

Abstract
Therapeutic antibodies have broad indications across diverse disease states, such as oncology, autoimmune diseases, and infectious diseases. New research continues to identify antibodies with therapeutic potential as well as methods to improve upon endogenous antibodies and to design antibodies de novo. On April 27–30, 2022, experts in antibody research across academia and industry met for the Keystone symposium “Antibodies as Drugs” to present the state-of-the-art in antibody therapeutics, repertoires and deep learning, bispecific antibodies, and engineering.
Keywords: allergies, antibodies, antibody design, bispecific antibodies, SARS-CoV-2, therapeutic antibodies
INTRODUCTION
Therapeutic antibodies represent a large portion of biologic drugs, with indications across oncology, autoimmune diseases, and infectious diseases. As antibody therapies become more widely available, finding ways to improve their efficacy, while minimizing resistance, is increasingly important. In addition, designing antibodies with new and improved functions, such as bispecific antibodies, as well as harnessing the unique features of non-IgG antibodies has the potential to unlock new modalities for antibody treatments.
On April 27–30, 2022, experts in antibody research across academia and industry met for the Keystone symposium “Antibodies as Drugs” to discuss recent progress in the development and use of therapeutic antibodies. Researchers presented work characterizing SARS-CoV-2 antibodies and how they contribute to immunity, noting that neutralization does not always lead to disease protection, and isolating antibodies from individuals infected with various pathogens, including malaria, HIV-1, and SARS-CoV-2, not only as a starting point for therapeutic antibodies but also to inform vaccination strategies. Several speakers also described how patient-derived antibodies from other conditions, such as autoimmune disorders and allergies, can inform treatment strategies. Another key theme throughout the meeting was the rise of bispecific antibodies, especially in oncology. Speakers discussed new technologies and approaches to develop bispecific antibodies and presented new data on several novel bispecific antibodies. Finally, several speakers described the use of computational methods in antibody design, predicting antibody–epitope interactions, and identifying new antibodies with improved properties that are inaccessible to in vivo approaches.
KEYNOTE ADDRESS: CHARACTERIZATION OF SARS-COV-2 ANTIBODIES FROM THE CORONAVIRUS IMMUNOTHERAPY CONSORTIUM
Erica Ollmann Saphire from the La Jolla Institute for Immunology opened the meeting with the keynote address on the characterization of SARS-CoV-2 antibodies by the Coronavirus Immunotherapy Consortium (CoVIC). CoVIC has acquired approximately 400 antibodies from across industry, academic, and government labs. Their goal is to independently analyze these antibodies and identify potentially effective antibody cocktails that are amenable to deployment in low-to-middle-income countries. Another key goal is to generate lasting resources for the field. With that in mind, the Consortium has made all their data freely available.
One of the unique aspects of CoVIC is that antibodies come from a wide range of sources, including SARS-CoV-1 survivors, SARS-CoV-2 survivors, vaccinees, nanobodies, and engineered multivalent formats. Antibodies are submitted by various companies and groups to CoVIC and distributed to experts in the field to conduct various analyses in parallel, including binding, competition, neutralization, immune function assays, structure analyses, and in vivo studies.
One of the key tasks of CoVIC is to identify an antibody cocktail as a potential therapeutic. Toward that goal, researchers have conducted fine-resolution epitope mapping using competition analyses via surface plasmon resonance to group antibodies. They identified seven major antibody categories, which can be further subdivided into subcategories based on their epitope in the receptor-binding domain (RBD) of the spike protein. Saphire showed how these categories are predictive of antibody function and can provide information on the antibody binding footprint, variant susceptibility, binding mode, and neutralization capacity. Saphire foresees creating an antibody kit for researchers to conduct competition analyses with their antibody of interest to understand the binding mode of their antibody without the need for structural analyses. They built a competition grid of 400 antibodies to help inform antibody cocktails by pairing antibodies that do not compete with each other. Saphire focused on 50 antibodies that represent a range of sources, germlines, and categories. She showed that certain antibody groups are more susceptible to escape by the beta and gamma SARS-CoV-2 variants, with some outliers that actually show increased neutralization. In general, there appeared to be a tradeoff between neutralization potency and breadth.1 In vivo analyses in a mouse model conducted by the Texas Biomedical Research Institute indicate that neutralization is necessary but not sufficient for protection. Saphire caveated this observation with the fact that many of the antibodies in their study were likely chosen based on their neutralization activity. Additional studies are needed to understand how antibodies may confer protection via other mechanisms.
Finally, Saphire described efforts to reengineer the SARS-CoV-2 spike protein as a potentially more effective vaccine. They have developed a more native-like stable spike protein that induces a higher-titer, longer-lasting neutralizing antibody response than the S-2P variant used in current vaccines.
THERAPEUTIC ANTIBODIES FROM PATIENTS
Identifying beneficial antibodies from autoimmune polyendocrinopathy candidiasis ecto-dermal dystrophy patients
Adrian C. Hayday from the Francis Crick Institute described the potentially therapeutic benefits of naturally occurring antibodies in patients with autoimmune polyendocrinopathy candidiasis ecto-dermal dystrophy (APECED), a rare condition that results from loss-of-function mutations in the autoimmune regulator protein, the primary regulator of central tolerance. Patients with APECED typically have autoimmune-related issues owing to the proliferation of autoreactive T cells. Hayday showed, however, that these patients also have high-affinity-neutralizing autoantibodies that may have beneficial effects. He focused on anticytokine antibodies in patients with APECED.2 Work by others has shown that these antibodies are biologically active; thus, antibodies against IL-17 and IL-22 were capable of neutralizing host innate immune protective mechanisms,3 and APECED patients with pre-existing antibodies to type I interferons are reportedly at higher risk for severe COVID-19 disease.4 Self-reactive antibodies also offer potentially beneficial effects, including an increased potential to mount an antitumor immune response.5
Hayday focused on the potential for anti-interferon antibodies to protect against autoimmune diseases. Many patients with APECED have autoreactive profiles associated with type 1 diabetes yet do not develop the disease. In particular, those with potent neutralizing antibodies against cytokines implicated in type 1 diabetes are less likely to have the disease.6 He suggested that antibodies that neutralize a target cytokine associated with a disease may provide protection against that disease. Hayday’s group has characterized the anti-interferon antibodies in patients with APECED and showed that they are very high affinity and specific for the native epitope conformation.2 One such anti-interferon antibody, 19D11, is being investigated as a therapy for select autoimmune rheumatic diseases. Initial results show that 19D11 potently neutralizes interferon-alpha activity and has a good initial safety profile in nonhuman primate (NHP) models.7 Given the high affinity of these antibodies, Hayday also foresees the potential to use them in diagnostic assessments and patient stratification.8
Developing hyperimmune IgG from convalescent plasma
Timothy D. Wiltshire from the Mayo Clinic described the development of hyperimmune IgG from convalescent plasma to treat COVID-19. Convalescent plasma was a common therapy for those with the severe disease early in the pandemic before effective antivirals and vaccines were available. The US FDA approved an expanded access program for convalescent plasma (the US Convalescent Plasma Program) that ultimately collected plasma from more than 100,000 individuals and treated almost 95,000 patients.15 Wiltshire has been involved in developing hyperimmune IgG from convalescent plasma as a therapy for patients with severe COVID-19. Compared to convalescent plasma, hyperimmune IgG does not require blood type matching, is highly concentrated, and can be administered at a defined, consistent dose. The Mayo Clinic has set up a GMP facility to produce and prove the purity, identity, and potency of the hyperimmune IgG. Recent in vitro data show that hyperimmune IgG preparations obtained from donors prior to the Delta wave have neutralization activity against Delta and Omicron variants, likely owing to its polyclonal nature.16 Given that effective therapies and vaccines are now widespread, the role of hyperimmune IgG will likely be in immunocompromised patients. Wiltshire presented a case study of a patient with refractory COVID-19 who had been hospitalized multiple times over the course of 1 year and continued to test positive via PCR despite treatment with antivirals and convalescent plasma. Treatment with a high-titer antibody eventually resulted in PCR negativity and discharge from the hospital.17
Identification of a broadly reactive antibody against Streptococcus pneumoniae
Jarrod J. Mousa from the University of Georgia described research on generating a broadly reactive antipneumococcal antibody treatment. While current vaccines are effective against pneumococcal disease, they only contain a small fraction of the more than 100 serotypes of Streptococcus pneumoniae. The current disease is primarily driven by serotypes not included in vaccines or by serotypes for which vaccines are ineffective. Mousa’s group has isolated and characterized monoclonal antibodies that target the conserved bacterial surface protein, PhtD. They identified several binding epitopes within PhtD and showed that anti-PhtD antibodies are broadly reactive against S. pneumoniae serotypes and can protect against infection when given prophylactically or as a treatment in mice.18 Antibodies that target the conserved N-terminus of PhtD are also reactive against other Pht family members. Mousa showed that administration of a single PhtD antibody prolongs survival in mice coinfected with influenza virus and S. pneumoniae. Combining different antibodies that target different epitopes provided synergistic protection.19
NEW TARGETS AND TOOLS
IgG FcRn blockade therapeutics
David P. Humphreys from UCB Pharma discussed the rationale and data for blocking FcRn to treat autoimmunediseases. FcRn is a receptor expressed at the placental barrier, vascular endothelium, and mucosal epithelium that binds to and recycles IgG antibodies and other proteins like albumin. The binding affinity of FcRn for IgG is pH dependent as titration of key histidine residues in the binding site impacts affinity.20 Humphreys advised researchers to consider the impact on FcRn binding when engineering the Fc of antibodies as mutations can enhance or decrease FcRn binding and impact the antibody’s half-life. Several rare autoimmune disorders, such as myasthenia gravis21 and immune thrombocytopenic purpura (ITP),22 are driven by IgG autoantibodies. Developing individual drugs for each one would be costly and unfeasible. Therefore, several companies are pursuing FcRn blockade, given its potential to provide benefits in multiple diseases by decreasing autoantibody half-life.23 Humphreys showed how FcRn antagonists differ in characteristics, including their Fc sequences, impact on serum albumin levels, target binding affinity and valency. However, it is currently unclear whether any of these differences translate into functional or clinical differences. One FcRn antagonist, efgartigimod, was approved by the US FDA in 2021. Efgartigimod is an engineered IgG1 Fc antibody fragment used for the treatment of myasthenia gravis. Additionally, Humphreys showed published data on an anti-FcRn antibody UCB Pharma is developing, rozanolixizumab. In both mice (transgenic for human FcRn) and humans, rozanolixizumab resulted in IgG clearance while having no meaningful effect on IgM, A, E, or D concentrations or albumin levels. Rozanolixizumab is being investigated in patients with generalized myasthenia gravis and other autoimmune disorders.24–26
Enhancing IgG hexamerization in therapeutic antibodies
Esther C. W. Breij from Genmab described the company’s HexaBody® technology and how it is being applied to develop novel antibody-based therapies. The HexaBody platform enhances IgG antibody hexamerization via a single point mutation in the IgG Fc domain.27 IgG hexamerization has been shown to be important for the binding of complement protein C1q to membrane-bound antibodies, which initiates the complement cascade.28,29 HexaBody molecules show enhanced complement-dependent cytotoxicity (CDC) while maintaining many of the features of wild-type IgG, such as FcγR binding and pharmacokinetic characteristics.27 Breij described three HexaBody molecules under development for hematologic and solid cancers. The first one, HexaBody-CD38, targets CD38, which is expressed on malignant plasma cells in multiple myeloma (MM). HexaBody-CD38 shows higher CDC activity than the anti-CD38 antibody daratumumab (currently approved for multiple indications in MM) in tumor cell lines as well as in malignant plasma cells from MM patient bone marrow samples. Its CDC activity is dependent on the hexamerization-enhancing mutation.30,31 The second one is a bispecific antibody that targets two nonoverlapping epitopes in CD37, which is highly expressed on mature B cells and B cell malignancies. Current anti-CD37 antibodies do not elicit CDC. Breij showed that both the hexamerization-enhancing mutation and dual epitope targeting unlock or enhance CDC when introduced separately and show a synergistic effect on CDC when combined in a single bispecific antibody.32 DuoHexaBody-CD37 consistently demonstrated CDC activity in samples from patients with B cell malignancies.33 The first-in-human trial in patients with relapsed/refractory B cell non-Hodgkin’s lymphoma is underway.34
Finally, Breij described the use of HexaBody to initiate non-CDC mechanisms, namely the activation of clustering-dependent membrane receptors. HexaBody-DR5/DR5 targets DR5, a death receptor that initiates apoptosis upon hyperclustering. Similar to DuoHexaBody-CD37, the combination of enhanced hexamerization and dual epitope targeting was found to be elicit effector functions most efficiently. However, HexaBody-DR5/DR5 is a combination of two DR5-specific HexaBody molecules (rather than a bispecific antibody with a hexamerization-enhancing mutation) that targets two nonoverlapping DR5 epitopes; while hexamerization-enhanced monoclonal DR5 antibodies already showed improved potency compared to wild-type antibodies, the combination of the two hexamerization-enhanced antibodies induced apoptosis more potently.35
Quantifying plasma cell repertoires in ITP and SARS-CoV-2
Pierre Bruhns from the Institut Pasteur Paris presented work on mapping the autoimmune plasma cell repertoire in patients with ITP, an autoimmune disorder in which autoreactive antibodies against surface proteins found on platelets induce platelet phagocytosis and destruction, resulting in an increased risk of bleeding.36 Bruhns’s group has mapped the distribution of antibody-secreting cells and quantified antibody affinity in patients with ITP using DropMap, a droplet microfluidic technique that measures antibody secretion and affinity for their antigen at the single-cell level.37 He showed that autoreactive antibody-secreting cells were present in the spleen, blood, and bone marrow of ITP patients. All three compartments produced autoreactive antibodies with low, medium, and high affinities, though there was significant variability between patients in both the level of antibody-producing cells and the distribution of antibody affinity. Treatment with the anti-CD38 antibody daratumumab for the depletion of antibody-secreting cells led to ITP remission in some patients, but no others.38 Bruhns’s group, in collaboration with Matthieu Mahévas’ group (Institut Necker Enfants Malades, Paris, France) is now investigating clonal relationships between high-affinity autoreactive cells from different organs in these patients. Matteo Broketa from Bruhns’s group presented unpublished work using DropMap to characterize the affinities of SARS-CoV-2 RBD-specific antibody-secreting cells after vaccination, in collaboration with the Mahévas group.
Leveraging antitumor antibodies for therapeutics
Genta Furuya from the University of Tokyo described the potential of tumor-resident antibodies as anticancer therapies. Characterization of the global landscape of the tumor-infiltrating Ig repertoire has identified sulfated glycosaminoglycan (GAG) as a major antigen in the tumor microenvironment.39 Furuya presented unpublished data further profiling the type of GAG found in tumors and identifying anti-GAG antibodies as potential therapeutic agents.
Considering immune memory in SARS-CoV-2 vaccine design
Blake M. Hauser from Aaron Schmidt’s lab at the Ragon Institute of MGH, MIT, and Harvard/Harvard Medical School presented work on rationally designed immunogens to enable immune focusing after SARS-CoV-2 exposure. Much of the vaccine research for SARS-CoV-2 has been in a virus-naive population. However, as the virus continues to spread and individuals continue to get vaccinated, future efforts must consider the effect of immune imprinting, or pre-existing immunity, on the immune response to other coronaviruses. Hauser designed several immunogens to focus the serum antibody response to the ACE2 receptor-binding motif (RBM) and the non-RBM portion of the RBD of the SARS-CoV-2 spike protein. To focus the serum antibody response toward the RBM, they created immunogens composed of the SARS-CoV-2 RBM grafted onto other coronavirus scaffolds or masked the non-RBM portion of the RBD via hyperglycosylation. Additionally, they engineered immunization regimens that used hyperglycosylation to mask the RBM of the SARS-CoV-2 RBD or that preferentially presented the non-RBM portion of the RBD, with the goal of focusing the antibody response toward the non-RBM portion of the RBD. Immunizing mice pre-exposed to wild-type SARS-CoV-2 spike with these different RBM-focusing and non-RBM-focusing immunogens showed that broad, potent neutralization of related sarbecoviruses could be achieved by boosting with RBM-focusing immunogens within the context of pre-existing immunity to SARS-CoV-2. Hauser also stressed that the protein engineering strategies used in this study can be adapted to other viral glycoproteins to target conserved epitopes.40
ANTIBODIES FROM WITHIN: VACCINATION AND DELIVERY TECHNOLOGIES
Matthieu Mahévas from INSERM presented data on tracking spike-specific memory B cells after infection with or vaccination against SARS-CoV-2. Data from early in the pandemic questioned the longevity of memory B cells after infection based on observations that SARS-CoV-2 disrupts germinal center reactions in patients with severe disease.41 To better understand whether SARS-CoV-2 compromises the generation of long-lived, affinity-matured B cells, Mahévas’s group conducted a longitudinal study tracking spike-specific B cells in patients with mild and severe COVID-19. He showed that early in infection, the immune response consists of two synchronous B cell responses: near germline B cell clones that recognize SARS-CoV-2 and pre-existing highly mutated memory B cells that are cross-reactive against seasonal coronaviruses. Over time, spike-specific B cells accumulate within the mature memory B cell compartment and are stable up to 6 months post-infection.42–44 Mahévas also showed how the diversity of the memory B cell pool changes over time and whether it provides protection against emerging SARS-CoV-2 variants. In a separate longitudinal cohort study, vaccination elicited robust expansion of RBD-specific memory B cells; however, the diversity of the B cell pool was maintained. The resulting B cell pool contained a high percentage of high-affinity memory B cells and neutralizing memory B cells against wild-type virus. While the percentage of clones against variants was decreased, the diversity of the memory B cell pool was sufficient such that some high-affinity clones against the variants were present as well, thus leading to a strong yet incomplete loss of neutralization against variants of concern.45
The Moderna COVID-19 vaccine: 1 year of human experience
Andrea Carfi from Moderna presented recent data for the Moderna mRNA vaccine, mRNA-1273 (Spikevax®), and described the company’s efforts to develop combination vaccines to address waning immunity and variants of concern. Early data from the mRNA-1273 clinical program showed that the vaccine elicits a strong T cell response and a neutralizing antibody response that persists through 6 months after boosting.46 Prior to Omicron, mRNA-1273 vaccination continued to provide robust protection against symptomatic disease as variants emerged, even though breakthrough infections increased.47–49 However, real-world data show that vaccine effectiveness against Omicron infection is significantly reduced, providing only ~5% protection after 1 year.48 Moderna is testing several boosting strategies to increase protection against new variants like Omicron. Carfi described a trial assessing a third dose of the current mRNA-1273 vaccine. In the study, a booster dose (third dose) increased neutralizing antibody titers against Omicron, but levels declined disproportionately faster than neutralizing antibodies against the wild-type virus.50 Three-dose vaccine effectiveness against infection also decreased quickly against Omicron, ending at approximately 50% 2 months after the last dose.48 Importantly, protection against severe disease and hospitalization remained high.
In a second boosting strategy, Moderna is evaluating the impact of the third dose of a new bi-valent vaccine (mRNA-1273.211) composed of mRNA-1273 and a mRNA encoding for the SARS-CoV-2 beta spike protein. The bi-valent vaccine elicited higher levels of binding and neutralizing antibodies against all tested variants, including Omicron, than the same dose of mRNA-1273. These data suggest that a strategy consisting of mRNA encoding for multiple SARS-CoV-2 spike sequences might provide better protection than vaccinating with a single variant.
Finally, Moderna is also developing a next-generation vaccine that encodes for portions of the spike protein critical for neutralization (mRNA-1283). A phase 1 study showed that mRNA-1283 elicits comparable neutralizing antibody levels of mRNA-1273 against ancestral SARS-CoV-2. This new vaccine may allow for dose-sparing and simpler storing conditions.
The antibody response to secondary dengue infection
Tineke Cantaert from the Institut Pasteur Cambodia discussed antidengue antibody responses that affect the severity of dengue fever. The majority of the 400 million cases of dengue virus infection are asymptomatic; but approximately 25% of infected individuals develop dengue fever.51,52 Patients are at higher risk of developing the severe disease if they had been previously infected with a different serotype of the dengue virus. This mismatch between the infecting serotype and the memory adaptive immune response is thought to result from cross-reactive antibodies and/or cross-reactive T cells, which could lead to the immunopathology associated with dengue fever.51 However, since less than 5% of patients with pre-existing antidengue immunity develop severe disease, there must be additional susceptibility factors determining disease severity. Cantaert described a longitudinal study of patients with dengue fever and their close contacts to identify factors associated with asymptomatic infection. They identified differences in the immune response between asymptomatic and hospitalized patients during the acute phase of infection. Asymptomatic individuals showed increases in antigen presentation pathways, T cell receptor (TCR) and BCR signaling, T cell apoptosis, and FcγRIIb signaling, while hospitalized patients showed increases in plasmablast development.53 They confirmed that direct dengue virus infection of B cells can increase their differentiation into plasmablasts.54 They also found that patients with severe dengue fever have increased IgG1 afucosylation, compared with asymptomatic patients. These afucosylated antibodies are elicited upon secondary dengue virus exposure, and their levels are predictive of disease severity during secondary infection. Cantaert noted that IgG1 afucosylation, combined with dengue virus immune status, can provide an accurate determinant for disease susceptibility and clinical severity.55
Anti-HIV and antimalarial antibodies elicited by vaccination
Peter D. Kwong from the Vaccine Research Center, NIAID discussed efforts to characterize anti-HIV and antimalarial vaccine-mediated antibodies. With regard to malaria, the dominant antigen is PfCSP, the most abundant antigen present during the sporozoite stage of the pathogen life cycle. Treatment with the human antibody CIS43, which targets the junctional epitope within PfCSP, can protect against disease for up to 9 months.56,57 Improving upon CIS43 has been challenging due to the repeat nature of the antigen and the lack of high-throughput assays to assess antibody function. Kwong and Prabhanshu Tripathi, also at the Vaccine Research Center, NIAID, described a knockin mouse model to increase the potency of CIS43. In brief, mice are engineered such that their B cells express inferred human germline CIS43 BCRs. The mice are then immunized with the junctional epitope and variant CIS43 antibodies are isolated and characterized.58 Tripathi showed that this method elicited highly protective antibodies, one of which, m43.151, provides approximately three-fold better protection than CIS43. They also observed that antibodies with higher affinity to the junctional peptide demonstrated higher protection. To improve the potency of CIS43 even further, they focused on increasing affinity to the junctional peptide using various platforms, which resulted in antibodies that were, in mice, 10-fold more potent than CIS43.58 The group is continuing to identify other antibody properties that correlate with increased protection to ultimately develop prophylactic antibody treatments that provide long-term protection at low doses.
Kwong continued by describing other work on characterizing HIV-neutralizing antibodies elicited by vaccination and the goal of generating an effective HIV vaccine. Vaccine-elicited antibodies have lower potency and breadth than naturally elicited antibodies because the latter have had many years to develop in chronically infected patients. The lineage of one such broadly neutralizing antibody, VRC01, was mapped over 15 years of sampling, demonstrating how a single B cell can evolve such that a diverse collection of broadly neutralizing antibodies has been generated.59 Kwong showed how the Vaccine Research Center is using antibodies identified from HIV-infected donors to identify, by working backward, high-quality immunogens to use in vaccines.60,61 Structures of several antibodies in complex with the HIV envelope protein have identified 20 antibody classes that cluster as six epitopebinding categories.62 As potential vaccine targets, they are focusing on glycan-free epitopes within the viral fusion peptide and CD4. Kwong showed that vaccine-elicited antibodies targeting these epitopes have a high breadth and diverse recognition modalities.
THE RISE OF BISPECIFICS
Increasing tumor selectivity with bispecific antibodies
Jonathan H. Davis from Invenra described the company’s two lead bispecific antibodies and their B-Body® Platform and SNIPER® Targeted Bispecifics. The B-Body platform creates high-quality IgG-like bispecific antibodies with properties similar to monoclonal antibodies, including excellent CMC behavior and yields (Figure 1A,B). The SNIPER approach enables precision targeting of two targets via a bispecific antibody to reduce toxicity and off-target effects while broadening the therapeutic window. These SNIPERs are designed to optimize monovalent binding to two separate targets, which ensures that they will preferentially bind to cells that express both targets. Davis described two SNIPER programs in preclinical development.
FIGURE 1.
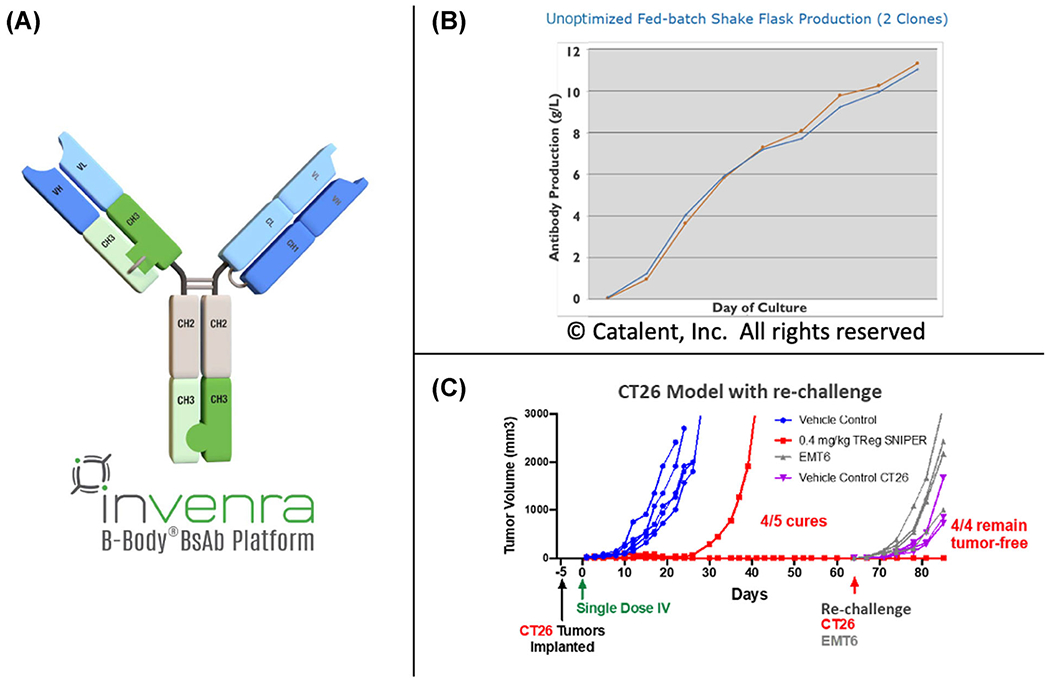
(A) Format of a B-Body. CH3 domains with mutations to drive heterodimerization replace one CH1/CL pair. (B) Nonoptimized production of several bispecific B-Body clones is 11 g/L. The final purified yield (also nonoptimized) was 5–6 g/L. (C) Treatment with the murine surrogate of a Treg SNIPER™ cures 4/5 mice at the lowest dose of 0.4 mg/kg dosed once. Higher doses cured all mice.
The first bispecific antibody was designed to deplete tumor regulatory T cells (Treg cells) by targeting a pair of receptors found together primarily on these cells, one at a high and one at a moderate level. They identified antibodies that target each receptor individually and generated a bispecific antibody that shows strong antibody-dependent cellular toxicity (ADCC) toward cells that express both targets. In mouse models, the murine surrogate Treg SNIPER bispecific showed a promising ability to selectively deplete tumor Treg cells while leaving normal levels of Tregs in theperiphery (Figure 1C). The second bispecific antibody was developed to target GD2, which is highly expressed on multiple tumors, and B7-H3, a T cell coinhibitory receptor. Monoclonal antibodies against GD2, an approved treatment for neuroblastoma, show antitumor activity but also cause severe pain in patients due to nonselective targeting of GD2-expression neurons. By using a SNIPER bispecific to preferentially target cells that express both GD2 and B7-H3, Invenra hopes to decrease the toxicity associated with targeting either receptor alone, while increasing the therapeutic window compared to current agents. They created a bispecific antibody by modulating the affinity of an existing anti-GD2 antibody and screening for B7-H3 antibodies to identify synergistic binders. Davis showed that the resulting candidate improved the specificity of ADCC compared to the marketed anti-GD2 antibody dinutuximab. He stressed that synergistic binding is key to developing bispecific antibodies that selectively target cells that express both targets. The geometry of the antibody binding on the cell surface is also important, as the second epitope must be accessible to the antibody when it is engaged with the first. Finding antibodies with the right binding affinities is dependent on the relative expression levels of the two targets. For targets that are expressed at roughly equal levels, antibody affinities can both be moderate, with synergy mostly dependent on geometry. For targets in which one is highly expressed compared to the other, reducing the affinity of the highly expressed target is key to achieving synergistic binding to cells expressing both targets.
Targeting tumor-infiltrating T cells with bispecific antibodies
Neil Brewis from F-star Therapeutics described an OX40/CD137 bispecific antibody, FS120, under development as an immuno-oncology treatment. Both CD137 and OX40 are expressed on tumor-infiltrating lymphocytes (TILs) and are involved in proliferation, activation, and survival. Monoclonal antibodies against OX40 have shown limited activity, while anti-CD137 antibodies are limited owing to associated liver toxicity. F-star hopes that targeting both receptors simultaneously will increase the safety and efficacy of targeting either one individually. FS120 is a tetravalent bispecific antibody that incorporates OX40 binding in the Fc region and CD137-specific Fabs. Brewis showed that FS120 binds human CD137 and OX40 simultaneously with subnanomolar affinity. In mice, an FS120 surrogate stimulated T cell activity and proliferation, exhibited strong antitumor activity, protected against rechallenge, and showed synergistic activity with an anti-PD-1 antibody. Engaging both targets was required for the activity. FS120 activated CD4+ and CD8+ T cells as well as NK cells and shows some potential to decrease Treg cell activity. This combined agonism approach was also shown to generate super-effector CD8+ T cells that clonally expand to greater levels, survive longer, and produce a greater quantity of cytokines, compared with T cells stimulated with an agonist of either costimulatory receptor individually.63 As OX40 and CD137 levels are normally low in the periphery, it is expected that FS120 will have a broad therapeutic window and selectively activate tumor-infiltrating T cells. F-star is evaluating which cancers show coexpression of CD137 and OX40 to inform the clinical program of FS120, and a phase 1 trial is underway in patients with advanced malignancies.
IgM-based bispecific antibodies
Bruce Keyt from IGM Biosciences described the company’s development of IgM-based bispecific antibodies. IgM antibodies exhibit high avidity given their ability to form multimeric structures with cell surface targets.64,65 Keyt focused on IGM-2323, a bispecific antibody that irreversibly binds to CD20 (a common marker of lymphoma cells) and engages CD3 on T cells. IGM-2323 shows superior binding to CD20+ cells and outcompetes a bivalent anti-CD20 IgG. IGM-2323 can kill lymphoma cells via two distinct mechanisms: CDC, which requires higher levels of antigen, and T cell-dependent cellular cytotoxicity, which functions at low antigen levels. Keyt noted that the increased avidity of IgM antibodies enables them to bind to cells that express low levels of a target; in the case of CD20, this is important as many patients who receive anti-CD20 antibodies as treatment develop resistance because of the downregulation of CD20. The superior binding ability and low cytotoxicity of IGM-2323, compared with IgG antibodies, may provide benefits in patients who are resistant to anti-CD20 therapy.65 And because IgM antibodies have a decreased potential to elevate cytokine levels, IGM-2323 may have an improved safety profile compared to IgG antibodies.66 In support of this, in a phase 1 trial in patients with relapsed/refractory B cell non-Hodgkin lymphoma, IGM-2323 demonstrated a 60% response rate with a low incidence of cytokine release syndrome.67 Phase 2 trials are underway.
Targeting γδ T cells with Gammabodies™
Felix L. Fennemann from LAVA Therapeutics described the company’s work to retarget γδ T cells, a subtype of T cells that have innate immunity features. They have a natural tendency to home to-, recognize and kill tumor cells. Higher levels of γδ T cells have been associated with better prognosis and increased survival in cancer patients.68,69 Via their Gammabody™ platform, Lava generates bispecific VHH-based antibodies that selectively target the γδ chain of cells expressing the Vγ9Vδ2 TCR and tumor-associated cell-surface molecules. They have developed a bispecific Gammabody™, LAVA-1223, that targets the TCR and EGFR, the latter being aberrantly (over-) expressed in many cancers and a target of several cancer therapies.70,71 Fennemann showed that LAVA-1223 binds to primary Vγ9Vδ2 T cells and is highly specific for EGFR in human cell lines. It induces potent, EGFR-dependent, Vγ9Vδ2 T cell activation and tumor cell lysis in various colorectal cancer cell lines with a good tolerability profile in NHPs.
Characterization of SARS-CoV-2 antibodies
Vincent Dussupt from Walter Reed Army Institute of Research discussed the characterization of monoclonal antibodies from SARS-CoV-2 patients. They isolated several high neutralization and binding antibodies that target the RBD and NTD. Dussupt showed that NTD-directed neutralizing antibodies have the unique ability to activate complement, while most RBD antibodies do not. In mice, all neutralizing antibodies were able to protect against disease. NTD antibodies provided protection at lower doses than RBD antibodies. Combining the two types of antibodies resulted in increased protection and suppression of the SARS-CoV-2 viral load. When assessed against SARS-CoV-2 variants of concern, most NTD antibodies lost their neutralization activity, but several RBD antibodies maintained high activity.72
REPERTOIRES, LEARNING, AND IN SILICO DESIGN
Keynote address: Computational modeling and design of antibody function
Arvind Sivasubramanian from Adimab discussed the company’s approach to using computational modeling to design bispecific antibodies. During his talk, Sivasubramanian focused on efforts to model Fab heterodimerization to increase cognate light chain–heavy chain pairing in bispecific antibodies. In an IgG-like bispecific antibody made up of two heavy chains and two light chains, there are 10 possible four-chain combinations. Combinations featuring heavy chain homodimers can be eliminated by using CH3 heterodimerization substitutions; however, avoiding the formation of light chain–heavy chain mismatches has been an active area of research. Data from Adimab show that there is a broad distribution of desired and undesired pairings for different bispecific antibodies, and it is difficult to predict whether a given heavy and light chain will pair correctly.
Sivasubramanian described one of Adimab’s solutions to optimize cognate light chain-heavy chain pairing. The approach consists of computationally designing novel hydrogen-bond networks via amino acid substitutions at the CH1:CK interface to encourage preferential pairing between the desired light chain–heavy chain pairs (positive states), while disfavoring amino acid substitutions that improve the interface between the mispairs (negative states). Computational designs with the largest energetic gap between the positive and negative states were selected for experimental testing. Sivasubramanian showed that designing only one of the CH1:CK interfaces in an IgG was not sufficient to eliminate the mispaired species. Using these single-interface designs as a starting point to design both CH1:CK interfaces, however, resulted in bispecific antibodies with a cognate pairing of more than 95% in IgGs that incorporate arbitrary pairs of wild-type variable domains. Adimab also conducted structural and biophysical analyses and found that the Fab domain orientations and CDR conformations, antigen binding affinity, and developability properties of the designed antibodies are similar to the wild-type forms.
De novo design of epitope-specific antibodies
Philip M. Kim from the University of Toronto discussed his group’s approach to computationally design protein–peptide binding and how they have applied this to antibody–epitope pairs. Kim’s de novo protein design algorithm incorporates three key principles.73 The first, transfer learning, addresses an important limitation for protein–peptide interactions, namely that there are few relevant structures in the Protein Data Bank (PDB). Transfer learning compensates for this data scarcity by pretraining the model on larger, similar datasets. In the case of protein–peptide interactions, this larger dataset consists of protein–fragment protein complexes. After this initial training, the model is then fine-tuned using the smaller, more relevant dataset. The second principle is incorporating graph networks to represent three-dimensional protein structures in a way that is unaffected by translation or rotation. The third principle, called attention, is a method to teach the algorithm long-range dependencies.73 Kim showed how they have applied deep learning to de novo protein design using the 100,000 structures and 70,000,000 sequences from PDB. Their deep graph neural network, ProteinSolver (design.proteinsolver.org), has accurately designed novel proteins and is available for researchers.
With regard to antibody design, Kim argued that computational methods can address many of the limitations of conventional protein engineering as they can systematically design the antigen-binding interface while considering other features important for stability, developability, and scaled-up manufacturing. He showed that for several known antigen–antibody pairs, much of the antigenic surface is not probed by known antibodies, leaving a lot of room for antibody design. Kim showed proof-of-concept data in which they redesigned Fabs to target novel epitopes on PD-L1 and other targets.
In silico improvement of an antimalarial antibody
Reda Rawi from the NIAID described the in silico improvement of the antimalarial antibody CIS43. Above, Kwong and Tripathi discussed an in vivo mouse model to improve the protection of CIS43 and make it a more viable candidate as a prophylactic treatment. One observation from that study was that antibodies with higher binding affinity to the junction peptide within the PfCSP antigen afforded higher protection.58
Rawi and his group developed a novel computational approach that uses residue–residue interaction energies to improve binding affinity to the junction peptide in the hopes of creating more potent antibodies. Initially, mutations were only allowed within a given radius of the junction peptide. Each amino acid in the antibody within this radius was mutated to each of the 20 amino acids and then the model estimated binding energies for single– and double–point mutations. Rawi’s group selected several antibodies to test experimentally. Among these was a new best-in-class antibody, P3-43, that showed good malarial protection in in vivo models. While experimental results showed that antibody–antigen binding affinity strongly correlated with in silico binding energies, suggesting that the model was successfully modeling the intended interaction, the correlation between binding affinity and protection was not universal.74 Rawi’s group is improving upon their in silico pipeline by devising ways to introduce mutations throughout the Fab region, not just around the antigen-binding site, and to select for mutations that improve other features, such as heavy–light chain interactions, solubility, half-life, and aggregation. As for P3-43, the group is moving forward with testing its ability to protect in a mosquito bite challenge and to assess its developability.
Predicting epitope binding
Eve Richardson from Charlotte Deane’s group at the University of Oxford presented the lab’s approaches to predicting epitope binding using structural modeling and deep learning. In general, the methods rely on comparing uncharacterized antibody sequences to a database of known antibody–epitope pairs and use structural and sequence similarity to predict epitopes for the uncharacterized antibodies. Richardson described three methods the lab has developed. The first, paratyping, groups antibodies based on sequence similarity within the predicted antigen-binding site. This method showed 84% precision when grouping antibodies that bind to Pertussis toxoid and identified new antibodies with different IGHV and IGHJ genes that would not normally be grouped looking at clonotype alone.75 The second method, AbLigity, combines paratyping with structural information. This method was shown to group antibodies to the same epitope with 95% precision.76 Finally, the third method, SPACE, groups antibodies using predicted structural information. When applied to the Coronavirus Antibody Database, SPACE accurately identified functionally similar, yet sequence diverse, antibodies.77 The three methods demonstrate that structural information derived by computational modeling and predicted paratype can be used separately or together to group sequence-distinct antibodies that bind to the same epitope.
Computational-directed improvements in developability and immunogenicity for therapeutic antibodies
Daniel Leventhal from Generate Biomedicines discussed the company’s approach to designing therapeutic antibodies using machine learning. Developing therapeutic biologics requires not only considering the drug’s efficacy but also manufacturability or developability, which includes stability, lack of aggregation, and expression levels, as well as immunogenicity. Generate Biomedicines has developed computational predictors for a variety of critical attributes to incorporate these considerations earlier in the drug development process. In this way, they hope to limit the attrition rate of promising effective drugs as they progress to later stages in the clinical pipeline. As a proof-of-concept demonstration, Leventhal described how the company has applied computational design approaches which utilize these predictors to improve the antibody bococizumab, which was discontinued during phase 3 clinical evaluation due to high immunogenicity and poor developability.78 Leventhal showed that redesigning bococizumab to optimize either developability (i.e., reduced self-association) or immunogenicity (reduced MHC II presentation) could improve upon these features. However, improving only one feature often resulted in making the other feature worse. Focusing on both simultaneously in the design phase showed that developability and immunogenicity could be co-optimized without compromising antigen binding. Leventhal hopes that computational design approaches like this can be broadly applied to candidate protein therapeutics early in development to maximize their probability of success during clinical testing and beyond.
Characterization of a new influenza B–directed antibody
Rachael M. Wolters from James Crowe’s lab at Vanderbilt University presented unpublished data on the characterization of an influenza B neuraminidase–directed antibody. Other influenza B antineuraminidase antibodies have been identified and characterized.79 Wolters described the breadth, function, and protective potential of a new human neuraminidase-directed antibody isolated from a naturally infected donor in 2020.
NOVEL THERAPEUTIC ANTIBODIES IN THE CLINIC
CD137 × PDL-1 bispecific antibody for tumor-selective T cell stimulation
Cecile A. W. Geuijen from Merus described the company’s bispecific antibody, MCLA-145, that targets CD137 and PD-L1. Merus focuses on the development of bispecific and trispecific antibodies for oncology indications. MCLA-145 was designed to activate TILs by inducing CD137, a costimulatory receptor expressed on antigen-experienced T cells that activate T cell activity, and antagonizing PD-L1, an immunosuppressive receptor often expressed on tumor cells. In preclinical models, combining a CD137 agonist and a PD-L1 antagonist synergistically enhanced tumor cell killing.80,81 Trimerization of CD137 is integral for optimal activity.
The idea behind a CD137 × PD-L1 bispecific antibody is that in the immunosuppressive environment of the tumor microenvironment, where PD-L1 is high, binding of MCLA-145 to PD-L1 will facilitate CD137 clustering and tumor-specific activation. Geuijen showed that MCLA-145 blocks the ligands of both CD137 and PD-1, and it increases antigen-specific CD8+ T cell priming and differentiation while overcoming Treg cell and myeloid cell suppressor activities. In mouse models, MCLA-145 increased CD8+ T cell frequency in the tumor and decreased the frequency of PD-L1-expressing monocytes. Additional data showed that binding of MCLA-145 results in the translocation of PD-L1 and CD137 toward a contact zone between antigen-experienced T cells that express CD137, and PD-L1–expressing tumor cells or APCs. This interaction activates CD137 and promotes the release of cytokines that induce T cell proliferation and activity.82
CD28 bispecific antibodies as costimulatory agents
Matthew A. Sleeman from Regeneron described the company’s development of costimulatory CD28 bispecific antibodies for the treatment of cancer. Regeneron’s VelocImmune mouse platform can be engineered to generate potent antibodies that use a single “universal” light chain. The pairing of two different antibodies with a universal light chain generates bispecific antibodies that eliminate the problem of mispairing with different variable heavy and light chains. In addition, Regeneron bispecific platform incorporates a substitution in the Fc domain that introduces asymmetric protein A binding, which allows for selective isolation of the heterodimeric bispecific antibodies once paired. Regenerons rational for combining immune-oncology agents is predicated on two concepts: whether a tumor is immunologically “hot” (i.e., some evidence of TILs) or “cold” (has noTILs). Immunologically hot tumors suggest that endogenous cancer or neo-antigens are presented to T cells but that their activation is suppressed, often via activation of the PD-L1 pathway. The use of anti-PD1 or PDL1 antibodies has revolutionized cancer care over the last decade; however, durable response rates still need to be improved. Therefore, the combination of an anti-PD-1, to release the brakes on T cells, with a costimulatory bispecific antibody targeting CD28 and a tumor-specific antigen, to act as the accelerator pedal, would be expected to enhance tumor-specific killing. In immunologically cold tumors, a different approach is being pursued that replicates antigen presentation via bispecific antibodies that target a tumor-specific antigen and CD3 on the T cell to generate what is described as signal 1, while also providing costimulation of the T cell (signal 2) via a bispecific antibody specific for a tumor-specific antigen and CD28.
While the above is an oversimplification of tumor biology, Regeneron has taken these broad concepts to help design therapeutic combinations that can enhance antitumor immunity. In one example, Regeneron developed a bispecific antibody to target a common antigen in prostate cancer, PSMA, and the costimulatory molecule CD28. The combination treatment with this bispecific antibody, PSMA × CD28, with an anti-PD-1 agent promoted tumor clearance and enhanced survival in mice compared with either therapy alone. Furthermore, immune profiling of tumor-infiltrating CD8+ T cells showed that anti-PD-1 treatment alone expanded effector T cells, but the combination treatment resulted in a greater proportion of cells with a memory-like phenotype.83
In the second example, Regeneron pursued a strategy by generating bispecific antibodies with specificity for MUC16 × CD28 and MUC16 × CD3 for the treatment of ovarian cancer. In in vitro studies, the costimulatory signal alone resulted in no activity, while the combination of the CD28 and CD3 bispecifics enhanced T cell activation, proliferation, and cytokine release.84 These examples demonstrate that providing costimulation using bispecifics activating CD28 can provide the crucial second signal to activate T cells either in combination with bispecifics that target CD3 or with PD1 checkpoint inhibitors. Moreover, these data demonstrate a novel technology platform that can harness the power of bispecific antibodies generated for a wide variety of different tumor antigens and cancers. Clinical studies are now ongoing evaluating these antibodies in patients with prostate or ovarian cancer.
Translating human antibodies for the treatment of allergies and neurological disorders
Niccolo Pengo from Mabylon AG described their efforts to translate human antibodies for the treatment of allergies and neurological disorders. Pengo started by describing Mabylon’s approach to identifying antibodies that recognize peanut epitopes and thereby block interactions between allergens and IgE antibodies, thus preventing an allergic reaction. Mabylon has created a cocktail of antibodies that recognize major nonoverlapping peanut epitopes from a cohort of approximately 100 individuals with peanut allergies. Pengo showed that one antibody cocktail, MY006, binds with high affinity to the major peanut allergens, competes with patient IgE antibodies, and inhibits basophil degranulation; in mouse models, MY006 prevented anaphylaxis and allergic reactions. Mabylon is also working to reformat the antibodies contained in MY006 into a multispecific format to simplify manufacturing and production. The company is using a similar antibody discovery pipeline to develop passive immunotherapy for pollen allergies.
Pengo also described Mabylon’s efforts to identify antibodies that target protein aggregates implicated in neurodegenerative diseases. Because there is not easy way to identify people who have raised an antibody response to these targets, Mabylon has partnered with the University Hospital Zurich to screen tens of thousands of volunteers for antibodies. Pengo focused on TDP-43, a protein implicated in ALS and other proteinopathies; Mabylon has identified anti-TDP-43 antibodies from a high-throughput screen of 60,000 individuals that preferentially bind the aggregated form of TDP-43. They are testing these antibodies in preclinical settings.
Broadly neutralizing Ebola antibodies
Francesca Rose Donnellan from Simon Draper’s lab at the University of Oxford presented unpublished work on the generation and characterization of synergistic broadly neutralizing antibodies that target the Ebola virus glycoprotein.
ACKNOWLEDGMENTS
T. Cantaert is an HHMI/Welcome Trust international research scholar (208710/Z/17/Z) and is supported by NIH grant U01AI151758–01.
Footnotes
COMPETING INTERESTS
N. Pengo is an employee of Mabylon AG. Outside of the work described herein, P. Bruhns received consulting fees from Regeneron Pharmaceuticals.
REFERENCES
- 1.Hastie KM, Li H, Bedinger D, Schendel SL, Dennison SM, Li K, Rayaprolu V, Yu X, Mann C, Zandonatti M, Diaz Avalos R, Zyla D, Buck T, Hui S, Shaffer K, Hariharan C, Yin J, Olmedillas E, Enriquez A, … Saphire EO (2021). Defining variant-resistant epitopes targeted by SARS-CoV-2 antibodies: A Global Consortium study. Science, 374, 472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meyer S, Woodward M, Hertel C, Vlaicu P, Haque Y, Kärner J, Macagno A, Onuoha SC, Fishman D, Peterson H, Metsküla K, Uibo R, Jäntti K, Hokynar K, Wolff ASB, Krohn K, Ranki A, Peterson P, Kisand K, … Peet A (2016). AIRE-deficient patients harbor unique high-affinity disease-ameliorating autoantibodies. Cell, 166, 582–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kisand K, Link M, Wolff ASB, Meager A, Tserel L, Org T, Murumägi A, Uibo R, Willcox N, Trebusak Podkrajsek K, Battelino T, Lobell A, Kämpe O, Lima K, Meloni A, Ergun-Longmire B, Maclaren NK, Perheentupa J, Krohn KJE, Peterson P (2008). Interferon autoantibodies associated with AIRE deficiency decrease the expression of IFN-stimulated genes. Blood, 112, 2657–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann H-H, Zhang Y, Dorgham K, Philippot Q, Rosain J, Béziat V, Manry J, Shaw E, Haljasmägi L, Peterson P, Lorenzo L, Bizien L, Trouillet-Assant S, Dobbs K, De Jesus AA, … Casanova JL (2020). Autoantibodies against type I IFNs in patients with life-threatening COVID-19.Science, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Träger U, Sierro S, Djordjevic G, Bouzo B, Khandwala S, Meloni A, Mortensen M, & Simon AK (2012). The immune response to melanoma is limited by thymic selection of self-antigens. PLoS One, 7, e35005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hertel C, Fishman D, Lorenc A, Ranki A, Krohn K, Peterson P, Kisand K, & Hayday A (2019). Response to comment on “AIRE-deficient patients harbor unique high-affinity disease-ameliorating autoantibodies.” eLife, 8, e45826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duguet F, Ortega-Ferreira C, Fould B, Darville H, Berger S, Chomel A, Leclerc G, Kisand K, Haljasmägi L, Hayday AC, Desvaux E, Nony E, Moingeon P, & De Ceuninck F (2021). S95021, a novel selective and pan-neutralizing anti interferon alpha (IFN-α) monoclonal antibody as a candidate treatment for selected autoimmune rheumatic diseases. Journal of Translational Autoimmunity, 4, 100093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodero MP, Decalf J, Bondet V, Hunt D, Rice GI, Werneke S, Mcglasson SL, Alyanakian M-A, Bader-Meunier B, Barnerias C, Bellon N, Belot A, Bodemer C, Briggs TA, Desguerre I, Frémond M-L, Hully M, Van Den Maagdenberg AMJM, Melki I, … Duffy D. (2017). Detection of interferon alpha protein reveals differential levels and cellular sources in disease. Journal of Experimental Medicine, 214, 1547–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tabula Muris Consortium; Overall coordination; Logistical coordination; Organ collection and processing; Library preparation and sequencing; Computational data analysis; Cell type annotation; Writing group; Supplemental text writing group; Principal investigators. ((2018). Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature, 562, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tabula Muris Consortium. (2020). A single cell transcriptomic atlas characterizes aging tissues in the mouse. Nature, 580, 590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Janssens J, De Waegeneer M, Kolluru SS, Davie K, Gardeux V, Saelens W, David FPA, Brbić M, Spanier K, Leskovec J, Mclaughlin CN, Xie Q, Jones RC, Brueckner K, Shim J, Tattikota SG, Schnorrer F, Rust K, … Zinzen RP (2022). Fly Cell Atlas: A single-nucleus transcriptomic atlas of the adult fruit fly. Science, 375, eabk2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.The Tabula Microcebus Consortium, Ezran C, Liu S, Chang S, Ming J, Botvinnik O, Penland L, Tarashansky A, de Morree A, Travaglini J, Hasegawa K, Sin H, Sit R, Okamoto J, Sinha R, Zhang Y, Karanewsky CJ, Pendleton JL, Morri M, Perret M, … Krasnow MA (2021). Tabula Microcebus: A transcriptomic cell atlas of mouse lemur, an emerging primate model organism. bioRxiv, 2021.12.12.469460. [Google Scholar]
- 13.Tabula Sapiens Consortium*, Jones RC, Karkanias J, Krasnow MA, Pisco AO, Quake SR, Salzman J, Yosef N, Bulthaup B, Brown P, Harper W, Hemenez M, Ponnusamy R, Salehi A, Sanagavarapu BA, Spallino E, Aaron KA, Concepcion W, Gardner JM, … Wyss-Coray T. (2022). The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science, 376, eabl4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Croote D, Darmanis S, Nadeau KC, & Quake SR (2018). High-affinity allergen-specific human antibodies cloned from single IgE B cell transcriptomes. Science, 362, 1306–1309. [DOI] [PubMed] [Google Scholar]
- 15.Senefeld JW, Johnson PW, Kunze KL, Bloch EM, Van Helmond N, Golafshar MA, Klassen SA, Klompas AM, Sexton MA, Diaz Soto JC, Grossman BJ, Tobian AAR, Goel R, Wiggins CC, Bruno KA, Van Buskirk CM, Stubbs JR, Winters JL, Casadevall A, … Fairweather D (2021). Access to and safety of COVID-19 convalescent plasma in the United States Expanded Access Program: A national registry study. PLoS Medicine, 18, e1003872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li M, Beck EJ, Laeyendecker O, Eby Y, Tobian AAR, Caturegli P, Wouters C, Chiklis GR, Block W, Mckie RO, Joyner MJ, Wiltshire TD, Dietz AB, Gniadek TJ, Shapiro AJ, Yarava A, Lane K, Hanley DF, Bloch EM, … Sullivan DJ (2022). Convalescent plasma with a high level of virus-specific antibody effectively neutralizes SARS-CoV-2 variants of concern. Blood Advances, 6, 3678–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ordaya EE, Abu Saleh OM, Stubbs JR, & Joyner MJ (2022). Vax-plasma in patients with refractory COVID-19. Mayo Clinic Proceedings, 97, 186–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang J, Gingerich AD, Royer F, Paschall AV, Pena-Briseno A, Avci FY, & Mousa JJ (2021). Broadly reactive human monoclonal antibodies targeting the pneumococcal histidine triad protein protect against fatal pneumococcal infection. Infection and Immunity, 89, e00747–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gingerich AD, Royer F, McCormick AL, Scasny A, Vidal JE, & Mousa JJ (2022). Synergistic protection against secondary pneumococcal infection by human monoclonal antibodies targeting distinct epitopes. bioRxiv, 2022.04.20.488872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’Hooghe L, Chalmers AD, Heywood S, & Whitley P (2017). Cell surface dynamics and cellular distribution of endogenous FcRn. PLoS One, 12, e0182695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Arora Y, & Levin K (2013). Myasthenia gravis: Newer therapies offer sustained improvement. Cleveland Clinic Journal of Medicine, 80, 711–721. [DOI] [PubMed] [Google Scholar]
- 22.Chang M, Nakagawa PA, Williams SA, Schwartz MR, Imfeld KL, Buzby JS, & Nugent DJ (2003). Immune thrombocytopenic purpura (ITP) plasma and purified ITP monoclonal autoantibodies inhibit megakaryocytopoiesis in vitro. Blood, 102, 887–895. [DOI] [PubMed] [Google Scholar]
- 23.Shock A, Humphreys D, & Nimmerjahn F (2020). Dissecting the mechanism of action of intravenous immunoglobulin in human autoimmune disease: Lessons from therapeutic modalities targeting Fcγ receptors. Journal of Allergy and Clinical Immunology, 146, 492–500. [DOI] [PubMed] [Google Scholar]
- 24.Smith B, Kiessling A, Lledo-Garcia R, Dixon KL, Christodoulou L, Catley MC, Atherfold P, D’Hooghe LE, Finney H, Greenslade K, Hailu H, Kevorkian L, Lightwood D, Meier C, Munro R, Qureshi O, Sarkar K, Shaw SP, Tewari R, … Brennan FR (2018). Generation and characterization of a high affinity anti-human FcRn antibody, rozanolixizumab, and the effects of different molecular formats on the reduction of plasma IgG concentration. mAbs, 10, 1111–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kiessling P, Lledo-Garcia R, Watanabe S, Langdon G, Tran D, Bari M , Christodoulou L, Jones E, Price G, Smith B, Brennan F, White I, & Jolles S (2017). The FcRn inhibitor rozanolixizumab reduces human serum IgG concentration: A randomized phase 1 study. Science Translational Medicine, 9, eaan1208. [DOI] [PubMed] [Google Scholar]
- 26.Robak T, Kaźmierczak M, Jarque I, Musteata V, Treliński J, Cooper N , Kiessling P, Massow U, Woltering F, Snipes R, Ke J, Langdon G, Bussel JB, & Jolles S (2020). Phase 2 multiple-dose study of an FcRn inhibitor, rozanolixizumab, in patients with primary immune thrombocytopenia. Blood Advances, 4, 4136–4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Jong RN, Beurskens FJ, Verploegen S, Strumane K, Van Kampen MD, Voorhorst M, Horstman W, Engelberts PJ, Oostindie SC, Wang G, Heck AJR, Schuurman J, & Parren PWHI (2016). A novel platform for the potentiation of therapeutic antibodies based on antigen-dependent formation of IgG hexamers at the cell surface. PLoS Biology, 14, e1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trouw LA, Pickering MC, & Blom AM (2017). The complement system as a potential therapeutic target in rheumatic disease. Nature Reviews Rheumatology, 13, 538–547. [DOI] [PubMed] [Google Scholar]
- 29.Diebolder CA, Beurskens FJ, De Jong RN, Koning RI, Strumane K, Lindorfer MA, Voorhorst M, Ugurlar D, Rosati S, Heck AJR, Van De Winkel JGJ, Wilson IA, Koster AJ, Taylor RP, Ollmann Saphire E, Burton DR, Schuurman J, Gros P, & Parren PWHI (2014). Complement is activated by IgG hexamers assembled at the cell surface. Science, 343, 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Goeij BE, Janmaat ML, Andringa G, Kil L, Van Kessel B, Frerichs KA, Lingnau A, Freidig A, Mutis T, Sasser AK, Breij ECW, Van De Donk NWCJ, Ahmadi T, & Satijn D (2019). Hexabody-CD38, a novel CD38 antibody with a hexamerization enhancing mutation, demonstrates enhanced complement-dependent cytotoxicity and shows potent anti-tumor activity in preclinical models of hematological malignancies. Blood, 134, 3106. [Google Scholar]
- 31.Hiemstra I, Janmaat ML, Boross P, van den Brakel J, Hagen W, Dooremalen S, Bosgra S, De Goeij BECG, Andringa G, Kil L, Sasser AK, Ahmadi T, Satijn D, & Breij ECW (2020). Preclinical anti-tumor activity of hexabody-CD38 in patient-derived B cell lymphoma and acute myeloid leukemia xenograft models. Blood, 136, 19–20. [Google Scholar]
- 32.Oostindie SC, Van Der Horst HJ, Kil LP, Strumane K, Overdijk MB, Van Den Brink EN, Van Den Brakel JHN, Rademaker HJ, Van Kessel B, Van Den Noort J, Chamuleau MED, Mutis T, Lindorfer MA, Taylor RP, Schuurman J, Parren PWHI, Beurskens FJ, & Breij ECW (2020). DuoHexaBody-CD37®, a novel biparatopic CD37 antibody with enhanced Fc-mediated hexamerization as a potential therapy for B-cell malignancies. Blood Cancer Journal, 10, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Der Horst HJ, Oostindie SC, Cillessen SAGM, Gelderloos AT, Overdijk MB, Nijhof IS, Zweegman S, Chamuleau MED, Breij ECW, & Mutis T (2021). Potent preclinical efficacy of DuoHexaBody-CD37 in B-cell malignancies. HemaSphere, 5, e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Genmab. (2021). Safety and efficacy of GEN3009 (DuoHexaBody®-CD37) in relapsed or refractory B-cell non-Hodgkin lymphoma-A first-in-human, open-label, phase 1/2a dose escalation trial with dose expansion cohorts. clinicaltrials.gov. [Google Scholar]
- 35.Overdijk MB, Strumane K, Beurskens FJ, Ortiz Buijsse A, Vermot-Desroches C, Vuillermoz BS, Kroes T, De Jong B, Hoevenaars N, Hibbert RG, Lingnau A, Forssmann U, Schuurman J, Parren PWHI, De Jong RN, & Breij ECW (2020). Dual epitope targeting and enhanced hexamerization by DR5 antibodies as a novel approach to induce potent antitumor activity through DR5 agonism. Molecular Cancer Therapeutics, 19, 2126–2138. [DOI] [PubMed] [Google Scholar]
- 36.Chaturvedi S, Arnold DM, & Mccrae KR (2018). Splenectomy for immune thrombocytopenia: Down but not out. Blood, 131, 1172–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bounab Y, Eyer K, Dixneuf S, Rybczynska M, Chauvel C, Mistretta M, Tran T, Aymerich N, Chenon G, Llitjos JF, Venet F, Monneret G, Gillespie IA, Cortez P, Moucadel V, Pachot A, Troesch A, Leissner P, Textoris J, … Védrine C (2020). Dynamic single-cell phenotyping of immune cells using the microfluidic platform DropMap. Nature Protocols, 15, 2920–2955. [DOI] [PubMed] [Google Scholar]
- 38.Canales-Herrerias P, Crickx E, Broketa M, Sokal A, Chenon G, Azzaoui I, Vandenberghe A, Perima A, Iannascoli B, Richard-Le Goff O, Castrillon C, Mottet G, Sterlin D, Robbins A, Michel M, England P, Millot GA, Eyer K, Baudry J, … Bruhns P (2022). High-affinity autoreactive plasma cells disseminate through multiple organs in patients with immune thrombocytopenic purpura. Journal of Clinical Investigation, 132, e153580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katoh H, Komura D, Konishi H, Suzuki R, Yamamoto A, Kakiuchi M, Sato R, Ushiku T, Yamamoto S, Tatsuno K, Oshima T, Nomura S, Seto Y, Fukayama M, Aburatani H, & Ishikawa S (2017). Immunogenetic profiling for gastric cancers identifies sulfated glycosaminoglycans as major and functional B cell antigens in human malignancies. Cell Reports, 20, 1073–1087. [DOI] [PubMed] [Google Scholar]
- 40.Hauser BM, Sangesland M, St Denis KJ, Lam EC, Case JB, Windsor IW, Feldman J, Caradonna TM, Kannegieter T, Diamond MS, Balazs AB, Lingwood D, & Schmidt AG (2022). Rationally designed immunogens enable immune focusing following SARS-CoV-2 spike imprinting. Cell Reports, 38, 110561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaneko N, Kuo H-H, Boucau J, Farmer JR, Allard-Chamard H, Mahajan VS, Piechocka-Trocha A, Lefteri K, Osborn M, Bals J, Bartsch YC, Bonheur N, Caradonna TM, Chevalier J, Chowdhury F, Diefenbach TJ, Einkauf K, Fallon J, Feldman J, … Pillai S (2020). Loss of Bcl-6-expressing T follicular helper cells and germinal centers in COVID-19. Cell, 183, 143–157.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sokal A, Chappert P, Barba-Spaeth G, Roeser A, Fourati S, Azzaoui I, Vandenberghe A, Fernandez I, Meola A, Bouvier-Alias M, Crickx E, Beldi-Ferchiou A, Hue S, Languille L, Michel M, Baloul S, Noizat-Pirenne F, Luka M, … Mahévas M (2021). Maturation and persistence of the anti-SARS-CoV-2 memory B cell response. Cell, 184, 1201–1213.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaebler C, Wang Z, Lorenzi JCC, Muecksch F, Finkin S, Tokuyama M, Cho A, Jankovic M, Schaefer-Babajew D, Oliveira TY, Cipolla M, Viant C, Barnes CO, Bram Y, Breton G, Hägglöf T , Mendoza P, Hurley A, Turroja M, … Nussenzweig MC (2021). Evolution of antibody immunity to SARS-CoV-2. Nature, 591, 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakharkar M, Rappazzo CG, Wieland-Alter WF, Hsieh C-L, Wrapp D, Esterman ES, Kaku CI, Wec AZ, Geoghegan JC, Mclellan JS, Connor RI, Wright PF, & Walker LM (2021). Prolonged evolution of the human B cell response to SARS-CoV-2 infection. Science Immunology, 6, eabg6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sokal A, Barba-Spaeth G, Fernández I, Broketa M, Azzaoui I, De La Selle A, Vandenberghe A, Fourati S, Roeser A, Meola A, Bouvier-Alias M, Crickx E, Languille L, Michel M, Godeau B, Gallien S, Melica G, Nguyen Y, Zarrouk V, … Mahévas M (2021). mRNA vaccination of naive and COVID-19-recovered individuals elicits potent memory B cells that recognize SARS-CoV-2 variants. Immunity, 54, 2893–2907.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doria-Rose N, Suthar MS, Makowski M, O’Connell S, Mcdermott AB, Flach B, Ledgerwood JE, Mascola JR, Graham BS, Lin BC, O’Dell S, Schmidt SD, Widge AT, Edara V-V, Anderson EJ, Lai L, Floyd K, Rouphael NG, Zarnitsyna V, … Kunwar P (2021). Antibody persistence through 6 months after the second dose of mRNA-1273 vaccine for Covid-19. New England Journal of Medicine, 384, 2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.El Sahly HM, Baden LR, Essink B, Doblecki-Lewis S, Martin JM, Anderson EJ, Campbell TB, Clark J, Jackson LA, Fichtenbaum CJ, Zervos M, Rankin B, Eder F, Feldman G, Kennelly C, Han-Conrad L, Levin M, Neuzil KM, Corey L, … Miller J (2021). Efficacy of the mRNA-1273 SARS-CoV-2 vaccine at completion of blinded phase. New England Journal of Medicine, 385, 1774–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tseng HF, Ackerson BK, Luo Y, Sy LS, Talarico CA, Tian Y, Bruxvoort KJ, Tubert JE, Florea A, Ku JH, Lee GS, Choi SK, Takhar HS, Aragones M, & Qian L (2022). Effectiveness of mRNA-1273 against SARS-CoV-2 Omicron and Delta variants. Nature Medicine, 28, 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baden LR, Sahly HME, Essink B, Follmann D, Neuzil KM, August A, Clouting H, Fortier G, Deng W, Han S, Zhao X, Leav B, Talarico C, Girard B, Paila YD, Tomassini JE, Schödel F, Pajon R, Zhou H, … Miller J (2021). Covid-19 in the phase 3 trial of mRNA-1273 during the Delta-variant surge. bioRxiv, 2021.09.17.21263624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pajon R, Doria-Rose NA, Shen X, Schmidt SD, O’Dell S, Mcdanal C, Feng W, Tong J, Eaton A, Maglinao M, Tang H, Manning KE, Edara V-V, Lai L, Ellis M, Moore KM, Floyd K, Foster SL, Posavad CM, … Montefiori DC (2022). SARS-CoV-2 Omicron variant neutralization after mRNA-1273 booster vaccination. New England Journal of Medicine, 386, 1088–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.St John AL, & Rathore APS (2019). Adaptive immune responses to primary and secondary dengue virus infections. Nature Reviews Immunology, 19, 218–230. [DOI] [PubMed] [Google Scholar]
- 52.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GRW, Simmons CP, Scott TW, Farrar JJ, & Hay SI (2013). The global distribution and burden of dengue. Nature, 496, 504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simon-Lorière E, Duong V, Tawfik A, Ung S, Ly S, Casadémont I, Prot M, Courtejoie N, Bleakley K, Buchy P, Tarantola A, Dussart P, Cantaert T, & Sakuntabhai A (2017). Increased adaptive immune responses and proper feedback regulation protect against clinical dengue. Science Translational Medicine, 9, eaal5088. [DOI] [PubMed] [Google Scholar]
- 54.Upasani V, Vo HTM, Auerswald H, Laurent D, Heng S, Duong V, Rodenhuis-Zybert IA, Dussart P, & Cantaert T (2020). Direct infection of B cells by dengue virus modulates B cell responses in a Cambodian Pediatric Cohort. Frontiers in Immunology, 11, 594813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bournazos S, Vo HTM, Duong V, Auerswald H, Ly S, Sakuntabhai A, Dussart P, Cantaert T, & Ravetch JV (2021). Antibody fucosylation predicts disease severity in secondary dengue infection. Science, 372, 1102–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kisalu NK, Idris AH, Weidle C, Flores-Garcia Y, Flynn BJ, Sack BK, Murphy S, Schön A, Freire E, Francica JR, Miller AB, Gregory J, March S, Liao H-X, Haynes BF, Wiehe K, Trama AM, Saunders KO, Gladden MA, … Seder RA (2018). A human monoclonal antibody prevents malaria infection by targeting a new site of vulnerability on the parasite. Nature Medicine, 24, 408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gaudinski MR, Berkowitz NM, Idris AH, Coates EE, Holman LA, Mendoza F, Gordon IJ, Plummer SH, Trofymenko O, Hu Z, Campos Chagas A, O’Connell S, Basappa M, Douek N, Narpala SR, Barry CR, Widge AT, Hicks R, Awan SF, … Seder RA (2021). A monoclonal antibody for malaria prevention. New England Journal of Medicine, 385, 803–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kratochvil S, Shen C-H, Lin Y-C, Xu K, Nair U, Da Silva Pereira L, Tripathi P, Arnold J, Chuang G-Y, Melzi E, Schön A, Zhang B, Dillon M, Bonilla B, Flynn BJ, Kirsch KH, Kisalu NK, Kiyuka PK, Liu T, … Batista FD (2021). Vaccination in a humanized mouse model elicits highly protective PfCSP-targeting anti-malarial antibodies. Immunity, 54, 2859–2876.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu X, Zhang Z, Schramm CA, Joyce MG, Kwon YD, Zhou T, Sheng Z, Zhang B, O’Dell S, Mckee K, Georgiev IS, Chuang G-Y, Longo NS, Lynch RM, Saunders KO, Soto C, Srivatsan S, Yang Y, Bailer RT, … Young A (2015). Maturation and diversity of the VRC01-antibody lineage over 15 years of chronic HIV-1 infection. Cell, 161, 470–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burton DR (2002). Antibodies, viruses and vaccines. Nature Reviews Immunology, 2, 706–713. [DOI] [PubMed] [Google Scholar]
- 61.Kwong PD, & Mascola JR (2018). HIV-1 vaccines based on antibody identification, B cell ontogeny, and epitope structure. Immunity, 48, 855–871. [DOI] [PubMed] [Google Scholar]
- 62.Chuang G-Y, Zhou J, Acharya P, Rawi R, Shen CH, Sheng Z, Zhang B, Zhou T, Bailer RT, Dandey VP, Doria-Rose NA, Louder MK, McKee K, Mascola JR, Shapiro L, & Kwong PD (2019). Structural survey of broadly neutralizing antibodies targeting the HIV-1 Env trimer delineates epitope categories and characteristics of recognition. Structure (London: England, 1993), 27, 196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gaspar M, Pravin J, Rodrigues L, Uhlenbroich S, Everett KL, Wollerton F, Morrow M, Tuna M, & Brewis N (2020).CD137/OX40 bispecific antibody induces potent antitumor activity that is dependent on target coengagement. Cancer Immunology Research, 8, 781–793. [DOI] [PubMed] [Google Scholar]
- 64.Hiramoto E, Tsutsumi A, Suzuki R, Matsuoka S, Arai S, Kikkawa M, & Miyazaki T (2018). The IgM pentamer is an asymmetric pentagon with an open groove that binds the AIM protein. Science Advances, 4, eaau1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Keyt BA, Baliga R, Sinclair AM, Carroll SF, & Peterson MS (2020). Structure, function, and therapeutic use of IgM antibodies. Antibodies, 9, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sun LL, Ellerman D, Mathieu M, Hristopoulos M, Chen X, Li Y, Yan X, Clark R, Reyes A, Stefanich E, Mai E, Young J, Johnson C, Huseni M, Wang X, Chen Y, Wang P, Wang H, Dybdal N, … Ebens AJ (2015). Anti-CD20/CD3 T cell-dependent bispecific antibody for the treatment of B cell malignancies. Science Translational Medicine, 7, 287ra70. [DOI] [PubMed] [Google Scholar]
- 67.Budde E. (2021). A phase 1 dose escalation study of IGM-2323, a novel anti-CD20 x anti-CD3 IgM T cell engager (TCE) in patients with advanced B-cell malignancies. Blood, 138, 132. [Google Scholar]
- 68.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, Diehn M, West RB, Plevritis SK, & Alizadeh AA (2015). The prognostic landscape of genes and infiltrating immune cells across human cancers. Nature Medicine, 21, 938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tosolini M, Pont F, Poupot M, Vergez F, Nicolau-Travers M-L, Vermijlen D, Sarry J-E, Dieli F, & Fournié J-J (2017). Assessment of tumor-infiltrating TCRVγ9Vδ2 γδ lymphocyte abundance by deconvolution of human cancers microarrays. Oncoimmunology, 6, e1284723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.De Bruin RCG, Lougheed SM, Van Der Kruk L, Stam AG, Hooijberg E, Roovers RC, Van Bergen En Henegouwen PMP, Verheul HMW, De Gruijl TD, & Van Der Vliet HJ (2016). Highly specific and potently activating Vγ9Vδ2-T cell specific nanobodies for diagnostic and therapeutic applications. Clinical Immunology, 169, 128–138. [DOI] [PubMed] [Google Scholar]
- 71.De Bruin RCG, Veluchamy JP, Lougheed SM, Schneiders FL, Lopez-Lastra S, Lameris R, Stam AG, Sebestyen Z, Kuball J, Molthoff CFM, Hooijberg E, Roovers RC, Santo JPD, Van Bergen En Henegouwen PMP, Verheul HMW, De Gruijl TD, & Van Der Vliet HJ (2017). A bispecific nanobody approach to leverage the potent and widely applicable tumor cytolytic capacity of Vγ9Vδ2-T cells. Oncoimmunology, 7, e1375641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dussupt V, Sankhala RS, Mendez-Rivera L, Townsley SM, Schmidt F, Wieczorek L, Lal KG, Donofrio GC, Tran U, Jackson ND, Zaky WI, Zemil M, Tritsch SR, Chen W-H, Martinez EJ, Ahmed A, Choe M, Chang WC, Hajduczki A, … Krebs SJ (2021). Low-dose in vivo protection and neutralization across SARS-CoV-2 variants by monoclonal antibody combinations. Nature Immunology, 22, 1503–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Abdin O, Nim S, Wen H, & Kim PM (2022). PepNN: A deep attention model for the identification of peptide binding sites. Communications Biology, 5, 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reveiz M, Tripathi P, Pereira LDS, Kiyuka P, Liu T, Zhang B, Yang Y, Bonilla BG, Dillon M, Lee M, Shen C-H, Schön A, Kratochvil S, Batista FD, Idris AH, Seder RA, Kwong PD, & Rawi R (2022). In silico improvement of highly protective anti-malarial antibodies. bioRxiv, 2022.04.08.487687. [Google Scholar]
- 75.Richardson E, Galson JD, Kellam P, Kelly DF, Smith SE, Palser A, Watson S, & Deane CM (2021). A computational method for immune repertoire mining that identifies novel binders from different clonotypes, demonstrated by identifying anti-pertussis toxoid antibodies. mAbs, 13, 1869406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wong WK, Robinson SA, Bujotzek A, Georges G, Lewis AP, Shi J, Snowden J, Taddese B, & Deane CM (2021). Ab-Ligity: Identifying sequence-dissimilar antibodies that bind to the same epitope. mAbs, 13, 1873478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Robinson SA, Raybould MIJ, Schneider C, Wong WK, Marks C, & Deane CM (2021). Epitope profiling using computational structural modelling demonstrated on coronavirus-binding antibodies. PLoS Computational Biology, 17, e1009675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li T, Jiang S, Ni B, Cui Q, Liu Q, & Zhao H (2019). Discontinued drugs for the treatment of cardiovascular disease from 2016 to 2018. International Journal of Molecular Sciences, 20, 4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Madsen A, Dai Y-N, Mcmahon M, Schmitz AJ, Turner JS, Tan J, Lei T, Alsoussi WB, Strohmeier S, Amor M, Mohammed BM, Mudd PA, Simon V, Cox RJ, Fremont DH, Krammer F, & Ellebedy AH (2020). Human antibodies targeting influenza B virus neuraminidase active site are broadly protective. Immunity, 53, 852–863.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, Hanada K-I, Almeida JR, Darko S, Douek DC, Yang JC, & Rosenberg SA (2014). PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. Journal of Clinical Investigation, 124, 2246–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Horton BL, Williams JB, Cabanov A, Spranger S, & Gajewski TF (2018). Intratumoral CD8+ T-cell apoptosis is a major component of T-cell dysfunction and impedes antitumor immunity. Cancer Immunology Research, 6, 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Geuijen C, Tacken P, Wang L-C, Klooster R, Van Loo PF, Zhou J, Mondal A, Liu Y-B, Kramer A, Condamine T, Volgina A, Hendriks LJA, Van Der Maaden H, Rovers E, Engels S, Fransen F, Den Blanken-Smit R, Zondag-Van Der Zande V, … Throsby M (2021). A human CD137×PD-L1 bispecific antibody promotes anti-tumor immunity via context-dependent T cell costimulation and checkpoint blockade. Nature Communications, 12, 4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Waite JC, Wang B, Haber L, Hermann A, Ullman E, Ye X, Dudgeon D, Slim R, Ajithdoss DK, Godin SJ, Ramos I, Wu Q, Oswald E, Poon P, Golubov J, Grote D, Stella J, Pawashe A, Finney J, … Skokos D (2020). Tumor-targeted CD28 bispecific antibodies enhance the antitumor efficacy of PD-1 immunotherapy. Science Translational Medicine, 12, eaba2325. [DOI] [PubMed] [Google Scholar]
- 84.Skokos D, Waite JC, Haber L, Crawford A, Hermann A, Ullman E, Slim R, Godin S, Ajithdoss D, Ye X, Wang B, Wu Q, Ramos I, Pawashe A, Canova L, Vazzana K, Ram P, Herlihy E, Ahmed H, … Yancopoulos GD (2020). A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Science Translational Medicine, 12, eaaw7888. [DOI] [PubMed] [Google Scholar]