

Summary
Dysregulation and accelerated activation of the alternative pathway (AP) of complement is known to cause or accentuate several pathologic conditions in which kidney injury leads to the appearance of hematuria and proteinuria and ultimately to the development of chronic renal failure. Multiple genetic and acquired defects involving plasma‐ and membrane‐associated proteins are probably necessary to impair the protection of host tissues and to confer a significant predisposition to AP‐mediated kidney diseases. This review aims to explore how our current understanding will make it possible to identify the mechanisms that underlie AP‐mediated kidney diseases and to discuss the available clinical evidence that supports complement‐directed therapies. Although the value of limiting uncontrolled complement activation has long been recognized, incorporating complement‐targeted treatments into clinical use has proved challenging. Availability of anti‐complement therapy has dramatically transformed the outcome of atypical hemolytic uremic syndrome, one of the most severe kidney diseases. Innovative drugs that directly counteract AP dysregulation have also opened new perspectives for the management of other kidney diseases in which complement activation is involved. However, gained experience indicates that the choice of drug should be tailored to each patient's characteristics, including clinical, histologic, genetic, and biochemical parameters. Successfully treating patients requires further research in the field and close collaboration between clinicians and researchers who have special expertise in the complement system.
Keywords: alternative pathway of complement, complement inactivating agents, complement system, glomerular diseases, rare kidney diseases
1. INTRODUCTION
The complement system, a central part of the innate immunity that serves as a first line of defense against foreign and altered host cells, is an extremely effective cell‐killing and inflammation‐provoking pathway. However, complement activation is a double‐edged sword because uncontrolled stimulation can be highly detrimental to host tissues. 1 , 2 , 3 In order to avoid self‐damage, a plethora of inhibitory mechanisms are known to prevent overwhelming activation at all stages of the complement cascade. The alternative pathway (AP) of complement is particularly significant for survival against invading pathogens and can be triggered by several other conditions, such as trauma, surgery, or pregnancy. Inappropriate AP activation may be damaging to the kidney, where the deposition of activated complement fragments from plasma in glomeruli and/or activated complement fragments locally produced may contribute to tissue injury. 4 Unlike the classic and lectin pathways, AP—the oldest evolutionary pathway of the system—is constantly self‐activated by the slow and spontaneous hydrolysis of C3, a process known as tick‐over, 5 and plays a vital role in amplifying complement activation. As a result, the AP is permanently active at a low level, enabling continuous monitoring of the body for disease‐causing pathogens and host processes. 1 , 2 , 6 Complement factor B (CFB) is a key component of this process (Figure 1). The cleavage product Bb combines with C3b to form C3 convertase (C3bBb) to cleave C3, which forms a positive feedback loop to continuously activate the AP. 7 , 8 The convertase complexes dissociate spontaneously in a few minutes, a process that is critical to prevent autologous tissue injury. Dysregulation of AP can occur as a result of acquired or genetically driven pathological events, both of which can lead to erroneous activation or insufficient control of pathway signaling. Complement and complement regulatory molecules may act in concert in a sophisticated interacting protein network, and multiple defects involving plasma‐ and membrane‐associated proteins are probably necessary to impair the protection of host tissues and to confer a significant predisposition to AP‐mediated kidney diseases.
FIGURE 1.
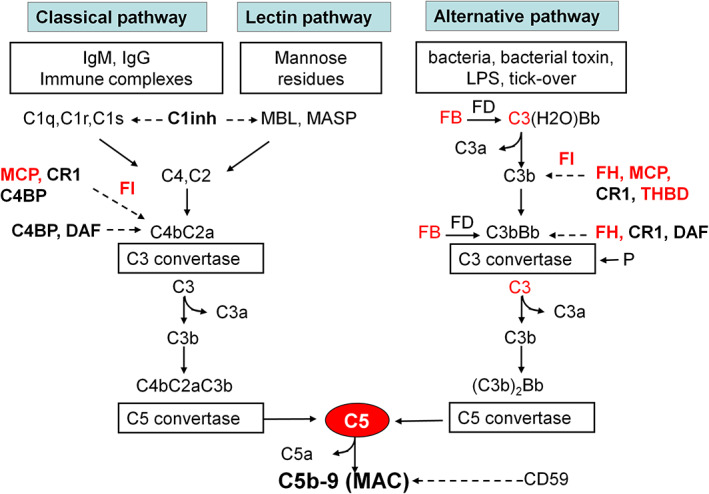
The complement cascade. Schematic overview of the complement cascade, illustrating the three activation pathways (classical, lectin, and alternative) with the C3 convertase complexes of the classical, lectin, and the alternative pathway and the common terminal pathway that leads to C5 cleavage and the formation of the anaphylatoxin C5a and of the membrane attack complex, composed of C5b, C6, C7, C8, and many copies of C9. The classical pathway is triggered by the binding of C1q to antibody‐antigen complexes. The lectin pathway is similar to the classical pathway but is activated by the binding of mannose‐binding lectin (MBL) to mannose residues, which activates mannose‐binding lectin serine peptidase (MASP) proteins. In contrast, the alternative pathway is continuously activated in plasma by low‐grade hydrolysis of C3 (C3H2O, tick‐over). The latter binds factor B, to form a C3(H2O)B complex. Factor D cleaves factor B to form the alternative pathway initiation C3 convertase that cleaves C3 to C3b. The activation is then amplified by the covalent binding of a small amount of C3b to hydroxyl groups on cell surface carbohydrates and proteins of target cells, such as bacterial cells. This C3b binds factor B, to form the amplification loop C3 convertase C3bBb. C3b also binds to the C3 convertase, forming the C5 convertase enzyme C3b2Bb. The alternative pathway is highly regulated to prevent non‐specific damage to host cells and limit the deposition of complement on the surface of pathogens. This fine regulation occurs through a number of membrane‐anchored and fluid‐phase regulators. Bold text denotes complement‐regulatory molecules; red text denotes proteins with genetic defects that have been associated with aHUS and/or IC‐MPGN/C3G. Abbreviations and definitions: C1inh, C1 inhibitor (inactivates C1r and C1s, MASP‐1 and MASP‐2); FB, complement factor B; FD, complement factor D; FH, complement factor H (binds C3b, exerts cofactor activity for FI‐mediated C3b cleavage, prevents the formation of the alternative pathway C3 convertase, and destabilizes (decay accelerating activity) the alternative pathway C3 and C5 convertases); FI, complement factor I (degrades C3b and C4b, aided by cofactors); C4BP, C4b‐binding protein (binds to C4b and has decay accelerating activity for the classical pathway C3 convertase and cofactor activity for FI‐mediated C4b cleavage); CD59, protectin (with vitronectin and clusterin, prevents C5b‐9 formation); CR1, complement receptor 1 (has decay accelerating activity as well as cofactor activity for FI‐mediated C3b and C4b cleavage); DAF, decay accelerating factor (has decay accelerating activity on C3/C5 convertases of the classical and alternative pathways); MCP, membrane cofactor protein (exerts cofactor activity for FI‐mediated C3b cleavage); P, properdin (the only positive regulator in the complement system, it stabilizes the alternative pathway C3 convertase); THBD, thrombomodulin (increases FH cofactor activity, activates procarboxypeptidase B‐mediated C3a and C5a inactivation).
Interest in the complement system has been boosted in the past 20 years by the discovery that rare severe kidney diseases, including atypical hemolytic uremic syndrome (aHUS) and C3 glomerulopathy/immune complex‐associated membranoproliferative glomerulonephritis (C3G/IC‐MPGN) spectrum, are disorders of AP regulation. 9 , 10 AP plays a primary pathogenetic role in both conditions; however, inappropriate or prolonged AP activation resulting in renal damage has been observed in several kidney diseases. This review aims to explore how our current understanding of systems biology, genetic, and clinical diagnostics will make it possible to identify the complex mechanisms that underlie AP‐mediated kidney diseases and to discuss the available clinical evidence that supports complement inhibition.
2. TWO PROTOTYPICAL COMPLEMENT‐MEDIATED KIDNEY DISEASES
2.1. Atypical Hemolytic uremic syndrome
2.1.1. Clinical manifestation and diagnosis
Hemolytic uremic syndrome (HUS) is a rare disease characterized by microangiopathic hemolytic anemia, thrombocytopenia, and renal function impairment caused by platelet thrombi in the microcirculation of the kidney and other organs. The most common form in children is associated with infection by certain strains of Escherichia coli, which produce Shiga‐like toxins (STEC‐HUS). Atypical HUS (aHUS)—the term has historically been used to define any HUS not caused by STEC‐HUS—accounts for about 10% of all cases and has a poor prognosis compared with the most common form of STEC‐HUS in children. 11 The estimated incidence of aHUS is one in 500,000 people per year in the United States. 12 Ultra‐rare recessive forms are associated with genetically determined cobalamin C or diacylglycerol kinase 3 (DGKE) deficiency. 13 , 14 , 15 In more recent classifications, improved by a greater understanding of pathogenetic mechanisms, the term "primary aHUS" is increasingly used when an underlying abnormality of the AP is strongly suspected and other causes of secondary aHUS have been ruled out. 16 In affected patients, dysregulation and accelerated activation of the AP can occur either through inherited or de novo abnormalities in the complement genes or through acquired autoantibodies to complement proteins. The onset of primary aHUS ranges from the neonatal period to adulthood. In many patients with an underlying complement “risk factor,” presentation in later life is consistent with the need for an environmental trigger. 17 , 18 A wide variety of triggers have been identified, including common viral and bacterial infections, transplants, drugs, autoimmune conditions, and pregnancy, 19 with a lifelong risk of recurrent episodes of aHUS in some patients. Environmental hits are likely to induce endothelial perturbation and complement activation, which in healthy individuals are self‐limiting as a result of multiple, redundant, regulatory mechanisms. 20 An individual with genetic abnormalities that affect complement regulation is otherwise particularly vulnerable to complement attack. Once the complement cascade is activated beyond a critical threshold, C3b formation and deposition occur on the vascular endothelium, which leads to further complement activation through the self‐amplifying loop of the alternative pathway, culminating in microangiopathic injury and thrombosis. Downstream clinical manifestations of aHUS can include impaired renal function up to end‐stage renal failure (ESRF), extrarenal organ damage, or death. 21 Extrarenal manifestations are reported in up to 20% of patients with aHUS. It is unclear whether these manifestations are a direct consequence of complement activation, thrombotic microangiopathy (TMA), or other factors, such as severe hypertension and uremia. 16 Before the introduction of complement inhibition therapy, up to 50% of aHUS cases progressed to ESRF or developed irreversible brain damage, and 25% died during the acute phase of the disease. 11 , 22
As with most TMAs, laboratory findings in aHUS can include hemolytic anemia, fragmented red blood cells, thrombocytopenia, and elevated levels of lactate dehydrogenase (LDH). Once routine biochemical and hematological analysis has demonstrated a TMA, investigations should focus on determining the underlying etiology and excluding other diagnoses. The first requirement is to measure ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) activity to diagnose or rule out thrombotic thrombocytopenic purpura (TTP). Investigating STEC‐HUS should be routine in all patients with presumed aHUS, as approximately 5% of STEC‐HUS cases involve no prodromal diarrhea, whereas 30% of complement‐mediated aHUS cases feature concurrent diarrhea or gastroenteritis. 23 Serum or plasma levels of complement proteins should be measured before treatment in all patients with primary aHUS. However, it should be noted that levels of C3 and soluble C5b‐9 [sC5b‐9 or membrane attack complex (MAC) or terminal complement complex, Figure 1] may be normal in a substantial fraction of patients with aHUS, even during the acute phase. 24 This profile is due to the distinctive dysregulation pattern of AP in aHUS, which mainly affects complement activation on cellular surfaces, rather than in the fluid phase. To reproduce this peculiar condition, an ex vivo assay was set up in which human microvascular endothelial cells (HMEC‐1), either in a resting condition or preactivated with adenosine 5'‐diphosphate (ADP) to mimic an in vivo trigger, were incubated with control serum or serum from aHUS patients. At the end of the incubation, C3b deposition and C5b‐9 formation were quantified using immunofluorescence and confocal microscopy. 25 Acute aHUS serum, but not serum from patients who were in remission, caused wider C3 and C5b‐9 deposits than control serum on unstimulated cells. On ADP‐activated cells, sera from 84% and 100% of patients who were in remission also induced excessive C3 and C5b‐9 deposits. In line with these results, the evaluation of C5b‐9 deposition on HMEC‐1 can be helpful in diagnosing aHUS (during the acute phase or in remission). 24
Primary aHUS is associated with a high rate of recurrence and poor outcomes after kidney transplantation. Notably, depending on the determinants of AP dysregulation involved, the risk of recurrence varies greatly, highlighting the importance of undertaking etiological investigations prior to kidney transplantation. 26
2.1.2. Genetic and acquired determinants of alternative pathway dysregulation
Atypical HUS is a genetically heterogeneous condition. In 40%–60% of patients with aHUS, genetic abnormalities that affect the complement regulatory proteins and the components of the alternative pathway C3 convertase have been identified through targeted sequencing and duplication/deletion detection. 11 , 27 , 28 , 29 , 30 , 31 The diagnosis of primary aHUS is established in a proband with aHUS by identifying a likely pathogenic variant(s) (LPVs) in one or more of the genes known to be associated with genetic aHUS. Less than 20% of cases are considered familial. 32 All other patients have no family history of the disease (sporadic aHUS), and most inherited the complement abnormality from an unaffected parent. Indeed, the majority of complement LPVs confer susceptibility to the development of aHUS and are heterozygous in aHUS patients. 33
Complement factor H (CFH) is a serum glycoprotein that regulates the function of the AP in the fluid phase and on cellular surfaces (Figure 1). It binds to C3b, accelerates the decay of the alternative pathway convertase C3bBb, and also acts as a cofactor for complement factor I, another C3b inhibitor, in the proteolytic inactivation of C3b, generating iC3b. 34 , 35 Abnormalities in CFH gene are the most commonly observed in patients with aHUS and have been documented in 20%‐30% of cases. Both homozygous and heterozygous CFH gene LPVs predispose to the development of aHUS. 28 , 35 , 36 , 37 , 38 , 39 , 40 , 41 Structural and functional characterization support the hypothesis that patients with aHUS and a CFH defect have a specific dysfunction in the protection of cellular surfaces from AP activation. 42 , 43 In patients with CFH mutations and normal levels of plasma CFH, the authors postulated that the mutation disrupted the function of the protein.
Complement factor I (CFI) is a plasma glycoprotein composed of two polypeptide chains linked by disulfide bonds. Both the light and heavy chains of factor I are encoded by the CFI gene. 44 The light chain contains the serine protease domain, which is responsible for cleaving and inactivating C4b and C3b. 45 Heterozygous mutations in the CFI gene, leading to factor I deficiency or dysfunction, have been identified in patients with aHUS. 17 , 46
Membrane cofactor protein (MCP or CD46 antigen), a transmembrane glycoprotein that is highly expressed in all tissues on endothelial cells and on all circulating cells, with the exception of erythrocytes, regulates both the alternative and classical complement pathways, acting as a cofactor for factor I to degrade C3b and C4b and to prevent C3 activation on cell surfaces. 47 , 48 In patients with aHUS, heterozygous, homozygous, compound heterozygote LPVs, and heterozygous deletion in the MCP gene have been described. 17 , 49 , 50 These mutations resulted in either reduced protein expression or impaired C3b binding capability. The penetrance of aHUS among subjects with MCP mutations is incomplete, and 25% of patients had combined mutations in other complement genes. 31
Factor B is essential in defending against encapsulated bacteria, and thus, individuals with factor B deficiency are prone to infection with Neisseria meningitidis and Streptococcus pneumoniae. 51 Conversely, overactive factor B can lead to excessive complement activation via the alternative pathway, resulting in kidney damage. Gain‐of‐function variants in CFB are rare and in some cases associated with low C3 levels in patient sera, 52 , 53 , 54 , 55 indicating complement activation in vivo. Mutations in the CFB gene have been shown to increase factor B binding affinity to C3b, thereby stabilizing the C3bBb convertase 56 and enhancing resistance to factor H‐mediated decay acceleration. 55 , 57 Notably, not all CFB rare variants have been shown to induce complement activation and not all individuals who carry CFB rare variants associated with aHUS develop the disease, even if circulating C3 levels are low. 53 , 56 , 58
C3 plays several important biologic roles in the classical, alternative, and lectin activation pathways (Figure 1). 59 Of the nine LPVs identified in C3 in 11 probands with aHUS, five resulted in a gain‐of‐function with resistance to degradation by MCP and CFI, and two resulted in haploinsufficiency. Family history, when available, showed incomplete penetrance. 60
Thrombomodulin (THBD) is an endothelial cell surface glycoprotein that forms a 1:1 complex with the coagulation factor thrombin, acting as an antithrombotic factor. Functional studies in vitro demonstrated that THBD also bind to C3b and factor H and negatively regulates complement by accelerating factor I‐mediated inactivation of C3b. Moreover, THBH promotes activation of the plasma procarboxypeptidase B, which in turn inactivates the anaphylatoxins C3a and C5a. 61 Impairing protection against complement activation, heterozygous LPVs in the THBD gene may contribute to the development of aHUS. Decreased serum C3 levels with C4 within normal limits are consistent with AP activation in reported cases. 61
Factor H‐related (FHR) proteins are emerging complement modulators and amplifiers that play different roles in the complement cascade. The human CFH–CFHR gene cluster is located on chromosome 1q32 in the regulators of complement activation region. The five CFHR genes are positioned downstream of the CFH gene and are arranged in the order CFHR3, CFHR1, CFHR4, CFHR2, and CFHR5. The five FHR proteins share structural homology and functions with each other and with factor H. This cluster represents an unstable, dynamic chromosomal region and a hotspot for structural rearrangements. 62 In patients with aHUS, deletions/insertions of chromosomal segments that result in hybrid genes, homozygous deletions of the CFHR3‐CFHR1 or CFHR1‐CFHR4 gene segments and rare CFHR gene variants have been described. 62 , 63 , 64 Homozygous deletion of CFHR3/CFHR1 is often associated with the formation of anti‐factor H autoantibodies (FHAA), which have been identified as acquired drivers of complement dysregulation in aHUS. 65 FHAA‐related aHUS is an unique subgroup of aHUS that can occur at any age, but is most prevalent in the pediatric population. These patients develop autoantibodies that bind to the C‐terminus of factor H, thus impairing the interaction of factor H with the cell surface and, consequently, its interaction with surface‐bound C3b, causing dysregulation and overactivity of the complement pathway. Further studies are needed to fully elucidate the complex genetic and environmental factors underlying FHAA‐related aHUS and to establish whether the combination of FHAA with LPVs in complement genes or other risk factors influences disease outcome and response to treatments. 30 , 66
It is not only the LPVs of gene coding for complement proteins but also other genetic susceptibility factors, such as the risk haplotypes (polymorphisms) that may increase the risk of TMA and as such contribute to the development of aHUS. 67 , 68 , 69 Single‐nucleotide polymorphisms (SNPs) such as common susceptibility variants in the CFH and MCP genes are strongly associated with aHUS. 39 , 70 , 71 Frémeaux‐Bacchi and colleagues 71 examined SNPs in both the CFH and the MCP genes in two large aHUS cohorts. In both cohorts, there was an association between aHUS and both CFH and MCP alleles. Furthermore, CFH and MCP haplotypes were significantly different in aHUS patients compared with controls. The results suggested that there are naturally occurring susceptibility factors in CFH and MCP genes for the development of aHUS. A characteristic feature of both CFH‐ and MCP‐associated aHUS is reduced penetrance and variable inheritance.
2.1.3. Therapy and monitoring
Plasma therapy, including plasma exchange and infusion (PE/PI), has been the mainstay of aHUS treatment for many years, despite the lack of controlled trials and high‐quality evidence for its efficacy. Even today, when targeted therapy with complement inhibitor is not available, plasma therapy remains the only approach with near‐complete global availability and is an important treatment for aHUS. Plasma therapy should be started as soon as aHUS is suspected and continued until the resolution of TMA. In individuals who respond, plasma exchange can be withdrawn gradually, although a significant proportion of patients requires continued treatment to maintain remission. 72 , 73 Historical cohort data show that response to plasma therapy is in part related to the genetic background of the treated individual. 33 Following the introduction of plasma therapy, the mortality rate of aHUS decreased, but hematological manifestations of the disease normalize only transiently and these treatments do not affect the underlying causative factors. Therefore, a recurrence of aHUS is likely in patients treated with PE/PI and some may no longer respond after long‐term therapy. 74 , 75 Within 1 year of an aHUS diagnosis, up to 65% of patients who receive plasma therapy sustain permanent kidney damage, develop ESRF, or die. 17
Eculizumab, a humanized anti‐C5 monoclonal antibody, was the first medication approved for treating aHUS in 2011. It is recommended as first‐line therapy for both adult and pediatric patients with a confirmed diagnosis of aHUS. By binding with high affinity to C5, eculizumab blocks the formation of C5a and the C5b‐9 cell membrane attack complex (Figure 1), leaving earlier functions of the complement system (opsonization and immune clearance) intact. Treatment with eculizumab has led to the inhibition of complement‐mediated TMA and the improvement and maintenance of kidney function in several clinical studies. 16 , 76 , 77 , 78 The efficacy and safety of eculizumab for the treatment of aHUS were firstly demonstrated in two prospective, open‐label, phase 2 trials, 79 one involving patients with clinical evidence of progressive TMA and the other involving patients with long disease duration, chronic kidney disease, and prolonged PE/PI. The data indicate that terminal complement blockade with eculizumab inhibits complement‐mediated TMA, decreases the need for TMA‐related intervention, significantly improves the platelet count and renal function across patient groups, and is associated with substantial kidney function recovery. An aHUS‐predisposing complement mutation is not required to begin treatment, since the drug is considered effective regardless of the presence of known complement mutations. 72 A systematic review that considered 15 studies involving 940 pediatric patients with aHUS treated with eculizumab confirms that the treatment resulted in a satisfactory response, with improvements in kidney function and hematological parameters for most patients. However, most studies were observational and had small sample sizes. 80
Eculizumab has been shown to induce remission of acute episodes of aHUS when administered early after the onset of the disease, 16 , 81 , 82 but can also successfully be used as a prophylactic treatment to prevent post‐transplantation aHUS recurrence in individuals who are at a moderate to high risk of recurrence. 83 , 84 , 85 , 86 Specifically, individuals with pathogenic variants in C3, CFB, and CFH or those who have the CFH/CFHR1 hybrid allele are considered to be at high risk for disease recurrence, whereas those carrying CFH antibodies, pathogenic variants in CFI, variants of uncertain significance, and/or no identified pathogenic variants are considered at moderate risk for disease recurrence. 84
Ravulizumab, a more recent humanized monoclonal antibody that targets the same epitope on the C5 protein as eculizumab, has also shown promising results in aHUS. This drug was engineered from eculizumab to have a longer half‐life, resulting in an infusion rate of every 8 weeks instead of every 2 weeks, as is the case with eculizumab. The phase 3 single‐arm study (NCT02949128) involving 58 adult patients with aHUS showed that ravulizumab induces a complete TMA remission in 53.6% of patients within 26 weeks. An improvement in renal function was observed in 68% of patients, and dialysis weaning was achieved in 58% of patients who were on dialysis at baseline. 87
At variance with eculizumab and ravulizumab, which are administered by intravenous infusion, crovalimab, another long‐acting C5 inhibitor, is administered subcutaneously. This drug will be examined in a phase 3 study (COMMUTE‐a and COMMUTE‐p) with adults or pediatric patients with aHUS (Table 1).
TABLE 1.
Ongoing interventional clinical trials targeting AP in kidney diseases
| ID number | Acronym or other ID number | Targeted protein | Status | Conditions | Interventions | Phase | Number of patients | Age |
|---|---|---|---|---|---|---|---|---|
| NCT04859608 | EspacECU | C5 | Recruiting | aHUS | Eculizumab | 4 | 80 | 18 y and older |
| NCT04958265 | COMMUTE‐p | C5 | Recruiting | aHUS | Crovalimab | 3 | 35 | 28 d to 17 y |
| NCT04861259 | COMMUTE‐a | C5 | Recruiting | aHUS | Crovalimab | 3 | 90 | 12 y and older |
| NCT04889430 | APPELHUS | Factor B | Recruiting | aHUS | Iptacopan | 3 | 50 | 18 y and older |
| NCT03131219 | ALXN1210‐aHUS‐312 | C5 | Active, not recruiting | aHUS | Ravulizumab | 3 | 31 | up to 17 y |
| NCT02949128 | ALXN1210‐aHUS‐311 | C5 | Active, not recruiting | aHUS | Ravulizumab | 3 | 58 | 12 y and older |
| NCT05067127 | VALIANT | C3 | Recruiting | C3G, IC‐MPGN, post‐Tx C3G or IC‐MPGN recurrence | Pegcetacoplan | 3 | 90 | 12 y and older |
| NCT04572854 | NOBLE | C3 | Recruiting | post‐Tx C3G or IC‐MPGN recurrence | Pegcetacoplan | 2 | 12 | 18 y and older |
| NCT03955445 |
LNP023 Extension study |
Factor B | Recruiting | C3G, post‐Tx C3G recurrence | Iptacopan | 2 | 95 | 18 y and older |
| NCT04817618 | APPEAR‐C3G | Factor B | Recruiting | C3G | Iptacopan | 3 | 68 | 18–60 years |
| NCT03453619 | APL2‐201 | C3 | Active, not recruiting | C3G, IgAN, LN, MN | Pegcetacoplan | 2 | 21 | 18 y and older |
| NCT05162066 | RENEW | Factor D | Active, not recruiting | C3G, IgAN, MN | BCX9930 | 2 | 42 | 18 y and older |
| NCT05097989 | ALXN2050‐NEPH‐201 | Factor D | Recruiting | IgAN, LN | ALXN2050 | 2 | 126 | 18–75 years |
| NCT04154787 | CLNP023D12201 | Factor B | Recruiting | MN | Iptacopan or Rituximab | 2 | 52 | 18 y and older |
| NCT03841448 | ALN‐CC5‐005 | C5 | Active, not recruiting | IgAN | Cemdisiran | 2 | 31 | 18–65 years |
| NCT04578834 | APPLAUSE‐IgAN | Factor B | Recruiting | IgAN | Iptacopan | 3 | 450 | 18 y and older |
| NCT04557462 | LNP023 Rollover Extension Program (REP) | Factor B | Recruiting | IgAN | Iptacopan | 3 | 410 | 18 y and older |
| NCT04564339 | SANCTUARY | C5 | Recruiting | IgAN, LN | Ravulizumab | 2 | 120 | 18–75 years |
| NCT05268289 | CLNP023K12201 | Factor B | Recruiting | LN | Iptacopan | 2 | 240 | 18 y and older |
Note: Retrieved from ClinicalTrials.gov database 1 September, 2022 https://clinicaltrials.gov/ct2/home
Strategy: aHUS OR Glomerulonephritis; Recruiting/Active, not recruiting; Interventional Studies; Phase 2, 3, 4; selected for drugs targeting alternative or terminal pathway of complement.
Abbreviations: aHUS, atypical hemolytic uremic syndrome; AP, alternative pathway of complement; C3G, C3 glomerulopathy; IC‐MPGN, immune complex‐associated membranoproliferative glomerulonephritis; IgAN, IgA nephropathy; LN, Lupus nephritis; MN, membranous nephropathy; Tx, transplant.
C5 inhibition is associated with increased susceptibility to Neisseria infections (including disseminated gonococcal infections) and with the potential risk of other infections, particularly those caused by encapsulated bacteria, including Streptococcus pneumoniae and Hemophilus influenzae type b (Hib), as well as Aspergillus in immunocompromised and neutropenic patients. Therefore, anti‐meningococcal, anti‐pneumococcal, and anti‐Hib vaccinations should be administered at least 2 weeks before the start of treatment, and antibiotic prophylaxis may be considered for the overall period of anti‐complement treatment in selected cases. 16
Platelet count and serum LDH concentration are the most sensitive laboratory markers for monitoring response to treatment. In patients with aHUS, lifelong anti‐C5 treatment was initially recommended, based on the assumption that patients with aHUS have continuous, systemic complement activation and are hence at high risk of relapse in case of treatment discontinuation. However, there is no definite evidence to support this assumption. 88 When and how to discontinue C5 inhibition treatment remain unresolved questions. Several retrospective series 89 , 90 , 91 , 92 and one more recent prospective trial 93 indicate that the presence of complement gene pathogenic variants and a previous history of recurrent disease are the main factors associated with a high risk of aHUS relapse after C5 blockade cessation. On the other hand, in some patients the dose of eculizumab was reduced over time, the interval between infusions extended, or treatment even stopped, without disease recurrences. Microscopic hematuria is one of the earliest markers of disease recurrence and prompt detection of microscopic hematuria by regular (twice weekly) urine dipstick analyses at home has been reported as a sensitive (admittedly non‐specific) approach for the prompt diagnosis of disease recurrence. 94 The diagnosis, however, must be confirmed by the subsequent detection of thrombocytopenia along with fragmented erythrocytes in the peripheral blood smear and other markers of microangiopathic hemolysis, including increased serum LDH levels and undetectable haptoglobin. The aim of discontinuing eculizumab therapy is primarily to protect patients against the risk of meningococcal infection, to which patients with complement deficiency are exposed because of their diminished capacity for complement‐mediated lysis of capsulated bacteria. 95 , 96 In a 10‐year observational study reflecting 28,518 patient‐years of cumulative exposure to eculizumab for PNH and aHUS treatment, the incidence of meningococcal infections was 0.25 per 100 patient‐years. Almost all cases occurred in patients who had received meningococcal vaccination, although not against all serotypes of Neisseria meningitides. 97 Antibiotic prophylaxis may prevent meningococcal infection but carries the risk of resistant bacterial strains emerging. Thus, because neither vaccines nor antibiotic prophylaxis guarantee full protection against meningococcal infection, treatment discontinuation—under close patient monitoring—could be a valuable option for patients with aHUS who are on chronic eculizumab therapy and are at low risk of disease recurrence. Eculizumab discontinuation is also proposed to minimize the intravenous infusion treatment impact on patients' quality of life. The current treatment regimen may be burdensome for individuals in terms of visits to the hospital. Venous access may also be difficult for these patients, in particular children, which can cause discomfort and prolong the time needed for infusion. Moreover, intravenous infusion may become more difficult over time because of progressive exhaustion of venous vascular accesses.
Besides being used for diagnostic purposes, the previously described ex vivo assay of complement activation on endothelial cells (2.1.1 section) 25 can also be useful to monitor the efficacy of eculizumab therapy in aHUS. A study that included 121 patients with aHUS 98 showed that the ex vivo test on ADP‐activated endothelium showed complement dysregulation in all patients who were not treated with eculizumab or plasma, independently of disease activity, while the test on unstimulated endothelium was positive only in those with active disease. Serum‐induced C5b‐9 deposits on activated and unstimulated endothelial cells normalized during eculizumab treatment. During eculizumab tapering/discontinuation, all patients who experienced relapses had elevated C5b‐9 deposits on unstimulated endothelium, compared to only 6% of those who remained in remission. The detection of serum‐induced complement deposition on resting endothelial cells highlights and possibly predicts relapses after eculizumab discontinuation. The ex vivo endothelial assay could therefore be an advance over previous complement activity assays, moving toward personalized complement inhibitor therapy in aHUS.
A phase 4 study is ongoing with the aim to improve efficiency of eculizumab administration based on therapeutic drug monitoring. A personalized spacing of eculizumab infusions, using a pharmacokinetic population model to estimate eculizumab concentration, will be compared to the usual administration scheme (NCT04859608, EspacECU, Table 1).
2.2. C3 glomerulopathy and immune complex‐associated membranoproliferative glomerulonephritis
2.2.1. Classification and clinical manifestation
Membranoproliferative glomerulonephritis (MPGN), also known as mesangiocapillary glomerulonephritis, is a pattern of glomerular injury observed in kidney biopsies, with characteristic light microscopic changes: mesangial hypercellularity, endocapillary proliferation, and duplication—double contours of the glomerular basement membrane (GBM). 99 The histopathologic finding of MPGN is one of the most challenging, since it does not refer to a specific disease but may instead be the result of different etiologies.
In 2011, a classification on the basis of immunofluorescence (IF) was proposed 100 , 101 that divides MPGN into (1) (C3G), characterized by dominant glomerular C3 deposition (at least two orders of intensity stronger than any other immune reactant) and little or no immunoglobulin (Ig) deposition and (2) immune complex‐associated MPGN (IC‐MPGN), with significant glomerular Ig and complement deposition. Through electron microscopy (EM), C3G may be further classified into dense deposit disease (DDD), with highly electron‐dense deposits in the GBM, and C3 glomerulonephritis (C3GN), with mesangial, subendothelial, subepithelial, and intramembranous deposits, but without the typical electron‐dense deposits of DDD. The term C3G is also used to define non‐specific alterations or other proliferative patterns that share C3‐dominant glomerular staining. 102 Careful evaluation can help identify an underlying cause in C3G or IC‐MPGN cases. When chronic infections, autoimmune diseases, or paraprotein‐related kidney diseases are ruled out, and a clear underlying etiology cannot be identified, C3G and IC‐MPGN are considered primary or idiopathic.
The current classification is based on the assumption that C3G arises from genetic and/or acquired abnormalities in the control of the AP, whereas IC‐MPGN, also termed immune complex glomerulonephritis (ICGN) in the most recent version of the Kidney Disease Improving Global Outcomes (KDIGO) Guidelines, 103 derives from the deposition of IC that trigger the classical complement pathway. The pathogenesis of these rare nephropathies is, however, more complex, and the role of AP activation in primary IC‐MPGN has also been clearly documented. 104 , 105 Among patients who underwent repeated biopsies, 40% had different IF staining patterns on the initial and follow‐up biopsies and 17% exhibited a shift from C3G to IC‐MPGN or vice versa. 106 Some children may present at onset with a biopsy characterized by proliferative GN and an IC‐MPGN pattern with co‐dominant C3 and Ig staining, with a subsequent biopsy showing dominant C3. 107 This likely relates to IC induced by infections or other triggers that initiate the disease in patients who have an underlying AP abnormality, and C3‐dominance may become evident following classical pathway inactivation after the resolution of infection or after immunosuppressive therapy. Recently, it has been shown that in three of 11 individuals initially diagnosed with IC‐MPGN, the diagnosis changed to C3GN following a second biopsy. 108 These findings are consistent with the hypothesis that during the course of C3G, there may be episodes of IC deposition, possibly triggered by infections.
Acquired drivers of disease include autoantibodies—referred to as nephritic factors—that stabilize the alternative pathway C3 and/or C5 convertase (C3NeF or C5NeFs).
Through an unsupervised cluster analysis in a cohort of 173 C3G and IC‐MPGN patients, we explored whether they could be divided into relatively homogeneous groups. 104 This approach, which places patients with many commonalities close together so that each individual cluster has greater homogeneity than the whole, has previously enabled the identification of disease subtypes of Parkinson's disease, Alzheimer's disease, asthma, and other conditions. 109 , 110 , 111 , 112 , 113 , 114 In the analysis, 34 histologic, biochemical, genetic, and clinical features that were available at disease onset were included. Four clusters were identified, indicating the existence of distinct disease entities characterized by specific pathogenetic mechanisms (Figure 2). Clusters 1‐2 included patients with fluid‐phase AP activation at both the C3 and C5 levels, highlighted by low serum C3 and high plasma levels of sC5b‐9, but those in cluster 2 also exhibited markers of activation of the classical pathway in the biopsy (C1q and IgG staining) and the highest prevalence of nephrotic syndrome. Patients in cluster 3 had fluid‐phase AP activation, mainly at the C3 level, and highly electron‐dense deposits in the GBM. Finally, cluster 4 was characterized by solid phase‐restricted complement activation with glomerular C3 deposits and a normal complement profile in the blood and had the highest risk of ESRF. In this regard, clusters did better at predicting renal survival than the conventional classification into IC‐MPGN, DDD, and C3GN. 115 Notably, while a large majority of DDD patients fell into cluster 3, C3GN and IC‐MPGN patients were distributed among clusters, reinforcing the overlap between C3GN and IC‐MPGN and the heterogeneity of the two histologic groups. Genetic and acquired complement abnormalities were highly prevalent in clusters 1‐3 but rare in cluster 4 (Figure 2). Further analysis revealed that variants affecting C3 and CFB, and C5NeFs were more prevalent in clusters 1 and 2, whereas cluster 3 patients had a higher prevalence of C3NeFs and of heterozygous CFH variants compared with the other clusters. 104 , 116 The cluster analysis approach was subsequently validated by an independent group in another cohort of 92 C3G and IC‐MPGN patients, with similar results. 117
FIGURE 2.
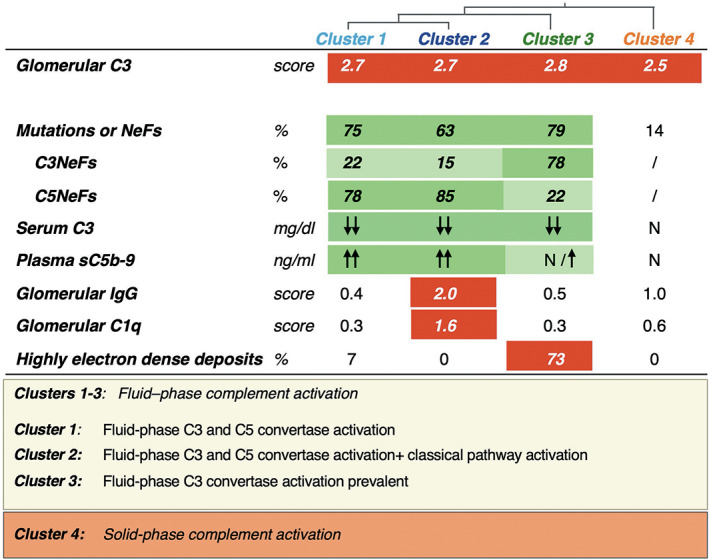
Characteristics of the 4 clusters obtained through unsupervised cluster analysis. Clusters 1 to 3 showed evidence of fluid‐phase alternative pathway activation and a high prevalence of complement gene abnormalities (mutations) and/or nephritic factors (NeFs). In clusters 1 and 2, AP activation occurs both at the C3 and C5 levels, as documented by low serum C3 and high levels of sC5b‐9. Cluster 2 is distinguished by the fact that these patients also have signs of activation of the classical pathway (Ig and C1q staining in biopsy). In cluster 3, fluid‐phase C3 convertase activity predominates over C5 convertase activity, as shown by mostly normal sC5b‐9 levels. Most of these patients have highly electron‐dense deposits in the glomerular basement membrane. Most patients in clusters 1 and 2 carried NeFs that stabilized both the C3 and the C5 convertases, whereas NeFs stabilizing the C3 convertase only were mainly found in cluster 3. Cluster 4 patients separated from the others since they have normal serum C3 levels but intense C3 staining in the kidney, indicating solid phase AP activation in the kidney. N: normal value. Green rectangles highlight abnormalities in circulating blood, and red rectangles highlight glomerular abnormalities. Reprinted from Noris M, Daina E, Remuzzi G. Membranoproliferative glomerulonephritis: no longer the same disease and may need very different treatment. Nephrol Dial Transplant. 2021 Oct 1:gfab281. doi: 10.1093/ndt/gfab281
Primary C3G and IC‐MPGN are rare, with an estimated prevalence of 1.2–1.6 per million in Europe. 118 The clinical picture is characterized by a variety of symptoms, ranging from mild disease with asymptomatic microscopic hematuria and/or proteinuria, to severe disease with nephritic or nephrotic syndrome (NS) and renal function impairment. In general, outcomes are poor. Relevant prognostic factors, reported in both adults and children, include NS at onset and a higher proportion of sclerotic glomeruli and crescents in kidney biopsies. 119 The predictive value of the histological features regarding disease outcome has recently been documented. 108 In a large cohort of C3G and IC‐MPGN patients, the risk of progression to kidney failure was associated with estimated glomerular filtration rate (GFR) and proteinuria at the time of biopsy, cellular/fibrocellular crescents, segmental sclerosis, and interstitial fibrosis/tubular atrophy scores.
The risk of ESRF is similar for patients with C3G and IC‐MPGN (4%–41% vs. 9%–41%). 104 , 120 Compared with other forms of GN, patients with ESRF have comparable rates of survival when they are on dialysis and following kidney transplantation, but significantly higher rates of allograft loss due to disease recurrence (54%–60% in C3G vs. 43% in IC‐MPGN). 120
2.2.2. Genetic and acquired determinants of alternative pathway dysregulation
Genetic and acquired abnormalities associated with dysregulation of the AP are found in around 50‐70% of patients with primary C3G/IC‐MPGN. These include LPVs that affect complement regulators, mainly factor H, or the two components of the alternative pathway C3 convertase, C3 and factor B; structural variants in CFH‐CFHRs genes; common susceptibility variants, and/or acquired abnormalities. Notably, the percentages of primary C3G and IC‐MPGN patients carrying genetic and/or acquired AP abnormalities were comparable. 104 , 119 , 120
Several reports have described LPVs in complement components and regulators, such as C3, CFB, CFH, MCP, CFI, and THBD. 102 , 119 , 121 Compound heterozygosity 122 and homozygous 40 and heterozygous 123 LPVs in the CFH gene associated with factor H deficiency have been reported. Gain‐of‐function LPVs in the CFB gene have been demonstrated in C3G and IC‐MPGN patients. 119 , 124 , 125
Rearrangements that affect the genes that encode the five FHR proteins and lead to internal duplications, deletions, or hybrid genes that result in the deregulation of FH activity were initially described in C3G patients; 126 however, more recently, they have been associated with primary IC‐MPGN as well. 127
Common susceptibility variants in the CFH and CFHR5 genes have been associated with MPGN, 128 strengthening the hypothesis that complement control plays a role in the pathogenesis of the disease.
Acquired drivers of disease include a heterogeneous group of nephritic factors (C3NeF or C5NeFs). They are the most commonly detected autoantibodies and recognize neoantigenic epitopes on C3bBb and C3b2Bb, the C3 and C5 convertases of the alternative pathway, respectively. 129 , 130 , 131 , 132 In the presence of C3NeFs and C5NeFs, the half‐lives of C3 and C5 convertase lengthen. Persistent cleavage of C3 drives down serum concentrations of C3 and increases serum concentrations of its cleavage products. 116
Inhibitory FHAA 16 , 127 , 130 , 133 , 134 and activating anti‐factor B or anti‐C3b autoantibodies have been observed in individuals with C3G. 9 As a general rule, acquired drivers extend the half‐life and stabilize the C3 convertase, which leads to persistent AP activation in the fluid phase. 135
2.2.3. Complement inhibition: ongoing studies and therapeutic perspectives
The optimal treatment for primary C3G and IC‐MPGN has not been established yet, and there are no approved drugs for the affected patients. Immunosuppressive therapy is often prescribed, although the choice of drug and duration of treatment is based on retrospective analyses, limited case series, and observational studies, rather than randomized controlled intervention trials. The recently released KDIGO Guideline for the Management of Glomerular Diseases 103 recommends the usual supportive measures (low‐salt diet, treatment of hypertension, reduction of proteinuria with angiotensin inhibition, and treatment of dyslipidemia) together with immunosuppression in the setting of moderate to severe disease (initially with mycophenolate mofetil plus glucocorticoids, and if this fails, with eculizumab).
Eculizumab has been employed in single patients or small series of patients with primary C3G and IC‐MPGN. The hypothesis is that by inhibiting the cleavage of C5, thereby precluding the formation of C5a and C5b‐9 (Figure 1), the drug might protect the kidneys from complement‐mediated damage. Published data suggest that high levels of serum sC5b‐9 before treatment may predict a better response. 136 Based on this consideration, we evaluated the effect of eculizumab in the context of a sequential off‐on‐off‐on design in ten patients (six with primary IC‐MPGN and four with primary C3GN) with nephrotic‐range proteinuria and high plasma sC5b‐9 levels. 137 The finding that sC5b‐9 plasma levels were fully normalized by eculizumab in all subjects, whereas proteinuria decreased in only three patients, is consistent with evidence that disease activity, at least in some patients, is only partially mediated by the activation of the terminal complement pathway. It could also be assumed that in non‐responder patients, the activation of other upstream C3‐convertase‐dependent pathways, which cannot be blocked by eculizumab, may cause kidney damage despite the inhibition of the terminal pathway. The heterogeneous response to eculizumab treatment could be related to the extent of terminal complement activation, which may vary substantially from patient to patient. 104
The oral C5aR1 antagonist CCX168 (avacopan), recently approved by the US Food and Drug Administration (FDA) for adjunctive treatment of anti‐neutrophil cytoplasmic autoantibody (ANCA)‐associated vasculitis, has been investigated in a phase 2 trial with C3G patients (ACCOLADE; NCT03301467). C5a is a potent anaphylatoxin that, by interacting with the C5aR1 receptor, increases vascular permeability and induces oxidative bursting and the release of pro‐inflammatory cytokines in myeloid cells, as well as having chemotactic properties for myeloid and lymphoid cells. 138 Avacopan blocks the interaction of C5a with C5aR1 and potentially exerts an anti‐inflammatory effect. Preliminary results from the ACCOLADE study 139 showed that there was an increase in the C3G Disease Chronicity Scores despite treatment with avacopan. Nonetheless, investigators noted differences in disease progression compared with placebo, suggesting that avacopan had at least a partial effect on attenuating C3G progression. Compared to eculizumab, avacopan does not affect the formation of the terminal complement complex, that plays a key role in controlling infections caused by encapsulated bacteria, such as Neisseria meningitidis. However, terminal pathway inhibitors like eculizumab or avacopan do not affect complement activation upstream of C5 and do not prevent the formation of C3 activation fragments and their accumulation in glomerular immune deposits of C3G and IC‐MPGN patients. The future therapeutic landscape for C3G/IC‐MPGN seems more encouraging thanks to new complement inhibitor drugs that directly counteract AP dysregulation.
Agents that target the AP are thus being tested in C3G and IC‐MPGN. In this regard, ACH‐0144471 (danicopan) 140 is a small, orally active inhibitor of factor D. Factor D is a serine protease, mainly produced by adipose tissue, that catalyzes the cleavage of factor B, a rate‐limiting step that converts the inactive enzyme proconvertase C3bB into the active C3 convertase C3bBb of the alternative pathway (Figure 1). By inhibiting factor D activity, danicopan specifically targets the control point of the complement cascade amplification loop, blocking C3 convertase formation and, therefore, significantly reducing the production of C3 cleavage products (C3 fragments) and downstream MAC formation. An open‐label phase 2 study in PNH documented the effective inhibition of hemolysis at week 24 in 12 patients with an inadequate response to eculizumab. In particular, the addition of danicopan resulted in a mean increase in hemoglobin of 2.4 g/dl and a clinically significant reduction in transfusion needs vs baseline in patients who were transfusion‐dependent on eculizumab. 141 Two proof‐of‐concept phase 2 studies with this factor D inhibitor, a randomized placebo‐controlled phase 2 study in C3G (NCT03369236) and a single‐arm phase 2 study with C3G or IC‐MPGN patients (NCT03459443), have been conducted. The manuscripts describing these results are in press, but the company has stated that they will halt development of the drug for C3G and IC‐MPGN, citing that the phase 2 study data showed a suboptimal clinical response, due to an insufficient pharmacokinetic and pharmacodynamic response and incomplete inhibition of the AP. 142 In vitro and in vivo studies indicated that a very high degree of factor D inhibition (likely more than 95%) needs to be achieved to efficiently block the AP. 143 These findings are particularly relevant to C3G and IC‐MPGN patients, who may require an even higher degree of factor D inhibition, since they suffer from hyperactivity of the AP due to C3 convertase dysregulation. In addition, circulating levels of factor D are dependent on kidney function, since factor D is filtered through the glomerulus and catabolized in the proximal renal tubule. 144 An inverse correlation between plasma factor D levels and creatinine clearance has been reported in patients with various renal diseases. 145 Preliminary results from the two clinical trials of danicopan in C3G and IC‐MPGN confirmed an inverse correlation between factor D levels and renal function, so that patients with renal impairment had higher than normal factor D levels, 146 which represents another hurdle for efficient AP inhibition in these conditions. A phase 1 clinical study has been planned (NCT04623710), involving healthy subjects and three cohorts of patients with severe, moderate, or mild impairment of renal function, respectively, to determine the effect of renal dysfunction on the pharmacokinetics and pharmacodynamics of the new, more potent factor D inhibitor ALXN2050. The drug is also currently being evaluated in a phase 2 study in PNH (NCT04170023). An open‐label, multicenter, proof‐of‐concept phase 2 study is ongoing to evaluate the safety, tolerability, and therapeutic potential of another factor D inhibitor (BCX9930) 147 administered for 24 weeks to adult participants with either C3G, IgA nephropathy, or membranous nephropathy (NCT05162066, RENEW, Table 1).
Another AP inhibitory drug is iptacopan (LNP023) a small, orally active molecule that binds to factor B. It does not prevent the formation of the C3 convertase, but it specifically inhibits C3 convertase enzymatic activity, blocking the conversion of C3 to C3b (Figure 1) and the activation of the amplification loop. In turn, this blockade prevents downstream generation of the AP C5 convertase, without affecting the activity of the classical/lectin pathway's C5 convertase. 148 In vitro, iptacopan inhibited complement activation in sera from C3G patients and inhibited the activity of the C3 convertase stabilized by C3NeFs isolated from C3G sera. 148 An open‐label non‐randomized phase 2 study on the efficacy, safety, and tolerability of iptacopan in patients with C3G on the native kidney or after transplant has been carried out (NCT03832114), and the long‐term extension study is ongoing (NCT03955445). Preliminary results from 12 adult patients with biopsy‐proven native C3G who received iptacopan for 12 weeks are available. Iptacopan inhibited AP activity, and plasma C3 levels recovered, with complete normalization in five of seven tested patients at 12 weeks. 149 Most importantly, urinary protein excretion fell by 49% at 12 weeks and renal function stabilized. The treatment was well‐tolerated, with no treatment‐emergent severe adverse events. A multicenter, randomized, double‐blind, placebo‐controlled phase 3 study on the efficacy and safety of iptacopan in C3G is ongoing (NCT04817618, Table 1).
Drugs that target C3 are also under clinical development. Specifically, APL‐2 (pegcetacoplan), a synthetic cyclic peptide conjugated to a polyethylene glycol polymer, binds to C3 and inhibits C3 activation from all three pathways. In addition, APL‐2 binds to C3b and prevents the activity of the C3 and C5 convertases (Figure 1). Pegcetacoplan was approved by the FDA in May 2021 for treating adult patients with PNH, 150 thereby further expanding the list of approved treatment options that target the complement system. The safety and efficacy of pegcetacoplan has been investigated in a phase 2 open‐label study (NCT03453619) involving patients with different glomerulopathies, including C3G. Preliminary results from C3G patients were presented at the 2020 ASN meeting. 151 Of the eight recruited patients, two non‐compliant patients were excluded from the analysis. The other six experienced an increase in serum C3 and a decrease in plasma sC5b‐9 levels, indicating that pegcetacoplan was able to modulate complement hyperactivity in C3G, both at the C3 and C5 level. During treatment, there was a trend toward a reduction in proteinuria (mean reduction of 24‐h urinary proteins at day 84: 50%) and an increase in serum albumin. A phase 2 open‐label randomized study is ongoing to evaluate the safety and efficacy of twice‐weekly subcutaneous doses of pegcetacoplan in the post‐transplant recurrence of C3G or IC‐MPGN (NCT04572854, NOBLE, Table 1). 152 The phase 3 randomized, placebo‐controlled, double‐blinded study in patients with a diagnosis of primary C3G or IC‐MPGN (with or without previous renal transplant) has recently been initiated (NCT05067127, VALIANT, Table 1). Of note, this is so far the only study opened to adolescent patients.
3. OTHER KIDNEY DISEASES ASSOCIATED WITH ALTERNATIVE COMPLEMENT PATHWAY DYSREGULATION
Dysregulation of the AP is known to cause or accentuate different inflammatory diseases in which glomerular injury leads to the appearance of hematuria and proteinuria and ultimately to the development of progressive chronic kidney disease. Experimental and clinical evidence is reported for each condition, with particular focus on the occurrence of AP dysregulation along with classical and lectin pathways involvement. Although the role of the AP is likely less prevalent in most of these conditions, growing data support careful evaluation of drugs specifically targeting AP in clinical trials (Table 1).
3.1. Membranous nephropathy
(MN) is one of the most common causes of NS in Caucasian, non‐diabetic adults, with estimated annual incidence rates of 2‐17 per million in Europe and 10‐12 per million in North America. 153 The disease can affect individuals of all ages, with a mean age of diagnosis of 50–60 years, 154 and a male‐to‐female ratio of 2:1. 155 MN is morphologically characterized by the deposition of IgG, the relevant antigens and complement components in the subepithelial space of the glomerular capillary wall, with variable degrees of GBM thickening. Despite there being a common histopathological pattern, MN is a heterogeneous disease, which occurs either in the absence of an associated disease (80% of cases) or in association with clinical conditions, such as hepatitis virus infection, systemic lupus erythematosus, malignancies, or drug toxicity, thereby classified into so‐called primary and secondary MN, respectively. 156 Heterogeneity is also highlighted by the variable clinical course. On average, one‐third of patients experience spontaneous remission, usually within the first 2 years of presentation. 157 , 158 The other two‐thirds of patients can be divided equally into those who maintain variable levels of proteinuria and stable long‐term kidney function and those who progress to ESRF. 159
Advances over the last two decades have shown that primary MN is a kidney‐specific autoimmune disease induced by autoantibodies specific to podocyte antigens, such as M‐type phospholipase A2 receptor (PLA2R) and thrombospondin type‐1 domain‐containing protein 7A (THSD7A), which have been identified in about 70% and less than 5% of adult patients, respectively. 160 , 161 More recently, several other proteins, such as contactin 1, semaphorin 3B, transforming growth factor‐β receptor 3, and netrin G1, have been characterized as potential autoantigens in primary MN. 162
Our understanding of the pathophysiology of MN largely comes from studies that used the rat model of Heymann nephritis induced by antibodies against the podocyte membrane protein megalin. In this experimental model, local complement activation by subepithelial immune complexes with subsequent podocyte damage through C5b‐9 is a major effector mechanism of proteinuria. 163 , 164 Nevertheless, since megalin is not expressed on human podocytes, it does not work as a disease mediator in patients with MN. Recently, murine models of PLA2R‐ 165 and THSD7A‐associated MN 166 , 167 have been developed, but they have not yet convincingly demonstrated the pathogenic relevance of complement activation.
The involvement of the complement system in patients with MN is based on the consistent presence of C3 and C5b‐9 alongside IgG in subepithelial deposits. 168 , 169 However, the exact contribution and clinical significance of the individual activation pathways remains a matter of investigation. PLA2R and THSD7A autoantibodies are predominantly of the IgG4 subclass, 170 , 171 , 172 , 173 which is unable to bind C1q and activate the classical complement pathway. 174 , 175 Accordingly, in kidney biopsies from patients with MN, glomerular staining for C1q is generally weak, 176 while staining for mannose‐binding lectin (MBL) and C4d is commonly positive, consistent with the activation of the lectin pathway. 177 , 178 , 179 Moreover, in PLA2R‐associated MN, altered glycosylation of IgG4 autoantibodies was found to promote binding of MBL and complement activation via the lectin pathway, leading to sublethal injury to human podocytes in culture. 180 On the other hand, cases of PLA2R‐associated MN have been reported in patients with complete MBL deficiency, with complement activation mainly induced by the alternative pathway, as determined based on glomerular deposition of factor B and properdin. 181 The activation of the AP has also been confirmed by mass spectrometry analyses of laser capture microdissected glomeruli from patients with PLA2R‐associated MN, which showed low levels of factor B and properdin, along with the accumulation of factor H and FHR proteins. 182 The pathogenic relevance of the AP was supported by findings in a mouse model of MN, where the lack of factor B prevented glomerular deposition of C5b‐9 and protected against albuminuria development. 183 In line with this, THSD7A immune complexes predominantly containing IgG4 have been found to activate complement in vitro via the alternative pathway, albeit only at a high surface density. 184 Another in vitro study showed that inhibition of the classical and lectin pathways significantly decreased complement‐mediated cytotoxicity induced by anti‐PLA2R antibodies, suggesting that the alternative pathway plays a limited role in complement activation. 185 It is conceivable that the low amounts of non‐IgG4 autoantibodies, which were found to be predominant in the early stage of immune deposition, 186 are sufficient to initiate complement activation by the classical pathway, which is followed by amplification through the alternative pathway. 187
In MN, alternative pathway activation may also occur independently of immune complexes, due to local complement dysregulation. In particular, the loss of heparan sulfate chains from the glomerular basal membrane, which has been observed in human and experimental MN, 183 , 188 , 189 could lead to impaired recruitment of factor H, the major inhibitor of the AP in plasma. It has also been posited that FHAA, which have been reported in a small subset of patients with primary MN, 190 , 191 may contribute to the activation of the AP. However, these antibodies were not identified in all the MN cohorts tested, and their presence did not correlate with worse disease outcome. 190 , 192 Thus, even when FHAA are produced in patients with primary MN, they are unlikely to play a significant role in the development of severe forms of the disease.
Collectively, the available evidence suggests that each of the three complement pathways may be active to different extents in patients with MN, but in most cases none appear to be exclusive or indispensable for disease initiation and progression.
3.2. Anti‐neutrophil cytoplasmic antibody‐associated vasculitis
Anti‐neutrophil cytoplasmic autoantibody (ANCA)‐associated vasculitides (AAV) are a group of systemic autoimmune diseases characterized by necrotizing inflammation of small vessels and the common presence of circulating autoantibodies against neutrophil primary granule proteins, especially proteinase 3 (PR3) and myeloperoxidase (MPO). 193 They comprise granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA). 194 Most patients with GPA have ANCA directed to PR3 (PR3‐ANCA), while those with MPA are predominantly MPO‐ANCA positive. 195 The global incidence of AAV was estimated to be 17.2 per million person‐year and the prevalence of 198 per million persons. 196 Incidence rates increase with age, and are marginally higher in males. 197 Although any organ and tissue can be involved in AAV, the kidneys and lung, which are rich in small vessels, are the most frequently and severely affected, with rapidly progressive glomerulonephritis and diffuse alveolar hemorrhage being major threats.
Histologically, renal involvement is characterized by necrotizing crescentic glomerulonephritis with little, if any, immunoglobulin and complement deposition in the glomeruli. 198 These findings, along with the observation that hypocomplementemia is rare in patients with AAV, previously led to the assumption that the complement system was minimally involved in the pathogenesis of these conditions. Over the past 15 years, however, studies using a mouse model of MPO‐ANCA vasculitis have suggested that complement plays a critical role in the development of AAV. 199 In this model, MPO‐deficient mice are immunized with purified murine MPO, and the subsequently produced autoantibodies are passively transferred into wildtype recipients, resulting in crescentic glomerulonephritis and vasculitis. When recipient mice were deficient in C5 or factor B, or pretreated with a C5‐inhibiting monoclonal antibody, no disease developed, while C4 deficiency did not have any protective effects, suggesting that complement activation via the alternative pathway is involved in the pathogenesis of AAV. 199 , 200 Intriguingly, C5aR1 blockage or deficiency protected against ANCA‐induced necrotizing and crescentic glomerulonephritis in mice, whereas C6 deficiency did not, pointing to the anaphylatoxin C5a, and not C5b‐9, as a pathogenic mediator of experimental AAV. 201 , 202 Indeed, the interaction between C5a and neutrophil C5aR1 was found to cause an amplification loop for ANCA‐induced neutrophil activation. 201 Consistent with this, another study showed that neutrophils, primed by cytokines or coagulation factors, were able to activate the AP on their membrane, leading to the release of C5a and further amplification of the inflammatory response. 203
Despite the usual absence of immune complex deposits, positive staining for C5b‐9, C3d, factor B, and properdin has been documented in kidney biopsies from patients with AAV. 204 , 205 The finding that factor B colocalized with C5b‐9 in active glomerular lesions suggests that activation of the AP could lead to kidney damage. 204 Further studies showed that properdin staining was associated with the proportion of cellular crescents and proteinuria levels, while the glomerular deposition of Bb, the active subunit of factor B, correlated with the percentage of total crescents observed in the kidney biopsy. 205 , 206 Likewise, plasma levels of Bb are closely associated with disease activity, including the proportion of crescents documented in renal biopsies, the erythrocyte sedimentation rate, and the Birmingham Vasculitis Activity Score. 207 The involvement of alternative pathway regulators in AAV has also been investigated. In particular, plasma levels of factor H were reported to be inversely associated with disease activity and with the proportion of total crescents and cellular crescents in kidney biopsy specimens. 208 Further research has shown that factor H from AAV patients is generally less effective in binding and regulating C3b, and in the protection of cells against complement damage. 209 Notably, two SNPs in CFH which were reported to be strongly associated with the risk of developing age‐related macular degeneration (ie, I62V, rs800292 and Y402H, rs1061170) were identified in some patients with AAV. 210 , 211 Whether such genetic variants directly account for the impaired functional activities of factor H observed in AAV patients remains ill defined. Moreover, in vitro evidence indicates that MPO, which can be released from neutrophils following activation by ANCA, binds to and inhibits the regulatory activity of factor H. 212 Thus, quantitative deficiency or functional impairment of factor H may be related to the development of AAV.
Together, these findings highlight the importance of complement activation through the alternative pathway in the pathogenesis of AAV.
3.3. Acute postinfectious glomerulonephritis
Acute postinfectious glomerulonephritis (APIGN) is a glomerular disease that occurs as a result of host response to an extrarenal infection. The classic example is poststreptococcal glomerulonephritis caused by specific nephritogenic strains of group A β‐hemolytic Streptococci in the setting of an infection of the pharynx or skin. 213 APIGN most commonly affects children, but it can also develop in adults, especially in patients who are older than 60. 214 Clinically, the disease presents with hematuria, proteinuria, hypertension, low serum C3 levels, and a variable degree of kidney function impairment. Although the prognosis for patients with APIGN is good overall, it has—rarely—been associated with chronic C3 consumption, persistent proteinuria, and even progression to ESRF. 213 , 215 There is considerable overlap in the clinical, biochemical, and histopathologic features of APIGN and C3G at onset, making a differential diagnosis challenging. 216
The pathogenesis of APIGN is thought to be the result of the glomerular deposition of immune complexes, either formed in situ or in the circulation, against Streptococcus bacteria antigens, with secondary complement activation, as shown by bright C3 staining on immunofluorescence microscopy. In spite of a robust antibody response to bacterial antigens, the activation of the classical complement pathway is inhibited by chemokine‐binding evasins secreted by Streptococcus bacteria, 217 and by proteins of the streptococcal surface, which bind a C4b‐binding protein. 218 , 219 In fact, the finding that a large majority of patients with APIGN have decreased serum C3 levels and normal C4 levels during the acute phase of the disease suggests that there is a selective activation of the AP. 220 The glomerular presence of properdin and the observation that C3 deposition may precede or occur without that of immunoglobulins also point to alternative pathway activation. 221 Moreover, streptococcal components have been found to activate this pathway in vitro. 222 , 223 Further research provided evidence of AP involvement in the pathogenesis of APIGN. In particular, most patients presenting with an atypical disease course, characterized by persistent proteinuria and hematuria, were found to have underlying abnormalities of the AP, including LPVs in genes that encode for complement‐regulating proteins and/or antibodies to the C3 convertase. 224 More recently, autoantibodies against factor B have been identified in 31 out of 34 children with APIGN. 225 At disease onset the anti‐factor B antibody titer, which decreased over time, correlated inversely with plasma C3 levels and directly with soluble C5b‐9 levels. In functional studies, anti‐factor B antibodies isolated from the patients enhanced the activity of the alternative pathway C3 convertase. 225 It remains to be established whether these antibodies are the actual drivers of alternative pathway activation and of kidney disease in APIGN, or if complement activation occurs before their appearance. 226
3.4. IgA nephropathy
IgAN is the most common primary glomerulonephritis worldwide, with the highest prevalence in Eastern Asia. The incidence has been estimated at 2–10 per 100,000 person per year and peaks during the second and third decades of life. 227 The clinical course of IgAN is heterogeneous: after 20 years of follow‐up following diagnosis, up to 40% of patients will have reached ESRF, but 20% of patients will have preserved renal function. 228 The pathogenesis of IgA is believed to follow a multi‐hit process involving the production of abnormal galactose‐deficient IgA1, which leads to the formation of anti‐galactose‐deficient IgA1 autoantibodies and the deposition of IgA1‐containing immune complexes in the mesangium, resulting in glomerular inflammation and kidney injury. 229 In addition to IgA1 deposition, IgAN is characterized by glomerular deposits of C3, properdin, C4d, MBL, and C5b‐9, whereas C1q is typically absent, suggesting a predominant involvement of the alternative and the lectin pathways in this disease. 230 Consistent with this, the ability of human polymeric IgA to activate the AP in vitro has been demonstrated. 231 Furthermore, in a rat model of IgA‐mediated glomerular inflammation, polymeric (but not monomeric) IgA triggered mesangial deposition of C3, whereas C4 and C1q were not detectable in the glomeruli. 232 These findings suggest that IgA polymerization plays a critical role in inducing the activation of the AP. In patients with IgAN, plasma levels of Ba, the smaller activation fragment of factor B, were found to be higher compared with those who had focal and segmental glomerulosclerosis or healthy controls. 233 Moreover, in IgAN patients plasma Ba levels correlated directly with circulating C3a concentrations and the degree of proteinuria, and inversely with estimated GFR, 233 suggesting a relationship between AP activation and the clinical severity of the disease.
Several lines of evidence point to the involvement of complement FHR proteins, which have been shown to antagonize factor H activity, in the pathogenesis of IgAN. In particular, genome‐wide association studies have identified a SNP within the CFH gene (ie, rs6677604) that is closely associated with a deletion polymorphism of the CFHR3 and CFHR1 genes (delCFHR3‐R1), whose presence was robustly associated with protection against IgAN. 234 , 235 Across populations worldwide, delCFHR3‐R1 frequency showed marked differences in a pattern inverse to that of disease prevalence. 235 In a Chinese cohort of IgAN patients, the protective delCFHR3‐R1 allele was associated with reduced mesangial C3 deposition, higher circulating levels of factor H, and lower C3a concentrations. 236 Moreover, rare CFHR5 gene variants were found to contribute to the genetic susceptibility to IgAN. 237 Two independent studies showed that circulating CFHR1 levels and the CFHR1/factor H ratio, as an index of the relative abundance of dysregulating and regulating proteins, were higher in IgAN patients than in healthy controls, and associated with more rapid disease progression irrespective of the delCFHR3‐R1 allele carriage. 238 , 239 Circulating levels of CFHR5 were also found to be higher in IgAN patients than in healthy controls in two large cohort studies and correlated with histologic markers of kidney injury. 238 , 240 Remarkably, glomerular deposition of CFHR5 has been observed in kidney biopsies from IgAN patients, along with complement‐activating products, 241 , 242 and associated with disease progression. 241 Together, these findings suggest that CFHR1 and CFHR5 may contribute to the pathogenesis of IgAN by impairing factor H‐dependent regulation of the AP, thereby influencing the severity of glomerular inflammation and injury.
3.5. Lupus nephritis
Systemic lupus erythematosus (SLE) is a chronic multisystem autoimmune disease of unknown etiology characterized by the loss of immune tolerance to endogenous nuclear and cellular antigens, which can lead to the injury of several organ and tissues. 243 The overall global incidence ranges from 1.5 to 11 cases per 100,000 person per year, with the global prevalence reported as ranging from 13 to 7,713.5 cases per 100,000 individuals. 244 Possible reasons for this wide discrepancy are discussed in ( 244 ). Lupus nephritis is one of the most severe manifestations of SLE, which develops in up to 60% of patients during the disease course, more commonly in individuals of African American, Hispanic, or Asian ethnicity who are younger and male. 245 The clinical presentation is highly variable, ranging from asymptomatic proteinuria and/or hematuria to rapid and progressive loss of renal function from glomerulonephritis. The risk of ESRF at ten and 15 years from LN diagnosis has been estimated at 17% and 22%, respectively. 246
The activation of the classical complement pathway, triggered by the interaction of C1q with immune complexes, has been recognized as an important mechanism in the pathogenesis of LN. 247 , 248 Nonetheless, several lines of experimental and clinical evidence point to the involvement of AP activation in the development or worsening of kidney injury. In particular, in MRL‐lpr mice, an animal model of LN, genetic deficiency of either factor B or factor D protected against glomerulonephritis. 249 , 250 Consistent with this, a reduction in factor B expression, achieved by antisense oligonucleotides, ameliorated kidney histopathology, reduced glomerular C3 deposition and proteinuria in two different mouse models of LN, MRL‐lpr and NZB/W F1. 251 In the same experimental models, treatment with a selective alternative pathway inhibitor consisting of a fragment of complement receptor 2 linked to the N‐terminal region of factor H (CR2‐fH), but not total complement inhibition, reduced glomerulonephritis. 252 , 253 The authors of these studies have postulated that the benefits of selectively inhibiting the AP may be related, at least in part, to the relative contributions of the alternative pathway versus the classical pathway in the handling of circulating immune complexes and apoptotic cells. Both the alternative and the classical pathways are involved in the clearance of immune complexes. 254 Therefore, it was hypothesized that inhibiting both pathways has the potential to increase circulating immune complex levels and exacerbate disease. The classical pathway also plays an important role in the clearance of apoptotic cells, which have been posited to provide a source of autoantigens responsible for driving antibody production in SLE. 255 Another study showed that treatment with a soluble Fc fusion protein of the complement receptor of the immunoglobulin subfamily (CRIg‐Fc), an intrinsic inhibitor of alternative pathway activation that binds to C3b, thereby blocking the formation of C5 convertase, significantly reduced proteinuria, kidney inflammation, and glomerular deposition of C3 and IgG in MRL‐lpr mice. 256 In patients with LN, the hypothesis that the AP contributes to the development of kidney damage is supported by the observation that reduced plasma levels of C3, but not C4, were independently associated with renal flare. 257 A large cohort study found lower plasma levels of C1q and C3, along with higher concentrations of Bb, C3a, C5a, and sC5b‐9 in patients with active LN compared to those in remission. 258 Moreover, Bb and C5b‐9 colocalized in the glomeruli of LN patients, further suggesting that AP activation may participate in complement‐mediated renal tissue injury. 258 Another study showed that patients with glomerular deposition of factor B and factor H had more severe interstitial fibrosis, while those with positive properdin staining exhibited higher urinary protein excretion. 259 Furthermore, LN patients with kidney biopsies showing glomerular deposition of C3 without C1q and C4, as an index of alternative pathway‐limited complement activation, had poorer response rates to one‐year immunosuppressive therapy and were more likely to experience renal disease progression. 260 Interestingly, a transcriptomic analysis found higher C3 and factor D expression in renal biopsies from patients with LN during flare than normal kidney controls. 261 6 months after induction therapy with corticosteroids, combined with either mycophenolate mofetil or cyclophosphamide, C3 and factor D expression further increased in the kidneys of patients who did not respond to treatment, but remained stable in those who achieved a complete clinical response. 261
The role of AP regulators in the pathogenesis of LN has also been investigated. In MRL‐lpr mice, a genetic deficiency of factor H accelerated the development of lupus nephritis and reduced animal survival. 262 At the clinical level, plasma levels of factor H were found to be significantly lower in patients with LN than in those with SLE without clinical evidence of renal involvement or healthy controls and inversely correlated with SLE disease activity index and renal activity index scores. 263 Moreover, some biofunctions of factor H, including binding activity to C3d, cell protection from complement‐mediated lysis and clearance of apoptotic cells, were reported to be impaired in about half of the patients with active LN. 264 Therefore, quantitative deficiency or dysfunction of factor H may play a role in the development of LN. Together, the available evidence suggests that uncontrolled complement activation, especially through the alternative pathway, promotes kidney injury in LN.
4. CONCLUSIONS
Despite its evolutionary role in survival and defense against infection, the complement system can be a prominent mediator and/or amplifier of the pathogenesis of many serious diseases, including kidney diseases. The success of eculizumab in the treatment of PNH has kindled the pharmaceutical industry's interest in the clinical development of inhibitors that target the complement system at various levels. 265 , 266 Complement inhibition has dramatically transformed the outcome of aHUS, one of the most severe kidney diseases. 88 The availability of complement‐directed therapies has also opened promising new perspectives for the management of several other kidney diseases in which complement activation is involved to a variable extent. Although the value of inhibiting AP‐mediated kidney diseases has long been recognized, incorporating complement‐targeted drugs into clinical use has proved challenging. Numerous drugs that interfere with AP activity have recently been developed and are currently undergoing testing. At least 19 clinical trials in this context are now registered (Table 1). However, clinical trials to test new therapeutics are difficult to carry out due to the rarity of these diseases. In addition, because each drug may act only on specific subgroups of patients, its effect on the overall population will likely be diluted and heavily influenced by the heterogeneity of these diseases. 3
In this regard, there is a need in clinical settings not only to make prognoses but also to assist in decision‐making regarding the most appropriate therapeutic agents. 267 When developing novel treatments for complement‐driven diseases, it is important to consider which component of the cascade may be the most appropriate target. For example, although inhibition of C5 impedes the C5a and MAC formation, this inhibition does not block the pro‐inflammatory and opsonization actions of C3, because C5 acts downstream of C3 as part of the terminal cascade (Figure 1). Therefore, anti‐C5 therapy may have limited effects in diseases where the involvement of C3 is prevalent in the pathogenesis. In addition to considerations regarding the proper target, it is also important to understand what dosages to use to optimize treatment efficacy. For example, responses to factor D inhibition may vary greatly, depending on the degree of AP dysregulation and on factor D levels. Specific in vitro and ex vivo tests are needed to verify the potential responsiveness of each patient to a given complement inhibitor drug and evaluate the dose associated with the maximal effect. The ex vivo assays used to evaluate alternative pathway C3 convertase activity 116 can, for instance, be considered a tool for monitoring patients treated with factor D inhibitor to understand whether they may benefit from the drug and, if so, to establish the effective dose. Ideally, the choice of drug should be tailored to each patient's individual characteristics, including clinical, histologic, and biochemical parameters and genetic and acquired complement abnormalities. Drugs need to not only be highly selective and potent but also be associated with minimal adverse effects and sustainable treatment costs. 268 Experience with PNH seems to show that drugs that act at different levels of the complement cascade can be administered in combination in a beneficial manner and with manageable toxicity. 141 The risks of new anti‐complement agents remain to be quantified, and it should be taken into account that drugs that target complement, either by blocking the AP or by more broadly inhibiting C3 or C5, may have greater effects on reducing the patient's defenses against bacterial infections. Successfully treating patients requires further research in the field and close collaboration between the clinicians and researchers who have an interest and special expertise in the complement system.
CONFLICT OF INTEREST
Giuseppe Remuzzi has consultancy agreements with Boehringer Inghelheim, Janssen Pharmaceuticals, Akebia Therapeutics, Alexion Pharmaceuticals, Alnylam, Inception Science Canada and BioCryst Pharmaceuticals. The funding sources had no role in writing the review or in the decision to submit the paper for publication.
ACKNOWLEDGMENTS
The authors are grateful to Kerstin Mierke for editing the manuscript. They also thank Dr. Norberto Perico and Dr. Marina Noris for helpful discussions. Open access funding provided by BIBLIOSAN.
This work was partially supported by the Italian Ministero della Salute (RF‐2016‐02361720); by Fondazione Regionale per la Ricerca Biomedica (Regione Lombardia), Project ERAPERMED2020‐151, GA 779282; and by Progetto DDD Onlus—Associazione per la lotta alla DDD (Milan, Italy).
Daina E, Cortinovis M, Remuzzi G. Kidney diseases. Immunol Rev. 2023;313:239‐261. doi: 10.1111/imr.13167
This article is part of a series of reviews covering The Alternative Pathway or Amplification Loop of Complement appearing in Volume 313 of Immunological Reviews.
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- 1. Merle NS, Church SE, Fremeaux‐Bacchi V, Roumenina LT. Complement system part I ‐ molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262. doi: 10.3389/fimmu.2015.00262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Merle NS, Noe R, Halbwachs‐Mecarelli L, Fremeaux‐Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. 2015;6:257. doi: 10.3389/fimmu.2015.00257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol. 2018;14(1):26‐47. doi: 10.1038/nrneph.2017.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. D'Agati VD, Bomback AS. In search of C3G tissue biomarkers. Kidney Int Rep. 2019;4(10):1359‐1361. doi: 10.1016/j.ekir.2019.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pangburn MK, Müller‐Eberhard HJ. Initiation of the alternative complement pathway due to spontaneous hydrolysis of the thioester of C3. Ann N Y Acad Sci. 1983;421:291‐298. doi: 10.1111/j.1749-6632.1983.tb18116.x [DOI] [PubMed] [Google Scholar]
- 6. Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20(1):34‐50. doi: 10.1038/cr.2009.139 [DOI] [PubMed] [Google Scholar]
- 7. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785‐797. doi: 10.1038/ni.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dobó J, Kocsis A, Gál P. Be on target: strategies of targeting alternative and lectin pathway components in complement‐mediated diseases. Front Immunol. 2018;9:1851. doi: 10.3389/fimmu.2018.01851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marinozzi MC, Roumenina LT, Chauvet S, et al. Anti‐factor B and anti‐C3b autoantibodies in C3 glomerulopathy and Ig‐associated membranoproliferative GN. J Am Soc Nephrol. 2017;28(5):1603‐1613. doi: 10.1681/ASN.2016030343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Noris M, Remuzzi G. Glomerular diseases dependent on complement activation, including atypical hemolytic uremic syndrome, membranoproliferative glomerulonephritis, and C3 glomerulopathy: core curriculum 2015. Am J Kidney Dis. 2015;66(2):359‐375. doi: 10.1053/j.ajkd.2015.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Noris M, Remuzzi G. Atypical hemolytic‐uremic syndrome. N Engl J Med. 2009;361(17):1676‐1687. doi: 10.1056/NEJMra0902814 [DOI] [PubMed] [Google Scholar]
- 12. https://ghr.nlm.nih.gov/condition/atypical‐hemolytic‐uremic‐syndrome Accessed September 15. 2022.
- 13. Huemer M, Scholl‐Bürgi S, Hadaya K, et al. Three new cases of late‐onset cblC defect and review of the literature illustrating when to consider inborn errors of metabolism beyond infancy. Orphanet J Rare Dis. 2014;9:161. doi: 10.1186/s13023-014-0161-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lemaire M, Frémeaux‐Bacchi V, Schaefer F, et al. Recessive mutations in DGKE cause atypical hemolytic‐uremic syndrome. Nat Genet. 2013;45(5):531‐536. doi: 10.1038/ng.2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mele C, Lemaire M, Iatropoulos P, et al. Characterization of a new DGKE intronic mutation in genetically unsolved cases of familial atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2015;10(6):1011‐1019. doi: 10.2215/CJN.08520814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goodship THJ, Cook HT, Fakhouri F, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: improving GLOBAL Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017;91(3):539‐551. doi: 10.1016/j.kint.2016.10.005 [DOI] [PubMed] [Google Scholar]
- 17. Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108(4):1267‐1279. doi: 10.1182/blood-2005-10-007252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Feitz WJC, van de Kar NCAJ, Orth‐Höller D, van den Heuvel LPJW, Licht C. The genetics of atypical hemolytic uremic syndrome. Med Genet. 2018;30(4):400‐409. doi: 10.1007/s11825-018-0216-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin Nephrol. 2013;33(6):508‐530. doi: 10.1016/j.semnephrol.2013.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Noris M, Mescia F, Remuzzi G. STEC‐HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol. 2012;8(11):622‐633. doi: 10.1038/nrneph.2012.195 [DOI] [PubMed] [Google Scholar]
- 21. Fremeaux‐Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8(4):554‐562. doi: 10.2215/CJN.04760512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kaplan BS, Meyers KE, Schulman SL. The pathogenesis and treatment of hemolytic uremic syndrome. J Am Soc Nephrol. 1998;9(6):1126‐1133. doi: 10.1681/ASN.V961126 [DOI] [PubMed] [Google Scholar]
- 23. Gerber A, Karch H, Allerberger F, Verweyen HM, Zimmerhackl LB. Clinical course and the role of shiga toxin‐producing Escherichia coli infection in the hemolytic‐uremic syndrome in pediatric patients, 1997‐2000, in Germany and Austria: a prospective study. J Infect Dis. 2002;186(4):493‐500. doi: 10.1086/341940 [DOI] [PubMed] [Google Scholar]
- 24. Afshar‐Kharghan V. COMPLEMENTing the diagnosis of aHUS. Blood. 2014;124(11):1699‐1700. doi: 10.1182/blood-2014-07-590356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Noris M, Galbusera M, Gastoldi S, et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood. 2014;124(11):1715‐1726. doi: 10.1182/blood-2014-02-558296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zuber J, Le Quintrec M, Morris H, Frémeaux‐Bacchi V, Loirat C, Legendre C. Targeted strategies in the prevention and management of atypical HUS recurrence after kidney transplantation. Transplant Rev (Orlando). 2013;27(4):117‐125. doi: 10.1016/j.trre.2013.07.003 [DOI] [PubMed] [Google Scholar]
- 27. Bu F, Borsa N, Gianluigi A, Smith RJH. Familial atypical hemolytic uremic syndrome: a review of its genetic and clinical aspects. Clin Dev Immunol. 2012;2012:370426. doi: 10.1155/2012/370426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Warwicker P, Goodship TH, Donne RL, et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998;53(4):836‐844. doi: 10.1111/j.1523-1755.1998.00824.x [DOI] [PubMed] [Google Scholar]
- 29. Fakhouri F, Zuber J, Frémeaux‐Bacchi V, Loirat C. Haemolytic uraemic syndrome. Lancet. 2017;390(10095):681‐696. doi: 10.1016/S0140-6736(17)30062-4 [DOI] [PubMed] [Google Scholar]
- 30. Valoti E, Alberti M, Iatropoulos P, et al. Rare functional variants in complement genes and anti‐FH autoantibodies‐associated aHUS. Front Immunol. 2019;10:853. doi: 10.3389/fimmu.2019.00853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bresin E, Rurali E, Caprioli J, et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol. 2013;24(3):475‐486. doi: 10.1681/ASN.2012090884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sellier‐Leclerc AL, Fremeaux‐Bacchi V, Dragon‐Durey MA, et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2007;18(8):2392‐2400. doi: 10.1681/ASN.2006080811 [DOI] [PubMed] [Google Scholar]
- 33. Noris M, Bresin E, Mele C, Remuzzi G. Genetic Atypical Hemolytic‐Uremic Syndrome. In: Adam MP, Everman DB, Mirzaa GM, et al., eds. GeneReviews®. University of Washington; 1993. Accessed September 13, 2022. http://www.ncbi.nlm.nih.gov/books/NBK1367/ [PubMed] [Google Scholar]
- 34. Ault BH. Factor H and the pathogenesis of renal diseases. Pediatr Nephrol. 2000;14(10–11):1045‐1053. doi: 10.1007/s004670050069 [DOI] [PubMed] [Google Scholar]
- 35. Pérez‐Caballero D, González‐Rubio C, Gallardo ME, et al. Clustering of missense mutations in the C‐terminal region of factor H in atypical hemolytic uremic syndrome. Am J Hum Genet. 2001;68(2):478‐484. doi: 10.1086/318201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rougier N, Kazatchkine MD, Rougier JP, et al. Human complement factor H deficiency associated with hemolytic uremic syndrome. J Am Soc Nephrol. 1998;9(12):2318‐2326. doi: 10.1681/ASN.V9122318 [DOI] [PubMed] [Google Scholar]
- 37. Richards A, Buddles MR, Donne RL, et al. Factor H mutations in hemolytic uremic syndrome cluster in exons 18‐20, a domain important for host cell recognition. Am J Hum Genet. 2001;68(2):485‐490. doi: 10.1086/318203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Neumann HPH, Salzmann M, Bohnert‐Iwan B, et al. Haemolytic uraemic syndrome and mutations of the factor H gene: a registry‐based study of German speaking countries. J Med Genet. 2003;40(9):676‐681. doi: 10.1136/jmg.40.9.676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Caprioli J, Castelletti F, Bucchioni S, et al. Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C‐257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet. 2003;12(24):3385‐3395. doi: 10.1093/hmg/ddg363 [DOI] [PubMed] [Google Scholar]
- 40. Dragon‐Durey MA, Frémeaux‐Bacchi V, Loirat C, et al. Heterozygous and homozygous factor h deficiencies associated with hemolytic uremic syndrome or membranoproliferative glomerulonephritis: report and genetic analysis of 16 cases. J Am Soc Nephrol. 2004;15(3):787‐795. doi: 10.1097/01.asn.0000115702.28859.a7 [DOI] [PubMed] [Google Scholar]
- 41. Saunders RE, Goodship THJ, Zipfel PF, Perkins SJ. An interactive web database of factor H‐associated hemolytic uremic syndrome mutations: insights into the structural consequences of disease‐associated mutations. Hum Mutat. 2006;27(1):21‐30. doi: 10.1002/humu.20268 [DOI] [PubMed] [Google Scholar]
- 42. Pangburn MK. Cutting edge: localization of the host recognition functions of complement factor H at the carboxyl‐terminal: implications for hemolytic uremic syndrome. J Immunol. 2002;169(9):4702‐4706. doi: 10.4049/jimmunol.169.9.4702 [DOI] [PubMed] [Google Scholar]
- 43. Sánchez‐Corral P, Pérez‐Caballero D, Huarte O, et al. Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome. Am J Hum Genet. 2002;71(6):1285‐1295. doi: 10.1086/344515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Catterall CF, Lyons A, Sim RB, Day AJ, Harris TJ. Characterization of primary amino acid sequence of human complement control protein factor I from an analysis of cDNA clones. Biochem J. 1987;242(3):849‐856. doi: 10.1042/bj2420849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vyse TJ, Bates GP, Walport MJ, Morley BJ. The organization of the human complement factor I gene (IF): a member of the serine protease gene family. Genomics. 1994;24(1):90‐98. doi: 10.1006/geno.1994.1585 [DOI] [PubMed] [Google Scholar]
- 46. Fremeaux‐Bacchi V, Dragon‐Durey MA, Blouin J, et al. Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet. 2004;41(6):e84. doi: 10.1136/jmg.2004.019083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lublin DM, Liszewski MK, Post TW, et al. Molecular cloning and chromosomal localization of human membrane cofactor protein (MCP). Evidence for inclusion in the multigene family of complement‐regulatory proteins. J Exp Med. 1988;168(1):181‐194. doi: 10.1084/jem.168.1.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liszewski MK, Leung M, Cui W, et al. Dissecting sites important for complement regulatory activity in membrane cofactor protein (MCP; CD46). J Biol Chem. 2000;275(48):37692‐37701. doi: 10.1074/jbc.M004650200 [DOI] [PubMed] [Google Scholar]
- 49. Richards A, Kemp EJ, Liszewski MK, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci U S A. 2003;100(22):12966‐12971. doi: 10.1073/pnas.2135497100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Noris M, Brioschi S, Caprioli J, et al. Familial haemolytic uraemic syndrome and an MCP mutation. Lancet. 2003;362(9395):1542‐1547. doi: 10.1016/S0140-6736(03)14742-3 [DOI] [PubMed] [Google Scholar]
- 51. Slade C, Bosco J, Unglik G, Bleasel K, Nagel M, Winship I. Deficiency in complement factor B. N Engl J Med. 2013;369(17):1667‐1669. doi: 10.1056/NEJMc1306326 [DOI] [PubMed] [Google Scholar]
- 52. Tawadrous H, Maga T, Sharma J, Kupferman J, Smith RJH, Schoeneman M. A novel mutation in the complement factor B gene (CFB) and atypical hemolytic uremic syndrome. Pediatr Nephrol. 2010;25(5):947‐951. doi: 10.1007/s00467-009-1415-3 [DOI] [PubMed] [Google Scholar]
- 53. Békássy ZD, Kristoffersson AC, Cronqvist M, et al. Eculizumab in an anephric patient with atypical haemolytic uraemic syndrome and advanced vascular lesions. Nephrol Dial Transplant. 2013;28(11):2899‐2907. doi: 10.1093/ndt/gft340 [DOI] [PubMed] [Google Scholar]
- 54. Funato M, Uemura O, Ushijima K, et al. A complement factor B mutation in a large kindred with atypical hemolytic uremic syndrome. J Clin Immunol. 2014;34(6):691‐695. doi: 10.1007/s10875-014-0058-8 [DOI] [PubMed] [Google Scholar]
- 55. Zhang Y, Kremsdorf RA, Sperati CJ, et al. Mutation of complement factor B causing massive fluid‐phase dysregulation of the alternative complement pathway can result in atypical hemolytic uremic syndrome. Kidney Int. 2020;98(5):1265‐1274. doi: 10.1016/j.kint.2020.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Marinozzi MC, Vergoz L, Rybkine T, et al. Complement factor B mutations in atypical hemolytic uremic syndrome‐disease‐relevant or benign? J Am Soc Nephrol. 2014;25(9):2053‐2065. doi: 10.1681/ASN.2013070796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Roumenina LT, Jablonski M, Hue C, et al. Hyperfunctional C3 convertase leads to complement deposition on endothelial cells and contributes to atypical hemolytic uremic syndrome. Blood. 2009;114(13):2837‐2845. doi: 10.1182/blood-2009-01-197640 [DOI] [PubMed] [Google Scholar]
- 58. Goicoechea de Jorge E, Harris CL, Esparza‐Gordillo J, et al. Gain‐of‐function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci U S A. 2007;104(1):240‐245. doi: 10.1073/pnas.0603420103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Reis S, Falcão DA, Isaac L. Clinical aspects and molecular basis of primary deficiencies of complement component C3 and its regulatory proteins factor I and factor H. Scand J Immunol. 2006;63(3):155‐168. doi: 10.1111/j.1365-3083.2006.01729.x [DOI] [PubMed] [Google Scholar]
- 60. Frémeaux‐Bacchi V, Miller EC, Liszewski MK, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. 2008;112(13):4948‐4952. doi: 10.1182/blood-2008-01-133702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Delvaeye M, Noris M, De Vriese A, et al. Thrombomodulin mutations in atypical hemolytic‐uremic syndrome. N Engl J Med. 2009;361(4):345‐357. doi: 10.1056/NEJMoa0810739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zipfel PF, Wiech T, Stea ED, Skerka C. CFHR gene variations provide insights in the pathogenesis of the kidney diseases atypical hemolytic uremic syndrome and C3 glomerulopathy. J Am Soc Nephrol. 2020;31(2):241‐256. doi: 10.1681/ASN.2019050515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Abarrategui‐Garrido C, Martínez‐Barricarte R, López‐Trascasa M, de Córdoba SR, Sánchez‐Corral P. Characterization of complement factor H‐related (CFHR) proteins in plasma reveals novel genetic variations of CFHR1 associated with atypical hemolytic uremic syndrome. Blood. 2009;114(19):4261‐4271. doi: 10.1182/blood-2009-05-223834 [DOI] [PubMed] [Google Scholar]
- 64. Bernabéu‐Herrero ME, Jiménez‐Alcázar M, Anter J, et al. Complement factor H, FHR‐3 and FHR‐1 variants associate in an extended haplotype conferring increased risk of atypical hemolytic uremic syndrome. Mol Immunol. 2015;67(2 Pt B):276‐286. doi: 10.1016/j.molimm.2015.06.021 [DOI] [PubMed] [Google Scholar]
- 65. Blanc C, Roumenina LT, Ashraf Y, et al. Overall neutralization of complement factor H by autoantibodies in the acute phase of the autoimmune form of atypical hemolytic uremic syndrome. J Immunol. 2012;189(7):3528‐3537. doi: 10.4049/jimmunol.1200679 [DOI] [PubMed] [Google Scholar]
- 66. Raina R, Mangat G, Hong G, et al. Anti‐factor H antibody and its role in atypical hemolytic uremic syndrome. Front Immunol. 2022;13:931210. doi: 10.3389/fimmu.2022.931210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844‐1859. doi: 10.2215/CJN.02210310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rodríguez de Córdoba S, Hidalgo MS, Pinto S, Tortajada A. Genetics of atypical hemolytic uremic syndrome (aHUS). Semin Thromb Hemost. 2014;40(4):422‐430. doi: 10.1055/s-0034-1375296 [DOI] [PubMed] [Google Scholar]
- 69. Arjona E, Huerta A, Goicoechea de Jorge E, Rodríguez de Córdoba S. Familial risk of developing atypical hemolytic‐uremic syndrome. Blood. 2020;136(13):1558‐1561. doi: 10.1182/blood.2020006931 [DOI] [PubMed] [Google Scholar]
- 70. Esparza‐Gordillo J, Goicoechea de Jorge E, Buil A, et al. Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet. 2005;14(5):703‐712. doi: 10.1093/hmg/ddi066 [DOI] [PubMed] [Google Scholar]
- 71. Fremeaux‐Bacchi V, Kemp EJ, Goodship JA, et al. The development of atypical haemolytic‐uraemic syndrome is influenced by susceptibility factors in factor H and membrane cofactor protein: evidence from two independent cohorts. J Med Genet. 2005;42(11):852‐856. doi: 10.1136/jmg.2005.030783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Loirat C, Fakhouri F, Ariceta G, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2016;31(1):15‐39. doi: 10.1007/s00467-015-3076-8 [DOI] [PubMed] [Google Scholar]
- 73. Taylor CM, Machin S, Wigmore SJ. Goodship THJ, working party from the Renal Association, the British Committee for Standards in Haematology and the British Transplantation Society. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol. 2010;148(1):37‐47. doi: 10.1111/j.1365-2141.2009.07916.x [DOI] [PubMed] [Google Scholar]
- 74. Loirat C, Garnier A, Sellier‐Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost. 2010;36(6):673‐681. doi: 10.1055/s-0030-1262890 [DOI] [PubMed] [Google Scholar]
- 75. Nathanson S, Ulinski T, Frémeaux‐Bacchi V, Deschênes G. Secondary failure of plasma therapy in factor H deficiency. Pediatr Nephrol. 2006;21(11):1769‐1771. doi: 10.1007/s00467-006-0237-9 [DOI] [PubMed] [Google Scholar]
- 76. Rathbone J, Kaltenthaler E, Richards A, Tappenden P, Bessey A, Cantrell A. A systematic review of eculizumab for atypical haemolytic uraemic syndrome (aHUS). BMJ Open. 2013;3(11):e003573. doi: 10.1136/bmjopen-2013-003573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Greenbaum LA, Fila M, Ardissino G, et al. Eculizumab is a safe and effective treatment in pediatric patients with atypical hemolytic uremic syndrome. Kidney Int. 2016;89(3):701‐711. doi: 10.1016/j.kint.2015.11.026 [DOI] [PubMed] [Google Scholar]
- 78. Fakhouri F, Hourmant M, Campistol JM, et al. Terminal complement inhibitor eculizumab in adult patients with atypical hemolytic uremic syndrome: a single‐arm, open‐label trial. Am J Kidney Dis. 2016;68(1):84‐93. doi: 10.1053/j.ajkd.2015.12.034 [DOI] [PubMed] [Google Scholar]
- 79. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic‐uremic syndrome. N Engl J Med. 2013;368(23):2169‐2181. doi: 10.1056/NEJMoa1208981 [DOI] [PubMed] [Google Scholar]
- 80. de Souza RM, Correa BHM, Melo PHM, et al. The treatment of atypical hemolytic uremic syndrome with eculizumab in pediatric patients: a systematic review. Pediatr Nephrol. 2022;2. doi: 10.1007/s00467-022-05683-2 [DOI] [PubMed] [Google Scholar]
- 81. Zuber J, Fakhouri F, Roumenina LT, Loirat C, Frémeaux‐Bacchi V. French Study Group for aHUS/C3G. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. 2012;8(11):643‐657. doi: 10.1038/nrneph.2012.214 [DOI] [PubMed] [Google Scholar]
- 82. Fakhouri F, Frémeaux‐Bacchi V, Loirat C. Atypical hemolytic uremic syndrome: from the rediscovery of complement to targeted therapy. Eur J Intern Med. 2013;24(6):492‐495. doi: 10.1016/j.ejim.2013.05.008 [DOI] [PubMed] [Google Scholar]
- 83. Nester C, Stewart Z, Myers D, et al. Pre‐emptive eculizumab and plasmapheresis for renal transplant in atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2011;6(6):1488‐1494. doi: 10.2215/CJN.10181110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zuber J, Le Quintrec M, Krid S, et al. Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant. 2012;12(12):3337‐3354. doi: 10.1111/j.1600-6143.2012.04252.x [DOI] [PubMed] [Google Scholar]
- 85. Weitz M, Amon O, Bassler D, Koenigsrainer A, Nadalin S. Prophylactic eculizumab prior to kidney transplantation for atypical hemolytic uremic syndrome. Pediatr Nephrol. 2011;26(8):1325‐1329. doi: 10.1007/s00467-011-1879-9 [DOI] [PubMed] [Google Scholar]
- 86. Krid S, Roumenina LT, Beury D, et al. Renal transplantation under prophylactic eculizumab in atypical hemolytic uremic syndrome with CFH/CFHR1 hybrid protein. Am J Transplant. 2012;12(7):1938‐1944. doi: 10.1111/j.1600-6143.2012.04051.x [DOI] [PubMed] [Google Scholar]
- 87. Rondeau E, Scully M, Ariceta G, et al. The long‐acting C5 inhibitor, Ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment. Kidney Int. 2020;97(6):1287‐1296. doi: 10.1016/j.kint.2020.01.035 [DOI] [PubMed] [Google Scholar]
- 88. Fakhouri F, Schwotzer N, Golshayan D, Frémeaux‐Bacchi V. The rational use of complement inhibitors in kidney diseases. Kidney Int Rep. 2022;7(6):1165‐1178. doi: 10.1016/j.ekir.2022.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fakhouri F, Fila M, Provôt F, et al. Pathogenic variants in complement genes and risk of atypical hemolytic uremic syndrome relapse after eculizumab discontinuation. Clin J Am Soc Nephrol. 2017;12(1):50‐59. doi: 10.2215/CJN.06440616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ardissino G, Possenti I, Tel F, Testa S, Salardi S, Ladisa V. Discontinuation of eculizumab treatment in atypical hemolytic uremic syndrome: an update. Am J Kidney Dis. 2015;66(1):172‐173. doi: 10.1053/j.ajkd.2015.04.010 [DOI] [PubMed] [Google Scholar]
- 91. Merrill SA, Brittingham ZD, Yuan X, Moliterno AR, Sperati CJ, Brodsky RA. Eculizumab cessation in atypical hemolytic uremic syndrome. Blood. 2017;130(3):368‐372. doi: 10.1182/blood-2017-02-770214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wetzels JFM, van de Kar NCAJ. Discontinuation of eculizumab maintenance treatment for atypical hemolytic uremic syndrome. Am J Kidney Dis. 2015;65(2):342. doi: 10.1053/j.ajkd.2014.04.039 [DOI] [PubMed] [Google Scholar]
- 93. Fakhouri F, Fila M, Hummel A, et al. Eculizumab discontinuation in children and adults with atypical hemolytic‐uremic syndrome: a prospective multicenter study. Blood. 2021;137(18):2438‐2449. doi: 10.1182/blood.2020009280 [DOI] [PubMed] [Google Scholar]
- 94. Ardissino G, Testa S, Possenti I, et al. Discontinuation of eculizumab maintenance treatment for atypical hemolytic uremic syndrome: a report of 10 cases. Am J Kidney Dis. 2014;64(4):633‐637. doi: 10.1053/j.ajkd.2014.01.434 [DOI] [PubMed] [Google Scholar]
- 95. Struijk GH, Bouts AHM, Rijkers GT, Kuin EAC, ten Berge IJM, Bemelman FJ. Meningococcal sepsis complicating eculizumab treatment despite prior vaccination. Am J Transplant. 2013;13(3):819‐820. doi: 10.1111/ajt.12032 [DOI] [PubMed] [Google Scholar]
- 96. Ram S, Lewis LA, Rice PA. Infections of people with complement deficiencies and patients who have undergone splenectomy. Clin Microbiol Rev. 2010;23(4):740‐780. doi: 10.1128/CMR.00048-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Socié G, Caby‐Tosi MP, Marantz JL, et al. Eculizumab in paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome: 10‐year pharmacovigilance analysis. Br J Haematol. 2019;185(2):297‐310. doi: 10.1111/bjh.15790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Galbusera M, Noris M, Gastoldi S, et al. An ex vivo test of complement activation on endothelium for individualized eculizumab therapy in hemolytic uremic syndrome. Am J Kidney Dis. 2019;74(1):56‐72. doi: 10.1053/j.ajkd.2018.11.012 [DOI] [PubMed] [Google Scholar]
- 99. Cook HT, Pickering MC. Histopathology of MPGN and C3 glomerulopathies. Nat Rev Nephrol. 2015;11(1):14‐22. doi: 10.1038/nrneph.2014.217 [DOI] [PubMed] [Google Scholar]
- 100. Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis: pathogenetic heterogeneity and proposal for a new classification. Semin Nephrol. 2011;31(4):341‐348. doi: 10.1016/j.semnephrol.2011.06.005 [DOI] [PubMed] [Google Scholar]
- 101. Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis‐‐a new look at an old entity. N Engl J Med. 2012;366(12):1119‐1131. doi: 10.1056/NEJMra1108178 [DOI] [PubMed] [Google Scholar]
- 102. Pickering MC, D'Agati VD, Nester CM, et al. C3 glomerulopathy: consensus report. Kidney Int. 2013;84(6):1079‐1089. doi: 10.1038/ki.2013.377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Rovin BH, Adler SG, Barratt J, et al. Executive summary of the KDIGO 2021 guideline for the management of glomerular diseases. Kidney Int. 2021;100(4):753‐779. doi: 10.1016/j.kint.2021.05.015 [DOI] [PubMed] [Google Scholar]
- 104. Iatropoulos P, Daina E, Curreri M, et al. Cluster analysis identifies distinct pathogenetic patterns in C3 glomerulopathies/immune complex‐mediated membranoproliferative GN. J Am Soc Nephrol. 2018;29(1):283‐294. doi: 10.1681/ASN.2017030258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Fakhouri F, Le Quintrec M, Frémeaux‐Bacchi V. Practical management of C3 glomerulopathy and Ig‐mediated MPGN: facts and uncertainties. Kidney Int. 2020;98(5):1135‐1148. doi: 10.1016/j.kint.2020.05.053 [DOI] [PubMed] [Google Scholar]
- 106. Hou J, Markowitz GS, Bomback AS, et al. Toward a working definition of C3 glomerulopathy by immunofluorescence. Kidney Int. 2014;85(2):450‐456. doi: 10.1038/ki.2013.340 [DOI] [PubMed] [Google Scholar]
- 107. Vivarelli M, van de Kar N, Labbadia R, Diomedi‐Camassei F, Thurman JM. A clinical approach to children with C3 glomerulopathy. Pediatr Nephrol. 2022;37(3):521‐535. doi: 10.1007/s00467-021-05088-7 [DOI] [PubMed] [Google Scholar]
- 108. Lomax‐Browne HJ, Medjeral‐Thomas NR, Barbour SJ, et al. Association of histologic parameters with outcome in C3 Glomerulopathy and idiopathic immunoglobulin‐associated membranoproliferative glomerulonephritis. Clin J Am Soc Nephrol. 2022;17(7):994‐1007. doi: 10.2215/CJN.16801221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. van Rooden SM, Heiser WJ, Kok JN, Verbaan D, van Hilten JJ, Marinus J. The identification of Parkinson's disease subtypes using cluster analysis: a systematic review. Mov Disord. 2010;25(8):969‐978. doi: 10.1002/mds.23116 [DOI] [PubMed] [Google Scholar]
- 110. Vogt W, Nagel D. Cluster analysis in diagnosis. Clin Chem. 1992;38(2):182‐198. [PubMed] [Google Scholar]
- 111. Lötvall J, Akdis CA, Bacharier LB, et al. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol. 2011;127(2):355‐360. doi: 10.1016/j.jaci.2010.11.037 [DOI] [PubMed] [Google Scholar]
- 112. Burgel PR, Paillasseur JL, Caillaud D, et al. Clinical COPD phenotypes: a novel approach using principal component and cluster analyses. Eur Respir J. 2010;36(3):531‐539. doi: 10.1183/09031936.00175109 [DOI] [PubMed] [Google Scholar]
- 113. Howrylak JA, Fuhlbrigge AL, Strunk RC, et al. Classification of childhood asthma phenotypes and long‐term clinical responses to inhaled anti‐inflammatory medications. J Allergy Clin Immunol. 2014;133(5):1289‐1300, 1300.e1–12. doi: 10.1016/j.jaci.2014.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Alashwal H, El Halaby M, Crouse JJ, Abdalla A, Moustafa AA. The application of unsupervised clustering methods to Alzheimer's disease. Front Comput Neurosci. 2019;13:31. doi: 10.3389/fncom.2019.00031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Cook HT, Pickering MC. clusters not classifications: making sense of complement‐mediated kidney injury. J Am Soc Nephrol. 2018;29(1):9‐12. doi: 10.1681/ASN.2017111183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Donadelli R, Pulieri P, Piras R, et al. Unraveling the molecular mechanisms underlying complement dysregulation by nephritic factors in C3G and IC‐MPGN. Front Immunol. 2018;9:2329. doi: 10.3389/fimmu.2018.02329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Garam N, Prohászka Z, Szilágyi Á, et al. Validation of distinct pathogenic patterns in a cohort of membranoproliferative glomerulonephritis patients by cluster analysis. Clin Kidney J. 2020;13(2):225‐234. doi: 10.1093/ckj/sfz073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Prevalence of rare diseases: Bibliographic data: Orphanet Report Series , Rare Diseases collection. Diseases listed in alphabetical order. 2021.http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_diseases.pdf.Accessed September 15, 2022.
- 119. Iatropoulos P, Noris M, Mele C, et al. Complement gene variants determine the risk of immunoglobulin‐associated MPGN and C3 glomerulopathy and predict long‐term renal outcome. Mol Immunol. 2016;71:131‐142. doi: 10.1016/j.molimm.2016.01.010 [DOI] [PubMed] [Google Scholar]
- 120. Servais A, Noël LH, Roumenina LT, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82(4):454‐464. doi: 10.1038/ki.2012.63 [DOI] [PubMed] [Google Scholar]
- 121. Martínez‐Barricarte R, Heurich M, Valdes‐Cañedo F, et al. Human C3 mutation reveals a mechanism of dense deposit disease pathogenesis and provides insights into complement activation and regulation. J Clin Invest. 2010;120(10):3702‐3712. doi: 10.1172/JCI43343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ault BH, Schmidt BZ, Fowler NL, et al. Human factor H deficiency. Mutations in framework cysteine residues and block in H protein secretion and intracellular catabolism. J Biol Chem. 1997;272(40):25168‐25175. doi: 10.1074/jbc.272.40.25168 [DOI] [PubMed] [Google Scholar]
- 123. Servais A, Frémeaux‐Bacchi V, Lequintrec M, et al. Primary glomerulonephritis with isolated C3 deposits: a new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet. 2007;44(3):193‐199. doi: 10.1136/jmg.2006.045328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Imamura H, Konomoto T, Tanaka E, et al. Familial C3 glomerulonephritis associated with mutations in the gene for complement factor B. Nephrol Dial Transplant. 2015;30(5):862‐864. doi: 10.1093/ndt/gfv054 [DOI] [PubMed] [Google Scholar]
- 125. Osborne AJ, Breno M, Borsa NG, et al. Statistical validation of rare complement variants provides insights into the molecular basis of atypical hemolytic uremic syndrome and C3 glomerulopathy. J Immunol. 2018;200(7):2464‐2478. doi: 10.4049/jimmunol.1701695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Józsi M, Tortajada A, Uzonyi B, Goicoechea de Jorge E, Rodríguez de Córdoba S. Factor H‐related proteins determine complement‐activating surfaces. Trends Immunol. 2015;36(6):374‐384. doi: 10.1016/j.it.2015.04.008 [DOI] [PubMed] [Google Scholar]
- 127. Piras R, Breno M, Valoti E, et al. CFH and CFHR copy number variations in c3 glomerulopathy and immune complex‐mediated membranoproliferative glomerulonephritis. Front Genet. 2021;12:670727. doi: 10.3389/fgene.2021.670727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Abrera‐Abeleda MA, Nishimura C, Smith JLH, et al. Variations in the complement regulatory genes factor H (CFH) and factor H related 5 (CFHR5) are associated with membranoproliferative glomerulonephritis type II (dense deposit disease). J Med Genet. 2006;43(7):582‐589. doi: 10.1136/jmg.2005.038315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Paixão‐Cavalcante D, López‐Trascasa M, Skattum L, et al. Sensitive and specific assays for C3 nephritic factors clarify mechanisms underlying complement dysregulation. Kidney Int. 2012;82(10):1084‐1092. doi: 10.1038/ki.2012.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Zhang Y, Meyer NC, Wang K, et al. Causes of alternative pathway dysregulation in dense deposit disease. Clin J Am Soc Nephrol. 2012;7(2):265‐274. doi: 10.2215/CJN.07900811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Nester CM, Smith RJ. Diagnosis and treatment of C3 glomerulopathy. Clin Nephrol. 2013;80(6):395‐403. doi: 10.5414/CN108057 [DOI] [PubMed] [Google Scholar]
- 132. Nicolas C, Vuiblet V, Baudouin V, et al. C3 nephritic factor associated with C3 glomerulopathy in children. Pediatr Nephrol. 2014;29(1):85‐94. doi: 10.1007/s00467-013-2605-6 [DOI] [PubMed] [Google Scholar]
- 133. Durey MAD, Sinha A, Togarsimalemath SK, Bagga A. Anti‐complement‐factor H‐associated glomerulopathies. Nat Rev Nephrol. 2016;12(9):563‐578. doi: 10.1038/nrneph.2016.99 [DOI] [PubMed] [Google Scholar]
- 134. Blanc C, Togarsimalemath SK, Chauvet S, et al. Anti‐factor H autoantibodies in C3 glomerulopathies and in atypical hemolytic uremic syndrome: one target, two diseases. J Immunol. 2015;194(11):5129‐5138. doi: 10.4049/jimmunol.1402770 [DOI] [PubMed] [Google Scholar]
- 135. Noris M, Remuzzi G. Genetics of immune‐mediated glomerular diseases: focus on complement. Semin Nephrol. 2017;37(5):447‐463. doi: 10.1016/j.semnephrol.2017.05.018 [DOI] [PubMed] [Google Scholar]
- 136. Bomback AS, Smith RJ, Barile GR, et al. Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol. 2012;7(5):748‐756. doi: 10.2215/CJN.12901211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ruggenenti P, Daina E, Gennarini A, et al. C5 convertase blockade in membranoproliferative glomerulonephritis: a single‐arm clinical trial. Am J Kidney Dis. 2019;74(2):224‐238. doi: 10.1053/j.ajkd.2018.12.046 [DOI] [PubMed] [Google Scholar]
- 138. Ort M, Dingemanse J, van den Anker J, Kaufmann P. Treatment of rare inflammatory kidney diseases: drugs targeting the terminal complement pathway. Front Immunol. 2020;11:599417. doi: 10.3389/fimmu.2020.599417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Bomback A, Herlitz LC, Yue H, et al. Effect of avacopan, a selective C5a receptor inhibitor, on C3G histologic index of disease chronicity. Kidney Int Rep. 2022;7(2):S47‐S48 [Abstract]. doi: 10.1016/j.ekir.2022.01.124 [DOI] [Google Scholar]
- 140. Wiles JA, Galvan MD, Podos SD, Geffner M, Huang M. Discovery and development of the oral complement factor d inhibitor danicopan (ACH‐4471). Curr Med Chem. 2020;27(25):4165‐4180. doi: 10.2174/0929867326666191001130342 [DOI] [PubMed] [Google Scholar]
- 141. Kulasekararaj AG, Risitano AM, Maciejewski JP, et al. Phase 2 study of danicopan in patients with paroxysmal nocturnal hemoglobinuria with an inadequate response to eculizumab. Blood. 2021;138(20):1928‐1938. doi: 10.1182/blood.2021011388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. https://medcitynews.com/2020/07/alexion‐drops‐kidney‐disease‐program‐for‐drug‐that‐was‐part‐of‐930m‐achillion‐buyout‐last‐year. Accessed September 15, 2022.
- 143. Wu X, Hutson I, Akk AM, et al. Contribution of adipose‐derived factor D/adipsin to complement alternative pathway activation: lessons from lipodystrophy. J Immunol. 2018;200(8):2786‐2797. doi: 10.4049/jimmunol.1701668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Sanders PW, Volanakis JE, Rostand SG, Galla JH. Human complement protein D catabolism by the rat kidney. J Clin Invest. 1986;77(4):1299‐1304. doi: 10.1172/JCI112434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Pascual M, Steiger G, Estreicher J, Macon K, Volanakis JE, Schifferli JA. Metabolism of complement factor D in renal failure. Kidney Int. 1988;34(4):529‐536. doi: 10.1038/ki.1988.214 [DOI] [PubMed] [Google Scholar]
- 146. Nester C, Podos S, Hogan J, et al. Clinical and biomarker characteristics of Patients with C3G or IC‐MPGN enrolled in two Phase II studies investigating the factor D Inhibitor danicopan. Nephrol Dial Transplant. 2021;36(Suppl 1):i49 [Abstract]. doi: 10.1093/ndt/gfab092.004 [DOI] [Google Scholar]
- 147. Nester C, Nast C, Appel G, et al. Evaluating BCX9930, an oral factor D inhibitor for treatment of complement‐mediated kidney disease: a proof‐of‐concept study (RENEW). Kidney Int Rep. 2022;7(6):S457‐S458 [Abstract]. doi: 10.1016/j.ekir.2022.04.067 [DOI] [Google Scholar]
- 148. Schubart A, Anderson K, Mainolfi N, et al. Small‐molecule factor B inhibitor for the treatment of complement‐mediated diseases. Proc Natl Acad Sci U S A. 2019;116(16):7926‐7931. doi: 10.1073/pnas.1820892116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Wong EKS, Praga M, Nester C, et al. Iptacopan (LNP023): a novel oral complement Alternative pathway factor B inhibitor safely and effectively stabilises eGFR in C3 glomerulopathy. Nephrol Dial Transplant. 2021;36(Suppl 1):i25 [Abstract]. doi: 10.1093/ndt/gfab121.005 [DOI] [Google Scholar]
- 150. Hoy SM. Pegcetacoplan: first approval. Drugs. 2021;81(12):1423‐1430. doi: 10.1007/s40265-021-01560-8 [DOI] [PubMed] [Google Scholar]
- 151. Dixon BP, Greenbaum LA, Huang L, et al. C3 inhibition with pegcetacoplan targets the underlying disease process of c3 glomerulopathy (C3G) and improves proteinuria. J Am Soc Nephrol. 2020; Kidney week 2020 [Abstract:PO1852]. [Google Scholar]
- 152. Remuzzi G, Dixon B, Fakhouri F, et al. Phase 3, randomized, multicenter study to evaluate the efficacy and safety of pegcetacoplan in treatment of C3G or IC‐MPGN. Kidney Int Rep. 2022;7(6):S459 [Abstract]. doi: 10.1016/j.ekir.2022.04.070 [DOI] [Google Scholar]
- 153. Ronco P, Beck L, Debiec H, et al. Membranous nephropathy. Nat Rev Dis Primers. 2021;7(1):69. doi: 10.1038/s41572-021-00303-z [DOI] [PubMed] [Google Scholar]
- 154. Rychlík I, Jancová E, Tesar V, et al. The Czech registry of renal biopsies. Occurrence of renal diseases in the years 1994‐2000. Nephrol Dial Transplant. 2004;19(12):3040‐3049. doi: 10.1093/ndt/gfh521 [DOI] [PubMed] [Google Scholar]
- 155. Hogan SL, Muller KE, Jennette JC, Falk RJ. A review of therapeutic studies of idiopathic membranous glomerulopathy. Am J Kidney Dis. 1995;25(6):862‐875. doi: 10.1016/0272-6386(95)90568-5 [DOI] [PubMed] [Google Scholar]
- 156. Ronco P, Plaisier E, Debiec H. Advances in Membranous Nephropathy. J Clin Med. 2021;10(4):607. doi: 10.3390/jcm10040607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Polanco N, Gutiérrez E, Covarsí A, et al. Spontaneous remission of nephrotic syndrome in idiopathic membranous nephropathy. J Am Soc Nephrol. 2010;21(4):697‐704. doi: 10.1681/ASN.2009080861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Polanco N, Gutiérrez E, Rivera F, et al. Spontaneous remission of nephrotic syndrome in membranous nephropathy with chronic renal impairment. Nephrol Dial Transplant. 2012;27(1):231‐234. doi: 10.1093/ndt/gfr285 [DOI] [PubMed] [Google Scholar]
- 159. Glassock RJ. Diagnosis and natural course of membranous nephropathy. Semin Nephrol. 2003;23(4):324‐332. doi: 10.1016/s0270-9295(03)00049-4 [DOI] [PubMed] [Google Scholar]
- 160. Beck LH, Bonegio RGB, Lambeau G, et al. M‐type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361(1):11‐21. doi: 10.1056/NEJMoa0810457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Tomas NM, Beck LH, Meyer‐Schwesinger C, et al. Thrombospondin type‐1 domain‐containing 7A in idiopathic membranous nephropathy. N Engl J Med. 2014;371(24):2277‐2287. doi: 10.1056/NEJMoa1409354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Hoxha E, Reinhard L, Stahl RAK. Membranous nephropathy: new pathogenic mechanisms and their clinical implications. Nat Rev Nephrol. 2022;18(7):466‐478. doi: 10.1038/s41581-022-00564-1 [DOI] [PubMed] [Google Scholar]
- 163. Baker PJ, Ochi RF, Schulze M, Johnson RJ, Campbell C, Couser WG. Depletion of C6 prevents development of proteinuria in experimental membranous nephropathy in rats. Am J Pathol. 1989;135(1):185‐194. [PMC free article] [PubMed] [Google Scholar]
- 164. Cybulsky AV, Rennke HG, Feintzeig ID, Salant DJ. Complement‐induced glomerular epithelial cell injury. Role of the membrane attack complex in rat membranous nephropathy. J Clin Invest. 1986;77(4):1096‐1107. doi: 10.1172/JCI112408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Meyer‐Schwesinger C, Tomas NM, Dehde S, et al. A novel mouse model of phospholipase A2 receptor 1‐associated membranous nephropathy mimics podocyte injury in patients. Kidney Int. 2020;97(5):913‐919. doi: 10.1016/j.kint.2019.10.022 [DOI] [PubMed] [Google Scholar]
- 166. Tomas NM, Meyer‐Schwesinger C, von Spiegel H, et al. A heterologous model of thrombospondin type 1 domain‐containing 7A‐associated membranous nephropathy. J Am Soc Nephrol. 2017;28(11):3262‐3277. doi: 10.1681/ASN.2017010030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Tomas NM, Hoxha E, Reinicke AT, et al. Autoantibodies against thrombospondin type 1 domain‐containing 7A induce membranous nephropathy. J Clin Invest. 2016;126(7):2519‐2532. doi: 10.1172/JCI85265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Stangou MJ, Marinaki S, Papachristou E, et al. Histological grading in primary membranous nephropathy is essential for clinical management and predicts outcome of patients. Histopathology. 2019;75(5):660‐671. doi: 10.1111/his.13955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Zhang XD, Cui Z, Zhang MF, et al. Clinical implications of pathological features of primary membranous nephropathy. BMC Nephrol. 2018;19(1):215. doi: 10.1186/s12882-018-1011-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Ma H, Sandor DG, Beck LH. The role of complement in membranous nephropathy. Semin Nephrol. 2013;33(6):531‐542. doi: 10.1016/j.semnephrol.2013.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Doi T, Mayumi M, Kanatsu K, Suehiro F, Hamashima Y. Distribution of IgG subclasses in membranous nephropathy. Clin Exp Immunol. 1984;58(1):57‐62. [PMC free article] [PubMed] [Google Scholar]
- 172. Larsen CP, Messias NC, Silva FG, Messias E, Walker PD. Determination of primary versus secondary membranous glomerulopathy utilizing phospholipase A2 receptor staining in renal biopsies. Mod Pathol. 2013;26(5):709‐715. doi: 10.1038/modpathol.2012.207 [DOI] [PubMed] [Google Scholar]
- 173. Zaghrini C, Seitz‐Polski B, Justino J, et al. Novel ELISA for thrombospondin type 1 domain‐containing 7A autoantibodies in membranous nephropathy. Kidney Int. 2019;95(3):666‐679. doi: 10.1016/j.kint.2018.10.024 [DOI] [PubMed] [Google Scholar]
- 174. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520. doi: 10.3389/fimmu.2014.00520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Tao MH, Canfield SM, Morrison SL. The differential ability of human IgG1 and IgG4 to activate complement is determined by the COOH‐terminal sequence of the CH2 domain. J Exp Med. 1991;173(4):1025‐1028. doi: 10.1084/jem.173.4.1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Hayashi N, Akiyama S, Okuyama H, et al. Clinicopathological characteristics of M‐type phospholipase A2 receptor (PLA2R)‐related membranous nephropathy in Japanese. Clin Exp Nephrol. 2015;19(5):797‐803. doi: 10.1007/s10157-014-1064-0 [DOI] [PubMed] [Google Scholar]
- 177. Lhotta K, Würzner R, König P. Glomerular deposition of mannose‐binding lectin in human glomerulonephritis. Nephrol Dial Transplant. 1999;14(4):881‐886. doi: 10.1093/ndt/14.4.881 [DOI] [PubMed] [Google Scholar]
- 178. Segawa Y, Hisano S, Matsushita M, et al. IgG subclasses and complement pathway in segmental and global membranous nephropathy. Pediatr Nephrol. 2010;25(6):1091‐1099. doi: 10.1007/s00467-009-1439-8 [DOI] [PubMed] [Google Scholar]
- 179. Hayashi N, Okada K, Matsui Y, et al. Glomerular mannose‐binding lectin deposition in intrinsic antigen‐related membranous nephropathy. Nephrol Dial Transplant. 2018;33(5):832‐840. doi: 10.1093/ndt/gfx235 [DOI] [PubMed] [Google Scholar]
- 180. Haddad G, Lorenzen JM, Ma H, et al. Altered glycosylation of IgG4 promotes lectin complement pathway activation in anti‐PLA2R1‐associated membranous nephropathy. J Clin Invest. 2021;131(5):140453. doi: 10.1172/JCI140453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Bally S, Debiec H, Ponard D, et al. Phospholipase A2 receptor‐related membranous nephropathy and mannan‐binding lectin deficiency. J Am Soc Nephrol. 2016;27(12):3539‐3544. doi: 10.1681/ASN.2015101155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Ravindran A, Madden B, Charlesworth MC, et al. Proteomic analysis of complement proteins in membranous nephropathy. Kidney Int Rep. 2020;5(5):618‐626. doi: 10.1016/j.ekir.2020.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Luo W, Olaru F, Miner JH, et al. Alternative pathway is essential for glomerular complement activation and proteinuria in a mouse model of membranous nephropathy. Front Immunol. 2018;9:1433. doi: 10.3389/fimmu.2018.01433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Manral P, Caza TN, Storey AJ, Beck LH, Borza DB. The alternative pathway is necessary and sufficient for complement activation by anti‐THSD7A autoantibodies, which are predominantly IgG4 in membranous nephropathy. Front Immunol. 2022;13:952235. doi: 10.3389/fimmu.2022.952235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Lateb M, Ouahmi H, Payré C, et al. Anti‐PLA2R1 antibodies containing sera induce in vitro cytotoxicity mediated by complement activation. J Immunol Res. 2019;2019:1324804. doi: 10.1155/2019/1324804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Huang CC, Lehman A, Albawardi A, et al. IgG subclass staining in renal biopsies with membranous glomerulonephritis indicates subclass switch during disease progression. Mod Pathol. 2013;26(6):799‐805. doi: 10.1038/modpathol.2012.237 [DOI] [PubMed] [Google Scholar]
- 187. Borza DB, Rana T, Olaru F, et al. Alternative pathway amplifies complement activation by human anti‐PLA2R antibodies in membranous nephropathy. J Am Soc Nephrol. 2014;25:66A [Abstract: FR‐Or087]. [Google Scholar]
- 188. Raats CJ, Luca ME, Bakker MA, et al. Reduction in glomerular heparan sulfate correlates with complement deposition and albuminuria in active Heymann nephritis. J Am Soc Nephrol. 1999;10(8):1689‐1699. doi: 10.1681/ASN.V1081689 [DOI] [PubMed] [Google Scholar]
- 189. Borza DB. Alternative pathway dysregulation and the conundrum of complement activation by IgG4 immune complexes in membranous nephropathy. Front Immunol. 2016;7:157. doi: 10.3389/fimmu.2016.00157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Sethi A, Miao J, Willrich MAV, et al. Limited significance of antifactor H antibodies in patients with membranous nephropathy. Clin J Am Soc Nephrol. 2021;16(6):939‐941. doi: 10.2215/CJN.16631020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Seikrit C, Ronco P, Debiec H. Factor H autoantibodies and membranous nephropathy. N Engl J Med. 2018;379(25):2479‐2481. doi: 10.1056/NEJMc1805857 [DOI] [PubMed] [Google Scholar]
- 192. Valoti E, Noris M, Remuzzi G. More about factor H autoantibodies in membranous nephropathy. N Engl J Med. 2019;381(16):1590‐1592. doi: 10.1056/NEJMc1905608 [DOI] [PubMed] [Google Scholar]
- 193. Kitching AR, Anders HJ, Basu N, et al. ANCA‐associated vasculitis. Nat Rev Dis Primers. 2020;6(1):71. doi: 10.1038/s41572-020-0204-y [DOI] [PubMed] [Google Scholar]
- 194. Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1‐11. doi: 10.1002/art.37715 [DOI] [PubMed] [Google Scholar]
- 195. Jennette JC, Nachman PH. ANCA glomerulonephritis and vasculitis. Clin J Am Soc Nephrol. 2017;12(10):1680‐1691. doi: 10.2215/CJN.02500317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Redondo‐Rodriguez R, Mena‐Vázquez N, Cabezas‐Lucena AM, Manrique‐Arija S, Mucientes A, Fernández‐Nebro A. Systematic review and metaanalysis of worldwide incidence and prevalence of antineutrophil cytoplasmic antibody (ANCA) associated vasculitis. J Clin Med. 2022;11(9):2573. doi: 10.3390/jcm11092573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Pearce FA, Lanyon PC, Grainge MJ, et al. Incidence of ANCA‐associated vasculitis in a UK mixed ethnicity population. Rheumatology (Oxford). 2016;55(9):1656‐1663. doi: 10.1093/rheumatology/kew232 [DOI] [PubMed] [Google Scholar]
- 198. Geetha D, Jefferson JA. ANCA‐associated vasculitis: core curriculum 2020. Am J Kidney Dis. 2020;75(1):124‐137. doi: 10.1053/j.ajkd.2019.04.031 [DOI] [PubMed] [Google Scholar]
- 199. Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti‐neutrophil cytoplasmic autoantibodies. Am J Pathol. 2007;170(1):52‐64. doi: 10.2353/ajpath.2007.060573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Huugen D, van Esch A, Xiao H, et al. Inhibition of complement factor C5 protects against anti‐myeloperoxidase antibody‐mediated glomerulonephritis in mice. Kidney Int. 2007;71(7):646‐654. doi: 10.1038/sj.ki.5002103 [DOI] [PubMed] [Google Scholar]
- 201. Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA‐induced glomerulonephritis. J Am Soc Nephrol. 2009;20(2):289‐298. doi: 10.1681/ASN.2008050497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Xiao H, Dairaghi DJ, Powers JP, et al. C5a receptor (CD88) blockade protects against MPO‐ANCA GN. J Am Soc Nephrol. 2014;25(2):225‐231. doi: 10.1681/ASN.2013020143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Camous L, Roumenina L, Bigot S, et al. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood. 2011;117(4):1340‐1349. doi: 10.1182/blood-2010-05-283564 [DOI] [PubMed] [Google Scholar]
- 204. Xing GQ, Chen M, Liu G, et al. Complement activation is involved in renal damage in human antineutrophil cytoplasmic autoantibody associated pauci‐immune vasculitis. J Clin Immunol. 2009;29(3):282‐291. doi: 10.1007/s10875-008-9268-2 [DOI] [PubMed] [Google Scholar]
- 205. Hilhorst M, van Paassen P, van Rie H, et al. Complement in ANCA‐associated glomerulonephritis. Nephrol Dial Transplant. 2017;32(8):1302‐1313. doi: 10.1093/ndt/gfv288 [DOI] [PubMed] [Google Scholar]
- 206. Gou SJ, Yuan J, Wang C, Zhao MH, Chen M. Alternative complement pathway activation products in urine and kidneys of patients with ANCA‐associated GN. Clin J Am Soc Nephrol. 2013;8(11):1884‐1891. doi: 10.2215/CJN.02790313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Gou SJ, Yuan J, Chen M, Yu F, Zhao MH. Circulating complement activation in patients with anti‐neutrophil cytoplasmic antibody‐associated vasculitis. Kidney Int. 2013;83(1):129‐137. doi: 10.1038/ki.2012.313 [DOI] [PubMed] [Google Scholar]
- 208. Chen SF, Wang FM, Li ZY, Yu F, Zhao MH, Chen M. Plasma complement factor H is associated with disease activity of patients with ANCA‐associated vasculitis. Arthritis Res Ther. 2015;17:129. doi: 10.1186/s13075-015-0656-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Chen SF, Wang FM, Li ZY, Yu F, Chen M, Zhao MH. The functional activities of complement factor H are impaired in patients with ANCA‐positive vasculitis. Clin Immunol. 2017;175:41‐50. doi: 10.1016/j.clim.2016.11.013 [DOI] [PubMed] [Google Scholar]
- 210. Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age‐related macular degeneration. Proc Natl Acad Sci U S A. 2005;102(20):7227‐7232. doi: 10.1073/pnas.0501536102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age‐related macular degeneration. Science. 2005;308(5720):419‐421. doi: 10.1126/science.1110359 [DOI] [PubMed] [Google Scholar]
- 212. Chen SF, Wang FM, Li ZY, Yu F, Chen M, Zhao MH. Myeloperoxidase influences the complement regulatory activity of complement factor H. Rheumatology (Oxford). 2018;57(12):2213‐2224. doi: 10.1093/rheumatology/kex529 [DOI] [PubMed] [Google Scholar]
- 213. Rodriguez‐Iturbe B, Musser JM. The current state of poststreptococcal glomerulonephritis. J Am Soc Nephrol. 2008;19(10):1855‐1864. doi: 10.1681/ASN.2008010092 [DOI] [PubMed] [Google Scholar]
- 214. Nasr SH, Markowitz GS, Stokes MB, Said SM, Valeri AM, D'Agati VD. Acute postinfectious glomerulonephritis in the modern era: experience with 86 adults and review of the literature. Medicine (Baltimore). 2008;87(1):21‐32. doi: 10.1097/md.0b013e318161b0fc [DOI] [PubMed] [Google Scholar]
- 215. Stratta P, Musetti C, Barreca A, Mazzucco G. New trends of an old disease: the acute post infectious glomerulonephritis at the beginning of the new millenium. J Nephrol. 2014;27(3):229‐239. doi: 10.1007/s40620-013-0018-z [DOI] [PubMed] [Google Scholar]
- 216. Smith RJH, Appel GB, Blom AM, et al. C3 glomerulopathy ‐ understanding a rare complement‐driven renal disease. Nat Rev Nephrol. 2019;15(3):129‐143. doi: 10.1038/s41581-018-0107-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Laabei M, Ermert D. Catch me if you can: streptococcus pyogenes complement evasion strategies. J Innate Immun. 2019;11(1):3‐12. doi: 10.1159/000492944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Thern A, Stenberg L, Dahlbäck B, Lindahl G. Ig‐binding surface proteins of Streptococcus pyogenes also bind human C4b‐binding protein (C4BP), a regulatory component of the complement system. J Immunol. 1995;154(1):375‐386. [PubMed] [Google Scholar]
- 219. Pérez‐Caballero D, García‐Laorden I, Cortés G, Wessels MR, de Córdoba SR, Albertí S. Interaction between complement regulators and Streptococcus pyogenes: binding of C4b‐binding protein and factor H/factor H‐like protein 1 to M18 strains involves two different cell surface molecules. J Immunol. 2004;173(11):6899‐6904. doi: 10.4049/jimmunol.173.11.6899 [DOI] [PubMed] [Google Scholar]
- 220. Endre ZH, Pussell BA, Charlesworth JA, Coovadia HM, Seedat YK. C3 metabolism in acute glomerulonephritis: implications for sites of complement activation. Kidney Int. 1984;25(6):937‐941. doi: 10.1038/ki.1984.113 [DOI] [PubMed] [Google Scholar]
- 221. Nordstrand A, Norgren M, Holm SE. Pathogenic mechanism of acute post‐streptococcal glomerulonephritis. Scand J Infect Dis. 1999;31(6):523‐537. doi: 10.1080/00365549950164382 [DOI] [PubMed] [Google Scholar]
- 222. Yoshizawa N, Yamakami K, Fujino M, et al. Nephritis‐associated plasmin receptor and acute poststreptococcal glomerulonephritis: characterization of the antigen and associated immune response. J Am Soc Nephrol. 2004;15(7):1785‐1793. doi: 10.1097/01.asn.0000130624.94920.6b [DOI] [PubMed] [Google Scholar]
- 223. Hummell DS, Swift AJ, Tomasz A, Winkelstein JA. Activation of the alternative complement pathway by pneumococcal lipoteichoic acid. Infect Immun. 1985;47(2):384‐387. doi: 10.1128/iai.47.2.384-387.1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Sethi S, Fervenza FC, Zhang Y, et al. Atypical postinfectious glomerulonephritis is associated with abnormalities in the alternative pathway of complement. Kidney Int. 2013;83(2):293‐299. doi: 10.1038/ki.2012.384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Chauvet S, Berthaud R, Devriese M, et al. Anti‐factor B antibodies and acute postinfectious GN in children. J Am Soc Nephrol. 2020;31(4):829‐840. doi: 10.1681/ASN.2019080851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Noris M, Remuzzi G. Challenges in understanding acute postinfectious glomerulonephritis: are anti‐factor b autoantibodies the answer? J Am Soc Nephrol. 2020;31(4):670‐672. doi: 10.1681/ASN.2020020168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Rajasekaran A, Julian BA, Rizk DV. IgA nephropathy: an interesting autoimmune kidney disease. Am J Med Sci. 2021;361(2):176‐194. doi: 10.1016/j.amjms.2020.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. D'Amico G. Natural history of idiopathic IgA nephropathy and factors predictive of disease outcome. Semin Nephrol. 2004;24(3):179‐196. doi: 10.1016/j.semnephrol.2004.01.001 [DOI] [PubMed] [Google Scholar]
- 229. Gutiérrez E, Carvaca‐Fontán F, Luzardo L, Morales E, Alonso M, Praga M. A personalized update on IgA nephropathy: a new vision and new future challenges. Nephron. 2020;144(11):555‐571. doi: 10.1159/000509997 [DOI] [PubMed] [Google Scholar]
- 230. Medjeral‐Thomas NR, Cook HT, Pickering MC. Complement activation in IgA nephropathy. Semin Immunopathol. 2021;43(5):679‐690. doi: 10.1007/s00281-021-00882-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Hiemstra PS, Gorter A, Stuurman ME, Van Es LA, Daha MR. Activation of the alternative pathway of complement by human serum IgA. Eur J Immunol. 1987;17(3):321‐326. doi: 10.1002/eji.1830170304 [DOI] [PubMed] [Google Scholar]
- 232. Stad RK, Bruijn JA, van Gijlswijk‐Janssen DJ, van Es LA, Daha MR. An acute model for IgA‐mediated glomerular inflammation in rats induced by monoclonal polymeric rat IgA antibodies. Clin Exp Immunol. 1993;92(3):514‐521. doi: 10.1111/j.1365-2249.1993.tb03430.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Chiu YL, Lin WC, Shu KH, et al. Alternative complement pathway is activated and associated with galactose‐deficient IgA1 antibody in IgA nephropathy patients. Front Immunol. 2021;12:638309. doi: 10.3389/fimmu.2021.638309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Gharavi AG, Kiryluk K, Choi M, et al. Genome‐wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011;43(4):321‐327. doi: 10.1038/ng.787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Kiryluk K, Li Y, Sanna‐Cherchi S, et al. Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS Genet. 2012;8(6):e1002765. doi: 10.1371/journal.pgen.1002765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Zhu L, Zhai YL, Wang FM, et al. Variants in complement factor H and complement factor H‐related protein genes, CFHR3 and CFHR1, affect complement activation in IgA nephropathy. J Am Soc Nephrol. 2015;26(5):1195‐1204. doi: 10.1681/ASN.2014010096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Zhai YL, Meng SJ, Zhu L, et al. Rare variants in the complement factor H‐related protein 5 gene contribute to genetic susceptibility to IgA nephropathy. J Am Soc Nephrol. 2016;27(9):2894‐2905. doi: 10.1681/ASN.2015010012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Medjeral‐Thomas NR, Lomax‐Browne HJ, Beckwith H, et al. Circulating complement factor H‐related proteins 1 and 5 correlate with disease activity in IgA nephropathy. Kidney Int. 2017;92(4):942‐952. doi: 10.1016/j.kint.2017.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Tortajada A, Gutiérrez E, Goicoechea de Jorge E, et al. Elevated factor H‐related protein 1 and factor H pathogenic variants decrease complement regulation in IgA nephropathy. Kidney Int. 2017;92(4):953‐963. doi: 10.1016/j.kint.2017.03.041 [DOI] [PubMed] [Google Scholar]
- 240. Zhu L, Guo WY, Shi SF, et al. Circulating complement factor H‐related protein 5 levels contribute to development and progression of IgA nephropathy. Kidney Int. 2018;94(1):150‐158. doi: 10.1016/j.kint.2018.02.023 [DOI] [PubMed] [Google Scholar]
- 241. Medjeral‐Thomas NR, Troldborg A, Constantinou N, et al. Progressive IgA nephropathy is associated with low circulating mannan‐binding lectin‐associated serine protease‐3 (MASP‐3) and increased glomerular factor H‐related protein‐5 (FHR5) deposition. Kidney Int Rep. 2018;3(2):426‐438. doi: 10.1016/j.ekir.2017.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Guo WY, Sun LJ, Dong HR, et al. Glomerular complement factor H‐related protein 5 is associated with histologic injury in immunoglobulin A nephropathy. Kidney Int Rep. 2021;6(2):404‐413. doi: 10.1016/j.ekir.2020.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365(22):2110‐2121. doi: 10.1056/NEJMra1100359 [DOI] [PubMed] [Google Scholar]
- 244. Barber MRW, Drenkard C, Falasinnu T, et al. Global epidemiology of systemic lupus erythematosus. Nat Rev Rheumatol. 2021;17(9):515‐532. doi: 10.1038/s41584-021-00668-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Anders HJ, Saxena R, Zhao MH, Parodis I, Salmon JE, Mohan C. Lupus nephritis. Nat Rev Dis Primers. 2020;6(1):7. doi: 10.1038/s41572-019-0141-9 [DOI] [PubMed] [Google Scholar]
- 246. Tektonidou MG, Dasgupta A, Ward MM. Risk of end‐stage renal disease in patients with lupus nephritis, 1971‐2015: a systematic review and bayesian meta‐analysis. Arthritis Rheumatol. 2016;68(6):1432‐1441. doi: 10.1002/art.39594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Karp DR. Complement and systemic lupus erythematosus. Curr Opin Rheumatol. 2005;17(5):538‐542. doi: 10.1097/01.bor.0000172799.03379.86 [DOI] [PubMed] [Google Scholar]
- 248. Bomback AS, Markowitz GS, Appel GB. Complement‐mediated glomerular diseases: a tale of 3 pathways. Kidney Int Rep. 2016;1(3):148‐155. doi: 10.1016/j.ekir.2016.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Watanabe H, Garnier G, Circolo A, et al. Modulation of renal disease in MRL/lpr mice genetically deficient in the alternative complement pathway factor B. J Immunol. 2000;164(2):786‐794. doi: 10.4049/jimmunol.164.2.786 [DOI] [PubMed] [Google Scholar]
- 250. Elliott MK, Jarmi T, Ruiz P, Xu Y, Holers VM, Gilkeson GS. Effects of complement factor D deficiency on the renal disease of MRL/lpr mice. Kidney Int. 2004;65(1):129‐138. doi: 10.1111/j.1523-1755.2004.00371.x [DOI] [PubMed] [Google Scholar]
- 251. Grossman TR, Hettrick LA, Johnson RB, et al. Inhibition of the alternative complement pathway by antisense oligonucleotides targeting complement factor B improves lupus nephritis in mice. Immunobiology. 2016;221(6):701‐708. doi: 10.1016/j.imbio.2015.08.001 [DOI] [PubMed] [Google Scholar]
- 252. Sekine H, Ruiz P, Gilkeson GS, Tomlinson S. The dual role of complement in the progression of renal disease in NZB/W F(1) mice and alternative pathway inhibition. Mol Immunol. 2011;49(1–2):317‐323. doi: 10.1016/j.molimm.2011.09.015 [DOI] [PubMed] [Google Scholar]
- 253. Sekine H, Kinser TTH, Qiao F, et al. The benefit of targeted and selective inhibition of the alternative complement pathway for modulating autoimmunity and renal disease in MRL/lpr mice. Arthritis Rheum. 2011;63(4):1076‐1085. doi: 10.1002/art.30222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Whakey K. Complement and immune complex disease. In: Whakey K, ed. Complement in health and disease. MTP Press; 1987:163‐183. [Google Scholar]
- 255. Manderson AP, Botto M, Walport MJ. The role of complement in the development of systemic lupus erythematosus. Annu Rev Immunol. 2004;22:431‐456. doi: 10.1146/annurev.immunol.22.012703.104549 [DOI] [PubMed] [Google Scholar]
- 256. Lieberman LA, Mizui M, Nalbandian A, Bossé R, Crispín JC, Tsokos GC. Complement receptor of the immunoglobulin superfamily reduces murine lupus nephritis and cutaneous disease. Clin Immunol. 2015;160(2):286‐291. doi: 10.1016/j.clim.2015.05.006 [DOI] [PubMed] [Google Scholar]
- 257. Birmingham DJ, Irshaid F, Nagaraja HN, et al. The complex nature of serum C3 and C4 as biomarkers of lupus renal flare. Lupus. 2010;19(11):1272‐1280. doi: 10.1177/0961203310371154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Song D, Guo WY, Wang FM, et al. Complement alternative pathway′s activation in patients with lupus nephritis. Am J Med Sci. 2017;353(3):247‐257. doi: 10.1016/j.amjms.2017.01.005 [DOI] [PubMed] [Google Scholar]
- 259. Sato N, Ohsawa I, Nagamachi S, et al. Significance of glomerular activation of the alternative pathway and lectin pathway in lupus nephritis. Lupus. 2011;20(13):1378‐1386. doi: 10.1177/0961203311415561 [DOI] [PubMed] [Google Scholar]
- 260. Kim H, Kim T, Kim M, et al. Activation of the alternative complement pathway predicts renal outcome in patients with lupus nephritis. Lupus. 2020;29(8):862‐871. doi: 10.1177/0961203320925165 [DOI] [PubMed] [Google Scholar]
- 261. Parikh SV, Malvar A, Song H, et al. Molecular imaging of the kidney in lupus nephritis to characterize response to treatment. Transl Res. 2017;182:1‐13. doi: 10.1016/j.trsl.2016.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Bao L, Haas M, Quigg RJ. Complement factor H deficiency accelerates development of lupus nephritis. J Am Soc Nephrol. 2011;22(2):285‐295. doi: 10.1681/ASN.2010060647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Wang FM, Yu F, Tan Y, Song D, Hui ZM. Serum complement factor H is associated with clinical and pathological activities of patients with lupus nephritis. Rheumatology (Oxford). 2012;51(12):2269‐2277. doi: 10.1093/rheumatology/kes218 [DOI] [PubMed] [Google Scholar]
- 264. Wang FM, Song D, Pang Y, Song Y, Yu F, Zhao MH. The dysfunctions of complement factor H in lupus nephritis. Lupus. 2016;25(12):1328‐1340. doi: 10.1177/0961203316642307 [DOI] [PubMed] [Google Scholar]
- 265. Zelek WM, Xie L, Morgan BP, Harris CL. Compendium of current complement therapeutics. Mol Immunol. 2019;114:341‐352. doi: 10.1016/j.molimm.2019.07.030 [DOI] [PubMed] [Google Scholar]
- 266. Ricklin D, Lambris JD. New milestones ahead in complement‐targeted therapy. Semin Immunol. 2016;28(3):208‐222. doi: 10.1016/j.smim.2016.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Caravaca‐Fontán F, Praga M. Prognostication for C3 glomerulopathy and idiopathic immunoglobulin‐associated membranoproliferative glomerulonephritis. Clin J Am Soc Nephrol. 2022;17(7):945‐948. doi: 10.2215/CJN.05490522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Ricklin D, Lambris JD. Complement‐targeted therapeutics. Nat Biotechnol. 2007;25(11):1265‐1275. doi: 10.1038/nbt1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.