

Abstract
The late‐stage site‐selective derivatisation of peptides has many potential applications in structure‐activity relationship studies and postsynthetic modification or conjugation of bioactive compounds. The development of orthogonal methods for C−H functionalisation is crucial for such peptide derivatisation. Among them, biocatalytic methods are increasingly attracting attention. Tryptophan halogenases emerged as valuable catalysts to functionalise tryptophan (Trp), while direct enzyme‐catalysed halogenation of synthetic peptides is yet unprecedented. Here, it is reported that the Trp 6‐halogenase Thal accepts a wide range of amides and peptides containing a Trp moiety. Increasing the sequence length and reaction optimisation made bromination of pentapeptides feasible with good turnovers and a broad sequence scope, while regioselectivity turned out to be sequence dependent. Comparison of X‐ray single crystal structures of Thal in complex with d‐Trp and a dipeptide revealed a significantly altered binding mode for the peptide. The viability of this bioorthogonal approach was exemplified by halogenation of a cyclic RGD peptide.
Keywords: Biocatalysis, bioconjugation, late-stage C−H activation, RGD peptides, tryptophan halogenase
Late‐stage halogenation of peptides has become feasible using a highly flexible halogenase that catalyses bromination of a wide range of amides and peptides. Upon optimization studies, even longer peptides carrying a terminal tryptophan residue were reasonably accepted leading to high conversions and remarkable selectivity. This novel bioorthogonal approach was exemplified by halogenating an RGD peptide derivative in the final step.

Introduction
Selective derivatisation of peptides and proteins is of long‐standing interest in medicinal chemistry to study and manipulate biochemical and cellular functions. [1] Yet, the limited diversity of functionalities natively found in biomolecules restricts strategies towards bioorthogonal modification. Whilst selective pressure incorporation and the expanded genetic code are viable approaches for recombinant incorporation of noncanonical amino acids into proteins, [2] shorter peptides are usually synthesised in a modular fashion using solution or solid phase approaches enabling insertion of non‐canonical residues.
However, typical drawbacks of such early‐stage approaches include the synthesis of building blocks due to the requirement of protecting groups or unwanted side effects like decomposition or epimerisation. In contrast, late‐stage modification (either postsynthetic or posttranslational) provides an attractive alternative: Non‐modified peptide precursors are assembled by well‐established synthetic methodologies in the first step. Subsequent site‐selective residue functionalisation is then applied on the final peptide sequence to provide access to noncanonical handles for conjugation or divergent synthesis of compound arrays. For example, different approaches of metal‐catalysed C−H activation have proven useful in this area. Tryptophan (Trp, 1) is an ideal candidate for derivatisation, e. g., arylation, alkenylation or alkylation, due to its low natural abundance and exceptional electronic properties.[ 3 , 4 , 5 , 6 ]
Generally, late‐stage functionalisation must proceed regio‐ and chemoselectively on highly functionalised peptides without the necessity of protecting or directing groups. [7] It is hence not surprising that biocatalysts harbouring an inherent bioorthogonality, high specificity, and compatibility with aqueous solution, are excellent tools for late‐stage modification that receive increasing attention. [8] Several enzymatic reactions, e. g., transpeptidation using sortase, template‐driven ligations or Cα‐formylglycine formation, are being used to obtain peptide conjugates.[ 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 ] Nevertheless, biocatalytic strategies enabling late‐stage C−H activation and functionalisation of peptides have rarely been evolved until now.
In this context, aryl halogenation provides an attractive target transformation as the halogen handle offers enhanced metabolic stability and the potential of further diversification by manifold cross‐coupling reactions. Flavin‐dependent tryptophan halogenases catalyse the regioselective transformation of a C−H bond of the indole moiety of Trp into a C−Cl or C−Br moiety, using O2, FADH2, and halide salts involving an electrophilic HOX species formed within the enzyme.[ 17 , 18 , 19 ] Several groups focused on broadening the synthetic utility of flavin‐dependent halogenases using directed evolution, immobilisation, cascade development and genome mining.[ 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 ] Flavin‐dependent halogenases involved in non‐ribosomal peptide synthesis often specifically act on substrates tethered to a peptidyl carrier protein making these enzymes incompatible for synthetic use.[ 28 , 29 ] Recently, an extraordinary Trp halogenase, MibH, capable of lanthipeptide halogenation, was characterised. [30] Despite the lack of a carrier protein, free Trp or slight variations of the substrate sequence were not accepted by MibH diminishing its synthetic utility.
Current approaches to obtain halogenated peptides mainly rely on early‐stage synthesis of the halogenated amino acid that is subsequently employed as a modular building block to assemble the peptide of interest (Scheme 1A). We described the biocatalytic synthesis of bromotryptophan used as a building block in peptide synthesis to perform side‐chain‐to‐tail cyclisation by cross‐coupling.[ 31 , 32 ] Goss and co‐workers introduced a chemogenetic approach employing in vivo halogenation of Trp for the biosynthesis of a natural peptide analogue.[ 33 , 34 ] In general, the necessity of host engineering along with insufficient acceptance and difficult product isolation of the noncanonical substrates hamper its broad applicability in vivo.
Scheme 1.
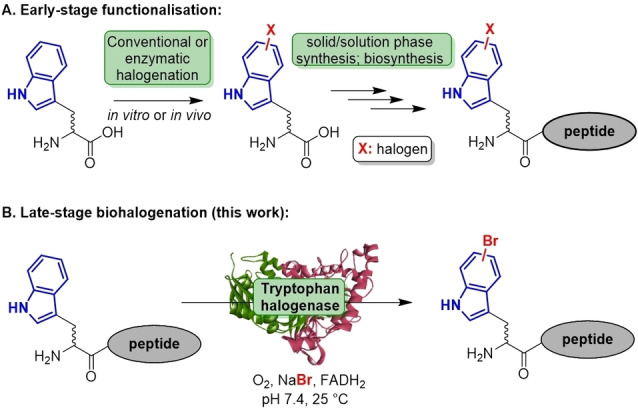
(A.) Approaches towards peptide functionalisation exemplified by comparison of early‐ vs. late‐stage halogenation. (B.) Biocatalytic approach presented herein.
To circumvent these bottlenecks, we envisioned developing a bioorthogonal platform towards late‐stage peptide halogenation under biocompatible conditions (Scheme 1B). By catalyst screening along with substrate design and optimisation studies, flavin‐dependent halogenases proved to be useful catalysts for the specific halogenation of peptide substrates. X‐ray single crystal structure determination also provided further insights into the binding of bulkier non‐native substrates.
Results and Discussion
An array of Trp derivatives was designed to assess the ability of halogenases to accommodate peptides rather than free Trp. According to different crystal structures, the substrate is tightly secluded in the active site forming crucial interactions to the enzyme scaffold.[ 17 , 35 ] Hence, modification of the Trp α‐amino group might not be accepted by Trp halogenases, as it undergoes an essential salt bridge with a nearby Glu residue (RebH, Thal: E461; Figure S1). Instead, the carboxy group is supposed to be more amenable to steric modifications. Based on these considerations, aliphatic Trp N‐carboxamides with varying chain length were synthesised and tested against a library of in‐house flavin‐dependent halogenases. [36]
Substrate (1 mm) was added to cell‐free extracts of halogenases along with concomitant cofactor regeneration in aqueous buffer (pH 7.4) at 25 °C (Scheme 2). By using this setup different in‐house flavin‐dependent halogenases were screened against an initial panel of ten substrates. The halogenases Xcc_1333 and Xcc_4156 from Xanthomonas campestris, [37] that were previously shown to catalyse bromination of indoles did not halogenate 2–5, whereas homologue Xcc_4345 gave traces of brominated Trp amides 3, 4 and 5 (<2 % each) as detected by MS.
Scheme 2.
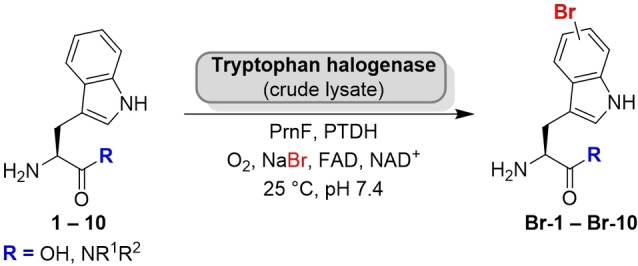
Bromination of C‐terminally substituted tryptophan derivatives (PrnF: flavin reductase; PTDH: phosphite dehydrogenase). The substrate scope of different halogenases towards Trp N‐alkyl amides was examined (results shown in Table 1).
In contrast, Trp halogenases, in particular the C6‐halogenase Thal, showed notable conversion in the screening assay, e. g., substrate 3 (R=NHMe) was quantitatively brominated. Even an extended chain length was tolerated (Table 1). In general, primary Trp amides turned out to be well‐accepted substrates; with the exception of the Trp 5‐halogenase PyrH that barely converted the non‐native Trp substrates (0–2 %). Brominated products were formed in reasonable quantities both by Thal and RebH, albeit with strongly differing total turnover. Conversions by RebH ranged from 10–20 % in many cases. The thermostable variant Thal‐GR is more limited in its substrate scope being less tolerant to more sterically demanding substrates. The high specificity might be related to its active site mutation S359G that was shown before to have an impact on substrate preference. [38] Among all the enzymes screened, wild type Thal turned out to be the most promiscuous enzyme. Even bulkier amides were brominated in good to high conversions reaching more than 90 % in some cases. Moreover, a slight difference between linear and branched chains was observed when comparing 7 with 8, for example. However, derivative 10, a constitutional isomer of 7, was barely halogenated, probably due to a steric clash caused by the bulkier tertiary amide in comparison to the single chain isomers.
Table 1.
Screening of Trp halogenases against a panel of different Trp N‐alkyl amides applying halogenase lysate according to Scheme 2 (50 % v/v halogenase lysate, 1.0 mm substrate, cofactor regeneration, 25 C, 20 h).
|
|
|
Conversion (HPLC)/% |
|||
|
Substrate |
R |
PyrH (C5)[a] |
Thal (C6)[a] |
Thal‐GR (C6)[a] |
RebH (C7)[a] |
|---|---|---|---|---|---|
|
l‐1 |
oh |
62 |
>99 |
>99 |
>99 |
|
2 |
NH2 |
2 |
>99 |
9 |
>99 |
|
3 |
NHMe |
0 |
>99 |
48 |
14 |
|
4 |
NHEt |
0 |
>99 |
16 |
10 |
|
5 |
NHnPr |
0 |
>99 |
5 |
14 |
|
6 |
NHiPr |
0 |
>99 |
1 |
9 |
|
7 |
NHnBu |
2 |
91 |
8 |
16 |
|
8 |
NHiBu |
2 |
88 |
4 |
19 |
|
9 |
1‐piperidinyl |
0 |
65 |
6 |
1 |
|
10 |
NEt2 |
0 |
9 |
0 |
3 |
[a] Regioselectivity towards native substrate l‐1.
The promising results for Thal prompted us to move on to peptide‐based substrates rather than aliphatic amides. Unlike the compounds tested before, biotransformations containing Trp dipeptides suffered from substantial hydrolysis of the substrate. Consequently, bromotryptophan predominated as an undesired reaction product due to proteases present in the cell extract that presumably superseded peptide halogenation.
To overcome this shortcoming, Thal was purified by affinity chromatography prior to screening further potential peptide substrates. The presence of flavin reductase PrnF together with an excess of NADH were essential to avoid cofactor limitations. Apart from the aforementioned alkyl amides, dipeptides were brominated with moderate to high conversions as evident by LC–MS (Scheme 3). Substrates comprising either Gly or Ala next to the Trp residue were reasonably brominated, whereas Thal turned out to be reluctant towards Pro‐containing peptides 13 and 14. Furthermore, 14 was also prone to a side reaction under biotransformation conditions suggesting the formation of a diketopiperazine (14 b). 14 b occurred as the dominant species along with its brominated derivative (Br‐14 b) when conducting reactions in cell‐free extracts (Scheme S2, Figure S2). In addition, β‐alanine (βAla) and γ‐aminobutyrate (GABA) were also tolerated as C‐terminal residues. Noteworthy, peptides carrying a d‐Trp (17–21) were readily brominated by Thal suggesting a high flexibility with respect to Trp configuration. Moderate bromination of tripeptide 22 suggested that an increase in sequence length did not severely affect the substrate scope.
Scheme 3.

Substrate scope of Thal (purified enzyme) analysed by LC–MS suggests a broad tolerance of different Trp alkyl amides and peptides. A. General reaction scheme. B. Secondary and tertiary Trp N‐alkyl amides are readily converted by Thal. C. Bromination of dipeptides containing l‐Trp at the N‐terminus. D. Peptides carrying a d‐Trp residue indicate a wide tolerance of residues attached to the carboxy group. Apart from various dipeptides, 22 is also brominated by Thal. General reaction conditions: 0.5 mm substrate, 95 μm Thal, 2.5 U mL−1 PrnF, 1.0 μm FAD, 50 mm NADH, 30 mm NaBr, phosphate buffer pH 7.4. Conversions mentioned in brackets refer to LC–MS data.
Although a broad substrate flexibility of Thal was confirmed, conversions were only low to moderate. It was considered that this might be due to insufficient cofactor regeneration, as the availability of reducing equivalents is restricted by undesired futile cycles that affect overall conversion. Accordingly, employing phosphite dehydrogenase that was previously utilised for NADH regeneration in halogenation of Trp turned out to be efficient in this case and led to a considerable increase of total turnovers (Figure S3). [39]
Thal's broad peptide scope prompted to apply this approach towards more sophisticated peptides. An array of sequences up to pentapeptides was synthesised and bromination was assayed by LC–MS (Figure 1A). Thal accepted all peptides within this extended panel (Table 2). An increase of chain length did not have a strong impact on conversion, still yielding between 47 and 83 %. Examining the progress of bromination of Ala peptide 27 indicated the steady increase of product over time (Figure 1B). After 240 min 71 % of substrate was converted, which slightly improved to 75 % over 20 h. The ESI(+) mass spectrum unequivocally confirmed presence of the brominated peptide, Br‐27, as the dominant species (Figure 1C).
Figure 1.

(A) General scheme of Thal‐catalysed peptide halogenation. Results are shown in Table 2. (B) RP‐HPLC diagram monitoring the reaction progress of bromination of 27 (3.3 min, blue) into Br‐27 (4.4 min, red). (C) MS spectra (ESI+) corresponding to signals highlighted in HPLC trace in (B) identify substrate (3.3 min, blue) and product (4.4 min, red). The isotope pattern indicates bromination.
Table 2.
Thal‐catalysed bromination of various peptides carrying an N‐terminal Trp residue as displayed in Figure 1A (0.5 mm substrate, 95 μm Thal, cofactor regeneration system).
|
Substrate |
Sequence |
Conv.[a]/% |
|
|---|---|---|---|
|
11 |
l‐Trp |
‐Gly‐[b] |
79 |
|
17 |
d‐Trp |
‐Gly‐ |
99 |
|
12 |
l‐Trp |
‐Ala‐[b] |
84 |
|
19 |
d‐Trp |
‐Ala‐[b] |
99 |
|
23 |
l‐Trp |
‐Gly‐Gly‐[b] |
53 |
|
22 |
d‐Trp |
‐Gly‐Gly‐ |
71 |
|
24 |
l‐Trp |
‐Gly‐Gly‐Gly‐[b] |
47 |
|
25 |
d‐Trp |
‐Gly‐Gly‐Gly‐ |
83 |
|
26 |
l‐Trp |
‐Ala‐Gly‐Gly‐[b] |
41 |
|
27 |
d‐Trp |
‐Ala‐Gly‐Gly‐ |
75 |
|
28 |
d‐Trp |
‐Gly‐Gly‐Gly‐Gly‐ |
52 |
[a] Determined by LC–MS. [b] Addition of protease inhibitors preventing substrate hydrolysis.
By adding a mixture of protease inhibitors, proteolytic cleavage of peptides with l‐Trp was successfully overcome, hence providing sufficient flexibility in terms of configuration of Trp residue (Figure S4). Briefly, after optimising the concentration of inhibitor additives, successful bromination of l‐Trp peptides, i. e., 11, 12, 23, 24, 26, became feasible without significant hydrolysis, thus making further efforts on enzyme purification unnecessary. However, bromination of different l‐Trp peptides revealed significantly lower conversions than their epimers as obvious for 24 and 25, for example, hence suggesting a preference for d‐configuration at the N‐terminus. Having established a method applicable to bromination of peptides with N‐terminal l‐ and d‐Trp, examination of sequence space amenable to Thal‐catalysed halogenation was of particular interest to investigate the viability of this approach (Figure 2A).
Figure 2.

(A) Bromination of different peptides with variation of the residue proximal to Trp (red frame) to investigate sequence specificity. (B) TTN determination for representative tetrapeptides according to the general sequence depicted in (A). Hydrophobic side chains as well as Ser are preferred moieties proximal to Trp.
Our initial results had indicated that Pro residues adjacent to Trp disfavour bromination (Scheme 3C). Representative sequences were synthesised, and total turnover numbers (TTNs) determined for each substrate (Figure 2B). The data unveiled a pronounced impact of proximal residues on efficiency of Trp halogenation, suggesting that Thal especially favours hydrophobic residues as indicated by Phe and Val giving the highest TTNs so far. It is even surprising that a smaller Gly residue adjacent to Trp gives a TTN about two times lower than Val or Phe.
Moreover, Ser has a positive impact on turnover compared to other residues. The preference for bulkier side chains was unexpected, since the native substrate is usually deeply buried in the active site. In contrast, Thal is reluctant to Lys near the Trp residue. Likewise, Asp severely affects bromination with a negligible TTN<2 indicating that both polarity and steric effects influence conversion. Alteration of the Trp position within the sequence exemplified for H‐GwGG‐NH2 resulted in traces of brominated product, indicating that positioning Trp at the N‐terminus is essential (Scheme S2, Figure S5).
To corroborate formation of halogenated peptides, reactions were performed on a milligram scale. Biotransformations were conducted with 15–40 μmol of substrate loading, applying a cell‐free extract of Thal or purified halogenase along with reaction components for concomitant cofactor regeneration in a total volume of 10–50 mL. As before, reactions went smoothly and gave solely the brominated peptides that were isolated by RP‐HPLC. Using this setup, four different brominated peptides, Br‐25, Br‐29 Br‐30, and Br‐34 were isolated. Bromotryptophan was unequivocally identified within the peptides. Surprisingly, it was observed by NMR that the regioselectivity was influenced by the substrate sequence: For peptide 25, the C6‐brominated peptide expected for Thal was present as the main isomer, but also C5‐bromination occurred (24 %, Table 3, Figure S6). Presumably, the peptidyl moiety causes a looser binding of the Trp side chain within the active site, which could be responsible for a flip of the indole plane, thus favouring a second isomer. In contrast, pronounced C6 selectivity was observed for peptides 29, 30 and 34. This indicates that both the nature of the enzyme and substrate features are crucial for regioselectivity.
Table 3.
Regioselectivity of peptide halogenation derived from 1H‐NMR data. Strict C6‐selectivity is achieved for bulkier residues next to the Trp moiety.
|
|
|
Regioisomers/%[a] |
|
|
Substrate |
Sequence |
C5/% |
C6/% |
|---|---|---|---|
|
25 |
H‐d‐Trp‐Gly‐Gly‐Gly‐NH2 |
24 |
76 |
|
29 |
H‐d‐Trp‐Val‐Gly‐Gly‐NH2 |
not found |
100 |
|
30 |
H‐d‐Trp‐Ser‐Gly‐Gly‐NH2 |
7 |
93 |
|
34 |
H‐d‐Trp‐Phe‐Gly‐Gly‐NH2 |
not found |
100 |
[a] Percentages refer to ratio of isomer signals calculated from integrals of the NMR signals.
Interested in the interactions between Thal and the peptide substrates, we envisaged obtaining a crystal structure of the enzyme‐substrate complex. As no structure for Thal with d‐1 was available, we initially investigated the Thal:d‐Trp complex to dissect how the Trp configuration impacts interactions in the active site.
The orientation of d‐Trp (d‐1) in the active site of Thal differs only slightly from that of l‐Trp (l‐1).[ 35 , 40 ] Both indole moieties are oriented in the same plane, yet slightly rotated. Notably, the distance between C6 and the ϵ‐amino group of the catalytic K79 remains the same (3.74 Å and 3.76 Å) (Figure S7). Likewise, the backbone nitrogen and the oxygen atoms of the carboxy groups superpose down to ∼0.5 Å. The greatest distance of about 1 Å occurs between the Cβ atoms of d‐1 and l‐1. d‐Trp:Thal forms an additional H‐bond between the backbone nitrogen and the hydroxy group of Y454, whereas it misses an H‐bond formed between the l‐Trp carboxy group and the hydroxy group of Y455 (Figure S8). Side chains of active site residues superpose well, except for the rings of H110 and F465 that are tilted by about 20 to compensate the changed distance towards the substrate. To get an objective measurement, whether the chirality of tryptophan was placed correctly a geometry analysis was performed by replacing l‐1 in chain A of l‐Trp‐Thal (PDB: 6H44) with d‐1 and vice versa for d‐Trp‐Thal (PDB: 8AD7) and refining each structure to convergence. Despite a good fit to the electron density, the ligand geometry (distortions of bond lengths and bond angles) and the clashes with the protein for the wrongly placed ligand are worse compared to the original structures (Table S1), overall confirming the correctness of the placed chirality.
Furthermore, the crystal structure of the dipeptide H‐d‐Trp‐Ser‐OH in complex with Thal was successfully resolved (PDB: 8AD8, Figure S9). A bromination assay with this substrate was conducted as described, only with lower enzyme loading at 5 mol %, and a conversion of 86 % was observed. Semi‐preparative bromination followed by NMR‐analysis indicated 75 % C6‐bromination. A part (residues 452–455) of the substrate‐binding loop that closes the active site in the presence of l‐1 or d‐1 is disordered and L114 moves away from the substrate leaving the dipeptide accessible to solvent.
This may facilitate C‐terminal extension of the substrate peptide with additional residues sticking out of the active site. The dipeptide backbone forms polar contacts with residues F465 to G469 (Figure S10). Surprisingly, the Trp residue of the dipeptide assumes a completely different position than l‐1 or d‐1 alone leading to a significantly longer distance between the C6 of the indole moiety and the ϵNH2 of K79 (6.81 Å instead of 3.75 Å) (Figure 3A). Such long distances are not unprecedented in flavin‐dependent halogenases. Even longer distances between ϵNH2 of the catalytic lysine and the halogenated carbon are present in multiple structures of MalA’ in complex with different native substrates. [41] Previously, Zhu et al. postulated that the regioselectivity of tryptophan halogenases is dominated by the fact that the catalytically preferred position of the indole moiety has the shortest distance to K79 (Thal). [42] Since this distance is very long in the Thal:peptide complex, we speculated that this is not the only mechanism crucial for regioselective halogenation of the dipeptide. Therefore, we replaced the ligand by its chlorination products in silico and analysed the impact of Cl substituents on steric conflicts in positions C5, C6, and C7 (Figure 3B). The placement of both C5‐ and C7‐chlorinated dipeptides resulted in multiple clashes with surrounding residues of the protein, which did not occur for the preferred C6 position. Thus, both the distance to K79 and steric conflicts of the potential reaction products may influence the regioselectivity of tryptophan halogenases.
Figure 3.

(A) Orientation of d‐Trp‐Ser (DWS) in Thal. Superposition of bound d‐Trp‐Ser (yellow carbons; from PDB: 8AD8) and l‐Trp (light blue carbons, from PDB: 6H44) within the active site. The different position of the indole moiety leads to a longer distance between C6 and the ϵNH2 of the catalytic lysine. (B) Steric conflicts of potential chlorination positions. Replacement of the ligand d‐Trp‐Ser by its potential chlorination products leads to clashes (dotted orange lines, lengths in Å) of the chlorine (green) at position C5 and C7 with surrounding residues. Chlorination of the catalytically preferred C6 position does not cause clashes. d‐5‐Chloro‐Trp‐Ser, d‐6‐chloro‐Trp‐Ser and d‐7‐chloro‐Trp‐Ser were superimposed onto d‐Trp‐Ser (carbon atoms in yellow).
Finally, we strived for employing this biocatalytic method in halogenation of a multifunctional oligopeptide. The cyclopeptide c(RGDfK), a derivative of the integrin ligand Cilengitide, offers a modification site using its Lys ϵ‐amino group that is widely applied to attach fluorophores or cytotoxic drugs to integrin ligands.[ 43 , 44 , 45 ] We sought to link the Thal substrate recognition sequence wVGG to the cyclic moiety c(RGDfK), thus providing a model peptide that should be halogenated in the final step using our newly established method. 31 was synthesised in analogy to a procedure described before. [46] Subsequently, enzymatic halogenation was carried out by applying the optimised reaction conditions (Figure 4). Thal recognised the Trp residue present in the linker motif and readily catalysed its bromination. RP‐HPLC analysis clearly proved successful late‐stage functionalisation indicating that the reaction went to completion after 180 min. Rapid turnover of the cyclic peptide even suggests additional interactions between the cyclopeptide and the enzyme that might exert a positive effect on substrate binding. The brominated RGD peptide was unambiguously identified by HR‐MS. Control experiments containing 31, cofactors and auxiliary enzymes, either lacking halogenase or containing heat‐inactivated protein, gave no signs of product formation (Figure S11). Thus, a non‐enzymatic transformation could be completely ruled out, hence proving that Thal is capable of late‐stage halogenation of multi‐functionalised peptides.
Figure 4.

(A) Bioorthogonal halogenation of RGD peptide 31. The cyclic peptide carrying a sequence motif (wVGG‐) attached to a Lys side chain is recognised by Thal and thus leads to selective halogenation of the d‐Trp residue within the oligopeptide. (B) RP‐HPLC diagram (280 nm trace) monitoring the reaction course indicates depletion of 31, while the signal of the brominated peptide increases. (C) HR‐MS (ESI+) spectrum corresponding to the HPLC signal marked in B. gives evidence for characteristic [M+H]+ and [M+2H]2+ (monoisotopic, m/z calc.: 569.725) signals. The insert on the right side displays an enlargement of the [M+H]+ signal identifying the brominated RGD peptide from the characteristic isotope pattern, m/z [M+H]+ (C49H68BrN15O12H+), calc.: 1138.443 (85 %), 1139.446 (50 %), 1140.442 (100 %), 1141.444 (54 %), 1142.446 (17 %).
Conclusion
In the presented study we envisaged developing an enzymatic method for direct halogenation of peptides. Catalyst screening, reaction and substrate optimisation unveiled that the Trp 6‐halogenases Thal is capable of halogenating Trp residues within various peptide sequences in good conversions. It was demonstrated that Thal is highly promiscuous and therefore accepts a broad array of substrate sequences carrying either l‐ or d‐Trp at the N‐terminus. In addition, the neighbouring residue is crucial for retaining C6‐selectivity, giving a preference for Val, Phe, or Ser in C‐terminal direction to Trp. Furthermore, the crystal structure of Thal in complex with a dipeptide is presented herein. While the structure of Thal with d‐1 resembles that of l‐1, [35] binding of H‐d‐Trp‐Ser‐NH2 led to pronounced differences in the binding mode. Less tight binding of the peptides into the active site compared to the amino acid alone suggests additional factors crucial for regioselectivity.
Overall, our results open up a straightforward approach to introduce the aryl halide motif into free peptides in aqueous buffer. A favourable Thal recognition motif containing a terminal Trp that is linked to a peptide of interest can facilitate orthogonal introduction of the halogen handle. To the best of our knowledge, application of enzymatic late‐stage halogenation on peptides is unprecedented, which offers various applications in the field of peptide modification. Compared with previously reported early precursor synthesis, this biocatalytic approach gives efficient and reliable access to halogenated peptides whilst avoiding laborious and yield limiting protection‐deprotection routes to obtain halogenated amino acids. The enzymatic introduction of a noncanonical handle in the last step triggers further diversification, for example, by cross‐coupling reactions. In ongoing investigations, we strive to expand this novel methodology by means of enzyme engineering to strengthen the utility of peptide halogenation towards a widely applicable tool for bioorthogonal late‐stage halogenation.
Experimental Section
Materials
Chemicals and solvents were obtained from commercial suppliers Sigma‐Aldrich, Acros Organics and TCI Chemicals in highest purity available for analytical applications (p. a.). pCIBhis‐PrnF encoding the flavin reductase was kindly donated by Prof. Dr. Karl‐Heinz van Pée. Plasmid pGro7 for chaperone co‐expression was obtained from TaKaRa. Tryptophan amide was purchased from Acros Organics. HisTALON metal affinity resin (TaKaRa) was used for protein purification. Halt Protease Inhibitor Cocktail, EDTA free, 100×, was purchased from Fisher Scientific (catalogue no. #87785).
Analytical methods
For HPLC separation coupled with simultaneous mass spectrometric analysis (LC–MS) samples were separated on an Agilent 1200 HPLC system (Agilent Technologies, Santa Clara, CA, USA) equipped with an autosampler, pump, column oven and diode array detector. LC flow was directed to an Agilent 6220 ESI time‐of‐flight mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) in extended dynamic range mode equipped with a Dual‐ESI source, operating with a spray voltage of 2.5 kV. Nitrogen generated by a nitrogen generator NGM 11 served as both the nebulizer gas and the drying gas. Spectra were recorded in range from m/z 100–2500 with an accumulation of 2 spectra per second. Data were analysed with the MassHunter Workstation Acquisition B.04.00 software provided by Agilent Technologies and applied for averaging several single spectra. Method: Eluent A: 94.9 % water/5 % acetonitrile/0.1 % formic acid; eluent B: 94.9 % acetonitrile/5 % water/0.1 % formic acid; column: Thermo Scientific, Hypersil Gold C18, 3 μm, 150×2.1 mm. Gradient (flow rate 0.30 mL min−1): 0→98 % B, 10 min; 98 % B isocratic, 1 min; 98→2% B, 0.5 min; 2 % B isocratic, 3.5 min.
High‐resolution mass spectra (HRMS) were recorded on a 6220 ESI time‐of‐flight mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) equipped with Agilent 1200 LC, Hypersil Gold C18 (50×2.1 mm, 1.9 μm) column using a linear gradient from 0 % to 98 % B over 4 minutes. Eluent A: 94.9 % water/5 % acetonitrile/0.1 % formic acid; eluent B: 94.9 % acetonitrile/5 % water/0.1 % formic acid. External calibration using Agilent tuning mix was performed prior to measurements.
NMR spectra were recorded on a Bruker Avance NEO 600 (1H: 600 MHz, 13C: 151 MHz) or a Bruker Avance 500HD spectrometer (1H: 500 MHz, 13C: 126 MHz). Chemical shifts were referenced to residual non‐deuterated solvent signal (DMSO‐d 6: 1H: 2.50 ppm/D2O: 4.79 ppm).
Halogenase expression and purification
Flavin‐dependent halogenases were encoded on pET28a vectors for heterologous expression in E. coli BL21 (DE3) as previously described.[ 24 , 37 , 47 , 48 ] Briefly, 1.5 L LB‐media containing kanamycin (60 mg L−1) and chloramphenicol (50 mg L−1) was inoculated with 30 mL of grown preculture of E. coli BL21 (DE3) pGro7 pET28a‐RebH, ‐Thal or ‐PyrH, respectively. The main culture was cultivated at 37 °C until OD600 nm=0.5 was reached. After the cooling the expression culture down to 25 °C overexpression was induced by addition of 0.1 mm IPTG and 2 g L−1 l‐arabinose. Cells were shaken at 150 rpm for 20 h, harvested by centrifugation (3220×g, 60 min, 4 °C), washed with 40 mL 0.1 m Na2HPO4 (pH 7.4) and stored at −20 °C. Procedures for the expression and purification of auxiliary enzymes (flavin reductase PrnF and phosphite dehydrogenase PTDH) can be found in the Supporting Information.
The cell pellet was resuspended in 30 mL 50 mm Na2HPO4 (pH 7.4)/300 mm NaCl and lysed by French‐Press (3 passages @ 1100 bar cell pressure). DNAseI from bovine (Applichem) was added and cell debris subsequently removed by centrifugation (12000×g, 30 min, 4 °C). The resulting lysate was either directly used for biotransformations or subsequently purified by immobilised ion metal affinity chromatography (IMAC).
The supernatant was filtered through a 0.45 μm Whatman filter and the cleared lysate was loaded on pre‐equilibrated HisTALON affinity resin (ca. 1.5 mL bed volume) in a glass column equipped with a frit. The suspension was inverted for 60 min at 4 °C to enable sufficient protein binding. Subsequently, non‐bound protein was removed by successive washing with (i) 10 mL of 50 mm Na2HPO4 (pH 7.4)/300 mm NaCl and (ii) 10 mL buffer supplemented with 10 mm imidazole. During each washing step the mixture was inverted again for 10 min at 4 °C. The halogenase was eluted with 50 mM Na2HPO4 (pH 7.4)/300 mm NaCl/300 mm imidazole collecting ca. 10 fractions of 0.5 mL. Fractions containing His6‐tagged halogenase as determined by Nano Drop (absorbance at 280 nm) and SDS PAGE were pooled and dialysed against 15 mm Na2HPO4/30 mm NaBr (pH 7.4) overnight at 4 °C resulting in approx. 25 mg of protein per litre of expression culture.
Determination of protein structure
For the determination of d‐Trp‐Thal structure, Thal was expressed, purified and crystallised as described.[ 35 , 40 ] The ligand d‐Trp was not soaked but co‐crystallised by adding 5 mm d‐Trp to the protein solution. For the d‐Trp‐Ser‐Thal structure, Thal was expressed and purified as described for Thal‐RebH5. [35] Purified Thal was crystallised using the sitting drop vapor‐diffusion method at 20 °C with a drop ratio of 1 : 1 by mixing a 15 mg mL−1 protein solution in buffer (10 mm Tris pH 7.4, 50 mm NaCl, 1 mm DTT) with an equal volume of reservoir solution (0.1 m bicine pH 9.0, 1.6 m K2HPO4/KH2PO4). Crystals appeared as hexagonal prisms within 5–7 days. Thal crystals containing the d‐Trp‐Ser dipeptide were obtained by soaking Thal crystals in reservoir solution additionally containing 20 mm d‐Trp‐Ser×TFA for 60 min. For cryoprotection, the crystals were transferred to reservoir solution supplemented with 30 % glycerol and 12 mm dipeptide at 20 °C before flash cooling in liquid nitrogen.
All data were collected at a temperature of 100 K. Data of d‐Trp‐Thal were collected at beamline P13 operated by EMBL Hamburg at the PETRA III storage ring at DESY, Hamburg, Germany. [49] Measurements of d‐Trp‐Ser‐Thal were carried out by Frank Lennartz from Manfred Weiss’ team at the BL14.2. beamline at the BESSY II electron storage ring operated by the Helmholtz‐Zentrum Berlin für Materialien und Energie. [50] The data sets were processed with XDS and scaled with XSCALE. [51] The structures were determined by molecular replacement using the programme Phaser [52] and chain A of the apo‐Thal structure (PDB ID 6H43) as search model. The structures were improved by modelling in COOT [53] and restrained refinement in Refmac5 [54] using non‐crystallographic symmetry restraints. Structures were built to near completion before placing ligands into difference density. A summary of data processing is shown in Table S2. The structure and restraints for the d‐Trp‐Ser dipeptide and its chlorination products were generated in eLBOW using the AM1 QM method. [55] The occupancies of all ligands were refined. The figures of the final structures were generated using PyMOL. The coordinates and structure factors of the d‐Trp‐Thal and d‐Trp‐Ser‐Thal structures were deposited in the Protein Data Bank with accession codes 8AD7 and 8AD8, respectively.
Screening of halogenases against tryptophan N‐alkyl amides
Assays were performed in a total volume of 100 μL in microtiter plates or 1.5 mL reaction vials and incubated at 25 °C for 20 h. In a buffered solution (50 mm Na2HPO4, pH 7.4) 50 μL lysate was mixed with 1 mm amide (TFA salt), 10 μm FAD, 50 mm sodium phosphite (pH 7,4) and 30 mm NaBr. Reactions were initiated by addition of lysate containing overexpressed halogenase, 1 mm NAD+ and sealed with AeraSeal film (Excel Scientific) for subsequent incubation at 25 °C for 20 h. The reactions were stopped by adding 100 μL MeOH and precipitate was spun down (10,000×g, 10 min). 50 μL of supernatant was further diluted with 150 μL water and analysed via LC–MS.
Enzymatic bromination of peptides
Biotransformations were performed in an aqueous buffered solution (50 mm Na2HPO4, pH 7.4) in a total volume of 100 μL. 0.5 mm of peptide substrate was mixed with 90 μm halogenase Thal (purified via IMAC), 10 μm FAD and 2.5 U mL−1 PrnF (purified via IMAC). NADH was added in a final concentration of 10 mm to initiate the reaction. The biotransformation mixtures were incubated in a shaking incubator (500 rpm) for 20 h at 25 °C. Reactions were stopped by addition of MeOH and spun down (10,000×g, 10 min). 50 μL of supernatant was diluted with 150 μL water to analysed by LC–MS.
Enzymatic bromination of peptides using Thal with concomitant NADH regeneration
Biotransformations were performed in an aqueous buffered solution (50 mm Na2HPO4, pH 7.4) in a total volume of 100 μL applying a final concentration of 0.5 mm of peptide, 90 μm halogenase Thal (purified via IMAC), 10 μm FAD, 30 mm NaBr and 2.5 U mL−1 PrnF (purified via IMAC). To enable in‐situ‐regeneration of NADH, 2 U mL−1 PTDH (lysate) and 1 mm NAD+ along with 50 mm Na2HPO3 (pH 7.4) were added to the biotransformation. To suppress proteolysis of peptides with an l‐tryptophan residue, protease inhibitor cocktail was added to the reaction mixture to reach a total volumetric ratio of 3 % (v/v).
After incubating the biotransformation for 20 h at 25 °C (500 rpm) the reactions were stopped by addition of MeOH and the resultant precipitate was spun down (10,000×g, 10 min). 50 μL of supernatant was diluted with 150 μL water and analysed by LC–MS. The dipeptide H‐d‐Trp‐Ser‐OH was assayed accordingly, only applying a lower halogenase loading of 50 μm.
Small preparative‐scale halogenations for determination of regioselectivity
A cell pellet of E. coli BL21(DE3) pGro7 pET28‐Thal containing overexpressed Thal was resuspended in 20 mL of aqueous buffer (50 mM Na2HPO4, 30 mM NaBr, pH 7.4) and lysed by French‐Press (3 passages @ 1100 bar cell pressure). Cell lysate was cleared by centrifugation for 30 min (12000×g, 4 °C) and the supernatant was provided in a 100 mL Erlenmeyer flask. To the halogenase lysate, 15–40 μm peptide substrate, PrnF (2.5 U mL−1), PTDH (2 U mL−1), 0.1 mm NAD+ , 10 μm FAD, 50 mm sodium phosphite, 30 mm NaBr and 15 mm Na2HPO4 (pH 7,4) were added in a total volume of 30–80 mL. Upon incubation for 20 h at 25 °C in a shaking incubator precipitate was removed by centrifugation (10000×g, 30 min, 4 °C), the crude product was freeze dried and purified by preparative RP‐HPLC on a Merck‐Hitachi preparative HPLC system (controller: D7000, pump: L7150, detector: L7420, absorbance monitored at 220 nm) equipped with a Macherey‐Nagel Nucleosil 100–7 C18 column (7 μm, 250×10 mm). A linear gradient from 0→75 % MeCN over 45 min was applied to isolate brominated peptide. Resultant product‐containing fractions were pooled, freeze‐dried and analysed by NMR and HRMS.
Synthetic procedures
Procedures on substrate synthesis and analytical data can be found in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgments
Support by Deutsche Forschungsgemeinschaft (SE 609/19‐1, NI 694/10‐1) is gratefully acknowledged. The authors thank Dr. Isabell Kemker for helpful advice on the synthesis of RGD peptides. The synchrotron data of d‐Trp‐Thal were collected at beamline P13 operated by EMBL Hamburg at the PETRA III storage ring (DESY, Hamburg, Germany). We would like to thank Dr. Isabel Bento and Saravanan Panneerselvam for the assistance in using the beamline. We thank the Helmholtz‐Zentrum für Materialien und Energie for the allocation synchrotron radiation beamtime. We are particularly thankful to Frank Lennartz and Dr. Manfred Weiss for performing data collection at BESSY BL14.2 as service during the COVID19 lockdown. We thankfully acknowledge the financial support by HZB (travel grant BESSY 2 MX: 17). Open Access funding enabled and organized by Projekt DEAL.
Schnepel C., Moritzer A.-C., Gäfe S., Montua N., Minges H., Nieß A., Niemann H. H., Sewald N., ChemBioChem 2023, 24, e202200569.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Sletten E. M., Bertozzi C. R., Angew. Chem. Int. Ed. 2009, 48, 6974–6998; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 7108–7133. [Google Scholar]
- 2. Wang L., Schultz P. G., Angew. Chem. Int. Ed. 2005, 44, 34–66; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 34–68. [Google Scholar]
- 3. Gruß H., Sewald N., Chem. Eur. J. 2020, 26, 5328–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ruiz-Rodríguez J., Albericio F., Lavilla R., Chem. Eur. J. 2010, 16, 1124–1127. [DOI] [PubMed] [Google Scholar]
- 5. Kaplaneris N., Rogge T., Yin R., Wang H., Sirvinskaite G., Ackermann L., Angew. Chem. Int. Ed. 2019, 58, 3476–3480; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3514–3518. [Google Scholar]
- 6. Schischko A., Kaplaneris N., Rogge T., Sirvinskaite G., Son J., Ackermann L., Nat. Commun. 2019, 10, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang W., Lorion M. M., Shah J., Kapdi A. R., Ackermann L., Angew. Chem. Int. Ed. 2018, 57, 14700–14717; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14912–14930. [Google Scholar]
- 8. Romero E., Jones B. S., Hogg B. N., Casamajo A. R., Hayes M. A., Flitsch S. L., Turner N. J., Schnepel C., Angew. Chem. Int. Ed. 2021, 60, 16824–6855; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 16962-16993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lotze J., Reinhardt U., Seitz O., Beck-Sickinger A. G., Mol. BioSyst. 2016, 12, 1731–1745. [DOI] [PubMed] [Google Scholar]
- 10. Rachel N. M., Toulouse J. L., Pelletier J. N., Bioconjugate Chem. 2017, 28, 2518–2523. [DOI] [PubMed] [Google Scholar]
- 11. Dennler P., Chiotellis A., Fischer E., Brégeon D., Belmant C., Gauthier L., Lhospice F., Romagne F., Schibli R., Bioconjugate Chem. 2014, 25, 569–578. [DOI] [PubMed] [Google Scholar]
- 12. Dai X., Böker A., Glebe U., RSC Adv. 2019, 9, 4700–4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Piotukh K., Geltinger B., Heinrich N., Gerth F., Beyermann M., Freund C., Schwarzer D., J. Am. Chem. Soc. 2011, 133, 17536–17539. [DOI] [PubMed] [Google Scholar]
- 14. Brauckhoff N., Hahne G., Yeh J. T.-H., Grossmann T. N., Angew. Chem. Int. Ed. 2014, 53, 4337–4340; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4425–4429. [Google Scholar]
- 15. Hudak J. E., Barfield R. M., de Hart G. W., Grob P., Nogales E., Bertozzi C. R., Rabuka D., Angew. Chem. Int. Ed. 2012, 51, 4161–4165; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4237–4241. [Google Scholar]
- 16. Krüger T., Weiland S., Falck G., Gerlach M., Boschanski M., Alam S., Müller K. M., Dierks T., Sewald N., Angew. Chem. Int. Ed. 2018, 57, 7245–7249; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7365–7369. [Google Scholar]
- 17. Dong C., Flecks S., Unversucht S., Haupt C., van Pée K. - H., Naismith J. H., Science 2005, 309, 2216–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yeh E., Blasiak L. C., Koglin A., Drennan C. L., Walsh C. T., Biochemistry 2007, 46, 1284–1292. [DOI] [PubMed] [Google Scholar]
- 19. Flecks S., Patallo E. P., Zhu X., Ernyei A. J., Seifert G., Schneider A., Dong C., Naismith J. H., van Pée K. - H., Angew. Chem. Int. Ed. 2008, 47, 9533–9536; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 9676–9679. [Google Scholar]
- 20. Payne J. T., Andorfer M. C., Lewis J. C., Angew. Chem. Int. Ed. 2013, 52, 5271–5274; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5379–5382. [Google Scholar]
- 21. Schnepel C., Minges H., Frese M., Sewald N., Angew. Chem. Int. Ed. 2016, 55, 14159–14163; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14365–14369. [Google Scholar]
- 22. Latham J., Henry J.-M., Sharif H. H., Menon B. R. K., Shepherd S. A., Greaney M. F., Micklefield J., Nat. Commun. 2016, 7, 11873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fisher B. F., Snodgrass H. M., Jones K. A., Andorfer M. C., Lewis J. C., ACS Cent. Sci. 2019, 5, 1844–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Neubauer P. R., Widmann C., Wibberg D., Schröder L., Frese M., Kottke T., Kalinowski J., Niemann H. H., Sewald N., PLoS One 2018, 13, e0196797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gkotsi D. S., Ludewig H., Sharma S. V., Connolly J. A., Dhaliwal J., Wang Y., Unsworth W. P., Taylor R. J. K., McLachlan M. M. W., Shanahan S., Naismith J. H., Goss R. J. M., Nat. Chem. 2019, 11, 1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Latham J., Brandenburger E., Shepherd S. A., Menon B. R. K., Micklefield J., Chem. Rev. 2018, 118, 232–269. [DOI] [PubMed] [Google Scholar]
- 27. Prakinee K., Phintha A., Visitsatthawong S., Lawan N., Sucharitakul J., Kantiwiriyawanitch C., Damborsky J., Chitnumsub P., van Pée K. - H., Chaiyen P., Nat. Catal. 2022, 5, 534–544. [Google Scholar]
- 28. Schmartz P. C., Zerbe K., Abou-Hadeed K., Robinson J. A., Org. Biomol. Chem. 2014, 12, 5574–5577. [DOI] [PubMed] [Google Scholar]
- 29. Kittilä T., Kittel C., Tailhades J., Butz D., Schoppet M., Büttner A., Goode R. J. A., Schittenhelm R. B., van Pée K. - H., Süssmuth R. D., Wohlleben W., Cryle M. J., Stegmann E., Chem. Sci. 2017, 8, 5992–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ortega M. A., Cogan D. P., Mukherjee S., Garg N., Li B., Thibodeaux G. N., Maffioli S. I., Donadio S., Sosio M., Escano J., Smith L., Nair S. K., van der Donk W. A., ACS Chem. Biol. 2017, 12, 548–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kemker I., Schnepel C., Schröder D. C., Marion A., Sewald N., J. Med. Chem. 2019, 62, 7417–7430. [DOI] [PubMed] [Google Scholar]
- 32. Dachwitz S., Scharkowski B., Sewald N., Chem. Eur. J. 2021, 27, 18043–18046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sharma S. V., Tong X., Pubill-Ulldemolins C., Cartmell C., Bogosyan E. J. A., Rackham E. J., Marelli E., Hamed R. B., Goss R. J. M., Nat. Commun. 2017, 8, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roy A. D., Grüschow S., Cairns N., Goss R. J. M., J. Am. Chem. Soc. 2010, 132, 12243–12245. [DOI] [PubMed] [Google Scholar]
- 35. Moritzer A.-C., Minges H., Prior T., Frese M., Sewald N., Niemann H. H., J. Biol. Chem. 2019, 294, 2529–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zacharie B., Connolly T. P., Penney C. L., J. Org. Chem. 1995, 60, 7072–7074. [Google Scholar]
- 37. Ismail M., Frese M., Patschkowski T., Ortseifen V., Niehaus K., Sewald N., Adv. Synth. Catal. 2019, 361, 2475–2486. [Google Scholar]
- 38. Minges H., Schnepel C., Böttcher D., Weiß M. S., Sproß J., Bornscheuer U. T., Sewald N., ChemCatChem 2020, 12, 818–831. [Google Scholar]
- 39. Schnepel C., Dodero V. I., Sewald N., Chem. Eur. J. 2021, 27, 5404–5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moritzer A.-C., Niemann H. H., Protein Sci. 2019, 28, 2112–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fraley A. E., Garcia-Borràs M., Tripathi A., Khare D., Mercado-Marin E. V., Tran H., Dan Q., Webb G. P., Watts K. R., Crews P., Sarpong R., Williams R. M., Smith J. L., Houk K. N., Sherman D. H., J. Am. Chem. Soc. 2017, 139, 12060–12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhu X., De Laurentis W., Leang K., Herrmann J., Ihlefeld K., van Pée K. - H., Naismith J. H., J. Mol. Biol. 2009, 391, 74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Haubner R., Gratias R., Diefenbach B., Goodman S. L., Jonczyk A., Kessler H., J. Am. Chem. Soc. 1996, 118, 7461–7472. [Google Scholar]
- 44. Dechantsreiter M. A., Planker E., Mathä B., Lohof E., Hölzemann G., Jonczyk A., Goodman S. L., Kessler H., J. Med. Chem. 1999, 42, 3033–3040. [DOI] [PubMed] [Google Scholar]
- 45. Nahrwold M., Weiß C., Bogner T., Mertink F., Conradi J., Sammet B., Palmisano R., Royo Gracia S., Preuße T., Sewald N., J. Med. Chem. 2013, 56, 1853–1864. [DOI] [PubMed] [Google Scholar]
- 46. Kemker I., Feiner R. C., Müller K. M., Sewald N., ChemBioChem 2020, 21, 496–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Frese M., Guzowska P. H., Voß H., Sewald N., ChemCatChem 2014, 6, 1270–1276. [Google Scholar]
- 48. Frese M., Schnepel C., Minges H., Voß H., Feiner R., Sewald N., ChemCatChem 2016, 8, 1799–1803. [Google Scholar]
- 49. Cianci M., Bourenkov G., Pompidor G., Karpics I., Kallio J., Bento I., Roessle M., Cipriani F., Fiedler S., Schneider T. R., J. Synchrotron Radiat. 2017, 24, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mueller U., Förster R., Hellmig M., Huschmann F. U., Kastner A., Malecki P., Pühringer S., Röwer M., Sparta K., Steffien M., Ühlein M., Wilk P., Weiss M. S., Eur. Phys. J. Plus 2015, 130, 141. [Google Scholar]
- 51. Kabsch W., Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Emsley P., Lohkamp B., Scott W. G., Cowtan K., Acta Crystallogr. Sect. D 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kovalevskiy O., Nicholls R. A., Long F., Carlon A., Murshudov G. N., Acta Crystallogr. Sect. Struct. Biol. 2018, 74, 215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moriarty N. W., Grosse-Kunstleve R. W., Adams P. D., Acta Crystallogr. Sect. D 2009, 65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.