

Abstract
HYPONATREMIA (SERUM SODIUM LEVEL LESS THAN 134 MMOL/L) is a common electrolyte disturbance. Its high prevalence and potential neurologic sequelae make a logical and rigorous differential diagnosis mandatory before any therapeutic intervention. A history of concurrent illness and medication use as well as the assessment of extracellular volume status on physical examination may provide useful clues as to the pathogenesis of hyponatremia. Measurement of the effective serum tonicity (serum osmolality less serum urea level) is the first step in the laboratory evaluation. In patients with normal or elevated effective serum osmolality (280 mOsm/kg or greater), pseudohyponatremia should be excluded. In the hypo-osmolar state (serum osmolality less than 280 mOsm/kg), urine osmolality is used to determine whether water excretion is normal or impaired. A urine osmolality value of less than 100 mOsm/kg indicates complete and appropriate suppression of antidiuretic hormone secretion. A urine sodium level less than 20 mmol/L is indicative of hypovolemia, whereas a level greater than 40 mmol/L is suggestive of the syndrome of inappropriate antidiuretic hormone secretion. Levels of hormones (thyroid-stimulating hormone and cortisol) and arterial blood gases should be determined in difficult cases of hyponatremia.
Hyponatremia (serum sodium level less than 134 mmol/L) is a common electrolyte disturbance occurring in a broad spectrum of patients, from asymptomatic to critically ill.1,2 There are serious neurologic sequelae associated with hyponatremia and its treatment. Therefore, a logical, rigorous differential diagnosis is mandatory before therapy can be begun.3,4 Since hyponatremia is caused primarily by the retention of solute-free water, its cause encompasses disorders associated with limitation in water excretion.5 The principal causes of hyponatremia are summarized in Table 1.
Table 1

As with other electrolyte abnormalities, the history and physical examination can provide important clues toward the correct diagnosis. In most cases the initial laboratory evaluation includes measurement of serum osmolality and urine osmolality (by osmometer if available), urine sodium concentration and serum levels of other electrolytes (potassium, chloride and bicarbonate) as well as serum concentrations of urea, glucose, uric acid, total proteins and triglycerides. In addition, determination of serum levels of thyroid-stimulating hormone and cortisol is important to exclude any associated endocrinopathy (Table 2, Fig. 1). Measurement of arterial blood gases is also useful in the differential diagnosis of hyponatremia, particularly in patients with abnormal serum bicarbonate concentrations.
Table 2

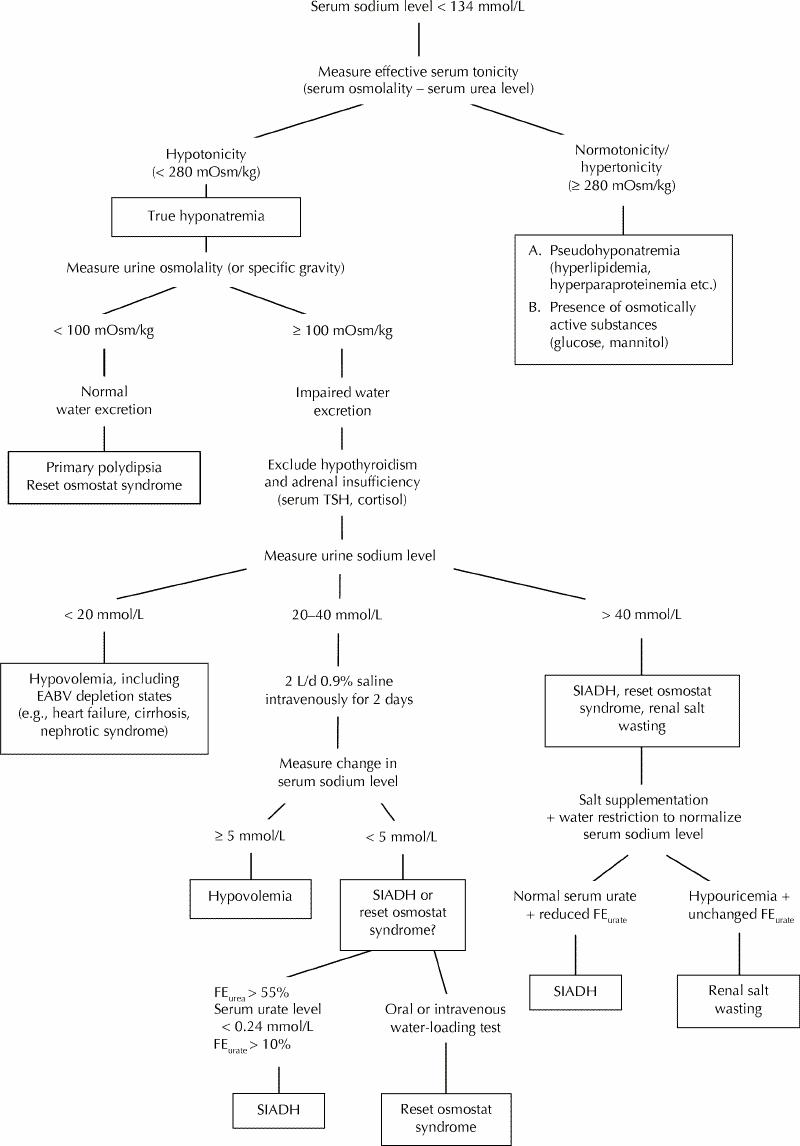
Fig. 1: Clinical diagnostic algorithm for hyponatremia. TSH = thyroid-stimulating hormone, EABV = effective arterial blood volume, SIADH = syndrome of inappropriate secretion of antidiuretic hormone, FE = fractional excretion.
The step-by-step diagnostic evaluation of hyponatremia is shown in Fig. 1.
Serum osmolality
The initial approach to the hyponatremic patient is to measure the serum osmolality to determine whether the hyponatremia represents a true hypo-osmolar state.1,2,5 Although urea contributes to the absolute value of serum osmolality measured with an osmometer, it does not hold water within the extracellular space because of its membrane permeability.1 Urea is an ineffective osmole and does not contribute to the effective serum osmolality (tonicity). Thus, in a patient with hyponatremia, normal or elevated effective serum osmolality (measured as serum osmolality less serum urea level in millimoles per litre) suggests the presence of either pseudohyponatremia (due to hyperparaproteinemia or hypertriglyceridemia) or increased concentrations of other osmoles, such as glucose and mannitol.1,6,7
In the context of marked hyperlipidemia (hypertriglyceridemia) and lactescent serum, lipids occupy space in the volume of serum, leading to lower readings in the concentrations of sodium and free water per litre of serum. However, the physiologically significant serum water and sodium and serum osmolality remain unaffected. Newer methods using ion-selective electrodes in the measurement of serum electrolytes may avoid this problem.7,8
In the presence of osmotically active substances (e.g., patients with hyperglycemia or those receiving mannitol infusions), an increase in serum osmolality is observed, which results in movement of water out of the cells and subsequently a reduction of the serum sodium level by dilution. It has been calculated that every increase of 3.4 mmol/L in the serum glucose level will draw enough water out of the cells to reduce the serum sodium concentration by 1 mmol/L (i.e., a decrease of 1.6 mmol/L in the serum sodium level per increase of 5.6 mmol/L in the glucose level).6,9 However, recent evidence suggests that the hyperglycemia-induced decrease in sodium concentration is considerably higher than the “standard” correction factor of 1.6, especially when glucose levels are greater than 22.2 mmol/L.10 Hillier and colleagues10 have proposed that a correction factor of 2.4 mmol/L is a better overall estimate of the association between sodium levels and glucose levels.
Urine osmolality
If a hypo-osmolar state is confirmed, the next step is to determine whether the ability of the kidneys to dilute the urine is intact by measuring urine osmolality.11 The normal response of the kidney is to elaborate maximally dilute urine (urine osmolality less than 100 mOsm/kg, specific gravity 1.003 or less). If the urine is maximally dilute, it indicates that antidiuretic hormone (ADH) secretion is completely and appropriately suppressed, a finding seen in patients with primary polydipsia or reset osmostat syndrome.5,12 Hyponatremia is unlikely to develop in the setting of an intact urine-diluting mechanism.13,14,15 However, it may occur in the rare case of patients who ingest large amounts of water (in isolated instances more than 10 to 15 L/d12). In these cases a tendency to hyponatremia will be unmasked and enhanced by a concurrent (even mild) impairment in water excretion.13,14,15 This occurs in the setting of central nervous system dysfunction and in patients receiving antipsychotic agents, or it may be due to nausea or stress-induced ADH secretion. Another unusual situation, in which modest amounts of fluid intake can lead to hyponatremia even when the urine-diluting ability is intact, is observed in cases of extremely reduced solute intake, in which the ability to excrete water is reduced by a poor dietary intake.14 This phenomenon has been described in patients with chronic alcoholism and is often referred to as “beer potomania syndrome” but has also been reported in patients with extremely limited intake of solid foods.13,14,15
Decreased urine osmolality is also observed in some patients with reset osmostat syndrome, when water intake reduces the serum osmolality below the new threshold for ADH release.16 Reset osmostat syndrome (discussed in a separate section later in this article) is a variant of the syndrome of inappropriate ADH secretion (SIADH) and is present in about one-third of patients with SIADH.
Urine sodium concentration
In patients with hyponatremia and inappropriately concentrated urine, it is particularly important to assess the effective arterial blood volume.11 A decreased volume is by far the most common cause of hyponatremia in everyday clinical practice.
Hypovolemia can be determined clinically by the presence of postural changes in blood pressure and pulse rate. Measurement of urine electrolyte levels is also extremely useful in the assessment of effective arterial blood volume, since patients with volume depletion exhibit low urinary excretion of sodium and chloride (sodium level less than 20 mmol/L, chloride level less than 20 mmol/L).2,11,17,18 A urine sodium level less than 20 mmol/L in hypovolemia is relevant if renal salt wasting does not exist.19 On the other hand, an increased urine sodium level is usually observed in patients with euvolemic hyponatremia (urinary sodium level greater than 40 mmol/L). However, the urine sodium concentration in euvolemic hyponatremia may be less than 20 mmol/L when dietary sodium intake is low.
An increased urine sodium concentration (greater than 40 mmol/L) is also found in sodium-wasting conditions, such as recent diuretic therapy, certain renal parenchymal diseases, adrenal insufficiency and metabolic alkalosis (Table 3).11,17,18 Hyponatremia occurs almost exclusively with thiazide (and, less commonly, loop) diuretics, which act in the distal tubule, interfering only with urine dilution.20 Several mechanisms have been postulated, including hypovolemia-stimulated ADH release, interference with urine dilution in the cortical diluting segment, and an alteration in osmoreceptor sensitivity and thirst regulation mediated by potassium depletion.20
Table 3

The ability of the kidney to both conserve sodium and dilute the urine is impaired in patients with progressive renal disease owing to the associated osmotic diuresis.11 However, the capacity of water excretion is relatively maintained in mild to moderate renal disease, and water retention and hyponatremia are seen only when the glomerular filtration rate falls to very low levels.11 For instance, when the glomerular filtration rate has fallen to about 5 mL/min, only about 1.5 L/24 h can be excreted as water. Hence, a patient drinking in excess of that amount will be at risk for water retention and hyponatremia. In patients with chronic renal failure, salt wasting seems to occur only after acute reductions in salt intake and is reversible or preventable if salt intake is gradually reduced over time.21
It is of interest that metabolic alkalosis represents one of the conditions in which volume depletion may not be associated with decreased urine sodium concentrations.11,22 This condition is seen primarily in patients presenting with vomiting or even chronic metabolic alkalosis. In this clinical setting (i.e., active vomiting) a disequilibrium state exists in which the filtered load of sodium bicarbonate exceeds the reabsorptive capacity of the proximal tubule, which leads to increased delivery of sodium bicarbonate distally.11,22 Bicarbonate acts as a nonreabsorbable anion in this situation, resulting in an obligatory sodium bicarbonate loss in the urine. Once vomiting has ceased and the patient has reached an equilibrium state, the urine sodium level will be low since the capacity of the proximal tubule for sodium bicarbonate reabsorption now equals the filtered load.11,22
In the context of metabolic alkalosis related to volume depletion, urinary chloride excretion is low (less than 10 to 20 mmol/L) owing to increased renal reabsorption of chloride. Therefore, determination of the urine chloride level may be of help, and this index is sometimes preferred to the urine sodium level as a measure of extracellular volume.22
In patients with equivocal findings (i.e., urine sodium level 20 to 40 mmol/L), the response of serum sodium and its fractional excretion (FENa+) to the administration of normal saline (1 to 2 L/d for 1 to 2 days) can be used to establish a correct diagnosis. An increase of less than 5 mmol/L in serum sodium levels and an increase of greater than 0.5% in FENa+ is highly suggestive of SIADH, whereas the opposite (an increase in serum sodium levels greater than 5 mmol/L and an increase of less than 0.5% in FENa+) is evident in hypovolemia.17,23
Urea and uric acid levels
Serum composition can also be used to assess the effective arterial blood volume. In fact, urea is particularly sensitive to hypovolemia. Thus, in patients with a normal serum creatinine level, an increase in serum urea levels (and therefore in the urea:creatinine ratio) suggests hypovolemia, whereas a decrease in serum urea levels is indicative of an increase in extracellular volume.11,24 In fact, a low serum urea concentration is frequently reported in patients with SIADH, in whom the volume is somewhat expanded.25,26,27 Musch and associates23 found that hyponatremic patients presenting with decreased FENa+ (less than 0.5%) combined with decreased fractional excretion of urea (less than 55%) responded successfully to the administration of normal saline, signifying a decrease in effective arterial blood volume (Fig. 1).
Similarly, serum uric acid levels may be used in the differential diagnosis of hyponatremia.19,25,26,27,28 It has been reported that patients with hypovolemia tend to exhibit increased serum uric acid levels (greater than 0.3 mmol/L).25 In contrast, in patients with SIADH, serum uric acid levels are actually depressed (less than 0.24 mmol/L).19,28 This decrease in serum uric acid levels typically results from an increase in urate excretion (fractional excretion of urate greater than 10%). Hyperuricuria has been attributed to water retention and the resultant expansion of serum volume, which, in turn, leads to reduced sodium and urate reabsorption.19,25,26,27,28 However, it remains questionable whether volume expansion per se is a major determinant that causes a significant increase in urate excretion rates in humans.29 In fact, there is evidence that hypouricemia related to high urate clearance may also be encountered in some hypovolemic patients who exhibit cerebral salt-wasting syndrome30 (this syndrome is discussed in a separate section later in this article).
Hormonal disorders
It is of interest that the serum sodium concentration may be low (by as much as 130 mmol/L) in pregnant women owing to human chorionic gonadotropin-induced release of a hormone (relaxin) that is associated with a downward resetting of serum osmolality.31
Hyponatremia can occur in the setting of adrenal (primary or secondary) insufficiency and hypothyroidism.32,33 Therefore, serum levels of thyroid-stimulating hormone and random cortisol should be determined in confusing cases of hyponatremia and before a diagnosis of SIADH is made. Glucocorticoid deficiency increases water permeability in the collecting tubules. Elevated ADH levels have also been found in patients with glucocorticoid deficiency.32 In patients with hypothyroidism, both ADH-mediated and intrarenal mechanisms have been implicated in the pathogenesis of hyponatremia.33
Acid-base status and potassium homeostasis
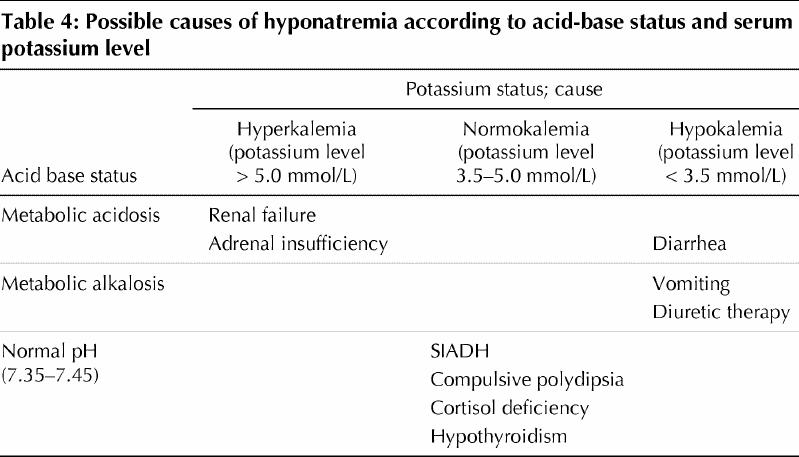
Acid-base status and potassium balance should be evaluated in certain cases of hyponatremia (Table 4).11,34,35 For example, the presence of metabolic acidosis and hyperkalemia is suggestive of renal function impairment or adrenal insufficiency, whereas a diarrheal syndrome is usually associated with metabolic acidosis and hypokalemia. Excessive vomiting or the use of diuretics may result in hyponatremia in association with metabolic alkalosis and hypokalemia. However, acid-base status and potassium balance are not disturbed in SIADH.34,35
Table 4
Special issues
Volume depletion versus syndrome of inappropriate antidiuretic hormone secretion
Hyponatremia is the most common electrolyte abnormality in the general hospital population (seen in 2% of patients in hospital).2,5,24 In the everyday clinical setting, hypovolemia and SIADH are 2 clinical entities of major significance associated with low serum sodium levels.11 It is therefore useful to be able to differentiate between these conditions by applying simple diagnostic tests and manoeuvres.11,24 Clinical findings, such as postural changes in blood pressure and pulse rate, together with laboratory evidence of hemoconcentration (e.g., high hematocrit and serum total protein values) suggest a hypovolemic state. Furthermore, volume-depleted patients exhibit more concentrated urine (urine osmolality greater than 450 mOsm/kg) and lower urinary sodium excretion (urine sodium level less than 20 mmol/L and FENa+ less than 1%) than do patients with SIADH (urine osmolality greater than 100 mOsm/kg, urine sodium level greater than 40 mmol/L and FENa+ greater than 1%). In addition, prerenal azotemia (serum urea:creatinine ratio greater than 0.17) and hyperuricemia (serum uric acid level greater than 0.3 mmol/L) are often present in patients with hypovolemic hyponatremia.19,28 In contrast, patients with SIADH commonly exhibit low serum uric acid levels (less than 0.24 mmol/L) associated with increased fractional excretion of urate (greater than 10%).19,28 Finally, the response of serum sodium and FENa+ to the administration of normal saline (1 to 2 L/d for 2 days) can be used to establish the correct diagnosis. Patients with hypovolemia respond with an increase of more than 5 mmol/L in serum sodium level together with an increase of less than 0.5% in FENa+; the opposite is expected in patients with SIADH.23
Reset osmostat syndrome
Reset osmostat syndrome may be identified as a cause of chronic hyponatremia in several conditions, such as pregnancy, hypovolemic states, quadriplegia, psychosis and other chronic debilitating illnesses, including tuberculosis, encephalitis, malignant disorders and malnutrition.16,36,37 The suggested mechanism is a downward resetting of the serum osmolality level at which the osmoreceptors control ADH secretion.37 Patients with reset osmostat syndrome commonly exhibit normovolemic hyponatremia, exhibit normal adrenal, renal and thyroid function and no evidence of cardiac or hepatic disease, exhibit normal excretion of a standard (10 to 15 mL/kg given orally or intravenously) water load (i.e., excretion of more than 80% within 4 hours), exhibit an intact urine-diluting ability, achieving essentially normal maximal urine dilution (serum osmolality less than 100 mOsm/kg) after an oral water-loading test, and retain the ability to concentrate urine at a serum osmolality level above the reset level. These features constitute the diagnostic criteria of the syndrome. Thus, the diagnosis of reset osmostat syndrome relies on the response to water restriction and the presence of an intact urine-diluting ability, which result in an increase in urine osmolality, since slight elevation of serum sodium levels will stimulate ADH release. Patients with reset osmostat syndrome have normal osmoreceptor responses to changes in serum osmolality, although the threshold for ADH release is reduced. Consequently, the serum sodium levels are below normal but stable (usually between 125 and 130 mmol/L), since the ability to excrete water is maintained.16,37
Cerebral salt-wasting syndrome versus syndrome of inappropriate antidiuretic hormone secretion
Cerebral salt-wasting syndrome has been described in patients with intracranial disease (mainly those with subarachnoid hemorrhage) who manifest hyponatremia and meet some of the diagnostic criteria of SIADH.38 Cerebral salt-wasting syndrome is not only regarded as a distinct clinical entity but is also considered by some, particularly neurosurgeons, to be more common than SIADH.39,40,41 However, in most of these patients there is evidence of extracellular fluid volume depletion, which stimulates ADH release.38,42,43 In this respect, the high urine sodium concentration seems to be due to urine sodium wasting and not to volume expansion. Although the exact mechanism of natriuresis is unknown, it has been suggested that a brain natriuretic peptide (atrial natriuretic peptide and endogenous ouabain have also been incriminated44) is released and causes an increase in sodium excretion and in urine volume.11,38 The natriuretic factor has been identified as a protein and has been partially characterized.44,45 Work is underway to isolate and identify this natriuretic factor. A natriuretic factor with characteristics similar to those observed in patients with intracranial disease has been reported in patients with Alzheimer's disease.45
The changes in uric acid homeostasis are particularly useful in differentiating between SIADH and cerebral salt-wasting syndrome.28,44 Uric acid levels are depressed in patients with SIADH,19,25,26,27 whereas in volume-depleted patients with hyponatremia uric acid levels are normal or slightly increased in the presence of intact renal tubular function.44 However, uric acid levels are depressed when there is renal salt wasting. In fact, serum uric acid levels in cerebral salt-wasting syndrome tend to be unexpectedly low owing to a tubular transport abnormality for uric acid.38,44 Thus, hypouricemia and high urine levels of uric acid may be common features of intracranial disease in general.44,46 Hypouricemia and increased renal uric acid excretion have been noted in patients with Alzheimer's disease even in the absence of hyponatremia.38,43,47 There is also evidence that hypouricemia and hyponatremia coexist in patients with AIDS.44 The course of hypouricemia after reversal of hyponatremia has been proposed as a useful diagnostic tool in distinguishing between SIADH and cerebral salt-wasting syndrome.28 Correction of the serum sodium concentration leads to normalization of uric acid handling by the kidney in SIADH, but in cerebral salt-wasting syndrome hypouricemia and increased renal uric acid excretion persist.38,44
The demonstration of a natriuretic factor in the serum of patients with persistent hypouricemia, hyperuricuria (fractional excretion of urate greater than 10%) and possible renal salt wasting has raised the possibility that the tubule transport defect for sodium and urate might be due to a single circulating plasma protein.29,45 It has been postulated that the natriuretic factor is present in patients with renal salt wasting but not in those with SIADH.44 In clinical practice the identification of this factor would be valuable in differentiating renal salt wasting from SIADH before appropriate therapy is started. Since renal salt wasting appears to be more common than SIADH in patients with intracranial disease, distinguishing between these 2 situations would help prevent potential complications resulting from inappropriate treatment.45
Footnotes
This article has been peer reviewed.
Contributors: All 3 authors contributed substantially to the manuscript. They were all involved in drafting and revising the article, and all approved the final version of the manuscript.
Competing interests: None declared.
Correspondence to: Dr. Moses S. Elisaf, Department of Internal Medicine, University of Ioannina Medical School, GR 451 10 Ioannina, Greece; fax +30 651 097016
References
- 1.Rose BD. New approach to disturbances in the plasma sodium concentration. Am J Med 1986;81:1033-40. [DOI] [PubMed]
- 2.Kumar S, Beri T. Sodium. Lancet 1998;352:220-8. [DOI] [PubMed]
- 3.Gross P, Hensen J. Evaluation of hyponatremia: Is there a rational approach? Nephrol Dial Transplant 1995;10:1789-91. [PubMed]
- 4.Fraser CL, Arieff AI. Epidemiology, pathophysiology, and management of hyponatremic encephalopathy. Am J Med 1997;102:67-77. [DOI] [PubMed]
- 5.Adrogue HJ, Madias NE. Hyponatremia. N Engl J Med 2000;342:1581-9. [DOI] [PubMed]
- 6.Mange K, Matsuura D, Cizman B, Soto H, Ziyadeh FN, Goldfarb S, et al. Language guiding therapy: the case of dehydration versus volume depletion. Ann Intern Med 1997;127:848-53. [DOI] [PubMed]
- 7.Weisberg LS. Pseudohyponatremia. A reappraisal. Am J Med 1989;89:315-8. [DOI] [PubMed]
- 8.Maas AHJ, Sigaard-Andersen O, Weisberg HF, Zijlstra WG. Ion-selective electrodes for sodium and potassium: a new problem of what is measured and what should be reported. Clin Chem 1985;31:482-5. [PubMed]
- 9.Katz MA. Hyperglycemia-induced hyponatremia: calculation of expected serum sodium depression. N Engl J Med 1973;289:843-4. [DOI] [PubMed]
- 10.Hillier TA, Abbott RD, Barrett EJ. Hyponatremia: evaluating the correction factor for hyperglycemia. Am J Med 1999;106:399-403. [DOI] [PubMed]
- 11.Rose BD. Hypoosmolal states — hyponatremia. In: Rose BD, Post TW, editors. Clinical physiology of acid-base and electrolyte disorders. 5th ed. New York: MacGraw Hill; 2001. p. 696-745.
- 12.Gillum DM, Linas SL. Water intoxication in a psychotic patient with normal water excretion. Am J Med 1984;77:773-4. [DOI] [PubMed]
- 13.Hilden T, Svendsen TL. Electrolyte disturbances in beer drinkers. A specific “hypoosmolality syndrome.” Lancet 1975;7928:245-6. [DOI] [PubMed]
- 14.Thaler SM, Teitelbaum I, Berl T. “Beer potomania” in non-beer drinkers: effect of dietary low solute intake. Am J Kidney Dis 1998;31:1028-31. [DOI] [PubMed]
- 15.Liamis GL, Milionis HJ, Rizos EC, Siamopoulos KC, Elisaf MS. Mechanisms of hyponatraemia in alcohol patients. Alcohol Alcohol 2000;35:612-6. [DOI] [PubMed]
- 16.Lipschutz JH, Arieff AI. Reset osmostat in a healthy patient. Ann Intern Med 1994;120:574-6. [DOI] [PubMed]
- 17.Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med 1987;83:905-8. [DOI] [PubMed]
- 18.Milionis HJ, Elisaf MS. Hyponatraemia in a patient with hepatic cirrhosis. Nephrol Dial Transplant 1999;14:787-9. [DOI] [PubMed]
- 19.Beck LH. Hypouricemia in the syndrome of inappropriate secretion of antidiuretic hormone. N Engl J Med 1979;301:528-30. [DOI] [PubMed]
- 20.Sonnenberck M, Friedlander Y, Rosin AJ. Diuretic induced severe hyponatremia: review and analysis of 129 reported patients. Chest 1993;193:601-6. [DOI] [PubMed]
- 21.Danovitch GM, Bourgoignie J, Bricker NS. Reversibility of the “salt-losing” tendency of chronic renal failure. N Engl J Med 1977;296:14-9. [DOI] [PubMed]
- 22.Sherman RA, Eisinger RP. The use (and misuse) of urinary sodium and chloride measurements. JAMA 1982;247:3121-4. [PubMed]
- 23.Musch W, Thimpont J, Vandervelde D, Verhaeverbeke I, Berghmans T, Decaux G. Combined fractional excretion of sodium and urea better predicts response to saline in hyponatremia than do usual clinical and biochemical parameters. Am J Med 1995;99:348-55. [DOI] [PubMed]
- 24.Elisaf MS, Milionis HJ, Siamopoulos KC. Electrolyte abnormalities in elderly patients admitted to a general medical ward. Geriatr Nephrol Urol 1991;7:73-9. [DOI] [PubMed]
- 25.Decaux G, Schlesser M, Coffernils M, Prospert F, Namias B, Brimioulle S, et al. Uric acid, anion gap and urea concentration in the diagnostic approach to hyponatremia. Clin Nephrol 1994;42(2):102-8. [PubMed]
- 26.Elisaf MS, Milionis HJ, Drosos AA. Hyponatremia due to inappropriate secretion of antidiuretic hormone in a patient with systemic lupus erythematosus. Clin Exp Rheumatol 1999;17:223-6. [PubMed]
- 27.Liamis G, Elisaf M. Syndrome of inappropriate antidiuresis associated with multiple sclerosis. J Neurol Sci 2000;172:38-40. [DOI] [PubMed]
- 28.Maesaka JK. An expanded view of SIADH, hyponatremia and hypouricemia. Clin Nephrol 1996;46:79-83. [PubMed]
- 29.Maesaka JK, Fishbane S. Regulation of renal urate excretion: a critical review. Am J Kidney Dis 1998;32:917-33. [DOI] [PubMed]
- 30.Tanneau RS, Moal MC, Rouhart F, Dueymes JM, Bourbigot B. Hypouricemia with high urate clearance in hyponatremia: Is it always a clue for increased effective volemia? Clin Nephrol 1995;44:128. [PubMed]
- 31.Davison JM, Shiells EA, Phillips PR, Lindheimer MD. Influence of humoral and volume factors on altered osmoregulation of normal human pregnancy. Am J Physiol 1990;258(4 Pt 2):F900-7. [DOI] [PubMed]
- 32.Linas SL, Berl T, Robertson GL, Aisenbrey GA, Schrier RW, Anderson RJ. Role of vasopressin in the impaired water excretion of glucocorticoid deficiency. Kidney Int 1980;18:58-67. [DOI] [PubMed]
- 33.Hanna FW, Scanlon MF. Hyponatraemia, hypothyroidism and role of arginine-vasopressin. Lancet 1997;350:755-6. [DOI] [PubMed]
- 34.Graber M, Corish D. The electrolytes in hyponatremia. Am J Kidney Dis 1991; 18:527-45. [DOI] [PubMed]
- 35.Decaux G, Crenier L, Namias B, Gervy C, Soupart A. Normal acid-base equilibrium in acute hyponatremia and mixed alkalosis in chronic hyponatremia induced by arginine vasopressin or 1-deamino-8-D-arginine vasopressin in rats. J Lab Clin Med 1994;123:892-8. [PubMed]
- 36.Elisaf MS, Milionis HJ, Siamopoulos KC. Chronic hyponatremia secondary to reset osmostat in a patient with advanced ovarian cancer. J Exp Clin Cancer Res 1996;15:313-4.
- 37.Elisaf MS, Konstantinides A, Siamopoulos KC. Chronic hyponatremia due to reset osmostat in a patient with colon cancer. Am J Nephrol 1996;16:349-51. [DOI] [PubMed]
- 38.Palmer BF. Hyponatraemia in a neurosurgical patient: syndrome of inappropriate antidiuretic hormone secretion versus cerebral salt wasting. Nephrol Dial Transplant 2000;15:262-8. [DOI] [PubMed]
- 39.Nelson PB, Seif SM, Maroon JC, Robinson GA. Hyponatremia in intracranial disease: perhaps not the syndrome of inappropriate secretion of antidiuretic hormone (SIADH). J Neurosurg 1981;55:938-41. [DOI] [PubMed]
- 40.Wijdick EFM, Vermeulen M, ten Haaf JA, Hijdra A, Bakker WH, van Gijn J. Volume depletion and natriuresis in patients with a ruptured intracranial aneurysm. Ann Neurol 1985;18:211-6. [DOI] [PubMed]
- 41.Sivakumar V, Rajshekhar V, Chandy MJ. Management of neurosurgical patients with hyponatremia and natriuresis. Neurosurgery 1994;43:269-74. [DOI] [PubMed]
- 42.Al-Mufti H, Arieff AI. Hyponatremia due to cerebral salt-wasting syndrome. Combined cerebral and distal tubular lesion. Am J Med 1984;77:740-6. [DOI] [PubMed]
- 43.Tanneau RA, Pennec YL, Jouquan J, Le Menn G. Cerebral salt wasting in elderly persons [letter]. Ann Intern Med 1987;107:120. [DOI] [PubMed]
- 44.Maesaka JK, Gupta S, Fishbane S. Cerebral salt-wasting syndrome: Does it exist? Nephron 1999;82:100-9. [DOI] [PubMed]
- 45.Maesaka JK, Wolf-Klein G, Piccione JM, Ma CM. Hypouricemia, abnormal renal tubular urate transport, and plasma natriuretic factor(s) in patients with Alzheimer's disease. J Am Geriatr Soc 1993;41:501-6. [DOI] [PubMed]
- 46.Maesaka JK, Venkatesan J, Piccione JM, Decker R, Dreisbach AW, Wetherington JD. Abnormal urate transport in patients with intracranial disease. Am J Kidney Dis 1992;19:10-5. [DOI] [PubMed]
- 47.Tang WW, Kaptein EM, Feinstein EI, Massry SG. Hyponatremia in hospitalized patients with the acquired immunodeficiency syndrome (AIDS) and the AIDS-related complex. Am J Med 1993;94:169-74. [DOI] [PubMed]