

Abstract
Blood‐based markers (BBMs) have recently shown promise to revolutionize the diagnostic and prognostic work‐up of Alzheimer's disease (AD), as well as to improve the design of interventional trials. Here we discuss in detail further research needed to be performed before widespread use of BBMs. We already now recommend use of BBMs as (pre‐)screeners to identify individuals likely to have AD pathological changes for inclusion in trials evaluating disease‐modifying therapies, provided the AD status is confirmed with positron emission tomography (PET) or cerebrospinal fluid (CSF) testing. We also encourage studying longitudinal BBM changes in ongoing as well as future interventional trials. However, BBMs should not yet be used as primary endpoints in pivotal trials. Further, we recommend to cautiously start using BBMs in specialized memory clinics as part of the diagnostic work‐up of patients with cognitive symptoms and the results should be confirmed whenever possible with CSF or PET. Additional data are needed before use of BBMs as stand‐alone diagnostic AD markers, or before considering use in primary care.
Keywords: Alzheimer's disease, appropriate use recommendations, blood‐based biomarkers, diagnosis, prognosis
1. INTRODUCTION
Blood‐based markers (BBMs) have recently shown promise to revolutionize the diagnostic and prognostic work‐up of Alzheimer's disease (AD), as well as to improve the design of interventional trials. We here aim to provide appropriate use recommendations for use of these BBMs in clinical practice and trials. To this aim, we discuss the current need for biomarkers; we briefly summarize the state‐of‐the‐art of results for the most promising BBMs; and, more importantly, we define research priorities needed to fill significant knowledge gaps. Finally, we describe the consensus appropriate use recommendations defined by this expert group for use of BBMs in the clinic as well in trials.
2. THE CURRENT NEEDS FOR BLOOD‐BASED AD BIOMARKERS
2.1. Clinical practice
Approximately 25% to 30% of patients with a clinical diagnosis of AD dementia are misdiagnosed when assessed at specialized dementia clinics, and the accuracy of clinical diagnosis is similar or even lower for other dementias, including frontotemporal dementia (FTD), dementia with Lewy bodies (DLB), and vascular dementia. 1 , 2 , 3 However, most patients with cognitive or behavioral symptoms are managed in primary care where the misdiagnosis is even higher. Fifty percent to 70% of symptomatic patients with AD are not recognized or correctly diagnosed in primary care today, because routine cognitive screening is not performed and there is a lack of easily accessible, time‐ and cost‐effective, and accurate diagnostic tools. 4 The problem is even worse in early stages of the disease, that is, in patients without dementia who have either subjective cognitive decline (SCD) or mild cognitive impairment (MCI). Further, clinicopathological studies highlight that the match between clinical phenotype and biology/neuropathology in neurodegenerative dementias is imperfect. 4 Such studies also note that the diseases have a preclinical prodrome during which symptoms may be absent or very mild and non‐specific despite active neuropathological processes. 4
Methods for individualized prognosis of progression from SCD and MCI to AD dementia are also largely lacking. Timely and accurate diagnosis of AD goes beyond providing patients with diagnostic and prognostic information. It extends to optimization of treatment strategies (e.g., with symptomatic cholinesterase inhibitors or possibly novel anti–amyloid beta [Aβ] therapies) and providing appropriate care. Misdiagnosis leads to unnecessary care‐seeking and costly investigations due to diagnostic uncertainty. The established cerebrospinal fluid (CSF) and positron emission tomography (PET) measures have excellent diagnostic properties, but they are less useful outside very specialized clinics due to limited accessibility, invasiveness (e.g., CSF measures require a lumbar puncture, and PET requires infusion of stable isotopes and exposure to radiation), contraindications (e.g., anticoagulant medication might prohibit lumbar puncture) and high costs (PET is expensive and not universally covered by health insurance). 4 This precludes use of CSF and PET biomarkers in most primary and secondary care settings worldwide. Thus, a major benefit of the use of BBMs in screening for AD pathology, or diagnosis, is that the collection of blood is less invasive and likely less costly than CSF or neuroimaging markers, and more feasible at the primary care levels where most individuals will present with cognitive symptoms. 4 , 5 Although the development of BBMs has been previously hindered by insufficient analytical sensitivities, recent studies suggest promising results using easily accessible and potentially scalable BBM tests. 4 , 5 For example, primarily in specialized memory clinics, blood‐based AD biomarkers have been shown to differentiate AD dementia from dementia caused by other neurodegenerative disorders with accuracies non‐inferior to CSF and PET biomarkers, and to predict future development of AD dementia in non‐demented patients with cognitive complaints. 4
2.2. Clinical trials
When targeting upstream pathologies, such as Aβ pathology, therapies will likely be more effective during the early preclinical (“pre‐symptomatic”) stages before manifest and irreversible neurodegeneration has already occurred. It is also possible that certain pathologies (e.g., Aβ pathology) might trigger downstream events (e.g., spread of neocortical tau aggregates and synaptic degeneration), in which the latter eventually becomes independent from the initiating event. 6 Therefore, diagnostic biomarkers identifying AD pathology before the onset of overt clinical symptoms are needed to recruit suitable individuals with early disease to clinical trials. 4 , 5 Today, clinical trials typically use Aβ‐PET or CSF to screen for preclinical AD in cognitively normal individuals. However, the very high costs and low accessibility (especially in more diverse socioeconomic settings), and high number of screen failures (i.e., individuals who turn out to have normal PET or CSF results) make this approach very challenging. Consequently, clinical trials in this early stage of the disease have been hampered by the difficulty to recruit large numbers of participants across diverse settings. For example, it took the A4 (Anti‐Amyloid Treatment in Asymptomatic Alzheimer's) trial, which was the first phase 3 trial in preclinical AD, 3.5 years and > 4000 amyloid PET scans to identify and randomize 1169 participants with elevated brain amyloid. Therefore, it is very likely that blood‐based AD biomarkers will be increasingly used to identify those more likely to have pre‐symptomatic AD, who then can undergo PET or CSF measurements to confirm preclinical AD before entering the trial. 4 , 5 Even for clinical trials involving prodromal or dementia due to AD, blood biomarkers may substantially reduce the cost of screening and time to fully enroll the trial. Further, there is a need for BBMs to study drug target engagement or pharmacodynamic drug effects on downstream disease processes like neurodegeneration or neuroinflammation. 4 As an example, in other neurological diseases, like multiple sclerosis (MS), spinal muscular atrophy, and human immunodeficiency virus (HIV)‐associated neurocognitive dysfunction, plasma neurofilament light (NfL) concentration decreases in response to disease‐modifying treatment as a sign of reduced neurodegeneration. 7 , 8 , 9
RESEARCH IN CONTEXT
Systematic Review: The authors reviewed the literature using conventional (e.g., PubMed) sources and meeting abstracts and presentations. We found that in the last few years the number of publications on Alzheimer's disease (AD)‐associated blood‐based biomarkers (BBMs) has increased dramatically, showing great promise for especially plasma phosphorylated tau, amyloid beta, glial fibrillary acidic protein, and neurofilament light for future use in both clinical practice and trials. However, few prospective studies have investigated the implementation of such BBMs in more heterogeneous populations. These pertinent works are appropriately cited.
Interpretation: In the current review we recommend use of AD‐associated BBMs as (pre‐)screeners in trials and cautious introduction of BBMs in clinical practice, provided AD status is confirmed whenever possible using cerebrospinal fluid or positron emission tomography.
Future Directions: In the current review, we summarize prioritized research needed to be performed before widespread use of AD‐associated BBMs in clinical trials and practice, including different types of preanalytical, analytical, and real‐world clinical studies.
3. WHAT BBMS ARE AVAILABLE TODAY AND HOW DO THEY PERFORM?
3.1. Plasma Aβ42/Aβ40
CSF Aβ42/Aβ40 is a robust biomarker for cerebral Aβ pathology, with a clear bimodal distribution and a small gray zone of results close to the cut‐point for positivity; a low ratio reflects selective depletion of Aβ42 from the CSF due to deposition in the growing plaques. 4 , 10 Similar reductions in Aβ42/Aβ40 can be seen in plasma. Several immunoprecipitation mass spectrometry (IP‐MS) methods have been developed in which Aβ from plasma is extracted and subjected to MS‐based quantification. Using such methods, clear group‐level reductions in plasma Aβ42/Aβ40 levels are observed in amyloid PET‐positive compared to PET‐negative people. 11 , 12 , 13 Further, immunochemical tests for plasma Aβ42/Aβ40 have been developed that are easier to implement in regular clinical chemistry laboratories. 14 However, in one head‐to‐head comparison, measurement of the Aβ42/Aβ40 ratios with the immunoassays exhibited lower diagnostic performance for detection of Aβ pathology compared to certain IP‐MS methods (the areas under the curve ranged between 0.69 and 0.78 for the different immunoassays compared to 0.86 for the best‐performing IP‐MS method). 15 An important feature of plasma Aβ42/Aβ40 is that the levels are fully changed already during the pre‐symptomatic disease stages; this is the reason this biomarker, like CSF Aβ42/Aβ40, can identify Aβ pathology in cognitively unimpaired (CU) people with accuracies as high as those observed in cognitively impaired individuals. 14
The major problem with plasma Aβ42/Aβ40 as an Aβ pathology test is the small fold change between Aβ‐positive and Aβ‐negative individuals (an 8%–15% reduction compared to 40%–60% in CSF). 4 , 15 The likely explanation is that the Aβ pathology‐related reduction in plasma Aβ42/Aβ40 occurs on top of peripherally present Aβ, because Aβ produced in extracerebral tissues are conceivably not affected by brain Aβ pathology. Consequently, plasma Aβ42/Aβ40 is a less robust brain Aβ pathology biomarker than CSF Aβ42/Aβ40 for biological reasons. 16 It is challenging to standardize and maintain stability of this type of test over time in clinical laboratory practice with the rigor needed to reliably detect the small difference between Aβ‐positive and ‐negative individuals. Stringent pre‐analytical and analytical protocols can help mitigate the robustness issue with promising results, 17 and there is today one clinical‐grade IP‐MS test for plasma Aβ42/Aβ40 available. However, given the biological reasons for lack of robustness, we should also continue to look for other blood biomarkers and/or combinations with other measures (other markers or demographics, see below) to detect Aβ pathology.
Research priorities (Table 1):
Biomarker development studies to make plasma Aβ tests that reflect the central nervous system (CNS)‐specific fraction of the peptide (e.g., by quantifying CNS‐specific forms of Aβ, or by isolating CNS‐specific vesicles) or have a greater fold change between Aβ‐positive and ‐negative individuals (e.g., C‐terminally extended Aβ species, misfolded Aβ, or CNS‐specific post‐translational modifications).
Real‐world studies on the robustness of plasma Aβ42/Aβ40 as a diagnostic test for cerebral Aβ pathology, incorporating the full range of potential pre‐analytical and analytical sources of variation, for example, by repeated samplings of the same individuals over a restricted time window. What are the impacts of such variations on the biomarker results and clinical decisions? What is the total allowable error for plasma Aβ tests to be clinically useful?
Real‐world studies in diverse populations in which the study participants undergo reference standard assessment of Aβ pathology (amyloid PET, CSF Aβ42/Aβ40, or neuropathology) to validate the results.
Better understanding of the longitudinal intra‐individual biological and disease‐associated variability and potential impact of medical comorbidities and concomitant medications.
TABLE 1.
Research priorities
| Aβ |
| Brain‐specific plasma Aβ tests: Biomarker development studies to make plasma Aβ tests that reflect the CNS‐specific fraction of the peptide (e.g., by quantifying CNS‐specific forms of Aβ, or by isolating CNS‐specific vesicles) or have a greater fold change between Aβ‐positive and ‐negative individuals (e.g., C‐terminally extended Aβ species, misfolded Aβ, or CNS‐specific post‐translational modifications). |
| Clinical robustness of plasma Aβ42/Aβ40: Real‐world studies on the robustness of plasma Aβ42/Aβ40 as a diagnostic test for cerebral Aβ pathology, incorporating the full range of potential pre‐analytical and analytical sources of variation, for example, by repeated samplings of the same individuals over a restricted time window. What are the impacts of such variations on the biomarker results and clinical decisions? What is the total allowable error for plasma Aβ tests to be clinically useful? |
| P‐tau |
| Different plasma p‐tau isoforms: Head‐to‐head studies comparing the performance of different p‐tau isoform tests in different clinical contexts and disease stages, using relevant reference standards. |
| Fully automated p‐tau assays. Development and validation of novel fully automated assays for different p‐tau markers (which has already been developed for Aβ42/Aβ40, NfL, and GFAP). |
| Longitudinal change in plasma p‐tau: Longitudinal studies of different pP‐tau variants examining biomarker stability and what a clinically relevant change in the concentration is. Further, establish associations between longitudinal changes in different p‐tau variants with changes in tau‐PET, imaging measures of neurodegeneration, and cognition. |
| NfL |
| Change in NfL in relation to imaging markers of neurodegeneration: Longitudinal studies examining the diagnostic performance of plasma NfL for detection of neurodegeneration in different age groups and diseases, using, for example, longitudinal MRI and FDG PET as biomarkers for neurodegeneration. Such longitudinal studies should also determine the intra‐individual biomarker stability of plasma NfL. |
| Brain‐specific isoforms of neurofilaments: The development of assays for CNS‐specific neurofilaments. |
| GFAP |
| Plasma GFAP versus neuropathology: Validation of the plasma GFAP against neuropathology to understand its relationship with disease mechanisms and key pathological substrates of the better separation for plasma (serum) versus CSF when it comes to AD. |
| Further characterization of plasma GFAP: Studies examining the diagnostic performance of plasma GFAP for detection of astrocytic activation in different age groups and in different clinical contexts using reference standard tests for neurodegeneration and AD pathology. |
| All |
| Diverse and representative populations: Real‐world studies in diverse and representative populations in which the study participants undergo reference standard assessment of Aβ and tau pathophysiology (preferably with neuropathological confirmation, or at least PET and CSF measures of Aβ and tau pathological changes) and evaluation of the causes of false positive and negative plasma biomarker outcomes. |
| Establishing the best plasma biomarker combinations: Establish the optimal combinations of plasma biomarkers in each clinical scenario when only using the top‐performing assays for all of the included BBMs (and when using relevant reference standards). |
| Compare the best plasma biomarker combinations versus CSF and PET: Compare the clinical performance of the best blood biomarker combinations to the clinical performance of the best CSF and/or PET biomarkers whenever possible to better understand when and how BBM can substitute for CSF and PET. |
| Improved diagnostic work‐up: Determine whether the plasma biomarker combinations improve the more basic clinical assessments done in most clinics today (i.e., assessment done without advanced CSF and PET assessments), and whether the addition of other easily accessible tests (such as brief cognitive tests or genetics) improve the diagnostic and prognostic work‐up even further. |
| Interpretation of biomarker results: Develop tools for interpretation of the results and for communication to the physician as well as to the patients. Define the different types of information that can be obtained, for example, diagnostic as well as prognostic information. |
| Potential confounders: Studies examining clinical confounders and biological factors, including race and ethnicity, peripheral neuropathies and other neurologic diseases, BMI, and kidney disease and the relative effects on the clinical performance of plasma Aβ42/Aβ40, p‐tau, NfL, and GFAP in large cohorts. |
| Biological variation: Study of other variables relevant for real‐world implementation, such as biological variation over time (intra‐day and between‐day variation) in individuals with different conditions. |
| Pre‐analytical protocol: Studies to refine the pre‐analytical protocols even further for the most relevant plasma biomarkers of today. For all new plasma biomarkers, the pre‐analytical variation should be defined, and for this preferably, as central and accessible biorepository is available. |
| Assay standardization. The development of candidate reference materials and methods for the most promising plasma AD biomarker assays. |
| Total random error: Study the total random error of a biomarker result by repeatedly collecting blood and repeatedly analyzing a certain biomarker over a shorter time period in ≈40 to 50 individuals (with and without AD). |
| Systematic error: To determine systematic error of a certain assay or platform by analyzing samples from the same pools of plasma over extended time periods and in different laboratories. |
| Clinical robustness: To determine clinical robustness in a real‐world setting by using the assay in extended prospective studies over 1 to 3 years where (1) pre‐defined cut offs are used, (2) samples are analyzed continuously over the study period (e.g., on a daily or weekly basis rather than in single batches), and (3) an appropriate reference standard is used (like Aβ PET). |
| Best markers as (pre‐)screeners: Studies determining which combinations of BBMs are consistently most optimal for detection of AD pathology changes in either preclinical (asymptomatic) or symptomatic AD when using the best assays for each biomarker. |
| Prospective validation of use of BBMs as (pre‐)screeners: Prospective studies with sample‐to‐sample analyses over extended time periods (a couple of years) and with a valid outcome (like Aβ‐PET, tau‐PET, or CSF AD biomarkers) to determine whether the clinical robustness of a BBM (or combination of BBMs) over time is high enough for use as pre‐screeners in clinical trial settings. |
| BBMs replacing PET or CSF when determining AD status for inclusion in trials: Determining whether use of conservative cut‐offs for certain BBMs (or combinations of BBMs) can result in valid predictions of the presence of AD pathological changes (>90%–95%), and thereby PET or CSF would not be needed for inclusions in AD trials of individuals with clearly abnormal BBMs. However, studies would also need to define the gray zone (with uncertain BBM results) where PET or CSF is needed for accurate detection of AD‐status. |
| Longitudinal plasma p‐tau, Aβ42/Aβ40, NfL, and GFAP levels in observational trials: Longitudinal, large‐scale, and diverse observational trials should determine the longitudinal changes in BBM over time, and whether such changes relate to other changes in more established biomarkers (such as MRI, CSF, and PET) and changes in clinically relevant outcomes (e.g., cognition, motor function, and activities of daily living). |
| Longitudinal plasma p‐tau, NfL, and GFAP levels in clinical trials: Study plasma p‐tau, NfL, and GFAP as exploratory outcome markers in different clinical trials to establish whether drug‐induced reductions in biomarker concentrations associated with clinically beneficial outcomes. |
| Developing and validating BBMs in specialized memory clinics: Evaluation of BBMs in diverse (real life) secondary and tertiary memory clinic populations should be done prospectively, using predefined cut‐offs, and using relevant and accurate reference standards. Further, we need to identify the optimal combinations of easily accessible and time‐/cost‐effective biomarkers and tests in memory clinic settings and study whether they outperform the clinical assessments used today in most such secondary and tertiary clinics. We should also study whether certain BBMs (or combinations with other easily accessible diagnostic methods) perform non‐inferior to CSF and PET, that is, can certain BBM‐based algorithms be used alone to support an AD diagnosis, or should they only be used as a gatekeeper to CSF/PET. |
| Improved patient management in memory clinics: Determine whether addition of BBMs to standard clinical assessments and imaging improves the diagnosis and significantly alters the management of the patients. |
| Developing and validating BBMs in primary care: Perform prospective studies in primary care settings, including representative and diverse populations with cognitive symptoms, where BBMs and brief cognitive tests and other easily accessible methods are performed in the primary care setting. However, the reference standard must be of high quality and preferably include CSF or PET for AD. |
| Combinations of BBM with other easily accessible and scalable tools in primary care: Identify the optimal combinations of easily accessible and time‐/cost‐effective biomarkers and tests in this setting (e.g., combining BBMs with digital cognitive tests). |
| Improved patient management in primary care: Study whether BBMs outperform what is already available today in primary care (standard of care today), and if they also improve diagnosis and management (including treatment decisions and referrals to memory clinics). |
Abbreviations: Aβ, amyloid beta; AD, Alzheimer's disease; BBM, blood‐based biomarkers; BMI, body mass index; CNS, central nervous system; CSF, cerebrospinal fluid; FDG, fluorodeoxyglucose; GFAP, glial fibrillary acidic protein; MRI, magnetic resonance imaging; NfL, neurofilament light; PET, positron emission tomography; p‐tau, phosphorylated tau.
3.2. Plasma phosphorylated tau
Several research groups have developed very sensitive phosphorylated tau (p‐tau) assays for use as blood biomarkers for AD, including assays for tau phosphorylated at amino acid 181 (p‐tau181), 217 (p‐tau217), or 231 (p‐tau231). It should be noted that all available p‐tau assays measure phospho‐forms of tau using antibodies that are directed to the N‐terminus or mid‐domain of the protein, because these forms of tau are present at much higher concentrations in biofluids than full‐length or C‐terminal tau due to proteolytic processing of tau in the release process of the molecule from neurons into biofluids. 18 Neuropathology‐based studies have shown that plasma p‐tau levels are related to both the density of Aβ plaques and tau tangles 19 and that levels of different plasma p‐tau variants (i.e., p‐tau181, p‐tau217, and p‐tau231) can differentiate between cases with significant AD brain pathology from those without. 20 , 21 , 22 , 23 , 24 , 25 Importantly, increased plasma levels of these p‐tau variants have specifically been observed in AD and not in other tauopathies, including primary age‐related tauopathy, progressive supranuclear palsy, corticobasal degeneration, or Pick's disease. It is presently unclear how well p‐tau markers in biofluids detect tau pathophysiology related to tangles, neuritic threads, tau filaments decorating neuritic plaques, and tau‐containing granulovacuolar degeneration bodies, but all of these may contribute to or associate with the p‐tau increase seen in AD. 19 , 26 In several large‐scale clinic‐based studies, plasma p‐tau has been shown to accurately separate AD dementia from other neurodegenerative diseases with high diagnostic accuracy. 20 , 22 , 23 , 24 , 27 , 28 Plasma p‐tau levels are increased ≈250% to 600% in AD dementia compared to the levels observed in non‐AD neurodegenerative disease; the largest relative increases in AD dementia are often observed for p‐tau217. 22 , 28 In the memory clinic setting, the diagnostic performance of plasma p‐tau217 has been shown to be similar to both CSF biomarkers and tau‐PET imaging. 22 Further, in patients with MCI, both plasma p‐tau181 and p‐tau217 have been shown to accurately predict future cognitive decline and conversion to AD dementia in the subsequent 2 to 6 years. 20 , 29 , 30 , 31 The comparison of different plasma p‐tau variants to detect AD neuropathologic changes in CU individuals is ongoing. Several studies have shown that plasma p‐tau181, p‐tau217, and p‐tau231 start to change when Aβ‐PET becomes abnormal, and some studies suggest that p‐tau231 might be changing slightly earlier than the other p‐tau markers. 24 , 32 A few studies also show that plasma p‐tau can predict subsequent cognitive decline and worsening of fibrillar tau pathology in CU individuals. 29 , 33 , 34 , 35
There are currently several high‐performing plasma p‐tau immunochemical assays with similar performance, which bodes well for successful clinical implementation, but there are also commonly used assays with lower performance according to head‐to‐head comparisons. 22 , 28 , 36 , 37 There is currently one assay that has been granted a Breakthrough Device designation by the US Food and Drug Administration (FDA) as an aid in diagnostic evaluation of AD (p‐tau181 27 ), and additional tests are in clinical development. Considerations for further research include establishment of factors influencing biological, pre‐analytical, and analytical variation and potential confounders, such as co‐occurrence of cerebrovascular and cardiovascular diseases and performance in more diverse populations.
Plasma p‐tau levels increase gradually over time in early stages of AD, which may relate to the number of AD‐affected neurons that still manage to synthesize and secrete tau, and especially p‐tau217 shows increase during both the preclinical and prodromal stages of the disease. 38 An emerging use of plasma p‐tau is to detect and monitor effects on tau pathophysiology by anti‐Aβ antibodies in clinical trials. During the Alzheimer's Association International Conference 2021, reduced plasma p‐tau217 concentration in response to donanemab treatment were reported (unpublished results), and similar results have been shown for aducanumab (in this case p‐tau181 reduction). 39
Research priorities (Table 1):
Head‐to‐head studies comparing the performance of different p‐tau isoforms in different clinical contexts and across disease stages. In such studies, it is important to note differences in the used platforms and materials (e.g., antibodies), and MS‐based methods for simultaneous detection of different p‐tau variants might be an advantage in this setting.
Development and validation of novel fully automated assays for different p‐tau markers (which has already been developed for Aβ42/Aβ40, NfL, and glial fibrillary acidic protein [GFAP]).
Longitudinal studies of different p‐tau variants examining intra‐individual biomarker stability and what a clinically relevant change in the concentration is. Further, establish associations between longitudinal changes in different p‐tau variants with changes in tau‐PET, imaging measures of neurodegeneration, and cognition.
Real‐world studies in diverse populations in which the study participants undergo reference standard assessment of Aβ and tau pathophysiology (preferably with neuropathological confirmation, or at least PET measures of Aβ and tau pathological changes) and evaluation of the causes of false positives and negatives.
Study plasma p‐tau as an exploratory outcome marker in clinical trials to establish whether drug‐induced reductions in biomarker concentrations associated with clinically beneficial outcomes.
3.3. Plasma NfL
For many years, CSF NfL has been used as a neuroaxonal injury marker. The highest NfL concentrations in CSF and blood are seen in amyotrophic lateral sclerosis (ALS), FTD, atypical parkinsonian disorders, MS, and HIV‐associated neurocognitive dysfunction. 40 , 41 A more modest elevation compared to age‐matched controls is seen in AD. The biomarker can be measured in both CSF and plasma (or serum), and virtually all CSF findings have been replicated in blood with sensitive assays. 40 , 42 Familial AD mutation carriers show a gradual change in blood NfL levels ≈1 decade before expected clinical onset, which probably marks the onset of neurodegeneration, and the higher the increase, the more rapid clinical disease progression. 43 , 44 In sporadic AD, there are associations of increased plasma NfL concentration with Aβ and tau positivity, as well as with longitudinal neurodegeneration as determined by magnetic resonance imaging (MRI); however, this is mainly visible at more advanced dementia stages. Moreover, there is a larger overlap between different AD disease stages than in familial AD, 45 most likely due to the multitude of different age‐related and neurodegenerative changes that may result in NfL increase in people older than 70 years of age. Importantly, NfL has a strong age relationship. This is likely the most important challenge when considering how to use the test clinically. In anti‐Aβ antibody trials, attenuated increases of CSF NfL have been reported, 46 , 47 but whether this is seen in blood as well is currently unknown. A positive feature of this markers is its strong pre‐analytical robustness, thus being unaffected by common variations in sample handling before analyses. 48 More data on biomarker performance in diverse populations, as well as on biological variation (e.g., influence of renal function, body mass index [BMI], and peripheral neuropathy), are also needed. Several laboratories around the globe are already analyzing plasma NfL in clinical laboratory practice.
Research priorities (Table 1):
Longitudinal studies examining the diagnostic performance of plasma NfL for detection of neurodegeneration in different age groups and diseases, using, for example, longitudinal MRI and fluorodeoxyglucose PET as biomarkers for neurodegeneration. Such longitudinal studies should also determine the intra‐individual biomarker stability of plasma NfL.
The development of assays for CNS‐specific neurofilaments.
Studies examining clinical confounders and biological factors across diverse populations, including peripheral neuropathies, BMI, and kidney disease and the relative effects on the clinical diagnostic and prognostic performance in large cohorts.
Examine plasma NfL as an exploratory outcome marker in clinical trials to establish whether drug‐induced reductions in biomarker concentrations associate with clinically beneficial outcomes.
3.4. Plasma GFAP
In AD, glial activation appears to be a reaction to Aβ pathology, for example, to degrade Aβ or to counteract induced excitotoxity or to supplement energy. For glial biomarkers, blood tests are difficult, due to high extra‐cerebral expression of many of the proteins, for example, in macrophages, making the blood tests less reflective of brain changes. However, one biomarker shows promise in this context: GFAP. The strongest expression of this protein is seen in brain astrocytes, and its blood concentration is strongly reflective of Aβ accumulation in the brain. 49 , 50 , 51 , 52 The association with Aβ pathology appears stronger for plasma GFAP than CSF GFAP, and plasma GFAP appears to be specific to Aβ pathology because it is not associated with fibrillar tau pathology when adjusting for Aβ pathology. 49 Although GFAP is likely not AD‐specific, the magnitude of change in non‐AD neurodegenerative diseases, except for progranulin (GRN) mutation‐related FTD, is relatively small compared to AD. 53 , 54 Studies have shown that plasma GFAP levels can predict subsequent cognitive change and AD dementia in patients with MCI 52 and cognitive decline in CU subjects. 55 , 56 Mild traumatic brain injury and cerebrovascular insults are important potential confounders. 57 , 58 Clinical‐grade assays for plasma GFAP exist but more studies are needed in a memory clinic context.
Research priorities (Table 1):
Validation of the plasma GFAP against neuropathology to understand its relationship with disease mechanisms and key pathological substrates of the better separation for plasma (serum) versus CSF when it comes to AD.
Studies examining the diagnostic performance of plasma GFAP for detection of astrocytic activation in different age groups and in different clinical contexts using reference standard tests for neurodegeneration and AD pathology.
Studies examining pre‐analytical confounders and biological factors, including peripheral neuropathy and kidney disease.
Study plasma GFAP as an exploratory outcome marker in different clinical trials to establish whether drug‐induced reductions in biomarker concentrations associated with clinically beneficial outcomes.
Studies examining clinical confounders and biological factors across diverse populations, including peripheral neuropathies, BMI, and kidney disease and the relative effects on the clinical diagnostic and prognostic performance in large cohorts
3.5. Combination of BBMs
Plasma biomarkers can be combined with each other or with other easily accessible tests to increase clinical performance. Several studies have investigated this topic, and prototype online algorithms have been developed for certain clinical scenarios in which different biomarkers are combined with clinical and demographic variables to obtain individualized outcomes. 30 , 31 , 59 For example, p‐tau might be combined with Aβ42/Aβ40 ratio to be able to detect range of amyloid levels and add to the prediction of cognitive decline. However, as mentioned above, certain plasma Aβ assays and p‐tau assays perform less optimally than others. The type of assays used to quantify a certain biomarker (e.g., Aβ42/Aβ40) in such analyses are important to notice.
3.5.1. Detecting amyloid pathology in CU and MCI
Plasma Aβ42/Aβ40 is the most studied biomarker for cerebral Aβ pathology in both CU and MCI individuals. 4 Many studies have consistently shown the added value of combining plasma Aβ42/Aβ40 with apolipoprotein E (APOE) genotype in this setting, 14 , 15 , 17 but this is somewhat controversial, because APOE genotype is an inborn risk indicator rather than a biomarker for Aβ pathology (the CSF Aβ tests work independently of APOE genotype to detect cerebral Aβ pathology 60 ). Evidence indicates that that p‐tau 59 and/or GFAP 55 might also add independent information. The addition of plasma p‐tau to plasma Aβ42/Aβ40 might have most added value in MCI compared to CU, because the plasma p‐tau levels increase with disease progression, but also because the performance of plasma Aβ42/Aβ40 might be slightly lower in MCI than in CU. 59
3.5.2. Distinguishing AD dementia from other dementias
In the dementia stage, high‐performing plasma p‐tau assays may be good enough on their own to differentiate AD dementia from other dementias, with similar performance as CSF and amyloid or tau PET markers. 22 As mentioned, plasma p‐tau217 performs slightly better than the other plasma p‐tau variants for AD diagnosis, likely because of the relatively large increase of this marker in the dementia stage of AD compared to other p‐tau isoforms. 22 , 28
In patients with dementia, a high plasma NfL value together with a normal p‐tau value, might indicate that the underlying etiology is a non‐AD dementia with substantial axonal degeneration like FTD or corticobasal degeneration. 4 , 10 , 23
3.5.3. Predicting development of AD dementia in non‐demented individuals
When using high‐performing assays, both plasma p‐tau181 and p‐tau217 have consistently been shown to be accurate markers when predicting future development of AD dementia in symptomatic patients with either MCI or SCD. 20 , 29 , 30 , 31 Similar results have been obtained for p‐tau231 24 and for GFAP. 52 , 56 The value of also adding plasma NfL or plasma Aβ42/Aβ40 is much lower and therefore less certain in this particular setting. 30 , 31 However, when plasma p‐tau is combined with other easily accessible methods, including APOE genotype and brief cognitive tests, the predictive algorithm performs as accurately as CSF‐based algorithms and clearly outperforms the prediction made by dementia experts. 30
3.5.4. Predicting global cognitive decline in non‐demented individuals
There is also a need to predict future cognitive decline at an individual level, which is not only caused by AD pathological changes. In CU individuals a combination of plasma p‐tau, Aβ42/Aβ40, and NfL was associated with subsequent cognitive decline. 33 However, in patients with MCI a combination of plasma p‐tau and NfL was found to predict decline in global cognition. 31
Research priorities (Table 1):
Study the optimal combinations of plasma biomarkers in each clinical scenario when only using high‐performing and analytically validated assays for all the included biomarkers.
Compare the clinical performance of the best blood biomarker combinations to the clinical performance of the best CSF and/or PET biomarkers whenever possible.
Determine whether the plasma biomarker combinations improve the more basic clinical assessments done in most clinics today (i.e., assessment done without advanced CSF and PET assessments), and whether the addition of other easily accessible tests (such as brief cognitive tests or genetics) improve the diagnostic and prognostic work‐up even further.
Develop tools for interpretation of the results and for communication and education to the physician as well as to the patients. Define the different types of information that can be obtained, for example, diagnostic as well as prognostic information.
4. IDENTIFY KEY STEPS NEEDED TO BE TAKEN BEFORE WIDESPREAD USE OF BBMS IN GENERAL
Assay validation against clinically relevant reference standards
Once a test has been analytically validated (meaning that the test specifically measures what it is supposed to measure with a high enough precision and dynamic range, see Andreasson et al. 61 for details), the diagnostic performance in clinically relevant settings needs to be established, preferably in relation to neuropathology and/or reference/gold standard tests. 19 , 20 , 21 , 22 , 23 , 24 However, novel plasma biomarkers can also be validated against other in vivo methods that reflect AD pathology (although not optimal, this approach may give an acceptable traceability chain toward neuropathology). Aβ‐PET is the most widely used reference standard given the fact that this method has a high agreement with a presence of Aβ plaques in the brain, and it was the only FDA‐approved measure of Aβ pathology until recently. 4 However, Aβ‐PET and CSF Aβ42‐based ratios (CSF Aβ42/Aβ40 or Aβ42/p‐tau) are largely interchangeable to determine the Aβ status 4 and CSF analysis is therefore commonly used as a valid reference standard instead of Aβ‐PET, with the first CSF Aβ42/Aβ40 in vitro diagnostic test cleared by the FDA in 2022. Emerging data have also shown that tau‐PET imaging reflects fibrillar tau pathology, 4 which is why this method might be used as a reference standard in addition to CSF or PET markers of Aβ pathology. In general, one should be careful and whenever possible not use clinic‐based syndrome diagnosis as reference standard that is not confirmed using either relevant in vivo biomarkers (e.g., certain PET and CSF methods) or neuropathology. Further, any clinic‐based diagnosis must have been established blinded to the outcome of the index test being evaluated.
Another endpoint that is important and relevant for patients and clinical trialists is cognitive decline. Important questions include: What is an individual's probability of cognitive decline and conversion to dementia over a certain time? How many years of relatively good function are ahead? Therefore, a better understanding of prognosis for a given biomarker level at the individual level is needed. Among CU populations, cognitive composites similar to the Preclinical Alzheimer's Cognitive Composite (PACC) are often used to detect a relevant cognitive change over time in research and clinical trial settings, 62 and in cognitively impaired populations (e.g., prodromal AD and mild AD dementia) the Clinical Dementia Rating scale (CDR), Alzheimer's Disease Assessment Scale‐Cognitive Subscale (ADAS‐cog), or Mini‐Mental State Examination (MMSE) are often used. When using progression to a certain type of dementia as outcome, the clinic‐based diagnosis must again have been established blinded to the outcome of the test being evaluated, and as mentioned above it is preferred that a clinic‐based dementia diagnosis (e.g., AD dementia) is confirmed using either validated in vivo biomarkers (e.g., certain PET and CSF methods) or neuropathology.
Research priorities (Table 1):
Perform head‐to‐head comparisons of different plasma biomarker assays when using relevant reference standards.
Establish the most optimal combinations of easily accessible biomarkers when using appropriate reference standards.
Establish cut‐points relevant for the different contexts of use.
Pre‐analytical protocol
The AD CSF biomarker analysis experience underscored the relevance of not only analytical, but also pre‐analytical, standardization. 63 , 64 Variation was partially caused by the aggregation‐prone nature of AD‐relevant proteins, especially Aβ, making them stick to certain plastics or aggregate in vitro. With this background, studies have started to define the effect of pre‐analytical variation and define protocols for sample handling of blood, plasma, and serum. In a large multi‐center study, the effects of frequently present variations in existing cohorts were evaluated and variables deemed relevant by experts on a range of plasma biomarkers. 48 The following variables were studied: (1) type of tubes used to collect the blood, (2) time between sample collection and centrifugation (for plasma), (3) centrifugation parameters (for plasma), (4) time from sample collection to storage in a freezer, (5) temperature of samples during the different processing steps, (6) the aliquot size used for storage in freezer, and (7) number of freeze/thaw cycles. After analyses of Aβ42/Aβ40, p‐tau181, total tau, GFAP and NfL, an easy‐to‐use standardized operating procedure for plasma handling was established. 48 We recommend using this protocol for collection of blood in both research settings and clinical trials and practice. Further, we note that it is important that a consensus protocol for pre‐analytical handling of blood samples is constructed in such way that it can be used outside specialized settings to enable widespread implementation globally. For example, according to the current protocol samples can be stored in a refrigerator (at 2–8°C) for 24 hours before centrifugation, and another 24 hours before being either analyzed or frozen. 48
Research priorities (Table 1):
Studies to refine the pre‐analytical protocols even further for the most relevant plasma biomarkers of today.
For all new plasma biomarkers, the pre‐analytical variation should be defined; a central and accessible biorepository would facilitate this work.
Study of other variables relevant for real‐world implementation, such as biological variation over time (intra‐day and between‐day variation) in individuals with different conditions.
4.1. Clinical‐grade assays
For real‐world implementation in clinical laboratories, it is critically important to have access to high precision in vitro diagnostic assays, and currently several such assays are developed for BBMs. This means that assays need to be optimized and analytically validated, but additionally must be produced in a way that guarantees analytical stability in the measurements (a low lot‐to‐lot variation and bias), which is the responsibility of the assay providers. However, in‐house validation of novel assays, as well as longitudinal assay performance monitoring through internal and external quality control programs, is essential as well, and guidelines for such validation and monitoring for AD biomarkers have been developed. 61 For the CSF biomarker assays, batch and inter‐laboratory variation were major sources of variation. 65 , 66 With the advent of several plasma biomarker assays by several commercial providers, the need for certified reference materials and methods to standardize the assays to each other has become urgent . 10 To achieve this, bodies such as the International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) and the Joint Committee for Traceability in Laboratory Medicine (JCTLM), entitled to certify such methods, should ideally be involved in the development process. Such standardization is crucial to directly compare results between different studies, and ultimately to define global reference limits and cut‐offs. 10 In the short term, such cut‐offs will be assay‐specific. These can be verified locally before implementation. Assay performance over time must then be monitored to ensure that the cut‐offs remain valid.
Research priorities (Table 1):
Expansion of the Alzheimer's Association Global Biomarker Standardization Consortium (GBSC) Quality Control (QC) Program for CSF biomarkers to plasma.
The development of certified reference materials and methods for the most promising plasma AD biomarker assays.
4.2. Non–AD‐associated factors affecting biomarker concentrations
Certain (confounding) factors, which are not associated with the actual neurodegenerative disease, may affect BBM concentrations. Such factors affecting CSF biomarker levels have been widely studied where, for example, older age is strongly associated with higher levels of NfL, even when correcting for known disease pathologies in the brain. 67 , 68 However, blood levels of these biomarkers might also be related to other factors, such as BMI, kidney disease, and comorbidities such as peripheral neuropathies. 5 For example, low BMI (low blood volume), cardiovascular disease, and impaired kidney function might all associate with higher blood levels of, for example, NfL and p‐tau, 69 , 70 , 71 , 72 , 73 but the clinical relevance of these effects is uncertain. 74 The influence of such covariates is ideally defined in well‐characterized, large, and diverse population‐based studies, especially if we target implementation in the normal population, but also in clinic‐based cohorts. However, it remains to be defined whether the cumulative effects of these confounders significantly affect the final biomarker results, beyond analytical and other sources of pre‐analytical variation. Further, a very relevant factor that might affect plasma biomarker levels is ethnicity, which is only beginning to be explored. 5 Studies report contradictory results regarding lower plasma levels of p‐tau in, for example Black or Latin American populations. 75 , 76 , 77 Observed differences may have been dependent on differences in comorbidities, socioeconomic, and educational factors, 76 and whether these factors have different effects at every disease stage is currently unknown. Thus, it is relevant to further study and consider potential confounding factors, such as socioeconomic factors next to other medical factors, for example, kidney function and BMI.
Research priorities (Table 1):
Studies examining factors that may affect the interpretation of BBMs are needed in large and diverse populations.
4.3. Clinical robustness
As mentioned above, several factors might induce variability in BBM values. There are both random and systematic errors. 78 Random errors include random fluctuations of the marker in blood and uncontrollable factors related to blood collection and analysis (e.g., intra‐assay variability). The total random error can be determined when performing test–retest analyses, that is, when collecting blood and performing the biomarker assay repeatedly close in time for the same individuals, using the same protocol for blood collection, pre‐analytical handling, and an identical assay set‐up. Systematic error can be related to systematic differences in biomarker levels in blood collected in the morning compared to evening, when using LiHep plasma instead of EDTA plasma, 48 or when changing the conditions of the assay (e.g., a new lot of antibodies or calibrators). The systematic error for a particular assay can be difficult to determine and often requires stability measurements over extended time periods using the same samples. Both random and systematic error can result in false classification of individuals, especially to those with biomarker values close to the predefined diagnostic cutoffs used for a certain assay. 48 To use a biomarker for which the values in the disease group are very close to the values in the normal group would require very low random and systematic errors to be clinically robust. As mentioned above, plasma Aβ42/Aβ40 is reduced by only 8% to 15% in AD, which is why the “total allowable error” for a clinically robust plasma Aβ42/Aβ40 assay is likely to be < 3% to 5%. For plasma p‐tau, the total allowable error is much higher considering that this marker is increased 250% to 650% in symptomatic AD.
FIGURE 1.
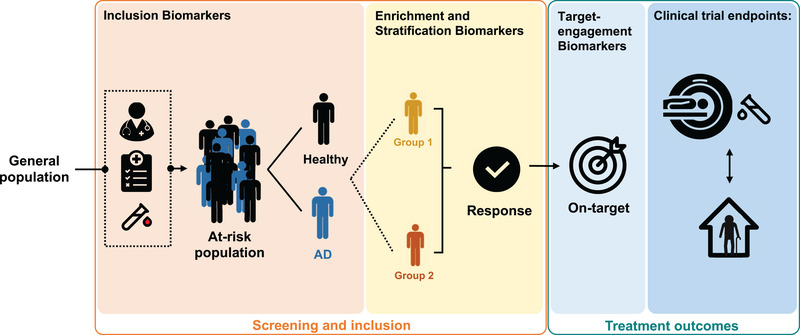
Potential future use of blood‐based biomarkers in clinical trials. Biomarker applications in clinical trials for Alzheimer's disease (AD) can be useful for screening, inclusion, and treatment outcomes. The various purposes of each type are indicated in the columns. Screening and inclusion: Clinical trials depend on correct grouping of subjects for the right treatments, therefore various biomarkers and inclusion criteria can be used. Categorization of subjects can be done using risk/susceptibility biomarkers (indicate the potential for developing the disease), diagnostic biomarkers (detect or confirm the presence of a disease), stratification markers (such as age or genetic risk factors, for example apolipoprotein E ε4, for which strata are expected to have a stronger response to treatment), and predictive biomarkers (determine who might benefit from and respond to a particular treatment). Treatment outcomes: Once the clinical trials have begun, response and outcomes for the subjects can be monitored by measuring the effects on the target protein of the drug as well by biomarkers as endpoints of the clinical trials; the holy grail is to use biomarkers as surrogate endpoints that predict the clinical endpoints
Research priorities (Table 1):
Study the total random error of a biomarker assay by repeatedly collecting blood and repeatedly analyzing a certain biomarker over a shorter time in ≈40 to 50 individuals (with and without AD).
To determine systematic error of a certain assay by analyzing samples from the same pools of plasma over extended time periods and in different laboratories.
To determine clinical robustness in a real‐world setting by using the assay in extended prospective studies over 1 to 3 years during which (1) pre‐defined cut‐offs are used, (2) samples are analyzed continuously over the study period (e.g., on a daily or weekly basis rather than in single batches), and (3) an appropriate reference standard is used (such as Aβ PET).
5. THE USE OF BBMS IN CLINICAL TRIALS
As depicted in Figure 1, BBMs might potentially be used to improve the design of clinical trials in many ways, including identification of individuals with the disease and use as surrogate endpoints predicting clinical efficacy.
5.1. The use of BBMs as a (pre‐)screening step in clinical trials
Currently the use of BBMs as a (pre‐)screener for trials is only possible in AD‐focused trials because of lack of specific BBMs for other neurodegenerative dementias. 4 Approximately 15% to 30% of CU individuals >60 years of age exhibit cerebral accumulation of Aβ pathology changes. 79 , 80 Consequently, it takes a lot of resources to identify individuals with preclinical AD for intervention trials when using Aβ PET or CSF AD biomarkers to screen cognitively healthy populations to identify individuals with asymptomatic AD pathology. 4 Several preliminary studies have suggested that using BBMs as (pre‐)screener, with only those with abnormal BBM levels undergoing PET or CSF, might substantially reduce the costs and the time needed for recruitment of study participants to preclinical AD trials. For example, it has been suggested that prescreening with a combination of plasma Aβ42/Aβ40 and APOE genotype can substantially reduce the number of Aβ‐PET scans needed to identify individuals with preclinical AD for trials. 14 , 81 In fact, an IP‐MS method for quantification of plasma Aβ42/Aβ40 11 is currently evaluated in a prospective fashion as a (pre‐)screener for detection of Aβ‐PET abnormality in the AHEAD 3‐45 trial evaluating the effects of lecanemab in preclinical AD (an anti‐amyloid immunotherapy; NCT04468659). Further, a plasma p‐tau217 assay 22 is used to identify individuals with preclinical AD in the TRAILBLAZER‐ALZ3 trial evaluating the clinical effects of donanemab (an anti‐amyloid immunotherapy) in a preclinical AD population (NCT05026866). However, it is still unclear if combinations of certain BBMs, like Aβ42/Aβ40 and p‐tau, 59 might reduce the costs even further when used as pre‐screeners in preclinical AD trials.
It is important to consider the ramifications of disclosing biomarker results to individuals who are currently asymptomatic, as there is not yet sufficient information to make accurate predictions at the individual level as to exact risk and timeframe of developing the symptoms of AD. Current prevention trials, such as the A4 Study, have disclosed amyloid PET scan results to >4000 individuals, using a carefully developed process of screening and education prior to disclosure, with clear language regarding the uncertainty of individual prediction. 82 With the use of BBMs, trials will hopefully reach more diverse communities. As there is currently less information, especially in diverse populations, regarding the optimal cut‐offs and predictive value of plasma biomarkers in asymptomatic individuals, it will be important to be circumspect in providing results to participants. Further, the process of disclosing AD BBM results must likely be further developed to work optimally also in populations with lower education level.
BBMs could potentially also be used to reduce costs for screening of symptomatic individuals with either prodromal AD or AD dementia for interventional trials, but because the prevalence of AD pathology is much higher in populations fulfilling the clinical criteria for amnestic MCI or mild AD dementia (≈50%–80% 79 , 83 , 84 ), the cost benefit for using BBM as a (pre‐)screener (with subsequent confirmation by either CSF or PET analysis, as we recommend) will be less obvious compared to in preclinical AD trials. As mentioned above, plasma p‐tau may be the most promising BBM for symptomatic AD, 22 but might be combined with, for example, APOE genotype or Aβ42/Aβ40 during the prodromal disease stages. 30
Further studies are needed to study whether certain BBMs in the future can be used as stand‐alone biomarkers, without confirmation of using CSF or PET, when including individuals with presumed preclinical AD in trials. The required positive predictive values of such BBMs should likely be > 90% to 95% in studies evaluating novel disease‐modifying therapies. Erroneously including study participants without AD in such intervention trials could result in ethical issues, including (1) disclosing incorrect information to an individual about AD status (see above), (2) subjecting individuals without AD to potential harmful therapies, and (3) reducing the statistical power to detect an effect of the treatment. That said, BBMs might be used as stand‐alone biomarkers identifying individuals at increased risk of having AD pathological changes in interventional studies with less risk of side effects such as lifestyle interventions.
Plasma biomarkers like NfL are also beginning to be used as screeners in trials including some genetic neurodegenerative diseases, for example, to sustain inclusion in trials targeting FTD associated with mutations in GRN or MAPT. In addition, plasma NfL increase may be used as a biomarker for detection onset of neurodegeneration in these mutation carriers who are about to enter the clinical phase of the disease. Finally, plasma AD biomarkers like p‐tau can be used to rule out concomitant AD pathology in individuals recruited to trials for non‐AD neurodegenerative diseases.
Research priorities (Table 1):
Studies determining which combinations of BBMs are consistently most optimal for detection of AD pathology changes in either preclinical (asymptomatic) or symptomatic AD when using the best assays for each biomarker.
Prospective studies with sample‐to‐sample analyses over extended time periods (a couple of years) and with a valid outcome (like Aβ‐PET, tau‐PET, or CSF AD biomarkers) to determine whether the clinical robustness of a BBM (or combination of BBMs) over time is high enough for use as pre‐screeners in clinical trial settings.
Determining whether use of conservative cut‐offs for certain BBMs (or combinations of BBMs) can result in valid predictions of the presence of AD pathological changes (> 90%–95%), and thereby PET or CSF would not be needed for inclusions in AD trials of individuals with clearly abnormal BBMs. However, studies would also need to define the gray zone (with uncertain BBM results) where PET or CSF is needed for accurate detection of AD status.
Recommendations for current use of BBMs as (pre‐)screeners in AD trials (Table 2):
BBMs, especially plasma Aβ42/Aβ40 and p‐tau assays with established thresholds, can already now be used as a first screening step in AD trials evaluating potential disease‐modifying therapies, provided the AD status is confirmed with PET or CSF in the participants with abnormal BBM outcomes before final inclusion in the trials.
In the future, it might be that only participants with uncertain BBM outcomes (e.g., biomarker results close to the cut‐off for positivity) need to undergo PET and CSF to confirm a positive AD status, and that those with clearly abnormal BBMs can enter the trial without such evaluations (i.e., if longitudinal PET or CSF assessments are not used as outcome measures in the trial). However, additional data are needed to determine whether the BBMs have high enough positive predictive values to serve as stand‐alone biomarkers for trial inclusion.
In non‐AD trials, BBMs (especially plasma Aβ42/Aβ40 and p‐tau assays with established thresholds) can be used to exclude patients likely having AD co‐pathology.
TABLE 2.
Recommendations of the use of AD‐associated BBMs in clinical trials and practice
| Biomarkers as a first screening step in clinical trials: |
| (1) BBMs, especially plasma Aβ42/Aβ40 and p‐tau assays with established thresholds, can already now be used as a first screening step in AD trials evaluating potential disease‐modifying therapies, provided the AD status is confirmed with PET or CSF in the participants with abnormal BBM outcomes before final inclusion in the trials. |
| (2) In the future, it might be that only participants with uncertain BBM outcomes (e.g., biomarker results close to the cut‐off for positivity) need to undergo PET and CSF to confirm a positive AD status, and that those with clearly abnormal BBMs can enter the trial without such evaluations (i.e., if longitudinal PET or CSF assessments are not used as outcome measures in the trial). However, additional data are needed to determine whether the BBMs have high enough positive predictive values to serve as stand‐alone biomarkers for trial inclusion. |
| (3) In non‐AD trials, BBMs (especially plasma Aβ42/Aβ40 and p‐tau assays with established thresholds) can be used to exclude patients likely having AD co‐pathology. |
| Surrogate biomarkers in clinical trials: |
| (4) BBMs can be used as exploratory outcomes in most clinical trials in AD and other neurodegenerative dementias. BBMs need further validation before they are used as primary endpoints in pivotal trials. BBMs could be used to inform decisions in clinical trials with adaptive design. |
| Use of BBMs in specialized memory clinic settings: |
| (5) BBMs (with established thresholds) should currently only be used in symptomatic patients at specialist clinics and the results should be confirmed whenever possible with CSF or PET. Additional data are needed before use of BBMs as stand‐alone diagnostic markers. |
| Use of BBMs in primary care: |
| (6) Additional data are needed for use of BBMs in primary care. |
Abbreviations: Aβ, amyloid beta; AD, Alzheimer's disease; BBM, blood‐based biomarkers; CSF, cerebrospinal fluid; PET, positron emission tomography; p‐tau, phosphorylated tau.
5.2. The use of BBM as a surrogate endpoint in trials
A challenging potential use of BBMs is as surrogate endpoints in clinical trials of disease‐modifying drug candidates. The basic idea is that a treatment‐induced change in a biomarker would reliably predict a beneficial clinical outcome, and that this prediction would be strong enough to replace the measurement of the clinical outcome as the endpoint in the trial. 85 The use of surrogate endpoint biomarkers in trials will likely be especially important in trials evaluating the effects of interventions in participants with preclinical (asymptomatic) AD for which very large and long‐term studies are needed when using a clinical outcome such as cognitive function. 86 The FDA suggests that surrogate endpoints can be classified into three groups depending on the level of clinical validation. First, a “candidate surrogate endpoint” is a biomarker that needs further study in observational studies and/or clinical trials. Second, a “reasonably likely surrogate endpoint” is a marker “supported by strong mechanistic and/or epidemiologic rationale, but the amount of clinical data available is not sufficient to show that they are a validated surrogate endpoint.” 87 Such a biomarker can be used to support the FDA's Accelerated Approval program, and today Aβ‐PET belongs to this category, but no other AD‐related biomarker. 87 Third, a “validated surrogate endpoint” should exhibit “a clear mechanistic rationale and clinical data providing strong evidence that an effect on the surrogate endpoint predicts a specific clinical benefit” and can consequently be used as a primary outcome in pivotal trials. 87 Today, no biomarker related to neurodegenerative diseases belongs to this category. Even though BBMs should today not be used as primary endpoints in pivotal trials, this does not preclude the use of certain BBMs for decision making in clinical trials with adaptive design, for which they could be used to inform decisions on continuing a trial or not (where the primary outcome is still a clinical outcome).
There are several observational studies showing that longitudinal changes in plasma NfL are related to change in brain atrophy and cognitive outcomes in, for example, AD, 45 MS, 8 and ALS. 88 Further, effective disease‐modifying treatment in, for example, MS and spinal muscular atrophy reduce NfL levels, and such reductions are associated with the clinical efficacy of the intervention. 89 , 90 Recently, plasma p‐tau has been shown to increase over time in AD, and such increases relate to brain atrophy and cognitive decline. 19 More importantly, as mentioned above, anti‐Aβ immunotherapies reliably induce reductions in plasma p‐tau levels that are correlated with slower worsening of the disease, 39 and one can speculate that plasma p‐tau might even be used in the future to monitor individual treatment responses to anti‐Aβ therapies in the clinic.
Research priorities (Table 1):
Longitudinal, large‐scale, and diverse observational trials should determine the longitudinal changes in BBM over time, and whether such changes relate to other changes in more established biomarkers (such as MRI, CSF, and PET) and changes in clinically relevant outcomes (e.g., cognition, motor function, and activities of daily living).
Study longitudinal changes of BBMs in samples collected from different interventional trials and correlate treatment‐related changes in BBMs with changes in clinical outcomes. Further, study the dynamics of the biomarker levels in response to treatment and the time needed to see a significant effect.
Recommendations for current use of BBMs as surrogate endpoints in trials (Table 2):
BBMs can be used as exploratory outcomes in most clinical trials in AD and other neurodegenerative dementias. BBMs need further validation before they are used as primary endpoints in pivotal trials. BBMs could be used to inform decisions in clinical trials with adaptive design.
6. THE USE OF BBM IN CLINICAL PRACTICE
6.1. The use of BBMs in specialized memory clinic settings
Appropriate use criteria for clinical use of AD biomarkers were published in 2013 for Aβ‐PET (currently being updated) and in 2018 for CSF AD biomarkers. 91 , 92 Both sets of criteria indicate that these biomarkers can be used in patients with cognitive impairment (i.e., MCI or dementia) to differentiate AD from other dementia disorders. Further, the CSF criteria also suggest using AD biomarkers in patients with SCD at increased risk of AD, but there is an ongoing debate on the clinical use of biomarkers in patients without objective cognitive impairment such as in SCD. 93 Naturally, patients with cognitive symptoms should undergo relevant clinical assessments and structural brain imaging, and these CSF or PET biomarkers should only be used when AD is a possible diagnosis and when such a diagnosis will alter the management of the patient. That is, BBM will always be an adjunct to, not a substitute for, a thorough clinical evaluation and the clinician should always interpret the biomarker findings in the context of the clinical symptomology of the patient.
As mentioned above, several studies have shown that certain BBMs, especially plasma p‐tau, can be used with accuracy in both secondary and tertiary memory clinics to (1) differentiate AD dementia from other neurodegenerative diseases, 20 , 22 , 23 , 24 , 27 , 28 (2) detect AD pathology in patients with MCI, 22 , 27 and (3) predict development of AD dementia in patients with SCD or MCI. 20 , 29 , 30 , 31 However, much more research is needed before widespread use of such BBM in the clinical practice of specialized memory clinics.
Research priorities (Table 1):
Evaluation of BBMs in diverse (real‐life) secondary and tertiary memory clinic populations. This should be done prospectively, using predefined cut‐offs, and using relevant and accurate reference standards.
Identify the optimal combinations of easily accessible and time‐/cost‐effective biomarkers and tests in memory clinic settings and study whether they outperform the clinical assessments used today in most such secondary and tertiary clinics.
Study whether certain BBMs (or combinations with other easily accessible diagnostic methods) perform non‐inferior to CSF and PET, that is can certain BBM‐based algorithms be used alone to support an AD diagnosis, or should they only be used as a gatekeeper to CSF/PET.
Determine whether addition of BBMs to standard clinical assessments and imaging improves the diagnosis and significantly alters the management of the patients.
Recommendations for current use of BBMs in specialized memory clinic settings (Table 2):
BBMs (with established thresholds) should currently only be used in symptomatic patients at specialist clinics and the results should be confirmed whenever possible with CSF or PET. Additional data are needed before use of BBMs as stand‐alone diagnostic markers.
6.2. The use of BBMs in primary care settings
In most countries patients with cognitive symptoms are primarily diagnosed and treated in primary care. However, the diagnostic work‐up today in primary care is very challenging, because the assessments often just include unspecific cognitive tests or questionnaires, basic blood tests (e.g., thyroid‐stimulating hormone, and vitamin B12) to rule out other causes of cognitive impairment, and sometimes a structural image of the brain. Few studies with validated reference standards have been performed in primary care to study how well these tests can differentiate between different dementia disorders. However, in most countries the frequency of missed or delayed diagnosis of dementia is very high. 94 Consequently, there is a great need for accurate BBM‐based diagnostic and prognostic algorithms that can substantially improve the diagnostic work‐up of AD (Figure 2).
FIGURE 2.
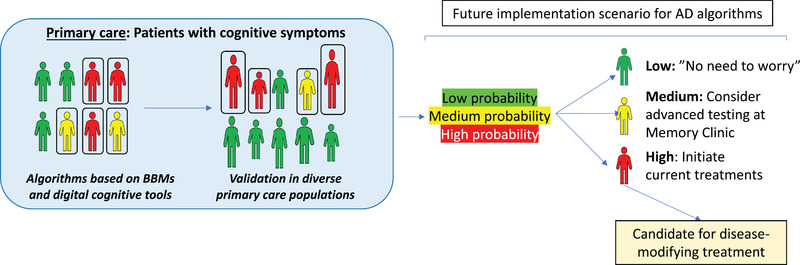
Potential future use of blood‐based biomarkers in primary care. In primary care we need easy and accurate methods to be able to identify different underlying neurodegenerative diseases in patients with cognitive complaints. Ideally blood‐based biomarkers together with clinical assessments could be used to determine the patient‐level probability of having a neurodegenerative disease like Alzheimer's disease (AD), which would improve patient management, including decisions regarding treatment or referrals to specialized clinics. However, it is very important that novel diagnostic algorithms (based on blood‐based biomarkers) are prospectively validated against relevant reference standards in large and diverse primary care populations before implementation in clinical practice
No studies have yet extensively evaluated BBMs for neurodegenerative diseases in primary care. Results obtained from secondary or tertiary memory clinics cannot be directly translated to the primary care setting. The prevalence of neurodegenerative diseases is lower in the primary care than specialized memory settings. Further, the patient population with cognitive symptoms in primary care, especially at older ages, is much more heterogenous with more frequent comorbidities (e.g., diabetes, cardiovascular diseases, kidney disease, depression, etc.) and co‐pathologies (e.g., cerebrovascular disease), and more diverse socioeconomic backgrounds. Consequently, well‐performed BBM studies in diverse primary care populations are needed. Such studies should also evaluate the impact of BBM on the diagnostic accuracy and any change in patient management. We need to establish whether novel BBMs might potentially improve the identification of patients with low likelihood of having a neurodegenerative disease underlying their cognitive symptoms, for which other causes should be considered and managed in the primary care setting (e.g., depression, anti‐cholinergic treatments). We also need to understand if they can identify those with a high likelihood of having a neurodegenerative disease who might receive adequate treatment already in the primary care setting or those who would need referral to a specialized clinic.
Once validated, education packages regarding when to use the biomarkers, what they represent, how to interpret the results in the context of comorbidities, and what to do with the results need to be developed in close collaboration between primary care physicians, dementia specialists, communication experts, and patient representatives (What does the patient want to know? what are the optimal tools for communication of results to patients?). A potential future scenario is that a set of the BBMs discussed above could become available to primary care physicians as part of their health monitoring toolbox, even for use in asymptomatic individuals. This potential use is currently premature and not supported by the classical World Health Organization criteria for screening. 95 Similarly, general population screenings and direct‐to‐consumer tests are not recommended.
Research priorities (Table 1):
Perform prospective studies in primary care settings, including representative and diverse populations with cognitive symptoms, where BBMs and brief cognitive tests and other easily accessible methods are performed in the primary care setting. However, the reference standard must be of high quality and preferably include CSF or PET for AD.
Identify the optimal combinations of easily accessible and time‐/cost‐effective biomarkers and tests in this setting (e.g., combining BBMs with digital cognitive tests).
Study whether BBMs outperform what is already available today in primary care (standard of care today), and if they also improve diagnosis and management (including treatment decisions and referrals to memory clinics).
Recommendations for current use of BBMs as in primary care (Table 2):
Additional data are needed for use of BBMs in primary care.
7. CONCLUDING REMARKS
Blood‐based biomarkers for AD are already now improving the design of clinical trials, and they are very likely to revolutionize the diagnostic work‐up of AD in the future. That said, the implementation of such markers in trials and practice must be done in a careful and controlled way not to accidentally cause more harm than good. Much more research is therefore needed before widespread clinical use of BBMs as we have outlined above. Such research is also needed before the community can establish Appropriate Use Criteria for clinical use of BBMs, which is a prerequisite for general use of such markers in the clinic. However, the acquired experience from implementation of CSF AD biomarkers and Aβ‐PET in many countries will ensure rapid validation of relevant BBMs in the first contexts of use, including trials and specialized memory clinics. The implementation of BBMs in primary care will likely take much longer, because relevant and high‐quality research studies on AD‐related BBMs in this setting are very few, but hopefully more prospective studies will be launched in the coming years using relevant and accurate reference standards. Finally, we must also develop and validate easily accessible and scalable biomarkers for non‐AD neurodegenerative disorders, such as synucleinopathies, TAR DNA‐binding protein 43, 3R tauopathies, and 4R tauopathies.
CONFLICTS OF INTEREST
Author disclosures are available in the supporting information.
Supporting information
SUPPORTING INFORMATION
ACKNOWLEDGMENTS
O.H. has acquired research support (for the institution) from ADx, AVID Radiopharmaceuticals, Biogen, Eli Lilly, Eisai, Fujirebio, GE Healthcare, Pfizer, and Roche. In the past 2 years, he has received consultancy/speaker fees from Amylyx, Alzpath, BioArctic, Biogen, Cerveau, Fujirebio, Genentech, Novartis, Roche, and Siemens. H.Z. has served on scientific advisory boards and/or as a consultant for Abbvie, Alector, Annexon, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, Nervgen, Novo Nordisk, Pinteon Therapeutics, Red Abbey Labs, Passage Bio, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave; has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche; and is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). C.E.T. has a collaboration contract with ADx Neurosciences, Quanterix, and Eli Lilly; performed contract research or received grants from AC‐Immune, Axon Neurosciences, Biogen, Brainstorm Therapeutics, Celgene, EIP Pharma, Eisai, PeopleBio, Roche, Toyama, Vivoryon. She serves on editorial boards of Medidact Neurologie/Springer, Alzheimer Research and Therapy, Neurology: Neuroimmunology & Neuroinflammation, and is editor of a Neuromethods book published by Springer. A.L.B. is supported by the NIH (U19AG063911, U54NS092089, R01AG038791, U01AG045390), Tau Research Consortium, Bluefield Project to Cure FTD, University of California Cures AD Program, Association for Frontotemporal Degeneration, CBD Solutions, Alzheimer's Drug Discovery Foundation, and the Alzheimer's Association. He receives research support from Biogen, Eisai, and Regeneron. He has served as a paid consultant for AGTC, Alector, Arkuda, Arvinas, AZTherapeutics, Denali, GSK, Humana, Oligomerix, Oscotec, Roche, Stealth, Third Rock, Transposon, Third Rock Ventures, TrueBinding, and Wave. R.A.S. is supported by NIH (P01AG036694; R01AG053798; R01AG054029; U24AG057437; R01AG061848; and R01AG063689), Alzheimer's Association, Fidelity, and GHR Foundation. She receives research support from Eli Lilly and Co., and Eisai. She has served as a paid consultant for AC Immune, Alynam, Genentech, Ionis, Janssen, NervGen, Oligomerix, Prothena, and Shioniogi. G.D.R. is supported by NIH (P30‐AG062422, R35 AG072362, U01‐AG057195, R21 P0545190), Alzheimer's Association (ZEN‐21‐848216), American College of Radiology, Rainwater Charitable Foundation, and the Alliance for Therapeutics in Neurodegeneration. He receives research support from Avid Radiopharmaceuticals, GE Healthcare, Life Molecular Imaging, and Genentech. He has served as a paid consultant for Eisai, Eli Lilly, Johnson & Johnson, and Roche. He is an Associate Editor for JAMA Neurology. R.M.E. is a full‐time employee of the Alzheimer's Association. M.C.C. is a full‐time employee of the Alzheimer's Association, and has a family member in the USC neuroscience graduate program.
Hansson O, Edelmayer RM, Boxer AL, et al. The Alzheimer's Association appropriate use recommendations for blood biomarkers in Alzheimer's disease. Alzheimer's Dement. 2022;18:2669–2686. 10.1002/alz.12756
REFERENCES
- 1. Knopman DS, DeKosky ST, Cummings JL, et al. Practice parameter: diagnosis of dementia (an evidence‐based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001;56(9):1143‐1153. 10.1212/wnl.56.9.1143 [DOI] [PubMed] [Google Scholar]
- 2. Rizzo G, Arcuti S, Copetti M, et al. Accuracy of clinical diagnosis of dementia with Lewy bodies: a systematic review and meta‐analysis. J Neurol Neurosurg Psychiatry. 2018;89(4):358‐366. 10.1136/jnnp-2017-316844 [DOI] [PubMed] [Google Scholar]
- 3. Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005‐2010. J Neuropathol Exp Neurol. 2012;71(4):266‐273. 10.1097/NEN.0b013e31824b211b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hansson O. Biomarkers for neurodegenerative diseases. Nat Med. 2021;27(6):954‐963. 10.1038/s41591-021-01382-x [DOI] [PubMed] [Google Scholar]
- 5. Teunissen CE, Verberk IMW, Thijssen EH, et al. Blood‐based biomarkers for Alzheimer's disease: towards clinical implementation. Lancet Neurol. 2022;21(1):66‐77. 10.1016/S1474-4422(21)00361-6 [DOI] [PubMed] [Google Scholar]
- 6. Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10(9):698‐712. 10.1038/nrd3505 [DOI] [PubMed] [Google Scholar]
- 7. Alves CRR, Petrillo M, Spellman R, et al. Implications of circulating neurofilamentsfor spinal muscular atrophytreatment early in life: a case series. Mol Ther Methods Clin Dev. 2021;23:524‐538. 10.1016/j.omtm.2021.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Disanto G, Barro C, Benkert P, et al. Serum Neurofilament light: a biomarker of neuronal damage in multiple sclerosis. Ann Neurol. 2017;81(6):857‐870. 10.1002/ana.24954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Anderson AM, Easley KA, Kasher N, et al. Neurofilament light chain in blood is negatively associated with neuropsychological performance in HIV‐infected adults and declines with initiation of antiretroviral therapy. J Neurovirol. 2018;24(6):695‐701. 10.1007/s13365-018-0664-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zetterberg H, Blennow K. Moving fluid biomarkers for Alzheimer's disease from research tools to routine clinical diagnostics. Mol Neurodegener. 2021;16(1):10. 10.1186/s13024-021-00430-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ovod V, Ramsey KN, Mawuenyega KG, et al. Amyloid beta concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement. 2017;13(8):841‐849. 10.1016/j.jalz.2017.06.2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nakamura A, Kaneko N, Villemagne VL, et al. High performance plasma amyloid‐beta biomarkers for Alzheimer's disease. Nature. 2018;554(7691):249‐254. 10.1038/nature25456 [DOI] [PubMed] [Google Scholar]
- 13. Keshavan A, Pannee J, Karikari TK, et al. Population‐based blood screening for preclinical Alzheimer's disease in a British birth cohort at age 70. Brain. 2021;144(2):434‐449. 10.1093/brain/awaa403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Palmqvist S, Janelidze S, Stomrud E, et al. Performance of fully automated plasma assays as screening tests for Alzheimer disease‐related beta‐amyloid status. JAMA Neurol. 2019;76(9):1060‐1069. 10.1001/jamaneurol.2019.1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Janelidze S, Teunissen CE, Zetterberg H, et al. Head‐to‐head comparison of 8 plasma amyloid‐beta 42/40 assays in Alzheimer disease. JAMA Neurol. 2021;78(11):1375‐1382. 10.1001/jamaneurol.2021.3180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Benedet AL, Brum WS, Hansson O, et al. The accuracy and robustness of plasma biomarker models for amyloid PET positivity. Alzheimers Res Ther. 2022;14(1):26. 10.1186/s13195-021-00942-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Y, Schindler SE, Bollinger JG, et al. Validation of Plasma amyloid‐beta 42/40 for detecting Alzheimer disease amyloid plaques. Neurology. 2022;98(7):e688‐e699. 10.1212/WNL.0000000000013211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sato C, Barthelemy NR, Mawuenyega KG, et al. Tau kinetics in neurons and the human central nervous system. Neuron. 2018;98(4):861‐864. 10.1016/j.neuron.2018.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mattsson‐Carlgren N, Janelidze S, Bateman RJ, et al. Soluble P‐tau217 reflects amyloid and tau pathology and mediates the association of amyloid with tau. EMBO Mol Med. 2021;13(6):e14022. 10.15252/emmm.202114022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Janelidze S, Mattsson N, Palmqvist S, et al. Plasma P‐tau181 in Alzheimer's disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer's dementia. Nat Med. 2020;26(3):379‐386. 10.1038/s41591-020-0755-1 [DOI] [PubMed] [Google Scholar]
- 21. Lantero Rodriguez J, Karikari TK, Suarez‐Calvet M, et al. Plasma p‐tau181 accurately predicts Alzheimer's disease pathology at least 8 years prior to post‐mortem and improves the clinical characterisation of cognitive decline. Acta Neuropathol. 2020;140(3):267‐278. 10.1007/s00401-020-02195-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Palmqvist S, Janelidze S, Quiroz YT, et al. Discriminative accuracy of plasma phospho‐tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA. 2020;324(8):772‐781. 10.1001/jama.2020.12134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thijssen EH, La Joie R, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer's disease and frontotemporal lobar degeneration. Nat Med. 2020;26(3):387‐397. 10.1038/s41591-020-0762-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ashton NJ, Pascoal TA, Karikari TK, et al. Plasma p‐tau231: a new biomarker for incipient Alzheimer's disease pathology. Acta Neuropathol. 2021;141(5):709‐724. 10.1007/s00401-021-02275-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mielke MM, Hagen CE, Xu J, et al. Plasma phospho‐tau181 increases with Alzheimer's disease clinical severity and is associated with tau‐ and amyloid‐positron emission tomography. Alzheimers Dement. 2018;14(8):989‐997. 10.1016/j.jalz.2018.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wennstrom M, Janelidze S, Nilsson KPR, et al. Cellular localization of p‐tau217 in brain and its association with p‐tau217 plasma levels. Acta Neuropathol Commun. 2022;10(1):3. 10.1186/s40478-021-01307-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karikari TK, Pascoal TA, Ashton NJ, et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer's disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020;19(5):422‐433. 10.1016/S1474-4422(20)30071-5 [DOI] [PubMed] [Google Scholar]
- 28. Thijssen EH, La Joie R, Strom A, et al. Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer's disease and frontotemporal lobar degeneration: a retrospective diagnostic performance study. Lancet Neurol. 2021;20(9):739‐752. 10.1016/S1474-4422(21)00214-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karikari TK, Benedet AL, Ashton NJ, et al. Diagnostic performance and prediction of clinical progression of plasma phospho‐tau181 in the Alzheimer's Disease Neuroimaging Initiative. Mol Psychiatry. 2021;26(2):429‐442. 10.1038/s41380-020-00923-z [DOI] [PubMed] [Google Scholar]
- 30. Palmqvist S, Tideman P, Cullen N, et al. Prediction of future Alzheimer's disease dementia using plasma phospho‐tau combined with other accessible measures. Nat Med. 2021;27(6):1034‐1042. 10.1038/s41591-021-01348-z [DOI] [PubMed] [Google Scholar]
- 31. Cullen N, Leuzy A, Palmqvist S, et al. Individualized prognosis of cognitive decline and dementia in mild cognitive impairment based on plasma biomarker combinations. Nature Aging. 2021;1(1): 114‐123. 10.1038/s43587-020-00003-5 [DOI] [PubMed] [Google Scholar]
- 32. Palmqvist S, Insel PS, Stomrud E, et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer's disease. EMBO Mol Med. 2019;11(12):e11170. 10.15252/emmm.201911170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cullen NC, Leuzy A, Janelidze S, et al. Plasma biomarkers of Alzheimer's disease improve prediction of cognitive decline in cognitively unimpaired elderly populations. Nat Commun. 2021;12(1):3555. 10.1038/s41467-021-23746-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leuzy A, Smith R, Cullen NC, et al. Biomarker‐Based prediction of longitudinal tau positron emission tomography in Alzheimer disease. JAMA Neurol. 2021. 10.1001/jamaneurol.2021.4654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pereira JB, Janelidze S, Stomrud E, et al. Plasma markers predict changes in amyloid, tau, atrophy and cognition in non‐demented subjects. Brain. 2021;144(9):2826‐2836. 10.1093/brain/awab163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bayoumy S, Verberk IMW, den Dulk B, et al. Clinical and analytical comparison of six Simoa assays for plasma P‐tau isoforms P‐tau181, P‐tau217, and P‐tau231. Alzheimers Res Ther. 2021;13(1):198. 10.1186/s13195-021-00939-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mielke MM, Frank RD, Dage JL, et al. Comparison of plasma phosphorylated tau species with amyloid and tau positron emission tomography, neurodegeneration, vascular pathology, and cognitive outcomes. JAMA Neurol. 2021;78(9):1108‐1117. 10.1001/jamaneurol.2021.2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mattsson‐Carlgren N, Janelidze S, Palmqvist S, et al. Longitudinal plasma p‐tau217 is increased in early stages of Alzheimer's disease. Brain. 2020;143(11):3234‐3241. 10.1093/brain/awaa286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Budd Haeberlein S, Aisen PS, Barkhof F, et al. Two randomized phase 3 studies of aducanumab in early Alzheimer's disease. J Prev Alzheimers Dis. 2022;9(2):197‐210. 10.14283/jpad.2022.30 [DOI] [PubMed] [Google Scholar]
- 40. Ashton NJ, Janelidze S, Al Khleifat A, et al. A multicentre validation study of the diagnostic value of plasma neurofilament light. Nat Commun. 2021;12(1):3400. 10.1038/s41467-021-23620-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bridel C, van Wieringen WN, Zetterberg H, et al. Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta‐analysis. JAMA Neurol. 2019;76(9):1035‐1048. 10.1001/jamaneurol.2019.1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gaetani L, Blennow K, Calabresi P, Di Filippo M, Parnetti L, Zetterberg H. Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry. 2019;90(8):870‐881. 10.1136/jnnp-2018-320106 [DOI] [PubMed] [Google Scholar]
- 43. Preische O, Schultz SA, Apel A, et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer's disease. Nat Med. 2019;25(2):277‐283. 10.1038/s41591-018-0304-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weston PSJ, Poole T, O'Connor A, et al. Longitudinal measurement of serum neurofilament light in presymptomatic familial Alzheimer's disease. Alzheimers Res Ther. 2019;11(1):19. 10.1186/s13195-019-0472-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mattsson N, Andreasson U, Zetterberg H, Blennow K. Alzheimer's Disease Neuroimaging I. Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2017;74(5):557‐566. 10.1001/jamaneurol.2016.6117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Salloway S, Farlow M, McDade E, et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer's disease. Nat Med. 2021;27(7):1187‐1196. 10.1038/s41591-021-01369-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Swanson CJ, Zhang Y, Dhadda S, et al. A randomized, double‐blind, phase 2b proof‐of‐concept clinical trial in early Alzheimer's disease with lecanemab, an anti‐Abeta protofibril antibody. Alzheimers Res Ther. 2021;13(1):80. 10.1186/s13195-021-00813-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Verberk IMW, Misdorp EO, Koelewijn J, et al. Characterization of pre‐analytical sample handling effects on a panel of Alzheimer's disease‐related blood‐based biomarkers: results from the Standardization of Alzheimer's Blood Biomarkers (SABB) working group. Alzheimers Dement. 2022;18(8):1484‐1497. 10.1002/alz.12510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pereira JB, Janelidze S, Smith R, et al. Plasma GFAP is an early marker of amyloid‐beta but not tau pathology in Alzheimer's disease. Brain. 2021;144(11):3505‐3516. 10.1093/brain/awab223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Benedet AL, Mila‐Aloma M, Vrillon A, et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 2021;78(12):1471‐1483. 10.1001/jamaneurol.2021.3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chatterjee P, Pedrini S, Ashton NJ, et al. Diagnostic and prognostic plasma biomarkers for preclinical Alzheimer's disease. Alzheimers Dement. 2022;18(6):1141‐1154. 10.1002/alz.12447 [DOI] [PubMed] [Google Scholar]
- 52. Cicognola C, Janelidze S, Hertze J, et al. Plasma glial fibrillary acidic protein detects Alzheimer pathology and predicts future conversion to Alzheimer dementia in patients with mild cognitive impairment. Alzheimers Res Ther. 2021;13(1):68. 10.1186/s13195-021-00804-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Heller C, Foiani MS, Moore K, et al. Plasma glial fibrillary acidic protein is raised in progranulin‐associated frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2020;91(3):263‐270. 10.1136/jnnp-2019-321954 [DOI] [PubMed] [Google Scholar]
- 54. Shir D, Graff‐Radford J, Hofrenning EI, et al. Association of plasma glial fibrillary acidic protein (GFAP) with neuroimaging of Alzheimer's disease and vascular pathology. Alzheimers Dement (Amst). 2022;14(1):e12291. 10.1002/dad2.12291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Verberk IMW, Thijssen E, Koelewijn J, et al. Combination of plasma amyloid beta(1‐42/1‐40) and glial fibrillary acidic protein strongly associates with cerebral amyloid pathology. Alzheimers Res Ther. 2020;12(1):118. 10.1186/s13195-020-00682-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Verberk IMW, Laarhuis MB, van den Bosch KA, et al. Serum markers glial fibrillary acidic protein and neurofilament light for prognosis and monitoring in cognitively normal older people: a prospective memory clinic‐based cohort study. Lancet Healthy Longevity. 2021;2(2):E87‐E95. 10.1016/S2666-7568(20)30061-1 [DOI] [PubMed] [Google Scholar]
- 57. Laverse E, Guo T, Zimmerman K, et al. Plasma glial fibrillary acidic protein and neurofilament light chain, but not tau, are biomarkers of sports‐related mild traumatic brain injury. Brain Commun. 2020;2(2):fcaa137. 10.1093/braincomms/fcaa137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mattila OS, Ashton NJ, Blennow K, et al. Ultra‐early differential diagnosis of acute cerebral ischemia and hemorrhagic stroke by measuring the prehospital release rate of GFAP. Clin Chem. 2021;67(10):1361‐1372. 10.1093/clinchem/hvab128 [DOI] [PubMed] [Google Scholar]
- 59. Janelidze S, Palmqvist S, Leuzy A, et al. Detecting amyloid positivity in early Alzheimer's disease using combinations of plasma Abeta42/Abeta40 and p‐tau. Alzheimers Dement. 2021;18(2):283‐293. 10.1002/alz.12395 [DOI] [PubMed] [Google Scholar]
- 60. Lautner R, Palmqvist S, Hansson O. APOE genotype and the diagnostic accuracy of CSF biomarkers for Alzheimer disease. JAMA Psychiatry. 2014;71(10):1183‐1191. 10.1001/jamapsychiatry.2014.1060 [DOI] [PubMed] [Google Scholar]
- 61. Andreasson U, Perret‐Liaudet A, van Waalwijk van Doorn LJ, et al. A practical guide to immunoassay method validation. Front Neurol. 2015;6:179. 10.3389/fneur.2015.00179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Papp KV, Rentz DM, Orlovsky I, Sperling RA, Mormino EC. Optimizing the preclinical Alzheimer's cognitive composite with semantic processing: the PACC5. Alzheimers Dement (N Y). 2017;3(4):668‐677. 10.1016/j.trci.2017.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hansson O, Mikulskis A, Fagan AM, et al. The impact of preanalytical variables on measuring cerebrospinal fluid biomarkers for Alzheimer's disease diagnosis: a review. Alzheimers Dement. 2018;14(10):1313‐1333. 10.1016/j.jalz.2018.05.008 [DOI] [PubMed] [Google Scholar]
- 64. Hansson O, Batrla R, Brix B, et al. The Alzheimer's Association international guidelines for handling of cerebrospinal fluid for routine clinical measurements of amyloid beta and tau. Alzheimers Dement. 2021;17(9):1575‐1582. 10.1002/alz.12316 [DOI] [PubMed] [Google Scholar]
- 65. Mattsson N, Andreasson U, Persson S, et al. CSF biomarker variability in the Alzheimer's Association quality control program. Alzheimers Dement. 2013;9(3):251‐261. 10.1016/j.jalz.2013.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tijms BM, Willemse EAJ, Zwan MD, et al. Unbiased approach to counteract upward drift in cerebrospinal fluid amyloid‐beta 1‐42 analysis results. Clin Chem. 2018;64(3):576‐585. 10.1373/clinchem.2017.281055 [DOI] [PubMed] [Google Scholar]
- 67. Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. 2018;14(10):577‐589. 10.1038/s41582-018-0058-z [DOI] [PubMed] [Google Scholar]
- 68. Mielke MM, Syrjanen JA, Blennow K, et al. Comparison of variables associated with cerebrospinal fluid neurofilament, total‐tau, and neurogranin. Alzheimers Dement. 2019;15(11):1437‐1447. 10.1016/j.jalz.2019.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Syrjanen JA, Campbell MR, Algeciras‐Schimnich A, et al. Associations of amyloid and neurodegeneration plasma biomarkers with comorbidities. Alzheimers Dement. 2022;18(6):1128‐1140. 10.1002/alz.12466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ticau S, Sridharan GV, Tsour S, et al. Neurofilament light chain as a biomarker of hereditary transthyretin‐mediated amyloidosis. Neurology. 2021;96(3):e412‐e422. 10.1212/WNL.0000000000011090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ladang A, Kovacs S, Lengele L, et al. Neurofilament light chain concentration in an aging population. Aging Clin Exp Res. 2022;34(2):331‐339. 10.1007/s40520-021-02054-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Beydoun MA, Noren Hooten N, Maldonado AI, et al. Body mass index and allostatic load are directly associated with longitudinal increase in plasma neurofilament light among urban middle‐aged adults. J Nutr. 2022;152(2):535‐549. 10.1093/jn/nxab381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mielke MM, Dage JL, Frank RD, et al. Performance of plasma phosphorylated tau 181 and 217 in the community. Nat Med. 2022. 10.1038/s41591-022-01822-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Binette AP, Janelidze S, Cullen N, et al. Confounding factors of Alzheimer's disease plasma biomarkers and their impact on clinical performance. medRxiv. 2022. 10.1101/2022.05.30.22275718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Brickman AM, Manly JJ, Honig LS, et al. Correlation of plasma and neuroimaging biomarkers in Alzheimer's disease. Ann Clin Transl Neurol. 2022;9(5):756‐761. 10.1002/acn3.51529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Windon C, Iaccarino L, Mundada N, et al. Comparison of plasma and CSF biomarkers across ethnoracial groups in the ADNI. Alzheimers Dement (Amst). 2022;14(1):e12315. 10.1002/dad2.12315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schindler SE, Karikari TK, Ashton NJ, et al. Effect of race on prediction of brain amyloidosis by plasma abeta42/abeta40, phosphorylated tau, and neurofilament light. Neurology. 2022. 10.1212/WNL.0000000000200358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dybkaer R. Result, error and uncertainty. Scand J Clin Lab Invest. 1995;55(2):97‐118. 10.3109/00365519509089602 [DOI] [PubMed] [Google Scholar]
- 79. Jansen WJ, Ossenkoppele R, Knol DL, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta‐analysis. JAMA. 2015;313(19):1924‐1938. 10.1001/jama.2015.4668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Insel PS, Hansson O, Mattsson‐Carlgren N. Association Between apolipoprotein E epsilon2 vs epsilon4, age, and beta‐Amyloid in adults without cognitive impairment. JAMA Neurol. 2021;78(2):229‐235. 10.1001/jamaneurol.2020.3780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Verberk IMW, Slot RE, Verfaillie SCJ, et al. Plasma amyloid as prescreener for the earliest Alzheimer pathological changes. Ann Neurol. 2018;84(5):648‐658. 10.1002/ana.25334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Grill JD, Raman R, Ernstrom K, et al. Short‐term psychological outcomes of disclosing amyloid imaging results to research participants who do not have cognitive impairment. JAMA Neurol. 2020;77(12):1504‐1513. 10.1001/jamaneurol.2020.2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild‐to‐moderate Alzheimer's disease. N Engl J Med. 2014;370(4):322‐333. 10.1056/NEJMoa1304839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sevigny J, Suhy J, Chiao P, et al. Amyloid PET screening for enrichment of early‐stage alzheimer disease clinical trials: experience in a phase 1b clinical trial. Alzheimer Dis Assoc Disord. 2016;30(1):1‐7. 10.1097/WAD.0000000000000144 [DOI] [PubMed] [Google Scholar]
- 85. Fleming TR, Powers JH. Biomarkers and surrogate endpoints in clinical trials. Stat Med. 2012;31(25):2973‐2984. 10.1002/sim.5403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Insel PS, Weiner M, Mackin RS, et al. Determining clinically meaningful decline in preclinical Alzheimer disease. Neurology. 2019;93(4):e322‐e333. 10.1212/WNL.0000000000007831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Administration USFaD . https://wwwfdagov/drugs/development‐resources/surrogate‐endpoint‐resources‐drug‐and‐biologic‐development
- 88. Benatar M, Zhang L, Wang L, et al. Validation of serum neurofilaments as prognostic and potential pharmacodynamic biomarkers for ALS. Neurology. 2020;95(1):e59‐e69. 10.1212/WNL.0000000000009559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Delcoigne B, Manouchehrinia A, Barro C, et al. Blood neurofilament light levels segregate treatment effects in multiple sclerosis. Neurology. 2020;94(11):e1201‐e1212. 10.1212/WNL.0000000000009097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Olsson B, Alberg L, Cullen NC, et al. NFL is a marker of treatment response in children with SMA treated with nusinersen. J Neurol. 2019;266(9): 2129‐2136. 10.1007/s00415-019-09389-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Johnson KA, Minoshima S, Bohnen NI, et al. Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer's Association. Alzheimers Dement. 2013;9(1):e1‐e16. 10.1016/j.jalz.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Shaw LM, Arias J, Blennow K, et al. Appropriate use criteria for lumbar puncture and cerebrospinal fluid testing in the diagnosis of Alzheimer's disease. Alzheimers Dement. 2018;14(11):1505‐1521. 10.1016/j.jalz.2018.07.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Dubois B, Villain N, Frisoni GB, et al. Clinical diagnosis of Alzheimer's disease: recommendations of the International Working Group. Lancet Neurol. 2021;20(6):484‐496. 10.1016/S1474-4422(21)00066-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bradford A, Kunik ME, Schulz P, Williams SP, Singh H. Missed and delayed diagnosis of dementia in primary care: prevalence and contributing factors. Alzheimer Dis Assoc Disord. 2009;23(4):306‐314. 10.1097/WAD.0b013e3181a6bebc [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Wilson JMG JG . Principles and practice of screening for disease. WHO http://wwwwhoint/bulletin/volumes/86/4/07‐050112BPpdf 1968.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING INFORMATION