

Abstract
As a recently discovered DNA repair enzyme, tyrosyl-DNA phosphodiesterase 1 (TDP1) removes topoisomerase IB (TOP1)-mediated DNA protein cross-links. Inhibiting TDP1 can potentiate the cytotoxicity of TOP1 inhibitors and overcome cancer cell resistance to TOP1 inhibitors. On the basis of our previous study, herein we report the synthesis of benzophenanthridinone derivatives as TOP1 and TDP1 inhibitors. Seven compounds (C2, C4, C5, C7, C8, C12, and C14) showed a robust TOP1 inhibitory activity (+++ or +++ +), and four compounds (A13, C12, C13, and C26) showed a TDP1 inhibition (half-maximal inhibitory concentration values of 15 or 19 μM). We also show that the dual TOP1 and TDP1 inhibitor C12 induces both cellular TOP1cc, TDP1cc formation and DNA damage, resulting in cancer cell apoptosis at a sub-micromolar concentration. In addition, C12 showed an enhanced activity in drug-resistant MCF-7/TDP1 cancer cells and was synergistic with topotecan in both MCF-7 and MCF-7/TDP1 cells.
Graphical Abstract
INTRODUCTION
DNA topoisomerase IB (TOP1) plays an essential role in DNA replication, transcription, and genomic stability by relaxing supercoiled DNA.1–3 During this process, TOP1 cleaves a single strand of DNA to form a transient covalent complex (TOP1cc) that allows supercoils to be dissipated.3 TOP1cc can be trapped by TOP1 poisons, such as camptothecin (CPT) derivatives that intercalate between the DNA base pairs at the cleavage site thereby preventing further religation of the broken strand, generating double-strand breaks and causing cell death.4–6 DNA damage produced by TOP1 poisons can be repaired through several pathways, including homologous recombination, cell cycle checkpoint signaling, and tyrosyl-DNA phosphodiesterase 1 (TDP1)-dependent pathways.3,7–9 TDP1 is a member of the phospholipase D superfamily and can specifically hydrolyze the 3′-phosphotyrosyl bond between the nicked DNA and peptide derived from TOP1cc to generate DNA breaks with 3′-phosphates and 5′-hydroxyl ends,10–13 following its recruitment by poly(ADPribose) polymerase (PARP1) activation.14,15 The resulting DNA breaks can be further repaired by PNKP, XRCC1, DNA polymerase β, and DNA ligase III.16,17 In this pathway, the TDP1-catalyzed hydrolysis of the 3′-phosphotyrosyl bond is a key step for initiating repair of the TOP1-mediated DNA damage, which would result in cancer cell resistance to therapeutic drugs, such as the TOP1 poisons topotecan, irinotecan, 10-hydroxycamptothecin, belotecan, enhertu, and trodelvy.6 Indeed, TDP1-overexpressing cells are resistant to TOP1 inhibitors.18 Conversely, cells with a low TDP1/TOP1 ratio tend to be sensitive to TOP1 inhibitors,19 as well as TDP1-deficient cells and TDP1 knockout mice, which show hypersensitivity to camptothecin.13,20–24 Accordingly, several TDP1 inhibitors have been reported to show a synergistic activity to camptothecin derivatives both in vitro and in vivo.25–30 Therefore, TDP1 is a rational target for the development of anticancer drugs.16,31
In addition, TDP1 also hydrolyzes a broad range of 3′-end blocking lesions generated by DNA oxidation and alkylation as well as 5′-phosphotyrosyl bonds derived from DNA topoisomerase II (TOP2)-mediated DNA damage, implying TDP1 has a broader role in the maintenance of genomic stability.20,21,31–34
Because of the unique physiological functions of TOP1 and TDP1, the discovery of their inhibitors has become a focus of attention. Despite the numerous TOP1 inhibitors and several TDP1 inhibitors that have been described,26,27,35–37 only a few dual inhibitors have been described in the literature.30,38–42 The indenoisoquinoline derivatives were the first reported dual TOP1 and TDP1 inhibitors.40,41 The representative bis-(indenoisoquinoline) inhibitor (Figure 1) shows a strong TOP1 inhibition equal to that of camptothecin and TDP1 inhibition with a half-maximal inhibitory concentration (IC50) value of 1.5 μM.41 In our effort to discover dual TOP1 and TDP1 inhibitors, two chalcone derivatives (Figure 1) have been recently identified from the roots of Isodon ternifolius Kudo with moderate dual TOP1 and TDP1 inhibition.42 In addition, oxynitidine derivatives have been found to have TOP1 and TDP1 inhibitory activities.25,43 And the oxynitidine TDP1 inhibitors exhibited a synergistic effect with CPT in MCF-7 cells. However, the dual inhibitors showed good TOP1 inhibition but low TDP1 inhibition. For example, NTD-96 (Figure 1) showed a TOP1 inhibitory activity of +++ and only a 12% TDP1 inhibitory activity at 100 μM concentration.25
Figure 1.

Reported dual TOP1/TDP1 inhibitors.
Molecular modeling shows two hydrogen bonds between the oxygen atoms of the methylenedioxy group of NTD-96, and the residues of TOP1, Thr718 (3.6 Å), and Asn722 (3.7 Å) were observed, implying this methylenedioxy group as playing a key role in the interaction between TOP1 and the inhibitor.25 Additionally, the position and number of the methoxy or methylenedioxy group affects the biological activity of the natural benzo[c]phenanthridine alkaloids, such as sanguinarine,44–46 chelerythrine,45,47 macarpine,48 fagaridine,49,50 and fagaronine (Figure S1, Supporting Information).51,52 These observations encouraged us to study the effect of the position and number of methoxy and methylenedioxy group as well as the displacement by a hydroxy or halogen group on TOP1 and TDP1 inhibitory activities. Therefore, three series of benzophenanthridinone derivatives were designed. On the basis of the structure of NTD-96, Series A analogues was designed with or without a single substituent in the A-, C-, or D-ring; Series B derivatives include two substituents or one methylenedioxy group in the A- or D-ring; and in Series C, multiple substituents were introduced in the A- and D-rings. Herein, we report the synthesis, TOP1 and TDP1 inhibitory activities, and the anticancer activity of these three series of novel benzophenanthridinone derivatives.
RESULTS AND DISCUSSION
Chemistry.
According to our previously reported method,25,53,54 the Series A analogues were synthesized and outlined in Pathway I (Scheme 1). The obtained Schiff’s base 2 by the coupling reaction of 2-bromobenzaldehyde 1 with N,N-dimethylaminoethylamine reacted with alkyne 3 to give the lactam intermediate 4 under Ni-based catalysis. Following the Swern oxidation of the hydroxyl group of 4, cyclization reaction under acid conditions gave the target products A1–A3, A5, A8–A10. From the intermediate 4ab′, the cyclization reaction gave two products, namely, A2 with a 1-methoxy substituent and A5 with a 3-methoxy substituent, at the same time. Also, two products A8 and A9 were obtained at the same time from the cyclization of 4ca′. The 2-bromo derivative A3 reacted with sodium methoxide to give the target A4 with a 2-methoxy substituent. Surprisingly, the cyclization reaction would be hampered if the R1 group was a 3- or 6-methoxy group or if the R2 group was a 2′-methoxy group on the intermediate 4. As a result, target compounds A6, A7, and A11 could not be prepared through Pathway I (Scheme 1). Therefore, we designed another synthetic route, Pathway II in Scheme 1. 2-Bromobenzoic acid 5 was amidated with naphthylamine 6 to provide the amide intermediate 7. Following the nucleophilic substitution with 2-dimethylaminoethyl chloride, a Heck cyclization reaction under Pd-based catalysis gave the target compounds A6, A7, and A11-A13.55
Scheme 1.

Synthesis of A1–A13a
aReagents and conditions: (a) NH2(CH2)2NMe2, MeOH, rt. (b) (i) N2, Ni(cod)2, P(o-Tol)3, MeCN, 80 °C; (ii) CsOH, K3[Fe(CN)6], MeOH, H2O, 80 °C. (c) (i) (COCl)2, DMSO, TEA, DCM, −60 °C; (ii) conc. hydrochloric acid, AcOH, 65 °C. (d) Na, CuI, MeOH, DMF, rt-110 °C. (e) (i) SOCl2, reflux; (ii) TEA, DCM, rt. (f) (i) Cl(CH2)2NMe2/HCl, NaH, DMF, 85 °C; (ii) N2, Pd(OAc)2, P(o-tol)3, Ag2CO3, DMF, 150 °C.
Similarly, Series B (B1–B8) and C (C1–C30) analogues were also synthesized through these two pathways. As shown in Scheme 2, compounds B1–B8 with a dimethoxy, difluoro, or methylenedioxy substituent at the A- or D-ring were synthesized.
Scheme 2.

Synthesis of B1–B8a
aReagents and conditions: (a) NH2 (CH2) NMe2, MeOH, rt. (b) (i) N2, Ni(cod)2, P(o-Tol)3, MeCN, 80 °C; (ii) CsOH, K3[Fe(CN)6], MeOH, H2O, 80 °C. (c) (i) (COCl)2, DMSO, TEA, DCM, −60 °C; (ii) conc. hydrochloric acid, AcOH, 65 °C. (d) (i) SOCl2, reflux; (ii) TEA, DCM, rt. (e) (i) Cl(CH2)2NMe2/HCl, NaH, DMF, 85 °C; (ii) N2, Pd(OAc)2, P(o-tol)3, Ag2CO3, DMF, 150 °C.
The syntheses of compounds C1–C30 were shown in Scheme 3, and their structures were outlined in Table 2. For the synthesis of the target compounds (C12, C13, C18, C19, C20, C22, C28, C29, and C30) with a hydroxyl substituent, the benzyl-protected materials (7qe′, 7re′, 4fd′, 4gd′, 4hd′, 7ue′, 4ef′, 4eg′, and 4eh′) were used. In Pathway I (Scheme 3), the benzyl groups were removed during the cyclization step in an acid condition to give the products C18–C20, and C28–C30. In Pathway II, following the Pd-catalyzed cyclization, the benzyl groups were removed with concentrated hydrochloric acid. From compounds 7re′ and 7ue′, the deprotection reaction gave the target products C13 and C22, respectively. However, we did not get the deprotection product using compound 7qe′; the methyl group at the 7-position was removed along with the benzyl group to give compound C12.
Scheme 3.

Synthesis of C1–C30a
aReagents and conditions: (a) NH2 (CH2) NMe2, MeOH, rt. (b) (i) N2, Ni(cod)2, P(o-Tol)3, MeCN, 80 °C; (ii) CsOH, K3[Fe(CN)6], MeOH, H2O, 80 °C. (c) (i) (COCl)2, DMSO, TEA, DCM, −60 °C; (ii) conc. hydrochloric acid, AcOH, 65 °C. (d) (i) SOCl2, reflux; (ii) TEA, DCM, rt. (e) (i) Cl(CH2)2NMe2/HCl, NaH, DMF, 85 °C; (ii) N2, Pd(OAc)2, P(o-tol)3, Ag2CO3, DMF, 150 °C. (f) For C12, C13 and C22, concentration hydrochloric acid, AcOH, 65 °C.
Table 2.
Structures of C1–C30 and Their TOP1 and TDP1 Inhibition, and Cytotoxicity
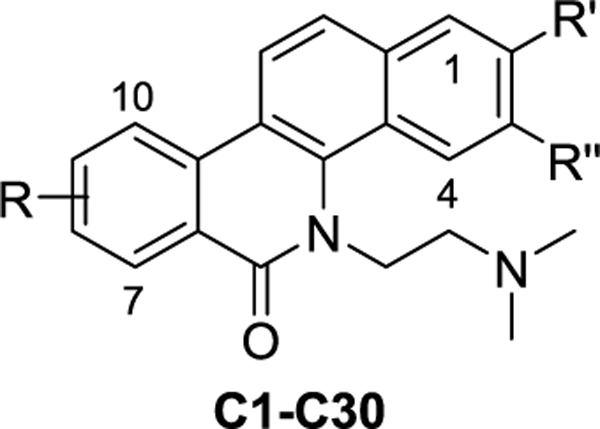
| |||||||
|---|---|---|---|---|---|---|---|
| GI50 ± SDa (μM) | |||||||
|
|
|||||||
| Cpd | R, R′, R′′ | TOP1 inhibitionb | TDP1 inhibitionc | HCT-116 | DU145 | MCF-7 | A549 |
| CPT | /d | ++++ | NDf | 0.050 ± 0.016 | 0.21 ± 0.07 | 0.35 ± 0.04 | 0.31 ± 0.03 |
| NTD-96 | 2,3-OCH2O-8, 9-diOMe | +++ | 12% | 0.14 ± 0.018 | 0.078 ± 0.007 | 0.20 ± 0.03 | 0.32 ± 0.04 |
| C1 | 2,3-OCH2O-7-OMe | + | 0 | 1.13 ± 0.03 | 0.86 ± 0.05 | 1.51 ± 0.22 | 6.66 ± 0.08 |
| C2 | 2,3-OCH2O-8-F | +++ | 0 | 0.93 ± 0.13 | 0.28 ± 0.04 | 1.44 ± 0.21 | 7.95 ± 0.37 |
| C3 | 2,3-OCH2O-8-Br | 0 | 0 | 10.12 ± 0.17 | 11.68 ± 0.83 | 5.44 ± 0.50 | 6.87 ± 1.11 |
| C4 | 2,3-OCH2O-8-NO2 | +++ | 0 | 0.24 ± 0.07 | 0.91 ± 0.08 | 0.57 ± 0.01 | 2.40 ± 0.36 |
| C5 | 2,3-OCH2O-8-OMe | ++++ | 0 | 1.18 ± 0.11 | 0.32 ± 0.01 | 2.99 ± 0.08 | 3.13 ± 0.82 |
| C6 | 2,3-OCH2O-9-F | ++ | 0 | 4.35 ± 0.78 | 3.82 ± 0.95 | 10.30 ± 2.05 | 9.27 ± 0.32 |
| C7 | 2,3-OCH2O-9-Cl | +++ | 0 | 2.92 ± 0.78 | 0.52 ± 0.04 | 10.18 ± 0.32 | 2.28 ± 0.88 |
| C8 | 2,3-OCH2O-9-OMe | +++ | 0 | 0.14 ± 0.02 | 0.32 ± 0.01 | 0.20 ± 0.04 | 1.61 ± 0.06 |
| C9 | 2,3-OCH2O-9-CF3 | ++ | 0 | 5.21 ± 0.28 | 1.00 ± 0.03 | 12.74 ± 0.62 | 1.49 ± 0.14 |
| C10 | 2,3-OCH2O-7,8-OCH2O | + | 27 ± 0.51 | 8.41 ± 0.21 | 4.13 ± 0.89 | 8.54 ± 0.91 | 12.21 ± 0.59 |
| C11 | 2,3-OCH2O-7,8-diOMe | 0 | 0 | 13.41 ± 0.73 | 40.21 ± 0.42 | 21.76 ± 7.69 | 12.81 ± 0.83 |
| C12 | 2,3-OCH2O-7,8-diOH | +++ | 19 ± 0.91 | 1.44 ± 0.28 | 0.80 ± 0.07 | 0.77 ± 0.10 | 1.11 ± 0.02 |
| C13 | 2,3-OCH2O-7-OH, 8-OMe | + | 15 ± 0.31 | 4.38 ± 0.65 | 0.44 ± 0.08 | 0.53 ± 0.08 | 3.09 ± 1.17 |
| C14 | 2,3-OCH2O-8, 9-diF | +++ | 0 | 2.79 ± 0.14 | 1.22 ± 0.01 | 3.24 ± 0.10 | 10.35 ± 0.17 |
| C15 | 2,3-OCH2O-8,9-OCH2O | + | 49% | 4.21 ± 0.87 | 3.27 ± 0.84 | 6.95 ± 1.92 | 1.86 ± 0.93 |
| C16 | 2,3-OCH2O-8-OMe, 9-Oi-Pr | + | 0 | 0.90 ± 0.02 | 6.61 ± 0.30 | 2.79 ± 0.20 | 3.35 ± 0.40 |
| C17 | 2,3-OCH2O-8-Oi-Pr, 9-OMe | 0 | 0 | 41.56 ± 4.86 | 7.88 ± 1.18 | 42.20 ± 2.06 | 34.71 ± 2.26 |
| C18 | 2,3-OCH2O-8-OMe, 9-OH | + | 0 | 9.35 ± 0.63 | 1.14 ± 0.45 | >100 | >100 |
| C19 | 2,3-OCH2O-8-OH, 9-OMe | + | 53 ± 1.5 | 0.63 ± 0.15 | 3.11 ± 2.70 | 6.26 ± 3.01 | 3.93 ± 0.73 |
| C20 | 2,3-OCH2O-8, 9-diOH | + | 0 | 8.48 ± 0.33 | >100 | 93.35 ± 3.04 | 84.94 ± 1.82 |
| C21 | 2,3-OCH2O-9, 10-diOMe | 0 | 0 | 26.17 ± 0.98 | 71.73 ± 0.83 | 24.40 ± 6.28 | 40.45 ± 0.95 |
| C22 | 2,3-OCH2O-9-OMe, 10-OH | 0 | 0 | 11.31 ± 0.44 | 11.25 ± 0.71 | >100 | 2.79 ± 0.35 |
| C23 | 2,3-diOMe 7,8-OCH2O− | + | 0 | 11.56 ± 1.67 | 15.78 ± 4.46 | 24.63 ± 5.96 | 23.74 ± 1.89 |
| C24 | 2,3,7,8-tetraOMe | 0 | 0 | >100 | 2.90 ± 0.83 | 7.22 ± 2.37 | 12.71 ± 3.61 |
| C25 | 2,3-diOMe 8,9-OCH2O− | 0/+ | 54 ± 1.0 | 10.15 ± 0.35 | 0.26 ± 0.05 | 10.72 ± 0.35 | 40.35 ± 1.96 |
| C26 | 2,3,8,9-tetraOMe | 0/+ | 19 ± 0.35 | 9.93 ± 0.33 | 25.43 ± 0.39 | 0.35 ± 0.02 | 1.53 ± 1.10 |
| C27 | 2,3,9,10-tetraOMe | 0 | 0 | >100 | >100 | >100 | 66.61 ± 1.92 |
| C28 | 2-OMe, 3-OH 8,9-diOMe | 0/+ | 0 | 12.88 ± 0.42 | 41.67 ± 0.75 | 2.70 ± 0.72 | 10.81 ± 0.81 |
| C29 | 2-OH, 3-OMe 8,9-diOMe | + | 44% | 1.21 ± 0.64 | 0.29 ± 0.09 | 1.66 ± 0.23 | 1.76 ± 0.39 |
| C30 | 2,3-diOH 8,9-diOMe | ++ | 0 | 26.19 ± 2.70 | 15.40 ± 0.52 | 3.62 ± 0.38 | 8.47 ± 0.76 |
The cytotoxicity GI50 values were defined as the concentrations corresponding to 50% cell growth inhibition and obtained from MTT assay.
The TOP1 cleavage inhibitory activity of the compounds was semiquantitatively expressed relative to CPT at 1 μM as follows: 0, no inhibition; 0/+, less than 20%; +, between 20% and 50% activity; ++, between 50% and 75% activity; +++, between 75% and 95% activity; ++++, equal activity.
The TDP1 inhibition, determined by using a fluorescence assay, was expressed as the percentage inhibition (%) of the compounds at 100 μM concentration or the IC50 value (μM, mean ± SD). Every experiment was repeated at least three times independently.
“/” means “inapplicable”.
ND means not determined.
Finally, altogether, fifty-one novel methoxy-, methylenedioxy-, hydroxy-, halo-, and nitro-substituted benzophenanthridinone derivatives were synthesized. Their structures were characterized through high-resolution mass spectrometry (HRMS) and NMR spectra.
TOP1 Inhibition.
All synthesized compounds were tested for TOP1 inhibition at 100, 10, 1, and 0.1 μM concentrations with the TOP1-mediated DNA cleavage assay.56 The TOP1 inhibitory activity, summarized in Tables 1 and 2, was semiquantitatively expressed relative to the TOP1 inhibition of camptothecin (CPT) at 1 μM: 0, no inhibition; 0/+, less than 20%; +, between 20% and 50% activity; ++, between 50% and 75% activity; +++, between 75% and 95% activity; ++++, equal activity to camptothecin at 1 μM concentration. Compound A1, the benzophenanthridinone scaffold with a 5-dimethylaminoethyl substituent, showed a low TOP1 inhibitory activity of + (Table 1). The addition of a single methoxy group in the A-, C-, and D-rings did not increase the TOP1 inhibition. Furthermore, the analogues (A6, A7, A11, and A12) with a methoxy group at the 4-, 7-, 10-, and 11-positions lost TOP1 inhibition. Similarly, the introduction of two methoxy groups or a methylenedioxy group in the A- and D-rings (Series B) did not increase the TOP1 inhibition (Table 1). The analogue B5 with 8,9-difluoro substituents showed an increased TOP1 inhibition of ++.
Table 1.
TOP1 and TDP1 Inhibition, and Cytotoxicity of A1–A13 and B1–B8
| GI50 ± SDc (μM) | ||||||
|---|---|---|---|---|---|---|
|
|
||||||
| Cpd | TOP1 inhibitiona | TDP1 inhibitionb | HCT-116 | DU145 | MCF-7 | A549 |
| CPT | ++++ | NDd | 0.050 ± 0.016 | 0.21 ± 0.07 | 0.35 ± 0.04 | 0.31 ± 0.03 |
| NTD-96 | +++ | 12% | 0.14 ± 0.018 | 0.078 ± 0.007 | 0.20 ± 0.03 | 0.32 ± 0.04 |
| A1 | + | 0 | 1.27 ± 0.13 | 25.21 ± 0.06 | 14.49 ± 1.33 | 10.98 ± 0.95 |
| A2 | + | 0 | 6.79 ± 0.59 | 10.88 ± 0.38 | 13.01 ± 3.11 | 11.86 ± 2.08 |
| A3 | + | 0 | 3.22 ± 0.19 | 15.39 ± 0.24 | 83.26 ± 5.42 | 2.61 ± 0.39 |
| A4 | + | 0 | 5.66 ± 1.24 | 24.98 ± 0.95 | 13.20 ± 0.61 | 4.96 ± 0.68 |
| A5 | + | 0 | 6.92 ± 0.28 | 27.15 ± 0.66 | 11.67 ± 0.61 | 11.95 ± 0.84 |
| A6 | 0 | 0 | 94.05 ± 1.02 | 3.53 ± 1.97 | 91.30 ± 5.94 | 65.14 ± 8.90 |
| A7 | 0 | 22% | 12.18 ± 1.27 | 60.27 ± 0.96 | 54.67 ± 4.87 | 12.18 ± 1.54 |
| A8 | + | 57% | 0.78 ± 0.11 | 15.57 ± 0.90 | 63.56 ± 13.88 | 2.75 ± 0.11 |
| A9 | + | 0 | 2.18 ± 0.13 | 3.77 ± 0.02 | 9.71 ± 2.44 | 0.94 ± 0.06 |
| A10 | + | 42% | 6.55 ± 0.61 | 12.34 ± 0.27 | 2.28 ± 0.66 | 6.41 ± 0.34 |
| A11 | 0 | 0 | 13.24 ± 2.11 | 18.17 ± 0.29 | 7.26 ± 1.89 | 17.05 ± 0.92 |
| A12 | 0 | 0 | 9.84 ± 0.87 | 6.72 ± 0.22 | 21.69 ± 4.61 | 17.72 ± 1.70 |
| A13 | 0/+ | 15 ± 2.2 | 6.93 ± 0.45 | 1.88 ± 0.17 | 2.65 ± 0.15 | 6.68 ± 0.31 |
| B1 | + | 0 | 2.01 ± 0.11 | 9.94 ± 0.65 | 8.85 ± 1.15 | 0.79 ± 0.08 |
| B2 | + | 26 ± 0.75 | 6.34 ± 0.88 | 17.38 ± 0.10 | 10.98 ± 0.71 | 12.31 ± 0.73 |
| B3 | + | 12% | 49.77 ± 6.57 | 68.10 ± 18.74 | 5.20 ± 1.65 | 13.07 ± 2.82 |
| B4 | 0 | 0 | 12.90 ± 0.63 | 30.97 ± 0.52 | 10.61 ± 2.00 | 8.58 ± 0.55 |
| B5 | ++ | 0 | 11.10 ± 0.27 | 2.04 ± 0.59 | 81.31 ± 2.65 | 65.53 ± 7.45 |
| B6 | + | 49% | 4.17 ± 1.15 | 20.46 ± 0.49 | 2.73 ± 0.83 | 3.78 ± 0.44 |
| B7 | + | 15% | 1.26 ± 0.02 | 1.41 ± 0.59 | 0.53 ± 0.07 | 1.22 ± 0.14 |
| B8 | 0 | 0 | 9.76 ± 0.66 | 4.51 ± 0.72 | 2.26 ± 0.06 | 2.33 ± 0.76 |
The TOP1 cleavage inhibitory activity of the compounds was semiquantitatively expressed relative to CPT at 1 μM as follows: 0, no inhibition; 0/+, less than 20%; +, between 20% and 50% activity; ++, between 50% and 75% activity; +++, between 75% and 95% activity; ++++, equal activity.
The TDP1 inhibition, determined by using a fluorescence assay, was expressed as the percentage inhibition (%) of the compounds at 100 μM concentration or the IC50 value (μM, mean ± SD). Every experiment was repeated at least three times independently.
The cytotoxicity GI50 values were defined as the concentrations corresponding to a 50% cell growth inhibition and obtained from an MTT assay.
“ND” means “not determined”.
For the Series C analogues (Table 2), the introduction of a methylenedioxy group at the 2,3-position and simultaneously one or two substituents in the A-ring increased the TOP1 inhibition significantly. The most potent inhibitor C5 with 2,3-methylenedioxy and 8-methoxy groups exhibited an equal potency (++++) to that of CPT. With the presence of a 2,3-methylenedioxy group, the compounds with 8-fluoro (C2), 8-nitro(C4), 9-chloro (C7), or 9-methoxy (C8) groups as well as with 7,8-dihydroxy (C12) or 8,9-difluoro groups (C14) exhibited the strong TOP1 inhibitory activity of +++, implying the important role of the 2,3-methylenedioxy group for TOP1 inhibition. Conversely, the compounds with a methoxy or hydroxy group substituted at the 2- and 3-positions (C23–C29) showed the limited TOP1 inhibitory potency of 0 to +, except for compound C30 with a moderate TOP1 inhibition of ++. Bigger substituents at the 8- or 9-positions were unfavorable for TOP1 potency. The compounds with 9-isopropoxy (C16) and 8-isopropoxy (C17) showed a low TOP1 inhibition. Surprisingly, the presence of a methoxy group at the 7- or 10-positon strongly reduced the TOP1 inhibitory activity, and compounds A7, A11, B4, B8, C11, C21, C24, and C27 had no detectable TOP1 inhibitory activity. The representative cleavage gels of C2, C4, C5, C7, C8, and C12 (Figure 2) show that the cleavage sites induced by the novel TOP1 inhibitors are similar to that of the indenoisoquinoline control LMP744 (Figure S2, Supporting Information) but not to that of CPT, which is consistent with the effects of NTD-96.25
Figure 2.

Representative gels of the TOP1-mediated DNA cleavage assay. Lane 1, DNA alone; lane 2, DNA and TOP1; lane 3, DNA and TOP1 with CPT (1 μM); lane 4, DNA and TOP1 with LMP744 (1 μM); lanes 5–28, DNA and TOP1 with the tested compounds at 0.1, 1.0, 10, and 100 μM concentrations, respectively. The arrows and numbers at left indicate the cleavage site positions.
To evaluate the ability of the synthesized novel inhibitor to induce cellular TOP1cc, C12 was selected for the immune-complex of enzyme (ICE) to DNA assay in MCF-7 cancer cells. As shown in Figure 3A, C12 induced cellular TOP1cc in a dose-dependent manner, similar to the positive control topotecan (TPT), implying that TOP1 is the cellular target of C12.
Figure 3.

(A) Induction of TOP1–DNA covalent cleavage complexes by in vivo complex of enzyme assay in MCF-7 cells. Lane 1, untreated control; lanes 2 and 3, cells treated with TPT at 25 and 50 μM, respectively; lanes 4–6, cells treated with C12 at 25, 50, and 100 μM, respectively. (B) Hypothetical binding mode of C12 in the ternary TOP1–DNA–drug cleavage complex (PDB: 1K4T). (C) Induction of TDP1-DNA covalent complex by ICE assay in MCF-7 and MCF-7/TDP1 cells. Lane 1, untreated control; lanes 2–4, cells treated with TPT (10 μM) and C12 at 10 and 100 μM, respectively; lanes 5 and 6, cells cotreated with TPT (10 μM) and C12 (10 and 100 μM).
The TOP2 relaxing assay (Figure S3, Supporting Information) indicated that C12 did not have an inhibitory activity to TOP2 at a 25 μM concentration.
To evaluate the rationale for TOP1 poisoning and the molecular binding mode of the synthesized novel inhibitors within the TOP1-DNA complex, molecular modeling was conducted on compound C12. A hypothetical binding mode was built by using in silico docking from the X-ray crystal of TOP1-DNA-ligand (PDB ID: 1K4T)57 according to our reported method.25 As shown in Figure 3B, the benzophenanthridinone scaffold of C12 intercalated in the DNA break and stacked with the +1 and −1 base pairs flanking the DNA cleavage site. Similar to the binding mode of NTD-96 with the TOP1-DNA complex,25 the 5-dimethylaminoethyl substituent extended into the minor groove of the DNA. Two hydrogen bonds (3.1 and 3.2 Å) between the lactam oxygen atom and the side-chain nitrogen of Arg364 were observed, implying the important role of hydrogen-bond receptor to the TOP1 inhibition potency. Also, two hydrogen bonds between the 7-hydroxy group and a base pair (2.9 Å) and between an oxygen atom in the dioxole ring and Asn722 (3.2 Å) might contribute to the trapping of TOP1cc, implying the important role of the dioxole ring for TOP1 inhibition. For the 2,3-dimethoxy-substituted derivative, there is no hydrogen bond observed between the compound and TOP1. The hypothetical binding mode of C26 (Figure S4, Supporting Information) indicated that the distance between the oxygen atom of the 3-methoxy group and Asn722 was 4.0 Å and that the distance between the lactam oxygen atom and Arg364 was 4.3 Å, possibly due to the steric bulk of 2,3-dimethoxy groups. This observation is consistent with the structure–activity relationship.
TDP1 Inhibition.
The TDP1 inhibition of the synthesized compounds was evaluated through a fluorescence assay, using a quenched fluorescent single-stranded oligonucleotide (5′-FAM-AGGATCTAAAAGACTT-BHQ-3′) as substrate.58 The compounds were first tested at 100 μM concentration, and the percentage inhibition was calculated. Compounds A13, B2, C10, C12, C13, C19, C25, and C26, with over 50% percentage inhibition, were further tested to determine their IC50 values, defined as the concentration of compound that inhibits 50% of enzyme activity. As shown in Tables 1 and 2, compound C12 showed both TOP1 (+++) and TDP1 inhibitory activity (IC50 = 19 ± 0.91 μM). A13 and C13 showed the most potent TDP1 inhibition, both with the IC50 value of 15 μM. However, their TOP1 inhibitory activities were low (0/+ and +). Similarly, C26 also showed a good TDP1 inhibition (19 ± 0.35 μM) and a low TOP1 inhibition (0/+). The most potent TOP1 inhibitor was C5 (++++) as well as C2, C4, C7, C8, and C14, which show a good TOP1 inhibition (+++) lacking a detectable TDP1 inhibitory activity.
It has been proposed that a transient enzyme-DNA covalent complex intermediate (TDP1cc) forms during the TDP1 function.21,59 To assess whether C12 can induce the formation of cellular TDP1cc, ICE assays were conducted in MCF-7 and TDP1-overexpressing MCF-7 (MCF-7/TDP1) cancer cells, established by the transfection of parental MCF-7 cells. As shown in Figure 3C, C12 induced the formation of cellular TDP1cc in MCF-7 cells at a high concentration (100 μM). Although there was no obvious TDP1cc formation observed after being incubated with the TOP1 inhibitor TPT or C12 alone at low concentration (10 μM), TDP1cc significantly increased after being coincubated with TPT and C12 (both at 10 and 100 μM) in MCF-7 cells. Similarly, in MCF-7/TDP1 cells, C12 could induce the formation of cellular TDP1cc, even at a low concentration (10 μM). After being coincubated with TPT and C12, cellular TDP1cc significantly increased. These results indicated that the benzophenanthridinone TDP1 inhibitor C12 could induce the formation of cellular TDP1cc in both MCF-7 and MCF-7/TDP1 cells.
Cytotoxicity Assays.
The cytotoxicity of the synthesized compounds was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay against four human tumor cell lines, including colon cancer (HCT116), prostate cancer (DU-145), breast cancer (MCF-7), and nonsmall cell lung cancer (A549) cell lines. The cells were treated for 72 h in a five-dose assay at concentrations ranging from 0.01 to 100 μM. The GI50 values, defined as the concentrations corresponding to 50% cell growth inhibition, were summarized in Tables 1 and 2.
As shown in Table 1, Series A and B analogues, which showed a TOP1 inhibitory activity ranging from 0 to ++, showed cytotoxicity with GI50 values at high and low micromolar levels. Only two Series A analogues showed a high cytotoxicity at a sub-micromolar level (A8: 0.78 μM for HCT-116 and A9: 0.94 μM for A549) as well as two Series B analogues (B1: 0.79 μM for A549 and B7: 0.53 μM for MCF-7). Compared with Series A and B analogues, the multialkoxy-substituted derivatives (Series C) showed a marked cytotoxicity against the four human cancer cells, which is consistent with their high TOP1 inhibition potency. The dual TOP1/TDP1 inhibitor C12 (TOP1: +++, TDP1:19 μM) showed good cytotoxicity against the four human cancer cell lines with GI50 values of 1.44 μM for HCT-116, 1.11 μM for A549, 0.80 μM for DU145, and 0.77 μM for MCF-7. The dual inhibitor C13 (TOP1: +, TDP1:15 μM) also showed good cytotoxicity with GI50 values between 0.44 and 4.38 μM. Although the most potent TOP1 inhibitor C5 (++++) exhibited high cytotoxicity against DU145 cells (GI50 = 0.32 ± 0.01 μM), it had a lower cytotoxicity than NTD-96 (+++), which might be due to the low cellular permeability of C5. Other analogues C2, C4, C7, and C8 with good TOP1 inhibition (+++) also showed a high cytotoxicity with GI50 values at a sub-micromolar range. Although C1, C19, C25, C26, and C29 showed a low TOP1 potency, they also showed a high cytotoxicity possibly due to their effect on other cellular targets. Among them, C8 showed the highest cytotoxicity for HCT-116 (0.14 μM) and MCF-7 (0.20 μM) as well as C25 for DU145 (0.26 μM) and B1 for A549 (0.79 μM).
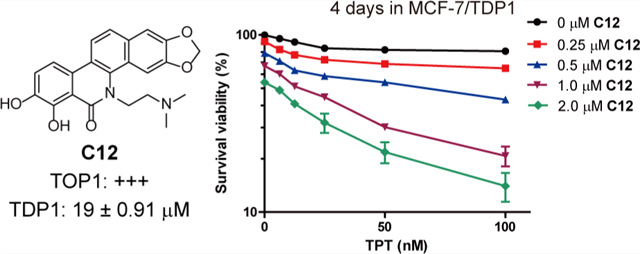
The cytotoxicity of C12 was further evaluated in MCF-7/ TDP1 cells. As shown in Table 3, compared with the parental cells MCF-7, the MCF-7/TDP1 cells exhibited a significant resistance to the well-known TOP1 inhibitors CPT (9.2-fold) and TPT (21.2-fold), as well as to our oxynitidine TOP1 inhibitor NTD-96 (19.9-fold), implying TDP1 overexpression results in a cellular resistance to TOP1 inhibitors. However, MCF-7/TDP1 cells showed a decreased resistance to C12 (3.4-fold), indicating that the TDP1 inhibition might reverse the resistance of MCF-7/TDP1 cells to TOP1 inhibitors.
Table 3.
Cytotoxicity of C12 in Drug-Resistant MCF-7 Cells
| GI50 ± SDa (μM) | |||
|---|---|---|---|
|
|
|||
| Cpd | MCF-7 | MCF-7/TDP1 | resistance indexb |
| CPT | 0.35 ± 0.04 | 3.21 ± 0.17 | 9.2 |
| TPT | 0.35 ± 0.01 | 7.42 ± 0.24 | 21.2 |
| NTD-96 | 0.20 ± 0.03 | 3.98 ± 0.9 | 19.9 |
| C12 | 0.77 ± 0.10 | 2.59 ± 0.31 | 3.4 |
GI50 values (mean ± SD) were defined as the concentrations of compounds that resulted in a 50% cell growth inhibition and obtained from an MTT assay. Every experiment was repeated at least three times.
The resistance index was calculated by dividing the GI50 of MCF-7/TDP1 cells by the GI50 of MCF-7 cells.
Synergistic Effects of C12 with TPT.
In our previous work, the oxynitidine TDP1 inhibitors were shown to have synergistic effects with TOP1 inhibitors in MCF-7 cancer cells.25 To assess whether a dual TOP1 and TDP1 inhibition could act synergistically with TOP1 inhibitors, the combined effects of C12 with TPT were tested in MCF-7 and MCF-7/ TDP1 cells. As shown in Figure 4A, after an incubation for 96 h at 37 °C, the cytotoxicity of TPT against MCF-7 cells significantly increased in the presence of C12 in a dose-dependent manner. At a 50 nM concentration, ~23% of cells were killed by TPT alone, and ~87% of cells were killed after being coincubated with 2 μM of C12. The combination index (CI) values were calculated and are shown in Figure 4B. The CI values of the combination treatment of TPT and C12 fell in the range of 0.1–0.9, and four doses showed strong synergistic effects (CI < 0.3). Similarly, C12 also showed a synergistic effect with TPT in MCF-7/TDP1 cells (Figure 4C). After a 96 h incubation, ~19% of MCF-7/TDP1 cells were killed by 100 nM TPT alone, and ~86% of cells were killed when being coincubated with 2 μM of C12. Figure 4D indicated that the CI values of the combination treatment of TPT and C12 for MCF-7/TDP1 cells also fell in the range of 0.1–0.9, and six doses showed strong synergistic effects (CI < 0.3). These results indicated that the benzophenanthridinone dual TOP1 and TDP1 inhibitor also showed a synergistic effect with the TOP1 inhibitor TPT in both MCF-7 and MCF-7/TDP1 cells.
Figure 4.

Synergistic effect and CI of C12 with TPT in the MCF-7 (A, B) and MCF-7/TDP1 (C, D) cells. The cells were coincubated for 96 h.
DNA Damage and Apoptosis Induced by C12.
To evaluate the DNA damage in response to the dual TOP1 and TDP1 inhibitor C12 in cancer cells, drug-induced γH2AX foci were tested by an immunofluorescence assay in both MCF-7 and MCF-7/TDP1 cells using TPT as a positive control. Similar to TPT, C12 induced γH2AX foci in MCF-7 cells in a dose-dependent manner (Figure 5A,B), implying the ability of C12 to induce cellular DNA damage due to the trapping of cellular TOP1cc. In MCF-7/TDP1 cells, the number of γH2AX foci induced by TPT decreased by 34% relative to wild-type (WT) MCF-7 cells, implying the ability of TDP1 to process TOP1cc.8,9 Although the number of γH2AX foci induced by C12 in MCF-7/TDP1 cells was also reduced, the decrease (22% and 28% at 1 and 2 μM, respectively) was less when compared with TPT. In addition, a combined treatment with C12 and TPT significantly increased the number of γH2AX foci in both MCF-7 and MCF-7/TDP1 cells (Figure 5A,B). In addition, C12 reduced the number of γH2AX foci induced by TPT (14% and 18% vs 34% for TPT alone), consistent with the TDP1 inhibition by C12. These results indicated that C12 effectively induced cellular DNA damage but also increased the ability of TPT to induce DNA damage in both MCF-7 and MCF-7/TDP1 cells, possibly due to its dual TOP1 and TDP1 inhibitory activities.
Figure 5.

(A) Histone γH2AX foci (red) induced by TPT and C12 alone or combined treatment in the MCF-7 and MCF-7/TDP1 cells. The cells were treated with drugs for 6 h at the indicated concentrations. DNA was stained with DAPI (blue). (B) Quantification of the number of γH2AX foci on the figure (A). Every experiment was repeated at least three times independently. (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, and (****) P < 0.0001. (C) Flow cytometry histograms of apoptosis in MCF-7 cells induced by C12 at 1, 2, and 4 μM, respectively.
To evaluate the induction of cell apoptosis by C12, flow cytometry assays were conducted. After an incubation with C12 for 24 h, apoptotic MCF-7 cells were detected, and C12 significantly induced MCF-7 cell apoptosis in a dose-dependent manner (Figure 5C). Approximately 74.99% (26.53% early apoptotic cells and 48.46% late apoptotic cells) were scored as apoptotic after a 24 h incubation with C12 at 4 μM concentration.
CONCLUSION
Herein three series of benzophenanthridinone derivatives (Series A, B, and C) with methoxy, methylenedioxy, hydroxy, and halo substituents in A-, C-, and D-rings were synthesized and evaluated for TOP1 and TDP1 inhibitory activities and cytotoxicity against four human cancer cell lines (HCT116, MCF-7, DU-145, and A549 cell lines). Enzyme-based assays indicated that C5 with an 8-methoxy substituent showed the highest TOP1 inhibitory potency (++++) equal to CPT, and six compounds C2, C4, C7, C8, C12, and C14 showed the strong TOP1 inhibitory activity of +++. Four compounds A13 (15 μM), C12 (19 μM), C13 (15 μM), and C26 (19 μM) also showed TDP1 inhibitory activity. Several compounds showed a high cytotoxicity in human cancer cells with GI50 values at a sub-micromolar concentration range. C8 with good TOP1 inhibition (+++) showed the highest cytotoxicity for HCT-116 (0.14 μM) and MCF-7 (0.20 μM). The dual TOP1 and TDP1 inhibitor C12 showed good cytotoxicity against these four human cancer cell lines with GI50 values ranging from 0.77 to 1.44 μM and enhanced activity in the drug-resistant MCF-7/ TDP1 cancer cells, possibly due to the dual TOP1 and TDP1 inhibition. C12 also showed synergy in combination with topotecan in both MCF-7 and MCF-7/TDP1 cells. C12 induced both cellular TOP1cc and TDP1cc in ICE assays. Additionally, C12 not only induced DNA damage but also enhanced topotecan-induced DNA damage in both MCF-7 and MCF-7/TDP1 cells, resulting in cancer cell apoptosis. This work suggests that the benzophenanthridinone derivatives deserve to be further investigated and potentially developed as dual TOP1 and TDP1 inhibitors.
EXPERIMENTAL SECTION
General Experiments.
All chemicals were purchased from local commercial suppliers and used without further purification unless otherwise specified. All the solvents were of an analytical reagent grade. Nuclear magnetic resonance spectra were recorded on a Bruker BioSpin GmbH spectrometer at 400 or 500 MHz using tetramethylsilane (TMS) as the internal reference. Melting points were determined in slides on an X-5 micro melting point apparatus without being corrected. Mass spectra were recorded on a Shimadzu LCMS-2020 instrument with an electrospray ionization (ESI) mass selective detector, and HRMS were recorded on a Shimadzu LCMS-IT-TOF mass spectrometer. The synthesized compounds were purified through column chromatography with silica gel (200–300 mesh) purchased from Qingdao Haiyang Chemical Co. Ltd. All compounds tested for biological activities were analyzed by high-performance liquid chromatography (HPLC), and their purities were more than 95%. The analysis condition was detection at 220 nm, 1.0 mL/min flow rate, and a linear gradient from 55% to 15% phosphatebuffered solution (PBS) buffer (pH 3) and 45%–85% methanol (MeOH) in 30 min.
General Procedure for the Synthesis of Schiff’s Base Intermediates (2).
The Schiff’s base intermediates were prepared from the reaction of 2-bromobenzaldehyde derivatives and N,N-dimethylaminoethylamine according to our reported method.25 The intermediates were used for the next synthesis without further purification.
General Procedure for the Synthesis of Lactam Intermediates (4).
The intermediate 4 was prepared from the reaction of the Schiff’s base 2 and the acetylene material 3 according to our reported method.25
General Procedure for the Synthesis of Compounds of Series A, B, and C from Lactams 4 (Pathway I).
The target products were prepared from the intermediate 4 according to our reported method.25 Briefly, the intermediate 4 (0.5 mmol) in a solution of freshly distilled CH2Cl2 (8 mL) was oxidized by dimethyl sulfoxide (DMSO) (365 μL, 5 mmol) and (COCl)2 (215 μL, 2.5 mmol) at −60 °C for 30 min. And then Et3N (1.5 mL, 10 mmol) was added dropwise to the reaction solution. The reaction solution was brought to room temperature (rt) and stirred for 2 h. The reaction was quenched by the addition of water (1 mL) at 0 °C. The organic layer was concentrated under reduced pressure, and the resulting residue was purified by silica gel column chromatography using dichloromethane (DCM)/methanol (40:1) as eluent. The obtained intermediate was added to a solution of acetic acid (AcOH) (10 mL) and concentrated hydrochloric acid (1.0 mL) in a 50 mL round-bottomed flask. The reaction solution was stirred at 65 °C overnight. Then, the reaction solution was adjusted to pH 7 with a saturated sodium bicarbonate solution and extracted with CH2Cl2 (3 × 20 mL). The combined organic layer was washed with saturated saline (3 × 30 mL), dried (MgSO4), and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography using chloroform/acetone (5:1) as eluent to give the target products.
Procedure for the Synthesis of Compound A4.
Under a nitrogen atmosphere, Na (280 mg, 12.2 mmol) was carefully added to absolute methanol (5 mL). The reaction solution was stirred at room temperature for 1 h. Then, the mixture of A3 (20 mg, 0.05 mmol) and CuI (3.8 mg, 0.02 mmol) in dimethylformamide (DMF) (2 mL) was added to the reaction solution. The resulting solution was stirred and heated under reflux overnight. The reaction was quenched by the addition of water (10 mL). The resulting solution was extracted with CH2Cl2 (3 × 20 mL). The combined organic layer was washed with saturated saline (3 × 30 mL), dried (MgSO4), and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography using dichloromethane/methanol (40:1) as eluent to give A4.
General Procedure for the Synthesis of Amide Intermediates (7).
The solution of SOCl2 (5 mL) and 2-bromobenzoic acid (1 mmol) was stirred in a 25 mL round-bottomed flask at 65 °C for 2 h. The excess SOCl2 was removed by reduced pressure. The solution of the resulting residue in freshly distilled CH2Cl2 (10 mL) was added dropwise to a solution of naphthylamine (0.8 mmol) and trimethylamine (0.1 mL) in freshly distilled CH2Cl2 (10 mL). The reaction solution was stirred at room temperature for 12 h, and then the reaction was quenched by the addition of a saturated sodium bicarbonate solution (1 mL). The resulting solution was washed with saturated saline (3 × 20 mL). The organic layer was dried (MgSO4) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using ethyl acetate (EtOAc)/ hexane (1:1) as eluent to give the amide intermediates 7.
General Procedure for the Synthesis of Compounds of Series A, B, and C from Amide 7 (Pathway II).
To the solution of intermediate 7 (0.5 mmol) in dry DMF (2 mL), NaH (60% dispersion in mineral oil, 80 mg, 2 mmol) was added. The reaction solution was stirred at room temperature for 1 h. Then, a solution of 2-dimethylaminoethyl chloride in dry DMF (2 mL) was added dropwise. The reaction solution was stirred at 85 °C overnight. The reaction was quenched by an addition of icy water. The resulting solution was extracted with ethyl acetate (3 × 20 mL). The combined organic layer was washed with saturated saline (3 × 20 mL), dried (MgSO4), and then concentrated under reduced pressure. Pd(OAc)2 (9 mg, 0.04 mmol), P(o-tol)3 (24 mg, 0.08 mmol; o-tol = ortho-toluene), and Ag2CO3 (220 mg, 0.8 mmol) were added to a solution of the resulting residue in dry DMF (10 mL). The reaction solution was stirred at 150 °C for 3 h under nitrogen atmosphere. After the mixture cooled to room temperature, saturated saline (20 mL) was added to the reaction solution. The resulting solution was extracted with ethyl acetate (3 × 20 mL). The combined organic layer was washed with saturated saline (3 × 20 mL), dried (MgSO4), and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography using dichloromethane/methanol (30:1) as eluent to give the target products and the benzyl-protected compounds of C12, C13, and C22.
Procedure for the Synthesis of Compounds C12, C13, and C22.
The benzyl-protected compounds of C12, C13, and C22 were deprotected as follows: The corresponding residue was added to the solution of concentrated hydrochloric acid (0.2 mL) and acetic acid (2 mL) in a 25 mL round-bottomed flask. The reaction solution was stirred at 65 °C overnight and then adjusted to pH 7 with a saturated NaHCO3 aqueous solution. The resulting solution was extracted with CH2Cl2 (3 × 20 mL). The combined organic layer was washed with saturated saline (3 × 30 mL), dried (MgSO4), and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography using dichloromethane/methanol (20:1) as eluent to give the target products.
TOP1-Mediated DNA Cleavage Assay.
A 3′-[32P]-labeled 117 base pair (bp) DNA oligonucleotide was prepared as previously described.56 A radiolabeled DNA substrate (~2 nM) was incubated with recombinant TOP1 in 20 μL of reaction buffer (10 mM Tris-HCl pH 7.5, 50 mM KCl, 5 mM MgCl2, 0.1 mM ethylenediaminetetraacetic acid (EDTA), and 15 mg/mL bovine serum albumin (BSA)) at 25 °C for 20 min in the presence of various concentrations of the tested compounds. The reactions were terminated by adding sodium dodecyl sulfate (SDS) (0.5% final concentration), followed by the addition of two volumes of loading dye (80% formamide, 10 mM sodium hydroxide, 1 mM sodium EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue). Aliquots of each reaction mixture were subjected to 20% denaturing polyacrylamide gel electrophoresis (PAGE). Gels were dried and visualized by using a phosphoimager and ImageQuant software (Molecular Dynamics). Cleavage sites are numbered to reflect actual sites on the 117 bp oligonucleotide.
TDP1 Inhibition Assay.
The TDP1 fluorescence assay was conducted according to the reported method.58 Briefly, a linear oligonucleotide labeled with the donor fluorophore 6-carboxyfluorescein (FAM) and Black Hole Quencher (BHQ), 5′-FAM-AGGATCTAAAAGACTT-BHQ-3′, was designed as a linear quenched fluorescent substrate. TDP1 solution (20 μL/well of purified TDP1 (100 nM) in 10 mM Tris-HCl, pH 7.5, 50 mM KCl, 1 mM EDTA, 1 mM dithiothreitol (DTT)) was dispensed into wells of a white 384-well plate (NEST). The tested compound solution in DMSO (5 μL) was pinned into assay plates and incubated at room temperature for 30 min. During this time, the plates were read by a Flash multimode reader (Molecular Devices) at Ex485/Em510 nm to identify false-positive compounds that had autofluorescence. The linear oligonucleotide substrate (25 μL, 35 nM) was dispensed into the wells to start the reaction. The whole plate was immediately read five times using a kinetic read on the Flash multimode reader (Molecular Devices) (Ex485/Em510 nm). The TDP1 percentage inhibition of the tested compounds was calculated by comparing the rate of increase in fluorescence throughout the time for the compound-treated wells to that of DMSO control wells.
The inhibition of TDP1 was also conducted with gel-based assays as described.60 Briefly, 1 nM of the DNA substrate (5′-Cy5-GATCTAAAAGACTT-pY-3′) was incubated with 10 pM recombinant TDP1 with a serial dilution of compound for 15 min at room temperature in a TDP1 reaction buffer (50 mM Tris HCl, pH 7.5, 80 mM KCl, 2 mM EDTA, 1 mM DTT, 40 μg/mL BSA, and 0.01% Tween-20). Reactions were stopped by adding an equal volume of loading buffer (99.5% formamide, 5 mM EDTA). Samples were then subjected to a 20% denaturing PAGE gel followed by gel scanning using a Typhoon FLA 9500 scanner (GE Healthcare). The IC50 of TDP1 inhibitors was calculated by comparing the percentage of cleavage product (5′Cy5-GATCTAAAAGACTT-p-3′) to DMSO control.
Cell Culture and MTT Assay.
The cancer cells were grown in GIBCO RPMI 1640 medium or Dulbecco’s Modified Eagle’s Medium (DMEM) complemented with 10% fetal bovine serum (FBS) at 37 °C with 5% CO2. For the drug treatment experiments, cells were treated with compounds and collected for various times in different experiments.
For the cytotoxicity measurements by MTT assay, the cancer cells were treated with the compounds (predissolved in DMSO) at a five-dose assay ranging from 0.01 to 100 μM concentration as reported previously.25 The percent inhibition of viability for each concentration of the compounds was calculated relative to the control, and the GI50 values were estimated by nonlinear regression analysis (GraphPad Prism).
For the drug combination experiments, human breast cancer MCF-7 cells and MCF-7/TDP1 cells were incubated with topotecan and the tested compounds for 96 h at 37 °C, and then they were measured by an MTT assay.
Immunodetection of Cellular TOP1cc and TDP1cc.
The ICE assays for the cellular enzyme–DNA adduct were performed according to the reported method.25,33 Briefly, mid log phase MCF-7 or MCF-7/TDP1 cells were incubated with drugs at the indicated concentration for 1 h, and then the cells were lysed with DNAzol reagent (1 mL) at 25 °C for 30 min. Ethanol (0.5 mL, 100%) was subsequently added and mixed with the lysate. The solution was incubated at −20 °C overnight. The genomic DNA was collected by centrifugation (12 000 rpm) at 25 °C for 10 min and washed with 75% ethanol. The precipitated DNA was dissolved in NaOH (8 mM, 0.2 mL). The pH value was adjusted to 7.2 by adding 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (1 M). After centrifugation, the supernatant was used to quantify the DNA concentration. DNA (2 μg) was dissolved in 30 μL of a NaH2PO4 buffer (25 mM, pH 6.5) and then loaded onto nitrocellulose membranes. Membranes were incubated with rabbit monoclonal to human TOP1 (Abcam, 1:1000) or TDP1 (Abcam, 1:1000) at 4 °C overnight, and then they were incubated with the appropriate horseradish peroxidase (HRP) conjugated secondary antibodies (Cell Signaling Technology, 1:3000) at room temperature for 1 h. Reactive dots were detected using an Immobilon Western Chemiluminescent HRP substrate (Millipore).
Immunofluorescence Assay.
Immunofluorescence staining and confocal microscopy were performed. After being incubated with the tested compounds for 6 h at 37 °C, the cells (MCF-7 and MCF-7/ TDP1 cells) were fixed with 4% paraformaldehyde for 10 min, then permeabilized with 0.5% Triton X-100/PBS at 37 °C for 30 min, and finally blocked with 5% goat serum/PBS at 37 °C for 3 h. The protocol was followed with the γH2AX antibody (No. 9718, Cell Signaling Technology) at 4 °C overnight. The glass coverslips were washed six times with blocking buffer and were then incubated with antirabbit Alexa 488-conjugated antibody (A21206, Life Technology), and 2 μg/mL of 4,6-diamidino-2-phenylindole (DAPI, Invitrogen) was diluted in 5% goat serum/PBS at 37 °C for 2 h. The dishes were again washed six times with blocking buffer, and then, digital images were recorded using an FV3000 microscope (Olympus) and analyzed with FV31S-SW software.
Apoptosis Detection.
MCF-7 cells (2 × 105) were planted in a culture dish and treated with C12 at the indicated concentration for 24 h. The apoptosis detection was applied using the fluorescein isothiocyanate (FITC) Annexin V/PI apoptosis Detection Kit (KeyGEN). The collected cells were resuspended in 500 μL of binding buffer and added with 5 μL of FITC Annexin V and propidium iodide (PI) dyes. The samples were slowly blended and incubated at 25 °C for 15 min away from light. Finally, the fluorescence-positive cells were quantified by flow cytometry (EPICS XL).
Molecular Modeling.
The PDB file (PDB ID: 1K4T)57 for the X-ray crystal structure of the TOP1-DNA-ligand complex was obtained, cleaned, and inspected for errors and missing residues. Hydrogens were added, and the water molecules and the ligand were deleted. The ternary complex ligand centroid coordinates for docking were defined using the ligand in the complex structure as the center of the binding pocket. The compounds were constructed and optimized using ChemDraw and saved in SDF file formats and were corrected and minimized using MOE. The top 30 docking poses per ligand were inspected visually following the docking runs. Energy minimizations were performed for selected ligand poses. The AMBER force field was utilized within the MOE for energy minimization.
Supplementary Material
ACKNOWLEDGMENTS
We wish to thank Dr. H.-B. Chen, Sun Yat-Sen University (Shenzhen), for the gift of MCF-7/TDP1 cells. This work was supported by the Guangdong Basic and Applied Basic Research Foundation (No. 2019A1515011317), Guangzhou Basic and Applied Basic Research Project (No. 202002030312), and the Guangdong Provincial Key Laboratory of Construction Foundation (No. 2017B030314030). W.W., K.A., and Y.P. are supported by the Intramural Program of the National Cancer Institute (Center for Cancer Research), National Institutes of Health, Bethesda, Maryland, United States (Z01 BC 006150–19).
ABBREVIATIONS USED
- TOP1
topoisomerase IB
- TOP1cc
TOP1–DNA cleavage complex
- TDP1
tyrosyl DNA–phosphodiesterase 1
- TDP1cc
TDP1-DNA covalent complex
- TOP2
topoisomerase II
- CPT
camptothecin
- TPT
topotecan
- ICE
immunocomplex of enzyme to DNA
- NMR
nuclear magnetic resonance
- HRMS
high-resolution mass spectra
- HPLC
high performance liquid chromatography
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-di-phenyl tetrazolium bromide
- γH2AX
phosphorylated histone H2AX
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00318.
Molecular formula strings (CSV)
Characterization data for the intermediates and the target products, the structures of benzo[c]-phenanthridine alkaloid, and LMP744 (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.1c00318
The authors declare no competing financial interest.
Contributor Information
De-Xuan Hu, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China.
Wen-Lin Tang, School of Pharmaceutical Sciences, Sun Yatsen University, Guangzhou 510006, China.
Yu Zhang, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China.
Hao Yang, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China.
Wenjie Wang, Developmental Therapeutics Branch and Laboratory of Molecular Pharmacology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda 20892, Maryland, United States.
Keli Agama, Developmental Therapeutics Branch and Laboratory of Molecular Pharmacology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda 20892, Maryland, United States.
Yves Pommier, Developmental Therapeutics Branch and Laboratory of Molecular Pharmacology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda 20892, Maryland, United States.
Lin-Kun An, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China; Guangdong Provincial Key Laboratory of New Drug Design and Evaluation, Guangzhou 510006, China.
REFERENCES
- (1).Champoux JJ DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem 2001, 70, 369–413. [DOI] [PubMed] [Google Scholar]
- (2).Wang JC Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol 2002, 3, 430–440. [DOI] [PubMed] [Google Scholar]
- (3).Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [DOI] [PubMed] [Google Scholar]
- (4).Pommier Y; Leo E; Zhang H; Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol 2010, 17, 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pommier Y; Marchand C. Interfacial inhibitors: targeting macromolecular complexes. Nat. Rev. Drug Discovery 2012, 11, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Thomas A; Pommier Y. Targeting topoisomerase I in the era of precision medicine. Clin. Cancer Res 2019, 25, 6581–6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Maede Y; Shimizu H; Fukushima T; Kogame T; Nakamura T; Miki T; Takeda S; Pommier Y; Murai J. Differential and common DNA repair pathways for topoisomerase I- and II-targeted drugs in a genetic DT40 repair cell screen panel. Mol. Cancer Ther 2014, 13, 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sun Y; Saha LK; Saha S; Jo U; Pommier Y. Debulking of topoisomerase DNA-protein crosslinks (TOP-DPC) by the proteasome, non-proteasomal and non-proteolytic pathways. DNA Repair 2020, 94, 102926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sun Y; Saha S; Wang W; Saha LK; Huang SN; Pommier Y. Excision repair of topoisomerase DNA-protein crosslinks (TOPDPC). DNA Repair 2020, 89, 102837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Yang SW; Burgin AB Jr.; Huizenga BN; Robertson CA; Yao KC; Nash HA A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc. Natl. Acad. Sci. U. S. A 1996, 93, 11534–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Pouliot JJ; Robertson CA; Nash HA Pathways for repair of topoisomerase I covalent complexes in Saccharomyces cerevisiae. Genes Cells 2001, 6, 677–687. [DOI] [PubMed] [Google Scholar]
- (12).Interthal H; Pouliot JJ; Champoux JJ The tyrosyl-DNA phosphodiesterase tdp1 is a member of the phospholipase D superfamily. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 12009–12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Debethune L; Kohlhagen G; Grandas A; Pommier Y. Processing of nucleopeptides mimicking the topoisomerase I-DNA covalent complex by tyrosyl-DNA phosphodiesterase. Nucleic Acids Res. 2002, 30, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhang YW; Regairaz M; Seiler JA; Agama KK; Doroshow JH; Pommier Y. Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res. 2011, 39, 3607–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Das BB; Antony S; Gupta S; Dexheimer TS; Redon CE; Garfield S; Shiloh Y; Pommier Y. Optimal function of the DNA repair enzyme tdp1 requires its phosphorylation by ATM and/or DNA-PK. EMBO J. 2009, 28, 3667–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dexheimer TS; Antony S; Marchand C; Pommier Y. Tyrosyl-DNA phosphodiesterase as a target for anticancer therapy. Anti-Cancer Agents Med. Chem 2008, 8, 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Saha LK; Wakasugi M; Akter S; Prasad R; Wilson SH; Shimizu N; Sasanuma H; Huang SN; Agama K; Pommier Y; Matsunaga T; Hirota K; Iwai S; Nakazawa Y; Ogi T; Takeda S. Topoisomerase I-driven repair of UV-induced damage in NER-deficient cells. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 14412–14420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Barthelmes HU; Habermeyer M; Christensen MO; Mielke C; Interthal H; Pouliot JJ; Boege F; Marko D. TDP1 overexpression in human cells counteracts DNA damage mediated by topoisomerases I and II. J. Biol. Chem 2004, 279, 55618–55625. [DOI] [PubMed] [Google Scholar]
- (19).Meisenberg C; Ward SE; Schmid P; El-Khamisy SF TDP1/TOP1 ratio as a promising indicator for the response of small cell lung cancer to topotecan. J. Cancer Sci. Ther 2014, 6, 258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Murai J; Huang SY; Das BB; Dexheimer TS; Takeda S; Pommier Y. Tyrosyl-DNA phosphodiesterase 1 (TDP1) repairs DNA damage induced by topoisomerases I and II and base alkylation in vertebrate cells. J. Biol. Chem 2012, 287, 12848–12857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Interthal H; Chen HJ; Kehl-Fie TE; Zotzmann J; Leppard JB; Champoux JJ SCAN1 mutant tdp1 accumulates the enzyme-DNA intermediate and causes camptothecin hypersensitivity. EMBO J. 2005, 24, 2224–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hawkins AJ; Subler MA; Akopiants K; Wiley JL; Taylor SM; Rice AC; Windle JJ; Valerie K; Povirk LF In vitro complementation of tdp1 deficiency indicates a stabilized enzyme-DNA adduct from tyrosyl but not glycolate lesions as a consequence of the SCAN1 mutation. DNA Repair 2009, 8, 654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Katyal S; El-Khamisy SF; Russell HR; Li Y; Ju L; Caldecott KW; McKinnon PJ TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J. 2007, 26, 4720–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hirano R; Interthal H; Huang C; Nakamura T; Deguchi K; Choi K; Bhattacharjee MB; Arimura K; Umehara F; Izumo S; Northrop JL; Salih MA; Inoue K; Armstrong DL; Champoux JJ; Takashima H; Boerkoel CF Spinocerebellar ataxia with axonal neuropathy: consequence of a tdp1 recessive neomorphic mutation? EMBO J. 2007, 26, 4732–4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhang XR; Wang HW; Tang WL; Zhang Y; Yang H; Hu DX; Ravji A; Marchand C; Kiselev E; Ofori-Atta K; Agama K; Pommier Y; An LK Discovery, synthesis, and evaluation of oxynitidine derivatives as dual inhibitors of DNA topoisomerase IB (TOP1) and tyrosyl-DNA phosphodiesterase 1 (TDP1), and potential antitumor agents. J. Med. Chem 2018, 61, 9908–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Komarova AO; Drenichev MS; Dyrkheeva NS; Kulikova IV; Oslovsky VE; Zakharova OD; Zakharenko AL; Mikhailov SN; Lavrik OI Novel group of tyrosyl-DNA-phosphodiesterase 1 inhibitors based on disaccharide nucleosides as drug prototypes for anti-cancer therapy. J. Enzyme Inhib. Med. Chem 2018, 33, 1415–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zakharenko A; Luzina O; Koval O; Nilov D; Gushchina I; Dyrkheeva N; Svedas V; Salakhutdinov N; Lavrik O. Tyrosyl-DNA Phosphodiesterase 1 Inhibitors: Usnic acid enamines enhance the cytotoxic effect of camptothecin. J. Nat. Prod 2016, 79, 2961–2967. [DOI] [PubMed] [Google Scholar]
- (28).Kovaleva K; Oleshko O; Mamontova E; Yarovaya O; Zakharova O; Zakharenko A; Kononova A; Dyrkheeva N; Cheresiz S; Pokrovsky A; Lavrik O; Salakhutdinov N. Dehydroabietylamine ureas and thioureas as tyrosyl-DNA phosphodiesterase 1 inhibitors that enhance the antitumor effect of Temozolomide on glioblastoma cells. J. Nat. Prod 2019, 82, 2443–2450. [DOI] [PubMed] [Google Scholar]
- (29).Gladkova ED; Nechepurenko IV; Bredikhin RA; Chepanova AA; Zakharenko AL; Luzina OA; Ilina ES; Dyrkheeva NS; Mamontova EM; Anarbaev RO; Reynisson J; Volcho KP; Salakhutdinov NF; Lavrik OI The first berberine-based inhibitors of tyrosyl-DNA phosphodiesterase 1 (Tdp1), an important DNA repair enzyme. Int. J. Mol. Sci 2020, 21, 7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zakharenko AL; Luzina OA; Sokolov DN; Kaledin VI; Nikolin VP; Popova NA; Patel J; Zakharova OD; Chepanova AA; Zafar A; Reynisson J; Leung E; Leung IKH; Volcho KP; Salakhutdinov NF; Lavrik OI Novel tyrosyl-DNA phosphodiesterase 1 inhibitors enhance the therapeutic impact of topotecan on in vivo tumor models. Eur. J. Med. Chem 2019, 161, 581–593. [DOI] [PubMed] [Google Scholar]
- (31).Comeaux EQ; van Waardenburg RC Tyrosyl-DNA phosphodiesterase I resolves both naturally and chemically induced DNA adducts and its potential as a therapeutic target. Drug Metab. Rev 2014, 46, 494–507. [DOI] [PubMed] [Google Scholar]
- (32).Chiang SC; Meagher M; Kassouf N; Hafezparast M; McKinnon PJ; Haywood R; El-Khamisy SF Mitochondrial protein-linked DNA breaks perturb mitochondrial gene transcription and trigger free radical-induced DNA damage. Science Adv. 2017, 3, No. e1602506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Huang SY; Murai J; Dalla Rosa I; Dexheimer TS; Naumova A; Gmeiner WH; Pommier Y. TDP1 repairs nuclear and mitochondrial DNA damage induced by chain-terminating anticancer and antiviral nucleoside analogs. Nucleic Acids Res. 2013, 41, 7793–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Huang SN; Pommier Y; Marchand C. Tyrosyl-DNA phosphodiesterase 1 (Tdp1) inhibitors. Expert Opin. Ther. Pat 2011, 21, 1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Takagi M; Ueda JY; Hwang JH; Hashimoto J; Izumikawa M; Murakami H; Sekido Y; Shin-Ya K. Tyrosyl-DNA phosphodiesterase 1 inhibitor from an anamorphic fungus. J. Nat. Prod 2012, 75, 764–767. [DOI] [PubMed] [Google Scholar]
- (36).Laev SS; Salakhutdinov NF; Lavrik OI Tyrosyl-DNA phosphodiesterase inhibitors: progress and potential. Bioorg. Med. Chem 2016, 24, 5017–5027. [DOI] [PubMed] [Google Scholar]
- (37).Baglini E; Salerno S; Barresi E; Robello M; Da Settimo F; Taliani S; Marini AM Multiple topoisomerase I (TopoI), topoisomerase II (TopoII) and tyrosyl-DNA phosphodiesterase (TDP) inhibitors in the development of anticancer drugs. Eur. J. Pharm. Sci 2021, 156, 105594. [DOI] [PubMed] [Google Scholar]
- (38).Zakharenko A; Dyrkheeva N; Lavrik O. Dual DNA topoisomerase 1 and tyrosyl-DNA phosphodiesterase 1 inhibition for improved anticancer activity. Med. Res. Rev 2019, 39, 1427–1441. [DOI] [PubMed] [Google Scholar]
- (39).Cardamone F; Pizzi S; Iacovelli F; Falconi M; Desideri A. Virtual screening for the development of dual-inhibitors targeting topoisomerase IB and tyrosyl-DNA phosphodiesterase 1. Curr. Drug Targets 2017, 18, 544–555. [DOI] [PubMed] [Google Scholar]
- (40).Nguyen TX; Morrell A; Conda-Sheridan M; Marchand C; Agama K; Bermingam A; Stephen AG; Chergui A; Naumova A; Fisher R; O’Keefe BR; Pommier Y; Cushman M. Synthesis and biological evaluation of the first dual tyrosyl-DNA phosphodiesterase I (Tdp1)-topoisomerase I (Top1) inhibitors. J. Med. Chem 2012, 55, 4457–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Wang P; Elsayed MSA; Plescia CB; Ravji A; Redon CE; Kiselev E; Marchand C; Zeleznik O; Agama K; Pommier Y; Cushman M. Synthesis and biological evaluation of the first triple inhibitors of human topoisomerase 1, tyrosyl-DNA phosphodiesterase 1 (Tdp1), and tyrosyl-DNA phosphodiesterase 2 (Tdp2). J. Med. Chem 2017, 60, 3275–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zhang HL; Zhang Y; Yan XL; Xiao LG; Hu DX; Yu Q; An LK Secondary metabolites from Isodon ternifolius (D. Don) Kudo and their anticancer activity as DNA topoisomerase IB and tyrosyl-DNA phosphodiesterase 1 inhibitors. Bioorg. Med. Chem 2020, 28, 115527. [DOI] [PubMed] [Google Scholar]
- (43).Tang WL; Zhang Y; Hu DX; Yang H; Yu Q; Chen JW; Agama K; Pommier Y; An LK Synthesis and biological evaluation of 5-aminoethyl benzophenanthridone derivatives as DNA topoisomerase IB inhibitors. Eur. J. Med. Chem 2019, 178, 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Achkar IW; Mraiche F; Mohammad RM; Uddin S. Anticancer potential of sanguinarine for various human malignancies. Future Med. Chem 2017, 9, 933–950. [DOI] [PubMed] [Google Scholar]
- (45).Walterova D; Ulrichova J; Valka I; Vicar J; Vavreckova C; Taborska E; Harjrader RJ; Meyer DL; Cerna H; Simanek V. Benzo[c]phenanthridine alkaloids sanguinarine and chelerythrine: biological activities and dental care applications. Acta Univ. Palacki. Olomuc. Fac. Med 1995, 139, 7–16. [PubMed] [Google Scholar]
- (46).Zielinska S; Jezierska-Domaradzka A; Wojciak-Kosior M; Sowa I; Junka A; Matkowski AM Greater celandine’s ups and downs-21 centuries of medicinal uses of chelidonium majus from the viewpoint of today’s pharmacology. Front. Pharmacol 2018, 9, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Tanahashi T; Zenk MH New hydroxylated benzo[c]-phenanthridine alkaloids from Eschscholtzia californica cell suspension cultures. J. Nat. Prod 1990, 53, 579–586. [DOI] [PubMed] [Google Scholar]
- (48).Sato H; Yamada R; Yanagihara M; Okuzawa H; Iwata H; Kurosawa A; Ichinomiya S; Suzuki R; Okabe H; Yano T; Kumamoto T; Suzuki N; Ishikawa T; Ueno K. New 2-aryl-1,4-naphthoquinone-1-oxime methyl ether compound induces microtubule depolymerization and subsequent apoptosis. J. Pharmacol. Sci 2012, 118, 467–478. [DOI] [PubMed] [Google Scholar]
- (49).Nakanishi T; Suzuki M. Revision of the structure of fagaridine based on the comparison of UV and NMR data of synthetic compounds. J. Nat. Prod 1998, 61, 1263–1267. [DOI] [PubMed] [Google Scholar]
- (50).Castillo D; Sauvain M; Rivaud M; Jullian V. In vitro and in vivo activity of benzo[c]phenanthridines against Leishmania amazonensis. Planta Med. 2014, 80, 902–906. [DOI] [PubMed] [Google Scholar]
- (51).Stermitz FR; Gillespie JP; Amoros LG; Romero R; Stermitz TA; Larson KA; Earl S; Ogg JE Synthesis and biological activity of some antitumor benzophenanthridinium salts. J. Med. Chem 1975, 18, 708–713. [DOI] [PubMed] [Google Scholar]
- (52).Larsen AK; Grondard L; Couprie J; Desoize B; Comoe L; Jardillier JC; Riou JF The antileukemic alkaloid fagaronine is an inhibitor of DNA topoisomerases I and II. Biochem. Pharmacol 1993, 46, 1403–1412. [DOI] [PubMed] [Google Scholar]
- (53).An LK; Zhang XR; Wang HW; Pommier Y; Kiselev E; Ravji A; Agama K. Preparation of oxynitidine derivatives useful as dual inhibitors of DNA topoisomerase IB (TOP1) and tyrosyl-DNA phosphodiesterase 1 (TDP1). U.S. Patent WO 2020023700 A2, Jan 30, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).An LK; Zhang XR; Wang HW; Tang WL; Hu DX; Zhang Y. Preparation of fused ring compounds as antitumor agents. China Patent CN 110759963 A, Feb 7, 2020. [Google Scholar]
- (55).Ruchelman AL; Singh SK; Wu X; Ray A; Yang JM; Li TK; Liu A; Liu LF; LaVoie EJ Diaza- and triazachrysenes: potent topoisomerase-targeting agents with exceptional antitumor activity against the human tumor xenograft, MDA-MB-435. Bioorg. Med. Chem. Lett 2002, 12, 3333–3336. [DOI] [PubMed] [Google Scholar]
- (56).Dexheimer TS; Pommier Y. DNA cleavage assay for the identification of topoisomerase I inhibitors. Nat. Protoc 2008, 3, 1736–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Staker BL; Hjerrild K; Feese MD; Behnke CA; Burgin AB Jr.; Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 15387–15392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Dean RA; Fam HK; An J; Choi K; Shimizu Y; Jones SJ; Boerkoel CF; Interthal H; Pfeifer TA Identification of a putative tdp1 inhibitor (CD00509) by in vitro and cell-based assays. J. Biomol. Screening 2014, 19, 1372–1382. [DOI] [PubMed] [Google Scholar]
- (59).Davies DR; Interthal H; Champoux JJ; Hol WG Insights into substrate binding and catalytic mechanism of human tyrosyl-DNA phosphodiesterase (Tdp1) from vanadate and tungstate-inhibited structures. J. Mol. Biol 2002, 324, 917–932. [DOI] [PubMed] [Google Scholar]
- (60).Zhao XZ; Kiselev E; Lountos GT; Wang W; Tropea JE; Needle D; Hilimire TA; Schneekloth JS; Waugh DS; Pommier Y; Burke TR Small molecule microarray identifies inhibitors of tyrosyl-DNA phosphodiesterase 1 that simultaneously access the catalytic pocket and two substrate binding sites. Chem. Sci 2021, 12, 3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.