

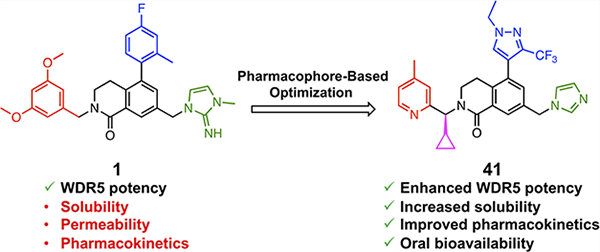
Abstract
WD repeat domain 5 (WDR5) is a nuclear scaffolding protein that forms many biologically important multiprotein complexes. The WIN site of WDR5 represents a promising pharmacological target in a variety of human cancers. Here, we describe the optimization of our initial WDR5 WIN-site inhibitor using a structure-guided pharmacophore-based convergent strategy to improve its druglike properties and pharmacokinetic profile. The core of the previous lead remained constant while a focused SAR effort on the three pharmacophore units was combined to generate a new in vivo lead series. Importantly, this new series of compounds has picomolar binding affinity, improved cellular antiproliferative activity and selectivity, and increased kinetic aqueous solubility. They also exhibit a desirable oral pharmacokinetic profile with manageable intravenous clearance and high oral bioavailability. Thus, these new leads are useful probes toward studying the effects of WDR5 inhibition.
GRAPHICAL ABSTRACT
INTRODUCTION
WD repeat domain 5 (WDR5) is a highly conserved member of the WD40-repeat protein family with a characteristic circular barrel-shaped 7-bladed β-propeller structure.1–3 WDR5 is a core scaffolding component that contains two major binding interfaces, the WDR5 binding motif (WBM) and WDR5 interaction motif (WIN) sites, on opposite sides to accommodate a large variety of partner proteins to form multiple protein complexes that are essential in a wide range of biological activities, most of which occur in the nucleus.4–12 WDR5 has been extensively studied for its role in the activity of MLL1 histone methyltransferase (HMT) complexes, in which MLL1 binds on the WIN site using a conserved arginine in the MLL/SET proteins as an anchor.7–9,11,13–17 The complex also contains at least three other evolutionarily conserved proteins (retinoblastoma binding protein 5 (RBBP5), ASH2L, and DPY30) binding through WIN-site-independent interactions and catalyzes histone H3 lysine 4 (H3K4) di- and tri-methylation (H3K4me2, me3).8,14 All other MLL/SET (MLL2–4, SETd1A, and SETd1B) family proteins can form similar assemblies by engaging WDR5 on the WIN site through the same interactions but are less dependent on this interaction for catalytic activity than MLL1.7,14–17 In addition, WDR5-containing nonspecific lethal (NSL) and Ada2-containing (ATAC) histone acetyltransferase (HAT) complexes carry out acetylation of histone H4 at lysine 16 (H4K16).13
WDR5 is also an essential cofactor for the MYC family of oncoprotein transcription factors. WDR5 is tethered to chromatin via the WIN site and recruits MYC to chromatin at tumor-critical target genes via a direct interaction of the WBM site of WDR5 with the conserved MYC Box IIIb element of MYC.18–20 A MYC mutant, with impaired binding to WDR5, thus exhibits reduced binding of MYC to chromatin and loss of oncogenic potential in vivo, further supporting the role of WDR5 as a critical cofactor for MYC proteins in malignant gene expression programs.20 In addition, aberrant overexpression of WDR5 can be found in a variety of aggressive cancers including bladder,21 breast,22 colorectal,23 gastric,24 pancreatic,25 prostate,26,27 neuroblastoma,28 head neck squamous cell carcinoma,29 liver,30 and various leukemias,31,32 and is often associated with poor prognoses.22,29,32 Therefore, inhibition of WDR5’s multifaceted role in human malignancies has emerged as an attractive potential anticancer therapy.4,10,18,33 The activity of WDR5 WIN site inhibition is mediated by displacement of WDR5 from chromatin at ribosomal protein genes (RPGs), which leads to an induction of nucleolar stress and activation of p53-dependent apoptosis.34,35 The small-molecule-mediated displacement of WDR5 also displaced the oncoprotein transcription factor MYC at these same RPGs, highlighting that MYC proteins may be one source of multi-factorial vulnerability to these agents.34,35
Structurally diverse WDR5 WIN-site inhibitors have been discovered (Figure 1), and their binding interactions at the WIN site have been confirmed by X-ray co-crystal structures.34–41 The mechanistic proof of concept was demonstrated by MM-589, a first-generation macrocyclic guanidine side chain containing peptidomimetic, that mimics MLL-WDR5 interactions within the WIN site.36 The probe was reported to exhibit subnanomolar binding affinity at the WIN site, potent functional inhibitory activity with a low nanomolar IC50 in the MLL1 HMT assay, and antiproliferative activities in MLL-fusion cancer cell lines.36 OICR-9429 was the first nonpeptide WIN-site inhibitor with a reported Kd of 30 nM, which was discovered from a high-throughput screen hit.37,38 The X-ray co-crystal structure of OICR-9429 bound to WDR5 revealed the methyl-piperazine moiety occupied the arginine pocket of the WIN site by mimicking the guanidine side chain of R3765 in the MLL peptide.37,38 Because OICR-9429 exhibits druglike properties, its scaffold has become a platform for further optimization of on-target potency and physicochemical properties by several groups. DDO-209339 and DDO-221340 are recent examples in this class that exhibit antiproliferative activities in MV4:11 cancer cells with reported GI50 values of ~10 μM. These compounds were also tested in an MV4:11 mouse xenograft model and showed marginal in vivo tumor growth inhibition by intraperitoneal injection (ip) and oral (po) dosing, respectively, despite their limited cellular potency.39,40 In addition, several WDR5 proteolysis-targeting chimeras (PROTACs) were designed by linking known WIN-site ligands to E3 ligase binding moieties, pomalidomide (CRBN) or VHL.42,43 These PROTACs caused reduction of the WDR5 protein expression level in cancer cells via E3 ligase mediated selective WDR5 degradation. The most advanced WDR5 PROTAC MS67 also demonstrated in vivo tumor growth inhibition in mouse models.43
Figure 1.

Chemical structures and in vitro profiles of representative WDR5 WIN-site inhibitors and WDR5 PROTAC. Compound 1 is also known as 16 in our previous report.35
We have also discovered a series of highly potent WDR5 inhibitors using fragment-based methods and structure-based design.34,35,41 Compound 1 contains a novel conformationally rigid 3,4-dihydroisoquinolin-1(2H)-one core that elicits a hydrogen-bond interaction with the backbone NH of C261 and provides ideal vectors to access key binding subsites in the WIN site.35 Compound 1 occupies the MLL peptide residue binding subsites, S2, S4, and S7, with the P2 imidazole-imine to mimic the guanidine side chain, 4-fluoro-2-methylphenyl P4, and 3,5-dimethoxybenzyl P7 pharmacophore units, respectively.35 This small-molecule probe exhibits a binding affinity below the theoretical detection limit of our binding assay (Ki < 20 pM) and potent antiproliferative activity in MV4:11 cells (GI50 = 38 nM) with 210-fold cellular selectivity assessed by the GI50 ratio between WDR5 insensitive K562 and sensitive MV4:11 cell lines.35 Compound 1 also caused the displacement of the WDR5 protein from chromatin and triggered transcriptional repression of WDR5 bound genes leading to p53 induction at a significantly lower concentration compared to the previous lead.35
Despite 1 having the highest in vitro on-target potency among the reported WDR5 WIN-site inhibitors, it is not an ideal chemical probe to demonstrate the therapeutic potential of WDR5 inhibitors in in vivo models due to its suboptimal physicochemical properties. Compound 1 contains three hydrophobic phenyl moieties, which limit its aqueous solubility. In addition, the probe shows very low systemic exposure in mice by oral dosing, and the imidazole-imine “warhead” that mimics the guanidine side chain is suspected to be a main contributor to poor permeability. Our cellular assay protocol requires 3–5 days of incubation with WIN-site inhibitors to deliver an antiproliferative effect, which suggests repeated doses of a WIN-site inhibitor are required to maintain a pharmacologically relevant WDR5 occupancy to be efficacious in vivo. Therefore, the discovery of a potent WDR5 WIN-site inhibitor with high oral bioavailability is critical to develop an effective and practical therapeutic agent. Here, we report our continued efforts toward that objective, building upon our recently reported compound 1. Our efforts led to the discovery of a new class of WDR5 inhibitors with enhanced potency and physicochemical properties using a pharmacophore-based convergent SAR strategy and X-ray structure-guided modifications. The new series of inhibitors exhibit significantly enhanced selective antiproliferative activity in WDR5 sensitive cells, aqueous solubility, and high oral bioavailability in mice.
RESULTS AND DISCUSSION
Compound 1 Profile and Pharmacophore-Based Convergent Optimization Strategy.
The physicochemical properties of our previous chemical probe 1 were profiled by in vitro assays and showed marginal kinetic aqueous solubility (29 μM at pH 7.4, 1% DMSO) and poor passive permeability (Pe < 0.6 × 10−6 cm/s) in the parallel artificial membrane permeability assay (PAMPA). Compound 1 also exhibited a suboptimal pharmacokinetic (PK) profile in mice including high intravenous (iv) clearance (78 mL/min/kg at 3 mg/kg) and an oral exposure below the limit of quantitation when dosed orally (3 mg/kg). In addition, signs of severe acute toxicity, such as reduced locomotive activity, lowered body temperature, and lethality, were observed when compound 1 was dosed (3 mg/kg) by an iv route. These data suggest that future WDR5 inhibitors would require significant improvement in druglike properties, in vivo PK and safety profiles from compound 1 while maintaining its high on-target potency to be used as an effective in vivo probe and a potential therapeutic agent.
The structure of compound 1 can be divided into four subunits (P2, P4, P7, and core) according to their binding subsite alignments with residues 3764–3773 (ARAEVHLRKS) of MLL1 in the WDR5 WIN site.44 The X-ray co-crystal structure of 1 bound to WDR5 (Figure 2) revealed that the conformationally rigid 3,4-dihydroisoquinolin-1(2H)-one core was positioned vertically in the center of the WIN site and provided the appropriate exit vectors for the P2, P4, and P7 pharmacophore units to extend to the S2, S4, and S7 binding subsites, respectively.35 The binding pose of the core unit was stabilized through a hydrogen-bond interaction between the core carbonyl oxygen and the backbone NH of C261 and a T-shaped π–π stacking interaction between the core phenyl moiety and the side chain of F133 as shown in Figure 2A,B. The P2 imidazole-imine “warhead” is the most critical element for binding affinity and selectivity by mimicking the guanidine side chain of R3765 in the MLL peptide.35 The imidazole portion of P2 is positioned between the two benzyl side chains of F133 and F263 to elicit a sandwiched π–π stacking interaction. In addition, the imine moiety is located within a hydrogen-bond distance to the backbone carbonyl of C261, and the interaction will be further strengthened by the formation of the positively charged iminium ion at physiological pH (calculated pKa = 11).35 The 4-fluoro-2-methylphenyl P4 moiety adopts an orthogonal conformation to the core unit to fully occupy the hydrophobic S4 subsite (Figure 2D). Conversely, the S7 binding site is a small hydrophobic cavity that can accommodate only half of the 3,5-dimethoxyphenyl P7 group (Figure 2E).
Figure 2.

X-ray co-crystal structure of 1 bound to the WDR5 WIN site (PDB ID: 6UCS).35 (A) Compound 1 (yellow carbon-capped sticks) bound to WDR5 represented as semitransparent electrostatic potential surface with labeled S2, S4, and S7 binding regions. (B) Key H-bond and π–π stacking binding interactions (red dashed lines) of the core unit and denoted WDR5 residues (green sticks). (C) Key H-bond and π–π stacking binding interactions (red dashed lines) of the P2 unit in the S2 subsite and denoted WDR5 residues (green sticks). (D) Binding interactions of the P4 unit represented as space-filling dots in the S4 subsite (gray surface). (E) Binding interactions of the P7 unit represented as space-filling dots in the S7 subsite (gray surface).
Based on the above structural information of the binding interactions of each subunit at the corresponding binding site, a pharmacophore-based convergent optimization strategy was devised as shown in Figure 3. The 3,4-dihydroisoquinolin-1(2H)-one core served as a chemically stable scaffold that provided proper vectors to nearby binding subsites and was kept constant throughout the study. Efficient convergent synthetic methodologies were developed to facilitate SAR efforts, and a focused structure-guided SAR development was performed on each subunit. All optimized subunits were then combined into a single molecule to retain all desirable attributes.
Figure 3.

Profile overview of 1 and considerations for the pharmacophore-based optimization strategy.
The S2 subsite in the WDR5 WIN site was evolved to accommodate the positively charged guanidinium side chain of the R3765 residue.14 The corresponding P2 moiety would be the most important anchor unit for the binding potency and may tolerate only a small subset of chemical matter that can maintain the critical binding interactions at the site. For this reason, OICR-9429 mimicked the electronic character of the natural ligand specificity with the methyl-piperazinium P2 moiety expecting similar binding interactions at the site. However, the WIN site surface representation of the MLL1 peptide 3764–3773 (ARAEVHLRKS) and OICR-9429 bound WDR5 X-ray structures showed the binding site assumed different bound conformations (Figure 4A,B). Inspection of both co-crystal structures revealed that the side chains of F133 and F263 in the S2 subsite and F149 in the S7 subsite of the OICR-9429 bound structure underwent significant conformational changes from the MLL1 peptide-binding pose to accommodate the chair shaped methyl-piperazinium P2 moiety shown in Figure 4C. The conformational change of F149 appeared to be synchronized with the conformational rearrangement of F133 to relieve steric strain. Conversely, the P2 imidazole-imine of compound 1 mimicked the flat geometry and electronic character of the guanidinium side chain to preserve the original protein conformation in the WIN site (Figure 4D). These structural analyses suggest that the S2 subsite may be conformationally flexible, and its changes may redefine the shape of the entire WIN site. Considering the results of our previous reports34,35 we would conduct a focused SAR effort using P2 moieties that could mimic the interactions of the imidazole-imine of 1.
Figure 4.

(A) Surface representation of the WDR5 WIN site (PDB ID: 3EG6)14 when bound to MLL1 peptide (magenta carbon-capped lines and sticks (P2 residue)) with labeled S2, S4, and S7 binding regions. (B) Surface representation of the WDR5 WIN site (PDB ID: 4QL1)38 when bound to OICR-9429 (cyan carbon-capped lines and sticks (S2 binding moiety)) with labeled S2, S4, and S7 binding regions. (C) Overlay of MLL1 peptide (magenta) and OICR-9429 (cyan) bound WDR5 protein structures in cartoon representation. The side chains of WDR5 residues F133, F149, and F263 of both structures are represented as sticks to demonstrate conformational changes. (D) Overlay of MLL1 peptide (magenta) and compound 1 (green) bound WDR5 protein structures in cartoon representation. The side chains of WDR5 residues F133, F149, and F263 for both structures are represented as sticks to demonstrate retention of binding poses.
Compared to the narrow derivatization requirements of the S2 subsite, the S4 and S7 subsites present a greater opportunity to optimize the druglike properties of WDR5 WIN-site inhibitors. The S4 and S7 subsites are more exposed with higher conformational flexibility than the S2 subsite allowing a range of chemical matter to be used as potential pharmacophore units in each site. Therefore, broad derivatization of the P4 and P7 units would be better suited to optimize the physicochemical properties as well as the binding affinity of WDR5 inhibitors. All three pharmacophore units of 1 are responsible for the suboptimal physicochemical and PK properties of the compound. The P2 imidazole-imine is suspected to be the main contributor to the poor PAMPA permeability of 1 due to its pH-dependent basicity profile and its replacement with a neutral pharmacophore is expected to improve the passive permeability of WDR5 inhibitors. The hydrophobic P4 and P7 phenyl moieties of 1 contribute to its limited aqueous solubility and can be replaced with more druglike heterocycles.
On-target potency of all newly prepared WDR5 inhibitors was profiled initially using a time-resolved fluorescence energy transfer (TR-FRET) assay to determine the competitive binding affinity (Ki) against the MLL1 derived FITC-peptide probe.34,35 WDR5 inhibition-mediated cellular efficacy was assessed in MLL-fusion cancer cell lines MV4:11 and MOLM-13, which express wild-type p53. The K562 leukemia cell line, which is null for p53 and refractory to WIN site inhibition, was used as an insensitive control. The half-maximal growth inhibition (GI50) ratio between K562 and MV4:11 serves as a cellular selectivity measure to ascertain primary cellular selectivity.34,35 Following optimization of the three pharmacophore units, druglike properties of select compounds were further profiled by kinetic aqueous solubility assays and in vivo PK studies in mice. The results of these studies led to the discovery of a new lead series of WDR5 inhibitors.
Optimization of the P4 Pharmacophore for Imidazole-Imine Series WDR5 Inhibitors.
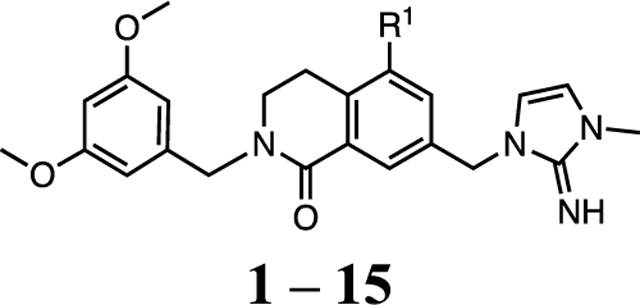
The 4-fluoro-2-methylphenyl P4 moiety of 1 is the most hydrophobic pharmacophore unit and contributes to its limited aqueous solubility. Therefore, we searched for new effective P4 moieties by employing more desirable heteroaryl groups with reduced hydrophobicity in 1. For rapid SAR development, we developed a new synthetic route (Scheme 1), in which the P4 moieties were installed in the last step of the synthesis. We used the calculated partition coefficient (cLogP) to monitor the predicted hydrophobicity of the compounds as substitutions were made to the P4 unit. The binding affinity of 1 to WDR5 was below the theoretical detection limit of the TR-FRET assay (Ki < 20 pM) and substitutions to the P4 unit maintained the tight picomolar binding affinity to WDR5 (Table 1). Compound 1 had a cLogP of 4.8, and substitution of the 2-methyl for the 2-methoxy group in 2 lowered the hydrophobicity of the compound. However, with the 2-methoxy group a 5-fold loss in antiproliferative activity was observed in both the MV4:11 and MOLM-13 cell lines. The phenol of 3 was tolerated better than methoxy but resulted in a 2-fold loss in cellular activity compared to 1. A series of 3-pyridyl P4 units with desirable substitutions from the phenyl series were then investigated to determine the effect of a heteroaryl group with increased hydration potential on the cellular potency of the compounds. The significant loss of binding and cellular activity for 4 served as a benchmark for the new series. The increased size of the P4 group using the 2-methoxy and 2,5-dimethyl substitutions for 5 and 6 recovered the cellular activity by 2-fold from compound 4. Introduction of the strong electron-withdrawing 2-trifluoromethyl group exhibited 10-fold enhanced cellular potency compared to 4, but compound 7 still had 3-fold weaker activity compared to 1. The 4-pyridyl P4 units in 8 and 9 improved the cellular activity by 5- and 3-fold compared to regioisomers 4 and 6, respectively. However, the expected activity boost was not observed for 9 compared to 8. The lack of improved cellular activity for the pyridyl series led to the investigation of other heterocycles for the P4 unit.
Scheme 1.

Synthesis of Imidazole-Imine Series Compounds 2–15a
aR1 is defined in Table 1. Conditions: (a) 3,5-dimethoxybenzylamine, NaBH(OAc)3, CH2Cl2, rt, overnight, then 1,4-dioxane, 110 °C, 99%; (b) phenyl triflimide, i-Pr2NEt, THF/CH2Cl2, rt, overnight, 79%; (c) B2Pin2, KOAc, PdCl2(dppf)·CH2Cl2, 1,4-dioxane, 100 °C overnight, 99%; (d) CuBr2, MeOH/H2O, 80 °C, 90%; (e) LiBHEt3 (1 M THF), THF, 0 °C, 1 h, 41%; (f) PBr3, CH2Cl2, 0 °C, 42%; (g) 1-methyl-1H-imidazol-2-amine hydrochloride, i-Pr2NEt, MeCN, 60 °C, overnight, quant.; (h) R1–B(OH)2 or R1–B(Pin), PdCl2(dppf)·CH2Cl2, K2CO3, 1,4-dioxane/H2O, 80 °C, overnight, 20–70%.
Table 1.
SAR Profile of P4 Imidazole-Imine Series
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| compnd | R1 = | cLogP |
Ki (nM) TR-FRETa |
Cell Proliferation Assays GI50(nM)a |
Selectivityb |
||
| WDR5 | MV4:11 | MOLM-13 | K562 | K562/MV4:11 | |||
|
| |||||||
| 1 c |
|
4.8 | <0.02 | 38 ± 9.0 | 78 ± 13 | 8000 ± 4100 | 210 |
| 2 |
|
4.0 | 0.033 ± 0.005 | 210 ± 19 | 420 ± 30 | 18000 ± 2800 | 86 |
| 3 |
|
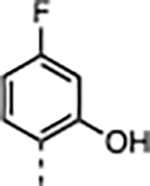
3.7 | 0.035 ± 0.005 | 88 ± 20 | 220 ± 23 | >30000 | >340 |
| 4 |
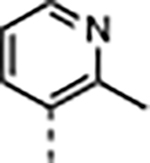
|
3.2 | 0.036 ± 0.005 | 1200 ± 310 | 3900 ± 410 | >30000 | >25 |
| 5 |
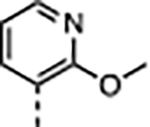
|
2.7 | 0.055 ± 0.005 | 560 ± 84 | 1600 ± 200 | >30000 | >54 |
| 6 |
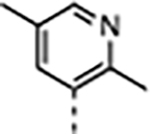
|
3.7 | 0.022 ± 0.001 | 640 ± 120 | 2400 ± 240 | >30000 | >47 |
| 7 |
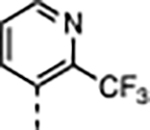
|
3.9 | <0.02 | 120 ± 59 | 290 ± 170 | >29000 | >240 |
| 8 |
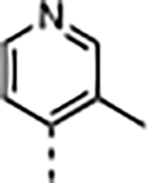
|
3.2 | 0.029 ± 0.002 | 230 ± 52 | 640 ± 120 | >30000 | >130 |
| 9 |
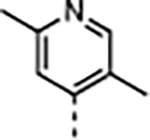
|
3.7 | <0.02 | 200 ± 52 | 560 ± 76 | >30000 | >150 |
| 10 |
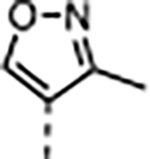
|
2.4 | 0.046 ± 0.002 | 850 ± 240 | 2100 ± 410 | >30000 | >35 |
| 11 |
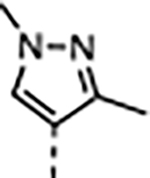
|
2.6 | 0.023 ± 0.001 | 210 ± 49 | 570 ± 85 | >30000 | >140 |
| 12 |
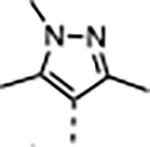
|
2.5 | 0.21 ± 0.008 | 19000 ± 3000 | >30000 | >30000 | >2 |
| 13 |
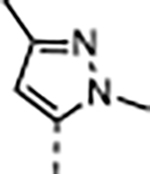
|
3.1 | 0.025 ± 0.003 | 85 ± 17 | 270 ± 31 | >30000 | >350 |
| 14 |
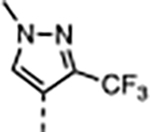
|
3.5 | <0.02 | 39 ± 9.0 | 110 ± 7.8 | >30000 | >770 |
| 15 |
|
4.0 | <0.02 | 36 ± 11 | 110 ± 9.8 | >30000 | >830 |
TR-FRET Ki, and cell proliferation GI50 values represent four independent replicate determinations ± standard deviation.
Selectivity is defined a GI50,K562/GI50,MV4:11 and is used to generally distinguish between on- and off-target inhibition mechanisms.
The data for 1 were reported in our previous report.35
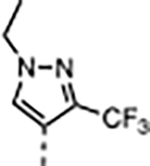
A series of five-membered heterocycles were profiled as WDR5 inhibitors keeping the similar substitution pattern as the previous series. Compound 10 with the 3-methylisoxazol-4-yl P4 served as a benchmark for this new series. The 1,3-dimethyl-1H-pyrazol-4-yl P4 of 11 contributed to the observed 2-fold increase in binding affinity and 4-fold increase in cellular activity. The additional methyl substitution for 12 was not tolerated and disrupted the binding of the compound to WDR5. Compound 13 with the 1,3-dimethyl-1H-pyrazol-5-yl P4 had a similar binding affinity and a 2-fold increase in cellular activity compared to 11. The trifluoromethyl in 14 and 15 gave a 5-fold increase in cellular activity compared to 11 and were comparable to the binding affinity and cellular activity of 1. The cLogP and selectivity of 14 and 15 were both improved over 1. The 1-methyl- and 1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl) P4 units were good replacements for the 4-fluoro-2-methylphenyl P4 unit and were kept in the further optimization of the compounds.
Optimization of the P2 Pharmacophore with Nonbasic Warheads.
A few representative compounds in Table 1 were tested in the PAMPA permeability assay and showed no improvement from compound 1, which suggests replacement of the P2 imidazole-imine is necessary to improve the poor physicochemical properties of the compounds. Our optimization efforts on the P2 moiety were then focused on five-membered heteroaryl groups that remain neutral at physiological pH to enhance the passive permeability while maintaining the flat geometry to form the key sandwiched π–π stacking interactions with F133 and F263 in the S2 subsite to preserve the binding conformation of the WIN site and potency of 1. We had previously discovered that N-linked imidazole was a hit in our early fragment screen.34 A synthetic route was developed from common intermediate 42 to install the P2 moiety in the last step of the sequence, which is depicted in Scheme 2. Compound 16 (Table 2) with the imidazole P2 unit maintained good potency against WDR5 sensitive cell lines by exhibiting only a 2-fold loss in activity compared to 1 and 14 with the P2 imidazole-imine. A series of N-linked azoles were investigated, and 17–19 showed significantly reduced binding affinity and cellular potency. This result suggests that the position of the nitrogen atoms and the electrostatic property of the heteroaryl groups were critical for engaging proper binding interactions in the S2 subsite. The imidazole P2 moiety was then further diversified by adding small hydrophobic substituents at the 2-position. The 2-methyl imidazole of 20 increased the cellular activity and was comparable to the potency of 1 and 14. However, larger alkyl substituents at the 2-position of the imidazole P2 unit in 21–23 were less tolerated in the S2 subsite. Withdrawing electron density away from the imidazole ring with the 2-difluoromethyl group of 24 was also detrimental to the binding interaction. The imidazole and 2-methyl imidazole were found to be good replacements for the imidazole-imine and were used in the further optimization of the WDR5 inhibitors. Finally, compound 20 with the 2-methyl imidazole was profiled in the PAMPA assay and exhibited a 10-fold enhanced passive permeability (Pe = 5.3 × 10−6 cm/s) compared to 1 that further justified our optimization strategy.
Scheme 2.

Synthesis of Nonbasic Warhead Series Compounds 16–24a
aR2 is defined in Table 2. Conditions: (a) (1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)boronic acid, PdCl2(dppf)·CH2Cl2, K2CO3, 1,4-dioxane/H2O, 90 °C, overnight, 85%; (b) LiBHEt3 (1 M THF), THF, 0 °C, 1 h, 95%; (c) PBr3, CH2Cl2, 0 °C, 90%; (d) R2–azole, MeCN, 50 °C, overnight, 45–60%.
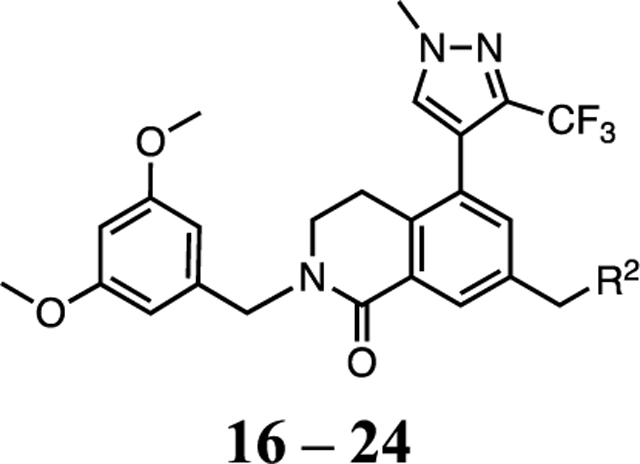
Table 2.
SAR Profile of Nonbasic Warhead Series
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| compnd | R2 = | cLogP |
Ki (nM) TR-FRETa |
Cell Proliferation Assays GI50 (nM)a |
Selectivityb |
||
| WDR5 | MV4:11 | MOLM-13 | K562 | K562/MV4:11 | |||
|
| |||||||
| 16 |
|
2.9 | <0.02 | 59 ± 5.6 | 180 ± 51 | 9100 ± 2100 | 150 |
| 17 |
|
3.9 | >6.7 | 22000 ± 6000 | 20000 ± 5400 | >30000 | >1 |
| 18 |
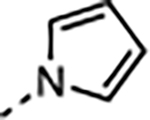
|
4.4 | >6.7 | 15000 ± 4000 | 15000 ± 3600 | >30000 | >2 |
| 19 |
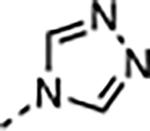
|
1.6 | 6.5 ± 1.7 | 16000 ± 1300 | >30000 | >30000 | >2 |
| 20 |
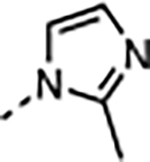
|
3.1 | <0.02 | 32 ± 13 | 110 ± 33 | 3700 ± 2500 | 120 |
| 21 |
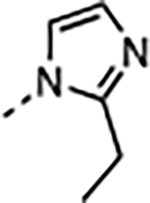
|
3.7 | 0.022 ± 0.001 | 94 ± 10 | 240 ± 53 | 7500 ± 1300 | 80 |
| 22 |
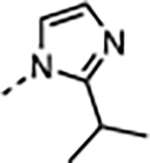
|
4.1 | 3.4 ± 0.17 | 4700 ± 500 | 7700 ± 2000 | 16000 ± 3100 | 3 |
| 23 |
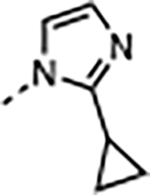
|
3.6 | 0.028 ± 0.002 | 240 ± 64 | 710 ± 140 | 8500 ± 2000 | 35 |
| 24 |
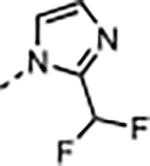
|
3.1 | >6.7 | 12000 ± 2800 | 20000 ± 4900 | >30000 | >3 |
TR-FRET Ki and cell proliferation GI50 values represent four independent replicate determinations ± standard deviation.
Selectivity is defined as GI50,K562/GI50,MV4:11 and is used to generally distinguish between on- and off-target inhibition mechanisms.
The X-ray co-crystal structure of 20 bound to WDR5 was obtained to understand binding interactions of the new P4 and P2 units. Compound 20 was positioned in the WIN site in a similar manner as 1 by accessing all key binding subsites (Figure 5A). The 3,4-dihydroisoquinolin-1(2H)-one core maintained the same T-shaped π–π stacking with F133 and H-bond between the carbonyl oxygen and the backbone NH of C261 to provide stable exit vectors to the S2, S4, and S7 subsites as previously described. The optimized 1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl) P4 unit bound tightly to the S4 subsite within van der Waals close contacts. The electron-withdrawing and hydrophobic nature of the trifluoromethyl substituent may balance both the hydration potential and overall hydrophobicity of the polar pyrazole pharmacophore unit to avoid significant loss of binding affinity due to increased desolvation energy. Indeed, the 1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl) P4 unit of 14 reduced its cLogP by 1.3 without a loss of potency compared to the corresponding 4-fluoro-2-methylphenyl of 1. The position of the N-methyl group showed that a slightly larger group may be accommodated, such as the N-ethyl group in 15, without a steric clash. Overall, the binding conformation of 20 was nearly superimposable with 1 including the newly optimized P4 and P2 units (Figure 5B). The SAR of the N-linked azoles showed that only imidazole and 2-methyl imidazole were able to maintain comparable binding affinity and antiproliferative activity to the corresponding P2 imidazole-imine of 14. The X-ray structure revealed that the 2-methyl imidazole of 20 bound deeply in the S2 subsite to maintain strong sandwiched π–π stacking interactions with F133 and F263 as denoted by the red dotted lines in Figure 5C, which was consistent with the P2 imidazole-imine of compound 1. Although the 2-methyl imidazole lacked the favorable direct H-bond found in the P2 imidazole-imine to the backbone carbonyl of C261, the effect did not cause a loss of potency for 20. Finally, the binding conformation of the three key phenylalanine residues, which defines the shape of the WIN site, was successfully preserved by 20 as shown in Figure 5D.
Figure 5.

X-ray co-crystal structure of 20 bound to WDR5 (PDB ID: 7U9Y). (A) Compound 20 (orange carbon-capped sticks) bound to WDR5 represented as a semitransparent electrostatic potential surface with labeled S2, S4, and S7 binding regions. (B) Overlay of 20 (orange sticks) and 1 (yellow sticks; PDB ID: 6UCS).35 (C) Key π–π stacking binding interactions (red dashed lines) of the 2-methyl imidazole P2 unit of 20 in the S2 subsite with denoted WDR5 residues. (D) Overlay of MLL1 peptide (magenta sticks, PDB ID: 3EG6),14 1 (green sticks, PDB ID: 6UCS),35 and 20 (yellow sticks) bound WDR5 protein structures in cartoon representation. The side chains of WDR5 residues F133, F149, and F263 for the structures are represented as sticks to demonstrate retention of binding poses.
Optimization of the P7 Pharmacophore Unit for 2-Methyl Imidazole WDR5 Inhibitors.
The next phase of optimization was for the P7 unit. X-ray structures of both 1 and 20 bound to WDR5 suggested that the solvent-exposed portion of the P7 unit can be modulated to optimize the physicochemical properties of the compound without a dramatic effect on the potency of the compound. To reduce the hydrophobicity of the P7 unit the 3,5-dimethoxyphenyl of 1 was replaced by the pyridyl moieties in 25–33 (Table 3) using the synthetic route depicted in Scheme 3. The P2 and P4 units of 20 were kept constant while the N-methyl or N-ethyl group at the R3-position were both investigated. Depending on the substitution of the P7 unit the cLogP values of 25–32 were reduced compared to 20 and supported our design of the new P7 unit. The unsubstituted 2-pyridylmethyl containing compound 25 exhibited significantly reduced binding affinity and cellular potency compared to 20 due to improper binding in the S7 subsite. We introduced a methyl group at various positions of the 2-pyridylmethyl group (26–28) to determine a positional preference of substitution for the S7 subsite. The 6-methyl group of 26 was beneficial to enhance both binding affinity and cellular potency in the WDR5 sensitive cell lines by 6- and 9-fold from 25, respectively. In addition, the 4-methyl substitution of 27 had an optimized interaction in the S7 subsite to regain all lost activity of 25 and showed comparable on-target potency to 20 with a reduced cLogP. However, the 3-methyl group of 28 was not tolerated in the subsite and led to a weaker potency than 25. The regioisomeric 3-methyl-4-pyridyl analog 29 showed that repositioning the nitrogen atom was unfavorable and the 4-methyl-2-pyridylmethyl group of 27 provided the best scaffold for the P7 pharmacophore. We then expanded the SAR of the 4-methyl group of 27 by introducing slightly larger groups. Both 4-methoxy 30 and 4-ethyl 31 were well tolerated and exhibited similar binding affinity and cellular potency compared to the parent 27. Compounds 32 and 33 containing the N-ethyl R3 group maintain a similar potency profile to 30 and 31, and these groups can be interchanged as needed to modulate the properties or PK profiles of future compounds. It is also noteworthy that all potent WDR5 inhibitors in the series (27, 30–34) exhibited enhanced cellular selectivity because each compound had significantly reduced antiproliferative activity in the WDR5 insensitive K562 cell line compared to the parent 20. In summary, the 2-pyridylmethyl group properly substituted at the 4-position is an effective P7 pharmacophore unit for both potency and druglike properties.
Table 3.
SAR Profile of the P7 Pyridylmethyl Series WDR5 Inhibitors
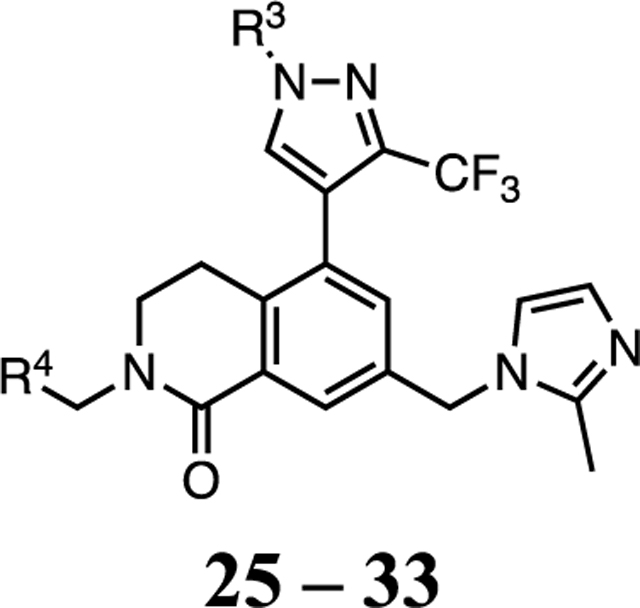
| ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| compnd | R3 = | R4 = | cLogP |
Ki (nM) TR-FRETa |
Cell Proliferation Assays GI50 (nM)a |
Selectivityb |
||
| WDR5 | MV4:11 | MOLM-13 | K562 | K562/MV4:11 | ||||
|
| ||||||||
| 25 | Me |
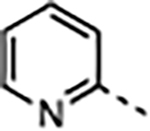
|
1.8 | 0.23 ± 0.03 | 1500 ± 160 | 3100 ± 200 | >30000 | >20 |
| 26 | Me |
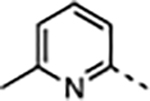
|
2.3 | 0.039 ± 0.005 | 170 ± 53 | 330 ± 120 | 13000 ± 2500 | 76 |
| 27 | Me |
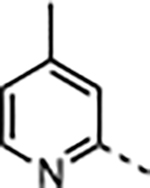
|
2.3 | 0.020 ± 0.004 | 23 ± 9.1 | 33 ± 12 | 3600 ± 310 | 160 |
| 28 | Me |
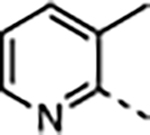
|
2.3 | 0.59 ± 0.19 | 3000 ± 840 | 4700 ± 790 | 22000 ± 6500 | 7 |
| 29 | Me |
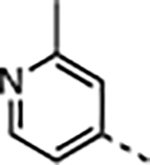
|
2.3 | 0.064 ± 0.009 | 270 ± 94 | 550 ± 200 | 13000 ± 7100 | 48 |
| 30 | Me |
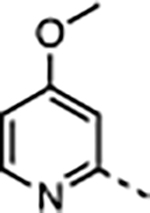
|
1.7 | <0.02 | 24 ± 8.6 | 39 ± 14 | 9500 ± 4800 | 400 |
| 31 | Me |
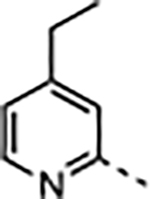
|
2.8 | <0.02 | 16 ± 7.3 | 25 ± 9.6 | 4100 ± 1300 | 260 |
| 32 | Et |
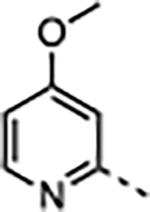
|
2.3 | <0.02 | 34 ± 18 | 59 ± 30 | 13000 ± 10000 | 380 |
| 33 | Et |
|
3.4 | <0.02 | 24 ± 8.2 | 43 ± 15 | 5600 ± 1100 | 230 |
TR-FRET Ki and cell proliferation GI50 values represent four independent replicate determinations ± standard deviation.
Selectivity is defined as GI50,K562/GI50,MV4:11 and is used to generally distinguish between on- and off-target inhibition mechanisms.
Scheme 3.

Synthesis of Pyridyl Series Compounds 25–33a
aR3 and R4 are defined in Table 3. Synthesis of 58 was reported in Scheme S1. Conditions: (a) R3-boronic acid, PdCl2(dppf)·CH2Cl2, K2CO3, 1,4-dioxane/H2O, 90 °C, overnight, 90%; (b) TFA, anisole, CH2Cl2, rt, overnight, 87%; (c) LiBHEt3 (1 M THF), THF, 0 °C, 1 h, quant.; (d) PBr3, CH2Cl2, 0 °C, quant.; (e) 2-methyl-1H-imidazole, MeCN, 50 °C, overnight, 70%; (f) R4-pyridylmethyl halide, NaH, DMF, 0 °C, 2 h, 25–71%.
Optimization of the α-Substituted 2-Pyridyl Series WDR5 Inhibitors to Improve Metabolic Stability.
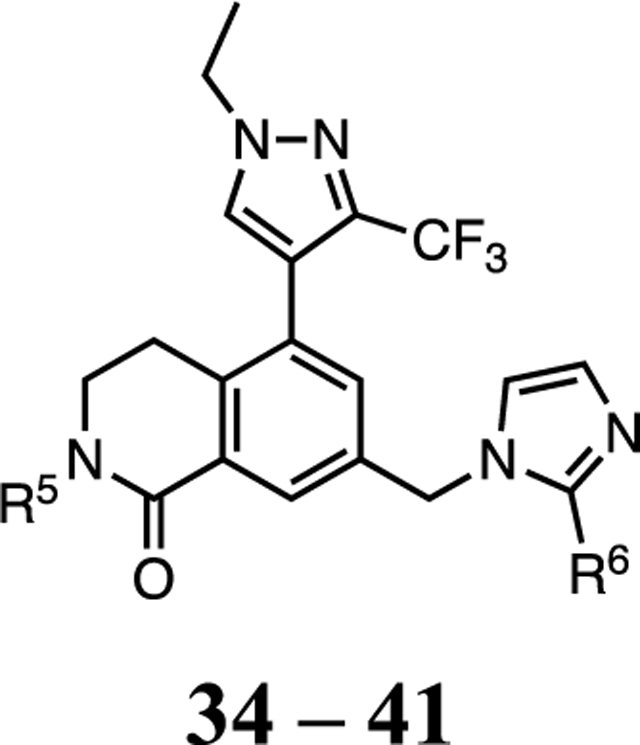
The final phase of optimization sought to take the WDR5 inhibitor with newly optimized P2, P4, and P7 pharmacophore units and identify what elements were necessary to produce a compound that exhibited an improved PK profile. The metabolic stability of 30 was tested in rat liver microsomes (RLM) and the predicted clearance was CLint = 220 μL/min/mg with a half-life of t1/2 = 3.2 min. The benzylic position of the P7 unit can be a potential metabolic soft spot. One possibility to improve the metabolic stability of the compound was to develop a series of inhibitors with α-substituted P7 units. From the X-ray crystal structure of 20, we predicted that substitution of the solvent-exposed benzylic position with small alkyl groups would be possible, and the (S)-enantiomer would maintain the optimal binding pose of 20. To assess the effect of the size of the benzylic substitution, methyl and cyclopropyl groups were chosen, and all designed compounds showed acceptable cLogP values. Optically pure α-substituted pyridin-2-ylmethanamine P7 units were prepared, and representative syntheses of 34–41 (Table 4) were shown in Scheme 4. The X-ray co-crystal structure of 37 determined its absolute configuration when bound to WDR5 (Figure 6). Compound 34 was observed to have a 3-fold improvement in cellular activity in the WDR5 sensitive cell lines compared to 32. The metabolic stability of 34 was also tested in RLM and the predicted clearance was CLint = 60 μL/min/mg with a half-life of t1/2 = 12 min, nearly a 4-fold improvement in both parameters. As the X-ray structure predicted, the (R)-methyl enantiomer 35 showed a 5-fold weaker cellular activity compared to 34. Compounds 36 and 37 featuring the (S)-cyclopropyl group were also tolerated and exhibited similar in vitro potency compared to 34. The imidazole P2 unit replaced the 2-methyl imidazole in 38 and maintained a similar cellular activity with a 2-fold enhanced metabolic stability in RLM (CLint = 31 μL/min/mg with a half-life of t1/2 = 22 min) compared to 34. This may suggest that unsubstituted imidazole can be a preferred P2 unit to deliver robust in vivo efficacy. Unsubstituted imidazole series compounds 38–41 also exhibited a parallel SAR pattern to corresponding 2-methyl-imidazole analogues 34–37, suggesting same binding poses for both series. Finally, all (S)-enantiomers in Table 4 showed a 2- to 3-fold increase in kinetic solubility due to all modifications made throughout our study compared to the initial probe 1. Consequently, future in vivo studies by oral dosing are more feasible with the improved aqueous solubility for these compounds.
Table 4.
SAR Profile of the α-Substituted 2-Pyridyl Series WDR5 Inhibitors
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| compnd | R5 = | R6 = | cLogP | Kinetic Solubility (μM) |
Ki (nM) TR-FRETa |
Cell Proliferation Assays GI50 (nM)a |
Selectivityb |
||
| WDR5 | MV4:11 | MOLM-13 | K562 | K562/MV4:11 | |||||
|
| |||||||||
| 34 |
|
Me | 2.6 | 80 | <0.02 | 11 ± 6.1 | 26 ± 8.8 | 2200 ± 2300 | 200 |
| 35 |
|
Me | 2.6 | n.d. | 0.029 ± 0.002 | 39 ± 9.0 | 110 ± 23 | 7800 ± 2700 | 200 |
| 36 |
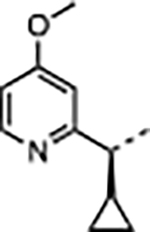
|
Me | 3.0 | 65 | <0.02 | 15 ± 4.9 | 36 ± 11 | 2600 ± 1500 | 170 |
| 37 |
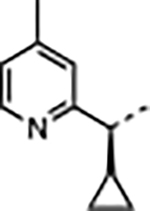
|
Me | 3.6 | 66 | <0.02 | 17 ± 6.6 | 43 ± 9.0 | 2500 ± 2100 | 150 |
| 38 |
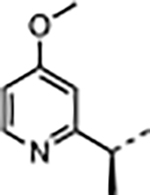
|
H | 2.3 | 86 | <0.02 | 9.7 ± 5.6 | 19 ± 6.2 | 1900 ± 1800 | 200 |
| 39 |
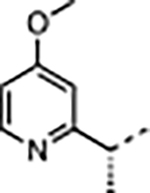
|
H | 2.3 | n.d. | 0.028 ± 0.002 | 39 ± 7.4 | 100 ± 23 | 12000 ± 3600 | 310 |
| 40 |
|
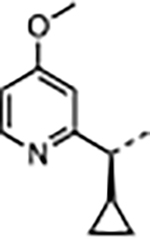
H | 2.7 | 55 | <0.02 | 19 ± 5.2 | 37 ± 8.1 | 1500 ± 860 | 79 |
| 41 |
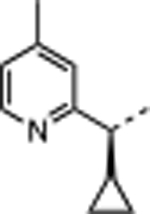
|
H | 3.3 | 60 | <0.02 | 13 ± 5.6 | 27 ± 4.5 | 3700 ± 3100 | 290 |
TR-FRET Ki, and cell proliferation GI50 values represent four independent replicate determinations ± standard deviation.
Selectivity is defined as GI50,K562/GI50,MV4:11 and is used to generally distinguish between on- and off-target inhibition mechanisms. Not determined = n.d.
Scheme 4.

Synthesis of Compounds 34, 35, 38, and 39a
aR6 is defined in Table 4. Conditions: (a) (S)- or (R)-2-methylpropane-2-sulfinamide, Cs2CO3, CH2Cl2, rt, overnight, 65%; (b) MeMgBr (3.4 M 2-MeTHF), THF, −78 °C, 84–92%; (c) HCl (4 M 1,4-dioxane), THF, 94%—quant.; (d) chiral amine, i-Pr2NEt, NaBH(OAc)3, CH2Cl2, rt, overnight, then 1,4-dioxane, 110 °C, 84%; (e) phenyl triflimide, i-Pr2NEt, THF/CH2Cl2, rt, overnight, 93%; (f) (1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)boronic acid, Pd(PPh3)4, Na2CO3, 1,4-dioxane/H2O, 80 °C, 96%; (g) LiBH4 (2 M THF) or LiBHEt3 (1 M THF), THF, 0 °C, 1 h, 72–85%; (h) CH3SO2Cl, i-Pr2NEt, CH2Cl2, 0 °C, then LiBr, THF, reflux, 53% or PBr3, CH2Cl2, 0 °C, 98%; (i) R6-imidazole, MeCN, 50 °C, overnight, 73%.
Figure 6.

X-ray co-crystal structure of 37 bound to WDR5 (PDB ID: 7UAS). (A) Compound 37 (white carbon-capped sticks) bound to WDR5 represented as semitransparent electrostatic potential surface with labeled S2, S4, and S7 binding regions. (B) Binding interactions of the (S)-1-cyclopropyl-1-(4-methylpyridine-2-yl)methyl P7 unit in the S7 subsite, pro(R)-H represented as a light orange ball. (C) Overlay of 20 (orange sticks) and 37 (white sticks).
To understand the binding mode of the α-substituted 2-pyridylmethyl P7 unit, the X-ray co-crystal structure of compound 37 bound to WDR5 was obtained. Compound 37 bound to WDR5 using the same key binding interactions as 20 (Figure 6A). The (S)-1-cyclopropyl-1-(4-methylpyridine-2-yl)methyl P7 unit bound in the S7 subsite as predicted (Figure 6B). The (S)-cyclopropyl group pointed out of the binding interface. Conversely, the pro(R)-hydrogen pointed toward the protein surface; therefore, compounds containing the opposite enantiomer, such as 35 and 39, would be forced to adopt a different binding conformation for the P7 moiety to avoid a steric clash, which caused a loss of binding affinity for these compounds. The overlaid WDR5 bound confirmations of 20 and 37 in Figure 6C are nearly superimposable for all pharmacophore units including the new P4 and P7 units.
Pharmacokinetic Profile of WDR5 Inhibitors.
PK properties of representative new WDR5 inhibitors were profiled in CD-1 mice in preparation for studying the effect of WDR5 inhibition in an in vivo setting. The compounds were dosed in CD-1 mice at 3 mg/kg iv and 10 mg/kg po and the plasma concentrations of each compound were monitored for 24 h. To obtain a practical oral therapeutic agent, a compound would need to have sufficient oral exposure to reach a sustained pharmacologically relevant target occupancy during a dosing period. We focused on the structural modifications that improved the suboptimal physicochemical properties and poor PK profile (high iv clearance and no oral bioavailability) of compound 1 while maintaining its high on-target potency. The passive permeability of the series was greatly improved by replacing the troublesome P2 imidazole-imine with an imidazole or 2-methyimidazole moiety. Optimization of P4 and P7 units also resulted in improved kinetic aqueous solubility and in vitro RLM stability. Consequently, we were gratified to observe oral bioavailability from all tested compounds, and 41 showed the highest oral exposure (AUC0,inf) of the compounds (Table 5). In general, lower iv clearance was accompanied by an increase in oral exposure and may be due to lower first-pass metabolism. Compounds containing the (S)-cyclopropyl at the benzylic position of the P7 unit (36, 37, 40, 41) showed significantly reduced iv clearance compared to the corresponding (S)-methyl analogs (34 and 38). In addition, WDR5 inhibitors containing the unsubstituted imidazole P2 unit (38, 40, 41) were superior oral agents to the corresponding 2-methyl-imidazole analogs (34, 36, 37) by achieving 4 to 7-fold higher oral exposures at the same dose level. Finally, all tested compounds were well tolerated in mice by both iv and po dosing and showed no clinical sign of abnormalities suggesting an enhanced safety profile for the series.
Table 5.
In Vivo PK Profile in CD-1 Micea
| 3 mg/kg iv |
10 mg/kg po |
||||||
|---|---|---|---|---|---|---|---|
| compnd | CL (mL/min/kg) | AUC0,inf (h·ng/mL) | Vss (L/kg) | Cmax (ng/mL) | AUC0,inf (h·ng/mL) | t1/2 (h) | F% |
| 1 b | 78 | 642 | 16 | n.c. | n.c. | n.c. | n.c. |
| 34 | 118 | 429 | 3.8 | 279 | 397 | 1.2 | 28 |
| 36 | 41 | 1240 | 1.6 | 652 | 508 | 1.4 | 12 |
| 37 | 25 | 2075 | 1.1 | 1083 | 1000 | 1.2 | 15 |
| 38 | 76 | 707 | 2.3 | 2013 | 1712 | 1.2 | 73 |
| 40 | 20 | 2570 | 0.7 | 2767 | 3626 | 1.2 | 42 |
| 41 | 26 | 1951 | 1.6 | 2083 | 3984 | 1.3 | 61 |
Pharmacokinetic parameter values represent the average of three animals.
The iv PK parameter values are for one surviving animal due to the lethality of 1. The oral exposure of 1 was below the limit of quantitation when administered orally at 3 mg/kg. Not calculated = n.c.
Chemical Synthesis.
Synthetic routes were designed around two objectives, each route would start from the same common intermediate 42, and, where feasible, the route would be arranged to install the pharmacophore of interest in the final synthetic step. The preparation of compounds 2–15 (Scheme 1) began from hemiacetal 42, which was prepared in three steps from a previously reported protocol.45 Compound 42 underwent reductive amination with 3,5-dimethoxybenzyl-amine to form the bicyclic dihydroisoquinolinone 43. Triflate 44 was synthesized using phenyl triflimide. The triflate was replaced by a bromide in two synthetic steps to allow the Suzuki–Miyaura cross-coupling with R1-boronic acids or R1-pinacol boronate esters to take place in the final synthetic step. Boronate pinacol ester 45 was prepared through Miyaura borylation of triflate 44 and was converted to bromide 46 using copper(II) bromide. The methyl ester of 46 was reduced to benzyl alcohol 47, which was brominated with phosphorus tribromide to yield alkyl bromide 48. Nucleophilic substitution of 48 with 1-methyl-1H-imidazol-2-amine hydrochloride gave compound 49 in excellent yield (>99%). Compound 49 underwent Suzuki–Miyaura cross-coupling with R1-boronic acids or R1-pinacol boronate esters to yield 2–15.
Based on the P4 SAR results, 1-methyl-3-(trifluoromethyl)-1H-pyrazol was chosen as the replacement P4 pharmacophore. The P2 pharmacophore was optimized by investigating a series of nonbasic azole compounds 16–24 (Scheme 2). To reduce the number of synthetic steps triflate 44 underwent Suzuki–Miyaura cross-coupling with (1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)boronic acid to yield 50. The methyl ester of 50 was reduced to benzyl alcohol 51 and was brominated to yield 52. Compound 52 underwent nucleophilic substitution with R2-azoles to yield compounds 16–24.
To optimize the P7 pharmacophore, lactam intermediates 57 and 58 were alkylated with R4-pyridylmethyl halides in the final synthetic step to produce compounds 25–33 (Scheme 3). Triflate 53 was synthesized through a previously reported synthetic route35 and underwent Suzuki–Miyaura cross-coupling with a pyrazole boronic acid to yield 54. The 2,4-dimethoxybenzyl group was removed under acidic conditions to yield lactam 55. The methyl ester of 55 was reduced and then brominated to yield 56. Nucleophilic substitution of 56 with 2-methyl-1H-imidazole produced lactam 57. Lactam 58 was synthesized using similar synthetic steps and is reported in the Supporting Information (Scheme S1). Compounds 57 and 58 were alkylated with R4-pyridylmethyl halides to yield 25–33.
Chiral α-substituted 34, 35, 38, and 39 (Scheme 4) were synthesized through the installation of chiral α-methyl benzylic amines 61 and 64 in the initial synthetic step. Enantiopure benzylic amines were synthesized in three steps beginning with the condensation of 4-methoxypicolinaldehyde with the appropriate Ellman’s chiral auxiliary, (S)- or (R)- 2-methylpropane-2-sulfinamide, to yield sulfinimines 59 and 62, respectively. The sulfinimines underwent diastereoselective addition with methyl magnesium bromide to yield sulfinamides 60 and 63, which were purified by normal-phase chromatography. Chiral α-methyl benzylic amines 61 and 64 were obtained as hydrochloride salts following deprotection of the chiral auxiliary under acidic conditions. Hemiacetal 42 underwent reductive amination with chiral α-methyl benzylic amines 61 and 64 to yield 65 and 66, respectively. Triflates 67 and 68 were synthesized using phenyl triflimide, and underwent Suzuki–Miyaura cross-coupling with (1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)boronic acid to yield 69 and 70. The methyl esters of 69 and 70 were reduced to benzyl alcohols 71 and 72 and were brominated to yield 73 and 74. Compounds 73 and 74 underwent nucleophilic substitution with 2-methyl-1H-imidazole or 1H-imidazole to yield compounds 34, 35, 38, and 39. Compounds 36, 37, 40, and 41 were synthesized by the same synthetic route and experimental details are reported in the Supporting Information (Scheme S2).
CONCLUSIONS
We describe the pharmacophore-based optimization of WDR5 inhibitors containing the 3,4-dihydroisoquinolin-1(2H)-one core. Compound 1 was previously discovered to have robust in vitro on-target potency against WDR5 sensitive cell lines; however, suboptimal physicochemical properties of specific pharmacophore units prevented the probe from having a desirable oral PK profile that would be expected to produce antitumor efficacy in an in vivo setting. To address the limitations of 1, we used a pharmacophore-based convergent SAR and X-ray structure-guided modification strategy to discover a new class of WDR5 inhibitors with enhanced potency and physicochemical properties. We focused our SAR effort on individual P2, P4, and P7 pharmacophore units and sequentially optimized each unit. The P2 imidazole-imine was found to be the main contributor to the poor cellular permeability of 1 due to its pH-dependent profile. The P2 imidazole-imine was replaced by the N-linked imidazole series. The X-ray structure of 20 confirmed that the 2-methyl imidazole P2 unit maintained the critical binding interactions in the S2 subsite that were originally observed with 1, which were critical to preserving the original binding mode to the WDR5 WIN site. Although compound 30 with optimized P2, P4, and P7 pharmacophores exhibited enhanced on-target potency, and improved physicochemical properties compared to the initial probe 1, its poor RLM metabolic stability became a concern for further development. We recognized the benzylic position of the P7 unit to be a metabolic soft spot and developed a series of α-substituted P7 units to resolve the issue. This strategy turned out to be effective as the (S)-methyl enantiomer of 34 showed increased RLM metabolic stability and oral bioavailability in mice. However, additional optimization was needed to reduce the high iv clearance of 34, which was unable to achieve a desired high oral exposure due to rapid first-pass metabolism. Finally, the (S)-cyclopropyl P7 and unsubstituted imidazole P2 in 40 and 41 significantly improved the PK profile including iv clearance and oral exposure and maintained potent in vitro on-target potency.
In summary, we have detailed the successful optimization of WDR5 inhibitors using a structure-guided and pharmacophore-based optimization strategy. The new in vivo leads 40 and 41 display potent in vitro potency and suitable PK profiles following oral dosing. Future studies will be aimed at optimizing potency and PK properties of a new lead series to demonstrate WDR5 inhibition-mediated in vivo efficacy in mouse xenograft tumor models by oral dosing and safety for the mechanism of action.
EXPERIMENTAL SECTION
General Chemistry.
All chemical reagents and reaction solvents were purchased from commercial suppliers and used as received. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker AVIII-HD 400 MHz spectrometer and processed using Bruker TopSpin 3.6.1. For 1H NMR spectra, chemical shifts are reported in parts per million (ppm) and are reported relative to residual nondeuterated solvent signals. Coupling constants are reported in hertz (Hz). The following abbreviations (or a combination, thereof) are used to describe splitting patterns: s, singlet; d, doublet; t, triplet; q, quartet; p, pentet; m, multiplet; br, broad. All final compounds were of >95% purity as measured by analytical reversed-phase HPLC. Analytical HPLC was performed on an Agilent 1200 series system with UV detection at 214 and 254 nm, along with evaporative light scattering detection (ELSD). Low-resolution mass spectra were obtained on an Agilent 6140 mass spectrometer with electrospray ionization (ESI). LCMS experiments were performed with the following parameters. Method 1: Phenomenex Kinetex 2.6 μm XB-C18 100 Å LC column (30 mm × 2.1 mm); 1.1 min gradient, 7–95% MeCN in H2O, and 0.1% TFA. Method 2: Phenomenex Kinetex 2.6 μm XB-C18 100 Å LC column (50 mm × 2.1 mm); 2 min gradient, 5–95% MeCN in H2O, and 0.1% TFA. Analytical thin-layer chromatography (TLC) was performed on Kieselgel 60 F254 glass plates precoated with a 0.25 mm thickness of silica gel. TLC plates were visualized with UV light and iodine. Silica gel chromatography was performed using a Teledyne ISCO Combiflash Rf system, eluting with varying concentrations of EtOAc in hexanes or MeOH in CH2Cl2. Preparative reversed-phase HPLC was performed on a Gilson HPLC equipped with a Phenomenex Kinetex C18 column, using varying concentrations of MeCN in H2O, and 0.1% TFA. Solvents for reactions, extraction, and washing were of ACS grade, and solvents for chromatography were of HPLC grade. Compounds 1, 42, and 53 were prepared according to previously reported protocols.35,45
General Procedure A: Reductive Amination.
Dimethyl 2-hydroxy-2,3-dihydrobenzofuran-4,6-dicarboxylate 42 (1 equiv) and an amine (1.5 equiv) were dissolved in CH2Cl2 and stirred at 30 °C for 30 min. Then sodium triacetoxyborohydride (2 equiv) was added and the reaction was stirred at 30 °C for 3 h. The reaction mixture was concentrated and dissolved in 1,4-dioxane and heated at 110 °C overnight. Saturated aqueous NaHCO3 was added, and the mixture was extracted with CH2Cl2. The combined organic phases were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The compound was used without further purification.
General Procedure B: Triflation of Phenol.
Phenyl triflimide (1.2 equiv) was added to a solution of phenol (1 equiv) and an amine (2.5 equiv) in THF/CH2Cl2 (5:1) at 23 °C and stirred for 14 h. Saturated aqueous NaHCO3 was added, and the mixture was extracted with CH2Cl2. The combined organic phases were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by flash chromatography (Combi-flash Rf, EtOAc/hexanes = 0–50% gradient).
General Procedure C: Reduction of Methyl Ester.
A solution of lithium triethylborohydride (1 M THF, 3 equiv) or lithium borohydride (2 M THF) was added dropwise to a solution of methyl ester (1 equiv) in THF at 0 °C. The reaction was stirred for 40 min, then quenched with saturated aqueous NaHCO3. The mixture was extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash chromatography (Combi-flash Rf, EtOAc/hexanes = 0–100% gradient).
General Procedure D: Bromination of Benzyl Alcohol.
PBr3 (2 equiv) was added to a solution of benzyl alcohol (1 equiv) in CH2Cl2 at 0 °C. The reaction was stirred and slowly warmed to room temperature. Saturated aqueous NaHCO3 was added, and the mixture was extracted with CH2Cl2. The combined organic layers were dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash chromatography (Combi-flash Rf, EtOAc/hexanes = 0–30% gradient).
General Procedure E: Suzuki–Miyaura Cross-Coupling.
The representative aryl halide or aryl triflate (1 equiv), aryl boronic acid or pinacol ester (2 equiv), K2CO3 (2 equiv), and Pd catalyst (0.05 equiv) were dissolved in 1,4-dioxane/H2O (4:1). The reaction mixture was placed under an argon atmosphere heated to 80–90 °C for 2–14 h and then cooled to room temperature. The reaction mixture was diluted with water and extracted with CH2Cl2. The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Intermediate compounds were purified by flash chromatography (Combi-flash Rf, EtOAc/hexanes = 0–100% gradient). Final compounds were purified by preparative reversed-phase HPLC (Phenomenex Gemini C18, MeCN/H2O = 15–70%, 0.1% TFA) followed by neutralization with saturated aqueous NaHCO3.
General Procedure F: Displacement of Benzyl Bromide with Amine.
R2-azole (3–4 equiv) was added to a solution of benzyl bromide (1 equiv) in acetonitrile at 23 °C. The reaction mixture was stirred for 12 h at 50 °C, then cooled to ambient temperature, filtered, and concentrated. Intermediate compounds were purified by flash chromatography (Combi-flash Rf, MeOH/CH2Cl2 = 0–10% gradient). Final compounds were purified by preparative reversed-phase HPLC (Phenomenex Gemini C18, MeCN/H2O = 15–70% gradient, 0.1% TFA) followed by neutralization with saturated aqueous NaHCO3.
General Procedure G: Lactam Alkylation.
Lactam (1 equiv) was dissolved in DMF and cooled to 0 °C. Sodium hydride (1.2 equiv) was added, and the reaction was stirred for 15 min. R4-halide (1.2 equiv) was added. The reaction was stirred and allowed to warm to room temperature and then water was added, and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by reversed-phase HPLC (Phenomenex Gemini C18, MeCN/H2O = 15–70% gradient, 0.1% TFA) followed by neutralization with saturated aqueous NaHCO3.
2-(3,5-Dimethoxybenzyl)-5-(4-fluoro-2-methoxyphenyl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (2).
General Procedure E was followed using 49 (50 mg, 0.1 mmol, 1 equiv), (4-fluoro-2-methoxyphenyl)-boronic acid (35 mg, 0.21 mmol, 2 equiv), potassium carbonate (36 mg, 0.26 mmol, 2.5 equiv), and PdCl2(dppf)·CH2Cl2 (4.2 mg, 0.005 mmol, 0.05 equiv) to obtain the title compound (34 mg, 0.064 mmol, 62% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.87 (d, J = 1.3 Hz, 1H), 7.31 (d, J = 1.7 Hz, 1H), 7.17 (dd, J = 8.3, 7.0 Hz, 1H), 7.02 (dd, J = 11.4, 2.4 Hz, 1H), 6.86 (td, J = 8.4, 2.4 Hz, 1H), 6.65 (s, 1H), 6.54 (s, 1H), 6.43 (d, J = 2.2 Hz, 2H), 6.40 (t, J = 2.2 Hz, 1H), 4.88 (s, 2H), 4.62 (s, 2H), 3.72 (s, 3H), 3.71 (s, 6H), 3.37–3.33 (m, 2H), 3.17 (s, 3H), 2.72–2.64 (m, 1H), 2.54–2.51 (m, 1H); LCMS (method 2, ESI): >95%, Rt = 1.647 min, m/z = 531.1 [M + H]+.
2-(3,5-Dimethoxybenzyl)-5-(4-fluoro-2-hydroxyphenyl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (3).
General Procedure E was followed using 49 (50 mg, 0.1 mmol, 1 equiv), (4-fluoro-2-hydroxyphenyl)-boronic acid (32 mg, 0.21 mmol, 2 equiv), potassium carbonate (36 mg, 0.26 mmol, 2.5 equiv), and PdCl2(dppf)·CH2Cl2 (4.2 mg, 0.005 mmol, 0.05 equiv) to obtain the title compound (10.4 mg, 0.020 mmol, 20% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.87 (d, J = 1.2 Hz, 1H), 7.40 (s, 1H), 7.07 (d, J = 2.4 Hz, 1H), 6.98 (t, J = 7.9 Hz, 1H), 6.92 (d, J = 2.4 Hz, 1H), 6.60 (d, J = 11.7 Hz, 1H), 6.44–6.38 (m, 5H), 5.11 (s, 2H), 4.61 (s, 2H), 3.69 (s, 6H), 3.42 (s, 3H), 3.34 (t, J = 6.5 Hz, 2H), 2.85–2.65 (brs, 2H); LCMS (method 2, ESI): >95%, Rt = 1.576 min, m/z = 517.0 [M + H]+.
2-(3,5-Dimethoxybenzyl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-5-(2-methylpyridin-3-yl)-3,4-dihydroisoquinolin-1(2H)-one (4).
General Procedure E was followed using 49 (50 mg, 0.1 mmol, 1 equiv), (2-methylpyridin-3-yl)boronic acid (28 mg, 0.21 mmol, 2 equiv), potassium carbonate (36 mg, 0.26 mmol, 2.5 equiv), and PdCl2(dppf)·CH2Cl2 (4.2 mg, 0.005 mmol, 0.05 equiv) to obtain the title compound (8.8 mg, 0.018 mmol, 17% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.49 (dd, J = 4.8, 1.7 Hz, 1H), 7.94 (d, J = 1.7 Hz, 1H), 7.55 (dd, J = 7.6, 1.7 Hz, 1H), 7.35 (d, J = 1.8 Hz, 1H), 7.29 (dd, J = 7.6, 4.9 Hz, 1H), 6.61 (d, J = 2.6 Hz, 1H), 6.50 (d, J = 2.6 Hz, 1H), 6.44 (d, J = 2.2 Hz, 2H), 6.40 (t, J = 2.2 Hz, 1H), 4.90 (s, 2H), 4.62 (dd, J = 40.8, 14.9 Hz, 2H), 3.70 (s, 6H), 3.39 (t, J = 6.6 Hz, 2H), 3.16 (s, 3H), 2.67–2.59 (m, 1H), 2.49–2.44 (m, 1H), 2.22 (s, 3H); LCMS (method 2, ESI): >95%, Rt = 1.252 min, m/z = 498.1 [M + H]+.
2-(3,5-Dimethoxybenzyl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-5-(2-methoxypyridin-3-yl)-3,4-dihydroisoquinolin-1(2H)-one (5).
General Procedure E was followed using 49 (50 mg, 0.1 mmol, 1 equiv), 2-methoxypyridine-3-boronic acid hydrate (35 mg, 0.21 mmol, 2 equiv), potassium carbonate (36 mg, 0.26 mmol, 2.5 equiv), and PdCl2(dppf)·CH2Cl2 (4.2 mg, 0.005 mmol, 0.05 equiv) to obtain the title compound (36.8 mg, 0.072 mmol, 70% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.23 (dd, J = 5.0, 1.9 Hz, 1H), 7.91 (s, 1H), 7.62 (dd, J = 7.2, 1.9 Hz, 1H), 7.38 (s, 1H), 7.11 (dd, J = 7.2, 5.0 Hz, 1H), 6.67 (s, 1H), 6.56 (s, 1H), 6.43 (d, J = 2.2 Hz, 2H), 6.40 (t, J = 2.2 Hz, 1H), 4.89 (s, 2H), 4.63 (s, 2H), 3.81 (s, 3H), 3.71 (s, 6H), 3.37 (t, J = 6.5 Hz, 2H), 3.18 (s, 3H), 2.75–2.65 (m, 2H); LCMS (method 2, ESI): >95%, Rt = 1.507 min, m/z = 514.1 [M + H]+.
2-(3,5-Dimethoxybenzyl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-5-(2-(trifluoromethyl)pyridin-3-yl)-3,4-dihydroisoquinolin-1(2H)-one (6).
General Procedure E was followed using 49 (50 mg, 0.1 mmol, 1 equiv), (2-(trifluoromethyl)pyridin-3-yl)boronic acid (39 mg, 0.21 mmol, 2 equiv), potassium carbonate (36 mg, 0.26 mmol, 2.5 equiv), and PdCl2(dppf)·CH2Cl2 (4.2 mg, 0.005 mmol, 0.05 equiv) to obtain the title compound (8.5 mg, 0.015 mmol, 15% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.82 (dd, J = 4.7, 1.3 Hz, 1H), 8.07 (d, J = 1.9 Hz, 1H), 7.96 (s, 1H), 7.94 (d, J = 1.1 Hz, 1H), 7.82 (dd, J = 7.8, 4.7 Hz, 1H), 7.41 (d, J = 1.5 Hz, 1H), 7.13 (d, J = 2.5 Hz, 1H), 7.06 (d, J = 2.5 Hz, 1H), 6.43 (d, J = 2.2 Hz, 2H), 6.41 (t, J = 2.2 Hz, 1H), 5.19 (dd, J = 20.2, 15.8 Hz, 2H), 4.63 (dd, J = 73.8, 14.9 Hz, 2H), 3.71 (s, 6H), 3.45 (s, 3H), 3.44–3.40 (m, 1H), 3.38–3.31 (m, 1H), 2.70–2.63 (m, 1H), 2.44–2.37 (m, 1H); LCMS (method 2, ESI): >95%, Rt = 1.543 min, m/z = 552.0 [M + H]+.
2-(3,5-Dimethoxybenzyl)-5-(2,5-dimethylpyridin-3-yl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (7).
General Procedure E was followed using 49 (30 mg, 0.062 mmol, 1 equiv), (2,5-dimethylpyridin-3-yl)boronic acid (19 mg, 0.12 mmol, 2 equiv), potassium carbonate (21 mg, 0.15 mmol, 2.5 equiv), and PdCl2(dppf)·CH2Cl2 (2.5 mg, 0.003 mmol, 0.05 equiv) to obtain the title compound (14.2 mg, 0.028 mmol, 45% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.32 (s, 1H), 7.92 (s, 1H), 7.37 (s, 1H), 7.32 (s, 1H), 6.60 (d, J = 2.2 Hz, 1H), 6.50 (d, J = 2.2 Hz, 1H), 6.44 (d, J = 1.9 Hz, 2H), 6.40 (t, J = 2.0 Hz, 1H), 4.87 (s, 2H), 4.62 (dd, J = 45.4, 14.9 Hz, 2H), 3.71 (s, 6H), 3.39 (t, J = 6.6 Hz, 2H), 3.15 (s, 3H), 2.67–2.59 (m, 1H), 2.54–2.46 (m, 1H), 2.28 (s, 3H), 2.16 (s, 3H); LCMS (method 2, ESI): >95%, Rt = 1.271 min, m/z = 512.1 [M + H]+.
2-(3,5-Dimethoxybenzyl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-5-(3-methylpyridin-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (8).
General Procedure E was followed using 49 (50 mg, 0.10 mmol, 1 equiv), (3-methylpyridin-4-yl)boronic acid (28 mg, 0.21 mmol, 2 equiv), potassium carbonate (36 mg, 0.26 mmol, 2.5 equiv), and PdCl2(dppf)·CH2Cl2 (4.2 mg, 0.005 mmol, 0.05 equiv) to obtain the title compound (25 mg, 0.05 mmol, 49% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.54 (s, 1H), 8.44 (d, J = 4.9 Hz, 1H), 7.94 (d, J = 1.8 Hz, 1H), 7.31 (d, J = 1.7 Hz, 1H), 7.17 (d, J = 4.9 Hz, 1H), 6.58 (d, J = 2.2 Hz, 1H), 6.47 (d, J = 2.4 Hz, 1H), 6.44 (d, J = 2.2 Hz, 2H), 6.40 (t, J = 2.2 Hz, 1H), 4.86 (s, 2H), 4.62 (dd, J = 34.2, 14.9 Hz, 2H), 3.70 (s, 6H), 3.39 (t, J = 6.6 Hz, 2H), 3.13 (s, 3H), 2.70–2.61 (m, 1H), 2.48–2.41 (m, 1H), 2.03 (s, 3H); LCMS (method 2, ESI): >95%, Rt = 1.224 min, m/z = 498.1 [M + H]+.
2-(3,5-Dimethoxybenzyl)-5-(2,5-dimethylpyridin-4-yl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (9).
General Procedure E was followed using 49 (30 mg, 0.062 mmol, 1 equiv), (2,5-dimethylpyridin-4-yl)boronic acid (19 mg, 0.12 mmol, 2 equiv), potassium carbonate (21 mg, 0.15 mmol, 2.5 equiv), and PdCl2(dppf)·CH2Cl2 (2.5 mg, 0.003 mmol, 0.05 equiv) to obtain the title compound (12 mg, 0.023 mmol, 38% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.34 (s, 1H), 7.93 (s, 1H), 7.28 (s, 1H), 7.03 (s, 1H), 6.57 (s, 1H), 6.47 (s, 1H), 6.44 (s, 2H), 6.40 (s, 1H), 4.85 (s, 2H), 4.62 (dd, J = 38.8, 15.2 Hz, 2H), 3.71 (s, 6H), 3.39 (t, J = 6.6 Hz, 2H), 3.13 (s, 3H), 2.67–2.61 (m, 1H), 2.48–2.55 (m, 1H), 2.44 (s, 3H), 1.98 (s, 3H); LCMS (method 2, ESI): >95%, Rt = 1.274 min, m/z = 512.1 [M + H]+.
2-(3,5-Dimethoxybenzyl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-5-(3-methylisoxazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (10).
General Procedure E was followed using 49 (30 mg, 0.062 mmol, 1 equiv), (3-methylisoxazol-4-yl)boronic acid (16 mg, 0.12 mmol, 2 equiv), potassium carbonate (21 mg, 0.15 mmol, 2.5 equiv), and PdCl2(dppf)·CH2Cl2 (2.5 mg, 0.003 mmol, 0.05 equiv) to obtain the title compound (13.3 mg, 0.027 mmol, 44% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.98 (s, 1H), 7.94 (s, 1H), 7.47 (s, 1H), 6.67 (s, 1H), 6.56 (s, 1H), 6.44 (s, 2H), 6.41 (s, 1H), 4.91 (s, 2H), 4.63 (s, 2H), 3.71 (s, 6H), 3.41 (t, J = 6.5 Hz, 2H), 3.18 (s, 3H), 2.81 (t, J = 6.4 Hz, 2H), 2.19 (s, 3H); LCMS (method 2, ESI): >95%, Rt = 1.472 min, m/z = 488.1 [M + H]+.
2-(3,5-Dimethoxybenzyl)-5-(1,3-dimethyl-1H-pyrazol-4-yl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (11).
General Procedure E was followed using 49 (50 mg, 0.10 mmol, 1 equiv), 1,3-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (46 mg, 0.21 mmol, 2 equiv), potassium carbonate (36 mg, 0.26 mmol, 2.5 equiv), and PdCl2(dppf)·CH2Cl2 (4.2 mg, 0.005 mmol, 0.05 equiv) to obtain the title compound (31.1 mg, 0.062 mmol, 60% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.86 (d, J = 1.7 Hz, 1H), 7.71 (s, 1H), 7.36 (d, J = 1.7 Hz, 1H), 6.85 (s, 1H), 6.74 (s, 1H), 6.44 (d, J = 2.2 Hz, 2H), 6.41 (t, J = 2.2 Hz, 1H), 5.01 (s, 2H), 4.62 (s, 2H), 3.78 (s, 3H), 3.71 (s, 6H), 3.38 (t, J = 6.5 Hz, 2H), 3.30 (s, 3H), 2.81 (t, J = 6.2 Hz, 2H), 2.07 (s, 3H); LCMS (method 2, ESI): >95%, Rt = 1.433 min, m/z = 501.1 [M + H]+.
2-(3,5-Dimethoxybenzyl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-5-(1,3,5-trimethyl-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (12).
General Procedure E was followed using 49 (50 mg, 0.10 mmol, 1 equiv), (1,3,5-trimethyl-1H-pyrazol-4-yl)boronic acid (32 mg, 0.21 mmol, 2 equiv), potassium carbonate (36 mg, 0.26 mmol, 2.5 equiv), and PdCl2(dppf) CH2Cl2 (4.2 mg, 0.005 mmol, 0.05 equiv) to obtain the title compound (7 mg, 0.01 mmol, 10% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.95 (s, 1H), 7.92 (d, J = 1.9 Hz, 1H), 7.26 (d, J = 2.0 Hz, 1H), 7.19 (d, J = 2.5 Hz, 1H), 7.07 (d, J = 2.5 Hz, 1H), 6.44 (d, J = 2.2 Hz, 2H), 6.41 (t, J = 2.2 Hz, 1H), 5.17 (s, 2H), 4.63 (s, 2H), 3.71 (s, 6H), 3.69 (s, 3H), 3.46 (s, 3H), 3.39 (t, J = 6.5 Hz, 2H), 2.65 (t, J = 6.5 Hz, 2H), 2.00 (s, 3H), 1.92 (s, 3H); LCMS (method 2, ESI): >95%, Rt = 1.381 min, m/z = 515.1 [M + H]+.
2-(3,5-Dimethoxybenzyl)-5-(1,3-dimethyl-1H-pyrazol-5-yl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (13).
General Procedure E was followed using 49 (50 mg, 0.10 mmol, 1 equiv), (1,3-dimethyl-1H-pyrazol-5-yl)boronic acid (29 mg, 0.21 mmol, 2 equiv), potassium carbonate (36 mg, 0.26 mmol, 2.5 equiv), and PdCl2(dppf)·CH2Cl2 (4.2 mg, 0.005 mmol, 0.05 equiv) to obtain the title compound (34.3 mg, 0.069 mmol, 67% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.96 (d, J = 1.8 Hz, 1H), 7.42 (d, J = 1.8 Hz, 1H), 6.57 (d, J = 2.5 Hz, 1H), 6.45 (s, 1H), 6.44 (d, J = 2.2 Hz, 2H), 6.40 (t, J = 2.2 Hz, 1H), 6.10 (s, 1H), 4.85 (s, 2H), 4.63 (s, 2H), 3.71 (s, 6H), 3.53 (s, 3H), 3.41 (t, J = 6.5 Hz, 2H), 3.12 (s, 3H), 2.74 (t, J = 6.5 Hz, 2H), 2.17 (s, 3H); LCMS (method 2, ESI): >95%, Rt = 1.448 min, m/z = 501.1 [M + H]+.
2-(3,5-Dimethoxybenzyl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (14).
General Procedure E was followed using 49 (60 mg, 0.12 mmol, 1 equiv), (1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)boronic acid (48 mg, 0.25 mmol, 2 equiv), potassium carbonate (43 mg, 0.31 mmol, 2.5 equiv), and PdCl2(dppf)·CH2Cl2 (5.0 mg, 0.006 mmol, 0.05 equiv) to obtain the title compound (48 mg, 0.087 mmol, 70% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.03 (s, 1H), 7.93 (d, J = 1.8 Hz, 1H), 7.31 (d, J = 1.5 Hz, 1H), 6.65 (s, 1H), 6.60 (s, 1H), 6.44 (d, J = 2.2 Hz, 2H), 6.41 (t, J = 2.2 Hz, 1H), 4.92 (s, 2H), 4.62 (s, 2H), 3.95 (s, 3H), 3.71 (s, 6H), 3.39 (t, J = 6.5 Hz, 2H), 3.20 (s, 3H), 2.71 (t, J = 6.5 Hz, 2H); LCMS (method 2, ESI): >95%, Rt = 1.545 min, m/z = 555.1 [M + H]+.
2-(3,5-Dimethoxybenzyl)-5-(1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (15).
General Procedure E was followed using 49 (50 mg, 0.10 mmol, 1 equiv), (1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)boronic acid (43 mg, 0.21 mmol, 2 equiv), potassium carbonate (36 mg, 0.26 mmol, 2.5 equiv), and PdCl2(dppf)·CH2Cl2 (4.2 mg, 0.005 mmol, 0.05 equiv) to obtain the title compound (39.7 mg, 0.070 mmol, 68% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.09 (s, 1H), 7.91 (d, J = 1.8 Hz, 1H), 7.34 (d, J = 1.4 Hz, 1H), 6.56 (s, 1H), 6.51 (s, 1H), 6.44 (d, J = 2.2 Hz, 2H), 6.41 (t, J = 2.2 Hz, 1H), 4.86 (s, 2H), 4.62 (s, 2H), 4.24 (q, J = 7.3 Hz, 2H), 3.71 (s, 6H), 3.39 (t, J = 6.5 Hz, 2H), 3.15 (s, 3H), 2.71 (t, J = 6.5 Hz, 2H), 1.43 (t, J = 7.3 Hz, 3H); LCMS (method 2, ESI): >95%, Rt = 1.611 min, m/z = 569.1 [M + H]+.
7-((1H-Imidazol-1-yl)methyl)-2-(3,5-dimethoxybenzyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (16).
General Procedure F was followed using 52 (50.0 mg, 0.093 mmol, 1 equiv) and 1H-imidazole (25.3 mg, 0.37 mmol, 4 equiv), to obtain the title compound (27.3 mg, 52.0 μmol, 56% yield). 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 2.0 Hz, 1H), 7.55 (s, 1H), 7.29 (s, 1H), 7.11–7.05 (m, 2H), 6.92 (s, 1H), 6.45 (d, J = 2.3 Hz, 2H), 6.37 (t, J = 2.2 Hz, 1H), 5.15 (s, 2H), 4.70 (s, 2H), 3.97 (s, 3H), 3.76 (s, 6H), 3.40 (t, J = 6.5 Hz, 2H), 2.72 (t, J = 6.5 Hz, 2H); LCMS (method 2, ESI): >95%. Rt = 1.537 min, m/z = 526.0 [M + H]+.
7-((1H-Pyrazol-1-yl)methyl)-2-(3,5-dimethoxybenzyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (17).
General Procedure F was followed using 52 (50 mg, 0.093 mmol, 1 equiv), and 1H-pyrazole (25.3 mg, 0.37 mmol, 4 equiv) to obtain the title compound (29.2 mg, 0.056 mmol, 60% yield). 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 2.0 Hz, 1H), 7.53 (d, J = 1.9 Hz, 1H), 7.43 (d, J = 2.3 Hz, 1H), 7.30 (s, 1H), 7.16 (d, J = 2.0 Hz, 1H), 6.45 (d, J = 2.2 Hz, 2H), 6.36 (t, J = 2.3 Hz, 1H), 6.31–6.25 (m, 1H), 5.36 (s, 2H), 4.70 (s, 2H), 3.97 (s, 3H), 3.76 (s, 6H), 3.38 (t, J = 6.5 Hz, 2H), 2.70 (t, J = 6.5 Hz, 2H); LCMS (method 2, ESI): >95%, Rt = 1.789 min, m/z = 526.0 [M + H]+.
7-((1H-Pyrrol-1-yl)methyl)-2-(3,5-dimethoxybenzyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (18).
Sodium hydride (8.9 mg, 0.37 mmol, 4 equiv) was added to a solution of 1H-pyrrole (24.9 mg, 0.37 mmol, 4 equiv) in DMF (3 mL) at 0 °C in an ice bath. After the reaction mixture was stirred for 10 min, a solution of 52 (50.0 mg, 0.093 mmol, 1 equiv) in DMF (2 mL) was added. Then the mixture was stirred at 25 °C for 12 h. Saturated aqueous NaHCO3 was added and the mixture was extracted with CH2Cl2. The combined organic phases were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by preparative reversed-phase HPLC (Phenomenex Gemini C18, MeCN/H2O = 15–70% gradient, 0.1% TFA) followed by neutralization with saturated aqueous NaHCO3 to obtain the title compound (22.4 mg, 0.043 mmol, 46% yield). 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 1.9 Hz, 1H), 7.29 (d, J = 1.1 Hz, 1H), 7.06 (d, J = 2.0 Hz, 1H), 6.69 (t, J = 2.1 Hz, 2H), 6.46 (d, J = 2.3 Hz, 2H), 6.37 (t, J = 2.3 Hz, 1H), 6.17 (t, J = 2.1 Hz, 2H), 5.10 (s, 2H), 4.70 (s, 2H), 3.96 (s, 3H), 3.76 (s, 6H), 3.43–3.36 (m, 2H), 2.70 (t, J = 6.5 Hz, 2H); LCMS (method 2, ESI): >95%, Rt = 1.963 min, m/z = 525.1 [M + H]+.
7-((4H-1,2,4-Triazol-4-yl)methyl)-2-(3,5-dimethoxybenzyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (19).
General Procedure F was followed using 52 (50 mg, 0.093 mmol, 1 equiv), and 1H-1,2,4-triazole (25.6 mg, 0.37 mmol, 4 equiv) to obtain the title compound (15.6 mg, 0.030 mmol, 32% yield). 1H NMR (400 MHz, CDCl3) δ 8.19 (s, 2H), 8.14 (d, J = 2.0 Hz, 1H), 7.32 (s, 1H), 7.11 (d, J = 2.0 Hz, 1H), 6.45 (d, J = 2.3 Hz, 2H), 6.37 (t, J = 2.3 Hz, 1H), 5.23 (s, 2H), 4.71 (s, 2H), 3.98 (s, 3H), 3.76 (s, 6H), 3.41 (t, J = 6.6 Hz, 2H), 2.74 (t, J = 6.6 Hz, 2H); LCMS (method 2, ESI): >95%, Rt = 1.570 min, m/z = 527.1 [M + H]+.
2-(3,5-Dimethoxybenzyl)-7-((2-methyl-1H-imidazol-1-yl)methyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (20).
General Procedure F was followed using 52 (50 mg, 0.093 mmol, 1 equiv), and 2-methyl-1H-imidazole (30.5 mg, 0.37 mmol, 4 equiv) to obtain the title compound (26.1 mg, 0.048 mmol, 52% yield). 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 2.0 Hz, 1H), 7.28 (s, 1H), 6.95–6.89 (m, 2H), 6.85 (s, 1H), 6.46 (d, J = 2.3 Hz, 2H), 6.37 (t, J = 2.3 Hz, 1H), 5.09 (s, 2H), 4.70 (s, 2H), 3.97 (s, 3H), 3.76 (s, 6H), 3.40 (t, J = 6.5 Hz, 2H), 2.71 (t, J = 6.5 Hz, 2H), 2.34 (s, 3H); LCMS (method 2, ESI): >95%, Rt = 1.551 min, m/z = 540.1 [M + H]+.
2-(3,5-Dimethoxybenzyl)-7-((2-ethyl-1H-imidazol-1-yl)methyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (21).
General Procedure F was followed using 52 (50 mg, 0.093 mmol, 1 equiv), and 2-ethyl-1H-imidazole (35.7 mg, 0.37 mmol, 4 equiv) to obtain the title compound (28.2 mg, 0.051 mmol, 55% yield). 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 2.0 Hz, 1H), 7.28 (s, 1H), 7.00–6.96 (m, 1H), 6.90 (d, J = 2.0 Hz, 1H), 6.87–6.83 (m, 1H), 6.45 (d, J = 2.3 Hz, 2H), 6.37 (t, J = 2.3 Hz, 1H), 5.10 (s, 2H), 4.70 (s, 2H), 3.97 (s, 3H), 3.76 (s, 6H), 3.39 (t, J = 6.5 Hz, 2H), 2.71 (t, J = 6.5 Hz, 2H), 2.63 (q, J = 7.5 Hz, 2H), 1.27 (d, J = 7.5 Hz, 3H); LCMS (method 2, ESI): >95%, Rt = 1.570 min, m/z = 554.1 [M + H]+.
2-(3,5-Dimethoxybenzyl)-7-((2-isopropyl-1H-imidazol-1-yl)methyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (22).
General Procedure F was followed using 52 (50 mg, 0.093 mmol, 1 equiv), and 2-isopropyl-1H-imidazole (40.9 mg, 0.37 mmol, 4 equiv) to obtain the title compound (26.4 mg, 0.047 mmol, 50% yield). 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 2.0 Hz, 1H), 7.29 (s, 1H), 7.01 (d, J = 1.3 Hz, 1H), 6.90 (d, J = 2.0 Hz, 1H), 6.82 (d, J = 1.3 Hz, 1H), 6.48 (d, J = 2.2 Hz, 2H), 6.39 (t, J = 2.3 Hz, 1H), 5.16 (s, 2H), 4.73 (s, 2H), 3.99 (s, 3H), 3.79 (s, 6H), 3.42 (t, J = 6.5 Hz, 2H), 2.98 (p, J = 6.8 Hz, 1H), 2.73 (t, J = 6.5 Hz, 2H), 1.29 (s, 3H), 1.27 (s, 3H); LCMS (method 2, ESI): >95%, Rt = 1.602 min, m/z = 568.0 [M + H]+.
7-((2-Cyclopropyl-1H-imidazol-1-yl)methyl)-2-(3,5-dimethoxybenzyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (23).
General Procedure F was followed using 52 (50 mg, 0.093 mmol, 1 equiv), and 2-cyclopropyl-1H-imidazole (40.1 mg, 0.37 mmol, 4 equiv) to obtain the title compound (30.0 mg, 0.053 mmol, 57% yield). 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 2.0 Hz, 1H), 7.28 (d, J = 1.1 Hz, 1H), 6.97 (d, J = 2.0 Hz, 1H), 6.89 (d, J = 1.3 Hz, 1H), 6.83 (d, J = 1.4 Hz, 1H), 6.46 (d, J = 2.3 Hz, 2H), 6.37 (t, J = 2.3 Hz, 1H), 5.23 (s, 2H), 4.71 (s, 2H), 3.97 (s, 3H), 3.76 (s, 6H), 3.40 (t, J = 6.5 Hz, 2H), 2.71 (t, J = 6.5 Hz, 2H), 1.71 (tt, J = 8.2, 5.0 Hz, 1H), 0.98–0.94 (m, 2H), 0.90–0.86 (m, 2H); LCMS (method 2, ESI): >95%, Rt = 1.596 min, m/z = 566.1 [M + H]+.
7-((2-(Difluoromethyl)-1H-imidazol-1-yl)methyl)-2-(3,5-dimethoxybenzyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (24).
General Procedure F was followed using 52 (50 mg, 0.093 mmol, 1 equiv), 2-(difluoromethyl)-1H-imidazole (43.9 mg, 0.37 mmol, 4 equiv), potassium carbonate (25.7 mg, 0.19 mmol, 2 equiv), and sodium iodide (6.9 mg, 0.046 mmol, 0.5 equiv) to obtain the title compound (26.2 mg, 0.046 mmol, 49% yield). 1H NMR (400 MHz, CDCl3) δ 8.12 (s, 1H), 7.30 (s, 1H), 7.08 (d, J = 17.1 Hz, 2H), 6.96 (s, 1H), 6.76 (t, J = 52.7 Hz, 1H), 6.48–6.42 (m, 2H), 6.37 (s, 1H), 5.35 (s, 2H), 4.70 (s, 2H), 3.97 (s, 3H), 3.76 (s, 6H), 3.40 (t, J = 6.5 Hz, 2H), 2.72 (t, J = 6.5 Hz, 2H); LCMS (method 2, ESI): >95%, Rt = 1.666 min, m/z = 576.0 [M + H]+.
7-((2-Methyl-1H-imidazol-1-yl)methyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-2-(pyridin-2-ylmethyl)-3,4-dihydroisoquinolin-1(2H)-one (25).
General Procedure G was followed using 57 (20 mg, 0.051 mmol, 1 equiv), and 2-(bromomethyl)pyridine (7.9 mg, 0.0463 mmol, 1.2 equiv) to obtain the title compound (10.1 mg, 0.021 mmol, 41% yield). 1H NMR (400 MHz, CD3OD) δ 8.71 (d, J = 5.2 Hz, 1H), 8.28 (td, J = 8.0, 1.6 Hz, 1H), 7.97 (d, J = 2.0 Hz, 1H), 7.83 (s, 1H), 7.81 (s, 1H), 7.74 (t, J = 6.4 Hz, 1H), 7.56 (d, J = 2.0 Hz, 1H), 7.51 (d, J = 2.0 Hz, 1H), 7.42 (d, J = 1.6 Hz, 1H), 5.50 (s, 2H), 5.03 (s, 2H), 4.03 (s, 3H), 3.72 (t, J = 6.8 Hz, 2H), 2.97 (t, J = 6.4 Hz, 2H), 2.64 (s, 3H); LCMS (method 2, ESI): >95%, Rt = 1.109 min, m/z = 481.0 [M + H]+.
7-((2-Methyl-1H-imidazol-1-yl)methyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-2-((6-methylpyridin-3-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (26).
General Procedure G was followed using 57 (20 mg, 0.051 mmol, 1 equiv), and 2-(chloromethyl)-6-methylpyridine (8.0 mg, 0.0565 mmol, 1.1 equiv) to obtain the title compound (14 mg, 0.028 mmol, 55% yield). 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 1.6 Hz, 1H), 7.56–7.52 (m, 1H), 7.30 (s, 1H), 7.17 (d, J = 7.6 Hz, 1H), 7.06 (d, J = 7.6 Hz, 1H), 6.95–6.92 (m, 2H), 6.86 (s, 1H), 5.09 (s, 2H), 4.86 (s, 2H), 3.98 (s, 3H), 3.56 (t, J = 6.8 Hz, 2H), 2.77 (t, J = 6.8 Hz, 2H), 2.53 (s, 3H), 2.35 (s, 3H); LCMS (method 2, ESI): >95%, Rt = 1.282 min, m/z = 495.0 [M + H]+.
7-((2-Methyl-1H-imidazol-1-yl)methyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-2-((4-methylpyridin-2-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (27).
General Procedure G was followed using 57 (10 mg, 0.0256 mmol, 1 equiv), and 2-(bromomethyl)-4-methylpyridine (5.2 mg, 0.028 mmol, 1.1 equiv) to obtain the title compound (3.7 mg, 0.007 mmol, 29% yield). 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 5.2 Hz, 1H), 8.07 (d, J = 1.6 Hz, 1H), 7.33 (s, 1H), 7.20 (s, 1H), 7.08 (s, 1H), 7.02 (d, J = 4.8 Hz, 1H), 6.96 (d, J = 1.6 Hz, 1H), 6.93 (d, J = 1.6 Hz, 1H), 5.14 (s, 2H), 4.84 (s, 2H), 4.00 (s, 3H), 3.59 (t, J = 6.4 Hz, 2H), 2.78 (t, J = 6.4 Hz, 2H), 2.52 (s, 3H), 2.33 (s, 3H); LCMS (method 2, ESI): >95%, Rt = 1.091 min, m/z = 495.0 [M + H]+.
7-((2-Methyl-1H-imidazol-1-yl)methyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-2-((3-methylpyridin-2-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (28).
General Procedure G was followed using 57 (20 mg, 0.0514 mmol, 1 equiv), and 2-(chloromethyl)-3-methylpyridine (8.0 mg, 0.0565 mmol, 1.1 equiv) to obtain the title compound (13 mg, 0.026 mmol, 51% yield). 1H NMR (400 MHz, CD3OD) δ 8.52 (d, J = 5.6 Hz, 1H), 8.20–8.13 (m, 1H), 7.96 (d, J = 2.4 Hz, 1H), 7.82 (s, 1H), 7.71–7.66 (m, 1H), 7.55 (d, J = 2.0 Hz, 1H), 7.51 (d, J = 2.0 Hz, 1H), 7.42 (s, 1H), 5.50 (s, 2H), 5.03 (d, J = 4.4 Hz, 2H), 4.02 (s, 3H), 3.70 (t, J = 6.4 Hz, 2H), 2.97 (t, J = 4.4 Hz, 2H), 2.64 (s, 3H), 2.55 (s, 3H); LCMS (method 2, ESI): >95%, Rt = 1.302 min, m/z = 495.0 [M + H]+.
7-((2-Methyl-1H-imidazol-1-yl)methyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-2-((2-methylpyridin-4-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (29).
General Procedure G was followed using 57 (20 mg, 0.026 mmol, 1 equiv), and 4-(bromomethyl)-2-methylpyridine (5.2 mg, 0.0283 mmol, 1.1 equiv) to obtain the title compound (13 mg, 0.017 mmol, 65% yield). 1H NMR (400 MHz, CDCl3) δ 8.45 (d, J = 5.2 Hz, 1H), 8.07 (d, J = 2.0 Hz, 1H), 7.30 (s, 1H), 7.09 (s, 1H), 7.04 (d, J = 4.8 Hz, 1H), 6.95–6.94 (m, 2H), 6.86 (d, J = 1.2 Hz, 1H), 5.10 (s, 2H), 4.73 (s, 2H), 3.98 (s, 3H), 3.42 (t, J = 6.8 Hz, 2H), 2.76 (t, J = 6.8 Hz, 2H), 2.54 (s, 3H), 2.35 (s, 3H); LCMS (method 2, ESI): >95%, Rt = 1.136 min, m/z = 495.0 [M + H]+.
2-((4-Methoxypyridin-2-yl)methyl)-7-((2-methyl-1H-imidazol-1-yl)methyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (30).
General Procedure G was followed using 57 (50 mg, 0.13 mmol, 1 equiv), and 2-(chloromethyl)-4-methoxypyridine (22 mg, 0.14 mmol, 1.1 equiv) to obtain the title compound (15.6 mg, 0.031 mmol, 25% yield). 1H NMR (400 MHz, CDCl3) δ 8.35 (d, J = 5.8 Hz, 1H), 8.07 (d, J = 1.9 Hz, 1H), 7.34 (s, 1H), 7.18 (d, J = 2.2 Hz, 1H), 6.97 (s, 2H), 6.92 (d, J = 2.1 Hz, 1H), 6.75 (dd, J = 5.7, 2.2 Hz, 1H), 5.17 (s, 2H), 4.84 (s, 2H), 4.01 (s, 3H), 3.85 (s, 3H), 3.62 (t, J = 6.6 Hz, 2H), 2.81 (t, J = 6.6 Hz, 2H), 2.64 (s, 3H); LCMS (method 2, ESI): >95%, Rt = 1.159 min, m/z = 511.0 [M + H]+.
2-((4-Ethylpyridin-2-yl)methyl)-7-((2-methyl-1H-imidazol-1-yl)methyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (31).
General Procedure G was followed using 57 (50 mg, 0.13 mmol, 1 equiv), and 2-(chloromethyl)-4-ethylpyridine (22 mg, 0.14 mmol, 1.1 equiv) to obtain the title compound (46.2 mg, 0.091 mmol, 71% yield). 1H NMR (400 MHz, CDCl3) δ 8.40 (d, J = 5.1 Hz, 1H), 8.07 (s, 1H), 7.31 (s, 1H), 7.21 (s, 1H), 7.05–7.04 (m, 2H), 6.94 (d, J = 1.2 Hz, 1H), 6.87 (s, 1H), 5.13 (s, 2H), 4.87 (s, 2H), 3.99 (s, 3H), 3.59 (t, J = 6.6 Hz, 2H), 2.77 (t, J = 6.5 Hz, 2H), 2.63 (q, J = 7.6 Hz, 2H), 2.48 (s, 3H), 1.23 (t, J = 7.6 Hz, 3H); LCMS (method 2, ESI): >95%, Rt = 1.186 min, m/z = 509.1 [M + H]+.
5-(1-Ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-2-((4-methoxypyridin-2-yl)methyl)-7-((2-methyl-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (32).
General Procedure G was followed using 58 (50 mg, 0.12 mmol, 1 equiv), and 2-(chloromethyl)-4-methoxypyridine (21 mg, 0.14 mmol, 1.1 equiv) to obtain the title compound (45.5 mg, 0.087 mmol, 70% yield). 1H NMR (400 MHz, CDCl3) δ 8.35 (d, J = 5.6 Hz, 1H), 8.06 (s, 1H), 7.35 (s, 1H), 7.16–7.06 (m, 1H), 6.97 (s, 1H), 6.91 (s, 2H), 6.74 (s, 1H), 5.13 (s, 2H), 4.84 (s, 2H), 4.25 (q, J = 7.2 Hz, 2H), 3.84 (s, 3H), 3.86 (brs, 2H), 2.79 (brs, 2H), 2.52 (brs, 3H), 1.56 (t, J = 7.3 Hz, 3H); LCMS (method 2, ESI): >95%, Rt = 1.201 min, m/z = 525.0 [M + H]+.
5-(1-Ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-2-((4-ethylpyridin-2-yl)methyl)-7-((2-methyl-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (33).
General Procedure G was followed using 58 (50 mg, 0.12 mmol, 1 equiv), and 2-(chloromethyl)-4-ethylpyridine (21 mg, 0.14 mmol, 1.1 equiv) to obtain the title compound (46 mg, 0.088 mmol, 71% yield). 1H NMR (400 MHz, CDCl3) δ 8.40 (d, J = 5.1 Hz, 1H), 8.07 (d, J = 1.9 Hz, 1H), 7.34 (s, 1H), 7.21 (s, 1H), 7.07 (s, 1H), 7.04 (dd, J = 5.1, 1.5 Hz, 1H), 6.96 (d, J = 1.9 Hz, 1H), 6.92 (s, 1H), 5.13 (s, 2H), 4.86 (s, 2H), 4.25 (q, J = 7.3 Hz, 2H), 3.59 (t, J = 6.6 Hz, 2H), 2.77 (t, J = 6.6 Hz, 2H), 2.63 (q, J = 7.6 Hz, 2H), 2.51 (s, 3H), 1.56 (t, J = 7.3 Hz, 3H), 1.23 (t, J = 7.6 Hz, 3H); LCMS (method 2, ESI): >95%, Rt = 1.271 min, m/z = 523.0 [M + H]+.
(S)-5-(1-Ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-2-(1-(4-methoxypyridin-2-yl)ethyl)-7-((2-methyl-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (34).
General Procedure F was followed using 73 (60 mg, 0.11 mmol, 1 equiv) and 2-methyl-1H-imidazole (18 mg, 0.22 mmol, 2 equiv) to obtain the title compound (21 mg, 0.04 mmol, 35% yield). 1H NMR (400 MHz, CDCl3) δ 8.39 (d, J = 5.4 Hz, 1H), 8.06 (d, J = 1.4 Hz, 1H), 7.31 (s, 1H), 6.97–6.96 (m, 2H), 6.93 (d, J = 2.4 Hz, 1H), 6.88 (d, J = 1.4 Hz, 1H), 6.74 (dd, J = 5.4, 1.4 Hz, 1H), 6.12 (q, J = 7.1 Hz, 1H), 5.11 (s, 2H), 4.25 (q, J = 7.4 Hz, 2H), 3.85 (s, 3H), 3.53–3.46 (m, 1H), 3.40–3.33 (m, 1H), 2.73–2.60 (m, 2H), 2.38 (s, 3H), 1.64 (t, J = 7.1 Hz, 3H), 1.57 (t, J = 7.4 Hz, 3H); LCMS (method 1, ESI): >95%, Rt = 1.04 min, m/z = 538.9 [M + H]+.
(R)-5-(1-Ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-2-(1-(4-methoxypyridin-2-yl)ethyl)-7-((2-methyl-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (35).
General Procedure F was followed using 74 (50 mg, 0.093 mmol, 1 equiv) and 2-methyl-1H-imidazole (31 mg, 0.37 mmol, 4 equiv) to obtain the title compound (36.6 mg, 0.068 mmol, 73% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.35–8.33 (m, 1H), 8.07 (s, 1H), 7.78 (s, 1H), 7.13 (s, 1H), 7.12 (s, 1H), 6.89–7.87 (m, 2H), 6.76 (s, 1H), 5.87 (q, J = 7.0 Hz, 1H), 5.22 (s, 2H), 4.23 (q, J = 7.2 Hz, 2H), 3.81 (s, 3H), 3.43–3.35 (m, 1H), 3.29–3.21 (m, 1H), 2.66 (t, J = 6.3 Hz, 2H), 2.20 (s, 3H), 1.51 (d, J = 7.1 Hz, 3H), 1.42 (t, J = 7.3 Hz, 3H); LCMS (method 2, ESI): >95%, Rt = 1.244 min, m/z = 539.1 [M + H]+.
(S)-2-(Cyclopropyl(4-methoxypyridin-2-yl)methyl)-5-(1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-7-((2-methyl-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (36).
General Procedure F was followed using S18 (150 mg, 0.27 mmol, 1 equiv) and 2-methyl-1H-imidazole (44 mg, 0.54 mmol, 2 equiv) to obtain the title compound (23 mg, 0.04 mmol, 15% yield). 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 5.7 Hz, 1H), 8.00 (d, J = 1.2 Hz, 1H), 7.30 (s, 1H), 6.97 (d, J = 2.3 Hz, 1H), 6.93 (s, 2H), 6.85 (s, 1H), 6.71 (dd, J = 5.7, 2.4 Hz, 1H), 5.07 (s, 2H), 5.04 (d, J = 10.2 Hz, 1H), 4.23 (q, J = 7.3 Hz, 2H), 3.83 (s, 3H), 3.75–3.62 (m, 2H), 2.77–2.64 (m, 2H), 2.34 (s, 3H), 1.55 (t, J = 7.3 Hz, 3H), 0.81–0.75 (m, 1H), 0.64–0.58 (m, 1H), 0.55–0.48 (m, 2H); LCMS (method 2, ESI): >95%, Rt = 0.78 min, m/z = 565.0 [M + H]+.
(S)-2-(Cyclopropyl(4-methylpyridin-2-yl)methyl)-5-(1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-7-((2-methyl-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (37).
General Procedure F was followed using S19 (102 mg, 0.19 mmol, 1 equiv) and 2-methyl-1H-imidazole (46 mg, 0.56 mmol, 3 equiv) to obtain the title compound (67.0 mg, 0.12 mmol, 65% yield). 1H NMR (400 MHz, CD3OD) δ 8.58 (d, J = 6.0 Hz, 1H), 8.03 (s, 1H), 7.93 (d, J = 2.0 Hz, 1H), 7.87 (s, 1H), 7.74 (d, J = 5.6 Hz, 1H), 7.54 (d, J = 2.0 Hz, 1H), 7.51 (d, J = 2.0 Hz, 1H), 7.42 (d, J = 1.2 Hz, 1H), 5.49 (s, 2H), 4.90 (s, 1H), 4.32 (q, J = 7.2 Hz, 2H), 3.86–3.80 (m, 1H), 3.75–3.68 (m, 1H), 2.96 (t, J = 6.4 Hz, 2H), 2.66 (s, 3H), 2.63 (s, 3H), 1.71–1.62 (m, 1H), 1.55 (t, J = 7.2 Hz, 3H), 0.99–0.95 (m, 1H), 0.87–0.82 (m, 1H), 0.70–0.63 (m, 2H); LCMS (method 2, ESI): >95%, Rt = 1.197 min, m/z = 549.0 [M + H]+.
(S)-7-((1H-Imidazol-1-yl)methyl)-5-(1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-2-(1-(4-methoxypyridin-2-yl)ethyl)-3,4-dihydroisoquinolin-1(2H)-one (38).
General Procedure F was followed using 73 (50 mg, 0.09 mmol, 1 equiv) and 1H-imidazole (12 mg, 0.18 mmol, 2 equiv) to obtain the title compound (8 mg, 0.015 mmol, 16% yield). 1H NMR (400 MHz, CDCl3) δ 8.36 (d, J = 5.8 Hz, 1H), 8.09 (d, J = 1.7 Hz, 1H), 7.57 (s, 1H), 7.31 (s, 1H), 7.09 (s, 2H), 6.92 (s, 1H), 6.91 (d, J = 2.4 Hz, 1H), 6.72 (dd, J = 5.7, 2.4 Hz, 1H), 6.10 (q, J = 7.0 Hz, 1H), 5.15 (s, 2H), 4.23 (q, J = 7.3 Hz, 2H), 3.82 (s, 3H), 3.51–3.44 (m, 1H), 3.30–3.32 (m, 1H), 2.72–2.58 (m, 2H), 1.62 (d, J = 7.1 Hz, 3H), 1.54 (t, J = 7.3 Hz, 3H); LCMS (method 2, ESI): >95%, Rt = 1.04 min, m/z = 525.0 [M + H]+.
(R)-7-((1H-Imidazol-1-yl)methyl)-5-(1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-2-(1-(4-methoxypyridin-2-yl)ethyl)-3,4-dihydroisoquinolin-1(2H)-one (39).
General Procedure F was followed using 74 (50 mg, 0.093 mmol, 1 equiv) and 1H-imidazole (25 mg, 0.37 mmol, 4 equiv) to obtain the title compound (35.5 mg, 0.068 mmol, 73% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.34 (d, J = 6.6 Hz, 1H), 8.07 (s, 1H), 7.89 (s, 1H), 7.76 (s, 1H), 7.30 (s, 1H), 7.18 (s, 1H), 6.90–6.87 (m, 3H), 5.87 (q, J = 7.2 Hz, 1H), 5.26 (s, 2H), 4.24 (q, J = 7.3 Hz, 2H), 3.81 (s, 3H), 3.40–3.35 (m, 1H), 3.27–3.20 (m, 1H), 2.65 (t, J = 6.5 Hz, 2H), 1.51 (d, J = 7.1 Hz, 3H), 1.42 (t, J = 7.3 Hz, 3H); LCMS (method 2, ESI): Rt = 1.216 min, m/z = 525.1 [M + H]+.
(S)-7-((1H-Imidazol-1-yl)methyl)-2-(cyclopropyl(4-methoxypyridin-2-yl)methyl)-5-(1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (40).
General Procedure F was followed using S18 (150 mg, 0.27 mmol, 1 equiv) and 1H-imidazole (37 mg, 0.54 mmol, 2 equiv) to obtain the title compound (24 mg, 0.04 mmol, 16% yield). 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 5.7 Hz, 1H), 8.05 (s, 1H), 7.54 (s, 1H), 7.31 (s, 1H), 7.07 (s, 2H), 6.97 (d, J = 2.3 Hz, 1H), 6.91 (s, 1H), 6.72 (dd, J = 2.4 Hz, 1H), 5.13 (s, 2H), 5.04 (d, J = 10.2 Hz, 1H), 4.24 (q, J = 7.3 Hz, 2H), 3.84 (s, 3H), 3.76–3.62 (m, 2H), 2.76 –2.65 (m, 2H), 1.55 (t, J = 7.3 Hz, 3H), 0.81–0.75 (m, 1H), 0.64–0.58 (m, 1H), 0.55–0.48 (m, 2H); LCMS (method 2, ESI): >95%, Rt = 1.20 min, m/z = 551.5 [M + H]+.
(S)-7-((1H-Imidazol-1-yl)methyl)-2-(cyclopropyl(4-methylpyridin-2-yl)methyl)-5-(1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (41).
General Procedure F was followed using S19 (30 mg, 0.055 mmol, 1 equiv) and 1H-imidazole (11 mg, 0.17 mmol, 3 equiv) to obtain the title compound (7.8 mg, 0.015 mmol, 27% yield). 1H NMR (400 MHz, CDCl3) δ 8.41 (d, J = 5.2 Hz, 1H), 8.06 (d, J = 2.0 Hz, 1H), 7.53 (s, 1H), 7.31 (s, 1H), 7.24 (s, 1H), 7.08 (d, J = 2.4 Hz, 2H), 7.01 (d, J = 4.4 Hz, 1H), 6.91 (s, 1H), 5.13 (s, 2H), 5.07 (d, J = 10.0 Hz, 1H), 4.25 (q, J = 7.2 Hz, 2H), 3.76–3.63 (m, 2H), 2.76–2.65 (m, 2H), 2.33 (s, 3H), 1.66–1.62 (m, 1H), 1.55 (t, J = 7.2 Hz, 3H), 0.80–0.75 (m, 1H), 0.63–0.59 (m, 1H), 0.54–0.51 (m, 2H); LCMS (method 2, ESI): >95%, Rt = 1.306 min, m/z = 535.0 [M + H]+.
Methyl 2-(3,5-dimethoxybenzyl)-5-hydroxy-1-oxo-1,2,3,4-tetra-hydroisoquinoline-7-carboxylate (43).
General Procedure A was followed using 42 (15.0 g, 59.5 mmol, 1 equiv), 3,5-dimethoxybenzylamine (13.4 mL, 89.2 mmol, 1.5 equiv), and sodium triacetoxyborohydride (25.2 g, 118.9 mmol, 2 equiv) to obtain the title compound (22 g, 59.2 mmol, 99% yield). 1H NMR (400 MHz, CDCl3) δ 8.81 (d, J = 1.6 Hz, 1H), 8.03 (d, J = 1.6 Hz, 1H), 6.45 (d, J = 2.3 Hz, 2H), 6.38 (t, J = 2.3 Hz, 1H), 4.72 (s, 2H), 3.95 (s, 3H), 3.76 (s, 6H), 3.53 (t, J = 6.6 Hz, 2H), 3.06 (t, J = 6.6 Hz, 2H); LCMS (method 2, ESI): Rt = 1.624 min, m/z = 372.1 [M + H]+.
Methyl 2-(3,5-dimethoxybenzyl)-1-oxo-5-(((trifluoromethyl)-sulfonyl)oxy)-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (44).
General Procedure B was followed using 43 (15.0 g, 40.4 mmol, 1 equiv), phenyl triflimide (17 g, 48.5 mmol, 1.2 equiv), and N,N-diisopropylethylamine (18 mL, 101.0 mmol, 2.5 equiv) to obtain the title compound (16.1 g, 31.9 mmol, 79% yield). 1H NMR (400 MHz, CDCl3) δ 8.83 (d, J = 1.6 Hz, 1H), 8.04 (d, J = 1.6 Hz, 1H), 6.47 (d, J = 2.3 Hz, 2H), 6.39 (t, J = 2.3 Hz, 1H), 4.73 (s, 2H), 3.96 (s, 3H), 3.77 (s, 6H), 3.54 (t, J = 6.6 Hz, 2H), 3.07 (t, J = 6.6 Hz, 2H); LCMS (method 2, ESI): Rt = 2.069 min, m/z = 504.0 [M + H]+.
Methyl 2-(3,5-dimethoxybenzyl)-1-oxo-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (45).
Compound 44 (7.55 g, 15.0 mmol, 1 equiv), bis-(pinacolato)diboron (7.61 g, 30.0 mmol, 2 equiv), KOAc (4.41 g, 45.0 mmol, 3 equiv), and PdCl2(dppf)·CH2Cl2 (0.612 g, 0.75 mmol) were dissolved in 1,4-dioxane (40 mL) and placed under an Ar atmosphere. The reaction mixture was stirred at 100 °C overnight. Brine was added to the cooled mixture and extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash chromatography (Combi-flash Rf, EtOAc/hexanes = 0–50% gradient) to afford the title compound (7.15 g, 14.9 mmol, 99% yield). 1H NMR (400 MHz, CDCl3) δ 8.87 (d, J = 2.0 Hz, 1H), 8.57 (d, J = 2.0 Hz, 1H), 6.48 (d, J = 2.3 Hz, 2H), 6.37 (t, J = 2.3 Hz, 1H), 4.73 (s, 2H), 3.93 (s, 3H), 3.76 (s, 6H), 3.47 (t, J = 6.6 Hz, 2H), 3.32 (t, J = 6.6 Hz, 2H), 1.33 (s, 12H); LCMS (method 2, ESI): Rt = 2.200 min, m/z = 482.1 [M + H]+.
Methyl 5-bromo-2-(3,5-dimethoxybenzyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (46).
A mixture of 45 (6.68 g, 13.9 mmol, 1 equiv), copper(II) bromide (9.30 g, 41.6 mmol, 3 equiv) and MeOH/H2O (1:1 v/v, 160 mL) was stirred and refluxed at 80 °C. The mixture was cooled to room temperature and diluted with EtOAc. The layers were separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash chromatography (Combi-flash Rf, EtOAc/hexanes = 0–50% gradient) to afford the title compound (5.43 g, 12.5 mmol, 90% yield). 1H NMR (400 MHz, CDCl3) δ 8.75 (d, J = 1.7 Hz, 1H), 8.33 (d, J = 1.7 Hz, 1H), 6.46 (d, J = 2.3 Hz, 2H), 6.38 (t, J = 2.3 Hz, 1H), 4.72 (s, 2H), 3.94 (s, 3H), 3.77 (s, 6H), 3.51 (t, J = 6.7 Hz, 2H), 3.07 (t, J = 6.7 Hz, 2H); LCMS (method 2, ESI): Rt = 1.971 min, m/z = 433.9 [M + H]+.
5-Bromo-2-(3,5-dimethoxybenzyl)-7-(hydroxymethyl)-3,4-dihydroisoquinolin-1(2H)-one (47).
General Procedure C was followed using 46 (5.43 g, 12.5 mmol, 1 equiv) to obtain the title compound (2.07 g, 5.1 mmol, 41% yield). 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 1.6 Hz, 1H), 7.74 (d, J = 1.7 Hz, 1H), 6.46 (d, J = 2.3 Hz, 2H), 6.38 (t, J = 2.3 Hz, 1H), 4.73 (d, J = 6.0 Hz, 2H), 4.72 (s, 2H), 3.82 (s, 1H), 3.77 (s, 6H), 3.49 (t, J = 6.7 Hz, 2H), 3.02 (t, J = 6.7 Hz, 2H); LCMS (method 2, ESI): Rt = 1.686 min, m/z = 406.0 [M + H]+.
5-Bromo-7-(bromomethyl)-2-(3,5-dimethoxybenzyl)-3,4-dihydroisoquinolin-1(2H)-one (48).
General Procedure C was followed using 47 (2.07 g, 5.10 mmol, 1 equiv) to obtain the title compound (992 mg, 2.11 mmol, 42% yield). 1H NMR (400 MHz, CDCl3) δ 8.14 (d, J = 1.9 Hz, 1H), 7.72 (d, J = 1.9 Hz, 1H), 6.46 (d, J = 2.3 Hz, 2H), 6.38 (t, J = 2.3 Hz, 1H), 4.71 (s, 2H), 4.46 (s, 2H), 3.77 (s, 6H), 3.49 (t, J = 6.7 Hz, 2H), 3.02 (t, J = 6.7 Hz, 2H); LCMS (method 2, ESI): Rt = 2.010 min, m/z = 467.8 [M + H]+.
5-Bromo-2-(3,5-dimethoxybenzyl)-7-((2-imino-3-methyl-2,3-dihydro-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (49).
1-Methyl-1H-imidazol-2-amine hydrochloride (847 mg, 6.34 mmol, 3 equiv) was added to a solution of 48 (992 mg, 2.11 mmol, 1 equiv), and N,N-diisopropylethylamine (1.5 mL, 8.46 mmol, 4 equiv) in acetonitrile (18 mL) at 23 °C. The reaction mixture was stirred for 12 h at 60 °C, then cooled to ambient temperature and concentrated under reduced pressure. Saturated aqueous NaHCO3 was added and the mixture was extracted with CH2Cl2. The combined organic layers were dried over MgSO4 and concentrated under reduced pressure to provide the title compound (1.03 g, 2.11 mmol, quant.), which was used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 1.6 Hz, 1H), 7.77 (d, J = 1.7 Hz, 1H), 6.74 (d, J = 2.5 Hz, 1H), 6.63 (d, J = 2.5 Hz, 1H), 6.44 (d, J = 2.2 Hz, 2H), 6.41 (t, J = 2.2 Hz, 1H), 4.91 (s, 2H), 4.62 (s, 2H), 3.71 (s, 6H), 3.50 (t, J = 6.7 Hz, 2H), 3.22 (s, 3H), 2.97 (t, J = 6.6 Hz, 2H); LCMS (method 2, ESI): Rt = 1.518 min, m/z = 484.9 [M + H]+.
Methyl 2-(3,5-dimethoxybenzyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (50).
General Procedure E was followed using 44 (7.0 g, 13.9 mmol, 1 equiv), (1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)boronic acid (4.1 g, 20.9 mmol, 1.5 equiv), potassium carbonate (4.8 g, 34.8 mmol, 2.5 equiv), and PdCl2(dppf) (814.0 mg, 1.2 mmol, 0.08 equiv) to obtain the title compound (5.95 g, 11.8 mmol, 85% yield). 1H NMR (400 MHz, CDCl3) δ 8.77 (d, J = 1.9 Hz, 1H), 7.95 (d, J = 1.9 Hz, 1H), 7.33–7.27 (m, 1H), 6.40 (d, J = 2.3 Hz, 2H), 6.30 (t, J = 2.3 Hz, 1H), 4.66 (s, 2H), 3.94 (s, 3H), 3.86 (s, 3H), 3.70 (s, 6H), 3.35 (t, J = 6.5 Hz, 2H), 2.71 (t, J = 6.5 Hz, 2H); LCMS (method 2, ESI): Rt = 1.878 min, m/z = 504.0 [M + H]+.
2-(3,5-Dimethoxybenzyl)-7-(hydroxymethyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (51).
General Procedure C was followed using 50 (4.0 g, 7.9 mmol, 1 equiv) to obtain the title compound (3.6 g, 7.5 mmol, 95% yield). 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 1.9 Hz, 1H), 7.37 (d, J = 1.8 Hz, 1H), 7.33 (d, J = 1.0 Hz, 1H), 6.43 (d, J = 2.3 Hz, 2H), 6.34 (t, J = 2.3 Hz, 1H), 4.67 (d, J = 1.3 Hz, 4H), 3.95 (s, 3H), 3.72 (s, 6H), 3.39–3.32 (m, 2H), 2.69 (t, J = 6.6 Hz, 2H); LCMS (method 2, ESI): Rt = 1.645 min, m/z = 476.0 [M + H]+.
7-(Bromomethyl)-2-(3,5-dimethoxybenzyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (52).
General Procedure D was followed using 51 (1.0 g, 2.1 mmol, 1 equiv) to obtain the title compound (1.07 g, 1.9 mmol, 90% yield). The residue was purified by flash chromatography (Combi-flash Rf, MeOH/CH2Cl2 = 0–10% gradient). 1H NMR (400 MHz, CDCl3) δ 8.22 (s, J = 2.0 Hz, 1H), 7.39 (d, J = 2.0 Hz, 1H), 7.38–7.35 (m, 1H), 6.46 (d, J = 2.3 Hz, 2H), 6.37 (t, J = 2.3 Hz, 1H), 4.71 (s, 2H), 4.52 (s, 2H), 4.00 (s, 3H), 3.76 (d, J = 2.2 Hz, 6H), 3.40 (t, J = 6.6 Hz, 2H), 2.72 (t, J = 6.5 Hz, 2H); LCMS (method 2, ESI): Rt = 1.965 min, m/z = 537.9 [M + H]+.
Methyl 2-(2,4-dimethoxybenzyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (54).
General Procedure E was followed using 53 (12.3 g, 24.3 mmol, 1 equiv), (1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)boronic acid (7.1 g, 36.4 mmol, 1.5 equiv), potassium carbonate (8.4 g, 60.8 mmol, 2.5 equiv), and PdCl2(dppf) (890 mg, 1.2 mmol, 0.05 equiv) to obtain the title compound (11.0 g, 21.8 mmol, 90% yield). 1H NMR (400 MHz, CDCl3) δ 8.81 (d, J = 1.9 Hz, 1H), 7.99 (d, J = 1.8 Hz, 1H), 7.36 (d, J = 1.1 Hz, 1H), 7.31–7.27 (m, 1H), 6.45 (dd, J = 6.2, 2.5 Hz, 2H), 4.73 (s, 2H), 4.00 (s, 3H), 3.92 (s, 3H), 3.80 (s, 3H), 3.79 (s, 3H), 3.46 (t, J = 6.6 Hz, 2H), 2.75 (t, J = 6.5 Hz, 2H); LCMS (method 2, ESI): Rt = 1.072 min, m/z = 504.4 [M + H]+.
Methyl 5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (55).
Anisole (24 mL, 218 mmol, 5 equiv) was added to a solution of 54 (22.0 g, 43.7 mmol, 1 equiv) in CH2Cl2 (50 mL) and TFA (100 mL). The reaction was stirred at room temperature overnight and then concentrated under reduced pressure. The residue was dissolved in EtOAc and washed with saturated aqueous NaHCO3. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash chromatography (Combi-flash Rf, EtOAc/hexanes = 0–100% gradient followed by MeOH/CH2Cl2 = 0–10% gradient) to provide the title compound (13.5 g, 38.2 mmol, 87% yield). 1H NMR (400 MHz, CDCl3) δ 8.77 (d, J = 1.6 Hz, 1H), 8.05 (d, J = 1.6 Hz, 1H), 7.40 (s, 1H), 6.25 (s, 1H), 4.03 (s, 3H), 3.92 (s, 3H), 3.51 (td, J = 6.5, 2.8 Hz, 2H), 2.84 (t, J = 6.5 Hz, 1H); LCMS (method 2, ESI): Rt = 1.363 min, m/z = 354.1 [M + H]+.
7-(Bromomethyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (56).
General Procedure C was followed using 55 (457 mg, 1.3 mmol, 1 equiv) to obtain 7-(hydroxymethyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (420 mg, 1.3 mmol, quant.), which was used without further purification. LCMS (method 2, ESI): Rt = 1.145 min, m/z = 326.1 [M + H]+. Then General Procedure D was followed using 7-(hydroxymethyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (420 mg, 1.3 mmol, 1 equiv) to obtain the title compound (440 mg, 1.3 mmol, quant.), which was used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 1.7 Hz, 1H), 7.42 (d, J = 1.9 Hz, 1H), 7.40 (s, 1H), 4.51 (s, 2H), 4.02 (s, 3H), 3.48 (td, J = 6.5, 2.7 Hz, 2H), 2.79 (t, J = 6.5 Hz, 2H); LCMS (ESI): m/z = 387.9 [M + H]+.
7-((2-Methyl-1H-imidazol-1-yl)methyl)-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-3,4-dihydroisoquinolin-1(2H)-one (57).
General Procedure F was followed using 56 (1.0 g, 2.6 mmol, 1 equiv) and 2-methyl-1H-imidazole (846 mg, 10.3 mmol, 4 equiv), to obtain the title compound (700 mg, 1.8 mmol, 70% yield). 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 2.0 Hz, 1H), 7.32 (s, 1H), 6.95 (d, J = 1.4 Hz, 1H), 6.94 (d, J = 2.0 Hz, 1H), 6.85 (d, J = 1.4 Hz, 1H), 5.99 (s, 1H), 5.09 (s, 2H), 4.00 (s, 3H), 3.48 (td, J = 6.5, 2.8 Hz, 2H), 2.79 (t, J = 6.5 Hz, 2H), 2.35 (s, 3H); LCMS (method 2, ESI): Rt = 0.973 min, m/z = 390.0 [M + H]+.
5-(1-Ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-7-((2-methyl-1H-imidazol-1-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (58).
General Procedure F was followed using S3 (909 mg, 2.3 mmol, 1 equiv) and 2-methyl-1H-imidazole (557 mg, 6.8 mmol, 3 equiv), to obtain the title compound (522 mg, 1.3 mmol, 57% yield). 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 2.1 Hz, 1H), 7.34 (d, J = 1.1 Hz, 1H), 6.96 (d, J = 2.0 Hz, 1H), 6.94 (d, J = 1.4 Hz, 1H), 6.85 (d, J = 1.4 Hz, 1H), 5.97 (s, 1H), 5.09 (s, 2H), 4.26 (q, J = 7.3 Hz, 2H), 3.48 (td, J = 6.5, 2.8 Hz, 2H), 2.78 (t, J = 6.5 Hz, 2H), 2.34 (s, 3H), 1.58 (t, J = 7.3 Hz, 3H); LCMS (ESI): m/z = 404.0 [M + H]+.
(S,E)-N-((4-Methoxypyridin-2-yl)methylene)-2-methylpropane-2-sulfinamide (59).
Cesium carbonate (1.0 g, 3.1 mmol, 1.5 equiv) was added to a solution of (S)-2-methylpropane-2-sulfinamide (0.28 g, 2.1 mmol, 1 equiv) and 4-methoxypicolinaldehyde (0.25 g, 2.1 mmol, 1 equiv) in 10 mL of CH2Cl2. The reaction was stirred at rt for 16 h. The reaction was filtered through celite and concentrated to dryness. The residue was purified by flash chromatography (Combi-flash Rf, EtOAc/hexanes = 5–75% gradient) to afford the title compound (0.38 g, 1.60 mmol, 77% yield). 1H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H), 8.58 (d, J = 5.8 Hz, 1H), 7.55 (d, J = 2.6 Hz, 1H), 6.94 (dd, J = 5.8. 2.6 Hz, 1H), 3.94 (s, 3H), 1.31 (s, 9H); LCMS (method 2, ESI): Rt = 0.85 min, m/z = 241.2 [M + H]+.
(S)-N-((S)-1-(4-Methoxypyridin-2-yl)ethyl)-2-methylpropane-2-sulfinamide (60).
A 3.4 M solution of methyl magnesium bromide in 2-methyltetrahydrofuran (35 mL, 118.2 mmol, 2.5 equiv) was added dropwise to a −78 °C solution of 59 (11.36 g, 47.3 mmol, 1 equiv) in 350 mL of THF. After 30 min, the reaction was quenched by addition of 30 mL of saturated aqueous NH4Cl solution. This was stirred for 30 min and then filtered. The filtrate was extracted with EtOAc, the combined organic layers were dried with MgSO4, filtered, and concentrated under reduced pressure. The major diastereomer was isolated by flash chromatography (Combi-flash Rf, EtOAc/hexanes = 0–100% gradient) to afford the title compound (10.12 g, 39.5 mmol, 84% yield). 1H NMR (400 MHz, CDCl3) δ 8.36 (d, J = 5.7 Hz, 1H), 6.81 (d, J = 2.4 Hz, 1H), 6.70 (dd, J = 5.8, 2.5 Hz, 1H), 4.74 (d, J = 5.2 Hz, 1H), 4.52 (p, J = 6.4 Hz, 1H), 3.84 (s, 3H), 1.49 (d, J = 6.7 Hz, 3H), 1.25 (s, 9H); LCMS (method 2, ESI): Rt = 0.176 min, m/z = 257.1 [M + H]+.
(S)-1-(4-Methoxypyridin-2-yl)ethan-1-amine hydrochloride (61).
A 4.0 M solution of HCl in dioxane (39 mL, 157.7 mmol, 4 equiv) was added to a solution of 60 (10.1 g, 39.4 mmol, 1 equiv) in 400 mL of THF. The reaction was stirred at room temperature for 45 min and followed by TLC. The reaction mixture was concentrated under reduced pressure to provide the title compound (5.62 g, 36.9 mmol, 94% yield), which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 8.70 (d, J = 6.9 Hz, 1H), 7.72 (d, J = 2.6 Hz, 1H), 7.56 (dd, J = 6.9, 2.6 Hz, 1H), 4.85 (q, J = 7.0 Hz, 1H), 4.20 (s, 3H), 1.79 (d, J = 7.0 Hz, 3H); LCMS (method 2, ESI): m/z = 153.2 [M + H]+.
(R,E)-N-((4-Methoxypyridin-2-yl)methylene)-2-methylpropane-2-sulfinamide (62).
Cesium carbonate (17.82 g, 54.7 mmol, 1.5 equiv) was added to a solution of (R)-2-methylpropane-2-sulfinamide (4.86 g, 40.1 mmol, 1.1 equiv) and 4-methoxypicolinaldehyde (5.00 g, 36.5 mmol, 1 equiv) in 200 mL of CH2Cl2. The reaction was stirred at rt for 16 h. Water was added to the mixture and extracted with CH2Cl2. The combined organic layers were dried with over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (Combi-flash Rf, EtOAc/hexanes = 0–40% gradient) to afford the title compound (5.67 g, 23.6 mmol, 65% yield). 1H NMR (400 MHz, CDCl3) δ 8.66 (s, 1H), 8.56 (d, J = 5.7 Hz, 1H), 7.52 (d, J = 2.5 Hz, 1H), 6.92 (dd, J = 5.7, 2.5 Hz, 1H), 3.92 (s, 3H), 1.28 (s, 9H); LCMS (method 2, ESI): Rt = 0.345 min, m/z = 241.1 [M + H]+.
(R)-N-((R)-1-(4-Methoxypyridin-2-yl)ethyl)-2-methylpropane-2-sulfinamide (63).
A 3.4 M solution of methyl magnesium bromide in 2-methyltetrahydrofuran (6.4 mL, 21.8 mmol, 1.5 equiv) was added dropwise to a −78 °C solution of 62 (3.50 g, 14.6 mmol, 1 equiv) in 100 mL of THF. After 30 min, the reaction was quenched by addition of 30 mL of saturated aqueous NH4Cl solution. This was stirred for 30 min and then filtered. The filtrate was extracted with EtOAc, the combined organic layers were dried with MgSO4, filtered, and concentrated under reduced pressure. The major diastereomer was isolated by flash chromatography (Combi-flash Rf, EtOAc/hexanes = 0–100% gradient) to afford the title compound (3.4278 g, 13.4 mmol, 92% yield). 1H NMR (400 MHz, CDCl3) δ 8.35 (d, J = 5.8 Hz, 1H), 6.81 (d, J = 2.3 Hz, 1H), 6.70 (dd, J = 5.7, 2.4 Hz, 1H), 4.78 (d, J = 4.9 Hz, 1H), 4.51 (p, 6.4 Hz, 1H), 3.84 (s, 3H), 1.50 (d, J = 6.7 Hz, 3H), 1.25 (s, 9H); LCMS (method 2, ESI): Rt = 0.355 min, m/z = 257.1 [M + H]+.
(R)-1-(4-Methoxypyridin-2-yl)ethan-1-amine hydrochloride (64).
A 4.0 M solution of HCl in dioxane (33.9 mL, 135 mmol, 10 equiv) was added to a solution of 63 (3.48 g, 13.6 mmol, 1 equiv) in 100 mL of THF. The reaction was stirred at room temperature for 45 min and followed by TLC. The reaction mixture was concentrated under reduced pressure to provide the title compound (2.67 g, 14.2 mmol, quant.), which was used without further purification. 1H NMR (400 MHz, DMSO-d6) δ 8.84 (brs, 3H), 8.59 (brs, 1H), 7.60 (brs, 1H), 7.26 (brs, 1H), 4.63 (brs, 1H), 3.98 (brs, 3H), 1.56 (brs, 3H); LCMS (method 2, ESI): Rt = 0.259 min, m/z = 153.2 [M + H]+.
Methyl (S)-5-hydroxy-2-(1-(4-methoxypyridin-2-yl)ethyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (65).
General Procedure A was followed using 42 (5.3 g, 21.0 mmol, 1 equiv), 61 (7.1 g, 31.5 mmol, 1.5 equiv), 2,4,6-trimethylpyridine (8.5 mL, 63 mmol, 3 equiv), and sodium cyanoborohydride (4.0 g, 63 mmol, 3 equiv) to obtain the crude title compound (7.5 g, 21.0 mmol, quant.). 1H NMR (400 MHz, CDCl3) δ 8.43 (dd, J = 5.9, 2.6 Hz, 1H), 8.26–8.24 (m, 1H), 7.61 (d, J = 1.5 Hz, 1H), 7.02–7.00 (m, 1H), 6.83–6.80 (m, 1H), 6.04–6.00 (m, 1H), 3.88–3.86 (m, 6H), 3.65–3.56 (m, 1H), 3.47–3.43 (m, 1H), 3.02–2.93 (m, 1H), 2.90–2.81 (m, 1H), 1.69 (d, J = 7.0 Hz, 3H); LCMS (method 2, ESI): m/z = 357.1 [M + H]+.
Methyl (R)-5-hydroxy-2-(1-(4-methoxypyridin-2-yl)ethyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (66).
General Procedure A was followed using 42 (1.0 g, 3.96 mmol, 1 equiv), 64 (1.12 g, 5.95 mmol, 1.5 equiv), N,N-diisopropylethylamine (2.1 mL, 11.9 mmol, 3 equiv) and sodium triacetoxyborohydride (2.52 g, 11.9 mmol, 3 equiv) to obtain the title compound (1.18 g, 3.31 mmol, 84% yield). 1H NMR (400 MHz, CDCl3) δ 8.43 (dd, J = 5.9, 2.6 Hz, 1H), 8.26–8.24 (m, 1H), 7.61 (d, J = 1.5 Hz, 1H), 7.02–7.00 (m, 1H), 6.83–6.80 (m, 1H), 6.04–6.00 (m, 1H), 3.88–3.86 (m, 6H), 3.65–3.56 (m, 1H), 3.47–3.43 (m, 1H), 3.02–2.93 (m, 1H), 2.90–2.81 (m, 1H), 1.69 (d, J = 7.0 Hz, 3H); LCMS (method 2, ESI): Rt = 1.190 min, m/z = 357.1 [M + H]+.
Methyl (S)-2-(1-(4-methoxypyridin-2-yl)ethyl)-1-oxo-5-(((trifluoromethyl)sulfonyl)oxy)-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (67).
General Procedure B was followed using 65 (7.5 g, 21.0 mmol, 1 equiv), phenyl triflimide (11.2 g, 31.5 mmol, 1.5 equiv), and triethylamine (8.8 mL, 63 mmol, 3 equiv) to obtain the title compound (10.1 g, 20.7 mmol, 98% yield). 1H NMR (400 MHz, CDCl3) δ 8.83 (d, J = 1.9 Hz, 1H), 8.41 (d, J = 5.8 Hz, 1H), 8.05 (d, J = 1.9 Hz, 1H), 6.93 (d, J = 2.7 Hz, 1H), 6.77 (dd, J = 5.8. 2.7 Hz, 1H), 6.11 (q, J = 7.2 Hz, 1H), 3.98 (s, 3H), 3.86 (s, 3H), 3.67–3.54 (m, 2H), 3.11–2.94 (m, 2H), 1.67 (d, J = 7.2 Hz, 3H); LCMS (method 2, ESI): Rt = 1.34 min, m/z = 488.8 [M + H]+.
Methyl (R)-2-(1-(4-methoxypyridin-2-yl)ethyl)-1-oxo-5-(((trifluoromethyl)sulfonyl)oxy)-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (68).
General Procedure B was followed using 66 (1.18 g, 3.31 mmol, 1 equiv), phenyl triflimide (1.42 g, 3.97 mmol, 1.2 equiv), and N,N-diisopropylethylamine (1.4 mL, 8.82 mmol, 2.5 equiv) to obtain the title compound (1.50 g, 3.08 mmol, 93% yield). 1H NMR (400 MHz, CDCl3) δ 8.80 (s, 1H), 8.39 (d, J = 5.8 Hz, 1H), 8.02 (s, 1H), 6.92 (s, 1H), 6.76 (dd, J = 5.7 Hz, 2.1 Hz, 1H), 6.06 (q, J = 6.9 Hz, 1H), 3.96 (s, 3H), 3.84 (s, 3H), 3.68–3.54 (m, 2H), 3.11–2.93 (m, 2H), 1.67 (d, J = 7.1 Hz, 3H); LCMS (method 2, ESI): Rt = 1.497 min, m/z = 489.0 [M + H]+.
Methyl (S)-5-(1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-2-(1-(4-methoxypyridin-2-yl)ethyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (69).
General Procedure E was followed using 67 (6 g, 12.3 mmol, 1 equiv), (1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)boronic acid (3.1 g, 14.8 mmol, 1.2 equiv), potassium carbonate (4.2 g, 30.7 mmol, 2.5 equiv), and tetrakis(triphenylphosphine)-palladium(0) (710 mg, 0.61 mmol, 0.05 equiv) to obtain the title compound (4.4 g, 8.8 mmol, 72% yield). 1H NMR (400 MHz, CDCl3) δ 8.80 (d, J = 1.9 Hz, 1H), 8.37 (d, J = 5.8 Hz, 1H), 8.02 (d, J = 1.9 Hz, 1H), 7.37 (s, 1H), 6.93 (d, J = 2.7 Hz, 1H), 6.73 (dd, J = 5.8. 2.7 Hz, 1H), 6.10 (q, J = 7.2 Hz, 1H), 4.25 (q, J = 7.4 Hz, 2H), 3.92 (s, 3H), 3.83 (s, 3H), 3.53–3.47 (m, 1H), 3.40–3.34 (m, 1H), 2.78–2.64 (m, 2H), 1.67 (d, J = 7.2 Hz, 3H), 1.56 (t, J = 7.4 Hz, 3H); LCMS (method 2, ESI): Rt = 1.33 min, m/z = 502.9 [M + H]+.
Methyl (R)-5-(1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-2-(1-(4-methoxypyridin-2-yl)ethyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-7-carboxylate (70).
General Procedure E was followed using 68 (500 mg, 1.02 mmol, 1 equiv), (1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)boronic acid (277 mg, 1.33 mmol, 1.3 equiv), sodium carbonate (271 mg, 2.56 mmol, 2.5 equiv), and tetrakis-(triphenylphosphine)palladium(0) (59.1 mg, 0.51 mmol, 0.05 equiv) to obtain the title compound (491 mg, 0.98 mmol, 96% yield). 1H NMR (400 MHz, CDCl3) δ 8.79 (d, J = 1.6 Hz, 1H), 8.37 (d, J = 5.8 Hz, 1H), 8.01 (d, J = 1.7 Hz, 1H), 7.37 (s, 1H), 6.95 (d, J = 2.2 Hz, 1H), 6.74 (dd, J = 5.8, 2.4 Hz, 1H), 6.07 (q, J = 6.8 Hz, 1H), 4.26 (q, J = 7.3 Hz, 2H), 3.92 (s, 3H), 3.84 (s, 3H), 3.56–3.49 (m, 1H), 3.43–3.37 (m, 1H), 2.81–2.66 (m, 2H), 1.64 (d, J = 7.1 Hz, 3H), 1.56 (t, J = 7.3 Hz, 3H); LCMS (method 2, ESI): Rt = 1.444 min, m/z = 503.1 [M + H]+.
(S)-5-(1-Ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-7-(hydroxymethyl)-2-(1-(4-methoxypyridin-2-yl)ethyl)-3,4-dihydroisoquinolin-1(2H)-one (71).
General Procedure C was followed using 69 (4.4 g, 8.8 mmol, 1 equiv) and a lithium borohydride solution (2 M THF, 44.0 mL, 88.0 mmol, 10 equiv) to obtain the title compound (3.0 g, 6.3 mmol, 72% yield). 1H NMR (400 MHz, CDCl3) δ 8.35 (d, J = 5.8 Hz, 1H), 8.14 (d, J = 1.7 Hz, 1H), 7.39 (d, J = 1.5 Hz, 1H), 7.34 (s, 1H), 6.91 (d, J = 2.7 Hz, 1H), 6.71 (dd, J = 5.8. 2.7 Hz, 1H), 6.11 (q, J = 7.2 Hz, 1H), 4.73 (s, 2H), 4.25 (q, J = 7.4 Hz, 2H), 3.82 (s, 3H), 3.47–3.42 (m, 1H), 3.34–3.27 (m, 1H), 2.70–2.60 (m, 2H), 1.61 (d, J = 7.2 Hz, 3H), 1.54 (t, J = 7.4 Hz, 3H); LCMS (method 2, ESI): Rt = 1.16 min, m/z = 475.0 [M + H]+.
(R)-5-(1-Ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-7-(hydroxymethyl)-2-(1-(4-methoxypyridin-2-yl)ethyl)-3,4-dihydroisoquinolin-1(2H)-one (72).
General Procedure C was followed using 70 (491 mg, 0.98 mmol, 1 equiv) and a lithium triethylborohydride solution (1 M THF, 2.9 mL, 2.93 mmol, 3 equiv) to obtain the title compound (394 mg, 0.83 mmol, 85% yield). 1H NMR (400 MHz, CDCl3) δ 8.42–8.39 (m, 1H), 8.09 (d, J = 6.4 Hz, 1H), 7.40 (s, 1H), 7.36 (s, 1H), 7.07–7.00 (m, 1H), 6.85–6.78 (m, 1H), 5.99–5.86 (m, 1H), 4.74–4.72 (m, 2H), 4.25 (q, J = 7.3 Hz, 2H), 3.91–3.86 (m, 3H), 3.65–3.41 (m, 2H), 2.86–2.66 (m, 2H), 1.77–1.66 (m, 3H), 1.56 (t, J = 7.3 Hz, 3H); LCMS (method 2, ESI): Rt = 1.319 min, m/z = 475.0 [M + H]+.
(S)-7-(Bromomethyl)-5-(1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-2-(1-(4-methoxypyridin-2-yl)ethyl)-3,4-dihydroisoquinolin-1(2H)-one (73).
A solution of 71 (1.35 g, 2.9 mmol, 1 equiv) in CH2Cl2 (30 mL) was cooled in an ice bath. N,N-Diisopropylethylamine (1 mL, 5.8 mmol, 2 equiv) and methanesulfonyl chloride (0.33 mL, 4.3 mmol, 1.5 equiv) were added, and the reaction was stirred for 1 h. The reaction was diluted with saturated aqueous NaHCO3, and the layers were separated. The organic layer was dried over MgSO4 and concentrated under reduced pressure. This material was dissolved in 20 mL of THF. Lithium bromide (0.62 g, 7.10 mmol, 2.5 equiv) was added, and the reaction mixture was refluxed for 1 h. The reaction was concentrated under reduced pressure, and the crude product was diluted with EtOAc and washed with water. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash chromatography (Combi-flash Rf, EtOAc/hexanes = 0–100% gradient) to obtain the title compound (0.8 g, 1.5 mmol, 53% yield). 1H NMR (400 MHz, CDCl3) δ 8.36 (d, J = 5.8 Hz, 1H), 8.19 (d, J = 1.7 Hz, 1H), 7.39 (d, J = 1.5 Hz, 1H), 7.36 (s, 1H), 6.91 (d, J = 2.7 Hz, 1H), 6.72 (dd, J = 5.8. 2.7 Hz, 1H), 6.11 (q, J = 7.2 Hz, 1H), 4.51 (s, 2H), 4.25 (q, J = 7.4 Hz, 2H), 3.82 (s, 3H), 3.50–3.44 (m 1H), 3.33–3.30 (m, 1H), 2.71–2.58 (m, 2H), 1.61 (d, J = 7.2 Hz, 3H), 1.54 (t, J = 7.4 Hz, 3H); LCMS (method 2, ESI): Rt = 1.42 min, m/z = 536.8 [M + H]+.
(R)-7-(Bromomethyl)-5-(1-ethyl-3-(trifluoromethyl)-1H-pyrazol-4-yl)-2-(1-(4-methoxypyridin-2-yl)ethyl)-3,4-dihydroisoquinolin-1(2H)-one (74).
General Procedure D was followed using 72 (390 mg, 0.82 mmol, 1 equiv) to obtain the title compound (431 mg, 0.80 mmol, 98% yield), which was used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 8.36 (d, J = 5.8 Hz, 1H), 8.18 (d, J = 1.9 Hz, 1H), 7.38 (d, J = 1.9 Hz, 1H), 7.36 (s, 1H), 6.91 (d, J = 2.4 Hz, 1H), 6.72 (dd, J = 5.8, 2.4 Hz, 1H), 6.10 (q, J = 7.1 Hz, 1H), 4.51 (s, 2H), 4.24 (q, J = 7.3 Hz, 2H), 3.82 (s, 3H), 3.56–3.40 (m, 1H), 3.36–3.28 (m, 1H), 2.73–2.58 (m, 2H), 1.61 (d, J = 7.1 Hz, 3H), 1.55 (t, J = 7.3 Hz, 3H); LCMS (method 2, ESI): Rt = 1.568 min, m/z = 537.0 [M + H]+.
Protein Expression and Purification.
Human WDR5 (aa: 22–334) was cloned into a modified pET27 vector (pBG104) with a 6×His-SUMO tag present at the N terminus. The plasmid was then transformed into E. coli BL21-Gold (DE3) cells. LB starter (100 mL) was used to inoculate a 10 L fermentation culture (BioFlo 415, New Brunswick Scientific), grown at 37 °C. Fermentation growth media contained KH2PO4 (4 g/L), K2HPO4 (6 g/L), Na2SO4 (2 g/L), K2SO4 (1 g/L), NaCl (0.5 g/L), Yeast Extract (5 g/L), glycerol (2 mL/L), Antifoam (0.2 mL/L), 5% LB medium, glucose (25 g/L), MgCl2 (2 mM), CaCl2 (0.1 mM), NH4Cl (2.5 g/L), and Kanamycin (50 μg/mL). When the cell density reached OD600 = 2.0, the temperature was lowered to 25 °C, and WDR5 expression induced by treatment with 1 mM isopropyl-β-d-thioga-lactoside (IPTG) overnight. Cell pellets were collected, dissolved in lysis buffer containing 1× PBS plus 300 mM NaCl, 20 mM imidazole, 5 mM BME, and 10% glycerol, and lysed by homogenization (APV-2000, APV). The lysate was cleared by centrifugation, filtered, and then applied to the Ni-column (140 mL, ProBond, Invitrogen). Bound protein was eluted using an imidazole gradient (0–500 mM). The His-SUMO-tag was cleaved by SUMO protease during dialysis and subsequently eliminated through a second Ni-column. WDR5 protein was then purified by size exclusion chromatography (HiLoad 26/60, Superdex 75, GE Healthcare) using crystallization buffer consisting of 20 mM HEPES, pH 7.0, 250 mM NaCl, and 5 mM DTT. The purity of protein was checked using SDS-PAGE. Purified WDR5 was then concentrated to 10 mg/mL and was stored at −80 °C.
Protein Crystallization, Data Collection, and Structure Refinement.
WDR5 apo- and co-crystals were obtained at 18 °C using the sitting drop vapor diffusion method. The crystallization condition was 0.1 M Bis-Tris or HEPES or HCl-Tris pH 6.0–8.0, 0.2 M ammonium acetate, 20–30% PEG3350. Compound 20 was crystallized under these conditions supplemented with 0.05% benzamidine hydrochloride. Crystals were flash-frozen directly in liquid nitrogen. Diffraction data were collected on the Life Sciences Collaborative Access Team (LS-CAT) 21-ID-D and G beamlines at the Advanced Photon Source (APS) at Argonne National Laboratory. Diffraction data were indexed, integrated, and scaled using HKL2000.46 Molecular replacement was applied using Phaser as implemented in CCP447 using a published structure (PDB code 3EG6). Refinement of the structural models was performed using PHENIX48 along with rounds of manual model building in COOT.49 All structure images were prepared with PyMOL. A summary of the final refinement statistics for structures including compounds 20 and 37 can be found in Table S1.
TR-FRET Competition Assay.
The 10-mer-Thr-FAM (AR-TEVHLRKS-(Ahx-Ahx)(Lys-(5-FAM)))44 peptide was purchased from GeneScript and used without additional purification. TR-FRET emissions were recorded on a BioTek Cytation3 instrument.
For the 10-mer-Thr-FAM peptide TR-FRET assay, LanthaScreen Elite Tb-anti His antibody (Tb-Ab) was purchased from Thermo-Fisher and used at 1 nM. The 10-mer-Thr-FAM peptide was used at 150 nM, while WDR5-His-SUMO tag protein was used at 2 nM. The working assay buffer composition was modified to pH 7.2 (1× phosphate buffered saline, 300 mM NaCl, 0.5 mM TCEP, 0.1% CHAPS). Stock compounds were dispensed to a white, flat-bottom OptiPlate plate (PerkinElmer) using an Echo Liquid Handler. An 11-point, semi-log dilution scheme with a top concentration of 1 μM (0.010 nM low concentration) was used with a final volume of 20 μL. Both the top concentration and the dilution scheme were adjusted to fit the anticipated potency of the compounds. The range of Ki values was calculated using the above probe concentration and assay conditions. The calculated upper and lower Ki limits were ~6.7 ± 0.010 and 0.020 ± 0.010 nM, respectively. Positive control wells (0% displacement) consisted of His-SUMO-WDR5 and 10-mer-Thr-FAM probe/Tb antibody mix occupying column 24, while negative control wells (100% displacement) consisted of the 10-mer-Thr-FAM probe/terbium antibody mix alone occupy column 1. The assay performed with an average Z′ value of 0.7 and was found to be tolerant to up to 5% DMSO, but routinely screened at 1% DMSO.
For IC50 determinations, plates were covered, shielded from light, and incubated for 1 h at room temperature with rocking. For the TR-FRET assay, measurement plates were excited at a wavelength of 340 nm and emission wavelengths of 495 and 520 nm were used. The ratio of the 520/495 wavelengths was used to assess the degree of the FRET signal and resulting peptide displacement. TR-FRET plate positive control wells include column 24 containing 10-mer-Thr-FAM peptide, His-SUMO-WDR5, and Tb-anti-His antibody to measure the maximum signal from the FRET response. The 520/495 emission ratio (TR-FRET) was used to calculate an IC50 (inhibitor concentration at which 50% of the bound peptide is displaced) by fitting the inhibition data using XLFit software (Guilford, U.K.) to single-site binding model. This was converted into a binding inhibition/displacement constant (Ki) using the formula50
where is the concentration of the free inhibitor at 50% inhibition, is the concentration of the free labeled ligand at 50% inhibition, is the concentration of the free protein at 0% inhibition, and represents the dissociation constant of the 10-mer-Thr-FAM probe. The reported values represent four independent replicate determinations ± standard deviation.
Cellular Proliferation Assays.
Cell proliferation was assayed using the Promega CellTiter-Glo Luminescent Kit in white, opaque, flat-bottomed 384-well plates. Two hundred cells were seeded per well for all cell lines in IMDM (K562, MV4:11) or RPMI (MOLM-13) supplemented with 10% fetal bovine serum. The cells were treated with 0.3% DMSO vehicle-only or 22 twofold dilutions of WDR5 inhibitors with a top concentration of 30 μM. The final DMSO concentration was 0.3% in all compound treatment experiments and at least two biological replicates were performed. The total volume of cells with inhibitor was 32 μL per well. Sterile media (30 μL) was added to all of the empty wells around the edge of the plate to prevent evaporation. Plates were incubated at 37 °C, 5% CO2 for 5 days. At the end of incubation, the plates were allowed to reach room temperature before adding 16 μL of CellTiter-Glo reagent per well. Plates were incubated at room temperature, covered from light, for 15 min before the luminescence was measured using the CellTiter-Glo protocol on a Cytation3 plate reader (BioTek, Winooski, VT). The raw luminescence values were normalized to the DMSO vehicle-only wells and PRISM software or XLfit (IDBS, Guildford, United Kingdom) was used to generate GI50 values. The reported GI50 values represent four independent replicate determinations ± standard deviation.
PAMPA Assay.
The passive permeability of 1 and 20 was assessed using a PAMPA assay performed at Curia Global, Inc. (Albany, NY). The artificial membrane was added to the acceptor well and overlaid with phosphate-buffered saline at pH 7.4. The compound (100 μM) was added to the donor plate, which was placed in the acceptor plate and incubated for 5 h at 20 °C. Verapamil was used as a positive control. Compound concentrations were determined by UV–vis absorbance, and the effective permeability (Pe, cm/s) was calculated. Two replicates were obtained, and average Pe values are reported.
Kinetic Solubility Assay.
The solubilities of 34, 36, 37, 38, 40, and 41 were assessed using a kinetic solubility assay performed at Curia Global, Inc. (Albany, NY). The tested compound (100 μM) was incubated for 1 h in 0.1 M phosphate-buffered saline (pH 7.4) and 0.1% DMSO at ambient room temperature. The dissolved concentration was determined by UV–vis absorbance. Verapamil and tamoxifen were used as controls. Two replicates were obtained, and average soluble concentrations (μM) are reported.
Microsomal Intrinsic Clearance Assay.
The metabolic stabilities of 30, 34, and 38 were assessed using a microsomal intrinsic clearance assay performed at Curia Global, Inc. (Albany, NY). The tested compound (1 μM) was incubated with 1 mg/mL of pooled rat liver microsomes, in phosphate buffer at 37 °C. Samples were taken at 0, 5, 10, 15, 30, and 45 min and analyzed by LCMS. Testosterone was used as the positive control. Two replicates were obtained, and average CLint and half-life values are reported.
In Vivo Pharmacokinetic Studies in Rodent.
All in vivo PK assessments were performed by Pharmaron, Inc. (Beijing, P. R. China). The studies were performed according to guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of Pharmaron following the guidance of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). Compound 1 was administered to male CD-1 mice at 3 mg/kg iv (n = 3) and 3 mg/kg po (n = 3) and formulated as follows: iv, 0.6 mg/mL solution in DMSO, PEG400, and saline (v/v/v, 5/50/45); po, 0.3 mg/mL solution in DMSO and 20% captisol in DI water (v/v, 10/90). Compounds 34, 36, 37, 38, 40, and 41 were administered to male CD-1 mice at 3 mg/kg iv (n = 3) and 10 mg/kg po (n = 3). Compounds 34, 36, 37, 38, and 40 were formulated as follows: iv, 0.6 mg/mL solution in DMSO, PEG400, and saline (v/v/v, 5/35/60); po, 1 mg/mL solution in DMSO and 20% captisol in DI water (v/v, 10/90). Compound 41 was formulated as 0.6 and 1 mg/mL solutions in ethanol, tocopherol poly(ethylene glycol) succinate (TPGS), PEG400 and water (v/v/v/v, 5/5/30/60) for iv and po dosing, respectively. Animals given 1 (3 mg/kg iv) experienced signs of severe acute toxicity, such as reduced locomotive activity, lowered body temperature, and lethality. The animals that were administered 34, 36, 37, 38, 40, and 41 behaved normally during the study and showed no adverse effects. Blood samples were collected from all mice at 5, 15, 30 min and 1, 2, 4, 8, and 24 h post-dose. Plasma samples were separated from blood by centrifugation and analyzed by LC-MS/MS.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the Vanderbilt High-Throughput Screening Core facility for compound management services and Vanderbilt University Biomolecular NMR Facility for use of Bruker NMR spectrometers, which receives support from an NIH SIG Grant (1S-10RR025677-01) and Vanderbilt University matching funds. The authors also thank contributing and supporting members of Leidos Biomedical Research at Frederick National Labs for Cancer Research, Curia Global, Inc. and Pharmaron, Inc. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by the Argonne National Laboratory, was supported by U.S. Department of Energy [Contract No. DE-AC02-06CH11357]. Use of the Life Sciences Collaborative Access Team Sector 21 was supported by the Michigan Economic Development Corporation and Michigan Technology Tri- Corridor Grant [grant number 085P1000817].
Funding
This work was generously supported in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Chemical Biology Consortium Contract No. HHSN261200800001E (to S.W.F.), The Vanderbilt Ingram Cancer Center Support grant (NIH: CA68485), CA236733 (to S.W.F. and W.P.T.), CA200709 (to W.P.T.), and startup funds to S.W.F. provided by Vanderbilt University. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
ABBREVIATIONS
- ALL
acute lymphoid leukemia
- AML
acute myeloid leukemia
- ATAC
Ada2-containing
- AUCinf
area under the plasma concentration–time curve from time 0 to infinity
- CRBN
pomalidomide
- GI50
half-maximal growth inhibition
- HAT
histone acetyltransferase
- HMT
histone methyltransferase
- H3K4
histone H3 lysine 4
- histone H3K4me2
me3, H3 lysine 4 di- and tri-methylation
- H4K16
histone H4 lysine 16
- MLL
mixed-lineage leukemia
- NSL
WDR5-containing nonspecific lethal
- PROTACs
proteolysis-targeting chimeras
- RBBP5
retinoblastoma binding protein 5
- RLM
rat liver microsomes
- RPGs
ribosomal protein genes
- TR-FRET
time-resolved fluorescence energy transfer
- WBM
WDR5 binding motif
- WDR5
WD repeat domain 5 protein
- WIN
WDR5 interaction motif
Footnotes
The authors declare the following competing financial interest(s): K.B.T., K.M.M., J.J.M., J.T., J.A., T.L., and S.W.F. are inventors on a patent application that was filed to protect this series of WDR5 inhibitors. All other authors declare no competing interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00195.
Experimental procedures and characterization of 58, S18, and S19; X-ray data collection and refinement statistics; and LCMS traces for 1, 34, 36, 37, 38, 40, and 41 (PDF)
SMILE table of compound structures (CSV)
Accession Codes
Atom coordinates and structure factors for WDR5–ligand complexes can be accessed in the PDB via the following accession codes: 7U9Y, compound 20; 7UAS, compound 37. The authors will release the atomic coordinates upon article publication.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.2c00195
Contributor Information
Kevin B. Teuscher, Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee 37232-0146, United States
Kenneth M. Meyers, Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee 37232-0146, United States
Qiangqiang Wei, Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee 37232-0146, United States.
Jonathan J. Mills, Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee 37232-0146, United States
Jianhua Tian, Molecular Design and Synthesis Center, Vanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, Tennessee 37232-0142, United States.
Joseph Alvarado, Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee 37232-0146, United States.
Jiqing Sai, Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee 37232-0146, United States.
Mayme Van Meveren, Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee 37232-0146, United States.
Taylor M. South, Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee 37232-0146, United States
Tyson A. Rietz, Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee 37232-0146, United States
Bin Zhao, Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee 37232-0146, United States.
William J. Moore, Leidos Biomedical Research, Frederick National Laboratory for Cancer Research, Frederick, Maryland 21701-4907, United States
Gordon M. Stott, Leidos Biomedical Research, Frederick National Laboratory for Cancer Research, Frederick, Maryland 21701-4907, United States
William P. Tansey, Department of Biochemistry and Department of Cell and Developmental Biology, Vanderbilt, University School of Medicine, Nashville, Tennessee 37232-0146, United States
Taekyu Lee, Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee 37232-0146, United States.
Stephen W. Fesik, Department of Biochemistry and Department of Pharmacology, Vanderbilt University School of Medicine, Nashville, Tennessee 37232-0146, United States Department of Chemistry, Vanderbilt University, Nashville, Tennessee 37232-0142, United States.
REFERENCES
- (1).Smith TF; Gaitatzes C; Saxena K; Neer EJ The WD Repeat: A Common Architecture for Diverse Functions. Trends Biochem. Sci. 1999, 24, 181–185. [DOI] [PubMed] [Google Scholar]
- (2).Stirnimann CU; Petsalaki E; Russell RB; Müller, C. W. WD40 Proteins Propel Cellular Networks. Trends Biochem. Sci. 2010, 35, 565–574. [DOI] [PubMed] [Google Scholar]
- (3).Xu C; Min J Structure and Function of WD40 Domain Proteins. Protein Cell 2011, 2, 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Guarnaccia AD; Tansey WP Moonlighting with WDR5: A Cellular Multitasker. J. Clin. Med. 2018, 7, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Couture JF; Collazo E; Trievel RC Molecular Recognition of Histone H3 by the WD40 Protein WDR5. Nat. Struct. Mol. Biol. 2006, 13, 698–703. [DOI] [PubMed] [Google Scholar]
- (6).Li D; Roberts R WD-Repeat Proteins: Structure Characteristics, Biological Function, and Their Involvement in Human Diseases. Cell. Mol. Life Sci. 2001, 58, 2085–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Patel A; Vought VE; Dharmarajan V; Cosgrove MS A Conserved Arginine-Containing Motif Crucial for the Assembly and Enzymatic Activity of the Mixed Lineage Leukemia Protein-1 Core Complex. J. Biol. Chem. 2008, 283, 32162–32175. [DOI] [PubMed] [Google Scholar]
- (8).Odho Z; Southall SM; Wilson JR Characterization of a Novel WDR5-Binding Site That Recruits RbBP5 through a Conserved Motif to Enhance Methylation of Histone H3 Lysine 4 by Mixed Lineage Leukemia Protein-1. J. Biol. Chem. 2010, 285, 32967–32976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Han Z; Guo L; Wang H; Shen Y; Deng XW; Chai J Structural Basis for the Specific Recognition of Methylated Histone H3 Lysine 4 by the WD-40 Protein WDR5. Mol. Cell 2006, 22, 137–144. [DOI] [PubMed] [Google Scholar]
- (10).Chen X; Xu J; Wang X; Long G; You Q; Guo X Targeting WD Repeat-Containing Protein 5 (WDR5): A Medicinal Chemistry Perspective. J. Med. Chem. 2021, 64, 10537–10556. [DOI] [PubMed] [Google Scholar]
- (11).Li Y; Han J; Zhang Y; Cao F; Liu Z; Li S; Wu J; Hu C; Wang Y; Shuai J; Chen J; Cao L; Li D; Shi P; Tian C; Zhang J; Dou Y; Li G; Chen Y; Lei M Structural Basis for Activity Regulation of MLL Family Methyltransferases. Nature 2016, 530, 447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jain BP; Pandey S WD40 Repeat Proteins: Signalling Scaffold with Diverse Functions. Protein J. 2018, 37, 391–406. [DOI] [PubMed] [Google Scholar]
- (13).Wu M; Shu HB MLL1/WDR5 Complex in Leukemogenesis and Epigenetic Regulation. Chin. J. Cancer 2011, 30, 240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Patel A; Dharmarajan V; Cosgrove MS Structure of WDR5 Bound to Mixed Lineage Leukemia Protein-1 Peptide. J. Biol. Chem. 2008, 283, 32158–32161. [DOI] [PubMed] [Google Scholar]
- (15).Hess JL MLL: A Histone Methyltransferase Disrupted in Leukemia. Trends Mol. Med. 2004, 10, 500–507. [DOI] [PubMed] [Google Scholar]
- (16).Dharmarajan V; Lee J-H; Patel A; Skalnik DG; Cosgrove MS Structural Basis for WDR5 Interaction (Win) Motif Recognition in Human SET1 Family Histone Methyltransferases. J. Biol. Chem. 2012, 287, 27275–27289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Alicea-Velázquez NL; Shinsky SA; Loh DM; Lee JH; Skalnik DG; Cosgrove MS Targeted Disruption of the Interaction between WD-40 Repeat Protein 5 (WDR5) and Mixed Lineage Leukemia (MLL)/SET1 Family Proteins Specifically Inhibits MLL1 and SETd1A Methyltransferase Complexes. J. Biol. Chem. 2016, 291, 22357–22372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Thomas LR; Adams CM; Fesik SW; Eischen CM; Tansey WP Targeting MYC through WDR5. Mol. Cell. Oncol. 2020, 7, No. 1709388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Thomas LR; Wang Q; Grieb BC; Phan J; Foshage AM; Sun Q; Olejniczak ET; Clark T; Dey S; Lorey S; Alicie B; Howard GC; Cawthon B; Ess KC; Eischen CM; Zhao Z; Fesik SW; Tansey WP Interaction with WDR5 Promotes Target Gene Recognition and Tumorigenesis by MYC. Mol. Cell 2015, 58, 440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Thomas LR; Adams CM; Wang J; Weissmiller AM; Creighton J; Lorey SL; Liu Q; Fesik SW; Eischen CM; Tansey WP Interaction of the Oncoprotein Transcription Factor MYC with Its Chromatin Cofactor WDR5 Is Essential for Tumor Maintenance. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 25260–25268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chen X; Xie W; Gu P; Cai Q; Wang B; Xie Y; Dong W; He W; Zhong G; Lin T; Huang J Upregulated WDR5 Promotes Proliferation, Self-Renewal and Chemoresistance in Bladder Cancer via Mediating H3K4 Trimethylation. Sci. Rep. 2015, 5, No. 8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Dai X; Guo W; Zhan C; Liu X; Bai Z; Yang Y WDR5 Expression Is Prognostic of Breast Cancer Outcome. PLoS One 2015, 10, No. e0124964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tan X; Chen S; Wu J; Lin J; Pan C; Ying X; Pan Z; Qiu L; Liu R; Geng R; Huang W PI3K/AKT-Mediated Upregulation of WDR5 Promotes Colorectal Cancer Metastasis by Directly Targeting ZNF407. Cell Death Dis. 2017, 8, No. e2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sun W; Guo F; Liu M Up-Regulated WDR5 Promotes Gastric Cancer Formation by Induced Cyclin D1 Expression. J. Cell. Biochem. 2018, 119, 3304–3316. [DOI] [PubMed] [Google Scholar]
- (25).Carugo A; Genovese G; Seth S; Nezi L; Rose JL; Bossi D; Cicalese A; Shah PK; Viale A; Pettazzoni PF; Akdemir KC; Bristow CA; Robinson FS; Tepper J; Sanchez N; Gupta S; Estecio MR; Giuliani V; Dellino GI; Riva L; Yao W; Di Francesco ME; Green T; D’Alesio C; Corti D; Kang Y; Jones P; Wang H; Fleming JB; Maitra A; Pelicci PG; Chin L; DePinho RA; Lanfrancone L; Heffernan TP; Draetta GF In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell Rep. 2016, 16, 133–147. [DOI] [PubMed] [Google Scholar]
- (26).Malek R; Gajula RP; Williams RD; Nghiem B; Simons BW; Nugent K; Wang H; Taparra K; Lemtiri-Chlieh G; Yoon AR; True L; An SS; DeWeese TL; Ross AE; Schaeffer EM; Pienta KJ; Hurley PJ; Morrissey C; Tran PT TWIST1-WDR5- Hottip Regulates Hoxa9 Chromatin to Facilitate Prostate Cancer Metastasis. Cancer Res. 2017, 77, 3181–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Tran PT; Gajula R; Williams R; Malek R; Nugent K; Walker A; Chettiar S; Wang H; Taparra K; Cades J; Herman J A Twist1-MLL-WDR5-HOTTIP Complex Regulates HOXA9 Chromatin to Facilitate Metastasis of Prostate Cancer. Int. J. Radiat. Oncol. 2014, 90, S177. [Google Scholar]
- (28).Sun Y; Bell JL; Carter D; Gherardi S; Poulos RC; Milazzo G; Wong JWH; Al-Awar R; Tee AE; Liu PY; Liu B; Atmadibrata B; Wong M; Trahair T; Zhao Q; Shohet JM; Haupt Y; Schulte JH; Brown PJ; Arrowsmith CH; Vedadi M; MacKenzie KL; Huttelmaier S; Perini G; Marshall GM; Braithwaite A; Liu T WDR5 Supports an N-Myc Transcriptional Complex That Drives a Protumorigenic Gene Expression Signature in Neuroblastoma. Cancer Res. 2015, 75, 5143–5154. [DOI] [PubMed] [Google Scholar]
- (29).Wu Y; Diao P; Li Z; Zhang W; Wang D; Wang Y; Cheng J Overexpression of WD Repeat Domain 5 Associates with Aggressive Clinicopathological Features and Unfavorable Prognosis in Head Neck Squamous Cell Carcinoma. J. Oral Pathol. Med. 2018, 47, 502–510. [DOI] [PubMed] [Google Scholar]
- (30).Cui Z; Li H; Liang F; Mu C; Mu Y; Zhang X; Liu J Effect of High WDR5 Expression on the Hepatocellular Carcinoma Prognosis. Oncol. Lett. 2018, 15, 7864–7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ge Z; Song EJ; Kawasawa YI; Li J; Dovat S; Song C WDR5 High Expression and Its Effect on Tumorigenesis in Leukemia. Oncotarget 2016, 7, 37740–37754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ge Z; Song E; Li J; Dovat S; Song C Clinical Significance of WDR5 High Expression and Its Effect on Tumorigenesis in Adult Leukemia. Blood 2015, 126, 3657. [Google Scholar]
- (33).Lu K; Tao H; Si X; Chen Q The Histone H3 Lysine 4 Presenter WDR5 as an Oncogenic Protein and Novel Epigenetic Target in Cancer. Front. Oncol. 2018, 8, 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Aho ER; Wang J; Gogliotti RD; Howard GC; Phan J; Acharya P; Macdonald JD; Cheng K; Lorey SL; Lu B; Wenzel S; Foshage AM; Alvarado J; Wang F; Shaw JG; Zhao B; Weissmiller AM; Thomas LR; Vakoc CR; Hall MD; Hiebert SW; Liu Q; Stauffer SR; Fesik SW; Tansey WP Displacement of WDR5 from Chromatin by a WIN Site Inhibitor with Picomolar Affinity. Cell Rep. 2019, 26, 2916–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Tian J; Teuscher KB; Aho ER; Alvarado JR; Mills JJ; Meyers KM; Gogliotti RD; Han C; Macdonald JD; Sai J; Shaw JG; Sensintaffar JL; Zhao B; Rietz TA; Thomas LR; Payne WG; Moore WJ; Stott GM; Kondo J; Inoue M; Coffey RJ; Tansey WP; Stauffer SR; Lee T; Fesik SW Discovery and Structure-Based Optimization of Potent and Selective WD Repeat Domain 5 (WDR5) Inhibitors Containing a Dihydroi-soquinolinone Bicyclic Core. J. Med. Chem. 2020, 63, 656–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Karatas H; Li Y; Liu L; Ji J; Lee S; Chen Y; Yang J; Huang L; Bernard D; Xu J; Townsend EC; Cao F; Ran X; Li X; Wen B; Sun D; Stuckey JA; Lei M; Dou Y; Wang S Discovery of a Highly Potent, Cell-Permeable Macrocyclic Peptidomimetic (MM-589) Targeting the WD Repeat Domain 5 Protein (WDR5)−Mixed Lineage Leukemia (MLL) Protein–Protein Interaction. J. Med. Chem. 2017, 60, 4818–4839. [DOI] [PubMed] [Google Scholar]
- (37).Grebien F; Vedadi M; Getlik M; Giambruno R; Grover A; Avellino R; Skucha A; Vittori S; Kuznetsova E; Smil D; Barsyte-Lovejoy D; Li F; Poda G; Schapira M; Wu H; Dong A; Senisterra G; Stukalov A; Huber KVM; Schönegger A; Marcellus R; Bilban M; Bock C; Brown PJ; Zuber J; Bennett KL; Al-awar R; Delwel R; Nerlov C; Arrowsmith CH; Superti-Furga G Pharmacological Targeting of the Wdr5-MLL Interaction in C/EBPα N-Terminal Leukemia. Nat. Chem. Biol. 2015, 11, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Getlik M; Smil D; Zepeda-Velázquez C; Bolshan Y; Poda G; Wu H; Dong A; Kuznetsova E; Marcellus R; Senisterra G; Dombrovski L; Hajian T; Kiyota T; Schapira M; Arrowsmith CH; Brown PJ; Vedadi M; Al-awar R Structure-Based Optimization of a Small Molecule Antagonist of the Interaction Between WD Repeat-Containing Protein 5 (WDR5) and Mixed-Lineage Leukemia 1 (MLL1). J. Med. Chem. 2016, 59, 2478–2496. [DOI] [PubMed] [Google Scholar]
- (39).Chen W; Chen X; Li D; Wang X; Long G; Jiang Z; You Q; Guo X Discovery of a Potent MLL1 and WDR5 Protein-Protein Interaction Inhibitor with in Vivo Antitumor Activity. Eur. J. Med. Chem. 2021, 223, No. 113667. [DOI] [PubMed] [Google Scholar]
- (40).Chen W; Chen X; Li D; Zhou J; Jiang Z; You Q; Guo X Discovery of DDO-2213 as a Potent and Orally Bioavailable Inhibitor of the WDR5-Mixed Lineage Leukemia 1 Protein-Protein Interaction for the Treatment of MLL Fusion Leukemia. J. Med. Chem. 2021, 64, 8221–8245. [DOI] [PubMed] [Google Scholar]
- (41).Wang F; Jeon KO; Salovich JM; Macdonald JD; Alvarado J; Gogliotti RD; Phan J; Olejniczak ET; Sun Q; Wang S; Camper D; Yuh JP; Shaw JG; Sai J; Rossanese OW; Tansey WP; Stauffer SR; Fesik SW Discovery of Potent 2-Aryl-6,7-Dihydro-5 H -Pyrrolo[1,2- a]Imidazoles as WDR5-WIN-Site Inhibitors Using Fragment-Based Methods and Structure-Based Design. J. Med. Chem. 2018, 61, 5623–5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Dölle A; Adhikari B; Krämer A; Weckesser J; Berner N; Berger LM; Diebold M; Szewczyk MM; Barsyte-Lovejoy D; Arrowsmith CH; Gebel J; Löhr F; Dötsch V; Eilers M; Heinzlmeir S; Kuster B; Sotriffer C; Wolf E; Knapp S Design, Synthesis, and Evaluation of WD-Repeat-Containing Protein 5 (WDR5) Degraders. J. Med. Chem. 2021, 64, 10682–10710. [DOI] [PubMed] [Google Scholar]
- (43).Yu X; Li D; Kottur J; Shen Y; Kim HS; Park KS; Tsai YH; Gong W; Wang J; Suzuki K; Parker J; Herring L; Kaniskan HÜ; Cai L; Jain R; Liu J; Aggarwal AK; Wang GG; Jin J A Selective WDR5 Degrader Inhibits Acute Myeloid Leukemia in Patient-Derived Mouse Models. Sci. Transl. Med. 2021, 13, No. eabj1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Karatas H; Townsend EC; Bernard D; Dou Y; Wang S Analysis of the Binding of Mixed Lineage Leukemia 1 (MLL1) and Histone 3 Peptides to WD Repeat Domain 5 (WDR5) for the Design of Inhibitors of the MLL1–WDR5 Interaction. J. Med. Chem. 2010, 53, 5179–5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bia H; Bailey S; Bhumralkar DR; Bi FC; Guo F; He M; Humphries PS; Ling AL; Lou J; Nukul S; Zhou R Fused Phenyl Amido Heterocyclic Compounds. U.S. Patent US7,842,713, November 30, 2010. [Google Scholar]
- (46).Otwinowski Z; Minor W Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (47).Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AGW; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the CCP 4 Suite and Current Developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Adams PD; Grosse-Kunstleve RW; Hung L-W; Ioerger TR; McCoy AJ; Moriarty NW; Read RJ; Sacchettini JC; Sauter NK; Terwilliger TC PHENIX: Building New Software for Automated Crystallographic Structure Determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2002, 58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- (49).Emsley P; Cowtan K Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (50).Nikolovska-Coleska Z; Wang R; Fang X; Pan H; Tomita Y; Li P; Roller PP; Krajewski K; Saito NG; Stuckey JA; Wang S Development and Optimization of a Binding Assay for the XIAP BIR3 Domain Using Fluorescence Polarization. Anal. Biochem. 2004, 332, 261–273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.