

Abstract
Triple‐negative breast cancer (TNBC) is the most aggressive breast cancer subtype and lacks specific targeted therapeutic agents. The current mechanistic evidence from cell‐based studies suggests that the matricellular protein SPARC has a tumor‐promoting role in TNBC; however, data on the clinical relevance of SPARC expression/secretion by tumor and stromal cells in TNBC are limited. Here, we analyzed by immunohistochemistry the prognostic value of tumor and stromal cell SPARC expression in 148 patients with non‐metastatic TNBC and long follow‐up (median: 5.4 years). We also quantified PD‐L1 and PD‐1 expression. We detected SPARC expression in tumor cells (42.4%), cancer‐associated fibroblasts (CAFs; 88.1%), tumor‐associated macrophages (77.1%), endothelial cells (75.2%) and tumor‐infiltrating lymphocytes (9.8%). Recurrence‐free survival was significantly lower in patients with SPARC‐expressing CAFs. Multivariate analysis showed that SPARC expression in CAFs was an independent prognostic factor. We also detected tumor and stromal cell SPARC expression in TNBC cytosols, and in patient‐derived xenografts and cell lines. Furthermore, we analyzed publicly available single‐cell mRNA sequencing data and found that in TNBC, SPARC is expressed by different CAF subpopulations, including myofibroblasts and inflammatory fibroblasts that are involved in tumor‐related processes. We then showed that fibroblast‐secreted SPARC had a tumor‐promoting role by inhibiting TNBC cell adhesion and stimulating their motility and invasiveness. Overall, our study demonstrates that SPARC expression in CAFs is an independent prognostic marker of poor outcome in TNBC. Patients with SPARC‐expressing CAFs could be eligible for anti‐SPARC targeted therapy.
Keywords: CAF, osteonectin, single‐cell mRNA sequencing, SPARC, TNBC
What's new?
In vitro evidence suggests that the matricellular protein SPARC has a tumor‐promoting role in triple‐negative breast cancer (TNBC). However, the clinical relevance of SPARC in triple‐negative breast cancer remains unclear. Here, the authors analyzed the prognostic value of tumor and stromal SPARC in 148 patients with non‐metastatic TNBC. SPARC was most often expressed by myofibroblasts and inflammatory cancer‐associated fibroblasts (CAFs), and fibroblast‐secreted SPARC exhibited a tumor‐promoting role in TNBC. Moreover, SPARC expression in CAFs was an independent prognostic marker of poor outcome. This study points to CAF‐derived SPARC as a potential novel therapeutic target in triple‐negative breast cancer.
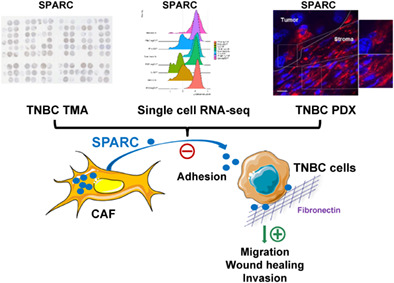
1. INTRODUCTION
Triple‐negative breast cancers (TNBC) are defined by the lack of estrogen receptor (ER), progesterone receptor (PR) and HER2 expression/amplification. TNBC represent 15% of all breast cancers. 1 Despite surgery, adjuvant chemotherapy and radiotherapy, TNBC prognosis is poor, mainly due to the disease heterogeneity and lack of specific therapeutic targets. TNBC is characterized by its unique tumor microenvironment that differs from that of other breast cancer subtypes and promotes cancer cell proliferation, angiogenesis and drug resistance, while inhibiting apoptosis and tumor immune suppression. 2 TNBC microenvironment components, such as transformed extracellular matrix, soluble factors, immune cells and re‐programmed fibroblasts, hamper the host antitumor response and helps tumor progression and metastasis formation. In TNBC, stroma heterogeneity remains poorly understood, thus limiting the development of stromal cell‐targeted therapies.
In the tumor microenvironment, heterogeneous populations of fibroblast‐like cells, collectively termed cancer‐associated fibroblasts (CAFs), are key players in the multicellular, stroma‐dependent alterations that contribute to cancer initiation and progression. 3 However, not all CAFs are tumor supportive. 4 For instance, normal fibroblasts have been shown to suppress tumor formation. 5 In breast cancer, CAF abundance has been associated with aggressive adenocarcinomas and predicts disease recurrence. 6 , 7 In TNBC, recent single‐cell RNA sequencing (scRNA‐seq) studies highlighted a considerable CAF heterogeneity. The CAF subpopulations that contribute to immune suppression, inflammation and chemoresistance are now increasingly better characterized. 8 , 9 , 10 , 11 In breast cancer, tumor‐associated macrophages (TAMs) are the most abundant inflammatory cells, and are typically M2‐polarized cells with suppressive capacity 12 linked to their enzymatic activities and anti‐inflammatory cytokine production. 13 TAMs support tumor progression and metastasis formation by blocking the anti‐tumor immunity and by secreting factors that promote angiogenesis and epithelial‐to‐mesenchymal transition. 12 High M2‐polarized TAM levels are associated with poorer TNBC outcome. 14 Tumor‐infiltrating lymphocytes (TILs) constitutes a robust and independent prognostic marker in TNBC treated with (neo)adjuvant chemotherapy. 15 , 16 TILs are associated with improved disease‐free and overall survival (OS) rates in TNBC. 17 Programmed cell death (PD‐1) (a CD‐28‐CTLA‐4 family member) is an immune check‐point receptor expressed by immune cells that contributes to the immune tolerance of self‐antigens by peripheral T cells. PD‐L1 (one of its ligand) is expressed by immune cells, epithelial breast cancer cells and TILs. Activation of the PD‐1‐PD‐L1 pathway specifically inhibits T‐cell activation, and is one of the mechanisms that allow cancer cells to escape the antitumor immune response. 18 It is thought that TNBC are more immunogenic than other breast cancers. Indeed, the available evidence indicates that in TNBC, PD‐L1 expression is more frequent (up to 60%) than in other breast cancers, and that PD‐L1 tumor expression is positively associated with stromal TILs. 19
The matricellular protein Secreted Protein Acidic and Rich in Cysteine (SPARC; also known as osteonectin or basement membrane 40, BM40) is a Ca2+‐binding glycoprotein that regulates extracellular matrix assembly and deposition, growth factor signaling and cell‐stroma interactions. 20 , 21 , 22 , 23 In cancer, SPARC is mainly secreted by neighboring stromal cells, and to a lower extent by tumor cells. 24 , 25 , 26 SPARC plays oncogenic or tumor‐suppressive roles, depending on the cancer type. 27 , 28 In breast cancer, SPARC has been associated with worse prognosis and has pro‐tumor functions. 25 , 29 , 30 , 31 , 32 , 33 , 34 In TNBC cells, SPARC stimulates their migration and invasion, 35 and promotes MMP‐2 activation, thereby contributing to the proteolytic cascades associated with tumor invasion. 36 Moreover, SPARC stimulates tumor growth and lung colonization after grafting of mouse 4T1 and LM3 TNBC cells in syngeneic mice by promoting cell cycling and expansion of myeloid‐derived suppressor cells. 33 Conversely, SPARC transfection in high‐grade isogenic breast cancer cells reduces tumor rate, and favors epithelial‐to‐mesenchymal transition and the formation of a highly immunosuppressive microenvironment composed of immune cells, such as myeloid‐derived suppressor cells. 31 Some in vitro studies in which SPARC was overexpressed or silenced in cancer cells showed its inhibitory effect on cancer cell motility, invasion and proliferation. 37 , 38 , 39 In TNBC, mechanistic cell‐based studies support a tumor‐promoting role, 40 suggesting that SPARC could be a candidate stromal therapeutic target.
The aim of this study was to evaluate SPARC expression in tumor and stromal cells, their prognostic value, and correlation with fibrosis, TAM infiltration, TIL density, PD‐L1 and PD‐1 levels in a large series of patients with non‐metastatic TNBC. The objective was to identify a TNBC subgroup with worse prognosis and eligible for stroma‐targeted therapy focused on extracellular matrix proteins.
2. MATERIALS AND METHODS
2.1. Antibodies and reagents
The rabbit polyclonal anti‐SPARC (15274‐1‐AP) and the mouse monoclonal anti‐periostin (clone No 1A11A3) antibodies were purchased from Proteintech. The mouse monoclonal anti‐SPARC (clone AON‐5031, sc‐73 472) and the mouse monoclonal anti‐HSC70 (clone B‐6, sc‐7298) antibodies were purchased from Santa Cruz Biotechnology. The mouse monoclonal anti‐tubulin antibody (clone 236‐10 501, #A11126) was from Thermo Fisher Scientific. The mouse monoclonal anti‐cytokeratin 5/6 antibody (clone 6D5/16 B4) was from Dako. The mouse monoclonal anti‐epidermal growth factor receptor (EGFR) antibody (clone 31G7) was from inVitroGen. The mouse monoclonal anti‐PD‐1 (clone MRQ‐22), and the mouse monoclonal anti‐CD163 (clone 10D6) antibodies were from BioSB. The rabbit monoclonal anti‐PD‐L1 (clone SP142) was from Roche. The horse anti‐mouse immunoglobulin G (IgG)‐horseradish peroxidase (#7076), and goat anti‐rabbit IgG‐HRP (#7074 S) secondary antibodies were from Cell Signaling Technology. The donkey anti‐goat HRP conjugated antibody (FT‐1I7890) was from Interchim. The Alexa Fluor 488‐conjugated anti‐rabbit IgG (#Ab150077) was purchased from Abcam, and the Alexa Fluor 594‐conjugated anti‐mouse IgG (711‐585‐152) from ImmunoResearch Laboratories. Hoechst 33342 (#FP‐BB1340) was from Interchim FluoProbes.
2.2. Patients and tumor samples
TNBC tissue micro‐arrays (TMAs) included tissue samples from 148 patients with unifocal, unilateral, non‐metastatic TNBC who underwent surgery at Montpellier Cancer Institute between 2002 and 2012. All patients were informed before surgery that their surgical specimens may be used for research purposes. Patients did not receive neoadjuvant chemotherapy before surgery. ER and PR negativity were defined as <10% expression by immunohistochemistry (IHC), and HER2 negativity was defined as IHC 0/1+ or 2+ and negative by fluorescent/chromogenic hybridization in situ. The study approval for patient‐derived xenografts (PDXs) was previously published. 41
2.3. Construction of TNBC TMAs
Tumor tissue blocks with enough material at gross inspection were selected from the Biological Resource Center. The presence of tumor tissue in sections was evaluated by a pathologist after hematoxylin‐eosin‐saffron (HES) staining of few sections. Two representative tumor areas were identified on each slide from which two malignant cores (1 mm in diameter) were extracted with a manual arraying instrument (Manual Tissue Arrayer 1, Beecher Instruments, Sun Prairie, WI, USA). After arraying completion, 4 μm sections were cut from the TMA blocks. One section was stained with HES and the others were used for IHC.
2.4. TMA IHC
TMA sections were incubated with antibodies against SPARC (clone AON‐5031), cytokeratin 5/6 (clone 6D5/16 B4), EGFR (clone 31G7), PD‐1 (clone MRQ‐22), PD‐L1 (clone SP142) and CD163 (clone 10D6) on a Autostainer Link48 platform (Dako) using the EnVision FLEX system (Dako) for signal amplification and diaminobenzidine tetrahydrochloride as chromogen. TMA sections were analyzed independently by two trained observers both blinded to the clinicopathological characteristics and patient outcomes. In case of disagreement, sections were revised by a third observer to reach a consensus. Results from duplicate cores, when available, were averaged. Basal‐like phenotype was defined by cytokeratin 5/6 and/or EGFR expression (>10% of tumor cells). SPARC signal in cancer cells was scored as negative (<1% of stained cells), or positive (≥ 1% of stained cells). SPARC signal in CAFs, TAMs, endothelial cells and TILs was scored as negative (<50% of stained cells), or positive (≥50% of stained cells). SPARC signal in normal epithelial breast tissue samples (N) was compared with the paired tumor sample (T) and scored as lower (N < T), equal (=), or higher (N ≥ T). TIL density (peritumoral and intratumoral) was evaluated on HE‐stained sections, and was scored as: 0 (no TILs), 1 (rare TILs), 2 (moderate infiltrate, fewer TILs than tumor cells) and 3 (diffuse infiltrate, more TILs than tumor cells). Fibrosis was evaluated on HE‐stained sections, and was scored as: 0 (no CAF), >20%, 20%‐50%, >50% of fibrosis. PD‐1 expression by TILs was scored as follows: not evaluable (no TILs), 0 (no stained TIL), 1 (<10% of stained TILs), 2 (10‐50% of stained TILs) and 3 (>50% of stained TILs). PD‐L1 expression in tumor cells was considered positive if detected in ≥1% of cells. TAM density was scored in CD163‐stained sections and compared with the TIL density: 0 (no TAM), 1 (rare TAMs), 2 (moderate infiltrate, fewer TAMs than TILs) and 3 (diffuse infiltrate, more TAMs than TILs).
2.5. Immunofluorescence analysis
Paraffin‐embedded PDX tissue sections were deparaffined, rehydrated, rinsed and saturated in PBS with 5% fetal calf serum (FCS) at 4°C overnight. Sections were incubated with 1.2 μg/ml anti‐SPARC rabbit polyclonal antibody (15274‐1‐AP) and 5 μg/ml anti‐periostin mouse monoclonal antibody (1A11A3), followed by incubation with AlexaFluor 488‐conjugated anti‐rabbit IgG and AlexaFluor 594‐conjugated anti‐mouse IgG (1/400), respectively. Nuclei were stained with 0.5 μg/ml Hoechst 33342. Sections were imaged with a 63× Plan‐Apochromat objective on z stacks with a Zeiss Axio Imager light microscope equipped with Apotome to eliminate out‐of‐focus fluorescence.
2.6. TNBC cytosols, cell lines, conditioned medium and western blotting
TNBC cytosols were previously prepared and frozen. 42 The MDA‐MB‐453 (RRID:CVCL_0418), MDA‐MB‐436 (RRID:CVCL_0623), MDA‐MB‐468 (RRID:CVCL_0419), Hs578T (RRID:CVCL_0332), BT‐549 (RRID:CVCL_1092) and HCC1806 (RRID:CVCL_1258) TNBC cell lines were obtained from SIRIC Montpellier Cancer. The SUM159 (RRID:CVCL_5423) TNBC cell line was from Asterand (Bioscience, UK). The MDA‐MB‐231 (RRID:CVCL_0062), TNBC cell line was previously described. 43 Human mammary fibroblasts (HMFs) were provided by J. Loncarek and J. Piette (CRCL Val d'Aurelle‐Paul Lamarque, Montpellier, France), 44 THP1 (RRID:CVCL_0006) monocytes by L. Gros (IRCM, Montpellier), and primary human umbilical vein endothelial cells (HUVECs) by M. Villalba (IRMB, Montpellier). Cell lines were cultured in DMEM with 10% FCS (EuroBio), except the SUM159 cell line (RPMI with 10% FCS) and the THP1 cell line (RPMI with 10% decomplemented FCS, 10 mM HEPES, 1 mM sodium pyruvate and 50 μM β‐mercaptoethanol). THP1 monocytes were differentiated into M0 macrophages by exposure to phorbol 12‐myristate 13‐acetate (100 ng/ml; Sigma Aldrich) for 48 h. Then, cells became adherent and the medium was replaced with fresh medium supplemented with interleukin‐4 (20 ng/ml) for 24 h to induce differentiation of M0 macrophages to M2‐polarized macrophages. The M2‐polarized THP1 phenotype was validated by analyzing CD206 expression by RT‐qPCR (Supplementary Materials and Methods). All experiments were performed with mycoplasma‐free cells. All cell lines were authenticated by short tandem repeat profiling within the last 3 years of their use. For western blotting experiments, cell lysates were prepared in lysis buffer (50 mM HEPES [pH 7.5], 150 mM NaCl, 10% glycerol, 1% Triton X‐100, 1.5 mM MgCl2, 1 mM EGTA) containing cOmplete Protease Inhibitor Cocktail (Roche, Switzerland), and centrifuged at 13000×g for 10 min. The corresponding conditioned media were centrifuged at 500×g for 5 min. Proteins from whole cytosols (20 μg) or cell lysates (30 μg) and conditioned media (40 μl) were separated on 13.5% SDS‐PAGE and analyzed by immunoblotting with the anti‐SPARC (clone AON‐5031) and anti‐tubulin antibodies using standard techniques. To prepare conditioned medium, HMFs were grown to 90% confluence in DMEM complemented with 10% FCS. Following washes with phenol red‐ and serum‐free medium to remove serum proteins, cells were incubated in DMEM buffered with 50 mM HEPES [pH 7.5] and without FCS for 24 h. Medium was harvested, and centrifuged at 1000 rpm for 5 min, followed or not by SPARC depletion. Briefly, HMF conditioned medium was incubated with 5 μg of monoclonal anti‐human SPARC antibody (clone AON‐5031, sc‐73 472) overnight, and pre‐absorbed to protein G‐agarose at 4°C. Then conditioned medium (SPARC‐immunodepleted or not) was filtered using 0.22 μm filters to eliminate cell debris. Cleared HMF conditioned medium (HFM CM) was collected and added to MDA‐MB‐231 cells for in vitro functional assays. SPARC immunodepletion was confirmed by western blotting.
2.7. ScRNA‐seq data meta‐analysis
To evaluate SPARC expression in different cell subtypes, previously published scRNA‐seq data were used. The first study included five patients with TNBC, 9 the second included six patients with TNBC, 10 and the third included eight patients with luminal and TNBC tumors. 11 Aligned 10× Genomics (Pleasanton, CA, USA) NGS data, obtained from the public archives (European Nucleotide Archive accession code PRJEB35405, Gene Expression Omnibus database accession code GSE118390 and European Genome‐Phenome Archive accession number AS00001004031), were loaded in R (4.0) and processed using the Seurat 3.4 package and default parameters. 45 Individual cell populations were annotated as published in the original scRNA‐seq study 9 , 10 , 11 with minor modifications when appropriate. To take into account CAF heterogeneity in the study by Karaayvaz et al, 10 the clearly different CAF populations, which were merged in this previous analysis, were named CAF‐A, CAF‐B and CAF‐C.
2.8. Cell adhesion, migration and invasion assays
MDA‐MB‐231 cell adhesion was assessed as previously described. 40 Briefly, 96‐well plates were coated with fibronectin (10 μg/ml; sc‐29 011; Santa Cruz Biotechnology) at 4°C overnight, and saturated with 1% bovine serum albumin (BSA) in PBS. MDA‐MB‐231 cells were detached with HyQTase (HyClone), washed in DMEM without FCS, and 5 10 4 cells were then plated and incubated in serum‐free HMF CM (SPARC‐immunodepleted or not) at 37°C for 30 min. Non‐adherent cells were removed by flotation on a dense Percoll solution containing 3.33% NaCl (1.10 g/L), and adherent cells were fixed (10% [vol/vol] glutaraldehyde) using the buoyancy method. 46 Cells were stained with 0.1% crystal violet, and absorbance was measured at 570 nm. For migration and invasion assays, 8‐μm pore Transwell inserts (polyvinyl pyrrolidone‐free polycarbonate filters) in 24‐well plates (Corning Inc., Corning, NY) were coated with 10 μg/ml fibronectin (500 ng) (migration assays) or Matrigel (100 μg, Corning) (invasion assays) at 4°C for 24 h. MDA‐MB‐231 cells were plated (5 × 104 cells/well) in serum‐free HMF CM (SPARC‐immunodepleted or not) on the coated insert in the upper chamber. In these different assays, DMEM supplemented with 10% FCS was used as chemoattractant in the bottom chamber. After 16 h, non‐migrating/non‐invading cells on the apical side of each insert were scraped off with a cotton swab, and migration and invasion were analyzed with two methods: (1) migrating/invading cells were fixed in methanol, stained with 0.1% crystal violet for 30 min, rinsed in water, and imaged with an optical microscope. Two images of the pre‐set field per insert were captured (×100); (2) migrating/invading cells were incubated with 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT; 5 mg/ml, 1/10 volume; Sigma‐Aldrich) added to the culture medium at 37°C for 4 h. Then, the culture medium/MTT solution was removed and centrifuged at 10000 rpm for 5 min. After centrifugation, cell pellets were suspended in DMSO. Concomitantly, 300 μl of DMSO was added to each well and thoroughly mixed for 5 min. The optical density values of stained cells (cell pellet and corresponding well) were measured using a microplate reader at 570 nm.
2.9. Wound healing assay by live cell imaging
Before each experiment, MDA‐MB‐231 cells were grown to confluence in 96‐well plates in a standard CO2 incubator. The 96‐pin IncuCyte WoundMaker was used to simultaneously create precise and reproducible wounds by gently removing cells from the confluent monolayer. After washing, serum‐free HMF CM (SPARC‐immunodepleted or not) was added, plates were placed in the IncuCyte device and cell monolayers were scanned every hour. Wound width, wound confluence and relative wound density were calculated using user‐informed algorithms that are part of the IncuCyte software package. These algorithms identify the wound region and provide visual representations of the segmentation parameters.
2.10. Tumor spheroids
To generate tumor spheroids, 5 × 103 MDA‐MB‐231 cells/well were seeded in 150 μl complete medium in ultra‐low attachment 96‐well plates (Corning 96‐well Clear Round Bottom Ultra‐Low Attachment Microplate, NY). Plates were centrifuged at 1000 rpm for 10 min, and 3 days later each spheroid was embedded in collagen gel that included 1× DMEM, penicillin and streptomycin, 2% of SPARC‐immunodepleted FCS, 3.75 g/L sodium bicarbonate, 20 mM Hepes, 1 mg/ml rat collagen I and 1.5 mM NaOH (qsp 150 μl/well in H2O). After 30 min at 37°C, serum‐free HMF CM (SPARC‐immunodepleted or not) was added on the spheroid‐containing polymerized collagen gel. MDA‐MB‐231 cell invasion area was analyzed in representative images with ImageJ.
2.11. Statistical analyses
Continuous variables were reported using medians and range and compared using the Kruskal‐Wallis test. Categorical variables were reported as numbers of observations and frequencies, and compared using the Pearson's chi‐square test or Fisher's exact test (if appropriate). All tests were two‐sided and P values <.05 were considered significant. Relapse‐free survival (RFS) and OS were estimated using the Kaplan‐Meier method and compared with the Log‐rank test. RFS was defined as the time between the date of the first histology analysis and the date of the first recurrence at any site. OS was defined as the time between the date of the first histology analysis and the date of death from any cause. Multivariate analyses were performed using Cox proportional hazard models (the P value of the likelihood ratio test is reported). Hazard ratios (HR) are given with their 95% confidence interval (CI). All statistical analyses were performed with the STATA 16.0 software (StatCorp, College Station, TX).
3. RESULTS
3.1. In TNBC, SPARC is expressed in stromal and tumor cells
To determine SPARC expression in TNBC (tumor and stroma), TMAs were generated using samples from 148 patients with TNBC (Table 1). Their median age was 61.5 years (range 30.2‐98.6), and 68.2% of them received adjuvant chemotherapy. Most TNBC (52.7%) were pT2, and 60.8% pN0. Moreover, 85.5% of tumors were ductal carcinomas, 6.9% lobular carcinomas, and 7.6% other histological types; 11% of tumors were classified as Scarff‐Bloom‐Richardson histological grade 1‐2. A basal‐like phenotype was observed in 61.9% of samples, and 66.9% of tumors expressed PD‐L1. In 51.7% of tumors, TAMs were more abundant than TILs, and > 20% of fibrosis was observed in 74.4% of tumors. SPARC expression (>50% of stained cells) in CAFs, TAMs, endothelial cells and TILs was detected in 88.1%, 77.1%, 75.2% and 9.8% of TNBC samples, respectively (Figure 1A, B and Table 1). SPARC staining in tumor cells (>1% stained tumor cells) was observed in 42.4% of TNBC samples (Figure 1A, Table 1). In 80% of samples, SPARC expression was lower in the adjacent normal breast tissue than in the tumor tissue (Figure 1A, C).
TABLE 1.
Clinicopathological characteristics of the whole TNBC population and SPARC expression status in cancer and stromal cells
| Clinical and tumor characteristics | Whole population (N = 148) | Clinical and tumor characteristics | Whole population (N = 148) |
|---|---|---|---|
| SPARC expression in TAMs | |||
| Age (years), median [min‐max] | 61.5 [30.2‐98.6] | Negative | 27 (22.9%) |
| <55 years | 51 (34.5%) | Positive | 91 (77.1%) |
| ≥55 years | 97 (65.5%) | Missing | 30 |
| Tumor size | SPARC expression in endothelial cells | ||
| T1 | 52 (35.1%) | Negative | 27 (24.8%) |
| T2 | 78 (52.7%) | Positive | 82 (75.2%) |
| T3/T4 | 18 (12.2%) | Missing | 39 |
| Nodal status | SPARC expression in TILs | ||
| N− | 90 (60.8%) | Negative | 74 (90.2%) |
| N+ | 58 (39.2%) | Positive | 8 (9.8%) |
| Missing | 66 | ||
| Histological grade (SBR) | TIL density | ||
| 1‐2 | 16 (11.0%) | [0‐1] | 42 (29.6%) |
| 3 | 130 (89.0%) | >1 | 100 (70.4%) |
| Missing | 2 | Missing | 6 |
| Histology | PD‐L1 expression in tumor cells | ||
| Ductal | 124 (85.5%) | <1% | 45 (33.1%) |
| Lobular | 10 (6.9%) | ≥1% | 91 (66.9%) |
| Other | 11 (7.6%) | Missing | 12 |
| Missing | 3 | ||
| Adjuvant chemotherapy | PD‐L1 expression in TILs | ||
| No | 47 (31.8%) | 0 | 20 (14.9%) |
| Yes | 101 (68.2%) | [0‐10] | 32 (23.9%) |
| [10‐50] | 40 (29.9%) | ||
| ≥ 50 | 42 (31.3%) | ||
| Missing | 14 | ||
| Basal‐like phenotype | PD1 expression in TILs | ||
| ≤10% | 56 (38.1%) | 0 | 18 (12.9%) |
| Basal | 91 (61.9%) | <10 | 30 (21.3%) |
| Missing | 1 | [10‐50] | 74 (52.9%) |
| ≥50 | 18 (12.9%) | ||
| Missing | 8 | ||
| SPARC expression in tumor cells | Fibrosis | ||
| Negative | 76 (57.6%) | 0 | 4 (3.0%) |
| Positive | 56 (42.4%) | < 20% | 31 (22.6%) |
| Missing | 16 | 20%‐50% | 27 (19.7%) |
| >50% | 75 (54.7%) | ||
| Missing | 11 | ||
| SPARC expression in CAFs | TAMs (inflammation) | ||
| Negative | 15 (11.9%) | 0/1 | 25 (17.5%) |
| Positive | 111 (88.1%) | 2 | 44 (30.8%) |
| Missing | 22 | 3 | 74 (51.7%) |
| Missing | 5 |
Abbreviations: CAFs, cancer‐associated fibroblasts; SBR, Scarff‐Bloom‐Richardson; TAMs, tumor‐associated macrophages; TILs, tumor‐infiltrating lymphocytes.
FIGURE 1.
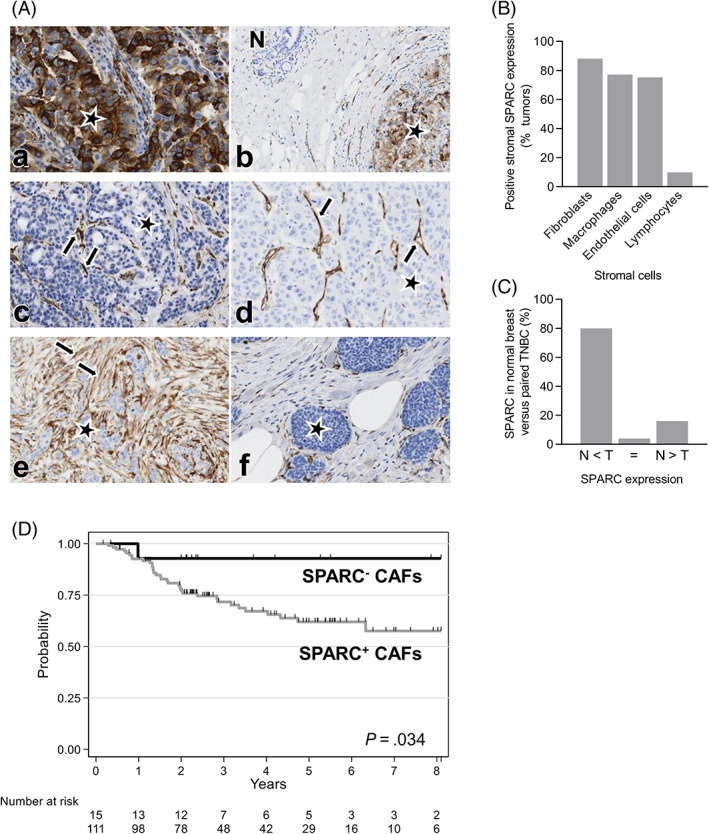
SPARC is a biomarker in TNBC and its expression in CAFs predicts RFS in TNBC. (A) Representative images of TNBC tissue sections showing SPARC expression in cancer cells, CAFs, TAMs, endothelial cells, and in normal breast. SPARC expression was analyzed in a TNBC TMA (n = 148 samples) by IHC using an anti‐SPARC antibody (clone AON‐5031). (a) SPARC expression in tumor cells. (b) Absence of SPARC expression in the adjacent normal breast tissue (N). (c) SPARC expression in TAMs. (d) SPARC expression in endothelial cells. (e) SPARC expression in CAFs. (f) Absence of SPARC expression in CAFs. SPARC scoring in cancer cells: positive (>1% of stained cells), negative (<1% of stained cells). SPARC scoring in stromal cells: positive (>50% of stained cells), negative (<50% of stained cells). Magnification ×200. Stars: tumor cells; arrows: SPARC staining. (B) Quantification of SPARC expression in TNBC stroma. Percentage of TNBC samples with positive SPARC signal (>50% of stained cells) in the indicated stromal cell types. N = 148 samples. (C) Quantification of SPARC expression in normal breast. Percentage of normal breast tissue samples in which SPARC expression was lower (N < T), similar (=) or higher (N > T) than in the adjacent TNBC. T, tumor; N, normal breast; n = 50 samples. (D) Relapse‐free survival according to SPARC expression status in CAFs. Patients with TNBC were divided in two subgroups according to SPARC expression in CAFs: SPARC+ CAFs and SPARC− CAFs
3.2. SPARC expression in CAFs predicts RFS in patients with TNBC
As SPARC was expressed in the tumor and stromal compartments, its prognostic value was then evaluated. The median follow‐up time was 5.4 years (range [0.1‐14.3]). Local or regional recurrence occurred in 10 (7%) patients, and metastases (alone or with loco‐regional recurrence) in 32 (22.5%) patients. RFS was not different in patients with SPARC‐positive (SPARC+) and SPARC‐negative (SPARC−) tumor cells (Table 2 and Figure S1). Conversely, RFS was lower in patients with SPARC+ than SPARC− CAFs (HR = 5.09, 95% CI [0.70‐37.18], P = .034) (Table 2 and Figure 1D). Moreover, RFS tended to be better in patients with SPARC+ than SPARC− TAMs (HR = 0.52, 95% CI [0.25‐1.07], P = .088) (Table 2 and Figure S2). SPARC expression status in endothelial cells (Figure S3) and TILs (Figure S4) did not have any prognostic value (Table 2). In univariate analysis, tumor size, nodal status, adjuvant chemotherapy and SPARC expression in CAFs were correlated with RFS (Table 2). In multivariate analysis, only nodal status (HR = 2.96, 95% CI [1.48‐5.94], P = .001), adjuvant chemotherapy (HR = 0.35, 95% CI [0.18‐0.68], P = .002) and SPARC expression in CAFs (HR = 6.17, 95% CI [0.84‐45.2], P = .015) were independent prognostic factors of RFS (Table 2). During the follow‐up, 46 (31.1%) patients died among whom 11 (7.4%) without any TNBC recurrence. In univariate analysis, age (P = .027), tumor size (P < .001), nodal status (P = .002) and adjuvant chemotherapy (P = .006) were associated with OS (Table S1). In multivariate analysis, only tumor size (P = .05), nodal status (P = .008) and adjuvant chemotherapy (P < .001) were independent prognostic factors of OS (Table S1). Patients with SPARC+ CAFs (n = 111, 88.1%) were younger (38.7% vs 6.7%; P = .018) and tended to have ductal tumors (88.0% vs 73.3%; P = .08) compared with patients with SPARC− CAFs (Table S2). In addition, SPARC+ TAMs and SPARC+ endothelial cells were detected more frequently in patients with SPARC+ than SPARC− CAFs (80.6% vs 41.7%, P = .007, and 78.0% vs 50%, P = .026, respectively) (Table S2). Fibrosis (>50%) was significantly less frequent in patients with SPARC+ than SPARC− CAFs (48.6% vs 80%; P = .028) (Table S2). PD‐L1 expression (>50%) in TILs was more frequently detected in patients with SPARC+ than SPARC− CAFs (34.8% vs 15.4%; P = .049) (Table S2). TIL density, PD‐L1 expression in tumor cells and PD‐1 expression in TILs were not significantly different between patients with SPARC+ and SPARC− CAFs (Table S2).
TABLE 2.
Univariate and multivariate Cox proportional hazard models to identify prognostic factors of recurrence‐free survival (RFS) in TNBC
| Clinical and tumor characteristics | Univariate analysis | Multivariate analysis |
|---|---|---|
| HR 95% CI | HR 95% CI | |
| N = 148 | N = 126 | |
| Age | N = 148 | |
| <55 years | 1 | |
| ≥55 years | 1.52 [0.77‐3.03] | |
| P = .214 | ||
| Tumor size | N = 148 | |
| T1 | 1 | |
| T2 | 1.67 [0.74‐3.75] | |
| T3/T4 | 5.08 [2.07‐12.47] | |
| P = .002 | ||
| Nodal status | N = 148 | |
| N− | 1 | 1 |
| N+ | 2.77 [1.49‐5.14] | 2.96 [1.48‐5.94] |
| P = .001 | P = .001 | |
| Histological grade (SBR) | N = 146 | |
| 1‐2 | 1 | |
| 3 | 0.82 [0.36‐1.85] | |
| P = .645 | ||
| Histology | N = 145 | |
| Ductal | 1 | |
| Lobular | 1.51 [0.59‐3.86] | |
| Other | 0.77 [0.19‐3.21] | |
| P = .651 | ||
| Adjuvant chemotherapy | N = 148 | |
| No | 1 | 1 |
| Yes | 0.43 [0.24‐0.78] | 0.35 [0.18‐0.68] |
| P = .007 | P = .002 | |
| Basal‐like phenotype | N = 147 | |
| Yes | 1 | |
| No | 1.55 [0.85‐2.83] | |
| P = .152 | ||
| SPARC expression in tumor cells | N = 132 | |
| Negative | 1 | |
| Positive | 0.84 [0.44‐1.62] | |
| P = .599 | ||
| SPARC expression in CAFs | N = 126 | |
| Negative | 1 | 1 |
| Positive | 5.09 [0.70‐37.18] | 6.17 [0.84‐45.2] |
| P = .034 | P = .015 | |
| SPARC expression in TAMs | N = 118 | |
| Negative | 1 | |
| Positive | 0.52 [0.25–1.07] | |
| P = .088 | ||
| SPARC expression in endothelial cells | N = 109 | |
| Negative | 1 | |
| Positive | 0.59 [0.29‐1.21] | |
| P = .165 | ||
| SPARC expression in TILs | N = 82 | |
| Negative | 1 | |
| Positive | 0.81 [0.19‐3.46] | |
| P = .769 | ||
| TIL density | N = 142 | |
| [0‐1] | 1 | |
| >1 | 0.92 [0.48‐1.77] | |
| P = .807 | ||
| PD‐L1 expression in tumor cells | N = 136 | |
| <1% | 1 | |
| ≥1% | 0.74 [0.39‐1.40] | |
| P = .360 | ||
| PD‐L1 expression in TILs | N = 134 | |
| 0 | 1 | |
| [0‐50] | 2.20 [0.66‐7.40] | |
| ≥50 | 2.12 [0.60‐7.52] | |
| P = .356 | ||
| PD1 expression in TILs | N = 140 | |
| 0 | 1 | |
| [0‐50] | 1.28 [0.46‐3.64] | |
| ≥50 | 0.80 [0.20‐3.21] | |
| P = .593 | ||
| Fibrosis | N = 137 | |
| ≤50% | 1 | |
| >50% | 0.98 [0.52‐1.83] | |
| P = .948 | ||
| TAMs (inflammation) | N = 143 | |
| 0/1 | 1 | |
| 2 | 1.97 [0.78‐4.96] | |
| 3 | 1.14 [0.46‐2.86] | |
| P = .180 |
Note: p value in bold, statistically significant.
Abbreviations: CAFs, cancer‐associated fibroblasts; CI, confidence interval; HR, hazard ratio; SBR, Scarff‐Bloom‐Richardson; TAMs, tumor‐associated macrophages; TILs, tumor‐infiltrating lymphocytes.
3.3. SPARC expression in TNBC cytosols, PDX and cell lines
To further validate SPARC expression in TNBC, its expression was assessed in the cytosols of 30 primary TNBC samples by western blot analysis. SPARC protein was detected in all cytosols and SPARC cleaved fragments in about 30% of samples (Figure 2A). SPARC protein expression and localization were then examined in two TNBC PDXs (PDX B1995 and PDX B3977). 41 SPARC was localized in stromal cells, including CAFs, in the extracellular matrix and in some tumor cells (Figure 2B). Next, SPARC expression and secretion were analyzed in TNBC and stromal cell lines. SPARC was expressed and secreted by three of the eight TNBC cell lines tested (SUM159, Hs578T, BT‐549) that exhibit a basal‐like phenotype (Figure 2C). SPARC was also expressed and secreted by HMFs, and to a lesser extent by HUVECs and M2‐polarized THP1 macrophages (Figure 2D and Figure S5).
FIGURE 2.
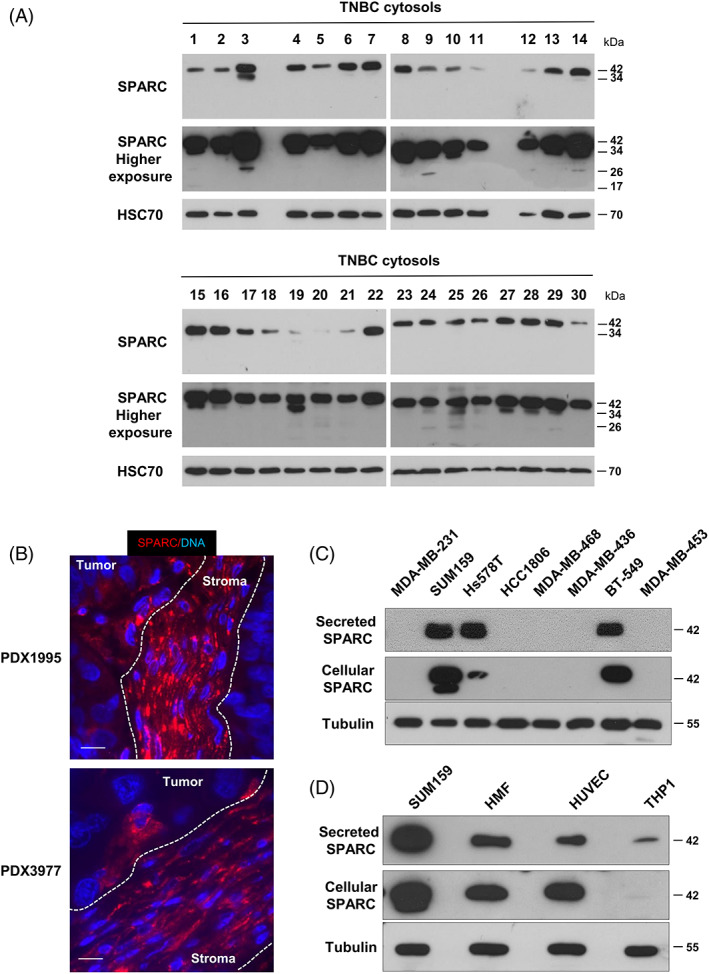
SPARC expression in TNBC cytosols, PDX, and cell lines. (A) SPARC expression in TNBC cytosols. SPARC expression was determined in 30 cytosols from primary TNBC samples. Whole cytosols (20 μg proteins) were analyzed by 13.5% SDS‐PAGE and immunoblotting with an anti‐SPARC antibody (clone AON‐5031). A higher exposure of SPARC is shown. HSC70 (clone B‐6) was used as loading control. (B) SPARC expression and localization in TNBC PDX. PDX B1995 and PDX B3977 sections were incubated with an anti‐SPARC polyclonal antibody (15274‐1‐AP) (red). Nuclei were stained with Hoechst 33342 (blue). Scale bar, 10 μm. (C) SPARC expression and secretion in TNBC cell lines. Whole cell extracts (30 μg proteins) and serum‐free 24 h conditioned media (40 μl) from the indicated TNBC cell lines were separated on 13.5% SDS‐PAGE and analyzed by immunoblotting with an anti‐SPARC (clone AON‐5031) antibody. Tubulin was used as loading control. (D) SPARC expression and secretion in stromal cell lines. Whole cell extracts (30 μg proteins) and serum‐free 24 h conditioned media (40 μl) from the indicated cell lines were separated on 13.5% SDS‐PAGE and analyzed by immunoblotting with an anti‐SPARC (clone AON‐5031) antibody. Tubulin was used as loading control
3.4. SPARC is expressed in different CAF subsets
Based on the finding that SPARC expression in CAFs predicts RFS in TNBC, SPARC expression in different CAF subpopulations was thoroughly investigated through meta‐analysis of recently published scRNA‐seq data from patients with TNBC. 9 , 10 , 11 In the first dataset (n = 5 patients with TNBC), 9 the t‐distributed Stochastic neighbor embedding (tSNE) technique identified 20 different cell populations, including two fibroblastic cell populations, the first with features of myofibroblasts (myCAFs), and the second with an inflammatory phenotype (iCAFs) characterized by high expression of growth factors and immunomodulatory molecules (Figure 3A). The scRNA‐seq data analysis 9 showed that SPARC mRNA was strongly expressed in myCAFs and iCAFs, as well as POSTN (the gene encoding periostin, a CAF‐secreted protein that promotes cancer progression and chemoresistance) 47 (Figure 3B). SPARC was also detected in perivascular endothelial cells, myoepithelial cells and basal cancer cells 9 (Figure 3B, Figure S6), in accordance with our TMA analysis (Table 1). In the second scRNA‐seq dataset (n = 6 patients with TNBC), 10 high SPARC and POSTN mRNA levels were detected in three distinct CAF subtypes, in endothelial cells, M2‐polarized macrophages and cancer cells (where expression varied in function of the patient) (Figure S7), consistent with our TMA data (Table 1). As these two meta‐analysis indicated that SPARC was expressed in different CAF subtypes, another scRNA‐seq dataset (n = 8 patients with breast cancer) that identified different myCAF and iCAF clusters was analyzed. 11 SPARC and POSTN mRNAs were detected mainly in myCAFs (ECM‐myCAF, TGFβ‐myCAF, Wound‐myCAF, IFNαβ‐myCAF, Acto‐myCAF clusters) and also in iCAFs (IFNγ‐iCAF, IL‐iCAF, detox‐iCAF clusters) (Figure S8). Altogether, this meta‐analysis highlighted that SPARC mRNA is expressed by different CAF subtypes, including myofibroblasts and inflammatory‐like CAFs involved in different tumor‐related processes, such as matrix remodeling, inflammation and resistance to therapy in TNBC. 9 , 11 To complement the scRNA‐seq findings, the localization of SPARC and periostin was investigated in the TNBC PDX B1995 microenvironment. Co‐labeling with anti‐SPARC and anti‐periostin antibodies showed that SPARC (in green) partially co‐localized with periostin (in red) in CAFs at the cancer cell‐stromal interface (Figure S9).
FIGURE 3.
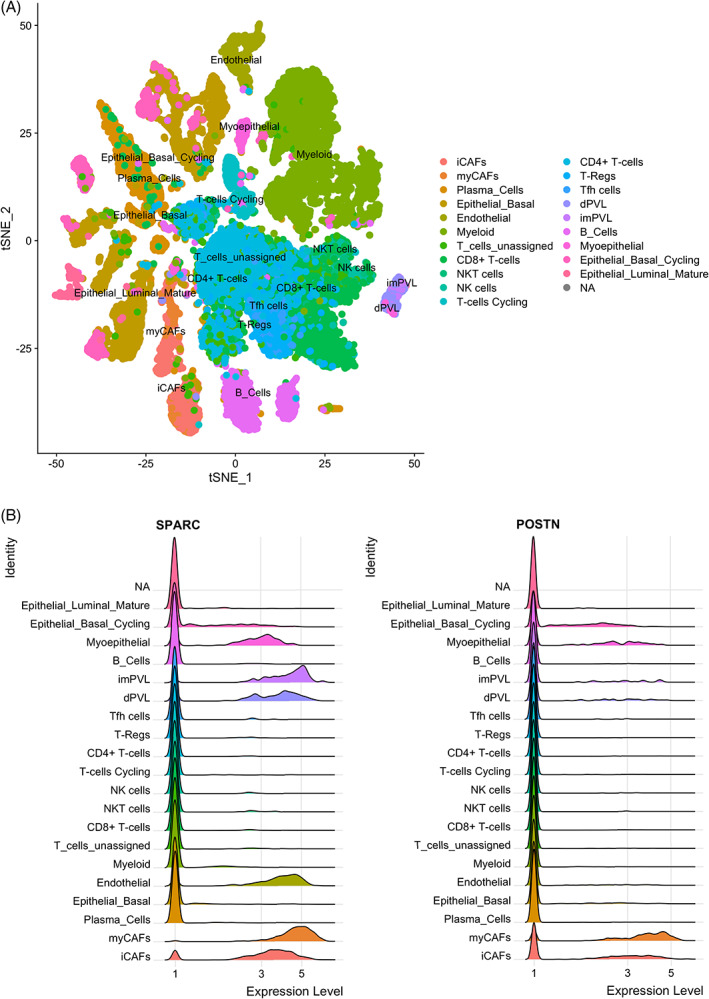
Expression of SPARC and POSTN mRNAs in TNBC by single‐cell RNA‐seq data analysis. (A) Cell populations. Twenty cell populations were identified by analysis of the previously published single‐cell RNA‐seq dataset PRJEB35405 that included five patients with TNBC, according to. 9 (B) SPARC and POSTN mRNA expression. Relative expression of SPARC and POSTN mRNA in each of the 20 populations identified by single‐cell RNA‐seq analysis, according to. 9 MyCAFs, myofibroblast‐like CAFs; iCAFs, inflammatory‐like CAFs; endothelial, endothelial cells; dPVL, differentiated perivascular‐like cells; imPVL, immature perivascular‐like cells; myoepithelial, myoepithelial cells; epithelial basal cycling, cancer cells
3.5. Fibroblast‐secreted SPARC affects TNBC cell adhesion, migration and invasion
To obtain some insights into the pathophysiological relevance of SPARC+ CAFs in TNBC, the effects on TNBC cell adhesion, motility, wound healing and invasiveness of SPARC‐secreting HMF CM were investigated (Figure S10). The adhesion of MDA‐MB‐231 cells on fibronectin was reduced by 1.3‐fold (P < .001) after incubation with HMF CM compared with SPARC‐immunodepleted HMF CM (Figure 4A). Cell motility analysis in Boyden chambers showed that 88% of MDA‐MB‐231 cells passed through the fibronectin‐coated filters after incubation with HMF CM (Figure 4B). Motility was reduced by 2.3‐fold when cells were incubated with SPARC‐immunodepleted CM (Figure 4B; P < .01). Moreover, wound healing was significantly faster in MDA‐MB‐231 cells incubated with HMF CM than with SPARC‐immunodepleted CM: wound closure was nearly complete after 16 h in the presence of HMF CM (Figure 4C). Lastly, MDA‐MB‐231 cell invasion through Matrigel‐coated filters in Boyden chambers was 1.6‐fold higher in the presence of HMF CM than SPARC‐immunodepleted CM (Figure 4D; P < .05). The capacity of HMF‐secreted SPARC to enhance MDA‐MB‐231 cell invasion was confirmed in a tumor spheroid assay (Figure 4E). MDA‐MB‐231 tumor spheroid invasiveness at day 3 was 3.4‐fold higher in the presence of HMF CM than SPARC‐immunodepleted CM (Figure 4E; P < .01). Thus, HMF‐secreted SPARC inhibits adhesion and promotes motility, wound healing and invasion of MDA‐MB‐231 TNBC cells, highlighting its pro‐tumor role.
FIGURE 4.
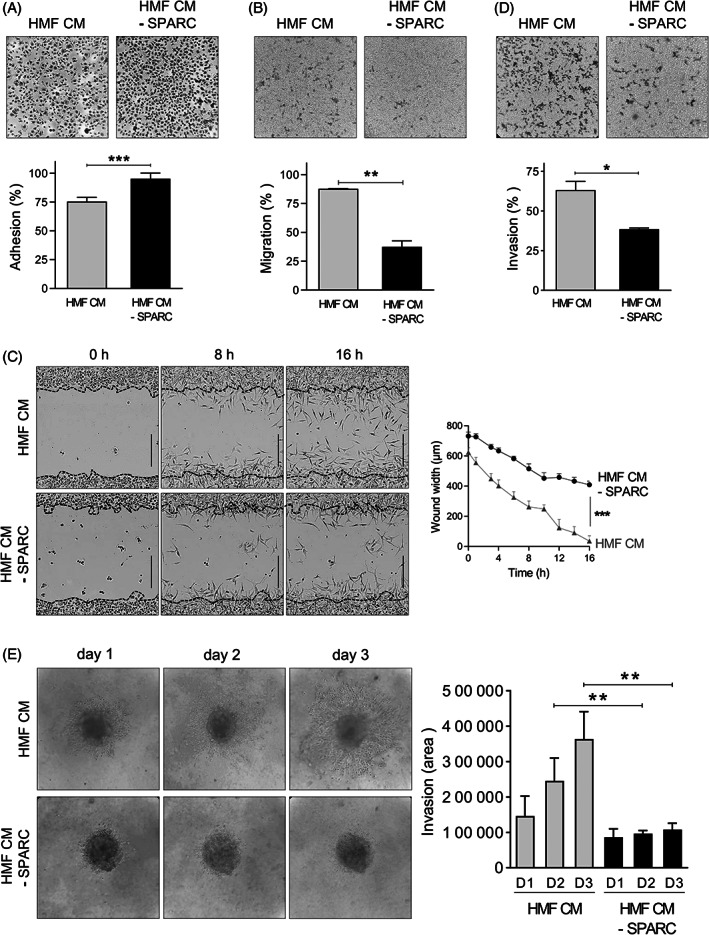
Effects of fibroblast‐secreted SPARC on TNBC cell adhesion, migration and invasion. (A) Cell adhesion. MDA‐MB‐231 cells were let to adhere on a fibronectin matrix in the presence of HMF conditioned medium (HMF CM) or SPARC‐immunodepleted HMF CM (HMF CM—SPARC) for 30 min. Upper panels, representative images of adherent cells stained with crystal violet. Lower panel, adhesion was quantified at 570 nm. Data are the mean (% of seeded cells) ± SD (n = 5); ***P < .001 (Student's t test). Similar results were obtained in three independent experiments. (B) Cell migration. MDA‐MB‐231 cells were let to migrate for 16 h on a fibronectin matrix in the presence of HMF CM or SPARC‐immunodepleted HMF CM (HMF CM ‐ SPARC). Upper panels, representative images of migrating cells stained with crystal violet. Lower panels, quantification of migrating MTT‐stained cells (absorbance was read at 570 nm). Data are the mean (% of seeded cells) ± SD (n = 3); **P < .01 (Student's t test). Similar results were obtained in three independent experiments. (C) Cell migration induced by wound healing. MDA‐MB‐231 sub‐confluent cell layers were wounded using the 96‐well IncuCyte scratch wound assay. Left panels, representative images of MDA‐MB‐231 wound healing over time (t = 0 h, t = 6 h, t = 16 h) in the presence of HMF CM or SPARC‐immunodepleted HMF CM (HMF CM ‐ SPARC). In the left panels, the initial scratch wound is delimited by the dashed lines. Bars, 400 μm. Right panel, wound healing (wound width, in μm) in the presence of HMF CM or SPARC‐immunodepleted HMF CM (HMF CM ‐ SPARC) was quantified over time. The data are the mean ± SD (n = 3); ***P < .001 (Student's t test). Similar results were obtained in another independent experiment. (D) Cell invasion. MDA‐MB‐231 cells were let to invade on a Matrigel matrix in the presence of HMF CM or SPARC‐immunodepleted HMF CM (HMF CM ‐ SPARC) for 16 h. Upper panels, representative images of invading cells stained with crystal violet. Lower panels, invading cells were stained with MTT and quantified at 570 nm. Data are the mean (% of seeded cells) ± SD (n = 3); ***P < .001 (Student's t test). Similar results were obtained in three independent experiments. (E) Cell invasion in tumor spheroid assay. MDA‐MB‐231 tumor spheroids embedded in collagen I gel were let to invade in the presence of HMF CM or SPARC‐immunodepleted HMF CM (HMF CM ‐ SPARC) for 3 days. Left panels, representative images of invading MDA‐MB‐231 cells. Right panel, the invading MDA‐MB‐231 cell area was quantified using Image J. Data are the mean ± SD (n = 5); **P < .01 (Student's t test).
4. DISCUSSION
Here, we showed that in TNBC, SPARC is expressed in both tumor and stromal cells, and that its expression in CAFs independently predicts RFS in patients with TNBC. Previous studies reported that SPARC is overexpressed in TNBC compared with other breast cancer molecular subtypes. 48 , 49 In our study using IHC, SPARC expression in tumor cells was detected in 42% of TNBC samples, in agreement with previous literature data (SPARC expression in 37 to 52% of TNBC). 32 , 48 , 49 However, SPARC expression in TNBC has never been correlated with clinicopathological parameters, such as age, histopathologic grade, tumor size and lymph node metastasis. 32 , 48 Watkins et al reported that in breast cancer, SPARC is detected more frequently in ductal carcinomas. 30 Similarly, we found that ductal carcinoma tended to be more frequent in patients with SPARC+ CAFs, and that patients with TNBC with SPARC+ CAFs were often younger. 50 SPARC (mRNA or protein) overexpression prognostic value is controversial in TNBC. High SPARC expression in TNBC has been associated with poor prognosis in some studies, 32 , 34 , 51 and with better prognosis in another. 48 We recently showed that high SPARC mRNA expression (n = 225 patients with TNBC) tends to be associated with shorter RFS using an on line survival tool. 40 , 52 In our current TNBC population, SPARC expression by tumor cells was not associated with RFS or OS. Studies using IHC reported that SPARC expression in tumor cells was associated with prognosis. 32 , 53 Here, we found that SPARC was mainly expressed by stromal cells, including CAFs, and that its expression in CAFs was an independent prognostic factor of poor RFS in TNBC. In patients with SPARC+ CAFs, TILs more frequently expressed PD‐L1, suggesting the interest to specifically evaluate the benefit of combining anti‐PD1 or ‐PD‐L1 with anti‐SPARC targeted therapies in this TNBC subgroup. Moreover, fibrosis was less frequent in TNBC samples with SPARC+ CAFs, suggesting a better drug accessibility in this TNBC subgroup. 54 Other studies 50 reported a frequent SPARC stromal expression, but none, to our knowledge, evaluated its prognostic value or determined SPARC expression in the different stromal cell types.
Here, we observed the presence of SPARC cleaved fragments in about 30% of TNBC cytosols. The anti‐SPARC antibody (clone AON‐5031) used for IHC recognizes full‐length SPARC and also some SPARC N‐terminal fragments. Therefore, the prognostic value of SPARC expression in CAFs in TNBC described in the present study could be explained by the activity of the full‐length protein and also of some of its cleaved fragments. SPARC includes three different structural and functional modules: the N‐terminal acidic domain, the follistatin‐like domain and the C‐terminal extracellular Ca2+ binding domain. 22 SPARC biological activity can be modulated by limited proteolysis, leading to the unmasking of distinct or amplified biological functions compared with those of the full‐length protein. 21 , 55 Matrix metalloproteinases (MMP‐1, −2, −3, −9 and − 13) cleave SPARC in vitro in its N‐terminal acid domain and in its extracellular Ca2+ binding domain, releasing fragments that have higher affinity for collagens and that modulate cell‐cell and cell‐matrix extracellular interactions in the tumor microenvironment. 56 Moreover, MMP‐3‐mediated SPARC cleavage in vitro produces fragments that affect angiogenesis. 57 Cleavage of SPARC extracellular Ca2+ binding domain by MMP‐8 and MMP‐13 has been detected in the serum of patients with lung cancer, suggesting their presence also in vivo. 58 Similarly, cathepsin K cleaves SPARC in vitro and in vivo in its N‐terminal acid domain and in its extracellular Ca2+ binding domain in mice harboring prostate cancer bone metastases. 59 We recently reported that secreted SPARC is cleaved by cathepsin D in TNBC, releasing a 9‐kDa SPARC fragment with enhanced oncogenic properties. 40
The meta‐analysis of previously published scRNA‐seq datasets 8 , 9 , 10 , 11 showed that SPARC is expressed by different CAF subsets in TNBC. CAFs are the most abundant stromal cells in many cancers, including TNBC, and they are a phenotypically heterogeneous population, generally described as having a myofibroblastic phenotype (ie, secretory and contractile cells that express α‐SMA). Recently, it was found that fibroblast heterogeneity occurs in breast cancers and in TNBC. 8 , 9 , 10 , 11 Two myofibroblastic subsets (CAF‐S1 and CAF‐S4) differentially accumulate in TNBC. 8 CAF‐S1 cells promote an immunosuppressive microenvironment, 8 whereas CAF‐S4 cells have pro‐metastatic function. 60 More recently, a scRNA‐seq approach in breast cancer identified eight clusters within the immunosuppressive CAF‐S1 subset, subdivided in myofibroblast‐like and inflammatory‐like CAFs. 11 Another scRNA‐seq‐based study identified myofibroblast‐like and inflammatory‐like CAFs with immunomodulatory properties in TNBC. 9 By reanalyzing these scRNA‐seq datasets, 9 , 10 , 11 we noticed that SPARC mRNA was expressed by different CAF subsets, especially myofibroblast‐like and inflammatory‐like CAFs, as well as POSTN, a gene encoding periostin, a protein that is secreted by CAFs with pro‐tumor activity in breast cancer. 47 We then confirmed that SPARC and periostin (partially) co‐localize in CAFs within the TNBC PDX microenvironment. Future studies will determine whether SPARC participates in the homeostasis of these different CAF subpopulations in TNBC, and whether SPARC has a different prognostic value when expressed in the different CAF subgroups in TNBC.
In TNBC, CAFs regulate a number of tumor‐promoting processes, including motility and invasion, drug resistance, inflammation and immunosuppression. 8 , 9 , 60 , 61 , 62 Our results showed that SPARC secreted by fibroblasts acts directly on TNBC cells by inhibiting their adhesion and promoting/facilitating their motility and invasiveness. It has been reported that SPARC regulates signaling pathways that influence epithelial‐to‐mesenchymal transition, cell adhesion, motility and invasiveness of cancer cells. 23 , 63 Moreover, SPARC activation of the ERK and AKT downstream signaling pathways modulates cancer cell adhesion, motility and invasion. 64 SPARC can bind directly to integrin receptors (αvβ1, αvβ3 and αvβ5), resulting in activation of the intracellular kinase Akt, the focal adhesion kinase FAK and the integrin‐related kinase ILK. 65 , 66 Future mechanistic studies should decipher the signaling pathways affected by CAF‐secreted SPARC in TNBC cells. All these findings suggest that SPARC may be a therapeutic target in TNBC. Drugs that target CAFs have emerged as an important option for improving cancer therapies, and targeting CAF‐derived extracellular matrix proteins has been proposed as an innovative anti‐stromal therapy. 67 Our work strongly suggests that CAF‐derived SPARC also may be a promising candidate for anti‐stromal therapy.
5. CONCLUSION
In this series, almost 88.1% of TNBC harbored SPARC+ CAFs and displayed distinct clinicopathological characteristics. SPARC expression in CAFs independently predicted worse RFS. This biomarker could be useful to identify a specific TNBC subgroup with worse prognosis. Furthermore, SPARC was expressed by different CAF subpopulations in TNBC, and fibroblast‐secreted SPARC exhibited pro‐tumor functions. Our results could have therapeutic implications for future anti‐SPARC+ CAF targeted therapy.
Abbreviations
- BM40
basement membrane 40
- BSA
bovine serum albumin
- CAFs
cancer‐associated fibroblasts
- Cath‐D
Cathepsin D
- CI
confidence interval
- CM
conditioned medium
- dPVL
differentiated perivascular‐like cells
- ECM
extracellular matrix
- EGFR
epithelial growth factor receptor
- ER
estrogen receptor
- FCS
fetal calf serum
- HES
hematoxylin‐eosin‐safranin
- HMF
human mammary fibroblast
- HR
hazard ratios
- HUVECs
human umbilical vein endothelial cells
- iCAFs
inflammatory‐like CAFs
- IFN
interferon
- IHC
immunohistochemistry
- IL
interleukin
- myCAFs
myofibroblasts‐like CAFs
- OS
overall survival
- PD‐1
programmed cell death 1
- PD‐L1
programmed cell death ligand 1
- PDX
patient‐derived xenograft
- PR
progesterone receptor
- PVL
immature perivascular‐like cells
- RFS
relapse‐free survival
- scRNA‐seq
single‐cell RNA sequencing
- SPARC
secreted protein acidic and rich in cysteine
- TAMs
tumor‐associated macrophages
- TILs
tumor‐infiltrating lymphocytes
- TMA
tissue microarray
- TNBC
triple negative breast cancer
- TNF
tumor necrosis factors
- tSNE
t‐distributed stochastic neighbor embedding
AUTHOR CONTRIBUTIONS
The work reported in the paper has been performed by the authors, unless clearly specified in the text. Lindsay B. Alcaraz, Aude Mallavialle, Andrei Turtoi, Pascal Roger, Séverine Guiu, Emmanuelle Liaudet‐Coopman designed the experiments and prepared the manuscript. Lindsay B. Alcaraz, Aude Mallavialle, Florence Boissière‐Michot, Hanane Mansouri, Joelle Simony‐Lafontaine, Andrei Turtoi, Pascal Roger performed the experiments. Lindsay B. Alcaraz, Aude Mallavialle, Caroline Mollevi, Florence Boissière‐Michot, William Jacot, Joelle Simony‐Lafontaine, Andrei Turtoi, Pascal Roger, Séverine Guiu, Emmanuelle Liaudet‐Coopman provided material and analyzed data. Lindsay B. Alcaraz, Aude Mallavialle, Caroline Mollevi, Florence Boissière‐Michot, Joelle Simony‐Lafontaine, Valérie Laurent‐Matha, Thierry Chardès, Andrei Turtoi, Pascal Roger, Séverine Guiu, Emmanuelle Liaudet‐Coopman analyzed data and proof‐read and finalized the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS STATEMENT
For TNBC cytosols, patient samples were processed according to the French Public Health Code (law n°2004‐800, articles L. 1243‐4 and R. 1243‐61), and the biological resources center has been authorized (authorization number: AC‐2008‐700; Val d'Aurelle, ICM, Montpellier) to deliver human samples for scientific research. TNBC samples were provided by the Biological Resource Center (Biobank number BB‐0033‐00059) after approval by the Montpellier Cancer Institute Institutional Review Board (ID number ICM‐CORT‐2016‐04), following the French Ethics and Legal regulations for the patients' information and consent. All patients were informed before surgery that their surgical specimens may be used for research purposes.
Supporting information
APPENDIX S1 Supporting information
ACKNOWLEDGEMENTS
This work was supported by a public grant overseen by the French National Research Agency (ANR) as part of the “Investissements d'Avenir” program (reference: ANR‐10‐LABX‐53‐01), SIRIC Montpellier Cancer Grant INCa_Inserm_DGOS_12553, University of Montpellier, the associations “Ligue Régionale du Gard”, “Ligue Régionale de l'Herault”, “Ligue Régionale de la Charente Maritime”, and “Association pour la Recherche sur le Cancer” (ARC). We thank Prof Marc Ychou (Director of ICM, Montpellier, France) for the ICM financial support for Dr Lindsay Alcaraz' salary. Dr Andrei Turtoi is thankful to Mr. Guillaume Tosato for R teaching. The authors acknowledge the Biological Resources Center from Montpellier Cancer Institute (n° BB‐0033‐00059).
Alcaraz LB, Mallavialle A, Mollevi C, et al. SPARC in cancer‐associated fibroblasts is an independent poor prognostic factor in non‐metastatic triple‐negative breast cancer and exhibits pro‐tumor activity. Int J Cancer. 2023;152(6):1243‐1258. doi: 10.1002/ijc.34345
Pascal Roger, Séverine Guiu and Emmanuelle Liaudet‐Coopman have contributed equally to this work.
Funding information French National Research Agency, Grant/Award Number: ANR‐10‐LABX‐53‐01; ICM Biobank, Grant/Award Number: BB‐0033‐00059
Contributor Information
Lindsay B. Alcaraz, Email: lindsay.alcaraz@inserm.fr.
Emmanuelle Liaudet‐Coopman, Email: emmanuelle.liaudet-coopman@inserm.fr.
DATA AVAILABILITY STATEMENT
Data sources and handling of publicly available datasets are described in the Materials and Methods. The R script used in the current study to generate Figures 3, S6, S7 and S8 was deposited in a public database: https://github.com/DirtyHarry80/BreastCanceR. Further information is available from the corresponding author upon request.
REFERENCES
- 1. Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple‐negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13:674‐690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zheng J, Gao P. Toward normalization of the tumor microenvironment for cancer therapy. Integr Cancer Ther. 2019;18:1534735419862352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gascard P, Tlsty TD. Carcinoma‐associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 2016;30:1002‐1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chiavarina B, Turtoi A. Collaborative and defensive fibroblasts in tumor progression and therapy resistance. Curr Med Chem. 2017;24:2846‐2859. [DOI] [PubMed] [Google Scholar]
- 5. Dotto GP, Weinberg RA, Ariza A. Malignant transformation of mouse primary keratinocytes by Harvey sarcoma virus and its modulation by surrounding normal cells. Proc Natl Acad Sci U S A. 1988;85:6389‐6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Toullec A, Gerald D, Despouy G, et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol Med. 2010;2:211‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Benyahia Z, Dussault N, Cayol M, et al. Stromal fibroblasts present in breast carcinomas promote tumor growth and angiogenesis through adrenomedullin secretion. Oncotarget. 2017;8:15744‐15762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Costa A, Kieffer Y, Scholer‐Dahirel A, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. 2018;33(463–79):e10. [DOI] [PubMed] [Google Scholar]
- 9. Wu SZ, Roden DL, Wang C, et al. Stromal cell diversity associated with immune evasion in human triple‐negative breast cancer. EMBO J. 2020;39:e104063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Karaayvaz M, Cristea S, Gillespie SM, et al. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single‐cell RNA‐seq. Nat Commun. 2018;9:3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kieffer Y, Hocine HR, Gentric G, et al. Single‐cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov. 2020;10:1330‐1351. [DOI] [PubMed] [Google Scholar]
- 12. Tariq M, Zhang J, Liang G, Ding L, He Q, Yang B. Macrophage polarization: anti‐cancer strategies to target tumor‐associated macrophage in breast cancer. J Cell Biochem. 2017;118:2484‐2501. [DOI] [PubMed] [Google Scholar]
- 13. Fuxe J, Karlsson MC. TGF‐beta‐induced epithelial‐mesenchymal transition: a link between cancer and inflammation. Semin Cancer Biol. 2012;22:455‐461. [DOI] [PubMed] [Google Scholar]
- 14. Jiang K, Dong M, Li C, Sheng J. Unraveling heterogeneity of tumor cells and microenvironment and its clinical implications for triple negative breast cancer. Front Oncol. 2021;11:557477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Loi S, Michiels S, Salgado R, et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: results from the FinHER trial. Ann Oncol. 2014;25:1544‐1550. [DOI] [PubMed] [Google Scholar]
- 16. Loi S, Sirtaine N, Piette F, et al. Prognostic and predictive value of tumor‐infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node‐positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin‐based chemotherapy: BIG 02‐98. J Clin Oncol. 2013;31:860‐867. [DOI] [PubMed] [Google Scholar]
- 17. Denkert C, von Minckwitz G, Darb‐Esfahani S, et al. Tumour‐infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018;19:40‐50. [DOI] [PubMed] [Google Scholar]
- 18. Dong H, Strome SE, Salomao DR, et al. Tumor‐associated B7‐H1 promotes T‐cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793‐800. [DOI] [PubMed] [Google Scholar]
- 19. Miglietta F, Griguolo G, Guarneri V, Dieci MV. Programmed cell death ligand 1 in breast cancer: technical aspects, prognostic implications, and predictive value. Oncologist. 2019;24:e1055‐e1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sage EH, Bornstein P. Extracellular proteins that modulate cell‐matrix interactions. SPARC, tenascin, and thrombospondin. J Biol Chem. 1991;266:14831‐14834. [PubMed] [Google Scholar]
- 21. Lane TF, Iruela‐Arispe ML, Johnson RS, Sage EH. SPARC is a source of copper‐binding peptides that stimulate angiogenesis. J Cell Biol. 1994;125:929‐943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brekken RA, Sage EH. SPARC, a matricellular protein: at the crossroads of cell‐matrix. Matrix Biol. 2000;19:569‐580. [DOI] [PubMed] [Google Scholar]
- 23. Murphy‐Ullrich JE, Sage EH. Revisiting the matricellular concept. Matrix Biol. 2014;37:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tichet M, Prod'Homme V, Fenouille N, et al. Tumour‐derived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signalling to promote metastasis. Nat Commun. 2015;6:6993. [DOI] [PubMed] [Google Scholar]
- 25. Hsiao YH, Lien HC, Hwa HL, Kuo WH, Chang KJ, Hsieh FJ. SPARC (osteonectin) in breast tumors of different histologic types and its role in the outcome of invasive ductal carcinoma. Breast J. 2010;16:305‐308. [DOI] [PubMed] [Google Scholar]
- 26. Barth PJ, Moll R, Ramaswamy A. Stromal remodeling and SPARC (secreted protein acid rich in cysteine) expression in invasive ductal carcinomas of the breast. Virchows Archiv. 2005;446:532‐536. [DOI] [PubMed] [Google Scholar]
- 27. Podhajcer OL, Benedetti LG, Girotti MR, Prada F, Salvatierra E, Llera AS. The role of the matricellular protein SPARC in the dynamic interaction between the tumor and the host. Cancer Metastasis Rev. 2008;27:691‐705. [DOI] [PubMed] [Google Scholar]
- 28. Nagaraju GP, Dontula R, El‐Rayes BF, Lakka SS. Molecular mechanisms underlying the divergent roles of SPARC in human carcinogenesis. Carcinogenesis. 2014;35:967‐973. [DOI] [PubMed] [Google Scholar]
- 29. Briggs J, Chamboredon S, Castellazzi M, Kerry JA, Bos TJ. Transcriptional upregulation of SPARC, in response to c‐Jun overexpression, contributes to increased motility and invasion of MCF7 breast cancer cells. Oncogene. 2002;21:7077‐7091. [DOI] [PubMed] [Google Scholar]
- 30. Watkins G, Douglas‐Jones A, Bryce R, Mansel RE, Jiang WG. Increased levels of SPARC (osteonectin) in human breast cancer tissues and its association with clinical outcomes. Prostaglandins Leukot Essent Fatty Acids. 2005;72:267‐272. [DOI] [PubMed] [Google Scholar]
- 31. Sangaletti S, Tripodo C, Santangelo A, et al. Mesenchymal transition of high‐grade breast carcinomas depends on extracellular matrix control of myeloid suppressor cell activity. Cell Rep. 2016;17:233‐248. [DOI] [PubMed] [Google Scholar]
- 32. Zhu A, Yuan P, Du F, et al. SPARC overexpression in primary tumors correlates with disease recurrence and overall survival in patients with triple negative breast cancer. Oncotarget. 2016;7:76628‐76634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guttlein LN, Benedetti LG, Fresno C, et al. Predictive outcomes for HER2‐enriched cancer using growth and metastasis signatures driven by SPARC. Mol Cancer Res. 2017;15:304‐316. [DOI] [PubMed] [Google Scholar]
- 34. McQuerry JA, Jenkins DF, Yost SE, et al. Pathway activity profiling of growth factor receptor network and stemness pathways differentiates metaplastic breast cancer histological subtypes. BMC Cancer. 2019;19:881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Campo McKnight DA, Sosnoski DM, Koblinski JE, Gay CV. Roles of osteonectin in the migration of breast cancer cells into bone. J Cell Biochem. 2006;97:288‐302. [DOI] [PubMed] [Google Scholar]
- 36. Gilles C, Bassuk JA, Pulyaeva H, Sage EH, Foidart JM, Thompson EW. SPARC/osteonectin induces matrix metalloproteinase 2 activation in human breast cancer cell lines. Cancer Res. 1998;58:5529‐5536. [PubMed] [Google Scholar]
- 37. Dhanesuan N, Sharp JA, Blick T, Price JT, Thompson EW. Doxycycline‐inducible expression of SPARC/Osteonectin/BM40 in MDA‐MB‐231 human breast cancer cells results in growth inhibition. Breast Cancer Res Treat. 2002;75:73‐85. [DOI] [PubMed] [Google Scholar]
- 38. Ma J, Gao S, Xie X, et al. SPARC inhibits breast cancer bone metastasis and may be a clinical therapeutic target. Oncol Lett. 2017;14:5876‐5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Koblinski JE, Kaplan‐Singer BR, VanOsdol SJ, et al. Endogenous osteonectin/SPARC/BM‐40 expression inhibits MDA‐MB‐231 breast cancer cell metastasis. Cancer Res. 2005;65:7370‐7377. [DOI] [PubMed] [Google Scholar]
- 40. Alcaraz LB, Mallavialle A, David T, et al. A 9‐kDa matricellular SPARC fragment released by cathepsin D exhibits pro‐tumor activity in the triple‐negative breast cancer microenvironment. Theranostics. 2021;11:6173‐6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. du Manoir S, Orsetti B, Bras‐Goncalves R, et al. Breast tumor PDXs are genetically plastic and correspond to a subset of aggressive cancers prone to relapse. Mol Oncol. 2014;8:431‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saadoun H, Lamy PJ, Thezenas S, et al. Prognostic impact of the inclusion of uPA/PAI‐1 tumor levels in the current adjuvant treatment decision‐making for early breast cancer. Future Oncol. 2014;10:195‐209. [DOI] [PubMed] [Google Scholar]
- 43. Berchem G, Glondu M, Gleizes M, et al. Cathepsin‐D affects multiple tumor progression steps in vivo: proliferation, angiogenesis and apoptosis. Oncogene. 2002;21:5951‐5955. [DOI] [PubMed] [Google Scholar]
- 44. Laurent‐Matha V, Maruani‐Herrmann S, Prebois C, et al. Catalytically inactive human cathepsin D triggers fibroblast invasive growth. J Cell Biol. 2005;168:489‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single‐cell data. Cell. 2019;177:1888‐1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Goodwin AE, Pauli BU. A new adhesion assay using buoyancy to remove non‐adherent cells. J Immunol Methods. 1995;187:213‐219. [DOI] [PubMed] [Google Scholar]
- 47. Oo KK, Kamolhan T, Soni A, et al. Development of an engineered peptide antagonist against periostin to overcome doxorubicin resistance in breast cancer. BMC Cancer. 2021;21:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lindner JL, Loibl S, Denkert C, et al. Expression of secreted protein acidic and rich in cysteine (SPARC) in breast cancer and response to neoadjuvant chemotherapy. Ann Oncol. 2015;26:95‐100. [DOI] [PubMed] [Google Scholar]
- 49. Park SY, Lee HE, Li H, Shipitsin M, Gelman R, Polyak K. Heterogeneity for stem cell‐related markers according to tumor subtype and histologic stage in breast cancer. Clin Cancer Res. 2010;16:876‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Witkiewicz AK, Freydin B, Chervoneva I, et al. Stromal CD10 and SPARC expression in ductal carcinoma in situ (DCIS) patients predicts disease recurrence. Cancer Biol Ther. 2010;10:391‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Azim HA Jr, Singhal S, Ignatiadis M, et al. Association between SPARC mRNA expression, prognosis and response to neoadjuvant chemotherapy in early breast cancer: a pooled in‐silico analysis. PLoS One. 2013;8:e62451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gyorffy B, Lanczky A, Eklund AC, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123:725‐731. [DOI] [PubMed] [Google Scholar]
- 53. Nagai MA, Gerhard R, Fregnani JH, et al. Prognostic value of NDRG1 and SPARC protein expression in breast cancer patients. Breast Cancer Res Treat. 2011;126:1‐14. [DOI] [PubMed] [Google Scholar]
- 54. Takai K, Le A, Weaver VM, Werb Z. Targeting the cancer‐associated fibroblasts as a treatment in triple‐negative breast cancer. Oncotarget. 2016;7:82889‐82901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tseng C, Kolonin MG. Proteolytic isoforms of SPARC induce adipose stromal cell mobilization in obesity. Stem Cells. 2016;34:174‐190. [DOI] [PubMed] [Google Scholar]
- 56. Sasaki T, Gohring W, Mann K, et al. Limited cleavage of extracellular matrix protein BM‐40 by matrix metalloproteinases increases its affinity for collagens. J Biol Chem. 1997;272:9237‐9243. [DOI] [PubMed] [Google Scholar]
- 57. Sage EH, Reed M, Funk SE, et al. Cleavage of the matricellular protein SPARC by matrix metalloproteinase 3 produces polypeptides that influence angiogenesis. J Biol Chem. 2003;278:37849‐37857. [DOI] [PubMed] [Google Scholar]
- 58. Kehlet SN, Manon‐Jensen T, Sun S, et al. A fragment of SPARC reflecting increased collagen affinity shows pathological relevance in lung cancer: implications of a new collagen chaperone function of SPARC. Cancer Biol Ther. 2018;19:904‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Podgorski I, Linebaugh BE, Koblinski JE, et al. Bone marrow‐derived cathepsin K cleaves SPARC in bone metastasis. Am J Pathol. 2009;175:1255‐1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pelon F, Bourachot B, Kieffer Y, et al. Cancer‐associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat Commun. 2020;11:404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Straussman R, Morikawa T, Shee K, et al. Tumour micro‐environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP‐expressing carcinoma‐associated fibroblasts synergizes with anti‐PD‐L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. 2013;110:20212‐20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gerarduzzi C, Hartmann U, Leask A, Drobetsky E. The matrix revolution: matricellular proteins and restructuring of the cancer microenvironment. Cancer Res. 2020;80:2705‐2717. [DOI] [PubMed] [Google Scholar]
- 64. Thomas SL, Alam R, Lemke N, Schultz LR, Gutierrez JA, Rempel SA. PTEN augments SPARC suppression of proliferation and inhibits SPARC‐induced migration by suppressing SHC‐RAF‐ERK and AKT signaling. Neuro Oncol. 2010;12:941‐955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nie J, Chang B, Traktuev DO, et al. IFATS collection: combinatorial peptides identify alpha5beta1 integrin as a receptor for the matricellular protein SPARC on adipose stromal cells. Stem Cells. 2008;26:2735‐2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shi Q, Bao S, Song L, et al. Targeting SPARC expression decreases glioma cellular survival and invasion associated with reduced activities of FAK and ILK kinases. Oncogene. 2007;26:4084‐4094. [DOI] [PubMed] [Google Scholar]
- 67. Mhaidly R, Mechta‐Grigoriou F. Role of cancer‐associated fibroblast subpopulations in immune infiltration, as a new means of treatment in cancer. Immunol Rev. 2021;302:259‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1 Supporting information
Data Availability Statement
Data sources and handling of publicly available datasets are described in the Materials and Methods. The R script used in the current study to generate Figures 3, S6, S7 and S8 was deposited in a public database: https://github.com/DirtyHarry80/BreastCanceR. Further information is available from the corresponding author upon request.