

Abstract
Age‐related skeletal muscle atrophy or sarcopenia is a significant societal problem that is becoming amplified as the world's population continues to increase. The regeneration of damaged skeletal muscle is mediated by muscle stem cells, but in old age muscle stem cells become functionally attenuated. The molecular mechanisms that govern muscle stem cell aging encompass changes across multiple regulatory layers and are integrated by the three‐dimensional organization of the genome. To quantitatively understand how hierarchical chromatin architecture changes during muscle stem cell aging, we generated 3D chromatin conformation maps (Hi‐C) and integrated these datasets with multi‐omic (chromatin accessibility and transcriptome) profiles from bulk populations and single cells. We observed that muscle stem cells display static behavior at global scales of chromatin organization during aging and extensive rewiring of local contacts at finer scales that were associated with variations in transcription factor binding and aberrant gene expression. These data provide insights into genome topology as a regulator of molecular function in stem cell aging.
Keywords: aging, chromatin architecture, muscle stem cells
The regeneration of damaged skeletal muscle is mediated by muscle stem cells, but in old age muscle stem cells become functionally attenuated. To quantitatively understand how hierarchical chromatin architecture changes during muscle stem cell aging, 3D chromatin conformation maps (Hi‐C) were generated and these datasets were integrated with multi‐omic (chromatin accessibility and transcriptome) profiles from bulk populations and single cells. Old aged muscle stem cells displayed static behavior at global scales of chromatin organization but extensive rewiring of local contacts at finer scales that were associated with variations in transcription factor binding and aberrant gene expression.
Abbreviations
- ATAC
Assay for Transposase‐Accessible Chromatin
- CCAN
cis‐co‐accessible network
- FACS
fluorescent activated cell sorting
- kb
kilobase
- MuSC
muscle stem cell
- Mb
megabase
- TAD
topologically associated domains
- TF
transcription factor
- 3D
three‐dimensional
1. INTRODUCTION
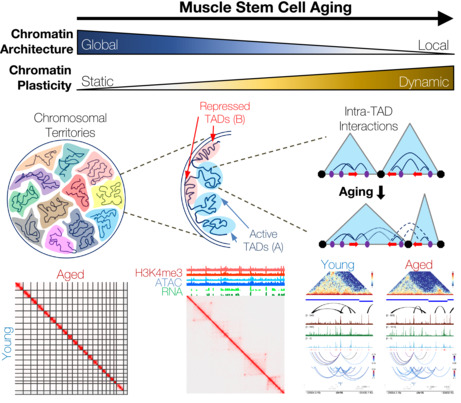
The growing population over the age of 65 presents significant healthcare challenges (Hung et al., 2011) including reduced mobility and increased frailty, which is nominally associated with declines in the volume, health, and repair of skeletal muscle. Skeletal muscle is regenerated by a population of resident muscle stem cells (MuSCs; Abou‐Khalil et al., 2015; Wang & Rudnicki, 2012) that decrease in number and function with age (Blau et al., 2015; Shcherbina et al., 2020). The molecular mechanisms that govern MuSC dysfunction in old age encompass changes across multiple inter‐connected molecular systems (López‐Otín et al., 2013; Ren et al., 2017; Singh et al., 2019) which collectively converge to drive aberrant chromatin packaging and three‐dimensional folding of the genome (Aguilar & Craighead, 2013). 3D genomic packaging of chromatin plays a primary role in regulating cellular functions by physically contacting distal regulatory elements such as enhancers with genes (Yu & Ren, 2017), facilitating changes in transcription (Dixon et al., 2015), yet how this architecture is modified in MuSCs in old age remains unknown (Bianconi & Mozzetta, 2022).
To probe how old age induces changes in nuclear organization in MuSCs, we performed in situ chromosome conformation capture followed by sequencing (Hi‐C) on MuSCs isolated from young and aged skeletal murine muscles. We integrated the Hi‐C maps with gene expression (RNA‐seq), chromatin modifications associated with promoters (H3K4me3), and chromatin accessibility (ATAC‐seq; Shcherbina et al., 2020) profiles, and observed stable chromatin architecture at the level of chromatin compartments. These results contrasted with topologically associated domains (TADs) and chromatin loops that displayed dynamic restructuring and encompassed genes associated with cell cycle regulation, maintenance of quiescence, and cellular stress. To increase the resolution of our approach, we generated single‐cell ATAC‐seq profiles of young and aged MuSCs and integrated these datasets with age‐matched single‐cell RNA‐seq datasets. This approach revealed extensive rewiring within chromatin hubs at the level of enhancer‐promoter contacts that was linked to alterations in gene expression. Together, this work represents a rich multi‐omic framework that provides insights into the regulation of pathological gene expression programs that attenuate MuSC regenerative potential in old age.
2. RESULTS
2.1. Profiling of global 3D genome organization in muscle stem cells during aging
To understand how MuSC genome organization is modified in aging, hind limb muscles (tibialis anterior, gastrocnemius, extensor digitorum longus, quadriceps) were isolated from young (3 months) and aged (24–26 months) mice. Fluorescent activated cell sorting (FACS) with both negative (Sca‐1−, CD45−, Mac‐1−, Ter‐119‐, CD31−) and positive surface markers (CXCR4+, β1‐integrin+) was used to isolate MuSCs (Aguilar et al., 2016; Yang et al., 2021) and genome‐wide chromatin interactions were profiled through in situ Hi‐C15 in biological duplicates from 75–100 k MuSCs (Figure 1a). The resulting proximity dataset replicates were processed with Juicer (Durand, Shamim, et al., 2016), yielding highly reproducible contact maps (>0.98 Pearson correlation, Figure S1A,B). Pooled replicates comprised ~1.86 x 108 chromosomal contacts for young MuSCs and ~ 1.56 x 108 chromosomal contacts for aged MuSCs, of which ~63% were long‐range (>20 kb) intra‐chromosomal cis‐interactions (Figure S1C). The Hi‐C matrices exhibited sufficient sequencing depth to reveal chromatin structures such as topologically associated domains (TADs) and chromatin loops with up to 5 kb resolution under visual inspection in JuiceBox (Durand, Robinson, et al., 2016; Figure 1b). The contact maps were segmented into ~1 Mb regions associated with euchromatic (A) and repressive (B) chromatin compartments by analyzing the first eigenvector of the Pearson correlation matrices at 100 kb resolution (Lieberman‐Aiden et al., 2009; Figure S1D). This analysis partitioned the genome into “A” and “B” compartments in a 52.5/47.5 ratio in both young and aged MuSCs (Figure S1E) with similar compartmentalization strengths (Flyamer et al., 2017) as measured by the average contact enrichment within and between compartments (Figure S1F). We observed minimal re‐arrangement of A/B compartments in old age, with <5% of the genome transitioning between compartments (Figures 1c,d). Consistent with previous findings, “A” compartments displayed increased chromatin accessibility (Shcherbina et al., 2020; ATAC‐seq (Buenrostro et al., 2013), Figure 1e) and activating H3K4me3 signals (Liu et al., 2013) compared to repressive “B” compartments, and aged MuSCs displayed increased chromatin accessibility within static “A” compartments (Figure S1G). RNA‐seq datasets (Shcherbina et al., 2020) of age‐matched MuSCs agreed with “A” and “B” chromatin compartment assignments, whereby genes within “A” compartments showed increased expression relative to genes within “B” compartments (2059 differentially expressed genes in “A”; 154 in “B”, Figure 1f). In addition, only the static “A” compartment displayed consistent changes in expression in old age MuSCs. Annotation of genes in the static “A” compartment revealed upregulation of transcripts in young MuSCs associated with cell‐cycle checkpoints (Sousa‐Victor et al., 2022; Cenpn, Ccne1, Cdc23), SUMOylation of chromatin organizing proteins (Cubeñas‐Potts & Matunis, 2013; Pias1/2, Satb2, Rnf2), and metabolism supportive of quiescence (Relaix et al., 2021), including fatty acid beta‐oxidation (Mecr, Hadha/b, acyl‐CoA dehydrogenase family) and the citric acid cycle (Pdk1/2, Ldha, Pdha1). In contrast, aged MuSCs showed increased expression in electron transport chain activity (Relaix et al., 2021; NADH:ubiquinone oxidoreductase family) and response to interferon‐beta (Ifnar2, Igtp, Irf1; Figure S1H). Genes in the static “B” compartment showed upregulation of G‐protein coupled receptor (GPCR) activity (Plppr1, Npy, Adra1b) in aged MuSCs, which has been linked to stem cell fate regulation (Kobayashi et al., 2010), while genes expressed in young MuSCs were related to Rho GTPase activity (Eliazer et al., 2019; Kann et al., 2022; Arhgap28/44, Pik3c3, Fgd4) and cell migration (Cdh13, Actc1, Vegfc). Summing these results shows minimal plasticity in global chromatin architecture during MuSC aging, suggesting that changes in MuSC expression with old age may be conferred through altered local interactions in open chromatin compartments.
FIGURE 1.
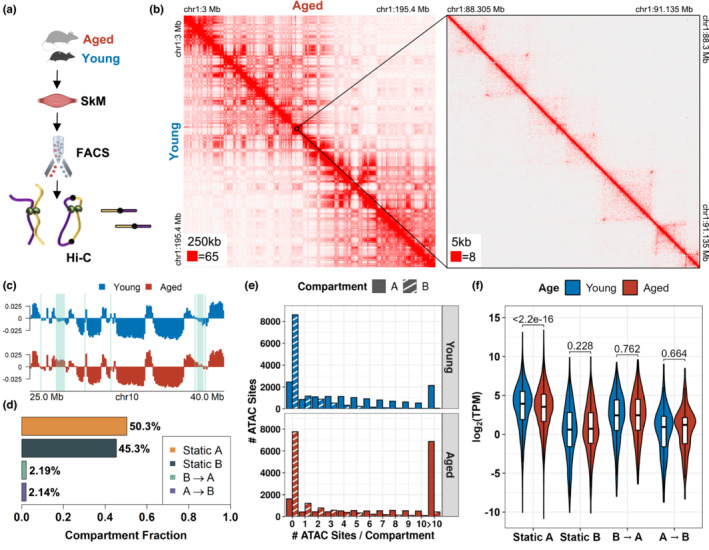
Global changes in 3D genome organization are largely static during muscle stem cell aging. (a) Schematic of experiment. (b) Normalized contact heatmaps of young (lower triangle) versus aged (upper triangle) MuSCs for chr1 at 250 kb resolution. A zoomed‐in region is shown at 5 kb resolution. The maximum color map value for pixels is shown in the bottom left corner of each heatmap. (c) Representative A/B compartment signal showing changes between young and aged MuSCs at 100 kb resolution. Compartment switches are highlighted in green. (d) Quantification of 100 kb bins that switch compartments (Young to Aged). (e) Distribution of ATAC‐seq peaks in A and B compartments. (f) Gene expression in log2(TPM) per A/B compartment in young and aged MuSCs. All statistical comparisons are unpaired Mann–Whitney U‐tests.
2.2. Local chromatin architecture is altered during muscle stem cell aging
To investigate local changes in chromatin topology, we characterized TADs in 40 kb‐resolution normalized contact matrices using HiCExplorer (Wolff et al., 2020). We identified 2,824 and 2,709 TADs in young and aged MuSCs, respectively (Figure 2a, Figure S2A). Integration of ATAC‐seq and H3K4me3 signals revealed enrichments within TAD domains in an “A” compartment‐dependent manner (Figure 2b,c, Figure S2B). Consistent with previous reports (Dixon et al., 2012), TAD boundaries were enriched for CTCF motifs (Figure S2C), gene promoters, and transcription termination sites (Figures S2D,E), and expression of housekeeping genes was enriched at stable TAD boundaries (Figure S2F). TADs showed extensive restructuring with old age, and TAD boundaries that were lost or gained with age were observed to rarely switch compartments from A → B or B → A (Figure 2d), and displayed reductions in both insulation strength (i.e., increased TAD separation scores; Figures S2G) and gene expression (Figure S2H) relative to stable boundaries. TAD rearrangements were classified into shifts, splits, merges, and indeterminate rearrangements comprising some combination of the previous three classifications (Figures 2c, Figures S2I,J). These rearrangements did not associate with changes in gene expression within TAD domains or at TAD boundaries, but boundaries that were gained with age (unique in aged MuSCs) were repositioned further from all nearby gene promoters compared to stable boundaries (Fisher's test p‐value <2.2 e‐16, Figure S2k). Stable boundaries and those that were lost with age (unique in young MuSCs) were stationed at similar distances to gene promoters, indicating that TADs are potentially restructured during aging to create more space within TAD domains for regulatory interactions.
FIGURE 2.
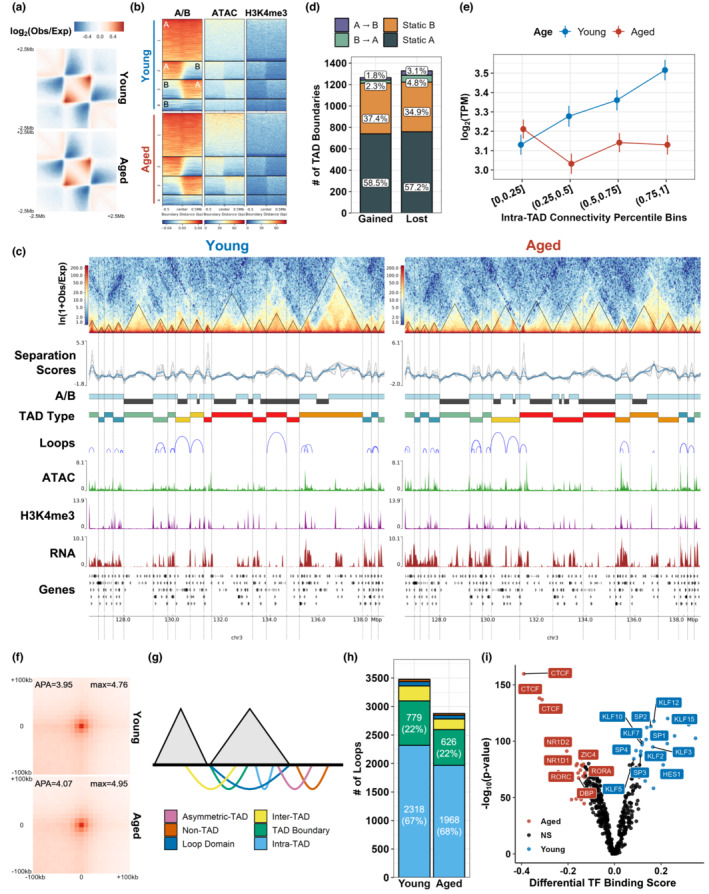
Muscle stem cells exhibit age‐dependent changes in topologically associated domains (TADs) and chromatin loops. (a) Aggregate contact heatmaps at 40 kb resolution over a 5 Mb region centered on contact domains (TADs). (b) Distributions of ATAC‐seq and H3K4me3 signals (RPKM) across a 1 Mb region centered on TAD boundaries. K‐means clustering (4 clusters) of the A/B compartment signal classified the boundaries by whether they fell within A/B compartments or divided compartment switching regions. (c) Representative contact heatmaps log1p(observed/expected), TAD separation score tracks, A/B compartments (A, blue; B, gray), TAD domains, TAD boundaries (vertical lines and triangles), chromatin loops, ATAC‐seq and H3K4me3 fold‐change signal tracks, and gene expression in RPKM for young and aged MuSCs. TAD domains are colored by TAD rearrangement type. (d) Distribution of TAD boundaries gained in aged and lost in young across A/B compartment groups. (e) Gene expression (mean ± SEM) in log2(TPM) against binned intra‐TAD connectivity percentiles. (f) Aggregate Peak Analysis (APA) of chromatin loops. (g) Diagram and (h) quantitation of chromatin loop classifications. (i) Volcano plot of inferred binding activity of expressed (TPM >1) transcription factors (TFs) in sites of accessible chromatin at loop anchors. The top 5% differentially bound TFs are colored in red (aged) and blue (young).
To further understand age‐dependent changes within TADs, we quantified the degree to which each TAD associates with itself by calculating intra‐TAD connectivity, the mean number of reads per intra‐TAD interaction as a fraction of reads for all cis‐interactions with flanking TADs (Dixon et al., 2015; Krijger et al., 2016; Figure S2L). Intra‐TAD connectivity complements TAD separation scores by assessing TAD compartmentalization rather than inter‐TAD insulation. Intra‐TAD connectivity was enriched in open “A” compartments (Figure S2M), was positively correlated with activating chromatin features in both young and aged MuSCs (Figure S2N), and increased with age (Figure S2O). Increased intra‐TAD connectivity was also strongly associated with increased gene expression in young MuSCs, but showed decreased association in old age (Figure 2e). For example, genes related to cell cycle regulation (Anapc1/4, Cdc25a, Mcm6, Prim2) and lipid metabolism (Acly, Ipmk, Sgms2, Aasdh, Pik3c3, Pi4k2b) were encompassed by TADs with strong intra‐TAD connectivity (top 20%) in both young and aged MuSCs, but showed significantly upregulated gene expression in young MuSCs compared to aged. In further support of this observation, we observed that the average distance between significant contacts within each TAD (degree of disorder (Lin et al., 2022)) increased with age, indicating decreased cooperation between transcription regulatory complexes (Figure S2P). Summing these results indicate that in aged MuSCs, TADs self‐interact in a stronger manner and alter TAD boundaries (Barrington et al., 2019), but chromatin folding within aged TADs is less organized and unable to drive coherent changes in gene expression.
2.3. Chromatin loops form differential gene regulatory units with age
The changes within TADs during aging suggest that interactions between gene promoters and their cognate enhancers may be altered in aged MuSCs. To further explore contact domains between genes and their regulatory elements, we called chromatin loops using Hi‐C computational unbiased peak search (HiCCUPS; Rao et al., 2014). We identified 3,478 and 2,877 chromatin loops in young and aged MuSCs (Figure S3A), respectively. High‐scoring aggregate peak analysis (APA) plots confirmed the accuracy of the loop calls and revealed similar aggregate contact strengths across age (Figure 2f). Approximately 90% of chromatin looping was constrained within individual TADs across aging (Figure 2g,h) and annotation of the loop anchors revealed enrichments for promoters, distal regulatory elements, and CTCF motifs (Figure S3B‐D), with distal regulatory elements frequently connected at one anchor with a promoter or another regulatory element at the other (Figure S3E). Additionally, H3K4me3 levels and chromatin accessibility at promoters were enhanced when connected to distal regulatory elements enriched with the same chromatin features (Figure S3F). Gene expression within loop domains was also upregulated in young and aged MuSCs (Figure S3G), in line with previous observations that chromatin looping within TADs protects gene expression patterns from aberrant promoter‐enhancer contacts that may form due to heightened intra‐TAD connectivity (Dowen et al., 2014). Stable loops in static A compartments contained genes related to Wnt signaling (Brack et al., 2008; von Maltzahn et al., 2012) and MuSC self‐renewal through fibroblast growth factor receptor (FGFR) signaling (Pawlikowski et al., 2017; ERK MAPK, PI3K/Akt, and Raf/Ras/MAPK signaling) and epidermal growth factor receptor (EGFR) signaling (Feige et al., 2021; Wang et al., 2019; Figure S3H). Loops that were lost with age in static A compartments were enriched for genes related to lamellipodium organization and vascular endothelial growth factor (VEGF) signaling, which has been shown to promote MuSC quiescence through interactions with niche vascular cells (Verma et al., 2018; Figure S3H). Vegfa was significantly upregulated in young MuSCs (Figure S3I), demonstrating a link between chromatin looping and quiescence maintenance.
To characterize the factors underlying expression changes associated with looping, we inferred differential binding of expressed transcription factors (TFs) at accessible sites within loop anchors using TF footprinting (Bentsen et al., 2020; Figure 2i, Figure S3J). We observed that young loop anchors were enriched for Notch‐related TFs (Fukada et al., 2011; Fukuda et al., 2019; Noguchi et al., 2019; Hes1) and multiple members of the Krüppel‐like factors (Klf) and Specificity Factors (Sp) families, which have been shown to contribute to quiescence, chromatin organization, and regulation of proliferation (Black et al., 2001; Hayashi et al., 2016; Jones et al., 2020; Wang et al., 2016). In contrast, aged loop anchors showed enrichments for CTCF, Zic4, and TFs that regulate the circadian clock (Nr1d1/2, Rora/c, Dbp), which synchronizes pathways that are critical for MuSC homeostasis such as autophagy and responses to cell stress (Benitah & Welz, 2020), and have been recently shown to modulate chromatin topology (Kim et al., 2018; Mermet et al., 2018). Summing these results suggest aged MuSCs lose chromatin loops and TF binding that are associated with pathways that participate in maintenance of quiescence and cellular stress.
2.4. Single muscle stem cell Multi‐Omic sequencing shows variation in chromatin hubs during aging
Recent evidence suggests that cohesin‐based extrusion of chromatin loops is short‐lived (Gabriele et al., 2022) and significant cellular heterogeneity exists at the level of promoter‐enhancer contacts (Arrastia et al., 2022). To increase the resolution of our analysis and further understand patterns of co‐accessible sites within TADs, we performed single‐cell ATAC‐seq (Buenrostro et al., 2015) on mononucleated cells isolated from young (3 months) and aged (28–29 months) hind limb muscles (Figure 3a). We collected 24,930 cells (10,603 in young and 14,327 in aged) that passed quality control and filtering thresholds (Table S1, Figure S4A‐C). Using a matrix of contiguous genomic tiles, we projected the datasets into low‐dimensional space by iterative latent semantic indexing (Becht et al., 2019), integrated them using Harmony (Korsunsky et al., 2019), and clustered the cells using the ArchR package (Granja et al., 2021). We annotated each cluster by calculating gene‐activity scores (Pliner et al., 2018), a metric derived from chromatin accessibility proximal to genes, and comparing scores at cell type marker genes with integrated single‐cell transcriptomes from young and aged MuSCs that we previously generated (Larouche et al., 2021) and age‐matched Tabula Muris Senis (Almanzar et al., 2020) datasets (Figure S4D‐G). This joint marker‐based identification approach revealed similar cell types observed in other single‐cell skeletal muscle atlases (Dell'Orso et al., 2019; Larouche et al., 2022) with differentially accessible sites in each cell type (Figure 3b, Figure S4H). For example, cells in the MuSC cluster were strongly enriched for gene activity scores and integrated gene expression for Pax7 and Sdc4 compared to other cell types (Figures 3c, Figure S4D,G).
FIGURE 3.
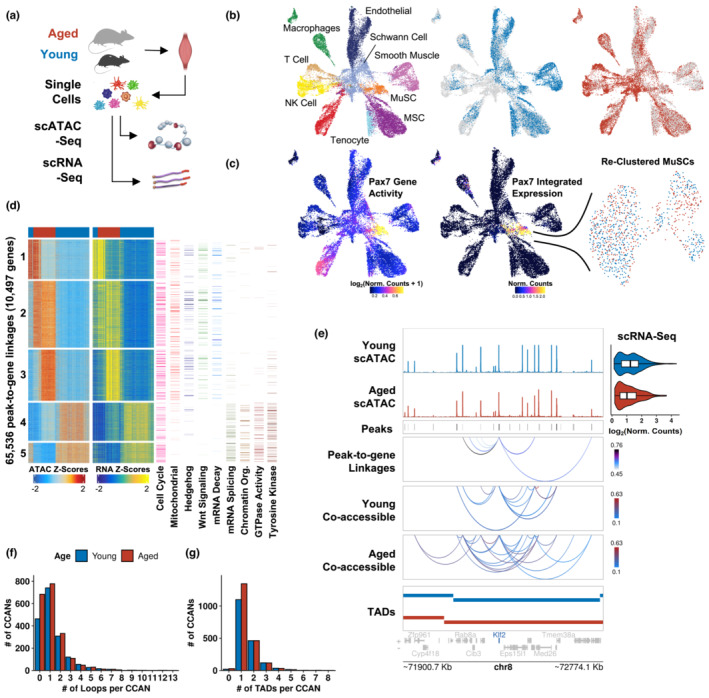
Single‐cell multi‐omic analysis of gene regulatory dynamics during muscle stem cell aging. (a) Schematic of single‐cell ATAC‐seq (scATAC‐seq) dataset generation and integration with single‐cell RNA‐seq (scRNA‐seq) datasets. (b) scATAC UMAP embeddings colored by cell type and age. (c) scATAC UMAP embeddings colored by Pax7 gene activity scores and linked gene expression from integrated scRNA datasets. The identified MuSC cluster was reclustered and colored by age. (d) Row‐scaled heatmaps of statistically significant peak‐to‐gene linkages. Each row represents either chromatin accessibility at a distal site (left) or expression of the target gene (right). The columns represent cell aggregates colored by age. K‐means clustering (5 clusters) reveals distinct regulatory networks between young and aged MuSCs. Heatmaps of representative Reactome pathways enriched in each cluster are shown to the right. (e) scATAC signal tracks, peaks, and peak‐to‐gene linkages for Hes1 colored by correlation scores. Cis‐co‐accessibility of distal sites involved in these linkages are shown below colored by co‐accessibility scores. TADs encompassing the locus are shown at the bottom. Expression from integrated single cell RNA datasets are shown as violin plots. (f) and (g) Histograms of the number of CCANs constrained within individual (f) loops and (g) TADs.
To identify putative distal regulatory elements, we called peaks in each cell type cluster using MACS2 (Zhang et al., 2008) and iteratively created a union set of 54,399 peaks in ArchR. Most peaks lay within promoters (54%), with 17% and 20% laying in distal and intronic regions, respectively (Figure S5A). The MuSCs were then separately clustered (Table S1, Figure S4B,C) and MuSC‐specific gene regulatory networks were identified by linking variation in chromatin accessibility at gene‐distal sites with differences in local gene expression. This analysis revealed 65,536 significant peak‐to‐gene linkages with positive regulatory effects representing potential enhancer‐gene interactions involving 23,227 peaks and 10,497 genes across young and aged MuSCs (FDR < 1 e‐4, correlation > 0.45; Figure 3d). All linkages were predominantly confined to an individual TAD, indicating that the detected positive regulatory interactions between distal elements and their target genes are spatially restricted (Figure S5A). Clustering the linkages via k‐means clustering revealed distinct cis‐regulatory interactions between young and aged MuSCs (Figure S5B), and annotation of clustered linkages revealed molecular processes that we previously identified in the static “A” chromatin compartment. For example, clusters 1, 2, and 3, contained linkages enriched in aged MuSCs that regulated mitochondrial translation (Mitochondrial ribosomal protein families), respiratory electron transport (mitochondrial ATP synthase and NADH:ubiquinone oxidoreductase families), and DNA repair (Ogg1, Mutyh, Mbd4), while clusters 4 and 5 were dominated by young MuSCs and showed strong enrichment for genes that participate in chromatin organization (Myc (Kieffer‐Kwon et al., 2017), AT‐rich interactive domain (ARID) family, Histone lysine demethylase & methyltransferase families) and signaling by Rho GTPases (Rac2, Rock1, Wnt4; Figure 3d). All clusters contained linkages that regulate cell cycle checkpoints, and the first three clusters contained regulatory networks that both promoted (Dhh, Smo, Gas1) and inhibited (Ptch1, Prkaca, Gsk3b) Hedgehog signaling, which has been recently shown to support the regenerative capacity of MuSCs and is attenuated with aging (Palla et al., 2022). Clusters dominated by aged MuSCs were also enriched for genes related to non‐canonical Wnt signaling (Fzd7, Fzd2, Rhoa, Rac1; Lacour et al., 2017; Le Grand et al., 2009; von Maltzahn et al., 2012), and protein trafficking through anterograde and retrograde transport between the endoplasmic reticulum and the Golgi body. Clusters enriched in young MuSCs were marked by genes that participate in signaling by receptor tyrosine kinases (fibroblast growth factors, VEGFA, EGFR), which are known to regulate MuSC homeostasis (Hindi & Kumar, 2016; Pawlikowski et al., 2017; Verma et al., 2018), as well as Notch signaling (Notch1, Maml1/2/3, Dtx2) and ubiquitination‐proteosome activity (Ubc, Uba52, Rtf1), which are known to support MuSC quiescence (Fujimaki et al., 2018; Kitajima et al., 2018; Noguchi et al., 2019). Clustering also revealed differences in linkages regulating RNA homeostasis whereby mRNA splicing was uniquely enriched in young MuSCs (splicing factors, spliceosome components, ribonucleoproteins, DEAD box proteins), while mRNA decay was marked in aged MuSCs (Zfp36l1, exosome complex genes, U6 snRNA‐associated Sm‐like protein genes), which regulates myogenic gene expression (Hausburg et al., 2015; Relaix et al., 2021) and is associated with the aging phenotype (Deschênes & Chabot, 2017; Harries, 2022; Stegeman & Weake, 2017; Stoeger et al., 2022).
Next, we investigated whether transcriptional differences between young and aged MuSCs could be explained by differences in connectivity with cis‐regulatory networks. We identified patterns of cis‐co‐accessibility between distal regulatory elements and promoters using Cicero (Pliner et al., 2018), yielding 39,038 and 39,984 pairs of co‐accessible sites in young and aged MuSCs, respectively (Figure S5C). Consistent with previous reports (Pliner et al., 2018; Sinnamon et al., 2019; Trevino et al., 2021), co‐accessible sites were more likely to be linked within the same TAD across co‐accessibility thresholds compared to distance‐matched peaks in separate TADs (Figure S5D). Similarly, we observed that ~60% of sites within the average peak‐to‐gene linkage were co‐accessible with each other, providing additional evidence that the linkages are part of spatially distinct networks. For example, linkages regulating differentially expressed genes, including Klf2, a regulator of muscle fusion (Sunadome et al., 2011) that was significantly upregulated in young MuSCs and differentially marked by young chromatin loop anchors, showed differences in co‐accessibility patterns, highlighting how variability in cis‐regulatory networks can drive differential gene expression (Figure 3e).
To identify co‐regulated chromatin hubs, we clustered the co‐accessible sites into cis‐co‐accessibility networks (CCANs), yielding 1746 and 1965 CCANs in young and aged MuSCs, respectively. CCANs were primarily constrained within individual loops (42.2% in young, 38.9% in aged) and TADs (62.5% in young, 67.0% in aged, Figures 3f,g). We next applied a maximum weighted bipartite matching algorithm (Pliner et al., 2018) that identified 1,173 pairs of stable CCANs during aging. These CCANs accounted for >85% of all co‐accessible sites in each dataset (Figure S5E) and encompassed ~50% of all differentially expressed genes. Furthermore, we observed that the average matched CCAN shared only 40.3% of its constituent sites between young and aged MuSCs (Figure S5F) and that differential gene expression within these networks was significantly upregulated in young (Figure S5G). These results indicate that age‐dependent expression changes are driven by co‐accessible sites leaving or joining chromatin hubs. For example, the matched CCANs containing Chd5, a core member of the nucleosome remodeling and deacetylase (NuRD) complex (Sreenivasan et al., 2021) that was enriched in young peak‐to‐gene linkages, and Ndufc1, a member of the NADH:ubiquinone oxidoreductase family upregulated in aged MuSCs, revealed altered connectivity with each gene body (Figure S5H,I). Together, these findings reveal extensive rewiring of connections between promoters and distal regulatory elements within chromatin hubs during aging that produce changes in gene expression. Changes in these interaction networks occurred primarily within individual TADs, which concentrate chromatin features that are supportive of gene expression, and chromatin loops, which insulate interactions and are associated with distinct TF networks.
3. DISCUSSION
Aging encompasses declines in the functionality of multiple pathways (Brunet et al., 2022; Goodell & Rando, 2015), yet how these different systems converge to modify genome organization in tissue resident stem cells has not been explored. To address this knowledge gap, we used a multi‐omic approach to evaluate each level of chromatin architecture in young and aged MuSCs. We first gleaned how global genome structure is largely static in old age, and >95% of A/B compartments are invariant between young and aged MuSCs. These results are consistent with hematopoietic and neural development (Bonev et al., 2017; Chen et al., 2019), whereby stem cells display strong differences in phenotype but highly similar chromosomal compartments. In contrast to static global organization, multiple types of TAD re‐arrangements, increased inter‐TAD interactions, and altered enhancer‐promoter contacts within open TADs were observed in aged MuSCs. Shifting of TADs displayed minimal impacts on gene expression, which is in line with previous studies that showed global loss of TAD boundaries through CTCF and/or cohesin deletion produced only mild perturbations to global gene expression (Nora et al., 2017; Rao et al., 2017; Schwarzer et al., 2017; Wutz et al., 2017). Given that different systems such as in‐vitro stem cell differentiation (Dixon et al., 2015) and acquisition of fibroblast senescence (Chandra et al., 2015; Criscione et al., 2016; Zhang et al., 2021) also show similar principles of stable global chromatin architecture, our results suggest that local alterations in genome folding within TADs are the primary drivers of changes in MuSC gene expression and associated age‐related regenerative dysfunction.
Chromatin looping plays a key role in regulation of gene expression and occurs through a cohesin‐based extrusion process that is stalled by DNA‐bound CTCF proteins positioned in a “convergent” orientation (Gabriele et al., 2022; Rao et al., 2014). We detected a decrease in the number of chromatin loops in aged MuSCs and analysis of TF motifs at loop anchors revealed a loss in notch‐related and Krüppel‐like TFs (Hsieh et al., 2017) that are associated with MuSC quiescence and chromatin stability (Black et al., 2001; Hayashi et al., 2016; Jones et al., 2020; Wang et al., 2016). Notch has previously been demonstrated to regulate targets by repositioning enhancers (Petrovic et al., 2019) and promoting increased interactions in enhancer “cliques”, which is consistent with our results in which cell cycle inhibitors and quiescence‐related genes display enhanced contacts. The loss of these TF interactions in old age and enriched CTCF binding suggest that aging may promote alterations in the formation and stability of extruded loops. This result is consistent with increased average distance between significant contacts or increased degree of disorder (Lin et al., 2022) in old age. As a consequence of loop dysregulation in old age, interactions between promoters and their concomitant regulatory elements are modified, driving alterations in TF binding and the affinity of distal regulatory elements for their target genes and associated expression levels. While a greater understanding of loop stability is needed, our results suggest MuSCs in old age may prematurely activate to generate ATP (Vian et al., 2018) to maintain or recreate lost loop domains.
Developmental maturation of stem cells has been shown to be driven by the combinatorial action of multiple enhancers, resulting in increased enhancer–promoter interactions of specific genes (Cai et al., 2020; Chen et al., 2019). Our results are consistent with this stem cell continuum, showing that old age results in alterations of promoter‐enhancer elements with accessible sites frequently leaving or joining chromatin hubs that maintain chromatin organization and quiescence. Recent studies have shown roles for MyoD (Wang et al., 2022), Fos (Barutcu et al., 2022), and Myc (He et al., 2021) as chromatin‐remodeling TFs that regulate pro‐myogenic programs in culture through altered chromatin looping near myogenic genes. Consistent with these studies, we found unique peak‐to‐gene linkages regulating Myc expression in young MuSCs and significant enrichments of TFs with similar binding sites to the Fos family in aged loop anchors, including JunD and cyclic AMP responsive element binding protein 1 (Creb1), a family member of activating transcription factor 1 (Atf1; Mayr & Montminy, 2001) known to regulate MuSC activation (Li & Fan, 2017; Stewart et al., 2011). Our findings show that rewiring of chromatin within TADs and loops underpins aberrant gene expression programs associated with pathological aging. Future work in this area will resolve additional structural features such as TAD stripes (Vian et al., 2018) and interactions with other nuclear structures such as speckles and nucleoli.
4. MATERIALS & METHODS
| Reagent or resource | Source | Identifier |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Dispase II (activity ≥0.5 units/mg solid) | Sigma | D4693‐1G |
| Collagenase Type II | Life Technologies | 17101015 |
| DMEM, high glucose, pyruvate | Life Technologies | 11995065 |
| Ham's F‐10 Nutrient Mix | Life Technologies | 11550043 |
| HBSS, no calcium, no magnesium, no phenol red | Life Technologies | 14175145 |
| Saline Phosphate Buffered Gibco PBS (Phosphate Buffered Saline) Solution pH 7.4 No phenol red no sodium pyruvate | Thermo Fisher |
10010023 |
| Fetal bovine serum | Life Technologies | 10437028 |
| Digitonin | Fisher Scientific | BN2006 |
| Trizma hydrochloride Solution, pH 7.4 | Sigma Aldrich | T2194 |
| Sodium chloride Solution, 5 M | Sigma Aldrich | 59222C |
| Magnesium chloride solution, 1 M | Sigma Aldrich | M1028 |
| Nonidet P40 substitute | Sigma Aldrich | 492018‐50ML |
| Sigma protector RNase inhibitor | Sigma Aldrich | 3335402001 |
| DL‐DTT | Sigma Aldrich | 646563 |
| 7‐AAD | Biolegend | 420403 |
| MACS BSA stock solution | Miltenyi | 130‐091‐376 |
| New England Biolabs, Inc. supplier diversity partner BSA‐molecular biology grade ‐ 12 mg | Fisher Scientific |
NC0506695 |
| Tween‐20 | BioRad | 1662404 |
| 0.5 M EDTA | Qiagen | 79306 |
| Formaldehyde, 37% by weight (with preservative/certified ACS) | Fisher Chemical | F79‐500 |
| KAPA HiFi HotStart Library Amplification Kit with Primer Mix | Fisher Scientific |
50–196‐5224 |
| Agencourt AMPure XP | Beckman Coulter | A63881 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher | Q32851 |
| Antibodies | ||
| APC anti‐Mouse Ly‐6A/E (Sca‐1), clone: D7, isotype: Rat IgG2a, κ | BioLegend | 108112 |
| APC anti‐Mouse CD45, clone: 30‐F11, isotype: Rat IgG2b, κ | BioLegend | 103112 |
| APC anti‐Mouse/Human CD11b, clone: M1/70. Isotype: Rat IgG2b, κ | BioLegend | 101212 |
| APC anti‐Mouse TER‐119, clone: TER‐119, isotype: Rat IgG2b, κ | BioLegend | 116212 |
| PE anti‐Mouse/Rat CD29, clone: HMβ1‐1, isotype: Armenian Hamster IgG | BioLegend | 102208 |
| Biotin Rat Anti‐Mouse CD184, clone: 2B11/CXCR4 (RUO), isotype: Rat IgG2b, κ, lot # 6336587 | BD Bioscience | 551968 |
| Streptavidin PE‐Cyanine7, lot # 4290713 | eBioscience | 25–4317‐82 |
| 4′,6‐Diamidino‐2‐Phenylindole, Dihydrochloride (DAPI), FluoroPure™ grade | Invitrogen | D21490 |
|
PC anti‐mouse Ly‐6A/E (Sca‐1), clone: D7, isotype: Rat IgG2a, κ |
Biolegend | BioLegend 108112; RRID:AB_313349 |
| Critical commercial assays | ||
| Arima Hi‐C + Kit | Arima Genomics | 202012‐1415 |
| Accel‐NGS® 2 S Plus DNA library kit | Swift Biosciences | 21024 |
| 2 S Set A single indexed adapters | Swift Biosciences | 26148 |
| Chromium next GEM Single Cell ATAC Reagent Kit (v1.1) | 10x Genomics | PN‐1000175 |
| Experimental models: Organisms/Strains | ||
| Pax7CreER/+;Rosa26TdTomato/+ mice (24–29 months) | University of Michigan | Jackson stock 017763 crossed with stock 007676 |
| Pax7CreER/+;Rosa26TdTomato/+ mice (3–4 months) | University of Michigan | Jackson stock 017763 crossed with stock 007676 |
| Deposited data | ||
| Aging muscle stem Cell ATAC‐seq and RNA‐seq datasets |
Shcherbina et al. (2020), Cell Reports |
GSE121589 |
| Aging muscle stem cell single cell RNA‐seq datasets |
Larouche et al. (2021), eLife |
GSE165978 |
| Tabula Muris Senis Single cell droplet RNA‐seq datasets |
Almanzar et al. (2020), Nature |
GSE149590 |
| Aging muscle stem cell Hi‐C datasets | This Manuscript | GSE214047 |
| Software | ||
| Gene annotation | https://www.gencodegenes.org/mouse/release_M25.html | Gencode vM25 |
| Juicer | Durand, Shamim, et al. (2016) | Juicer Tools v1.22.01 |
| Juicebox | Durand, Robinson, et al. (2016) | Juicebox v1.11.08 |
| FAN‐C | Kruse et al. (2020) | FAN‐C v0.9.21 |
| HiCExplorer | Wolff et al. (2020) | HiCExplorer v3.7.2 |
| pyGenomeTracks | Ramírez et al. (2018); Lopez‐Delisle et al. (2021) | pyGenomeTracks v3.6 |
| MDkNN | Lin et al. (2022) | MDkNN v0.0.1 |
| Plotgardener | Kramer et al. (2022) | Plotgardener v1.2.10 |
| TOBIAS | Bentsen et al. (2020) | TOBIAS v0.13.3 |
| Cell Ranger ATAC | 10x Genomics | Cell Ranger ATAC v2.1.0 |
| Seurat | Stuart et al. (2019); Bioconductor | Seurat v4.1.1 |
| DoubletFinder | McGinnis et al. (2019) | DoubletFinder v2.0.3 |
| MACS2 | Zhang et al. (2008) | MACS2 v2.1.1 |
| GenomicInteractions | Harmston et al. (2015); Bioconductor | GenomicInteractions v1.28.0 |
| GenomicRanges | Lawrence et al. (2013); Bioconductor | GenomicRanges v1.46.1 |
| JASPAR TF Motifs | Castro‐Mondragon et al. (2022) | JASPAR CORE 2022 |
| ArchR | Granja et al. (2021) | ArchR v1.0.2 |
| Cicero | Pliner et al. (2018) | Cicero v1.3.6 |
| WebGestalt | Liao et al. (2019) | WebGestalt 2019 |
| ChIPseeker | Yu et al. (2015) | ChIPseeker v1.30.3 |
4.1. Hi‐C library preparation and sequencing
4.1.1. Animals
Young (4 months) and aged (24–26 months) Pax7CreER/+;Rosa26TdTomato/+ female mice were obtained from a breeding colony at the University of Michigan (UM). All mice were fed normal chow ad libitum and housed on a 12:12 h light–dark cycle under UM veterinary staff supervision. All procedures were approved by the University Committee on the Use and Care of Animals at UM and were in accordance with the U.S. National Institute of Health (NIH).
4.1.2. Isolation and crosslinking of muscle stem cells
MuSCs were isolated by FACS as previously described (Shcherbina et al., 2020). In brief, hind limb muscles were harvested from mice using sterile surgical tools. Muscle tissue was minced and digested in 20 ml of a digestion solution (2.5 U/mL Dispase II and 0.2% [~5500 U/mL] Collagenase Type II in Dulbecco's modified Eagle medium [DMEM]). Samples were incubated at 37°C for 60 min. Once the digestion was complete, 20 ml of F10 media containing 20% heat inactivated FBS was added into each sample to inactivate enzyme activity. The solution was then filtered through a 70 μm cell strainer into a new 50 ml conical tube and centrifuged at 350× g for 5 min. Subsequently, the protocol for crosslinking low input mammalian cells was followed, as written in the Arima‐HiC kit workflow provided by Arima Genomics Inc. Briefly, the pellets were re‐suspended in 1× PBS and 37% formaldehyde was added to obtain a final concentration of 2% formaldehyde. The samples were inverted and incubated at RT for 10 min. Arima‐Hi‐C Stop Solution 1 was added to samples, and they incubated for an additional 5 min. The samples were then placed on ice to incubate for 15 min. Cells were pelleted by centrifugation for 5 min at 500× g, the supernatant was discarded, and cells were re‐suspended in staining media (2% heat‐inactivated FBS, 2 mM EDTA in Hank's buffered salt solution) and antibody cocktail containing Sca‐1:APC (1:400), CD45:APC (1:400), CD11b:APC (1:400), Ter119:APC (1:400), CD29/β1‐integrin:PE (1:200), and CD184/CXCR‐4: BIOTIN (1:100) and incubated for 30 min on ice in the dark. Cells and antibodies were diluted in 3 mL of staining solution, centrifuged at 350× g for 5 min, and supernatants discarded. Pellets were re‐suspended in 200 μL staining solution containing PECy7:STREPTAVIDIN (1:100) and incubated on ice for 20 min in the dark. Again, samples were diluted in 3 mL staining solution, centrifuged, supernatants discarded, and pellets re‐suspended in 200 μL staining buffer. Samples were filtered through 70 μm cell strainers before the FACS.
4.1.3. Preparation of Hi‐C libraries & sequencing
In situ Hi‐C was performed using the Arima‐HiC Protocol. Approximately 75,000–100,000 sorted muscle cells were crosslinked, permeabilized and chromatin digested as specified by the manufacturer in biological replicates. Restriction fragment ends were then labeled with biotinylated nucleotides and proximal DNA ligated, followed by reversal of cross‐links. The DNA was then sheared using Diagenode's Bioruptor sonicator for 4 cycles of 15 s on, 90 s off, then briefly centrifuged. An additional 4 cycles of sonication were performed after centrifugation. The resultant DNA was size‐selected using AMPure XP beads followed by verification with the Arima Quality Control 2 assay. Biotinylated junctions were then magnetically isolated with streptavidin coated beads, followed by end‐repair, A‐tailing, sequencing adaptor ligation, and PCR amplification. The resultant libraries were then size‐selected using AMPure XP beads and sequenced with 150 bp paired‐end reads at a depth of 750 million reads per library on an Illumina NovaSeq flow cell.
4.2. Hi‐C data preprocessing
Sequence quality metrics were checked in all samples using FastQC (v0.11.9, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) before downstream processing. Read adapters were trimmed using Cutadapt (Martin, 2011) (v2.6). Only trimmed reads >20 bp were retained and 25 bases were trimmed from the 3′ ends. Paired‐end reads for each replicate were then split and each mate was aligned as single‐end reads to the mouse reference genome (mm10) using BWA aln and samse (v0.7.17‐r1188). Uniquely mapped reads classified as “optimal” or “suboptimal” by BWA with a maximum edit distance of three and MAPQ ≥ 30 were retained. Paired mates were then re‐joined and duplicate reads removed. Each chromosome was split into 5 kb bins and the number of interactions in each bin was counted. Interactions were eliminated if they occurred on the same fragment (i.e., overlapping reads on the same chromosome) or if the distance between the start coordinates of each read pair was <1 kb. All remaining reads were kept if they participated in at least one interaction. The resulting paired contact maps were converted to .hic files using the pre command in Juicer Tools (Durand, Shamim, et al., 2016; Durand, Robinson, et al., 2016; v1.22.01) with a file of Arima restriction sites created with the generate_site_positions.py script. The .hic file was created with the following 9 base‐pair‐delimited resolutions: 2.5 mb, 1 mb, 500 kb, 250 kb, 100 kb, 50 kb, 40 kb, 25 kb, 10 kb, and 5 kb.
4.3. Hi‐C quality control
Pairwise Pearson correlations were computed between Knight‐Ruiz‐normalized replicates at 250 kb‐resolution with HiCExplorer (Wolff et al., 2020; v3.7.2) after converting .hic files to .cool files using the hicConvertFormat command. All replicates showed Pearson correlations <0.98 and replicates were pooled for downstream processing. The fraction of inter‐chromosomal interactions in pooled replicates was ~23% while ~63% of interactions occurred more than 20 kb apart and ~14% of interactions occurred less than 20 kb apart, indicating good quality libraries. Replicates also showed the same distributions of count enrichment at different genomic ranges as shown by the hicPlotDistVsCounts command with a maximum distance from the diagonal of 30 Mb.
4.4. A/B compartment analysis
A/B compartments were identified from the first eigenvectors of the Pearson correlation matrices of 100 kb‐resolution contact matrices using the “compartments” command in the FAN‐C100 (v0.9.21) package. The sign of the first eigenvector was oriented by the average GC content of each domain such that domains with higher GC content were assigned positive signs (A compartment) while domains with lower GC content were assigned negative signs (B compartment). The compartment eigenvector BED files were then exported from FAN‐C and analyzed in R to identify compartments that were static or shifted between young and aged MuSCs. Saddle plots were generated using as an additional output of the “compartments” command.
4.5. TAD calling and characterization
4.5.1. Identification of TADs
TADs were called from Knight‐Ruiz‐normalized contact matrices at several resolutions (10, 40, 100, 250, and 500 kb) and FDR thresholds (0.1, 0.05, 0.01, 0.005, and 0.001) using the “hicFindTADs” command in the HiCExplorer package with default parameters. The final set of TADs and associated boundaries (40 kb resolution, FDR < 0.01) was selected by visual inspection with pyGenomeTracks(Lopez‐Delisle et al., 2021; Ramírez et al., 2018; v3.6) and comparisons of the size and number of TADs at each pair of parameters. Aggregate contact matrices around TAD domains were plotted using FAN‐C aggregate.
4.5.2. Calculation of Intra‐TAD connectivity
Intra‐TAD connectivity was calculated as the mean number of reads per intra‐TAD interaction as a fraction of reads for all cis‐interactions with flanking TADs. The number of contacts within TADs and between TADs was collected using the “hicInterIntraTAD” command in HiCExplorer.
4.5.3. Degree of disorder calculations
The degree of disorder (DoD) was calculated using the MDkNN (Lin et al., 2022) package (v0.0.1). Calculations were performed on Knight‐Ruiz‐normalized contact matrices at 10 kb resolution with the recommended parameters (k = 3, ww = 5, pw = 2, top = 0.7, ratio = 0.05, gap = 0.2).
4.6. Chromatin loop analysis
4.6.1. Loop calling
Chromatin loops were identified using the HiCCUPS algorithm (Rao et al., 2014) with default parameters for medium resolution maps (hiccups ‐m 512 ‐c (all chromosomes) ‐r 5000,10,000,25,000 ‐k KR ‐f 0.1,0.1,0.1 ‐p 4,2,1 ‐i 7,5,3 ‐t 0.02,1.5,1.75,2 ‐d 20,000,20,000,50,000 /path/to/hic/file path/to/loops/files). The algorithm was run in a Google Colaboratory notebook with a GPU hardware accelerator. The merged loop list across all resolutions (i.e., 5 kb, 10 kb, 25 kb) were used for downstream analyses.
4.6.2. Aggregate peak analysis (APA)
Aggregate peak analysis (Rao et al., 2014) was used with default settings to evaluate focal contact enrichment at merged loops. We summarized the genome‐wide APA analysis using the ratio of the central pixel to the mean of the mean of the pixels in the lower left corner. For visualization, we used the genome‐wide normalized APA matrices at 10 kb resolution. In these matrices, each submatrix was normalized by its mean such that the mean of the submatrix was 1.
4.7. Feature annotation and gene set enrichment analysis
TAD boundaries and loop anchors were annotated for genomic features using ChIPseeker (v1.30.3; Yu et al., 2015) with a promoter region of ±1 kb. Overlapping genomic annotations were resolved in order of decreasing priority as follows: Promoter, 5UTR, 3UTR, Exon, Intron, Downstream, Intergenic. Gene ontology (GO) and Reactome term enrichments were performed using over‐representation (ORA) and gene set enrichment (GSEA) analysis (Subramanian et al., 2005) using WebGestalt 2019 (Liao et al., 2019). Tested gene sets were limited to those containing between 5 and 2000 genes. Significant term enrichment thresholds were set at FDR < 0.05. For GSEA, genes were ranked according to the signal to noise metric (Subramanian et al., 2005).
4.8. CTCF motif finding
CTCF motifs were identified across the mm10 genome using scanMotifGenomeWide.pl from the HOMER suite (Heinz et al., 2010) with the following HOMER position weight matrix (PWM) for CTCF:
| >ANAGTGCCACCTGGTGGCCA | CTCF(Zf)/CD4 + ‐CTCF‐ChIP‐Seq (Barski_et_al. 2007)/Homer,BestGuess:CTCF(Zf)/CD4 + ‐CTCF‐ChIP‐Seq (Barski_et_al. 2007)/Homer(1.000) | 8.704837 | ‐6.281855 e+03 | 0 | 15000.0,4645.0,2877.22765.0,0.00 e+00 |
| 0.447 | 0.221 | 0.181 | 0.151 | ||
| 0.037037037037037 | 0.34034034034034 | 0.236236236236236 | 0.386386386386386 | ||
| 0.499 | 0.061 | 0.33 | 0.11 | ||
| 0.033033033033033 | 0.377377377377377 | 0.528528528528528 | 0.0610610610610611 | ||
| 0.023023023023023 | 0.378378378378378 | 0.005005005005005 | 0.593593593593594 | ||
| 0.061 | 0.005 | 0.887 | 0.047 | ||
| 0.0790790790790791 | 0.905905905905906 | 0.005005005005005 | 0.01001001001001 | ||
| 0.002 | 0.994 | 0.001 | 0.003 | ||
| 0.501501501501502 | 0.475475475475475 | 0.00700700700700701 | 0.016016016016016 | ||
| 0.002 | 0.527 | 0.004 | 0.467 | ||
| 0.003 | 0.995 | 0.001 | 0.001 | ||
| 0.03 | 0.036 | 0.004 | 0.93 | ||
| 0.382 | 0.042 | 0.446 | 0.13 | ||
| 0.02 | 0.273 | 0.686 | 0.021 | ||
| 0.047 | 0.039 | 0.014 | 0.9 | ||
| 0.002 | 0.001 | 0.995 | 0.002 | ||
| 0.04 | 0.034 | 0.873 | 0.053 | ||
| 0.161161161161161 | 0.527527527527528 | 0.0610610610610611 | 0.25025025025025 | ||
| 0.277 | 0.428 | 0.117 | 0.178 | ||
| 0.541 | 0.092 | 0.267 | 0.1 | ||
CTCF motifs were enumerated in 5 kb windows across loop domains and in 20 kb windows across TAD domains. Loop and TAD domains were divided into 40 bins and the average number of motifs was plotted within each bin.
4.9. Integration with RNA‐Sequencing
4.9.1. Gene expression quantification
Paired‐end RNA‐seq data from MuSCs (Shcherbina et al., 2020) was aligned to the mm10 reference genome with the STAR algorithm (STAR_2.5.0a; Dobin et al., 2013) using default parameters. RSEM (Li & Dewey, 2011) quantification was applied to the aligned reads. The full set of flags used in the STAR command is as follows:
STAR ‐‐genomeLoad NoSharedMemory ‐‐outFilterMultimapNmax 20 ‐‐alignSJoverhangMin 8 ‐‐alignSJDBoverhangMin 1 ‐‐outFilterMismatchNmax 999 ‐‐outFilterMismatchNoverReadLmax 0.04 ‐‐alignIntronMin 20 ‐‐alignIntronMax 1000000 ‐‐alignMatesGapMax 1000000 ‐‐outSAMunmapped Within ‐‐outFilterType BySJout ‐‐outSAMattributes NH HI AS NM MD ‐‐outSAMtype BAM SortedByCoordinate ‐‐quantMode TranscriptomeSAM ‐‐sjdbScore 1 ‐‐limitBAMsortRAM 60000000000 ‐‐twopassMode Basic ‐‐twopass1readsN ‐1.
4.9.2. Gene expression analysis
Gene expression analysis between young and aged samples was performed by limma (Ritchie et al., 2015) analysis in R. The Expected Counts from RSEM were transformed to counts per million using the voom (Law et al., 2014) R package with a design formula: Count~Age, with Age = {Young, Aged}. Surrogate variable analysis was performed with the SVA package (Leek et al., 2012) using a null model of voom$E ~ 1, and a design matrix of voom$E ~ Day+Age. Contributions from the surrogate variables were quantified and removed from the voom$E data matrix. Pairwise Pearson and Spearman correlation values were computed between all sva‐corrected replicates. Any replicate that had r < 0.9 with other replicates for a given sample was excluded from further analysis. Expression analyses were performed on known genes filtered to those with TPM (transcript per million) values greater than 0 in at least one sample. Differentially expressed genes were identified as those with log2(fold‐change) > 1.5 and adjusted p‐values < 0.05. Compartments, loop anchors, and TADs were annotated with mm10 genes (GRCm38.p6 GENCODE M25) by identifying genes whose promoter regions (TSS ± 1 kb) intersected each set of regions.
4.10. Integration with ATAC‐seq and H3K4me3 ChIP‐seq datasets
4.10.1. ATAC‐seq data processing and analysis
ATAC‐seq datasets from young and aged MuSCs were collected previously (Shcherbina et al., 2020). The ATAC‐seq samples were analyzed with the ENCODE ATAC‐seq processing pipeline (Maher, 2012; https://github.com/ENCODE‐DCC/atac‐seq‐pipeline, version 1.1.7). Read adapters were trimmed with the cutadapt algorithm (Martin, 2011) and aligned to the mm10 reference genome using Bowtie2 (Langmead & Salzberg, 2012). Duplicates were then marked using Picard (https://broadinstitute.github.io/picard/) and removed from the aligned reads using SAMtools (Li et al., 2009). The resulting BAM files were filtered to remove unmapped or unpaired reads and reads with MAPQ scores below 30. The MACS2 (Zhang et al., 2008) peak caller was used to call peaks from the aligned ATAC‐seq samples and the naive overlap peak set from all replicates for a given sample was used for downstream processing.
4.10.2. H3K4me3 ChIP‐seq data processing and analysis
FASTQ files for H3K4me3 ChIP‐seq datasets from young and aged MuSCs were obtained from Liu et al., 2013 and processed with the ENCODE ChIP‐seq processing pipeline (Maher, 2012; https://github.com/ENCODE‐DCC/chip‐seq‐pipeline2, version 2.1.1) using the “histone” option. Paired‐end read adapters were trimmed using Trimmomatic (Bolger et al., 2014) and aligned to the mm10 reference genome using Bowtie2 (Langmead & Salzberg, 2012). Duplicates were then marked with Picard (https://broadinstitute.github.io/picard/) and removed with SAMtools (Li et al., 2009). The resulting BAM files were filtered to remove unmapped or unpaired reads and reads with MAPQ scores below 30.
4.10.3. Signal track coverage
RPKM‐normalized signal tracks were generated from merged ATAC‐seq and H3K4me3 bam alignments using the “bamCoverage" command in the deepTools suite (v.3.3.0; Ramírez et al., 2016) with a bin size of 1. Reads with the 780 SAM flag and mapping qualities <30 were excluded. The mean signals in chromatin compartment groups, TADs, and chromatin loop anchor were calculated using the “multiBigWigSummary” command in deepTools. Blacklisted regions (Amemiya et al., 2019) were excluded from these calculations.
4.10.4. Clustering of signals across TAD boundaries by compartment
The mean signal of the first eigenvector of the Hi‐C Pearson correlation matrix was calculated in 10 kb‐bins across 1 Mb regions flanking TAD boundaries using the “computeMatrix” command in the deepTools suite. K‐means clustering of the resulting matrices into four clusters using the “plotHeatmap” command partitioned the TAD boundaries into those that lay within A/B compartments or spanned A/B compartment switches. Matrices generated from ATAC‐seq and H3K4me3 fold‐change signals in the same regions per cluster were plotted as heatmaps.
4.10.5. Pearson correlation of ATAC‐seq and H3K4me3 signals with TADs
Mean fold‐change signals from ATAC‐seq and H3K4me3 data were calculated in 1 kb bins within TAD domains scaled to 15 kb regions and in flanking 15 kb regions using the computeMatrix command. Pearson correlation between the matrix columns for each signal was calculated in R, producing 45x45 matrices. Heatmaps were then generated for each correlation matrix.
4.10.6. Transcription factor footprint analysis
TF motifs were downloaded from the JASPAR CORE 2022 database (Castro‐Mondragon et al., 2022) and filtered to those expressed (TPM ≥ 1) in our bulk RNA‐seq datasets. Heterodimer motifs were considered expressed if at least one partner was expressed.
All footprint analyses within chromatin loop anchors were performed using the TOBIAS toolkit (v0.13.3; Bentsen et al., 2020). The bam mm10 alignments from the ENCODE ATAC‐seq pipeline were corrected for Tn5 enzymatic bias using the “ATACorrect” function. Blacklisted regions (Amemiya et al., 2019) were excluded from the bias estimations. To perform differential TF binding analysis, a union set of ATAC peaks was generated across young and aged MuSCs using the bedtools merge command. Continuous footprinting scores for the young and aged bias‐corrected ATAC signals were then calculated across the union peak set using the “ScoreBigwig” function. Estimates of TF binding positions were calculated using the “BINDetect” function in the subset of union ATAC peaks that lay within merged loop anchors. Significant TFs were identified as those above the 95th percentile or below the 5th percentile of differential binding scores, or those above the 95th percentile of −log10(p‐values).
4.11. Generation and processing of skeletal muscle single cell ATAC and RNA datasets
4.11.1. Single cell RNA datasets
Single cell RNA libraries (10x Genomics) from FACS‐isolated MuSCs were previously generated from uninjured young (2–3 months) and aged (22–24 months) hindlimb muscles (Larouche et al., 2021). We downloaded a pre‐processed droplet single‐cell RNA dataset from limb muscle generated by the Tabula Muris Senis (TMS) atlas (Almanzar et al., 2020) and converted it to a Seurat (v4.1.1; Hao et al., 2021) object. The 3‐month and 24‐month data from the TMS object were used for downstream analyses.
Tabula Muris Senis datasets were considered pre‐processed and exempt from further quality control filtering. Our previously generated MuSC datasets were filtered to retain high‐quality cells expressing >500 unique molecular identifiers (UMIs), between 300 and 4,000 genes, <10% mitochondrial reads, and gene complexities (log10(# genes)/log10(# UMIs)) > 0.8.
4.11.2. Integration of single cell RNA datasets
All datasets were separately log‐normalized and scaled to 10,000 using the “NormalizeData” function, and feature selection was performed using the “vst” method in the “FindVariableFeatures” function with the number of top variable features set to 2000. Contributions from the number of UMIs per cell were regressed out. We additionally regressed out contributions from the percentage of mitochondrial reads in our previously generated datasets. We performed doublet analysis using DoubletFinder (v.2.0.3; McGinnis et al., 2019) with a 4.8% expected doublet rate and found no clear evidence of doublets in any dataset. The datasets were then integrated using the canonical correlation analysis (CCA) implementation in Seurat with default parameters.
The integrated dataset was re‐scaled and principal component analysis (PCA) was performed on the 2000 most variable genes. The number of PCs used to represent the dataset was quantitatively determined by identifying the minimum between the last PC where the change in variation was more than 0.1% and the last PC associated with <5% of variation that also accounted for >90% of the cumulative variation in the dataset (33 PCs). These PCs were used to compute the nearest neighbor graph (FindNeighbors) and were visualized using the “uwot” implementation of uniform manifold approximation and projection (UMAP; Becht et al., 2019) with min.dist = 0.5, n.neighbors = 30, and the “cosine” distance metric. Cells were clustered using the Louvain clustering algorithm implemented by Seurats “FindCluster” function with a resolution of 0.1.
4.11.3. Single cell RNA cell type annotation
Differentially expressed genes between clusters in the integrated dataset were identified using the “FindAllMarkers” function in Seurat with a Wilcoxon test to compare genes with >0.25 log2(fold‐change) that were expressed in >10% of cells in each cluster. Clusters were annotated by cell type according to differentially expressed marker genes. Expression of marker genes was verified in each dataset within the integrated object using the “FindConservedMarkers” function with the same parameters.
4.11.4. Skeletal muscle nuclei extraction and preparation of single cell ATAC libraries
Young (3 months old) and aged (28–29 months old) Pax7CreER/+;Rosa26TdTomato/+ female mice were obtained from a breeding colony at UM. All mice were fed normal chow ad libitum and housed on a 12:12 h light–dark cycle under UM veterinary staff supervision. All procedures were approved by the University Committee on the Use and Care of Animals at UM and the Institutional Animal Care and Committee and were in accordance with the U.S. National Institute of Health (NIH).
Mouse hindlimb muscles were extracted and placed into separate petri dishes containing ice‐cold PBS. Using surgical scissors, muscle tissues were minced and placed into 20 mL of digestion buffer (DMEM with Collagenase type II (0.2%) and Dispase II (2.5 U/mL)) per mouse. Samples were placed on a shaker in a 37°C incubator for 1.5 h and mixed by pipette every 30 min. The enzymes were then inactivated by addition of 20 mL 20% heat‐inactivated fetal bovine serum (HI‐FBS) in Ham's F10 media. The solution was passed through a 70 μm cell strainers, centrifuged, washed in PBS containing 3% BSA, and nuclei isolation was performed according to 10× Demonstrated Protocol CG000375 Revision B starting at step 1.1c. Nuclei from two age‐matched mice were pooled prior to sorting, at step 1.1 n, and 7‐AAD+ nuclei were sorted on a Sony MA900 or MoFlo Astrios 3. Sorted nuclei were then permeabilized according to the same protocol. In total, 10,000 permeabilized nuclei were loaded onto the 10x Genomics Chromium single cell controller and single nuclei were captured into nanoliter‐scale gel bead‐in‐emulsions (GEMs). Single nuclei ATAC library constructions were performed using the ATAC‐seq NextGEM kit (10× Genomics). All libraries were submitted for 51 x 51bp paired‐end sequencing on a NovaSeq 6000 with 50,000 targeted reads per cell.
4.11.5. Single cell ATAC processing
Raw sequencing data were demultiplexed and converted to FASTQ files using the “bcl‐convert” command (Illumina, v3.9.3) and aligned to the mm10 reference genome (refdata‐cellranger‐arc‐mm10‐2020‐A‐2.0.0) using the “cellranger‐atac count” command (10× Genomics, cellranger‐atac‐2.1.0).
scATAC fragment files from all samples were processed simultaneously using the ArchR package (v1.0.2). Low‐quality cells were removed based on transcription start site (TSS) enrichment (>8), minimum number of unique fragments per cell (>1000), and the ratio of fragments in blacklisted (Amemiya et al., 2019) regions (<0.05). Doublets were filtered using the “addDoubletScores” and “filterDoublets” functions with default parameters (7.96% of total cells removed). Next, a cell‐feature matrix of 500 bp genome‐wide tiles was used to create a low‐dimensional representation of the dataset through an iterative latent semantic indexing (LSI) approach (“addIterativeLSI” function; 4 iterations; 30 LSI dimensions; 15,000 variable features; 0.05, 0.1, and 0.2 clustering resolutions). Batch effects were corrected using Harmony (“addHarmony” with default parameters; Korsunsky et al., 2019) and LSI dimensions that were highly correlated with sequencing depth were excluded from downstream analyses (Pearson correlation > 0.75). Cells were visualized using the “uwot” implementation of UMAP (Becht et al., 2019) embeddings using Harmony‐corrected dimensions with the cosine metric, nNeighbors = 30, and minDist = 0.5 (“addUMAP”), and cells were clustered using the FindClusters function in Seurat (v4.1.1; Stuart et al., 2019) with resolution 0.2 (“addClusters”).
4.11.6. Single cell ATAC cell type annotation and imputed gene expression
Prior to labeling scATAC clusters with scRNA annotations, gene activity scores were inferred from scATAC data using the “addGeneScoreMatrix” function in ArchR. These scores predict gene expression levels based on the accessibility of nearby regulatory elements. We identified marker genes for each scATAC cluster based on gene activity scores using a Wilcoxon test with the “getMarkerFeatures” function. A suitable null background for this analysis was corrected for biases from TSS enrichment and the log10(# of unique fragments) per cell. Cell types were tentatively assigned to clusters based on these marker genes (FDR ≤ 0.01, log2(fold‐change) ≥ 1.25), including MuSCs, which were marked by high Pax7 and Myod1 gene activity scores. To visualize these scores on UMAP embeddings, we smoothed gene activity scores across neighboring cells using MAGIC (Markov affinity‐based graph imputation of cells) imputation (van Dijk et al., 2018). We then used Seurat's CCA implementation to assign the most similar scRNA cell to each scATAC cell by comparing the scATAC gene activity and scRNA gene expression matrices (“addGeneIntegrationMatrix”), effectively transferring scRNA cell type labels to the scATAC cells. Gene expression profiles from scRNA cells were assigned to the corresponding scATAC cells. This integration was constrained to ensure that the putative scATAC MuSC cluster aligned with the scRNA MuSC cluster. scATAC clusters were then merged according to cell type annotation.
4.11.7. Single cell ATAC peak calling
To overcome the sparsity of scATAC data, we generated pseudo‐bulk replicates for each cell type cluster (“addGroupCoverages”) and pseudo‐bulk peak calling was performed using MACS2 (Zhang et al., 2008) (“addReproduciblePeakSet”). Peaks were merged into a union set in ArchR to create a single peak matrix (“addPeakMatrix”) for the entire scATAC dataset. Differential peak accessibility (FDR ≤ 0.1, log2(fold‐change) ≥ 1) across cell types was verified using the “getMarkerFeatures” function with the same parameters as the analysis performed for gene activity scores. A heatmap of marker peaks was plotted using the “plotMarkerHeatmap” function.
4.12. Multi‐omic profiling of muscle stem cells
4.12.1. Reclustering of MuSCs
The MuSC cluster of the scATAC dataset with linked scRNA expression was isolated and reclustered using the same iterative LSI procedure described previously (“addIterativeLSI”) with two iterations and 500 bp genome‐wide tiles, followed by Harmony correction. Pseudo‐bulk replicates were generated for young and aged MuSCs and a unified pseudo‐bulk peak set was identified using MACS2 (Zhang et al., 2008) and the ArchR merging procedure.
4.12.2. Identification and clustering of Peak‐to‐Gene linkages
Peak‐to‐gene correlation analysis was performed to identify putative gene‐enhancer regulatory linkages from scATAC and scRNA data. This was accomplished with the “addPeak2GeneLinks” function in ArchR with the first 30 LSI dimensions and a maximum distance between gene promoters and the center of accessible sites of 500 kb. This function requires the generation of low‐overlapping cell aggregates to overcome the sparsity of scATAC data. We slightly modified the cell aggregation step of ArchR's implementation such that aggregates were composed solely of young or aged MuSCs. This modification allowed us to compute peak‐to‐gene correlations across the MuSC cluster while retaining age‐specific contributions. The resulting linkages were filtered (correlation > 0.45, FDR < 1 e‐4) for downstream analyses. Linkages were clustered into 5 k‐means clusters and visualized using the “plotPeak2GeneHeatmap” function. Groups of linkages dominated by aged or young MuSCs could thus be visually identified.
4.12.3. Gene set annotation of Peak‐to‐Gene linkages
Enriched GO and Reactome terms for linked genes in each cluster were analyzed using over‐representation analysis in WebGestalt 2019 (Liao et al., 2019) with a FDR threshold of 0.1. Gene sets were limited to those containing between 5 and 2000 genes. Genes in representative enriched Reactome terms per cluster were aggregated and shown as heatmap annotations (Figure 3d). The parents of selected terms were related to cell cycle regulation (R‐MMU‐1640170), mitochondrial activity (R‐MMU‐1430728, R‐MMU‐1852241, R‐MMU‐382551), hedgehog signaling (R‐MMU‐162582), mRNA decay (R‐MMU‐8953854), mRNA splicing (R‐MMU‐72203), Wnt signaling (R‐MMU‐162582), chromatin organization (R‐MMU‐4839726), Rho GTPase activity (R‐MMU‐162582), and Receptor Tyrosine Kinase (RTK) signaling (R‐MMU‐162582).
Chromatin organization (R‐MMU‐4839726, R‐MMU‐3247509), SUMOylation of proteins (R‐MMU‐4570464, R‐MMU‐2990846), cell cycle regulation (R‐MMU‐69206, R‐MMU‐1640170, R‐MMU‐69278, R‐MMU‐69275, R‐MMU‐174184, R‐MMU‐69231, R‐MMU‐68882), Hedgehog signaling (R‐MMU‐5358351, R‐MMU‐5610787, R‐MMU‐5632684, R‐MMU‐5635838), and mitochondrial activity (R‐MMU‐5389840, R‐MMU‐611105, R‐MMU‐163210, R‐MMU‐8949215).
4.12.4. Generation of cis‐co‐accessibility networks (CCANs)
We used the Cicero (Pliner et al., 2018) package (v1.3.6) to generate cis‐co‐accessibility maps of regulatory elements and gene promoters. Young and aged MuSCs were separately reclustered using the peak matrix from the MuSC cluster and UMAP embeddings were generated for each group. The peak matrix for each group was binarized and converted to cell_data_set objects using the “make_atac_cds” function in Cicero. Peaks containing at least one Tn5 insertion were identified and size factors were estimated using the “detect_genes” and “estimate_size_factors” functions in Monocle3 (Cao et al., 2019). The resulting objects were combined with the previously determined UMAP coordinates to create CDS objects for Cicero using the “make_cicero_cds” function in Cicero. The number of cells per aggregate (“k” parameter) was selected as the maximum number of cells such that the median number of cells shared between aggregates was 0 and no more than 10% of cells were shared between paired aggregates on average (k = 40 for young and aged). This yielded 320,645 and 356,194 pairs of accessible sites with positive co‐accessibility scores in young and aged MuSCs, respectively, which we filtered using a co‐accessibility score of 0.1. Next, we generated the cis‐co‐accessibility maps using the “run_cicero” function with window = 5e5 and sample_num = 100. Linked sites were annotated for genomic features using the ChIPseeker package (v1.30.3; Yu et al., 2015). We identified cis‐co‐accessibility networks (CCANs) in young and aged maps using the “generate_ccans” function with a co‐accessibility cutoff of 0.1, which maximized the number of CCANs across both maps. We used the maxmatching package (v0.1.0) in R to identify stable CCANs such that the total number of co‐accessible elements between pairs of CCANs was maximized. To identify differences in connectivity within matched CCANs, we found CCANs in merged young and aged datasets (k = 50) with the same parameters and found CCANs that matched CCANs matched between young and aged datasets.
4.12.5. Enrichment of cis‐co‐accessible sites within TADs
Pairs of cis‐co‐accessible sites were grouped by the distance between them into bins of 25 kb. We calculated the fold enrichment of sites within TADs per distance bin as the ratio between the number of pairs within an individual TAD and the number of pairs that lay in different TADs. A null background comparison was performing this same analysis on sites that were shuffled in each distance bin and chromosome such that links were randomly assigned between sites. This procedure was repeated 500 times per distance bin.
4.12.6. Visualization of peak‐to‐gene linkages and Cicero connections
Integrated visualizations of single cell ATAC tracks, peak‐to‐gene linkages, and cis‐co‐accessible sites were made using the Plotgardener (Kramer et al., 2022) package (v1.2.0). ATAC signal tracks were normalized to the number of reads in TSS regions to account for differences in sequencing depth and sample quality. The heights of linkages and cis‐co‐accessible sites are scaled to the distance between anchors.
AUTHOR CONTRIBUTIONS
J.L., K.M.S., and P.M.F. performed experiments. B.A.Y. analyzed data. C.A.A. designed the experiments. S.C.J.P. contributed reagents / tools. B.A.Y., and C.A.A. wrote the manuscript.
CONFLICT OF INTEREST
The authors declare no competing interests.
DATA ACCESSION CODE
Supporting information
Appendix S1
ACKNOWLEDGMENTS
The authors thank Arima Genomics for assistance with Hi‐C, and the University of Michigan DNA Sequencing Core for assistance with sequencing. The authors also thank Anna Shcherbina, and Jie Liu for insights into bioinformatics analysis, and members of the Parker and Aguilar laboratories. Research reported in this publication was supported by a National Science Foundation CAREER award (2045977), partially supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number P30 AR069620 (CAA), the 3 M Foundation (CAA), American Federation for Aging Research Grant for Junior Faculty (CAA), the University of Michigan Geriatrics Center and National Institute of Aging under award number P30 AG024824 (CAA), the Department of Defense and Congressionally Directed Medical Research Program W81XWH2010336 and W81XWH2110491 (C.A.A.), Defense Advanced Research Projects Agency (DARPA) “BETR” award D20AC0002 (C.A.A.) awarded by the U.S. Department of the Interior (DOI), Interior Business Center, and the National Institute of Biomedical Imaging and Bioengineering Training Award T32 EB005582 (BAY). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Yang, B. A. , Larouche, J. A. , Sabin, K. M. , Fraczek, P. M. , Parker, S. C. J. , & Aguilar, C. A. (2023). Three‐dimensional chromatin re‐organization during muscle stem cell aging. Aging Cell, 22, e13789. 10.1111/acel.13789
DATA AVAILABILITY STATEMENT
Our data has been uploaded onto the Gene Expression Omnibus (GEO) and the number is listed in the manuscript.
REFERENCES
- Abou‐Khalil, R. , Yang, F. , Lieu, S. , Julien, A. , Perry, J. , Pereira, C. , Relaix, F. , Miclau, T. , Marcucio, R. , & Colnot, C. (2015). Role of muscle stem cells during skeletal regeneration. Stem Cells, 33, 1501–1511. [DOI] [PubMed] [Google Scholar]
- Aguilar, C. A. , & Craighead, H. G. (2013). Micro‐ and nanoscale devices for the investigation of epigenetics and chromatin dynamics. Nature Nanotechnology, 8, 709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar, C. A. , Pop, R. , Shcherbina, A. , Watts, A. , Matheny, R. W., Jr. , Cacchiarelli, D. , Han, W. M. , Shin, E. , Nakhai, S. A. , Jang, Y. C. , Carrigan, C. T. , Gifford, C. A. , Kottke, M. A. , Cesana, M. , Lee, J. , Urso, M. L. , & Meissner, A. (2016). Transcriptional and chromatin dynamics of muscle regeneration after severe trauma. Stem Cell Reports, 7, 983–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almanzar, N. , Antony, J. , Baghel, A. S. , Bakerman, I. , Bansal, I. , Barres, B. A. , Beachy, P. A. , Berdnik, D. , Bilen, B. , Brownfield, D. , Cain, C. , Chan, C. K. F. , Chen, M. B. , Clarke, M. F. , Conley, S. D. , Darmanis, S. , Demers, A. , Demir, K. , de Morree, A. , … Zou, J. (2020). A single‐cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature, 583, 590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amemiya, H. M. , Kundaje, A. , & Boyle, A. P. (2019). The ENCODE blacklist: Identification of problematic regions of the genome. Scientific Reports, 9, 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrastia, M. V. , Jachowicz, J. W. , Ollikainen, N. , Curtis, M. S. , Lai, C. , Quinodoz, S. A. , Selck, D. A. , Ismagilov, R. F. , & Guttman, M. (2022). Single‐cell measurement of higher‐order 3D genome organization with scSPRITE. Nature Biotechnology, 40, 64–73. [DOI] [PubMed] [Google Scholar]
- Barrington, C. , Georgopoulou, D. , Pezic, D. , Varsally, W. , Herrero, J. , & Hadjur, S. (2019). Enhancer accessibility and CTCF occupancy underlie asymmetric TAD architecture and cell type specific genome topology. Nature Communications, 10, 2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski, A. , Cuddapah, S. , Cui, K. , Roh, T.‐Y. , Schones, D. E. , Wang, Z. , Wei, G. , Chepelev, I. , & Zhao, K. (2007). High‐Resolution Profiling of Histone Methylations in the Human Genome. Cell, 129, 823–837. 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Barutcu, A. R. , Elizalde, G. , Gonzalez, A. E. , Soni, K. , Rinn, J. L. , Wagers, A. J. , & Almada, A. E. (2022). Prolonged FOS activity disrupts a global myogenic transcriptional program by altering 3D chromatin architecture in primary muscle progenitor cells. Skeletal Muscle, 12, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht, E. , McInnes, L. , Healy, J. , Dutertre, C. A. , Kwok, I. W. H. , Ng, L. G. , Ginhoux, F. , & Newell, E. W. (2019). Dimensionality reduction for visualizing single‐cell data using UMAP. Nature Biotechnology, 37, 38–44. [DOI] [PubMed] [Google Scholar]
- Benitah, S. A. , & Welz, P.‐S. (2020). Circadian regulation of adult stem cell homeostasis and aging. Cell Stem Cell, 26, 817–831. [DOI] [PubMed] [Google Scholar]
- Bentsen, M. , Goymann, P. , Schultheis, H. , Klee, K. , Petrova, A. , Wiegandt, R. , Fust, A. , Preussner, J. , Kuenne, C. , Braun, T. , Kim, J. , & Looso, M. (2020). ATAC‐seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nature Communications, 11, 4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianconi, V. , & Mozzetta, C. (2022). Epigenetic control of muscle stem cells: Time for a new dimension. Trends in Genetics, 38, 501–513. [DOI] [PubMed] [Google Scholar]
- Black, A. R. , Black, J. D. , & Azizkhan‐Clifford, J. (2001). Sp1 and krüppel‐like factor family of transcription factors in cell growth regulation and cancer. Journal of Cellular Physiology, 188, 143–160. [DOI] [PubMed] [Google Scholar]
- Blau, H. M. , Cosgrove, B. D. , & Ho, A. T. V. (2015). The central role of muscle stem cells in regenerative failure with aging. Nature Medicine, 21, 854–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev, B. , Mendelson Cohen, N. , Szabo, Q. , Fritsch, L. , Papadopoulos, G. L. , Lubling, Y. , Xu, X. , Lv, X. , Hugnot, J. P. , Tanay, A. , & Cavalli, G. (2017). Multiscale 3D genome rewiring during mouse neural development. Cell, 171, 557–572.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack, A. S. , Conboy, I. M. , Conboy, M. J. , Shen, J. , & Rando, T. A. (2008). A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for Normal adult myogenesis. Cell Stem Cell, 2, 50–59. [DOI] [PubMed] [Google Scholar]
- Brunet, A. , Goodell, M. A. , & Rando, T. A. (2022). Ageing and rejuvenation of tissue stem cells and their niches. Nature Reviews. Molecular Cell Biology, 1–18, 45–62. 10.1038/s41580-022-00510-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro, J. D. , Giresi, P. G. , Zaba, L. C. , Chang, H. Y. , & Greenleaf, W. J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA‐binding proteins and nucleosome position. Nature Methods, 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro, J. D. , Wu, B. , Litzenburger, U. M. , Ruff, D. , Gonzales, M. L. , Snyder, M. P. , Chang, H. Y. , & Greenleaf, W. J. (2015). Single‐cell chromatin accessibility reveals principles of regulatory variation. Nature, 523, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, W. , Huang, J. , Zhu, Q. , Li, B. E. , Seruggia, D. , Zhou, P. , Nguyen, M. , Fujiwara, Y. , Xie, H. , Yang, Z. , Hong, D. , Ren, P. , Xu, J. , Pu, W. T. , Yuan, G. C. , & Orkin, S. H. (2020). Enhancer dependence of cell‐type–specific gene expression increases with developmental age. Proceedings of the National Academy of Sciences of the United States of America, 117, 21450–21458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, J. , Spielmann, M. , Qiu, X. , Huang, X. , Ibrahim, D. M. , Hill, A. J. , Zhang, F. , Mundlos, S. , Christiansen, L. , Steemers, F. J. , Trapnell, C. , & Shendure, J. (2019). The single‐cell transcriptional landscape of mammalian organogenesis. Nature, 566, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro‐Mondragon, J. A. , Riudavets‐Puig, R. , Rauluseviciute, I. , Berhanu Lemma, R. , Turchi, L. , Blanc‐Mathieu, R. , Lucas, J. , Boddie, P. , Khan, A. , Manosalva Pérez, N. , Fornes, O. , Leung, T. Y. , Aguirre, A. , Hammal, F. , Schmelter, D. , Baranasic, D. , Ballester, B. , Sandelin, A. , Lenhard, B. , … Mathelier, A. (2022). JASPAR 2022: The 9th release of the open‐access database of transcription factor binding profiles. Nucleic Acids Research, 50, D165–D173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra, T. , Ewels, P. A. , Schoenfelder, S. , Furlan‐Magaril, M. , Wingett, S. W. , Kirschner, K. , Thuret, J. Y. , Andrews, S. , Fraser, P. , & Reik, W. (2015). Global reorganization of the nuclear landscape in senescent cells. Cell Reports, 10, 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. , Yu, W. , Tober, J. , Gao, P. , He, B. , Lee, K. , Trieu, T. , Blobel, G. A. , Speck, N. A. , & Tan, K. (2019). Spatial genome re‐organization between fetal and adult hematopoietic stem cells. Cell Reports, 29, 4200–4211.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscione, S. W. , de Cecco, M. , Siranosian, B. , Zhang, Y. , Kreiling, J. A. , Sedivy, J. M. , & Neretti, N. (2016). Reorganization of chromosome architecture in replicative cellular senescence. Science Advances, 2, e1500882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubeñas‐Potts, C. , & Matunis, M. J. (2013). SUMO: A multifaceted modifier of chromatin structure and function. Developmental Cell, 24, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Orso, S. , Juan, A. H. , Ko, K. D. , Naz, F. , Perovanovic, J. , Gutierrez‐Cruz, G. , Feng, X. , & Sartorelli, V. (2019). Single cell analysis of adult mouse skeletal muscle stem cells in homeostatic and regenerative conditions. Development, 146, dev174177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschênes, M. , & Chabot, B. (2017). The emerging role of alternative splicing in senescence and aging. Aging Cell, 16, 918–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon, J. R. , Jung, I. , Selvaraj, S. , Shen, Y. , Antosiewicz‐Bourget, J. E. , Lee, A. Y. , Ye, Z. , Kim, A. , Rajagopal, N. , Xie, W. , Diao, Y. , Liang, J. , Zhao, H. , Lobanenkov, V. V. , Ecker, J. R. , Thomson, J. A. , & Ren, B. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature, 518, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon, J. R. , Selvaraj, S. , Yue, F. , Kim, A. , Li, Y. , Shen, Y. , Hu, M. , Liu, J. S. , & Ren, B. (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature, 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin, A. , Davis, C. A. , Schlesinger, F. , Drenkow, J. , Zaleski, C. , Jha, S. , Batut, P. , Chaisson, M. , & Gingeras, T. R. (2013). STAR: Ultrafast universal RNA‐seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowen, J. M. , Fan, Z. P. , Hnisz, D. , Ren, G. , Abraham, B. J. , Zhang, L. N. , Weintraub, A. S. , Schuijers, J. , Lee, T. I. , Zhao, K. , & Young, R. A. (2014). Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell, 159, 374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand, N. C. , Robinson, J. T. , Shamim, M. S. , Machol, I. , Mesirov, J. P. , Lander, E. S. , & Aiden, E. L. (2016). Juicebox provides a visualization system for hi‐C contact maps with unlimited zoom. Cell Systems, 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand, N. C. , Shamim, M. S. , Machol, I. , Rao, S. S. , Huntley, M. H. , Lander, E. S. , & Aiden, E. L. (2016). Juicer provides a one‐click system for analyzing loop‐resolution hi‐C experiments. Cell Systems, 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliazer, S. , Muncie, J. M. , Christensen, J. , Sun, X. , D'Urso, R. S. , Weaver, V. M. , & Brack, A. S. (2019). Wnt4 from the niche controls the Mechano‐properties and quiescent state of muscle stem cells. Cell Stem Cell, 25, 654–665.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige, P. , Tsai, E. C. , & Rudnicki, M. A. (2021). Analysis of human satellite cell dynamics on cultured adult skeletal muscle myofibers. Skeletal Muscle, 11, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyamer, I. M. , Gassler, J. , Imakaev, M. , Brandão, H. B. , Ulianov, S. V. , Abdennur, N. , Razin, S. V. , Mirny, L. A. , & Tachibana‐Konwalski, K. (2017). Single‐nucleus hi‐C reveals unique chromatin reorganization at oocyte‐to‐zygote transition. Nature, 544, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimaki, S. , Seko, D. , Kitajima, Y. , Yoshioka, K. , Tsuchiya, Y. , Masuda, S. , & Ono, Y. (2018). Notch1 and Notch2 coordinately regulate stem cell function in the quiescent and activated states of muscle satellite cells. Stem Cells, 36, 278–285. [DOI] [PubMed] [Google Scholar]
- Fukada, S. , Yamaguchi, M. , Kokubo, H. , Ogawa, R. , Uezumi, A. , Yoneda, T. , Matev, M. M. , Motohashi, N. , Ito, T. , Zolkiewska, A. , Johnson, R. L. , Saga, Y. , Miyagoe‐Suzuki, Y. , Tsujikawa, K. , Takeda, S. , & Yamamoto, H. (2011). Hesr1 and Hesr3 are essential to generate undifferentiated quiescent satellite cells and to maintain satellite cell numbers. Development, 138, 4609–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda, S. , Kaneshige, A. , Kaji, T. , Noguchi, Y. T. , Takemoto, Y. , Zhang, L. , Tsujikawa, K. , Kokubo, H. , Uezumi, A. , Maehara, K. , Harada, A. , Ohkawa, Y. , & Fukada, S. I. (2019). Sustained expression of HeyL is critical for the proliferation of muscle stem cells in overloaded muscle. eLife, 8, e48284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriele, M. , Brandão, H. B. , Grosse‐Holz, S. , Jha, A. , Dailey, G. M. , Cattoglio, C. , Hsieh, T. H. S. , Mirny, L. , Zechner, C. , & Hansen, A. S. (2022). Dynamics of CTCF‐ and cohesin‐mediated chromatin looping revealed by live‐cell imaging. Science, 376, 496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodell, M. A. , & Rando, T. A. (2015). Stem cells and healthy aging. Science, 350, 1199–1204. [DOI] [PubMed] [Google Scholar]
- Granja, J. M. , Corces, M. R. , Pierce, S. E. , Bagdatli, S. T. , Choudhry, H. , Chang, H. Y. , & Greenleaf, W. J. (2021). ArchR is a scalable software package for integrative single‐cell chromatin accessibility analysis. Nature Genetics, 53, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao, Y. , Hao, S. , Andersen‐Nissen, E. , Mauck, W. M., III , Zheng, S. , Butler, A. , Lee, M. J. , Wilk, A. J. , Darby, C. , Zager, M. , Hoffman, P. , Stoeckius, M. , Papalexi, E. , Mimitou, E. P. , Jain, J. , Srivastava, A. , Stuart, T. , Fleming, L. M. , Yeung, B. , … Satija, R. (2021). Integrated analysis of multimodal single‐cell data. Cell, 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmston, N. , Ing‐Simmons, E. , Perry, M. , Barešić, A. , & Lenhard, B. (2015). GenomicInteractions: An R/Bioconductor package for manipulating and investigating chromatin interaction data. BMC Genomics, 16, 963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harries, L. W. (2022). Dysregulated RNA processing and metabolism: A new hallmark of ageing and provocation for cellular senescence. The FEBS Journal. 10.1111/febs.16462 [DOI] [PubMed] [Google Scholar]
- Hausburg, M. A. , Doles, J. D. , Clement, S. L. , Cadwallader, A. B. , Hall, M. N. , Blackshear, P. J. , Lykke‐Andersen, J. , & Olwin, B. B. (2015). Post‐transcriptional regulation of satellite cell quiescence by TTP‐mediated mRNA decay. eLife, 4, e03390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, S. , Manabe, I. , Suzuki, Y. , Relaix, F. , & Oishi, Y. (2016). Klf5 regulates muscle differentiation by directly targeting muscle‐specific genes in cooperation with MyoD in mice. eLife, 5, e17462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, L. , Ding, Y. , Zhao, Y. , So, K. K. , Peng, X. L. , Li, Y. , Yuan, J. , He, Z. , Chen, X. , Sun, H. , & Wang, H. (2021). CRISPR/Cas9/AAV9‐mediated in vivo editing identifies MYC regulation of 3D genome in skeletal muscle stem cell. Stem Cell Reports, 16, 2442–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz, S. , Benner, C. , Spann, N. , Bertolino, E. , Lin, Y. C. , Laslo, P. , Cheng, J. X. , Murre, C. , Singh, H. , & Glass, C. K. (2010). Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Molecular Cell, 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindi, S. M. , & Kumar, A. (2016). TRAF6 regulates satellite stem cell self‐renewal and function during regenerative myogenesis. The Journal of Clinical Investigation, 126, 151–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh, P. N. , Zhou, G. , Yuan, Y. , Zhang, R. , Prosdocimo, D. A. , Sangwung, P. , Borton, A. H. , Boriushkin, E. , Hamik, A. , Fujioka, H. , Fealy, C. E. , Kirwan, J. P. , Peters, M. , Lu, Y. , Liao, X. , Ramírez‐Bergeron, D. , Feng, Z. , & Jain, M. K. (2017). A conserved KLF‐autophagy pathway modulates nematode lifespan and mammalian age‐associated vascular dysfunction. Nature Communications, 8, 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung, W. W. , Ross, J. S. , Boockvar, K. S. , & Siu, A. L. (2011). Recent trends in chronic disease, impairment and disability among older adults in the United States. BMC Geriatrics, 11, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, J. , Chen, Y. , Tiwari, M. , Li, J. , Ling, J. , & Sen, G. L. (2020). KLF3 mediates epidermal differentiation through the epigenomic writer CBP. iScience, 23, 101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann, A. P. , Hung, M. , Wang, W. , Nguyen, J. , Gilbert, P. M. , Wu, Z. , & Krauss, R. S. (2022). An injury‐responsive Rac‐to‐rho GTPase switch drives activation of muscle stem cells through rapid cytoskeletal remodeling. Cell Stem Cell, 29, 933–947.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer‐Kwon, K.‐R. , Nimura, K. , Rao, S. S. P. , Xu, J. , Jung, S. , Pekowska, A. , Dose, M. , Stevens, E. , Mathe, E. , Dong, P. , Huang, S. C. , Ricci, M. A. , Baranello, L. , Zheng, Y. , Tomassoni Ardori, F. , Resch, W. , Stavreva, D. , Nelson, S. , McAndrew, M. , … Casellas, R. (2017). Myc regulates chromatin decompaction and nuclear architecture during B cell activation. Molecular Cell, 67, 566–578.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y. H. , Marhon, S. A. , Zhang, Y. , Steger, D. J. , Won, K. J. , & Lazar, M. A. (2018). Rev‐erbα dynamically modulates chromatin looping to control circadian gene transcription. Science, 359, 1274–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajima, Y. , Suzuki, N. , Nunomiya, A. , Osana, S. , Yoshioka, K. , Tashiro, Y. , Takahashi, R. , Ono, Y. , Aoki, M. , & Nagatomi, R. (2018). The ubiquitin‐proteasome system is indispensable for the maintenance of muscle stem cells. Stem Cell Reports, 11, 1523–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, N. R. , Hawes, S. M. , Crook, J. M. , & Pébay, A. (2010). G‐protein coupled receptors in stem cell self‐renewal and differentiation. Stem Cell Reviews and Reports, 6, 351–366. [DOI] [PubMed] [Google Scholar]
- Korsunsky, I. , Millard, N. , Fan, J. , Slowikowski, K. , Zhang, F. , Wei, K. , Baglaenko, Y. , Brenner, M. , Loh, P. R. , & Raychaudhuri, S. (2019). Fast, sensitive and accurate integration of single‐cell data with harmony. Nature Methods, 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer, N. E. , Davis, E. S. , Wenger, C. D. , Deoudes, E. M. , Parker, S. M. , Love, M. I. , & Phanstiel, D. H. (2022). Plotgardener: cultivating precise multi‐panel figures in R. Bioinformatics, 38, 2042–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijger, P. H. L. , di Stefano, B. , de Wit, E. , Limone, F. , van Oevelen, C. , de Laat, W. , & Graf, T. (2016). Cell‐of‐origin‐specific 3D genome structure acquired during somatic cell reprogramming. Cell Stem Cell, 18, 597–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse, K. , Hug, C. B. , & Vaquerizas, J. M. (2020). FAN‐C: A feature‐rich framework for the analysis and visualisation of chromosome conformation capture data. Genome Biology, 21, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacour, F. , Vezin, E. , Bentzinger, C. F. , Sincennes, M. C. , Giordani, L. , Ferry, A. , Mitchell, R. , Patel, K. , Rudnicki, M. A. , Chaboissier, M. C. , Chassot, A. A. , & le Grand, F. (2017). R‐spondin1 controls muscle cell fusion through dual regulation of antagonistic Wnt signaling pathways. Cell Reports, 18, 2320–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , & Salzberg, S. L. (2012). Fast gapped‐read alignment with bowtie 2. Nature Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larouche, J. , Mohiuddin, M. , Choi, J. J. , Ulintz, P. J. , Fraczek, P. , Sabin, K. , Pitchiaya, S. , Kurpiers, S. J. , Castor‐Macias, J. , Liu, W. , Hastings, R. L. , Brown, L. A. , Markworth, J. F. , De Silva, K. , Levi, B. , Merajver, S. D. , Valdez, G. , Chakkalakal, J. V. , Jang, Y. C. , … Aguilar, C. A. (2021). Murine muscle stem cell response to perturbations of the neuromuscular junction are attenuated with aging. eLife, 10, e66749. https://elifesciences.org/articles/66749, 10.7554/eLife.66749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larouche, J. A. , Fraczek, P. M. , Kurpiers, S. J. , Yang, B. A. , Davis, C. , Castor‐Macias, J. A. , Sabin, K. , Anderson, S. , Harrer, J. , Hall, M. , Brooks, S. V. , Jang, Y. C. , Willett, N. , Shea, L. D. , & Aguilar, C. A. (2022). Neutrophil and natural killer cell imbalances prevent muscle stem cell–mediated regeneration following murine volumetric muscle loss. Proceedings of the National Academy of Sciences of the United States of America, 119, e2111445119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law, C. W. , Chen, Y. , Shi, W. , & Smyth, G. K. (2014). Voom: Precision weights unlock linear model analysis tools for RNA‐seq read counts. Genome Biology, 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, M. , Huber, W. , Pagès, H. , Aboyoun, P. , Carlson, M. , Gentleman, R. , Morgan, M. T. , & Carey, V. J. (2013). Software for computing and annotating genomic ranges. PLoS Computational Biology, 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grand, F. , Jones, A. E. , Seale, V. , Scimè, A. , & Rudnicki, M. A. (2009). Wnt7a activates the planar cell polarity pathway to drive the symmetric expansion of satellite stem cells. Cell Stem Cell, 4, 535–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek, J. T. , Johnson, W. E. , Parker, H. S. , Jaffe, A. E. , & Storey, J. D. (2012). The sva package for removing batch effects and other unwanted variation in high‐throughput experiments. Bioinformatics, 28, 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, B. , & Dewey, C. N. (2011). RSEM: Accurate transcript quantification from RNA‐seq data with or without a reference genome. BMC Bioinformatics, 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , Durbin, R. , & 1000 Genome Project Data Processing Subgroup . (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , & Fan, C.‐M. (2017). A CREB‐MPP7‐AMOT regulatory Axis controls muscle stem cell expansion and self‐renewal competence. Cell Reports, 21, 1253–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, Y. , Wang, J. , Jaehnig, E. J. , Shi, Z. , & Zhang, B. (2019). WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Research, 47, W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman‐Aiden, E. , van Berkum, N. L. , Williams, L. , Imakaev, M. , Ragoczy, T. , Telling, A. , Amit, I. , Lajoie, B. R. , Sabo, P. J. , Dorschner, M. O. , Sandstrom, R. , Bernstein, B. , Bender, M. A. , Groudine, M. , Gnirke, A. , Stamatoyannopoulos, J. , Mirny, L. A. , Lander, E. S. , & Dekker, J. (2009). Comprehensive mapping of long‐range interactions reveals folding principles of the human genome. Science, 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, D. , Xu, W. , Hong, P. , Wu, C. , Zhang, Z. , Zhang, S. , Xing, L. , Yang, B. , Zhou, W. , Xiao, Q. , Wang, J. , Wang, C. , He, Y. , Chen, X. , Cao, X. , Man, J. , Reheman, A. , Wu, X. , Hao, X. , … Cao, G. (2022). Decoding the spatial chromatin organization and dynamic epigenetic landscapes of macrophage cells during differentiation and immune activation. Nature Communications, 13, 5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L. , Cheung, T. H. , Charville, G. W. , Hurgo, B. M. C. , Leavitt, T. , Shih, J. , Brunet, A. , & Rando, T. A. (2013). Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Reports, 4, 189–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Delisle, L. , Rabbani, L. , Wolff, J. , Bhardwaj, V. , Backofen, R. , Grüning, B. , Ramírez, F. , & Manke, T. (2021). pyGenomeTracks: Reproducible plots for multivariate genomic datasets. Bioinformatics, 37, 422–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López‐Otín, C. , Blasco, M. A. , Partridge, L. , Serrano, M. , & Kroemer, G. (2013). The hallmarks of aging. Cell, 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher, B. (2012). ENCODE: The human encyclopaedia. Nature, 489, 46–48. [DOI] [PubMed] [Google Scholar]
- Martin, M. (2011). Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.Journal, 17, 10–12. [Google Scholar]
- Mayr, B. , & Montminy, M. (2001). Transcriptional regulation by the phosphorylation‐dependent factor CREB. Nature Reviews. Molecular Cell Biology, 2, 599–609. [DOI] [PubMed] [Google Scholar]
- McGinnis, C. S. , Murrow, L. M. , & Gartner, Z. J. (2019). DoubletFinder: Doublet detection in single‐cell RNA sequencing data using artificial nearest neighbors. Cell Systems, 8, 329–337.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermet, J. , Yeung, J. , Hurni, C. , Mauvoisin, D. , Gustafson, K. , Jouffe, C. , Nicolas, D. , Emmenegger, Y. , Gobet, C. , Franken, P. , Gachon, F. , & Naef, F. (2018). Clock‐dependent chromatin topology modulates circadian transcription and behavior. Genes & Development, 32, 347–358. 10.1101/gad.312397.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi, Y. , Nakamura, M. , Hino, N. , Nogami, J. , Tsuji, S. , Sato, T. , Zhang, L. , Tsujikawa, K. , Tanaka, T. , Izawa, K. , Okada, Y. , Doi, T. , Kokubo, H. , Harada, A. , Uezumi, A. , Gessler, M. , Ohkawa, Y. , & Fukada, S. I. (2019). Cell‐autonomous and redundant roles of Hey1 and HeyL in muscle stem cells: HeyL requires Hes1 to bind diverse DNA sites. Development, 146, dev163618. [DOI] [PubMed] [Google Scholar]
- Nora, E. P. , Goloborodko, A. , Valton, A. L. , Gibcus, J. H. , Uebersohn, A. , Abdennur, N. , Dekker, J. , Mirny, L. A. , & Bruneau, B. G. (2017). Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell, 169, 930–944.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palla, A. R. , Hilgendorf, K. I. , Yang, A. V. , Kerr, J. P. , Hinken, A. C. , Demeter, J. , Kraft, P. , Mooney, N. A. , Yucel, N. , Burns, D. M. , Wang, Y. X. , Jackson, P. K. , & Blau, H. M. (2022). Primary cilia on muscle stem cells are critical to maintain regenerative capacity and are lost during aging. Nature Communications, 13, 1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlikowski, B. , Vogler, T. O. , Gadek, K. , & Olwin, B. B. (2017). Regulation of skeletal muscle stem cells by fibroblast growth factors. Developmental Dynamics, 246, 359–367. [DOI] [PubMed] [Google Scholar]
- Petrovic, J. , Zhou, Y. , Fasolino, M. , Goldman, N. , Schwartz, G. W. , Mumbach, M. R. , Nguyen, S. C. , Rome, K. S. , Sela, Y. , Zapataro, Z. , Blacklow, S. C. , Kruhlak, M. J. , Shi, J. , Aster, J. C. , Joyce, E. F. , Little, S. C. , Vahedi, G. , Pear, W. S. , & Faryabi, R. B. (2019). Oncogenic notch promotes long‐range regulatory interactions within hyperconnected 3D cliques. Molecular Cell, 73, 1174–1190.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliner, H. A. , Packer, J. S. , McFaline‐Figueroa, J. L. , Cusanovich, D. A. , Daza, R. M. , Aghamirzaie, D. , Srivatsan, S. , Qiu, X. , Jackson, D. , Minkina, A. , Adey, A. C. , Steemers, F. J. , Shendure, J. , & Trapnell, C. (2018). Cicero predicts cis‐regulatory DNA interactions from single‐cell chromatin accessibility data. Molecular Cell, 71, 858–871.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez, F. , Bhardwaj, V. , Arrigoni, L. , Lam, K. C. , Grüning, B. A. , Villaveces, J. , Habermann, B. , Akhtar, A. , & Manke, T. (2018). High‐resolution TADs reveal DNA sequences underlying genome organization in flies. Nature Communications, 9, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez, F. , Ryan, D. P. , Grüning, B. , Bhardwaj, V. , Kilpert, F. , Richter, A. S. , Heyne, S. , Dündar, F. , & Manke, T. (2016). deepTools2: A next generation web server for deep‐sequencing data analysis. Nucleic Acids Research, 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, S. S. P. , Huang, S. C. , Glenn St Hilaire, B. , Engreitz, J. M. , Perez, E. M. , Kieffer‐Kwon, K. R. , Sanborn, A. L. , Johnstone, S. E. , Bascom, G. D. , Bochkov, I. D. , Huang, X. , Shamim, M. S. , Shin, J. , Turner, D. , Ye, Z. , Omer, A. D. , Robinson, J. T. , Schlick, T. , Bernstein, B. E. , … Aiden, E. L. (2017). Cohesin loss eliminates all loop domains. Cell, 171, 305–320.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, S. S. P. , Huntley, M. H. , Durand, N. C. , Stamenova, E. K. , Bochkov, I. D. , Robinson, J. T. , Sanborn, A. L. , Machol, I. , Omer, A. D. , Lander, E. S. , & Aiden, E. L. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relaix, F. , Bencze, M. , Borok, M. J. , der Vartanian, A. , Gattazzo, F. , Mademtzoglou, D. , Perez‐Diaz, S. , Prola, A. , Reyes‐Fernandez, P. C. , Rotini, A. , & Taglietti, V. (2021). Perspectives on skeletal muscle stem cells. Nature Communications, 12, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, R. , Ocampo, A. , Liu, G.‐H. , & Izpisua Belmonte, J. C. (2017). Regulation of stem cell aging by metabolism and epigenetics. Cell Metabolism, 26, 460–474. [DOI] [PubMed] [Google Scholar]
- Ritchie, M. E. , Phipson, B. , Wu, D. , Hu, Y. , Law, C. W. , Shi, W. , & Smyth, G. K. (2015). Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Research, 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer, W. , Abdennur, N. , Goloborodko, A. , Pekowska, A. , Fudenberg, G. , Loe‐Mie, Y. , Fonseca, N. A. , Huber, W. , Haering, C. H. , Mirny, L. , & Spitz, F. (2017). Two independent modes of chromatin organization revealed by cohesin removal. Nature, 551, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbina, A. , Larouche, J. , Fraczek, P. , Yang, B. A. , Brown, L. A. , Markworth, J. F. , Chung, C. H. , Khaliq, M. , de Silva, K. , Choi, J. J. , Fallahi‐Sichani, M. , Chandrasekaran, S. , Jang, Y. C. , Brooks, S. V. , & Aguilar, C. A. (2020). Dissecting murine muscle stem cell aging through regeneration using integrative genomic analysis. Cell Reports, 32, 107964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, P. P. , Demmitt, B. A. , Nath, R. D. , & Brunet, A. (2019). The genetics of aging: A vertebrate perspective. Cell, 177, 200–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnamon, J. R. , Torkenczy, K. A. , Linhoff, M. W. , Vitak, S. A. , Mulqueen, R. M. , Pliner, H. A. , Trapnell, C. , Steemers, F. J. , Mandel, G. , & Adey, A. C. (2019). The accessible chromatin landscape of the murine hippocampus at single‐cell resolution. Genome Research, 29, 857–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa‐Victor, P. , García‐Prat, L. , & Muñoz‐Cánoves, P. (2022). Control of satellite cell function in muscle regeneration and its disruption in ageing. Nature Reviews. Molecular Cell Biology, 23, 204–226. [DOI] [PubMed] [Google Scholar]
- Sreenivasan, K. , Rodríguez‐delaRosa, A. , Kim, J. , Mesquita, D. , Segalés, J. , Arco, P. G. D. , Espejo, I. , Ianni, A. , di Croce, L. , Relaix, F. , Redondo, J. M. , Braun, T. , Serrano, A. L. , Perdiguero, E. , & Muñoz‐Cánoves, P. (2021). CHD4 ensures stem cell lineage fidelity during skeletal muscle regeneration. Stem Cell Reports, 16, 2089–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegeman, R. , & Weake, V. M. (2017). Transcriptional signatures of aging. Journal of Molecular Biology, 429, 2427–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, R. , Flechner, L. , Montminy, M. , & Berdeaux, R. (2011). CREB is activated by muscle injury and promotes muscle regeneration. PLoS One, 6, e24714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeger, T. , Grant, R. A. , McQuattie‐Pimentel, A. C. , Anekalla, K. R. , Liu, S. S. , Tejedor‐Navarro, H. , Singer, B. D. , Abdala‐Valencia, H. , Schwake, M. , Tetreault, M. P. , Perlman, H. , Balch, W. E. , Chandel, N. S. , Ridge, K. M. , Sznajder, J. I. , Morimoto, R. I. , Misharin, A. V. , Budinger, G. R. S. , & Nunes Amaral, L. A. (2022). Aging is associated with a systemic length‐associated transcriptome imbalance. Nature Aging, 2, 1191–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart, T. , Butler, A. , Hoffman, P. , Hafemeister, C. , Papalexi, E. , Mauck, W. M., III , Hao, Y. , Stoeckius, M. , Smibert, P. , & Satija, R. (2019). Comprehensive integration of single‐cell data. Cell, 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian, A. , Tamayo, P. , Mootha, V. K. , Mukherjee, S. , Ebert, B. L. , Gillette, M. A. , Paulovich, A. , Pomeroy, S. L. , Golub, T. R. , Lander, E. S. , & Mesirov, J. P. (2005). Gene set enrichment analysis: A knowledge‐based approach for interpreting genome‐wide expression profiles. Proceedings of the National Academy of Sciences, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunadome, K. , Yamamoto, T. , Ebisuya, M. , Kondoh, K. , Sehara‐Fujisawa, A. , & Nishida, E. (2011). ERK5 regulates muscle cell fusion through Klf transcription factors. Developmental Cell, 20, 192–205. [DOI] [PubMed] [Google Scholar]
- Trevino, A. E. , Müller, F. , Andersen, J. , Sundaram, L. , Kathiria, A. , Shcherbina, A. , Farh, K. , Chang, H. Y. , Pașca, A. M. , Kundaje, A. , Pașca, S. P. , & Greenleaf, W. J. (2021). Chromatin and gene‐regulatory dynamics of the developing human cerebral cortex at single‐cell resolution. Cell, 184, 5053–5069.e23. [DOI] [PubMed] [Google Scholar]
- van Dijk, D. , Sharma, R. , Nainys, J. , Yim, K. , Kathail, P. , Carr, A. J. , Burdziak, C. , Moon, K. R. , Chaffer, C. L. , Pattabiraman, D. , Bierie, B. , Mazutis, L. , Wolf, G. , Krishnaswamy, S. , & Pe'er, D. (2018). Recovering gene interactions from single‐cell data using data diffusion. Cell, 174, 716–729.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma, M. , Asakura, Y. , Murakonda, B. S. R. , Pengo, T. , Latroche, C. , Chazaud, B. , McLoon, L. K. , & Asakura, A. (2018). Muscle satellite cell cross‐talk with a vascular niche maintains quiescence via VEGF and notch signaling. Cell Stem Cell, 23, 530–543.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vian, L. , Pękowska, A. , Rao, S. S. P. , Kieffer‐Kwon, K. R. , Jung, S. , Baranello, L. , Huang, S. C. , el Khattabi, L. , Dose, M. , Pruett, N. , Sanborn, A. L. , Canela, A. , Maman, Y. , Oksanen, A. , Resch, W. , Li, X. , Lee, B. , Kovalchuk, A. L. , Tang, Z. , … Casellas, R. (2018). The energetics and physiological impact of Cohesin extrusion. Cell, 173, 1165–1178.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Maltzahn, J. , Chang, N. C. , Bentzinger, C. F. , & Rudnicki, M. A. (2012). Wnt signaling in myogenesis. Trends in Cell Biology, 22, 602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R. , Chen, F. , Chen, Q. , Wan, X. , Shi, M. , Chen, A. K. , Ma, Z. , Li, G. , Wang, M. , Ying, Y. , Liu, Q. , Li, H. , Zhang, X. , Ma, J. , Zhong, J. , Chen, M. , Zhang, M. Q. , Zhang, Y. , Chen, Y. , & Zhu, D. (2022). MyoD is a 3D genome structure organizer for muscle cell identity. Nature Communications, 13, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Shen, Q. W. , Wang, J. , Zhang, Z. , Feng, F. , Chen, T. , Zhang, Y. , Wei, H. , Li, Z. , Wang, X. , & Wang, Y. (2016). KLF7 regulates satellite cell quiescence in response to extracellular signaling. Stem Cells, 34, 1310–1320. [DOI] [PubMed] [Google Scholar]
- Wang, Y. X. , Feige, P. , Brun, C. E. , Hekmatnejad, B. , Dumont, N. A. , Renaud, J. M. , Faulkes, S. , Guindon, D. E. , & Rudnicki, M. A. (2019). EGFR‐Aurka signaling rescues polarity and regeneration defects in dystrophin‐deficient muscle stem cells by increasing asymmetric divisions. Cell Stem Cell, 24, 419–432.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. X. , & Rudnicki, M. A. (2012). Satellite cells, the engines of muscle repair. Nature Reviews. Molecular Cell Biology, 13, 127–133. [DOI] [PubMed] [Google Scholar]
- Wolff, J. , Rabbani, L. , Gilsbach, R. , Richard, G. , Manke, T. , Backofen, R. , & Grüning, B. A. (2020). Galaxy HiCExplorer 3: A web server for reproducible hi‐C, capture hi‐C and single‐cell hi‐C data analysis, quality control and visualization. Nucleic Acids Research, 48, W177–W184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wutz, G. , Várnai, C. , Nagasaka, K. , Cisneros, D. A. , Stocsits, R. R. , Tang, W. , Schoenfelder, S. , Jessberger, G. , Muhar, M. , Hossain, M. J. , Walther, N. , Koch, B. , Kueblbeck, M. , Ellenberg, J. , Zuber, J. , Fraser, P. , & Peters, J. M. (2017). Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. The EMBO Journal, 36, 3573–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, B. A. , Castor‐Macias, J. , Fraczek, P. , Cornett, A. , Brown, L. A. , Kim, M. , Brooks, S. V. , Lombaert, I. M. A. , Lee, J. H. , & Aguilar, C. A. (2021). Sestrins regulate muscle stem cell metabolic homeostasis. Stem Cell Reports, 0, 2078–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, G. , Wang, L.‐G. , & He, Q.‐Y. (2015). ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics, 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Yu, M. , & Ren, B. (2017). The three‐dimensional organization of mammalian genomes. Annual Review of Cell and Developmental Biology, 33, 265–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Liu, X. , du, Z. , Wei, L. , Fang, H. , Dong, Q. , Niu, J. , Li, Y. , Gao, J. , Zhang, M. Q. , Xie, W. , & Wang, X. (2021). The loss of heterochromatin is associated with multiscale three‐dimensional genome reorganization and aberrant transcription during cellular senescence. Genome Research, 31, 1121–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Liu, T. , Meyer, C. A. , Eeckhoute, J. , Johnson, D. S. , Bernstein, B. E. , Nusbaum, C. , Myers, R. M. , Brown, M. , Li, W. , & Liu, X. S. (2008). Model‐based analysis of ChIP‐seq (MACS). Genome Biology, 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
Our data has been uploaded onto the Gene Expression Omnibus (GEO) and the number is listed in the manuscript.