

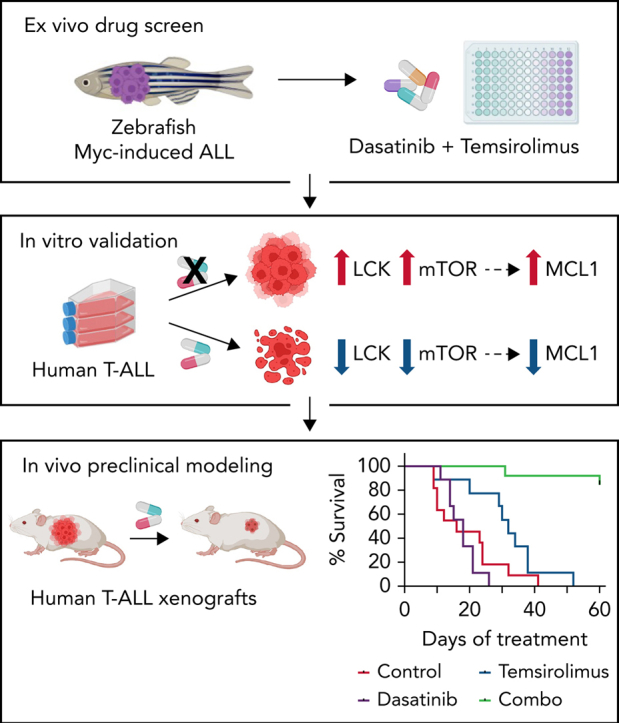
Abstract
Relapse and refractory T-cell acute lymphoblastic leukemia (T-ALL) has a poor prognosis, and new combination therapies are sorely needed. Here, we used an ex vivo high-throughput screening platform to identify drug combinations that kill zebrafish T-ALL and then validated top drug combinations for preclinical efficacy in human disease. This work uncovered potent drug synergies between AKT/mTORC1 (mammalian target of rapamycin complex 1) inhibitors and the general tyrosine kinase inhibitor dasatinib. Importantly, these same drug combinations effectively killed a subset of relapse and dexamethasone-resistant zebrafish T-ALL. Clinical trials are currently underway using the combination of mTORC1 inhibitor temsirolimus and dasatinib in other pediatric cancer indications, leading us to prioritize this therapy for preclinical testing. This combination effectively curbed T-ALL growth in human cell lines and primary human T-ALL and was well tolerated and effective in suppressing leukemia growth in patient-derived xenografts (PDX) grown in mice. Mechanistically, dasatinib inhibited phosphorylation and activation of the lymphocyte-specific protein tyrosine kinase (LCK) to blunt the T-cell receptor (TCR) signaling pathway, and when complexed with mTORC1 inhibition, induced potent T-ALL cell killing through reducing MCL-1 protein expression. In total, our work uncovered unexpected roles for the LCK kinase and its regulation of downstream TCR signaling in suppressing apoptosis and driving continued leukemia growth. Analysis of a wide array of primary human T-ALLs and PDXs grown in mice suggest that combination of temsirolimus and dasatinib treatment will be efficacious for a large fraction of human T-ALLs.
Visual Abstract
Key Points
-
•
Temsirolimus mTORC1 inhibitor and dasatinib kinase inhibitor synergize to kill a large fraction of xenograft and primary human T-ALL.
-
•
The LCK kinase, which is inhibited by dasatinib, has unexpected roles in regulating TCR signaling pathways to drive continued T-ALL growth.
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) affects thousands of children and adults each year, and its incidence is increasing in the United States.1 It is classified into at least 7 distinct molecular subtypes based on their differences in transcription factor activation and arrest at different stages of T-cell development.2, 3, 4 Despite the wide array of oncogenic drivers that initiate this disease, leukemic cells also rely on activation of common pathways for leukemic cell transformation and progression. For example, the phosphatidylinositol 3-kinase (PI3K)/AKT/mTORC1 (mammalian target of rapamycin complex 1) pathway is active in a large fraction of T-ALL and elevates cell proliferation, increases stem cell number, and is strongly associated with treatment resistance.10, 5, 6, 7, 8, 9 This pathway is also acquired during evolution of T-ALL as a resistance mechanism to dexamethasone corticosteroid therapy.8,11, 12, 13 Indeed, refractory and relapse disease, especially in the context of dexamethasone resistance, are the major challenge facing patients. Only 30% of children and less than 10% of adults with refractory and relapse disease will survive.1,10 Therefore, developing new treatments that efficiently kill refractory and relapse T-ALL is a top clinical imperative.
Lymphocyte-specific protein tyrosine kinase (LCK) is required for T-cell development and maturation.14,15 For example, LCK-null mice fail to develop mature T cells and arrest at the CD4−, CD8− (CD44−, CD25+, DN3 stage) stage of thymocyte maturation. These arrested lymphocytes are unable to signal through the T-cell receptor (TCR) and subsequently die in the absence of positive selection.16 Mature T lymphocytes also use LCK to regulate T-cell receptor-mediated responses to antigen, ultimately regulating downstream phosphorylation programs that drive differentiation, proliferation, and/or cytokine secretion.17,18 Despite important roles for LCK in regulating normal T-cell development and its necessity for downstream TCR phosphorylation signaling, its roles in T-ALL are not well understood. Indeed, T-ALLs are arrested during a wide array of T-cell maturation stages, universally express activated LCK irrespective of subtype or mutational spectrum, and yet, many T-ALLs lack functional, membrane localized pre-TCR and TCR.4,19, 20, 21, 22 Moreover, a subset of T-ALLs can sustain loss of LCK and proliferate well in culture,23, 24, 25 leading many to question the role of LCK and subsequent downstream TCR phosphorylation signaling as a driver of T-ALL survival and growth. By contrast, others have shown that glucocorticoid-resistant T-ALLs have high activity of LCK and yet high TCR signaling also kills T-ALL cells.26, 27, 28 Taken together, these seemingly opposing roles of the TCR/LCK signaling pathway in T-ALL have confused the field for decades.
Here, we identified dasatinib and temsirolimus as a potent combination therapy for the treatment of a wide array of human T-ALL, including dexamethasone-resistant disease. Dasatinib inhibits LCK and subsequently the downstream TCR signaling pathway. By contrast, temsirolimus blocks mTORC1, a well-known regulator of proliferation, stemness, and therapy resistance in T-ALL.8,10,12,13,29,30 These 2 pathways converge on the regulation of the MCL1 antiapoptotic gene. We also discovered an unexpected role of LCK in driving continued leukemia growth and suggest that, like maturation of normal T cells that undergo positive and negative selection, T-ALLs require just the right levels of downstream TCR signaling to sustain growth and survival.
Methods
Ex vivo drug screening using transgenic zebrafish ALL
Creation of mosaic CG1-strain transgenic animals, monitoring for ALL onset, and cell transplantation was performed as described (Massachusetts General Hospital (MGH) #2011N000127).31 The lck:mMyc and lck:mCherry transgenes used the 7.5-kb zebrafish lck promoter (Addgene plasmid 58891). Zebrafish ALL cells were harvested, plated into 96-well plates, incubated for 72 hours with drugs and analyzed by CellTiter-Glo assay. IC50 values were assigned using the GraphPad Prism software (GraphPad, San Diego, CA). Combination indexes were calculated using the nonconstant ratio method within the CalcuSyn software package (Biosoft, Cambridge, UK) and synergy assigned if the combination index was <1.0.
Human cell culture and in vitro drug treatments
A total of 1 × 104 to 4 × 104 human T-ALL cells were plated per well in a volume of 100 μL culture media in 96-well plates. Drugs were administered at a log2 or log4 dilution series. Cells were incubated at 37°C, 5% CO2, and cell viability/growth analyzed by CellTiter-Glo after 24 or 72 hours of treatment (Promega, Madison, WI). Combination indexes were calculated using the constant ratio method in the CalcuSyn software package. Cell cycle and proliferation were assessed by flow cytometry using the Click-It EdU Kit (Life Technologies, Carlsbad, CA) and apoptosis using the Annexin V Apoptosis Detection Kit APC (eBioscience, San Diego, CA). Western blot analysis and flow cytometry were completed essentially as described,27 with primary and secondary antibodies noted in the supplemental Methods available on the Blood Web site.
Mouse xenograft studies
Mouse xenografts used luciferase-labeled cell lines and patient-derived xenograft (PDX) models engrafted by tail-vein injection and were approved under animal protocol MGH #2009-P-002756. Cell line engrafted mice were treated after 15 days of engraftment, whereas PDXs were treated when animals contained ≥5% hCD45-positive cells in the peripheral blood. Dasatinib was orally administered (10 mg/kg per day, 80 mM citrate buffer pH 3.1)32 and temsirolimus 8 hours later by intraperitoneal injection (10 mg/kg per day).33 Survival analyses used the Kaplan-Meier log-rank (Mantel-Cox) test using the GraphPad Prism software.
At necropsy, marrow, spleen, and peripheral blood were analyzed for percent blasts and phospho-S6K and phospho-LCK expression by flow cytometry. A portion of the spleen was fixed in 4% paraformaldehyde for histopathologic examination that included hematoxylin and eosin staining, Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), and immunohistochemistry (IHC) for hCD45, pLCK (Y394), and pS6K (T389). The ratio of positively stained cells to unstained cells was calculated automatically in ImageJ34 from 3 separate areas of each spleen. Significance was calculated by unpaired t test.
Analysis of primary patient sample responses to combination therapy
Bone marrow and peripheral blood collection from patients was performed in accordance with the Declaration of Helsinki (Pirkanmaa Hospital District Ethical Committee, ETL code R13109 and ethical committee of Helsinki University Hospital, permits 239/13/03/00/2010 and 303/13/03/01/2011). Viably frozen mononuclear cells were suspended in 12.5% HS-5 conditioned media,35 and 5 × 103 live cells were seeded with MultiFlow FX.RAD (BioTek, Winooski, WT) into compound containing 384-well plates (25 μL). Cell viability was measured using CellTiter-Glo (Promega) and single drug sensitivity was analyzed using drug sensitivity scores (DSS).36 Synergy scores were assigned with CalcuSyn software, and SynergyFinder 2.0 software using the Loewe and ZIP reference models.37
Analysis of human T-ALL samples
Immunophenotype, TCR status, fusion genes, translocations, clinical features, T-ALL subtype, and mutational profiles of T-ALL cell lines were gathered from public databases (https://depmap.org and https://humantallcelllines.wordpress.com/) and the literature.38, 39, 40, 41, 42 Raw RNA-seq data of human T-ALL cell lines were acquired from National Center for Biotechnology Information (NCBI) SRA repository using sra-toolkit (v2.8.2-1) (GSE103046, GSE148522, and NCBI BioProject PRJNA523380).43, 44, 45
Primary patient samples were whole exome and mRNA sequenced and analyzed essentially as previously described.46, 47, 48 Whole genome sequencing, whole exome sequencing, and RNA-seq data of T-ALL patient samples are available in European Genome-phenome Archive under the accession number EGAS00001005945 or in OSFHOME with doi identifier 10.17605/OSF.IO/BK54U. Additional methods are provided as supplemental Material.
Results
Ex vivo screening identifies combination therapies that kill zebrafish T-ALL
To rapidly identify drug sensitivities in T- and B-ALL, we developed an ex vivo screening platform to assess drug-induced cell killing of zebrafish ALL (Figure 1A). The zebrafish ALL model has identified genetic pathways that drive cancer initiation and progression8,49, 50, 51, 52 and have aided in developing therapeutic strategies for human disease.8,53, 54, 55 The rag2:mMyc zebrafish model produces T-, B-, and mixed lineage leukemias,56, 57, 58, 59 whereas the new lck:mMyc transgenic model only develops CD4+/CD8+ clonal T-ALL (supplemental Figure 1). Leukemia cells were harvested from rag2:mMyc and lck:mMyc ALL, plated into 96-well plates (1 × 104 cells per well, purity > 90%), grown for 32 hours at 28.5°C, treated with dimethyl sulfoxide or compounds for 72 hours, and assessed for viability using CellTiter-Glo (Figure 1A). In total, 6 primary and 11 transplanted T-ALLs and 3 transplanted B-ALLs were assessed in the screen (Figures 1B). Ten US Food and Drug Administration–approved and investigational drugs were assessed singly for effects on killing zebrafish ALL, including drugs commonly used as frontline and maintenance therapy including corticosteroids dexamethasone and prednisolone, and common chemotherapy agents like vincristine, doxorubicin, and l-asparaginase and small molecules such as AKT inhibitor MK-2206 and the general tyrosine kinase inhibitor dasatinib. As may be expected, our screening approach revealed wide differences in ALL drug sensitivities and varied leukemia responses to single drugs (Figure 1B-C; supplemental Figure 2). In total, 7 of the 10 drugs killed T-ALL cells from at least 1 model, with the most robust responses observed with glucocorticoids and MK-2206 (Figure 1B-C). Interestingly, primary lck:mMyc leukemia #1 (denoted with orange triangle) and rag2:mMyc leukemia #9 (denoted with blue triangle) were initially killed by dexamethasone and MK-2206, but their corresponding transplanted ALLs were resistant to these drugs (orange and blue circles, respectively; Figure 1B). Similarly, transplanted T-ALLs arising from the same primary leukemia #8 (denoted with green circles) also exhibited differing therapy responses between engrafted leukemia clones. These results are consistent with both xenograft and zebrafish transplant experiments that show refractory T-ALL clones that drive relapse are selected following transplant and the extent to which clonal heterogeneity can drive therapy responses.8,11
Figure 1.

Ex vivo drug screening identifies synergies between dasatinib and AKT inhibitor in killing dexamethasone-resistant zebrafish T-ALL. (A) Schematic of experimental design. (B) Comparison of single and combination drug responses in primary (triangles) and transplanted ALL (circles). T-ALL samples are denoted by filled shapes, whereas B-ALL samples are represented as open shapes. Samples with the same color originate from the same primary leukemia (orange, blue, and green). (C) Representative dose-response curves for sensitive (red line) and insensitive ALLs (blue line) to single drug treatment with dexamethasone (Dex), MK-2206, or dasatinib (Das). (D) Dose-response curves following single drug and combination therapy for a representative, transplanted lck:cMyc T-ALL (L1.5). Green line highlights synergistic drug combinations. (E) Isobolograms normalized to the IC50 of each drug. Combination index average (CIaverage) derived from all experimental data points with <1 indicating synergy. Error bars ± standard error of the mean (SEM). n = 3 samples/data point (C-E). ∗P < .05; ∗∗P < .01 by Student 2-tailed t test.
Given the potent single agent efficacy of dexamethasone, MK-2206, and dasatinib in killing a subset of zebrafish ALLs, we next assessed the pairwise combinations of each of these therapies in killing ALL using our ex vivo screening platform. Marked synergies were seen for each of these combinations (Figure 1B,D-E) and was expected for dexamethasone and MK-2206 or dasatinib and dexamethasone combination therapies that had been reported previously.8,13,26,60 The most synergistic combination, dasatinib and MK-2206 (CIAverage = 0.10) was novel (Figure 1D-E) and had not been previously investigated in the context of T-ALL. Importantly, many samples did not respond to single dasatinib or MK-2206 treatment but were exquisitely sensitive to combination therapy (Figure 1B). In total, 4 of 9 dexamethasone-resistant zebrafish T-ALLs were effectively killed following combination treatment with dasatinib and MK-2206. Our data validate the utility of the high-throughput drug screening approach to identify potential new drug combinations for T-ALL, including dexamethasone-resistant leukemias.
Combination of dasatinib and AKT/mTOR inhibitors kill human T-ALL cells
We next assessed the efficiency of the combination of dasatinib and MK-2206 in human T-ALL, uncovering potent synergies of these drugs in killing Jurkat, MOLT-4, and PF-382 cells (supplemental Figure 3). Neither drug alone was able to efficiently kill any of these T-ALL models. MK-2206 has adverse effects in patients and is not used clinically; thus, we repeated our drug combination experiments using dasatinib along with the clinically available mTORC1 inhibitor temsirolimus in a larger panel of human T-ALL cell lines (Figure 2; supplemental Figure 4). Strong drug synergy in killing T-ALL was seen in 5 of 11 cell line models and included Jurkat, MOLT-4, PF-382, SUPT1, and DND41. This drug synergy was associated with elevated apoptosis and S-phase arrest (Figure 2). The remaining 6 no-synergistic T-ALL cell lines exhibited heterogeneity in responses similar to those found in zebrafish T-ALL (supplemental Figure 4B). There is an ongoing phase 1 clinical trial in children that complexes dasatinib and temsirolimus in advanced malignant solid tumors (NCT02389309), making this combination our top choice for prioritizing for further studies outlined below. These results show that cotargeting the AKT/mTOR pathway along with dasatinib leads to potent and synergistic cell killing in a large subset of human T-ALL by stalling the cell cycle and subsequently inducing cell death.
Figure 2.

Combination of dasatinib and temsirolimus mTOR inhibitor elicit potent cell cycle arrest and leukemia cell killing in human T-ALL. (A) Jurkat. (B) MOLT-4. (C) PF-382. Dose-response curves following single drug and combination therapy (i). Dasatinib and temsirolimus after 72 hours of treatment and assessed by CellTiter-Glo. Combination indexes for ED50 concentrations are shown. Green lines highlight synergistic combination treatments. Cell proliferation assessed by EdU/PI staining (ii). Apoptosis assessed by AnnexinV/PI staining (iii). Error bars ± SEM. n = 3 samples/data point. ∗P < .05; ∗∗P < .01; ∗∗∗P < .001 in comparing single vs combination treated cells (i) or in assessing differences in overall percentages of cells in S-phase or AnnexinV+/PI+ cells by Student 2-tailed t test (ii and iii).
LCK mediates the effect of dasatinib
Because dasatinib is a potent inhibitor of the LCK kinase in normal T cells and chimeric antigen receptor T cells,61, 62, 63 and LCK is required for TCR signaling,17,64,65 we next correlated the suppressed activity of the TCR/LCK pathway following combination treatment with dasatinib and either temsirolimus or MK-2206 (Figure 3A-B; supplemental Figure 3D). Dasatinib alone or in combination elicited potent suppression of LCK phosphorylation at the activating phospho-tyrosine Y394 in Jurkat, MOLT-4, and PF-382 cells. This led to the inhibition of many downstream components of the TCR signaling pathway, including loss of phosphorylation of ZAP70 and LAT, both of which are required downstream of LCK for efficient signaling. By contrast, neither temsirolimus nor MK-2206 treatment had any effect on LCK phosphorylation or subsequent downstream TCR signaling molecules, but instead effectively inhibited the phosphorylation of S6K at residue Y389. Dasatinib treatment also partially decreased AKT phosphorylation that is in line with inhibition of the costimulatory CD28 receptor that elicits low-level crosstalk from LCK to the PI3K pathway but had little effect on phosphorylation of S6K. Our results show that dasatinib treatment results in inhibition of downstream TCR signaling and strongly suggest that LCK kinase is the target of dasatinib in T-ALL.
Figure 3.

Dasatinib inhibits LCK phosphorylation and downstream TCR signaling and induces cell killing when cotreated with temsirolimus. (A) Schematic of TCR and PI3K/AKT/mTOR signaling pathways along with pathway inhibitors. (B) Western blot analysis of T-ALL cells following drug treatment (dasatinib at 100 nM, temsirolimus at 200 nM) for 30 minutes. Representative blot from 3 biological replicates. (C) Western blot showing expression of total LCK and ZAP70 in Jurkat and MOLT-4 wild-type, LCK-deficient (J.Cam1.6 and MOLT-4 clone 12) and ZAP70-deficient (P116) T-ALL cells. Histone H3 (H3) expression used as loading control. (D) Dose-response curves following single drug and combination therapy in LCK- or ZAP70-deficient cells after 72 hours of treatment assessed by CellTiter-Glo. Error bars ± SEM. n = 3 samples/data point. Not significant (ns) by Student 2-tailed t test.
To directly test whether LCK is the target of dasatinib, we next assessed drug effects in LCK-deficient Jurkat (J.Cam1.6) and 3 independent CRISPR-Cas9 edited LCK-null MOLT4 clones (Figure 3C-D; supplemental Figure 5). As expected, LCK knockout cells were efficiently killed by single treatment with MK-2206 AKT-inhibitor or temsirolimus mTOR-inhibitor (IC50 values provided in supplemental Table 1). LCK-null cells exhibited strong sensitization to single agent temsirolimus but lacked synergy when complexed with dasatinib (Figures 3D; supplemental Figure 5). ZAP70 is a kinase required for TCR pathway activity and is a downstream target of LCK. Thus, not surprisingly, ZAP70-deficient Jurkat (P116) cells were also exquisitely sensitive to MK2206 or temsirolimus monotherapy and exhibited complete abrogation of synergy with dasatinib (Figure 3D). LCK phosphorylation was attenuated by dasatinib in ZAP70-deficient T-ALL cells without altering cell viability (Figure 3D; supplemental Figure 5). These results indicate that dasatinib specifically inhibits the LCK signaling pathway and when complexed with inhibitors of AKT/mTOR pathway, potently kills human T-ALL.
Leukemic cells often suppress proapoptotic pathways as a mechanism for transformation and continued tumor maintenance.66, 67, 68 In both T-ALL and normal T-cell development, the antiapoptotic BCL-2 family of proteins are often upregulated to curb cell death69,70 and include specific regulation of BCL-2, BCL-xL and MCL-1. Hence, we examined their expression in T-ALL cell lines in response to mono- and combination therapies (Figure 4A). Although dasatinib monotherapy reduced BCL2 expression in MOLT4 and PF382 cells, and subsequently the phosphorylation of BCL2 (S70), which is required for the full antiapoptotic function of BCL2,71 similar responses were not observed in Jurkat cells (Figure 4A). Moreover, downregulation of BCL2 was not further potentiated by single or cotreatment with temsirolimus, suggesting that BCL-2 downregulation was not responsible for combination therapy-induced T-ALL cell killing. Expression of BCL-xL was not altered by single or combination therapies. By contrast, all 3 synergistic T-ALL cell lines potently suppressed MCL-1 protein expression following combination therapy. As would be predicted, if the TCR/LCK and mTORC1 pathways act in parallel and yet redundantly suppress apoptosis, single drug treatment with either temsirolimus or dasatinib had no impact on MCL1 expression in these models. Strikingly, CCRF-CEM and HPB-ALL cell lines that are not killed by combination dasatinib and temsirolimus also failed to downregulate MCL-1 protein expression following treatment (supplemental Figure 6C). These data again suggest that MCL-1 is the convergent downstream target of LCK and mTORC1 pathways in combination responsive cell lines and supports a model by which nonresponsive cell lines likely activate other pathways to maintain high levels of MCL-1 to support survival and growth. Finally, treatment with 2 MCL-1 inhibitors, AZD5991 and AMG-176, potently induced T-ALL cell killing in Jurkat, MOLT-4, and PF-382 cells that was not further increased by addition of either dasatinib or temsirolimus (Figure 4B-C; supplemental Figure 6A-B). These results are in keeping with potent responses of human T-ALL to MCL-1 monotherapy54,72 and lack of cell death in T-ALL cells treated with BCL2 inhibitor alone.54,73,74 These data again support a model where the drug combination of dasatinib and temsirolimus converges on MCL1 to downregulate its expression and potentiate cell death in a subset of human T-ALLs.
Figure 4.

Combination dasatinib and temsirolimus inhibits MCL-1 expression. (A) Western blot analysis of Jurkat, MOLT-4, and PF-382 T-ALL cells following drug treatment for 120 hours. Representative blot from 3 biological replicates. GAPDH expression used as loading control. (B) Dose-response curves following single drug and combination therapy. Dasatinib, temsirolimus, and MCL-1 inhibitor AZD5991 after 24 hours of treatment and assessed by CellTiter-Glo. Combination indexes for ED50 concentrations are shown (not significant if ≥0.5). (C) AZD5991 potently kills T-ALL cells after 4 days of treatment. AZD5991 (250 nM), dasatinib (500 nM), and temsirolimus (2 μM). Error bars ± SEM. n = 3 samples/data point. ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001 by Student 2-tailed t test.
Dasatinib and temsirolimus kill human T-ALL in mouse xenografts
To provide preclinical rationale for assessing temsirolimus and dasatinib combination therapy in patients, we next extended our analysis of therapy responses to PDX models and primary patient samples. Specifically, NOD/SCID/Il2gr-null mice (NSG) mice were engrafted with 4 PDX models and allowed to progress to have a high leukemia burden. T-ALL cells were isolated from the spleen and submitted to ex vivo drug testing. Following 72 hours of combination treatment, PDX T-ALL cells had reduced cell viability when assessed by CellTiter-Glo assay (Figure 5A), whereas single drug treatments had little effect. Importantly, all 4 PDX explant models responded to the combination therapy. Combination treatment resulted in diminished LCK and S6K phosphorylation in T-ALL cells following 30 minutes (S42512 and TA10) or 24 hours of drug treatment (24836 and 44179; Figure 5B), confirming on target drug responses in PDX explant cultures.
Figure 5.


Primary human T-ALL cells are killed by dasatinib and temsirolimus combination treatment. (A-B) Quantification of combination therapy responses in ex vivo treated patient-derived xenograft cells isolated directly from the spleen of leukemic mice. Dose-response curves following 72 hours of therapy treatment (A). Quantification of phosphorylated LCK+/S6K+ T-ALL cells for PDX 42512 and PDX TA10 following 30 minutes of drug treatment or 24 hours for PDX 44179 and PDX 24836 (B). Error bars denote ± standard deviation in panel A and SEM in panel B, n = 3 replicates per data point and green lines denote synergistic combination treatments. ∗P < .05; ∗∗P < .01; ∗∗∗P < .001 by 2-tailed Student t test. Not significant (ns). (C-D) Primary patient samples following ex vivo combination treatment for 48 hours (ALLT-323, ALLT-373, and ALLT-5221) or 72 hours (ALLT-379, ALLT-6787, and ALLT-7891). Dose-response curves with green lines denote synergistic drug responses (C, average across duplicate samples shown). Synergy plots showing responses of drugs over varied dosing (D). Synergy scores were assigned using the Loewe method and significance of >10 denoting marked synergy (ALLT-323, ALLT-379, ALLT-373, and ALLT-6787).
We next analyzed therapy responses from 9 pediatric and 5 adult T-ALL diagnostic samples (supplemental Table 2; Figure 5C-D). Following ex vivo treatment, 4 primary pediatric T-ALLs responded to combination therapy, 1 of which was resistant to dexamethasone-induced cell killing (ALLT-323; Figure 5C-D). By contrast, none of the adult T-ALLs tested showed similar responses to combination therapy. From analysis of drug responses in cell line models, patient-derived xenografts, and patient samples, we find that 45% of T-ALLs tested exhibited synergistic killing following combination dasatinib and temsirolimus treatment (n = 13 of 29; supplemental Figure 7C). Analysis of responses across T-ALL subtypes and association with specific genetic mutations failed to identify any obvious biomarkers or correlations in predicting therapy responses (supplemental Figure 7; supplemental Table 2), although there appeared to be a trend toward higher TCR/LCK pathway activity in samples that exhibited synergistic therapy responses (supplemental Figure 7B). These results are akin to those recently described in predicting transient dasatinib monotherapy responses in human T-ALL based on high TCR signaling component expression.60
We next assessed the in vivo efficacy of these drugs using mouse xenograft models. First, MOLT-4 and Jurkat cells were lentiviral transfected with a luciferase/dsRED2 reporter and engrafted into immune-deficient NSG mice by tail-vein IV injection. Once animals developed systemic disease by 15 days after engraftment, mice were randomized into vehicle control, single drug treatment, or combination treatment groups. As expected, vehicle-treated animals exhibited progressive disease that was associated with increased leukemia burden and resulted in early morbidity (Figure 6A-D; supplemental Figure 8A-C). Single-drug treatment also failed to efficiently curb human T-ALL growth in vivo. By contrast, combination therapy potently inhibited growth of both Jurkat and MOLT-4 leukemias grown in NSG mice. Mice treated with the drug combination also had reduced numbers of T-ALL cells in peripheral blood, bone marrow, and spleen after extended drug treatment (Figure 6E; supplemental Figure 8D). Flow cytometry also confirmed on-target inhibitory activity of dasatinib and temsirolimus in curbing pLCK and pS6K in human T-ALL cells obtained from the peripheral blood, bone marrow, and spleen of engrafted mice (supplemental Figure 8E). Histopathologic staining confirmed overall increases in apoptotic lymphoblasts in the spleen following combination therapy in both MOLT-4 and Jurkat engrafted animals at the end of treatment (Figure 6F-G; supplemental Figure 8F). Immunohistochemistry also confirmed a decrease of CD45+ cells in combination treated spleens and an overall diminution of fluorescence intensity for pS6K and pLCK in combination treated CD45+ T-ALL cells (Figures 6F,H; supplemental Figure 9).
Figure 6.

Combination of dasatinib and temsirolimus inhibit Jurkat T-ALL growth in mouse xenografts. (A) Schematic of experimental design. (B) Representative images of NSG mice engrafted with luciferase+/dsRED2+ Jurkat cells prior to the first day of treatment (day 0) or imaged after treatment on the days noted within each image panel. (C) Kaplan-Meier survival curves. Combination treated mice had significant survival benefit compared with nontreated or monotherapy-treated mice (∗P < .05; ∗∗P < .01, log-rank, Mantel-Cox test). (D) Average radiance of each individual mice measured by bioluminescence. Squares denote the last radiance measurement of moribund animals. Two of 6 combination-treated mice had undetectable leukemia burden at 60 days. ∗P < .05; ∗∗∗P < .001; by Tukey’s post hoc analysis. (E) Quantification of dsRed+ T-ALL cells by flow cytometry analysis of the spleen, bone marrow (BM), and peripheral blood from engrafted mice. Error bars equal ± SEM. ∗P < .05; ∗∗P < .01 by Tukey’s post hoc analysis. (F) Histopathologic analysis of spleens from control and combination therapy-treated mice. Hematoxylin and eosin (H&E), TUNEL, and co-immunohistochemistry for human CD45 (hCD45, FITC) along with either phospho-LCK or phospho-S6K (Alexa Fluor–594) and DAPI (blue). Scale bars equal 20 μm. Arrows show representative stained cells. (G) Quantification of IHC analysis of spleens denoting the total number of hCD45 T-ALL cells/3 mm2 across replicates (i) and TUNEL (ii). The average percentage of positive cells ± SEM is noted. (H) Quantification of phospho-LCK or phospho-S6K staining in CD45+ T-ALL cells found in the spleen based on IHC staining. Average intensity is denoted by black bars quantified across >3 animals per condition and 3 sections per spleen. More than 3000 cells were analyzed per condition (G-H). ∗∗P < .01; ∗∗∗∗P < .0001 by Student 2-tailed t test (G-H).
To validate the drug combination efficacy in killing patient-derived xenografts as the highest standard for preclinical evaluation, we next engrafted 2 PDXs into immunodeficient NSG mice by IV tail-vein injection (Figure 7A; PDX 44179 and PDX 24836). Animals were treated when ≥5% leukemic blasts were detected in the circulating blood by 15 to 20 days after engraftment. Mice were then randomized into treatment groups and treated for 8 weeks. Blood was analyzed weekly by cheek bleeds, and the amount of leukemia blasts were measured by flow cytometry and quantified as the percentage of human CD45-positive cells present in blood circulation (Figure 7A). Vehicle and monotherapy-treated mice exhibited rapid leukemia onset with high leukemia burden, whereas mice treated with the combination therapy had lower circulating leukemic blasts being found throughout the treatment (Figure 7B-C,G-H). Flow cytometry also confirmed that combination treated animals had lower percentages of CD45+ leukemic blasts in the peripheral blood, bone marrow, and spleen at the end of treatment, even compared with monotherapy-treated mice (Figure 7D,I). TUNEL staining confirmed overall increases in apoptotic lymphoblasts in the spleen following combination therapy in both PDX models (Figures 7E,J; supplemental Figure 10). We also confirmed on-target efficiency of temsirolimus and dasatinib by observing a diminution of fluorescence intensity of pS6K and pLCK in CD45+ T-ALL cells found in the spleen (Figure 7F,K; supplemental Figure 10). There were no adverse effects on overall health, survival, and average body weight between leukemic mice treated with control, single- or double-drug combination over the course of treatment (supplemental Figure 9B), suggesting that the drug combination was well tolerated in mice. These data confirm that dasatinib and temsirolimus kill a subset of patient-derived human T-ALL in vivo.
Figure 7.

Combination of dasatinib and temsirolimus suppresses T-ALL growth in patient-derived xenografts. (A) Schematic of experimental design. (B-F) PDX 441979. (G-K) PDX 24836. (B,G) Leukemia burden assessed by the percentage of CD45+ T-ALL in the peripheral blood of engrafted mice. Combination therapy curbed tumor growth compared with control treated mice. P values denote differences based on 1-way analysis of variance followed by Tukey post hoc test. (C,H) Kaplan-Meier survival curves. (∗P < .05; ∗∗P < .01; ∗∗∗P < .001, log-rank, Mantel-Cox test). (D,I) Flow cytometry quantification of hCD45+ T-ALL cells detected in the spleen, bone marrow (BM), and peripheral blood of engrafted mice at the end of treatment. Error bars equal ± SEM. (E,J) Quantification of spleen sections showing total number of hCD45 T-ALL cells/3 mm2 (i) and TUNEL (ii). Error bars denote ± standard deviation. (F,K) Quantification of phospho-LCK or phospho-S6K staining in CD45+ T-ALL cells found in the spleen based on IHC staining. Average intensity is denoted by black bars quantified across >3 animals per condition and 3 sections per spleen. More than 3000 cells were analyzed per condition. ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001; Student 2-tailed t test (D-F,I-K).
Discussion
We identified that dasatinib suppresses LCK kinase activation and subsequently turns off the TCR signaling pathway to halt cell cycle progression and induce T-ALL cell death. Because T-ALL cells often lack functional TCR75 and LCK-deficient T-ALL cell lines are viable,23 the role of LCK as a critical driver of T-ALL viability was unexpected. We and others have recently identified the exquisite dependency of T-ALL cells on achieving the right balance of TCR signaling to sustain growth. For example, Trinquand et al28 showed that hyperactive TCR signaling kills T-ALL cells, whereas we uncovered that the PRL3 phosphatase suppresses hyperactive T-cell phosphorylation signaling pathways to suppress apoptosis in leukemia cells.27 The results presented here starkly contrast with these studies and rather suggest a Goldilocks scenario where LCK and downstream TCR signaling must be sustained and fine-tuned to stimulate leukemia cell growth. Our data, along with others,60,76 support a model where dasatinib inhibits LCK function by blocking downstream signals from the TCR signaling complex, leading to suppressed proliferation and death in a large fraction of T-ALL. Yet, the dependency of MOLT-4 and PF-382 cells on LCK signaling was unexpected, as these cells lack intact and functional TCR.75,77 As recently suggested by our group,27 downstream phosphorylation of classic TCR signaling molecules can be regulated in a receptor agnostic manner in T-ALL and likely represents a new therapeutic vulnerability for a substantial fraction of human T-ALLs. These data raise the intriguing hypothesis that other tyrosine kinase receptors or downstream mutational activation of TCR pathway components activate LCK in a subset of T-ALL to drive continued leukemia growth and will surely be the subject of future studies.
Our work also uncovered potent drug synergies between dasatinib and temsirolimus in killing a subset of human T-ALL and specifically targeting LCK and S6K phosphorylation, respectively. Dasatinib and other tyrosine kinase inhibitors have been used for the treatment of Philadelphia chromosome positive B-ALL, NUP214-ABL1+ T-ALL, and ABL1 amplified T-ALL,78 suggesting the utility and well-tolerated application of dasatinib in the clinic. Yet, single drug treatment often leads to the acquisition of dasatinib-resistant mutations within the BCR-ABL1 kinase domain79 or the compensatory upregulation of Shp2/Ras/ERK and PI3K/AKT/mTOR pathways.80 Similar results were recently extended to non-ABL1 rearranged human T-ALL, where dasatinib monotherapy lead to only short-term responses in killing human T-ALL in vivo with a large majority of leukemias rapidly progressing on monotherapy.60,81 Additionally, a case report of 1 patient with refractory T-ALL treated with dasatinib monotherapy reported a relapse after few months of treatment.82 In the context of chronic myeloid leukemia, the dual PI3K/mTOR inhibitor NVP-BEZ235 resensitized resistant cells to TKI treatment and induced potent cell killing.80 These data suggest that ABL kinase and mTORC1 lie in the same linear pathway to drive sustained tumor growth in chronic myeloid leukemia. By contrast, our work in T-ALL has uncovered that the TCR/LCK signaling pathway and AKT/mTORC1 largely function in parallel and redundant pathways to inhibit apoptosis and sustain growth in part by modulating MCL-1 expression. In the setting of T-ALL, MCL-1 protein stability can be regulated by TCR signaling and acts independent of transcriptional changes in MCL1.83,84 MCL-1 protein stability is also regulated by growth factors, PI3K, and AKT in wide array of cancers, including human T-ALL.85 For example, chemical epistasis experiments have shown that PI3K inhibition regulates MCL-1 phosphorylation on S159 through a glycogen synthase kinase 3-dependent mechanism, resulting in ubiquitination and proteasomal degradation in human T-ALL.72,86 Future studies will focus on uncovering the precise mechanisms by which each pathway regulates MCL1 expression in T-ALL.
Combination of dasatinib and temsirolimus therapy killed 45% of human T-ALLs tested, was agnostic of T-ALL subtype or mutational spectrum, and killed dexamethasone-refractory zebrafish and human T-ALL. This drug combination was well tolerated over the 8 weeks of treatment with no adverse effects, animal losses, or significant changes in weight across all 4 models tested to date. Indeed, combination of dasatinib and everolimus, an mTOR inhibitor related to temsirolimus with similar in function, was recently evaluated in high-grade pediatric glioma patients and had limited toxicity that included rash, mucositis, and fatigue that did not require dose reduction. No significant adverse events reported.87 In total, our work now provides in vivo preclinical validation for combination of dasatinib and temsirolimus for treatment of aggressive and therapy-resistant T-ALL that will be likely useful for a large fraction of patients.
Finally, we demonstrate that coadministration of dasatinib and AKT/mTORC1 pathway inhibitors synergize to kill human T-ALL, likely in part by the convergent downregulation of the MCL-1, an antiapoptotic protein of the Bcl-2 family. Indeed MCL-1 is translationally upregulated by the mTORC1 pathway in B-cell lymphomas and is regulated at multiple stages of T-cell maturation and following LCK/TCR pathway activation.83,88 Interestingly, MCL-1 dependency is seen in a wide array of hematologic and solid malignancies, including T-ALL.54,72,89,90 However, direct targeting of MCL-1 as a monotherapy in hematologic malignancies results in serious side effects including cardiac toxicity.91 Moreover, MCL1 loss in mouse models results in severe hematologic defects,92,93 impaired neural development,94 and defects in synovial fibroblasts,95 suggesting a low-therapeutic window for the use of MCL-1 inhibitors in the setting of pediatric cancers. Rather, reducing MCL1 expression by targeting tissue-restricted pathways like TCR signaling along with mTORC1 may provide a better and more tractable therapeutic strategy to deploy clinically in T-ALL.
Ultimately, our studies provided new insights into LCK-mediated signaling effects on T-ALL survival and credential dasatinib and temsirolimus as a new combination therapy for killing pediatric T-ALL that should be considered for future clinical evaluation.
Conflict-of-interest disclosure: C.A.H. has received research funding from Celgene, KronosBio, Novartis, Oncopeptides, Orion Pharma, and the IMI2 projects HARMONY and HARMONY PLUS unrelated to this study. All remaining authors declare no competing financial interests.
Acknowledgments
The authors thank the Finnish IT Center for Science and UEF Bioinformatics Center, University of Eastern Finland, Finland, for computation resources; Eini Eskola for assistance in laboratory work; David Weinstock for kindly providing PDX samples; Tony Letai for advice on apoptotic pathways to analyze in treated T-ALLs; Brian Millet and Daniel O’Neill for thoughtful discussions; and the patients for providing samples for our studies. The FIMM Technology Center High Throughput Biomedicine Unit and Sequencing Laboratory are acknowledged for their expert technical support.
This work was supported by National Institutes of Health grant R01CA211734 (D.M.L.), the Massachusetts General Hospital Research Scholar Award (D.M.L.), Academy of Finland (O.L. and M.H., 321553; O.L., 310106), Cancer Foundation Finland (O.L., M.H., C.A.H.), Jane and Aatos Erkko Foundation (O.L., M.H.), Sigrid Juselius Foundation (O.L., M.H., C.A.H.), Aamu Foundation (O.L.), Finnish Hematology Association (S.Laukkanen), Finnish Blood Disease Research Foundation (S.Laukkanen), and the Competitive State Research Financing of the Expert Responsibility Area of Tampere University Hospital (O.L., 9V033 and 9X027). FIMM High Throughput Biomedicine Unit and Sequencing Laboratory are financially supported by the University of Helsinki (HiLife) and Biocenter Finland.
Authorship
Contribution: D.M.L., O.L., S. Laukkanen, and A.V. conceived the study. S. Loontiens, A.V., C.Y., L.O., D.D., A.H.J., E.J.A., L.W., J.R.A., K.M., A.A., and N.H. conducted experiments. S. Loontiens and J.Y. produced gateway plasmids. S.I, S.M., S.A., and S.P.G. analyzed sequencing data. C.Y., K.G., and M.K. advised in animal experiments. A.P. assisted with primary patient samples. T.R. advised with the experimental design and data analysis of primary patient samples. C.A.H. provided primary patient samples and advised with the data. M.H. advised with data. D.L. and O.L. supervised the study. All authors reviewed and accepted the manuscript.
Footnotes
Gene expression data is publicly available in European Genome-phenome Archive under the accession number EGAS00001005945.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Contributor Information
Olli Lohi, Email: olli.lohi@tuni.fi.
David M. Langenau, Email: dlangenau@mgh.harvard.edu.
Supplementary Material
References
- 1.Hao T, Li-Talley M, Buck A, Chen W. An emerging trend of rapid increase of leukemia but not all cancers in the aging population in the United States. Sci Rep. 2019;9(1) doi: 10.1038/s41598-019-48445-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Vlierberghe P, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest. 2012;122(10):3398–3406. doi: 10.1172/JCI61269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Girardi T, Vicente C, Cools J, De Keersmaecker K. The genetics and molecular biology of T-ALL. Blood. 2017;129(9):1113–1123. doi: 10.1182/blood-2016-10-706465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tremblay CS, Hoang T, Hoang T. Early T cell differentiation lessons from T-cell acute lymphoblastic leukemia. Prog Mol Biol Transl Sci. 2010;92(C):121–156. doi: 10.1016/S1877-1173(10)92006-1. [DOI] [PubMed] [Google Scholar]
- 5.Gutierrez A, Sanda T, Grebliunaite R, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009;114(3):647–650. doi: 10.1182/blood-2009-02-206722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lonetti A, Cappellini A, Bertaina A, et al. Improving nelarabine efficacy in T cell acute lymphoblastic leukemia by targeting aberrant PI3K/AKT/mTOR signaling pathway. J Hematol Oncol. 2016;9(1):114. doi: 10.1186/s13045-016-0344-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lynch JT, McEwen R, Crafter C, et al. Identification of differential PI3K pathway target dependencies in T-cell acute lymphoblastic leukemia through a large cancer cell panel screen. Oncotarget. 2016;7(16):22128–22139. doi: 10.18632/oncotarget.8031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blackburn JS, Liu S, Wilder JL, et al. Clonal evolution enhances leukemia-propagating cell frequency in T cell acute lymphoblastic leukemia through Akt/mTORC1 pathway activation. Cancer Cell. 2014;25(3):366–378. doi: 10.1016/j.ccr.2014.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, Easton J, Shao Y, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1211–1218. doi: 10.1038/ng.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Smedt R, Morscio J, Goossens S, Van Vlierberghe P. Targeting steroid resistance in T-cell acute lymphoblastic leukemia. Blood Rev. 2019;38 doi: 10.1016/j.blre.2019.100591. [DOI] [PubMed] [Google Scholar]
- 11.Clappier E, Gerby B, Sigaux F, et al. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J Exp Med. 2011;208(4):653–661. doi: 10.1084/jem.20110105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cantley AM, Welsch M, Ambesi-Impiombato A, et al. Small molecule that reverses dexamethasone resistance in T-cell acute lymphoblastic leukemia (T-ALL) ACS Med Chem Lett. 2014;5(7):754–759. doi: 10.1021/ml500044g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piovan E, Yu J, Tosello V, et al. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell. 2013;24(6):766–776. doi: 10.1016/j.ccr.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiang YJ, Hodes RJ. T-cell development is regulated by the coordinated function of proximal and distal Lck promoters active at different developmental stages. Eur J Immunol. 2016;46(10):2401–2408. doi: 10.1002/eji.201646440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 2004;23(48):7990–8000. doi: 10.1038/sj.onc.1208074. [DOI] [PubMed] [Google Scholar]
- 16.van Oers NSC, Lowin-Kropf B, Finlay D, Connolly K, Weiss A. Alpha beta T cell development is abolished in mice lacking both Lck and Fyn protein tyrosine kinases. Immunity. 1996;5(5):429–436. doi: 10.1016/s1074-7613(00)80499-9. [DOI] [PubMed] [Google Scholar]
- 17.Gascoigne NRJ, Casas J, Brzostek J, Rybakin V. Initiation of TCR phosphorylation and signal transduction. Front Immunol. 2011;2:72. doi: 10.3389/fimmu.2011.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Love PE, Hayes SM. ITAM-mediated signaling by the T-cell antigen receptor. Cold Spring Harb Perspect Biol. 2010;2(6):a002485. doi: 10.1101/cshperspect.a002485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cordo’ V, van der Zwet JCG, Canté-Barrett K, Pieters R, Meijerink JPP T-cell acute lymphoblastic leukemia: a roadmap to targeted therapies. Blood Cancer Discov. 2020;2(1):19–31. doi: 10.1158/2643-3230.BCD-20-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Dongen JJ, Krissansen GW, Wolvers-Tettero IL, et al. Cytoplasmic expression of the CD3 antigen as a diagnostic marker for immature T-cell malignancies. Blood. 1988;71(3):603–612. [PubMed] [Google Scholar]
- 21.Gocho Y, Yang JJ. Genetic defects in hematopoietic transcription factors and predisposition to acute lymphoblastic leukemia. Blood. 2019;134(10):793–797. doi: 10.1182/blood.2018852400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cordo’ V, Meijer MT, Hagelaar R, et al. Phosphoproteomic profiling of T cell acute lymphoblastic leukemia reveals targetable kinases and combination treatment strategies. Nat. Commun. 2022;13(1):1–13. doi: 10.1038/s41467-022-28682-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldsmith MA, Weiss A. Isolation and characterization of a T-lymphocyte somatic mutant with altered signal transduction by the antigen receptor. Proc Natl Acad Sci USA. 1987;84(19):6879–6883. doi: 10.1073/pnas.84.19.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Straus DB, Weiss A. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell. 1992;70(4):585–593. doi: 10.1016/0092-8674(92)90428-f. [DOI] [PubMed] [Google Scholar]
- 25.Vico-Barranco I, Arbulo-Echevarria MM, Serrano-García I, et al. A novel, LAT/Lck double deficient T cell subline J.CaM1.7 for combined analysis of early TCR signaling. Cells. 2021;10(2):1–20. doi: 10.3390/cells10020343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serafin V, Capuzzo G, Milani G, et al. Glucocorticoid resistance is reverted by LCK inhibition in pediatric T-cell acute lymphoblastic leukemia. Blood. 2017;130(25):2750–2761. doi: 10.1182/blood-2017-05-784603. [DOI] [PubMed] [Google Scholar]
- 27.Garcia EG, Veloso A, Oliveira ML, et al. PRL3 enhances T-cell acute lymphoblastic leukemia growth through suppressing T-cell signaling pathways and apoptosis. Leukemia. 2020;35(3):679–690. doi: 10.1038/s41375-020-0937-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trinquand A, Dos Santos NR, Tran Quang C, et al. Triggering the TCR developmental checkpoint activates a therapeutically targetable tumor suppressive pathway in T-cell leukemia. Cancer Discov. 2016;6(9):972–985. doi: 10.1158/2159-8290.CD-15-0675. [DOI] [PubMed] [Google Scholar]
- 29.Evangelisti C, Chiarini F, McCubrey JA, Martelli AM. Therapeutic targeting of mTOR in T-cell acute lymphoblastic leukemia: an update. Int J Mol Sci. 2018;19(7):E1878. doi: 10.3390/ijms19071878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rheingold SR, Tasian SK, Whitlock JA, et al. A phase 1 trial of temsirolimus and intensive re-induction chemotherapy for 2nd or greater relapse of acute lymphoblastic leukaemia: a Children’s Oncology Group study (ADVL1114) Br J Haematol. 2017;177(3):467–474. doi: 10.1111/bjh.14569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blackburn JS, Liu S, Langenau DM. Quantifying the frequency of tumor-propagating cells using limiting dilution cell transplantation in syngeneic zebrafish. J Vis Exp. 2011;53 doi: 10.3791/2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boulos N, Mulder HL, Calabrese CR, et al. Chemotherapeutic agents circumvent emergence of dasatinib-resistant BCR-ABL kinase mutations in a precise mouse model of Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. 2011;117(13):3585–3595. doi: 10.1182/blood-2010-08-301267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teachey DT, Obzut DA, Cooperman J, et al. The mTOR inhibitor CCI-779 induces apoptosis and inhibits growth in preclinical models of primary adult human ALL. Blood. 2006;107(3):1149–1155. doi: 10.1182/blood-2005-05-1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karjalainen R, Pemovska T, Popa M, et al. JAK1/2 and BCL2 inhibitors synergize to counteract bone marrow stromal cell-induced protection of AML. Blood. 2017;130(6):789–802. doi: 10.1182/blood-2016-02-699363. [DOI] [PubMed] [Google Scholar]
- 36.Yadav B, Pemovska T, Szwajda A, et al. Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci Rep. 2014;4(1):1–10. doi: 10.1038/srep05193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ianevski A, Giri AK, Aittokallio T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020;48(W1):W488–W493. doi: 10.1093/nar/gkaa216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagel S, Venturini L, Meyer C, et al. Multiple mechanisms induce ectopic expression of LYL1 in subsets of T-ALL cell lines. Leuk Res. 2010;34(4):521–528. doi: 10.1016/j.leukres.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 39.Kalender Atak Z, De Keersmaecker K, Gianfelici V, et al. High accuracy mutation detection in leukemia on a selected panel of cancer genes. PLoS One. 2012;7(6) doi: 10.1371/journal.pone.0038463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elwood NJ, Green AR, Melder A, Begley CG, Nicola N. The SCL protein displays cell-specific heterogeneity in size. Leukemia. 1994;8(1):106–114. [PubMed] [Google Scholar]
- 41.Drexler HG. Academic Press; 2000. The leukemia-lymphoma cell line factsbook. [Google Scholar]
- 42.Mansour MR, Abraham BJ, Anders L, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346(6215):1373–1377. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ngoc PCT, Tan SH, Tan TK, et al. Identification of novel lncRNAs regulated by the TAL1 complex in T-cell acute lymphoblastic leukemia. Leukemia. 2018;32(10):2138–2151. doi: 10.1038/s41375-018-0110-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaw TI, Dong L, Tian L, et al. Integrative network analysis reveals USP7 haploinsufficiency inhibits E-protein activity in pediatric T-lineage acute lymphoblastic leukemia (T-ALL) Sci Rep. 2021;11(1):1–12. doi: 10.1038/s41598-021-84647-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghandi M, Huang FW, Jané-Valbuena J, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569(7757):503–508. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koskela HLM, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366(20):1905–1913. doi: 10.1056/NEJMoa1114885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dufva O, Pölönen P, Brück O, et al. Immunogenomic landscape of hematological malignancies. Cancer Cell. 2020;38(3):424–428. doi: 10.1016/j.ccell.2020.08.019. [DOI] [PubMed] [Google Scholar]
- 48.Laukkanen S, Oksa L, Nikkilä A, et al. SIX6 is a TAL1-regulated transcription factor in T-ALL and associated with inferior outcome. Leuk Lymphoma. 2020;61(13):3089–3100. doi: 10.1080/10428194.2020.1804560. [DOI] [PubMed] [Google Scholar]
- 49.Blackburn JS, Liu S, Raiser DM, et al. Notch signaling expands a pre-malignant pool of T-cell acute lymphoblastic leukemia clones without affecting leukemia-propagating cell frequency. Leukemia. 2012;26(9):2069–2078. doi: 10.1038/leu.2012.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lobbardi R, Pinder J, Martinez-Pastor B, et al. TOX regulates growth, DNA repair, and genomic instability in T-cell acute lymphoblastic leukemia. Cancer Discov. 2017;7(11):1336–1353. doi: 10.1158/2159-8290.CD-17-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feng H, Stachura DL, White RM, et al. T-lymphoblastic lymphoma cells express high levels of BCL2, S1P1, and ICAM1, leading to a blockade of tumor cell intravasation. Cancer Cell. 2010;18(4):353–366. doi: 10.1016/j.ccr.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mansour MR, He S, Li Z, et al. JDP2: An oncogenic bZIP transcription factor in T cell acute lymphoblastic leukemia. J Exp Med. 2018;215(7):1929–1945. doi: 10.1084/jem.20170484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ridges S, Heaton WL, Joshi D, et al. Zebrafish screen identifies novel compound with selective toxicity against leukemia. Blood. 2012;119(24):5621–5631. doi: 10.1182/blood-2011-12-398818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Z, He S, Look AT. The MCL1-specific inhibitor S63845 acts synergistically with venetoclax/ABT-199 to induce apoptosis in T-cell acute lymphoblastic leukemia cells. Leukemia. 2019;33(1):262–266. doi: 10.1038/s41375-018-0201-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gutierrez A, Pan L, Groen RWJ, et al. Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J Clin Invest. 2014;124(2):644–655. doi: 10.1172/JCI65093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garcia EG, Iyer S, Garcia SP, et al. Cell of origin dictates aggression and stem cell number in acute lymphoblastic leukemia. Leukemia. 2018;32(8):1860–1865. doi: 10.1038/s41375-018-0130-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Borga C, Foster CA, Iyer S, Garcia SP, Langenau DM, Frazer JK. Molecularly distinct models of zebrafish Myc-induced B cell leukemia. Leukemia. 2019;33(2):559–562. doi: 10.1038/s41375-018-0328-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Langenau DM, Traver D, Ferrando AA, et al. Myc-induced T cell leukemia in transgenic zebrafish. Science. 2003;299(5608):887–890. doi: 10.1126/science.1080280. [DOI] [PubMed] [Google Scholar]
- 59.Borga C, Park G, Foster C, et al. Simultaneous B and T cell acute lymphoblastic leukemias in zebrafish driven by transgenic MYC: implications for oncogenesis and lymphopoiesis. Leukemia. 2019;33(2):333–347. doi: 10.1038/s41375-018-0226-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gocho Y, Liu J, Hu J, et al. Network-based systems pharmacology reveals heterogeneity in LCK and BCL2 signaling and therapeutic sensitivity of T-cell acute lymphoblastic leukemia. Nat Cancer. 2021;2(3):284–299. doi: 10.1038/s43018-020-00167-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schade AE, Schieven GL, Townsend R, et al. Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood. 2008;111(3):1366–1377. doi: 10.1182/blood-2007-04-084814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee KC, Ouwehand I, Giannini AL, Thomas NS, Dibb NJ, Bijlmakers MJ. Lck is a key target of imatinib and dasatinib in T-cell activation. Leukemia. 2010;24(4):896–900. doi: 10.1038/leu.2010.11. [DOI] [PubMed] [Google Scholar]
- 63.Suryadevara CM, Desai R, Farber SH, et al. Preventing Lck activation in CAR T cells confers Treg resistance but requires 4-1BB signaling for them to persist and treat solid tumors in nonlymphodepleted hosts. Clin Cancer Res. 2019;25(1):358–368. doi: 10.1158/1078-0432.CCR-18-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rossy J, Williamson DJ, Gaus K. How does the kinase Lck phosphorylate the T cell receptor? Spatial organization as a regulatory mechanism. Front Immunol. 2012;3:167. doi: 10.3389/fimmu.2012.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hwang JR, Byeon Y, Kim D, Park SG. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp Mol Med. 2020;52(5):750–761. doi: 10.1038/s12276-020-0435-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prokop A, Wieder T, Sturm I, et al. Relapse in childhood acute lymphoblastic leukemia is associated with a decrease of the Bax/Bcl-2 ratio and loss of spontaneous caspase-3 processing in vivo. Leukemia. 2000;14(9):1606–1613. doi: 10.1038/sj.leu.2401866. [DOI] [PubMed] [Google Scholar]
- 67.Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci USA. 2004;101(16):6164–6169. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bachmann PS, Piazza RG, Janes ME, et al. Epigenetic silencing of BIM in glucocorticoid poor-responsive pediatric acute lymphoblastic leukemia, and its reversal by histone deacetylase inhibition. Blood. 2010;116(16):3013–3022. doi: 10.1182/blood-2010-05-284968. [DOI] [PubMed] [Google Scholar]
- 69.Gratiot-Deans J, Merino R, Nuñez G, Turka LA. Bcl-2 expression during T-cell development: early loss and late return occur at specific stages of commitment to differentiation and survival. Proc Natl Acad Sci USA. 1994;91(22):10685–10689. doi: 10.1073/pnas.91.22.10685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peirs S, Matthijssens F, Goossens S, et al. ABT-199 mediated inhibition of BCL-2 as a novel therapeutic strategy in T-cell acute lymphoblastic leukemia. Blood. 2014;124(25):3738–3747. doi: 10.1182/blood-2014-05-574566. [DOI] [PubMed] [Google Scholar]
- 71.Ruvolo PP, Deng X, May WS. Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia. 2001;15(4):515–522. doi: 10.1038/sj.leu.2402090. [DOI] [PubMed] [Google Scholar]
- 72.Inuzuka H, Shaik S, Onoyama I, et al. SCFFBW7 regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471(7336):104–109. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Choudhary GS, Al-Harbi S, Mazumder S, et al. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015;6(1) doi: 10.1038/cddis.2014.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Suryani S, Carol H, Chonghaile TN, et al. Cell and molecular determinants of in vivo efficacy of the BH3 mimetic ABT-263 against pediatric acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2014;20(17):4520–4531. doi: 10.1158/1078-0432.CCR-14-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hara J, Benedict SH, Champagne E, Mak TW, Minden M, Gelfand EW. Comparison of T cell receptor alpha, beta, and gamma gene rearrangement and expression in T cell acute lymphoblastic leukemia. J Clin Invest. 1988;81(4):989–996. doi: 10.1172/JCI113453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Laukkanen S, Grönroos T, Pölönen P, et al. In silico and preclinical drug screening identifies dasatinib as a targeted therapy for T-ALL. Blood Cancer J. 2017;7(9) doi: 10.1038/bcj.2017.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sandberg Y, Verhaaf B, van Gastel-Mol EJ, et al. Human T-cell lines with well-defined T-cell receptor gene rearrangements as controls for the BIOMED-2 multiplex polymerase chain reaction tubes. Leukemia. 2007;21(2):230–237. doi: 10.1038/sj.leu.2404486. [DOI] [PubMed] [Google Scholar]
- 78.Crombet O, Lastrapes K, Zieske A, Morales-Arias J. Complete morphologic and molecular remission after introduction of dasatinib in the treatment of a pediatric patient with t-cell acute lymphoblastic leukemia and ABL1 amplification. Pediatr Blood Cancer. 2012;59(2):333–334. doi: 10.1002/pbc.23327. [DOI] [PubMed] [Google Scholar]
- 79.Soverini S, Colarossi S, Gnani A, et al. Resistance to dasatinib in Philadelphia-positive leukemia patients and the presence or the selection of mutations at residues 315 and 317 in the BCR-ABL kinase domain. Haematologica. 2007;92(3):401–404. doi: 10.3324/haematol.10822. [DOI] [PubMed] [Google Scholar]
- 80.Wöhrle FU, Halbach S, Aumann K, et al. Gab2 signaling in chronic myeloid leukemia cells confers resistance to multiple Bcr-Abl inhibitors. Leukemia. 2013;27(1):118–129. doi: 10.1038/leu.2012.222. [DOI] [PubMed] [Google Scholar]
- 81.Shi Y, Beckett MC, Blair HJ, et al. Phase II-like murine trial identifies synergy between dexamethasone and dasatinib in T-cell acute lymphoblastic leukemia. Haematologica. 2021;106(4):1056–1066. doi: 10.3324/haematol.2019.241026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Frismantas V, Dobay MP, Rinaldi A, et al. Ex vivo drug response profiling detects recurrent sensitivity patterns in drug-resistant acute lymphoblastic leukemia. Blood. 2017;129(11):e26–e37. doi: 10.1182/blood-2016-09-738070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dzhagalov I, Dunkle A, He Y.-W. The anti-apoptotic Bcl-2 family member Mcl-1 promotes T lymphocyte survival at multiple stages. J Immunol. 2008;181(1):521–528. doi: 10.4049/jimmunol.181.1.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wensveen FM, van Gisbergen KPJM, Derks IAM, et al. Apoptosis threshold set by Noxa and Mcl-1 after T cell activation regulates competitive selection of high-affinity clones. Immunity. 2010;32(6):754–765. doi: 10.1016/j.immuni.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 85.Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010;584(14):2981–2989. doi: 10.1016/j.febslet.2010.05.061. [DOI] [PubMed] [Google Scholar]
- 86.Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21(6):749–760. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 87.Miklja Z, Yadav VN, Cartaxo RT, et al. Everolimus improves the efficacy of dasatinib in PDGFRα-driven glioma. J Clin Invest. 2020;130(10):5313–5325. doi: 10.1172/JCI133310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mills JR, Hippo Y, Robert F, et al. mTORC1 promotes survival through translational control of Mcl-1. Proc Natl Acad Sci USA. 2008;105(31):10853–10858. doi: 10.1073/pnas.0804821105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Campbell KJ, Mason SM, Winder ML, et al. Breast cancer dependence on MCL-1 is due to its canonical anti-apoptotic function. Cell Death Differ. 2021;28(9):2589–2600. doi: 10.1038/s41418-021-00773-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nangia V, Siddiqui FM, Caenepeel S, et al. Exploiting MCL1 dependency with combination MEK + MCL1 inhibitors leads to induction of apoptosis and tumor regression in KRAS-mutant non-small cell lung cancer. Cancer Discov. 2018;8(12):1598–1613. doi: 10.1158/2159-8290.CD-18-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roberts AW, Wei AH, Huang DCS. BCL2 and MCL1 inhibitors for hematologic malignancies. Blood. 2021;138(13):1120–1136. doi: 10.1182/blood.2020006785. [DOI] [PubMed] [Google Scholar]
- 92.Opferman JT, Letai A, Beard C, et al. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426(6967):671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 93.Opferman JT, Iwasaki H, Ong CC, et al. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307(5712):1101–1104. doi: 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- 94.Arbour N, Vanderluit JL, Le Grand JN, et al. Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J Neurosci. 2008;28(24):6068–6078. doi: 10.1523/JNEUROSCI.4940-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu H, Eksarko P, Temkin V, et al. Mcl-1 is essential for the survival of synovial fibroblasts in rheumatoid arthritis. J Immunol. 2005;175(12):8337–8345. doi: 10.4049/jimmunol.175.12.8337. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.