

Abstract
Propionic acidemia (PA, OMIM 606054) is a devastating inborn error of metabolism arising from mutations that reduce the activity of the mitochondrial enzyme propionyl‐CoA carboxylase (PCC). The defects in PCC reduce the concentrations of nonesterified coenzyme A (CoASH), thus compromising mitochondrial function and disrupting intermediary metabolism. Here, we use a hypomorphic PA mouse model to test the effectiveness of BBP‐671 in correcting the metabolic imbalances in PA. BBP‐671 is a high‐affinity allosteric pantothenate kinase activator that counteracts feedback inhibition of the enzyme to increase the intracellular concentration of CoA. Liver CoASH and acetyl‐CoA are depressed in PA mice and BBP‐671 treatment normalizes the cellular concentrations of these two key cofactors. Hepatic propionyl‐CoA is also reduced by BBP‐671 leading to an improved intracellular C3:C2‐CoA ratio. Elevated plasma C3:C2‐carnitine ratio and methylcitrate, hallmark biomarkers of PA, are significantly reduced by BBP‐671. The large elevations of malate and α‐ketoglutarate in the urine of PA mice are biomarkers for compromised tricarboxylic acid cycle activity and BBP‐671 therapy reduces the amounts of both metabolites. Furthermore, the low survival of PA mice is restored to normal by BBP‐671. These data show that BBP‐671 relieves CoA sequestration, improves mitochondrial function, reduces plasma PA biomarkers, and extends the lifespan of PA mice, providing the preclinical foundation for the therapeutic potential of BBP‐671.
Keywords: metabolomics, propionic acidemia, propionyl‐CoA carboxylase, Coenzyme A, mitochondria, tricarboxcylic acid cycle

1. INTRODUCTION
Propionic acidemia (PA, OMIM 606054) is a devastating inborn error of metabolism with substantial morbidity and mortality. 1 PA arises from missense mutations in one of the mitochondrial propionyl‐CoA carboxylase (PCC) (E.C. 6.4.1.3) genes (PCCA or PCCB) that produce proteins with diminished catalytic activity, resulting in compromised catabolism of propionyl‐CoA (C3‐CoA). 2 , 3 , 4 , 5 , 6 Reduced PCC activity leads to the accumulation of intracellular C3‐CoA, 7 , 8 which is converted to propionyl‐carnitine (C3‐carnitine) that is released into the plasma and urine. 2 The severity of PA varies depending on the impact of the mutations on PCC catalytic activity 3 , 4 , 5 , 6 , 9 and the plasma C3:C2‐carnitine ratio is a key biomarker used to screen newborns. 10 C3‐carnitine is not considered a toxic metabolite but rather a mechanism to release non‐esterified (free) CoA (CoASH) and eliminate excess propionate from the body. 2 Methylcitrate formed by the condensation of C3‐CoA with oxaloacetate is another mechanism that releases CoASH from C3‐CoA. Methylcitrate is a weak inhibitor and is not a substrate for ATP:citrate lyase, aconitase, or other enzymes in acetyl‐CoA metabolism 11 , 12 and its elimination in urine functions to release CoASH in tissues and removes propionate from the body through its elimination in the urine. These metabolites are robust biomarkers for PA.
A recent analysis of a hypomorphic mouse model of PA suggests that reduced cellular CoASH triggers mitochondrial dysfunction and represents an underlying metabolic crisis in PA. 7 CoASH functions as a cofactor in numerous reactions in intermediary metabolism and is a critical substrate for two key steps in mitochondrial energy metabolism: pyruvate and α‐ketoglutarate dehydrogenases. The cellular total CoA concentration is controlled by the activities of three separately encoded pantothenate kinases (PANK1α,β; PANK2, and PANK3) that catalyze the first and rate‐controlling step in CoASH biosynthesis. 13 The analysis of mice following the chemical inhibition of these PANK activities shows that reduced tissue CoA leads to mitochondrial dysfunction highlighted by reduced hepatic fatty acid β‐oxidation capacity and impaired gluconeogenesis. 14 Analysis of Pank1 −/− mice shows that reduced liver CoASH prevents normal fuel switching to mitochondrial fatty acid oxidation during fasting. 15 Pank1 −/− Pank2 −/− and Pank1 −/− Syn‐Pank2 −/− mice are more severely affected and succumb to a postnatal metabolic crisis arising from compromised oxidative metabolism. 16 , 17 The TCA cycle is localized to the mitochondria and is the major oxidative energy‐generating process in mammalian tissues. In the PA mouse model, the elevation of C3‐CoA leads to severe deficiencies in cellular CoASH and acetyl‐CoA that are coupled to the copious elimination of tricarboxylic acid cycle (TCA) intermediates in the plasma and urine indicating compromised mitochondrial function. 7 Mitochondrial dysfunction is also indicated in PA based on the inhibition of pyruvate oxidation, 18 , 19 fatty acid oxidation, 19 , 20 ureagenesis, 21 , 22 and gluconeogenesis 21 , 22 , 23 in cell cultures treated with propionate. Compromised TCA cycle activity in human PA patients is indicated by the elimination of TCA cycle intermediates in urine. 11 , 24
There are no approved therapies to treat PA, and treatment for patients with this disease is often limited to restrictive low‐protein or synthetic diets intended to reduce C3‐CoA formation. 1 , 25 Human liver transplantation can lower the circulating concentrations of PA biomarkers and improve the quality of life, though transplant patients remain at risk of developing complications. 26 , 27 , 28 , 29 , 30 , 31 , 32 Gene or RNA therapies under active development reduce the concentrations of PA biomarkers in mouse models. 33 , 34 , 35 , 36 , 37 Small molecule therapeutic strategies are also under exploration. 38 , 39 PANK activity is potently inhibited by C3‐CoA and this inhibition is reversed by PZ‐3022, a representative member of a drug‐like series of allosteric PANK activators, called pantazines, that elevate tissue CoA, 40 , 41 attenuate the inhibition of PANK activity by C3‐CoA and improve mitochondrial function in a PA mouse model. 7 BBP‐671 is a pantazine that has been chemically optimized for pharmaceutical use and is under investigation in a clinical trial as a potential therapy for PA and methylmalonic acidemia (ClinicalTrials.gov Identifier: NCT04836494). Here, we validate the efficacy of BBP‐671 in a preclinical mouse model of PA. We find that BBP‐671 improves survival, substantially relieves hepatic CoA sequestration, reduces the plasma C3:C2‐carnitine ratio, and improves mitochondrial TCA cycle function in PA mice, thus providing direct evidence for the therapeutic potential of BBP‐671 for ameliorating the mitochondrial crisis in organic acidemias involving CoA sequestration.
2. METHODS
2.1. Analytical measurements
The mass spectrometry methods for quantitation of CoA, organic metabolites, and acyl carnitines in urine and plasma were performed as described. 7 CoA and CoA thioesters were obtained from Avanti Polar Lipids (Croda International); l‐(methyl‐d 9)carnitine, l‐(methyl‐d 3)acetyl‐carnitine, l‐(methyl‐d 3)propionyl‐carnitine, l‐(methyl‐d 3)butyryl‐carnitine, l‐(methyl‐d 9)isovaleryl‐carnitine, and Labeled Carnitine Standards Set B were obtained from Cambridge Isotope Labs and (1,2‐13C)acetyl‐CoA was obtained from Sigma‐Aldrich. The surface plasmon resonance binding studies with immobilized PANK3 and the thermal shift experiments were performed as described. 7 PANK activity assays were performed as described using purified recombinant human PANK1β, mature mitochondrial PANK2, or PANK3 proteins. 41 , 42 The background subtracted DPM values were fit to the Morrison equation (GraphPad software). The protein melting temperatures in the thermal melt curves were calculated from the average data with second order smoothing and 10 neighbors in GraphPad software. PANK activity measurements in the presence of BBP‐671 were performed as described. 7 The background subtracted DPM values were normalized to the maximum activity for each experiment which was fit to a variable slope dose response curve.
Where T is the maximum activity, IC50 is the 50% inhibition concentration, X is the concentration of propionyl‐CoA, and h is the Hill slope. The experiments showing how BBP‐671 alters the response of PANKs to C3‐CoA were performed using 1 μg of each PANK isoform plus BBP‐671 (PANK1β, 7.5 μM; PANK2, 3 μM; and PANK3, 2 μM) as described. 7 Human liver‐derived C3A (HepG2/C3A) cells (#CRL‐1074) were purchased from ATCC and maintained in DMEM supplemented with 2 mM glutamine, 10% fetal bovine serum (Atlanta Biologicals), 50 U/mL penicillin and 50 mg/ml streptomycin. C3A cells were grown to a density of 6–8 × 106 in 100‐mm petri dishes at 37°C in 5% CO2:95% air for 24 h following the addition of BBP‐671 dissolved in dimethylsulfoxide (0.1% final), which was present at the same concentration in control cultures. Total CoA in cells and tissues was measured as described. 43
2.2. BBP‐671
BBP‐671 was synthesized following the previously described route. 40 Purity was greater than 98% by liquid chromatography with ultraviolet/evaporative light‐scattering detection and mass spectrometry analysis (Figure S2). BBP‐671: 1‐(4‐[6‐chloropyridazin‐3‐yl]piperazin‐1‐yl)‐2‐(4‐cyclopropyl‐3‐fluorophenyl)ethan‐1‐one. The 1H NMR (500 MHz, chloroform‐d) δ 7.26 (d, J = 9.5 Hz, 1H), 7.00–6.82 (m, 4H), 3.93–3.70 (m, 4H), 3.59 (d, J = 24.8 Hz, 6H), 2.06 (td, J = 8.7, 4.4 Hz, 1H), 1.11–0.87 (m, 2H), 0.80–0.61 (m, 2H) (Figure S3). 13C NMR (126 MHz, CDCl3) δ 169.39, 162.87, 160.92, 158.76, 147.59, 133.36, 133.29, 129.54, 129.43, 129.04, 126.42, 126.38, 124.14, 124.12, 115.40, 115.32, 115.14, 45.35, 45.26, 44.65, 41.13, 40.28, 8.53, 8.49, 7.80. ESI‐MS (M + 1): 375.52 (Figure S3).
2.3. X‐ray crystallography
The structure of the PANK3•AMPPNP•Mg2+•BBP‐671 complex was determined as described. 7 Diffraction data were collected at the SER‐CAT beam line 22‐ID at the Advanced Photon Source and processed using HKL2000. 44 PHASER 45 was used to solve the structure by molecular replacement using the PANK3•AMPPNP•Mg2+structure (PDB ID: 6B3V). The structure was refined and optimized using PHENIX 46 and COOT, 47 and was validated using MolProbity. 48 The data collection and refinement statistics are presented in Table S1. Structures were rendered with PyMOL (version 2.5, Schrödinger, LLC).
2.4. BBP‐671 Pharmacokinetics and pharmacodynamics
Pharmacokinetic and pharmacodynamic parameters for BBP‐671 were evaluated in C57BL/6J mice. BBP‐671 formulated in 0.5% Methocel suspension was administered orally at 1 mg/kg dose. Blood samples were collected by cardiac puncture under light isoflurane anesthesia at 0.5, 1, 2, 4, 8, 12, and 24 h. Liver and brain tissues were also harvested at the same time points and immediately frozen in liquid nitrogen. There were three mice at each time point. The blood samples were collected with ethylenediaminetetraacetic acid (EDTA) as an anticoagulant. Plasma samples were separated by centrifugation of whole blood and stored below −70°C until analysis. All samples were processed for analysis by protein precipitation using acetonitrile. BBP‐671 was quantified by LC–MS/MS using a Shimadzu Prominence UFLC attached to a QTrap 4500 equipped with a Turbo V ion source (Sciex) as described. 41 The multiple reaction monitoring transitions for BBP‐671 and the warfarin internal standard were 375.2/199.1 and 309.1/163.0 m/z, respectively. Data were analyzed with MultiQuant 3.0.2 software (Sciex). Drug levels of BBP‐671 were evaluated in the plasma of male BALB/c mice following a single intravenous dose of 2 mg/kg or a single oral dose of 10 mg/kg (SAI LifeSciences Ltd., India). Plasma concentration versus time data were analyzed by non‐compartmental approaches using the Phoenix WinNonlin 8.0 software program. The following pharmacokinetic parameters are reported: t½, Tmax, C0, Cmax, AUClast, AUCinf, Cl, Vss, and %F (Table S2).
2.5. Animal experiments
C57BL/6J mice (age 8 weeks) were purchased from Jackson Laboratory. The genetic mouse model of propionic acidemia 49 was obtained as frozen sperm from Michael Barry (Mayo Clinic, Rochester Minnesota, USA). Although the mutant PCCA allele is not expressed in its native genetic context, this mouse has been used extensively to model PA disease. 33 , 35 , 36 , 50 , 51 , 52 , 53 Following re‐animation, mice were genotyped using Pcca, PCCA, and GFP primers as described. 7 Pcca −/− PCCA(A138T) tg/0 mice were crossed with Pcca +/− mice to maintain the transgene as a single copy in all study animals. Progeny distribution followed a Mendelian pattern of inheritance, and 1 in 8 pups had the desired Pcca −/− PCCA(A138T) tg/0 genotype. Matched wild‐type mice had a Pcca +/+ genotype. Animals were maintained on a purified chow diet with the nutritional balance typically found in rodent diets (17.9% protein, 62.4% carbohydrate, 6% fat), that lack soybean meal (Tables S3 and S4). Animal rooms were maintained at room temperature 72 ± 2 °F, humidity 50% ± 10%, and a 14‐h light/10‐h dark cycle with the dark cycle starting at 20:00. BBP‐671 was formulated with the diet by Envigo. Water was supplied ad libitum. WT mice had a slightly higher mean weight than the PA mice, 8 but the difference was not significant based on the number of animals used in this study (p > 0.5). Mice were euthanized, tissues collected, and flash frozen immediately with liquid nitrogen, and plasma was collected in EDTA then frozen. Urine was collected over a 24 h period from mice singly housed in metabolic cages, following prior acclimatization for 24 h. Multiple experimenters performed biochemical analyses and were blinded during data acquisition. Male and female PA mice were randomized into treatment groups without exclusions. Separate cohorts of PA pups received chow +/− BBP‐671 following weaning at postnatal day P19‐21 until the evaluation of disease parameters at P50.
2.6. Statistical analysis
Statistical tests were performed using GraphPad Prism software v9.1.2 (http://graphpad.com/scientific-software/prism/). An unpaired parametric t‐test was used when comparing two sample populations. The sample sizes, the means, and the p values are reported in the figures or legends.
3. RESULTS
3.1. Mechanism of BBP‐671 action
Pantazines are a series of related small molecules that bind mammalian PANKs and modulate their respective PANK activities. 40 Pantazines have been shown in preclinical mouse models to correct the metabolic imbalances associated with pantothenate kinase‐associated neurodegeneration 41 and PA. 7 PZ‐289141 possesses high PANK3 affinity, but it is also rapidly cleared from the body (Figure S1). 7 PZ‐3022 has improved metabolic stability, but has reduced PANK3 affinity (Figure S1). 7 BBP‐671 is a pantazine developed for pharmaceutical use in humans that combines high PANK3 affinity and high metabolic stability (Figure S1). BBP‐671 most closely matches the ideal chemical properties of effective drugs that target the human central nervous system compared to the prior pantazines (Figure S1). 54 , 55 Also, BBP‐671 has excellent chemical and physical stability at ambient temperatures, low hygroscopicity, and excellent compressibility suitable to the development of a solid oral dosage form. BBP‐671 is a potent binder of all three PANK isoforms as measured by its impact on PANK activity (Figure 1A). PANK3 and PANK2 are more sensitive to BBP‐671 than PANK1β. BBP‐671 is more potent on all PANK targets compared to PZ‐289141 and PZ‐3022. 7 Surface plasmon resonance was used to directly measure the binding of BBP‐671 to PANK3 (Figure 1B). The kD for BBP‐671 is 97 pM and the residence time on PANK3 is 21.4 min. BBP‐671 binding increases the thermal denaturation temperature of PANK3 illustrating the enhanced stability of the drug‐bound protein (Figure 1C). The X‐ray structure of the PANK3•ATP•Mg2+•BBP‐671 complex shows that the cyclopropyl group binds at the same location as the dimethyl group in pantothenate but the drug extends out of the substrate site to engage the dimer interface with the chlorine substituent (Figure 1D). The isopropyl moiety packs into a hydrophobic cavity created by a flexible flap that is disordered in the PANK3•ATP•Mg2+ complex 56 but becomes structured in the PANK3•AMPPNP•Mg2+•BBP‐671 complex (Figure 1D). The piperazine ring acts as a spacer to present the pyridazine ring to the opposite protomer to hydrogen bond with R306′ and engage W341′ by π–π stacking interactions. These interactions effectively crosslink the two PANK3 protomers accounting for the ability of BBP‐671 to stabilize the PANK3 structure.
FIGURE 1.
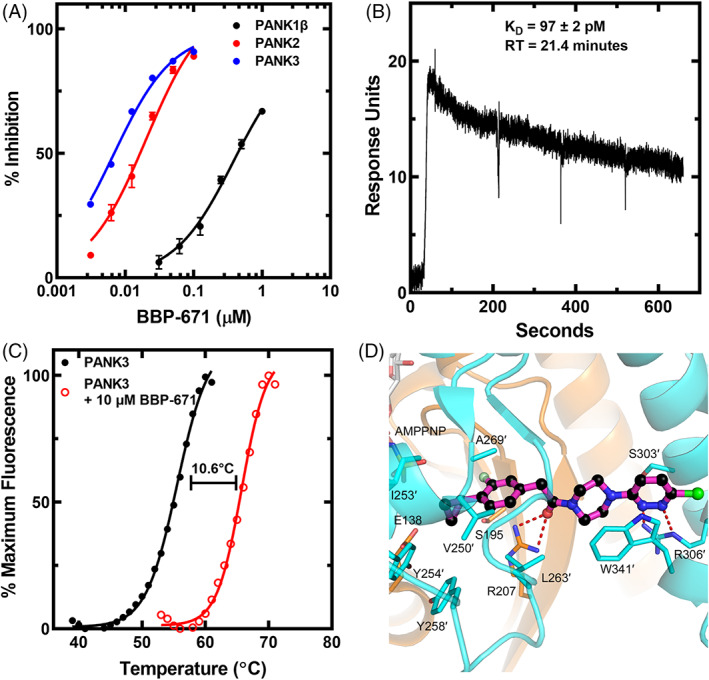
Properties of BBP‐671. (A) Binding of BBP‐671 to PANK1β, mature PANK2 and PANK3 measured using the PANK activity assay. The plotted data represent the mean ± SD from 4 data sets. The lines are the fit of the data to the Morrison equation using GraphPad software. The Ki's were: PANK1β, 102 ± 21.4 nM; PANK2, 2.02 ± 0.52 nM; and PANK3, 1.39 ± 0.33 nM. (B) Surface plasmon resonance analysis of BBP‐671 binding to PANK3 in the presence of 1 mM ATP•Mg2+. The KD and residence time were calculated as 97 ± 2 pM and 21.4 min, respectively. The data are the average of four triplicate experiments. (C) Thermal stability of PANK3 in the presence and absence of 10 μM BBP‐671 in buffer containing 2 mM ATP and 10 mM Mg2+. The data are an average of four experiments. (D) Crystal structure of the PanK3•AMPPNP•Mg2+•BBP‐671complex (PDBID: 7UE8). The PANK3 active site is formed by residues from both protomers (tan and cyan coloring) of the dimer, and the residues labeled with prime (′) are from the cyan protomer. BBP‐671 is magenta, fluorine is light green and chlorine is green. Hydrogen bonds are denoted as red dashes.
3.2. BBP‐671 elevates CoA
PANKs are allosteric enzymes where both protomers simultaneously switch between two distinct conformations: the active ATP•Mg2+ bound form and an inhibited acetyl‐CoA bound conformation (Figure 2A). 57 This feedback mechanism coupled with the relative expression levels of PANK isoforms determines the cellular concentration of total CoA. 14 , 58 , 59 In the normal catalytic cycle, ATP•Mg2+ cooperatively binds to the PANK dimer switching both protomers to the active, closed conformation and following catalysis, both of the active sites are empty. 57 Acetyl‐CoA can then bind to the apo‐enzyme switching its conformation to the inactive state. Sub‐saturating pantazine (BBP‐671) concentrations interrupt this normal catalytic cycle and feedback regulatory mechanism by binding to only one protomer, while maintaining the catalytic capacity of the opposite protomer (Figure 2A). 7 , 41 At the end of the catalytic cycle, the PANK3•BBP‐671 complex remains locked in its active conformation preventing acetyl‐CoA inhibition and allowing another cycle of catalysis to continue. This effect renders PANK3 dimers with a pantazine bound to only one protomer refractory to feedback inhibition by acetyl‐CoA (Figure 2A). Pantazines, including BBP‐671, thus overcome the feedback inhibition of PANKs by acetyl‐CoA and other CoA thioesters in cells to elevate CoA. 7 , 41
FIGURE 2.
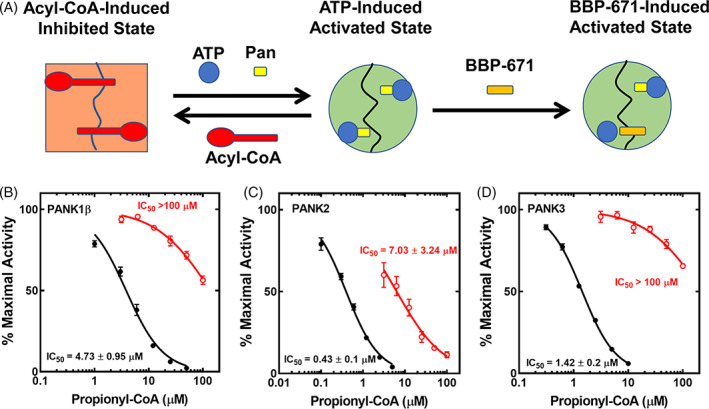
BBP‐671 renders PANK resistant to C3‐CoA inhibition in vitro. (A) Schematic diagram illustrating BBP‐671 mechanism of action. PANKs exist in two conformations: a catalytically inactive conformation that is stabilized by acyl‐CoA binding and a catalytically competent state that is stabilized by ATP binding. BBP‐671 binding stabilizes the active conformation by binding across the dimer interface and preventing acetyl‐CoA binding and inhibition. (B) Inhibition of PANK1β by C3‐CoA in the presence (red) and absence (black) of BBP‐671 as described in Methods. (C) Inhibition of PANK2 (the mature processed form of human PANK2) by C3‐CoA in the presence (red) and absence (black) of BBP‐671. (D) Inhibition of PANK3 by C3‐CoA in the presence (red) and absence (black) of BBP‐671. Data from four data sets represent the mean ± SD. Lines were fit using the variable slope equation with GraphPad software.
The ability of BBP‐671 to also act as an allosteric PANK3 activator in the presence of propionyl‐CoA (C3‐CoA) was tested in biochemical assays designed to mimic the mixture of ligands in cells (Figures 2B–D). The assays contained a purified PANK or a PANK plus a concentration of BBP‐671 that binds to approximately 25% of the total PANK protein. In the absence of BBP‐671, the activities of all three PANK isoforms are extinguished in a concentration‐dependent manner by C3‐CoA; however, all PANK isoforms are refractory to C3‐CoA inhibition when BBP‐671 is present in the radiochemical assay (Figures 2B–D). These data show that BBP‐671 functions like other pantazines to create a PANK catalytic cycle that is refractory to acyl‐CoA inhibition (Figure 2A). BBP‐671 dose–response experiments in cultured hepatic C3A cells illustrate the concentration‐dependent action of BBP‐671 as an allosteric activator and orthosteric inhibitor of PANKs (Figure 3A). At low concentrations, BBP‐671 elevated the intracellular CoA concentration and at higher concentrations this effect is diminished due to BBP‐671 binding to both protomers of the PANK dimer to block catalysis.
FIGURE 3.
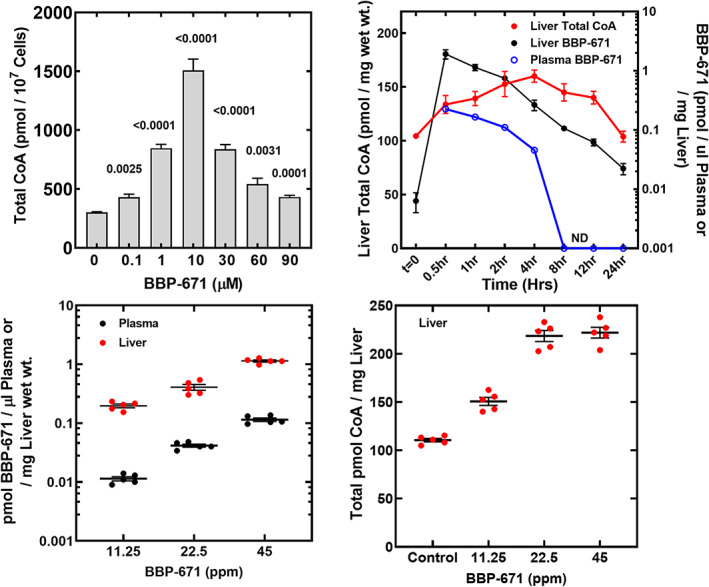
BBP‐671 elevates cellular CoA. (A) Effect of BBP‐671 concentrations on cellular CoA levels in cultured hepatocyte‐derived C3A cells (n = 4). (B) BBP‐671 (1 mg/kg) was administered to C57BL/6J male mice (three mice per time point) by oral gavage and at the indicated times the hepatic CoA concentrations, and plasma and liver BBP‐671 levels were measured. (C) Chow was formulated with either 11.25, 22.5, or 45 ppm BBP‐671. C57BL/6J male mice (five mice in each group) were maintained on the diets for 4 weeks, and BBP‐671 levels in plasma and liver were determined by mass spectrometry. (D) Total CoA was determined in the livers of the C57BL/6J mice maintained on the three BBP‐671 supplemented diets for 4 weeks. The data represent the mean ± SE.
3.3. BBP‐671 pharmacokinetics/pharmacodynamics
The pharmacokinetics of BBP‐671 were first evaluated in BALB/c mice. The half‐life for BBP‐671 in plasma was 0.7–0.96 h and it was 93.2% orally bioavailable (Table S2). The pharmacokinetic/pharmacodynamic analysis of BBP‐671 was performed with C57BL/6J mice to correlate plasma and liver drug levels with the effect of BBP‐671 on hepatic total CoA concentrations (Figure 3B). Animals were orally dosed with 1 mg/kg of BBP‐671 at time zero, and at the indicated times, groups of three mice were analyzed for drug and CoA levels. BBP‐671 rapidly entered the liver and the half‐life for BBP‐671 in liver tissue was longer than in plasma (Figure 3B). By 8 h, BBP‐671 levels in plasma were below detection but BBP‐671 remained detectable in liver 12 h later. The longer retention of BBP‐671 in tissues may be attributed to its high affinity and long residence time on PANK. CoA levels increased to the maximum at 4 h after drug delivery and CoA remained elevated until the 24 h time point. PANK is the first step in CoA biosynthesis and the lag time between the appearance of the drug and CoA elevation reflects the time required for phosphopantothenate, the PANK product, to be converted to CoA and accumulate in the liver. As the liver drug levels decrease, CoA concentrations gradually decrease as CoA turnover returns the liver to its pre‐treatment equilibrium. BBP‐671 is also brain penetrant and elevates the concentration of CoA in the brain (Figure S4).
BBP‐671 was formulated in the chow to examine the impact of longer‐term treatment on disease parameters in PA mice. Based on the affinity of BBP‐671 for PANK3, we formulated three diets containing either 11.25, 22.5, or 45 ppm of BBP‐671 to determine the proper BBP‐671 dosing to maximize liver CoA. Male C57BL/6 mice were fed the control and BBP‐671‐supplemented diets for 4 weeks. The plasma and liver BBP‐671 concentrations increase proportionally with the level of drug in the diet (Figure 3C). Hepatic CoA concentrations are elevated by 11.25 ppm BBP‐671 and reached a maximum elevation at 22.5 ppm BBP‐671 (Figure 3D). Although liver BBP‐671 concentrations were higher with the 45‐ppm diet, there was no further increase in the liver CoA concentration (Figure 3D). The steady‐state plasma concentration of BBP‐671 required to maximally elevate liver CoA was 42 nM (Figure 3C). BBP‐671 was also brain penetrant and elevated the total CoA concentration in brain (Figure S4). These data support the use of the diet containing 22.5 ppm BBP‐671 to elevate liver CoA and measure its impact on biomarkers in the PA mouse model.
3.4. BBP‐671 extends the lifespan and relieves CoA sequestration in PA mice
We placed cohorts of PA mice and their wild‐type littermate controls on a purified diet (Tables S3 and S4) or the same diet supplemented with 22.5 ppm BBP‐671. The PA mice had lower liver total CoA that was elevated by BBP‐671 treatment illustrating the effect of the drug in elevating total liver CoA (Figure S5). BBP‐671 was present in the plasma and livers of PA mice maintained on the BBP‐671 supplemented diet (Figure S5). BBP‐671 significantly increased the survival of the PA mice based on the Mantel‐Cox (p = 0.0036) and Gehan‐Breslow‐Wilcoxon (p = 0.0043) statistical tests (Figure 4A). Male and female PA mice have reduced hepatic concentrations of non‐esterified CoA (CoASH) and acetyl‐CoA, and significantly elevated C3‐CoA compared to their wild‐type littermates (Figure 4B), illustrating the CoA sequestration metabolic phenotype of the PA mouse. 7 CoA sequestration led to a markedly elevated C3:C2‐CoA ratio in liver (Figure 4B). BBP‐671 relieved this PA‐induced CoA metabolic phenotype by elevating hepatic CoASH and acetyl‐CoA concentrations to near wild‐type levels. C3‐CoA remained abnormally elevated compared to wild‐type littermates; however, total C3‐CoA was reduced substantially by BBP‐671 therapy (Figure 4B). The combination of higher C2‐CoA and lower C3‐CoA led to a significant reduction in the hepatic C3:C2‐CoA ratio. These data show that BBP‐671 promoted survival of the PA mouse model and relieved CoA sequestration in PA liver.
FIGURE 4.
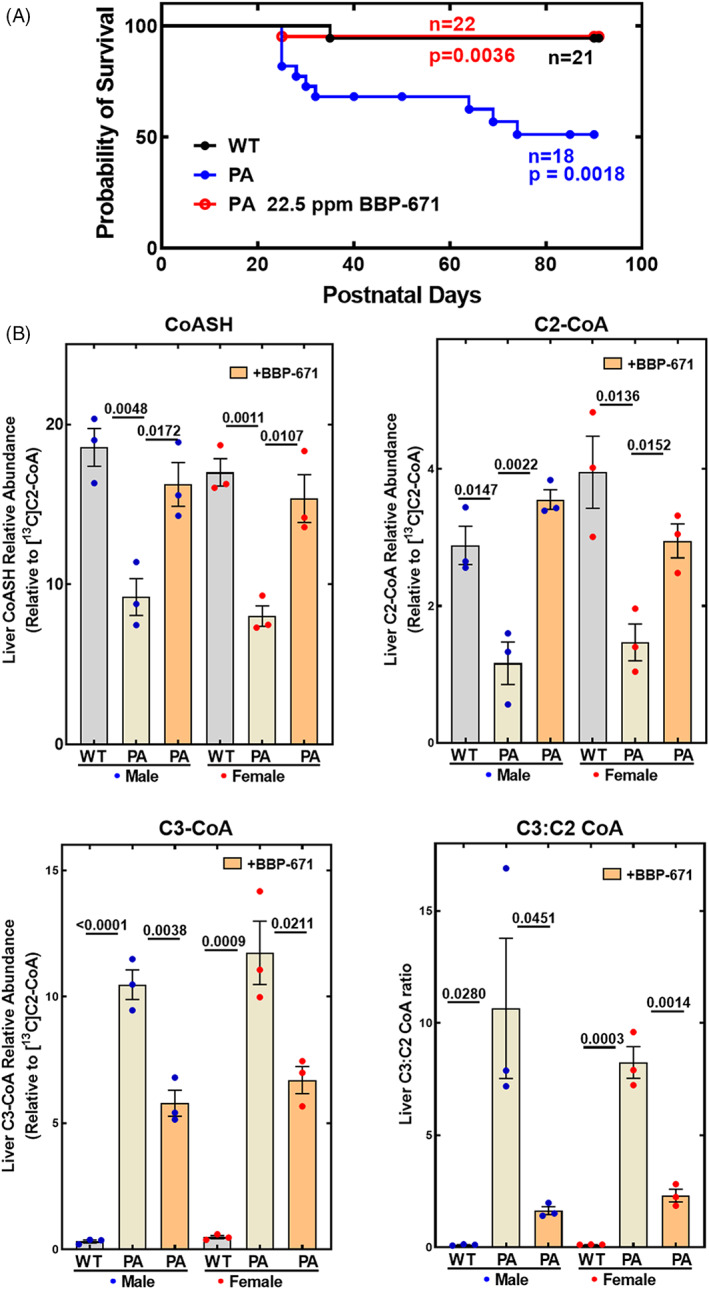
BBP‐671 prolongs survival and relieves CoA sequestration in PA mice. (A) The survival of wild‐type littermate control mice (n = 21: 11 male; 10 female), PA mice (n = 22: 11 male; 11 female), and PA mice maintained on BBP‐671‐supplemented chow (n = 18: 8 male; 10 female) are compared. (B) Nonesterified CoA (CoASH), acetyl‐CoA (C2‐CoA), propionyl‐CoA (C3‐CoA) and the C3:C2‐CoA ratio were measured by mass spectrometry in the livers of wild‐type and PA mice and are compared to the concentrations in PA mice maintained on BBP‐671 chow (n = 3). The data represent the mean ± SE. The p values are shown in the figure panels.
3.5. BBP‐671 reduces plasma and urine C3:C2‐carnitine ratios
The hallmark biomarker for PA disease is the plasma C3:C2‐carnitine ratio. Non‐esterified carnitine and C2‐carnitine are the most abundant species in the plasma of normal animals (Figure 5A). In the PA mice, plasma C2‐carnitine was significantly reduced. The plasma C3‐carnitine concentrations were low as expected in wild‐type mice, but in PA mice C3‐carnitine was greatly elevated (Figure 5A). The combination of low C2‐carnitine and high C3‐carnitine led to a massively elevated plasma C3:C2‐carnitine ratio (Figure 5A). BBP‐671 supplementation did not alter plasma carnitine but increased circulating C2‐carnitine concentrations and decreased plasma C3‐carnitine (Figure 5A). These effects combine to lower the plasma C3:C2‐carnitine ratio in BBP‐671‐treated mice.
FIGURE 5.
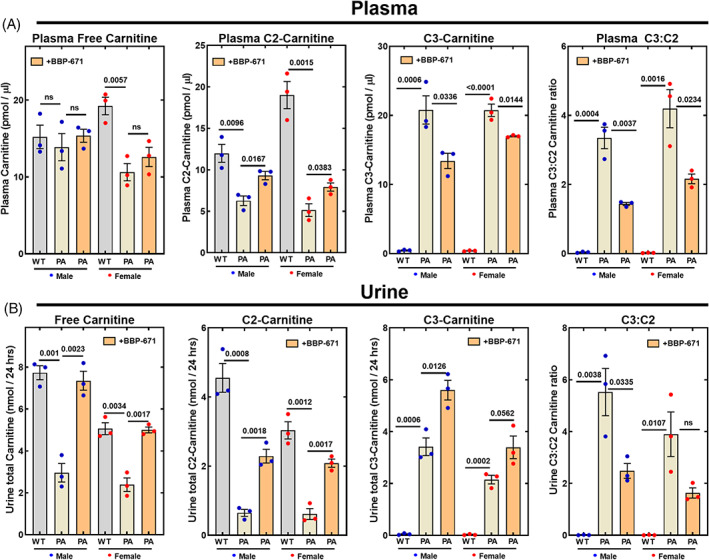
BBP‐671 reduces the C3:C2‐carnitine ratio in plasma and urine. Acyl‐carnitine concentrations and the C3:C2 ratios determined by mass spectrometry in male and female wild‐type and PA mice are compared to BBP‐671‐treated PA mice in (A) Plasma (n = 3); (B) Urine (n = 3). The data represent the mean ± SE. The p values are reported in each panel. ns means p > 0.05.
Carnitine in urine was significantly reduced in the PA mice and its concentration in urine was normalized by BBP‐671 (Figure 5B). C2‐carnitine was the second major excreted acyl‐carnitine in the urine of wild‐type mice that was reduced in the PA mice. The amount of C2‐carnitine in PA urine is substantially elevated by BBP‐671 (Figure 5B). C3‐carnitine was normally a minor constituent of urine but was a major acyl‐carnitine in PA mice, leading to an elevated C3:C2‐carnitine ratio in urine (Figure 5B). BBP‐671 treatment resulted in the elevation of both C2‐ and C3‐carnitines, but the net effect was lowering the C3:C2‐carnitine ratio in urine. These data show that the elevation of liver CoASH and C2‐CoA concentrations by BBP‐671 normalized the plasma C2‐carnitine concentrations, improved the plasma C3:C2‐carnitine ratio and enhanced the elimination of C3‐carnitine in the urine.
3.6. Impact of BBP‐671 on mitochondrial metabolites
Another hallmark of PA mice is the elevation of intermediates in the TCA cycle in plasma and urine. 7 , 8 The sentinel TCA cycle‐related biomarkers that are elevated in the PA mouse model are malate, methylcitrate, α‐ketoglutarate and methylmalonate. Although malate is not regularly measured in human PA, there are two reports that note PA patients have elevated TCA cycle intermediates, such as malate and α‐ketoglutarate, excreted in urine. 11 , 24 PA mice had 3‐fold elevation in plasma malate and an 80‐fold elevation in urinary malate (Figure 6A). The lesser increase in malate in the plasma compared to urine was consistent with the saturation of malate resorption in the kidneys, 60 a process that explains why TCA intermediates are more significantly elevated in urine than in plasma. BBP‐671 treatment did not significantly alter plasma malate concentrations in PA mice (Figure 6A); however, the copious amount of malate in the urine of PA mice was substantially reduced by BBP‐671, pointing to significant restoration of TCA cycle function (Figure 6B). Methylcitrate arises from citrate synthase in the TCA cycle using C3‐CoA as an alternate substrate instead of C2‐CoA. BBP‐671 reduced methylcitrate in plasma (Figure 6A), which was attributed to the change in the cellular C3:C2‐CoA ratio (Figure 4B). Although methylcitrate concentrations in plasma were lower, its formation remained an efficient mechanism for conversion of C3‐CoA to an inert urinary metabolite in BBP‐671‐treated mice (Figure 6B). The elevation of plasma methylmalonate in PA mice may seem counterintuitive, but the TCA cycle is so compromised in affected mice that the methylmalonyl‐CoA produced by the residual activity of mutant PCC cannot be efficiently metabolized further by a sluggish TCA cycle and is converted to methylmalonate by a thioesterase. 7 The reduction of methylmalonate in the plasma and urine of PA mice following BBP‐671 therapy (Figure 6A,B) was consistent with the restoration of mitochondrial TCA cycle function. α‐Ketoglutarate was another intermediate that was elevated in PA mice indicating that it also was not efficiently metabolized by the CoASH‐dependent α‐ketoglutarate dehydrogenase in the TCA cycle. 7 BBP‐671 treatment led to significant reductions in α‐ketoglutarate concentrations in both the plasma (Figure 6A) and urine (Figure 6B) indicating improved utilization of α‐ketoglutarate by the TCA cycle.
FIGURE 6.
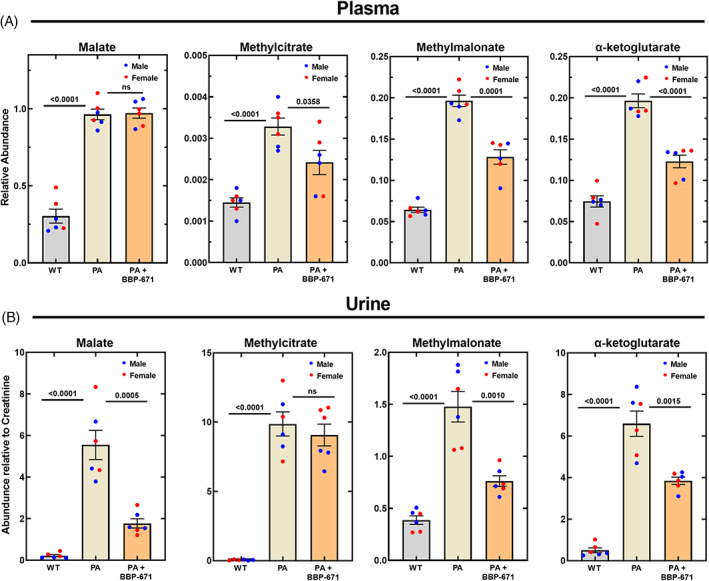
BBP‐671 reduces TCA cycle‐related metabolite accumulation in PA mice. Sentinel TCA metabolites were measured by mass spectrometry in wild‐type, PA mice, and PA mice treated with BBP‐671 in (A) Plasma metabolites relative to the internal standard; (B) Urine metabolites relative to creatinine. The male mice (n = 3) are blue dots and the female mice (n = 3) are red dots. The data represent the mean ± SE. The p values are shown. ns means p > 0.05. ns means p > 0.05.
4. DISCUSSION
BBP‐671 is a high affinity activator of all PANK isoforms and its effects on PA disease parameters are attributed to its ability to counteract the inhibition of PANKs by C3‐CoA, leading to the elevation of liver CoASH and C2‐CoA concentrations (Figure 7). CoASH is an essential substrate for two key steps in the TCA cycle, pyruvate and α‐ketoglutarate dehydrogenases, and when the concentrations of CoASH decrease, the cycle does not operate efficiently at these steps. The release of malate and α‐ketoglutarate into the plasma and urine of PA mice provides direct evidence that reduced TCA cycle activity is an important metabolic imbalance triggered by reduced PCC activity (Figure 7). BBP‐671 treatment significantly reduces malate in urine and α‐ketoglutarate concentrations in plasma and urine indicating that the relief of CoA sequestration improves TCA cycle activity and utilization of these intermediates. Analysis of PANK isoform knockout mice shows that compromised mitochondrial metabolism is a major consequence of insufficient cellular CoA, 14 , 15 , 16 , 17 , 61 and mitochondrial dysfunction is apparent in cell models of acidemia diseases. 18 , 19 , 20 , 21 , 22 , 23 , 62 , 63 Compromised TCA cycle activity in human PA patients is indicated by the elimination of TCA cycle intermediates in urine. 11 , 24 The concentrations of TCA cycle intermediates (malate and α‐ketoglutarate) in plasma or urine are not measured in standard clinical practice as biomarkers for mitochondrial function in PA, 64 but the solid connection between these metabolites and PA disease in mice and cell cultures suggests that TCA metabolites should be evaluated in human disease. Our data also indicate that the ability of BBP‐671 to normalize the PA biomarker, the plasma C3:C2‐carnitine ratio, is tied to mitochondrial function. C2‐CoA sits at the center of metabolism and depressed C2‐CoA coupled with elevated TCA cycle intermediates may disrupt intermediary metabolism at multiple points in light of their roles as metabolic substrates, 65 as regulatory ligands, 66 as redox regulators, 67 and in epigenetic control. 68 , 69 However, the survival benefit of BBP‐671 therapy points to CoASH sequestration and TCA cycle impairment as a major determinant of the metabolic imbalance associated with PA disease.
FIGURE 7.
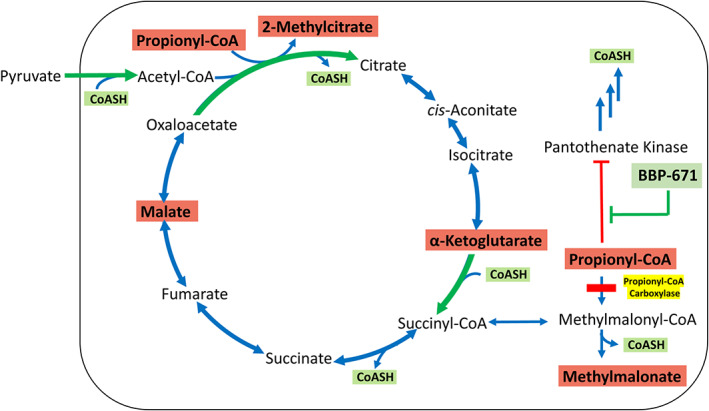
Role of CoA in the formation of TCA cycle‐related metabolites. There are two key, irreversible steps (green arrows) that depend on CoASH in the TCA cycle: pyruvate dehydrogenase and α‐ketoglutarate dehydrogenase. The block at propionyl‐CoA carboxylase (yellow) leads to the accumulation of C3‐CoA and the inhibition of PANK. The sequestration of CoA as C3‐CoA coupled with the inhibition of PANK leads to a cellular deficiency in CoASH. Acetyl‐CoA is also reduced due to the lack of CoASH to support pyruvate dehydrogenase activity. BBP‐671 counteracts that inhibition of PANK leading to an increase in CoASH and restoration of acetyl‐CoA and mitochondrial function. Metabolites that are significantly elevated in PA are shown in red boxes.
Although the impact of PA disease in liver has been extensively studied in the Pcca −/− PCCA(A138T) tg/0 mouse model, 7 , 33 , 35 , 36 , 50 , 51 , 52 , 53 , 70 understanding the impact of PCC mutations in other tissues is complicated in this model because the tissue‐selective transgene expression does not reflect the actual tissue‐specific expression of native PCC. 7 , 70 The amount of C3‐CoA produced and the extent of CoASH sequestration will vary depending on the normal PCC expression level in each individual cell type. The rate of CoASH biosynthesis is also governed in tissues by the expression levels of the three PANK isoforms, leading to the observed variations in tissue CoA concentrations. 71 Correcting liver metabolism with liver transplantation 26 , 27 , 28 , 29 , 30 , 31 , 32 or gene or RNA 33 , 34 , 35 , 36 , 37 therapy to restore liver PCC activity will not directly alter the extent of cellular CoASH sequestration or mitochondrial dysfunction in the other body tissues because CoA formation and mitochondrial metabolism are cell autonomous. PA patients have impaired neurocognitive development, 72 and as a small molecule that penetrates the brain, BBP‐671 has the potential to correct CoASH sequestration and defective TCA cycles in the brain and other affected tissues. Another small molecule approach to PA is treatment with 2,2‐dimethylbutyrate (HST5040), which enters cells and is converted to its CoA thioester derivative. 38 , 39 This treatment reduces C3‐CoA and its downstream metabolites by sequestering CoASH as the HST5040 thioester. However, there are animal toxicities that arise from exposure to xenobiotic carboxylic acids, notably valproic acid, that are directly attributed to CoASH sequestration into non‐metabolizable thioesters, 62 , 73 , 74 , 75 , 76 , 77 and the utility of HST5040 remains to be tested in a PA mouse model. Finally, BBP‐671 therapy may be beneficial in other organic acidemias, like methylmalonic acidemia, and β‐oxidation inborn errors of metabolism that are predicted to trigger the accumulation of a particular CoA thioester that would consequently sequester CoASH. 78 , 79 The accumulation of an acyl‐CoA PANK inhibitor in these diseases will trigger the same responses in liver as PA (PANK inhibition and CoASH depletion) that will depress the activity of the TCA cycle, energy production and the intracellular C2‐CoA concentration. Activation of PANK by BBP‐671 would be anticipated to restore the intracellular CoASH concentration and mitochondrial function in these diseases as well.
FUNDING INFORMATION
This work was supported by a research grant from CoA Therapeutics Inc., a BridgeBio Pharma, Inc., company, and ALSAC, St. Jude Children's Research Hospital.
CONFLICT OF INTEREST
S. Jackowski is a member of the Scientific Advisory Board of CoA Therapeutics and a former member of the Scientific and Medical Advisory Board of the NBIA Disorders Association. C.S., M.‐K.Y., R.E.L., C.O.R., and S.J. are co‐inventors of U.S. patent no. US10899734B2 (“Small molecule modulators of pantothenate kinase”) and S.J., C.O.R., R.E.L, C.S., R.T., and M.‐K. Y. are co‐inventors on the pending U.S. patent applications nos. 16/957996 (“Methods of treating disorders associated with CASTOR”) and 16/958022 (“Small molecule modulators of pantothenate kinases”).
ANIMAL RIGHTS
All procedures were performed according to protocols 323 and 556 as approved by the St. Jude Children's Research Hospital Institutional Animal Care and Use Committee.
Supporting information
Table S1 Data collection and Refinement Statistics
Table S2 BBP‐671 pharmacokinetics. Mean pharmacokinetic parameters of BBP‐671 in plasma following single intravenous or oral administration to male BALB/c mice a .
Table S3 Diet composition. (Envigo: TTD.170542)
Table S4 Selected Nutrient Information (Total kcal/g = 3.7)
Figure S1 Properties of BBP‐671. (A) A comparative summary of the sentinel pharmacokinetic properties of PZ‐28914, 41 PZ‐30227 and BBP‐671 (Table S2). (B) The mean values, preferred range and suggested limits on the chemical properties of the top 25 drugs used to treat neurological disease 54 , 55 compared to PZ‐2891, PZ‐3022 and BBP‐671 calculated using ChemDraw v21. PSA, polar surface area; HBD, hydrogen bond number; cLogP, partition coefficient between n‐octanol and water; MW, molecular weight.
Figure S2 Purity of BBP‐671. (A) Liquid chromatography‐mass spectrometric (LC–MS) analysis of BBP‐671 using evaporative light‐scattering detection (upper panel), ultraviolet detection (middle panel) or total LC–MS ion current (lower panel). (B) Mass spectrum of BBP‐671.
Figure S3 1H‐ and 13C‐NMR spectra of BBP‐671. (A) The proton NMR spectrum of BBP‐671. (B) The carbon‐13 NMR spectrum of BBP‐671.
Figure S4: PKPD of BBP‐671 in the forebrain. (A) BBP‐671 was administered at 1 mg/kg to C57BL/6J male mice (three mice per time point) by gavage and forebrain tissue was taken at indicated times (see Figure 2B). (B) Chow was formulated with 11.25, 22.5, or 45 ppm BBP‐671, and C57BL/6J male mice (five mice in each group) were maintained on the diets for 4 weeks, and BBP‐671 levels, and total CoA was determined in the forebrain. The data are mean and SE.
Figure S5 Total CoA and BBP‐671 levels. (A) Liver total CoA in wild‐type littermate controls was measured and compared to CoA in the liver of BBP‐671‐treated and untreated PA mice. (B) Plasma and liver BBP‐671 concentrations. There were 3 males (blue dots) and 3 females (red dots) in each group and the mice were maintained on chow formulated either with or without 22.5 ppm BBP‐671 as indicated. The data are mean ± SE.
ACKNOWLEDGMENTS
This work was supported by a research grant from CoA Therapeutics Inc., a BridgeBio Pharma, Inc., company, and ALSAC, St. Jude Children's Research Hospital. We thank Jina Wang, Karen Miller, Katie Creed and Lois Richmond for their expert technical assistance.
Subramanian C, Frank MW, Tangallapally R, et al. Relief of CoA sequestration and restoration of mitochondrial function in a mouse model of propionic acidemia. J Inherit Metab Dis. 2023;46(1):28‐42. doi: 10.1002/jimd.12570
Communicating Editor: Ronald JA Wanders
Funding information American Lebanese Syrian Associated Charitie, Grant/Award Number: N/A; CoA Therpaeutics
DATA AVAILABILITY STATEMENT
The data used in the preparation of the graphs is available from the corresponding author on reasonable request. The PANK3•ATP•Mg2+•BBP‐671 complex structure is deposited in the Protein Data Bank (PDBID: 7UE8).
REFERENCES
- 1. Baumgartner MR, Horster F, Dionisi‐Vici C, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis. 2014;9:130. doi: 10.1186/s13023-014-0130-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wongkittichote P, Mew A. N, Chapman KA Propionyl‐CoA carboxylase ‐ a review. Mol Genet Metab. 2017;122:145‐152. doi: 10.1016/j.ymgme.2017.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Clavero S, Martinez MA, Perez B, Perez‐Cerda C, Ugarte M. Desviat LR functional characterization of PCCA mutations causing propionic acidemia. Biochim Biophys Acta. 2002;1588:119‐125. [DOI] [PubMed] [Google Scholar]
- 4. Desviat LR, Perez B, Perez‐Cerda C, Rodriguez‐Pombo P, Clavero S. Ugarte M propionic acidemia: mutation update and functional and structural effects of the variant alleles. Mol Genet Metab. 2004;83:28‐37. doi: 10.1016/j.ymgme.2004.08.001 [DOI] [PubMed] [Google Scholar]
- 5. Al‐Hamed MH, Imtiaz F, Al‐Hassnan Z, et al. Spectrum of mutations underlying propionic acidemia and further insight into a genotype‐phenotype correlation for the common mutation in Saudi Arabia. Mol Genet Metab Rep. 2019;18:22‐29. doi: 10.1016/j.ymgmr.2018.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rivera‐Barahona A, Navarrete R, Garcia‐Rodriguez R, et al. Identification of 34 novel mutations in propionic acidemia: functional characterization of missense variants and phenotype associations. Mol Genet Metab. 2018;125:266‐275. doi: 10.1016/j.ymgme.2018.09.008 [DOI] [PubMed] [Google Scholar]
- 7. Subramanian C, Frank MW, Tangallapally R, et al. Pantothenate kinase activation relieves coenzyme a sequestration and improves mitochondrial function in mice with propionic acidemia. Sci Transl Med. 2021;13:eabf5965. doi: 10.1126/scitranslmed.abf5965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao C, Wang Y, Yang H, et al. Propionic acidemia in mice: liver acyl‐CoA levels and clinical course. Mol Genet Metab. 2022;135:47‐55. doi: 10.1016/j.ymgme.2021.11.011 [DOI] [PubMed] [Google Scholar]
- 9. Perez B, Desviat LR, Rodriguez‐Pombo P, et al. Propionic acidemia: identification of twenty‐four novel mutations in Europe and North America. Mol Genet Metab. 2003;78:59‐67. doi: 10.1016/s1096-7192(02)00197-x [DOI] [PubMed] [Google Scholar]
- 10. Turgeon CT, Magera MJ, Cuthbert CD, et al. Determination of total homocysteine, methylmalonic acid, and 2‐methylcitric acid in dried blood spots by tandem mass spectrometry. Clin Chem. 2010;56:1686‐1695. doi: 10.1373/clinchem.2010.148957 [DOI] [PubMed] [Google Scholar]
- 11. Ando T, Rasmussen K, Wright JM. Nyhan WL isolation and identification of methylcitrate, a major metabolic product of propionate in patients with propionic acidemia. J Biol Chem. 1972;247:2200‐2204. [PubMed] [Google Scholar]
- 12. Cheema‐Dhadli S, Leznoff CC. Halperin ML effect of 2‐methylcitrate on citrate metabolism: implications for the management of patients with propionic acidemia and methylmalonic aciduria. Pediatr Res. 1975;9:905‐908. doi: 10.1203/00006450-197512000-00008 [DOI] [PubMed] [Google Scholar]
- 13. Leonardi R, Zhang Y‐M, Rock CO. Jackowski S coenzyme a: Back in action. Prog Lipid Res. 2005;44:125‐153. doi: 10.1016/j.plipres.2005.04.001 [DOI] [PubMed] [Google Scholar]
- 14. Zhang YM, Chohnan S, Virga KG, et al. Chemical knockout of pantothenate kinase reveals the metabolic and genetic program responsible for hepatic coenzyme a homeostasis. Chem Biol. 2007;14:291‐302. doi: 10.1016/j.chembiol.2007.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leonardi R, Rehg JE, Rock CO. Jackowski S Pantothenate kinase 1 is required to support the metabolic transition from the fed to the fasted state. PLoS ONE. 2010;5:e11107. doi: 10.1371/journal.pone.0011107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garcia M, Leonardi R, Zhang YM, Rehg JE. Jackowski S germline deletion of pantothenate kinases 1 and 2 reveals the key roles for CoA in postnatal metabolism. PLoS One. 2012;7:e40871. doi: 10.1371/journal.pone.0040871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Subramanian C, Yao J, Frank MW, Rock CO. Jackowski S a pantothenate kinase‐deficient mouse model reveals a gene expression program associated with brain coenzyme a reduction. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165663. doi: 10.1016/j.bbadis.2020.165663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Patel TB, DeBuysere MS. Olson MS the effect of propionate on the regulation of the pyruvate dehydrogenase complex in the rat liver. Arch Biochem Biophys. 1983;220:405‐414. doi: 10.1016/0003-9861(83)90430-7 [DOI] [PubMed] [Google Scholar]
- 19. Brass EP, Fennessey PV. Miller LV inhibition of oxidative metabolism by propionic acid and its reversal by carnitine in isolated rat hepatocytes. Biochem J. 1986;236:131‐136. doi: 10.1042/bj2360131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Y, Christopher BA, Wilson KA, et al. Propionate‐induced changes in cardiac metabolism, notably CoA trapping, are not altered by L‐carnitine. Am J Physiol Endocrinol Metab. 2018;315:E622‐E633. doi: 10.1152/ajpendo.00081.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Walajtys‐Rode E, Coll KE. Williamson JR effects of branched chain alpha‐ketoacids on the metabolism of isolated rat liver cells. II. Interactions with gluconeogenesis and urea synthesis. J Biol Chem. 1979;254:11521‐11529. [PubMed] [Google Scholar]
- 22. Martin‐Requero A, Corkey BE, Cerdan S, Walajtys‐Rode E, Parrilla RL. Williamson JR interactions between α‐ketoisovalerate metabolism and the pathways of gluconeogenesis and urea synthesis in isolated hepatocytes. J Biol Chem. 1983;258:3673‐3681. [PubMed] [Google Scholar]
- 23. Brass EP. Effect of α‐ketobutyrate on palmitic acid and pyruvate metabolism in isolated rat hepatocytes. Biochim Biophys Acta. 1986;888:18‐24. doi: 10.1016/0167-4889(86)90065-0 [DOI] [PubMed] [Google Scholar]
- 24. Bergstrom T, Greter J, Levin AH, Steen G, Tryding N. Wass U Propionyl‐CoA carboxylase deficiency: case report, effect of low‐protein diet and identification of 3‐oxo‐2‐methylvaleric acid 3‐hydroxy‐2‐methylvaleric acid, and maleic acid in urine. Scand J Clin Lab Invest. 1981;41:117‐126. doi: 10.3109/00365518109092023 [DOI] [PubMed] [Google Scholar]
- 25. Jurecki E, Ueda K, Frazier D, et al. Nutrition management guideline for propionic acidemia: an evidence‐ and consensus‐based approach. Mol Genet Metab. 2019;126:341‐354. doi: 10.1016/j.ymgme.2019.02.007 [DOI] [PubMed] [Google Scholar]
- 26. Barshes NR, Vanatta JM, Patel AJ, et al. Evaluation and management of patients with propionic acidemia undergoing liver transplantation: a comprehensive review. Pediatr Transplant. 2006;10:773‐781. doi: 10.1111/j.1399-3046.2006.00569.x [DOI] [PubMed] [Google Scholar]
- 27. Quintero J, Molera C, Juamperez J, et al. The role of liver transplantation in propionic acidemia. Liver Transpl. 2018;24:1736‐1745. doi: 10.1002/lt.25344 [DOI] [PubMed] [Google Scholar]
- 28. Rela M, Battula N, Madanur M, et al. Auxiliary liver transplantation for propionic acidemia: a 10‐year follow‐up. Am J Transplant. 2007;7:2200‐2203. doi: 10.1111/j.1600-6143.2007.01899.x [DOI] [PubMed] [Google Scholar]
- 29. Kasahara M, Sakamoto S, Kanazawa H, et al. Living‐donor liver transplantation for propionic acidemia. Pediatr Transplant. 2012;16:230‐234. doi: 10.1111/j.1399-3046.2011.01607.x [DOI] [PubMed] [Google Scholar]
- 30. Vara R, Turner C, Mundy H, et al. Liver transplantation for propionic acidemia in children. Liver Transpl. 2011;17:661‐667. doi: 10.1002/lt.22279 [DOI] [PubMed] [Google Scholar]
- 31. Nagao M, Tanaka T, Morii M, Wakai S, Horikawa R. Kasahara M improved neurologic prognosis for a patient with propionic acidemia who received early living donor liver transplantation. Mol Genet Metab. 2013;108:25‐29. doi: 10.1016/j.ymgme.2012.10.022 [DOI] [PubMed] [Google Scholar]
- 32. Charbit‐Henrion F, Lacaille F, McKiernan P, et al. Early and late complications after liver transplantation for propionic acidemia in children: a two centers study. Am J Transplant. 2015;15:786‐791. doi: 10.1111/ajt.13027 [DOI] [PubMed] [Google Scholar]
- 33. Hofherr SE, Senac JS, Chen CY, Palmer DJ, Ng P. Barry MA short‐term rescue of neonatal lethality in a mouse model of propionic acidemia by gene therapy. Hum Gene Ther. 2009;20:169‐180. doi: 10.1089/hum.2008.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guenzel AJ, Collard R, Kraus JP, Matern D. Barry MA long‐term sex‐biased correction of circulating propionic acidemia disease markers by adeno‐associated virus vectors. Hum Gene Ther. 2015;26:153‐160. doi: 10.1089/hum.2014.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Collard R, Majtan T, Park I. Kraus JP import of TAT‐conjugated propionyl coenzyme a carboxylase using models of propionic acidemia. Mol Cell Biol. 2018;38:e00491‐e00417. doi: 10.1128/MCB.00491-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jiang L, Park JS, Yin L, et al. Dual mRNA therapy restores metabolic function in long‐term studies in mice with propionic acidemia. Nat Commun. 2020;11:5339. doi: 10.1038/s41467-020-19156-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Head PE, Myung S, Chen Y, et al. Aberrant methylmalonylation underlies methylmalonic acidemia and is attenuated by an engineered sirtuin. Sci Transl Med. 2022;14:eabn4772. doi: 10.1126/scitranslmed.abn4772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Armstrong AJ, Henke BR, Collado MS, et al. Identification of 2,2‐Dimethylbutanoic acid (HST5040), a clinical development candidate for the treatment of propionic Acidemia and Methylmalonic Acidemia. J Med Chem. 2021;64:5037‐5048. doi: 10.1021/acs.jmedchem.1c00124 [DOI] [PubMed] [Google Scholar]
- 39. Armstrong AJ, Collado MS, Henke BR, et al. A novel small molecule approach for the treatment of propionic and methylmalonic acidemias. Mol Genet Metab. 2021;133:71‐82. doi: 10.1016/j.ymgme.2021.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sharma LK, Yun MK, Subramanian C, et al. LipE guided discovery of isopropylphenyl pyridazines as pantothenate kinase modulators. Bioorg Med Chem. 2021;52:116504. doi: 10.1016/j.bmc.2021.116504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sharma LK, Subramanian C, Yun MK, et al. A therapeutic approach to pantothenate kinase associated neurodegeneration. Nat Commun. 2018;9:4399. doi: 10.1038/s41467-018-06703-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leonardi R, Zhang Y‐M, Yun M‐K, et al. Modulation of pantothenate kinase 3 activity by small molecules that interact with the substrate/allosteric regulatory domain. Chem Biol Drug des. 2010;17:892‐902. doi: 10.1016/j.chembiol.2010.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Frank MW, Subramanian C, Rock CO, Jackowski S. Quantification of coenzyme a in cells and tissues. J Vis Exp. 2019;151. doi: 10.3791/60182 [DOI] [PubMed] [Google Scholar]
- 44. Otwinowski Z. Minor W processing of X‐ray diffraction data collected in oscillation mode. Meth Enzymol. 1997;276:307‐326. doi: 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 45. McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32‐41. doi: 10.1107/S0907444906045975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Afonine PV, Grosse‐Kunstleve RW, Echols N, et al. Towards automated crystallographic structure refinement with phenix.Refine. Acta Crystallogr D Biol Crystallogr. 2012;68:352‐367. doi: 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Emsley P, Lohkamp B, Scott WG. Cowtan K features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486‐501. doi: 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen VB, Arendall WB 3rd, Headd JJ, et al. MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12‐21. doi: 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guenzel AJ, Hofherr SE, Hillestad M, et al. Generation of a hypomorphic model of propionic acidemia amenable to gene therapy testing. Mol Ther. 2013;21:1316‐1323. doi: 10.1038/mt.2013.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gallego‐Villar L, Rivera‐Barahona A, Cuevas‐Martin C, et al. In vivo evidence of mitochondrial dysfunction and altered redox homeostasis in a genetic mouse model of propionic acidemia: implications for the pathophysiology of this disorder. Free Radic Biol Med. 2016;96:1‐12. doi: 10.1016/j.freeradbiomed.2016.04.007 [DOI] [PubMed] [Google Scholar]
- 51. Rivera‐Barahona A, Alonso‐Barroso E, Perez B, Murphy MP, Richard E. Desviat LR treatment with antioxidants ameliorates oxidative damage in a mouse model of propionic acidemia. Mol Genet Metab. 2017;122:43‐50. doi: 10.1016/j.ymgme.2017.07.009 [DOI] [PubMed] [Google Scholar]
- 52. Rivera‐Barahona A, Fulgencio‐Covian A, Perez‐Cerda C, et al. Dysregulated miRNAs and their pathogenic implications for the neurometabolic disease propionic acidemia. Sci Rep. 2017;7:5727. doi: 10.1038/s41598-017-06420-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tamayo M, Fulgencio‐Covian A, Navarro‐Garcia JA, et al. Intracellular calcium mishandling leads to cardiac dysfunction and ventricular arrhythmias in a mouse model of propionic acidemia. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165586. doi: 10.1016/j.bbadis.2019.165586 [DOI] [PubMed] [Google Scholar]
- 54. Hitchcock SA. Pennington LD structure‐brain exposure relationships. J Med Chem. 2006;49:7559‐7583. doi: 10.1021/jm060642i [DOI] [PubMed] [Google Scholar]
- 55. Rankovic ZCNS. Drug design: balancing physicochemical properties for optimal brain exposure. J Med Chem. 2015;58:2584‐2608. doi: 10.1021/jm501535r [DOI] [PubMed] [Google Scholar]
- 56. Miller DJ, Zhang Y‐M, Subramanian C, Rock CO. White SW transcriptional regulation of membrane lipid homeostasis. Nat Struct Mol Biol. 2010;17:971‐975. doi: 10.1038/nsmb.1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Subramanian C, Yun MK, Yao J, et al. Allosteric regulation of mammalian pantothenate kinase. J Biol Chem. 2016;291:22302‐22314. doi: 10.1074/jbc.M116.748061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rock CO, Calder RB, Karim MA. Jackowski S Pantothenate kinase regulation of the intracellular concentration of coenzyme a. J Biol Chem. 2000;275:1377‐1383. doi: 10.1074/jbc.275.2.1377 [DOI] [PubMed] [Google Scholar]
- 59. Zhang Y‐M, Rock CO. Jackowski S feedback regulation of murine pantothenate kinase 3 by coenzyme a and coenzyme a thioesters. J Biol Chem. 2005;280:32594‐32601. doi: 10.1074/jbc.M506275200 [DOI] [PubMed] [Google Scholar]
- 60. Vishwakarma P. Lotspeich WD the excretion of I‐malic acid in relation to the tricarboxylic acid cycle in the kidney. J Clin Invest. 1959;38:414‐423. doi: 10.1172/JCI103816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Leonardi R, Rock CO. Jackowski S Pank1 deletion in leptin‐deficient mice reduces hyperglycaemia and hyperinsulinaemia and modifies global metabolism without affecting insulin resistance. Diabetologia. 2014;57:1466‐1475. doi: 10.1007/s00125-014-3245-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Brass EP. Overview of coenzyme a metabolism and its role in cellular toxicity. Chem Biol Interact. 1994;90:203‐214. [DOI] [PubMed] [Google Scholar]
- 63. Mitchell GA, Gauthier N, Lesimple A, Wang SP, Mamer O. Qureshi I hereditary and acquired diseases of acyl‐coenzyme a metabolism. Mol Genet Metab. 2008;94:4‐15. doi: 10.1016/j.ymgme.2007.12.005 [DOI] [PubMed] [Google Scholar]
- 64. Longo N, Sass JO, Jurecka A. Vockley J biomarkers for drug development in propionic and methylmalonic acidemias. J Inherit Metab Dis. 2022;45:132‐143. doi: 10.1002/jimd.12478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shi L. Tu BP acetyl‐CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol. 2015;33:125‐131. doi: 10.1016/j.ceb.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Martinez‐Reyes I. Chandel NS mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11:102. doi: 10.1038/s41467-019-13668-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gout I. Coenzyme a, protein CoAlation and redox regulation in mammalian cells. Biochem Soc Trans. 2018;46:721‐728. doi: 10.1042/BST20170506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kaelin WG Jr. McKnight SL influence of metabolism on epigenetics and disease. Cell. 2013;153:56‐69. doi: 10.1016/j.cell.2013.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chisolm DA. Weinmann AS connections between metabolism and epigenetics in programming cellular differentiation. Annu Rev Immunol. 2018;36:221‐246. doi: 10.1146/annurev-immunol-042617-053127 [DOI] [PubMed] [Google Scholar]
- 70. He W, Wang Y, Xie EJ, Barry MA. Zhang GF metabolic perturbations mediated by propionyl‐CoA accumulation in organs of mouse model of propionic acidemia. Mol Genet Metab. 2021;134:257‐266. doi: 10.1016/j.ymgme.2021.09.009 [DOI] [PubMed] [Google Scholar]
- 71. Dansie LE, Reeves S, Miller K, et al. Physiological roles of the pantothenate kinases. Biochem Soc Trans. 2014;42:1033‐1036. doi: 10.1042/BST20140096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Grunert SC, Mullerleile S, De Silva L, et al. Propionic acidemia: clinical course and outcome in 55 pediatric and adolescent patients. Orphanet J Rare Dis. 2013;8:6. doi: 10.1186/1750-1172-8-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. McCune SA, Durant PJ, Flanders LE. Harris RA inhibition of hepatic gluconeogenesis and lipogenesis by benzoic acid, p‐tert.‐butylbenzoic acid, and a structurally related hypolipidemic agent SC‐33459. Arch Biochem Biophys. 1982;214:124‐133. doi: 10.1016/0003-9861(82)90014-5 [DOI] [PubMed] [Google Scholar]
- 74. Silva MF, Aires CC, Luis PB, et al. Valproic acid metabolism and its effects on mitochondrial fatty acid oxidation: a review. J Inherit Metab Dis. 2008;31:205‐216. doi: 10.1007/s10545-008-0841-x [DOI] [PubMed] [Google Scholar]
- 75. Ponchaut S, van Hoof F, Veitch K. In vitro effects of valproate and valproate metabolites on mitochondrial oxidations. Relevance of CoA sequestration to the observed inhibitions. Biochem Pharmacol. 1992;43:2435‐2442. doi: 10.1016/0006-2952(92)90324-c [DOI] [PubMed] [Google Scholar]
- 76. Kesterson JW, Granneman GR. Machinist JM the hepatotoxicity of valproic acid and its metabolites in rats. I. Toxicologic, biochemical and histopathologic studies. Hepatology. 1984;4:1143‐1152. doi: 10.1002/hep.1840040609 [DOI] [PubMed] [Google Scholar]
- 77. Silva MF, Ruiter JP, Li J, et al. Synthesis and intramitochondrial levels of valproyl‐coenzyme a metabolites. Anal Biochem. 2001;290:60‐67. doi: 10.1006/abio.2000.4947 [DOI] [PubMed] [Google Scholar]
- 78. Yang H, Zhao C, Tang MC, et al. Inborn errors of mitochondrial acyl‐coenzyme a metabolism: acyl‐CoA biology meets the clinic. Mol Genet Metab. 2019;128:30‐44. doi: 10.1016/j.ymgme.2019.05.002 [DOI] [PubMed] [Google Scholar]
- 79. Yang H, Zhao C, Wang Y, Wang SP, Mitchell GA hereditary diseases of coenzyme a thioester metabolism. Biochem Soc Trans 2019;47:149–155. d 10.1042/BST20180423 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Data collection and Refinement Statistics
Table S2 BBP‐671 pharmacokinetics. Mean pharmacokinetic parameters of BBP‐671 in plasma following single intravenous or oral administration to male BALB/c mice a .
Table S3 Diet composition. (Envigo: TTD.170542)
Table S4 Selected Nutrient Information (Total kcal/g = 3.7)
Figure S1 Properties of BBP‐671. (A) A comparative summary of the sentinel pharmacokinetic properties of PZ‐28914, 41 PZ‐30227 and BBP‐671 (Table S2). (B) The mean values, preferred range and suggested limits on the chemical properties of the top 25 drugs used to treat neurological disease 54 , 55 compared to PZ‐2891, PZ‐3022 and BBP‐671 calculated using ChemDraw v21. PSA, polar surface area; HBD, hydrogen bond number; cLogP, partition coefficient between n‐octanol and water; MW, molecular weight.
Figure S2 Purity of BBP‐671. (A) Liquid chromatography‐mass spectrometric (LC–MS) analysis of BBP‐671 using evaporative light‐scattering detection (upper panel), ultraviolet detection (middle panel) or total LC–MS ion current (lower panel). (B) Mass spectrum of BBP‐671.
Figure S3 1H‐ and 13C‐NMR spectra of BBP‐671. (A) The proton NMR spectrum of BBP‐671. (B) The carbon‐13 NMR spectrum of BBP‐671.
Figure S4: PKPD of BBP‐671 in the forebrain. (A) BBP‐671 was administered at 1 mg/kg to C57BL/6J male mice (three mice per time point) by gavage and forebrain tissue was taken at indicated times (see Figure 2B). (B) Chow was formulated with 11.25, 22.5, or 45 ppm BBP‐671, and C57BL/6J male mice (five mice in each group) were maintained on the diets for 4 weeks, and BBP‐671 levels, and total CoA was determined in the forebrain. The data are mean and SE.
Figure S5 Total CoA and BBP‐671 levels. (A) Liver total CoA in wild‐type littermate controls was measured and compared to CoA in the liver of BBP‐671‐treated and untreated PA mice. (B) Plasma and liver BBP‐671 concentrations. There were 3 males (blue dots) and 3 females (red dots) in each group and the mice were maintained on chow formulated either with or without 22.5 ppm BBP‐671 as indicated. The data are mean ± SE.
Data Availability Statement
The data used in the preparation of the graphs is available from the corresponding author on reasonable request. The PANK3•ATP•Mg2+•BBP‐671 complex structure is deposited in the Protein Data Bank (PDBID: 7UE8).