

Abstract
lovo‐cel (bb1111; LentiGlobin for sickle cell disease [SCD]) gene therapy (GT) comprises autologous transplantation of hematopoietic stem and progenitor cells transduced with the BB305 lentiviral vector encoding a modified β‐globin gene (βA‐T87Q) to produce anti‐sickling hemoglobin (HbAT87Q). The efficacy and safety of lovo‐cel for SCD are being evaluated in the ongoing phase 1/2 HGB‐206 study (ClinicalTrials.gov: NCT02140554). The treatment process evolved over time, using learnings from outcomes in the initial patients to optimize lovo‐cel's benefit–risk profile. Following modest expression of HbAT87Q in the initial patients (Group A, n = 7), alterations were made to the treatment process for patients subsequently enrolled in Group B (n = 2, patients B1 and B2), including improvements to cell collection and lovo‐cel manufacturing. After 6 months, median Group A peripheral blood vector copy number (≥0.08 c/dg) and HbAT87Q levels (≥0.46 g/dL) were inadequate for substantial clinical effect but stable and sustained over 5.5 years; both markedly improved in Group B (patient B1: ≥0.53 c/dg and ≥2.69 g/dL; patient B2: ≥2.14 c/dg and ≥6.40 g/dL, respectively) and generated improved biologic and clinical efficacy in Group B, including higher total hemoglobin and decreased hemolysis. The safety of the lovo‐cel for SCD treatment regimen largely reflected the known side effects of HSPC collection, busulfan conditioning regimen, and underlying SCD; acute myeloid leukemia was observed in two patients in Group A and deemed unlikely related to insertional oncogenesis. Changes made during development of the lovo‐cel treatment process were associated with improved outcomes and provide lessons for future SCD GT studies.
Treatment process evolution. *The target TNC for BMH was ≥6 × 108/kg per patient. The Group B1 patient received lovo‐cel produced using both the original and refined manufacturing process, and the Group B2 patient received lovo‐cel produced using only the refined manufacturing process. Both Group B patients received lovo‐cel manufactured from HSPCs collected using BMH; however, the Group B2 patient also underwent rescue HSPC collection by plerixafor mobilization/apheresis for the exploratory evaluation of its safety and enhancement of the lovo‐cel manufacturing process. The Group B2 patient did not receive lovo‐cel manufactured from HSPCs collected using plerixafor mobilization/apheresis.
Figure adapted from N Engl J Med, Kanter, J. et al., Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease, 386, 617‐628. Copyright © 2022 Massachusetts Medical Society. Reprinted with permission.
AUC, area under the curve; BMH, bone marrow harvest; DP, drug product; HSPC, hematopoietic stem and progenitor cell; TNC, total nucleated cell count.

1. INTRODUCTION
Sickle cell disease (SCD) is a complex, progressive, and debilitating genetic disease caused by a single point mutation in the β‐globin gene. 1 , 2 Sickle hemoglobin (HbS) production and its subsequent polymerization causes red blood cell (RBC) sickling, which is responsible for recurrent vaso‐occlusive events, chronic hemolytic anemia, and progressive vasculopathy. These clinical manifestations of SCD are associated with reduced quality of life, significant morbidity, and early mortality. 1 , 2 , 3
Current management of SCD relies on disease‐modifying therapies that significantly reduce but do not eliminate symptoms or halt disease progression. 4 , 5 Human leukocyte antigen‐matched sibling allogeneic hematopoietic stem cell transplantation is potentially curative for SCD 3 , 6 , 7 , 8 ; however, it is associated with graft‐versus‐host disease (GVHD) and graft rejection among other causes of transplant‐related mortality. Furthermore, only 14%–20% of patients have matched sibling donors. 1 , 6 , 7 , 9 , 10
lovo‐cel (bb1111; LentiGlobin for SCD) gene therapy (GT) is administered by autologous hematopoietic stem and progenitor cells (HSPCs) transplantation. The autologous HSPCs are transduced with the BB305 lentiviral vector (LVV) encoding the modified β‐globin gene (βA‐T87Q), which results in the production of anti‐sickling hemoglobin (HbAT87Q). 11 , 12 By utilizing autologous transplantation, lovo‐cel all but eliminates two important limitations of allogeneic transplantation, GVHD and graft rejection, and may provide a curative treatment while abrogating the need for a well‐matched donor. HbAT87Q is a modified adult hemoglobin (HbA) specifically designed with a single amino acid substitution (threonine to glutamine at position 87; T87Q) to sterically inhibit HbS polymerization, while otherwise maintaining the same morphology and function as HbA (Figure S1). The T87Q mutation is traceable and enables differentiation of endogenous HbAT87Q production from transfused HbA, and the genomic location of BB305 LVV insertion sites can be precisely identified during safety monitoring via the long terminal repeat sequences.
Pre‐clinical studies have demonstrated that lovo‐cel‐mediated endogenous expression of βA‐T87Q strongly reduces HbS expression and inhibits HbS polymerization. 11 , 12 , 13 These data, together with the first SCD patient study of lovo‐cel treatment, 14 , 15 provide proof of concept and have guided the ongoing phase 1/2 HGB‐206 study, which is evaluating the efficacy and safety of lovo‐cel for SCD. Interim analysis of the pivotal HGB‐206 cohort (Group C) demonstrated the sustained production of HbAT87Q in 85% of RBCs, leading to near‐normalization of key hemolysis markers and the complete resolution of severe vaso‐occlusive events. 16 Here, we describe the evolution of the lovo‐cel treatment process in the initial cohorts (Groups A and B) of the HGB‐206 study and its impact on biologic and clinical outcomes. Importantly, these changes have laid the foundation for the current genetic therapies in SCD.
2. METHODS
2.1. Study design and overview
HGB‐206 is an ongoing phase 1/2, non‐randomized, open‐label, multi‐site, single‐dose clinical study of lovo‐cel for SCD and is the largest study of GT in SCD to date. HGB‐206 is being conducted at 11 sites across the United States in accordance with the Declaration of Helsinki and the International Conference on Harmonization guidelines for Good Clinical Practice. Enrolled patients completed a written informed consent form prior to screening. HGB‐206 was initiated as a phase 1 study to evaluate the safety, preliminary efficacy, and pharmacokinetics (PK)/pharmacodynamics (PD) of lovo‐cel, and to assess the risk–benefit profile in patients with SCD. The study has been subsequently updated to a phase 1/2 registrational study to evaluate efficacy endpoints (ClinicalTrials.gov: NCT02140554; Trial: HGB‐206).
The GT treatment process of lovo‐cel for SCD was modified during the study, leading to the retrospective designation of three sequential cohorts: Groups A, B, and C. Each cohort was determined by the specific treatment process used at that time. Here, we describe the evolution of the changes made in Groups A and B; data from the ongoing pivotal evaluation of Group C have been reported separately. 16 The lovo‐cel treatment process involves several steps: (1) pre‐collection preparation and HSPC collection, 17 , 18 (2) lovo‐cel manufacturing via the ex vivo transduction of autologous HSPCs with the BB305 LVV containing the βA‐T87Q transgene, 17 (3) myeloablative conditioning, 17 , 18 (4) lovo‐cel infusion, 18 , 19 and (5) engraftment of HSPCs leading to the production of HbAT87Q with subsequent follow‐up 20 , 21 (Figure 1A). Group A patients received lovo‐cel produced using the original manufacturing process, including lovo‐cel drug product (DP) that was developed from bone marrow (BM)‐harvested CD34+ HSPCs. Following durable but suboptimal expression of HbAT87Q in Group A, substantial changes were instituted in the lovo‐cel treatment process to optimize biologic and clinical outcomes and were assessed in Group B (Figure 1B). Subsequent patients enrolled into Group B received lovo‐cel produced using a refined manufacturing process in addition to other changes to the treatment protocol. Group B was further divided into Group B1, consisting of one patient who received lovo‐cel produced using both the original and refined manufacturing process, and Group B2, consisting of one patient who received lovo‐cel produced only by the refined manufacturing process (Figure 1B). Upon completion of a 2‐year follow‐up period, eligible patients from HGB‐206 are followed for an additional 13 years as part of a long‐term follow‐up study (LTF‐307; NCT04628585) and data from LTF‐307 are also reported here.
FIGURE 1.
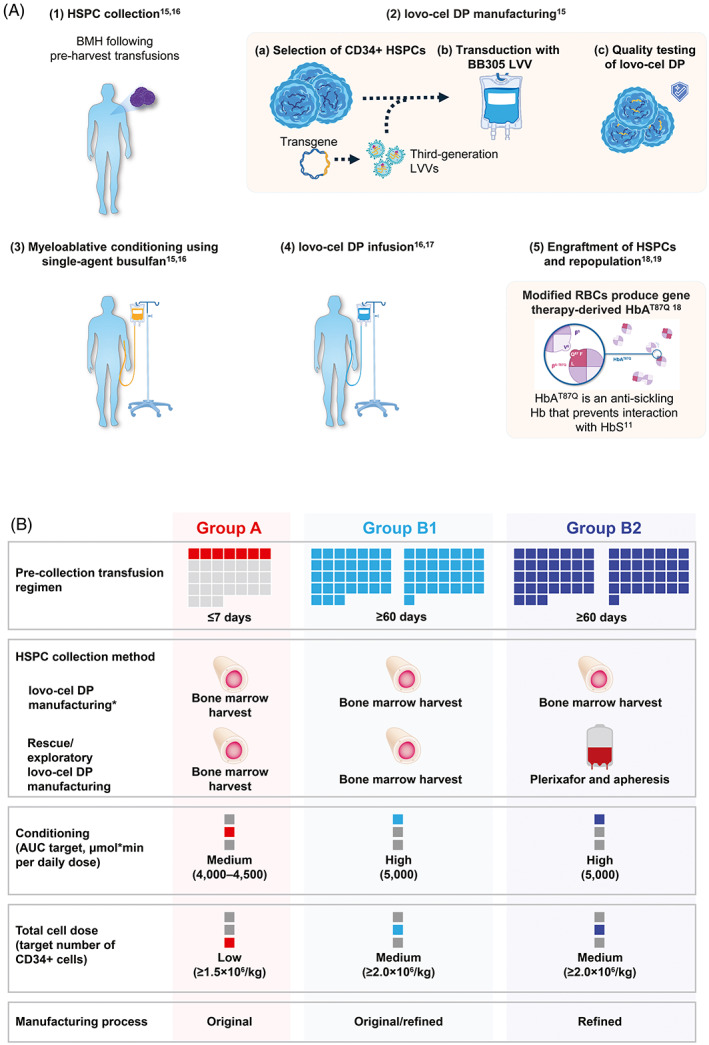
Overview of the lovo‐cel treatment process and HGB‐206 study design evolution. (A) Gene therapy treatment process of lovo‐cel for SCD. (B) Treatment process evolution. *The target TNC for BMH was ≥6 × 108/kg per patient. The Group B1 patient received lovo‐cel produced using both the original and refined manufacturing process, and the Group B2 patient received lovo‐cel produced using only the refined manufacturing process. Both Group B patients received lovo‐cel manufactured from HSPCs collected using BMH; however, the Group B2 patient also underwent rescue HSPC collection by plerixafor mobilization/apheresis for the exploratory evaluation of its safety and enhancement of the lovo‐cel manufacturing process. The Group B2 patient did not receive lovo‐cel manufactured from HSPCs collected using plerixafor mobilization/apheresis. Figure adapted from N Engl J Med, Kanter, J. et al., Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease, 386, 617–628. Copyright © 2022 Massachusetts Medical Society. Reprinted with permission. 16 AUC, area under the curve; BMH, bone marrow harvest; DP, drug product; HbAT87Q, Hb with modified β‐globin gene (βA‐T87Q); HbS, sickle Hb; HSPC, hematopoietic stem and progenitor cell; LVV, lentiviral vector; RBC, red blood cell; SCD, sickle cell disease; TNC, total nucleated cell count. [Color figure can be viewed at wileyonlinelibrary.com]
The methodology reported below is applicable to both Groups A and B, unless otherwise specified.
2.2. Patient eligibility
Patients were ≥18 years of age with a documented βS/βS, βS/β0, or βS/β+ genotype and a severe SCD diagnosis as judged by the investigator with either recurrent severe vaso‐occlusive crises (VOCs) (≥2 per year in the prior 2 years), acute chest syndrome (ACS) (≥2 in prior 2 years, with ≥1 episode in the prior year), history of overt stroke, or tricuspid regurgitant jet velocity (TRJV) >2.5 m/s. A severe VOC was defined as an episode of pain lasting >2 h and requiring care at a medical facility, including priapism episodes lasting >2 h requiring care at a medical facility, and patients also must have prior hydroxyurea (HU) intolerance or failure. Patients were also required to have a Karnofsky performance score of ≥60. Further details of the eligibility criteria are provided in Table S1.
2.2.1. Alterations to eligibility for Group B
Given the potential for regular transfusions to suppress dyserythropoiesis in the bone marrow, it was hypothesized that patients with severe SCD who were on a regular transfusion program may have improved outcomes with lovo‐cel compared with those not on a transfusion program. The eligibility criteria for Group B were therefore expanded to include patients who satisfied other clinical severity criteria prior to instituting chronic red cell therapy (CRCT), including ≥2 severe, recurrent VOCs per year in the 2 years before initiation of CRCT and/or ≥2 episodes of ACS with ≥1 episode in the year before initiation of CRCT. Further details of protocol changes to inclusion and exclusion criteria between groups are outlined in Table S2.
2.3. Pre‐harvest transfusion and cell collection
In Group A, simple or exchange packed RBC (pRBC) transfusions were required ≤7 days prior to BM harvest (BMH) to establish a mandatory Hb target of 10–12 g/dL and an HbS proportion of <30% of total Hb. A BMH volume of 15–20 ml/kg was required for both lovo‐cel manufacture and rescue aliquot of unmodified cells. For patients who could not obtain the necessary target cell dose, the harvest was repeated until sufficient cells were obtained. Patients were consented to have up to 30 ml of the collected BM used for studies exploring improvements to the lovo‐cel manufacturing process.
2.3.1. Alterations to pre‐harvest transfusion and cell collection for Group B
Group B patients underwent mandatory simple or exchange pRBC transfusions for ≥60 days prior to collection in order to reduce stress erythropoiesis. Both Group B patients received lovo‐cel manufactured from HSPCs collected using BMH; however, the Group B2 patient also underwent rescue HSPC collection by plerixafor mobilization/apheresis for the exploratory evaluation of its safety and enhancement of the lovo‐cel manufacturing process 22 (Table S2). The Group B2 patient did not receive lovo‐cel manufactured from HSPCs collected using plerixafor mobilization/apheresis.
2.4. lovo‐cel DP manufacturing
lovo‐cel was produced by transducing autologous HSPCs with the BB305 LVV encoding βA‐T87Q using an original manufacturing process in Group A that was subsequently refined in Group B to increase yield and improve transduction efficiency. 22
2.4.1. Alterations to lovo‐cel DP manufacturing for Group B
The Group B1 patient received one lovo‐cel DP lot that was manufactured using the original process, and another lot that was manufactured using the refined process. The Group B2 patient received two lovo‐cel DP lots that were both manufactured using the refined process.
2.5. Myeloablative conditioning and lovo‐cel DP infusion
Before treatment, myeloablative conditioning with single‐agent busulfan was administered to patients over 4 days at a starting dose of 3.2 mg/kg/day or 0.8 mg/kg every 6 h. The target busulfan area under the curve (AUC) was 4000–4500 μmol × min per daily dose in Group A. One week after the start of conditioning, and after a minimum 72‐h washout period, patients received a lovo‐cel infusion at a cell dose of ≥1.5 × 106 CD34+ cells/kg (study Day 1).
2.5.1. Alterations to myeloablative conditioning and lovo‐cel DP infusion for Group B
Given the insufficient engraftment of modified stem cells in Group A, the target busulfan AUC in Group B was increased to 5000 μmol × min per daily dose and PK assessments were required. Adequate washout was retrospectively confirmed, with measurements 48 and 72 h after the final dose of busulfan. Furthermore, the lovo‐cel DP cell dose was also increased for Group B to ≥2.0 × 106 CD34+ cells/kg based on published data suggesting that increased cell doses may promote improved clinical outcomes. 23
2.6. Study endpoints
2.6.1. Biologic efficacy
Peripheral blood (PB) samples were collected to determine functional βA‐T87Q‐globin expression, Hb proportions (HbS/HbAT87Q), and total non‐transfused Hb (HbS + HbF + HbA2 + HbAT87Q) using high‐performance liquid chromatography at multiple pre‐determined time points. Vector copy number (VCN) was measured in PB at the same time points using quantitative polymerase chain reaction. Correlation analyses assessing lovo‐cel DP VCN, 6‐month PB VCN, and 6‐month HbAT87Q levels were performed.
2.6.2. Clinical efficacy
The frequency of severe VOC + ACS after lovo‐cel infusion was compared with the 24 months prior to informed consent; VOCs and ACS were determined using the definitions outlined in the eligibility criteria. Other clinical outcome assessments included HU use, key hemolysis markers, and tests of cardiac function (TRJV and left ventricular ejection fraction [LVEF]).
2.6.3. Safety
Safety endpoints comprised successful neutrophil engraftment (absolute neutrophil count [ANC] ≥0.5 × 109/L for 3 days), platelet engraftment (platelets ≥50 × 109/L for 3 days without platelet transfusions), and the evaluation of adverse events (AEs). In all patients, the presence of replication‐competent lentivirus was monitored, and insertion sites were analyzed for clonal predominance. Additionally, any potential events of vector‐mediated insertional oncogenesis were assessed. Additional safety assessments included screening for irregular antibodies and evaluation of lymphocyte populations to assess immunologic reconstitution.
2.7. Sample size and statistical methods
Sample sizes for Groups A and B were not determined by formal statistical methods and all summary statistics were descriptive. The intent‐to‐treat (ITT) population included all patients who initiated any study procedures beginning with stem cell collection, and the transplant population included all patients who received lovo‐cel infusion. All efficacy endpoints were analyzed in the transplant population; safety endpoints were assessed in the ITT population for AEs related to cell collection, and in the transplant population for AEs related to conditioning or after lovo‐cel infusion.
3. RESULTS
3.1. Patient characteristics and disposition
Between February 2015 and January 2016, a total of 11 patients were enrolled in the HGB‐206 study. Of those, two patients withdrew (one withdrew consent and the other was withdrawn per investigator decision) and lovo‐cel treatment was administered to seven patients in Group A and two patients in Group B (Group B1, n = 1; Group B2, n = 1) (Figure S2). Patients were followed for a median of 61.5 months (min–max: 55.5–66.1) in Group A, 48.3 months in Group B1, and 44.4 months in Group B2, for a combined total of 43.5 patient‐years across Groups A and B, and we report data through to 17 February 2021. Data are reported as median (min–max) for Group A unless stated otherwise, and the values for the single patients in Group B1 and B2 are reported.
Patient characteristics were similar across all groups (Table 1). All patients had a βS/βS genotype. In the 2 years prior to informed consent, patients in Groups A, B1, and B2 had 4.5, 17.5, and 2.5 VOCs/year, respectively, and 1, 0, and 1 ACS/year, with elevated TRJV (>2.5 m/s) 24 , 25 in 1, 0, and 1 patient respectively. Two patients (28.6%) in Group A had a prior stroke and were receiving chronic pRBC transfusions at the time of consent. Six patients (85.7%) in Group A and both Group B patients (100%) had frequent or chronic pain and were receiving opioid medications. In the 3 months prior to informed consent, five patients (71.4%) in Group A and both Group B patients (100%) received HU.
TABLE 1.
Baseline demographic, and lovo‐cel treatment and drug.
| Characteristic | Group A (N = 7) | Group B1 (N = 1) | Group B2 (N = 1) |
|---|---|---|---|
| Patient characteristics | |||
| Age at ICF, year, median (min–max) | 26 (18–42) | 22 | 27 |
| Male, n (%) | 6 (85.7) | 1 | 1 |
| Race, n (%) | |||
| Black or African American | 7 (100) | 1 | 0 |
| Asian | 0 | 0 | 1 |
| Ethnicity, n (%) | |||
| Hispanic | 0 | 0 | 0 |
| Non‐Hispanic | 7 (100) | 1 | 1 |
| βS/βS genotype, n (%) | 7 (100) | 1 | 1 |
| SCD history | |||
| SCD history in 2 years prior to ICF | |||
| VOCs, events/year, median (min–max) | 4.5 (2.0–27.0) | 17.5 | 2.5 |
| ACS, events/year, median (min–max) | 1.0 (1.0–1.0) | 0 | 1.0 |
| Any pRBC transfusion prior to ICF, n (%) | 2 (28.6) | 1 | 1 |
| Any TRJV >2.5 m/s prior to ICF, n (%) a | 1 (14.3) | 0 | 1 |
| Any stroke prior to ICF, n (%) | 2 (28.6) | 0 | 0 |
| Opioids prior to ICF for acute/chronic pain, n (%) | 6 (85.7) | 1 | 1 |
| HU use 3 months prior to ICF, n (%) | 5 (71.4) | 1 | 1 |
| lovo‐cel treatment process characteristics | |||
| Number of BMHs, median (min–max) | 2 (1–4) | 3 | 2 |
| Busulfan AUC, μmol × min, median (min–max) | 4747 (4084–5290) | 5017 | 5256 |
| G‐CSF use post‐infusion, patients, n (%) | 3 (42.9) | 0 | 0 |
| Days to neutrophil engraftment, days, median (min–max) † | 22 (17–29) | 28 | 23 |
| Days to platelet engraftment, days, median (min–max) ‡ | 45 (29–63) | 54 | 31 |
| Hospitalization duration from conditioning to discharge, days, median (min–max) | 37 (29–54) | 46 | 36 |
| lovo‐cel drug product characteristics | |||
| Cell collection method | BMH | BMH | BMH |
| Manufacturing process | Original process | Both original and refined process § | Refined process § |
| lovo‐cel DP VCN, c/dg, median (min–max) | 0.6 (0.5–1.3) | 2.3 | 3.8 |
| %LVV+, median (min–max) | 27.7 (9.0–42.0) | 63.0 | 92.0 |
| Total CD34+, ×106 cells/kg, median (min–max) | 2.1 (1.6–5.1) | 2.2 | 3.2 |
| Total CD34hi/+, ×106 cells/kg, median (min–max) | 1.4 (1.1–2.8) | 1.3 | 2.7 |
| LT HSPCs, % CD34hi/+ HSPCs, median (min–max) | 58.0 (54.6–75.0) | 59.8 | 82.9 |
Abbreviations: ACS, acute chest syndrome; ANC, absolute neutrophil count; AUC, area under the curve; BMH, bone marrow harvest; c/dg, copies per diploid genome; DP, drug product; G‐CSF, granulocyte colony‐stimulating factor; HSPC, hematopoietic stem and progenitor cell; HU, hydroxyurea; ICF, informed consent form; LVV+, lentiviral vector positive; LT, long‐term; max, maximum; min, minimum; pRBC, packed red blood cell; SCD, sickle cell disease; TRJV, tricuspid regurgitant jet velocity; VCN, vector copy number; VOC, vaso‐occlusive crisis.
TRJV ≥2.5 m/s confers an increased risk for mortality, 24 , 25 and TRJV ≥2.5 m/s was reported in one Group A patient (>2.7 m/s) and the Group B2 patient (2.5 m/s).
Defined as the first day of ANC ≥0.5 × 109/L for three consecutive measurements on different days without receiving backup cells at any time during the neutropenic phase.
Defined as the first of three consecutive platelet measurements ≥50 × 109/L on different days without platelet transfusions for 7 days immediately preceding and during the evaluation period.
Patients in Group B were treated under an updated protocol with the Group B1 patient receiving lovo‐cel produced using both the original and refined manufacturing process, and the Group B2 patient receiving lovo‐cel produced using only the refined manufacturing process.
Group B patients had marked improvements in lovo‐cel DP and treatment characteristics compared with Group A (Table 1). Importantly, these improvements were greater in the Group B2 patient (lovo‐cel produced only using the refined manufacturing process) compared with the Group B1 patient (lovo‐cel produced using both the original and refined manufacturing process). Both Group B1 (5017 μmol × min) and B2 (5256 μmol × min) patients had higher busulfan dose AUC compared with Group A patients (4747 μmol × min). lovo‐cel DP VCN and percentage of LVV+ cells were substantially higher in Groups B2 (3.8 copies/diploid genome [c/dg]; 92%) and B1 (2.3 c/dg; 63%) patients compared with Group A (0.6 c/dg; 27.7%). The HSPC collection processes resulted in a substantial improvement in the quality and quantity of CD34+ cells in the Group B2 patients compared with the Group B1 patient and Group A patients (Table 1). These observations suggest that the collection process and modified manufacturing process improved lovo‐cel characteristics.
3.2. Biologic efficacy
PB VCN stabilized in all patients by 6 months after lovo‐cel infusion and remained stable through to the date of last follow‐up, suggesting that vector‐positive, long‐term repopulating stem cells are capable of sustaining long‐term erythropoiesis with HbAT87Q production (Figure 2A). From 6 months through last visit, there were marked improvements in biologic efficacy parameters in the Group B1 patient, and to a greater extent in the Group B2 patient, compared with Group A. First, PB VCN was ≥0.08, ≥0.53, and ≥2.14 c/dg from Month 6 through to last visit in Groups A, B1, and B2, respectively (Figure 2A); a similar trend was observed in the percentage of LVV+ cells over time (Figure S3). Second, the improved PB VCN generated an increase in HbAT87Q production of ≥0.46, ≥2.69, and ≥6.40 g/dL from Month 6 through to last visit in Groups A, B1, and B2 respectively (Figure 2B). Third, HbAT87Q expression was durable across Groups A, B1, and B2, contributing to 12%, 35%, and 54% of total Hb, respectively, at last visit (independent of transfusion) (Figure 2C). Fourth, increased HbAT87Q production reduced the HbS fraction to 69%, 55%, and 46% of total Hb in non‐transfused patients at last visit in Groups A, B1, and B2 respectively (Figure 2C). Finally, increased HbAT87Q production also corresponded with increased total Hb of ≥8.90, ≥9.80, and ≥12.30 g/dL from Month 6 through to last visit in Groups A, B1, and B2 respectively (Figure 2D). Of interest, HbF levels increased by as much as 1.42, 1.36, and 0.11 g/dL within 2–6 months after lovo‐cel infusion in Groups A, B1, and B2, respectively, and subsequently stabilized by ~12–18 months (Figure 2E).
FIGURE 2.
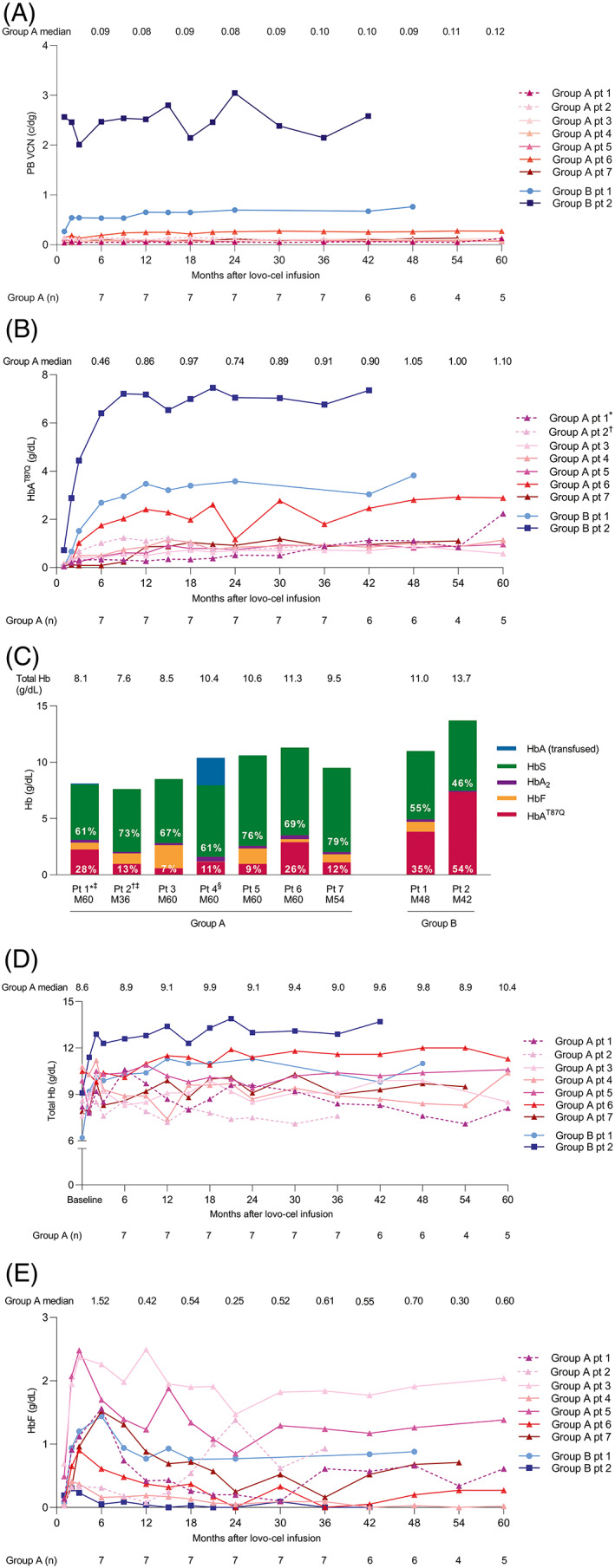
Hb fractions over time and at last visit following lovo‐cel infusion. (A) PB VCN. (B) HbAT87Q. (C) Hb fractions at last visit. (D) Total Hb. (E) HbF. Dashed lines indicate Group A patients 1 and 2 who, for an extended period post‐engraftment, were receiving regular transfusions and hydroxyurea; pRBC transfusion volume, day of transfusion, and transfusion type are reported in Table S3.*Patient diagnosed with AML in 2021. 27 †Patient diagnosed with myelodysplastic syndrome/AML in 2018 26 ; data for efficacy endpoints are shown up to last visit prior to diagnosis of malignancy. ‡Received regular transfusions and hydroxyurea for an extended period after lovo‐cel infusion, including the year prior to last visit; packed red blood cell transfusion volume, day of transfusion, and transfusion type are reported in Table S3. §Received a transfusion at last follow‐up. Total Hb baseline values (panel C) were defined as the average of the two most recent Hb assessments at/prior to screening; assessments were separated by >1 month, ≤24 months before informed consent, and excluded any patients who received a pRBC transfusion ≤3 months of each assessment; for patients receiving chronic transfusions, Hb values ≤24 months before the start of a regular transfusion program and separated by >1 month were used. AML, acute myeloid leukemia; c/dg, copies per diploid genome; Hb, hemoglobin; HbA, adult Hb; HbAT87Q, Hb with modified β‐globin gene (βA‐T87Q); HbF, fetal Hb; HbS, sickle Hb; M, month; pt, patient; PB, peripheral blood; pRBC, packed red blood cell; pt, patient; VCN, vector copy number. [Color figure can be viewed at wileyonlinelibrary.com]
Correlation analyses evaluated the relationships between lovo‐cel DP VCN, PB VCN, and HbAT87Q production. lovo‐cel DP VCN at infusion and PB VCN at Month 6 were highly correlated in Groups A and B (r = 0.93, p = 0.0003) (Figure S4A) suggesting long‐term engraftment and repopulation of transduced HSPCs. lovo‐cel DP VCN at infusion and HbAT87Q levels at Month 6 were also highly correlated (r = 0.90, p = 0.001) (Figure S4B), suggesting increased lovo‐cel DP βA‐T87Q transduction efficiency in Group B compared with Group A, with higher lovo‐cel DP VCN translating into higher production of HbAT87Q. Similarly, PB VCN and HbAT87Q levels at Month 6 were highly correlated (r = 0.99, p < 0.0001) (Figure S4C), demonstrating that higher PB VCN in Group B was associated with increased HbAT87Q production.
3.3. Clinical efficacy
Following lovo‐cel infusion, the median annualized VOC + ACS rate decreased from 5.0 (min–max: 2.5–27.5) to 0.7 (min–max: 0.0–21.1) in patients in Groups A and B with a history of VOC + ACS (n = 8) (Figure S5). This included an 82.6% reduction in Group A, a 79.1% reduction in the Group B1 patient, and a 100% reduction in the Group B2 patient. Some patients continued beyond 24 months in HGB‐206 while awaiting enrolment into the long‐term follow up study, LTF‐307; post‐follow‐up in HGB‐206, 3/7 patients in Group A and 1/2 patients in Group B experienced a VOE while in LTF‐307 with follow‐up to 66.1 months. There were no reports of stroke after lovo‐cel infusion, despite two patients in Group A with a history of stroke. Importantly, Group A patients 1 and 2 had an inadequate therapeutic response after lovo‐cel infusion due to low transduction efficiency, continued to receive regular transfusions (Table S3), and were restarted on HU after lovo‐cel treatment in an attempt to ameliorate SCD complications; no other patients received regular transfusions after lovo‐cel infusion.
Overall, key markers of hemolysis were reduced after lovo‐cel infusion in Groups A and B (Figure S6). Cardiac function assessments, including TRJV and LVEF, were stable after lovo‐cel infusion (Figure S7).
3.4. Safety
3.4.1. Bone marrow harvest
During the lovo‐cel treatment process, five patients (55.6%) in Group A and one patient (50.0%) in Group B who initiated stem cell collection had ≥1 AE attributed to BMH and classified as ≥Grade 3 (Table 2). All patients in Groups A and B who received lovo‐cel infusion had ≥1 AE attributed to conditioning that was classified as ≥Grade 3.
TABLE 2.
AEs during lovo‐cel treatment process.
| Number of patients with ≥1 AE, n (%) | Group A | Group B |
|---|---|---|
| Attributed to BMH a | N = 9 | N = 2 |
| Any AE | 9 (100.0) | 2 (100.0) |
| ≥Grade 3 AE | 5 (55.6) | 1 (50.0) |
| Serious AE | 2 (22.2) | 1 (50.0) |
| Attributed to conditioning † | N = 7 | N = 2 |
| Any AE | 7 (100.0) | 2 (100.0) |
| ≥Grade 3 AE | 7 (100.0) | 2 (100.0) |
| Serious AE | 3 (42.9) | 1 (50.0) |
| Post‐lovo‐cel infusion through to last visit † | N = 7 | N = 2 |
| Any AE | 7 (100.0) | 2 (100.0) |
| Any AE attributed by investigator to lovo‐cel infusion ‡ | 1 (14.3) | 1 (50.0) |
| ≥Grade 3 AE | 7 (100.0) | 2 (100.0) |
| Serious AE | 6 (85.7) | 2 (100.0) |
| ≥Grade 3 AE occurring in ≥2 Group A or ≥1 Group B patients | ||
| Thrombocytopenia | 6 (85.7) | 1 (50.0) |
| Neutropenia | 5 (71.5) | 2 (100.0) |
| Stomatitis | 5 (71.5) | 2 (100.0) |
| Febrile neutropenia | 4 (57.1) | 1 (50.0) |
| Sickle cell anemia with crisis | 4 (57.1) | 1 (50.0) |
| Pharyngeal inflammation | 2 (28.6) | 1 (50.0) |
| ACS | 2 (28.6) | 0 |
| AML § | 2 (28.6) | 0 |
| Bacteremia | 2 (28.6) | 0 |
| Pyrexia | 2 (28.6) | 0 |
| Leukopenia | 1 (14.3) | 2 (100.0) |
| Alanine aminotransferase increased | 1 (14.3) | 1 (50.0) |
| Aspartate aminotransferase increased | 1 (14.3) | 1 (50.0) |
| COVID‐19 pneumonia | 0 | 1 (50.0) |
| Dehydration | 0 | 1 (50.0) |
| Diarrhea | 0 | 1 (50.0) |
| Epistaxis | 0 | 1 (50.0) |
| Face edema | 0 | 1 (50.0) |
| Vomiting | 0 | 1 (50.0) |
| Serious AE occurring in ≥2 Group A or ≥1 Group B patients | ||
| Sickle cell anemia with crisis | 4 (57.1) | 1 (50.0) |
| ACS | 2 (28.6) | 0 |
| AML § | 2 (28.6) | 0 |
| Bacteremia | 2 (28.6) | 0 |
| Pyrexia | 2 (28.6) | 0 |
| COVID‐19 pneumonia | 0 | 1 (50.0) |
| Dehydration | 0 | 1 (50.0) |
| Diarrhea | 0 | 1 (50.0) |
| Face edema | 0 | 1 (50.0) |
| Vomiting | 0 | 1 (50.0) |
Note: Both Group B patients received lovo‐cel manufactured from HSPCs collected only using BMH; however, the Group B2 patient also underwent HSPC collection by plerixafor mobilization/apheresis for the exploratory evaluation of its safety and improvement of the lovo‐cel manufacturing process. The Group B2 patient did not receive any lovo‐cel manufactured from HSPCs collected using plerixafor mobilization/apheresis. The Group B2 patient had HSPCs collected using BMH on three separate occasions over 10 months, and on one occasion using plerixafor apheresis between the second and third BMH. AEs are ordered by frequency in Group A.
Abbreviations: ACS, acute chest syndrome; AE, adverse event; AML, acute myeloid leukemia; BMH, bone marrow harvest; COVID‐19, coronavirus disease 2019; HSPC, hematopoietic stem and progenitor cell.
Reported in intent‐to‐treat population.
Reported in the transplant population.
Two AEs were attributed by the investigator as possibly related to lovo‐cel infusion: a non‐serious event of hot flush and one case of AML.
3.4.2. Engraftment
Neutrophil engraftment (ANC ≥0.5 × 109/L for 3 consecutive days) was achieved at Days 22, 28, and 23, and platelet engraftment (platelet count ≥50 × 109/L for 3 consecutive days) was observed at Days 45, 54, and 31 for Groups A, B1, and B2 respectively (Table 1).
3.4.3. Adverse events
From lovo‐cel infusion through to last visit, all patients in Groups A and B had ≥1 AE classified as ≥Grade 3 and the most common were thrombocytopenia (85.7%), neutropenia (71.5%), and stomatitis (71.5%) in Group A, and leukopenia (100%), neutropenia (100%), and stomatitis (100%) in Group B (Table 2). Analysis of long‐term hematopoietic immune reconstitution demonstrated stabilization of CD4 levels (≥0.4 × 109/L) 3 months after infusion and rapid reconstitution of CD19 cells, without the need for immunomodulation, and may provide insight into when patients may resume normal activities.
Two cases of acute myeloid leukemia (AML) were reported in Group A. 26 , 27 The first case (Group A, patient 2) was initially diagnosed as myelodysplastic syndrome approximately 3 years after lovo‐cel infusion and subsequently progressed to AML that was fatal. 26 Further investigations revealed the absence of BB305 LVV integration in the CD34+ blast cell population and presence of monosomy 7. The case was assessed by the investigator as unlikely related to lovo‐cel for SCD and was attributed to the use of busulfan. A second case (Group A, patient 1) was diagnosed with AML and BB305 LVV+ blast cells 5.5 years after lovo‐cel infusion. 27 Analyses showed that BB305 LVV insertion in the blast cells was present in the VAMP4 gene, which has no known role in the development of AML or any cellular process related to oncogenesis. Genetic testing identified several known AML driver mutations in this patient's blast cells, including monosomy 7, suggesting a molecular basis for the observed case. The AML resulted in death and was deemed unlikely related to BB305 LVV insertion because of the insertion site location, low transgene expression in blast cells, and lack of effect on expression of surrounding genes. The case was assessed by the investigator as possibly related to lovo‐cel for SCD.
Genes of interest related to proto‐oncogenesis (MECOM and LMO2) 28 were not associated with the top 10 relative insertion site frequency in any patient (Figure S8). No unique insertion site was >13.2% at any time point in Group A (excluding Group A Patient 1 who was diagnosed with AML in 2021) and improvements to polyclonality were seen in Group B with no UIS >2.9% at any time point.
No cases of veno‐occlusive disease of the liver or graft failure were observed. No replication‐competent lentivirus was detected.
4. DISCUSSION
The GT treatment process of lovo‐cel for SCD used in HGB‐206 has substantially evolved through an intense data‐driven iterative process, achieving a progressive improvement in biologic and clinical outcomes. In the initial cohort (Group A) of the HGB‐206 study, modest levels of HbAT87Q were sustained over 5.5 years of follow‐up and associated with a moderate reduction in VOC + ACS. Substantial changes to the lovo‐cel treatment process in subsequent Group B patients, including cell collection and manufacturing, led to notable improvements in lovo‐cel characteristics, HbAT87Q production, and clinical outcomes. Together, these data culminated in an optimized lovo‐cel treatment process that is being evaluated in the ongoing pivotal Group C of the HGB‐206 study, which recently reported positive interim results in 35 patients with SCD. 16
Our initial experience with Group A suggested that higher HbAT87Q levels should expand clinical benefit and we identified several areas in the treatment process for improvement in Group B: (1) a transfusion regimen to reduce stress erythropoiesis and normalize BM prior to BMH; (2) increased busulfan target AUC based on current guidelines 29 to reduce hematopoietic contribution by residual untransduced progenitors; (3) improved stem cell collection technique to achieve higher cell doses to improve engraftment and polyclonal repopulation of the bone marrow; and (4) refinement of the manufacturing process to improve cell quality and yield with increased DP VCN and higher proportion of transduced HSPCs. Overall, these enhancements in Group B led to increased HSPC quality and yield that, together with increased engraftment of transduced cells, correlated with higher PB VCN and HbAT87Q production. Both Group B patients showed higher total Hb levels, reduced HbS, and decreased hemolysis. The Group B2 patient who received lovo‐cel manufactured using only the refined process had a 4‐fold/2‐fold greater PB VCN, twofold greater HbAT87Q level, and higher total Hb compared with the Group B1 patient who received lovo‐cel produced using both the original and refined manufacturing processes. In all patient groups, VOC + ACS cases were reduced following lovo‐cel infusion, and hematologic recovery was observed without graft failure. Additional exploration is needed to evaluate the effect of these changes on other SCD‐related complications. The pivotal evaluation of efficacy is taking place using a further refined protocol in Group C of the HGB‐206 study, which recently reported positive interim results. 16
HbF induction was markedly different between groups A and B. In Group A, HbF levels increased post‐infusion, later decreased and finally stabilized at approximately 12–18 months. This finding has also been observed after allogeneic transplantation and graft rejection 30 , 31 , 32 suggesting that elevated HbF may indicate increased stress erythropoiesis in some settings. These data also emphasize that the steady‐state, durable clinical effectiveness of GT for SCD is most accurately assessed only after HbF levels have stabilized post‐transplantation, and that individual variation in HbF levels should be included in the assessment of clinical outcomes. The high level of HbAT87Q and low level of HbF in the Group B2 patient suggest a potential relationship that merits further investigation.
The lovo‐cel treatment process comprises several steps; all patients in Groups A and B had ≥1 AE attributed to conditioning. There were two AEs possibly related to lovo‐cel infusion: a non‐serious event of hot flush and a serious event of AML. To date, two patients treated in Group A developed AML and following further investigation, both cases were deemed unlikely related to vector integration. 26 , 27 There are several potential risk factors for the development of AML in Group A patients, which are explored fully in our Case Report, 27 and are expected to be mitigated by improvements to the lovo‐cel treatment process in Groups B and C. Of note, the two patients who developed AML in Group A were treated with lovo‐cel produced from BM‐harvested HSPCs using the original version of the lovo‐cel manufacturing process, which resulted in a low cell dose with limited CD34hi/+ long‐term repopulating HSPCs. The lower cell dose contains fewer HSPC clones to repopulate the BM, thereby requiring more replication cycles, which increases proliferative stress. Individuals with SCD have a 2‐ to 11‐fold higher rate of hematologic malignancies compared with the general population, and it is also possible that persistent hemolysis, anemia, HU use, 33 , 34 , 35 and post‐treatment proliferative stress in these patients promoted driver mutations that caused AML. The majority of AEs and SAEs in the lovo‐cel for SCD clinical development program were unrelated to lovo‐cel and the safety of the lovo‐cel for SCD treatment regimen largely reflected the known side effects of HSPC collection and busulfan conditioning regimen.
The evolution of the lovo‐cel treatment process in Groups A and B subsequently informed the optimized protocol that is being evaluated in the ongoing pivotal Group C cohort. 16 In addition to the changes described above, several further protocol enhancements have been implemented in Group C. These updates include HSPC collection by plerixafor mobilization/apheresis to improve safety and yield of cell collection 22 and an increased cell dose to further improve cell engraftment and HbAT87Q production. These modifications have positively impacted the quality (CD34hi/+) and number of HSPCs for ex vivo manipulation, 22 , 36 which are expected to positively impact clinical benefit. This includes a reduction in hematopoietic and proliferative stresses to minimize the risk of post‐transplantation AML. Based on the data in Groups A and B, in which PB VCN and HbAT87Q levels stabilized at 6 months and persisted through to 5.5 years, we expect to see similar durability in Group C. The limited patient numbers and statistical inference from Groups A and B will be addressed in the larger Group C cohort of ≥35 patients, and the study has been updated from phase 1 to a phase 1/2 registrational study to reflect the efficacy endpoints being assessed in Group C.
Manufacturing and treatment process enhancements in Groups A and B were achieved through an intense data‐driven iterative process and led to marked improvements in biologic and clinical outcomes of lovo‐cel for SCD. These optimizations were expedited by the correlation of PK and PD biomarkers with lovo‐cel DP characteristics within weeks or months of treatment. The success of the HGB‐206 study evolution reaffirms the importance of developing DP analytics and on‐treatment biomarkers that accurately reflect the expected relationship between GT and disease physiology. Our data also underscore the importance of long‐term monitoring for genotoxicity in GT clinical trials and the utility of sequencing tools to assess the potential contribution of GT‐related mutagenesis. We hope that this study will model how future SCD GT studies can leverage the feedback loop between clinical data, PD/PK, and DP analytics to expedite the improvement of DP and ultimately clinical outcomes for patients with SCD.
AUTHOR CONTRIBUTIONS
Alexis A. Thompson, David Davidson, Julie Kanter, Jean‐Antoine Ribeil, John F. Tisdale, Mohammed Asmal, and Mark C. Walters designed the study. Alex Miller, David Davidson, Jean‐Antoine Ribeil, and Mohammed Asmal supervised the study. Alexis A. Thompson, John F. Tisdale, Julie Kanter, Mark C. Walters, Matthew Hsieh, and Naoya Uchida provided clinical data. Francis J. Pierciey Jr., John F. Tisdale, Melissa Bonner, Manfred Schmidt, and Philippe Leboulch performed research. Ruiting Guo performed statistical analysis. Philippe Leboulch is the patent holder to the βA‐T87Q globin gene, lovo‐cel BB305 vector, and packaging plasmids. All authors analyzed and interpreted data, as well as contributed to the drafting and critical revision of the manuscript and provided final approval of the submitted manuscript.
FUNDING INFORMATION
This study was supported by research funding from bluebird bio to the authors. Medical writing support was provided by Luke Burke, Ph.D. and Aisling Koning, Ph.D. (Synergy Medical Communications, UK); copy‐editing support was provided by Kyle Lambe (Synergy Medical Communications, UK) and Ketaki Kadam (bluebird bio). Medical writing support and copy‐editing support were also provided by Audrey W. Hou, PharmD (bluebird bio). This support was funded by bluebird bio.
CONFLICT OF INTEREST
Julie Kanter reports honoraria from Medscape, Guidepoint Global, GLG, Jeffries, Cowen, and Wells Fargo; served as a consultant for bluebird bio, Novartis, Graphite, Fulcrum, and Axcella Rx; served on scientific advisory boards for bluebird bio, Novartis, Forma, and Beam; and reports membership of the National Alliance of Sickle Cell Disease and SCDAA Medical and Research Advisory Board.
Alexis A. Thompson reports research funding from CRISPR/Vertex, Novartis, Bristol Myers Squibb, Baxalta, BioMarin, and bluebird bio; and served as a consultant for Bristol Myers Squibb, Beam, Agios, and bluebird bio.
Francis J. Pierciey Jr. reports employment and equity ownership from bluebird bio.
Philippe Leboulch reports equity ownership from bluebird bio and patent ownership for the βA‐T87Q gene, the lovo‐cel BB305 lentiviral vector, and packaging plasmids.
Manfred Schmidt reports employment from the German Cancer Research Center and equity ownership from GeneWerk GmbH.
Melissa Bonner reports employment and equity ownership from bluebird bio.
Ruiting Guo reports employment and equity ownership from bluebird bio.
Alex Miller reports employment and equity ownership from bluebird bio.
Jean‐Antoine Ribeil reports equity ownership from bluebird bio.
David Davidson reports compensation and equity ownership from bluebird bio.
Mohammed Asmal reports equity ownership from bluebird bio.
Mark C. Walters served as medical director for AllCells and as a consultant for AllCells, Ensoma, Vertex, and BioLabs.
The remaining authors declare no competing financial interests.
Supporting information
Table S1. Eligibility criteria for Groups A and B of the HGB‐206 study.
Table S2. Protocol evolution in Group A and Group B of the HGB‐206 study.
Table S3. pRBC transfusion day, type and volume in Group A patients 1 and 2 who received extended pRBC transfusion after lovo‐cel infusion.
Figure S1. HbAT87Q limits polymerization. HbAT87Q is a modified HbA specifically designed with a single amino acid change to give it anti‐sickling properties, while otherwise maintaining the same morphology and function as naturally occurring HbA. Modified red blood cells produce gene therapy‐derived HbAT87Q, sterically inhibiting polymerization with HbS and thus limiting sickling. 1 HbA, adult hemoglobin; HbAT87Q, Hb with modified β‐globin gene (βA‐T87Q); HbS, sickle hemoglobin.
Figure S2. Patient disposition in Groups A and B of the HGB‐206 study. After a 2‐year follow‐up period in HGB‐206, all 9 lovo‐cel‐infused patients enrolled into a 13‐year follow‐up study. Median follow‐up duration expressed as median (min–max) for Group A. Max, maximum; min, minimum.
Figure S3. Percentage of LVV+ cells over time. Percentage of LVV+ cells from BFU‐E colonies cultured from PB. *Patient diagnosed with AML in 2021. 2 †Patient diagnosed with myelodysplastic syndrome/acute myeloid leukemia in 2018 3 ; data for efficacy endpoints are shown up to last visit prior to diagnosis of malignancy. BFU‐E, burst forming unit‐erythroid; LVV, lentiviral vector; PB, peripheral blood; pt, patient.
Figure S4. Correlation analyses of lovo‐cel DP VCN at infusion, PB VCN at Month 6 after lovo‐cel infusion, and HbAT87Q level at Month 6 after lovo‐cel infusion. (A) lovo‐cel DP VCN at infusion versus PB VCN at Month 6. (B) lovo‐cel DP VCN at infusion versus HbAT87Q level at Month 6. (C) PB VCN at Month 6 versus HbAT87Q level at Month 6. The fitted curves in B and C are based on linear regression between HbAT87Q and transformed VCN using the following equation: VCN/(1 + VCN). c/dg, copies per diploid genome; DP, drug product; HbAT87Q, Hb with modified β‐globin gene (βA‐T87Q); PB, peripheral blood; pt, patient; VCN, vector copy number.
Figure S5. Rate of annualized VOC + ACS before and after lovo‐cel infusion. Patients with ≥4 VOC + ACS in 24 months prior to informed consent and ≥6 months of follow‐up after lovo‐cel infusion were included (N = 8; 6 from Group A, 2 from Group B). VOC was defined as an acute episode of pain due to small‐vessel obstruction. ACS was defined as an acute event with pneumonia‐like symptoms and the presence of a new pulmonary infiltrate. One patient did not meet the criteria for ≥4 VOC + ACS in 24 months prior to informed consent and had been enrolled based on their history of overt stroke prior to its removal from the inclusion criteria. Hatched area represents time period between informed consent and lovo‐cel infusion, during which VOC + ACS data were not reported because patients received pre‐harvest transfusions. *Patient diagnosed with AML in 2021. 27 †Patients received regular transfusions and hydroxyurea after lovo‐cel infusion; packed red blood cell transfusion volume, day of transfusion, and transfusion type are reported in Table S3. ‡Patient diagnosed with myelodysplastic syndrome/AML in 2018. 26 ACS, acute chest syndrome; AML, acute myeloid leukemia; HbAT87Q, Hb with modified β‐globin gene (βA‐T87Q); M, month; max, maximum; min, minimum; VOC, vaso‐occlusive crisis.
Figure S6. Key hemolysis markers over time. (A) Reticulocyte count. (B) Lactate dehydrogenase. (C) Indirect bilirubin. Dashed lines show patients who received regular transfusions and hydroxyurea for an extended period post‐engraftment. Gray area denotes upper and lower limit of normal. 4 , 5 *Patient diagnosed with AML in 2021. 2 †Patient diagnosed with myelodysplastic syndrome/AML in 2018 3 ; data for efficacy endpoints are shown up to last visit prior to diagnosis of malignancy. AML, acute myeloid leukemia; Pt, patient.
Figure S7. Cardiac function over time. (A) TRJV. (B) LVEF. TRJV data were not available for Group A patient 7. TRJV ≥2.5 m/s confers an increased risk for mortality. 6 , 7 *Patient diagnosed with AML in 2021. 2 †Patient diagnosed with myelodysplastic syndrome/AML in 2018 3 ; data for efficacy endpoints are shown up to last visit prior to diagnosis of malignancy. AML, acute myeloid leukemia; LVEF, left ventricular ejection fraction; pt, patient; TRJV, tricuspid regurgitant jet velocity.
Figure S8. Thermometer of top 10 unique insertion sites at last visit. All patients were assessed using S‐EPTS/LM‐PCR analysis with the exception of patient 2 in Group A who was assessed using (nr)LAM‐PCR. The most frequent (#1) UIS is presented in red through to the #10 UIS in purple. Last visit included unscheduled visits. *Patient diagnosed with AML in 2021 2 ; †Patient diagnosed with myelodysplastic syndrome/AML in 2018. 3 (nr)LAM‐PCR, non‐restrictive linear amplification‐mediated polymerase chain reaction; AML, acute myeloid leukemia; S‐EPTS/LM‐PCR, shearing extension primer tag selection ligation‐mediated polymerase chain reaction; UIS, unique insertion site.
ACKNOWLEDGMENTS
We thank the patients and caregivers, investigators, health care providers, research staff, and study and data coordinators who participated in this trial. Medical writing support was provided by Luke Burke, Ph.D. and Aisling Koning, Ph.D. (Synergy Medical Communications, UK); copy‐editing support was provided by Kyle Lambe (Synergy Medical Communications, UK) and Ketaki Kadam (bluebird bio). Medical writing support and copy‐editing support were also provided by Audrey W. Hou, PharmD (bluebird bio). This support was funded by bluebird bio. The authors wish to pay tribute to Dr Manfred Schmidt, both a friend and colleague, who provided invaluable contributions to the field of cell and gene therapy. Marina Cavazzana (Necker Children's Hospital and Paris Descartes‐Sorbonne Paris Cité University) provided contributions to the study's clinical development and protocol. The following authors have new academic/company affiliations: Jean‐Antoine Ribeil (Section of Hematology/Oncology, Boston University and Boston Medical Center, Sickle Cell Center, Boston University School of Medicine, Boston, MA); Mohammed Asmal (OrbiMed Advisors, New York, NY); David Davidson (Tessera Therapeutics, Cambridge, MA); Ruiting Guo (Moderna, Inc., Cambridge, MA). This study was supported by research funding from bluebird bio to the authors.
Kanter J, Thompson AA, Pierciey FJ Jr, et al. Lovo‐cel gene therapy for sickle cell disease: Treatment process evolution and outcomes in the initial groups of the HGB‐206 study. Am J Hematol. 2023;98(1):11‐22. doi: 10.1002/ajh.26741
Funding information bluebird bio
DATA AVAILABILITY STATEMENT
Appropriately de‐identified patient‐level data sets and supporting documents may be shared following attainment of applicable marketing approvals and consistent with criteria established by bluebird bio and/or industry best practices to maintain the privacy of study participants. The statistical analysis plan is included as a data supplement available with the online version of this article. For more information, please contact datasharing@bluebirdbio.com.
REFERENCES
- 1. Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4:18010. [DOI] [PubMed] [Google Scholar]
- 2. Sundd P, Gladwin MT, Novelli EM. Pathophysiology of sickle cell disease. Annu Rev Pathol. 2019;14:263‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bernaudin F, Socie G, Kuentz M, et al. Long‐term results of related myeloablative stem‐cell transplantation to cure sickle cell disease. Blood. 2007;110(7):2749‐2756. [DOI] [PubMed] [Google Scholar]
- 4. Platt OS, Orkin SH, Dover G, Beardsley GP, Miller B, Nathan DG. Hydroxyurea increases fetal hemoglobin production in sickle cell anemia. Trans Assoc Am Phys. 1984;97:268‐274. [PubMed] [Google Scholar]
- 5. Bradt PS, E., Synnott, P. , Chapman, R. , Rind, D.M. ; Pearson, S. Crizanlizumab, Voxelotor, and L‐glutamine for sickle cell disease: effectiveness and value. Institute for Clinical and Economic Review;2020.
- 6. Sheth S, Licursi M, Bhatia M. Sickle cell disease: time for a closer look at treatment options? Br J Haematol. 2013;162(4):455‐464. [DOI] [PubMed] [Google Scholar]
- 7. Walters MC, De Castro LM, Sullivan KM, et al. Indications and results of HLA‐identical sibling hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2016;22(2):207‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Walters MC, Hardy K, Edwards S, et al. Pulmonary, gonadal, and central nervous system status after bone marrow transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2010;16(2):263‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shenoy S. Hematopoietic stem cell transplantation for sickle cell disease: current practice and emerging trends. Hematology Am Soc Hematol Educ Program. 2011;2011:273‐279. [DOI] [PubMed] [Google Scholar]
- 10. Mahesri M, Schneeweiss S, Globe D, et al. Clinical outcomes following bone marrow transplantation in patients with sickle cell disease: a cohort study of US Medicaid enrollees. Eur J Haematol. 2021;106(2):273‐280. [DOI] [PubMed] [Google Scholar]
- 11. Pawliuk R, Westerman KA, Fabry ME, et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science. 2001;294(5550):2368‐2371. [DOI] [PubMed] [Google Scholar]
- 12. Takekoshi KJ, Oh YH, Westerman KW, London IM, Leboulch P. Retroviral transfer of a human beta‐globin/delta‐globin hybrid gene linked to beta locus control region hypersensitive site 2 aimed at the gene therapy of sickle cell disease. Proc Natl Acad Sci U S A. 1995;92(7):3014‐3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Demirci S, Gudmundsdottir B, Li Q, et al. betaT87Q‐globin gene therapy reduces sickle hemoglobin production, allowing for ex vivo anti‐sickling activity in human erythroid cells. Mol Ther Methods Clin Dev. 2020;17:912‐921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ribeil JA, Hacein‐Bey‐Abina S, Payen E, et al. Gene therapy in a patient with sickle cell disease. N Engl J Med. 2017;376(9):848‐855. [DOI] [PubMed] [Google Scholar]
- 15. Magrin E, Semeraro M, Hebert N, et al. Long‐term outcomes of lentiviral gene therapy for the β‐hemoglobinopathies: the HGB‐205 trial. Nat Med. 2022;28(1):81‐88. [DOI] [PubMed] [Google Scholar]
- 16. Kanter J, Walters MC, Krishnamurti L, et al. Biologic and clinical efficacy of LentiGlobin for sickle cell disease. N Engl J Med. 2021;386(7):617‐628. [DOI] [PubMed] [Google Scholar]
- 17. Negre O, Eggimann AV, Beuzard Y, et al. Gene therapy of the beta‐Hemoglobinopathies by Lentiviral transfer of the beta(a[T87Q])‐globin gene. Hum Gene Ther. 2016;27(2):148‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Morgan RA, Gray D, Lomova A, Kohn DB. Hematopoietic stem cell gene therapy: Progress and lessons learned. Cell Stem Cell. 2017;21(5):574‐590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Naldini L. Genetic engineering of hematopoiesis: current stage of clinical translation and future perspectives. EMBO Mol Med. 2019;11(3): e9958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bonner MKJ, Macari E, Lane R, et al. The relationships between target gene transduction, engraftment of HSCs and RBC physiology in sickle cell disease gene therapy. Blood. 2019;134:206. [Google Scholar]
- 21. FDA . FDA guidance for industry. Long term follow‐up after administration of human gene therapy products. 2020. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/long-term-follow-after-administration-human-gene-therapy-products (Accessed September 2022)
- 22. Tisdale JF, Pierciey FJ Jr, Bonner M, et al. Safety and feasibility of hematopoietic progenitor stem cell collection by mobilization with plerixafor followed by apheresis vs bone marrow harvest in patients with sickle cell disease in the multi‐center HGB‐206 trial. Am J Hematol. 2020;95(9):E239‐E242. [DOI] [PubMed] [Google Scholar]
- 23. Cavazzana MRJA, Payen E, Suarez F, et al. Outcomes of gene therapy for severe sickle disease and Beta‐thalassemia major via transplantation of autologous hematopoietic stem cells transduced ex vivo with a lentiviral Beta AT87Q‐globin vector. Blood. 2015;126:202. [Google Scholar]
- 24. Klings ES, Machado RF, Barst RJ, et al. An official American Thoracic Society clinical practice guideline: diagnosis, risk stratification, and management of pulmonary hypertension of sickle cell disease. Am J Respir Crit Care Med. 2014;189(6):727‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350(9):886‐895. [DOI] [PubMed] [Google Scholar]
- 26. Hsieh MM, Bonner M, Pierciey FJ, et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. 2020;4(9):2058‐2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goyal S, Tisdale J, Schmidt M, et al. Acute myeloid leukemia case after gene therapy for sickle cell disease. N Engl J Med. 2022;386(2):138‐147. [DOI] [PubMed] [Google Scholar]
- 28. Cavazzana‐Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human β‐thalassaemia. Nature. 2010;467(7313):318‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Andersson BS, Thall PF, Madden T, et al. Busulfan systemic exposure relative to regimen‐related toxicity and acute graft‐versus‐host disease: defining a therapeutic window for i.v. BuCy2 in chronic myelogenous leukemia. Biol Blood Marrow Transplant. 2002;8(9):477‐485. [DOI] [PubMed] [Google Scholar]
- 30. Ferster A, Corazza F, Vertongen F, et al. Transplanted sickle‐cell disease patients with autologous bone marrow recovery after graft failure develop increased levels of fetal haemoglobin which corrects disease severity. Br J Haematol. 1995;90(4):804‐808. [DOI] [PubMed] [Google Scholar]
- 31. Locatelli F, Maccario R, Comoli P, et al. Hematopoietic and immune recovery after transplantation of cord blood progenitor cells in children. Bone Marrow Transplant. 1996;18(6):1095‐1101. [PubMed] [Google Scholar]
- 32. Dover GJ, Boyer SH, Zinkham WH. Production of erythrocytes that contain fetal hemoglobin in anemia. Transient in vivo changes. J Clin Invest. 1979;63(2):173‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cuthbert D, Stein BL. Therapy‐associated leukemic transformation in myeloproliferative neoplasms ‐ what do we know? Best Pract Res Clin Haematol. 2019;32(1):65‐73. [DOI] [PubMed] [Google Scholar]
- 34. Brunson A, Keegan THM, Bang H, Mahajan A, Paulukonis S, Wun T. Increased risk of leukemia among sickle cell disease patients in California. Blood. 2017;130(13):1597‐1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seminog OO, Ogunlaja OI, Yeates D, Goldacre MJ. Risk of individual malignant neoplasms in patients with sickle cell disease: English national record linkage study. J R Soc Med. 2016;109(8):303‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leonard A, Bonifacino A, Dominical VM, et al. Bone marrow characterization in sickle cell disease: inflammation and stress erythropoiesis lead to suboptimal CD34 recovery. Br J Haematol. 2019;186(2):286‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Eligibility criteria for Groups A and B of the HGB‐206 study.
Table S2. Protocol evolution in Group A and Group B of the HGB‐206 study.
Table S3. pRBC transfusion day, type and volume in Group A patients 1 and 2 who received extended pRBC transfusion after lovo‐cel infusion.
Figure S1. HbAT87Q limits polymerization. HbAT87Q is a modified HbA specifically designed with a single amino acid change to give it anti‐sickling properties, while otherwise maintaining the same morphology and function as naturally occurring HbA. Modified red blood cells produce gene therapy‐derived HbAT87Q, sterically inhibiting polymerization with HbS and thus limiting sickling. 1 HbA, adult hemoglobin; HbAT87Q, Hb with modified β‐globin gene (βA‐T87Q); HbS, sickle hemoglobin.
Figure S2. Patient disposition in Groups A and B of the HGB‐206 study. After a 2‐year follow‐up period in HGB‐206, all 9 lovo‐cel‐infused patients enrolled into a 13‐year follow‐up study. Median follow‐up duration expressed as median (min–max) for Group A. Max, maximum; min, minimum.
Figure S3. Percentage of LVV+ cells over time. Percentage of LVV+ cells from BFU‐E colonies cultured from PB. *Patient diagnosed with AML in 2021. 2 †Patient diagnosed with myelodysplastic syndrome/acute myeloid leukemia in 2018 3 ; data for efficacy endpoints are shown up to last visit prior to diagnosis of malignancy. BFU‐E, burst forming unit‐erythroid; LVV, lentiviral vector; PB, peripheral blood; pt, patient.
Figure S4. Correlation analyses of lovo‐cel DP VCN at infusion, PB VCN at Month 6 after lovo‐cel infusion, and HbAT87Q level at Month 6 after lovo‐cel infusion. (A) lovo‐cel DP VCN at infusion versus PB VCN at Month 6. (B) lovo‐cel DP VCN at infusion versus HbAT87Q level at Month 6. (C) PB VCN at Month 6 versus HbAT87Q level at Month 6. The fitted curves in B and C are based on linear regression between HbAT87Q and transformed VCN using the following equation: VCN/(1 + VCN). c/dg, copies per diploid genome; DP, drug product; HbAT87Q, Hb with modified β‐globin gene (βA‐T87Q); PB, peripheral blood; pt, patient; VCN, vector copy number.
Figure S5. Rate of annualized VOC + ACS before and after lovo‐cel infusion. Patients with ≥4 VOC + ACS in 24 months prior to informed consent and ≥6 months of follow‐up after lovo‐cel infusion were included (N = 8; 6 from Group A, 2 from Group B). VOC was defined as an acute episode of pain due to small‐vessel obstruction. ACS was defined as an acute event with pneumonia‐like symptoms and the presence of a new pulmonary infiltrate. One patient did not meet the criteria for ≥4 VOC + ACS in 24 months prior to informed consent and had been enrolled based on their history of overt stroke prior to its removal from the inclusion criteria. Hatched area represents time period between informed consent and lovo‐cel infusion, during which VOC + ACS data were not reported because patients received pre‐harvest transfusions. *Patient diagnosed with AML in 2021. 27 †Patients received regular transfusions and hydroxyurea after lovo‐cel infusion; packed red blood cell transfusion volume, day of transfusion, and transfusion type are reported in Table S3. ‡Patient diagnosed with myelodysplastic syndrome/AML in 2018. 26 ACS, acute chest syndrome; AML, acute myeloid leukemia; HbAT87Q, Hb with modified β‐globin gene (βA‐T87Q); M, month; max, maximum; min, minimum; VOC, vaso‐occlusive crisis.
Figure S6. Key hemolysis markers over time. (A) Reticulocyte count. (B) Lactate dehydrogenase. (C) Indirect bilirubin. Dashed lines show patients who received regular transfusions and hydroxyurea for an extended period post‐engraftment. Gray area denotes upper and lower limit of normal. 4 , 5 *Patient diagnosed with AML in 2021. 2 †Patient diagnosed with myelodysplastic syndrome/AML in 2018 3 ; data for efficacy endpoints are shown up to last visit prior to diagnosis of malignancy. AML, acute myeloid leukemia; Pt, patient.
Figure S7. Cardiac function over time. (A) TRJV. (B) LVEF. TRJV data were not available for Group A patient 7. TRJV ≥2.5 m/s confers an increased risk for mortality. 6 , 7 *Patient diagnosed with AML in 2021. 2 †Patient diagnosed with myelodysplastic syndrome/AML in 2018 3 ; data for efficacy endpoints are shown up to last visit prior to diagnosis of malignancy. AML, acute myeloid leukemia; LVEF, left ventricular ejection fraction; pt, patient; TRJV, tricuspid regurgitant jet velocity.
Figure S8. Thermometer of top 10 unique insertion sites at last visit. All patients were assessed using S‐EPTS/LM‐PCR analysis with the exception of patient 2 in Group A who was assessed using (nr)LAM‐PCR. The most frequent (#1) UIS is presented in red through to the #10 UIS in purple. Last visit included unscheduled visits. *Patient diagnosed with AML in 2021 2 ; †Patient diagnosed with myelodysplastic syndrome/AML in 2018. 3 (nr)LAM‐PCR, non‐restrictive linear amplification‐mediated polymerase chain reaction; AML, acute myeloid leukemia; S‐EPTS/LM‐PCR, shearing extension primer tag selection ligation‐mediated polymerase chain reaction; UIS, unique insertion site.
Data Availability Statement
Appropriately de‐identified patient‐level data sets and supporting documents may be shared following attainment of applicable marketing approvals and consistent with criteria established by bluebird bio and/or industry best practices to maintain the privacy of study participants. The statistical analysis plan is included as a data supplement available with the online version of this article. For more information, please contact datasharing@bluebirdbio.com.