

Summary
Understanding chromosome recombination behavior in polyploidy species is key to advancing genetic discoveries. In blueberry, a tetraploid species, the line of evidences about its genetic behavior still remain poorly understood, owing to the inter‐specific, and inter‐ploidy admixture of its genome and lack of in depth genome‐wide inheritance and comparative structural studies.
Here we describe a new high‐quality, phased, chromosome‐scale genome of a diploid blueberry, clone W85. The genome was integrated with cytogenetics and high‐density, genetic maps representing six tetraploid blueberry cultivars, harboring different levels of wild genome admixture, to uncover recombination behavior and structural genome divergence across tetraploid and wild diploid species.
Analysis of chromosome inheritance and pairing demonstrated that tetraploid blueberry behaves as an autotetraploid with tetrasomic inheritance. Comparative analysis demonstrated the presence of a reciprocal, heterozygous, translocation spanning one homolog of chr‐6 and one of chr‐10 in the cultivar Draper. The translocation affects pairing and recombination of chromosomes 6 and 10. Besides the translocation detected in Draper, no other structural genomic divergences were detected across tetraploid cultivars and highly inter‐crossable wild diploid species.
These findings and resources will facilitate new genetic and comparative genomic studies in Vaccinium and the development of genomic assisted selection strategy for this crop.
Keywords: autopolyploid, blueberry (Vaccinium corymbosum L.), centromeric repeat, chromosomal translocation, chromosome structure, phased genome, polyploid genetic behavior
Introduction
Blueberries (Vaccinium spp.) are well‐recognized as a rich source of health‐promoting phytochemicals, which have contributed to a rapid increase in consumer demand and production over the past two decades (Mengist et al., 2020a). Domesticated only c. 100 yr ago, tetraploid highbush (V. corymbosum; HB) is the most widely grown blueberry, accounting for vast majority of the US blueberry production (Mengist et al., 2020b). During the last four decades, several HB cultivars have been released by incorporating traits associated with environmental adaptation from closely related wild diploid species (Retamales & Hancock, 2018). As a result of these breeding efforts, HB are classified based on chilling requirement and winter hardiness, into northern highbush (NHB) and southern highbush (SHB) blueberries. Development of these SHB and NHB varieties contributed to expansion of the geographic area of production of HB across the US and globally.
Development of marker‐assisted breeding (MAB) capacity to accelerate the selection process and ‘pyramid’ multiple traits are primary goals of blueberry breeding programs (Retamales & Hancock, 2018). To that end, in recent years genomic resources and genetic studies in blueberry have expanded. The first blueberry reference genome was developed using a wild diploid blueberry species, V. caesariense (clone W85‐20) a.k.a diploid V. corymbosum, since it is morphologically similar to HB (Bruederle & Vorsa, 1994; Gupta et al., 2015). Recently, a phased assembly of a tetraploid NHB cultivar Draper was released and is being used as a reference for multiple genetic and genomic studies (Colle et al., 2019). Despite these advances, the contiguity of the V. caesariense and Draper genome assemblies was still relatively low (contig N50 < 39 kb), and the V. caesariense genome was not assembled at the chromosome (chr) level.
Understanding the chromosome recombination behavior (disomic or polysomic inheritance) of polyploid species like HB is another critical step to optimize genetic studies and improve the efficiency of MAB. In HB, studies based on the segregation ratios of certain traits and a small number of molecular markers suggested that blueberry behaves as an autotetraploid (Qu & Hancock, 2001). However, some early cytogenetic studies suggested allopolyploid behavior (Jelenkovic & Hough, 1970). Recently, comparative genomic analysis in Draper unveiled potential difference among homologous chromosomes and signature of subgenome differentiation suggesting possible allopolyploid origin (Colle et al., 2019). Given the complex inter‐specific, and inter‐ploidy genetic admixture existing within this taxon, it is possible that full or partial allopolyploid genetic behavior could exist among blueberry cultivars. New high‐throughput genotyping and genomic resources developed for blueberry, with new statistical methods to study polyploidy genetics, have opened new opportunities to further address these questions. Despite these advances, the question of whether blueberry behaves as a fully autopolyploid, allopolyploid or a segmental allopolyploid remains unsolved. Additionally, chromosomal structural differences in tetraploid blueberry and their impact on chromosome inheritance remain to be assessed and verified.
To continue building upon recent advances in blueberry genetics and genomics, here we developed and used a high‐quality, phased, chromosome‐scale assembly of V. caesariense, cytogenetics and genetic linkage maps to investigate HB polyploid recombination behavior, and conservation of chromosomal structural features such as centromeric repeats and rearrangements, that can affect chromosome pairing and recombination in tetraploid blueberry.
Materials and Methods
Plant material, sequencing and genome assembly
Vaccinium caesariense clone, W85‐20 (referred as W85 hereafter), was used for whole‐genome sequencing (Supporting Information Methods S1). Sequences included 53 Gb PacBio data (80× physical coverage), 13 Gb (41.7× physical coverage) and 153 Gb (469.5× physical coverage) Illumina sequences from the Chicago and the Hi‐C libraries, respectively (Table S1). In addition, 13.3 Gb PacBio Iso‐Seq and 247 Gb Illumina sequencing data representing transcripts from multiple tissues including leaf, stem, flowers, flower buds and fruit tissues were generated (Table S1).
A de novo assembly was developed using Falcon assembler v.0.3.0 (Chin et al., 2016) and contigs were subsequently polished with Pilon v.1.24 (Walker et al., 2014) using the Hi‐C and Chicago Illumina short reads as input (Methods S2). Chicago and Hi‐C data were used for phasing the genome using Falcon Phase v.2.0 (Kronenberg et al., 2021). The resulting haplotype‐phased contigs were used with the Hi‐C and Chicago sequences for scaffolding with the HighRise pipeline (Putnam et al., 2016), independently for each haplotype. A linkage map representing the W85 genome, including 17 486 single‐nucleotide polymorphism (SNP) markers (Qi et al., 2021), was used to anchor the genome. Marker sequences were aligned against the W85 phased assemblies using Bwa‐aln v.0.7.17 (default parameters) (Li & Durbin, 2009). During this process, the Hi‐C and Chicago data interaction heat map in conjunction with the linkage map were used to identify and correct chimeric regions (Methods S2).
Repetitive sequence annotation and analysis
The repeat library and its annotation were built using Edta v.1.9.7 (Ou et al., 2019), which performed a de novo transposable element (TE) annotation by integrating structure‐ and homology‐based approaches. Default parameters were used in all cases except for the usage of the genome's CDS Fasta to aid in annotation, and the –sensitive 1, parameter that enables the use of RepeatModeler v.2.0 to identify any remaining TEs. The quality of the W85 assembly in terms of contiguity of repeat space was assessed using the Lai v.2.9 (Ou et al., 2018) deployed in the Ltr_retriever v.2.9 (Ou & Jiang, 2018) (Methods S3).
One million random read pairs of W85 (NCBI accession no. SRR837868) were analyzed using Tarean (Novák et al., 2017) to identify potential satellite DNA sequences (Methods S3).
Gene prediction and annotation
Gene prediction was performed using Maker v.3.01.03 (Cantarel et al., 2008) by integrating ab initio gene prediction and evidence‐based predictions and was performed independently on the two haplotypes. Illumina reads from transcriptome data (Table S1) were mapped to the W85 p0 and p1 assemblies using Star v.2.7.10a (Dobin et al., 2012), and StringTie (Pertea et al., 2016). The PacBio Iso‐Seq reads were processed using the IsoSeq3 pipeline (https://github.com/PacificBiosciences/IsoSeq ). High quality Iso‐Seq sequences were mapped against the W85 genome assembly using Gmap v.2021‐12‐17 (Wu & Watanabe, 2005). The resulting GFF3 files were used for gene model prediction and correction of the ab initio predictions.
The repeat library was used as the input for Maker to mask repetitive sequences. Ab initio gene prediction was performed using Augustus v.2.5.5 (Stanke & Waack, 2003; Stanke et al., 2006) and Snap v.2006‐07‐28 (Korf, 2004), that were trained using sets of high‐quality predicted genes, high‐quality Iso‐Seq full‐length transcripts and Illumina data (Table S1). Maker was then run‐in empirical mode with three iterations to identify the best gene models by integrating ab initio predictions and experimental evidence.
The quality and completeness of the predicted gene models were assessed using Busco v.5.3.2 (Manni et al., 2021) plant dataset (odb10). The putative function of the predicted genes was annotated using GenBank nonredundant (nr) protein and InterPro databases. Blastx was used to compare the predicted coding sequences (CDS) (e‐value ≤ 1e−10) with the nr database (Sayers et al., 2019). Blast2GO v.1.4.11 (Götz et al., 2008) was used to annotate the gene ontology (GO) terms of genes. The protein domains were annotated using InterProScan v.5.32.71.0 (Jones et al., 2014) based on all available protein databases. Resistance genes (R‐genes) were identified using Drago2 (Osuna‐Cruz et al., 2018). Infernal v.1.1.2 (Nawrocki et al., 2009) software was used for the in silico prediction of microRNAs (miRNAs) and small nuclear RNAs (snRNAs) in the assembled genome, implementing the cmsearch function against Rfam database (v.13.0).
Haplotype comparison and diversity analysis
To identify structural variants (SVs), SNPs and insertion/deletion polymorphism (indels) between the two haplotypes, syntenic blocks were identified using MCScanX v.1.1.11 (Wang et al., 2012). The homologous syntenic pairs were identified based on the following criteria: (1) paired regions must be on homologous haplotypes, (2) the length of each homologous haplotype should be less than three times the length of its counterpart and (3) aligned regions must cover over 50% of the whole region. One gene and its best homologous gene on the complementary haplotype were considered as homologous gene pair.
Single‐nucleotide polymorphisms and indels between homologous syntenic blocks were identified using lastz software (Harris, 2007) with the parameters ‐‐chain ‐‐format = diff ‐‐matchcount = 3000 ‐‐rdotplot ‐‐strand = plus/minus ‐‐ambiguous = n, and annotated using SnpEff v.5.1 (Cingolani et al., 2012).
For SVs identification, the two haplotypes of a chromosome were aligned using MUMmer4 (Marçais et al., 2018) with show‐diff function. Present Absent Variation (PAV) genes were identified as those genes located within as syntenic region, lacking a homolog at the complementary haplotype, while its surrounding genes had homologs that were collinear between two haplotypes. Gene enrichment analysis of PAV genes were analyzed using AgriGO v.2.0 (Tian et al., 2017) using a Fisher test and false discovery rate (FDR) < 0.05 and custom annotation.
Linkage map construction
Three mapping populations named DS × J (n = 196), R × A (n = 346) and D × B (n = 168), were used for linkage map construction. Draper (D) and Draper Selection‐44392 (hereafter DS) are NHB selections/cultivars, whereas Arlen (A), Jewel (J), Biloxi (B) and Reveille (R) are SHB cultivars with 10–60% of their genome represented by wild species (Methods S4). Total genomic DNA from individual plants was extracted using CTAB method (Panta et al., 2004). For DS × J and R × A genotyping was performed using a sequence capture technology at RAPiD Genomics (Gainesville, FL, USA) targeting 31 063, and 10 000 genomic regions (probes), respectively. For D × B, genotyping was performed using Genotyping by Sequencing (GBS) (Methods S4).
High‐quality reads were mapped against the W85_v2 p0 assembly using Bwa‐aligner v.0.7.17 (Li & Durbin, 2009). Uniquely mapped reads were used to call SNPs. For R × A and DS × J, SNP calling was carried out using Freebayes v.1.3.4 (Garrison & Marth, 2012), and filtered with the following criteria: (1) minimum mapping quality of 20; (2) mean depth of coverage of 50; (3) maximum missing data of 10% across SNPs and individuals; (4) only biallelic loci using vcftools v.0.1.16 (Danecek et al., 2011). For D × B, SNP calling was performed using the Tassel‐Gbs pipeline v.2 (Glaubitz et al., 2014), with a mean read depth of eight, maximum missing data of 20% across SNPs and only biallelic loci. The tetraploid allele dosages were called based on the read depth counts and ‘F1’ model using updog R package v.2.1.1 (Gerard et al., 2018). The genotypes were coded as 0 for nulliplex (AAAA), 1 for simplex (AAAB), 2 for duplex (AABB), 3 for triplex (ABBB), and 4 for quadruplex (BBBB).
The linkage maps were constructed per parent using PolymapR v.1.1.2 (Bourke et al., 2018) (Methods S4). The markers representing each set of four homologous chromosomes were combined to develop an integrated linkage map. Meiotic recombination rate was estimated using loess smoothing with a span of 0.4 using MareyMap v.1.3.6 (Rezvoy et al., 2007).
Estimation of double reduction, quadrivalent and preferential chromosome pairing
The rate of double reduction (DR) and quadrivalent chromosome pairing was estimated using TetraOrigin software (Zheng et al., 2016), implemented in Mathematica v.11 (Wolfram Research Inc., Champaign, IL, USA). For a given marker, the probability of DR rate was averaged over the number of offspring on parental meiosis. The quadrivalent chromosome pairing was calculated by dividing the number of offspring with the quadrivalent pairing by the total number of offspring in the mapping population.
Preferential pairing was tested based on linked simplex × nulliplex (S × N) markers in repulsion phase using polymapR (Bourke et al., 2018). The analysis was performed setting a minimum distance between markers at 0.25, 0.5 and 1 cM. This analysis established the basis of a binomial test (H0:rdisom ≥ 1/3), and corrected for multiple testing using the FDR with α = 0.05 (Benjamini & Hochberg, 1995).
tetraorigin (Zheng et al., 2016) was used to estimate the most likely bivalent pairing in each individual (Methods S5). To estimate preferential pairing using a bivalent pairing model, both bivalentPhasing and bivalentDecoding were set to ‘True’. A χ2 test was performed on the counts of each class on parental basis to examine deviations from one‐third (P < 0.001) as previously described (Bourke et al., 2017).
The meiotic pairing behavior of the 4× varieties Arlen, DS, Draper and Jewel was also evaluated cytologically, by examining at least 40 pollen mother cells at diakinesis‐metaphase I in each variety.
Fluorescence in situ hybridization (FISH) analysis
Selected satellites of W85 were analyzed by fluorescence in situ hybridization (FISH) using young flower buds of the W85, V. darrowii and of the tetraploids DS, Draper and Jewel, according to published procedures (Iovene et al., 2008). Oligonucleotide probes and PCR primers were designed using the consensus sequences of the repeats (Methods S6).
Results
Phased genome assembly revealed high allelic diversity
The W85_v2 assembled haplotypes span 703 Mb (phase 0, p0) and 643 Mb (phase 1, p1), accounting for 108% and 99% of the estimated haploid genome size (651 ± 58 Mb) (Costich et al., 1993), and with a contig N50 length of 424 and 370 kb, respectively (Tables 1, S2). The Hi‐C and Chicago sequences along with a W85 linkage map (Qi et al., 2021) were used to anchor > 97.1% of both p0 and p1 assembled haplotypes, and to correct chimeric sequences (Fig. S1; Tables S3, S4). After this process, the linkage map was collinear to the 12 chromosomes of each phase and the Hi‐C heatmap showed a uniform distribution of genomic interactions along the diagonal (Figs S2, S3).
Table 1.
Summary statistics for genome assembly and annotation.
| Phase 0 | Phase 1 | |
|---|---|---|
| Genome assembly | ||
| Estimated genome size (Mb) a | 651 ± 58 | 651 ± 58 |
| Assembled genome size (Mb) | 703 | 643 |
| Fraction of genome covered (%) | 108 | 99 |
| Scaftigs (contigs) | ||
| Number of sequences | 3234 | 3212 |
| N50 size (kb) | 425 | 370 |
| L50 (kb) | 464 | 480 |
| Maximum sequence length (kb) | 2844 | 2509 |
| Minimum sequence length (bp) | 416 | 77 |
| Scaffolds | ||
| Number of scaffold | 273 | 270 |
| N50 size (kb) | 56 354 | 52 472 |
| Maximum sequence length (kb) | 66 558 | 60 760 |
| Minimum sequence length (kb) | 13.32 | 5.99 |
| GC content (%) | 35.81 | 37.48 |
| Genome annotation | ||
| Repeat content (%) | 45.3 | 45.3 |
| Number of protein‐coding genes | 34 895 | 33 183 |
| Average gene length (bp) | 4524 | 4554 |
| Average exon length (bp) | 235 | 238 |
| Average intron length (bp) | 770 | 770 |
| Average number of exons per gene | 5.3 | 5.3 |
Over 99.7% Illumina and PacBio Iso‐Seq transcriptome sequences aligned to the p0 and p1 assemblies, respectively (Tables S5, S6). The Busco score was > 99% (Table S7) and the long terminal repeat (LTR) assembly index (LAI) was 13.3, which is within the range of high‐quality assembly (Ou et al., 2018). These results demonstrated that the W85_v2 genome assembly covers the majority of gene space and assembled complex repetitive elements (Fig. 1).
Fig. 1.
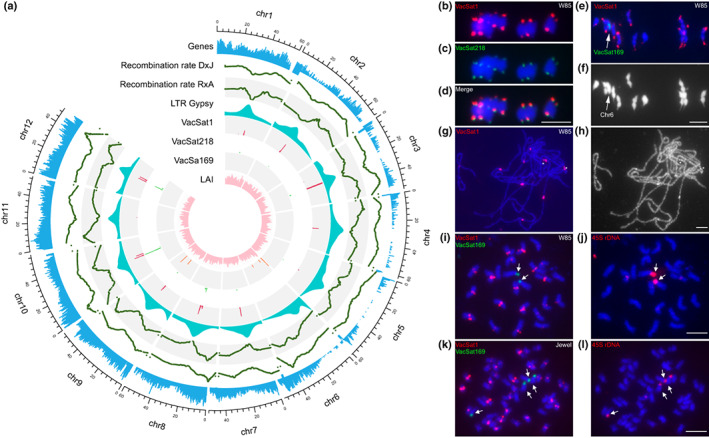
Chromosomal features of Vaccinium caesariense W85 genome (2n = 2x = 24) and representative V. corymbosum tetraploid blueberry cultivars (2n = 4x = 48). (a) Circular multi‐track plot illustrating the following chromosomal features: gene density, LTR/gypsy density, satellite repeat VacSat1, VacSat218 and VacSat169 and genome wide recombination rates estimated from two mapping populations (DS × J and R × A). The density of each feature was estimated considering a 200 kb window of W85_v2 genome assembly p0; b–l) Fluorescence in situ hybridization (FISH) of satellite repeats and 45S rDNA on the chromosomes of W85 (2n = 2x = 24) and Jewel (2n = 4x = 48). (b–d) Distribution of VacSat1 (b, red signals) and VacSat218 (c, green signals) on meiotic metaphase I chromosomes (in blue) of W85; (d) image merged from (b) and (c). (e, f) Localization of VacSat1 (red) and VacSat169 (green, arrow) on W85 meiotic metaphase I chromosomes (blue); (f) the chromosomes in (e) shown in gray scale to better visualize the location of VacSat1 on stretched terminal regions of most bivalents. (g) Localization of VacSat1 (red) on W85 meiotic pachytene chromosomes; (h) pachytene chromosomes in (g) shown in gray scale to enhance the visualization of the heterochromatic domains overlapped by VacSat1. Two chromosomes each had two adjacent VacSat1 signals separated by a short gap on unlabeled chromatin (asterisks). (i) Distribution of VacSat1 (red) and VacSat169 (green) on mitotic metaphase chromosomes (blue) of W85; VacSat169 is located on two chromosomes lacking VacSat1 signals (arrows); (j) the same chromosome plate used in (i) hybridized with 45S rDNA (red, arrows). (k) Localization of VacSat1 (red) and VacSat169 (green) on mitotic metaphase chromosomes of Jewel (2n = 4x = 48); VacSat169 is located on four chromosomes lacking VacSat1 signals (arrows); (l) the same chromosome plate used in (k) hybridized with 45S rDNA (red, arrows). Bar, 5 μm.
In the two haplotypes, 34 895 (p0) and 33 183 (p1) genes were predicted (Table S8) and > 97% were anchored to the 12 chromosomes. Over 95% of conserved orthologous angiosperm genes had a match with the predicted genes and > 97% were annotated (Tables S7, S9, S10). Repetitive sequences accounted for 45.3% of the genome (Table S11). The most abundant Class I TEs were long terminal repeat retrotransposons (LTR‐RTs), specifically the superfamily LTR/Gypsy, followed by LTR/Copia, while Tc1/Mariner DNA transposon was the most abundant class II TEs. Overall, the number and the structure of the predicted genes and fraction of repetitive sequences were similar to those predicted in the tetraploid Draper genome (32 139 genes/haplotype, 44.3% repeats) (Colle et al., 2019) (Table S12).
Haplotype comparison identified 372 syntenic blocks with an average intra‐genomic diversity of 1.66%, (Fig. 2). Between syntenic blocks, 3408 842 SNPs, 1188 547 indels, and 40 050 SVs (> 100 bp) were detected, and 358 and 75 SVs spanned > 100 kb and 1 Mb, respectively (Fig. S4; Table S13). Within syntenic blocks, 21 458 genes pairs (62% of all predicted genes) were identified as having syntenic orthologs on the two haplotypes, and 6157 genes were PAV genes. Among homologous pairs and PAV genes, 22 422 genes (52%) and 1521 genes (25%) harbored polymorphisms with moderate (e.g. missense variant) to high (e.g. stop gained) predicted impact effect on the protein coding regions (Cingolani et al., 2012), respectively (Table S14). PAV genes involved in defense and environmental adaption were enriched (Fig. S5), with c. 13% being R‐genes (Table S14).
Fig. 2.

Haplotype diversity in the Vaccinium caesariense W85 genome. The central blue bars represent the two haplotypes of chromosome 1. Haplotype on the left represent the phase 0 (p0) assembly and the one on the right represent the phase 1 (p1) assembly. The gray lines indicate paired allelic genes. The green, cyan, violet and red color bar plots indicate gene density, single‐nucleotide polymorphism density, indel density and the density of potential deleterious effect variants, respectively. All numbers were determined considering a 200 kb windows.
Satellite repeat analysis identified a putative centromeric repeat
Based on tandem repeat analysis using Tarean (Novák et al., 2017), four potential satellites (named VacSat1/Sat61/Sat218/Sat169), with different monomer length (from 101 to 480 bp), AT content (from > 80% to 45%) and genomic abundance were characterized (Methods S1; Table S15).
VacSat1 and VacSat218, with consensus monomer of 147 and 101 bp, respectively, represent variants of the same repeat family. Within PacBio reads, VacSat1 was organized in arrays of 147 bp adjacent monomers (Table S15). Conversely, VacSat218 motif occurred as few (< 5) monomers interspersed within VacSat1 arrays (Fig. S6). In silico and FISH mapping of VacSat1 and VacSat218 revealed their location in most W85 centromeric regions, which also displayed low recombination rate and high density of LTR Gypsy elements (Fig. 1a–h). At metaphase I, the hybridization signals of VacSat1/Sat218 were detected at the polar opposites of the bivalent chromosomes (Fig. 1b–f). These results indicated that most W85 centromeres are either directly associated with or immediately adjacent to VacSat1/Sat218 arrays. VacSat1 generated stronger FISH signals than VacSat218, reflecting their different abundance in the genome (Fig. S6). In addition, the intensity of VacSat1 FISH signals varied among chromosomes, with stronger signals in about half of the centromeres.
One chromosome pair, devoid of VacSat1/Sat218 sequences (Fig. 1a,i,j), was unambiguously identified as chr‐6 because it carries VacSat169 repeats at the end of the short arm (Fig. 1a,i,j). Bioinformatic and FISH analysis indicated that the VacSat169 repeats co‐localized with 18S‐25S rDNA (Figs 1a,i,j, S7). Analysis of PacBio reads revealed that VacSat169 is arranged in three to four adjacent monomers located in the intergenic spacer, close to the 5′ end of the 18S rRNA (Fig. S7). As expected, the region spanning VacSat169/rDNA on chr‐6 has a low recombination frequency (Fig. 1a) because the rDNA arrays are protected from meiotic recombination to ensure genome stability over generations (Sims et al., 2019).
VacSat61 was a repeat family located interstitially on most pseudomolecules with monomers of 235–238 bp and an underlying pattern of 115–118 bp sub‐monomers, suggesting the presence of a higher‐order repeat (HOR) (Methods S3; Fig. S8). FISH mapping of VacSat61 generated distinct signals on two to four chromosomes and weak signals on the rest (Fig. S8), confirming that this repeat family is less abundant than VacSat1. No clusters of telomeric repeats were detected by tarean. However, FISH using a typical plant telomeric motif (TTTAGGG)n labeled the ends of each W85 chromosome (Fig. S9). Overall, the FISH pattern of these repeats is consistent with their predicted location on the 12 pseudomolecules physical map, further supporting the quality of the W85_v2 genome assembly.
Centromeric repeat VacSat1 is conserved across diploid and tetraploid blueberry species
For comparison among highly inter‐fertile species belonging to Vaccinium section Cyanococcus, we evaluated the sequence diversity and distribution of the VacSat1 repeats between diploid species (V. darrowii and W85), and tetraploid HB cultivars, Draper and Jewel. Bilberry (V. myrtillus), belonging to the section V. myrtillus, was used as an outgroup.
The sequences of 300 random VacSat1 monomers from Draper, W85, V. darrowii and bilberry genomes were aligned (Colle et al., 2019; Wu et al., 2021). The monomers from the Draper, V. darrowii and W85 shared high similarity (> 99%) and could not be separated based on phylogenetic analysis (Fig. 3a,b). In contrast, bilberry monomers clustered separately and had a lower similarity (c. 97%) relative to W85, V. darrowii and Draper monomers.
Fig. 3.
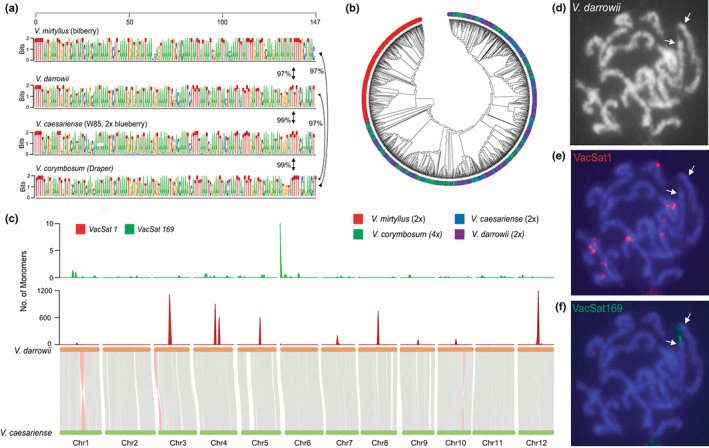
Comparative analysis of putative satellite centromeric repeat VacSat1 (147 bp monomer) in Vaccinium species. (a) VacSat1 sequence logo representing monomers extracted from four Vaccinium species, Draper (tetraploid, V. corymbosum), W85 (diploid, V. caesariense), bilberry (diploid, V. myrtillus) and evergreen blueberry (diploid, V. darrowii). Numbers next to the arrows indicate the average percent similarity of VacSat1 monomers across species estimated using the Maximum Composite Likelihood. (b) Phylogenetic analysis of VacSat1 monomer sequences inferred using the Neighbor‐Joining method implemented in Mega11 (Tamura et al., 2021). For the sequence logo and phylogenetic analysis, 300 VacSat1 monomer sequences/species were used. (c) Synteny analysis between W85_v2 against V. darrowii genome and distribution of VacSat1 and VacSat169 in V. darrowii physical maps (1 Mb windows). (d–f) Localization of VacSat1 and VacSat169 repeats on V. darrowii chromosomes using fluorescence in situ hybridization (FISH). (d) A somatic metaphase chromosomes of V. darrowii hybridized with (e) VacSat1 (red signals) and (f) VacSat169 (green). Arrows (in d and e) indicate the chromosomes with a terminal VacSat169 signal and no detectable VacSat1 repeats.
The FISH distribution of VacSat1 and VacSat169 in the tetraploids Jewel and DS was comparable to that of W85. Overall, the FISH signals in the tetraploids were twice as many as those observed for W85 (Figs 1k,l, S10). A minor difference in DS was a weaker VacSat169 signal on one chr‐6 homolog compared to the others, likely due to a lower amount of the repeat (Fig. S10a–c). Nevertheless, at metaphase I in both varieties, the four chromosomes with VacSat169 signals formed either two bivalents or a single quadrivalents (see also earlier; Fig. S10). Similarly, in silico and FISH analysis in V. darrowii highlighted a conserved and comparable distribution of VacSat1 and VacSat169 (Fig. 3c). These results demonstrated that VacSat1 is highly conserved among closely related species belonging to the Vaccinium section Cyanococcus.
Comparative analysis between W85 and tetraploid genomes highlights a reciprocal translocation in the Draper genome
For comparative structural analysis between the W85 and tetraploid genomes, two high‐density linkage maps were constructed using two F1 mapping populations named DS × J and R × A. The DS × J and R × A maps included 29 236 SNPs and 79 362 SNPs and spanned 1484 and 1499.9 cM, respectively (Fig. 4a,b; Tables S16, S17). The marker density across the eight homologs (h1–h8) of each linkage group was very high and evenly distributed (Figs 4a,b, S11, S12; Tables S16, S17).
Fig. 4.
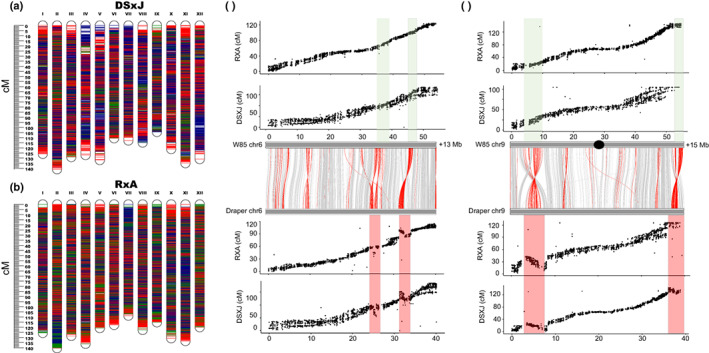
Development of ultra‐dense linkage maps and comparative analysis between Vaccinium caesariense diploid genome (W85) and V. corymbosum tetraploid genome (Draper). (a, b) Integrated linkage maps representing the DS × J (a) and R × A (b) mapping populations. Single‐nucleotide polymorphism positions are marked in red for markers representing Arlen (A) and Draper Selection‐44392 (DS), green for marker representing Reveille (R) and Jewel (J) and blue for markers that are common to the two parents. (c, d) x–y plots represent the collinearity between DS × J and R × A LG6 (c) and LG9 (d) with W85 and Draper chr‐6 and chr‐9, respectively. The gray bars represent the alignment between W85 and Draper chr‐6 (c) and W85 and Draper chr9 (d). Red lines between Draper and W85 chromosomes represent potential rearrangements or chimeric regions, gray line represent collinear regions. Transparent green boxes indicate rearranged regions that were collinear between W85 chr‐6 and chr‐9 assemblies (0) and the DS × J and R × A linkage maps. The same regions highlighted in transparent red boxes were not collinear with Draper chr‐6 and chr‐9. Those regions represent chimeric sequences in the Draper assembly rather that true chromosome rearrangements.
The W85 genome was highly collinear with linkage maps and the Draper genome (longest haplotypes, chr‐1–12 and W85 p0) (Fig. S13). Few minor rearrangements were identified on Draper chr‐2, ‐3, ‐5, ‐6, ‐7 and ‐9 and were largely located near centromeric and telomeric regions (Figs 4c,d, S13). Considering the high level of collinearity between the linkage maps and the W85_v2 genome, and the fact that the markers and the sequences were ordered independently from each other across these three resources (DS × J, R × A maps and W85_v2 genome), it is likely that the rearrangements observed in the Draper genome are the results of chimeric sequences.
Next, the comparative analysis with the Draper genome was expanded to all 48 phased chromosomes. Synteny analysis suggested the presence of a reciprocal, heterozygous, translocation between one homolog of chr‐6 and one of chr‐10 (Fig. 5a). Indeed, c. 14 Mb of a Draper chr‐6 homolog (chr‐30, hereafter, chr‐610) aligned with W85 chr‐10, while the rest of the chromosome (c. 26 Mb) mapped to W85 chr‐6 (Fig. 5a). Similarly, c. 4.8 Mb of a Draper chr‐10 homolog (chr 34, hereafter, chr‐106) mapped to the short arm end of W85 chr‐6 including the VacSat169 repeat region, whereas the rest of the chromosome (c. 33 Mb) mapped to W85 chr‐10 (Fig. 5a). Another Draper chr‐10 homolog (chr‐46) consisted only of a portion of W85 chr‐10, possibly due to either incomplete assembly or to a large chromosomal deletion. Otherwise, each of the remaining Draper chromosomes was highly collinear with only a single W85 chromosome.
Fig. 5.

Reciprocal inter‐chromosome translocation and its impact on chromosome pairing and recombination in Vaccinium corymbosum cv. Draper. (a) Synteny analysis between W85 (V. caesariense) chr‐6 and chr‐10 (p0) (gray bars) with Draper Chr 6 haplotypes (VaccDscaff6, 18, 30, 42) (blue bars) and chr‐10 haplotypes (VaccDscaff10, 22, 34 and 46) (orange bars). Blue and beige lines represent regions that were collinear. Violet lines represent regions that were translocated between W85 chr‐6 and chr‐10. Heat map with green and red shades in the W85 chr‐6 and chr‐10 represent the density (200 kb windows) of VacSat169 and VacSat1, respectively. Green and red dots in the Draper haplotypes represent the expected position of VacSat169 and VacSat1, respectively. (b, c) Fluorescence in situ hybridization (FISH) of VacSat1 (red) and VacSat169 (green) on mitotic metaphase chromosomes of Draper (2n = 4x = 48). VacSat169 signals were located on three chr‐6 homologs with no detectable VacSat1 signals and on the translocation chromosome chr‐106 carrying also VacSat1 repeats (arrows). (d–k) Meiotic pairing behavior of the four chromosomes carrying VacSat169 repeats (green signals) at diakinesis‐metaphase I in Draper. (d) Gray‐scale image of a Draper metaphase I cell with 24 bivalents; (e, inset) the same image with VacSat1 (red) and VacSat169 (green) FISH signals; the arrows (in d and e) indicate 6–6 bivalent, 6–610 bivalent and 106–10 bivalent. Chr‐610 has no detectable FISH signals of either probe, whereas chr‐106 has both VacSat169 and VacSat1 signals. (f) Gray‐scale image of a Draper metaphase I cell containing a tetravalent ring; (g) the same cell hybridized with VacSat1 and VacSat169 repeats. The arrows point to the tetravalent involving a chr‐6, chr‐610, a chr‐10, and chr‐106, and to a 6–6 bivalent. (h) Gray‐scale image of a Draper metaphase I cell containing a hexavalent chain. (i) The same cell hybridized with VacSat1 and VacSat169 repeats. The arrows indicate the hexavalent made of three chr‐6 homologs, chr‐106, and other two chromosomes. (j) Gray‐scale image of a Draper metaphase I cell containing a hexavalent chain; (k) the same cell with VacSat1 (red) and VacSat169 FISH signals. The arrows indicate a 6–6 bivalent, and the hexavalent made of one copy of chr‐6, chr‐106, and other four chromosomes. Bar, 5 μm. (l) Clustering of Draper single‐nucleotide polymorphism markers obtained from the D × B population into respective linkage groups based on logarithms of odds (LOD) score. Black circles represent markers grouped in linkage groups, and representing the four haplotypes. Lines between circles represent linkage between markers grouped in each LG. Orange and blue color inside the LGs representing chr‐6 and chr‐10, indicates the marker composition of each haplotype. Circles with orange and blue colors contain markers from both chr‐6 and chr‐10.
Cytogenetics analysis was used to validate the reciprocal translocation. Due to the rearrangement, the translocation chr‐610 should have lost its VacSat169 repeat array, which in turn, should be transposed to chr‐106 (Fig. 5a). Thus, chr‐610 should have no detectable FISH signals of either VacSat169 or VacSat1, whereas chr‐106 should harbor both VacSat169 and VacSat1 signals. Notably, in the other genotypes analyzed (W85, V. darrowii, DS and Jewel), none of the chr‐6 homologs had detectable VacSat1 signals (Figs 1i–l, S10). Using FISH mapping of VacSat169 and VacSat1 in Draper, we detected one chromosome with both terminal VacSat169 and interstitial VacSat1 signals. Our hypothesis is that this chromosome is the translocation chr‐106 (Fig. 5b,c). The three other chromosomes with a terminal VacSat169 site had no detectable VacSat1 repeats and correspond to three nonrearranged copies of chr‐6 (Fig. 5b,c). Overall, these results demonstrated the presence of an inter‐chromosomal translocation in the Draper genome.
The inter‐chromosomal translocation alters chr‐6 and chr‐10 meiotic behavior in the Draper genome
We were intrigued by how the four chromosomes with terminal VacSat169 signals pair during meiosis in Draper vs DS and Jewel. Therefore, we conducted FISH using VacSat169 and VacSat1 at diakinesis‐metaphase I stages. In the pollen mother cells of DS (n = 102) and Jewel (n = 49), the four chr‐6 homologs carrying VacSat169 formed either two bivalents (in > 90% of the cells) or a single tetravalent, whereas hexavalents were never detected (Fig. S10; Table S18). By contrast, in Draper, the chromosomes with the VacSat169 signals (that is, three homologs of chr‐6 and chr‐106 with both VacSat169 and VacSat1) formed various configurations, including hexavalents, involving a total of five, six and eight chromosomes in 18%, 78% and 3% of the cells (n = 130), respectively (Figs 5d–k, S14; Table S18). In particular, in > 60% of the cells (n = 130), two chr‐6 homologs formed a bivalent, whereas another chr‐6 formed a bivalent with a chromosome with no detectable FISH signals of either probe. We speculate that this chromosome with no signals is the translocation chr‐610. Finally, chr‐106 (with both VacSat169 and VacSat1 signals) formed a bivalent with a chromosome carrying a VacSat1 site, likely a chr‐10 homolog (106–10 bivalent; Figs 5d,e, S14a,b). In about 18% of the cells, chr‐106 appeared as a univalent, whereas two chr‐6 homologs formed a 6–6 bivalent, and another chr‐6 formed a bivalent (6–610 bivalent) or a trivalent, likely a 6–610–10 trivalent (Fig. S14c–e). About 8% of the cells contained a tetravalent (likely a 6–610–10–106) plus a 6–6 bivalent (Figs 5f–g, S14f). Notably, about 7% of the cells contained a hexavalent formation. In some cells, the hexavalent ring or chain was made of three chr‐6 homologs, plus chr‐106 (with both VacSat169 and VacSat1) and two other chromosomes (likely chr‐610 and a chr‐10 homolog; Figs 5h,i, S14g). In other cells, it involved a chr‐6, chr‐106 and other four chromosomes (likely chr‐610 and three chr‐10 homologs each with VacSat1 signals), whereas other two chr‐6 homologs formed a 6–6 bivalent (Figs 5j,k, S14h; Table S18). Interestingly, each chr‐10 had a detectable VacSat1 signal, suggesting that the partial copy of chr‐10 (chr‐46) in the Draper reference genome (Colle et al., 2019) is due to incomplete assembly rather than a chromosomal deletion.
The impact of the inter‐chromosomal translocation on chromosome pairing/recombination was also assessed in a bi‐parental mapping population named D × B derived from a cross between Draper (the translocation‐carrier) and Biloxi (Methods S4). Parent‐specific markers were used for clustering analysis, which is based on pairwise recombination frequency. At logarithms of odds (LOD) score 6, Biloxi specific markers clustered into 12 groups, and each group included four subgroups, representing the 12 blueberry chromosomes and four homologs, respectively (Fig. 5l). In contrast, Draper markers clustered into 11 groups, with 10 groups representing 10 chromosomes each with four homologs, and one group containing eight subgroups representing chr‐6 and chr‐10 (Fig. 5l). Among these eight subgroups, three subgroups comprised only chr‐6 markers, three comprised only chr‐10 markers and two comprised a mix of chr‐6 and chr‐10 markers. Increasing the LOD score to 9 did not change the clustering results. These results demonstrated a significant level of recombination between chr‐6 and chr‐10, which is not expected across nonhomologous chromosomes, unless they share some sequences due to a reciprocal translocation. After linkage analysis, three chr‐10 homologs had relatively low number of markers and the chr‐106 haplotype was not represented (Fig. S15; Table S19). For chr‐6, three homologs had normal number of markers and one haplotype had a mix of chr‐6 and chr‐10 markers and represented chr‐610.
Overall, these results demonstrated that the inter‐chromosomal translocation in the Draper genome altered its pairing, recombination and segregation behavior, and in turn, affected the linkage map construction (Fig. S16).
Blueberries behave as an autotetraploid with tetrasomic inheritance
The quadrivalent chromosome pairing behavior estimated using molecular markers varied between the DS × J, R × A and D × B mapping populations and, to some degree, between the parents. For the DS × J and R × A mapping populations, the quadrivalent formation ranged from 24% on DS chr‐7 to 53% on Arlen chr‐3 (Fig. 6a; Tables S20, S21). The degree of quadrivalent formation for D × B was relatively lower compared to R × A and DS × J ranging from 3% on Draper chr‐7 to 26.8% on Biloxi chr‐10 (Fig. 6a; Tables S22, S23).
Fig. 6.
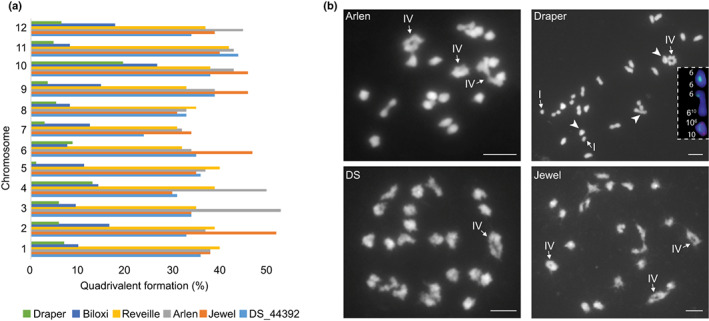
Chromosome pairing behavior evaluated using molecular markers and cytogenetic work in Vaccinium corymbosum cv. Draper, Biloxi, Arlen, Reveille, Jewel and selection Draper Selection‐44392 (DS). (a) Summary of quadrivalent formation (%) in parents of three linkage maps developed here. (b) Representative diakinesis‐metaphase I cells from the tetraploid blueberries Arlen containing two tetravalent rings, a tetravalent chain and 18 bivalents; Draper with a tetravalent ring, two univalents and 21 bivalents; chr‐6 homologs and translocation chromosomes paired as bivalent (arrowheads) and are shown in the inset with their fluorescence in situ hybridization (FISH) signals of VacSat169 (green) and VacSat1 (red); DS with a tetravalent ring and 22 bivalents; Jewel with three tetravalent rings and 18 bivalents. Arrows point to the tetravalents (IV) and univalent (I). Bar, 5 μm.
Overall, there were substantial quadrivalent chromosome pairing in DS × J and R × A mapping populations, as also indicated by the cytological analysis of three parental varieties at diakinesis‐metaphase I (Arlen, DS, and Jewel) (Figs 6b, S17; Table S24). In DS, 32% of the pollen mother cells at metaphase I (n = 68) contained up to two quadrivalents, whereas in 65% of the cells, the chromosomes formed bivalents exclusively (Fig. S17). Arlen and Jewel had a higher frequency of meiocytes with at least one and up to four quadrivalents (63%, n = 60 and 66%, n = 42, respectively). Unpaired chromosomes (univalents) occurred at various frequencies (from 4% in DS to up 48% in Jewel), sometimes in the same cells containing the quadrivalent (Fig. 6b; Table S24).
In addition, the rate of DR increased towards the telomere regions, reaching a maximum of 9% per parent at the telomeric regions for most of the chromosomes of the DS × J and R × A mapping populations (Figs S18, S19).
Preferential pairing based on pairs of closely‐linked S × N markers in the repulsion phase did not show significant deviations between closely linked S × N markers in DS × J (Figs S20, S21). However, results obtained for R × A revealed some significant (FDR adjusted P < 0.05) preferential pairing on Arlen chr‐7 (Fig. S22) and Reveille chr‐11 (Fig. S23). The preferential pairing was also investigated using a multi‐point approach, identity‐by‐descent probabilities for the population. For DS × J mapping population, we did not observe any significant deviations at both bivalent pairing and quadrivalent pairing models (Tables S20–S25). For the R × A mapping population, there was significant (P < 0.001) deviation from random pairing on Reveille chr‐1 at bivalent pairing model (Table S26) but not at quadrivalent pairing model (Table S21). Furthermore, the strength of the preferential pairing parameter was weak on Reveille chr‐1 (Table S21). For D × B, no significant preferential pairing was observed at both bivalent and quadrivalent models except for Biloxi at the bivalent model on chr‐9 (Tables S22, S23). Although there were a few indications of preferential pairing in R × A and D × B mapping populations, the results were not consistent between the different models (bivalent and quadrivalent) and the two methods. Overall, our results did not reveal strong enough evidence to demonstrate any preferential pairing in blueberry.
Discussion
High quality genomic resources provide novel insight into the structure of the blueberry genomes
Here we presented a high‐quality phased assembly of V. caesariense, also classified as diploid V. corymbosum, and that represents the closest diploid reference genome relative to the genome of cultivated tetraploid highbush blueberry (Bruederle & Vorsa, 1994). Compared with W85_v1 genome (Gupta et al., 2015), the W85_v2 assembly represents a 384‐ and 84‐fold reduction in the number of scaffolds and contigs, respectively, and a 161‐fold improvement in contiguity (N50 length) at the contig level (Table S2). In addition, it contains > 116% (378 Mb) extra known sequences (contig level), was anchored to chromosomes, and was phased. Compared to the Draper genome (Colle et al., 2019), the contiguity at the contig level increased about 10‐fold (Table S2) and comparison at the structural level with the W85_v2 genome highlighted some chimeric sequences on the Draper assembly. Also, multiple regions spanning > 1 Mb (Fig. S13) were absent in the longest Draper haplotype assembly (Dscaf 1–12) which resulted in a higher genome coverage and density of the linkage maps (Fig. 4).
The W85_v2 genome enabled the localization of regions enriched by low complexity sequences. Previous bioinformatic analyses identified VacSat1 as the most abundant satellite repeats in Vaccinium spp., (Sultana et al., 2020). Here, we provided the first evidence of the centromeric localization of this repeat on most W85 chromosomes. Future analyses of the blueberry centromeric chromatin could clarify whether there are chromosome‐specific centromeric sequences in blueberry (e.g. specific to the chromosomes lacking VacSat1), as reported in other species (Yang et al., 2018). Notably, VacSat1 sequence and distribution were conserved between tetraploid HB and the crossable wild diploid species belonging to the Vaccinium section Cyanococcus. This similarity, combined with the high level of collinearity detected across these genomes, might have contributed to the successful introgression of wild species from this taxon into the cultivated blueberry and to the chromosome stability in interspecific hybrids (Lyrene & Olmstead, 2012).
Finally, a comparison of the two haplotypes revealed a high level of heterozygosity represented by SNPs, indels and SVs, including PAV genes. Such a high level of heterozygosity can have the following multiple effects: (1) mask the deleterious effect of recessive alleles; (2) create allele‐specific expression or allelic imbalance; and (3) create new gene variants such as PAV. The high level of heterozygosity maintained in this outcrossing species might contributes to plant survival and environmental adaption, similar to what has been observed in other crops (Zhou et al., 2020).
Evidence and impact of a reciprocal translocation for blueberry genetic analysis and breeding
Chromosome translocations have played significant roles in trait diversity, speciation and evolution (Martin et al., 2020). Recent advances in sequencing and genotyping technologies have facilitated precise identification of chromosomal rearrangements, including reciprocal translocations in many crops, especially in species with disomic inheritance such as banana (Martin et al., 2020) and cotton (Yang et al., 2019). However, similar studies in species with polysomic inheritance like blueberry are very limited. This is partially due to difficulties in generating high‐quality linkage maps and genome assemblies for these species. Recently, a phased assembly of the tetraploid blueberry cultivar Draper, was released (Colle et al., 2019), and comparative analysis among Draper homologous and nonhomolog chromosomes revealed a possible heterozygous reciprocal translocation between one homolog of chr‐6 and chr‐10. However, presence of the translocation through cytogenetic evidence, its impact on chromosome pairing and recombination, and its presence in other tetraploid genomes were not previously evaluated.
In this study, comparative genomic, cytogenetic and linkage analysis demonstrated the presence of this reciprocal translocation, which formed two fused chromosomes, chr‐610 and chr‐106. The co‐localization between VacSat1 and VacSat169 repeats on the same chromosome is specific to the translocation chr‐106, making these repeats ideal cytogenetic markers for future studies.
The reciprocal translocation can have multiple effects on chromosome pairing and recombination. In bananas, the impact of heterozygous reciprocal translocations on chromosome recombination rate ranged from no effect to full suppression (Martin et al., 2020). In our cytogenetic analysis, we observed multiple abnormal chromosome pairings configurations including hexavalent. Therefore, the translocation affected the linkage map construction and the linkage relationships between chr‐6 and chr‐10, by causing ‘pseudo linkage’ near the translocation sites. One of the homologs of chr‐10 was not retained in the final linkage map, whereas the marker coverage in the other chr‐10 homolog was lower, indicating that this translocation may either limit recombination in the translocation sites or cause chromosome recombination frequencies that are not accounted for by available statistical models used for linkage analysis (Durrant et al., 2006; Preedy & Hackett, 2016). Also, approximately 20% of the chr‐106 was univalent, which may result in a lagging chromosome and gametes with no chr‐106. This phenomenon would impact GWAS (i.e. genome‐wide association study) analysis by generating a strong population structure (Farré et al., 2012), and it could also prevent the precise localization of quantitative trait locus (QTL) and distort/weaken statistical support for marker/trait associations. Studies also found that reciprocal translocation can affect gene expression (Harewood et al., 2010; Muramoto et al., 2018). Interestingly, comparative gene expression analysis among blueberry homologous chromosomes indicated that chr‐6 and chr‐10 had the most stable pattern of gene expression dominance across fruit development (Colle et al., 2019). Preliminary analysis of genes spanning the translocations indicated that biological processes such as cellular and metabolic processes were the most significantly enriched GO terms in both chr‐6 and chr‐10. Similarly, molecular function such as nucleic acid binding, RNA‐binding and hydrolase activity, and cell components including intracellular and organelles were the most enriched GO terms in both chromosomal regions (Fig. S24). Future work will focus on narrowing which genes and how their expression are affected by the translocation and how it affect their function. The reciprocal translocation can directly affect phenotypes and cell functionality, including pollen viability. Blueberries shed their pollen as permanent tetrads, which, in turn, represent the four products of single meiotic events. A preliminary evaluation of pollen viability using Alexander's stain indicated that over 70% of the pollen tetrads of Draper (n = 600) had all four grains viable, while tetrads with 3 : 4 and 0 : 4 viable : nonviable pollen grains were 22% and < 1%, respectively. Notably, the percent of total viable pollen grains was high (90%, n = 2400) and comparable to the estimates reported for other popular tetraploid blueberry cultivars (Krebs & Hancock, 1990). Although plants heterozygous for a translocation are expected to have a significant decrease of viable pollen (e.g. 50% observed in Arabidopsis thaliana) (Clark & Krysan, 2010), our preliminary data suggests that the translocation may not cause such a drastic reduction in tetraploid blueberry. Genetic studies for economically important traits in blueberry are rapidly expanding, and some QTL were mapped on chr‐6 and chr‐10 (Qi et al., 2021). This information opens opportunities to investigate how and if the translocation affects the inheritance of cold hardiness and other important phenotypes.
Multiple mechanisms could have contributed to the translocation including hybridization with wild species and polyploidization (Yang et al., 2019; Chen & Ni, 2006; Martin et al., 2020). In blueberry, hybridization with wild diploid species from the Vaccinium section Cyanococcus via 2n gametes has been used extensively for the introgression of multiple traits. For instance, up to 8% of the Draper genome derived from V. darrowii (1.6%), V. tenellum (0.4%) and V. angustifolium (6%) (Brevis et al., 2008). Inter‐specific hybridizations with species harboring the translocation could have introduced the translocation into cultivated germplasm. Our results indicated that the V. darrowii genome (Yu et al., 2021) is highly collinear with all W85 chromosomes (Fig. 3c), and that none of the V. darrowii chromosomes harbored both VacSat1 and VacSat169 as observed in Draper. These results exclude the role of V. darrowii in the introgression of the translocation.
The rate of incidence of this translocation across blueberry germplasm and its effect can have direct implications on breeding and genetic studies. This study did not support the presence of the reciprocal translocation in other HB cultivars such as Arlen, Reveille, Biloxi, Jewel and DS. However, the presence of heterozygous translocations in blueberry was already suspected by Vorsa (1990) who observed pentavalent formation in a triploid V. corymbosum (NC‐856‐1) that did not share a direct ancestor with Draper. This observation suggests that other genotypes carrying translocations are likely present in blueberry germplasm. However, since the translocation is heterozygous, gametes lacking the translocation can be produced, and fertilization between similar gametes could generate progenies that lack the translocation. This reduces the likelihood of its fixation within the blueberry germplasm.
Overall, the results presented here open opportunities for future genetic studies to evaluate the origin, distribution of the translocation in the blueberry germplasm and its impact on trait inheritance.
Evidence for autopolyploid genetic behaviors of blueberry
Understanding the polyploid origin (being autopolyploid or allopolyploid) and genetic behavior (disomic or tetrasomic), in a polyploid crop is critical to advancing genetic studies and breeding. Addressing these two questions in blueberry is a relatively complex task, for two reasons: (1) the Vaccinium section Cyanococcus is comprised of diploid, tetraploid and hexaploid distinct species, that can easily intercross producing completely interfertile hybrids (Lyrene et al., 2003); (2) multiple diploid species have been introgressed into blueberry cultivars, making HB (V. corymbosum) cultivars a highly diverse polyploid genetic system (Retamales & Hancock, 2018). The only study addressing the genetic behavior of tetraploid blueberry based on molecular markers (RAPD) indicated that blueberry behaves as an autopolyploid (Qu & Hancock, 1995, 2001). However, the number of markers used in this study was very limited (n = 31), and it was unknown what proportion/fraction of the genome was assessed. Here, we present the first and most comprehensive study that evaluated the polyploid behavior of tetraploid blueberry based on preferential pairing, quadrivalent formation and double reduction, using high‐density molecular markers spanning the entire genome paired with cytogenetic analysis. To account for potential inter‐cultivar variation, recombination behavior was evaluated in six cultivars, including SHB and NHB, all of which have a different fraction (10–60%) of their genomes introgressed from wild diploid species (Brevis et al., 2008).
No evidence of preferential pairing was found, demonstrating that the mode of inheritance of all chromosomes and all blueberry cultivars evaluated here is polysomic (tetrasomic), consistent with previous data based on RAPD markers (Qu & Hancock, 1995, 2001). The degree of quadrivalent formation was substantial, ranging from 24% to 53% for the DS × J and R × A mapping populations, and it was lower in D × B (between 3% and 27%). These results are comparable to that of potato, a certain autopolyploid species (Choudhary et al., 2020). Double‐reduction is a consequence of polysomic inheritance and occurs when sister chromatids segregate into the same gamete. Here DR rate, which was assessed for the first time in blueberry, was detected in all chromosomes, and it increased towards the telomeric regions consistent with what has been described in other autopolyploid species (Bourke et al., 2015).
Overall, we provided strong evidence that blueberry behaves as an autopolyploid during meiosis. Furthermore, we demonstrated the presence and the impact of a heterozygous reciprocal translocation in the Draper genome, which can affect genetic studies and breeding. Finally, comparative analyses among members of the Vaccinium section Cyanococcus highlighted that the structure of their chromosomes, including centromeric repeats, are highly conserved. These findings likely explain the complexity of this taxa characterized by extensive hybridization between ploidy/species and overlapping morphologies (Retamales & Hancock, 2018). This information will serve as a framework to extend comparative genome analysis within Vaccinium spp. and advance genomic‐assisted breeding in blueberry.
Competing interests
None declared.
Author contributions
M Iorizzo conceived the overall study. M Iorizzo, M Iovene, PPE, HB and MFM designed the study. M Iorizzo and HA coordinated W85 sequencing. HB and MFM performed genome assembly, gene prediction, and comparative analysis. MFM performed tetraploid linkage maps and genetic analysis. SJT, AEP and PPE coordinated and performed annotation of the repetitive sequences. M Iovene, DDP, and GC coordinated and performed satellite repeat and cytogenetic analysis. LJR and XQ coordinated and developed the diploid linkage map and provided plant material for sequencing. LB, DC, RJ and LG developed the D × B data. NVB, and TM provided material for cytogenetic work and DS × J material. MFM, M Iorizzo and M Iovene wrote the first draft of the manuscript. HB, DDP, GC, SJT, AEP, HA, XQ, TM, NVB, LG, RJ, DC, MAL, LB, PPE and LJR edited the manuscript. MFM and HB contributed equally to this work.
Supporting information
Fig. S1 Example of a chimeric sequence in the W85_v2 assembly.
Fig. S2 Collinearity between the high‐density linkage map representing the W85 genome and the longest 12 scaffolds of the W85_v2 genome assembly.
Fig. S3 Hi‐C heatmap.
Fig. S4 Haplotype diversity in the W85_v2 chromosomes.
Fig. S5 PAV gene's enrichment analysis.
Fig. S6 Distribution of VacSat218 repeats compared to VacSat1 on W85 chromosomes.
Fig. S7 Organization and distribution of VacSat169 and 45S rDNA in W85 genome.
Fig. S8 Organization and distribution of VacSat61 repeats in W85 genome.
Fig. S9 FISH mapping of the telomeric motif (TTTAGGG)n and VactSat1 repeat.
Fig. S10 Meiotic pairing of the four chr‐6 homologs in the tetraploid blueberries DS and Jewel.
Fig. S11 Distribution of markers across eight DS × J homologs.
Fig. S12 Distribution of markers across eight R × A homologs.
Fig. S13 Collinearity between DS × J and R × A LGs 1–12 and the corresponding chromosomes 1–12 in the W85 and Draper genome assemblies.
Fig. S14 Meiotic configurations of Draper chr‐6 homologs and translocation chr‐610 and chr‐106 at diakinesis‐metaphase I.
Fig. S15 Distribution of markers across eight homologs LGs representing chromosomes 1–12 (I–XII) of the DxB mapping population.
Fig. S16 Segregation of the chromosomes carrying VacSat169 repeats at anaphase I/telophase I of DS and Draper.
Fig. S17 Chromosome distribution diakinesis‐metaphase I.
Fig. S18 Double reduction analysis in DS × J.
Fig. S19 Double reduction analysis in R × A.
Fig. S20 Preferential pairing analysis in DS.
Fig. S21 Preferential pairing analysis in Jewel.
Fig. S22 Preferential pairing analysis in Arlen.
Fig. S23 Preferential pairing analysis in Reveille.
Fig. S24 Gene ontology (GO) enrichment analysis of genes located on the regions exchanged between chromosome 6 and chromosome 10.
Methods S1 Plant material collection, DNA and RNA extraction, and sequencing.
Methods S2 Genome assembly.
Methods S3 Repetitive sequence analysis.
Methods S4 Construction of linkage maps.
Methods S5 Estimation of a preferential pairing parameter.
Methods S6 Fluorescence in situ hybridization (FISH) analysis.
Table S1 Summary of the W85 genome and transcriptome sequences used in this study.
Table S2 Summary statistics for the new assembled W85 genome (W85_v2) and comparison with W85_v1 and tetraploid Draper genomes.
Table S3 Summary of the number of chimeric regions corrected by Hi‐Rise, Hi‐C and linkage map.
Table S4 W85 genome assembly organized by pseudomolecules (chromosomes) and two phases (p0 and p1).
Table S5 Summary statistics of PacBio Iso‐Seq sequences mapped on the W85_v2 genome (p0 and p1).
Table S6 Assessment of the gene space coverage based upon alignment of the RNA‐sequencing data obtained from different tissues and treatments, against the W85_v2 assembly (p0 and p1).
Table S7 Summary of Busco analysis using plant dataset (odb10).
Table S8 Summary of gene prediction in the W85_v2 genome assembly.
Table S9 Summary statistics of W85 genes annotation.
Table S10 Predicted resistance‐genes in the W85_v2 assembly (p0 and p1).
Table S11 Statistics of the repetitive sequences annotated de novo in W85_v2 genome assembly (p0 and p1).
Table S12 Statistics of the gene prediction features in W85_v2 genome, tetraploid blueberry genome Draper, Solanum lycopersicum, Arabidopsis thaliana, Daucus carota and Beta vulgaris.
Table S13 Summary of the number of single‐nucleotide polymorphisms, INDELs and SV identified between the two haplotypes of the 12 chromosomes.
Table S14 Summary of present and absent (PAV) genes across the p0 and p1 haplotypes.
Table S15 Sequence characteristics of putative satellite repeats identified in the diploid blueberry W85 through the tarean pipeline.
Table S16 Distribution of single‐nucleotide polymorphisms into genotype classes and summary of integrated map of DS × J mapping population.
Table S17 Distribution of single‐nucleotide polymorphisms into genotype classes and summary of integrated map of R × A mapping population.
Table S18 Frequency of different types of pairing among the chromosomes carrying terminal VacSat169 repeats in DS 4432 (DS), Jewel and Draper at diakinesis‐metaphase I.
Table S19 Summary of marker distribution across chromosomes in Draper × Biloxi (B × D) mapping population.
Table S20 Predicted quadrivalent pairing and preferential pairing under the full model for the Draper Selection‐44392 × J mapping population.
Table S21 Predicted quadrivalent pairing and preferential pairing under the full model for the R × A mapping population.
Table S22 Summary of preferential pairing at the bivalent model for the B × D mapping population.
Table S23 Predicted quadrivalent pairing and preferential pairing under the full model for the B × D mapping population.
Table S24 Frequency of meiocytes at diakinesis‐metaphase I containing exclusively bivalents (24 II), at least one multivalent (IV or VI), and/or univalent (univ) in blueberry varieties.
Table S25 Summary of preferential pairing at the bivalent model for the DS × J mapping population.
Table S26 Summary of preferential pairing at the bivalent model for the R × A mapping population.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
The authors are grateful to the North Carolina State University Bioinformatics Research Center for use of their high‐performance computing cluster. The authors thank Rosa Paparo for excellent technical support in the cytogenetic analysis and Dr James Hancock for providing background information about the pedigree of the DS × J mapping population. This research was supported by the Foundation for Food and Agriculture Research (FFAR) under award number 534667 and the National Institute of Food and Agriculture, United States Department of Agriculture (USDA), under award no. 2019‐51181‐30015, project ‘VacciniumCAP: Leveraging genetic and genomic resources to enable development of blueberry and cranberry cultivars with improved fruit quality attributes.’ M Iorizzo was also supported by the USDA National Institute of Food and Agriculture, Hatch project 1008691. PacBio sequencing was supported by North Carolina State University (NCSU), Plants for Human Health Institute (PHHI)‐Raleigh seed grant initiative. Genotyping of the DS × J population was funded by David H. Murdock Research Institute (DHMRI) (https://dhmri.org), project P2EP Core Project, blueberry mapping. PPE and SJT were supported by USDA Hatch project 1009804, USDA AFRI 2018‐67013‐27592 and MSU AgBioResearch. Thanks also to Dr Dorrie Main who managed the Genome Database for Vaccinium (GDV) with support from the USDA‐NIFA NRSP10 project. M Iovene was partially supported by the project BLUEPAINT (DBA.AD002.452).
We dedicate this article in Dr Chad Finn's memory. Dr Chad Finn served as a world‐renowned blueberry breeder, who released numerous blueberry cultivars as part of the USDA‐ARS and Oregon State University. As a passionate geneticist and resourceful collaborator, Dr Finn made significant contributions to advance genetic discoveries in this crop. The work presented here represents an example of his collaborative contributions to the discipline.
Contributor Information
Marina Iovene, Email: marina.iovene@ibbr.cnr.it.
Patrick P. Edger, Email: pedger@gmail.com.
Massimo Iorizzo, Email: miorizz@ncsu.edu.
Data availability
PacBio and Illumina (Hi‐C, Chicago and Hi‐seq RNA‐seq) raw data that were used for genome assembly and annotations are available at the National Center for Biotechnology Information (NCBI) under BioProject No. PRJNA755705. The W85_v2.0 genome assembly, annotation, sequences of the genes, transcripts and proteins, can be accessible through the Genome Database for Vaccinium (GDV, https://www.vaccinium.org/). Sequences (assembly and annotation files) are available under the subdirectory Blueberry/Genomes/W85‐20 v2.0 (https://www.vaccinium.org/bio_data/1659687). The genome is available to search within the GDV database, Blast, PathwayCyc and JBrowse. The linkage maps were made available through GDV under the Search/map search subdirectory.
References
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society: Series B (Methodological) 57: 289–300. [Google Scholar]
- Bourke PM, Arens P, Voorrips RE, Esselink GD, Koning‐Boucoiran CFS, van't Westende WPC, Santos Leonardo T, Wissink P, Zheng C, van Geest G et al. 2017. Partial preferential chromosome pairing is genotype dependent in tetraploid rose. The Plant Journal 90: 330–343. [DOI] [PubMed] [Google Scholar]
- Bourke PM, van Geest G, Voorrips RE, Jansen J, Kranenburg T, Shahin A, Visser RGF, Arens P, Smulders MJM, Maliepaard C. 2018. polymapr—linkage analysis and genetic map construction from F1 populations of outcrossing polyploids. Bioinformatics 34: 3496–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourke PM, Voorrips RE, Visser RGF, Maliepaard C. 2015. The double‐reduction landscape in tetraploid potato as revealed by a high‐density linkage map. Genetics 201: 853–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brevis PA, Bassil NV, Ballington JR, Hancock JF. 2008. Impact of wide hybridization on highbush blueberry breeding. Journal of the American Society for Horticultural Science 133: 427–437. [Google Scholar]
- Bruederle LP, Vorsa N. 1994. Genetic differentiation of diploid blueberry, Vaccinium sect. Cyanococcus (Ericaceae). Systematic Botany 19: 337–349. [Google Scholar]
- Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, Moore B, Holt C, Alvarado AS, Yandell M. 2008. Maker: an easy‐to‐use annotation pipeline designed for emerging model organism genomes. Genome Research 18: 188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, Ni Z. 2006. Mechanisms of genomic rearrangements and gene expression changes in plant polyploids. Bioessays 28: 240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin C‐S, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A, Dunn C, O'Malley R, Figueroa‐Balderas R, Morales‐Cruz A et al. 2016. Phased diploid genome assembly with single‐molecule real‐time sequencing. Nature Methods 13: 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A, Wright L, Ponce O, Chen J, Prashar A, Sanchez‐Moran E, Luo Z, Compton L. 2020. Varietal variation and chromosome behaviour during meiosis in Solanum tuberosum . Heredity 125: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark KA, Krysan PJ. 2010. Chromosomal translocations are a common phenomenon in Arabidopsis thaliana T‐DNA insertion lines. The Plant Journal 64: 990–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colle M, Leisner CP, Wai CM, Ou S, Bird KA, Wang J, Wisecaver JH, Yocca AE, Alger EI, Tang H et al. 2019. Haplotype‐phased genome and evolution of phytonutrient pathways of tetraploid blueberry. GigaScience 8: giz012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costich DE, Ortiz R, Meagher TR, Bruederle LP, Vorsa N. 1993. Determination of ploidy level and nuclear DNA content in blueberry by flow cytometry. Theoretical and Applied Genetics 86: 1001–1006. [DOI] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST et al. 2011. The variant call format and VCFtools . Bioinformatics 27: 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Zaleski C, Jha S, Gingeras TR, Batut P, Davis CA, Chaisson M, Schlesinger F, Drenkow J. 2012. Star: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant JD, Gardunia BW, Livingstone KD, Stevens MR, Jellen EN. 2006. An algorithm for analyzing linkages affected by heterozygous translocations: QuadMap . Journal of Heredity 97: 62–66. [DOI] [PubMed] [Google Scholar]
- Farré A, Cuadrado A, Lacasa‐Benito I, Cistué L, Schubert I, Comadran J, Jansen J, Romagosa I. 2012. Genetic characterization of a reciprocal translocation present in a widely grown barley variety. Molecular Breeding 30: 1109–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Marth G. 2012. Haplotype‐based variant detection from short‐read sequencing. arXiv: 1207.3907.
- Gerard D, Ferrão LFV, Garcia AAF, Stephens M. 2018. Genotyping polyploids from messy sequencing data. Genetics 210: 789–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q, Buckler ES. 2014. Tassel‐Gbs: a high capacity genotyping by sequencing analysis pipeline. PLoS ONE 9: e90346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz S, Garcia‐Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talón M, Dopazo J, Conesa A. 2008. High‐throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Research 36: 3420–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V, Estrada AD, Blakley I, Reid R, Patel K, Meyer MD, Andersen SU, Brown AF, Lila MA, Loraine AE. 2015. RNA‐seq analysis and annotation of a draft blueberry genome assembly identifies candidate genes involved in fruit ripening. GigaScience 4: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harewood L, Schütz F, Boyle S, Perry P, Delorenzi M, Bickmore WA, Reymond A. 2010. The effect of translocation‐induced nuclear reorganization on gene expression. Genome Research 20: 554–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS. 2007. Improved pairwise alignmnet of genomic DNA . PhD thesis, Pennsylvania State Univ. [WWW document] URL https://www.bx.psu.edu/~rsharris/rsharris_phd_thesis_2007.pdf [accessed 30 March 2021].
- Iovene M, Wielgus SM, Simon PW, Buell CR, Jiang J. 2008. Chromatin structure and physical mapping of chromosome 6 of potato and comparative analyses with tomato. Genetics 180: 1307–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelenkovic G, Hough LF. 1970. Chromosome associations in the first meiotic division in three tetraploid clones of Vaccinium corymbosum L. Canadian Journal of Genetics and Cytology 12: 316–324. [Google Scholar]
- Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G et al. 2014. InterProScan 5: genome‐scale protein function classification. Bioinformatics 30: 1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I. 2004. Gene finding in novel genomes. BMC Bioinformatics 5: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs SL, Hancock JF. 1990. Early‐acting inbreeding depression and reproductive success in the highbush blueberry, Vaccinium corymbosum L. Theoretical and Applied Genetics 79: 825–832. [DOI] [PubMed] [Google Scholar]
- Kronenberg ZN, Rhie A, Koren S, Concepcion GT, Peluso P, Munson KM, Porubsky D, Kuhn K, Mueller KA, Low WY et al. 2021. Extended haplotype‐phasing of long‐read de novo genome assemblies using Hi‐C. Nature Communications 12: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyrene PM, Olmstead JW. 2012. The use of inter‐sectional hybrids in blueberry breeding. International Journal of Fruit Science 12: 269–275. [Google Scholar]
- Lyrene PM, Vorsa N, Ballington JR. 2003. Polyploidy and sexual polyploidization in the genus Vaccinium . Euphytica 133: 27–36. [Google Scholar]
- Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM. 2021. Busco update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Molecular Biology and Evolution 38: 4647–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL, Zimin A. 2018. MUMmer4: a fast and versatile genome alignment system. PLoS Computational Biology 14: e1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Baurens FC, Hervouet C, Salmon F, Delos JM, Labadie K, Perdereau A, Mournet P, Blois L, Dupouy M et al. 2020. Chromosome reciprocal translocations have accompanied subspecies evolution in bananas. The Plant Journal 104: 1698–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengist MF, Burtch H, Debelo H, Pottorff M, Bostan H, Nunn C, Corbin S, Kay CD, Bassil N, Hummer K et al. 2020a. Development of a genetic framework to improve the efficiency of bioactive delivery from blueberry. Scientific Reports 10: 17311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengist MF, Grace MH, Xiong J, Kay CD, Bassil N, Hummer K, Ferruzzi MG, Lila MA, Iorizzo M. 2020b. Diversity in metabolites and fruit quality traits in blueberry enables ploidy and species differentiation and establishes a strategy for future genetic studies. Frontiers in Plant Science 11: 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramoto N, Oda A, Tanaka H, Nakamura T, Kugou K, Suda K, Kobayashi A, Yoneda S, Ikeuchi A, Sugimoto H et al. 2018. Phenotypic diversification by enhanced genome restructuring after induction of multiple DNA double‐strand breaks. Nature Communications 9: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki EP, Kolbe DL, Eddy SR. 2009. Infernal 1.0: inference of RNA alignments. Bioinformatics 25: 1335–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novák P, Ávila Robledillo L, Kobližková A, Vrbová I, Neumann P, Macas J. 2017. Tarean: a computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Research 45: e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osuna‐Cruz CM, Paytuvi‐Gallart A, Di Donato A, Sundesha V, Andolfo G, Cigliano RA, Sanseverino W, Ercolano MR. 2018. PRGdb 3.0: a comprehensive platform for prediction and analysis of plant disease resistance genes. Nucleic Acids Research 46: D1197–D1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou S, Chen J, Jiang N. 2018. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Research 46: e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou S, Jiang N. 2018. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiology 176: 1410–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou S, Su W, Liao Y, Chougule K, Agda JRA, Hellinga AJ, Lugo CSB, Elliott TA, Ware D, Peterson T et al. 2019. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biology 20: 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panta GR, Rowland LJ, Arora R, Ogden EL, Lim CC. 2004. Inheritance of cold hardiness and dehydrin genes in diploid mapping populations of blueberry. Journal of Crop Improvement 10: 37–52. [Google Scholar]
- Pellicer J, Leitch IJ. 2020. The plant DNA C‐values database (release 7.1): an updated online repository of plant genome size data for comparative studies. New Phytologist 226: 301–305. [DOI] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. 2016. Transcript‐level expression analysis of RNA‐seq experiments with HISAT, StringTie and Ballgown. Nature Protocols 11: 1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preedy KF, Hackett CA. 2016. A rapid marker ordering approach for high‐density genetic linkage maps in experimental autotetraploid populations using multidimensional scaling. Theoretical and Applied Genetics 129: 2117–2132. [DOI] [PubMed] [Google Scholar]
- Putnam NH, O'Connell BL, Stites JC, Rice BJ, Blanchette M, Calef R, Troll CJ, Fields A, Hartley PD, Sugnet CW et al. 2016. Chromosome‐scale shotgun assembly using an in vitro method for long‐range linkage. Genome Research 26: 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Ogden EL, Bostan H, Sargent DJ, Ward J, Gilbert J, Iorizzo M, Rowland LJ. 2021. High‐density linkage map construction and QTL identification in a diploid blueberry mapping population. Frontiers in Plant Science 12: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu L, Hancock JF. 1995. Nature of 2n gamete formation and mode of inheritance in interspecific hybrids of diploid Vaccinium darrowi and tetraploid V. corymbosum . Theoretical and Applied Genetics 91: 1309–1315. [DOI] [PubMed] [Google Scholar]
- Qu L, Hancock JF. 2001. Detecting and mapping repulsion‐phase linkage in polyploids with polysomic inheritance. Theoretical and Applied Genetics 103: 136–143. [Google Scholar]
- Retamales JB, Hancock JF. 2018. Blueberries. Wallington, UK: CABI. [Google Scholar]
- Rezvoy C, Charif D, Guéguen L, Marais GAB. 2007. MareyMap: an R‐based tool with graphical interface for estimating recombination rates. Bioinformatics 23: 2188–2189. [DOI] [PubMed] [Google Scholar]
- Sayers EW, Agarwala R, Bolton EE, Brister JR, Canese K, Clark K, Connor R, Fiorini N, Funk K, Hefferon T et al. 2019. Database resources of the National Center for Biotechnology Information. Nucleic Acids Research 47: D23–D28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims J, Copenhaver GP, Schlögelhofer P. 2019. Meiotic DNA repair in the nucleolus employs a nonhomologous end‐joining mechanism. Plant Cell 31: 2259–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B. 2006. Augustus: ab initio prediction of alternative transcripts. Nucleic Acids Research 34: W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Waack S. 2003. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19: ii215–ii225. [DOI] [PubMed] [Google Scholar]
- Sultana N, Menzel G, Heitkam T, Kojima KK, Bao W, Serçe S. 2020. Bioinformatic and molecular analysis of satellite repeat diversity in Vaccinium genomes. Genes 11: 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Kumar S. 2021. Mega11: molecular evolutionary genetics analysis v.11. Molecular Biology and Evolution 38: 3022–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian T, Liu Y, Yan H, You Q, Yi X, Du Z, Xu W, Su Z. 2017. agriGO v.2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Research 45: W122–W129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorsa N. 1990. Quantitative analysis of meiotic chromosome pairing in triploid blueberry: evidence for preferential pairing. Genome 33: 60–67. [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9: e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H et al. 2012. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Research 40: e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Deng C, Hilario E, Albert NW, Lafferty D, Grierson ERP, Plunkett BJ, Elborough C, Saei A, Günther CS et al. 2021. A chromosome‐scale assembly of the bilberry genome identifies a complex locus controlling berry anthocyanin composition. Molecular Ecology Resources 22: 345–360. [DOI] [PubMed] [Google Scholar]
- Wu TD, Watanabe CK. 2005. Gmap: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21: 1859–1875. [DOI] [PubMed] [Google Scholar]
- Yang X, Zhao H, Zhang T, Zeng Z, Zhang P, Zhu B, Han Y, Braz GT, Casler MD, Schmutz J et al. 2018. Amplification and adaptation of centromeric repeats in polyploid switchgrass species. New Phytologist 218: 1645–1657. [DOI] [PubMed] [Google Scholar]
- Yang Z, Ge X, Yang Z, Qin W, Sun G, Wang Z, Li Z, Liu J, Wu J, Wang Y et al. 2019. Extensive intraspecific gene order and gene structural variations in upland cotton cultivars. Nature Communications 10: 2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Hulse‐kemp AM, Babiker E, Staton M. 2021. High‐quality reference genome and annotation aids understanding of berry development for evergreen blueberry (Vaccinium darrowii). Horticulture Research 8: 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Voorrips RE, Jansen J, Hackett CA, Ho J, Bink MCAM. 2016. Probabilistic multilocus haplotype reconstruction in outcrossing tetraploids. Genetics 203: 119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Tang D, Huang W, Yang Z, Zhang Y, Hamilton JP, Visser RGF, Bachem CWB, Buell CR, Zhang Z et al. 2020. Haplotype‐resolved genome analyses of a heterozygous diploid potato. Nature Genetics 52: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Example of a chimeric sequence in the W85_v2 assembly.
Fig. S2 Collinearity between the high‐density linkage map representing the W85 genome and the longest 12 scaffolds of the W85_v2 genome assembly.
Fig. S3 Hi‐C heatmap.
Fig. S4 Haplotype diversity in the W85_v2 chromosomes.
Fig. S5 PAV gene's enrichment analysis.
Fig. S6 Distribution of VacSat218 repeats compared to VacSat1 on W85 chromosomes.
Fig. S7 Organization and distribution of VacSat169 and 45S rDNA in W85 genome.
Fig. S8 Organization and distribution of VacSat61 repeats in W85 genome.
Fig. S9 FISH mapping of the telomeric motif (TTTAGGG)n and VactSat1 repeat.
Fig. S10 Meiotic pairing of the four chr‐6 homologs in the tetraploid blueberries DS and Jewel.
Fig. S11 Distribution of markers across eight DS × J homologs.
Fig. S12 Distribution of markers across eight R × A homologs.
Fig. S13 Collinearity between DS × J and R × A LGs 1–12 and the corresponding chromosomes 1–12 in the W85 and Draper genome assemblies.
Fig. S14 Meiotic configurations of Draper chr‐6 homologs and translocation chr‐610 and chr‐106 at diakinesis‐metaphase I.
Fig. S15 Distribution of markers across eight homologs LGs representing chromosomes 1–12 (I–XII) of the DxB mapping population.
Fig. S16 Segregation of the chromosomes carrying VacSat169 repeats at anaphase I/telophase I of DS and Draper.
Fig. S17 Chromosome distribution diakinesis‐metaphase I.
Fig. S18 Double reduction analysis in DS × J.
Fig. S19 Double reduction analysis in R × A.
Fig. S20 Preferential pairing analysis in DS.
Fig. S21 Preferential pairing analysis in Jewel.
Fig. S22 Preferential pairing analysis in Arlen.
Fig. S23 Preferential pairing analysis in Reveille.
Fig. S24 Gene ontology (GO) enrichment analysis of genes located on the regions exchanged between chromosome 6 and chromosome 10.
Methods S1 Plant material collection, DNA and RNA extraction, and sequencing.
Methods S2 Genome assembly.
Methods S3 Repetitive sequence analysis.
Methods S4 Construction of linkage maps.
Methods S5 Estimation of a preferential pairing parameter.
Methods S6 Fluorescence in situ hybridization (FISH) analysis.
Table S1 Summary of the W85 genome and transcriptome sequences used in this study.
Table S2 Summary statistics for the new assembled W85 genome (W85_v2) and comparison with W85_v1 and tetraploid Draper genomes.
Table S3 Summary of the number of chimeric regions corrected by Hi‐Rise, Hi‐C and linkage map.
Table S4 W85 genome assembly organized by pseudomolecules (chromosomes) and two phases (p0 and p1).
Table S5 Summary statistics of PacBio Iso‐Seq sequences mapped on the W85_v2 genome (p0 and p1).
Table S6 Assessment of the gene space coverage based upon alignment of the RNA‐sequencing data obtained from different tissues and treatments, against the W85_v2 assembly (p0 and p1).
Table S7 Summary of Busco analysis using plant dataset (odb10).
Table S8 Summary of gene prediction in the W85_v2 genome assembly.
Table S9 Summary statistics of W85 genes annotation.
Table S10 Predicted resistance‐genes in the W85_v2 assembly (p0 and p1).
Table S11 Statistics of the repetitive sequences annotated de novo in W85_v2 genome assembly (p0 and p1).
Table S12 Statistics of the gene prediction features in W85_v2 genome, tetraploid blueberry genome Draper, Solanum lycopersicum, Arabidopsis thaliana, Daucus carota and Beta vulgaris.
Table S13 Summary of the number of single‐nucleotide polymorphisms, INDELs and SV identified between the two haplotypes of the 12 chromosomes.
Table S14 Summary of present and absent (PAV) genes across the p0 and p1 haplotypes.
Table S15 Sequence characteristics of putative satellite repeats identified in the diploid blueberry W85 through the tarean pipeline.
Table S16 Distribution of single‐nucleotide polymorphisms into genotype classes and summary of integrated map of DS × J mapping population.
Table S17 Distribution of single‐nucleotide polymorphisms into genotype classes and summary of integrated map of R × A mapping population.
Table S18 Frequency of different types of pairing among the chromosomes carrying terminal VacSat169 repeats in DS 4432 (DS), Jewel and Draper at diakinesis‐metaphase I.
Table S19 Summary of marker distribution across chromosomes in Draper × Biloxi (B × D) mapping population.
Table S20 Predicted quadrivalent pairing and preferential pairing under the full model for the Draper Selection‐44392 × J mapping population.
Table S21 Predicted quadrivalent pairing and preferential pairing under the full model for the R × A mapping population.
Table S22 Summary of preferential pairing at the bivalent model for the B × D mapping population.
Table S23 Predicted quadrivalent pairing and preferential pairing under the full model for the B × D mapping population.
Table S24 Frequency of meiocytes at diakinesis‐metaphase I containing exclusively bivalents (24 II), at least one multivalent (IV or VI), and/or univalent (univ) in blueberry varieties.
Table S25 Summary of preferential pairing at the bivalent model for the DS × J mapping population.
Table S26 Summary of preferential pairing at the bivalent model for the R × A mapping population.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
PacBio and Illumina (Hi‐C, Chicago and Hi‐seq RNA‐seq) raw data that were used for genome assembly and annotations are available at the National Center for Biotechnology Information (NCBI) under BioProject No. PRJNA755705. The W85_v2.0 genome assembly, annotation, sequences of the genes, transcripts and proteins, can be accessible through the Genome Database for Vaccinium (GDV, https://www.vaccinium.org/). Sequences (assembly and annotation files) are available under the subdirectory Blueberry/Genomes/W85‐20 v2.0 (https://www.vaccinium.org/bio_data/1659687). The genome is available to search within the GDV database, Blast, PathwayCyc and JBrowse. The linkage maps were made available through GDV under the Search/map search subdirectory.