

Abstract
Dysfunction of the p53 gene and the presence of the MDR1 gene are associated with many malignant tumors including endometrial cancer and are responsible for cancer therapeutic resistance and poor survival. Thus, there is a critical need to devise novel combinatorial therapies with multiple mechanisms of action to overcome drug resistance. Here, we report a new ciprofloxacin derivative (CIP2b) tested either alone or in combination with taxanes against four human endometrial cancer cell lines. In vitro studies revealed that a combination of paclitaxel + CIP2b had synergistic cytotoxic effects against MDR1-expressing type-II human endometrial cancer cells with loss-of-function p53 (Hec50co LOFp53). Enhanced antitumor effects were confirmed by substantial increases in caspase-3 expression, cell population shifts toward the G2/M phase, and reduction of cdc2 phosphorylation. It was found that CIP2b targets multiple pathways including the inhibition of MDR1, topoisomerase I, and topoisomerase II, as well as enhancing the effects of paclitaxel (PTX) on microtubule assembly. In vivo treatment with the combination of PTX + CIP2b also led to significantly increased accumulation of PTX in tumors (compared to CIP2b alone) and reduction in tumor growth. Enhanced in vivo cytotoxic effects were confirmed by histological and immunohistochemical examination of the tumor tissues. Complete blood count and blood biochemistry data confirmed the absence of any apparent off-target toxicity. Thus, combination therapy with PTX and CIP2b targeted multiple pathways and represents an approach that could result in improved tolerance and efficacy in patients with type-II endometrial cancer harboring the MDR1 gene and p53 mutations.
Keywords: MDR1 and p53 genes, Topoisomerases, Microtubules, Drug resistance, Drug accumulation, Molecular combinatorial therapy
1. Introduction
Endometrial cancer is a heterogeneous group of tumors with variable clinical behavior that influences survival [1, 2]. In contrast to most other types of cancer, the incidence and mortality rates among women with endometrial cancer are rising in the United States [3–5]. Based on the clinical features and pathogenesis, endometrial cancers have been conventionally classified into estrogen-dependent type I and estrogen-independent type II endometrial carcinomas [6]. However, the current clinical practice is moving towards classifying endometrial cancer into four molecular subtypes using the ProMisE (Proactive Molecular Risk Classifier for Endometrial Cancer) algorithm: (i) mismatch repair (MMR) deficiency, (ii) p53 mutation status, (iii) polymerase-E exonuclease domain (POLE) mutation status, and (iv) endometrial carcinomas with no specific molecular alteration [7]. Among these subtypes, patients with abnormalities in the tumor suppressor p53 gene have by far the worst overall prognosis and response to chemotherapy, which is the current standard therapy for aggressive endometrial tumors, and have the shortest survival [8, 9]. Dysfunction of p53 acquires oncogenic properties, such as metastasis, invasion, increased proliferation, and cell survival, and is associated with chemo‐ and radio-resistance which results in poor survival in endometrial cancer patients [10]. Mutant p53 proteins are detected in more than 90% of type II endometrial cancer whereas they are only found in 10–40% of type I endometrial cancer which is associated with a favorable prognosis and clinical outcomes [11]. Further, the standard chemotherapeutic regimen of paclitaxel (PTX) + carboplatin has multiple undesirable side effects (e.g., myelosuppression and peripheral neuropathy) that question the justification of its use in the clinic [12]. To reverse the alarming trend of increasing mortality in endometrial cancer, there is a critical need to devise novel combinatorial treatment strategies that enhance the efficacy of standard chemotherapy while simultaneously mitigating adverse events.
Taxanes (e.g., PTX) are a class of antimitotic cancer drugs that stabilize the microtubule dynamic through binding to the β-subunit of the tubulin heterodimer, thereby promoting microtubule assembly (polymerization) and inhibiting subsequent disassembly (depolymerization) [13]. Such alteration in microtubule dynamics prevents mitosis and induces apoptosis. Despite the ability of taxanes to treat multiple types of cancer, resistance is nevertheless one of the major reasons that contribute to high failure rates and relapses in cancer patients [14, 15]. Several approaches have been described in the literature aimed at re-sensitizing cancer cells to this class of chemotherapeutic agents. One approach is to target multidrug resistance protein-1 (MDR1), which is overexpressed in approximately 50% of cancer patients. Additionally, reports confirm a correlation between p53-deficiency and MDR1 overexpression [16, 17], which further aggravates drug resistance. Notably, the use of MDR1 inhibitors potentiates the efficacy of PTX by increasing its intracellular accumulation and improving its pharmacokinetics [18–20]. This leads to improved sensitivity of cancer cells to chemotherapeutics. For example, combined treatment with PTX and the MDR1 inhibitor, tariquidar, reversed chemoresistance in human ovarian cancer cells in vitro and in vivo [21]. Other approaches to re-sensitize cancer cells toward taxanes include targeting DNA Topoisomerases I and II (Topo I and II), which govern the topological structure of DNA during transcription, mitosis, and repair [22–24], and targeting microtubule assembly. Inhibitors of Topo I and II, such as camptothecin (CAM) analogs (e.g. irinotecan, Camptostar®) and etoposide (ETO, Vepesid®), respectively, are FDA-approved chemotherapeutics effective in treating several types of cancers and have been investigated to treat gynecological cancers, either alone or in combination with taxanes and other chemotherapeutics [25–28]. However, the side effects of these currently used inhibitors limit their widespread use in combinatorial therapies [29, 30]. Alternatively, drugs that stabilize microtubule assembly by promoting tubulin polymerization may synergize with PTX, a wellknown microtubule stabilizer, and augment its anticancer activity, especially when both agents bind to distinct tubulin-binding sites [31].
Although progress has been made in our understanding of the mechanisms that support chemoresistance, there is a lack of therapies aimed at collectively targeting these resistance pathways. Ideally, when combined with targeting MDR1, Topo I and II, and microtubule assembly, treatment with PTX should be more effective than if only one pathway is targeted. Moreover, using a cocktail of more than two drugs can be problematic due to the increased risk of toxic side effects. For example, Folfirinox, which is a cocktail of four drugs (leucovorin, fluorouracil, irinotecan, and oxaliplatin) has been used to treat pancreatic and colorectal cancers, but it is associated with increased toxicity such as febrile neutropenia [32, 33]. The potential antitumor effects of quinolones, such as ciprofloxacin (CIP, a well-tolerated antibacterial agent), have been recently uncovered and related to their ability to inhibit DNA replication and induce apoptosis. However, the cytotoxic activity against tumors was achieved at relatively high concentrations [34]. Therefore, chemical modifications are needed to tune and shift the activity from antimicrobial to antitumoral effects. Previous studies on quinolone antibiotics showed that N-substitutions can alter the biological profile, shifting it from antibacterial to tumoricidal [35–37]. In this current article, we identify and investigate a 7-(4-(N-substituted carbamoyl methyl) piperazin-1-yl) CIP derivative (CIP2b) (Fig. S1), revealing that it possesses anticancer activity; thus, potentially overcoming some of the limitations of taxanes while demonstrating no noticeable toxicity. We further describe the biological basis for the efficacy of CIP2b and the synergistic effect with PTX, which stems from enhanced cellular accumulation due to blocking the MDR pathway, inhibition of DNA replication, and stabilization of microtubule assembly (Scheme 1). When tested against four different human endometrial cancer cell lines, this derivative was also capable of synergizing with PTX to augment its efficacy by inducing cell death in vitro. Moreover, our study showed that CIP2b is safe and effective and synergizes with PTX to halt the progression of type II human endometrial cancer Hec50co cells with a loss-of-function mutation in p53 transcript (Hec50co LOFp53) in vivo. These findings hold the promise of a combined therapeutic strategy to treat type II endometrial cancer.
Scheme 1.
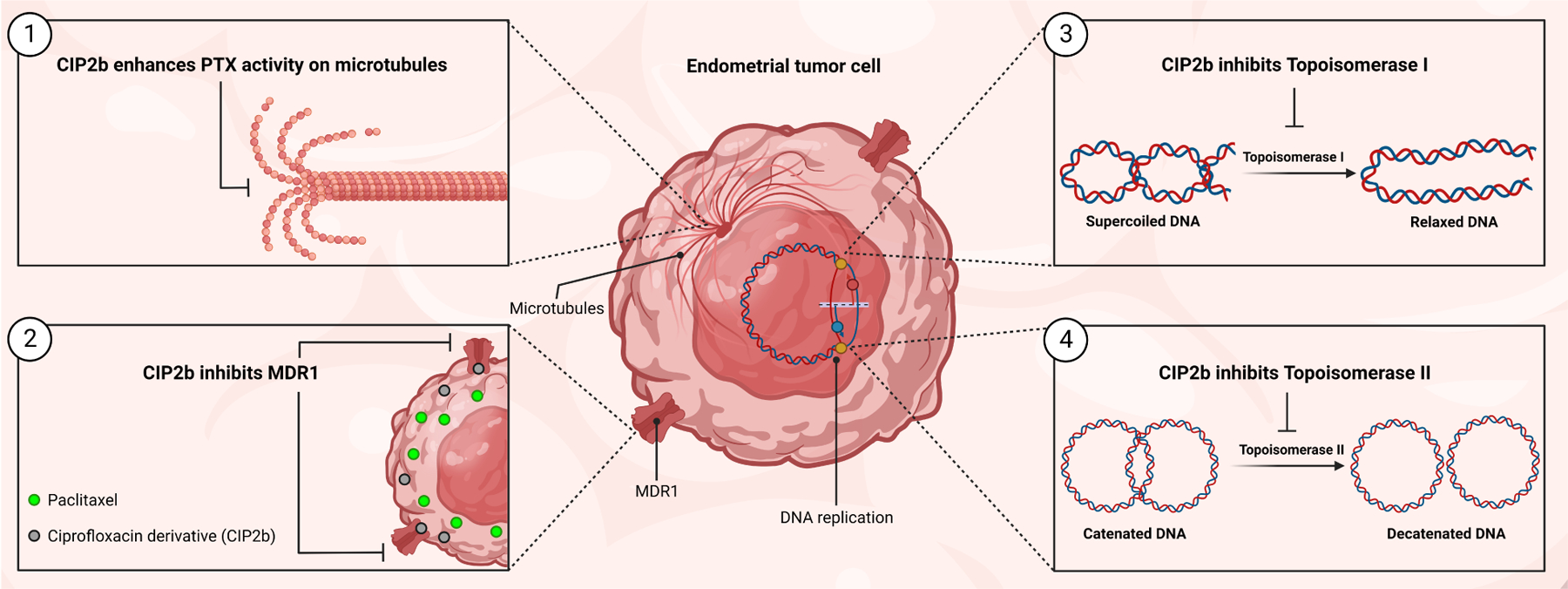
Schematic illustration of the four mechanisms of actions of the combinatorial therapy of PTX + CIP2b.
2. Materials and Methods
2.1. Materials
CAM (Cat. No. 208925), dimethyl sulfoxide (DMSO, Cat. No. D2438), DNase I from bovine pancreas (Cat. No. DN25), ETO (Cat. No. 341205), 7-hydroxy flavone (Cat. No. H4530), 2-mercaptoethanol (Cat. No. M6250), and anti-β-Actin mouse monoclonal antibody (Cat. No. A1978) were purchased from Sigma Aldrich (St. Louis, MO). Ciprofloxacin (Cat. No. 449620050), NP-40 Surfact-Amps™ 10% Detergent Solution (Cat. No. 28324), Collagenase Type IV (Cat. No. 17104019), Oregon Green™ 488 PTX (PTX-OG, Cat. No. P22310), propidium iodide 1 mg/mL (Cat. No. P3566), RNase A (DNase and protease-free) 10 mg/mL (Cat. No. EN0531), Tween-80 (Cat. No. BP338-500), UltraPure™ Ethidium Bromide 10 mg/mL (Cat. No. 15585011), 1X live cell imaging solution (Cat. No. A14291DJ), 4-well chambered coverglass with non-removable wells (Cat. No. 155383), and PageRuler Plus Prestained Protein Ladder 10–250 kDa (Cat. No. 26619) were purchased from ThermoFisher Scientific (Waltham, MA). Human Topoisomerase I (Cat. No. TG1015-1A) and II (Cat. No. TG1001-1A) assay kits were purchased from TopoGEN Inc. (Buena Vista, CO) while fluorescence-based tubulin polymerization assay kit (Cat. No. BK011P) and multiplex activity assay fluorometric kit (Cat. No. ab219915) were purchased from Cytoskeleton Inc. (Denver, CO) and Abcam (Waltham, MA), respectively. The CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) (Cat. No. G3581) was purchased from Promega (Madison, WI). Disruption beads, 2.3mm Zirconia/Silica Beads (Cat. No. 11079125z) were purchased from BioSpec (Bartlesville, OK). Paclitaxel (Cat. No. P-9600) and docetaxel (Cat. No. D-1000) powders were purchased from LC Laboratories (Woburn, MA) while paclitaxel solution (6 mg/mL, Cat. No. 70860-200-05) was purchased from Athenex, Inc. (Buffalo, NY). Anti-rabbit IgG, HRP-linked Antibody (Cat. No. 7074), cdc2 antibody (Cat. No. 77055), phospho-cdc2 (Tyr15) (10A11) Rabbit mAb (Cat. No. 4539), cdc25C (5H9) Rabbit mAb (Cat. No. 4688), 20X LumiGLO Reagent and 20X Peroxide (Cat. No. 7003) were purchased from Cell Signaling Technology (Danvers, MA). Albumin (BSA), bovine fraction V (Cat. No. A30075), and Tween 20 (Cat. No. P20370) were obtained from Research Products International (Mount Prospect, IL) while RIPA Lysis Buffer (Cat. No. sc-24948) and m-IgGκ BP-HRP antibody (Cat. No. sc-516102) were purchased from Santa Cruz Biotechnology (Dallas, TX). Trans-Blot Turbo Mini 0.2 μm Nitrocellulose Transfer Packs (Cat. No. 1704158), 4–20% Mini-PROTEAN TGX Stain-Free Protein Gels (Cat. No. 4568094), 10X Tris/Glycine Buffer (Cat. No. 1610734), and 10X Tris Buffered Saline (TBS) (Cat. No. 1706435) were purchased from Bio-Rad Laboratories (Hercules, CA). All other reagents, buffers, and solvents were at least of analytical grade and were used as received without further purification.
2.2. Synthesis and Characterization of CIP2b
Bromoacetyl bromide (BrCOCH2Br) dissolved in methylene chloride (30 mL) was added gradually to a stirred mixture of 4-chloroaniline (Cl-C6H4-NH2) dissolved in methylene chloride (20 mL), 1.3 mM potassium carbonate, and distilled water (100 mL). The mixture was stirred in a cold room (4°C) for approximately 2 hours, and this was followed by stirring at room temperature overnight. The organic layer was separated, washed with distilled water, and dried under vacuum to obtain the intermediate (Cl-C6H4-NH-CO-CH2-Br). Finally, 0.04 M triethanolamine was added to a mixture of CIP and intermediate (1:1) in 50 mL acetonitrile. The mixture was then heated under reflux for 5 hours. Next, the precipitate was filtered, washed with acetonitrile, and dried under a vacuum to obtain CIP2b. To verify the purity and characterize CIP2b, the obtained dry powder was examined using 1H NMR (Bruker Avance NEO-500, 500 MHZ) and mass spectrometry (Q Exactive Orbitrap Mass Spectrometer, Thermo Scientific). In 1H NMR and mass spectrometry analyses, CIP2b was dissolved in DMSO-d6 and acetonitrile, respectively. PTX and PTX + CIP2b (1:1) samples were also examined using 1H NMR to observe any changes in chemical shifts of both drugs before and after being combined.
2.3. Cell Lines and Cell Culturing Protocol
In this study, six different cancer cell lines were used, four of which were human endometrial cancer cell lines bearing different p53 mutations. The first cell line was the Ishikawa-H cell line, which was derived from a moderately differentiated type I human endometrioid adenocarcinoma and contained a wild-type p53 transcript (Ishikawa-H WTp53). The second cell line was the Hec50co cell line with a loss-of-function mutation in p53 transcript (Hec50co LOFp53), which was derived from poorly differentiated aggressive type-II adenocarcinoma with papillary serous sub-differentiation and carried a null p53 allele. The third cell line was the Hec50co cell line with a gain-of-function mutation in p53 R175H transcript (Hec50co GOFp53), which was a lab-derived from Hec50co cells transfected with a construct containing R175H-p53 using Lipofectamine 2000. The fourth cell line was KLE gain-of-function TP53 mutants, R175H (KLE GOFp53, purchased from ATCC, Manassas, VA), which was derived from a poorly differentiated endometrial carcinoma and subverts the tumor suppressor function of p53 into an oncomorphic p53 mutation (i.e., confers oncogenic activity). Ishikawa-H WTp53 and Hec50co LOFp53 cells were cultured in Dulbecco’s modified Eagle’s medium 1X (DMEM, Cat. No. 11965-092) supplemented with 1% penicillin/streptomycin (Pen/Strep, 100 U/mL, Cat. No. 15140122), 10% fetal bovine serum (FBS, Cat. No. S11150), and 0.01% sodium pyruvate (Cat. No. 11360-070) (ThermoFisher Scientific). Hec50co GOFp53 cells were grown in the same medium plus 0.08% geneticin, selective antibiotic (G418 Sulfate 50 mg/mL, Cat. No. 10131035, ThermoFisher Scientific), to main stable p53 R175H expression. KLE GOFp53 cells were passaged and expanded in Roswell Park Memorial Institute 1640 medium (RPMI 1640, Cat. No. 11875093, ThermoFisher Scientific) supplemented with 1% Pen/Strep and 10% FBS. Besides the four human EC type I and II cell lines, Lewis lung carcinoma-porcine kidney 1 cell lines, LLC-PK1-WT (wild-type), and LLC-PK1-MDR1 (overexpressing Multidrug Resistance Mutation 1 gene) were used in this study. These cell lines were cultured in DMEM supplemented with 1% Pen/Strep, 10% FBS, and 0.01% sodium pyruvate. For the trypsinization process, 0.25% Trypsin-EDTA 1X (Cat. No. 25200-056, ThermoFisher Scientific) was used. All cells were incubated and maintained in a Galaxy 170S CO2 humidified cell culture incubator (Eppendorf, Germany) set at 37°C under a 5% CO2 flow.
2.4. Cell Viability Assay Using Endometrial Cancer Cell Lines
Single treatments of PTX, CIP2b, and CIP (the parent compound) or a combination of PTX with CIP2b or PTX with CIP were tested against four different endometrial cancer cell lines. Briefly, Ishikawa-H WTp53, Hec50co LOFp53, and Hec50co GOFp53 cells were seeded at a density of 1500 cells per well in 96-well tissue culture plates (CELLTREAT Scientific Product, Pepperell, MA) and incubated at 37°C with 5% CO2 for in a complete medium. Since KLE are very slow-growing cells, they were seeded at a density of 6000 cells per well in 96-well plates. After 24 hours, the medium was removed and the cells were treated with either PTX alone at a concentration of 0.1, 1, 10, 20, 50, and 100 nM, or CIP2b alone at a concentration of 0.1, 1, 10, 20, 50, and 100 μM, or CIP alone at a concentration of 0.1, 1, 10, 20, 50, and 100 μM. CIP2b has poor water solubility (22.7 μg/mL) whereas its solubility in DMSO is > 1000-fold higher (28.1 mg/mL). Therefore, stock solutions of CIP2b and all other compounds were prepared in DMSO and then diluted in a complete medium. These treatments were added either as monotherapies or in the indicated combinations. After incubation with the treatments for 72 hours, the complete medium was removed and the MTS reagent mixed with the complete medium was added to the wells and incubated for approximately 3 hours. Finally, the absorbance was recorded at 490 nm using a Spectra Max plus 384 Microplate Spectrophotometer (Molecular Devices, Sunnyvale, CA). Percentage cell viability was expressed as the average absorbance of the test group relative to that of the control group (untreated cells). The contribution of plain medium with no cells to the absorbance value was also taken into the consideration by measuring the absorbance of cell-free culture media mixed with MTS reagent and subtracting the absorbance value from those of the treated wells (containing cells). Similarly, the cell viability assay was repeated to test other taxane compounds (docetaxel, DOC) either alone or in combination with either CIP2b or CIP against four different endometrial cancer cell lines. In this experiment, DOC was used with a concentration of 0.1, 1, 10, 20, 50, and 100 nM.
2.5. IC50 Values and Combination Index
The concentration that inhibited cell proliferation by 50% relative to the control (IC50) was calculated using GraphPad Prism (La Jolla, CA). Nonlinear regression curve fitting was performed to estimate the IC50 values. Synergism was detected following the Chou-Talalay method (CI < 1 indicates synergism, CI = 1 indicates an additive effect, and CI > 1 indicates antagonism) using CompuSyn software (Ver. 1.0).
2.6. Cell Cycle Assay Using Endometrial Cancer Cell Lines
To investigate the effect of the drug combination on the cell cycle of four different endometrial cancer cell lines, Ishikawa-H WTp53, Hec50co LOFp53, and Hec50co GOFp53 cells were seeded at 200 × 103 cells per well in 6-well plates while KLE GOFp53 cell cells were seeded at 800 × 103 for 24 hours in the complete medium. The medium was then removed and replaced with PTX (1, 5, and 10 nM), CIP2b (10 μM), or combinations of PTX with CIP2b, or with a fresh complete medium (untreated cells). Cells were incubated with the treatments for 24 hours, then the medium was removed, and the cells were washed twice with Dulbecco’s phosphate-buffered saline (DPBS 1X, ThermoFisher Scientific), trypsinized (0.3 mL/well), and collected by centrifugation (230 xg, 5 minutes). Cells were fixed using ice-cold 70% ethanol and stored at 4°C for 30 minutes, then incubated with 100 μg/mL RNase A in 1% v/v NP-40 Surfact-Amps® solution (0.5 mL/sample) for 30 minutes at 37°C. Propidium iodide (50 μg/mL, 0.5 mL/sample) was added prior to analysis by flow cytometry (FACScan, BD Biosciences) using CellQuest software (Ver. 3.3). The data were further analyzed by ModFit LT (Ver. 5.0) to calculate the percentage of cells in each phase of the cell cycle (i.e., G0/G1, S, G2/M).
2.7. Sequential versus Concurrent Treatment
Optimal combinatorial drug delivery in the clinic is controversial, and its application for cancer therapy is based on multiple factors such as cancer type and treatment used. Therefore, in this experiment, to better understand the dosing strategy, concurrent and sequential administration of PTX and CIP2b were assessed. To test concurrent and sequential administration of PTX and CIP2b, Hec50co LOFp53 cells were seeded at 1500 cells/well for 24 hours, as described in the cell viability assay. For sequential administration, the first treatment was added, and 4 hours later the second treatment was added. For the concurrent administration, both treatments were given at the same time. After approximately 72 hours of incubation, the medium was removed, and the MTS reagent mixed with the medium was added. Plates were incubated for about 3 hours, and the absorbance was recorded at 490 nm using a Spectra Max plus 384 Microplate Spectrophotometer. Percentage cell viability was expressed as the average absorbance of the test group relative to that of the control group (untreated cells). Similarly, IC50 was calculated using GraphPad Prism software.
2.8. Intracellular Accumulation of PTX Using Flow Cytometer
To evaluate the cellular accumulation of PTX in the presence or absence of CIP2b, Hec50co LOFp53 cells were seeded at a density of 200,000 cells per well in 6-well plates in a complete medium and incubated for 24 hours. Then, the medium was removed and a DMSO-based solution of fluorescently labeled PTX (PTX-OG, 0.1 mg/mL) was added at a concentration of 400 nM per well, in a final volume of 2 mL medium. The fluorescent label (Oregon Green™ 488) is attached to the 7-carbon of the paclitaxel, a strategy that permits selective binding of the probe to microtubules. CIP2b (in DMSO) was added at 4 or 40 μM, either alone or in combination with PTX-OG. After 2 hours, the medium was removed, and the cells were washed thoroughly with 1X DPBS, trypsinized, and collected by centrifugation (230 xg, 5 minutes). Cells were analyzed by flow cytometry (FACScan, Becton Dickinson, Franklin Lakes, NJ).
2.9. Intracellular Accumulation of PTX Using Confocal Microscopy
Intracellular accumulation of PTX co-delivered with CIP2b was also studied qualitatively in Hec50co LOFp53 cells using confocal microscopy. Hec50co LOFp53 cells were seeded in 4-well chambered coverglass with non-removable wells at a density of 3 × 104 cells/well in a volume of 0.8 mL complete medium and incubated for 24 hours. The medium was carefully aspirated, and cells were incubated with different treatments (400 nM PTX-OG, 40 μM CIP2b, or a combination of both compounds) for 2 hours. The medium was removed, and this was followed by washing twice with 1X Live Cell Imaging Solution (Cat. No. A14291DJ, ThermoFisher Scientific). Samples were visualized using Leica TCS SP8 STED confocal laser scanning microscope (Leica Microsystems Inc., Buffalo Grove, IL), and the confocal images were processed using the ImageJ-based Fiji software.
2.10. Molecular Docking Simulation
Three-dimensional (3D) crystallographic structures were retrieved from the RCSB Protein Data Bank (PDB) database (Table S2). Targets included: human Topo I (PDB ID: 1T8I), Topo II (PDB ID: 5GWK), MDR1 (PDB ID: 6QEX), and αβ-tubulin (PDB ID: 1JFF). Water molecules, bound antibodies, unnecessary ions, and other hetero atoms were removed from the structures using BIOVIA Discovery Studio Visualizer v20.1.0.19295 (Accelrys) program. The two-dimensional (2D) structure of the ligand CIP2b was drawn in ChemDraw (version 20) and converted to a 3D structure by Discovery Studio Visualizer. Energy minimization of the ligand was conducted by the Merck molecular force field 94 (MMFF94), and Gasteiger was the charge calculation method. Rigid docking was chosen by converting all rotatable bonds of the ligand to non-rotatable to minimize standard errors coming from the rotatable bonds during the docking simulation. The binding site was analyzed in Discovery Studio Visualizer for targeted site-specific docking and grid box parameters were obtained covering the target binding site. Missing residues, polar hydrogens, Kollman charges, and Gasteiger charges were added to the protein structures using AutoDock Tools (ADT), a free graphical user interface of MGL software packages v1.5.7rc1. Molecular docking simulation was conducted using AutoDock Vina (version 1.1.2). Lamarckian Genetic Algorithm (LGA) was used to find the greatest conformational space for the ligand with a population of 150 individuals. The maximum number of generations and evaluations was set at 27,000 and 2,500,000, respectively. The best-docked pose was selected based on the lowest binding energy. Simulation results were analyzed by Discovery Studio Visualizer and PyMOL version 2.3.
2.11. MDR1 Efflux Transporter
Overexpression of the MDR1 gene, reflected in the abundance of the MDR1 efflux pump, is one of the major reasons for PTX chemoresistance in cancer. In this experiment, LLC-PK1-WT and LLC-PK1-MDR1 cell lines were used. To compare the activity of the drug combination using LLC-PK1-WT and LLC-PK1-MDR1 cells, PTX intracellular accumulation was measured, as described above (PTX-OG). This was also followed by a cell viability assay (MTS) to assess the cytotoxic effects of the drug combination against LLC-PK1-WT and LLC-PK1-MDR1 cell lines, as described above.
2.12. Topoisomerase I Inhibition Assay
A plasmid-based Topo I assay was performed according to the manufacturer’s instructions. Briefly, 1 μL of the test compounds (PTX, CIP, CIP2b, or CAM) at 2 mM, was mixed with 2 μL of the reaction buffer (10X tris-glycine-SDS), and deionized water was added to make an 18 μL solution. This was followed by adding 1 μL of supercoiled DNA (scDNA) and 1 μL of human Topo I (10 U/μL) (final drug concentration was 100 μM). Also, scDNA ± Topo I was used as a control (i.e., not containing drugs). In addition, relaxed DNA was added as a control. Samples were incubated on a heating block (Isotemp 125D Digital Heat Block, ThermoFisher Scientific) set at 37°C for 30 minutes. The reaction was stopped by adding a 5X stop buffer. Samples were then loaded directly into the wells of a 1% agarose gel (Research Products International). The gel was run at 150 V until the dye front had traveled approximately 5 cm (run time was approximately 2 hours). The gel was stained with 1 μg/mL ethidium bromide in deionized water for 15 minutes, followed by de-staining (washing) with deionized water for 30 minutes before imaging. The image was taken using an Invitrogen iBright CL 1500 Imaging System (ThermoFisher Scientific). The intensity of the final bands in the gel was quantified using ImageJ, and the results are expressed as the percentage of Topo I inhibition.
2.13. Topoisomerase II Inhibition Assay
A plasmid-based Topo II assay was performed according to the manufacturer’s instructions. In this experiment, 1 μL of tested compounds (PTX, CIP, CIP2b, ETO) at 2 mM was mixed with 4 μL of the reaction buffer (mixture of buffer A (0.5 M Tris-HCl (pH 8), 1.50 M NaCl, 100 mM MgCl2, 5 mM dithiothreitol, 300 μg BSA/mL) and buffer B (10X ATP Buffer), and deionized water was added to make 18 μL solution. This was followed by adding 1 μL of catenated DNA (ctDNA) and 1 μL of human Topo IIα (8 U/μL) (final drug concentration was 100 μM). Additionally, decatenated DNA (de-ctDNA), ctDNA ±Topo II and relaxed DNA were used as controls (i.e., not containing drugs). Reactions were carried out, and samples were run on gels, processed, and imaged as in the Topo I assay. Results are expressed as the percentage of Topo II inhibition.
2.14. Tubulin Polymerization Assay
To test tubulin activity, a fluorescence-based tubulin polymerization assay was performed according to the manufacturer’s instructions. Briefly, stock solutions of PTX and CIP2b (in DMSO) were diluted with UltraPure™ water to make a final concentration of 3 μM. In addition, a 75 μM CaCl2 aqueous solution was used as a control in this experiment (to inhibit tubulin polymerization). Before starting the experiment, the 96-well plate and fluorimeter (SpectraMax M5 Microplate Reader) were pre-warmed to 37°C. The fluorimeter was set up for kinetic measurements every minute for 1 hour at excitation 360 nm and emission 450 nm. Samples and tubulin were added to a 96-well plate, and the plate was inserted into the plate reader. Kinetic measurements started immediately, and data were collected and analyzed.
2.15. Apoptosis and Caspase Activity
The expression of caspase 3, 8, and 9 was measured using a Multiplex Activity Assay Fluorometric Kit. Briefly, Hec50co cells were plated in 96-well plates at a density of 3000 cells/well in 100 μL of complete medium and incubated for 24 hours. After incubation, the medium was removed, and different drug concentrations either alone or in combination were added (PTX 5 and 10 nM; CIP2b 10, 25, and 50 μM). Cells were treated for 24 or 72 hours. Caspase kit reagents were added to the cells according to the manufacturer’s protocol and incubated at room temperature for 60 minutes. Fluorescence intensity (at excitation/emission values of 535/620 nm, 490/525 nm, and 370/450 nm for caspase-3, −8, and −9, respectively) was then measured using a SpectraMax M5 microplate reader (Molecular Devices).
2.16. Western Blotting Analysis
Hec50co LOFp53 cells were seeded at a density of 2 × 106/10 mL complete medium in Petri dishes and incubated at 37°C and 5% CO2 for 24 hours. Then, treatments were added (PTX or CIP2b or a combination of both), and the dishes were again incubated for another 24 hours. Next, dishes were placed on ice, and the medium was aspirated. Cells were then washed with ice-cold 1X DPBS. This was followed by adding an ice-cold RIPA Lysis Buffer (1 mL/Petri dish), and the cells were scraped off using a pre-cooled plastic cell scraper. Then, cell suspensions were gently transferred into pre-cooled 1.5 mL microcentrifuge Eppendorf tubes and incubated on ice for 30 minutes. After incubation, samples were spined down at 16,000 xg for 20 minutes at 4°C. Supernatants were collected in fresh pre-cooled 1.5 mL microcentrifuge Eppendorf tubes and stored at −20°C until analysis. Protein content in the samples was measured using Micro BCA™ Protein Assay Kit (Cat. No. 23235, ThermoFisher Scientific), as per the manufacturer’s instructions. For the Western Blotting staining protocol, 30 μg total proteins from each sample were loaded after being mixed with 6X Laemmli Sample Buffer (Tris-HCl 375 mM, 6% SDS,50% glycerol,0.045% bromphenol blue and 2.1M 2-mercaptoethanol).
2.17. Animals for in vivo Experiments
For experiments involving the human endometrial cancer xenograft model (Hec50co LOFp53), female athymic Nu/Nu mice (6–8 weeks, Charles River, Wilmington, MA) were used. For other experiments (e.g., pharmacokinetics, biodistribution, safety), female and/or male BALB/c mice (6–8 weeks, Jackson Laboratory, Bar Harbor, ME) were used. Male and female mice were separated into separate cages with no more than 5 mice per cage. Mice were kept under controlled temperature (23 ± 2°C) at the University of Iowa animal care facility. Mice were also monitored closely for any signs of distress or discomfort. Food was provided ad libitum, and mice were exposed to 12 hours of light/dark cycles. All animal experiments performed were approved by the University of Iowa Institutional Animal Care and Use Committee (IACUC). When anesthesia was needed, mice were injected IP with the ketamine-xylazine mixture (87.5 mg/kg ketamine and 12.5 mg/kg xylazine) prior to performing the experiment.
2.18. In vivo Antitumor Efficacy Study Using a Human Endometrial Cancer Xenograft Model
Female athymic Nu/Nu mice were subcutaneously challenged with Hec50co cells (1 × 106 cells/mouse). Cells were suspended in ice-cold serum-free 1X DMEM at a concentration of 1 × 107 cells/mL. Mice were then monitored for tumor growth. Tumor dimensions (length, width, and height) were measured using a digital caliper. When mice had palpable tumors (152 mm ±68 mm), they were randomly distributed into 3 treatment groups: DPBS (n=3), PTX (n=4), and PTX+CIP2b (n=4). Treatments were administered SC near the tumor site (i.e., peritumorally) in five doses on days 26, 30, 33, 37, and 40 post-tumor challenge. Each dose consisted of 200 μL of either PBS, 5 mg/kg PTX (Paclitaxel concentrate diluted in DPBS), or 5 mg/kg PTX + 5 mg/kg CIP2b (CIP2b in DPBS:Tween-80:ethanol 85:10:5). For CIP2b formulation, it was initially dissolved in a mixture of tween-80 and ethanol to prepare drug concentrate, and this was followed by dilution with DPBS prior to injection – the same concept was used in Taxotere, an injectable marketed product for DOC [38]. Body weights and tumor volumes (using Equation 1) were monitored twice a week for up to 2 weeks after the last dose (i.e., up to day 53 post-tumor challenge). On the last day of the study (day 53 post-tumor challenge), mice were euthanized, tumors were collected, and their weights were measured.
| Equation 1: |
2.19. In vivo PTX Accumulation in Xenograft Tumors
In this study, athymic Nu/Nu mice were challenged subcutaneously (SC) with 1 × 106 Hec50co tumor cells per mouse on day zero of the experiment. When the tumor grew to a palpable size range of 100–150 mm3, as measured by Equation 1, treatments were administered SC at the tumor site (peritumorally). Treatment groups (n = 3 per group) included: (i) DPBS, (ii) 0.1 mg/kg PTX-OG, and (iii) 0.1 mg/kg PTX-OG + 5 mg/kg CIP2b. After 24 hours, mice were euthanized, and tumors were collected and submerged in Hank’s Balanced Salt Solution (HBSS) with calcium and magnesium. Tumors were then taken out of HBSS (one at a time to maintain cell viability), placed on a Petri dish, and mechanically minced with a sterile scalpel and forceps into small pieces. Tumor pieces were transferred to 50 mL tubes containing 10 mL of the pre-warmed (37°C) sterile cell dissociation buffer (HBSS containing 50,000 U of collagenase IV and 100,000 U of DNase I). Samples were incubated at 37°C under continuous rotation at 300 rpm for 60 minutes in an incubator-shaker. Cell suspensions were filtered through a 70 μm cell strainer, then a 40 μm cell strainer to remove any aggregates. Subsequently, the filtrate (i.e., single-cell suspension) was centrifuged at 230 xg for 10 minutes. Cell pellets were collected and resuspended in Ammonium-Chloride-Potassium (ACK) lysing buffer (deionized water containing 1% w/v of NH4Cl, 0.125% w/v of KHCO3, 0.25% v/v of 5% EDTA; pH 7.4) to lyse red blood cells and incubated at room temperature for approximately 3 minutes. Cells were collected and washed twice with 1X DPBS to remove free drug (i.e., extracellular drug). Finally, cells were resuspended in a complete medium containing 25 μg/mL propidium iodide to evaluate the cell viability after processing the tumors into single-cell suspensions. Cell suspensions were acquired using a FACSCalibur flow cytometer, and data were analyzed using FlowJo software.
2.20. Immunohistochemistry Analysis
To further assess the antitumor activity of the tested treatments, immunohistochemistry (IHC) analysis was performed on the tumor tissues. Female athymic Nu/Nu mice were subcutaneously challenged with Hec50co cells (1 × 106 cells/mouse), as described above. When mice had palpable tumors (day 18 post-tumor challenge), they were randomly distributed into 3 treatment groups: PBS, PTX, and PTX + CIP2b, and treatments were administered. On day 50 post-tumor challenge, mice were euthanized, and tumors were collected and washed with 1X DPBS. Tumors were then stored overnight in a 70% v/v ethanol 200 proof (in NanoPure™ water) to rehydrate specimens. Subsequently, the ethanol was replaced by a 10% neutral buffered formalin until processing. Tumor specimens were processed into paraffin tissue blocks followed by sectioning and mounting onto microscope slides. Slides were stained (i) with hematoxylin and eosin (H&E) to reveal tumor microanatomy, (ii) for caspase 3 to assess apoptosis, and (iii) for β-tubulin to examine microtubule integrity. Stained samples were visualized using a CKX41 Inverted Microscope equipped with a DP70 Digital Camera System (Olympus, Japan). The scale bar was added using ImageJ.
2.21. Pharmacokinetics and Biodistribution of CIP2b
Female BALB/c mice were injected IV via the tail vein with a solution of CIP2b in DPBS:Tween-80:ethanol 85:10:5 either at a dose of 1.25 mg/kg or 3.75 mg/kg. After predetermined time points (5, 15, 30, 60, 300, and 1440 minutes), mice were anesthetized, and their blood was collected by cardiac puncture in heparinized tubes (Heparin sodium, 1,000 USP Units/mL, Hospira, Lake Forest, IL). Blood samples were centrifuged at 16,000 xg for 15 minutes at 4°C, and plasma (supernatant) was collected and stored at −80°C until further analysis. In addition, vital body organs (heart, lungs, liver, spleen, pancreas, and kidneys) were also collected from euthanized mice (cervical dislocation) and stored at −80°C for subsequent analysis.
To analyze plasma samples, 2 mL ethyl acetate and 15 μL of the internal standard (IS) solution (50 μg/mL of 7-hydroxy flavone in methanol) were added to 200 μL plasma, and the mixture was vortexed for 5 minutes, centrifuged (16,000 xg, 5 minutes), and separated. The process was repeated, and a total of 4 mL ethyl acetate per sample was completely evaporated under a steady nitrogen stream using TurboVap LV (Caliper LifeSciences) at room temperature for 90 minutes. Next, 150 μL of methanol and water (HPLC grade) mixture (1:1) were added to the residue, and the resultant dispersion was centrifuged (16,000 xg, 30 minutes). Finally, the clear supernatant was injected into the HPLC for CIP2b quantification.
To extract the CIP2b from the frozen organs, approximately 100–400 mg of organs were sliced and homogenized using a bead tissue homogenizer (Fisher Brand Bead Mill-4, Hampton, NH) in screw-capped 2-mL tubes containing ~25 zirconia beads and 25 μL of 1X DPBS each. The homogenate was then transferred to 15 mL tubes, and 2 mL of ethyl acetate and 15 μL of the IS solution were added. The mixture was vortexed for 5 minutes, and then the samples were centrifuged for 10 minutes to collect the supernatant. This process was repeated twice, and a total of 4 mL ethyl acetate per sample was collected, and then completely evaporated under a steady nitrogen stream using TurboVap LV at room temperature for 90 minutes. Next, 150 μL of methanol and water mixture (1:1) were added to the residue, and the resultant dispersion was centrifuged (16,000 xg, 30 minutes). Finally, the clear supernatant was injected into the HPLC for CIP2b quantification.
2.22. CIP2b Content Measurement Using HPLC
An HPLC system (Agilent Infinity 1100, Santa Clara, CA) equipped with a Waters Symmetry C18 column (Cat. No. WAT045905, 5 μm pore size, 4.6 mm × 150 mm, Milford, MA) was used in this experiment. A gradient elution method was used to measure CIP2b content in the plasma and organs. The mobile phase consisted of solution A (water + 0.1% v/v trifluoroacetic acid (TFA)) and solution B (methanol:acetonitrile 95:5 + 0.1% v/v TFA). The gradient elution schedule started with solution A:solution B 90:10 then changed gradually to 30:70 during the first 20 minutes, then remained at 30:70 for 2 more minutes, and finally changed gradually to 90:10 during the last 3 minutes of the HPLC run (total run time was 25 min per sample). The flow rate of mobile phases was 1 mL/ minute at room temperature and the injection volume was set to be 50 μL. The detection wavelength was set to 275 nm. A standard curve was constructed for each organ using relevant blank mice organ homogenates spiked with 15 μL of CIP2b standard solutions of different concentrations and 15 μL of the IS solution.
2.23. CIP2b Safety Profile
To assess CIP-2b toxicity, 15 female and 15 male BALB/c mice were randomly distributed into 5 groups as follows: (i) PBS, (ii) 1.25 mg/kg CIP2b, (iii) 3.75 mg/kg CIP2b, (iv) 5 mg/kg CIP2b, and (v) bank vehicle (DPBS:Tween-80:ethanol 85:10:5). Doses were administered three times/week for 1 week via IV injection. One week later, mice were anesthetized and the whole blood samples were collected via cardiac puncture. The half volume of the whole blood samples was used to perform a complete blood count while the remaining half of the whole blood samples were incubated for 30 minutes at room temperature, and this was followed by centrifugation for 15 minutes at 16,000 xg and 4°C to harvest serum for comprehensive blood chemistry analyses by measuring different enzyme levels and biochemical markers. In addition, vital body organs were collected, and their weight was measured.
2.24. Statistical Analysis
Student’s t-test, one-way ANOVA, or two-way ANOVA followed by a Tukey post hoc test was used to determine significant differences between test groups. The experimental results are presented as mean ± standard deviation. Statistical analysis was performed using Prism 9 (GraphPad Prism, La Jolla, CA). Values where P < 0.05 were considered statistically significant.
3. Results
3.1. Properties of CIP2b
The color of CIP2b is pale yellow and crystalline in nature. The designation of CIP2b peaks in the 1H NMR spectrum demonstrated the purity of the synthesized CIP2b (Fig. S2). The chemical shifts of CIP2b appeared at δ = 1.02–1.17 ppm (2H, cyclopropyl-H), δ = 1.91 ppm (2H, cyclopropyl-H), δ = 2.34–2.61 ppm (4H, piperazinyl-H), δ = 3.09 ppm (2H, −N−CH2−CO), δ = 3.15–3.25 ppm (4H, piperazinyl-H), δ = 3.66 ppm (1H, cyclopropyl-H), δ = 7.23 ppm (2H, Ar-H), δ = 7.42 ppm (1H, H-8), δ = 7.54 ppm (2H, Ar-H), δ = 7.76 ppm (1H, H-5), δ = 8.50 ppm (1H, H-2), δ = 9.76 ppm (1H, −NH−CO), and δ =15.04 ppm (1H, COOH). The 1H NMR spectrum of PTX was consistent with what has been reported in the literature (Fig. S3). In addition, the 1H NMR spectrum of PTX + CIP2b demonstrated unchanged chemical shifts compared to the spectra of the drugs alone (Fig. S4), suggesting that the level of interaction between PTX and CIP2b is limited to non-covalent interactions. CIP2b was also characterized by electrospray ionization high-resolution Orbitrap mass spectrometry whereby [M+H]+ parent ion was identified as m/z 499.15 in the positive ionization mode; indicating that the mass of CIP2b is 498.15 Da (Fig. S5).
3.2. Synergistic Cytotoxic Effects of Combinatorial Treatment
The cytotoxic effects of CIP2b and PTX combination therapy against four different endometrial cancer cell lines were studied to investigate the involvement of p53 mutational status. The results demonstrated that the drug combination of PTX + CIP2b produced cytotoxic effects in all four cell lines (Ishikawa-H WTp53, Hec50co LOFp53, Hec50co GOFp53, and KLE GOFp53) but with different magnitudes (Fig. 1). For example, incubation of Ishikawa-H WTp53 cells with PTX + different CIP2b concentrations resulted in marginal enhancement in the cytotoxic effects compared to PTX alone (Fig. 1a). In contrast, incubation of Hec50co LOFp53 cells with PTX + CIP2b combination resulted in greater cytotoxic effects in comparison to PTX alone (Fig. 1b). For example, 10 nM PTX + 10 μM CIP2b resulted in lower cell viability (8.3 ± 0.3%) compared to cell viabilities of 31.2 ± 4.1% and 99.6 ± 0.4% when 10 nM PTX and 10 μM CIP2b were used as a single treatment, respectively. Similar to Hec50co LOFp53 cells, improvements in cytotoxic effects were also observed when the drug combination was tested against Hec50co GOFp53 (Fig. 1c). Treating these cells with 10 nM PTX + 10 μM CIP2b decreased the cell viability to 9.0 ± 0.5% versus 17.9 ± 1.4% and 101.1 ± 1.0% upon using a single treatment of 10 nM PTX and 10 μM CIP2b, respectively. On the other hand, the combination treatment was less effective against the KLE GOFp53 cell line where adding 10 nM PTX + 10 μM CIP2b reduced the cell viability to only 58.1 ± 0.9% whereas 10 nM PTX and 10 μM CIP2b as a single treatment resulted in 63.5 ± 0.6% and 102.4 ± 3.0% cell viability, respectively (Fig. 1d). The cell viability of single and combination therapy using different concentrations was statistically compared (Fig. S6). Furthermore, the addition of CIP (parent compound of CIP2b) either alone or in combination with PTX showed no benefits in comparison to PTX alone in the four cell lines (Fig. 1e–1h). However, CIP2b alone exhibited cytotoxic effects against all tested cell lines (at concentrations > 10 μM) suggesting that the chemical modifications made to the CIP backbone to synthesize CIP2b improved anticancer properties compared to the parent compound. In addition, it was observed that even the use of a relatively low concentration of CIP2b with PTX improved the antitumor effects of PTX as evidenced by the significant drop in the IC50 (Fig. 1i–1l and Table S1). This was cell line-dependent since the decrease in the IC50 was only significant in the Hec50co LOFp53 and Hec50co GOFp53 cells treated with PTX + 1 μM CIP2b. Adding relatively high concentrations of CIP2b (10 and 20 μM) to PTX resulted in significantly enhanced cytotoxicity compared to PTX alone in all four cell lines. The reductions in the cell viabilities were likely synergistic since most of the combination index (CI) values of the concentrations tested were <1 (Fig. 1m–1p), especially for the Hec50co LOFp53 cell line. Similar synergistic cytotoxic effects were observed when docetaxel (DOC) + CIP2b combinatorial treatments were tested against the four different endometrial cancer cell lines (Fig. S7 and Table S1).
Fig. 1.
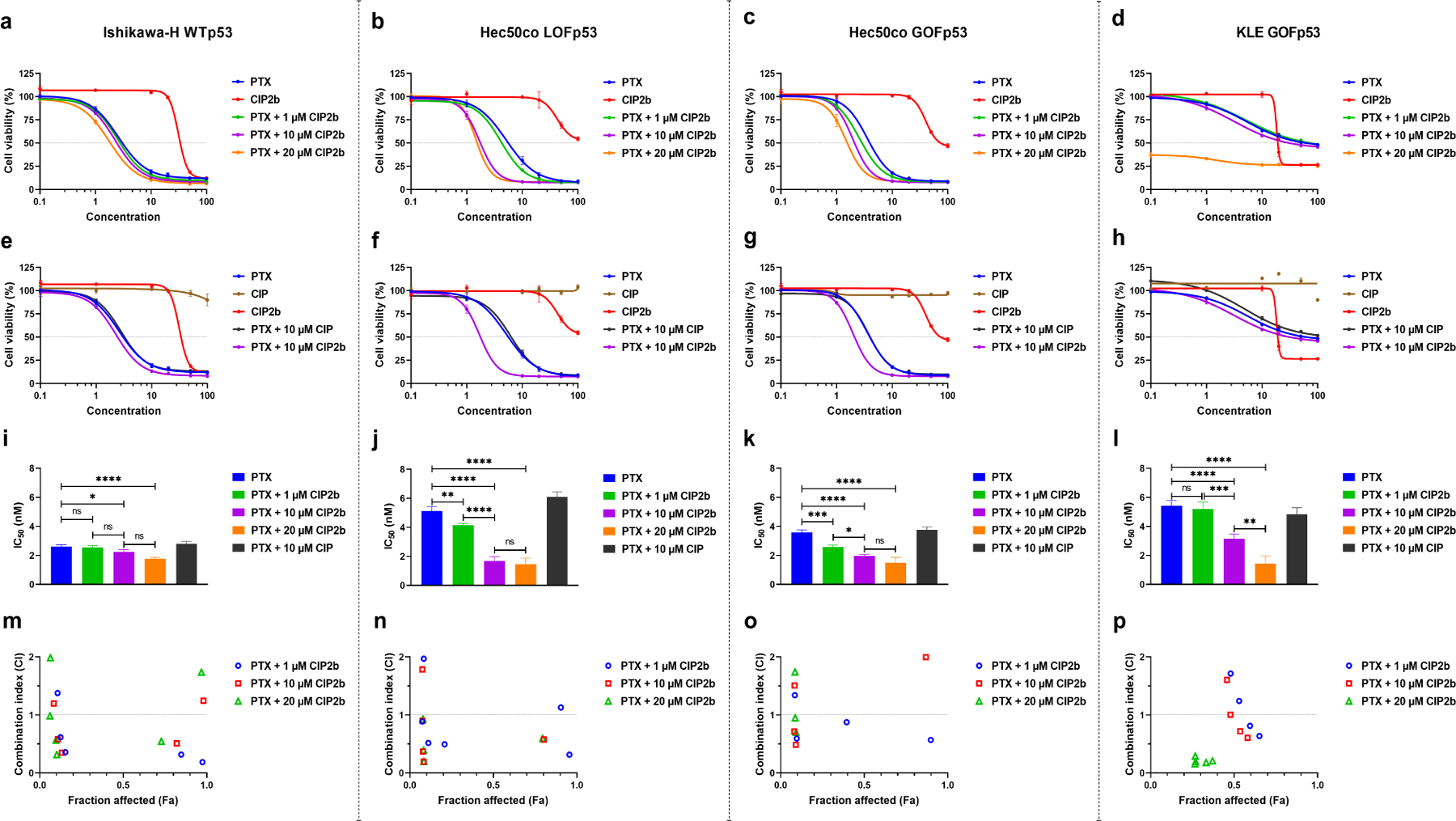
In vitro antitumor activity of PTX ± CIP2b combinatorial treatment against four different human endometrial cancer cell lines. (a–d) Cytotoxicity assay for indicated cell line following treatment for 72 hours with different concentrations of either PTX or CIP2b or different concentrations of PTX and fixed concentrations of CIP2b. (e–h), Cytotoxicity assay for indicated cell line following treatment for 72 hours with different concentrations of either PTX or CIP or CIP2b or different concentrations of PTX and fixed concentrations of either CIP or CIP2b. (i–l), Estimated IC50 values for indicated cell line following treatment with PTX ± CIP2b. (m–p), Cytotoxic synergy between PTX and CIP2b is calculated for indicated cell lines using the Combination Index (CI) method where CI values less than 1 indicate synergy. Data are plotted as mean ± SD (n=3). A one–way ANOVA with Tukey post hoc test was used for statistical analysis. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant.
3.3. Cell Cycle Arrest
Cell cycle analysis of four different endometrial cancer cell lines revealed that the effect of the treatments on the cell population in each cell cycle phase (G0/G1 phase, S phase, and G2/M phase) was cell line dependent (Fig. 2a–2d). DNA cell cycle results revealed that the combinatorial treatment of PTX + CIP2b shifted the Ishikawa-H WTp53 cell population marginally towards the G2/M phase compared to PTX alone (Fig. 2e). However, when PTX + CIP2b combination was tested against Hec50co LOFp53 (Fig. 2f) and Hec50co GOFp53 (Fig. 2g) cells, there was a clear change in the proportion of cells present in the different cell cycle phases. For instance, 10 nM PTX + 10 μM CIP2b arrested the vast majority of Hec50co LOFp53 (74.9 ± 2.1%) and Hec50co GOFp53 (83.7 ± 0.4%) cells in the G2/M phase in comparison to 10 nM PTX alone (43.7 ± 0.5 and 57.3 ± 1.1%, respectively). KLE GOFp53 cells showed minimal changes in cell population upon using the drug combination (Fig. 2h). This is likely due to KLE GOFp53 cells being slow growing, having a doubling time of 34–72 hours [39, 40]; and these cells were incubated with the treatments for only 24 hours in these cell cycle analyses. Also, it is worth noting that CIP2b alone did not affect the proportions of the cell populations (for all cell line lines) in the different cell cycle phases compared to untreated cells, and this was expected since the tested dose of CIP2b (10 μM) did not cause cytotoxic effects (Fig. 1).
Fig. 2.
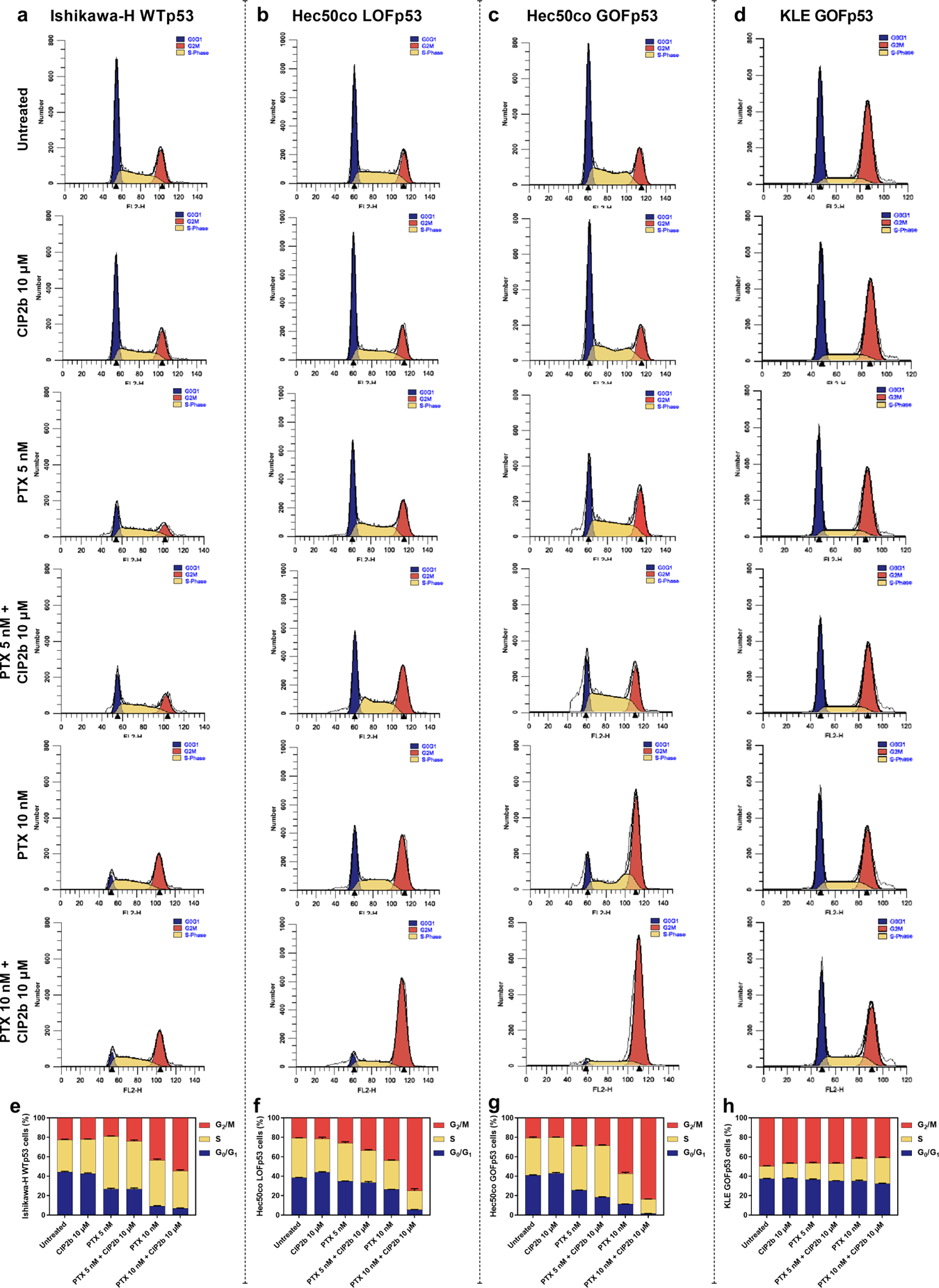
Role of the mitotic spindle checkpoint in the cellular response to the therapeutic combination of PTX + CIP2b following the cell cycle analysis using ModFit LT software. (a, b, c, and d) Histograms of Ishikawa–H WTp53, Hec50co LOFp53, Hec50co GOFp53, and KLE GOFp53 cells, respectively, with indicated treatments represent cell cycle phases: G0/G1, S, and G2/M. (e, f, g, and h) Distribution of Ishikawa–H WTp53, Hec50co LOFp53, Hec50co GOFp53, and KLE GOFp53 cells, respectively, in each stage of the cell cycle after incubation with the indicated treatment for 24 hours. Data are plotted as mean ± SD (n=3).
3.4. Mode of Administration
In this experiment, the effect of adding the drugs concurrently versus sequentially was studied using Hec50co LOFp53 cells. The results showed that concurrent administration was as effective as sequential administration (Fig. 3a). This was also supported by the statistically non-significant differences between IC50 values among the treatment groups (Fig. 3b). This means both concurrent and sequential administration of the drug combination were acceptable options. However, concurrent administration was chosen in all subsequent studies because it is more practical, especially when it comes to treatment administration in vivo compared to multiple injections required for the sequential strategy (compliance).
Fig. 3.
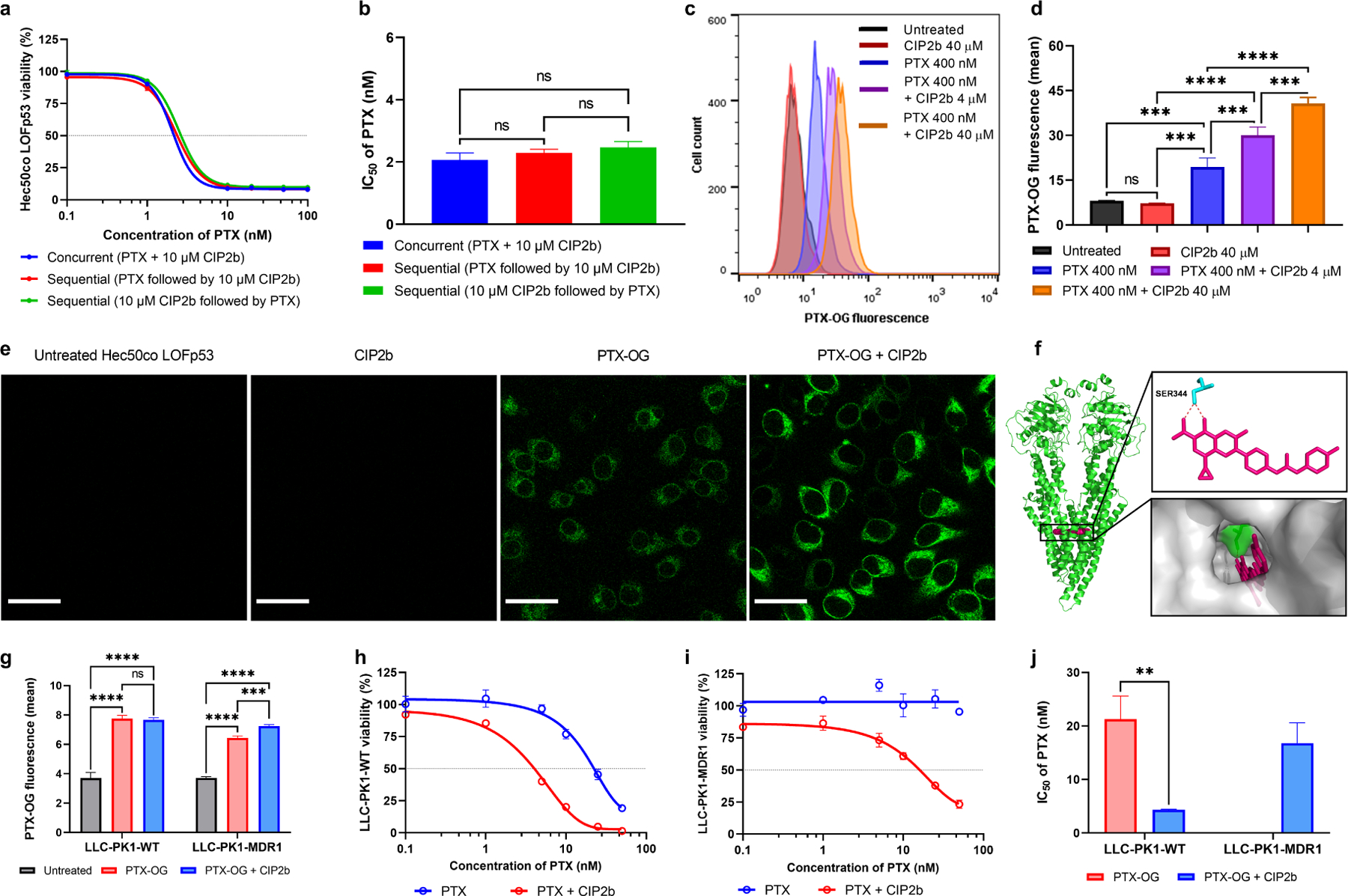
Effect of PTX + CIP2b combinatorial therapy on the MDR1. (a) Cytotoxicity assays of MDR1-expressing Hec50co LOFp53 endometrial cancer cells following treatment for 72 hours with different concentrations of PTX and fixed concentration of CIP2b (10 μM) using two different treatment strategies: concurrent treatment where both compounds were added to the cells at the same time vs sequential treatment where the PTX was added either after or before the CIP2b. (b) IC50 values of PTX following the addition of 10 μM CIP2b against MDR1–expressing Hec50co LOFp53 endometrial cancer cells. (c) Representative flow cytometric histograms of PTX–OG intracellular accumulation experiment. (d) Intracellular accumulation of PTX–OG (400 nM) in MDR1–expressing Hec50co LOFp53 cells in the presence or absence of CIP2b (4 or 40 μM) after 2 hours of treatment, as determined by flow cytometric analysis. (e) Confocal microscopy images of Hec50co cells cultured with PTX–OG ± CIP2b (scale bar = 30 μm). (f) Molecular interaction of CIP2b with human MDR1. CIP2b molecule is colored pink, the non–conventional long–distance hydrogen bond is colored red, and active site residues are colored cyan. Polar contact surfaces are presented as green while the target protein is presented as a transparent surface in gray. (g) Flow cytometric assay of the intracellular accumulation of PTX–OG (400 nM) in LLC–PK1–WT or LLC–PK1–MDR1 cells in the presence or absence of CIP2b (4 μM). (h and i) Percent cell survival following treatment of LLC–PK1–WT cells and LLC-PK1–MDR1 cells, respectively, for 72 hours with different concentrations of PTX in the presence or absence of 50 μM CIP2b. (j) IC50 values of PTX following the addition of 50 μM CIP2b against LLC–PK1–WT cells and LLC–PK1–MDR1 cells. Data are plotted as mean ± SD (n=3). A one–way (b and c) or two–way (g) ANOVA with Tukey post hoc test or one–sided t–test (j) were used for statistical analysis. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant.
3.5. Intracellular Accumulation of PTX
It was found that PTX (fluorescently labeled) was readily taken up by Hec50co LOFp53 cells as demonstrated by a significant increase in the fluorescence intensity when compared to untreated cells (Fig. 3c and 3d). However, co-delivery of CIP2b with PTX significantly enhanced the intracellular accumulation of PTX in a dose-dependent manner. Furthermore, this was qualitatively confirmed using confocal laser microscopy of Hec50co LOFp53 cells cultured with PTX in the absence or presence of CIP2b where cells incubated with drug combination exhibited brighter green fluorescence suggesting that greater PTX was accumulated intracellularly (Fig. 3e).
3.6. MDR1 Inhibition
The activity of CIP2b against the MDR1 gene was initially investigated in silico by exploring the molecular interaction between CIP2b and human MDR1 using molecular docking simulation. CIP2b is predicted to exhibit a strong binding affinity towards MDR1 with the binding free energy of −10.8 kcal/mol (Table S3 and Fig. S8). CIP2b is predicted to exhibit a strong binding affinity to the PTX binding site of the MDR1 (e.g., SER344 residue) (Fig. 3f). Given the enhanced uptake of PTX observed when delivered in combination with CIP2b (Fig. 3c–3e) and molecular docking simulation (Fig. 3f), the ability of CIP2b to inhibit MDR1-mediated efflux of PTX was further confirmed by exploring the in vitro intracellular accumulation of PTX. In order to confirm that the enhancement of uptake was related, at least in part, to MDR1, the intracellular accumulation of PTX in the presence of CIP2b in a cell line that overexpresses MDR1 gene (LLC-PK1-MDR1 cells) while using its wild type cells (LLC-PK1-WT cells) as control was evaluated. LLC-PK1-WT cells and their MDR1-overexpressing variant were treated with PTX ± CIP2b and compared the effects on intracellular accumulation of PTX. Flow cytometry-based cellular uptake analysis showed that CIP2b significantly enhanced the intracellular accumulation of PTX in MDR1-overexpressing cells whereas there was no difference between the two treatments in wild-type cells (Fig. 3g), suggesting CIP2b inhibits MDR1 function. Additionally, the cytotoxic activity of the drug combination was explored against these two cell lines. It was found that the combination of CIP2b and PTX was more cytotoxic against LLC-PK1-WT cells (PTX IC50 of 4.3 ± 0.1 nM) than PTX alone (PTX IC50 of 21.3 ± 4.3 nM) (Fig. 3h–1j). While PTX was ineffective up to the maximum concentration used (50 nM) against LLC-PK1-MDR1 cells (i.e., no IC50 reached for PTX), an IC50 of 16.8 ± 3.8 nM was achieved when PTX was combined with CIP2b, which is a promising finding confirming the inhibition of efflux pathway. Further confirmation of the enhanced cytotoxic effect of combining PTX and CIP2b is shown with bright-field microscopy images (Fig. S9). Despite the superiority in cytotoxicity observed in wild-type cells that received the combination of PTX + CIP2b (Fig. 3h), the absence of any advantage in PTX intercellular accumulation (Fig. 3g) suggests that CIP2b utilizes at least one other mechanism of action to enhance the antitumor activity of PTX.
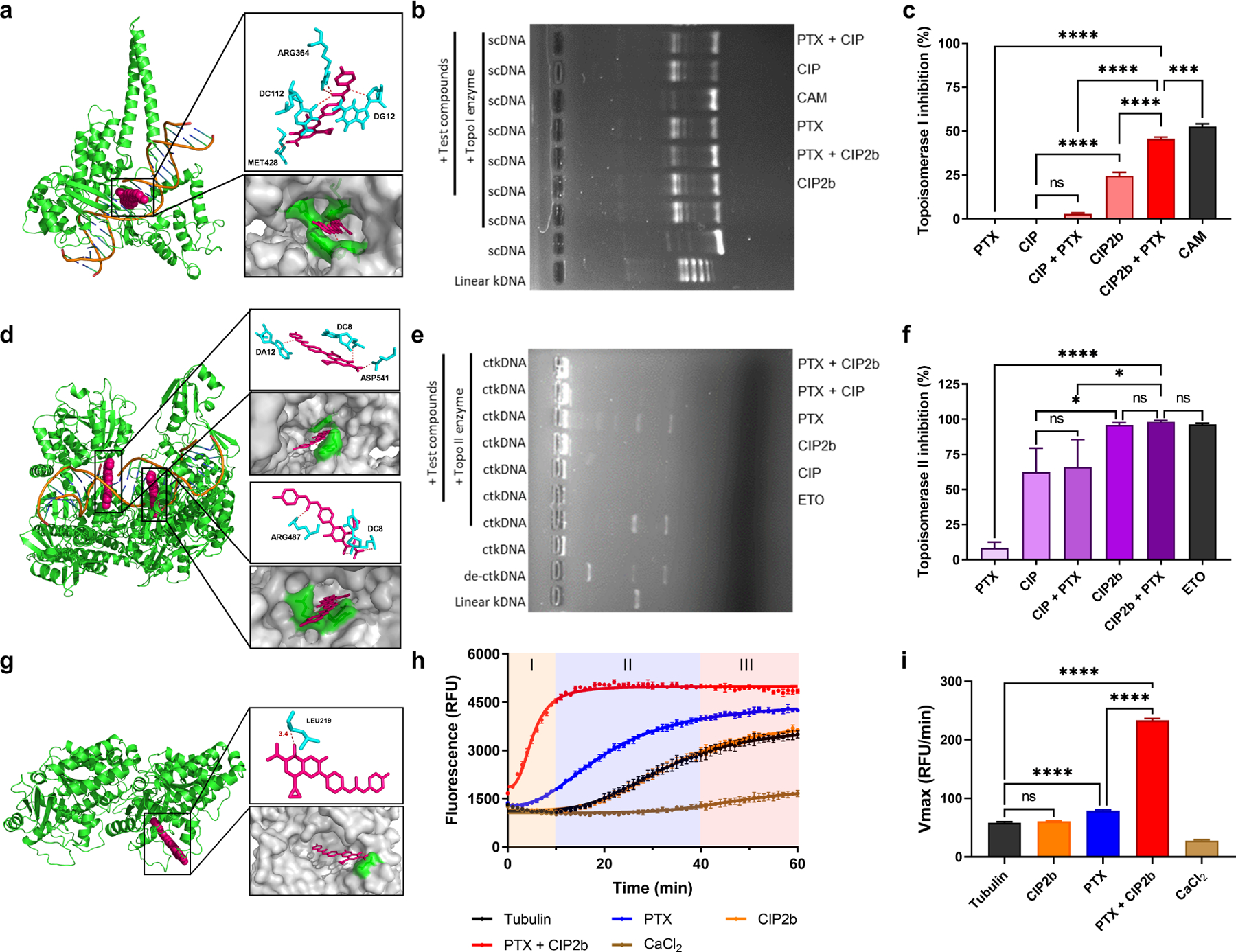
3.7. Inhibition of Topoisomerase I
Simulation of the molecular interaction of CIP2b with human Topo I showed that CIP2b is predicted to interact with various Topo I residues such as DC112 and ARG364 (Fig. 4a) with the binding free energy of −10.8 kcal/mol (Table S3 and Fig. S8). Topo I inhibitory activities of PTX, CIP2b, and their combination was also evaluated in vitro while CAM was used as a positive control. While PTX and CIP have no activity on Topo I, results obtained from the Topo I assay demonstrated that CIP2b alone provided some Topo I inhibitory activity (24.5 ± 1.6 %) (Fig. 4b and 4c). However, when PTX was combined with CIP2b, the magnitude of enzyme inhibition was further increased (45.6 ± 0.8 %), almost to the same degree as the positive control, CAM (52.6 ± 1.3 %), an inhibitor of Topo I activity. The exact mechanism underlying this potentiation is still not clear; however, one possible explanation is that PTX enhances the binding of CIP2b to Topo I.
Fig. 4.
Molecular mechanisms of action of the therapeutic combination of PTX + CIP2b. (a) Molecular interaction of CIP2b with human topoisomerase I. (b) Representative image of agarose gel electrophoresis showing inhibition of topoisomerase I. (c) Topoisomerase I inhibitory activity of CIP2b, CIP, PTX, or indicated combinations represented as a bar graph based on quantification of the band intensity in electrophoretic gels. (d) Molecular interaction of CIP2b with human topoisomerase II. (e) Representative image of agarose gel electrophoresis showing inhibition of topoisomerase II. (f) Topoisomerase II inhibitory activity of CIP2b, CIP, PTX, or indicated combinations represented as a bar graph based on quantification of the band intensity in electrophoretic gels. (g) Molecular interaction of CIP2b with tubulin dimers. (h) β–tubulin polymerization activity in the presence of PTX and CIP2b, alone or in combination. Tubulin and CaCl2 were used as controls. The three phases of polymerization are shown: I (nucleation), II (growth), and III (steady state). (i) Vmax values as an indicator of tubulin/ligand interactions. In all molecular docking graphs (a, d, and g), the CIP2b molecule is colored pink, the non–conventional long–distance hydrogen bond is colored red, and active site residues are colored cyan. Polar contact surfaces are presented as green, and the target protein is presented as a transparent surface in gray. Data are plotted as mean ± SD (n=3). A one–way ANOVA with Tukey post hoc test was used for statistical analysis. *, P < 0.05; ****, P < 0.0001; ns, nonsignificant.
3.8. Inhibition of Topoisomerase II
In silico molecular docking analysis also showed that CIP2b is predicted to have a strong binding affinity to human Topo II (binding free energy of ≤ −12 kcal/mol, Table S3 and Fig. S8) where CIP2b interacts with DC8, DA12, and ARG487, which represent the binding sites for ETO on Topo II (Fig. 4d). In addition, in vitro data showed that CIP exhibited partial inhibition of Topo II (62.3 ± 17.1%) (Fig. 4e and 4f), which is in line with recent findings [41]. Surprisingly, CIP2b alone achieved even stronger Topo II inhibition (96.1 ± 1.4%), similar to that of the positive control, ETO (96.3 ± 0.8%). PTX alone had a negligible effect on Topo II inhibition (8.3 ± 4.2%) and adding PTX to either CIP or CIP2b did not enhance the activity against Topo II compared to CIP and CIP2b alone. However, in the case of CIP2b, which could inhibit Topo II activity by almost 100% when used alone, a further significant increase by adding PTX was therefore unlikely.
3.9. Enhancement of Tubulin Polymerization
CIP2b is predicted to strongly interact with the PTX interaction site of β-tubulin in α- and β-tubulin dimers (binding energy −9.6 kcal/mol, Table S3 and Fig. S8) and stabilize the formed microtubules while interacting with different tubulin dimer residues such as LEU219 (Fig. 4g). Tubulin polymerization assay also indicated that PTX, as expected, increased the rate of tubulin polymerization, with an onset time of approximately 5 minutes versus 15 minutes when PTX was not added (Fig. 4h). Surprisingly, when CIP2b was combined with PTX, there was a marked enhancement in the rate of tubulin polymerization, and its onset was instantaneous. In comparison, CIP2b alone had no effect on tubulin polymerization. These results clearly demonstrate that CIP2b promotes the microtubule stabilization activity of PTX even though alone it has no effect on tubulin polymerization. The observed increased tubulin polymerization rate when CIP2b was added to PTX compared to PTX alone (Fig. 4i) suggests that CIP2b accelerates the kinetics of the binding of PTX to its specific sites in tubulin molecules.
3.10. Caspase Cascade
Up to this point, mechanisms by which CIP2b may be augmenting the cytotoxic activity of PTX on cancer cells were explored. To determine how these changes influence cell apoptosis, levels of activated caspases-3, −8, and −9 in Hec50co LOFp53 cells treated for 24 and 72 hours with different concentrations of PTX ± CIP2b were measured. In particular, 24 hours after treatment, there were no significant changes in the caspase-3 activity regardless of treatment type (Fig. 5a). Similarly, there was no significant difference in caspase-8 activity (Fig. 5b), indicating the absence of any involvement of the extrinsic pathway or death receptors. However, treatment of cells with 10 nM PTX resulted in a significant increase in caspase-9 activity whereas 5 nM PTX did not show a significant difference compared to the untreated group (Fig. 5c). Additionally, caspase-9 activity in CIP2b treated cells was significantly higher than the untreated group at all tested concentrations and in a concentration-dependent manner. Notably, there was a significant increase in caspase-9 activity in cells treated with 5 nM PTX combined with any of the CIP2b concentrations (i.e., 10, 25, 50 μM) relative to cells treated with 5 nM PTX alone. After 72 hours, cells treated with 5 nM or 10 nM PTX alone displayed significantly higher levels of caspase-3 activity compared to untreated cells, and these levels were further increased significantly when CIP2b was co-delivered with PTX (Fig. 5d). Treatment with CIP2b alone did not induce significant activation of caspase-3 after 72 hours even though it showed some activation of caspase-9 after 24 hours. In addition, none of the treatments induced any substantial changes in caspase-8 activation (Fig. 5e). Finally, at 72 hours following co-treatment with PTX and CIP2b, caspase-9 activation was not changed or was decreased (Fig. 5f) compared to the 24-hour measurements.
Fig. 5.
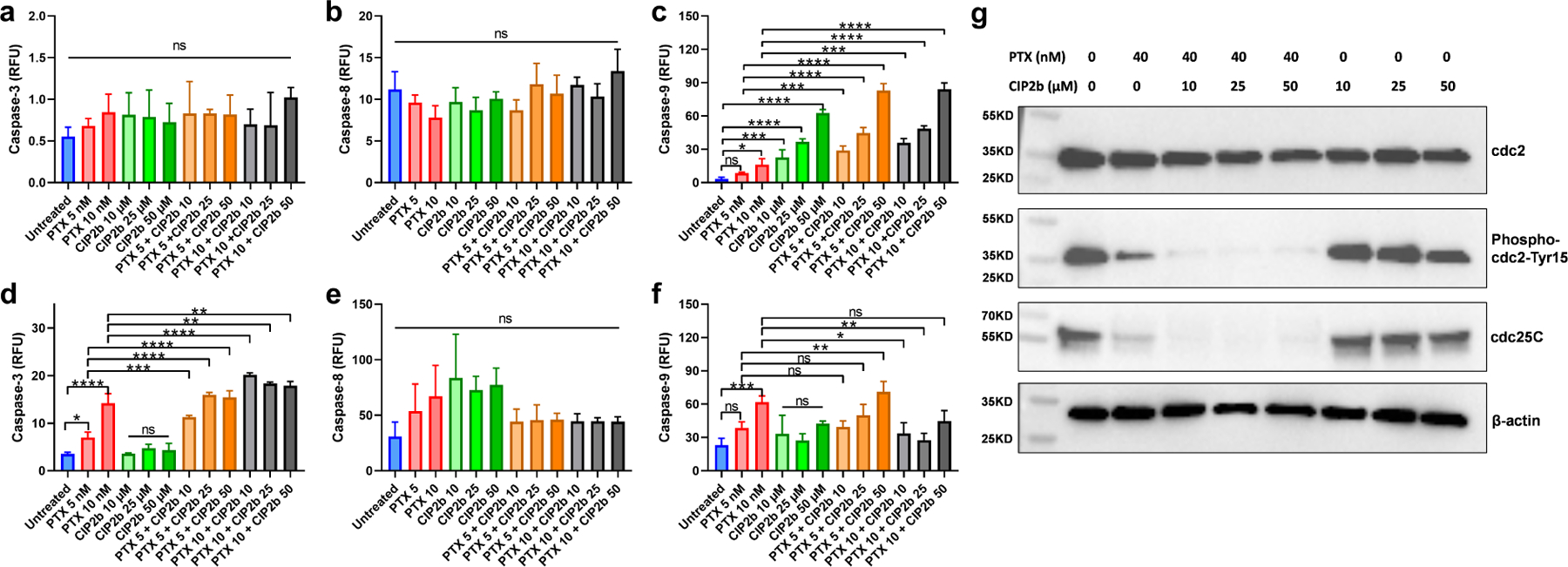
Caspase cascade and key G2/M phase regulators of Hec50co LOFp53 cells treated with PTX ± CIP2b. (a, b, and c) Expression of caspase–3, caspase–8, and caspase–9, respectively, in Hec50co LOFp53 cells incubated with the designated treatment for 24 hours. (d, e, and f), Expression of caspase–3, caspase–8, and caspase–9, respectively, in Hec50co LOFp53 cells incubated with the designated treatment for 72 hours. (g) Western blot analysis to evaluate the effect of PTX ± CIP2b on cell cycle regulatory proteins in Hec50co LOFp53 cells. The cell lysate was subjected to SDS–PAGE followed by transfer to nitrocellulose membranes. Membranes were probed with cdc25C, phospho-cdc2 Tyr 15, total cdc2, and β–actin antibodies, followed by incubation with corresponding horseradish peroxidase–conjugated secondary antibody, and visualized by an ECL detection system. Data are plotted as mean ± SD (n=3). A one–way ANOVA with Tukey post hoc test was used for statistical analysis. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant.
3.11. Cell Cycle Regulators
Lysates of Hec50co LOFp53 cells incubated with different treatments were initially processed to measure total protein content (Fig. S10) and then analyzed using Western blotting to detect the presence or absence of certain proteins. The drug combination of PTX + CIP2b was found to markedly arrest Hec50co LOFp53 in the G2/M phase (Fig. 2). Based on this finding, further experiments were performed to investigate the key G2/M phase regulators that the drug combination abrogates. Comparing single and combinatorial therapy, western blot analysis revealed that a combination of PTX and CIP2b inhibited the cdc25C phosphatase due to phosphorylation at Ser16 (Fig. 5g and Table S4). As a result, phosphorylation of cell division control 2 (cdc2) at Tyr15 was dramatically inhibited. Dephosphorylation of cdc2 at Tyr15 and activation of cdc2 ultimately led to G2/M checkpoint abrogation which prevented tumor cells from repairing their DNA damage and forced them into mitotic catastrophe and apoptosis.
3.12. In vivo Intracellular Accumulation of PTX in Hec50co LOFp53 Xenograft Tumors
The enhanced intracellular accumulation (Fig. 3c–3e) and increased cytotoxicity of PTX induced by CIP2b (Fig. 1) that were observed in vitro prompted further investigation as to whether these findings could also be translated in vivo. To test this, athymic Nu/Nu mice were challenged with Hec50co LOFp53 cells and the tumors were allowed to reach a considerable size (152.2 mm3). PTX uptake studies by Hec50co LOFp53 tumor cells in vivo were performed (Fig. 6a) and involved SC peritumoral administrations of PTX ± CIP2b. Mice treated with PTX alone possessed tumor cells that exhibited a subtle but non-significant increase in intracellular PTX accumulation compared to tumor cells from untreated control mice (Fig. 6b and 6c). However, co-administration of CIP2b and PTX significantly enhanced the intracellular accumulation of PTX in the tumor.
Fig. 6.
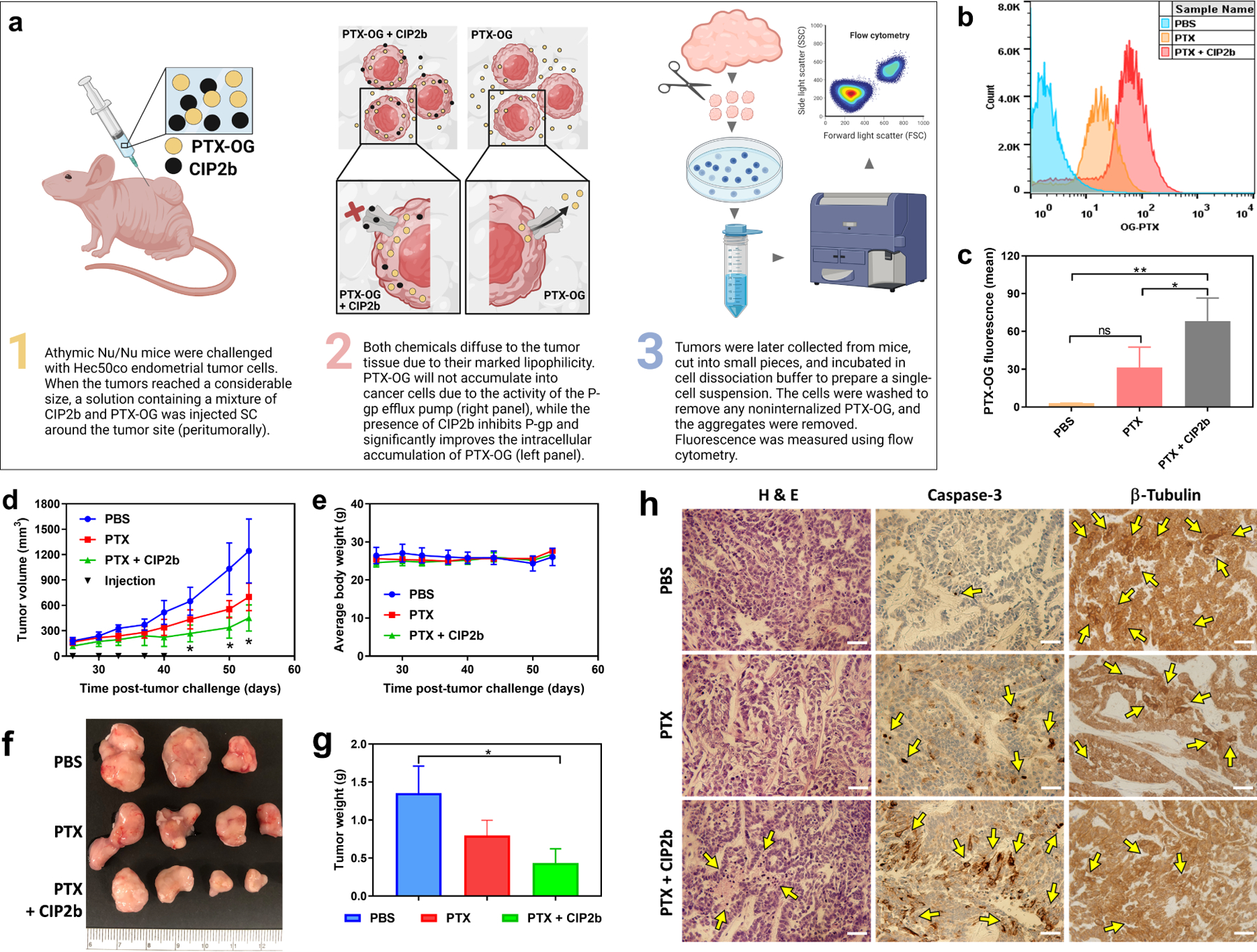
In vivo tumor accumulation and antitumor efficacy of PTX ± CIP2b. (a) Graphical representation of the in vivo PTX–OG intracellular accumulation study in the presence or absence of CIP2b. (b) Representative histograms of PTX–OG fluorescence in tumor–bearing mice treated with PTX ± CIP2b. (c) Flow cytometric analysis of PTX–OG in tumor cells (i.e., single–cell suspension) derived from mice challenged with Hec50co LOFp53 cells. (d) Hec50co LOFp53 tumor growth (human endometrial cancer xenografts) in athymic Nu/Nu mice following treatment with PBS, PTX, or PTX + CIP2b. (e) Body weight monitoring of the mice following their treatment with PBS, PTX, or PTX + CIP2b. (f) Gross examination of tumors collected from mice injected with the indicated treatments (day 53 post–tumor challenge). (g) Average tumor weights following mice euthanasia and tumor collection from mice injected with indicated treatments (day 53 post–tumor challenge). (h) Histological and immunohistochemical analysis of tumor tissues. Tumors were collected from representative mice treated with either PBS, PTX, or PTX + CIP2b. Sections were stained with H&E or antibodies against caspase 3 (indicating apoptosis) or β–tubulin (indicating microtubule assembly). Yellow arrows point out necrotic areas (H&E) or where antibodies showed abundant binding to their specific target. Scale bar = 500 μm. Data are plotted as mean ± SD (n=3–4). A one–way ANOVA with Tukey post hoc test (c) or one–sided t–test (d and g) were used for statistical analysis. *, P < 0.05; **, P < 0.01; ns, nonsignificant.
3.13. In vivo Antitumor Efficacy of PTX-CIP2b Against Human Endometrial Cancer Xenografts
The efficacy of combining PTX and CIP2b on tumor growth was then evaluated by injecting Hec50co LOFp53 tumor-bearing athymic Nu/Nu mice with PBS or PTX ± CIP2b. It was observed that mice in the PBS group harbored tumors that grew rapidly in comparison to the treated groups (Fig. 6d). Furthermore, mice treated with PTX alone had average tumor volumes that were smaller than those in control mice injected with PBS, albeit not significantly (P>0.05). In contrast, mice treated with the combination of PTX + CIP2b demonstrated the slowest tumor growth over time, and the average tumor volume on day 53 post-tumor challenge was 451.2 mm3, which was significantly smaller than that of the PBS group (1242.7 mm3; P<0.05). No significant statistical difference was observed between the PTX + CIP2b group and PTX alone group. Also, no difference in average body weight was observed among the different groups (Fig. 6e), suggesting the treatment was not toxic at the tested doses. On the last day of the study (day 53), tumors were collected and imaged (Fig. 6f). Additionally, the average weight of tumors from mice treated with PTX + CIP2b was significantly less than that of the mice in the PBS group (P<0.05, Fig. 6g), whereas the average weight of tumors from mice treated with PTX alone was not significantly different compared to the PBS group (P>0.05).
3.14. Immunohistochemical Analysis of Hec50co LOFp53 Tumors
Hec50co LOFp53 tumor tissues were fixed, embedded, and histologically as well as immunohistochemically examined. Compared to other treatment groups, H&E staining of tumors collected from mice treated with PTX + CIP2b showed broad necrotic areas and an abundance of pyknotic cells with condensed chromatin (Fig. 6h), indicating necrosis and/or apoptosis [42]. This demonstrates that combined treatment of PTX + CIP2b effectively induced cancer cell death inside tumors. Immunohistochemical staining of tumors collected from mice treated with PTX + CIP2b (versus PBS or PTX alone) further supported the efficacy of this treatment combination in inhibiting tumor progression. For example, tumors from mice treated with PTX + CIP2b possessed extensive areas of apoptosis, as determined by the expression of cleaved caspase-3, in comparison to the other treatment groups (Fig. 6h). Furthermore, tumor cells in the group that received PTX + CIP2b exhibited minimal expression of free unbound β-tubulin, suggesting that the drug combination efficiently targeted tubulin and disrupted microtubule assembly (Fig. 6h).
3.15. Pharmacokinetics and Biodistribution
To study the pharmacokinetics and biodistribution of CIP2b, two different doses (1.25 and 3.75 mg/kg) were tested (Fig. 7a). At the early time points (up to 5 hours following CIP2b administration), there was a gradual decrease in the CIP2b plasma levels associated with a marginal difference in the CIP2b plasma concentration in mice injected with 1.25 and 3.75 mg/kg CIP2b. However, as time passed (> 5 hours), CIP2b plasma concentration in mice injected with 3.75 mg/kg CIP2b was markedly higher than that of the 1.25 mg/kg CIP2b. Fitting pharmacokinetic models to the CIP2b plasma level data showed that the low dose of CIP2b was well-represented by the one-compartment model whereas plasma concentrations of CIP2b high dose were well-described by the two-compartment model (Fig. 7b). The enhancement in the CIP2b plasma concentration upon administration of the high dose of CIP2b was also reflected in the pharmacokinetic parameters (Table 1). For example, the predicted area under the plasma concentration-time curve (AUC0-inf) of CIP2b increased by 7.5% when 3.75 mg/kg CIP2b had been injected (6007.63 μg.min/mL) compared to 1.25 mg/kg CIP2b (5590.96 μg.min/mL). Also, administering a high dose of CIP2b prolonged the elimination t1/2 by approximately 14.6% (340.90 vs 390.53 min). CIP2b biodistribution results showed that when 3.75 mg/kg of CIP2b was administered, CIP2b preferentially accumulated in the lungs in comparison to 1.25 mg/kg of CIP2b (Fig. 7c and 7d), which could be interpreted as an advantage if the CIP2b were to be used to treat lung cancer. Also, 3.75 mg/kg of CIP2b is distributed to the spleen, and a lesser extent to other organs such as the liver, heart, kidney, and pancreas. On the other hand, when 1.25 mg/kg CIP2b was injected, there was a substantial decrease in the CIP2b accumulation in the lungs compared to 3.75 mg/kg CIP2b while CIP2b concentrations in the other organs were at the same levels.
Fig. 7.
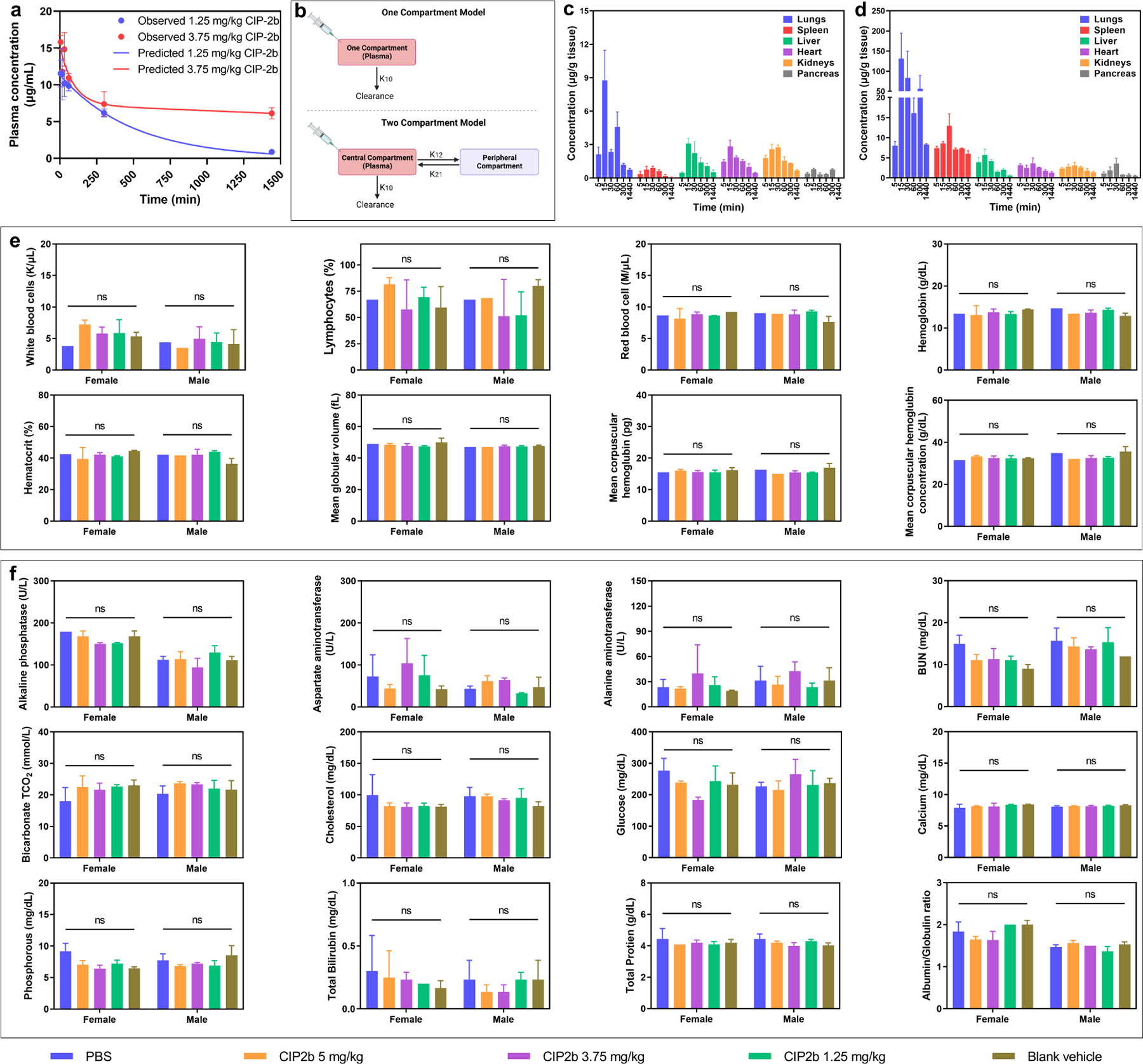
Pharmacokinetics, biodistribution, and safety profile of CIP2b. (a) Observed and predicted plasma CIP2b concentration versus time following IV administration of CIP2b into female BALB/c mice. (b) Schematic representation of the one‐ and two–compartment models for IV administration. K10 is the elimination rate constant while K12 and K21 are the first–order distribution rate constants depicting CIP2b transfer between the central and the peripheral compartments. (c and d) Biodistribution of CIP2b into the vital body organs following the IV injection of 1.25 and 3.75 mg/kg CIP2b, respectively. (e) Complete blood count (hematological data) of healthy female and male BALB/c mice following IV administration of CIP2b. (f) Enzyme levels and biochemical markers (serum data) of healthy female and male BALB/c mice following IV administration of CIP2b. Data are plotted as mean ± SD (n=3). A two–way ANOVA with the Tukey post hoc test was used for statistical analysis. ns, nonsignificant.
Table 1.
Pharmacokinetic parameters of CIP2b.
| Parameter | Unit | 1.25 mg/kg | 3.75 mg/kg |
|---|---|---|---|
| k10 | 1/min | 0.002033314 | 0.001993397 |
| k12 | 1/min | – | 0.003786634 |
| k21 | 1/min | – | 0.032530846 |
| t1/2 | Min | 340.8953098 | – |
| t1/2 alpha | Min | – | 18.97162136 |
| t1/2 beta | Min | – | 390.5326555 |
| C0 | μg/mL | 11.36818025 | 11.97559669 |
| V | (μg)/μg/mL) | 2.199120656 | 2.087578653 |
| Cl | (μg)/(μg/mL)/min | 0.004471503 | 0.004161373 |
| V2 | (μg)/(μg/mL) | – | 0.242996967 |
| Cl2 | (μg)/(μg/mL)/min | – | 0.007904897 |
| AUC0‐t | μg/mL*min | 5291.814850 | 5544.201937 |
| AUC0‐inf | μg/mL*min | 5590.961683 | 6007.633155 |
| AUMC | μg/mL*min2 | 2749679.532 | 3364573.355 |
| MRT | Min | 491.8079729 | 560.0497347 |
| Vss | μg/(μg/mL) | 2.199120656 | 2.330575620 |
3.16. Safety Profile of CIP2b
To assess the safety of the new drug molecule (CIP2b), female and male BALB/c mice were given various doses of CIP2b (1.25, 3.75, and 5 mg/kg). Then, whole blood samples were collected, sera samples were harvested, and major vital organs were gathered to measure their weight. Also, body weight was monitored over time. Using blood and serum samples, complete blood count and comprehensive blood chemistry analyses by measuring different enzyme levels and biochemical markers were carried out. Results showed that there were no significant differences in the parameters measured in the blood and serum samples between treatment groups (Fig. 7e and 7f). These data, combined with the absence of any significant change in the organ and body weight (Fig. S11), suggested the absence of any apparent toxicity; thereby, CIP2b is safe at the tested dose range (1.25–5 mg/kg).
4. Discussion
Chemoresistance is one of the major challenges associated with chemotherapies such as PTX [43]. Mechanisms of chemoresistance can involve tumor suppressor genes (e.g., TP53) and/or active transport efflux pumps (e.g., MDR1) [44]. Therefore, high doses and multiple administrations associated with each conventional single-drug therapy are generally required to overcome chemoresistance, however, this often results in the induction of detrimental side effects [45]. Over the past years, a combination therapy approach where two or more therapeutic agents are administered has been developed and clinically employed [46]. Chemotherapeutic-based combination approaches attracted great interest in clinical settings because of their superior efficacy profile compared to conventional single-drug treatment. For instance, the combination of PTX and carboplatin is used as a gold standard treatment for ovarian cancer [47, 48]. The rationale behind observed improvements in efficacy is that the combination approach could target different pathways and diminish the possibility of drug resistance; thus, potentiating the antitumor drug effect [49]. Concerning TP53 mutations, it has been recently found that the cell cycle of cancer cells with dysfunctional checkpoints as a result of p53 mutations can be arrested by using checkpoint inhibitors. More than 90% of type II endometrial cancer cases exhibit p53 mutations [11]. Endometrial cancer cells expressing LOFp53 can maintain the G2/M checkpoint in the cell cycle by activating the compensatory anti-apoptotic P38/MK2 pathway and its downstream components, which ultimately desensitizes cancer cells against chemotherapies. To overcome the emergence of chemoresistance and to combat the heterogeneity of tumors, chemotherapeutic cocktails are used. For instance, anticancer drugs are combined with Chk1/2 checkpoint inhibitors to force the tumor cells with damaged DNA to skip S and G2/M arrest and then enter mitosis which results in a mitotic catastrophe [50]. However, until now, the few tested treatments/trials have shown severe side effects with no or limited effectiveness [51, 52].
In this work, a combination of PTX + CIP2b has been developed to revert PTX resistance in p53-mutant and MDR1 gene-expressing type II human endometrial cancer (Hec50co LOFp53). Hec50co tumor cells have lost the expression of p53 as a result of deletion at the junction of exon 6 and intron 6–7 [53]. The dysfunction of p53 in type II human Hec50co cells is linked to chemotherapy resistance [54]. Our data showed that co-administration of CIP2b with PTX significantly improved the antitumor activity of PTX and resulted in synergistic cytotoxic effects against the Hec50co LOFp53 cells (Fig. 1). Similar synergistic cytotoxic effects were also observed when DOC + CIP2b combinatorial treatment was tested against Hec50co LOFp53 cells suggesting that taxanes can benefit from co-administration of CIP2b. Further investigation revealed that PTX + CIP2b markedly arrested Hec50co LOFp53 in the G2/M phase (Fig. 2). In general, activation of cdc2 regulates the entry of cells into mitosis (M phase). Dephosphorylation of cdc2 at tyrosine 15 (Tyr15) is one of the critical regulatory steps in activating cdc2 during the progression into mitosis (abrogates G2/M checkpoint). Whilst phosphorylation of cdc2 at Tyr15 results in inhibition of cdc2 (maintains G2/M checkpoint). Furthermore, cdc25C phosphatase is responsible for the removal of phosphates at Tyr15 (i.e., dephosphorylation) and subsequent activation of cdc2, which ultimately triggers the entry of cells into mitosis. Western blotting data indicated that the PTX + CIP2b drug combination activated cdc2 by reducing phosphorylation at Tyr15 (Fig. 5g). Besides key G2/M phase regulators, the PTX + CIP2b drug combination was also found to induce programmed cell death (Fig. 5a–f). Apoptosis can be triggered through two main pathways: (i) extrinsic pathway which is mediated by the activation of caspase-8 and (ii) intrinsic (mitochondrial) pathway which is mediated by the activation of caspase-9. Paclitaxel is known to induce intrinsic apoptosis by activating caspases-9, not caspase-8 [55]. Since caspase-9 is an initiator caspase [56], it was expected to increase in activity at relatively early time points and then decrease, as was primarily observed in our experiments. In contrast, caspase-3 is an executioner end-stage caspase [56], and its activity increases more slowly [57]. Even though CIP2b alone induced early activation of caspase-9 in Hec50co LOFp53 cells, this did not result in a corresponding rise in caspase-3 levels. This may be due to the activity of other anti-apoptotic proteins (e.g. XIAP or survivin) which may be the case for Hec50co LOFp53 cells [58]. This is probably why the addition of PTX significantly promoted the cytotoxic activity of CIP2b possibly via the reversal of these anti-apoptotic behaviors of Hec50co LOFp53 cells.
Our mechanistic studies revealed that the combinatorial therapy of PTX + CIP2b works on targets at least four pathways including, MDR1, Topo I, Topo II, and β tubulin. PTX is a substrate of MDR1, a membrane-bound efflux protein belonging to the ATP-binding cassette (ABC) family of transporters [18] whereas CIP is not a substrate of MDR1 when tested against LLC-PK1 cells [59]. Overexpression of MDR1 is linked with poor prognosis and low survival rates in cancer patients [21]. The MDR1-mediated efflux of PTX from cancer cells significantly reduces the intracellular accumulation of the drug and hence its antitumor activity. The use of third-generation MDR1 inhibitors (e.g. tariquidar, elacridar, zosuquidar) to combat chemoresistance against taxanes (e.g., PTX and DOC) in pre-clinical [21, 60] and clinical [61] studies is widely described; however, little to no additional benefits were observed [62–65]. To the best of our knowledge, very few or even no studies investigated the intracellular accumulation of MDR1 substrates in vivo in a tumor model when an MDR1 inhibitor is concomitantly or sequentially administered. CIP2b also fulfills most of the requirements for MDR1 substrates as it comprises 8 atoms of combined nitrogen and oxygen, has a molecular weight of 498.15 g/mol (Fig. S3), and has two acidic pKa values (5.4 and 6) [66, 67]. Both in vitro and in vivo findings demonstrated that CIP2b substantially enhanced PTX accumulation in tumor cells.
Besides MDR1 inhibition, results also showed that the drug combination of PTX + CIP2b markedly inhibited the activity of Topo I and II enzymes which play essential roles in DNA replication and transcription where they are responsible for removing tangled and knotted DNA that form during the cell division. Therefore, inhibition of these enzymes can result in increased DNA damage and subsequent elimination of these unwanted cells [68]. Topoisomerase inhibitors (e.g. topotecan and carboplatin) and PTX have been utilized in combination to improve anticancer efficacy and minimize chemo-resistance in metastatic endometrial cancer [69] whereas irinotecan and PTX have been similarly utilized for non-small cell lung cancer [70].
Another mechanism of action by which PTX + CIP2b functions is through inducing microtubule stability. Microtubules are one of the major cytoskeletal components and play an essential role in spindle formation generated by α and β tubulin polymerization. Since PTX is a well-known tubulin stabilizer (and consequently an inducer of apoptosis), it is critical to investigate whether this particular cytotoxic function of PTX is enhanced when CIP2b was added. PTX is known to enhance the rate of tubulin polymerization and stabilize formed microtubules, though both functions take place at different dynamics. Microtubule stabilization needs very few PTX molecules, in contrast to tubulin polymerization, which requires a stoichiometric ratio of 1:1 of PTX: tubulin [71]. PTX is thought to induce conformational changes in tubulin molecules which subsequently promotes their polymerization [71, 72] whereas CIP does not impact microtubule assembly in cells [73]. In vitro and in vivo data revealed that CIP2b enhanced the effects of PTX on β tubulin polymerization. Overall, these findings (inhibition of MDR1, Topo I, and Topo II, and microtubule stabilization) provide additional evidence that both drugs (CIP2b and PTX) promote the function of each other in a manner that potentially boosts antitumor potency.
In vivo PTX tumor accumulation study demonstrated that a significant increase in intratumoral PTX accumulation was observed in the tumors treated with the combination of PTX + CIP2b compared to treatment with PTX alone (Fig. 6c). The combined use of CIP2b with PTX, therefore, improved the overall antitumor activity of PTX by increasing the PTX concentrations within the tumor cell and produced more potent cell cytotoxicity as evident by the slower tumor growth in the mice given the drug combination (Fig. 6d). The importance of this observation lies in the potential of using lower PTX doses to achieve the same tumor accumulation and less collateral damage to other healthy tissues when CIP2b is co-administered with it, compared to those achieved with PTX alone. Although no significant statistical difference was observed between the PTX + CIP2b group and PTX alone group, the level of significance comparing the two groups suggested trends toward significance (Fig. 6d, P = 0.1139; Fig. 6g, P = 0.1178). Further optimization of the CIP2b:PTX ratio could improve the outcomes as suggested by the combination index results (Fig. 1n). In the in vivo studies, 1:1 CIP2b:PTX (5 mg/kg dose each) was used due to the limited solubility (0.5 mg/mL) of CIP2b in the vehicle (DPBS:Tween-80:ethanol 85:10:5). Enhanced antitumor activity of the drug combination was further supported by the histological and immunohistochemical examination of tumor tissues where mice treated with the drug combination had tumor tissues associated with substantial apoptotic cell death (Fig. 6h) as noted by the upregulation of caspase-3 expression. These in vivo efficacy findings are in line with the in vitro data.
In vivo pharmacokinetic and biodistribution studies showed that administering a high dose of CIP2b markedly delayed the elimination, prolonged circulation time, and shifted the elimination pharmacokinetics towards a biexponential 2-compartmental model (Fig. 7a). This is due to the several-fold accumulation of CIP2b in the lungs of mice when a higher dose was used (Fig. 7d), which eventually augmented the reliance of plasma elimination rate on the equilibrium kinetics between the lungs and plasma as the large drug amount in the lungs would act as a reservoir. As more drug is eliminated from the central compartment, more drug from the lungs, either free or bound to tissue proteins, intravasate back into the blood to restore tissue-to-plasma equilibrium, resulting in apparent slower elimination and more ‘sustained’ levels in the plasma. A similar pattern was also found in the spleen as well. This was clearly not the case when a smaller dose is used, as a mono-exponential elimination pattern was observed in the plasma, and no noticeable lung accumulation was found. Even though markedly higher pulmonary tissues-to-plasma concentration ratios were previously reported with other fluoroquinolones [74], more investigation is needed to explain this phenomenon. Currently, a polymeric nanoparticle-based combinatorial therapy for the targeted treatment of cancer is under development, and this aims to shift the biodistribution from healthy vital organs to tumor tissue. This can also allow for the administration of a high CIP2b:PTX ratio and overcome the solubility limitation of CIP2b in the surfactant-based vehicle.
5. Conclusions
In summary, CIP2b was shown to be a novel MDR1 inhibitor with potent inhibitory function against Topo I and II. When co-administered with PTX, CIP2b also enhanced the PTX stabilization effect on microtubules and resulted in synergistic cytotoxic effects against the MDR1-expressing Hec50co LOFp53 type II human endometrial cancer cells. Augmented antitumor properties were extensively characterized using different mechanistic studies to provide a better understanding of how combinatorial therapy functions. Synergistic cytotoxic effects observed in vitro were further confirmed by in vivo data using human endometrial cancer xenografts. These findings suggest this treatment combination can provide a more effective and better-tolerated therapeutic for patients with type II endometrial cancer.
Supplementary Material
Acknowledgments
This research was supported by the National Cancer Institute of the National Institutes of Health (Award Number P30CA086862) through the Holden Comprehensive Cancer Center at the University of Iowa. We thank Professors Kimberly Leslie and Erlio Gurpide for the kind gift of the human endometrial cancer cell lines (Ishikawa-H WTp53, Hec50co LOFp53, and Hec50co GOFp53). We also thank Professor John Markowitz (University of Florida) for the kind gift of the Lewis lung carcinoma-porcine kidney 1 cell lines (LLC-PK1-WT and LLC-PK1- MDR1). The authors also would like to acknowledge the use of the University of Iowa Central Microscopy Research Facility and the Flow Cytometry Facility.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Credit author statement
Conceptualization: S.O.A., Y.W.N., E.I.W., K.E., and A.K.S.; Methodology: S.O.A. and Y.W.N.; Validation: S.O.A.; Formal analysis: S.O.A. and Y.W.N.; Investigation: S.O.A., Y.W.N., E.I.W., S.S., and X.M.; Resources: S.O.A., Y.W.N., and H.H.M.; Data curation: S.O.A. and Y.W.N.; Writing – original draft: S.O.A.; Writing, review, and editing: S.O.A., Y.W.N., E.I.W., S.S., and A.K.S.; Visualization: S.O.A.; Supervision: G.A.A., S.Y., and A.K.S.; Project administration: S.O.A., Y.W.N., and A.K.S.; Funding acquisition: A.K.S.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Data availability
Data will be made available on request.
References
- [1].Mota A, Oltra SS, Selenica P, Moiola CP, Casas–Arozamena C, Lopez–Gil C, Diaz E, Gatius S, Ruiz–Miro M, Calvo A, Rojo–Sebastian A, Hurtado P, Pineiro R, Colas E, Gil–Moreno A, Reis–Filho JS, Muinelo–Romay L, Abal M, Matias–Guiu X, Weigelt B, Moreno–Bueno G, Intratumor genetic heterogeneity and clonal evolution to decode endometrial cancer progression, Oncogene 41(13) (2022) 1835–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gatius S, Cuevas D, Fernandez C, Roman–Canal B, Adamoli V, Piulats JM, Eritja N, Martin-Satue M, Moreno–Bueno G, Matias–Guiu X, Tumor Heterogeneity in Endometrial Carcinoma: Practical Consequences, Pathobiology 85(1–2) (2018) 35–40. [DOI] [PubMed] [Google Scholar]
- [3].Setiawan VW, Yang HP, Pike MC, McCann SE, Yu H, Xiang YB, Wolk A, Wentzensen N, Weiss NS, Webb PM, van den Brandt PA, van de Vijver K, Thompson PJ, Australian G National Endometrial Cancer Study, Strom BL, Spurdle AB, Soslow RA, Shu XO, Schairer C, Sacerdote C, Rohan TE, Robien K, Risch HA, Ricceri F, Rebbeck TR, Rastogi R, Prescott J, Polidoro S, Park Y, Olson SH, Moysich KB, Miller AB, McCullough ML, Matsuno RK, Magliocco AM, Lurie G, Lu L, Lissowska J, Liang X, Lacey JV Jr., Kolonel LN, Henderson BE, Hankinson SE, Hakansson N, Goodman MT, Gaudet MM, Garcia–Closas M, Friedenreich CM, Freudenheim JL, Doherty J, De Vivo I, Courneya KS, Cook LS, Chen C, Cerhan JR, Cai H, Brinton LA, Bernstein L, Anderson KE, Anton-Culver H, Schouten LJ, Horn–Ross PL, Type I and II endometrial cancers: have they different risk factors?, Journal of clinical oncology : official journal of the American Society of Clinical Oncology 31(20) (2013) 2607–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Murali R, Soslow RA, Weigelt B, Classification of endometrial carcinoma: more than two types, The Lancet. Oncology 15(7) (2014) e268–78. [DOI] [PubMed] [Google Scholar]
- [5].Lu KH, Broaddus RR, Endometrial Cancer, The New England journal of medicine 383(21) (2020) 2053–2064. [DOI] [PubMed] [Google Scholar]
- [6].Bokhman JV, Two pathogenetic types of endometrial carcinoma, Gynecol Oncol 15(1) (1983) 10–7. [DOI] [PubMed] [Google Scholar]
- [7].Talhouk A, Hoang LN, McConechy MK, Nakonechny Q, Leo J, Cheng A, Leung S, Yang W, Lum A, Kobel M, Lee CH, Soslow RA, Huntsman DG, Gilks CB, McAlpine JN, Molecular classification of endometrial carcinoma on diagnostic specimens is highly concordant with final hysterectomy: Earlier prognostic information to guide treatment, Gynecol Oncol 143(1) (2016) 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Stelloo E, Nout RA, Osse EM, Jurgenliemk–Schulz IJ, Jobsen JJ, Lutgens LC, van der Steen-Banasik EM, Nijman HW, Putter H, Bosse T, Creutzberg CL, Smit VT, Improved Risk Assessment by Integrating Molecular and Clinicopathological Factors in Early–stage Endometrial Cancer–Combined Analysis of the PORTEC Cohorts, Clin Cancer Res 22(16) (2016) 4215–24. [DOI] [PubMed] [Google Scholar]
- [9].Talhouk A, McConechy MK, Leung S, Yang W, Lum A, Senz J, Boyd N, Pike J, Anglesio M, Kwon JS, Karnezis AN, Huntsman DG, Gilks CB, McAlpine JN, Confirmation of ProMisE: A simple, genomics–based clinical classifier for endometrial cancer, Cancer 123(5) (2017) 802–813. [DOI] [PubMed] [Google Scholar]
- [10].Akiyama A, Minaguchi T, Fujieda K, Hosokawa Y, Nishida K, Shikama A, Tasaka N, Sakurai M, Ochi H, Satoh T, Abnormal accumulation of p53 predicts radioresistance leading to poor survival in patients with endometrial carcinoma, Oncol Lett 18(6) (2019) 5952–5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zheng W, Xiang L, Fadare O, Kong B, A proposed model for endometrial serous carcinogenesis, Am J Surg Pathol 35(1) (2011) e1–e14. [DOI] [PubMed] [Google Scholar]
- [12].Miller DS, Filiaci VL, Mannel RS, Cohn DE, Matsumoto T, Tewari KS, DiSilvestro P, Pearl ML, Argenta PA, Powell MA, Zweizig SL, Warshal DP, Hanjani P, Carney ME, Huang H, Cella D, Zaino R, Fleming GF, Carboplatin and Paclitaxel for Advanced Endometrial Cancer: Final Overall Survival and Adverse Event Analysis of a Phase III Trial (NRG Oncology/GOG0209), J Clin Oncol 38(33) (2020) 3841–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kampan NC, Madondo MT, McNally OM, Quinn M, Plebanski M, Paclitaxel and Its Evolving Role in the Management of Ovarian Cancer, Biomed Res Int 2015 (2015) 413076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Winer EP, Berry DA, Woolf S, Duggan D, Kornblith A, Harris LN, Michaelson RA, Kirshner JA, Fleming GF, Perry MC, Graham ML, Sharp SA, Keresztes R, Henderson IC, Hudis C, Muss H, Norton L, Failure of higher–dose paclitaxel to improve outcome in patients with metastatic breast cancer: cancer and leukemia group B trial 9342, J Clin Oncol 22(11) (2004) 2061–8. [DOI] [PubMed] [Google Scholar]
- [15].Yin S, Bhattacharya R, Cabral F, Human mutations that confer paclitaxel resistance, Mol Cancer Ther 9(2) (2010) 327–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Thottassery JV, Zambetti GP, Arimori K, Schuetz EG, Schuetz JD, p53–dependent regulation of MDR1 gene expression causes selective resistance to chemotherapeutic agents, Proceedings of the National Academy of Sciences of the United States of America 94(20) (1997) 11037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bush JA, Li G, Regulation of the Mdr1 isoforms in a p53–deficient mouse model, Carcinogenesis 23(10) (2002) 1603–7. [DOI] [PubMed] [Google Scholar]
- [18].De Vera AA, Gupta P, Lei Z, Liao D, Narayanan S, Teng Q, Reznik SE, Chen ZS, Immunooncology agent IPI–549 is a modulator of P–glycoprotein (P–gp, MDR1, ABCB1)–mediated multidrug resistance (MDR) in cancer: In vitro and in vivo, Cancer Lett 442 (2019) 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Callies S, de Alwis DP, Harris A, Vasey P, Beijnen JH, Schellens JH, Burgess M, Aarons L, A population pharmacokinetic model for paclitaxel in the presence of a novel P–gp modulator, Zosuquidar Trihydrochloride (LY335979), Br J Clin Pharmacol 56(1) (2003) 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chico I, Kang MH, Bergan R, Abraham J, Bakke S, Meadows B, Rutt A, Robey R, Choyke P, Merino M, Goldspiel B, Smith T, Steinberg S, Figg WD, Fojo T, Bates S, Phase I study of infusional paclitaxel in combination with the P–glycoprotein antagonist PSC 833, J Clin Oncol 19(3) (2001) 832–42. [DOI] [PubMed] [Google Scholar]
- [21].Zhang Y, Sriraman SK, Kenny HA, Luther E, Torchilin V, Lengyel E, Reversal of Chemoresistance in Ovarian Cancer by Co–Delivery of a P–Glycoprotein Inhibitor and Paclitaxel in a Liposomal Platform, Mol Cancer Ther 15(10) (2016) 2282–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Subba Rao AV, Vishnu Vardhan MV, Subba Reddy NV, Srinivasa Reddy T, Shaik SP, Bagul C, Kamal A, Synthesis and biological evaluation of imidazopyridinyl–1,3,4–oxadiazole conjugates as apoptosis inducers and topoisomerase IIalpha inhibitors, Bioorganic chemistry 69 (2016) 7–19. [DOI] [PubMed] [Google Scholar]
- [23].Ferraro C, Quemeneur L, Fournel S, Prigent AF, Revillard JP, Bonnefoy–Berard N, The topoisomerase inhibitors camptothecin and etoposide induce a CD95–independent apoptosis of activated peripheral lymphocytes, Cell death and differentiation 7(2) (2000) 197–206. [DOI] [PubMed] [Google Scholar]
- [24].Mohammed HHH, Abd El–Hafeez AA, Ebeid K, Mekkawy AI, Abourehab MAS, Wafa EI, Alhaj–Suliman SO, Salem AK, Ghosh P, Abuo–Rahma GEA, Hayallah AM, Abbas SH, New 1,2,3–triazole linked ciprofloxacin–chalcones induce DNA damage by inhibiting human topoisomerase I& II and tubulin polymerization, J Enzyme Inhib Med Chem 37(1) (2022) 1346–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Saif MW, Diasio RB, Edotecarin: a novel topoisomerase I inhibitor, Clinical colorectal cancer 5(1) (2005) 27–36. [DOI] [PubMed] [Google Scholar]
- [26].Frumovitz M, Munsell MF, Burzawa JK, Byers LA, Ramalingam P, Brown J, Coleman RL, Combination therapy with topotecan, paclitaxel, and bevacizumab improves progression–free survival in recurrent small cell neuroendocrine carcinoma of the cervix, Gynecologic oncology 144(1) (2017) 46–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Aoki D, Katsumata N, Nakanishi T, Kigawa J, Fujiwara K, Takehara K, Kamiura S, Hiura M, Hatae M, Sugiyama T, Ochiai K, Noda K, A phase II clinical trial of topotecan in Japanese patients with relapsed ovarian carcinoma, Japanese journal of clinical oncology 41(3) (2011) 320–7. [DOI] [PubMed] [Google Scholar]
- [28].Holloway RW, Treatment options for endometrial cancer: experience with topotecan, Gynecologic oncology 90(3 Pt 2) (2003) S28–33. [DOI] [PubMed] [Google Scholar]
- [29].Pendleton M, Lindsey RH Jr., Felix CA, Grimwade D, Osheroff N, Topoisomerase II and leukemia, Annals of the New York Academy of Sciences 1310 (2014) 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Oppegard LM, Delgado JL, Kulkarni CA, Towle TR, Hart DE, Williams BP, Lentz SRC, Norris BJ, Flory CM, Schumacher RJ, Murry DJ, Kerns RJ, Hiasa H, Novel N–1 substituted fluoroquinolones inhibit human topoisomerase I activity and exhibit anti–proliferative activity, Investigational new drugs 37(2) (2019) 378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rohena CC, Mooberry SL, Recent progress with microtubule stabilizers: new compounds, binding modes and cellular activities, Natural product reports 31(3) (2014) 335–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lambert A, Gavoille C, Conroy T, Current status on the place of FOLFIRINOX in metastatic pancreatic cancer and future directions, Therap Adv Gastroenterol 10(8) (2017) 631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Irisawa A, Takeno M, Watanabe K, Takahashi H, Mitsunaga S, Ikeda M, Incidence of and risk factors for severe neutropenia during treatment with the modified FOLFIRINOX therapy in patients with advanced pancreatic cancer, Sci Rep 12(1) (2022) 15574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chrzanowska A, Roszkowski P, Bielenica A, Olejarz W, Stepien K, Struga M, Anticancer and antimicrobial effects of novel ciprofloxacin fatty acids conjugates, Eur J Med Chem 185 (2020) 111810. [DOI] [PubMed] [Google Scholar]
- [35].MIRZAEI M, FOROUMADI A, Synthesis and In–vitro Antibacterial Activity of N–Piperazinyl Quinolone Derivatives with a 2–Thienyl Group, Pharmacy and Pharmacology Communications 6(8) (2000) 351–354. [Google Scholar]
- [36].Nieto MJ, Alovero FL, Manzo RH, Mazzieri MR, Benzenesulfonamide analogs of fluoroquinolones. Antibacterial activity and QSAR studies, Eur J Med Chem 40(4) (2005) 361–9. [DOI] [PubMed] [Google Scholar]
- [37].Rajabalian S, Foroumadi A, Shafiee A, Emami S, Functionalized N(2–oxyiminoethyl) piperazinyl quinolones as new cytotoxic agents, J Pharm Pharm Sci 10(2) (2007) 153–8. [PubMed] [Google Scholar]
- [38].Hart M, Acott S, Physical and chemical stability of Taxotere (docetaxel) one–vial (20 mg/ml) infusion solution following refrigerated storage, Ecancermedicalscience 4 (2010) 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Qu W, Zhao Y, Wang X, Qi Y, Zhou C, Hua Y, Hou J, Jiang SW, Culture characters, genetic background, estrogen/progesterone receptor expression, and tumorigenic activities of frequently used sixteen endometrial cancer cell lines, Clin Chim Acta 489 (2019) 225–232. [DOI] [PubMed] [Google Scholar]
- [40].Theisen ER, Gajiwala S, Bearss J, Sorna V, Sharma S, Janat–Amsbury M, Reversible inhibition of lysine specific demethylase 1 is a novel anti–tumor strategy for poorly differentiated endometrial carcinoma, BMC Cancer 14 (2014) 752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hangas A, Aasumets K, Kekalainen NJ, Paloheina M, Pohjoismaki JL, Gerhold JM, Goffart S, Ciprofloxacin impairs mitochondrial DNA replication initiation through inhibition of Topoisomerase 2, Nucleic acids research 46(18) (2018) 9625–9636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Elmore SA, Dixon D, Hailey JR, Harada T, Herbert RA, Maronpot RR, Nolte T, Rehg JE, Rittinghausen S, Rosol TJ, Satoh H, Vidal JD, Willard–Mack CL, Creasy DM, Recommendations from the INHAND Apoptosis/Necrosis Working Group, Toxicologic pathology 44(2) (2016) 173–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ramos A, Sadeghi S, Tabatabaeian H, Battling Chemoresistance in Cancer: Root Causes and Strategies to Uproot Them, Int J Mol Sci 22(17) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Alhaj–Suliman SO, Wafa EI, Salem AK, Engineering nanosystems to overcome barriers to cancer diagnosis and treatment, Adv Drug Deliv Rev 189 (2022) 114482. [DOI] [PubMed] [Google Scholar]
- [45].Gerrie AS, Power MM, Shepherd JD, Savage KJ, Sehn LH, Connors JM, Chemoresistance can be overcome with high–dose chemotherapy and autologous stem–cell transplantation for relapsed and refractory Hodgkin lymphoma, Ann Oncol 25(11) (2014) 2218–2223. [DOI] [PubMed] [Google Scholar]
- [46].Garcia–Fuente A, Vazquez F, Vieitez JM, Garcia Alonso FJ, Martin JI, Ferrer J, CISNE: An accurate description of dose–effect and synergism in combination therapies, Sci Rep 8(1) (2018) 4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ledermann JA, First–line treatment of ovarian cancer: questions and controversies to address, Ther Adv Med Oncol 10 (2018) 1758835918768232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Rose PG, First–Line Chemotherapy for Ovarian Cancer: Inferences From Recent Studies, Oncologist 21(11) (2016) 1286–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, Yeger H, Combination therapy in combating cancer, Oncotarget 8(23) (2017) 38022–38043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mc Gee MM, Targeting the Mitotic Catastrophe Signaling Pathway in Cancer, Mediators Inflamm 2015 (2015) 146282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sausville E, Lorusso P, Carducci M, Carter J, Quinn MF, Malburg L, Azad N, Cosgrove D, Knight R, Barker P, Zabludoff S, Agbo F, Oakes P, Senderowicz A, Phase I dose–escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors, Cancer Chemother Pharmacol 73(3) (2014) 539–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Scagliotti G, Kang JH, Smith D, Rosenberg R, Park K, Kim SW, Su WC, Boyd TE, Richards DA, Novello S, Hynes SM, Myrand SP, Lin J, Smyth EN, Wijayawardana S, Lin AB, Pinder–Schenck M, Phase II evaluation of LY2603618, a first–generation CHK1 inhibitor, in combination with pemetrexed in patients with advanced or metastatic non–small cell lung cancer, Invest New Drugs 34(5) (2016) 625–35. [DOI] [PubMed] [Google Scholar]
- [53].Albitar L, Pickett G, Morgan M, Davies S, Leslie KK, Models representing type I and type II human endometrial cancers: Ishikawa H and Hec50co cells, Gynecol Oncol 106(1) (2007) 52–64. [DOI] [PubMed] [Google Scholar]
- [54].Albitar L, Carter MB, Davies S, Leslie KK, Consequences of the loss of p53, RB1, and PTEN: relationship to gefitinib resistance in endometrial cancer, Gynecol Oncol 106(1) (2007) 94–104. [DOI] [PubMed] [Google Scholar]
- [55].Khing TM, Choi WS, Kim DM, Po WW, Thein W, Shin CY, Sohn UD, The effect of paclitaxel on apoptosis, autophagy and mitotic catastrophe in AGS cells, Sci Rep 11(1) (2021) 23490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mekkawy AI, Naguib YW, Alhaj–Suliman SO, Wafa EI, Ebeid K, Acri T, Salem AK, Paclitaxel anticancer activity is enhanced by the MEK 1/2 inhibitor PD98059 in vitro and by PD98059–loaded nanoparticles in BRAF(V600E) melanoma–bearing mice, Int J Pharm 606 (2021) 120876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bozec A, Ruffion A, Decaussin M, Andre J, Devonec M, Benahmed M, Mauduit C, Activation of caspases−3, −6, and −9 during finasteride treatment of benign prostatic hyperplasia, The Journal of clinical endocrinology and metabolism 90(1) (2005) 17–25. [DOI] [PubMed] [Google Scholar]
- [58].Meng X, Brachova P, Yang S, Xiong Z, Zhang Y, Thiel KW, Leslie KK, Knockdown of MTDH sensitizes endometrial cancer cells to cell death induction by death receptor ligand TRAIL and HDAC inhibitor LBH589 co–treatment, PloS one 6(6) (2011) e20920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Park MS, Okochi H, Benet LZ, Is Ciprofloxacin a Substrate of P–glycoprotein?, Arch Drug Inf 4(1) (2011) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Patil Y, Sadhukha T, Ma L, Panyam J, Nanoparticle–mediated simultaneous and targeted delivery of paclitaxel and tariquidar overcomes tumor drug resistance, Journal of controlled release : official journal of the Controlled Release Society 136(1) (2009) 21–9. [DOI] [PubMed] [Google Scholar]
- [61].Kelly RJ, Robey RW, Chen CC, Draper D, Luchenko V, Barnett D, Oldham RK, Caluag Z, Frye AR, Steinberg SM, Fojo T, Bates SE, A pharmacodynamic study of the P–glycoprotein antagonist CBT-1(R) in combination with paclitaxel in solid tumors, The oncologist 17(4) (2012) 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kelly RJ, Draper D, Chen CC, Robey RW, Figg WD, Piekarz RL, Chen X, Gardner ER, Balis FM, Venkatesan AM, Steinberg SM, Fojo T, Bates SE, A pharmacodynamic study of docetaxel in combination with the P–glycoprotein antagonist tariquidar (XR9576) in patients with lung, ovarian, and cervical cancer, Clin Cancer Res 17(3) (2011) 569–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Fox E, Widemann BC, Pastakia D, Chen CC, Yang SX, Cole D, Balis FM, Pharmacokinetic and pharmacodynamic study of tariquidar (XR9576), a P–glycoprotein inhibitor, in combination with doxorubicin, vinorelbine, or docetaxel in children and adolescents with refractory solid tumors, Cancer Chemother Pharmacol 76(6) (2015) 1273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Pusztai L, Wagner P, Ibrahim N, Rivera E, Theriault R, Booser D, Symmans FW, Wong F, Blumenschein G, Fleming DR, Rouzier R, Boniface G, Hortobagyi GN, Phase II study of tariquidar, a selective P–glycoprotein inhibitor, in patients with chemotherapy–resistant, advanced breast carcinoma, Cancer 104(4) (2005) 682–91. [DOI] [PubMed] [Google Scholar]
- [65].Ruff P, Vorobiof DA, Jordaan JP, Demetriou GS, Moodley SD, Nosworthy AL, Werner ID, Raats J, Burgess LJ, A randomized, placebo–controlled, double–blind phase 2 study of docetaxel compared to docetaxel plus zosuquidar (LY335979) in women with metastatic or locally recurrent breast cancer who have received one prior chemotherapy regimen, Cancer Chemother Pharmacol 64(4) (2009) 763–8. [DOI] [PubMed] [Google Scholar]
- [66].Didziapetris R, Japertas P, Avdeef A, Petrauskas A, Classification analysis of P–glycoprotein substrate specificity, Journal of drug targeting 11(7) (2003) 391–406. [DOI] [PubMed] [Google Scholar]
- [67].Singh S, Prasad NR, Chufan EE, Patel BA, Wang YJ, Chen ZS, Ambudkar SV, Talele TT, Design and synthesis of human ABCB1 (P–glycoprotein) inhibitors by peptide coupling of diverse chemical scaffolds on carboxyl and amino termini of (S)–valine–derived thiazole amino acid, Journal of medicinal chemistry 57(10) (2014) 4058–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Dang X, Ogbu SC, Zhao J, Nguyen LNT, Cao D, Nguyen LN, Khanal S, Schank M, Thakuri BKC, Wu XY, Morrison ZD, Zhang J, Li Z, El Gazzar M, Ning S, Wang L, Wang Z, Moorman JP, Yao ZQ, Inhibition of topoisomerase IIA (Top2alpha) induces telomeric DNA damage and T cell dysfunction during chronic viral infection, Cell Death Dis 11(3) (2020) 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Papadimitriou CA, Fountzilas G, Bafaloukos D, Bozas G, Kalofonos H, Pectasides D, Aravantinos G, Bamias A, Dimopoulos MA, Hellenic Co–operative Oncology G, Paclitaxel, topotecan, and carboplatin in metastatic endometrial cancinoma: a Hellenic Co–operative Oncology Group (HeCOG) study, Gynecologic oncology 111(1) (2008) 27–34. [DOI] [PubMed] [Google Scholar]
- [70].Stathopoulos GP, Dimitroulis J, Antoniou D, Katis C, Tsavdaridis D, Armenaki O, Marosis C, Michalopoulou P, Grigoratou T, Stathopoulos J, Front–line paclitaxel and irinotecan combination chemotherapy in advanced non–small–cell lung cancer: a phase I–II trial, British journal of cancer 93(10) (2005) 1106–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Jordan MA, Wilson L, Microtubules as a target for anticancer drugs, Nature reviews. Cancer 4(4) (2004) 253–65. [DOI] [PubMed] [Google Scholar]
- [72].Nogales E, Structural insights into microtubule function, Annual review of biochemistry 69 (2000) 277–302. [DOI] [PubMed] [Google Scholar]
- [73].Badal S, Her YF, Maher LJ 3rd, Nonantibiotic Effects of Fluoroquinolones in Mammalian Cells, J Biol Chem 290(36) (2015) 22287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Cao G, Zhu Y, Xie X, Chen Y, Yu J, Zhang J, Chen Z, Pang L, Zhang Y, Shi Y, Pharmacokinetics and pharmacodynamics of levofloxacin in bronchial mucosa and lung tissue of patients undergoing pulmonary operation, Exp Ther Med 20(1) (2020) 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.