

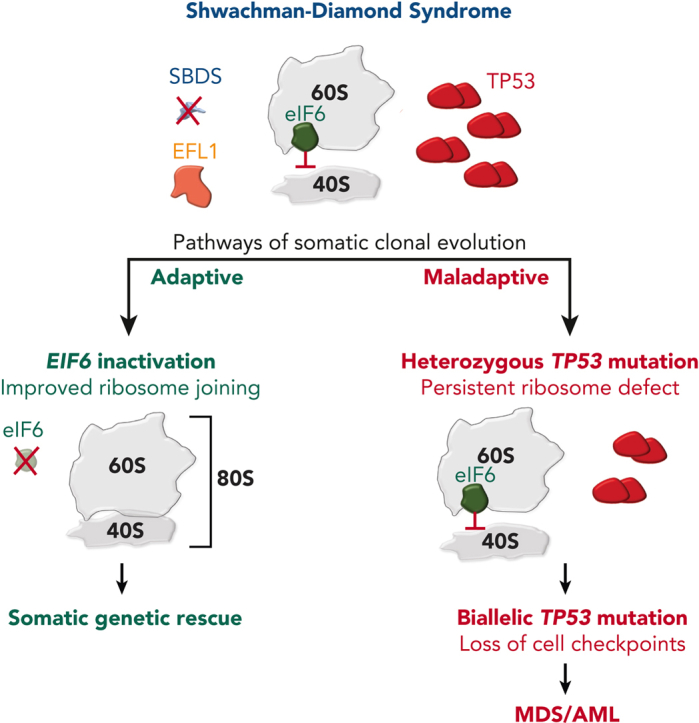
Visual Abstract
Abstract
Shwachman-Diamond syndrome (SDS) is an inherited multisystem ribosomopathy characterized by exocrine pancreatic deficiency, bone marrow failure, and predisposition to myeloid malignancies. The pathobiology of SDS results from impaired ribosomal maturation due to the deficiency of SBDS and the inability to evict the antiassociation factor eIF6 from the 60S ribosomal subunit. Clinical outcomes for patients with SDS who develop myeloid malignancies are extremely poor because of high treatment-related toxicities and a high rate of refractory disease/relapse even after allogeneic hematopoietic stem cell transplant (HSCT). Registry data indicate that outcomes are improved for patients with SDS who undergo routine bone marrow surveillance and receive an HSCT before developing an overt malignancy. However, the optimal approach to hematologic surveillance and the timing of HSCT for patients with SDS is not clearly established. Recent studies have elucidated distinct patterns of somatic blood mutations in patients with SDS that either alleviate the ribosome defect via somatic rescue (heterozygous EIF6 inactivation) or disrupt cellular checkpoints, resulting in increased leukemogenic potential (heterozygous TP53 inactivation). Genomic analysis revealed that most myeloid malignancies in patients with SDS have biallelic loss-of-function TP53 mutations. Single-cell DNA sequencing of SDS bone marrow samples can detect premalignant biallelic TP53-mutated clones before clinical diagnosis, suggesting that molecular surveillance may enhance the detection of incipient myeloid malignancies when HSCT may be most effective. Here, we review the clinical, genetic, and biologic features of SDS. In addition, we present evidence supporting the hematologic surveillance for patients with SDS that incorporates clinical, pathologic, and molecular data to risk stratify patients and prioritize transplant evaluation for patients with SDS with high-risk features.
Routine use of next-generation sequencing of hematologic malignancies has greatly expanded the representation of hereditary predisposition syndromes among patients with leukemia. Introduced by Associate Editor Mario Cazzola, this Review Series highlights 4 such genetic predisposition syndromes and provides strong support for the need to include germ line genetic testing for patients with myelodysplasia and myeloid leukemia.
Introduction
Shwachman-Diamond syndrome (SDS) is an inherited ribosomopathy characterized by exocrine pancreatic deficiency, bone marrow failure (BMF), and predisposition to myeloid malignancies.1, 2, 3 The majority of patients with SDS harbor biallelic germ line mutations in the SBDS gene,4 which is an essential and highly conserved component of the ribosome assembly pathway.5 Over several decades, studies have revealed important insights into ribosome biology, cellular consequences of SBDS deficiency, and molecular pathways of somatic clonal evolution that lead to malignant transformation.5, 6, 7, 8, 9, 10
Clinical manifestations of SDS are highly variable and can affect multiple organ systems.1,2,11 However, all patients with SDS share an increased risk of developing myeloid malignancies.12, 13, 14 Although many patients with SDS are diagnosed during childhood, subtle clinical phenotypes may delay the diagnosis of SDS until early adulthood.11 In an analysis of the Center for International Blood and Marrow Transplant Research (CIBMTR) myelodysplastic syndrome (MDS) database, 4% of young adults under the age of 40 years who received an allogeneic hematopoietic stem cell transplant (HSCT) were found to have clinically unrecognized SDS with biallelic germ line SBDS mutations.15 Clinical outcomes for patients with SDS with myeloid malignancies are exceptionally poor because of high treatment-related toxicities and high rates of refractory disease/relapse even with a HSCT.15,16 Therefore, a detailed understanding of the biological mechanisms of leukemogenesis in SDS may provide opportunities to detect the earliest evidence of an evolving myeloid malignancy when therapeutic intervention is most beneficial.
In this review, we provide an overview of SDS pathobiology, germ line genetics, and recent studies identifying distinct pathways of somatic clonal evolution in SDS that result in somatic rescue vs malignant transformation. Lastly, we provide a framework for hematologic surveillance that incorporates clinical, pathologic, and molecular data to inform clinical decision making before the development of overt myeloid malignancies.
Ribosome biogenesis and the pathobiology of SDS
Ribosomes are ribonucleoprotein complexes composed of ribosomal RNAs (rRNAs) and ribosomal proteins (r-proteins), which perform the essential process of translation of messenger RNA (mRNA) sequences to proteins.17 A human cell contains between 105 and 107 ribosomes that synthesize the diverse repertoire of proteins required for life.18,19 Ribosome biogenesis is highly conserved across all domains of life and represents the most energetically costly processes in actively dividing cells.5,17,19 Consequently, quality control mechanisms and regulatory checkpoints have evolved to ensure efficient ribosome maturation and coordinated translation as a response to changes in gene expression.19
Ribosome biogenesis begins in the nucleolus with transcription of the preribosomal RNA by RNA polymerase I.17 The nascent preribosomal RNA then undergoes extensive folding, biochemical modifications, and nucleolytic processing to generate the mature rRNA components that will eventually be incorporated into the large 60S and small 40S ribosomal subunits. Approximately 300 ribosome assembly factors and small nucleolar RNAs interact with the maturing pre-60S and pre-40S ribosomal subunits throughout transit from the nucleolus to the cytoplasm.5,17,19
The final maturation step of the nascent 60S ribosomal subunit requires the removal of the antiassociation factor eIF6 by the guanosine triphosphate hydrolase (GTPase) EFL1 and its essential cofactor SBDS (Figure 1).5,7 The mechanism of eIF6 eviction from the 60S subunit has been elucidated over the last 2 decades.7,20, 21, 22, 23, 24, 25, 26, 27 Specifically, eIF6 binds to the sarcin-ricin loop (SRL), uL3 (RpL3), uL14 (RpL23), and eL24 (RpL24) on the intersubunit interface of the nascent 60S subunit.7 Bound eIF6 sterically blocks the joining of the 40S ribosomal subunit.7 SBDS (Sdo1 in yeast) is composed of 3 highly conserved domains.28,29 The N-terminal domain I (amino acids 2-96) binds in close proximity to uL16 (RpL10), interacts with the peptidyl transferase center, and extends into the peptide-exit channel.7 Domain II (amino acids 97-170) is composed of a bundle of 3 alpha helices, and domain III (amino acids 171-250) interacts with the SRL and the P-stalk base.5,7,28 The binding of EFL1 to the 60S subunit triggers a 180° conformational rotation of SBDS domain III and the overall structural “accommodation” of EFL1 on the 60S intersubunit surface.5 Bound EFL1 competes with eIF6 for an overlapping binding site on the SRL, leading to the displacement of eIF6 from the 60S subunit.5,7 GTP hydrolysis then results in the dissociation of EFL1 and SBDS from the 60S subunit, which allows the joining of the 40S subunit to form the translationally competent 80S ribosome.7,22,23 After the termination of translation, the 60S and 40S ribosomal subunits dissociate in the cytoplasm, and the adenosine triphosphate–binding cassette protein ABCE1 sequesters the 40S subunit and prevents its rejoining with the 60S subunit.30, 31, 32 A recent study supports a model in which cytoplasmic eIF6 rebinds to mature 60S ribosome subunits to maintain them in an inactive state.9 Therefore, SBDS and EFL1 are required for both the initial maturation of 60S subunits and its subsequent reactivation via the removal of eIF6. For a comprehensive overview of SBDS and ribosome biogenesis, there are several reviews on the topic.5,17
Figure 1.

Defective ribosome subunit joining in SDS. The top panel shows a normal ribosome subunit joining that occurs after eviction of eIF6 by SBDS and EFL1. The bottom panel shows an impaired ribosome joining defect in SDS due to the inability to displace eIF6 from the 60S intersubunit surface.
In the context of SDS, SBDS deficiency results in impaired ribosome subunit joining due to the inability to remove eIF6 from the nascent 60S subunit.5,7 This defect in ribosome maturation leads to an impairment of translational capacity, reduced HSC fitness, and upregulation of TP53 activity.4,33, 34, 35 Increased translational stress leads to tissue-specific stem cell attrition through senescence or apoptosis, which can be rescued by TP53 knockout.15,36, 37, 38, 39, 40 Furthermore, data from cell line models suggest that SBDS deficiency can preferentially affect translation of specific mRNAs, such as CEBPA/B isoforms, essential for hematopoietic differentiation.41 In addition to a cell-intrinsic defect in HSCs, SBDS knockout within the bone marrow stroma of murine models has shown to detrimentally affect HSC function, and stromal cells from patients with SDS fail to sustain hematopoiesis in vitro.42, 43, 44, 45
SDS germ line genetics
Germ line genetic testing is an essential part of the diagnostic evaluation for SDS because clinical manifestations may be subtle and/or overlap with other BMF and immunologic/inflammatory disorders. Biallelic pathogenic mutations in SBDS are observed in 90% of patients with SDS.4,46 The 2 most common SBDS mutations occur within exon 2: c.258>+2T>C and c.183_184TA>CT.4,12, 13, 14 Both mutations result from gene conversion events involving the adjacent pseudogene SBDSP1 and result in markedly reduced or absent expression of functional SBDS protein.4,47 The c.258+2T>C mutation disrupts the 5′ splice site of intron 2 but allows some residual full-length SBDS to be translated, whereas the c.183_184TA>CT mutation introduces an in-frame stop codon at K62. Less common SBDS missense variants disrupt critical interactions with the rRNA within the 60S ribosomal subunit.2,7 Although the majority of SBDS mutations are inherited, ∼10% of SBDS mutations are de novo.
There are several unique challenges to the genetic testing of SDS. First, the SBDSP1 pseudogene shares 97% sequence identity with SBDS.4 This feature complicates mutation identification or estimation of the variant allele fraction (VAF) of SBDS variants because of misaligned reads to SBDSP1. Manual review of the sequencing data is often required to accurately determine the VAF of SBDS mutations. Among individuals with 2 SBDS mutations, it is essential to determine whether variants occur on the same (cis) or separate (trans) alleles.4,48 SDS only occurs when both alleles of the SBDS gene are altered, whereas a single heterozygous SBDS mutation (eg, c.258+2T>C) together with 1 wild-type SBDS allele is insufficient to cause disease.12 Targeted genetic testing of SBDS variants in parents is useful to distinguish between these possibilities. Lastly, if the clinical suspicion of SDS remains high in the absence of detected biallelic SBDS variants, then more extensive genetic testing is warranted to exclude larger structural variants that are missed on most clinical gene panels.12,49,50
Rare germ line variants in other genes involved in ribosome biology, such as EFL1, DNAJC21, and SRP54 cause conditions with overlapping clinical features of SDS (referred to here as SDS-like disorders), including BMF and exocrine pancreatic insufficiency. Homozygous germ line missense mutations in EFL1 have been reported for several patients.51, 52, 53, 54 Biallelic mutations in DNAJC21, which recycles the nuclear export protein proliferation-associated protein 2G4 (PA2G4),55 have been reported in several patients with an SDS-like phenotype.55,56 Most recently, germ line mutations in signal recognition particle 54 (SRP54), a conserved GTPase, have been described in patients with an autosomal dominant condition resembling features of SDS.57,58 SRP54 missense variants reduce protein stability and cotranslational protein secretion via the SRP.57, 58, 59 SRP54 mutations have also been reported to affect the noncanonical splicing of the transcription factor and unfolded protein response mediator XBP1.60
Clinical manifestations of SDS
The clinical manifestations of SDS are highly variable, may involve multiple organ systems, and affect patients across the age spectrum (Table 1). SDS is most commonly diagnosed during early childhood, when patients also present with cytopenias, BMF, recurrent infections, failure to thrive, and steatorrhea.1,61 Neutropenia is the most common blood count abnormality, and granulocyte colony stimulating factor may be required for managing patients with severe neutropenia. Exocrine pancreatic insufficiency (low pancreatic isoamylase and/or fecal elastase) is present in virtually all patients with biallelic SBDS mutations, but not all patients have clinical symptoms of steatorrhea and fat malabsorption. 61,62 Although pancreatic function may improve with age, patients remain with an increased risk of hematologic complications and malignancy.12, 13, 14 Elevated liver transaminases and hepatomegaly may be seen in young children with SDS and typically resolve without treatment.63 Skeletal dysplasia frequently occurs in SDS, including metaphyseal dysostosis, short flared ribs, delayed ossification, and osteopenia.64,65 Additional features of SDS include endocrine abnormalities, inflammatory complications including blepharitis and arthritis, a variety of congenital malformations, cardiac abnormalities, immunologic abnormalities, learning disabilities, and behavioral issues.1,61,66, 67, 68 Case reports of primary solid tumors have also been reported among patients with SDS,69, 70, 71 but further study of solid tumor risk in SDS is needed. Adults with SDS may have more subtle clinical manifestations and only be diagnosed during evaluation for pancreatic lipomatosis,72,73 unexplained cytopenias,12,14 or MDS.15,16 Please refer to several reviews of the clinical manifestations of SDS for additional details.2,3,61,74 The clinical spectra associated with mutations in EFL1, DNAJC21, and SRP54 are emerging, but data are still scant. Therefore, the remainder of this review will focus on patients with biallelic SBDS mutations.
Table 1.
Clinical manifestations and care of patients with SDS
| Hematologic | |
| Peripheral blood cytopenias | CBC with differential |
| Reticulocyte count | |
| BMF | Bone marrow aspirate and biopsy |
| Immunophenotypic analysis by flow cytometry | |
| Conventional karyotype and FISH | |
| Somatic myeloid NGS panel | |
| MDS/AML | Cytoreductive therapy as clinically indicated |
| HLA-typing and evaluation for stem cell transplant as indicated | |
| Immune dysregulation | Serum immunoglobulins (IgG, IgM, IgA, IgE) |
| Lymphocyte subsets (T-cell subsets, B cells, natural killer cells) | |
| Coagulopathy | Prothrombin time |
| Nonhematologic | |
| Exocrine pancreatic dysfunction | Serum pancreatic isoamylase |
| Fecal elastase | |
| Serum levels of vitamins A, D, E, and K, copper, and selenium | |
| Pancreatic enzyme supplementation | |
| Osteopenia/osteoporosis | Consultation with endocrinology as indicated |
| Short stature | Assessment of age-adjusted growth |
| Skeletal abnormalities | Consultation with orthopedics as indicated |
| Cognitive delay | Neurocognitive evaluation as indicated |
| Neurobehavioral conditions | Consultation with behavioral specialist or psychiatry as indicated |
| Genetics | |
| Germ line genetic testing | Consultation with experts in SDS genetics |
| Inherited BMF gene panelincluding SBDS, EFL1, DNAJC21, SRP54 |
CBC, complete blood count; HLA, human leukocyte antigen.
Predisposition to myeloid malignancies
Patients with SDS have an increased risk of developing myeloid malignancies because of the intrinsic fitness defect caused by SBDS deficiency. Although the risk of severe BMF is greatest in early life, the risk of myeloid malignancy increases progressively with age.12, 13, 14 This germ line predisposition was first reported in a small case series of patients with SDS who developed MDS or acute myeloid leukemia (AML).75,76 Subsequent longitudinal data from multiple international registries confirmed that SDS represents a germ line myeloid malignancy predisposition syndrome.12, 13, 14,16,66,77,78 In the French SDS registry, the incidence of myeloid malignancy was ∼30% in patients with SDS by the age of 30 years.13 The 20-year cumulative incidence of MDS/leukemia was 9.8% (95% confidence interval, 3.7-19.5) in the Italian SDS registry with a median follow-up of 12.6 years.14 Among patients in the North American SDS registry, 17% developed a myeloid malignancy at a median age of 18 years (range, 0.5-47 years).12,16 Collectively, these observations underscore the need for longitudinal hematologic surveillance of patients with SDS given the underlying germ line predisposition to myeloid malignancies and the poor clinical outcome after malignancy develops.
Prognosis and treatment of myeloid malignancies in patients with SDS
Long-term outcomes for patients who develop MDS and AML are very poor because of high-risk disease features, including TP53 mutations and complex karyotype,16 as well as a high rate of treatment-related toxicities.15 A retrospective analysis of 36 patients from the North American SDS registry reported a median survival of 7.7 years and 1 year for MDS and AML, respectively.16 The risk of developing myeloid malignancies has been most extensively characterized among patients with SBDS mutations, and the leukemia risk associated with germ line variants in EFL1, DNAJC21, and SRP54 is unknown. However, cases of myeloid malignancies have been reported in patients with DNAJC2179 and SRP54 mutations,80,81 suggesting that these patients may also have a predisposition to leukemia.
There is no standard treatment for MDS or AML in the context of SDS. Numerous treatment regimens are reported in the literature,16 however, no treatment approach has proven to be superior. Patients with SDS often have poor blood count recovery after chemotherapy. In addition, the intensity of chemotherapy and approach to HSCT must be balanced against the high risk of toxicity and mortality. In an analysis of the SDS registry, 54% of patients with MDS received allogeneic HSCT as upfront therapy compared with those with AML who all received cytoreductive chemotherapy.16 Ten of 12 patients with AML were treated with an anthracycline-based regimen, and 2 patients were treated with a hypomethylating agent (HMA).16 The combination of HMA with the BCL2 inhibitor venetoclax is a highly effective treatment for high-risk myeloid malignancies,82 but there is limited published data on the safety and efficacy of this regimen for patients with SDS. Novel therapies with potential efficacy in TP53-mutated MDS and AML are being evaluated in clinical trials. Given the predominance of TP53 somatic mutations, these agents may be efficacious in patients with SDS, but safety and efficacy data are currently lacking. Patients should be referred to open clinical trials available at the time of their malignancy diagnosis.
Allogeneic HSCT is the only potential curative therapy for BMF or myeloid malignancies in patients with SDS.83,84 A consistent finding from retrospective analyses is that long-term outcomes are significantly better for patients who had received transplantation for BMF than for patients with overt myeloid malignancies.16,83 Among 52 patients with SDS in the CIBMTR database, BMF and MDS/AML were the transplant indications for 39 and 13 patients, respectively. Overall survival (OS) at 5-years was 72% for patients with BMF compared with only 15% among those with MDS/AML. In a separate analysis from the Severe Aplastic Anemia Working Party of the European Society for Blood and Marrow Transplantation, survival outcomes were similarly improved for patients with BMF vs those with MDS or AML (5-year OS of 70.7% [BMF] vs 28.8% [MDS/AML]; P = .005), indicating that HSCT before the development of overt malignancy may be beneficial.16,83 Among adult patients with MDS in the CIBMTR database, the 2-year OS was 15% among patients with biallelic germ line SBDS mutations, and all of these patients had somatic TP53 mutations and died within 3 years of receiving the transplant because of disease relapse.15 The choice of conditioning regimen requires careful consideration because patients with SDS are at increased risk of transplant-related toxicity and nonrelapse mortality.16,83 Recently, the Severe Aplastic Anemia Working Party of the European Society for Blood and Marrow Transplantation published expert consensus recommendations advocating for the preferred use of bone marrow grafts and reduced-intensity conditioning regimens for patients with SDS and BMF to mitigate transplant morbidity and mortality.16,85 In addition, related individuals with a heterozygous SBDS mutation should not be excluded as potential stem cell donors because there is no clinical phenotype associated with SBDS carriers.16,83,85
Current published literature supports early consideration of HSCT for patients with SDS with evidence of an evolving myeloid malignancy. However, the risk of transplant-related toxicity and mortality presents a clinical dilemma for providers with respect to the necessity, timing, and approach to transplant in patients with SDS. Recent studies have identified clinical, pathologic, and genomic features predictive of imminent myeloid transformation in order to inform transplant approach and improve long-term outcomes. The following section reviews advances in our understanding of the molecular alterations that contribute to myeloid malignancy in SDS.
Pathways of somatic clonal evolution in SDS
Expansion of somatically mutated HSCs, termed clonal hematopoiesis (CH), is very common among patients with SDS.6,8,86 In contrast to age-associated CH mutations affecting DNA methylation pathways (eg, DNMT3A, TET2),87 CH in SDS is characterized by mutations that alleviate or bypass the intrinsic fitness defect of SBDS deficiency. An understanding of the biological effect and leukemogenic potential of somatic alterations in patients with SDS is an essential aspect of hematologic surveillance. The following section focuses exclusively on recurrent somatic alterations in patients with biallelic SBDS mutations, because there are limited data for patients with germ lines EFL1, DNAJC21, and SRP54.
Cytogenetics abnormalities
Isochromosome 7q (i[7][q10] or i7q) and deletion of the long arm of chromosome 20 (del[20][q11] or del20q) are the most recurrent cytogenetic abnormalities in patients with SDS.88, 89, 90, 91 Isochromosome 7 results in duplication of the SBDS gene (located at 7q11) and preferentially duplicates the hypomorphic c.258+2T>C mutation.92,93 The i7q clone is not associated with an increased risk of progression to MDS.94 However, other chromosome 7 abnormalities (eg, monosomy 7 and del7q) should raise a concern for progression toward myeloid malignancy.95,96 Similar to i7q, del20q is not associated with risk of progression to malignancy.97, 98, 99 Although the size of the deleted region is variable (range, 1.7-26.9 Mb),89 del20q in patients with SDS results in deletion of the EIF6 gene (located at 20q11), which may be an adaptive mechanism, as discussed in the next section.8
EIF6 mutations
Two groups recently identified recurrent somatic EIF6 mutation in patients with SDS.6,8 Complementary functional models from these studies demonstrated that EIF6 mutations lead to improved ribosome joining and translational efficiency in patients with SDS by 2 distinct mechanisms: (1) decreased eIF6 stability or (2) impaired binding of eIF6 to the 60S ribosomal subunit. Notably, the discovery of loss-of-function EIF6 mutations in patients with SDS recapitulates earlier work that characterized spontaneous mutations in Tif6 (yeast ortholog of EIF6) of Sdo1-deficient yeast models that rescued growth, ribosome subunit joining, and protein translation.21,27
In an analysis from the North American SDS registry of paired marrow and fibroblast samples collected longitudinally with annotated clinical outcomes, deep whole-exome sequencing identified recurrent somatic EIF6 mutations in a discovery cohort of 25 patients with SDS.6 Ultrasensitive error-corrected sequencing of a validation cohort of 110 patients with SDS and SDS-like disorders confirmed frequent acquisition of somatic EIF6 mutations that persisted from an early age in patients with biallelic SBDS mutations. The majority of mutations were missense substitutions that occurred throughout the coding region of EIF6 with several hotspots identified, including N106S and R96W. Structural modeling and functional studies showed that somatic EIF6 missense mutations cause reduced protein stability or impaired binding to the 60S ribosomal subunit. Expression of eIF6 N106S and shRNA knock down of EIF6 in SBDS-deficient human CD34+ cells improved myeloid and erythroid colony formation, improved the ribosome assembly defect, and increased protein translation consistent with somatic genetic rescue of hematopoietic function. In addition, single-cell DNA sequencing (scDNA-seq) revealed that EIF6 mutations occur in distinct clones. Importantly, somatic EIF6 mutations were not observed within the malignant clone of patients with SDS with myeloid malignancies, indicating EIF6-mutated clones have limited leukemogenic potential.
In a separate study conducted in the United Kingdom, a similar spectrum of somatic EIF6 missense and truncating mutations were identified among 40 patients with SDS.8 The authors also detected cytogenetic clones with interstitial deletions involving EIF6 and a balanced translocation resulting in the disruption of EIF6 on chromosome 20q. Molecular dynamic simulations predicted that EIF6 missense mutations disrupt critical hydrogen bonds within the tertiary structure and destabilize critical interactions with uL14 and eL24 in the 60S ribosomal subunit. Functional characterization of EIF6 missense mutations in human cell lines and a conditional Sdo1-knockout yeast model showed that somatic EIF6 missense mutations cause impaired protein stability and binding affinity to the 60S ribosomal subunit. In addition, the authors showed that EIF6 mutations rescued the larval lethality of an SBDS-deficient Drosophila model, demonstrating that eIF6 inactivation rescues the developmental impairment caused by SBDS deficiency.8
Taken together, both analyses support a model in which EIF6 inactivation in SBDS-deficient cells results in enhanced cellular fitness by indirect somatic genetic rescue. Moreover, the functional consequence of EIF6 mutations provides a strong biological rationale for the development of eIF6-targeted therapies for patients with SDS.
TP53 mutations
TP53 is an essential tumor suppressor gene located at 17p.13.1 with pleiotropic roles involving cell cycle regulation,100 apoptosis,101 ribosome stress,102 and DNA damage.103 Early studies found that TP53 expression was increased in the bone marrow of patients with SDS compared to patients with other forms of BMF.34,104 Subsequently, recurrent somatic TP53 mutations were detected in the bone marrow of patients with SDS without myeloid malignancies, indicating that SBDS deficiency is a strong selection pressure favoring HSC clones with TP53 mutations.86 Furthermore, the significant enrichment of TP53 mutations in patients with SDS with MDS15 suggested that somatic TP53 mutations are early initiating events that precede the development of a myeloid malignancy. This hypothesis was validated through analysis of serial samples of patients with SDS with and without myeloid malignancies from the North American SDS registry.6 Whole-exome sequencing of TP53-mutated myeloid malignancies demonstrated that all cases had biallelic TP53-inactivating mutations by 1 of 3 mechanisms: (1) deletion of the other TP53 allele on 17p, (2) copy number neutral loss of heterozygosity (CN-LOH) at 17p, or (3) acquisition of a second TP53 coding mutation in the other allele. Among patients with SDS without a myeloid malignancy, somatic TP53 mutations were identified in 45% of patients using ultrasensitive error-corrected next-generation sequencing (NGS) with a range of 0 to 15 mutations per patient.6 TP53 missense mutations clustered within the DNA-binding domain, whereas truncating variants occurred throughout the gene. TP53 knockdown in SBDS-deficient cells reduces p21 expression but does not ameliorate the ribosome joining defect. The distinct biological consequences of somatic TP53 and EIF6 mutations support a model of parallel pathways of somatic clonal evolution in SDS that either alleviate the ribosome defect via somatic rescue (EIF6 inactivation) or disrupt cellular checkpoints, resulting in increased leukemogenic potential (TP53 inactivation).6 Similar to EIF6 mutations, scDNA-seq confirmed that TP53 mutations are predominantly heterozygous and occur in independent clones.6 Importantly, heterozygous TP53-mutated clones can persist at low levels for years without progression to malignancy, indicating that heterozygous TP53 mutations are likely insufficient to cause malignant transformation. However, scDNA-seq can detect small biallelic TP53-mutated subclones in patients with SDS years before a clinical diagnosis of a myeloid malignancy (Figures 2 and 3). In the North American SDS registry analysis,6 a patient with SDS developed AML despite undergoing routine bone marrow surveillance with stable blood counts and no overt dysplasia or cytogenetic abnormalities. Sequencing of banked samples using scDNA-seq identified the biallelic TP53-mutated clone that would eventually transform to leukemia ∼4 years before the clinical diagnosis. This raises the appealing possibility of using scDNA-seq for the early clinical detection of low-level, premalignant subclones with biallelic TP53 mutations that would not be detected using bulk NGS.
Figure 2.

Pathways of somatic clonal evolution in SDS. Somatic blood mutations in TP53 (red) and EIF6 (green) are recurrent in patients with SDS and confer a selective advantage to HSC via distinct mechanisms. Inactivating EIF6 mutations relieve the fitness defect caused by SBDS deficiency by improving ribosome joining and translation efficiency. In contrast, heterozygous TP53 mutations improve HSC fitness by decreasing the cell checkpoint activation (eg, p21) but do not improve the underlying ribosome joining defect. Acquisition of a second TP53 mutation (ie, biallelic TP53-mutated clone) results in the complete loss of cellular checkpoints, genomic instability, and eventual progression to a myeloid malignancy.
Figure 3.

Clonal dynamics of somatic mutations in SDS. (A) Representative example of clinical NGS in a patient with SDS undergoing serial bone marrow examinations. TP53- and EIF6-mutated clones are colored red and green, respectively. Solid red line corresponds to the biallelic TP53 clone that eventually progresses to MDS. Light-blue bar highlights year 3 of bone marrow surveillance. (B) scDNA-seq at year 3 of surveillance. Each row represents a unique EIF6- and TP53-mutated clones with VAF as a percentage below. Each dot corresponds to a single cell belonging to a given clone. Heterozygous mutations have a VAF of ∼0.5. The TP53 mutation in clone 3 has a VAF of ∼1.0, indicating biallelic TP53 mutation. The biallelic TP53-mutated clone expands exponentially and represents the dominant clone at the time of MDS diagnosis. This scenario demonstrates the ability of scDNA-seq to reveal the clonal hierarchy of somatic mutations and identify high-risk TP53 mutations years before the clinical diagnosis of a myeloid malignancy. In addition, we provide a specific clinical example in the text describing a patient with SDS who was found to have a detectable biallelic TP53-mutated clone using scDNA-seq that eventually gave rise to leukemia despite no other concerning clinicopathologic or cytogenetic features during routine surveillance of bone marrow and blood counts a few months preceeding leukemia diagnosis.
Other recurrent somatic mutations
Several other genes are recurrently mutated in bone marrow cells of patients with SDS. Somatic mutations in PRPF8 and CSNK1A1 were identified in 12% and 6% of patients with SDS, respectively.6 The clinical significance of these gene mutations in patients with SDS remains to be determined. Furthermore, although TP53 inactivation is the dominant pathway of malignant transformation in SDS, cases of MDS and AML have been reported in the absence of somatic TP53 mutations.6 Therefore, a myeloid NGS gene panel is recommended over TP53 mutation testing alone for molecular surveillance.
Approach to hematologic surveillance in SDS
The abysmal prognosis of myeloid malignancy in SDS contrasted with the excellent outcomes of HSCT before a malignant transformation provides a compelling rationale for hematologic surveillance and early intervention for patients at high risk. The current approach to hematologic surveillance for patients with SDS includes peripheral blood count monitoring, serial bone marrow examinations, and molecular NGS data (Table 2). Peripheral blood counts of patients with SDS vary across the age spectrum and in response to critical stressors.12 Serial monitoring of patients’ CBC with differential every 3 to 6 months can identify consistent trends that may indicate a worsening bone marrow function. However, patients with SDS may not exhibit significant blood count changes before developing a myeloid malignancy,16 which underscores the need for routine bone marrow evaluation.
Table 2.
Recommended hematologic surveillance for patients with SDS
| Goal of surveillance: identify patients with high risk features before the development of overt myeloid malignancies | ||
|---|---|---|
| High risk features: | ||
| 1) Falling blood counts, particularly with increasing bone marrow cellularity | ||
| 2) Progressive bone marrow dysplasia (particularly in the erythroid lineage) | ||
| 3) Large and/or rapidly expanding TP53-mutated clones | ||
| 4) High-risk cytogenetic abnormalities (eg, -17p, -7/-7q, complex karyotype) |
| Clinical test | Frequency | Comments |
|---|---|---|
| Peripheral blood monitoring | Every 3-6 mo | |
| CBC with differential | CBC monitoring alone is inadequate for hematologic surveillance of patients with SDS | |
| Reticulocyte count | ||
| Bone marrow examination | Every 12 mo∗ | |
| Aspirate | Assessment of morphologic dysplasia and blast count | |
| Core biopsy | Evaluation of marrow cellularity, dysplasia, and blast count | |
| Flow cytometry | Characterization of immature and aberrant immunophenotypic myeloid populations | |
| Somatic NGS panel | Detection of pathogenic somatic mutations, particularly involving TP53 | |
| Conventional karyotype | Detection of clonal cytogenetic abnormalities (eg, i7q, del20q) | |
| FISH | Increases sensitivity of detecting cytogenetic abnormalities | |
| TP53 immunostain | Optional | Screen for TP53 missense mutations |
| Microarray | Optional | Detection of copy number alterations, particularly involving 17p |
A shorter interval between bone marrow biopsies may be indicated if HRFs are present.
Bone marrow evaluation should include a marrow aspirate, core biopsy, flow cytometry, conventional karyotype, fluorescence in situ hybridization (FISH), and somatic NGS. Frequent baseline bone marrow abnormalities in patients with SDS include overall marrow hypocellularity and dyspoiesis affecting the myeloid lineage with nuclear hyposegmentation and hypogranulation.16 High-risk features (HRFs) that should raise some concern for progression to MDS include multilineage dysplasia, particularly in the erythroid lineage, high-risk cytogenetic abnormalities, and increasing marrow cellularity despite falling peripheral blood counts. Based on the known leukemogenic potential of TP53 mutations, we also recommend somatic NGS as a component of routine hematologic surveillance for patients with SDS. If concerning bone marrow findings are identified, a follow-up marrow exam is helpful to determine whether changes are transient, persistent, or progressive. Additional studies including TP53 immunostain and microarray may be useful in assessing TP53 mutation clonal size and allelic state.105,106 Although additional data are needed, current data suggest that routine marrow surveillance can identify patients with HRFs having good transplant outcomes in a disease with a terrible prognosis after development of overt myeloid malignancy. Thoughtful discussion between clinicians experienced with SDS and patients/families is needed to weigh the risks and benefits of transplant vs continued monitoring.
There are several important considerations with respect to interpreting clinical NGS results in patients with SDS. First, scDNA-seq is not currently available clinically, and bulk NGS cannot reliably determine the allelic state of TP53-mutated clones. Therefore, we recommend integration of serial NGS data along with other clinicopathological parameters to identify patients with SDS with an exceptionally high risk of malignant transformation (Table 2). In the North American SDS registry analysis, the median VAF of TP53 mutations in patients without a myeloid malignancy was 0.0044 (range, 0.002-0.193), which is below the limit of detection of most clinical NGS assays. In our experience, somatic TP53 mutations detected by standard clinical NGS without error correction (limit of detection, 1%-5% VAF) and particularly mutations that expand over a short interval should raise concern for potential progression toward a myeloid malignancy. Bone marrow assessment of TP53 mutation clonal dynamics is preferred over peripheral blood for several reasons. Although peripheral blood NGS can reliably detect pathogenic mutations present in the bone marrow, this assessment does not allow integration of the other important variables obtained with bone marrow biopsy, including overall cellularity, blast count, cytogenetic abnormalities, and morphologic dysplasia. In addition, the VAF of TP53 mutations measured from peripheral blood can significantly underestimate the TP53-mutated clone size in the bone marrow, particularly for patients with SDS with severe neutropenia. In a study of 164 patients with hematologic malignancies, paired peripheral blood and bone marrow samples were analyzed for concordance using NGS.107 Despite a high degree of concordance for the detection of mutations (ie, presence or absence), the VAF in the peripheral blood was frequently less than the VAF in the bone marrow. For these reasons, we recommend serial bone marrow assessment for accurate and consistent assessment of the clonal burden and dynamics of TP53 somatic mutations in patients with SDS. Given the complexities involved in monitoring patients with SDS, we encourage patients and providers to contact SDS experts for additional resources and assistance with interpretation of NGS results.
Conclusions and future directions
Our understanding of SDS and its associated leukemia predisposition has increased dramatically since the first clinical description 50 years ago.108, 109, 110 The genetic basis of SDS attributes to germ line mutations in SBDS, and genetic testing is now integrated into the diagnostic evaluation of patients with a BMF or myeloid malignancy. SBDS deficiency is a powerful selection pressure that promotes both compensatory (EIF6 inactivation) and leukemogenic (TP53 inactivation) pathways of somatic clonal evolution in HSCs. This molecular understanding has revealed the significance of incorporating serial NGS into routine hematologic surveillance of patients with SDS. In particular, scDNA-seq is a promising tool that may enhance detection of high-risk TP53-mutated clones, and studies are currently underway. The survival rate is poor for patients with SDS who develop a myeloid malignancy; therefore, close monitoring and early referral for HSCT is recommended for patients with SDS with high-risk features. Similar biology-driven surveillance approaches may also be beneficial for other germ line leukemia predisposition syndromes.
Conflict-of-interest disclosure: C.R.R. has provided consulting for RenBio. A.S. declares no competing financial interests.
Acknowledgments
The authors thank Coleman Lindsley and Kasiani Myers for the critical reading of the manuscript. The authors also thank the patients and families affected with SDS who have contributed to our collective understanding of this rare disorder.
This work by A.S. and C.R.R. was supported by National Institutes of Health (NIH), National Institute of Diabetes and Digestive and Kidney Diseases grant 5RC2DK122533. C.R.R. is also supported by the Myeloid Malignancies SPORE Career Enhancement Program, Leukemia and Lymphoma Society Special Fellow Award, and the Edward P. Evans Fund for MDS Research.
Authorship
Contribution: A.S. and C.R.R. contributed equally to the text, figures, and tables of this manuscript.
References
- 1.Burroughs L, Woolfrey A, Shimamura A. Shwachman-Diamond syndrome: a review of the clinical presentation, molecular pathogenesis, diagnosis, and treatment. Hematol Oncol Clin North Am. 2009;23(2):233–248. doi: 10.1016/j.hoc.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Myers KC, Davies SM, Shimamura A. Clinical and molecular pathophysiology of Shwachman-Diamond syndrome: an update. Hematol Oncol Clin North Am. 2013;27(1):117–128. doi: 10.1016/j.hoc.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson AS, Myers KC. Diagnosis, treatment, and molecular pathology of Shwachman-Diamond syndrome. Hematol Oncol Clin North Am. 2018;32(4):687–700. doi: 10.1016/j.hoc.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 4.Boocock GRB, Morrison JA, Popovic M, et al. Mutations in SBDS are associated with Shwachman–Diamond syndrome. Nat Genet. 2002;33(1):97–101. doi: 10.1038/ng1062. [DOI] [PubMed] [Google Scholar]
- 5.Warren AJ. Molecular basis of the human ribosomopathy Shwachman-Diamond syndrome. Adv Biol Regul. 2018;67:109–127. doi: 10.1016/j.jbior.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kennedy AL, Myers KC, Bowman J, et al. Distinct genetic pathways define pre-malignant versus compensatory clonal hematopoiesis in Shwachman-Diamond syndrome. Nat Commun. 2021;12(1):1334. doi: 10.1038/s41467-021-21588-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weis F, Giudice E, Churcher M, et al. Mechanism of eIF6 release from the nascent 60S ribosomal subunit. Nat Struct Mol Biol. 2015;22(11):914–919. doi: 10.1038/nsmb.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan S, Kermasson L, Hilcenko C, et al. Somatic genetic rescue of a germline ribosome assembly defect. Nat Commun. 2021;12(1):5044. doi: 10.1038/s41467-021-24999-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaako P, Faille A, Tan S, et al. eIF6 rebinding dynamically couples ribosome maturation and translation. Nat Commun. 2022;13(1):1562. doi: 10.1038/s41467-022-29214-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai FD, Lindsley RC. Clonal hematopoiesis in the inherited bone marrow failure syndromes. Blood. 2020;136(14):1615–1622. doi: 10.1182/blood.2019000990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson AS, Giri N, Gianferante DM, et al. Shwachman Diamond syndrome: narrow genotypic spectrum and variable clinical features. Pediatr Res. 2022;92(6):1671–1680. doi: 10.1038/s41390-022-02009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furutani E, Liu S, Galvin A, et al. Hematologic complications with age in Shwachman-Diamond syndrome. Blood Adv. 2022;6(1):297–306. doi: 10.1182/bloodadvances.2021005539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donadieu J, Fenneteau O, Beaupain B, et al. Classification of and risk factors for hematologic complications in a French national cohort of 102 patients with Shwachman-Diamond syndrome. Haematologica. 2012;97(9):1312–1319. doi: 10.3324/haematol.2011.057489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cesaro S, Pegoraro A, Sainati L, et al. A prospective study of hematologic complications and long-term survival of Italian patients affected by Shwachman-Diamond syndrome. J Pediatr. 2020;219:196–201.e1. doi: 10.1016/j.jpeds.2019.12.041. [DOI] [PubMed] [Google Scholar]
- 15.Lindsley RC, Saber W, Mar BG, et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N Engl J Med. 2017;376(6):536–547. doi: 10.1056/NEJMoa1611604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Myers KC, Furutani E, Weller E, et al. Clinical features and outcomes of patients with Shwachman-Diamond syndrome and myelodysplastic syndrome or acute myeloid leukaemia: a multicentre, retrospective, cohort study. Lancet Haematol. 2020;7(3):e238–e246. doi: 10.1016/S2352-3026(19)30206-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baßler J, Hurt E. Eukaryotic ribosome assembly. Annu Rev Biochem. 2019;88:281–306. doi: 10.1146/annurev-biochem-013118-110817. [DOI] [PubMed] [Google Scholar]
- 18.Shi Z, Fujii K, Kovary KM, et al. Heterogeneous ribosomes preferentially translate distinct subpools of mRNAs genome-wide. Mol Cell. 2017;67(1):71–83.e7. doi: 10.1016/j.molcel.2017.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shore D, Albert B. Ribosome biogenesis and the cellular energy economy. Curr Biol. 2022;32(12):R611–R617. doi: 10.1016/j.cub.2022.04.083. [DOI] [PubMed] [Google Scholar]
- 20.Ceci M, Gaviraghi C, Gorrini C, et al. Release of eIF6 (p27BBP) from the 60S subunit allows 80S ribosome assembly. Nature. 2003;426(6966):579–584. doi: 10.1038/nature02160. [DOI] [PubMed] [Google Scholar]
- 21.Burwick N, Coats SA, Nakamura T, Shimamura A. Impaired ribosomal subunit association in Shwachman-Diamond syndrome. Blood. 2012;120(26):5143–5152. doi: 10.1182/blood-2012-04-420166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gartmann M, Blau M, Armache J-P, et al. Mechanism of eIF6-mediated inhibition of ribosomal subunit joining. J Biol Chem. 2010;285(20):14848–14851. doi: 10.1074/jbc.C109.096057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finch AJ, Hilcenko C, Basse N, et al. Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 2011;25(9):917–929. doi: 10.1101/gad.623011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bécam AM, Nasr F, Racki WJ, Zagulski M, Herbert CJ. Ria1p (Ynl163c), a protein similar to elongation factors 2, is involved in the biogenesis of the 60S subunit of the ribosome in Saccharomyces cerevisiae. Mol Genet Genomics. 2001;266(3):454–462. doi: 10.1007/s004380100548. [DOI] [PubMed] [Google Scholar]
- 25.Senger B, Lafontaine DL, Graindorge JS, et al. The nucle(ol)ar Tif6p and Efl1p are required for a late cytoplasmic step of ribosome synthesis. Mol Cell. 2001;8(6):1363–1373. doi: 10.1016/s1097-2765(01)00403-8. [DOI] [PubMed] [Google Scholar]
- 26.Wong CC, Traynor D, Basse N, Kay RR, Warren AJ. Defective ribosome assembly in Shwachman-Diamond syndrome. Blood. 2011;118(16):4305–4312. doi: 10.1182/blood-2011-06-353938. [DOI] [PubMed] [Google Scholar]
- 27.Menne TF, Goyenechea B, Sánchez-Puig N, et al. The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat Genet. 2007;39(4):486–495. doi: 10.1038/ng1994. [DOI] [PubMed] [Google Scholar]
- 28.Shammas C, Menne TF, Hilcenko C, et al. Structural and mutational analysis of the SBDS protein family. Insight into the leukemia-associated Shwachman-Diamond Syndrome. J Biol Chem. 2005;280(19):19221–19229. doi: 10.1074/jbc.M414656200. [DOI] [PubMed] [Google Scholar]
- 29.Savchenko A, Krogan N, Cort JR, et al. The Shwachman-Bodian-Diamond syndrome protein family is involved in RNA metabolism. J Biol Chem. 2005;280(19):19213–19220. doi: 10.1074/jbc.M414421200. [DOI] [PubMed] [Google Scholar]
- 30.Pisarev AV, Skabkin MA, Pisareva VP, et al. The role of ABCE1 in eukaryotic posttermination ribosomal recycling. Mol Cell. 2010;37(2):196–210. doi: 10.1016/j.molcel.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shoemaker CJ, Green R. Kinetic analysis reveals the ordered coupling of translation termination and ribosome recycling in yeast. Proc Natl Acad Sci U S A. 2011;108(51):E1392–E1398. doi: 10.1073/pnas.1113956108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heuer A, Gerovac M, Schmidt C, et al. Structure of the 40S-ABCE1 post-splitting complex in ribosome recycling and translation initiation. Nat Struct Mol Biol. 2017;24(5):453–460. doi: 10.1038/nsmb.3396. [DOI] [PubMed] [Google Scholar]
- 33.Sen S, Wang H, Nghiem CL, et al. The ribosome-related protein, SBDS, is critical for normal erythropoiesis. Blood. 2011;118(24):6407–6417. doi: 10.1182/blood-2011-02-335190. [DOI] [PubMed] [Google Scholar]
- 34.Dror Y. P53 protein overexpression in Shwachman-Diamond syndrome. Arch Pathol Lab Med. 2002;126(10):1157–1158. doi: 10.5858/2002-126-1157b-PPOISS. author reply 1158. [DOI] [PubMed] [Google Scholar]
- 35.Austin KM, Gupta ML, Jr., Coats SA, et al. Mitotic spindle destabilization and genomic instability in Shwachman-Diamond syndrome. J Clin Invest. 2008;118(4):1511–1518. doi: 10.1172/JCI33764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frattini A, Bolamperti S, Valli R, et al. Enhanced p53 levels are involved in the reduced mineralization capacity of osteoblasts derived from Shwachman-Diamond syndrome subjects. Int J Mol Sci. 2021;22(24):13331. doi: 10.3390/ijms222413331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamabata T, Umeda K, Kouzuki K, et al. Pluripotent stem cell model of Shwachman-Diamond syndrome reveals apoptotic predisposition of hemoangiogenic progenitors. Sci Rep. 2020;10(1):14859. doi: 10.1038/s41598-020-71844-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaako P, Debnath S, Olsson K, et al. Disruption of the 5S RNP-Mdm2 interaction significantly improves the erythroid defect in a mouse model for Diamond-Blackfan anemia. Leukemia. 2015;29(11):2221–2229. doi: 10.1038/leu.2015.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barlow JL, Drynan LF, Hewett DR, et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q–syndrome. Nat Med. 2010;16(1):59–66. doi: 10.1038/nm.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tourlakis ME, Zhang S, Ball HL, et al. In vivo senescence in the Sbds-deficient murine pancreas: cell-type specific consequences of translation insufficiency. PLoS Genet. 2015;11(6):e1005288. doi: 10.1371/journal.pgen.1005288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.In K, Zaini MA, Müller C, et al. Shwachman-Bodian-Diamond syndrome (SBDS) protein deficiency impairs translation re-initiation from C/EBPα and C/EBPβ mRNAs. Nucleic Acids Res. 2016;44(9):4134–4146. doi: 10.1093/nar/gkw005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raaijmakers MHGP, Mukherjee S, Guo S, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464(7290):852–857. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zambetti NA, Ping Z, Chen S, et al. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell. 2016;19(5):613–627. doi: 10.1016/j.stem.2016.08.021. [DOI] [PubMed] [Google Scholar]
- 44.Dror Y, Freedman MH. Shwachman-Diamond syndrome: An inherited preleukemic bone marrow failure disorder with aberrant hematopoietic progenitors and faulty marrow microenvironment. Blood. 1999;94(9):3048–3054. [PubMed] [Google Scholar]
- 45.Zha J, Kunselman LK, Xie HM, et al. Inducible SBDS deletion impairs bone marrow niche capacity to engraft donor bone marrow after transplantation. Blood Adv. 2022;6(1):108–120. doi: 10.1182/bloodadvances.2021004640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woloszynek JR, Rothbaum RJ, Rawls AS, et al. Mutations of the SBDS gene are present in most patients with Shwachman-Diamond syndrome. Blood. 2004;104(12):3588–3590. doi: 10.1182/blood-2004-04-1516. [DOI] [PubMed] [Google Scholar]
- 47.Austin KM, Leary RJ, Shimamura A. The Shwachman-Diamond SBDS protein localizes to the nucleolus. Blood. 2005;106(4):1253–1258. doi: 10.1182/blood-2005-02-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shimamura A. Aplastic anemia and clonal evolution: germ line and somatic genetics. Hematology Am Soc Hematol Educ Program. 2016;2016(1):74–82. doi: 10.1182/asheducation-2016.1.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carvalho CMB, Zuccherato LW, Williams CL, et al. Structural variation and missense mutation in SBDS associated with Shwachman-Diamond syndrome. BMC Med Genet. 2014;15:64. doi: 10.1186/1471-2350-15-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Costa E, Duque F, Oliveira J, et al. Identification of a novel AluSx-mediated deletion of exon 3 in the SBDS gene in a patient with Shwachman-Diamond syndrome. Blood Cells Mol Dis. 2007;39(1):96–101. doi: 10.1016/j.bcmd.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 51.Stepensky P, Chacón-Flores M, Kim KH, et al. Mutations in EFL1, an SBDS partner, are associated with infantile pancytopenia, exocrine pancreatic insufficiency and skeletal anomalies in aShwachman-Diamond like syndrome. J Med Genet. 2017;54(8):558–566. doi: 10.1136/jmedgenet-2016-104366. [DOI] [PubMed] [Google Scholar]
- 52.Tan QK-G, Cope H, Spillmann RC, et al. Further evidence for the involvement of EFL1 in a Shwachman–Diamond-like syndrome and expansion of the phenotypic features. Cold Spring Harb Mol Case Stud. 2018;4(5):a003046. doi: 10.1101/mcs.a003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan S, Kermasson L, Hoslin A, et al. EFL1 mutations impair eIF6 release to cause Shwachman-Diamond syndrome. Blood. 2019;134(3):277–290. doi: 10.1182/blood.2018893404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee S, Shin CH, Lee J, et al. Somatic uniparental disomy mitigates the most damaging EFL1 allele combination in Shwachman-Diamond syndrome. Blood. 2021;138(21):2117–2128. doi: 10.1182/blood.2021010913. [DOI] [PubMed] [Google Scholar]
- 55.Tummala H, Walne AJ, Williams M, et al. DNAJC21 mutations link a cancer-prone bone marrow failure syndrome to corruption in 60S ribosome subunit maturation. Am J Hum Genet. 2016;99(1):115–124. doi: 10.1016/j.ajhg.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dhanraj S, Matveev A, Li H, et al. Biallelic mutations in DNAJC21 cause Shwachman-Diamond syndrome. Blood. 2017;129(11):1557–1562. doi: 10.1182/blood-2016-08-735431. [DOI] [PubMed] [Google Scholar]
- 57.Bellanné-Chantelot C, Schmaltz-Panneau B, Marty C, et al. Mutations in the SRP54 gene cause severe congenital neutropenia as well as Shwachman-Diamond-like syndrome. Blood. 2018;132(12):1318–1331. doi: 10.1182/blood-2017-12-820308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carapito R, Konantz M, Paillard C, et al. Mutations in signal recognition particle SRP54 cause syndromic neutropenia with Shwachman-Diamond-like features. J Clin Invest. 2017;127(11):4090–4103. doi: 10.1172/JCI92876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Juaire KD, Lapouge K, Becker MMM, et al. Structural and functional impact of SRP54 mutations causing severe congenital neutropenia. Structure. 2021;29(1):15–28.e7. doi: 10.1016/j.str.2020.09.008. [DOI] [PubMed] [Google Scholar]
- 60.Schürch C, Schaefer T, Müller JS, et al. SRP54 mutations induce congenital neutropenia via dominant-negative effects on XBP1 splicing. Blood. 2021;137(10):1340–1352. doi: 10.1182/blood.2020008115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Myers KC, Bolyard AA, Otto B, et al. Variable clinical presentation of Shwachman–Diamond syndrome: Update from the North American Shwachman–Diamond syndrome registry. J Pediatr. 2014;164(4):866–870. doi: 10.1016/j.jpeds.2013.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ip WF, Dupuis A, Ellis L, et al. Serum pancreatic enzymes define the pancreatic phenotype in patients with Shwachman-Diamond syndrome. J Pediatr. 2002;141(2):259–265. doi: 10.1067/mpd.2002.125849. [DOI] [PubMed] [Google Scholar]
- 63.Toiviainen-Salo S, Durie PR, Numminen K, et al. The natural history of Shwachman-Diamond syndrome-associated liver disease from childhood to adulthood. J Pediatr. 2009;155(6):807–811.e2. doi: 10.1016/j.jpeds.2009.06.047. [DOI] [PubMed] [Google Scholar]
- 64.Toiviainen-Salo S, Mäyränpää MK, Durie PR, et al. Shwachman–Diamond syndrome is associated with low-turnover osteoporosis. Bone. 2007;41(6):965–972. doi: 10.1016/j.bone.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 65.Mäkitie O, Ellis L, Durie PR, et al. Skeletal phenotype in patients with Shwachman-Diamond syndrome and mutations in SBDS. Clin Genet. 2004;65(2):101–112. doi: 10.1111/j.0009-9163.2004.00198.x. [DOI] [PubMed] [Google Scholar]
- 66.Myers KC, Bolyard AA, Otto B, et al. Variable clinical presentation of Shwachman-Diamond syndrome: Update From the North-American Shwachman-Diamond syndrome registry. J Pediatr. 2014;164(4):866–870. doi: 10.1016/j.jpeds.2013.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Furutani E, Shah AS, Zhao Y, et al. Inflammatory manifestations in patients with Shwachman-Diamond syndrome: A novel phenotype. Am J Med Genet A. 2020;182(7):1754–1760. doi: 10.1002/ajmg.a.61593. [DOI] [PubMed] [Google Scholar]
- 68.Kerr EN, Ellis L, Dupuis A, Rommens JM, Durie PR. The behavioral phenotype of school-age children with shwachman diamond syndrome indicates neurocognitive dysfunction with loss of Shwachman-Bodian-Diamond syndrome gene function. J Pediatr. 2010;156(3):433–438. doi: 10.1016/j.jpeds.2009.09.026. [DOI] [PubMed] [Google Scholar]
- 69.Singh SA, Vlachos A, Morgenstern NJ, et al. Breast cancer in a case of Shwachman Diamond syndrome. Pediatr Blood Cancer. 2012;59(5):945–946. doi: 10.1002/pbc.24052. [DOI] [PubMed] [Google Scholar]
- 70.Sack JE, Kuchnir L, Demierre M-F. Dermatofibrosarcoma protuberans arising in the context of Shwachman-Diamond syndrome. Pediatr Dermatol. 2011;28(5):568–569. doi: 10.1111/j.1525-1470.2010.01244.x. [DOI] [PubMed] [Google Scholar]
- 71.Bou Mitri F, Beaupain B, Flejou J-F, et al. Shwachman-Diamond syndrome and solid tumors: Three new patients from the French Registry for Severe Chronic Neutropenia and literature review. Pediatr Blood Cancer. 2021;68(7):e29071. doi: 10.1002/pbc.29071. [DOI] [PubMed] [Google Scholar]
- 72.Robberecht E, Nachtegaele P, Van Rattinghe R, et al. Pancreatic lipomatosis in the Shwachman-Diamond syndrome. Identification by sonography and CT-scan. Pediatr Radiol. 1985;15(5):348–349. doi: 10.1007/BF02386774. [DOI] [PubMed] [Google Scholar]
- 73.MacMaster SA, Cummings TM. Computed tomography and ultrasonography findings for an adult with Shwachman syndrome and pancreatic lipomatosis. Can Assoc Radiol J. 1993;44(4):301–303. [PubMed] [Google Scholar]
- 74.Dror Y, Donadieu J, Koglmeier J, et al. Draft consensus guidelines for diagnosis and treatment of Shwachman-Diamond syndrome. Ann N Y Acad Sci. 2011;1242:40–55. doi: 10.1111/j.1749-6632.2011.06349.x. [DOI] [PubMed] [Google Scholar]
- 75.Dror Y, Squire J, Durie P, Freedman MH. Malignant myeloid transformation with isochromosome 7q in Shwachman-Diamond syndrome. Leukemia. 1998;12(10):1591–1595. doi: 10.1038/sj.leu.2401147. [DOI] [PubMed] [Google Scholar]
- 76.Dror Y, Freedman MH. Shwachman-diamond syndrome. Br J Haematol. 2002;118(3):701–713. doi: 10.1046/j.1365-2141.2002.03585.x. [DOI] [PubMed] [Google Scholar]
- 77.Donadieu J, Michel G, Merlin E, et al. Hematopoietic stem cell transplantation for Shwachman-Diamond syndrome: experience of the French neutropenia registry. Bone Marrow Transplant. 2005;36(9):787–792. doi: 10.1038/sj.bmt.1705141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hashmi SK, Allen C, Klaassen R, et al. Comparative analysis of Shwachman-Diamond syndrome to other inherited bone marrow failure syndromes and genotype-phenotype correlation. Clin Genet. 2011;79(5):448–458. doi: 10.1111/j.1399-0004.2010.01468.x. [DOI] [PubMed] [Google Scholar]
- 79.D’Amours G, Lopes F, Gauthier J, et al. Refining the phenotype associated with biallelic DNAJC21 mutations. Clin Genet. 2018;94(2):252–258. doi: 10.1111/cge.13370. [DOI] [PubMed] [Google Scholar]
- 80.Sabulski A, Grier DD, Myers KC, Davies SM, Rubinstein JD. Acute myeloid leukemia in SRP54-mutated congenital neutropenia. EJHaem. 2022;3(2):521–525. doi: 10.1002/jha2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Calvo C, Lainey E, Caye A, et al. Leukaemic transformation in a 10-year-old girl with SRP54 congenital neutropenia. Br J Haematol. 2022;198(6):1069–1072. doi: 10.1111/bjh.18334. [DOI] [PubMed] [Google Scholar]
- 82.DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617–629. doi: 10.1056/NEJMoa2012971. [DOI] [PubMed] [Google Scholar]
- 83.Cesaro S, Pillon M, Sauer M, et al. Long-term outcome after allogeneic hematopoietic stem cell transplantation for Shwachman-Diamond syndrome: a retrospective analysis and a review of the literature by the Severe Aplastic Anemia Working Party of the European Society for Blood and Marrow Transplantation (SAAWP-EBMT) Bone Marrow Transplant. 2020;55(9):1796–1809. doi: 10.1038/s41409-020-0863-z. [DOI] [PubMed] [Google Scholar]
- 84.Myers K, Hebert K, Antin J, et al. Hematopoietic stem cell transplantation for Shwachman-Diamond syndrome. Biol Blood Marrow Transplant. 2020;26(8):1446–1451. doi: 10.1016/j.bbmt.2020.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cesaro S, Donadieu J, Cipolli M, et al. Stem cell transplantation in patients affected by Shwachman-Diamond syndrome: Expert consensus and recommendations from the EBMT Severe Aplastic Anaemia Working Party. Transplant Cell Ther. 2022;28(10):637–649. doi: 10.1016/j.jtct.2022.07.010. [DOI] [PubMed] [Google Scholar]
- 86.Xia J, Miller CA, Baty J, et al. Somatic mutations and clonal hematopoiesis in congenital neutropenia. Blood. 2018;131(4):408–416. doi: 10.1182/blood-2017-08-801985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jaiswal S, Ebert BL. Clonal hematopoiesis in human aging and disease. Science. 2019;366(6465):eaan4673. doi: 10.1126/science.aan4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shimamura A. Shwachman-Diamond syndrome. Semin Hematol. 2006;43(3):178–188. doi: 10.1053/j.seminhematol.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 89.Khan AW, Kennedy B, Furutani E, et al. The frequent and clinically benign anomalies of chromosomes 7 and 20 in Shwachman-diamond syndrome may be subject to further clonal variations. Mol Cytogenet. 2021;14(1):54. doi: 10.1186/s13039-021-00575-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Valli R, Pressato B, Marletta C, et al. Different loss of material in recurrent chromosome 20 interstitial deletions in Shwachman-Diamond syndrome and in myeloid neoplasms. Mol Cytogenet. 2013;6(1):56. doi: 10.1186/1755-8166-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Parikh S, Perdigones N, Paessler M, et al. Acquired copy number neutral loss of heterozygosity of chromosome 7 associated with clonal haematopoiesis in a patient with Shwachman-Diamond syndrome. Br J Haematol. 2012;159(4):480–482. doi: 10.1111/bjh.12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maserati E, Minelli A, Olivieri C, et al. Isochromosome (7)(q10) in Shwachman syndrome without MDS/AML and role of chromosome 7 anomalies in myeloproliferative disorders. Cancer Genet Cytogenet. 2000;121(2):167–171. doi: 10.1016/s0165-4608(00)00246-6. [DOI] [PubMed] [Google Scholar]
- 93.Minelli A, Maserati E, Nicolis E, et al. The isochromosome i(7)(q10) carrying c.258+2t>c mutation of the SBDS gene does not promote development of myeloid malignancies in patients with Shwachman syndrome. Leukemia. 2009;23(4):708–711. doi: 10.1038/leu.2008.369. [DOI] [PubMed] [Google Scholar]
- 94.Mellink CHM, Alders M, van der Lelie H, Hennekam RHC, Kuijpers TW. SBDS mutations and isochromosome 7q in a patient with Shwachman-Diamond syndrome: no predisposition to malignant transformation? Cancer Genet Cytogenet. 2004;154(2):144–149. doi: 10.1016/j.cancergencyto.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 95.Inaba T, Honda H, Matsui H. The enigma of monosomy 7. Blood. 2018;131(26):2891–2898. doi: 10.1182/blood-2017-12-822262. [DOI] [PubMed] [Google Scholar]
- 96.Valli R, De Paoli E, Nacci L, et al. Novel recurrent chromosome anomalies in Shwachman-Diamond syndrome. Pediatr Blood Cancer. 2017;64(8):bc.26454. doi: 10.1002/pbc.26454. [DOI] [PubMed] [Google Scholar]
- 97.Maserati E, Pressato B, Valli R, et al. The route to development of myelodysplastic syndrome/acute myeloid leukaemia in Shwachman-Diamond syndrome: the role of ageing, karyotype instability, and acquired chromosome anomalies. Br J Haematol. 2009;145(2):190–197. doi: 10.1111/j.1365-2141.2009.07611.x. [DOI] [PubMed] [Google Scholar]
- 98.Pressato B, Valli R, Marletta C, et al. Deletion of chromosome 20 in bone marrow of patients with Shwachman-Diamond syndrome, loss of the EIF6 gene and benign prognosis. Br J Haematol. 2012;157(4):503–505. doi: 10.1111/j.1365-2141.2012.09033.x. [DOI] [PubMed] [Google Scholar]
- 99.Pressato B, Valli R, Marletta C, et al. Cytogenetic monitoring in Shwachman-Diamond syndrome: a note on clonal progression and a practical warning. J Pediatr Hematol Oncol. 2015;37(4):307–310. doi: 10.1097/MPH.0000000000000268. [DOI] [PubMed] [Google Scholar]
- 100.Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20(15):1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 101.Wu X, Deng Y. Bax and BH3-domain-only proteins in p53-mediated apoptosis. Front Biosci. 2002;7:d151–d156. doi: 10.2741/A772. [DOI] [PubMed] [Google Scholar]
- 102.Pestov DG, Strezoska Z, Lau LF. Evidence of p53-dependent cross-talk between ribosome biogenesis and the cell cycle: effects of nucleolar protein Bop1 on G(1)/S transition. Mol Cell Biol. 2001;21(13):4246–4255. doi: 10.1128/MCB.21.13.4246-4255.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nakagawa K, Taya Y, Tamai K, Yamaizumi M. Requirement of ATM in phosphorylation of the human p53 protein at serine 15 following DNA double-strand breaks. Mol Cell Biol. 1999;19(4):2828–2834. doi: 10.1128/mcb.19.4.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Elghetany MT, Alter BP. p53 protein overexpression in bone marrow biopsies of patients with Shwachman-Diamond syndrome has a prevalence similar to that of patients with refractory anemia. Arch Pathol Lab Med. 2002;126(4):452–455. doi: 10.5858/2002-126-0452-PPOIBM. [DOI] [PubMed] [Google Scholar]
- 105.McGraw KL, Nguyen J, Komrokji RS, et al. Immunohistochemical pattern of p53 is a measure of TP53 mutation burden and adverse clinical outcome in myelodysplastic syndromes and secondary acute myeloid leukemia. Haematologica. 2016;101(8):e320–e323. doi: 10.3324/haematol.2016.143214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Babushok DV, Xie HM, Roth JJ, et al. Single nucleotide polymorphism array analysis of bone marrow failure patients reveals characteristic patterns of genetic changes. Br J Haematol. 2014;164(1):73–82. doi: 10.1111/bjh.12603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lucas F, Michaels PD, Wang D, Kim AS. Mutational analysis of hematologic neoplasms in 164 paired peripheral blood and bone marrow samples by next-generation sequencing. Blood Adv. 2020;4(18):4362–4365. doi: 10.1182/bloodadvances.2020002306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nezelof C, Watchi M. [Lipomatous congenital hypoplasia of the exocrine pancreas in children. (2 cases and review of the literature)] Arch Fr Pediatr. 1961;18:1135–1172. [PubMed] [Google Scholar]
- 109.Shwachman H, Diamond LK, Oski FA, Khaw KT. The syndrome of pancreatic insufficiency and bone marrow dysfunction. J Pediatr. 1964;65:645–663. doi: 10.1016/s0022-3476(64)80150-5. [DOI] [PubMed] [Google Scholar]
- 110.Bodian M, Sheldon W, Lightwood R. Congenital hypoplasia of the exocrine pancreas. Acta Paediatr. 1964;53:282–293. doi: 10.1111/j.1651-2227.1964.tb07237.x. [DOI] [PubMed] [Google Scholar]