

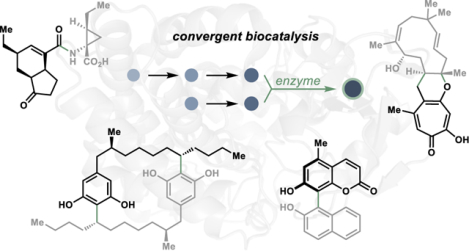
Abstract
Achieving convergent synthetic strategies has long been a gold standard in constructing complex molecular skeletons, allowing for the rapid generation of complexity in comparatively streamlined synthetic routes. Traditionally, biocatalysis has not played a prominent role in convergent laboratory synthesis, with the application of biocatalysts in convergent strategies primarily limited to the synthesis of chiral fragments. Although the use of enzymes to enable convergent synthetic approaches is relatively new and emerging, combining the efficiency of convergent transformations with the selectivity achievable through biocatalysis creates new opportunities for efficient synthetic strategies. This Perspective provides an overview of recent developments in biocatalytic strategies for convergent transformations and offers insights into the advantages of these methods compared to their small molecule-based counterparts.
Graphical Abstract
INTRODUCTION
Determining the major disconnections for a targeted campaign is an important exercise that governs the overall cost-effectiveness and atom economy in a multistep synthesis.1 As defined by Hendrickson in 1975,2 an ideal synthesis entails “only construction reactions involving no intermediary refunctionalisations, and leading directly to the target, not only its skeleton but also its correctly placed functionality.” Toward this goal of designing increasingly streamlined synthetic routes focused on skeletal construction, convergent synthesis has risen as a governing principle in synthetic design.2, 3 In convergent transformations, two independently synthesized fragments are stitched together to construct the core skeleton of the targeted product in a single, complexity-generating step.3 In contrast to linear sequences, in which a starting material undergoes a series of iterative chemical transformations to deliver a final product of interest, the convergent synthesis of small molecules offers advantages in reducing the overall step count of a sequence while increasing the overall yield (Figure 1a).4
Figure 1.
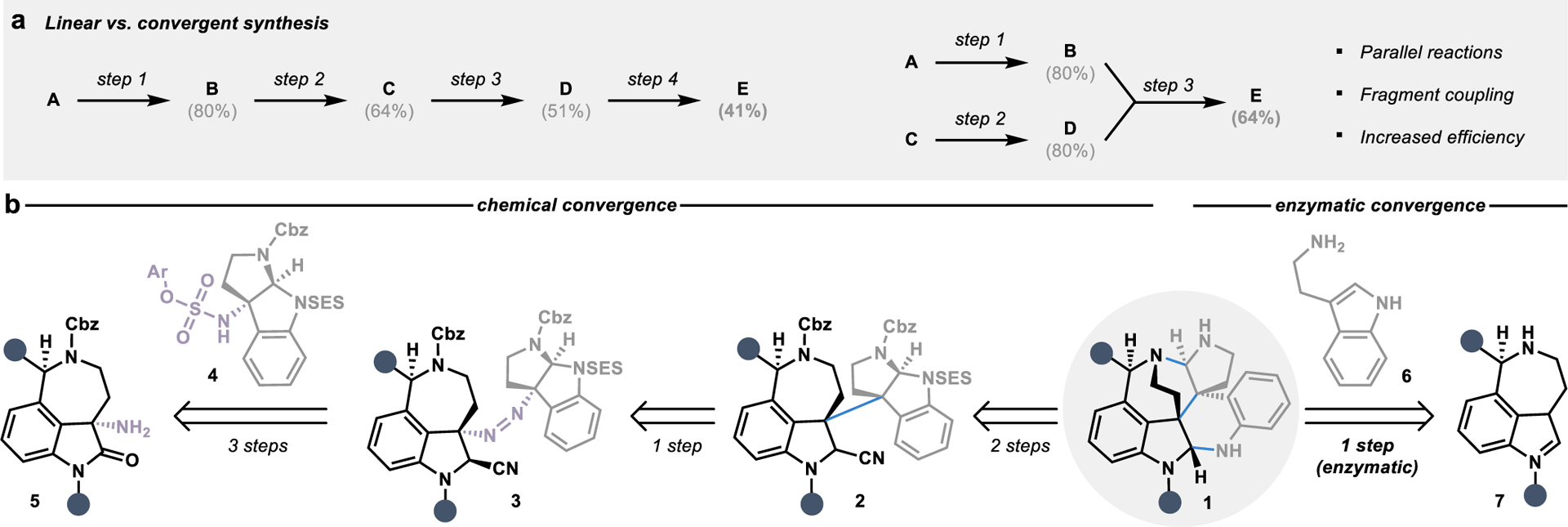
Biocatalysis in convergent synthesis. (a) Comparison of target product yields in hypothetical linear versus convergent synthetic routes. (b) Representative retrosynthesis of alkaloid natural product core 1 using chemical methods compared to enzymatic convergence with a cytochrome P450 enzyme shows transformative power of biocatalysis in convergent synthesis.
In the history of contemporary synthetic organic chemistry, a plethora of fragment coupling strategies using small molecule-based catalysts have been developed and employed in synthetic campaigns, ultimately reshaping the way we approach the construction of molecules. However, the advantages inherent to convergent synthetic strategies can be difficult to realize based on practical challenges encountered in their application, especially for guiding the selectivity of the fragment coupling event. For example, in the synthesis of the complex skeleton present in the communesin family of alkaloid natural products (see 1), Movassaghi and coworkers devised an elegant convergent route that employed a radical–radical coupling to form the central carbon–carbon bond uniting the two fragments in 2.5 However, to achieve selectivity in the convergent C–C bond-forming step, a detour to an intermediate diazene 3 was necessary, requiring additional steps for the installation of directing and protecting groups in the fragment synthesis and coupling.5 In contrast, a single cytochrome P450 enzyme can unite unprotected building blocks 6 and 7 with catalyst-controlled selectivity in the biosynthesis of communesin alkaloids, circumventing any requirement for intermediary functionalization steps (Figure 1b).6
The precision and efficiency offered by enzyme catalysis has propelled the incorporation of biocatalytic strategies into synthetic campaigns in academic and industrial spheres.7–9 Most biocatalytic reactions employed in multi-step synthesis have centered on individual functional group manipulations to access chiral building blocks or late-stage functionalization reactions.10–12 This trend can be observed in the role biocatalysis has historically played in convergent synthesis, with the utilization of enzymes for the synthesis of key fragments present in active pharmaceutical ingredients (Figure 2). Enzymatic kinetic resolutions dominated the field of biocatalysis in the 1990s and early 2000s, resulting in the development of robust strategies for the preparation of chiral building blocks,13–15 including the chiral amines present in amoxicillin (8)16 and draflazine (9).17, 18 The development of directed evolution-based strategies to tailor biocatalysts for a targeted transformation drastically expanded the application of biocatalysis in asymmetric catalysis.19 This new wave of biocatalysis brought the synthesis of chiral alcohols to the forefront of the field, with a wealth of catalysts capable of the stereoselective reduction of ketones, as demonstrated in the convergent synthesis of atorvastatin (10)20 and atazanavir (11).21, 22 Additionally, biocatalysis has played a significant role in oxidative desymmetrization reactions, enabling the synthesis of the key chiral fragments present in telaprevir (12)23 and islatravir (13).24
Figure 2.
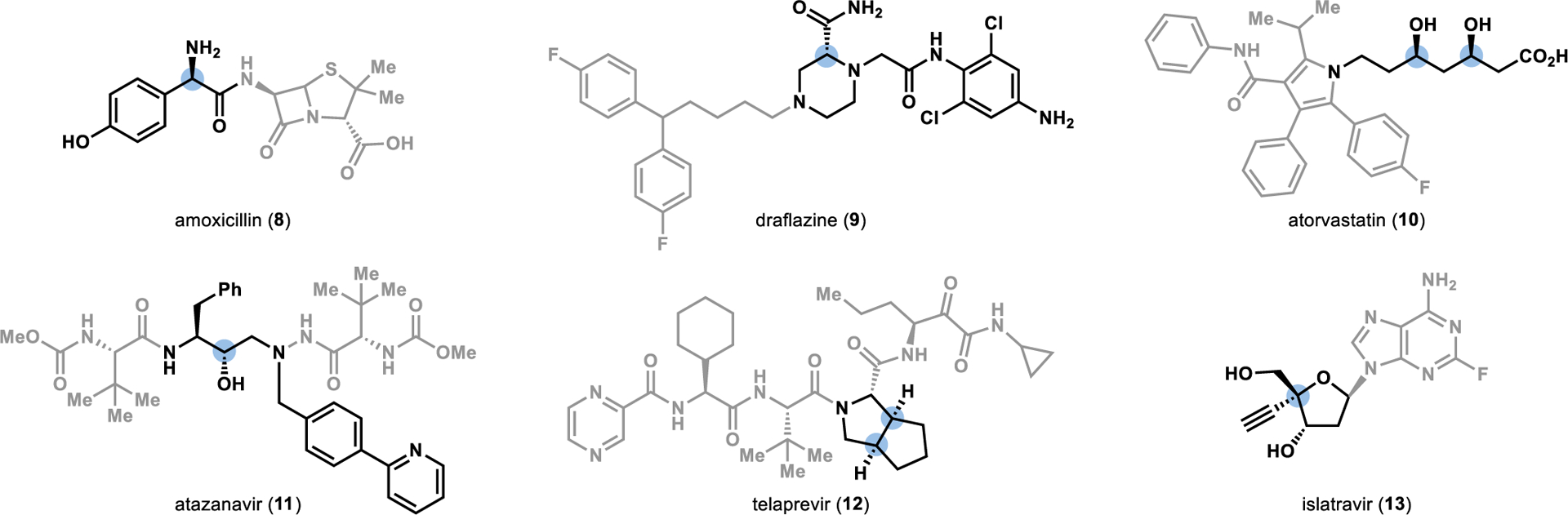
Biocatalytic synthesis of chiral fragments employed in convergent syntheses toward active pharmaceutical ingredients. The stereocenter set by an enzyme is highlighted in blue.
Although the extensive development of biocatalysts for functional group interconversions has increased the footprint of biocatalysis in the retrosynthetic playbook, biocatalysts are less commonly considered for constructing the core skeleton of molecules. This missed opportunity in biocatalysis is exemplified by the structural complexity of the molecular frameworks represented in natural products assembled through total enzymatic synthesis. Merging the selectivity offered by biocatalysis with the efficiency of convergent transformations holds the potential for increasingly streamlined strategies for complex molecule synthesis. This Perspective provides an overview of recent developments in biocatalytic strategies for convergent transformations. We provide insights into the development of convergent biocatalytic reactions, their incorporation in multistep synthesis, and probe the advantages convergent biocatalytic methods provide compared to their small molecule-based counterparts. As a non-comprehensive review, the goal of this Perspective is to discuss the current state of convergent biocatalysis and how this mode of synthetic convergence can impact the way we think about making molecules.
BIOCATALYTIC TOOLBOX FOR FRAGMENT COUPLING REACTIONS
The most well-studied biosynthetic pathways embrace linear blueprints with an iterative assembly of the core skeleton and functionality present in natural products.25–29 An example of this can be seen in the biosynthesis of the vast class of terpenoid natural products, in which simple isoprene building blocks are iteratively assembled and cyclized to provide structurally complex terpene scaffolds.25 Advances in DNA sequencing and bioinformatic technologies have fueled the discovery of biosynthetic enzymes from secondary metabolism, further illuminating Nature’s strategies for complex molecule synthesis.30 Among these strategies, a growing number of biosynthetic enzymes have been identified that break from the canonical linear blueprint for natural product synthesis by stitching together two or more building blocks in convergent transformations.31, 32 These enzymes offer the efficiency of convergent reactions with the precision of enzyme catalysis, thereby avoiding the traditionally accepted requirements for protecting groups and functional handles in synthetic planning. The transformative potential of these enzyme catalysts for convergent synthesis has been explored over the last decade, leading to promising advances in biocatalytic strategies for convergent C–N and C–C bond formation.
Convergent synthesis of unnatural nucleosides.
The earliest examples of enzymes applied as convergent biocatalysts can be found in the synthesis of carbohydrates.33 Aldolases are robust catalysts for the union of simple building blocks to form both natural and unnatural sugars with programmable stereoselectivity to rapidly build molecular complexity.34, 35 Additionally, glycosyltransferases can stitch together carbohydrates of varying complexity or perform highly site-selective glycosylations of complex molecules with the formation of a glycosidic bond.36–39
An excellent example of the power of both of these transformations can be found in the total synthesis of the HIV antiviral drug islatravir (18).24 Inspired by the bacterial nucleotide salvage pathway,40 Merck and Codexis engineered an enzymatic cascade to transform chiral aldehyde 14 into the unnatural nucleoside 18 in a single step (Figure 3a).24 The first step in this cascade utilized an aldolase (DERA) that natively catalyzes the reversible retro-aldol cleavage of a sugar into glyceraldehyde 3-phosphate and acetaldehyde. To reverse the direction of the aldol reaction, the aldolase was engineered to tolerate a large excesses of acetaldehyde, thereby driving the equilibrium toward the formation of the desired sugar 16. Once formed, sugar 16 was selectively phosphorylated by a phosphorylase (PPM) at the C5 position to provide the correct functional handle to allow for the ligation of the nucleobase 17 to the sugar to afford the unnatural nucleoside 18.
Figure 3.
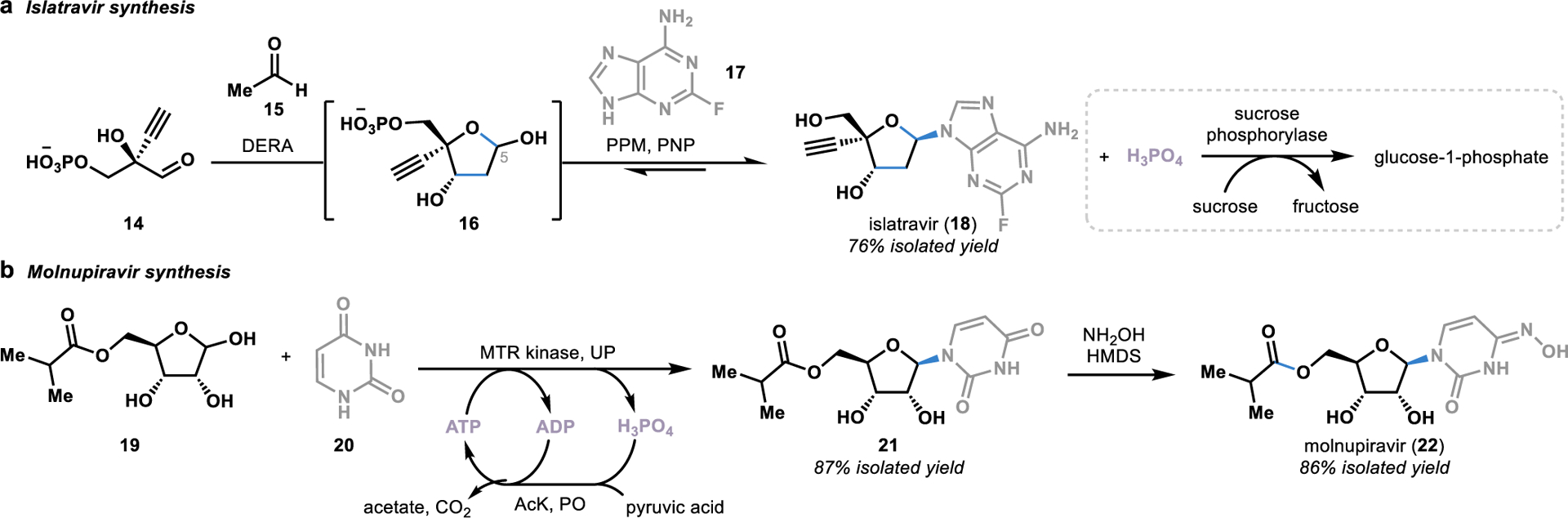
Convergent synthesis of antiviral drugs using reversible phosphorylases from nucleotide salvage pathways. DERA: deoxyribose 5-phosphate aldolase; PPM: phosphopentomutase; PNP: purine nucleotide phosphorylase; MTR kinase: 5-S-methylthioribose kinase; UP: uridine phosphorylase; PO: pyruvate oxidase; AcK: acetate kinase; HMDS: hexamethyldisilazane.
Nucleoside phosphorylases (such as PNP) typically catalyze the reversible cleavage of nucleosides to free the purine or pyrimidine base in nucleotide salvage pathways.40 Given the reversible nature of these reactions, nucleoside phosphorylases can be harnessed as convergent catalysts to synthesize nucleosides.40 However, for this convergent chemistry to be realized, a mechanism to drive the equilibrium toward the nucleoside formation must be incorporated to consume free inorganic phosphates released through the progression of the reaction. To accomplish this, Codexis and Merck incorporated sucrose phosphorylase into the cascade, allowing for the irreversible consumption of free phosphate present in the reaction by converting sucrose to glucose-1-phosphate and fructose.24
Another example of using a nucleoside phosphorylase from a nucleotide salvage pathway to synthesize an unnatural nucleoside was recently disclosed in the synthesis of the COVID-19 antiviral molnupiravir (22; Figure 3b).41 Similarly to the islatravir synthesis, Merck engineered an enzymatic cascade in which a phosphorylase (MTR kinase) added a phosphate group to ribose 19, enabling the ligation of uracil (20) to the ribose through the displacement of the phosphate to form the unnatural nucleoside 21. However, in contrast to the phosphate sequestration strategy employed in the islatravir (18) synthesis, Merck employed an ATP recycling strategy in which the free phosphate was converted to ATP. Not only did this prevent the undesired nucleoside cleavage reaction, but the generation of ATP also provided MTR kinase with the required phosphate source for the phosphorylation step, thereby alleviating the cost of supplying the reaction with stoichiometric amounts of ATP.
Directed evolution of the key enzymes involved in the syntheses of islatravir (18) and molnupiravir (22) led to substantial improvements in the efficiency of the synthesis of these unnatural nucleosides over alternative synthetic approaches. For example, a previous chemical synthesis of islatravir (18) from a chiral building block similar to 14 was achieved with a 17% total yield over ten steps,42 providing a stark contrast to the 76% yield provided by the enzymatic cascade that takes place in a single reaction vessel.24 Similarly, following installation of the oxime moiety onto nucleoside 21, molnupiravir (22) was accessed in only three steps in a 69% overall yield, representing a drastic improvement over the previously developed 10-step synthesis starting from ribose that afforded <10% yield using small molecule-based synthetic strategies.41
Altogether, these biocatalytic strategies present a robust tool in the retrosynthetic design of unnatural nucleosides, breaking from the necessity of protecting group toggling and the challenge of setting the anomeric stereochemistry hindering analogous chemical strategies. Moreover, the tunability of nucleoside phosphorylases through protein engineering positions this class of enzymes as powerful convergent catalysts for the ligation of increasingly complex sugars and nucleobases to access potent antiviral drugs.
Convergent amidation reactions.
Similar to glycosidic bonds, amide bonds are a common covalent link in biological systems, with a widespread presence in proteins, peptides, and secondary metabolites.43 Despite their ubiquitous presence in peptides and bioactive organic small molecules, traditional methods of constructing amide bonds often require the activation of the carboxylic acid motif using stoichiometric reagents and rely on protecting group strategies to achieve chemoselectivity.44 This often results in stoichiometric organic waste, posing significant challenges in amidation reactions, especially those carried out on an industrial scale.45
In an alternative approach to the complex ribosome machinery that performs iterative amidation reactions in the synthesis of proteins and peptides,46 Nature has evolved a catalog of biosynthetic enzymes that perform convergent amidation reactions in the synthesis of secondary metabolites (Figure 4a).47 This subset of enzymes are composed primarily of ligases called amide bond synthetases.48 These ATP-dependent enzymes mediate the tethering of adenosine monophosphate (AMP) to a substrate harboring a carboxylate. This creates a leaving group that allows for the substitution reaction with a free amine present on the second fragment, thereby forming the target amide linkage, as demonstrated in the convergent synthesis of the antidepressant moclobemide (30; Figure 4b).49
Figure 4.
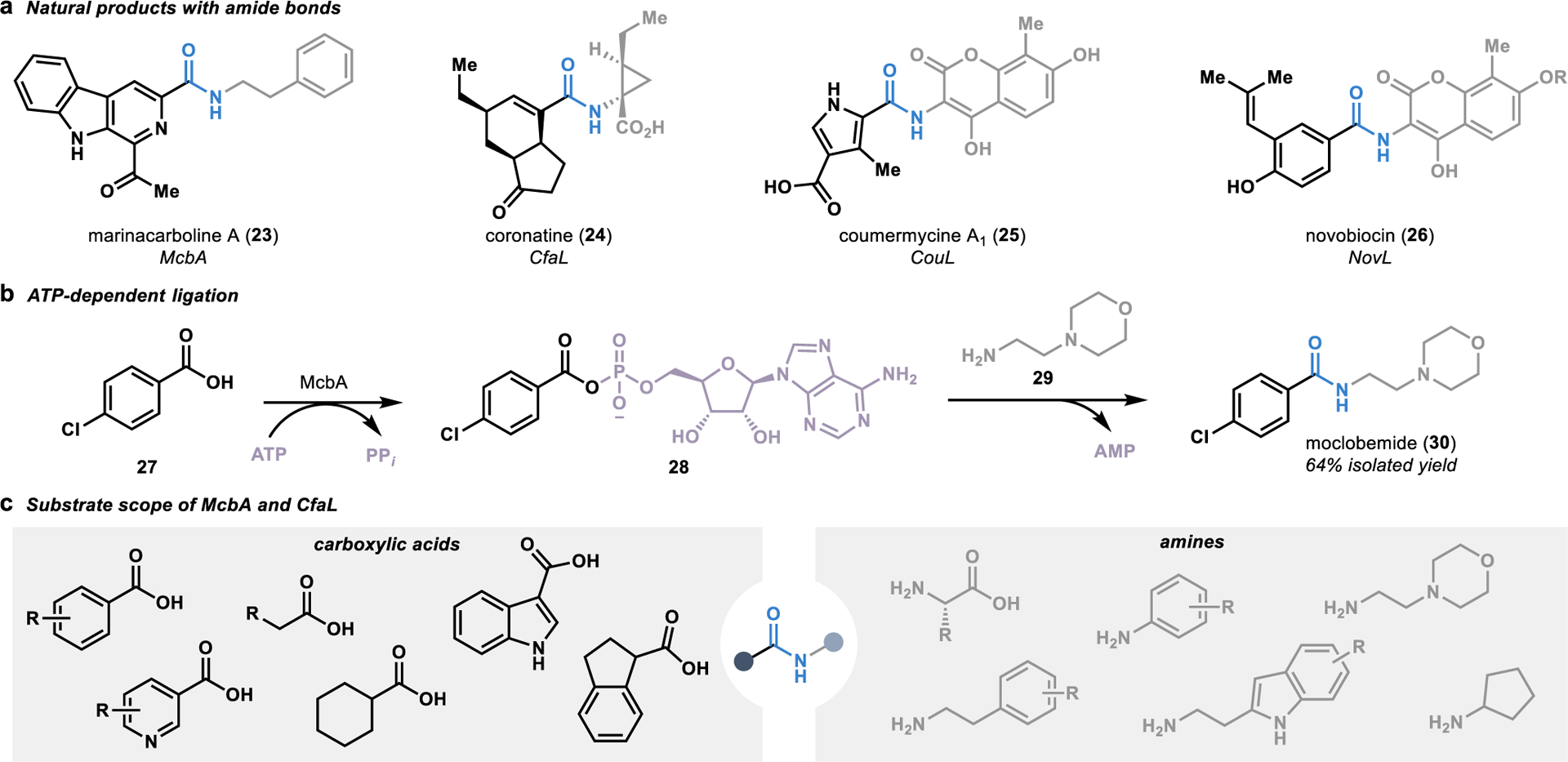
Convergent amide bond formation with ligases. (a) Amide bonds in natural products formed by ligases called amide bond synthetases. (b) Amide bond synthetases utilize ATP to activate a carboxylic acid fragment for nucleophilic attack by an amine fragment, as is demonstrated in the biocatalytic synthesis of moclobemide (30). (c) Several amide bond synthetases have demonstrated the ability to accept a wide range of carboxylic acid and amine fragments in biocatalytic amidation reactions.
Substrate scope profiling of several amide bond synthetases has demonstrated a broad substrate scope, with these ligases accepting a wide range of carboxylic acids and amines (Figure 4c).49–51 The most broadly studied ligase is McbA, an amide bond synthetase involved in the biosynthesis of marinacarbolines such as 23.50 Substrate scope profiling of McbA has demonstrated that it can be used as a versatile catalyst for amide coupling reactions, with the ability to accept a range of aromatic carboxylic acids and both aliphatic and aromatic amines.49, 51 The promiscuity of McbA was further validated by a crystal structure of the ligase, showing a flexible binding site with relatively few specific binding interactions. In addition to McbA, several CfaL orthologues associated with coronatine biosynthesis (24) were recently discovered to perform promiscuous amide couplings.52 Expanding on the chemistry achievable with McbA, the CfaL ligases accept both aromatic and aliphatic acids in reactions with various natural and unnatural amino acids serving as the amine coupling partner.
Altogether, this class of enzymes has demonstrated an ability to template a diverse array of carboxylic acids and amines in their active site for fragment coupling reactions. Significantly, these biocatalytic reactions offer the unique advantage of catalytic regeneration of the ATP activating reagent,49 thereby eliminating the requirement for stoichiometric activating reagents and organic waste inherent to many traditional approaches for amide bond formation.44 These factors, combined with the pivotal role amide bond formation plays in medicinal chemistry,53 poise amide bond synthetases as an attractive tool for convergent amide bond formation.
Convergent amination reactions.
The prevalence of chiral amine moieties in pharmaceutical agents has led to an intense interest in developing biocatalytic tools for stereoselective amination reactions.54 The majority of these biocatalytic strategies rely on transaminases that perform functional group interconversions to form chiral amine building blocks.55 However, several enzymes have been characterized which perform convergent amination reactions through the generation of imine intermediates. These imine intermediates are formed by condensing one fragment bearing a free amine and a secondary fragment bearing a carbonyl, forming an electrophilic species poised for nucleophilic attack (Figure 5a).
Figure 5.
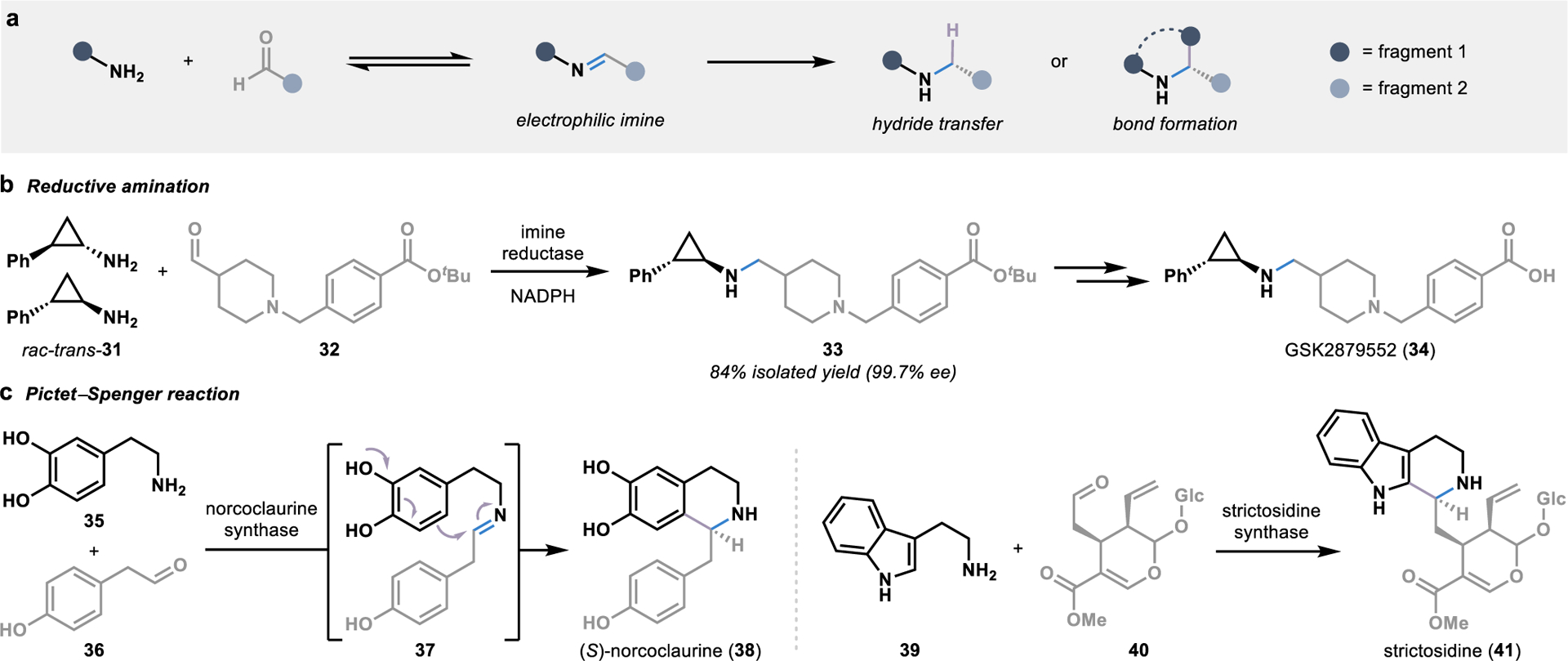
Biocatalytic strategies for convergent amination reactions. (a) The condensation of fragments bearing an amine and an aldehyde forms electrophilic imine intermediates poised for stereoselective functionalization by an enzyme. (b) Imine reductases utilize NADPH to stereoselectively delivery a hydride to an imine intermediate in the biocatalytic synthesis of chiral amines. (c) Pictet-Spenglerases harness imine intermediates in the biosynthesis of alkaloid metabolites, allowing for the intramolecular formation of a C-C bond through an electrophilic aromatic substitution reaction.
In one scenario, a hydride can be delivered to the imine intermediate in a reductive amination reaction, a transformation that has emerged as one of the leading approaches toward amine synthesis in the pharmaceutical industry.53 As an alternative to the highly reactive reducing agents typically required in chemical reductive aminations, NADPH can be used as a hydride source in imine reductase-catalyzed reductive aminations.55 The majority of biocatalytic strategies for reductive aminations have been developed with simple amine substrates; however, imine reductases can also unite two larger fragments through the formation of a C–N bond.56 An excellent example of this can be found in the asymmetric synthesis of GSK2879552 (34), a lysine-specific demethylase-1 inhibitor currently in clinical trials for the treatment of several cancers (Figure 5b).57 The previous synthesis of GSK2879552 (34) required a classic resolution of the racemic amine fragment 31 prior to reductive amination using stoichiometric sodium borohydride at low temperatures. To overcome issues with cost, the number of steps, solvent usage, and stoichiometric boron waste presented by the chemical process, a biocatalytic strategy for the formation of key intermediate 33 was explored.57 Specifically, an imine reductase was engineered to catalyze the reductive amination with the simultaneous kinetic resolution of the amine fragment 31, providing key intermediate 33 in >99% ee through a single step from racemic starting material 31.
As an alternative to enzymatic delivery of a hydride to an imine intermediate, Nature has evolved enzymes that exploit the electrophilicity of imine intermediates to achieve stereoselective cyclization reactions, such as Pictet–Spengler reactions, in the biosynthesis of various alkaloids including 38 and 41.58 The first enzyme-catalyzed Pictet–Spengler reaction was discovered in 1977,59 over sixty years after Amé Pictet and Theodor Spengler first discovered the abiotic version of this reaction.60 Since then, an ever-expanding collection of Pictet–Spenglerases have been characterized in plant secondary metabolite pathways, including noroclaurine and strictosidine synthase (Figure 5c).58 Following the intermolecular condensation of a β-arylethylamine and an aldehyde to form an imine intermediate (such as 37), an intramolecular C–C bond is formed through an electrophilic aromatic substitution reaction.61, 62 In addition to their natural reactivity, both noroclaurine and strictosidine synthase have demonstrated broad substrate scope, with the ability to couple a variety of fragments in the synthesis of diverse alkaloid products.63
Convergent alkylation reactions.
Beyond intramolecular C–C bond formation, such as those catalyzed by Pictet–Spenglerases, a growing number of biocatalysts capable of mediating intermolecular C–C bond formation have been developed.64 The majority of these biocatalytic reactions include late-stage C–H functionalization reactions catalyzed by transferases or the convergence of two small units catalyzed by lyases.64–66 The last several years have brought with them an expansion to this biocatalytic toolbox for intermolecular C–C bond formation, including the development of convergent alkylation reactions. These biocatalytic strategies have been developed with new-to-nature reactions and by harnessing complexity-generating biosynthetic enzymes in secondary metabolite pathways.
Radical coupling reactions offer new retrosynthetic disconnections in complex molecular synthesis compared to reactions based on ionic chemistry. However, the inherently high reactivity and frequently short lifetimes of radical intermediates create a significant challenge in controlling the chemo-, site- and stereoselectivity of radical transformations.67 Over the last several years, creative strategies pairing the radical chemistry initiated with photocatalysis and the selectivity achievable for reactions within an enzyme active site have been developed for stereoselective formation of C(sp3)–C(sp3) bonds through radical coupling reactions.68
Ene-reductases are flavin-dependent enzymes that naturally catalyze the asymmetric reduction of alkenes.69 In a typical alkene reduction, a hydride from NADPH is transferred to the flavin cofactor to form the reduced cofactor. The reduced flavin cofactor is then poised to deliver a hydride to an alkene substrate bound in the enzyme active site. However, this mechanism can be interceded by binding an α-halo carbonyl substrate (42) followed by excitation by visible light, thereby triggering a single electron transfer in the presence of the reduced flavin cofactor.70 This photoexcitation-promoted electron transfer results in the release of a halide anion, leaving behind intermediate 43 with a carbon-centered radical that is primed for a Giese-type addition with a second alkene substrate (44). Finally, a stereoselective hydrogen atom transfer from the semiquinone flavin cofactor (47) to radical intermediate 45 forms C–C coupled product 46, thereby completing the catalytic cycle. This photoenzymatic approach for the hydroalkylation of alkenes has been exploited for the versatile C–C coupling of various α-halo carbonyls and alkenes to from γ-stereogenic ketones, amides, and esters (Figure 6a).70–72
Figure 6.
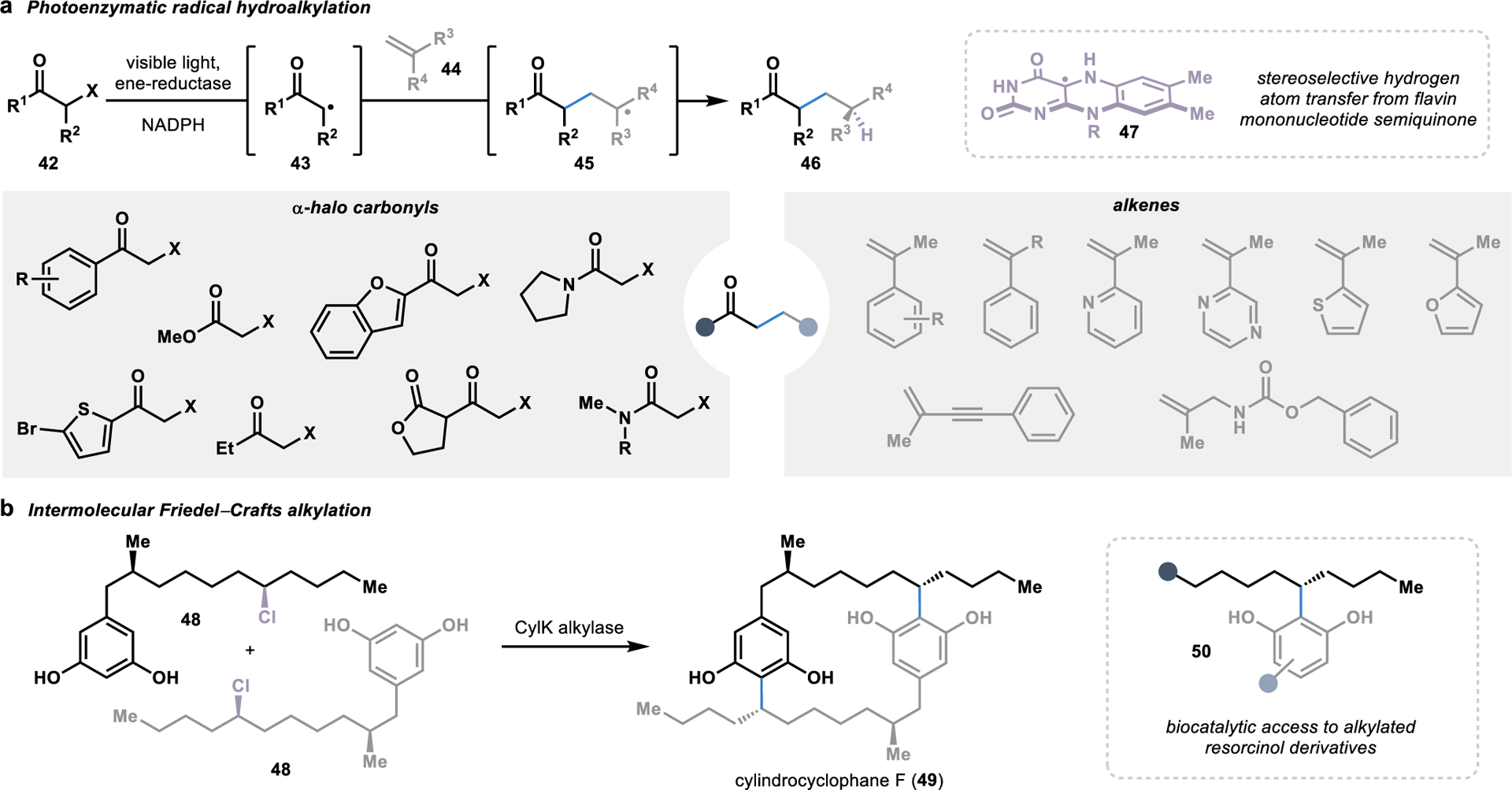
Biocatalytic strategies for intermolecular alkylation reactions. (a) In a photoenzymatic approach, ene-reductases can mediate a stereoselective hydrogen atom transfer from their flavin cofactor upon initiation of a radical hydroalkylation by visible light. (b) Alternatively, a Friedel-Crafts alkylase in the biosynthesis of dimeric cylindrocyclophane F (49) can alkylate resorcinol derivatives with aliphatic alkyl chains.
In addition to photoenzymatic hydroalkylation reactions, several strategies for convergent C–C bond formation through biocatalytic Friedel–Crafts alkylation reactions have been explored.73–75 Discovered in 1877 by Charles Friedel and James Mason Crafts, the Friedel–Crafts reaction has become an incredibly versatile reaction commonly used to accomplish alkylation and acylation reactions.76 Furthermore, a number of enzymes have been identified to perform intermolecular Friedel–Crafts reactions in secondary metabolite pathways, several of which have been harnessed as biocatalysts for C–C bond formation.64 Of particular interest in convergent biocatalysis, one of these biosynthetic enzymes performs a late-stage dimerization reaction to form the natural product cylindrocyclophane F (49).77 In a head-to-tail Friedel–Crafts alkylation, CylK catalyzes the site- and stereoselective dimerization of substrates harboring resorcinol and an alkyl halide (48; Figure 6b). Beyond the native reactivity, CylK can also catalyze a range of Friedel–Crafts alkylations with resorcinol derivatives and alkyl halide chains terminating in a hydroxyl group or extended chains bearing amides, thioesters, esters, and ketones.74
Intermolecular Diels–Alder reactions.
Like the Friedel–Crafts alkylation reaction, the Diels–Alder reaction represents another classic synthetic transformation in every synthetic chemist’s retrosynthetic playbook. In particular, the ubiquity of the cyclohexane skeleton in natural product structures has positioned the Diels–Alder reaction as a versatile transformation for convergent strategies toward complex molecules.78 It has inspired an intense interest in the discovery of biosynthetic enzymes that perform this reaction.79 Nevertheless, for many decades, the discovery of intermolecular Diels–Alderases eluded chemists, raising into question the fundamental existence of enzymes that have evolved explicitly for this function and spurring the development of artificial Diels–Alderase enzymes.
Artificial Diels–Alderases harness the acid-base activation and stereocontrol provided upon binding of a given diene and dienophile in a protein cavity for asymmetric cycloadditions (Figure 7a).80 Several artificial metalloenzymes have been designed to catalyze Diels–Alder reactions, often utilizing a copper species as a Lewis acid catalyst to promote the convergent cycloaddition.81 Alternatively, stabilization of the desired pericyclic transition state can be achieved by training catalytic antibodies to bind a synthetic transition state mimic82 or through the computational design of de novo enzymes.83
Figure 7.
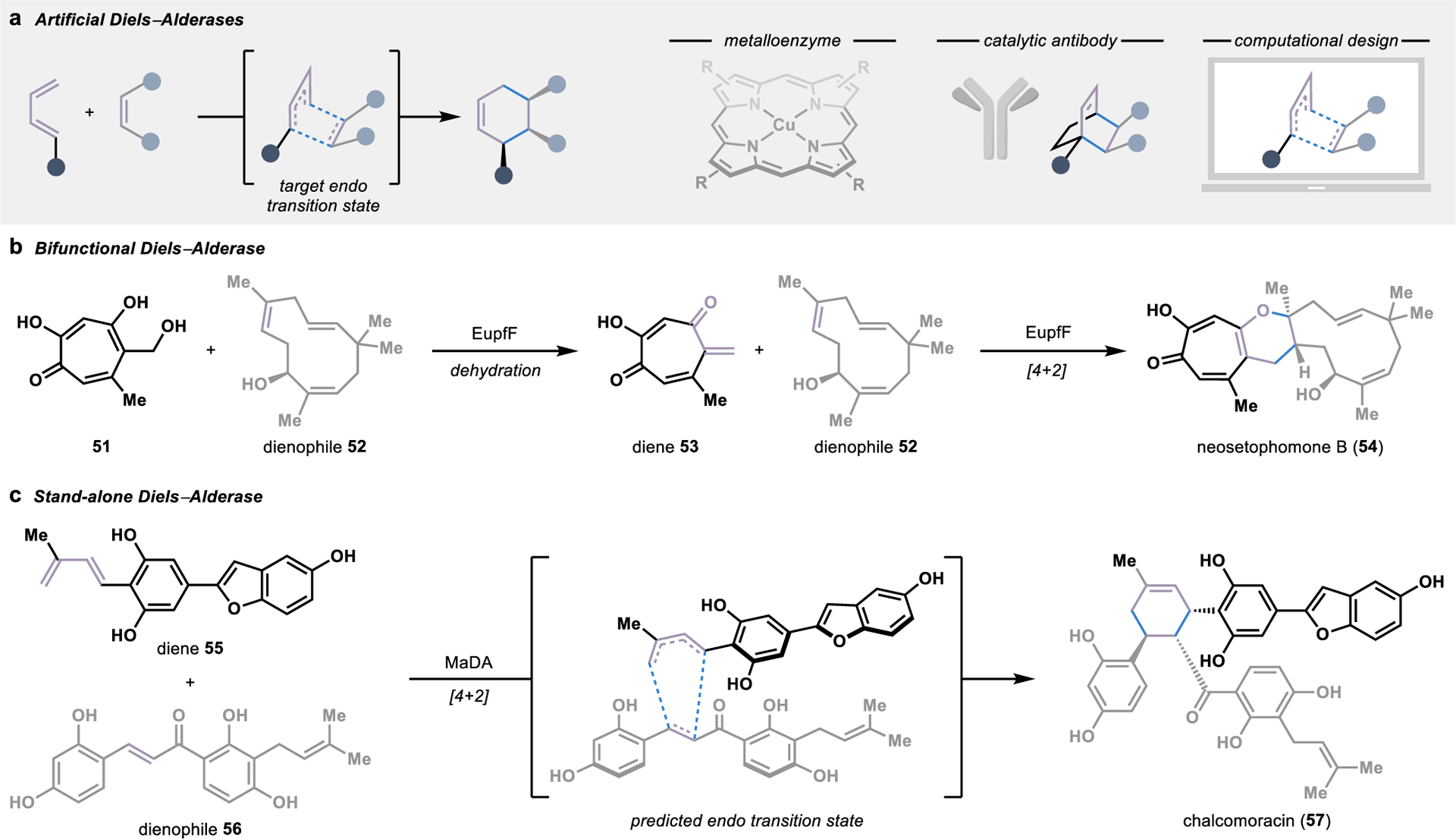
Biocatalytic strategies for intermolecular Diels–Alder reactions. (a) Artificial Diels–Alderases can be engineered by anchoring a metal cofactors into an enzyme cavity, training catalytic antibodies against a target transition state mimic, or computation design. (b) A bifunctional Diels–Alderase in the biosynthesis of neosetophomone B (54) catalyzes both the dehydration to form diene 53 and a [4+2] cycloaddition. (c) A stand-alone natural Diels–Alderase that catalyzes a concerted, intermolecular [4+2] cycloaddition was first discovered in the biosynthesis of chalcomoracin (57).
In addition to advances in the design of artificial enzymes, there have been several significant milestones in the discovery of biosynthetic enzymes performing Diels–Alder reactions. Of the biosynthetic Diels–Alderases identified to date, the majority evolved from divergent ancestors with no involvement in the catalysis of Diels–Alder reactions.84 As such, many of these biosynthetic enzymes are bifunctional, first catalyzing the preparation of one of the two substrates followed by stereoselective [4+2] cycloaddition within the enzyme active site.84 This can be observed in the biosynthesis of the tropolonic natural product neosetophomone B (54), in which EupfF first catalyzes the dehydration of a tropolone 51 to form the diene 53 required for a spontaneous [4+2] cycloaddition (Figure 7b).85 However, due to the bifunctional nature of these catalysts, the mechanism of their involvement in the cycloaddition is sometimes unclear, with only reactions proceeding through a concerted, synchronous pericyclic transition state constituting a true Diels–Alder reaction.79
The long-standing debate over the existence of biosynthetic enzymes specifically evolved to catalyze a Diels–Alder reaction was finally put to rest in 2011, when an enzyme evolved to perform a concerted, intramolecular [4+2] cycloaddition was identified.86 This landmark discovery led to the identification of additional stand-alone Diels–Alderases catalyzing intramolecular reactions through homology-based searches with its sequence. However, it wasn’t until almost ten years later that the first stand-alone Diels–Alderase catalyzing an intermolecular reaction was identified in the biosynthesis of the plant natural product chalcomoracin (57).87 This Diels–Alderase, MaDA, is a flavin-dependent enzyme that unites diene 5 and dienophile 56 in a [4+2] cycloaddition through a concerted, asynchronous mechanism (Figure 7c). Despite the absence of redox chemistry in the [4+2] cycloaddition, the flavin cofactor was identified to be essential for catalytic activity and is likely involved in hydrogen-bonding interactions with the substrate. Substrate scope analysis of the enzyme demonstrated promiscuous activity, with the ability to accept different combinations of unnatural dienes and dienophiles, ultimately allowing for the chemoenzymatic synthesis of various natural product derivatives.
The myriad of small molecule-based chiral ligands developed for asymmetric Diels–Alder reactions offers versatility that has yet to be accomplished using biocatalytic strategies.88 However, performing reactions within these protein environments, either using artificially-designed or natural enzymes, offers the unique advantage of further genetic manipulation to allow for designer catalysts with high levels of efficiency and selectivity. Moreover, the possibility for accessing isomeric scaffolds using Diels–Alderases with complementary selectivity compared to traditional synthetic methods positions this class of enzymes as potentially powerful tools for convergent synthesis.
Intermolecular oxidative coupling reactions.
A growing number of enzymes from secondary metabolite pathways, like the previously described CylK alkylase and Diels–Alderases, have been implicated in complexity-generating dimerization reactions.32 Convergent strategies such as these for the construction of natural products break from the canonical linear sequences invoked in many biosynthetic pathways and offer compelling new tools for convergent synthesis. Of the biosynthetic dimerization reactions characterized to date, one of the most prevalent modes of dimerization is through intermolecular oxidative coupling reactions.89
The proposed mechanism for oxidative dimerization reactions in Nature involves the abstraction of a hydrogen atom from a phenolic or indole-containing substrate followed by a radical C–C coupling or addition reaction, often with the formation of a biaryl bond (Figure 8a).89 Traditional synthetic strategies to construct biaryl bonds face fundamental challenges in controlling the selectivity of bond formation. For example, metal-catalyzed cross-couplings such as Suzuki and Negishi reactions require the prefunctionalization of both fragments to dictate the site of bond formation, thereby adding steps to a given synthetic campaign.90 Alternatively, the direct oxidative coupling of two C–H bonds drastically increases the efficiency of a convergent synthetic sequence, albeit often at the expense of selectivity in the bond-forming event.91 In contrast, enzymes mediating these transformations have the advantage of catalyst control over the site- and stereoselectivity of the oxidative coupling reaction by templating the two substrates in a specific orientation in the enzyme active site (Figure 8b).
Figure 8.
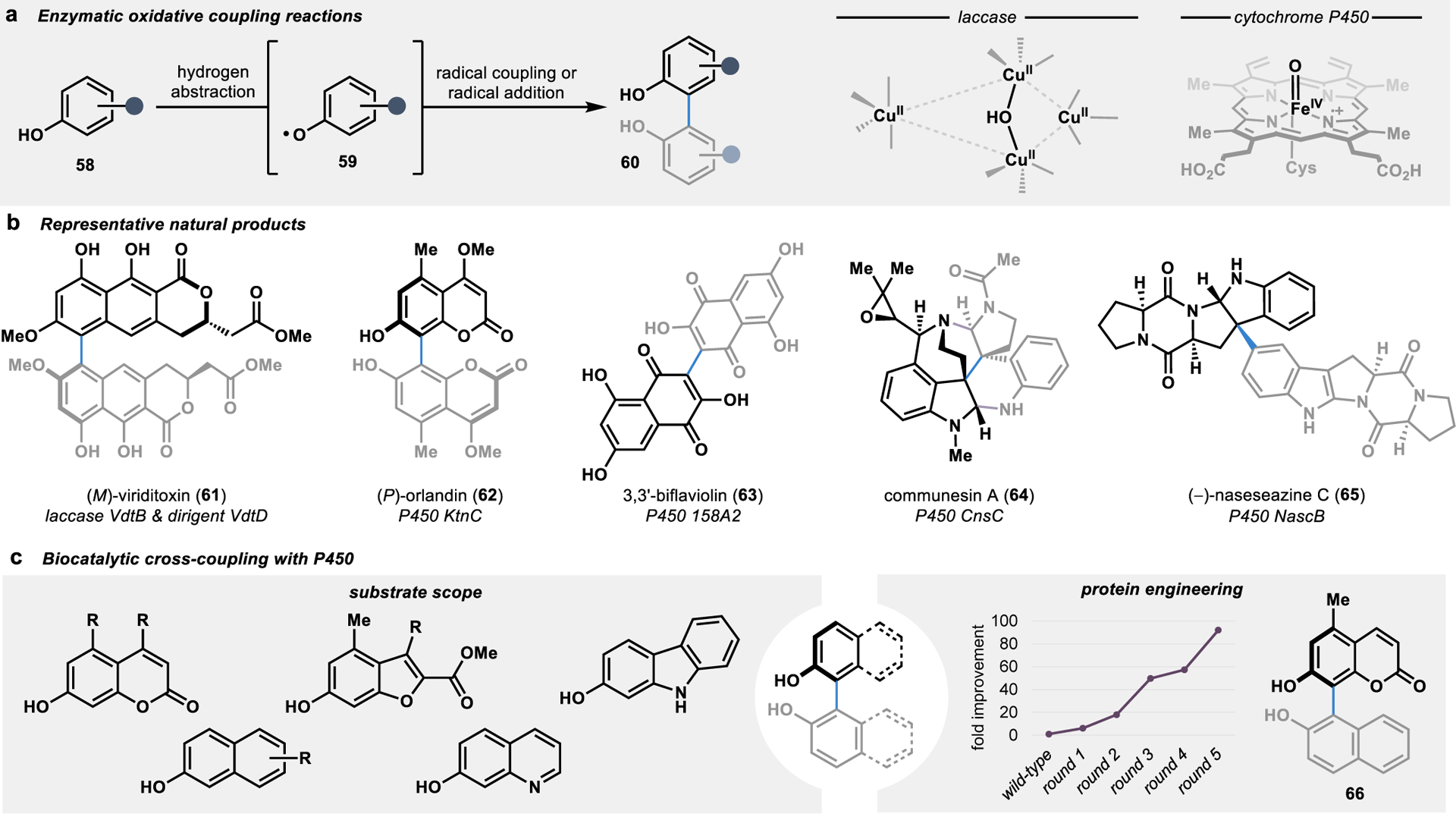
Biocatalytic C–C fragment coupling through oxidative coupling reactions. (a) Oxidative enzymes such as laccases and P450s can abstract a hydrogen from a phenolic substrate (58) to generate radical intermediate 59 for C–C bond formation. (b) Many biaryl and heterodimeric alkaloid natural products are formed through selective oxidative coupling reactions in fungal and bacterial biosynthetic pathways. (c) Biosynthetic P450s are highly tunable catalysts that can be harnessed for diverse biaryl coupling reactions.
One approach to oxidative dimerization in secondary metabolism involves enzymatic oxidation followed by rapid binding of the oxidized substrates to an auxiliary protein, wherein the templating of the two substrates in a specific orientation allows for a stereoselective radical–radical coupling reaction.92 These oxidations are typically performed by laccases, a class of multicopper oxidases that readily oxidize diverse phenolic substrates.93, 94 Laccase-catalyzed oxidations are ubiquitous in fungal and plant systems, such as in the biosynthesis of lignin, and have been harnessed for a variety of biotechnological applications.95 However, the application of laccases in asymmetric catalysis has been limited due to the scarcity of examples in which laccases exert control over the bond formation event following the initial oxidation.96 To overcome this limitation, auxiliary dirigent proteins can be harnessed to regain control over the selectivity of the bond formation event.89, 92, 97, 98 For example, in the convergent biosynthesis of viriditoxin (61), a late-stage oxidative dimerization catalyzed by a laccase and dirigent protein forms (P)-61 with excellent atroposelectivity (>95:5 er).98 In contrast, removal of the dirigent protein from the oxidative dimerization results in a complete loss in atroposelectivity, with the formation of a mixture of (P)-61 and (M)-61 in a 1:2 ratio.98
In addition to laccases, cytochrome P450 enzymes (P450s) have been identified as convergent catalysts in the biosynthesis of numerous dimeric natural products.32 However, unlike most laccases, these P450s exert control over both the oxidation and selectivity of the bond formation event.89 This catalyst-controlled selectivity has been demonstrated in the biosynthesis of an ever-expanding catalog of biaryl products from fungi and bacteria, with P450s catalyzing the selective dimerization of highly oxygenated tricyclic, coumarin, and naphthalene substrates (such as 62 and 63).89 In addition to the oxidative coupling of phenolic substrates, P450s have also been implicated in the oxidative coupling of substrates harboring indoles in the biosynthesis of alkaloids (such as 64 and 65).99–101
Recently, the potential application of P450s involved in the biosynthesis of biaryl metabolites was investigated as a tool for more general biocatalytic oxidative cross-coupling reactions.102 In particular, a P450 that naturally dimerizes a coumarin substrate to form bicoumarin 62 was identified to catalyze an array of coumarin cross-coupling reactions uniting a coumarin substrate with diverse phenolic substrates to form tetra-ortho-substituted biaryl scaffolds.102 Moreover, through the directed evolution of the P450, a catalyst with improved activity, site-selectivity, and atroposelectivity was engineered for the cross-coupling reaction to form biaryl 66 (Figure 8c). This tunability of P450 catalysts for oxidative cross-coupling reactions overcomes several limitations inherent to small molecule-based methods. For example, traditional oxidative cross-coupling methods often rely on the statistical distribution of individual substrate dimers and cross-coupling products in cases where blocking groups are not used.103, 104 Alternatively, small molecule-based methods for oxidative cross-coupling can be rendered selective, albeit at the expense of blocking groups.105, 106 Compared to the inherent limitations of small moleculebased catalysts for oxidative cross-coupling reactions, the developed biocatalytic approach provides a superior platform by providing a paradigm for catalyst-controlled selectivity and overcomes the requirements of blocking groups and limitations of substrate-controlled reaction outcomes.102
CONCLUSION & OUTLOOK
Until recently, the area of convergent biocatalysis remained largely untapped, preventing the full realization of the impact enzymes can offer to synthesize complex molecules.12 Encouragingly, the last decade has seen an increase in the development of biocatalytic strategies for C–N and C–C fragment coupling reactions. Nevertheless, these convergent biocatalytic strategies remain prohibitively underdeveloped compared to the vast repertoire of small molecule catalysts and reagents that mediate fragment couplings reactions. The further development of this category of biocatalysts can potentially change the retrosynthetic framework for fragment coupling reactions by circumventing traditionally accepted requirements for protecting groups and functional handles into synthetic planning. To realize this goal, it is imperative that significant efforts are spent in the discovery of biosynthetic enzymes from secondary metabolite pathways that perform fragment coupling reactions, particularly ones that can streamline bond constructions that are often difficult to achieve using traditional synthetic approaches. Additionally, continued development of biocatalytic strategies for bread-and-butter reactions in medicinal chemistry such as amide bond formation and reductive aminations.53, 107 Expanding the repertoire of biocatalytic strategies for these key transformations will provide more cost-effective and greener synthetic routes, especially considering the advantages of cofactor recycling in biocatalytic reactions offers compared to the often expensive and wasteful use of stoichiometric reagents in chemical transformations.108
The continued discovery of convergent biocatalytic equivalents for existing chemical methods for powerful bond constructions will expand the repertoire of biocatalytic convergence for use in the chemoenzymatic total synthesis of complex molecules. Traditional challenges associated with applying biocatalytic transformations in synthesis have included enzyme availability and limited substrate promiscuity, thus contributing to the slow adoption of biocatalytic methods within the mainstream synthetic community.109 However, the repertoire of biocatalytic methods has increased tremendously in recent years, and chemoenzymatic synthesis methods are taking up speed.8 To accelerate this trend, broader commercialization of enzymes is needed to increase their accessibility.
Moreover, to enable a more extensive adoption of biocatalytic methods to redefine the retrosynthesis of organic molecules, several innovations and developments are required in the field. A minimal connection is often made using enzymes as catalysts for routine chemical synthesis. This typically leaves a contemporary synthetic chemist believing that enzymes are only used for precise chemical transformations. Exposure to enzymatic chemistry should be included in routine undergraduate organic chemistry courses to expose potential future scientists to the use of biocatalytic methods for chemical synthesis.12 In parallel to the exposure of biocatalytic methods to undergraduate students, it is also crucial for biocatalytic methods to be broadly integrated with chemical platforms and databases routinely used for synthetic planning. An excellent example of this can be seen in the creation of RetroBioCat, an online database and synthetic planning tool focused on the implementation of biocatalytic cascades into retrosynthetic analysis.110 The inclusion of enzymatic methods in scientific solutions also necessitates the need to study and understand the factors contributing to the advantages and limitations of carrying out biocatalysis versus chemocatalysis for specific chemical transformations.
ACKNOWLEDGMENT
This work was supported by funds from the University of Michigan’s Life Sciences Institute and the Chemistry Department, Alfred P. Sloan Foundation, Research Corporation Cottrell Scholars program, and NIH R35 GM124880. L. E. Z. is grateful to the NIH National Center for Complementary & Integrative Health of the NIH F31AT010973. The authors thank Dr. Kendrick Smith for helpful discussion and feedback on this manuscript.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.McCowen SV; Doering NA; Sarpong R Retrosynthetic Strategies and Their Impact on Synthesis of Arcutane Natural Products. Chem. Sci 2020, 11, 7538–7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hendrickson JB Systematic Synthesis Design. IV. Numerical Codification of Construction Reactions. J. Am. Chem. Soc 1975, 97, 5784–5800. [Google Scholar]
- 3.Urabe D; Asaba T; Inoue M Convergent Strategies in Total Syntheses of Complex Terpenoids. Chem. Rev 2015, 115, 9207–9231. [DOI] [PubMed] [Google Scholar]
- 4.Smith MB, Synthetic Strategies. In Organic Synthesis (Third Edition), Smith MB, Ed. Academic Press: Oxford, 2010; pp 897–997. [Google Scholar]
- 5.Pompeo MM; Cheah JH; Movassaghi M Total Synthesis and Anti-cancer Activity of All Known Communesin Alkaloids and Related Derivatives. J. Am. Chem. Soc 2019, 141, 14411–14420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin H-C; McMahon TC; Patel A; Corsello M; Simon A; Xu W; Zhao M; Houk KN; Garg NK; Tang Y P450-Mediated Coupling of Indole Fragments to Forge Communesin and Unnatural Isomers. J. Am. Chem. Soc 2016, 138, 4002–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winkler CK; Schrittwieser JH; Kroutil W Power of Biocatalysis for Organic Synthesis. ACS Central Science 2021, 7, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakrabarty S; Romero EO; Pyser JB; Yazarians JA; Narayan ARH Chemoenzymatic Total Synthesis of Natural Products. Acc. Chem. Res 2021, 54, 1374–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu S; Snajdrova R; Moore JC; Baldenius K; Bornscheuer UT Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed 2021, 60, 88–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yi D; Bayer T; Badenhorst CPS; Wu S; Doerr M; Höhne M; Bornscheuer UT Recent Trends in Biocatalysis. Chem. Soc. Rev 2021, 50, 8003–8049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hönig M; Sondermann P; Turner NJ; Carreira EM Enantioselective Chemo- and Biocatalysis: Partners in Retrosynthesis. Angew. Chem. Int. Ed 2017, 56, 8942–8973. [DOI] [PubMed] [Google Scholar]
- 12.Turner NJ; O’Reilly E Biocatalytic Retrosynthesis. Nat. Chem. Biol 2013, 9, 285–288. [DOI] [PubMed] [Google Scholar]
- 13.Breuer M; Ditrich K; Habicher T; Hauer B; Keßeler M; Stürmer R; Zelinski T Industrial Methods for the Production of Optically Active Intermediates. Angew. Chem. Int. Ed 2004, 43, 788–824. [DOI] [PubMed] [Google Scholar]
- 14.Schmid A; Dordick JS; Hauer B; Kiener A; Wubbolts M; Witholt B Industrial Biocatalysis Today and Tomorrow. Nature 2001, 409, 258–268. [DOI] [PubMed] [Google Scholar]
- 15.Pàmies O; Bäckvall J-E Combined Metal Catalysis and Biocatalysis for an Efficient Deracemization Process. Curr. Opin. Biotechnol 2003, 14, 407–413. [DOI] [PubMed] [Google Scholar]
- 16.May O; Verseck S; Bommarius A; Drauz K Development of Dynamic Kinetic Resolution Processes for Biocatalytic Production of Natural and Nonnatural L-Amino Acids. Org. Process Res. Dev 2002, 6, 452–457. [Google Scholar]
- 17.Bruce M; St. Laurent D; Poindexter G; Monkovic I; Huang S; Balasubramanian N Kinetic Resolution of Piperazine-2-Carboxamide by Leucine Aminopeptidase. An Application in the Synthesis of the Nucleoside Transport Blocker (–)-Draflazine. Synth. Commun 1995, 25, 2673–2684. [Google Scholar]
- 18.Eichhorn E; Roduit J-P; Shaw N; Heinzmann K; Kiener A Preparation of (S)-Piperazine-2-Carboxylic Acid, (R)-Piperazine-2-Carboxylic Acid, and (S)-Piperidine-2-Carboxylic Acid by Kinetic Resolution of the Corresponding Racemic Carboxamides with Stereoselective Amidases in Whole Bacterial Cells. Tetrahedron Asymmetry 1997, 8, 2533–2536. [Google Scholar]
- 19.Bornscheuer UT; Huisman GW; Kazlauskas RJ; Lutz S; Moore JC; Robins K Engineering the Third Wave of Biocatalysis. Nature 2012, 485, 185–194. [DOI] [PubMed] [Google Scholar]
- 20.Ma SK; Gruber J; Davis C; Newman L; Gray D; Wang A; Grate J; Huisman GW; Sheldon RA A Green-by-Design Biocatalytic Process for Atorvastatin Intermediate. Green Chem. 2010, 12, 81–86. [Google Scholar]
- 21.de Miranda AS; Simon RC; Grischek B; de Paula GC; Horta BA; de Miranda LS; Kroutil W; Kappe CO; de Souza RO Chiral Chlorohydrins from the Biocatalyzed Reduction of Chloroketones: Chiral Building Blocks for Antiretroviral Drugs. ChemCatChem 2015, 7, 984–992. [Google Scholar]
- 22.Wu K; Zheng K; Xiong L; Yang Z; Jiang Z; Meng X; Shao L Efficient Synthesis of an Antiviral Drug Intermediate Using an Enhanced Short-Chain Dehydrogenase in an Aqueous-Organic Solvent System. Appl. Microbiol. Biotechnol 2019, 103, 4417–4427. [DOI] [PubMed] [Google Scholar]
- 23.Köhler V; Bailey KR; Znabet A; Raftery J; Helliwell M; Turner NJ Enantioselective Biocatalytic Oxidative Desymmetrization of Substituted Pyrrolidines. Angew. Chem 2010, 122, 2228–2230. [DOI] [PubMed] [Google Scholar]
- 24.Huffman MA; Fryszkowska A; Alvizo O; Borra-Garske M; Campos KR; Canada KA; Devine PN; Duan D; Forstater JH; Grosser ST Design of an In Vitro Biocatalytic Cascade for the Manufacture of Islatravir. Science 2019, 366, 1255–1259. [DOI] [PubMed] [Google Scholar]
- 25.Lange BM; Rujan T; Martin W; Croteau R Isoprenoid Biosynthesis: The Evolution of Two Ancient and Distinct Pathways Across Genomes. Proc. Natl. Acad. Sci 2000, 97, 13172–13177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Staunton J; Weissman KJ Polyketide Biosynthesis: A Millennium Review. Nat. Prod. Rep 2001, 18, 380–416. [DOI] [PubMed] [Google Scholar]
- 27.Maeda H; Dudareva N The Shikimate Pathway and Aromatic Amino Acid Biosynthesis in Plants. Annu. Rev. Plant Biol 2012, 63, 73–105. [DOI] [PubMed] [Google Scholar]
- 28.Tang M-C; Zou Y; Watanabe K; Walsh CT; Tang Y Oxidative Cyclization in Natural Product Biosynthesis. Chem. Rev 2017, 117, 5226–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen H; Du L Iterative Polyketide Biosynthesis by Modular Polyketide Synthases in Bacteria. Appl. Microbiol. Biotechnol 2016, 100, 541–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walsh CT; Fischbach MA Natural Products Version 2.0: Connecting Genes to Molecules. J. Am. Chem. Soc 2010, 132, 2469–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lian G; Yu B Naturally Occurring Dimers from Chemical Perspective. Chem. Biodiversity 2010, 7, 2660–2691. [DOI] [PubMed] [Google Scholar]
- 32.Liu J; Liu A; Hu Y Enzymatic Dimerization in the Biosynthetic Pathway of Microbial Natural Products. Nat. Prod. Rep 2021, 38, 1469–1505. [DOI] [PubMed] [Google Scholar]
- 33.Toone EJ; Whitesides GM, Enzymes in Carbohydrate Synthesis. ACS Publications: 1991; pp 1–22. [Google Scholar]
- 34.Wong CH; Whitesides GM Synthesis of Sugars by Aldolase-Catalyzed Condensation Reactions. J. Org. Chem 1983, 48, 3199–3205. [Google Scholar]
- 35.Li A; Cai L; Chen Z; Wang M; Wang N; Nakanishi H; Gao X-D; Li Z Recent Advances in the Synthesis of Rare Sugars Using DHAP-Dependent Aldolases. Carbohydr. Res 2017, 452, 108–115. [DOI] [PubMed] [Google Scholar]
- 36.Koeller KM; Wong C-H Synthesis of Complex Carbohydrates and Glycoconjugates: Enzyme-Based and Programmable One-Pot Strategies. Chem. Rev 2000, 100, 4465–4494. [DOI] [PubMed] [Google Scholar]
- 37.Li T-L; Liu Y-C; Lyu S-Y Combining Biocatalysis and Chemoselective Chemistries for Glycopeptide Antibiotics Modification. Curr. Opin. Chem. Biol 2012, 16, 170–178. [DOI] [PubMed] [Google Scholar]
- 38.Yamaguchi T; Amin MN; Toonstra C; Wang L-X Chemoenzymatic Synthesis and Receptor Binding of Mannose-6-Phosphate (M6P)-Containing Glycoprotein Ligands Reveal Unusual Structural Requirements for M6P Receptor Recognition. J. Am. Chem. Soc 2016, 138, 12472–12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amin MN; Huang W; Mizanur RM; Wang L-X Convergent Synthesis of Homogeneous Glc1Man9GlcNAc2-Protein and Derivatives as Ligands of Molecular Chaperones in Protein Quality Control. J. Am. Chem. Soc 2011, 133, 14404–14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mikhailopulo I; Miroshnikov A New Trends in Nucleoside Biotechnology. Acta Naturae 2010, 2, 36–58. [PMC free article] [PubMed] [Google Scholar]
- 41.McIntosh JA; Benkovics T; Silverman SM; Huffman MA; Kong J; Maligres PE; Itoh T; Yang H; Verma D; Pan W; Ho H-I; Vroom J; Knight AM; Hurtak JA; Klapars A; Fryszkowska A; Morris WJ; Strotman NA; Murphy GS; Maloney KM; Fier PS Engineered Ribosyl-1-Kinase Enables Concise Synthesis of Molnupiravir, An Antiviral for COVID-19. ACS Cent. Sci 2021, 7, 1980–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McLaughlin M; Kong J; Belyk KM; Chen B; Gibson AW; Keen SP; Lieberman DR; Milczek EM; Moore JC; Murray D Enantioselective Synthesis of 4′-Ethynyl-2-Fluoro-2′-Deoxyadenosine (EFdA) via Enzymatic Desymmetrization. Org. Lett 2017, 19, 926–929. [DOI] [PubMed] [Google Scholar]
- 43.The Amide Linkage: Structural Significance, Chemistry, Biochemistry and Material Science; Greenberg A, Breneman CM, Liebman JF, Eds.; Wiley: New York, 2000. [Google Scholar]
- 44.Pattabiraman VR; Bode JW Rethinking Amide Bond Synthesis. Nature 2011, 480, 471–479. [DOI] [PubMed] [Google Scholar]
- 45.Sabatini MT; Boulton LT; Sneddon HF; Sheppard TD A Green Chemistry Perspective on Catalytic Amide Bond Formation. Nat. Catal 2019, 2, 10–17. [Google Scholar]
- 46.Schmeing TM; Ramakrishnan V What Recent Ribosome Structures Have Revealed About the Mechanism of Translation. Nature 2009, 461, 1234–1242. [DOI] [PubMed] [Google Scholar]
- 47.Goswami A; Van Lanen SG Enzymatic Strategies and Biocatalysts for Amide Bond Formation: Tricks of the Trade Outside of the Ribosome. Mol. BioSys 2015, 11, 338–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winn M; Richardson SM; Campopiano DJ; Micklefield J Harnessing and Angineering Amide Bond Forming Ligases for the Synthesis of Amides. Curr. Opin. Chem. Biol 2020, 55, 77–85. [DOI] [PubMed] [Google Scholar]
- 49.Petchey MR; Rowlinson B; Lloyd RC; Fairlamb IJ; Grogan G Biocatalytic Synthesis of Moclobemide Using the Amide Bond Synthetase McbA Coupled with an ATP Recycling System. ACS Catal. 2020, 10, 4659–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ji C; Chen Q; Li Q; Huang H; Song Y; Ma J; Ju J Chemoenzymatic Synthesis of β-Carboline Derivatives using McbA, A New ATP-Dependent Amide Synthetase. Tetrahedron Lett. 2014, 55, 4901–4904. [Google Scholar]
- 51.Petchey M; Cuetos A; Rowlinson B; Dannevald S; Frese A; Sutton PW; Lovelock S; Lloyd RC; Fairlamb IJ; Grogan G The Broad Aryl Acid Specificity of the Amide Bond Synthetase McbA Suggests Potential for the Biocatalytic Synthesis of Amides. Angew. Chem. Int. Ed 2018, 57, 11584–11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Winn M; Rowlinson M; Wang F; Bering L; Francis D; Levy C; Micklefield J Discovery, Characterization and Engineering of Ligases for Amide Synthesis. Nature 2021, 593, 391–398. [DOI] [PubMed] [Google Scholar]
- 53.Roughley SD; Jordan AM The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]
- 54.Patil MD; Grogan G; Bommarius A; Yun H Oxidoreductase-Catalyzed Synthesis of Chiral Amines. ACS Catal. 2018, 8, 10985–11015. [Google Scholar]
- 55.Slabu I; Galman JL; Lloyd RC; Turner NJ Discovery, Engineering, and Synthetic Application of Transaminase Biocatalysts. ACS Catal. 2017, 7, 8263–8284. [Google Scholar]
- 56.Marshall JR; Yao P; Montgomery SL; Finnigan JD; Thorpe TW; Palmer RB; Mangas-Sanchez J; Duncan RA; Heath RS; Graham KM Screening and Characterization of a Diverse Panel of Metagenomic Imine Reductases for Biocatalytic Reductive Amination. Nat. Chem 2021, 13, 140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schober M; MacDermaid C; Ollis AA; Chang S; Khan D; Hosford J; Latham J; Ihnken LAF; Brown MJB; Fuerst D; Sanganee MJ; Roiban G-D Chiral Synthesis of LSD1 Inhibitor GSK2879552 Enabled by Directed Evolution of an Imine Reductase. Nat. Catal 2019, 2, 909–915. [Google Scholar]
- 58.Roddan R; Ward JM; Keep NH; Hailes HC Pictet–Spenglerases in Alkaloid Biosynthesis: Future Applications in Biocatalysis. Curr. Opin. Chem. Biol 2020, 55, 69–76. [DOI] [PubMed] [Google Scholar]
- 59.Stoeckigt J; Husson H; Kan-Fan C; Zenk M Cathenamine, A Central Intermediate in the Cell Free Biosynthesis of Ajmalicine and Related Indole Alkaloids. J. Am. Chem. Soc 1977, 164–166. [Google Scholar]
- 60.Stöckigt J; Antonchick AP; Wu F; Waldmann H The Pictet–Spengler Reaction in Nature and in Organic Chemistry. Angew. Chem. Int. Ed 2011, 50, 8538–8564. [DOI] [PubMed] [Google Scholar]
- 61.Maresh JJ; Giddings L-A; Friedrich A; Loris EA; Panjikar S; Trout BL; Stöckigt J; Peters B; O’Connor SE Strictosidine Synthase: Mechanism of a Pictet–Spengler Catalyzing Enzyme. J. Am. Chem. Soc 2008, 130, 710–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lichman BR; Gershater MC; Lamming ED; Pesnot T; Sula A; Keep NH; Hailes HC; Ward JM ‘Dopamine-First’ Mechanism Enables the Rational Engineering of the Norcoclaurine Synthase Aldehyde Activity Profile. FEBS J. 2015, 282, 1137–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cigan E; Eggbauer B; Schrittwieser JH; Kroutil W The Role of Biocatalysis in the Asymmetric Synthesis of Alkaloids–An Update. RSC Adv. 2021, 11, 28223–28270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zetzsche LE; Narayan ARH Broadening the Scope of Biocatalytic C–C Bond Formation. Nat. Rev. Chem 2020, 4, 334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmidt NG; Eger E; Kroutil W Building Bridges: Biocatalytic C–C-Bond Formation toward Multifunctional Products. ACS Catal. 2016, 6, 4286–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liang D-M; Liu J-H; Wu H; Wang B-B; Zhu H-J; Qiao J-J Glycosyltransferases: Mechanisms and Applications in Natural Product Development. Chem. Soc. Rev 2015, 44, 8350–8374. [DOI] [PubMed] [Google Scholar]
- 67.Yan M; Lo JC; Edwards JT; Baran PS Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc 2016, 138, 12692–12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grosheva D; Hyster TK Light-Driven Flavin-Based Biocatalysis. First Edition ed.; Wiley-VCH GmbH: 2021; 293–313. [Google Scholar]
- 69.Toogood HS; Scrutton NS Discovery, Characterization, Engineering, and Applications of Ene-Reductases for Industrial Biocatalysis. ACS Catal. 2018, 8, 3532–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang X; Wang B; Wang Y; Jiang G; Feng J; Zhao H Photoenzymatic Enantioselective Intermolecular Radical Hydroalkylation. Nature 2020, 584, 69–74. [DOI] [PubMed] [Google Scholar]
- 71.Page CG; Cooper SJ; DeHovitz JS; Oblinsky DG; Biegasiewicz KF; Antropow AH; Armbrust KW; Ellis JM; Hamann LG; Horn EJ; Oberg KM; Scholes GD; Hyster TK Quaternary Charge-Transfer Complex Enables Photoenzymatic Intermolecular Hydroalkylation of Olefins. J. Am. Chem. Soc 2020, 143, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fu H; Lam H; Emmanuel MA; Kim JH; Sandoval BA; Hyster TK Ground-State Electron Transfer as an Initiation Mechanism for Biocatalytic C–C Bond Forming Reactions. J. Am. Chem. Soc 2021, 143, 9622–9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Watkins-Dulaney E; Straathof S; Arnold F Tryptophan Synthase: Biocatalyst Extraordinaire. ChemBioChem 2021, 22, 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schultz EE; Braffman NR; Luescher MU; Hager HH; Balskus EP Biocatalytic Friedel–Crafts Alkylation Using a Promiscuous Biosynthetic Enzyme. Angew. Chem. Int. Ed 2019, 58, 3151–3155. [DOI] [PubMed] [Google Scholar]
- 75.Leveson-Gower RB; Zhou Z; Drienovská I; Roelfes G Unlocking Iminium Catalysis in Artificial Enzymes to Create a Friedel–Crafts Alkylase. ACS Catal. 2021, 11, 6763–6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rueping M; Nachtsheim BJ A Review of New Developments in the Friedel-Crafts Alkylation - From Green Chemistry to Asymmetric Catalysis. Beilstein J Org Chem 2010, 6, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nakamura H; Schultz EE; Balskus EP A New Strategy for Aromatic Ring Alkylation in Cylindrocyclophane Biosynthesis. Nat. Chem. Biol 2017, 13, 916–921. [DOI] [PubMed] [Google Scholar]
- 78.Nicolaou KC; Snyder SA; Montagnon T; Vassilikogiannakis G The Diels–Alder Reaction in Total Synthesis. Angew. Chem. Int. Ed 2002, 41, 1668–1698. [DOI] [PubMed] [Google Scholar]
- 79.Klas K; Tsukamoto S; Sherman DH; Williams RM Natural Diels–Alderases: Elusive and Irresistable. J. Org. Chem 2015, 80, 11672–11685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ghattas W; Mahy J-P; Réglier M; Simaan AJ Artificial Enzymes for Diels–Alder Reactions. ChemBioChem 2021, 22, 443–459. [DOI] [PubMed] [Google Scholar]
- 81.Reetz MT Artificial Metalloenzymes as Catalysts in Stereoselective Diels–Alder Reactions. Chem. Rec 2012, 12, 391–406. [DOI] [PubMed] [Google Scholar]
- 82.Hilvert D; Hill KW; Nared KD; Auditor MTM Antibody Catalysis of the Diels-Alder Reaction. J. Am. Chem. Soc 1989, 111, 9261–9262. [Google Scholar]
- 83.Siegel JB; Zanghellini A; Lovick HM; Kiss G; Lambert AR; Clair JLS; Gallaher JL; Hilvert D; Gelb MH; Stoddard BL Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diels-Alder Reaction. Science 2010, 329, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dan Q; Newmister SA; Klas KR; Fraley AE; McAfoos TJ; Somoza AD; Sunderhaus JD; Ye Y; Shende VV; Yu F Fungal Indole Alkaloid Biogenesis Through Evolution of a Bifunctional Reductase/Diels–Alderase. Nat. Chem. Biol 2019, 11, 972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen Q; Gao J; Jamieson C; Liu J; Ohashi M; Bai J; Yan D; Liu B; Che Y; Wang Y Enzymatic Intermolecular Hetero-Diels–Alder Reaction in the Biosynthesis of Tropolonic Sesquiterpenes. J. Am. Chem. Soc 2019, 141, 14052–14056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim HJ; Ruszczycky MW; Choi S -h.; Liu, Y.-n.; Liu, H.-w. Enzyme-Catalysed [4+2] Cycloaddition is a Key Step in the Biosynthesis of Spinosyn A. Nature 2011, 473, 109–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gao L; Su C; Du X; Wang R; Chen S; Zhou Y; Liu C; Liu X; Tian R; Zhang L FAD-Dependent Enzyme-Catalysed Intermolecular [4+2] Cycloaddition in Natural Product Biosynthesis. Nat. Chem 2020, 12, 620–628. [DOI] [PubMed] [Google Scholar]
- 88.Corey EJ Catalytic Enantioselective Diels–Alder Reactions: Methods, Mechanistic Fundamentals, Pathways, and Applications. Angew. Chem. Int. Ed 2002, 41, 1650–1667. [DOI] [PubMed] [Google Scholar]
- 89.Hüttel W; Müller M Regio- and Stereoselective Intermolecular Phenol Coupling Enzymes in Secondary Metabolite Biosynthesis. Nat. Prod. Rep 2021, 38, 1011–1043. [DOI] [PubMed] [Google Scholar]
- 90.Yin L; Liebscher J Carbon–Carbon Coupling Reactions Catalyzed by Heterogeneous Palladium Catalysts. Chem. Rev 2007, 107, 133–173. [DOI] [PubMed] [Google Scholar]
- 91.Kozlowski MC Oxidative Coupling in Complexity Building Transforms. Acc. Chem. Res 2017, 50, 638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pickel B; Schaller A Dirigent Proteins: Molecular Characteristics and Potential Biotechnological Applications. Appl. Microbiol. Biotechnol 2013, 97, 8427–8438. [DOI] [PubMed] [Google Scholar]
- 93.Jeon J-R; Baldrian P; Murugesan K; Chang Y-S Laccase-Catalysed Oxidations of Naturally Occurring Phenols: From In Vivo Biosynthetic Pathways to Green Synthetic Applications. Microb Biotechnol 2012, 5, 318–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Enguita FJ; Martins LO; Henriques AO; Carrondo MA Crystal Structure of a Bacterial Endospore Coat Component: A Laccase with Enhanced Thermostability Properties. J. Biol. Chem 2003, 278, 19416–19425. [DOI] [PubMed] [Google Scholar]
- 95.Mate DM; Alcalde M Laccase: A Multi-Purpose Biocatalyst at the Forefront of Biotechnology. Microb Biotechnol 2017, 10, 1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sagui F; Chirivì C; Fontana G; Nicotra S; Passarella D; Riva S; Danieli B Laccase-Catalyzed Coupling of Catharanthine and Vindoline: An Efficient Approach to the Bisindole Alkaloid Anhydrovinblastine. Tetrahedron 2009, 65, 312–317. [Google Scholar]
- 97.Kim SS; Sattely ES Dirigent Proteins Guide Asymmetric Heterocoupling for the Synthesis of Complex Natural Product Analogues. J. Am. Chem. Soc 2021, 143, 5011–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hu J; Li H; Chooi Y-H Fungal Dirigent Protein Controls the Stereoselectivity of Multicopper Oxidase-Catalyzed Phenol Coupling in Viriditoxin Biosynthesis. J. Am. Chem. Soc 2019, 141, 8068–8072. [DOI] [PubMed] [Google Scholar]
- 99.Guo Z; Li P; Chen G; Li C; Cao Z; Zhang Y; Ren J; Xiang H; Lin S; Ju J; Chen Y Design and Biosynthesis of Dimeric Alboflavusins with Biaryl Linkages via Regiospecific C–C Bond Coupling. J. Am. Chem. Soc 2018, 140, 18009–18015. [DOI] [PubMed] [Google Scholar]
- 100.Tian W; Sun C; Zheng M; Harmer JR; Yu M; Zhang Y; Peng H; Zhu D; Deng Z; Chen S-L Efficient Biosynthesis of Heterodimeric C 3-Aryl Pyrroloindoline Alkaloids. Nat. Commun 2018, 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shende VV; Khatri Y; Newmister SA; Sanders JN; Lindovska P; Yu F; Doyon TJ; Kim J; Houk K; Movassaghi M Structure and Function of NzeB, A Versatile C–C and C–N Bond-Forming Diketopiperazine Dimerase. J. Am. Chem. Soc 2020, 142, 17413–17424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zetzsche L; Yazarians J; Chakrabarty S; Hinze M; Lukowski A; Joyce L; Narayan A Biaryl Bond Formation Through Biocatalytic Oxidative Cross-Coupling Reactions. ChemRxiv 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Meesala Y; Wu H-L; Koteswararao B; Kuo T-S; Lee W-Z Aerobic Oxidative Coupling of 2-Naphthol Derivatives Catalyzed by a Hexanuclear Bis(μ-hydroxo)copper(II) Catalyst. Organometallics 2014, 33, 4385–4393. [Google Scholar]
- 104.Qu S; Greenhalgh MD; Smith AD Isothiourea-Catalysed Regioselective Acylative Kinetic Resolution of Axially Chiral Biaryl Diols. Chem. Eur. J 2019, 25, 2816–2823. [DOI] [PubMed] [Google Scholar]
- 105.Nieves-Quinones Y; Paniak TJ; Lee YE; Kim SM; Tcyrulnikov S; Kozlowski MC Chromium-Salen Catalyzed Cross-coupling of Phenols: Mechanism and Origin of the Selectivity. J. Am. Chem. Soc 2019, 141, 10016–10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bringmann G; Price Mortimer AJ; Keller PA; Gresser MJ; Garner J; Breuning M Atroposelective Synthesis of Axially Chiral Biaryl Compounds. Angew. Chem. Int. Ed 2005, 44, 5384–5427. [DOI] [PubMed] [Google Scholar]
- 107.Boström J; Brown DG; Young RJ; Keserü GM Expanding the Medicinal Chemistry Synthetic Toolbox. Nat. Rev. Drug. Discov 2018, 17, 709–727. [DOI] [PubMed] [Google Scholar]
- 108.Mordhorst S; Andexer JN Round, Round We Go–Strategies for Enzymatic Cofactor Regeneration. Nat. Prod. Rep 2020, 37, 1316–1333. [DOI] [PubMed] [Google Scholar]
- 109.Chakrabarty S; Wang Y; Perkins JC; Narayan ARH Scalable Biocatalytic C–H Oxyfunctionalization Reactions. Chem. Soc. Rev 2020, 49, 8137–8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Finnigan W; Hepworth LJ; Flitsch SL; Turner NJ RetroBioCat as a Computer-Aided Synthesis Planning Tool for Biocatalytic Reactions and Cascades. Nat. Catal 2021, 4, 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]