. A.; Long J. R. Overcoming Metastable CO2 Adsorption in a Bulky Diamine-Appended Metal-Organic Framework. J. Am. Chem. Soc. 2021, 143, 15258–15270. 10.1021/jacs.1c06434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drisdell W. S.; Poloni R.; McDonald T. M.; Pascal T. A.; Wan L. F.; Pemmaraju C.; Vlaisavljevich B.; Odoh S. O.; Neaton J. B.; Long J. R.; Prendergast D.; Kortright J. B. Probing the mechanism of CO2 capture in diamine-appended metal-organic frameworks using measured and simulated X-ray spectroscopy. Phys. Chem. Chem. Phys. 2015, 17, 21448–21457. 10.1039/c5cp02951a. [DOI] [PubMed] [Google Scholar]
- Kim E. J.; Siegelman R. L.; Jiang H. Z. H.; Forse A. C.; Lee J. H.; Martell J. D.; Milner P. J.; Falkowski J. M.; Neaton J. B.; Reimer J. A.; Weston S. C.; Long J. R. Cooperative carbon capture and steam regeneration with tetraamine-appended metal-organic frameworks. Science 2020, 369, 392–396. 10.1126/science.abb3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald T. M.; Lee W. R.; Mason J. A.; Wiers B. M.; Hong C. S.; Long J. R. Capture of Carbon Dioxide from Air and Flue Gas in the Alkylamine-Appended Metal-Organic Framework mmen-Mg-2(dobpdc). J. Am. Chem. Soc. 2012, 134, 7056–7065. 10.1021/ja300034j. [DOI] [PubMed] [Google Scholar]
- McDonald T. M.; Mason J. A.; Kong X. Q.; Bloch E. D.; Gygi D.; Dani A.; Crocellà V.; Giordanino F.; Odoh S. O.; Drisdell W. S.; Vlaisavljevich B.; Dzubak A. L.; Poloni R.; Schnell S. K.; Planas N.; Lee K.; Pascal T.; Wan L. W. F.; Prendergast D.; Neaton J. B.; Smit B.; Kortright J. B.; Gagliardi L.; Bordiga S.; Reimer J. A.; Long J. R. Cooperative insertion of CO2 in diamine-appended metal-organic frameworks. Nature 2015, 519, 303–308. 10.1038/nature14327. [DOI] [PubMed] [Google Scholar]
- Milner P. J.; Siegelman R. L.; Forse A. C.; Gonzalez M. I.; Runčevski T.; Martell J. D.; Reimer J. A.; Long J. R. A Diaminopropane-Appended Metal–Organic Framework Enabling Efficient CO2 Capture from Coal Flue Gas via a Mixed Adsorption Mechanism. J. Am. Chem. Soc. 2017, 139, 13541–13553. 10.1021/jacs.7b07612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegelman R. L.; McDonald T. M.; Gonzalez M. I.; Martell J. D.; Milner P. J.; Mason J. A.; Berger A. H.; Bhown A. S.; Long J. R. Controlling Cooperative CO2 Adsorption in Diamine-Appended Mg-2(dobpdc) Metal-Organic Frameworks. J. Am. Chem. Soc. 2017, 139, 10526–10538. 10.1021/jacs.7b05858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaikwad S.; Kim S. J.; Han S. CO2 Capture Using Amine-functionalized Bimetallic MIL-101 MOFs and Their Stability on Exposure to Humid Air and Acid Gases. Microporous Mesoporous Mater. 2019, 277, 253–260. 10.1016/j.micromeso.2018.11.001. [DOI] [Google Scholar]
- Gaikwad S.; Kim Y.; Gaikwad R.; Han S. Enhanced CO2 capture capacity of amine-functionalized MOF-177 metal organic framework. J. Environ. Chem. Eng. 2021, 9, 105523. 10.1016/j.jece.2021.105523. [DOI] [Google Scholar]
- Zhu J. J.; Wu L. B.; Bu Z. Y.; Jie S. Y.; Li B. G. Polyethylenimine-Grafted HKUST-Type MOF/PoIyHIPE Porous Composites (PEI@PGD-H) as Highly Efficient CO2 Adsorbents. Ind. Eng. Chem. Res. 2019, 58, 4257–4266. 10.1021/acs.iecr.9b00213. [DOI] [Google Scholar]
- Huang A. S.; Feng B. Facile synthesis of PEI-GO@ZIF-8 hybrid material for CO2 capture. Int. J. Hydrog. Energy 2018, 43, 2224–2231. 10.1016/j.ijhydene.2017.12.070. [DOI] [Google Scholar]
- Didas S. A.; Kulkarni A. R.; Sholl D. S.; Jones C. W. Role of Amine Structure on Carbon Dioxide Adsorption from Ultradilute Gas Streams such as Ambient Air. ChemSusChem 2012, 5, 2058–2064. 10.1002/cssc.201200196. [DOI] [PubMed] [Google Scholar]
- Watabe T.; Yogo K. Isotherms and isosteric heats of adsorption for CO2 in amine-functionalized mesoporous silicas. Sep. Purif. Technol. 2013, 120, 20–23. 10.1016/j.seppur.2013.09.011. [DOI] [Google Scholar]
- Bromberg L.; Diao Y.; Wu H.; Speakman S. A.; Hatton T. A. Chromium(III) Terephthalate Metal Organic Framework (MIL-101): HF-Free Synthesis, Structure, Polyoxometalate Composites, and Catalytic Properties. Chem. Mater. 2012, 24, 1664–1675. 10.1021/cm2034382. [DOI] [Google Scholar]
- Kosky P.; Balmer R.; Keat W.; Wise G.. Mechanical Engineering. In Exploring Engineering, 5th ed.; Kosky P., Balmer R., Keat W., Wise G., Eds.; Academic Press, 2021; Chapter 14, pp 317–340. [Google Scholar]
- Cvetanović R. J.; Amenomiya Y.. Application of a Temperature-Programmed Desorption Technique to Catalyst Studies. Advances in Catalysis; Eley D. D., Pines H., Weisz P. B., Eds.; Academic Press, 1967; Vol. 17, pp 103–149. [Google Scholar]
- Aziz B.; Hedin N.; Bacsik Z. Quantification of chemisorption and physisorption of carbon dioxide on porous silica modified by propylamines: Effect of amine density. Microporous Mesoporous Mater. 2012, 159, 42–49. 10.1016/j.micromeso.2012.04.007. [DOI] [Google Scholar]
- Bacsik Z.; Atluri R.; Garcia-Bennett A. E.; Hedin N. Temperature-Induced Uptake of CO2 and Formation of Carbamates in Mesocaged Silica Modified with n-Propylamines. Langmuir 2010, 26, 10013–10024. 10.1021/la1001495. [DOI] [PubMed] [Google Scholar]
- Donaldson T. L.; Nguyen Y. N. Carbon-Dioxide Reaction-Kinetics and Transport in Aqueous Amine Membranes. Ind. Eng. Chem. Fundam. 1980, 19, 260–266. 10.1021/i160075a005. [DOI] [Google Scholar]
- Yan J.; Yu Y.; Ma C.; Xiao J.; Xia Q. B.; Li Y. W.; Li Z. Adsorption Isotherms and Kinetics of Water Vapor on Novel Adsorbents MIL-101(Cr)@GO with Super-high Capacity. Appl. Therm. Eng. 2015, 84, 118–125. 10.1016/j.applthermaleng.2015.03.040. [DOI] [Google Scholar]
- Castro R. H. R.; Quach D. V. Analysis of Anhydrous and Hydrated Surface Energies of gamma-Al2O3 by Water Adsorption Microcalorimetry. J. Phys. Chem. C 2012, 116, 24726–24733. 10.1021/jp309319j. [DOI] [Google Scholar]
- Suprynowicz Z.; Rayss J.; Patrykiejew A. The influence of the water content on the adsorption properties of γ-Al2O3. Chromatographia 1978, 11, 742–746. 10.1007/bf02269111. [DOI] [Google Scholar]
- Yoo C. J.; Lee L. C.; Jones C. W. Probing Intramolecular versus Intermolecular CO2 Adsorption on Amine-Grafted SBA-15. Langmuir 2015, 31, 13350–13360. 10.1021/acs.langmuir.5b03657. [DOI] [PubMed] [Google Scholar]
- Yu J.; Chuang S. S. C. The Structure of Adsorbed Species on Immobilized Amines in CO2 Capture: An in Situ IR Study. Energy Fuels 2016, 30, 7579–7587. 10.1021/acs.energyfuels.6b01423. [DOI] [Google Scholar]
- Moon H. J.; Carrillo J. M.; Leisen J.; Sumpter B. G.; Osti N. C.; Tyagi M.; Jones C. W. Understanding the Impacts of Support-Polymer Interactions on the Dynamics of Poly(ethyleneimine) Confined in Mesoporous SBA-15. J. Am. Chem. Soc. 2022, 144, 11664–11675. 10.1021/jacs.2c03028. [DOI] [PubMed] [Google Scholar]
- Fu D.; Park Y.; Davis M. E. Zinc Containing Small-Pore Zeolites for Capture of Low Concentration Carbon Dioxide. Angew. Chem. 2022, 134, e202112916 10.1002/ange.202112916. [DOI] [PubMed] [Google Scholar]
- Zhai Y. X.; Chuang S. S. C. The Nature of Adsorbed Carbon Dioxide on Immobilized Amines during Carbon Dioxide Capture from Air and Simulated Flue Gas. Energy Technol. 2017, 5, 510–519. 10.1002/ente.201600685. [DOI] [Google Scholar]
- Yu J.; Chuang S. S. C. The Role of Water in CO2 Capture by Amine. Ind. Eng. Chem. Res. 2017, 56, 6337–6347. 10.1021/acs.iecr.7b00715. [DOI] [Google Scholar]
- Hedin N.; Bacsik Z. Perspectives on the adsorption of CO2 on amine-modified silica studied by infrared spectroscopy. Curr. Opin. Green Sustain. Chem. 2019, 16, 13–19. 10.1016/j.cogsc.2018.11.010. [DOI] [Google Scholar]
- Bacsik Z.; Ahlsten N.; Ziadi A.; Zhao G. Y.; Garcia-Bennett A. E.; Martín-Matute B.; Hedin N. Mechanisms and Kinetics for Sorption of CO2 on Bicontinuous Mesoporous Silica Modified with n-Propylamine. Langmuir 2011, 27, 11118–11128. 10.1021/la202033p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didas S. A.; Sakwa-Novak M. A.; Foo G. S.; Sievers C.; Jones C. W. Effect of Amine Surface Coverage on the Co-Adsorption of CO2 and Water: Spectral Deconvolution of Adsorbed Species. J. Phys. Chem. Lett. 2014, 5, 4194–4200. 10.1021/jz502032c. [DOI] [PubMed] [Google Scholar]
- Didas S. A.; Zhu R. S.; Brunelli N. A.; Sholl D. S.; Jones C. W. Thermal, Oxidative and CO2 Induced Degradation of Primary Amines Used for CO2 Capture: Effect of Alkyl Linker on Stability. J. Phys. Chem. C 2014, 118, 12302–12311. 10.1021/jp5025137. [DOI] [Google Scholar]
- Brunelli N. A.; Didas S. A.; Venkatasubbaiah K.; Jones C. W. Tuning Cooperativity by Controlling the Linker Length of Silica-Supported Amines in Catalysis and CO2 Capture. J. Am. Chem. Soc. 2012, 134, 13950–13953. 10.1021/ja305601g. [DOI] [PubMed] [Google Scholar]
- Zhao T.; Jeremias F.; Boldog I.; Nguyen B.; Henninger S. K.; Janiak C. High-yield, fluoride-free and large-scale synthesis of MIL-101(Cr). Dalton Trans. 2015, 44, 16791–16801. 10.1039/c5dt02625c. [DOI] [PubMed] [Google Scholar]
- Hong D. Y.; Hwang Y. K.; Serre C.; Férey G.; Chang J. S. Porous Chromium Terephthalate MIL-101 with Coordinatively Unsaturated Sites: Surface Functionalization, Encapsulation, Sorption and Catalysis. Adv. Funct. Mater. 2009, 19, 1537–1552. 10.1002/adfm.200801130. [DOI] [Google Scholar]
- Hwang Y. K.; Hong D. Y.; Chang J. S.; Jhung S. H.; Seo Y. K.; Kim J.; Vimont A.; Daturi M.; Serre C.; Férey G. Amine Grafting on Coordinatively Unsaturated Metal Centers of MOFs: Consequences for Catalysis and Metal Encapsulation. Angew. Chem. 2008, 47, 4144–4148. 10.1002/anie.200705998. [DOI] [PubMed] [Google Scholar]
- Peri J. B.; Hannan R. B. Surface Hydroxyl Groups on γ-Alumina. J. Phys. Chem. 1960, 64, 1526–1530. 10.1021/j100839a044. [DOI] [Google Scholar]
- Chen Y.; Zhang L. F. Surface Interaction model of γ-alumina-supported metal oxides. Catal. Lett. 1992, 12, 51–62. 10.1007/bf00767188. [DOI] [Google Scholar]
- Lagauche M.; Larmier K.; Jolimaitre E.; Barthelet K.; Chizallet C.; Favergeon L.; Pijolat M. Thermodynamic Characterization of the Hydroxyl Group on the gamma-Alumina Surface by the Energy Distribution Function. J. Phys. Chem. C 2017, 121, 16770–16782. 10.1021/acs.jpcc.7b02498. [DOI] [Google Scholar]
- Takeuchi M.; Martra G.; Coluccia S.; Anpo M. Investigations of the structure of H2O clusters adsorbed on TiO2 surfaces by near-infrared absorption spectroscopy. J. Phys. Chem. B 2005, 109, 7387–7391. 10.1021/jp040630d. [DOI] [PubMed] [Google Scholar]
- Vico S.; Palys B.; Buess-Herman C. Hydration of a polysulfone anion-exchange membrane studied by vibrational spectroscopy. Langmuir 2003, 19, 3282–3287. 10.1021/la026290+. [DOI] [Google Scholar]
- Hahn M. W.; Steib M.; Jentys A.; Lercher J. A. Mechanism and Kinetics of CO2 Adsorption on Surface Bonded Amines. J. Phys. Chem. C 2015, 119, 4126–4135. 10.1021/jp512001t. [DOI] [Google Scholar]
- Li K. J.; Kress J. D.; Mebane D. S. The Mechanism of CO2 Adsorption under Dry and Humid Conditions in Mesoporous Silica-Supported Amine Sorbents. J. Phys. Chem. C 2016, 120, 23683–23691. 10.1021/acs.jpcc.6b08808. [DOI] [Google Scholar]
- Young J.; García-Díez E.; Garcia S.; van der Spek M. The impact of binary water-CO2 isotherm models on the optimal performance of sorbent-based direct air capture processes. Energy Environ. Sci. 2021, 14, 5377–5394. 10.1039/d1ee01272j. [DOI] [Google Scholar]
- Liu Q.; Ning L.; Zheng S.; Tao M.; Shi Y.; He Y. Adsorption of Carbon Dioxide by MIL-101 (Cr): Regeneration Conditions and Influence of Flue Gas Contaminants. Sci. Rep. 2013, 3, 2916. 10.1038/srep02916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. J.; Chen C. H.; Shimon D.; Hayes S. E.; Sievers C.; Jones C. W. Effect of Humidity on the CO2 Adsorption of Tertiary Amine Grafted SBA-15. J. Phys. Chem. C 2017, 121, 23480–23487. 10.1021/acs.jpcc.7b07930. [DOI] [Google Scholar]
- Moore J. K.; Sakwa-Novak M. A.; Chaikittisilp W.; Mehta A. K.; Conradi M. S.; Jones C. W.; Hayes S. E. Characterization of a Mixture of CO2 Adsorption Products in Hyperbranched Aminosilica Adsorbents by 13C Solid-State NMR. Environ. Sci. Technol. 2015, 49, 13684–13691. 10.1021/acs.est.5b02930. [DOI] [PubMed] [Google Scholar]
- Chen C. H.; Shimon D.; Lee J. J.; Didas S. A.; Mehta A. K.; Sievers C.; Jones C. W.; Hayes S. E. Spectroscopic Characterization of Adsorbed 13CO2 on 3-Aminopropylsilyl-Modified SBA15 Mesoporous Silica. Environ. Sci. Technol. 2017, 51, 6553–6559. 10.1021/acs.est.6b06605. [DOI] [PubMed] [Google Scholar]
- Chen C. H.; Shimon D.; Lee J. J.; Mentink-Vigier F.; Hung I.; Sievers C.; Jones C. W.; Hayes S. E. The ″Missing″ Bicarbonate in CO2 Chemisorption Reactions on Solid Amine Sorbents. J. Am. Chem. Soc. 2018, 140, 8648–8651. 10.1021/jacs.8b04520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. H.; Sesti E. L.; Lee J. J.; Mentink-Vigier F.; Sievers C.; Jones C. W.; Hayes S. E. NMR Reveals Two Bicarbonate Environments in SBA15-Solid-Amine CO2 Sorbents. J. Phys. Chem. C 2021, 125, 16759–16765. 10.1021/acs.jpcc.1c04145. [DOI] [Google Scholar]
- Miller D. D.; Yu J.; Chuang S. S. C. Unraveling the Structure and Binding Energy of Adsorbed CO2/H2O on Amine Sorbents. J. Phys. Chem. C 2020, 124, 24677–24689. 10.1021/acs.jpcc.0c04942. [DOI] [Google Scholar]
- Yu J.; Zhai Y. X.; Chuang S. S. C. Water Enhancement in CO2 Capture by Amines: An Insight into CO2-H2O Interactions on Amine Films and Sorbents. Ind. Eng. Chem. Res. 2018, 57, 4052–4062. 10.1021/acs.iecr.7b05114. [DOI] [Google Scholar]
- Siegelman R. L.; Milner P. J.; Forse A. C.; Lee J. H.; Colwell K. A.; Neaton J. B.; Reimer J. A.; Weston S. C.; Long J. R. Water Enables Efficient CO2 Capture from Natural Gas Flue Emissions in an Oxidation-Resistant Diamine-Appended Metal-Organic Framework. J. Am. Chem. Soc. 2019, 141, 13171–13186. 10.1021/jacs.9b05567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darunte L. A.; Terada Y.; Murdock C. R.; Walton K. S.; Sholl D. S.; Jones C. W. Monolith-Supported Amine-Functionalized Mg2(dobpdc) Adsorbents for CO2 Capture. ACS Appl. Mater. Interfaces 2017, 9, 17042–17050. 10.1021/acsami.7b02035. [DOI] [PubMed] [Google Scholar]
- Sujan A. R.; Pang S. H.; Zhu G. H.; Jones C. W.; Lively R. P. Direct CO2 Capture from Air using Poly(ethylenimine)-Loaded Polymer/Silica Fiber Sorbents. ACS Sustain. Chem. Eng. 2019, 7, 5264–5273. 10.1021/acssuschemeng.8b06203. [DOI] [Google Scholar]
- DeWitt S. J. A.; Lively R. P. MIL-101(Cr) Polymeric Fiber Adsorbents for Sub-Ambient Post-Combustion CO2 Capture. Ind. Eng. Chem. Res. 2022, 61, 13612–13623. 10.1021/acs.iecr.2c01837. [DOI] [Google Scholar]
- Quan W. Y.; Holmes H. E.; Zhang F. Y.; Hamlett B. L.; Finn M. G.; Abney C. W.; Kapelewski M. T.; Weston S. C.; Lively R. P.; Koros W. J. Scalable Formation of Diamine-Appended Metal-Organic Framework Hollow Fiber Sorbents for Postcombustion CO2 Capture. JACS Au. 2022, 2, 1350–1358. 10.1021/jacsau.2c00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.