

Abstract
Activated cardiac fibroblasts are involved in both reparative wound healing and maladaptive cardiac fibrosis after myocardial infarction (MI). Recent evidence suggests that PU.1 inhibition can enable reprogramming of profibrotic fibroblasts to quiescent fibroblasts, leading to attenuation of pathologic fibrosis in several fibrosis models. The role of PU.1 in acute MI has not been tested. We designed a randomized, blinded study to evaluate whether DB1976, a PU.1 inhibitor, attenuates cardiac function deterioration and fibrosis in a murine model of MI. A total of 44 Ai9 periostin-Cre transgenic mice were subjected to 60 min of coronary occlusion followed by reperfusion. At 7 days after MI, 37 mice were randomly assigned to control (vehicle) or DB1976 treatment and followed for 2 weeks. Left ventricular ejection fraction (EF), assessed by echocardiography, did not differ between the two groups before or after treatment (final EF, 33.3±1.0% in control group and 31.2±1.3% in DB1976 group). Subgroup analysis of female and male mice showed the same results. There were no differences in cardiac scar (trichrome stain) and fibrosis (interstitial/perivascular collagen; picrosirius stain) between groups. Results from the per-protocol dataset (including mice with pre-treatment EF <35% only) were consistent with the full dataset. In conclusion, this randomized, blinded study demonstrates that DB1976, a PU.1 inhibitor, does not attenuate cardiac functional deterioration or cardiac fibrosis in a mouse model of MI caused by coronary occlusion/reperfusion.
Keywords: PU.1, periostin, myocardial infarction, cardiac function, fibrosis
Introduction
Cardiac fibroblasts are one of the most abundant cell types in the adult mammalian heart [1, 2]. Following myocardial injuries, especially myocardial infarction (MI), cardiomyocyte necrosis triggers the activation of fibroblasts that are involved in both reparative wound healing and maladaptive cardiac fibrosis [3–5]. On one hand, cardiac fibrosis and the collagen-rich cardiac scar have long been considered to be detrimental by impairing cardiac function; therefore, preventing or reducing the accumulation of collagen in the heart has been a major therapeutic goal [6, 7]. On the other hand, emerging evidence also suggests that cardiac fibroblasts are essential for protective reparative responses to myocardial injury and healing [3, 4, 6, 8].
Although the mechanisms that drive these functionally opposing profibrotic and reparative phenotypes of fibroblasts remain unclear [4, 9], a new therapeutic avenue that enables reprograming activated fibroblasts to modify their properties in cardiac scar formation has recently emerged [6, 9]. Among the potential candidates, PU.1, a member of the E26 transformation-specific family of transcription factors, has emerged as an interesting target because of its essential role in fibroblast polarization and induction of an extracellular matrix (ECM)-producing profibrotic phenotypic switch [9]. Studies have also shown that pharmacological or genetic inhibition of PU.1 leads to regression of pathologic fibrosis in various fibrosis models and across several organs, such as skin [9], liver [9], lung [9], kidney [9] and recently in the atrium of the heart [10]. However, whether PU.1 inhibition can attenuate fibrosis after MI and improve cardiac function remains unknown.
Endogenous periostin is specifically upregulated in activated cardiac fibroblasts after myocardial injury and stress, but not in quiescent cardiac fibroblasts, cardiomyocytes, vascular cells, or hematopoietic cells [11]. Therefore, periostin-Cre transgenic mice, in which Cre recombinase is driven by a 3.9-kb periostin promoter, have become a useful animal model in recent studies of fibrosis in heart disease [3, 4, 7, 8]. Being a profibrotic factor, periostin itself is also considered an attractive target for reducing cardiac fibrosis [7, 12]. It would be interesting to see whether PU.1 inhibition has beneficial effects on periostin-expressing activated cardiac fibroblasts and to elucidate the interaction between PU.1 and periostin in cardiac scar formation and healing after MI.
Despite decades of intensive work and innumerable publications that suggested that many compounds and interventions may potentially reduce or reverse cardiac fibrosis in preclinical research [6], clinical translation has been wanting [6, 13]. One of the factors that hinder clinical translation may be that methodological shortcomings and suboptimal reporting remain prevalent in preclinical cardiovascular research [14, 15]. To address this problem, we developed CAESAR, the first publicly available, multicenter consortium for rigorous preclinical studies of cardioprotection [16, 17]. CAESAR was based on the cardinal principles of randomization, investigator blinding, a priori exclusion criteria, strict protocol adherence, appropriate statistical analyses, and assessment of reproducibility [15–17]. CAESAR [15–21], and later the preclinical animal experiment guidelines [22], have promoted increased awareness of the need for rigor and reproducibility in designing experiments. The use of these principles has enabled us to provide rigorous evaluation of putative therapies [23–27].
In current study, we designed a randomized, blinded preclinical study to evaluate whether pharmacological PU.1 inhibition attenuates cardiac functional deterioration and fibrosis after MI in an ischemia/reperfusion mouse model and, if so, whether the mechanism involves periostin-expressing activated cardiac fibroblasts. The study was conducted in accordance with the CAESAR principles [15–19].
Methods
Animals
All animal procedures were approved by the University of Louisville Institutional Animal Care and Use Committee (protocol number:14034) and conformed with the Guide for the Care and Use of Laboratory Animals published by National Institutes of Health.
In order to study the fibrosis induced by periostin-expressing activated cardiac fibroblasts, we generated a mouse line by breeding Ai9 mice (Jackson Lab, stock number: 007905) with periostin-Cre transgenic mice (obtained from Dr. Simon J. Conway) [4, 11]. Ai9 and periostin-Cre double transgenic mice, male and female, 14–18 weeks old, were used in this study (Fig. 1A).
Fig. 1: Transgenic animals and study flowchart.
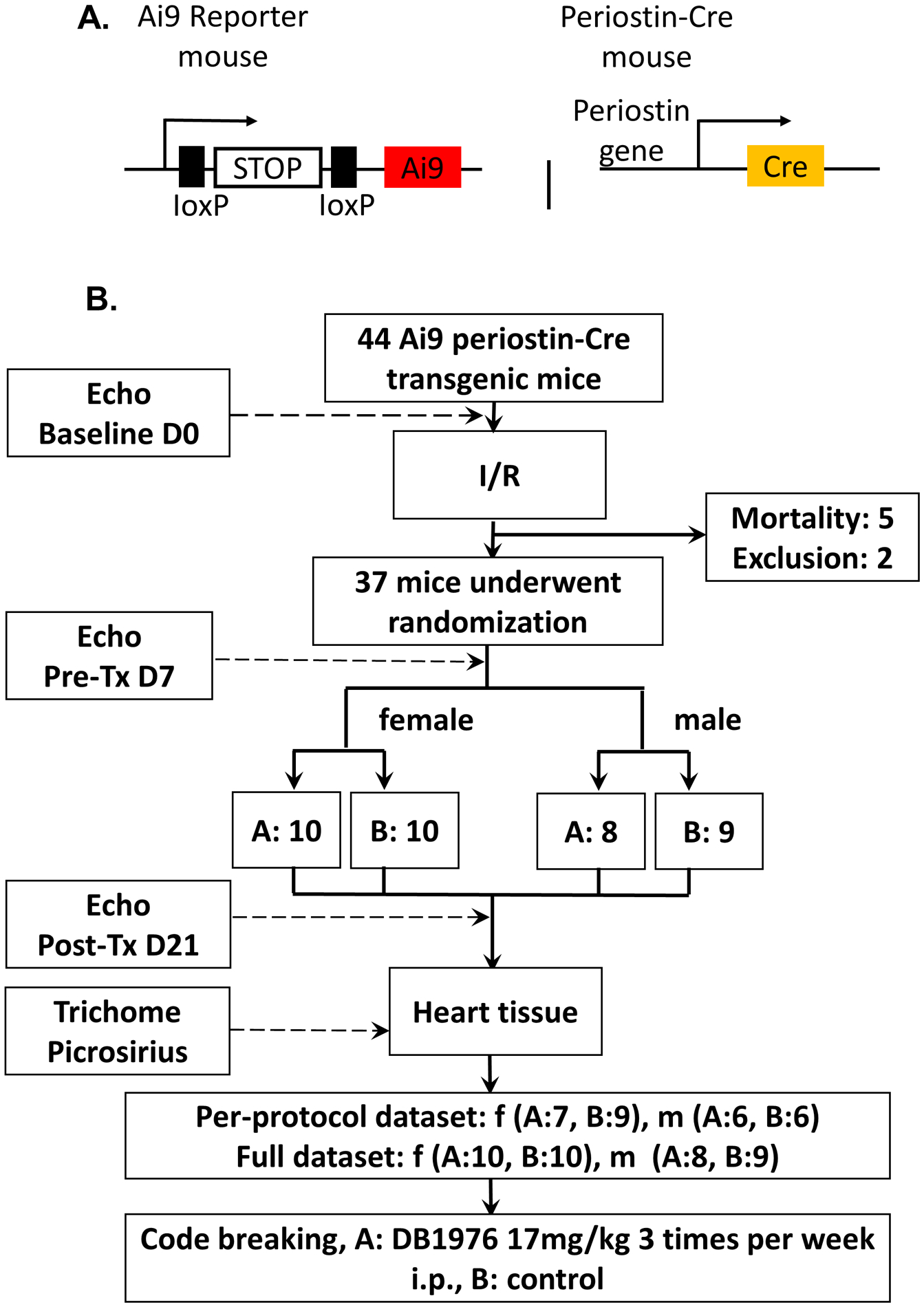
A. Schematic of double transgenic mouse line: Ai9 reporter mice were cross bred with periostin-Cre mice. The double transgenic mice were used in this study. B. Flowchart of the randomized blinded study. A total of 44 transgenic mice were enrolled. After baseline echocardiographic measurements, they were subjected to 60 min of coronary occlusion followed by reperfusion. Five mice died after surgery and 2 were excluded. The remaining 37 mice underwent a pre-treatment echocardiographic study 7 days after MI, were separated according to their gender, and then were randomly assigned to group A or B. There were 10 female mice in each group, 8 male mice in group A and 9 male mice in group B. Mice received intraperitoneal injection of either A or B solution at a dose of 2 μl/g body weight, 3 times a week for 2 weeks. After the final echocardiographic measurement (21 days after MI), the hearts were fixed for histology. The echocardiographic measurements and trichome and picrosirius stain images were analyzed by blinded researchers. After the results were presented as Per-protocol dataset (which includes only mice with pre-treatment EF <35%) and Full dataset (which includes all mice assigned to treatments) and submitted to the principal investigator, the group code was broken, revealing that group A was DB1976 while group B was control.
Preparation of PU.1 inhibitor
We used C₂₀H₁₆N₈Se (Acme Bioscience Inc, Lot: 1767115), also known as DB1976 [10, 28], as the PU.1 inhibitor. DB1976 competitively and potently inhibits PU.1 binding and strongly inhibits the PU.1/DNA complex with the high specificity of PU.1 for AT-rich flanking sequences of the 5′-GGAA-3′ core and minimal effects on other E26 transformation-specific transcription factors[9, 28]. To prepare a working solution with concentration of 8.5 mg/ml, DB1976 powder was diluted in electrolyzed S-water. The same volume of electrolyzed S-water served as control. To comply with the double blinded design, a green color DNA loading dye (Promega, Ref: G190A) was added in the control solution to match the green color of DB1976 solution. A total volume of 150 μl DB1976 or control solution was aliquoted into each 1ml vial, marked as “A” or “B”, and stored in −20°C freezer. On the date of injection, solutions were freshly made by thawing each vial at room temperature and sent to the animal lab. The remaining unused solution in the vial was discarded. A third-party person was in charge of preparing the solutions and hiding the group code until the final analyzed results were submitted to the principal investigator.
Mouse Model of Ischemia/reperfusion
The murine model of ischemia/reperfusion (I/R) was modified from the CAESAR study [16, 17]. Similar to previous experiments [16, 23, 29–36], after the heart was exposed by a lateral thoracotomy, an 8–0 nylon suture was passed under the left coronary artery approximately 2 mm below the left auricle and a nontraumatic balloon occluder was applied on the artery. Ischemia was induced by tightening and inflating the occluder. After 60 min of occlusion, the occluder was deflated and removed to achieve reperfusion. Rectal temperature was carefully monitored and maintained between 36.8 and 37.2°C throughout the experiment. The electrocardiogram (ECG) was continuously monitored during the experiment. Typical ischemic ECG changes (widened QRS and elevated ST segment) and the pale color of the ischemic myocardial region were used to confirm successful coronary occlusion. Animals whose ECG changes were atypical or did not persist throughout the ischemic period were excluded before treatment assignments [16, 23, 34].
Treatment protocol
The experiments were conducted in a randomized and double blinded fashion [15, 16, 23]. As illustrated in Fig. 1B, one week after MI, all surviving animals that met inclusion criteria were separated by gender, and then randomly assigned to two treatment groups, labeled initially as Group A or B, according to a random number list. Mice received intraperitoneal (i.p.) injection of either A or B solution at a dose of 2 μl/g body weight, 3 times a week for 2 weeks. The dose yields 17 mg/kg per injection of DB1976 or equal volume of control solution. At the end of the follow-up, after final echocardiographic measurements were acquired, the animals were sacrificed and the hearts fixed for histology. To maintain the blinded design, all enrolled mice were treated and followed throughout the study. All mice that finished the study were included in the full dataset, but only the mice that manifested reduced cardiac function before treatment (ejection fraction [EF] <35%) were included in the per-protocol (PP) dataset. Based on our previous studies [24–26, 35], 35% is an adequate EF cutoff point to evaluate the beneficial effect of treatments in this model. The group code was kept blinded to the surgeons, sonographer, pathology investigator and data analyzer. The code was broken in front of all parties by a third-party person after all the results were submitted to the principal investigator [15, 16].
Echocardiography
Transthoracic echocardiography was performed by a sonographer who was blinded to group assignment. Serial echocardiograms were obtained at baseline (before MI), pre-treatment (7 days after MI), and 2 weeks post-treatment (21 days after MI). A Vevo 2100 Imaging System (VisualSonics, Inc.) equipped with a 30-MHz transducer was used. Parasternal long axis views were acquired to measure left ventricle (LV) end-diastolic and end-systolic volume (EDV and ESV), stroke volume (SV), and EF. Three measurements were taken and averaged for each parameter. All echocardiography measurements were performed under isoflurane anesthesia (3% for induction and around 1% for maintenance). Body temperature was maintained at 37.0±0.2°C throughout the study. Echocardiographic images were analyzed offline by blinded observers using the Vevo 2100 workstation software. Left ventricular (LV) EF was calculated by using the formula: ((EDV-ESV)/EDV) × 100% [23, 26, 27, 29–31]. After all the echocardiographic results were analyzed, the results were presented as per-protocol (PP) dataset, which includes only mice with pre-treatment EF <35%, and full dataset, which includes all mice that completed the experiments.
Histologic analysis
The methods used for histologic analyses has been described [27, 29–31, 35, 37, 38]. Briefly, after the final echocardiogram, the hearts were arrested, excised, perfused by 10% neutral buffered formalin solution, and fixed in formalin for 24 h. The hearts were sectioned into three transverse slices of equal thickness from apex to base. The slices were weighted, and then subsequently embedded, sectioned, and mounted. Later, the sections were deparaffinized and rehydrated for Masson’s trichrome stain to determine scar size according to published protocols [35]. The risk region was defined as the transmural area between the furthest outer lateral edges of the scar. Viable myocardium in the risk area was determined as the difference between risk and scar areas. The cumulative weight of the three slices was considered as the weight of the whole heart. The LV mass in each slice was measured by multiplying the percentage of LV area/whole heart tissue area times the weight of each slice; the sum of three slices yields the total LV mass. The size of the scar was expressed as a percentage of the mass of the risk area with respect to the entire LV. Myocardial collagen content was quantitated on picrosirius red-stained heart images acquired under polarized light microscopy by determining collagen area per mm2 of risk region or nonrisk region. Images were acquired digitally and areas measured using NIKON software (NIS-Elements AR Analysis 4.13.05 64-bit). The investigator performing histologic analyses was blinded throughout the study.
Statistical analysis
Data are presented as mean ± SEM. All continuous data were analyzed with Student’s t-tests or one-way ANOVA for normally distributed data followed by unpaired Student’s t tests with the Bonferroni correction, or Kruskal–Wallis one-way analysis of variance on ranks for data that are not normally distributed, as appropriate. The chi-square Fisher exact test was used for rate comparison. A P value <0.05 was considered statistically significant. All statistical analyses were performed using the Sigma Stat software system [39–41].
The sample size calculation for two independent group means with one interaction factor (gender) was conducted via G*Power software [42]. The type I error rate was set at (α) =0.05, type II error rate (β) =0.1, and power (1-β) =0.9. Based on previous literature [10], the major endpoint, cardiac scar measured by trichrome stain in the control group and DB1976 group is expected to be 23.9±1.9% and 14.4±1.4%. Assuming that the effects of DB1976 are not different between male and female mice, the calculated minimum sample size in each group of each gender is 4. Minimum total sample size is 16. Assuming a 30% mortality and exclusion rate, the minimum sample size required is 16×130%=21.
Results
A total of 44 mice were originally enrolled for the I/R procedure. Five mice died after the procedure and before group assignment; another 2 mice were excluded because the ischemic ECG changes were atypical. The remaining 37 mice were separated according to their gender and then randomly assigned to two groups, originally labeled as Group A and B. There was no additional mortality during 2 weeks of treatment and follow-up. After all results were submitted to the principal investigator, the group code was broken, revealing that Group A was DB1976 and Group B was control. According to per-protocol exclusion criteria, 4 mice in the control group (1 female; 3 male) and 5 mice in the DB1976 group (3 female; 2 male) were excluded from the PP dataset because their pre-treatment EF was ≥35% (Fig. 1B). The age, body weight, and exclusion did not differ between the two groups, as detailed in Supplemental Table.1.
Echocardiographically-assessed cardiac function is not improved in DB1976-treated mice.
Echocardiographic studies were performed at baseline (before MI), pre-treatment (7 days after MI) and 2 weeks post-treatment (21 days after MI). As shown in Fig. 2, in both control and DB1976 groups, compared with baseline, average EF was reduced > 30 units at pre-treatment, suggesting a sizable MI model in these mice. EF was not significantly different between mice assigned to control or DB1976 group at either baseline or pre-treatment time point, indicating that the severity of pre-treatment LV dysfunction was comparable. During the 2 weeks of treatment, EF in both groups was basically unchanged. There were no significant differences in EF between groups at 2 weeks after treatment, with the final EF being 33.3±1.0% in the control group and 31.2±1.3% in DB1976 group (Fig. 2A and Supplemental Table 2). The net change (delta) in EF between pre-treatment and post-treatment also was not significantly different: 1.2±1.1% in control and −0.4±1.1 in DB1976-treated mice (Fig. 2B).
Fig. 2: Echocardiographically assessed cardiac function is not improved in DB1976 treated mice.
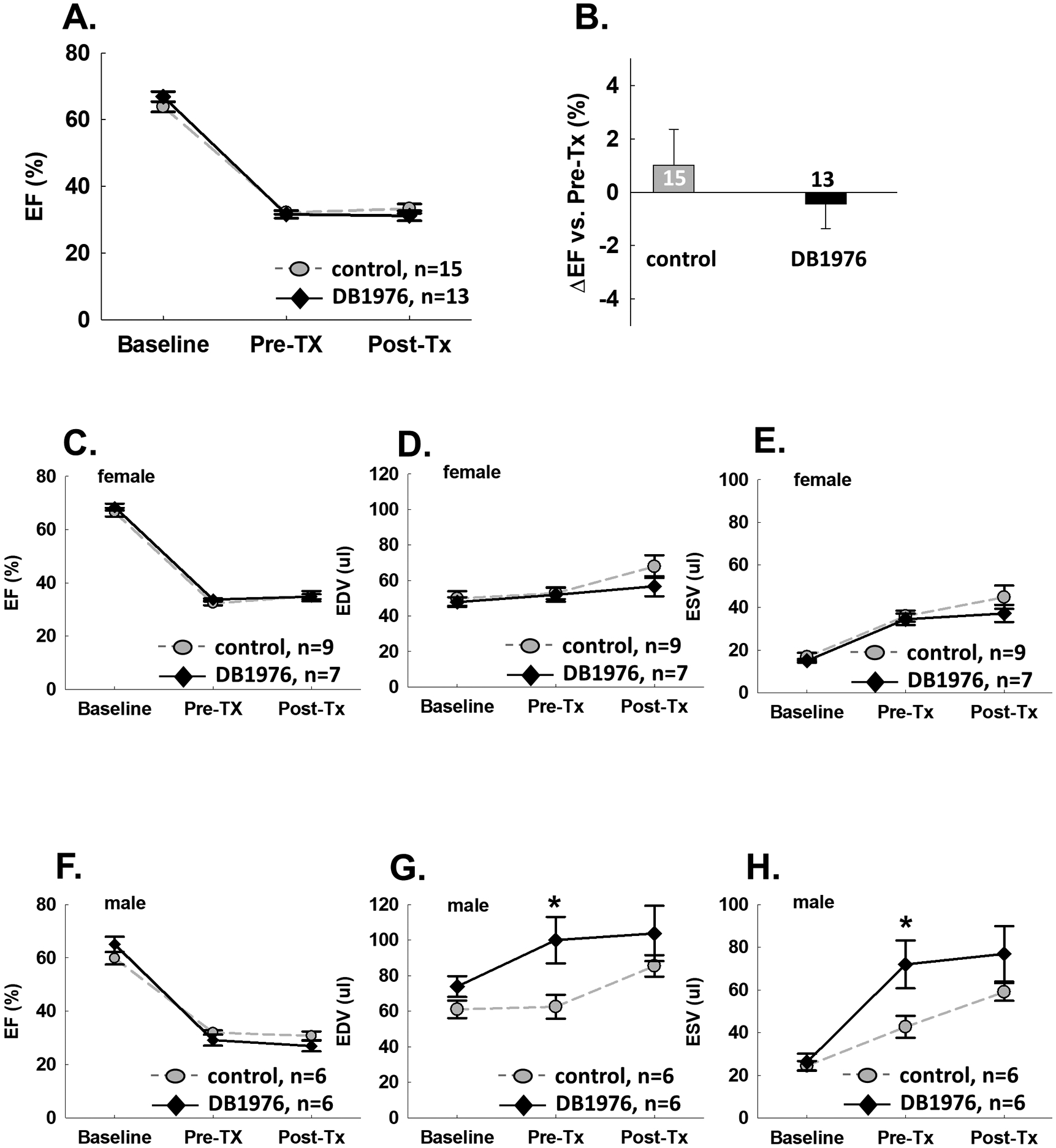
Serial echocardiographic studies were performed at baseline (before MI), pre-treatment (7 days after MI), and post-treatment (21 days after MI). Comparison between DB1976 and control groups. A. ejection fraction (EF); B. ΔEF (change at post-treatment compared with pre-treatment). Subgroup results from female mice. C. EF; D. LV end-diastolic volume (EDV); E. LV end-systolic volume (ESV). Subgroup results from male mice: F-H. Data are mean ± SEM. * P<0.05.
Since male mice have generally larger body and heart weight compared with female mice at the same age, subgroup analysis of cardiac function was performed according to gender. In female mice, there were no differences between control and DB1976 groups in EF, EDV or ESV at all 3 time points (Fig. 2C, 2D, 2E). In male mice, EF also demonstrated no difference between groups at all 3 time points (Fig. 2F). Interestingly, DB1976-treated male mice manifested more severe LV dilation than control mice before treatment: pre-treatment EDV was 100.0±13.1 μl vs. 62.5±6.7 μl (P<0.05). Although post-treatment EDV was not significantly different, 103.7±15.6 μl vs. 85.5±6.1 μl, the net increase of EDV after treatment was less in DB1976-treated male mice compared with control male mice (3.7±5.0 μl vs. 22.9±3.5 μl, P<0.05) (Fig. 2G). Detailed echocardiographic results can be found in Supplemental Table 2.
Scar size and cardiac fibrosis are not attenuated in DB1976-treated hearts.
The results of trichome stain analysis are summarized in Fig. 3. The LV mass (Fig. 3A) and risk region mass/LV mass ratio (Fig. 3B) were similar in both groups in either female or male mice, indicating that the size of the risk region was comparable. We observed no difference in scar size, measured either as scar/LV mass ratio or scar/risk region ratio, between the DB1976 group and the control group (Fig. 3C–3D). Cardiac fibrosis, measured by the amount of interstitial and perivascular collagen via picrosirius red stain analysis in both non-risk (noninfarcted) and risk regions was shown in Fig.3E–3F. There was no difference in cardiac fibrosis between the 2 groups in either risk region or noninfarcted region. Representative trichrome stained heart sections are shown in Fig. 3G (upper panel) and picrosirius red stained sections in Fig. 3G (lower panel).
Fig. 3: Cardiac scar and fibrosis are not attenuated in DB1976 treated hearts.
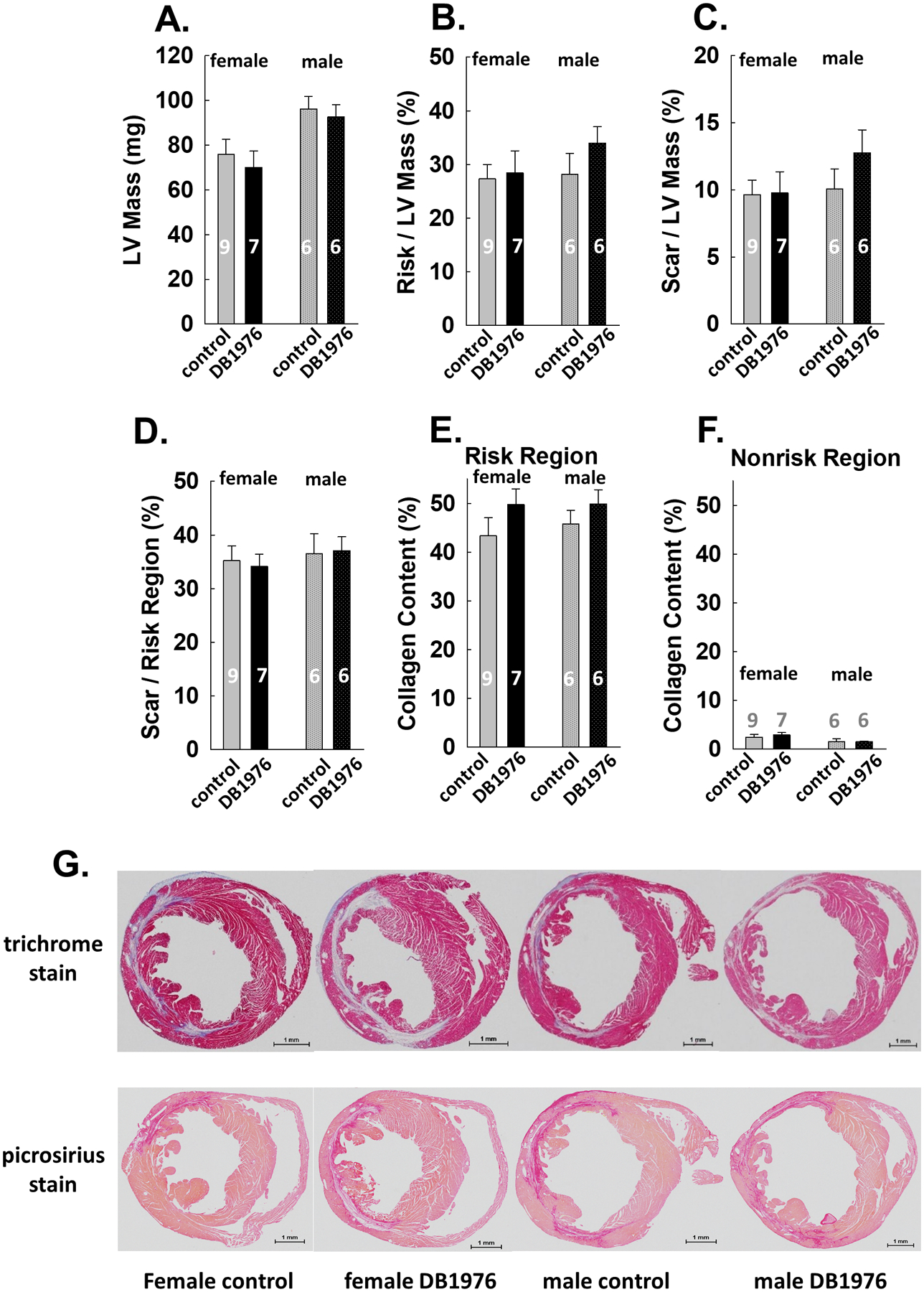
A. LV mass; B. ratio of risk region mass to LV mass; C. ratio of scar mass to LV mass. D. ratio of scar mass to risk region mass. E. Cardiac collagen content in risk region. F. Cardiac collagen content in nonrisk region. G. Representative trichrome staining (upper panel) and picrosirius red staining (lower panel) images from female and male mice in the control and DB1976 groups. Data are mean ± SEM. Animal numbers are shown in bars.
Comparison of results from PP dataset and full datasets.
The results presented above were from the PP dataset, which excluded mice with EF before treatment ≥35% (small infarcts). To eliminate any selective bias, we also performed a full dataset analysis that included all the mice assigned to either treatment and compared the results with those from the PP dataset. As shown in Fig. 4, the results from the full dataset were consistent with those from the PP dataset with respect to EF, EDV, ESV, trichome stain and picrosirius stain measurements. Detailed results from the full dataset can be found in Supplemental Table 3.
Fig. 4: Results from the full dataset are consistent with the PP dataset.
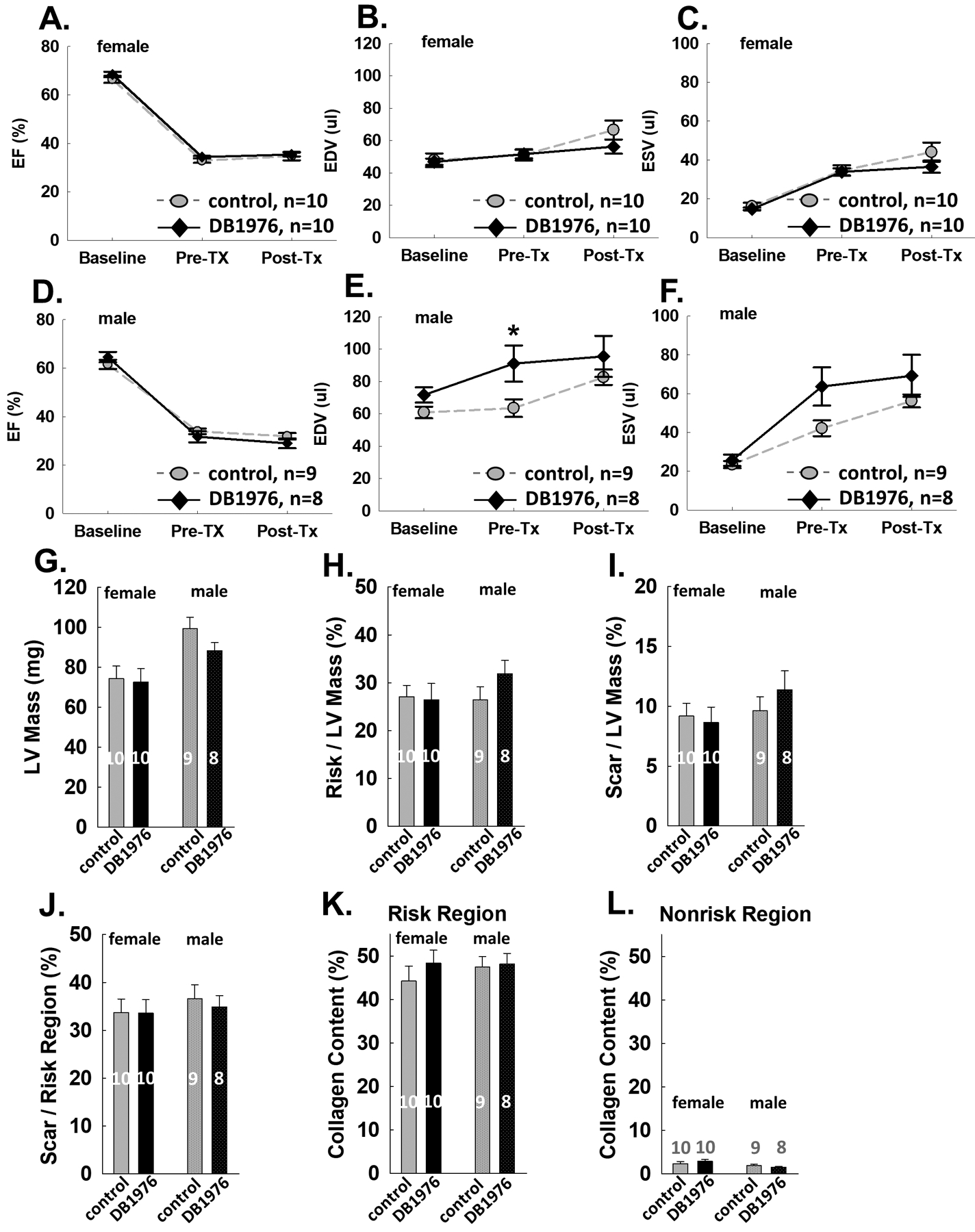
Echocardiographic results from female mice in the full dataset: A-C. Results from male mice in the full dataset: D-F. Trichrome results from the full dataset: G-J. Collagen results from the full data set: K-L. Data are mean ± SEM. Animal numbers are shown in bars. * P<0.05.
Discussion
Over the last decade, our understanding of the functions of cardiac fibroblasts has moved beyond their roles in heart structure and ECM generation, to their dual nature in adaptive and maladaptive responses after injury, and now further to the mechanism of their activation and their polarization to different functional phenotypes [6, 9, 13]. Owing to their capacity to be reprogrammed to alternative cell lineages, cardiac fibroblasts have become an attractive target to modulate cardiac scar formation and healing [13]. It has been reported that PU.1 inhibition can disrupt the fibrotic network and enable reprogramming of profibrotic fibroblasts to quiescent fibroblasts, leading to attenuation of pathologic fibrosis in bleomycin-induced skin, lung fibrosis [9], CCl4- induced liver fibrosis [9] and Ang II-induced atrial fibrosis models [10] in mice. To test whether PU.1 inhibition can reduce cardiac fibrosis and improve cardiac function after MI in mice, we conducted a rigorous randomized, blinded preclinical study. Our results show that DB1976, a PU.1 inhibitor, did not attenuate LV functional deterioration nor reduce cardiac fibrosis after MI. The results were consistent in both the PP dataset and the full dataset. To our knowledge, this is the first study to evaluate the efficacy of PU.1 inhibitor in a murine model of MI induced by coronary occlusion and reperfusion.
In the current study, we administered DB1976 at 7 days post-MI, at a dose of 17 mg/kg i.p. 3 times per week for 2 weeks. The rationale for starting the treatment 7 days post-MI but not immediately at reperfusion is based on the dual nature of cardiac fibroblasts in the reparative and profibrotic responses after MI. We wanted to intervene in the profibrotic phase of activated cardiac fibroblasts but not in the reparative phase after MI. It is known that in mice, the inflammatory phase lasts 3–4 days after MI [43], and the subsequent resolution and repair phase lasts 10–14 days [43]; however, chronic inflammation and incomplete healing can persist over the long term in large infarcts [43]. Genetic lineage tracing revealed that cardiac fibroblasts are activated and proliferate during the first week after MI in mice [5, 44], and differentiate into myofibroblasts, the major cell type that contributes to ECM and fibrotic scar formation, at 4–7 days post MI [1, 5, 44, 45]. Myofibroblasts, which express periostin and α-smooth muscle actin (αSMA), lose αSMA expression and the myofibroblast phenotype and transition to an alternative stage refractory to proliferation by 10–14 days after MI [44, 45]. The loss of the myofibroblast marker and phenotype precedes the reorganization of the ECM associated with scar maturation [45]. A growing body of evidence suggests that 7–10 days post-MI is a critical phase that marks a functional transition of activated myofibroblasts from preserving ventricular wall integrity to maladaptive collagen accumulation [4, 44, 45]. We hypothesized that this phase is an ideal time for PU.1 inhibition, with the goal of redirecting fibroblast polarization and enabling reprogramming of ECM-producing profibrotic fibroblasts to quiescent fibroblasts. Regarding the dose of DB1976, previous papers used 5 mg/kg/day i.p. [9, 10], or a higher dose of 17 mg/kg 3 times per week [28]. In the current study, we used the same higher dose that was proved to be effective in PU.1 inhibition [28]. Our results, obtained with a rigorous preclinical experiment, are basically “negative”. Before discussing conclusions regarding our original hypothesis, it is appropriate to examine the technical rigor of our study. Although awareness of the need for rigor in preclinical studies has increased in the cardiovascular community in recent years, shortcomings in study design remain prevalent [14, 15]. The major study design elements include power and sample size estimation, randomization, blinding, consideration of sex as a biological variable, and a priori exclusion criteria.
We estimated the sample size with a strict standard. The type I error rate was set at (α) =0.05, the type II error rate (β) =0.1, and the power (1-β) =0.9. Since there is no previous paper reporting PU.1 inhibition in a mouse model of MI, we used the results from Ang II-induced an atrial fibrosis mouse model [10] as reference. As detailed in Methods, considering gender as a control factor, we used a factorial design that can increase the power [46]. The total number of mice assigned to treatments was 37, more than 2 times the required minimum sample size of 16 mice. Therefore, we are confident that we have the power to draw firm conclusions regarding the sample size of this study.
We used a stratified randomization design to ensure that similar numbers of female or male mice were assigned to both treatment groups. The randomization design was hidden by a third-party person until code breaking. Attesting to the success of this randomization strategy, there were 10 female mice assigned to DB1976 or control, 8 male mice to DB1976, and 9 male mice to control. There was no difference between the 2 groups in echocardiographically measured pre-treatment EF or Trichrome measured risk/LV mass ratio.
Compliance with blinding was observed throughout the study. To blind the surgeons, sonographer and pathology investigator, the randomization list was kept by a third-party person and the treatment solutions were prepared in such a way that they could not be distinguished. The person analyzing data was blinded to group assignment. The group code was not revealed until all the results were submitted to the principal investigator.
Both female and male mice were included in the present study. The sex of the animal was considered as a control factor in the stratified randomization design. Subgroup analysis suggested that the results were consistent in the major endpoints (EF and scar) in female and male subgroups.
Beside exclusion of mice with atypical ECG during I/R surgery, we also used a preset exclusion criterion of pre-treatment EF ≥35%. To maintain blinding, echocardiographic analysis of all 3 time points was performed offline after the experiment was finished. Therefore, we did not know the pre-treatment EF of a mouse when it was assigned to a group. The mice with pre-treatment EF ≥35% were treated and followed through the study, but their results were excluded from the PP dataset. As shown above, the results from the PP dataset and the full dataset were consistent.
Based on the rigor of this study, we can confidently reject our original hypothesis. DB1976, a PU.1 inhibitor, did not attenuate cardiac function deterioration nor cardiac fibrosis after MI. These results suggest that PU.1 inhibition may not be a fruitful approach to modulating cardiac scar and tissue fibrosis after MI. Cardiac fibrosis after I/R is a complex process in which not only cardiac fibroblasts, but also cardiomyocytes and inflammatory and immune cells are involved [3, 4, 6, 47]. PU.1, which can regulate the expression of many myeloid and lymphoid genes, is required for natural killer cells [48] and monocytes/macrophages [49] differentiation. Defective natural killer cells and macrophages may cause impaired healing after MI. Hu et al. reported that the beneficial effect of PU.1 inhibition on Ang II-induced atrial fibrosis is through attenuation of TGF-β1/Smad3 expression [10]. Others have reported that TGF-β1/Smad3 is required for proper reparative cardiac fibrosis, and that the loss of TGF-β1/Smad3 in activated cardiac fibroblasts results in impaired scar remodeling and cardiac function [3, 4]. Therefore, depression of TGF-β1/Smad3 activity by PU.1 inhibition may result in impaired reparative cardiac fibrosis. Future work should use a more specific reprogramming approach aimed at targeting cardiac fibroblasts [3, 4, 6].
In conclusion, we present the results of a randomized, blinded preclinical study which demonstrates that DB1976, a PU.1 inhibitor does not attenuate cardiac function deterioration nor cardiac fibrosis in a mouse model of MI induced by coronary occlusion/reperfusion.
Supplementary Material
Acknowledgements
This work was supported in part by National Institutes of Health grants P01 HL078825 (RB).We thank Dr. Tamer Mohamed for providing animals and materials and input into protocol design.
Abbreviations
- MI
myocardial infarction
- I/R
ischemia/reperfusion
- ECG
electrocardiography
- LV
left ventricle
- EDV
end-diastolic volume
- ESV
end-systolic volume
- SV
stroke volume
- EF
ejection fraction
Footnotes
Conflicts of interest/Competing interests:The authors have no conflict of interest to declare.
Ethics declarations:All of the authors have approved that the submitted works are original and the paper has not been published and is not being considered for publication elsewhere.
Ethics approval: All animal procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Louisville Institutional Animal Care and Use Committee (protocol number:14034).
Consent for publication: All of the authors have approved the final version of this manuscript and have consented to the submission of this manuscript to the journal.
Availability of data and material:
All data and material are available from the corresponding author on reasonable request.
References
- 1.Tallquist MD and Molkentin JD (2017) Redefining the identity of cardiac fibroblasts. Nat Rev Cardiol 14:484–491. doi: 10.1038/nrcardio.2017.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Litvinukova M, Talavera-Lopez C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, Nadelmann ER, Roberts K, Tuck L, Fasouli ES, DeLaughter DM, McDonough B, Wakimoto H, Gorham JM, Samari S, Mahbubani KT, Saeb-Parsy K, Patone G, Boyle JJ, Zhang H, Zhang H, Viveiros A, Oudit GY, Bayraktar OA, Seidman JG, Seidman CE, Noseda M, Hubner N and Teichmann SA (2020) Cells of the adult human heart. Nature 588:466–472. doi: 10.1038/s41586-020-2797-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Villalobos E, Criollo A, Schiattarella GG, Altamirano F, French KM, May HI, Jiang N, Nguyen NUN, Romero D, Roa JC, Garcia L, Diaz-Araya G, Morselli E, Ferdous A, Conway SJ, Sadek HA, Gillette TG, Lavandero S and Hill JA (2019) Fibroblast Primary Cilia Are Required for Cardiac Fibrosis. Circulation 139:2342–2357. doi: 10.1161/CIRCULATIONAHA.117.028752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kong P, Shinde AV, Su Y, Russo I, Chen B, Saxena A, Conway SJ, Graff JM and Frangogiannis NG (2018) Opposing Actions of Fibroblast and Cardiomyocyte Smad3 Signaling in the Infarcted Myocardium. Circulation 137:707–724. doi: 10.1161/CIRCULATIONAHA.117.029622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, SC JL, Aronow BJ, Tallquist MD and Molkentin JD (2016) Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun 7:12260. doi: 10.1038/ncomms12260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gourdie RG, Dimmeler S and Kohl P (2016) Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat Rev Drug Discov 15:620–638. doi: 10.1038/nrd.2016.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaur H, Takefuji M, Ngai CY, Carvalho J, Bayer J, Wietelmann A, Poetsch A, Hoelper S, Conway SJ, Mollmann H, Looso M, Troidl C, Offermanns S and Wettschureck N (2016) Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice. Circ Res 118:1906–17. doi: 10.1161/CIRCRESAHA.116.308643 [DOI] [PubMed] [Google Scholar]
- 8.Takeda N, Manabe I, Uchino Y, Eguchi K, Matsumoto S, Nishimura S, Shindo T, Sano M, Otsu K, Snider P, Conway SJ and Nagai R (2010) Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J Clin Invest 120:254–65. doi: 10.1172/JCI40295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wohlfahrt T, Rauber S, Uebe S, Luber M, Soare A, Ekici A, Weber S, Matei AE, Chen CW, Maier C, Karouzakis E, Kiener HP, Pachera E, Dees C, Beyer C, Daniel C, Gelse K, Kremer AE, Naschberger E, Sturzl M, Butter F, Sticherling M, Finotto S, Kreuter A, Kaplan MH, Jungel A, Gay S, Nutt SL, Boykin DW, Poon GMK, Distler O, Schett G, Distler JHW and Ramming A (2019) PU.1 controls fibroblast polarization and tissue fibrosis. Nature 566:344–349. doi: 10.1038/s41586-019-0896-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu J, Zhang JJ, Li L, Wang SL, Yang HT, Fan XW, Zhang LM, Hu GL, Fu HX, Song WF, Yan LJ, Liu JJ, Wu JT and Kong B (2021) PU.1 inhibition attenuates atrial fibrosis and atrial fibrillation vulnerability induced by angiotensin-II by reducing TGF-beta1/Smads pathway activation. J Cell Mol Med 25:6746–6759. doi: 10.1111/jcmm.16678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snider P, Hinton RB, Moreno-Rodriguez RA, Wang J, Rogers R, Lindsley A, Li F, Ingram DA, Menick D, Field L, Firulli AB, Molkentin JD, Markwald R and Conway SJ (2008) Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circ Res 102:752–60. doi: 10.1161/CIRCRESAHA.107.159517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taniyama Y, Katsuragi N, Sanada F, Azuma J, Iekushi K, Koibuchi N, Okayama K, Ikeda-Iwabu Y, Muratsu J, Otsu R, Rakugi H and Morishita R (2016) Selective Blockade of Periostin Exon 17 Preserves Cardiac Performance in Acute Myocardial Infarction. Hypertension 67:356–61. doi: 10.1161/HYPERTENSIONAHA.115.06265 [DOI] [PubMed] [Google Scholar]
- 13.Furtado MB, Nim HT, Boyd SE and Rosenthal NA (2016) View from the heart: cardiac fibroblasts in development, scarring and regeneration. Development 143:387–97. doi: 10.1242/dev.120576 [DOI] [PubMed] [Google Scholar]
- 14.Jung RG, Stotts C, Makwana D, Motazedian P, Di Santo P, Goh CY, Verreault-Julien L, Simard T, Ramirez FD and Hibbert B (2021) Methodological Rigor in Preclinical Cardiovascular Research: Contemporary Performance of AHA Scientific Publications. Circ Res 129:887–889. doi: 10.1161/CIRCRESAHA.121.319921 [DOI] [PubMed] [Google Scholar]
- 15.Bolli R (2021) CAESAR’s legacy: a new era of rigor in preclinical studies of cardioprotection. Basic Res Cardiol 116:33. doi: 10.1007/s00395-021-00874-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones SP, Tang XL, Guo Y, Steenbergen C, Lefer DJ, Kukreja RC, Kong M, Li Q, Bhushan S, Zhu X, Du J, Nong Y, Stowers HL, Kondo K, Hunt GN, Goodchild TT, Orr A, Chang CC, Ockaili R, Salloum FN and Bolli R (2015) The NHLBI-sponsored Consortium for preclinicAl assESsment of cARdioprotective therapies (CAESAR): a new paradigm for rigorous, accurate, and reproducible evaluation of putative infarct-sparing interventions in mice, rabbits, and pigs. Circ Res 116:572–86. doi: 10.1161/CIRCRESAHA.116.305462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lefer DJ and Bolli R (2011) Development of an NIH consortium for preclinicAl AssESsment of CARdioprotective therapies (CAESAR): a paradigm shift in studies of infarct size limitation. J Cardiovasc Pharmacol Ther 16:332–9. doi: 10.1177/1074248411414155 [DOI] [PubMed] [Google Scholar]
- 18.Bolli R (2019) Paul Simpson and Scientific Rigor. Circ Res 124:194. doi: 10.1161/CIRCRESAHA.118.314621 [DOI] [PubMed] [Google Scholar]
- 19.Bolli R (2017) New Initiatives to Improve the Rigor and Reproducibility of Articles Published in Circulation Research. Circ Res 121:472–479. doi: 10.1161/CIRCRESAHA.117.311678 [DOI] [PubMed] [Google Scholar]
- 20.Bolli R (2015) Reflections on the Irreproducibility of Scientific Papers. Circ Res 117:665–6. doi: 10.1161/CIRCRESAHA.115.307496 [DOI] [PubMed] [Google Scholar]
- 21.Bolli R, Becker L, Gross G, Mentzer R Jr., Balshaw D, Lathrop DA and Ischemia NWGotToTfPtHf (2004) Myocardial protection at a crossroads: the need for translation into clinical therapy. Circ Res 95:125–34. doi: 10.1161/01.RES.0000137171.97172.d7 [DOI] [PubMed] [Google Scholar]
- 22.Lindsey ML, Bolli R, Canty JM Jr., Du XJ, Frangogiannis NG, Frantz S, Gourdie RG, Holmes JW, Jones SP, Kloner RA, Lefer DJ, Liao R, Murphy E, Ping P, Przyklenk K, Recchia FA, Schwartz Longacre L, Ripplinger CM, Van Eyk JE and Heusch G (2018) Guidelines for experimental models of myocardial ischemia and infarction. Am J Physiol Heart Circ Physiol 314:H812–H838. doi: 10.1152/ajpheart.00335.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nong Y, Guo Y, Gumpert A, Li Q, Tomlin A, Zhu X and Bolli R (2021) Single dose of synthetic microRNA-199a or microRNA-149 mimic does not improve cardiac function in a murine model of myocardial infarction. Mol Cell Biochem 476:4093–4106. doi: 10.1007/s11010-021-04227-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Q, Guo Y, Nong Y, Tomlin A, Gumpert A, Zhu X, Hassan SA and Bolli R (2021) Comparison of Repeated Doses of C-kit-Positive Cardiac Cells versus a Single Equivalent Combined Dose in a Murine Model of Chronic Ischemic Cardiomyopathy. Int J Mol Sci 22. doi: 10.3390/ijms22063145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo Y, Nong Y, Li Q, Tomlin A, Kahlon A, Gumpert A, Slezak J, Zhu X and Bolli R (2021) Comparison of One and Three Intraventricular Injections of Cardiac Progenitor Cells in a Murine Model of Chronic Ischemic Cardiomyopathy. Stem Cell Rev Rep 17:604–615. doi: 10.1007/s12015-020-10063-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Audam TN, Nong Y, Tomlin A, Jurkovic A, Li H, Zhu X, Long BW, Zheng YW, Weirick T, Brittian KR, Riggs DW, Gumpert A, Uchida S, Guo Y, Wysoczynski M and Jones SP (2020) Cardiac mesenchymal cells from failing and nonfailing hearts limit ventricular dilation when administered late after infarction. Am J Physiol Heart Circ Physiol 319:H109–H122. doi: 10.1152/ajpheart.00114.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wysoczynski M, Guo Y, Moore JBt, Muthusamy S, Li Q, Nasr M, Li H, Nong Y, Wu W, Tomlin AA, Zhu X, Hunt G, Gumpert AM, Book MJ, Khan A, Tang XL and Bolli R (2017) Myocardial Reparative Properties of Cardiac Mesenchymal Cells Isolated on the Basis of Adherence. J Am Coll Cardiol 69:1824–1838. doi: 10.1016/j.jacc.2017.01.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antony-Debre I, Paul A, Leite J, Mitchell K, Kim HM, Carvajal LA, Todorova TI, Huang K, Kumar A, Farahat AA, Bartholdy B, Narayanagari SR, Chen J, Ambesi-Impiombato A, Ferrando AA, Mantzaris I, Gavathiotis E, Verma A, Will B, Boykin DW, Wilson WD, Poon GM and Steidl U (2017) Pharmacological inhibition of the transcription factor PU.1 in leukemia. J Clin Invest 127:4297–4313. doi: 10.1172/JCI92504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehra P, Guo Y, Nong Y, Lorkiewicz P, Nasr M, Li Q, Muthusamy S, Bradley JA, Bhatnagar A, Wysoczynski M, Bolli R and Hill BG (2018) Cardiac mesenchymal cells from diabetic mice are ineffective for cell therapy-mediated myocardial repair. Basic Res Cardiol 113:46. doi: 10.1007/s00395-018-0703-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo Y, Wysoczynski M, Nong Y, Tomlin A, Zhu X, Gumpert AM, Nasr M, Muthusamy S, Li H, Book M, Khan A, Hong KU, Li Q and Bolli R (2017) Repeated doses of cardiac mesenchymal cells are therapeutically superior to a single dose in mice with old myocardial infarction. Basic Res Cardiol 112:18. doi: 10.1007/s00395-017-0606-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cai C, Guo Y, Teng L, Nong Y, Tan M, Book MJ, Zhu X, Wang XL, Du J, Wu WJ, Xie W, Hong KU, Li Q and Bolli R (2015) Preconditioning Human Cardiac Stem Cells with an HO-1 Inducer Exerts Beneficial Effects After Cell Transplantation in the Infarcted Murine Heart. Stem Cells 33:3596–607. doi: 10.1002/stem.2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo Y, Tukaye DN, Wu WJ, Zhu X, Book M, Tan W, Jones SP, Rokosh G, Narumiya S, Li Q and Bolli R (2012) The COX-2/PGI2 receptor axis plays an obligatory role in mediating the cardioprotection conferred by the late phase of ischemic preconditioning. PLoS One 7:e41178. doi: 10.1371/journal.pone.0041178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo Y, Flaherty MP, Wu WJ, Tan W, Zhu X, Li Q and Bolli R (2012) Genetic background, gender, age, body temperature, and arterial blood pH have a major impact on myocardial infarct size in the mouse and need to be carefully measured and/or taken into account: results of a comprehensive analysis of determinants of infarct size in 1,074 mice. Basic Res Cardiol 107:288. doi: 10.1007/s00395-012-0288-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo Y, Wu WJ, Qiu Y, Tang XL, Yang Z and Bolli R (1998) Demonstration of an early and a late phase of ischemic preconditioning in mice. Am J Physiol 275:H1375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo Y, Nong Y, Li Q, Tomlin A, Kahlon A, Gumpert A, Slezak J, Zhu X and Bolli R (2020) Comparison of One and Three Intraventricular Injections of Cardiac Progenitor Cells in a Murine Model of Chronic Ischemic Cardiomyopathy. Stem Cell Rev Rep. doi: 10.1007/s12015-020-10063-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nong Y, Guo Y, Tomlin A, Zhu X, Wysoczynski M, Li Q and Bolli R (2021) Echocardiography-guided percutaneous left ventricular intracavitary injection as a cell delivery approach in infarcted mice. Mol Cell Biochem 476:2135–2148. doi: 10.1007/s11010-021-04077-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hong KU, Guo Y, Li QH, Cao P, Al-Maqtari T, Vajravelu BN, Du J, Book MJ, Zhu X, Nong Y, Bhatnagar A and Bolli R (2014) c-kit+ Cardiac stem cells alleviate post-myocardial infarction left ventricular dysfunction despite poor engraftment and negligible retention in the recipient heart. PLoS One 9:e96725. doi: 10.1371/journal.pone.0096725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong KU, Li QH, Guo Y, Patton NS, Moktar A, Bhatnagar A and Bolli R (2013) A highly sensitive and accurate method to quantify absolute numbers of c-kit+ cardiac stem cells following transplantation in mice. Basic Res Cardiol 108:346. doi: 10.1007/s00395-013-0346-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Triana JF, Li XY, Jamaluddin U, Thornby JI and Bolli R (1991) Postischemic myocardial “stunning”. Identification of major differences between the open-chest and the conscious dog and evaluation of the oxygen radical hypothesis in the conscious dog. Circ Res 69:731–47. doi: 10.1161/01.res.69.3.731 [DOI] [PubMed] [Google Scholar]
- 40.Ping P, Zhang J, Huang S, Cao X, Tang XL, Li RC, Zheng YT, Qiu Y, Clerk A, Sugden P, Han J and Bolli R (1999) PKC-dependent activation of p46/p54 JNKs during ischemic preconditioning in conscious rabbits. Am J Physiol 277:H1771–85. doi: 10.1152/ajpheart.1999.277.5.H1771 [DOI] [PubMed] [Google Scholar]
- 41.Li XY, McCay PB, Zughaib M, Jeroudi MO, Triana JF and Bolli R (1993) Demonstration of free radical generation in the “stunned” myocardium in the conscious dog and identification of major differences between conscious and open-chest dogs. J Clin Invest 92:1025–41. doi: 10.1172/JCI116608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Faul F, Erdfelder E, Lang AG and Buchner A (2007) G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 39:175–91. doi: 10.3758/bf03193146 [DOI] [PubMed] [Google Scholar]
- 43.Prabhu SD and Frangogiannis NG (2016) The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ Res 119:91–112. doi: 10.1161/CIRCRESAHA.116.303577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Le Bras A (2018) Dynamics of fibroblast activation in the infarcted heart. Nat Rev Cardiol 15:379. doi: 10.1038/s41569-018-0025-9 [DOI] [PubMed] [Google Scholar]
- 45.Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC and Molkentin JD (2018) Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest 128:2127–2143. doi: 10.1172/JCI98215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaskill BN and Garner JP (2020) Power to the People: Power, Negative Results and Sample Size. J Am Assoc Lab Anim Sci 59:9–16. doi: 10.30802/AALAS-JAALAS-19-000042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wysoczynski M, Khan A and Bolli R (2018) New Paradigms in Cell Therapy: Repeated Dosing, Intravenous Delivery, Immunomodulatory Actions, and New Cell Types. Circ Res 123:138–158. doi: 10.1161/CIRCRESAHA.118.313251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Colucci F, Samson SI, DeKoter RP, Lantz O, Singh H and Di Santo JP (2001) Differential requirement for the transcription factor PU.1 in the generation of natural killer cells versus B and T cells. Blood 97:2625–32. doi: 10.1182/blood.v97.9.2625 [DOI] [PubMed] [Google Scholar]
- 49.Wan X, Chowdhury IH, Jie Z, Choudhuri S and Garg NJ (2019) Origin of Monocytes/Macrophages Contributing to Chronic Inflammation in Chagas Disease: SIRT1 Inhibition of FAK-NFkappaB-Dependent Proliferation and Proinflammatory Activation of Macrophages. Cells 9. doi: 10.3390/cells9010080 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and material are available from the corresponding author on reasonable request.