

Abstract
Cytochrome P450 (CYP) heme‐thiolate monooxygenases catalyze the hydroxylation of the C−H bonds of organic molecules. This reaction is initiated by a ferryl‐oxo heme radical cation (Cpd I). These enzymes can also catalyze sulfoxidation reactions and the ferric‐hydroperoxy complex (Cpd 0) and the Fe(III)‐H2O2 complex have been proposed as alternative oxidants for this transformation. To investigate this, the oxidation of 4‐alkylthiobenzoic acids and 4‐methoxybenzoic acid by the CYP199A4 enzyme from Rhodopseudomonas palustris HaA2 was compared using both monooxygenase and peroxygenase pathways. By examining mutants at the mechanistically important, conserved acid alcohol‐pair (D251N, T252A and T252E) the relative amounts of the reactive intermediates that would form in these reactions were disturbed. Substrate binding and X‐ray crystal structures helped to understand changes in the activity and enabled an attempt to evaluate whether multiple oxidants can participate in these reactions. In peroxygenase reactions the T252E mutant had higher activity towards sulfoxidation than O‐demethylation but in the monooxygenase reactions with the WT enzyme the activity of both reactions was similar. The peroxygenase activity of the T252A mutant was greater for sulfoxidation reactions than the WT enzyme, which is the reverse of the activity changes observed for O‐demethylation. The monooxygenase activity and coupling efficiency of sulfoxidation and oxidative demethylation were reduced by similar degrees with the T252A mutant. These observations infer that while Cpd I is required for O‐dealkylation, another oxidant may contribute to sulfoxidation. Based on the activity of the CYP199A4 mutants it is proposed that this is the Fe(III)‐H2O2 complex which would be more abundant in the peroxide‐driven reactions.
Keywords: enzyme mechanism, heme enzymes, oxygen activation, sulfoxidation, X-ray crystal structures
Engineering peroxygenase activity into a P450 monooxygenase through mutations of the oxygen activation machinery resulted in a higher activity towards sulfoxidation than O‐demethylation compared to the WT enzyme. This implies the existence of a second oxidant capable of catalyzing sulfoxidation which is proposed to be the Fe(III)‐H2O2 complex as proposed in theoretical work by Shaik.

Introduction
Cytochrome P450 (CYP) heme‐thiolate monooxygenases are most often associated with catalytic oxygenation of the C−H bonds of organic molecules. [1] This reaction is initiated by the active intermediate, so‐called compound I (Cpd I), a ferryl‐oxo heme radical cation.[ 1a , 2 ] Cpd I abstracts the hydrogen atom from the substrate, generating a Fe(IV)‐OH species (Compound II, Cpd II) and an organic radical.[ 2b , 2f , 3 ] These two species then recombine to yield the alcohol product. [3] Under ideal reaction conditions these two steps can merge into a dynamically coupled process. [4]
In the archetypal physiological, NADH/O2‐driven, P450 catalytic cycle, dioxygen activation requires two protons, sourced from the surrounding solvent, and two electrons. [5] The first electron is used to reduce the substrate‐bound heme to the ferrous form, enabling dioxygen binding. A second electron is then transferred (Scheme 1) and the resulting ferric‐peroxo anion is protonated on the distal oxygen. This protonation may be coupled to the electron transfer yielding the ferric‐hydroperoxy intermediate (Cpd 0) in a single step. [5] Another protonation on the distal oxygen triggers the rupturing of the O−O bond, generating Cpd I. [5]
Scheme 1.

The intermediates formed during the cytochrome P450 catalytic cycle (addition of dioxygen, two protons and two electrons, black) and the peroxide shunt mechanism (addition of hydrogen peroxide, blue). Potential active oxidants for sulfoxidation formed during CYP199A4 turnovers are highlighted (Fe(III)‐H2O2, Cpd 0 and Cpd I). Shaik and coworkers have also proposed that Cpd I could be formed from the Fe(III)‐H2O2 species in hydrogen peroxide‐driven reactions via an O−O homolysis/H‐atom abstraction mechanism.[ 2a , 6 ]
Both the ferric peroxo and Cpd 0 species (Scheme 1) have been proposed as alternative intermediates for specific P450‐catalyzed reactions such as C−C cleavage of α‐hydroxyketones, sulfoxidation and epoxidation.[ 6b , 7 , 8 ] For example, it was observed that the T252A mutant of P450cam (CYP101A1) catalyzed epoxidation (albeit less efficiently than the WT enzyme).[ 7a , 8b ] Similar results were observed for the equivalent mutants of other CYP enzymes strengthening the argument that Cpd 0 can mediate epoxidation. Despite this experimental evidence in favor of Cpd 0 in epoxidation, theoretical studies disputed Cpd 0’s involvement in this reaction and favored the use of Cpd I.[ 2a , 6c , 9 ] The Fe(III)‐H2O2 complex is an additional intermediate that has been proposed to mediate sulfoxidation.[ 6b , 10 ] Shaik has reported that the Fe(III)‐H2O2 complex would be a sluggish C−H hydroxylation catalyst [11] but should be eminently capable of catalyzing sulfoxidation. [6b] In fact, it was calculated to catalyze sulfoxidation more rapidly than it could transform into Cpd I. [6b] Cpd 0, which has also been proposed to be another oxidant in sulfoxidation, was calculated to be a weak oxidant.[ 6b , 7a , 9a , 10 , 12 ] Researchers conducting experimental research on sulfoxidation reactions have reached different conclusions favoring Cpd I or Cpd 0 being involved in this chemical transformation.[ 7a , 13 ]
Correct, timely delivery of the two protons involved in the CYP catalytic cycle is required for the efficient generation of Cpd I. A conserved acid‐alcohol pair of residues controls these protonation steps in the majority of P450 enzymes. Mutation of either residue of the acid‐alcohol pair impairs the enzyme's ability to catalyze hydroxylation reactions as it is less able to form Cpd I. [14] For example, the bacterial enzyme P450cam hydroxylates camphor to give 5‐exo‐hydroxycamphor with ∼100 % coupling efficiency. The threonine to alanine (T252A) mutant, described above, does not diminish the NADH oxidation rate but the reducing equivalents are almost exclusively used to generate H2O2.[ 14 , 15 , 16 ] It has been proposed that the hydrogen bond between Cpd 0 and the conserved threonine in the WT enzyme (Figure 1) stabilizes Cpd 0 and encourages protonation on the distal oxygen by increasing its proton affinity.[ 1b , 14 ] When T252 is mutated to alanine, conversion of Cpd 0 into Cpd I is interrupted, leading to accumulation of Cpd 0 and ultimately incorrect protonation on the proximal oxygen with release of H2O2 (a process termed uncoupling). [17]
Figure 1.
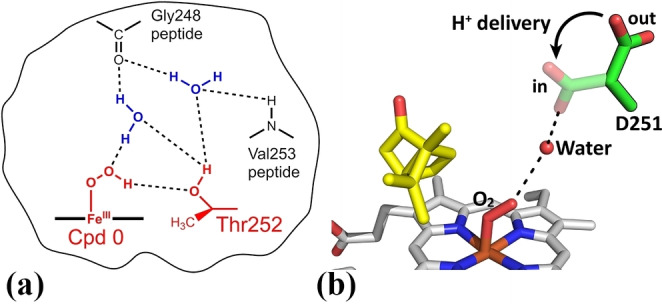
(a) In P450cam Thr252 is believed to accept a hydrogen bond from Cpd 0, which encourages protonation on the distal oxygen to generate Cpd I. [1b] (b) Asp251 is hypothesized to rotate in and out of the active site, delivering protons into the active site.[ 1b , 18 ] The potential in and out positions of the D251 residue have been shown here with the rotation into the active site to facilitate proton delivery highlighted by the arrow.
Mutation of the conserved aspartate 251 residue of P450cam to asparagine (D251N mutant), and the equivalent mutation in other P450 enzymes, results in a significant reduction in the rate of NADH oxidation (by a factor of 100 or more).[ 16b , 19 ] The role of this acidic residue is to deliver the protons required for oxygen activation (Figure 1).[ 1b , 18 ] Thus, mutation of D251 severely reduces the rate of the protonation steps with a potential build‐up of the ferric‐peroxo intermediate.[ 16b , 20 ] Therefore, in theory, the threonine‐to‐alanine and aspartate‐to‐asparagine mutants can be used to probe whether alternative oxidants to Cpd I participate in P450 reactions.[ 1b , 7a , 8b , 10 , 12a , 14 , 16b , 17 , 19 ]
The P450 enzyme CYP199A4, from Rhodopseudomonas palustris strain HaA2 binds para‐substituted benzoic acids with high affinity. The oxidation activity of the enzyme is usually restricted to the para‐substituent. [21] For example, the CYP199A4 enzyme has a high affinity for 4‐methoxybenzoic acid (K d=0.28 μM), and demethylates this substrate to form 4‐hydroxybenzoic acid with a product formation rate in excess of 1,000 nmol.(nmol‐CYP)−1.min−1. CYP199A4 also exhibits similarly high affinities for the closely related para‐substituted benzoic acid 4‐methylthiobenzoic acid. However, this substrate undergoes heteroatom oxidation rather than demethylation. [22] The CYP199A4 system is comprised of the P450 and two electron transfer partners: a [2Fe‐2S] ferredoxin (HaPux) and a flavin adenine dinucleotide containing ferredoxin reductase (HaPuR). The ferredoxin reductase and ferredoxin mediate electron transfer from NADH to CYP199A4, resulting in the high monooxygenase activities observed with these para‐substituted benzoic acids.[ 21e , 23 ] Several substrate‐bound CYP199A4 crystal structures have been solved. The benzoic acid moiety of these substrates is involved in a coordinated and highly specific set of interactions with the side chains of active site amino acids.[ 9b , 21c , 21d , 22 , 24 ] These result in the para‐substituent being held over the heme in an ideal position for oxidation.
The T252A and D251N mutants of CYP199A4 have been generated, studied and structurally characterized.[ 9b , 25 ] In contrast to the equivalent variant of P450cam, the T252A mutant of CYP199A4 displayed higher coupling efficiency. [25] Thus, mutation of the conserved threonine has a less of a deleterious effect on CYP199A4’s ability to form Cpd I although H2O2 was still generated from the main uncoupling pathway. [25] While the T252A mutant of CYP199A4 must still generate Cpd I, it does so less efficiently than the WT enzyme. This CYP199A4 variant did not significantly enhance epoxidation and differences with the WT enzyme may be related to changes at other steps of the catalytic cycle. [9b] The D251N mutant of CYP199A4 reduces the rate of proton transfer and results in low activity towards reactions such as hydroxylation, the O‐demethylation of 4‐methoxybenzoic acid and the epoxidation of 4‐vinylbenzoic acid. [25] This is in accord with what is observed with other CYP enzymes and implies a lack of reactivity of the ferric‐peroxo anion in these types of reactions.
The Fe(III)‐H2O2 intermediate must be generated during the H2O2‐driven peroxide shunt reactions (by initial binding of H2O2 to the ferric iron; Scheme 1). [26] This intermediate would only be formed during the native NADH/O2‐driven catalytic cycle when incorrect protonation on the proximal oxygen of Cpd 0 instead of the distal oxygen occurs. Recently the T252E mutant of CYP199A4 was shown to completely inhibit the monooxygenase pathway but promote peroxygenase activity. [27]
As different mutations of the CYP199A4 enzyme at the residues required for oxygen activation are available (T252A, T252E and D251N) and have been generated and studied for oxidative demethylation, this system is well suited for the investigation of other cytochrome P450‐catalyzed monooxygenase reactions. By performing H2O2‐driven reactions with the T252E mutant, we can assess whether the Fe(III)‐H2O2 intermediate is likely to participate in certain P450 reactions. The T252A and D251N variants can be used to assess the likely involvement of Cpd 0 or the ferric‐peroxo intermediate. Reactions which can be effectively mediated by the Fe(III)‐H2O2 species may be more efficiently catalyzed by the peroxygenase pathway in the T252E mutant than those requiring Cpd I formation. Here we compare the sulfoxidation of 4‐methyl‐ and 4‐ethyl‐thiobenzoic acid with wild‐type (WT) CYP199A4 and the T252E, T252A and D251N mutants (Figure 2). By comparing the activity of WT CYP199A4 and these mutants towards these different substrates in NADH‐supported and H2O2‐driven reactions, we can evaluate whether multiple oxidants participate in P450‐catalyzed sulfoxidation reactions.
Figure 2.

The para‐substituted benzoic acid substrates investigated with WT CYP199A4 and the T252E, D251N and T252A variants for NADH‐ and H2O2‐supported reactions.
Results
Substrate binding to the CYP199A4 mutants
The addition of 4‐methylthiobenzoic acid to the T252A mutant of CYP199A4 resulted in a 70 % shift from the low‐spin (LS) to the high‐spin (HS) ferric form, which is the same magnitude of change as observed in the WT enzyme (Table 1). In contrast, a significantly smaller shift (20 %) was induced in the D251N mutant. The addition of 4‐ethylthiobenzoic acid to D251N did not induce any discernible shift to the HS form (HS) but resulted in a small red‐shift of the Soret band (from ∼420 nm to ∼421 nm; Figure S1). Binding of the same substrate to the T252A mutant induced a 35 % shift to the HS form (Figure S1). This is greater than the 10 % shift to HS observed when it binds to the WT enzyme. [22] The binding affinity for 4‐methylthio‐ and 4‐ethylthio‐benzoic acids was highest with the D251N mutant followed by the T252A mutant, and both these were higher than the WT CYP199A4 (Figure S2, Table 1). Due to the small red‐shift of the Soret band observed with several of the substrate/enzyme combinations, the binding affinity could not be as accurately determined (Table 1). However, all of the substrates were bound tightly by all of the CYP199A4 variants.
Table 1.
Binding and in vitro NADH oxidation assay data for WT CYP199A4 and the D251N, T252A and T252E variants with 4‐methylthio‐ and 4‐ethylthio‐benzoic acids. The data for 4‐methoxybenzoic acid is shown for comparison.
|
Substrate/CYP199A4 BA benzoic acid |
Soret shift % HS |
K d [μM] |
NADH [min−1] |
PFR [min−1] |
Coupling [%] |
|---|---|---|---|---|---|
|
4‐methylthioBA/WT |
70 |
2.3±0.3 |
1260±20 |
∼1260 |
∼100 |
|
4‐methylthioBA/T252A |
70 |
1.4±0.1 |
1060±20 |
433±29 |
42±2 |
|
4‐methylthioBA/D251N |
20 |
0.83±0.08 |
10±1 |
3.5±0.1 |
34±2 |
|
4‐methylthioBA/T252E |
∼1.5 nm red shift[a] |
0.1±0.1 |
7±1 |
1±0.1 |
15±1 |
|
4‐ethylthioBA/WT |
10 %[b] |
0.99±0.05 |
139±0.4 |
138±0.9 |
100±1 |
|
4‐ethylthioBA/T252A |
35 % |
0.25±0.05 |
441±8 |
164±7 |
37±1 |
|
4‐ethylthioBA/D251N |
∼1 nm red shift[a]* |
0.12±0.25 |
7±1 |
1.1±0.1 |
15±1 |
|
4‐ethylthioBA/T252E |
∼2 nm red shift[a] |
0.02±0.06 |
6±1 |
0.2±0.02 |
4±1 |
|
4‐methoxyBA/WT[c] |
>95 |
0.28±0.01 |
1580±20 |
1440±30 |
91±1 |
|
4‐methoxyBA/T252A[c] |
>95 |
0.22±0.02 |
833±8 |
570±56 |
68±5 |
|
4‐methoxyBA/D251N[c] |
>95 |
0.17±0.01 |
7±0.8 |
4±1 |
59±3 |
|
4‐methoxyBA/T252E[c] |
5 |
0.4±0.1 |
9±0.5 |
0.8±0.1 |
10±1 |
[a] The magnitude of the red shifts observed are relatively small and given the sensitivity of the instrument are provided as approximate values. The relative magnitude of the reported red shifts are consistent over multiple measurements. * Addition of 4‐ethylthiobenzoic acid to D251N red‐shifted the Soret band from 420→421 nm and induced an unusual difference spectrum (Figures S1 and S4). [b] Addition of 4‐ethylthiobenzoic acid to WT CYP199A4 induced a 10 % type I shift to HS but also red‐shifted the Soret band from 418→419 nm. [22] [c] Data reported previously.[ 22 , 25 ]
Both sulfur‐containing substrates red‐shifted the absorbance maximum of the Soret band of the T252E variant by ∼1.5–∼2.0 nm (Figure S3). While these shifts are small clear changes were observed in the UV‐vis difference spectra (Figure S2). 4‐Ethylthiobenzoic acid induced a type II difference spectrum with a trough and a peak at 414 nm and 433 nm, respectively (Figure S2). In contrast, addition of 4‐methylthiobenzoic acid resulted in a type I shift (peak 376 nm and a trough at 416 nm) which was similar to the majority of those induced with the other CYP199A4 variants and despite the small red shift in the Soret band (Figure S2). The difference spectrum that arose when 4‐ethylthiobenzoic acid was added to the D251N mutant was more unusual (Figure S4). This was compared to spectra obtained on the addition of thioanisole and dithiothreitol to P450cam and found to be distinct (Figure S4, Figure S5, Table S1). [28] Overall, the binding of the alkylthiobenzoic acids to the CYP199A4 variants resulted in greater variation in the observed UV‐vis spectra of the enzyme inferring different changes in the environment of the heme with the different combinations (Table 1). However, there was evidence that each substrate was binding to the enzyme and that the binding affinity across the variants was similar (Table 1).
Oxidation of the thiobenzoic acids by the CYP199A4 mutants
The rate of the P450 catalytic cycle, as measured by the NADH oxidation rate, of the T252A mutant was 84 % of the rate observed with the WT enzyme for 4‐methylthiobenzoic acid (Table 1). Enzyme‐catalyzed oxidation of each substrate with T252A‐CYP199A4 showed formation of the same sulfoxide products as with the WT enzyme. However, the turnovers were less efficiently coupled, resulting in lower levels of product formation (Table 1). Oxidation reactions with the T252A mutant showed elevated levels of H2O2 formation, which accounted for the majority of uncoupled NADH equivalents. This is expected as removal of the threonine residue is thought to destabilize certain iron‐oxy intermediates and promote hydrogen peroxide uncoupling.[ 15 , 25 , 29 ] As with the oxidation of 4‐methoxybenzoic acid with the T252A variant the NADH equivalents not used for the sulfoxidation of 4‐methylthiobenzoic acid were mostly channeled into H2O2 generation (31 % and 38 %, respectively). [25]
In line with the greater spin‐state shift induced upon binding of 4‐ethylthiobenzoic acid to the T252A mutant (35 vs. 10 %), the NADH consumption rate was ∼3‐fold faster for this mutant compared to the WT enzyme (441 vs. 139 min−1; Table 1, Figure S6). The coupling efficiency was substantially lower for the T252A mutant than for the WT enzyme (37 vs. ∼100 %). Overall, the T252A mutant generated the sulfoxide of 4‐ethylthiobenzoic acid with a higher product formation rate (164 vs. 138 min−1; Table 1). A small amount of 4‐mercaptobenzoic acid was detected (∼2 % of the product in the T252A turnover and ∼0.7 % of the product in the WT turnover; Figure S7, Table S2).
The NADH oxidation activity of both the D251N and T252E mutants with each substrate was several orders of magnitude slower than that observed for the WT enzyme (Table 1, Figure S6, Figure S8). For both mutants, NADH was oxidized at a rate comparable to the NADH leak rate of the WT enzyme in the absence of substrate (≈9 min−1). The overall coupling efficiency and consequently the product formation activity towards sulfoxide formation was also greatly reduced with both the D251N and the T252E mutants. In each instance, the 4‐alkylsulfinylbenzoic acid was the major product. The levels of dealkylation product (4‐mercaptobenzoic acid) were negligible and no further oxidation metabolites were detected (Figure 3, Figure S7, Figure S9 and Figure S10).
Figure 3.

HPLC analysis of the NADH/O2‐driven oxidation of 4‐methylthiobenzoic acid by T252E (black) and WT CYP199A4 (blue). In red is a control reaction omitting the P450. 4‐Methylsulfinylbenzoic acid elutes at 4.7 min and 4‐mercaptobenzoic acid at 12.5 min. The substrate appears at 13.6 min. Negligible levels of 4‐mercaptobenzoic acid were detected. No sulfone was detected. Gradient: 20–95 % acetonitrile in H2O with 0.1 % TFA. Detection wavelength: 254 nm.
Comparison of the activity of H2O2‐shunt driven P450 catalyzed sulfoxidation reactions
Reactions were performed to assess the T252E mutant's ability to catalyze sulfoxidation using H2O2 as a surrogate oxygen donor (Table 2). Over a 20‐minute period, the T252E enzyme converted 114 μM of 4‐methoxybenzoic acid into 4‐hydroxybenzoic acid in reactions driven by 50 mM H2O2. The T252E mutant displayed markedly higher activity towards sulfoxidation of 4‐methylthiobenzoic acid than oxidation of 4‐methoxybenzoic acid. The quantity of 4‐methylsulfinylbenzoic acid detected after a 20‐minute reaction was 245±2 μM after thioether conversion into the sulfoxide by H2O2 alone was taken into account (Figure S11, Table S3). The average product formation rate over the 20‐min period was 1.9 and 4.1 μM (μM−P450)−1 min−1 for 4‐methoxybenzoic acid and 4‐methylthiobenzoic acid, respectively (Table S3).
Table 2.
Product generated by WT CYP199A4 and the T252E, D251N and T252A mutants using the H2O2‐shunt pathway during 240‐min reactions. The values given are mean ± SD, with n≥2.
|
Substrate |
Product concentration [μM] |
|||
|---|---|---|---|---|
|
BA benzoic acid |
T252E |
WT |
T252A |
D251N |
|
4‐methoxyBA |
379±7 |
156±2 |
89±3 |
55±1 |
|
4‐methylthioBA* |
417±17 |
40±3 |
83±1 |
7±8 |
|
4‐ethylthioBA* |
107±4 |
1±8 |
38±11 |
∼0 |
* Lower levels of H2O2 (6 mM) were used for the enzyme catalyzed oxidation of the thioethers compared to that (50 mM) used for 4‐methoxybenzoic acid turnovers. Note that the amount of product detected in the control reactions without P450 has been subtracted from the amount of product in the enzyme turnovers (See Table S4 for further details).
We next undertook H2O2‐driven reactions with the WT, T252E, D251N and T252A isoforms of CYP199A4 and their activity towards oxidative demethylation and sulfoxidation were compared over a longer time period (Table 2). Product formation was monitored over a 4 h period and a time course of product formation was constructed for each isoform (Figure S12–S15, Table S4). Given the propensity of the thioethers to be oxidized by H2O2 a lower concentration (6 mM) was used to minimize background oxidation to the sulfoxide. Control reactions with CYP101B1 or heat‐denatured T252E (sources of heme or free iron) or when the P450 enzyme was omitted entirely were undertaken. Importantly, only low levels of product (or no product in the case of oxidative demethylation) were detected in these control reactions (Figure S12, Table S4).
With the T252E mutant, peroxygenase mediated oxidation of 4‐methoxybenzoic acid produced more than twice the amount of 4‐hydroxybenzoic acid that was generated by the WT enzyme (final concentrations were 379 vs. 156 μM; Table 2). [27b] In contrast, the T252A and D251N mutants yielded considerably less product (89 and 55 μM, respectively; Table 2). While the T252E mutant was active for ∼4 h, the T252A and D251N mutants were readily inactivated and produced little additional product after the first 20 minutes of the reaction (Figure 4 and Figure S15).
Figure 4.

(a) Time course of 4‐methoxybenzoic acid conversion into 4‐hydroxybenzoic acid by CYP199A4 isoforms. (b) HPLC analysis of the amount of 4‐hydroxybenzoic acid product (RT=5.8 min) generated by the CYP199A4 isoforms over 240 minutes. (c) Time course of 4‐methylthiobenzoic acid conversion into the corresponding sulfoxide by CYP199A4 isoforms. (d) HPLC analysis of the amount of the 4‐methylsulfinylbenzoic acid product (RT=4.7 min) generated by the CYP199A4 isoforms over 240 minutes. No sulfone (sulfone RT=6.9 min) was detected. Note that in the HPLC analysis the internal standard and substrate peaks are not shown in the Figures above (see Figure S13 and S14 for further details and the full chromatograms).
The T252E mutant exhibited 10‐fold higher activity towards the sulfoxidation of 4‐methylthiobenzoic acid than the WT enzyme using 6 mM H2O2 (Figure 4, Table 2 and Figure S14). In control reactions omitting the P450, the presence of 6 mM H2O2 resulted in only slow sulfoxidation of 4‐methylthiobenzoic acid. Over a 4‐hour period, the T252E mutant generated an estimated 417 μM of sulfoxide, whereas the WT enzyme only formed 40 μM (Table 2 and Table S4). The sulfoxide concentration in the D251N reaction mixture was low (∼7 μM over the control at 4 hr) though at each time point there was always an excess of the sulfoxide when compared to the controls (Figure 4). The T252A mutant, on the other hand, displayed elevated activity towards sulfoxidation compared to the WT enzyme, yielding 83 μM sulfoxide (Table 2 and Figure 4).
Analogous results were obtained when we oxidized 4‐ethylthiobenzoic acid utilizing the peroxide shunt pathway (Figure S15). Again, the substrate was only slowly converted into the sulfoxide when exposed to 6 mM H2O2 in the absence of P450 (Table 2 and Table S4). The T252E mutant formed the most product (107 μM), while the T252A mutant generated less (38 μM sulfoxide) but the latter was again more active towards sulfoxidation than the WT enzyme. The concentration of sulfoxide in the WT and D251N reaction mixtures was roughly equal to that in control reactions lacking the P450 after 4 h; therefore, the product formed by these isoforms is reported as ∼1 μM and 0 μM, respectively. All CYP199A4 isoforms tested were less active towards oxidation of 4‐ethylthiobenzoic acid than 4‐methylthiobenzoic acid using H2O2.
Overall, in the H2O2‐driven peroxygenase reactions, the T252E mutant had higher activity towards sulfoxidation than O‐demethylation but in the NADH/O2‐supported monooxygenase reactions with the WT enzyme the activity of both was similar.
Crystal structures of the CYP199A4 T252E mutant in complex with 4‐methylthio‐ and 4‐ethylthio‐benzoic acid
Crystal structures were determined for the T252E mutant in complex with 4‐methylthiobenzoic acid and 4‐ethylthiobenzoic acid (Figure 5). The structures of both were determined at 1.66‐Å resolution (Table S5). The fold of the T252E mutant in complex with these substrates was similar to that of the WT enzyme and like the WT structures both contained a chloride anion (RMSD ∼0.4 Å, Figure S16, Figure S17).
Figure 5.

Crystal structures of the T252E mutant of CYP199A4 in complex with 4‐methylthiobenzoic acid (a; PDB code: 7TP6) and 4‐ethylthiobenzoic acid (b; PDB code: 7TP5). A 2mFo‐DFc composite omit map or feature‐enhanced map of the substrate, the heme aqua ligand (W1), the E252 side‐chain and the phenylalanine 298 residue is shown as gray mesh contoured at 1.5 or 1.0 σ (1.5 Å carve). The sulfur of each substrate interacts with the iron‐bound oxygen ligand (3.3 Å and 3.1 Å, respectively).
Neither substrate displaced the heme‐bound aqua ligand and there was an interaction between the aqua ligand and glutamate 252 residue. [27b] In the 4‐ethylthiobenzoic acid structure (Figure 5), the refined occupancy of the aqua ligand was 100 %, while in the 4‐methylthiobenzoic acid structure the occupancy was 80 % (Table S6). The Fe−O distance was similar in each structure (between 2.1–2.2 Å for the both 4‐thioethyl and 4‐thiomethyl substrates) and comparable to the Fe−O distance in the structure of T252E CYP199A4 with 4‐methoxybenzoic acid bound (Table S6). [27b] In both structures, the substrate sulfur is in close proximity (≤3.3 Å) to the heme‐bound oxygen donor and would be interacting with this ligand (Figure 5). [30] In the 4‐methylthiobenzoic acid structure, the sulfur is 3.3 Å from the aqua ligand, while in the 4‐ethylthiobenzoic acid structure the sulfur is closer (3.1 Å; Table S6). This set of interactions of the heme‐aqua ligand with the sulfur and the short Fe−O bond distance presumably contributes to the observed red‐shift of the Soret band upon addition of these substrates to this enzyme.
The positioning of these substrates in the active site of the T252E mutant was only subtly different to their positioning in the WT enzyme (Table S6 and Figure S18). The overlaid structures of WT CYP199A4 and the T252E mutant in complex with 4‐methylthio‐ and 4‐ethylthio‐benzoic acid demonstrate that in the T252E structures, the substrates are positioned further from the heme due to the presence of the heme aqua ligand (Figure S18). As a result of this shift, the methylthio moiety of 4‐methylthiobenzoic acid is nearer to the phenylalanine 298 residue (F298) in the T252E structure than in the WT structure and consequently the phenyl ring of F298 rotates to accommodate the methylthio moiety (Figure 5 and Figure S19a). When the larger 4‐ethylthiobenzoic acid binds to the T252E mutant, the ethylthio moiety displaces the side‐chain of F298, which moves away from the heme. An analogous movement is also observed in the structures of the WT enzyme when bulky ligands are bound.[ 22 , 24 ] The T252E mutation also alters the structure of the I‐helix including the position of the D251 residue (Figure S19b). [27b]
The X‐ray crystal structures of the D251N and T252A CYP199A4 mutants with 4‐methylthiobenzoic acid bound
We also determined the crystal structures for the T252A and D251N mutants in complex with 4‐methylthiobenzoic acid (Figure 6). The structures were determined at 1.88‐Å and 1.44‐Å resolution, respectively (Table S7). The fold of both mutants was similar to that of the WT enzyme bound to the same substrate (RMSD 0.167 Å for D251N and 0.437 Å for T252A, Figure S20, Figure S21). While electron density for a chloride anion could be found in the D251N variant this was absent in the T252A mutant structure (Figure S22).
Figure 6.

Crystal structure of 4‐methylthiobenzoic acid‐bound to the T252A (a) and D251N (b) variants (PDB codes: 8DYB and 7TQM). These were determined at 1.88‐Å and 1.44‐Å resolution, respectively. In green is the 4‐methylthiobenzoic acid substrate, in yellow are active‐site residues and in gray is the heme. Waters are displayed as red spheres; the occupancy of the heme‐bound aqua ligand in both (W1 and W126) is 100 %. A 2mFo‐DFc feature‐enhanced map of the substrate, heme‐bound aqua ligand, and the A252 or the N251 mutant residue is shown as gray mesh contoured at 1.0 σ (1.5‐Å carve radius). The F298 side chain in the D251N variant occupies multiple positions and the 2mFo‐DFc feature‐enhanced map is shown (see Figure S28 and S29 for further details). In purple is the chloride which caps the active‐site entrance (in D251N; it is absent in T252A).
The structure of the T252A variant has an additional water molecule in the active site (Figure 6). The occupancy of both the substrate and this water molecule is 100 % (Table S8). However, this water (W126) is loosely bound to the heme in the presence of the substrate (Fe−O distance 2.5 Å, Table S8). The water molecule is offset from the axis of the Fe−S bond with an S−Fe−OH2 angle of ∼164° (Table S9, Figure S23). As noted above an aqua ligand is present in the T252E variant, but no water is present in the structure of the substrate‐bound WT enzyme (Table S9, Figure S24). Both the WT and the T252A variant of CYP199A4 have a 70 % HS ferric component after the addition of 4‐methylthiobenzoic acid. The distance of the water ligand from the heme and its offset nature in the T252A variant suggest it has a weaker interaction with the ferric iron. This could result in the observation of a high‐spin heme.[ 9b , 31 ] In this structure the binding orientation and interactions of 4‐methylthiobenzoic acid within the active site were similar to those of the WT enzyme. The S atom of the substrate was 3.3 Å from W126 (Table S8). In common with other structures of the T252ACYP199A4 variant there were no significant changes in the conformation of the dioxygen binding groove. [25] However, there were changes in the position of the D251 and N255 residues (Figure S26).
Changes were observed in the structure of the D251N variant (Figure 6, Figure S27 and Figure S28). In this structure, there was a heme‐bound distal aqua ligand in addition to the substrate. The occupancy of both the substrate and the heme‐bound aqua ligand (W1) is 100 % (Table S8). The Fe‐OH2 (W1) bond length is shorter than in the T252A variant (2.3 Å vs. 2.5 Å, Table S9) and the S−Fe−OH2 angle was ∼169 °. The S atom of the substrate was 3.3 Å from W1 (Table S8). These observations are consistent with the lower HS state heme content upon the addition of substrate compared to the WT and the T252A variant (Table 1 and Figure S24). We note here that the heme conformation can also alter the spectroscopic properties of the heme. [32] However, across all the substrates we report here the heme conformation was similar. There are differences in how far the iron was in the plane of the heme (Figure S30 and Table S10). This displacement of the Fe was smaller for the 6‐coordinate heme structures compared to the 5‐coordinate WT structure (Table S10). However, there was not an obvious correlation between the extent of this displacement in the 6‐coordinate species with the distal Fe−O interaction (Table S9).
The mutated residue (N251) is present in two conformations (Figure 6). In one, the side chain has rotated into the active site (60 % occupancy). In the other conformation, N251 is pointing away from the active site (40 % occupancy; Figure S28, Figure S29, Figure S31). The electron density of the side chain of F298 also infers it is present in two conformations, which have occupancies of 46 % and 54 % (Figure 6). These two conformations are related by rotation of the F298 phenyl ring (Figure S27, Figure S28, Figure S29).
Overall, the structures of the substrate binding pockets across the WT enzyme and the three variants studied here are similar when bound to 4‐methoxybenzoic acid (Figure S32) and 4‐methylthiobenzoic acid (Figure S33). However, perhaps due to the different properties and large size of the sulfur atom more changes are observed in the heme coordination and the orientation of certain amino acid residues in the 4‐methylthiobenzoic acid‐bound set of structures relative to the 4‐methoxybenzoate structures (Figure S33). This highlights the flexible nature of the substrate‐binding pocket of these enzymes.
Enantioselectivity of NADH/O2‐ and H2O2‐driven sulfoxidation of 4‐ethylthiobenzoic acid
If a second oxidant other than Cpd I can mediate certain reactions such as sulfoxidation and epoxidation (e. g. Fe(III)‐H2O2 or Fe(III)‐OOH)[ 6b , 7a , 8a ], we would expect that the D251N, T252A and T252E mutants could display different enantioselectivity than the WT enzyme. This is because the active oxidants could be positioned differently relative to the substrate. [7a] Therefore, we measured the enantioselectivity of NADH‐supported and H2O2‐driven sulfoxidation by these isoforms. The crystal structures of these mutants above revealed that the positioning of the substrate within the active site is minimally altered by these mutations (Figure S33). Therefore, changes in enantioselectivity could be due to a change in the identity of the active oxidant.
Unfortunately, the enantioselectivity of CYP199A4‐catalyzed sulfoxidation of 4‐methylthiobenzoic acid could not be determined as the enantiomers could not be separated. [22] NADH‐supported sulfoxidation of 4‐ethylthiobenzoic acid catalyzed by WT CYP199A4 proceeds with high enantioselectivity (83 % ee, Table 3, Table S11) [22] . The major enantiomer was the (S)‐sulfoxide for all the mutants in both the H2O2‐driven and NADH‐supported reactions (Figure S34). [22] The measured enantioselectivity of NADH‐supported sulfoxidation of 4‐ethylthiobenzoic acid by the T252A mutant enzyme was 82 %. That of the D251N and T252E mutants was lower (38 % and 68 % ee, respectively, Table 3, Table S11, Figure S34).
Table 3.
Enantioselectivity of sulfoxidation of 4‐ethylthiobenzoic acid by WT CYP199A4 and the T252E, D251N and T252A mutants.
|
|
CYP199A4 isoform |
|||
|---|---|---|---|---|
|
ee of 4‐ethylsulfinylBA product (%) |
T252E |
WT |
T252A |
D251N |
|
NADH/O2‐driven reaction[a] |
68 % |
83 % |
82 % |
38 % |
[a] The enantioselectivity of the NADH/O2‐driven reaction. BA: benzoic acid.
Accurate measurement of the enantioselectivity of the H2O2‐driven reactions was difficult due to background oxidation of the thioether by H2O2 (Figure S32) which yields a racemic sulfoxide (Figure S35). The measured enantioselectivities were 18 % (T252E), 27 % (WT), 18 % (T252A) and 15 % ee (D251N), but these values would underestimate the true enantioselectivity of the enzyme‐catalyzed reactions due to background oxidation (Table S11).
Discussion
It has been reported that the T268A P450BM3 (CYP102A1) mutant (of the acid‐alcohol pair) was able to perform the sulfoxidation of thiafatty acids at the same rate and with the same coupling efficiency as the WT enzyme. [7a] It was also found that the sulfoxidation of thiafatty acids by P450BM3 occurred with opposite enantioselectivity to the hydroxylation of fatty acids, suggesting that an oxidant other than Cpd I could catalyze sulfoxidation. [7a] As Cpd 0 should be more abundant in this mutant and Cpd I formation would be hindered these results implied that sulfoxidation was mediated by Cpd 0 and not Cpd I in this mutant (although the results did not distinguish Cpd 0 from the Fe(III)‐H2O2 complex that is formed from Cpd 0). Here, the T252A mutant of CYP199A4 performed sulfoxidation of 4‐ethylthiobenzoic acid with a substantially lower coupling efficiency compared to the WT enzyme but with a higher NADH oxidation activity. The activity and the coupling efficiency of the T252A variant for oxidation of 4‐methoxybenzoic acid was substantially lower than those observed for the WT enzyme. The enantioselectivity of 4‐ethylsulfinylbenzoic acid formed was not altered when the T252A mutant was used. These results do not indicate that Cpd 0 is a highly efficient oxidant for sulfoxidation in this system and would be consistent with DFT studies, which suggested that Cpd 0 is a weak oxidant.[ 6a , 6b ]
In NADH‐supported reactions, the T252E mutant had minimal activity towards para‐substituted benzoic acid substrates with methylthio and ethylthio functional groups. The product formation rate ranged from 0 to ∼1 min−1. This confirms that the T252E variant is unable to operate efficiently via the normal catalytic cycle. During the H2O2‐driven reactions it is hypothesized that H2O2 binds to the heme iron to give Fe(III)‐H2O2, which can convert into Cpd I via Cpd 0 (Scheme 1). The ferric‐peroxo anion intermediate would be bypassed in these reactions. [33] Shaik has also proposed that the Fe(III)‐H2O2 complex can also convert into Cpd I via an O−O homolysis/H‐abstraction mechanism (Scheme 1). [6c] It was concluded that this is the mechanism by which Cpd I is generated in the peroxygenase enzyme P450SPα (and WT P450cam when it is shunted with H2O2).[ 6c , 26a ] Cpd I is required for hydroxylation and O‐dealkylation, but sulfoxidation may be supported by this earlier Fe(III)‐H2O2 complex that may be more abundant in the peroxide‐driven reactions.[ 8 , 10 , 34 ]
In NADH‐supported reactions, WT CYP199A4 is marginally more active towards O‐demethylation of 4‐methoxybenzoic acid than the sulfoxidation of 4‐methylthiobenzoic acid. In H2O2‐driven reactions, the T252E variant performed sulfoxidation considerably more efficiently than O‐demethylation. If sulfoxidation was solely mediated by Cpd I, we would not expect this reaction to have occurred more efficiently than O‐demethylation. [13] A plausible explanation for this is that the Fe(III)‐H2O2 complex is a second oxidant capable of catalyzing sulfoxidation as proposed by Shaik. [6b] Our results agree with earlier work which also found that in H2O2‐driven reactions the T268E mutant of P450BM3 performed sulfoxidation faster than epoxidation and C−H hydroxylation. [12b]
In the H2O2‐driven reactions WT CYP199A4 as well as the T252A and D251N mutants were both less active than the T252E mutant. The T252A mutant was also less active than the WT enzyme towards O‐demethylation. This implies that when the WT enzyme is shunted with H2O2, the conserved acid‐alcohol pair still has a role in generating the active catalytic species or in stabilizing the heme in the presence of hydrogen peroxide. In contrast, the T252A mutant was more active towards sulfoxidation of 4‐methylthio‐ and 4‐ethylthio‐benzoic acid than the WT enzyme, which also infers the involvement of a second oxidant in sulfoxidation. Fe(III)‐H2O2 would be expected to form in greater quantities in this variant compared to the WT enzyme as Cpd 0 requires protonation before H2O2 is released in the uncoupling pathway. This could be an alternative explanation for the results obtained with the T268A mutant of P450BM3 and thiafatty acids.
The reduced peroxygenase activity of the D251N mutant suggests that delivery of protons to the active site is still important. The low peroxygenase activity of the T252A variant agrees with previous studies that found substitution of the conserved threonine in WT P450cam and P450BM3 with aliphatic residues severely reduced their peroxygenase activity.[ 29 , 35 ] In H2O2‐supported reactions, WT P450cam was capable of hydroxylating camphor, whereas the T252A mutant had minimal activity. [29] Shaik proposed the greater peroxygenase activity of WT P450cam is due to the fact that in the T252A mutant the Fe(III)‐H2O2 complex is unstable and H2O2 would dissociate, whereas in the WT enzyme the threonine stabilizes the Fe(III)‐H2O2 species through hydrogen bonding interactions, so that it can convert into Cpd I. [6c] Ma et al. [35a] similarly found that substitution of the conserved threonine with valine in the F87A mutant of P450BM3 abolished this enzyme's ability to epoxidize styrene using H2O2. These results and those here emphasize that the conserved acid‐alcohol pair can be important when WT P450s are shunted using H2O2. When H2O2 binds to the iron, the Fe(III)‐H2O2 species may lose a proton to give Cpd 0. The threonine would then be required to stabilize Cpd 0. The aspartate would also be needed to shuttle protons into the active site to protonate Cpd 0 on the distal oxygen, which leads to O−O bond scission and formation of the active Cpd I species (Scheme 1).
In conclusion, in H2O2‐driven reactions, T252E displayed higher activity towards sulfoxidation than O‐demethylation and higher sulfoxidation activity that the WT enzyme. This implies the existence of a second oxidant capable of catalyzing sulfoxidation that is more abundant in these H2O2‐driven reactions. Based on comparisons with the activity of the T252A and the D251N mutants and the theoretical studies of others we propose that this is the Fe(III)‐H2O2 complex. [6b]
Experimental Section
Production and purification of CYP101B1, ferredoxin (HaPux), ferredoxin reductase (HaPuR), and WT, T252A, D251N and T252E CYP199A4: The materials and instruments used have been described previously.[ 27b , 36 ] HaPux, HaPuR, WT (wild‐type) CYP199A4 and the D251N and T252A mutants were prepared according to the published procedures.[ 25 , 36 ] The T252E mutant of CYP199A4 was produced and purified according to the previously described method [27b] ; this mutant was co‐expressed with ferrochelatase to improve heme incorporation [37] and the LB growth medium was supplemented with 2 mM ferric ammonium citrate during protein expression. WT CYP101B1, used for control reactions, was prepared according to the published procedure. [38]
Pyridine hemochromogen assay: P450 extinction coefficients were determined using the pyridine hemochromogen assay described previously.[27b],[39] The values for the WT enzyme and the D251N, T252A and T252E mutants were 125±3, 123±1, 120±4 and 119±2 mM−1 cm−1, respectively. The extinction coefficient determined from the CO binding assay was 122.6 mM−1 cm−1 and the literature value for WT CYP199A4 is 119 mM−1 cm−1. [40]
Substrate binding and spin‐state shift analysis: To determine the substrate binding affinity (K d) to the CYP199A4 isoforms, UV‐Vis titrations were performed as described previously using a Cary 60 UV‐vis spectrophotomerter (Agilent).[ 25 , 27b , 36 ] Spin‐state shifts induced by binding of substrates to the CYP199A4 isoforms were measured using a Cary 60 UV‐vis spectrophotomerter (Agilent). The spectra were recorded between 700 and 250 nm using a scan rate of 600 nm min−1 (and repeated at 60 nm min−1 to verify smaller wavelength shifts in the Soret maximum). [36]
X‐ray crystallography: X‐ray crystallography was performed as described previously.[ 27b , 36 ] X‐ray diffraction data for crystals of T252ECYP199A4 bound to 4‐methylthio‐ and 4‐ethylthio‐benzoic acid were obtained at 100 K on the MX1 beamline at the Australian Synchrotron. [41] The exposure time was 1 s, oscillation angle 1° and wavelength 0.9537 Å. iMosflm [42] was used to index and integrate the data, and the data were then scaled and merged using Aimless [43] (part of the CCP4 suite of programs). [44] X‐ray diffraction data for the crystal of D251NCYP199A4 bound to 4‐methylthiobenzoic acid were collected at 100 K on the MX2 beamline at the Australian Synchrotron. [45] Automatic data processing was carried out at the Australian synchrotron using xdsme and Aimless. [45] PhaserMR in the CCP4 suite of programs was used to solve the phase problem by the molecular replacement method, [46] using a high‐resolution CYP199A4 structure (PDB: 5UVB) as the search model. Coordinates and restraints for the ligand were generated using Phenix eLBOW [47] and the protein model was manually rebuilt in Coot. [48] Refinement was performed using phenix.refine. [49] Solvent was added automatically using phenix.refine and positive density in the anion binding site of CYP199A4 was modeled as a chloride ion. [21d] The Fe−S(Cys) and Fe‐OH2 bond lengths were not restrained during refinement. The occupancy of the substrate, alternative protein conformations and water ligand, if present, was refined using phenix.refine. Composite omit maps [50] and feature‐enhanced maps [51] were generated using Phenix to verify the presence of the ligand in the enzyme's active site. The validation tool MolProbity was used to assess the quality of the model before the structure was deposited into the Protein Data Bank (www.rcsb.org). [52] Images were generated using PyMOL. [53] Crystal structure analysis was performed with UCSF Chimera [54] , PyMOL [53] and Coot [48] . The ferryl oxygen of Compound I (Cpd I) was modeled 1.62 Å above the heme iron (along the axis defined by the Fe−S bond) in PyMOL using the “Create Atom Along Bond” script downloaded from https://pymolwiki.org/index.php/CreateAtom. The ferryl oxygen was modeled 1.62 Å above the heme iron based on computational studies which calculated that the Fe=O bond length of P450 Cpd I is 1.62 Å. [55] This is similar to the Fe=O bond length of P450 Cpd I measured using EXAFS (1.670 Å). [56]
In vitro NADH turnovers and quantification of H2O2 uncoupling: In vitro NADH‐supported CYP199A4 oxidation reactions were performed according to the procedure reported in the literature.[ 27b , 36 ] For the reactions with WT, T252A and D251N CYP199A4, an NADH concentration of ∼320 μM was added at the start of the reactions (corresponding to an initial NADH absorbance of ∼2.00 AU at 340 nm). These reactions were performed in the presence of catalase (to determine product formation) and the absence of catalase (to determine uncoupling due to H2O2 formation). The rate of NADH consumption by the T252E mutant in the presence of substrate was slow (equivalent to the leak rate in the absence of substrate (∼9 min−1)) and therefore half the quantity of NADH (∼160 μM NADH) was used in these reactions. NADH consumption rates reported for the T252E and D251N mutant are the initial rates over the first 10 minutes of the reaction. H2O2 generated via uncoupling during these in vitro NADH‐supported reactions was quantified using the horseradish peroxidase/phenol/4‐aminoantipyrine assay described previously.[ 36 , 57 ]
Oxidation products were quantified by HPLC as described previously.[ 27b , 36 ] A calibration curve could not be constructed for 4‐ethylsulfinylbenzoic acid as a pure sample of this compound was not available. 4‐Ethylsulfinylbenzoic acid generated in the P450 reactions was instead quantified using the calibration curve for 4‐methylsulfinylbenzoic acid.
Hydrogen peroxide driven turnovers: H2O2‐driven oxidation reactions with WT, T252E, T252A and D251N CYP199A4 were performed according to the published procedure. [27b] Reaction volumes were 600 μL and contained 1 mM substrate, 3 μM P450 and 50 mM H2O2 in 50 mM Tris‐HCl buffer (pH 7.4). Reactions were performed at 30 °C and 132 μL aliquots were quenched after 20 min by addition of 10 μL of 10 mg mL−1 bovine liver catalase. Internal standard (2 μL of 10 mM 9‐hydroxyfluorene in EtOH) and 66 μL of acetonitrile before analysis by HPLC. Thioethers can be oxidized to sulfoxides with H2O2. [58] For subsequent 4‐hour turnovers with the thioethers, 6 mM H2O2 was used instead of 50 mM to minimize background oxidation. At time points of 0, 20, 60, 120 and 240 min, aliquots of the reaction mixture (132 μL) were removed and quenched. For the 4‐methoxybenzoic acid reactions, a H2O2 concentration of 50 mM was used. Control reactions were performed which omitted the P450, or contained CYP101B1 or heat‐denatured T252ECYP199A4, according to the published procedure. [27b]
Enantioselective HPLC analysis: Chiral products generated by WT and mutant forms of CYP199A4 were analyzed using enantioselective HPLC. The 4‐ethylthiobenzoic acid turnovers were extracted with EtOAc (3×400 μL), and evaporated to dryness. Chiral analysis was performed after conversion of the compounds into the methyl esters with diazomethane. The column used was a cellulose carbamate coated silica column (OD−H, 10 μm, 250×4.6 mm, CHIRALCEL). The enantiomers were eluted with 5 % isopropyl alcohol in hexane at 0.8 mL min−1. The eluate was monitored at 270 nm.
Synthesis of 4‐ethylsulfinylbenzoic acid: To 4‐ethylthiobenzoic acid (1 mM) in Tris‐HCl buffer (50 mM, pH 7.4) was added 200 mM H2O2 (Scheme 2). [58] After 2 h the reaction was quenched with catalase before being prepared for HPLC analysis as described previously.[ 27b , 36 ]
Scheme 2.

The synthesis of 4‐ethylsulfinylbenzoic acid.
Abbreviations
CYP or P450, cytochrome P450; Cpd 0, compound 0, the ferric‐hydroperoxy intermediate; Cpd I, compound I; NADH, reduced nicotinamide adenine dinucleotide; LS, low‐spin; HS, high‐spin; TFA, trifluoroacetic acid; WT, wild‐type.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was supported by ARC grant DP140103229 (to J.J.D.V. and S.G.B.) and S.G.B. acknowledges the ARC for Future Fellowships (FT140100355). This research was undertaken in part using the MX1 and MX2 beamlines at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector. The authors also acknowledge the award of an Australian Government Research Training Program Scholarship to T.C. and L.R.C. (PhD) and M.N.P. (M. Phil and PhD). M.N.P. thanks the University of Adelaide for a Constance Fraser PhD Scholarship. This research was supported by an AINSE Ltd. Postgraduate Research Award (PGRA) to M.N.P. Open Access publishing facilitated by The University of Adelaide, as part of the Wiley ‐ The University of Adelaide agreement via the Council of Australian University Librarians.
Podgorski M. N., Coleman T., Churchman L. R., Bruning J. B., De Voss J. J., Bell S. G., Chem. Eur. J. 2022, 28, e202202428.
Contributor Information
Prof. James J. De Voss, Email: j.devoss@uq.edu.au.
Dr. Stephen G. Bell, Email: stephen.bell@adelaide.edu.au.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1.
- 1a. Ortiz de Montellano P. R., Chem. Rev. 2010, 110, 932–948; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Poulos T. L., Chem. Rev. 2014, 114, 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Ogliaro F., de Visser S. P., Cohen S., Sharma P. K., Shaik S., J. Am. Chem. Soc. 2002, 124, 2806–2817; [DOI] [PubMed] [Google Scholar]
- 2b. Rittle J., Green M. T., Science 2010, 330, 933–937; [DOI] [PubMed] [Google Scholar]
- 2c. Sharma P. K., De Visser S. P., Shaik S., J. Am. Chem. Soc. 2003, 125, 8698–8699; [DOI] [PubMed] [Google Scholar]
- 2d. Meunier B., de Visser S. P., Shaik S., Chem. Rev. 2004, 104, 3947–3980; [DOI] [PubMed] [Google Scholar]
- 2e. Shaik S., Cohen S., Wang Y., Chen H., Kumar D., Thiel W., Chem. Rev. 2010, 110, 949–1017; [DOI] [PubMed] [Google Scholar]
- 2f. Groves J. T., McClusky G. A., J. Am. Chem. Soc. 1976, 98, 859–861. [Google Scholar]
- 3. Huang X., Groves J. T., J. Biol. Inorg. Chem. 2017, 22, 185–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sarkar M. R., Houston S. D., Savage G. P., Williams C. M., Krenske E. H., Bell S. G., De Voss J. J., J. Am. Chem. Soc. 2019, 141, 19688–19699. [DOI] [PubMed] [Google Scholar]
- 5. Schlichting I., Berendzen J., Chu K., Stock A. M., Maves S.A., Benson D. E., Sweet R. M., Ringe D., Petsko G.A., Sligar S. G., Science 2000, 287, 1615–1622. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Li C., Zhang L., Zhang C., Hirao H., Wu W., Shaik S., Angew. Chem. Int. Ed. 2007, 46, 8168–8170; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 8316–8318; [Google Scholar]
- 6b. Wang B., Li C., Cho K. B., Nam W., Shaik S., J. Chem. Theory Comput. 2013, 9, 2519–2525; [DOI] [PubMed] [Google Scholar]
- 6c. Wang B., Li C., Dubey K. D., Shaik S., J. Am. Chem. Soc. 2015, 137, 7379–7390. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Cryle M. J., Voss J. J. D., Angew. Chem. Int. Ed. 2006, 45, 8221–8223; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 8401–8403; [Google Scholar]
- 7b.M. Akhtar, J.N. Wright in Acyl-Carbon Bond Cleaving Cytochrome P450 Enzymes: CYP17A1, CYP19A1 and CYP51A1, Vol. 851 Eds.: E. G. Hrycay and S. M. Bandiera), Springer International Publishing, Cham, Switzerland, 2015, pp. 107–130; [DOI] [PubMed]
- 7c. Akhtar M., Wright J.N., Adv. Exp. Med. Biol. 2015, 851, 107–130; [DOI] [PubMed] [Google Scholar]
- 7d. Gregory M. C., Denisov I. G., Grinkova Y. V., Khatri Y., Sligar S. G., J. Am. Chem. Soc. 2013, 135, 16245–16247; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7e. Mak P. J., Gregory M. C., Denisov I. G., Sligar S. G., Kincaid J. R., Proc. Natl. Acad. Sci. USA 2015, 112, 15856–15861; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7f. Yoshimoto F. K., Jung I. J., Goyal S., Gonzalez E., Guengerich F. P., J. Am. Chem. Soc. 2016, 138, 12124–12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Vaz A. D.N., McGinnity D. F., Coon M. J., Proc. Nat. Acad. of Sci. U. S.A. 1998, 95, 3555–3560; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Jin S., Makris T. M., Bryson T.A., Sligar S. G., Dawson J. H., J. Am. Chem. Soc. 2003, 125, 3406–3407. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Park M. J., Lee J., Suh Y., Kim J., Nam W., J. Am. Chem. Soc. 2006, 128, 2630–2634; [DOI] [PubMed] [Google Scholar]
- 9b. Coleman T., Kirk A. M., Chao R. R., Podgorski M.N., Harbort J. S., Churchman L. R., Bruning J. B., Bernhardt P. V., Harmer J. R., Krenske E. H., De Voss J. J., Bell S. G., ACS Catal. 2021, 11, 1995–2010. [Google Scholar]
- 10. Modi A. R., Dawson J. H. in Oxidizing Intermediates in P450 Catalysis: A Case for Multiple Oxidants, Eds.: Hrycay and S. M. Bandiera E. G.), Springer International Publishing, Cham, 2015, pp. 63–81. [DOI] [PubMed] [Google Scholar]
- 11. Derat E., Kumar D., Hirao H., Shaik S., J. Am. Chem. Soc. 2006, 128, 473–484. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Volz T. J., Rock D.A., Jones J. P., J. Am. Chem. Soc. 2002, 124, 9724–9725; [DOI] [PubMed] [Google Scholar]
- 12b. Dezvarei S., Shoji O., Watanabe Y., Bell S. G., Catal. Commun. 2019. [Google Scholar]
- 13. Wang J. B., Huang Q., Peng W., Wu P., Yu D., Chen B., Wang B., Reetz M. T., J. Am. Chem. Soc. 2020, 142, 2068–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nagano S., Poulos T. L., J. Biol. Chem. 2005, 280, 31659–31663. [DOI] [PubMed] [Google Scholar]
- 15. Imai M., Shimada H., Watanabe Y., Matsushima-Hibiya Y., Makino R., Koga H., Horiuchi T., Ishimura Y., Proc. Nat. Acad. Sci. USA 1989, 86, 7823–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Kimata Y., Shimada H., Hirose T., Ishimura Y., Biochem. Biophys. Res. Commun. 1995, 208, 96–102; [DOI] [PubMed] [Google Scholar]
- 16b. Stok J. E., Yamada S., Farlow A. J., Slessor K. E., De Voss J. J., Biochim. Biophys. Acta Proteins Proteomics 2013, 1834, 688–696. [DOI] [PubMed] [Google Scholar]
- 17. Dubey K. D., Wang B., Vajpai M., Shaik S., Chem. Sci. 2017, 8, 5335–5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Follmer A. H., Tripathi S., Poulos T. L., J. Am. Chem. Soc. 2019, 141, 2678–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vidakovic M., Sligar S. G., Li H., Poulos T. L., Biochemistry 1998, 37, 9211–9219. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Benson D. E., Suslick K. S., Sligar S. G., Biochemistry 1997, 36, 5104–5107; [DOI] [PubMed] [Google Scholar]
- 20b. Davydov R., Macdonald I. D. G., Makris T. M., Sligar S. G., Hoffman B. M., J. Am. Chem. Soc. 1999, 121, 10654–10655. [Google Scholar]
- 21.
- 21a. Bell S. G., Hoskins N., Xu F., Caprotti D., Rao Z., Wong L. L., Biochem. Biophys. Res. Commun. 2006, 342, 191–196; [DOI] [PubMed] [Google Scholar]
- 21b. Bell S. G., Tan A. B., Johnson E. O., Wong L. L., Mol. BioSyst. 2010, 6, 206–214; [DOI] [PubMed] [Google Scholar]
- 21c. Bell S. G., Yang W., Tan A. B., Zhou R., Johnson E. O., Zhang A., Zhou W., Rao Z., Wong L. L., Dalton Trans. 2012, 41, 8703–8714; [DOI] [PubMed] [Google Scholar]
- 21d. Bell S. G., Zhou R., Yang W., Tan A. B., Gentleman A. S., Wong L. L., Zhou W., Chemistry 2012, 18, 16677–16688; [DOI] [PubMed] [Google Scholar]
- 21e. Coleman T., Chao R. R., Bruning J. B., De Voss J., Bell S. G., RSC Adv. 2015, 5, 52007–52018; [Google Scholar]
- 21f. Chao R. R., De Voss J. J., Bell S. G., RSC Adv. 2016, 6, 55286–55297. [Google Scholar]
- 22. Coleman T., Wong S. H., Podgorski M.N., Bruning J. B., De Voss J. J., Bell S. G., ACS Catal. 2018, 5915–5927. [Google Scholar]
- 23. Coleman T., Chao R. R., De Voss J., Bell S. G., Biochim. Biophys. Acta Proteins Proteomics 2016, 1864, 667–675. [DOI] [PubMed] [Google Scholar]
- 24. Podgorski M.N., Harbort J. S., Coleman T., Stok J. E., Yorke J.A., Wong L. L., Bruning J. B., Bernhardt P. V., De Voss J. J., Harmer J. R., Bell S. G., Biochemistry 2020, 59, 1038–1050. [DOI] [PubMed] [Google Scholar]
- 25. Coleman T., Stok J. E., Podgorski M. N., Bruning J. B., De Voss J. J., Bell S. G., J. Biol. Inorg. Chem. 2020, 25, 583–596. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Ramanan R., Dubey K. D., Wang B., Mandal D., Shaik S., J. Am. Chem. Soc. 2016, 138, 6786–6797; [DOI] [PubMed] [Google Scholar]
- 26b. Chandrasena R. E. P., Vatsis K. P., Coon M. J., Hollenberg P. F., Newcomb M., J. Am. Chem. Soc. 2004, 126, 115–126. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Shoji O., Fujishiro T., Nishio K., Kano Y., Kimoto H., Chien S.-C., Onoda H., Muramatsu A., Tanaka S., Hori A., Sugimoto H., Shiro Y., Watanabe Y., Catal. Sci. Technol. 2016, 6, 5806–5811; [Google Scholar]
- 27b. Podgorski M.N., Harbort J. S., Lee J. H. Z., Nguyen G. T. H., Bruning J. B., Donald W.A., Bernhardt P. V., Harmer J. R., Bell S. G., ACS Catal. 2022, 1614–1625. [Google Scholar]
- 28. Nastainzcyk W., Ruf H. H., Ullrich V., Eur. J. Biochem. 1975, 60, 615–620. [DOI] [PubMed] [Google Scholar]
- 29. Martinis S.A., Atkins W. M., Stayton P. S., Sligar S. G., J. Am. Chem. Soc. 1989, 111, 9252–9253. [Google Scholar]
- 30. Lupyan D., Abramov Y. A., Sherman W., J. Comput.-Aided Mol. Des. 2012, 26, 1195–1205. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Loew G. H., Harris D. L., Chem. Rev. 2000, 100, 407–420; [DOI] [PubMed] [Google Scholar]
- 31b. Lochner M., Meuwly M., Woggon W.-D., Chem. Commun. 2003, 1330–1332; [DOI] [PubMed] [Google Scholar]
- 31c. Raag R., Poulos T. L., Biochemistry 1991, 30, 2674–2684. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Scheidt W. R., Li J., Sage J. T., Chem. Rev. 2017, 117, 12532–12563; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32b. Karunakaran V., Denisov I., Sligar S. G., Champion P. M., J. Phys. Chem. B 2011, 115, 5665–5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nagel R., Peters R. J., Angew. Chem. Int. Ed. 2018, 57, 6082–6085; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6190–6193. [Google Scholar]
- 34. Kells P. M., Ouellet H., Santos-Aberturas J., Aparicio J. F., Podust L. M., Chem. Biol. 2010, 17, 841–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.
- 35a. Ma N., Chen Z., Chen J., Chen J., Wang C., Zhou H., Yao L., Shoji O., Watanabe Y., Cong Z., Angew. Chem. Int. Ed. 2018, 57, 7628–7633; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7754–7759; [Google Scholar]
- 35b. Chen J., Kong F., Ma N., Zhao P., Liu C., Wang X., Cong Z., ACS Catal. 2019, 9, 7350–7355; [Google Scholar]
- 35c.P. C. Cirino in Laboratory Evolution of Cytochrome P450 Peroxygenase Activity Vol. Ph.D California Institute of Technology, Pasadena, California 2004.
- 36. Podgorski M.N., Coleman T., Chao R. R., De Voss J. J., Bruning J. B., Bell S. G., J. Inorg. Biochem. 2020, 203, 110913. [DOI] [PubMed] [Google Scholar]
- 37. Sudhamsu J., Kabir M., Airola M. V., Patel B.A., Yeh S.-R., Rousseau D. L., Crane B. R., Prot. Express. Purif. 2010, 73, 78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sarkar M. R., Lee J. H. Z., Bell S. G., ChemBioChem 2017, 18, 2119–2128. [DOI] [PubMed] [Google Scholar]
- 39.
- 39a. Berry E.A., Trumpower B. L., Anal. Biochem. 1987, 161, 1–15; [DOI] [PubMed] [Google Scholar]
- 39b. Barr I., Guo F., Bio-protocol 2015, 5, e1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bell S. G., Tan A. B. H., Johnson E. O. D., Wong L.-L., Mol. BioSyst. 2009, 6, 206–214. [DOI] [PubMed] [Google Scholar]
- 41. Cowieson N. P., Aragao D., Clift M., Ericsson D. J., Gee C., Harrop S. J., Mudie N., Panjikar S., Price J. R., Riboldi-Tunnicliffe A., Williamson R., Caradoc-Davies T., J. Synchrotron Radiat. 2015, 22, 187–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Battye T. G. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. W., Acta Crystallogr. Sect. D 2011, 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Evans P. R., Murshudov G.N., Acta Crystallogr. Sect. D 2013, 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G.N., Pannu N. S., Potterton E.A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Acta Crystallogr. Sect. D 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aragão D., Aishima J., Cherukuvada H., Clarken R., Clift M., Cowieson N. P., Ericsson D. J., Gee C. L., Macedo S., Mudie N., Panjikar S., Price J. R., Riboldi-Tunnicliffe A., Rostan R., Williamson R., Caradoc-Davies T. T., J. Synchrotron Radiat. 2018, 25, 885–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moriarty N. W., Grosse-Kunstleve R. W., Adams P. D., Acta Crystallogr. Sect. D 2009, 65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Emsley P., Lohkamp B., Scott W. G., Cowtan K., Acta Crystallogr. Sect. D 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.
- 49a. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D., Acta Crystallogr. Sect. D 2012, 68, 352–367; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49b. Adams P. D., Afonine P. V., Bunkoczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., Acta Crystallogr. Sect. D 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Adams P. D., Read R. J., Zwart P. H., Hung L.-W., Acta Crystallogr. Sect. D 2008, 64, 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Afonine P. V., Moriarty N. W., Mustyakimov M., Sobolev O. V., Terwilliger T. C., Turk D., Urzhumtsev A., Adams P. D., Acta Crystallogr. Sect. D 2015, 71, 646–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen V. B., W. B. Arendall, III , Headd J. J., Keedy D.A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., Acta Crystallogr. Sect. D 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schrodinger, LLC in The PyMOL Molecular Graphics System, Version 1.4.1, Vol. 2015.
- 54. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 55. Lonsdale R., Oláh J., Mulholland A. J., Harvey J.N., J. Am. Chem. Soc. 2011, 133, 15464–15474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Krest C. M., Silakov A., Rittle J., Yosca T. H., Onderko E. L., Calixto J. C., Green M. T., Nat. Chem. 2015, 7, 696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bell S. G., Hoskins N., Xu F., Caprotti D., Rao Z., Wong L.-L., Biochem. Biophys. Res. Commun. 2006, 342, 191–196. [DOI] [PubMed] [Google Scholar]
- 58.R. Bruckner, in Organic Mechanisms : Reactions, Stereochemistry and Synthesis, (ed. D. M. Harmata) Springer Berlin Heidelberg, Berlin/Heidelberg, 2010, pp 737–826.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.