

Abstract
We prepared a collection of complex cycloheptatriene-containing azetidine lactones by applying two key photochemical reactions: “aza-Yang” cyclization and Buchner carbene insertion into aromatic rings. While photolysis of phenacyl amines leads to a rapid charge transfer and elimination, we found that a simple protonation of the amine enables the formation of azetidinols as single diastereomers. We provide evidence, through ultrafast spectroscopy, for the electron transfer from free amines in the excited state. Further, we characterize aza-Yang reaction by establishing the dependence of initial reaction rates on rates of photon absorption. Unanticipated change in reactivity in morpholine analogs is explained through interactions with the tosylate anion. Buchner reaction proceeds with slight preference for one diastereomer over the other, and successful reaction requires electron-donating carbene-stabilizing substituents. Overall, sixteen compounds were prepared over seven steps. Guided by an increase in structural complexity, efforts such as this one extend reach of chemists into unexplored chemical space and provide useful quantities of new compounds for studies focused on their properties.
Graphical Abstract
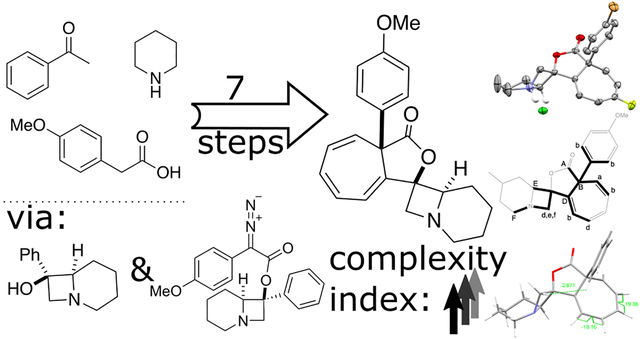
1. Introduction
Target-oriented organic synthesis is a well-codified endeavor. The stationary nature of the target (often a natural product of considerable structural complexity) and the finality of the effort when the target is reached have contributed to the utility of target-oriented synthesis for probing the capabilities of existing synthetic methods as well as the discovery of new ones, as various researchers execute different pathways en route to a target. Multiple measures of success exist in this realm.1 Surrounding the field of target-oriented synthesis lies the broader field of diversity-oriented synthesis in which the goal is not a singular target molecule, but rather entirely new molecules inhabiting unexplored chemical space.2 Searching for “newness” rather than a specific target molecule leaves one asking questions like the following: How to plan such a reaction sequence? How to know when to end? Which concessions to efficiency and selectivity are allowed and which ones are best avoided? For a given end product, what is the measure by which we can evaluate the sequence leading to it?
We have become proponents of using indices of structural complexity for guiding structural exploration of chemical space.3,4 Using these indices to monitor reactions conducted, or planned, accomplishes two goals: it favors the synthesis of new classes of structures, and it enables the discovery of new synthetic methods and/or the refining of existing ones in new contexts.
In addition to structural complexity, the “information density” of molecules, a notion recently introduced by Shenvi and Forli,5 may also help to define choices among different sequential steps in a non-targeted synthesis even more concisely. Information specified by the structure of one member from a group of isomeric molecules can be considered high. The goal of non-targeted synthesis is to minimize the number of atoms while maximizing the number of possible isomers! The synthetic challenge is to prepare selectively one isomer when many others are possible. Crucially for applications in the quest for bioactive compounds, a collection of such isomers, or closely related molecules, can become a powerful tool for probing the biology if paired with suitable assay for biological activity.6
In Figure 1 we present a sample non-targeted synthetic strategy aimed at constructing a collection of “non-natural” complex structures occupying new chemical space. The strategy is based on two key photochemical reactions that achieve a significant and rapid increase in structural complexity, and generate appealing motifs, i.e., rigidified polycyclic, spirocyclic or fused systems. As an example, we first made azetidinols through a modified Norrish-Yang cyclization that we dubbed “aza-Yang” (Figure 1A, aza-Yang).7 We then used the tertiary alcohol obtained in the aza-Yang product to install a group on which a carbene precursor could be introduced. Finally, an intramolecular Buchner reaction8,9 completes the sequence (Figure 1A, Buchner). This de-aromatization reaction achieves insertion of a carbene into a benzene ring carried through from the photochemical aza-Yang reaction, contributing to the overall high utilization of functionalities present in the starting materials. The products of this reaction sequence contain three stereocenters (one of which is all-carbon quaternary), 4-, 5-, 6-, and 7-membered rings, a tertiary amine, a γ-lactone, and a cycloheptatriene; overall, a lot of new functionality for just a few synthetic steps.
Figure 1:
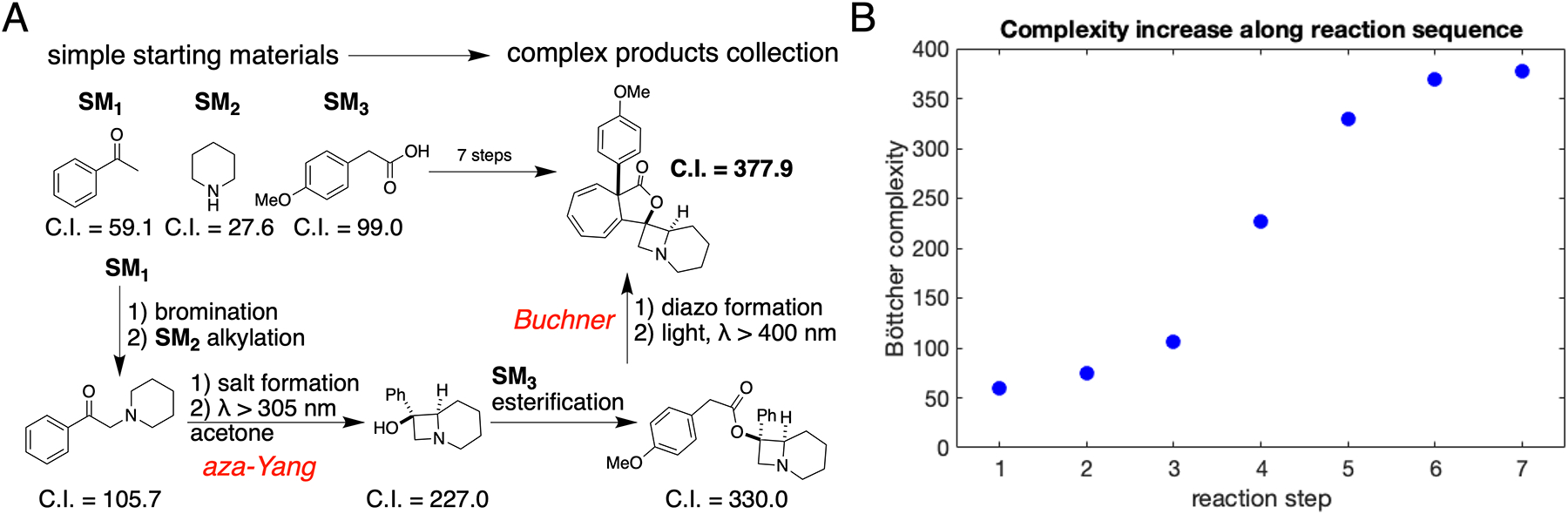
Chemical space exploration with complexity index as a guide. A) Sequence of synthetic manipulations used to create a collection of complex products from simple starting materials (SM1, SM2, and SM3,). Key photochemical reactions are labeled in red italic letters. B) Graphical representation of an increase in structural complexity index (C.I.) along the reaction sequence.
Tracking the increase in structural complexity4 across the sequence of seven reactions shows (Figure 1B) that the aza-Yang reaction doubles the complexity of the simple phenacyl-piperidine (C.I. 227.0 versus 105.7). A large increase in C.I. is obtained in the coupling step, (4 →5), but this is due to new atoms and connectivity being introduced when two molecules are joined together. The flattening of the complexity curve suggests a natural stop to the reaction sequence. Members of the final compound collection (Figure 9C) have C.I. in the range from 347 to 534. For perspective, raw Böttcher complexity indices of several bioactive compounds, such as penicillin, lovastatin, vincristine, and vancomycin are listed in Table 1 along with C.I. values normalized for molecular weight.
Figure 9:
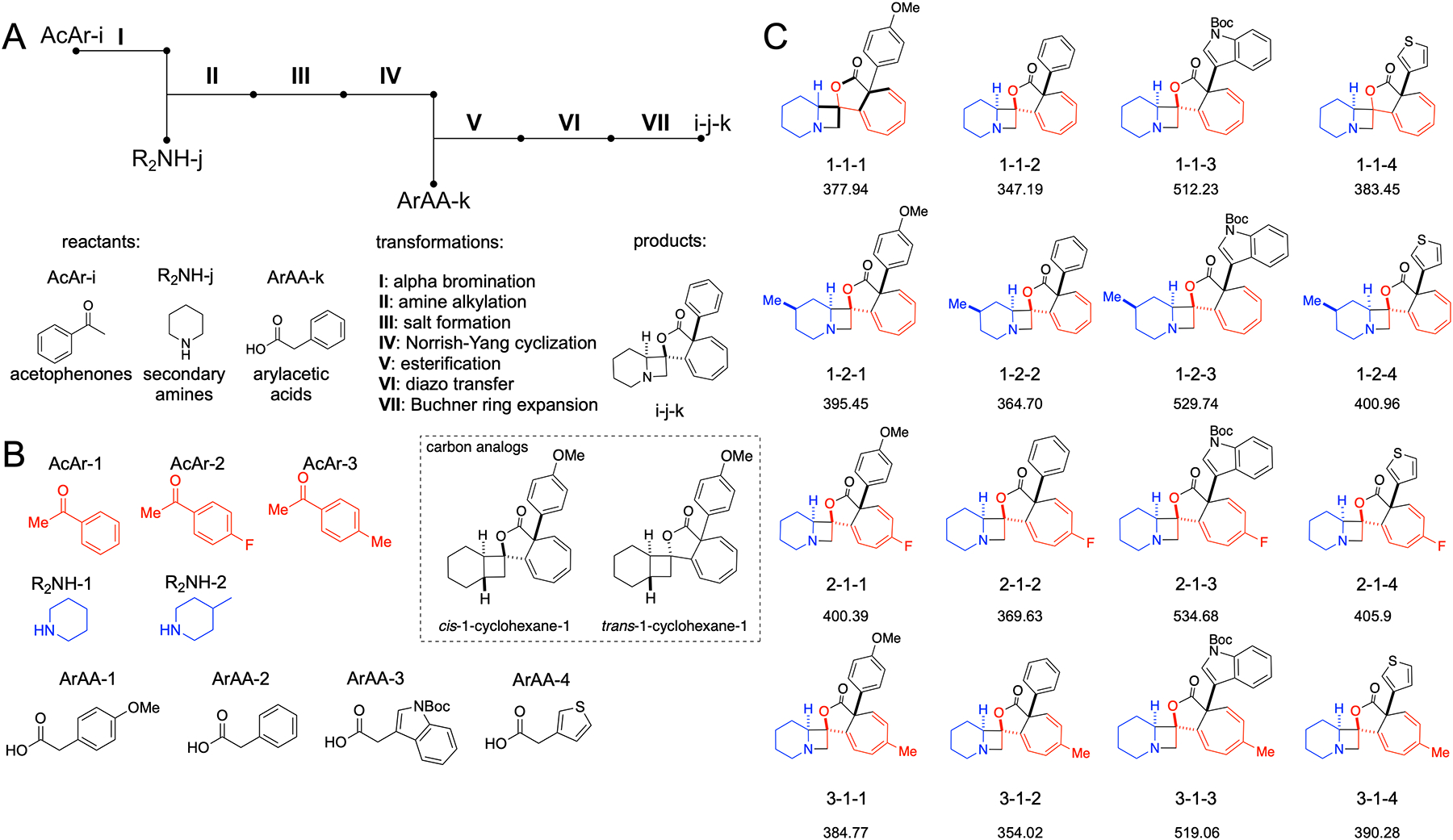
Library of cycloheptatriene azetidine lactones prepared by applying two photochemical reactions described above. A) Schematic route leading from acetophenones (AcAr), secondary amines (R2NH), and arylacetic acids (ArAA) consists of seven transformations (I-VII). B) Structures of three types of building blocks used. C) Structures of final compounds with names encoding the building blocks used to make them (acetophenone-amine-arylacetic acid.) In structure 1-1-1 bonds that are made in the process are bolded. Complexity indices are shown below the code. Two analogs of 1-1-1 containing cyclohexane instead of piperidine (N → CH edit)are shown in dashed box.
Table 1:
Computed Böttcher complexity indices (C.I.) of several arbitrarily chosen well-known bioactive molecules and of one of the members of compound library synthesized by us. Complexity normalized by molecular weight as a proxy for size of these molecules is also shown.
| name | C.I. | C.I./MW |
|---|---|---|
| penicillin | 382.24 | 1.15 |
| lovastatin | 410.31 | 1.01 |
| vincristine | 1010.31 | 1.22 |
| vancomycin | 1844.34 | 1.27 |
| 1-1-1, Figure 9C | 377.94 | 1.08 |
As mentioned, Figure 1 provides a simple example of a sequence leading to non-targeted products, which nevertheless resemble drug-like molecules and are likely to exhibit drug-like physico-chemical properties. The value of such molecules lies in their use as inputs to non-targeted screening for bioactivity. Just as in targeted synthesis, the path in Figure 1 raises all the usual questions about reaction mechanisms, selectivity, functional group tolerance, etc. Thus, by pursuing the non-targeted approach we do not depart from the themes of synthetic chemistry, and we also gain in accessing new chemical (and biological?) space.
2. Results & Discussion
2.1. Aza-Yang cyclization
2.1.1. General observations
The chemistry of excited states can provide a useful entry to new structural chemical space, but challenges in controlling the highly reactive intermediates often need to be addressed.
Norrish-Yang reactions produce strained, four-membered rings by converting the energy of absorbed photons to chemical energy through the interconversion of favorably oriented bonds.10,11 First, a carbonyl group (often an aryl ketone) absorbs a photon causing an electron to undergo an π*← n transition. This is followed by a singlet-to-triplet intersystem crossing. The triplet is sufficiently long lived to abstract a hydrogen radical from the γ carbon forming two carbon-centered radicals (one of them stabilized by the neighboring oxygen and the aromatic ring in the case of aryl ketones). Radical recombination of this intermediate forms the 4-membered ring.
To experimentally acquaint ourselves with a standard Norrish-Yang cyclization, we first irradiated an acetonitrile solution of cyclohexyl acetophenone, 1, with a point-source Hg-Xe arc lamp.12 We obtained a mixture of diastereomeric trans-fused cyclobutanols 2 and 3 (Figure 2A) as determined by X-ray diffraction of their p-bromobenzoic acid esters 4 and 5 (CCDC 2040104 and 2039100).
Figure 2:
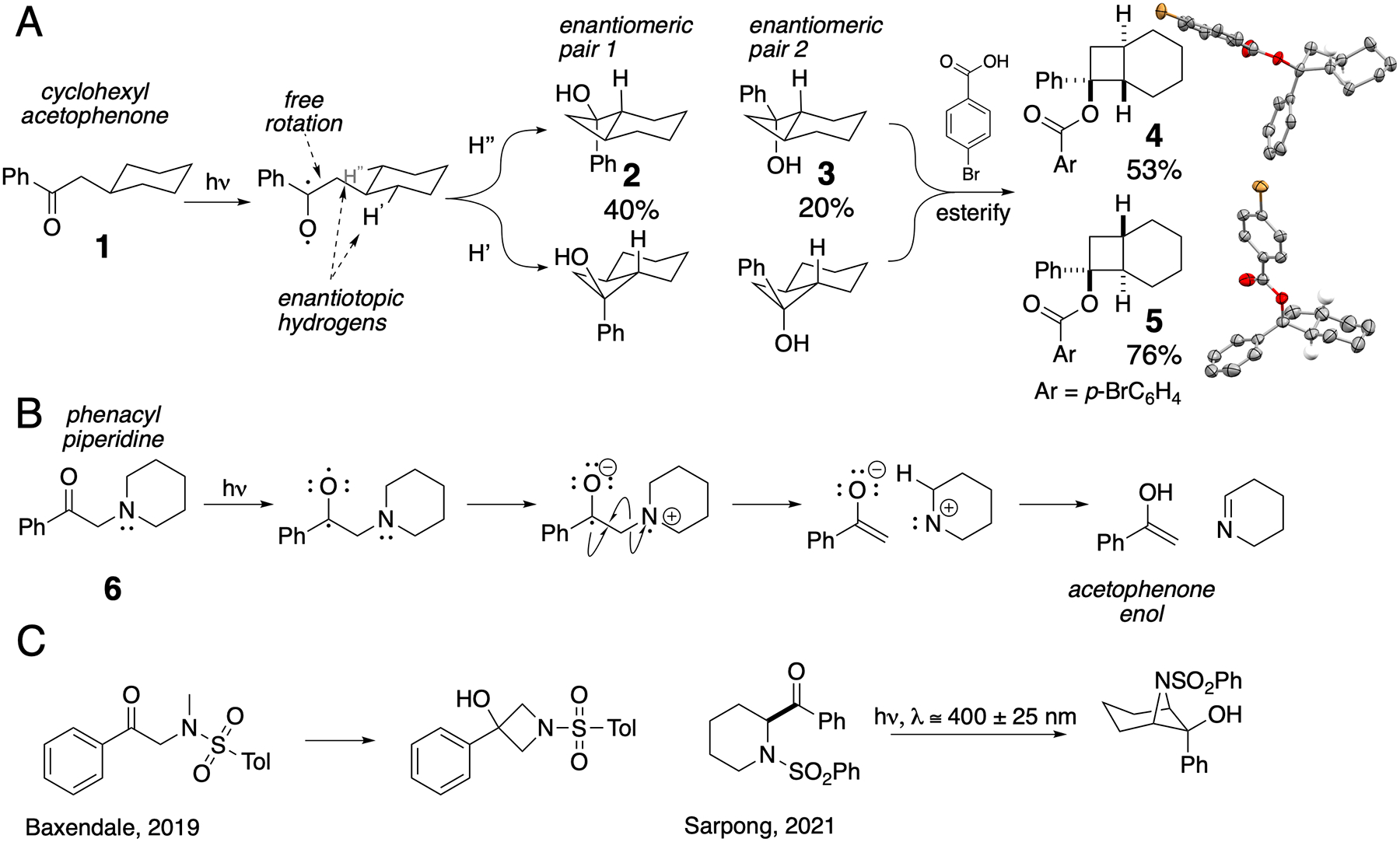
Norrish-Yang cyclization is a C–H functionalization reaction that produces strained four-membered rings. A) Cyclohexyl acetophenone, 1, is isomerized with ultraviolet light to diastereomeric cyclobutanols 2 and 3. Structures of their crystaline p-bromobenzoate esters are shown. B) Extension of this method with a C–H →N modification fails because a fast electron transfer leads to elimination instead of cyclization. C) Chemists previously addressed this issue by installing electron-withdrawing groups to “hide” the lone pair on a nitrogen.
A particularly useful variant of Norrish-Yang reaction would include a nitrogen atom at the β position of the photo-excitable ketone (such as 6 in Figure 2B) and would provide a route to azetidines in a simple manner.13 Azetidines have elicited recent interest as useful scaffolds for biologically active molecules.14–16 This approach fails, however, because of an increased electron density at the β position (a lone pair of electrons on nitrogen). More facile than γ H-radical abstraction is quenching of the triplet excited state through a rapid electron transfer from nitrogen to form a ketyl radical, which opens up a more favorable degradation pathway through the scission of the C–N bond, yielding methylketones and imine oligomers, or amines through complex reductive processes (Figure 2B).17,18 While undesirable in the context of azetidinol formation, generation of ketyl radical anions by photoinduced electron transfer has found numerous synthetic applications.19 One way to avoid it has been to protect the amine so that its lone pair engages in conjugation with other groups (e.g. by converting it to amides, carbamates, sulfonamides; Figure 2C),20–22 but this necessitates additional steps (introduction and removal of the electron-withdrawing group) and precludes the use of tertiary amines. Early studies by Aoyama and others identified the potential of this transformation23,24 and pointed to solid-state photochemistry as a way to control chemo-, regio-, and stereoselectivity in this reaction.25 Enantioenriched products have been obtained by performing these reactions in chiral hosts.26 Cyclic aminals used by Pedrosa and Andrés27 also likely avoid charge transfer through the anomeric delocalization of the nitrogen lone pair, as do aniline-derived substrates.28
The solution we present here is to conceal the lone pair through simple salt formation (Figure 3A). Thus, the tosylate salt of symmetric 6 dissolved in acetonitrile or acetone and irradiated at wavelengths that promote π*← n transition, produces racemic 7 (CCDC 2039099) as a single diastereomer with some elimination analogous to the one shown in Figure 2B. The tosylate anion which occupies one of the faces of this molecule (top face in the crystal structure shown in Figure 3B, CCDC 2051429) may be preventing the rotation around C–C bond which in cyclohexyl acetophenone 1 leads to diastereomeric mixture of products (Figure 2A). Larger phenyl group (A-value = 3)29 stays away from the anion, which places smaller hydroxy group (A-value = 0.87)29 trans from the bridgehead hydrogen. Interestingly, the final deprotonated amine has its lone pair cis to the bridgehead hydrogen.30 consistent with a Walden inversion on nitrogen upon deprotonation.
Figure 3:
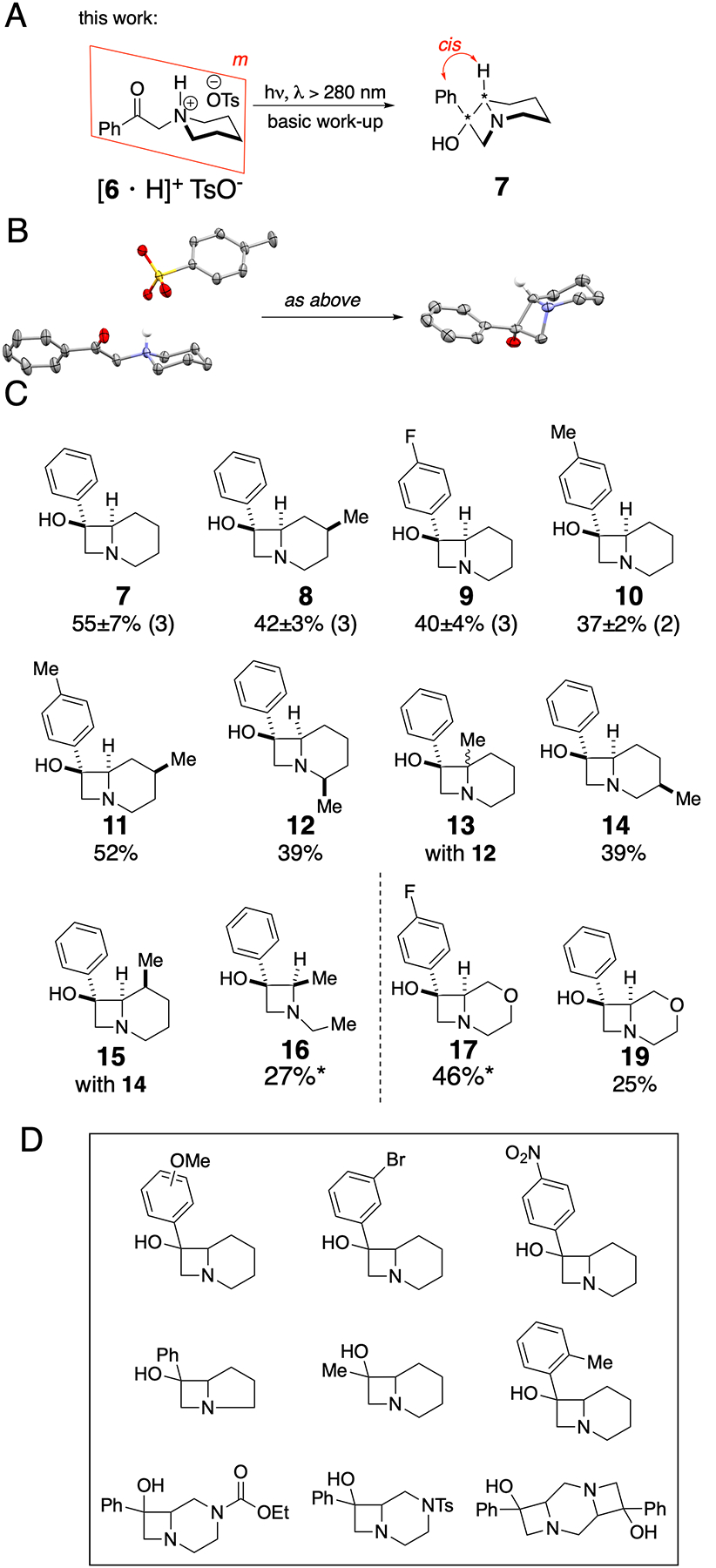
Simple protonation of nitrogen enables photochemical synthesis of azetidinols. A) Diastereomerically defined product 7 is obtained upon irradiation of an acetonitrile or acetone solution of a tosylate salt of phenacylpiperidine, 6. B) X-ray diffraction structures of the reactant and product above (carbon, gray; oxygen, red; sulfur, yellow; hydrogen, when shown, white). C) Successfully synthesized azetidinols. Isolated yields are given as averages and standard deviations with the number of repeated experiments in parentheses. Star signifies that the yield was calculated by comparing 1H NMR signals of the product to those of 1,3,5-trimethoxybenzene. D) Attempted, but unrealized products.
Following this protocol we successfully prepared azetidinols 7 – 17 and 19 (Figure 3C). Of these, we used 7 – 10 for further chemical elaboration into a compound collection. Compound 8 was obtained from a symmetric 4-methylpiperidine-containing substrate. The stereochemistry of the methyl substituent was established as endo (CCDC 2105274). Compounds 14 and 15 were both obtained from 3-methylpiperidine and co-crystallized as exo heterodimers (CCDC 2101761). Compounds 12 (CCDC 2126174) and 13 come from 2-methylpiperidine suggesting that the tertiary radical is not excluded from cyclization due to an increase in steric congestion.
We also encountered substrates that resisted azetidinol formation (Figure 3D) by either being inert or by degrading to complicated mixtures of products. For example, electronrich substitutents on the arylketones are known to preferentially undergo π*← π transition and are inert in Norrish-Yang reaction. Acetone derivatives are not suitable substrates for this reaction, nor are substrates where the geometry prevents hydrogen radical abstraction (such as in pyrrolidines or ortho-substituted acetophenones).31 If a substrate contains an aryl bromide, ultraviolet light generates aryl and bromine radicals which lead to complex mixtures of products.
In some amines tosylate salt formation (through nitrogen protonation) prior to photolysis does not allow azetidine formation. For example, simple change of the substrate from phenacylpiperidine, 6, to phenacylmorpholine 18, (Figure 4A) yielded neither the azetidinol nor the elimination products. We were surprised by this abrupt change in reactivity emanating from a somewhat remote oxygen in the ring. We wondered if subtle geometry changes in the substrate could account for this total lack of reactivity. To our surprise, the solid-state structures of the salts of these two analogs (Figure 4) revealed a stark difference in the positioning of the tosylate anion, which allowed us to formulate a hypothesis to explain the origin of this shift in reactivity. In 6 (CCDC 2051429), the tosylate is positioned “over” the saturated heterocycle and away from the benzoyl group, whereas in 18 (CCDC 2104641), it is hovering directly over the benzoyl group that is excited. In this configuration, the proximal tosylate, at a distance of 3 Å, is ideally positioned to accept the excess energy stored in the carbonyl excited state, thereby quenching it.
Figure 4:
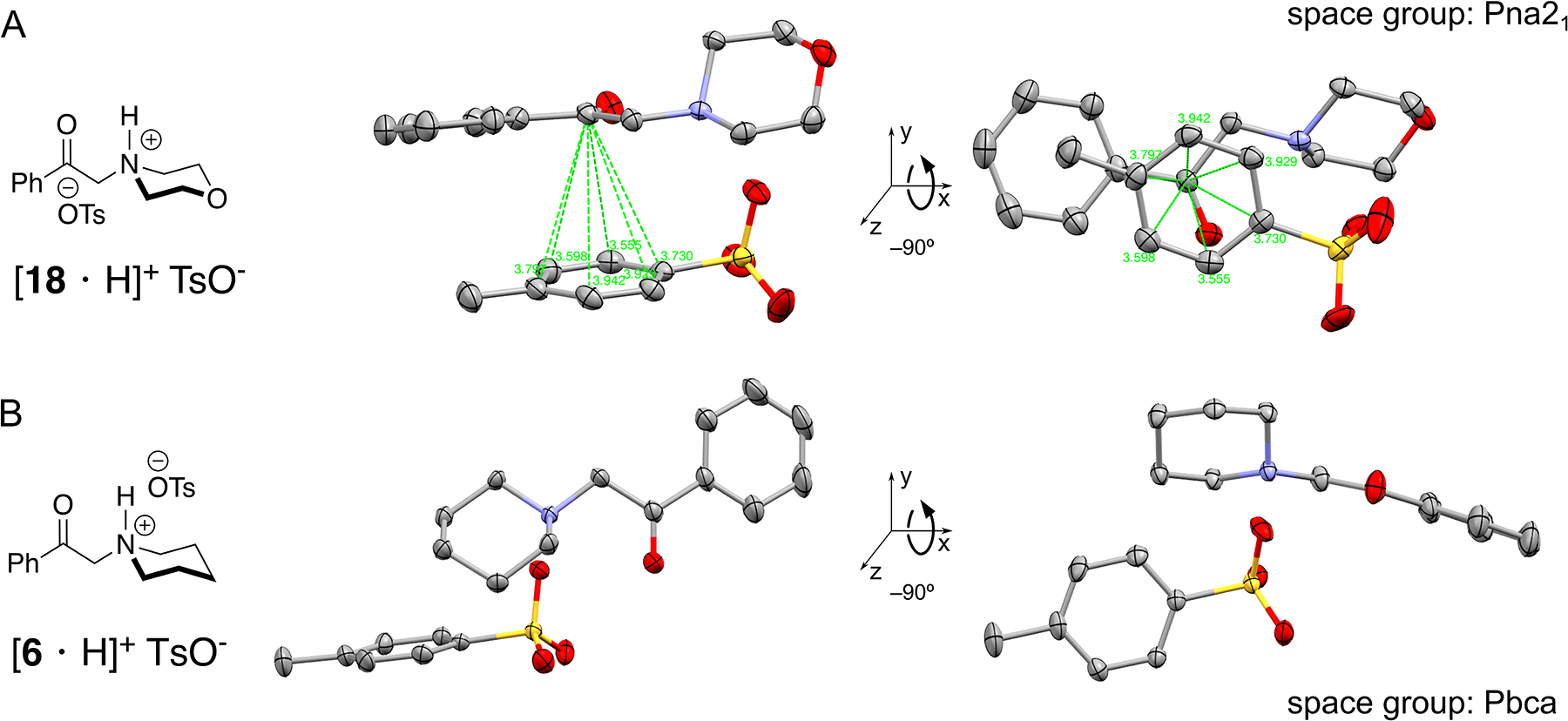
Tosylate salts can also be a liability in this photochemical reaction. We hypothesized that in the morpholine-containing substrate, [18H]+ TsO− (A), positioning of the tosylate parallel and proximal to the benzoyl group interferes with the subsequent γ hydrogen abstraction. The arrangement of the tosylate is quite different in the substrate that successfully cyclizes (B).
We tested this hypothesis by preparing a mesylate salt of the phenacyl morpholine. Indeed, the mesylate salt engaged in photochemical reactions and produced the expected azetidinol 19 (fluoro analog 17 CCDC 2127602). We also tested other acids (perchloric, acetic, formic, oxalic, and camphorsulfonic acid), but tosylate salts were most ideal either for the outcomes (minimizing the elimination and maximizing cyclization) or for the ease of manipulation (see Supporting Information Section 3.2 for quantitative results of these survey experiments).
2.1.2. Characterization of the aza-Yang excited state
Having empirically observed that free amines prefer elimination to cyclization, whereas protonated amines cyclize to azetidinols, we investigated whether differences in primary photochemical processes might explain why protonation works. For this we turned to ultrafast spectroscopy to reveal what happens to the excited state very soon after the absorption of a photon. We subjected solutions of seven compounds (Figure 5A) to pulsed irradiation with ultrafast laser light at 280 nm (corresponding to the π*← n excitation of the aryl ketone). A time-delayed probe pulse measured the change in absorption on the fs-ps timescale across the wavelength range from 334 nm to 684 nm. The transient absorption spectra reveal very different behavior for the free amines compared with the protonated tosylate salts. In the former, our analysis indicates that the excited state decays within tens of ps. For both 6 and 18, we observe two different time constants for the decay (9 ps and 59 ps for 6, and 10 ps and 115 ps for 18), which may be related to different steps in the dissociation reaction (Figure 5).
Figure 5:
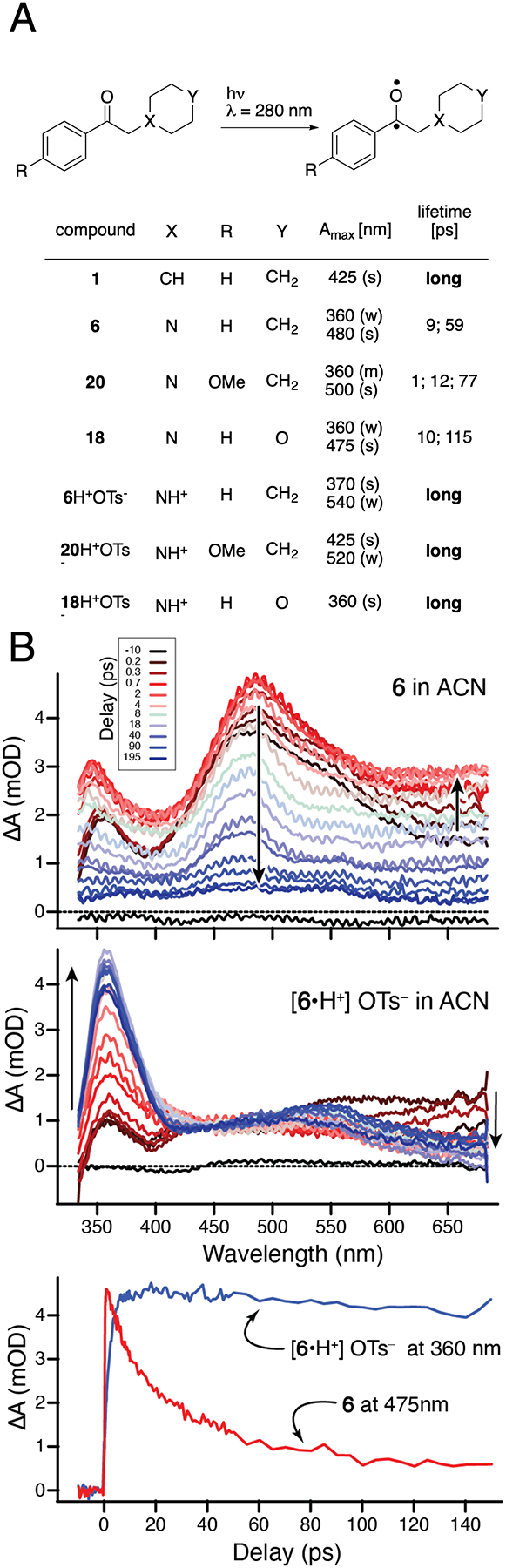
Ultrafast spectroscopy of the triplet excited state provides evidence for extending triplet state lifetime through protonation. A) The investigated molecules 6, 18, and 20 have short half-lives, whereas their tosylate salts have long half-lives, comparable to the lifetime of the C–H analog, 1. B) Excited state absorption spectra of 6 (top panel), [6H]+ TsO− (middle), and time evolution of absorption maxima at 360 nm and 475 nm (bottom).
In contrast to the rapid relaxation of the excited states in free amines, the excited states of the protonated amines undergo rapid intersystem crossing (ISC) in 2 ps, resulting in a triplet excited state with much longer lifetime of at least several nanoseconds, well beyond the range of our measurement.The rapid ISC and long-lived triplet state is similar to the excited state dynamics of acetophenone. In fact, for the tosylate salts of both 6 and 18 we observe a signature below 500 nm that decays with a lifetime of 300 fs that is reminiscent of the S2 →S1 decay recently reported for acetophenone,32 followed by 2 ps ISC to the triplet state. A similar time-scale for ISC is also observed in 20, although the triplet spectrum is shifted to longer wavelength by 60 nm, possibly indicating (π-π*) rather than (n -π*) configuration of the lowest triplet state for the methoxy compound (Supporting Information Section 9, Figure S26). The different character of the triplet state in 20 compared with 6 and 18 could explain the absence of the desired reaction in this molecule.
Overall, the ultrafast spectroscopy provides clear evidence that the electron transfer from the amine to carbonyl forming ketyl radical anion and an aminium radical is responsible for the quick disappearance of the excited states of 6, 18, and 20. The product of this electron transfer is then poised for homolytic cleavage of the carbon-nitrogen bond and elimination of acetophenone enol following proton abstraction and formation of tetrahydropyridine (Figure 2B). A more detailed analysis of the ultrafast spectroscopy of these compounds is forthcoming.
2.1.3. Determination of the quantum yield
The rate of a photochemical reaction depends on the characteristics of the light source (intensity, wavelengths), and the ability of a photo-substrate to absorb photons, in addition to the concentration of the reactants, and temperature.
Most commonly, the rate of the reaction is related to the rate of the absorption of photons in the reactor, Ia, through the expression , where [S] is the substrate concentration.33 Both the factor, β, and the exponent, n, are unknown, similar to the normal treatment of non-photochemical reaction. However, the rate of photon absorption varies along the length of the reactor as the light is gradually diminished by the absorbing species, and cannot be easily systematically varied to solve for β and n.
We studied the initial rates of the aza-Yang cyclization under four different conditions: 1) variable concentration of the substrate, keeping the nominal power output of the light source at 300 W, and using a high-pass, cut-off filter at 305 nm; 2) as for 1, but with the high-pass filter at 280 nm; 3) as for 1, but with a band-pass filter centered at 280 nm (with a 10 nm half-width); and 4) variable nominal power output with the concentration of the substrate constant at 200 μM and with the cut-off filter at 305 nm (Supporting Information Section 8, Tables S4 and S5).
We obtained initial bulk reaction rates under these conditions and compared them to the rate of photon absorption. For the initial rates of the aza-Yang reaction, we monitored the disappearance of the substrate with a UV-vis spectrophotometer by removing the reaction cell from the source light path every minute during the initial stages of the reaction and every 10 minutes later on. Making these measurements stopped the clock on the reaction progress, and we verified that the reaction doesn’t proceed in the dark. We undertook a factor analysis approach to de-noise the UV spectra obtained,34 and with the aid of calibration standards, infer the concentration of the substrate at different time points. Denoising was achieved by transforming the data through a fast Fourier transform and removing the high and low frequency terms which correspond to noise and background of measurements.35 To estimate the concentrations, the most optimal calibration matrix consisted of the substrate as a salt and p-toluenesulfonic acid, which was interestingly a suitable proxy for monitoring the appearance of the product as the azetidinol (tosylate salt) products do not have any characteristic absorption of their own other than the tosylate anion. As a rule, the rates of the appearance of the product are somewhat lower than the rates of the disappearance of the starting material (approximately 10–20%). To estimate the rate of photon absorption, we applied the following formula:
(where Eλ is the power of light source at wavelength λ, C is the concentration of the substrate, b is the reaction path length, NA is the Avogadro’s number, V is the reaction volume, h is the Planck’s constant, and c is the speed of light) using data on the irradiance of the light source from the manufacturer (Figure 6A). This data shows how the power of the source varies with wavelength for a constant irradiated area at a given distance. We needed to correct this data in two ways. First, the irradiance of the lamp is measured at 0.5 m while our reaction is at a 0.3 m distance from the source. This is easily accomplished by multiplying with 0.52/0.32 = 2.78. Second, the filters we used in the reaction change the irradiance. To correct for this we recorded the transmittance of the relevant filters in the range 200 nm to 800 nm and we multiplied irradiance at each wavelength with the transmittance (divided by 100) at that wavelength (Figure 6B). Finally, we needed to account for substrate’s absorbing characteristics, so we integrated the data on the substrate’s extinction coefficients (from 200 nm to 800 nm, Figure 6C) with the lamp irradiance data. Including the constants (Avogadro’s number, Planck’s constant, speed of light, and reaction cell volume) we arrive at the rate of the absorption of photons, Ia. Plotting these values for the rate of photon absorption against the initial rates obtained with different filter and substrate concentrations gives a linear dependence (Figure 6D).
Figure 6:
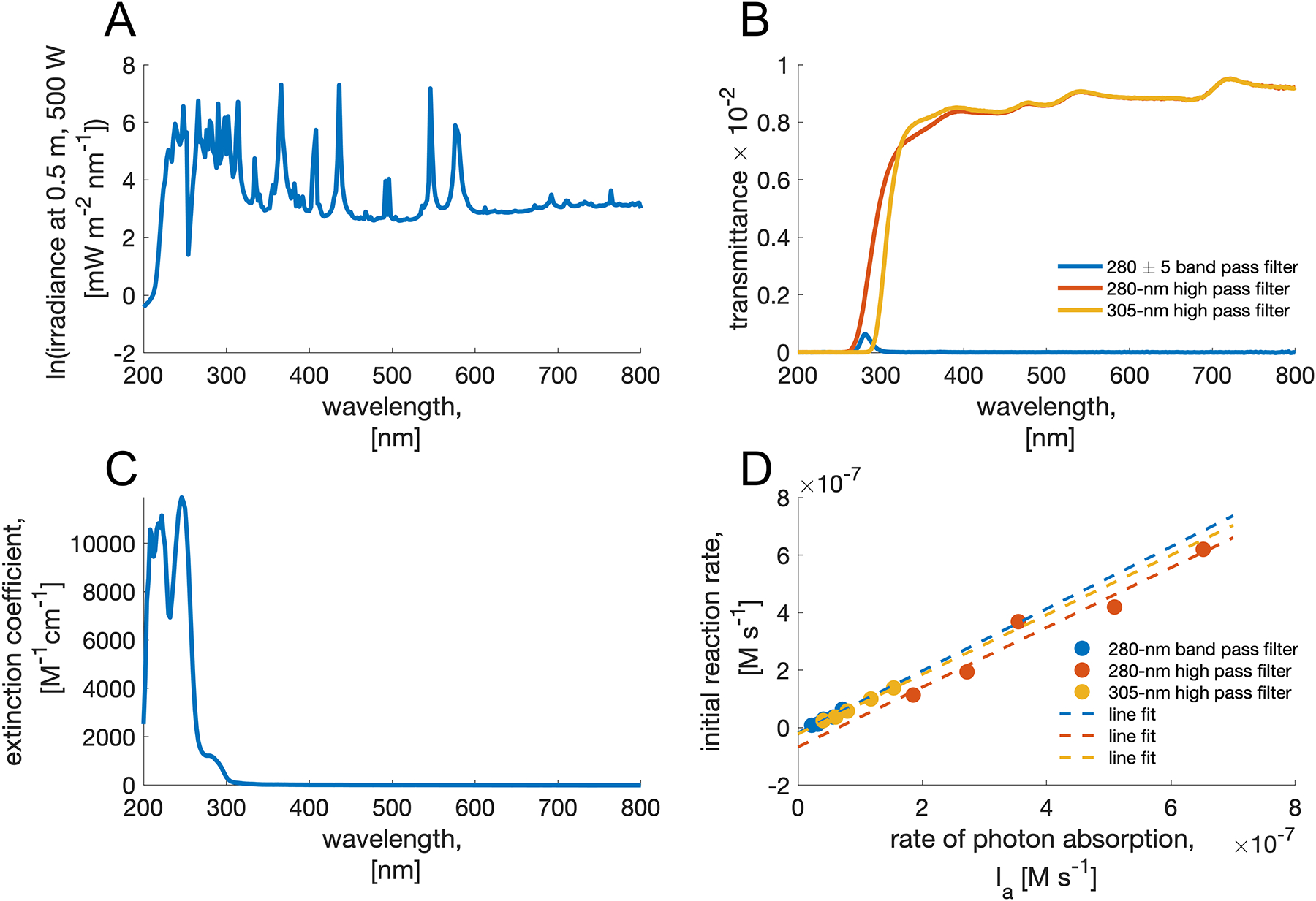
Rate of absorbed photons is directly proportional to the rate of aza-Yang reaction. A) Irradiance spectrum of the light source (Hg-Xe lamp) as provided by the manufacturer. B) Transmittance of 3 different filters used in determination of kinetic profile of the reaction. C) The extinction coefficients of [6H]+ TsO−. D) Linear relationship between Ia and reaction initial rates gives quantum yield of the reaction from the slope of fitted lines.
Because the dependence of initial rate on the rate of the absorbed photons follows linear and not exponential law (in, , where [S] is the substrate concentration), the ratio of local and measured rates approaches unity. This is likely due to low absorptivity for π*← n transition. The three sets of experiments (280 nm band pass, 280 nm high pass, and 305 nm high pass filters) all give similar slopes 1.078, 1.039, and 1.036 which correspond to the quantum yield Φ for the consumption of the substrate. Even though the absorption of the photons may be inefficient, it appears that once the photon is absorbed, the consumption of the substrate ensues.
2.2. Buchner reaction
Having established the reliable route to azetidinols, we sought to elaborate them structurally by coupling various arylacetic acids to the tertiary alcohol (created in the aza-Yang step), and then pairing this moiety with the aryl group (which had been introduced into the molecule as a chromophore for the absorption of the photon by aryl ketones) through a carbene insertion reaction.
Esterifications of the tertiary alcohols proceeded smoothly (keeping DMAP catalytic to avoid rearrangement of O-acyl to N -acyl urea which is incompetent in further coupling).36 Tertiary benzylic alcohols did not epimerize under the reaction conditions (cf. crystal CCDC 2122291). Aryl acetate esters were converted to diazo compounds by treatment with p-acetamidobenzene-sulfonylazide (p-ABSA) in the presence of diazabicycloundecane (DBU).37 The esters without the aryl group had to be first benzoylated at the α position and then treated with p-ABSA, which in one step converts the β-keto esters to diazo compounds with a loss of p-acetamidobenzensulfonamide. Unfortunately, these compounds were not suitable substrates for the Buchner ring expansion reaction because they underwent a faster hydride shift to give α, β-unsaturated esters.
It is interesting to note that the carbene can add over two diastereotopic positions on the aryl group (denoted with red letters a and b in Figure 7). This divergence leads to two diastereomeric norcaradienes which upon 6-electron electrocyclic opening of the cyclopropane produce two diastereomeric cycloheptatrienes. The opening of the cyclopropane needs to proceed disrotatotary outward, which implies that this is a thermal process. Modeling predicts nearly equal total energy of these diastereomers (0.12 kcal/mol) and about a 4 kcal/mol difference between norcaradienes and cycloheptatrienes. Experimentally, ratios of diastereomers ranged from 2:1 to 8:1 (Supporting Information Section 6, Table S3), but we were often unable to fully characterize the minor one. Contrary to what we observed, the computed energy difference would result in 1.2:1 ratio of diastereomers, which suggests that the origin of this moderate diastereoselectivity is kinetic in nature.
Figure 7:
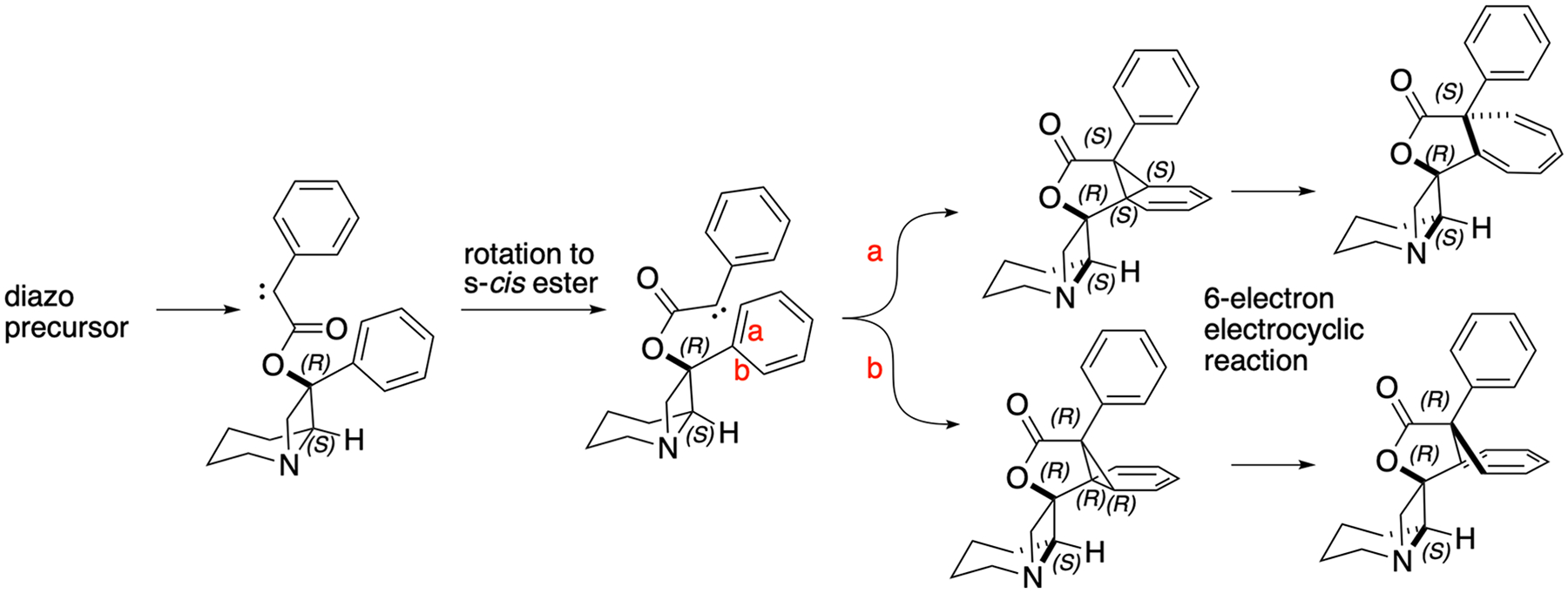
Carbene formed upon diazo group photolysis can add over two diastereotopic positions on a proximal aromatic group (red letters a and b). Diastereomeric norcaradienes are opened up through a 6-electron electrocyclic reaction to give diastereomeric cycloheptatrienes. Steric congestion allows only lightmediated carbene generation. Attempts to stabilize carbene through coordination with metals were not fruitful.
Infrared spectroscopy provided evidence for the formation of γ-lactones. Vibrational stretching frequency for γ-lactone carbonyl is 1780 cm−1 whereas less strained δ-lactones vibrate at 1760 cm−1. Stereochemistry of the all-carbon quaternary stereocenter was established through X-ray diffraction of a fluoro- and bromo-containing analog (Figure 8A) and through NOE experiments, by observed correlation between exo azetidine methylene proton and the protons at the ortho position on the aromatic ring (Supporting Information Section 6.21). Key observed HMBC correlations established connectivity for the carbon skeleton (Figure 8B). Cycloheptatrienes are not planar and coupling constants between vinylic protons provide further evidence for its formation. In addition to coupling of protons on a cis-double bond with J1,3 = 10 Hz, a second coupling constant of J1,3 = 6.2 Hz is also observed, consistent with coupling in cyclic polyenes where distortion of the cycloheptatriene ring from planarity leads to dihedral angles that deviate from 0°.38 These torsions are around 19° in computed models of both diastereomers (Figure 8C). Karplus-Altona equation predicts 7.2 Hz coupling between protons with this torsion.39,40 Analogous torsion from the crystal structure of a cycloheptatriene substituted with a fluorine atom is 25°, closer to 30° predicted from the coupling constant (Figure 8A).
Figure 8:
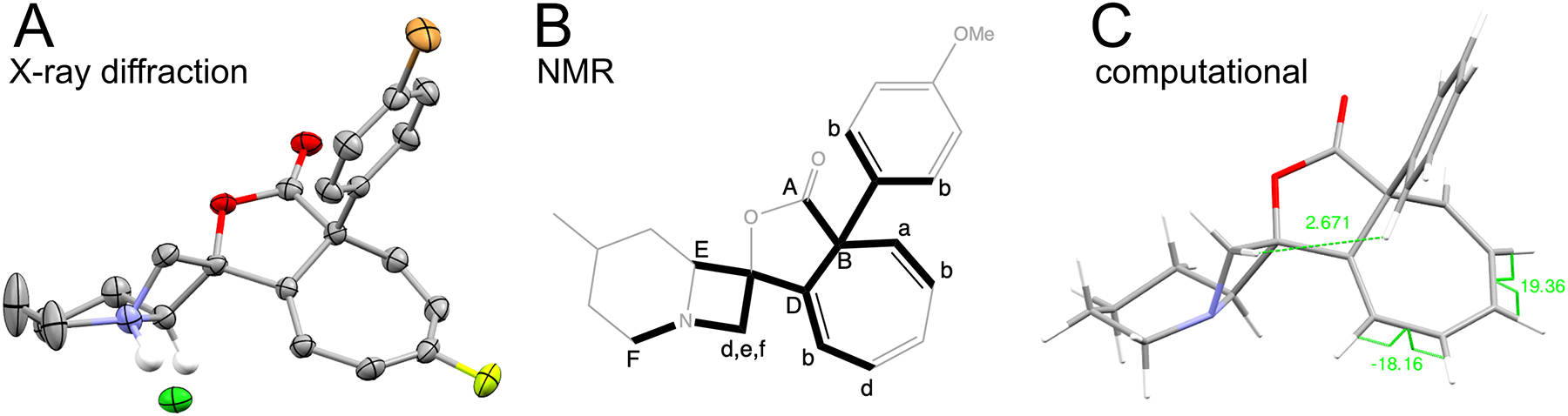
Three models of final structures. A) Crystal structure of a cycloheptatriene-containing azetidine lactone hydrochloride salt (CCDC 2126589). Carbon, gray; nitrogen, blue; oxygen, red; fluorine, yellow; chlorine, green; bromine, ocher; hydrogens, white (only the ones at azetidine fusion are shown). B) Example of key HMBC correlations found for compound 1-2-1 (Figure 9C) established the connectivity of the skeleton. In this scheme, the same letter is used at the origin (lower case for proton) and the terminus (upper case for carbon) when these nuclei “see” each other in HMBC experiment. Paths through three bonds (H–X–X–C) are bolded for the key HMBC relationships. C) Computed model of compound 1-1-2 (Figure 9C) with B3LYP DFT functional and Pople’s triple split basis set with six Gaussian functions. NOE interactions between exo methylene proton on azetidine and ortho protons on the aromatic ring at distance 2.67Å and the absence of the analogous “through-space” interaction from the methine azetidine proton suggest that the major diastereomer is the one shown. Dihedral angles in cycloheptatriene are also shown.
2.3. Collection synthesis
Ultimately, we constructed a small demonstration collection of 16 compounds based on the two key complexity-generating photochemical reactions described above. The building blocks used to produce the collection are three aryl ketones, two piperidines, and four arylacetic acids (3 × 2 × 4 = 24 would be the size of the full collection made with these building blocks; Figure 9B). The sequence of seven transformations (Figure 9A) applied to these reactants produces cycloheptatriene-containing azetidine lactones (Figure 9C) in quantities of several milligrams each and fully structurally characterized.
Noteworthy are the members of the compound collection made through insertion of in-doleacetic acid (indole nitrogen protected with a Boc group, column 3, Figure 9C), although their diazo precursors degrade more rapidly compared to the phenyl or thienyl counterparts. Using p-bromophenylacetic acid we observed the scission of the C–Br bond unless the spectrum of the light source was limited to the region of blue light. A high pass filter with a cut-off at 440 nm together with a solution of CuSO4 ·5 H2O (17 mM in 2.7 M ammonium hydroxide, 10 cm thickness) in front of the light beam improved the reaction somewhat, consistent with previous reports on using blue light for carbene generation.41 Even so, the reaction was low yielding and we excluded these molecules from the final collection even though they were useful for stereochemical disambiguation of the all-carbon quaternary stereocenter (cf. Figure 8A). While there is no systematic way to evaluate stability of compounds, we observed that these compounds remained intact in aqueous media at pH 8, and we employed 0.1 M solution of NaOH to desalt free amines without transforming the compounds through, for example, opening of the lactone ring.
3. Conclusion
We have provided an example of successful navigation to new regions of chemical space by selecting reactions that increase structural complexity along a reaction sequence. To accomplish this, we developed a convenient way to synthesize azetidinols and we arrived at a firm quantitative description of the key photochemical reaction. We suggest that efforts in this arena can expand chemical knowledge while providing useful quantities of new compounds for studies focused on their properties.
Supplementary Material
Acknowledgement
Part of this research was financially supported through an NIH COBRE Chemical Biology of Infectious Disease (P20GM113117) Pilot Project. This research was supported by the NIH Maximizing Access to Research Careers (MARC) grant T34GM136453 to the University of Kansas. NSF MRI grant CHE-0923449 was used to purchase the X-ray diffractometer and software used in this study. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number TG-CHE200120. CGE acknowledges support from the Individual Research and Development (IR/D) program while serving at the National Science Foundation.
We thank Dr. Cynthia Day (Wake Forest University) and Dr. Elizabeth Hillard (University of Kansas) for collecting the X-ray diffraction data for the compounds in this publication, and Dr. Victor Day for help with the disordered structures. We thank the NMR core lab scientists Dr. Sarah Neuenswander and Dr. Justin Douglas for acquiring the NOESY spectra. We also wish to thank Dr. Todd Williams and Mr. Larry Seib of the KU mass spectrometry laboratory for their efforts in acquiring the ESI spectra. LCT premier was purchased with support from NIH SIG S10 RR019398. Mr. Ambrosee X. Wilkinson’s help with synthesis of several starting materials is acknowledged. Prof. Emeritus Robert Hanzlik is credited with critical comments and suggestions during the preparation of this manuscript.
Footnotes
Supporting Information Available
Additional data supporting the claims made in this manuscript are available in the Supporting Information document. Analysis scripts, code to generate graphs, compound .sdf files with assigned NMR shifts, X-ray diffraction .cif files, and Gamess output files are available on the laboratory’s GitHub repository associated with this paper: https://github.com/boskovicgroup/aza_yang_buchner This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- (1).Nicolaou K; Vourloumis D; Winssinger N; Baran PS The art and science of total synthesis at the dawn of the twenty-first century. Angew. Chem. Int. Ed 2000, 39, 44–122. [PubMed] [Google Scholar]
- (2).Burke MD; Schreiber SL A planning strategy for diversity-oriented synthesis. Angew. Chem. Int. Ed 2004, 43, 46–58. [DOI] [PubMed] [Google Scholar]
- (3).Bertz SH The first general index of molecular complexity. J. Am. Chem. Soc 1981, 103, 3599–3601. [Google Scholar]
- (4).Böttcher T An Additive Definition of Molecular Complexity. J. Chem. Inf. Model 2016, 56, 462–470. [DOI] [PubMed] [Google Scholar]
- (5).Demoret RM; Baker MA; Ohtawa M; Chen S; Lam CC; Khom S; Roberto M; Forli S; Houk KN; Shenvi RA Synthetic, Mechanistic, and Biological Interrogation of Ginkgo biloba Chemical Space En Route to (−)-Bilobalide. J. Am. Chem. Soc 2020, 142, 18599–18618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wagner BK; Schreiber SL The power of sophisticated phenotypic screening and modern mechanism-of-action methods. Cell Chem. Biol 2016, 23, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Breslow R; Winnik MA Remote oxidation of unactivated methylene groups. J. Am. Chem. Soc 1969, 91, 3083–3084. [Google Scholar]
- (8).Buchner E; Curtius T Ueber die Einwirkung von Diazoessigäther auf aromatische Kohlenwasserstoffe. Ber. Dtsch. Chem. Ges 1885, 18, 2377–2379. [Google Scholar]
- (9).Fleming GS; Beeler AB Regioselective and Enantioselective Intermolecular Buchner Ring Expansions in Flow. Org. Lett 2017, 19, 5268–5271. [DOI] [PubMed] [Google Scholar]
- (10).Yang NC; Yang D-DH Photochemical Reactions Of Ketones In Solution. J. Am. Chem. Soc 1958, 80, 2913–2914. [Google Scholar]
- (11).Chen C The past, present, and future of the Yang reaction. Org. Biomol. Chem 2016, 14, 8641–8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Baum WA; Dunkelman L Ultraviolet Radiation of the High Pressure Xenon Arc. J. Opt. Soc. Am 1950, 40, 782–786. [Google Scholar]
- (13).Carreira EM; Fessard TC Four-membered ring-containing spirocycles: synthetic strategies and opportunities. Chem. Rev 2014, 114, 8257–8322. [DOI] [PubMed] [Google Scholar]
- (14).Becker MR; Wearing ER; Schindler CS Synthesis of azetidines via visible-light-mediated intermolecular [2+ 2] photocycloadditions. Nat. Chem 2020, 12, 898–905. [DOI] [PubMed] [Google Scholar]
- (15).Becker MR; Richardson AD; Schindler CS Functionalized azetidines via visible light-enabled aza Paternò-Büchi reactions. Nat. Commun 2019, 10, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Mitasev B; Yang J; Gusovsky F; Girish D; Khile AS; Balla VS; Vikram V; Vaddi A; Bathula S; Sugandham SR; Terli C; Kalla V; Rayaprolu PK; Talabhakthula RK; Gotoda M; Melillo B; Schreiber SL; Fang FG Crystallization-Based Synthetic Route to Antimalarial Agent BRD5018: Diazocene Ring Formation via a Staudinger-aza-Wittig Reaction on an Azetidine-Ribose Template. Org. Process Res. Dev 2021, [Google Scholar]
- (17).Padwa A; Eisenhardt W; Gruber R; Pashayan D Evidence for electron transfer in the photochemistry of π-π* triplet states. J. Am. Chem. Soc 1969, 91, 1857–1859. [Google Scholar]
- (18).Padwa A; Eisenhardt W; Gruber R; Pashayan D Electron transfer in the type II photoelimination of α-aminoacetophenones. J. Am. Chem. Soc 1971, 93, 6998–7005. [Google Scholar]
- (19).Cossy J; Belotti D Generation of ketyl radical anions by photoinduced electron transfer (PET) between ketones and amines. Synthetic applications. Tetrahedron 2006, 62, 6459–6470. [Google Scholar]
- (20).Ruggeri M; Dombrowski AW; Djuric SW; Baxendale IR Photochemical Flow Synthesis of 3-Hydroxyazetidines. ChemPhotoChem 2019, 3, 1212–1218. [Google Scholar]
- (21).Jurczyk J; Lux MC; Adpressa D; Kim SF; Lam Y.-h.; Yeung CS; Sarpong R Photomediated ring contraction of saturated heterocycles. Science 2021, 373, 1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Roberts CA; Park B; Xu L-P; Roque JB; Yeung CS; Musaev DG; Sarpong R; LaLonde RL Sequential Norrish–Yang Cyclization and C–C Cleavage/Cross-Coupling of a [4.1.0] Fused Saturated Azacycle. J. Org. Chem 2021, 86, 12436–12442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Aoyama H; Hasegawa T; Watabe M; Shiraishi H; Omote Y Photochemical reactions of N, N-disubstituted. alpha.-oxoamides. J. Org. Chem 1978, 43, 419–422. [Google Scholar]
- (24).Gramain J-C; Ouazzani-Chahdi L; Troin Y Systemes bicycliques azotes: une voie d’acces photochimique aux aza-1 bicyclo (n. 2.0) alcanes substitues. Tetrahedron Lett. 1981, 22, 3185–3188. [Google Scholar]
- (25).Aoyama H; Miyazaki K-I; Sakamoto M; Omote Y Solid state photoreaction of n, n-dialkylpyruvamides: inclusion complexes with deoxycholic acid or cyclodextrin. Tetrahedron 1987, 43, 1513–1518. [Google Scholar]
- (26).Toda F; Tanaka K; Kakinoki O; Kawakami T Highly enantioselective photocyclization of N-(aryloylmethyl)-. delta.-valerolactams. J. Org. Chem 1993, 58, 3783–3784. [Google Scholar]
- (27).Pedrosa R; Andrés C; Nieto J; del Pozo S Diastereoselective Yang photocyclization reactions in solution. Synthesis of enantiopure azetidin-3-ol derivatives. J. Org. Chem 2005, 70, 1408–1416. [DOI] [PubMed] [Google Scholar]
- (28).Hill J; Zakaria MM; Mumford D Photoreactions of α-amino ketones derived from heterocyclic secondary amines. Journal of the Chemical Society, Perkin Transactions 1 1983, 2455–2458. [Google Scholar]
- (29).Eliel EL; Wilen SH Stereochemistry of organic compounds; John Wiley & Sons, 1994. [Google Scholar]
- (30).Belostotskii AM; Markevich E Conformational dynamics in nitrogen-fused azabi-cycles. J. Org. Chem 2003, 68, 3055–3063. [DOI] [PubMed] [Google Scholar]
- (31).Horspool WM; Lenci F CRC Handbook of Organic Photochemistry and Photobiology, Volumes 1 & 2; CRC press, 2003; Chapter 52, pp 18,19. [Google Scholar]
- (32).Robertson PA; Bishop HM; Orr-Ewing AJ Tuning the Excited-State Dynamics of Acetophenone Using Metal Ions in Solution. J. Phys. Chem. Lett 2021, 12, 5473–5478. [DOI] [PubMed] [Google Scholar]
- (33).Calvert JG; Pitts JN Photochemistry. Wiley, New York: 1966, 8, 99. [Google Scholar]
- (34).Malinowski ER; Howery DG Factor analysis in chemistry; Wiley, 1980. [Google Scholar]
- (35).Weismüller JA; Chanady A Quantitative multicomponent analysis of complex mixtures by means of Full Spectrum quantitation and principal component analysis. TrAC, Trends Anal. Chem 1992, 11, 86–90. [Google Scholar]
- (36).Neises B; Steglich W Simple method for the esterification of carboxylic acids. Angew. Chem. Int. Ed 1978, 17, 522–524. [Google Scholar]
- (37).Baum JS; Shook DA; Davies HM; Smith HD Diazotransfer reactions with p-acetamidobenzenesulfonyl azide. Synth. Commun 1987, 17, 1709–1716. [Google Scholar]
- (38).Lambert JB; Durham LJ; Lepoutere P; Roberts JD Nuclear Magnetic Resonance Spectroscopy. Analysis and Temperature Dependence of the Spectra of Cycloheptatriene and 7, 7-Bistrifluoromethylcycloheptatriene1. J. Am. Chem. Soc 1965, 87, 3896–3899. [Google Scholar]
- (39).Karplus M Vicinal proton coupling in nuclear magnetic resonance. J. Am. Chem. Soc 1963, 85, 2870–2871. [Google Scholar]
- (40).Haasnoot C; de Leeuw FA; Altona C The relationship between proton-proton NMR coupling constants and substituent electronegativities—I: An empirical generalization of the Karplus equation. Tetrahedron 1980, 36, 2783–2792. [Google Scholar]
- (41).Jurberg ID; Davies HM Blue light-promoted photolysis of aryldiazoacetates. Chem. Sci 2018, 9, 5112–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.