

Abstract
Chromosomal DNA double-strand breaks (DSBs) are the effective lesion of radiotherapy and other clastogenic cancer therapeutics, and are also the initiating event of many approaches to gene editing. Ligation of the DSBs by end joining (EJ) pathways can restore the broken chromosome, but the repair junctions can have insertion/deletion (indel) mutations. The indel patterns resulting from DSB EJ are likely defined by the initial structure of the DNA ends, how the ends are processed and synapsed prior to ligation, and the factors that mediate the ligation step. In this review, we describe key factors that influence these steps of DSB EJ in mammalian cells, which is significant both for understanding mutagenesis resulting from clastogenic cancer therapeutics, and for developing approaches to manipulating gene editing outcomes.
Keywords: ATM, C-NHEJ, DNA polymerase theta, DNA-PKcs, Double-strand break repair, Gene editing
1. Introduction
Use of ionizing radiation (IR) remains a central approach to cancer treatment worldwide, with estimates of nearly half of cancer patients receiving radiotherapy during their course of treatment [1,2]. The primary effective lesions caused by IR are chromosomal DNA double-strand breaks (DSBs) [3,4]. Other clastogens (chromosomal-breaking agents), such as topoisomerase poisons, also remain cornerstones of cancer therapy [5]. Combining clastogens with treatments that target DSB repair could cause persistently broken chromosomes, chromosomal rearrangements, or other mutagenic events that could potentiate tumor cell death and improve efficacy [4,6,7]. Characterizing the mechanisms that define such mutagenic outcomes of DSB repair is critical for developing such therapeutic strategies [4,6], as it will reveal effective therapeutic targets and define tumor-specific vulnerabilities.
Understanding the mechanisms that underlie DSB repair outcomes is also central to gene editing. The adaptation of the Streptococcus pyogenes Cas9 nuclease paired with single guide RNAs to generate targeted DSBs has led to a transformation of molecular biology [8,9] and clinical applications for gene editing [10]. Characterizing how factors can be manipulated to affect DSB repair outcomes can provide insight for how to favor a desired gene editing event. Many gene editing studies focus on improving the frequency of homologous recombination (HR) events vs. end joining (EJ), and thereby introduce a specific sequence provided by an HR repair template [11]. However, manipulating the pattern of insertion/deletion (indel) mutations during EJ repair also has the potential to favor specific gene editing outcomes. Thus, the focus of this review is on factors that influence indel patterns during EJ in mammalian cells (Fig. 1), which is significant both for understanding mutagenesis resulting from clastogenic cancer therapeutics, and for developing approaches to manipulate gene editing outcomes.
Fig. 1.
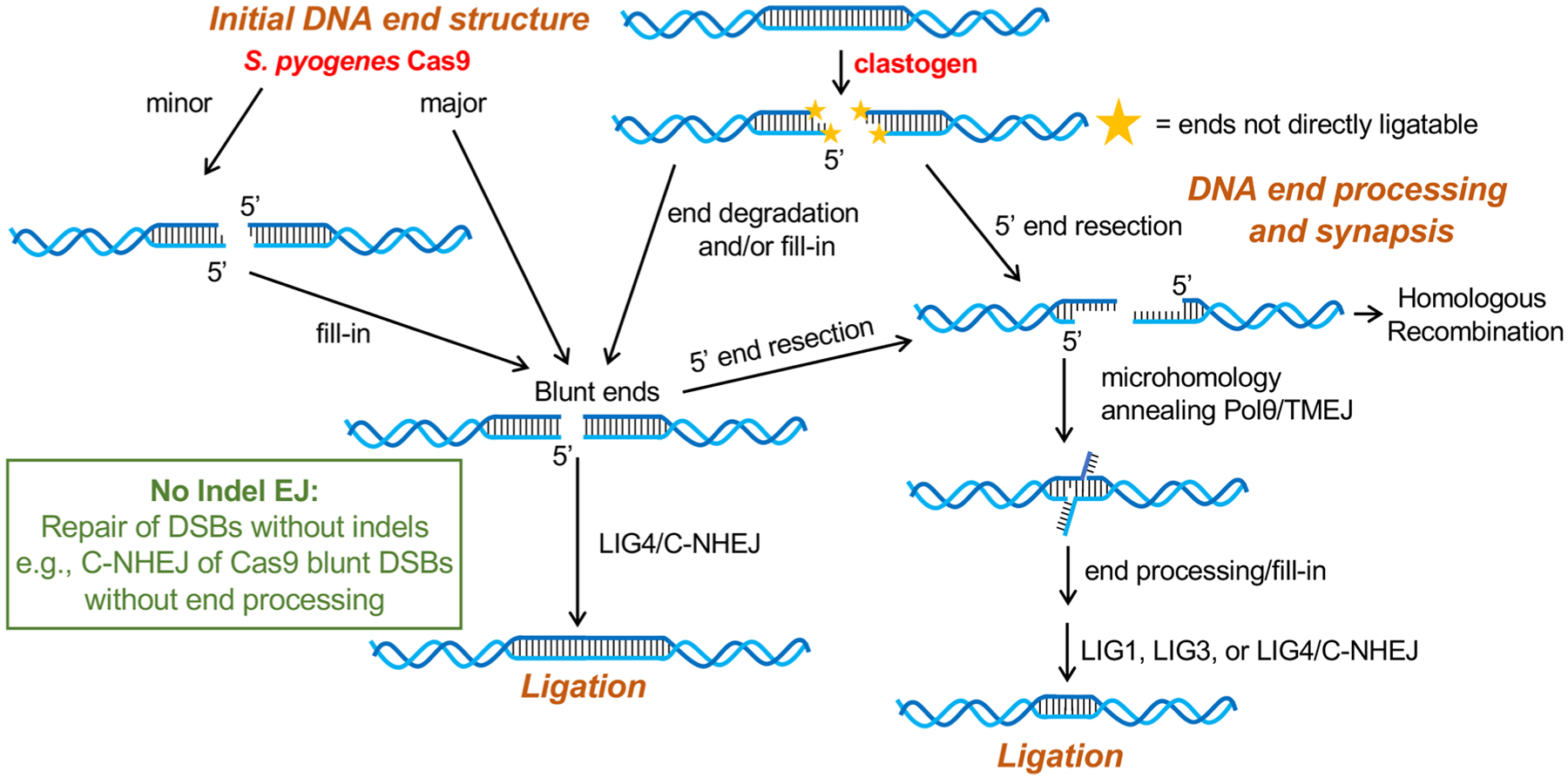
Conditions that influence indel outcomes from DSB EJ: structures of the DNA ends, processing and synapsis of the ends, and ligation. Shown are DSB ends induced by therapeutic clastogens that are often not directly ligatable before end processing, as well DSBs induced by site-specific nucleases (e.g., Streptococcus pyogenes Cas9) that can be readily ligated, but are nonetheless prone to end processing. Shown is a model that end processing to yield blunt ends subsequently requires the C-NHEJ complex (i.e., LIG4 and associated co-factors) for repair. In contrast, end resection that generates 3’ ssDNA with flanking extensive microhomology forms the substrate for Polθ-mediated end synapsis and fill-in synthesis, thereby generating DNA nicks that could be repaired by any of the three ligases (LIG1, LIG3, and LIG4). Shown in the box is the definition of No Indel EJ, which can be detected in genetic assays by examining repair of distal ends from two tandem Cas9 blunt-ended DSBs.
2. Initial DNA end structure of DSBs likely affects indel formation
Indel outcomes of DSB EJ are influenced by several conditions, including the initial structure of the DSB ends, how the DSB ends are processed and synapsed to generate an intermediate that is readily ligated, and the factors that mediate the ligation (Fig. 1). DSBs induced by IR are unlikely to be readily ligated, since such DSBs can involve ends with phosphoglycolate termini and other structures, such as lesions with base damage or abasic sites [12,13]. Similarly, DSBs induced by Topoisomerase II involve a DNA-protein crosslink [5]. As another example, DSB ends generated during V(D)J recombination involve open signal ends and hairpin coding ends, the latter of which are not readily ligated [14,15]. In these cases, such DSB ends need to be processed to generate ligatable ends, and if such processing involves loss or insertion of nucleotides, subsequent EJ repair will cause indel mutations.
In contrast, for Cas9 DSBs, the primary structure of such DSBs are blunt DNA ends [8,9,16–20], which do not require end processing to enable ligation. Measuring blunt EJ of single Cas9 DSBs is not feasible using genetic approaches, since this repair outcome is identical to the original sequence. However, by inducing two tandem Cas9 DSBs, it is possible to quantify repair of the distal blunt DNA ends without indel mutations, which our group has referred to as No Indel EJ (Fig. 1). Of course, such EJ causes the loss of the DNA fragment between the two tandem DSBs, but “No Indel EJ” refers to the notion that there are no indel mutations from the edges of each distal DSB end. Several studies have shown that No Indel EJ is frequent [16–20], indicating that precise/accurate repair of blunt DSBs induced by Cas9 is a robust repair outcome.
While Cas9 generally induces blunt ended DSBs, non-blunt DSBs with 5’ overhangs are a minor product, which can lead to insertion mutations (Fig. 1). Specifically, Cas9 can induce DSBs with 5’ overhangs, typically 1–2 nt long, which if filled-in to generate blunt DSBs prior to EJ, causes short insertion mutations [21–25]. Since fill-in of the 5’ overhangs prior to ligation produces an indel mutation rendering the target site resistant to further cycles of cutting and precise repair, such insertion mutation repair outcomes are frequently detected [21–25].
3. Roles of C-NHEJ and Polθ for end synapsis and ligation of distinct DNA end structures
EJ of blunt DSBs, either induced by Cas9 directly, or following fill-in of Cas9 5’ overhangs, has been shown to be highly dependent on canonical non-homologous end joining (C-NHEJ) [16–19,21–25]. The C-NHEJ complex involves DNA ligase 4 (LIG4) and associated co-factors, including XRCC4, the KU heterodimer, XLF, and DNA-PKcs. The C-NHEJ complex is proficient for blunt DSB EJ, because it can facilitate synapsis of blunt DNA ends in close proximity, and with LIG4 positioned to enable ligation [16–19,21,23–25]. Recent cryo-EM structures have demonstrated such synapsis with a short-range C-NHEJ complex [26,27]. Notably, this complex lacks DNA-PKcs [26,27]; and DNA-PKcs, along with the accessory C-NHEJ factor PAXX [28], have a less pronounced role on blunt DSB EJ vs. XLF [16,25] (see below).
In contrast, repair of DSBs leading to deletion mutations using extensive microhomology are largely independent of C-NHEJ, and are promoted by DNA polymerase theta (Polθ/POLQ). Polθ is an A-family DNA polymerase that can anneal short lengths of complementary ssDNA (i.e., microhomology) to initiate DNA synthesis, which has been referred to as theta-mediated EJ (TMEJ) [29–33]. Such Polθ-mediated DNA synthesis could establish an annealing intermediate with DNA nicks that could then be joined by any of the three DNA ligases (LIG1, LIG3, or LIG4) [34,35]. However, the length of microhomology, the length of the annealing intermediate, and the protein co-factors that mediate recruitment of each ligase to distinct DNA structures, each likely affect the relative ability for these ligases to facilitate repair. For example, use of 4 nt of microhomology to template an extension of 2 nt on each side of the DSB would generate an 8 nt annealing intermediate that may be readily ligated by any of the three ligases. In contrast, use of 1 nt of microhomology to bridge DNA ends may remain reliant on the C-NHEJ complex to form a stable synapsis intermediate for ligation.
The cut off for microhomology length that distinguishes dependence on C-NHEJ vs. Polθ supports this notion. By examining terminal microhomology use (i.e., microhomology at the edge of DSBs) for repair of Cas9 DSBs, C-NHEJ was shown to be required for events up to 2 nt of microhomology, and Polθ is important for events 4–6 nt [16,36]. In contrast, C-NHEJ and Polθ appear redundant for events with 3 nt of terminal microhomology [16]. Furthermore, combined loss of C-NHEJ (i.e., KU) and Polθ causes a marked reduction in EJ using 4 nt of microhomology that is either at the DSB termini or embedded from the edge of the DSB [37]. This finding is consistent with several studies that combined loss of C-NHEJ and Polθ cause a marked loss of Cas9 EJ events, as well as a substantial increase in radiosensitivity [29,38]. Another study examined microhomology usage for repair of a single Cas9 DSB, finding that Polθ can promote EJ events with ≥ 2 nt of microhomology that are embedded from the DSB, and also with a bias towards microhomology near the edge of the DSB [39]. In summary, C-NHEJ appears specifically required for EJ events that are not stabilized by an extensive annealing intermediate, whereas Polθ appears important to generate such an annealing intermediate to facilitate ligation that could be mediated by any of the three ligases.
Indels consistent with C-NHEJ and TMEJ have been observed from large-scale analysis of Cas9 DSBs in mammalian cells [40–42], as well as from sequencing of samples post-radiotherapy [43,44]. For instance, one study of thousands of different synthetic Cas9 cleavage sites found that the two most frequent repair outcomes are single base insertions (perhaps caused by staggered Cas9 DSBs as described above) and short (≥ 3 nt) deletions with microhomology [40]. This study also found that the proximity of matching microhomology favors the use of microhomology to cause a deletion mutation [40], which is consistent with TMEJ [39]. For secondary malignancies following IR treatment of both human patients and mouse models, both insertion and deletion mutations are found, with deletion mutations often showing evidence of microhomology [43,44]. Although, a study of post-radiotherapy gliomas found induction of small deletions that predominantly lacked microhomology, which is consistent with C-NHEJ [45]. Finally, thyroid cancers from individuals exposed to radiation due to the Chernobyl incident showed associations of radiation dose with deletion mutations irrespective of microhomology length [46], which is consistent with C-NHEJ playing a significant role in such indels. An important future direction will be to determine how the genetic landscape of individual tumors affects the relative contribution of C-NHEJ vs. TMEJ for DSB repair, which could guide personalized medicine approaches for development of radiosensitizers.
4. Processing of DNA breaks for C-NHEJ and TMEJ
The means of DSB end processing likely dictates whether C-NHEJ or Polθ/TMEJ are subsequently involved in repair of the ends, thereby affecting the indel outcome. Namely, end processing to yield blunt DSBs and/or ends with a very short annealing intermediate are likely repaired by C-NHEJ [16,36,47]. Blunt DSB ends could be generated from ends with damaged termini that are processed to remove the terminal bases, or DSB ends with short 5’ overhangs that are filled-in or degraded (Fig. 1). DSB ends with a short annealing intermediate (e.g., 1 bp of microhomology) can be generated by processing of ends by polymerases that add nucleotides to generate such microhomology, or nucleases that catalyze limited end degradation to reveal short microhomology. C-NHEJ is likely the major pathway of repair of such blunt DNA ends, or ends stabilized by short microhomology, which would then cause short indel mutations. Studies of extrachromosomal end joining substrates with diverse end structures supports this notion [47]. In contrast, if such DSB ends are processed into 3’ ssDNA to be substrates for Polθ, the size of the deletion mutation is likely governed by the position of microhomology that flanks the DSB.
This concept is supported by experiments with overexpression of the potent 3’ exonuclease TREX2 [48], which likely causes destabilization of 3’ ssDNA, thereby requiring C-NHEJ repair without an annealing intermediate. Our group first reported that co-expression of TREX2 markedly promotes deletion mutations while also suppressing EJ events with incorrect DSB ends (i.e., distal ends from two tandem DSBs) [49,50]. Furthermore, our group showed that the mutagenic events induced by TREX2 expression are dependent on C-NHEJ [51]. These studies used the I-SceI endonuclease to generate targeted chromosomal DSBs. Subsequently, another study showed that TREX2 expression could similarly promote mutagenic EJ with other nucleases (i.e., other homing endonucleases apart from I-SceI, transcription activator-like effector nucleases, and zinc-finger nucleases) [52], and our group showed that combining TREX2 with Cas9 also promotes mutagenic EJ [53], which has been confirmed in other studies [18,40,54–56]. As mentioned above, the deletions induced by expression of TREX2 with I-SceI are dependent on C-NHEJ, which is also the case for Cas9 [18]. Furthermore, such EJ events show low microhomology usage [18], which is consistent with TREX2 causing end degradation that is likely processed into blunt DSBs that are joined by C-NHEJ. Namely, TREX2 likely degrades 3’ ssDNA that is the substrate for annealing and fill-in synthesis via TMEJ, such that C-NHEJ is required for repair.
Accordingly, regulation of nucleases and polymerases (in addition to Polθ) to generate blunt ends, 3’ ssDNA, or other DSB end structures likely has a substantial effect on DSB indel patterns. DSB end processing to yield 3’ ssDNA that is the substrate for Polθ/TMEJ is referred to as end resection, which is also the initiating step of HR [57]. A central factor of end resection is CtIP, which forms a complex with the MRE11 nuclease [57]. CtIP is important not only for HR, but also mutagenic EJ events that show microhomology at the junctions [16,21,58,59]. It is unclear how CtIP-mediated resection is regulated to promote TMEJ vs. HR, although regions of CtIP that affect the extent of resection have been recently discovered [60]. Furthermore, the conditions that favor CtIP-mediated end resection are unclear, apart from cell cycle phase, since CtIP is activated by CDK phosphorylation events in S/G2 [57]. Other nucleases also likely function in concert with CtIP/MRE11 to influence the extent of 3’ ssDNA, and/or compete with CtIP to induce distinct end processing events [57]. Indeed, other nucleases have been implicated in end degradation during EJ, including Artemis, WRN, DNA2, FEN1, and Metnase [59,61–66].
The Artemis nuclease appears to have a central role in processing DNA ends for C-NHEJ, particularly for opening the hairpin coding ends generated during V(D)J recombination [66]. In contrast to CtIP/MRE11-dependent end resection that favors formation of 3’ ssDNA, Artemis can process DNA ends into distinct structures [66,67]. As examples, Artemis is proficient at processing 5’ overhangs into blunt DNA ends, and blunt DNA ends into 3’ overhangs [66,67]. In each case, it appears that Artemis is able to distort DNA ends into hairpin-like structures, and then cleave these structures [66,67]. Accordingly, Artemis is likely proficient at processing diverse DNA end structures induced by IR to yield ligatable ends. Consistent with this notion, Artemis is important for repair of IR-induced DSBs [68,69]. The influence of Artemis on indel patterns for Cas9 DSBs remains poorly understood, although there are some indications that Artemis can affect such repair outcomes. For one, overexpression of Artemis has been shown to promote mutagenic EJ of DSBs induced by Cas9 and other nucleases [52,55]. In addition, a recent study that evaluated how DNA repair factors affect Cas9 indel patterns showed that Artemis (DCLRE1C gene) appears to be important for a subset of deletions [22].
Apart from nucleases, several other enzymes can process DNA ends for ligation. For example, phosphodiesterases can directly process DNA ends with phosphoglycolate termini [12]. Furthermore, DNA polymerases, such as Pol λ, Pol μ, and terminal deoxynucleotidyl transferase (TdT) also appear to generate DNA ends for ligation via C-NHEJ [70]. For example, Pol λ appears important for fill-in of 5’ overhangs, based on studies with Cas9 staggered DSBs causing insertion mutations, as described above [22]. Pol μ and TdT also are important for generating ligatable ends by adding nucleotides to otherwise incompatible ends to facilitate short annealing intermediates for repair via C-NHEJ [70–72]. Furthermore, several of these polymerases show a bias for ribonucleotide addition that appears to generate optimal substrates for ligation via C-NHEJ [71]. In summary, DSB processing via nucleases, enzymes that directly repair DNA termini, and DNA polymerases have a significant influence on indel outcomes.
5. Influence of DNA-PKcs and ATM on end joining outcomes
The DNA damage response kinases DNA-PKcs and ATM also substantially affect indel outcomes by promoting and inhibiting C-NHEJ, respectively. Examining these kinases is significant both for understanding the mechanism of indel formation, and also because potent and selective small molecule inhibitors of these kinases are being developed for clinical use [73,74]. DNA-PKcs is the catalytic subunit of the DNA dependent protein kinase (DNA-PK), which is composed of DNA-PKcs and the KU heterodimer [14,75]. Recent cryo-EM structures have identified two different complexes with DNA-PKcs that bridge DNA ends: one with KU alone, and another that also includes the C-NHEJ factors XRCC4, LIG4, and XLF [26,27]. Both of these complexes show relatively long-range synapsis of the DNA ends, in that the ends are too far apart to facilitate ligation [26,27]. Such long range synapsis is also consistent with single molecule studies in Xenopus extracts [76]. Cryo-EM analysis also found a C-NHEJ complex bridging DNA ends without DNA-PKcs (i.e., with XRCC4, LIG4, and XLF) with the ends positioned for ligation [27]. Since such a short-range complex can form without DNA-PKcs, the requirement for DNA-PKcs for chromosomal EJ events has been unclear. Our group recently reported that DNA-PKcs and its kinase activity are less important than XLF for repair of blunt DSBs, but becomes critical when XLF-mediated end bridging interactions are weakened [25]. These findings support the notion that DNA-PKcs, and hence the long-range C-NHEJ complex, promotes blunt DNA end synapsis in a manner that is partially redundant with the bridging interactions mediated by XLF. In contrast, DNA-PKcs kinase activity appears important to suppress the size of deletion mutations in a manner independent of XLF [25].
Accordingly, DNA-PKcs may be more critical for regulating DNA end processing events vs. mediating the ligation step of C-NHEJ per se. For one, DNA-PKcs is an activator of the Artemis nuclease [15,66], and a cryo-EM study has shown that DNA-PKcs autophosphorylation in its ABCDE cluster causes a conformational change to enable access of Artemis to DNA ends [77]. Thus, DNA-PKcs and its kinase activity appear critical for mediating Artemis nuclease activity, which could also extend to other nucleases and DNA end processing factors. Along these lines, a study in Xenopus extracts found that at least some end processing events are restricted to the C-NHEJ short-range complex [78], such that transition to the short-range complex via DNA-PKcs autophosphorylation may be central to regulating such end processing. Furthermore, a recent preprint has shown evidence that distinct DNA-PKcs dimerization interfaces appear to have differential effects on nucleolytic vs. end-filling of DSB ends [79]. In addition, DNA-PKcs may be a key regulator of the MRN/CtIP nuclease complex (i.e., the MRE11 nuclease in complex with RAD50, NBS1, and CtIP) [80]. Namely, DNA ends bound to DNA-PK are efficient substrates for the MRN/CtIP complex to nick DNA on the side of DNA-PK that is distal to the DNA end, which appears important to initiate end resection [80]. Altogether, these studies indicate that DNA-PKcs is likely a central regulator of DSB end processing, such that defining the mechanisms that regulate DNA-PKcs to favor particular end processing events remains a central question for understanding EJ indel patterns.
The ATM kinase has been long associated with radioresistance, DSB repair, chromatin alterations at DSBs, and cell cycle checkpoints [81]. However, the influence of ATM on specific DSB repair outcomes has emerged relatively recently, with several studies indicating that ATM is a key negative regulator of C-NHEJ. For one, ATM was shown to suppress C-NHEJ events measured using the TREX2 approach described above [51]. Similarly, ATM appears to inhibit the hallmarks of C-NHEJ repair of Cas9 DSBs (i.e., No Indel EJ between two DSBs, and insertions likely due to staggered Cas9 DSB) [18,19,82]. Furthermore, in the context of one ended DSBs in DNA replication, ATM has been shown to suppress C-NHEJ factor recruitment, which appears critical for limiting toxic chromosomal rearrangements [83]. Similarly, chromosomal rearrangements in neurons that are induced by loss of ATM have been shown to be mediated by C-NHEJ (i.e., LIG4) [84]. In contrast, ATM is not obviously important for HR repair of DSBs [85,86], and breast cancers associated with ATM-deficiency show mutation/rearrangement signatures distinct from those of HR-deficient tumors [87].
There are hundreds of phosphorylation targets of the ATM kinase [81,88], but the specific targets that are important to suppress C-NHEJ are unclear. A central target of ATM is the histone variant H2AX, which is phosphorylated at residue 139 in the C-terminus (i.e., γH2AX) in chromatin that flanks DSBs [89]. Interestingly, similar to ATM deficiency, loss of H2AX also appears to cause an increase in C-NHEJ events (e.g., an increase on No Indel EJ of Cas9 DSBs) [18,90]. One working model is that the ATM kinase and H2AX are important for DSB end resection that favors EJ events with indel mutations (e.g., TMEJ). Consistent with this notion, ATM appears to modulate the function of CtIP [91]. As well, two factors regulated by ATM, the NBS1 subunit of the MRE11 complex and the RNF8 ubiquitin ligase, are also important to suppress No Indel EJ, and thereby promote EJ with indel mutations [37]. As another possibility, ATM could directly inhibit C-NHEJ, which is consistent with reports of ATM-mediated regulation of DNA-PKcs [92]. Furthermore, the Artemis nuclease is a kinase target of ATM [69].
ATM is also an important regulator of the Shieldin complex that includes 53BP1, but the impact of this complex on indel formation during EJ is unclear. Namely, while 53BP1 restricts end resection and HR [93], this function does not apparently have a major effect on the balance of No Indel EJ vs. EJ events using microhomology [37]. However, one study showed that 53BP1 suppresses complex indels that involve capture of ectopic DNA [94]. Also, a recent survey of how DNA damage response factors affect Cas9 indel patterns identified a subset of indel types affected by 53BP1, which appear to be similar to those affected by Artemis [22]. Further studies are needed to define how the 53BP1 and the rest of the Shieldin complex, including the ASTE1 nuclease [95], may affect indel formation during C-NHEJ and TMEJ. In addition, apart from indel patterns per se, 53BP1 has a substantial effect on long range EJ events, such as fusion of deprotected telomeres, and class switch recombination [93]. 53BP1 is proposed to promote DSB mobility to mediate such EJ events [96,97]. Similarly, ATM was also found to promote DSB mobility [98–100]. Specifically, in ATM deficient cells, dysfunctional telomeres failed to gain their maximal mobility [100], and inhibition of ATM in human cells resulted in a pronounced confinement of DSB mobility and reduced DSB clustering [98,99].
6. Concluding remarks and future directions
In summary, indel outcomes from chromosomal DSB EJ are affected by DNA end structure, DNA end processing and synapsis, and the regulation of the ligation step. Understanding the mechanisms that affect indel outcomes is relevant for defining the mechanisms of mutagenesis induced by radiotherapy and other clastogenic cancer therapeutics, as well as for developing approaches to manipulate gene editing outcomes. Several of the studies described above are based on examining indel patterns after induction of site-specific DSBs (e.g., Cas9), which is a powerful approach since the initiating lesion is precisely defined, and because examining such DSBs is significant for gene editing. However, a disadvantage of this approach is that nuclease-generated DSBs lack the complexity of other clastogenic events, such as radiotherapy. An important future direction would be to develop an approach generating targeted complex chromosomal DSBs, similar to what has been developed for causing oxidative damage at telomeres [101]. Another future direction is to define the mechanisms that regulate DSB end processing by distinct nucleases, particularly for CtIP/MRE11 vs. Artemis. Along these lines, it will be important to define how CtIP/MRE11-mediated resection is regulated to facilitate TMEJ vs. HR. Similarly, it will be interesting to define how the chromatin landscape prior to DSB induction, which appears to affect the efficiency of HR [102], might also affect EJ outcomes. In addition to defining regulation of DSB end processing, another key area of study is to define the mechanisms of end synapsis via C-NHEJ and TMEJ, and how such synapsis is affected by neighboring chromatin. Understanding mechanisms of end synapsis is significant, since such synapsis is likely critical for both timely repair of DSBs, as well as the suppression of EJ events between distal DSB ends (e.g., DSBs from different chromosomes), which can cause chromosomal rearrangements. Finally, understanding how ATM and DNA-PKcs affect each of these steps of EJ is important both for defining the mechanisms of indel formation, as well as for developing small molecule inhibitors of these kinases for clinical use.
Acknowledgements
We thank Ragini Bhargava for helpful discussions. This work was supported by the National Cancer Institute of the National Institutes of Health (USA): R01CA256989, R01CA240392 (J.M.S.).
Abbreviations:
- DSB
Double-Strand Break
- C-NHEJ
Canonical Non-Homologous End Joining
- DNA-PK
DNA dependent protein kinase
- EJ
End Joining
- HR
Homologous Recombination
- indel
insertion/deletion mutation
- IR
Ionizing Radiation
- TMEJ
theta-mediated end joining
Footnotes
Conflict of Interest
The authors declare that there are no conflicts of interest.
References
- [1].Pan HY, Haffty BG, Falit BP, Buchholz TA, Wilson LD, Hahn SM, Smith BD, Supply and demand for radiation oncology in the United States: updated projections for 2015 to 2025, Int. J. Radiat. Oncol. Biol. Phys 96 (2016) 493–500. [DOI] [PubMed] [Google Scholar]
- [2].Abdel-Wahab M, Gondhowiardjo SS, Rosa AA, Lievens Y, El-Haj N, Polo Rubio JA, Prajogi GB, Helgadottir H, Zubizarreta E, Meghzifene A, Ashraf V, Hahn S, Williams T, Gospodarowicz M, Global radiotherapy: current status and future directions-white paper, JCO Glob. Oncol 7 (2021) 827–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Iliakis G, The role of DNA double strand breaks in ionizing radiation-induced killing of eukaryotic cells, Bioessays 13 (1991) 641–648. [DOI] [PubMed] [Google Scholar]
- [4].Mladenov E, Magin S, Soni A, Iliakis G, DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy, Front. Oncol 3 (2013) 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Nitiss JL, Targeting DNA topoisomerase II in cancer chemotherapy, Nat. Rev. Cancer 9 (2009) 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Le QT, Shirato H, Giaccia AJ, Koong AC, Emerging treatment paradigms in radiation oncology, Clin. Cancer Res 21 (2015) 3393–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bakhoum SF, Cantley LC, The multifaceted role of chromosomal instability in cancer and its microenvironment, Cell 174 (2018) 1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E, A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity, Science 337 (2012) 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F, Multiplex genome engineering using CRISPR/Cas systems, Science 339 (2013) 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Urnov FD, Imagine CRISPR cures, Mol. Ther 29 (2021) 3103–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nambiar TS, Baudrier L, Billon P, Ciccia A, CRISPR-based genome editing through the lens of DNA repair, Mol. Cell 82 (2022) 348–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Menon V, Povirk LF, End-processing nucleases and phosphodiesterases: an elite supporting cast for the non-homologous end joining pathway of DNA double-strand break repair, DNA Repair 43 (2016) 57–68. [DOI] [PubMed] [Google Scholar]
- [13].Schipler A, Iliakis G, DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice, Nucleic Acids Res. 41 (2013) 7589–7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jackson SP, Jeggo PA, DNA double-strand break repair and V(D)J recombination: involvement of DNA-PK, Trends Biochem. Sci 20 (1995) 412–415. [DOI] [PubMed] [Google Scholar]
- [15].Ma Y, Pannicke U, Schwarz K, Lieber MR, Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination, Cell 108 (2002) 781–794. [DOI] [PubMed] [Google Scholar]
- [16].Bhargava R, Sandhu M, Muk S, Lee G, Vaidehi N, Stark JM, C-NHEJ without indels is robust and requires synergistic function of distinct XLF domains, Nat. Commun 9 (2018) 2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Eki R, She J, Parlak M, Benamar M, Du KP, Kumar P, Abbas T, A robust CRISPR-Cas9-based fluorescent reporter assay for the detection and quantification of DNA double-strand break repair, Nucleic Acids Res. 48 (2020), e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bhargava R, Carson CR, Lee G, Stark JM, Contribution of canonical nonhomologous end joining to chromosomal rearrangements is enhanced by ATM kinase deficiency, Proc. Natl. Acad. Sci. USA 114 (2017) 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bhargava R, Lopezcolorado FW, Tsai LJ, Stark JM, The canonical non-homologous end joining factor XLF promotes chromosomal deletion rearrangements in human cells, J. Biol. Chem 295 (2020) 125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nambiar TS, Billon P, Diedenhofen G, Hayward SB, Taglialatela A, Cai K, Huang JW, Leuzzi G, Cuella-Martin R, Palacios A, Gupta A, Egli D, Ciccia A, Stimulation of CRISPR-mediated homology-directed repair by an engineered RAD18 variant, Nat. Commun 10 (2019) 3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shou J, Li J, Liu Y, Wu Q, Precise and predictable CRISPR chromosomal rearrangements reveal principles of Cas9-mediated nucleotide insertion, Mol. Cell 71 (2018), 498–509 e494. [DOI] [PubMed] [Google Scholar]
- [22].Hussmann JA, Ling J, Ravisankar P, Yan J, Cirincione A, Xu A, Simpson D, Yang D, Bothmer A, Cotta-Ramusino C, Weissman JS, Adamson B, Mapping the genetic landscape of DNA double-strand break repair, Cell 184 (2021), 5653–5669 e5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lemos BR, Kaplan AC, Bae JE, Ferrazzoli AE, Kuo J, Anand RP, Waterman DP, Haber JE, CRISPR/Cas9 cleavages in budding yeast reveal templated insertions and strand-specific insertion/deletion profiles, Proc. Natl. Acad. Sci. USA 115 (2018) E2040–E2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Feng W, Simpson DA, Cho JE, Carvajal-Garcia J, Smith CM, Headley KM, Hathaway N, Ramsden DA, Gupta GP, Marker-free quantification of repair pathway utilization at Cas9-induced double-strand breaks, Nucleic Acids Res. 49 (2021) 5095–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cisneros-Aguirre M, Lopezcolorado FW, Tsai LJ, Bhargava R, Stark JM, The importance of DNAPKcs for blunt DNA end joining is magnified when XLF is weakened, Nat. Commun 13 (2021) 3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chaplin AK, Hardwick SW, Stavridi AK, Buehl CJ, Goff NJ, Ropars V, Liang S, De Oliveira TM, Chirgadze DY, Meek K, Charbonnier JB, Blundell TL, Cryo-EM of NHEJ supercomplexes provides insights into DNA repair, Mol. Cell 81 (2021), 3400–3409 e3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen S, Lee L, Naila T, Fishbain S, Wang A, Tomkinson AE, Lees-Miller SP, He Y, Structural basis of long-range to short-range synaptic transition in NHEJ, Nature 593 (2021) 294–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ochi T, Blackford AN, Coates J, Jhujh S, Mehmood S, Tamura N, Travers J, Wu Q, Draviam VM, Robinson CV, Blundell TL, Jackson SP, DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair, Science 347 (2015) 185–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wyatt DW, Feng W, Conlin MP, Yousefzadeh MJ, Roberts SA, Mieczkowski P, Wood RD, Gupta GP, Ramsden DA, Essential roles for polymerase theta-mediated end joining in the repair of chromosome breaks, Mol. Cell 63 (2016) 662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zahn KE, Averill AM, Aller P, Wood RD, Doublie S, Human DNA polymerase theta grasps the primer terminus to mediate DNA repair, Nat. Struct. Mol. Biol 22 (2015) 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kent T, Chandramouly G, McDevitt SM, Ozdemir AY, Pomerantz RT, Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta, Nat. Struct. Mol. Biol 22 (2015) 230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].van Bostelen I, van Schendel R, Romeijn R, Tijsterman M, Translesion synthesis polymerases are dispensable for C. elegans reproduction but suppress genome scarring by polymerase theta-mediated end joining, PLoS Genet. 16 (2020), e1008759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yu AM, McVey M, Synthesis-dependent microhomology-mediated end joining accounts for multiple types of repair junctions, Nucleic Acids Res. 38 (2010) 5706–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sallmyr A, Rashid I, Bhandari SK, Naila T, Tomkinson AE, Human DNA ligases in replication and repair, DNA Repair 93 (2020), 102908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Simsek D, Brunet E, Wong SY, Katyal S, Gao Y, McKinnon PJ, Lou J, Zhang L, Li J, Rebar EJ, Gregory PD, Holmes MC, Jasin M, DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation, PLoS Genet. 7 (2011), e1002080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kelso AA, Lopezcolorado FW, Bhargava R, Stark JM, Distinct roles of RAD52 and POLQ in chromosomal break repair and replication stress response, PLoS Genet. 15 (2019), e1008319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tsai LJ, Lopezcolorado FW, Bhargava R, Mendez-Dorantes C, Jahanshir E, Stark JM, RNF8 has both KU-dependent and independent roles in chromosomal break repair, Nucleic Acids Res. 48 (2020) 6032–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Schimmel J, Kool H, van Schendel R, Tijsterman M, Mutational signatures of non-homologous and polymerase theta-mediated end-joining in embryonic stem cells, EMBO J. 36 (2017) 3634–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Carvajal-Garcia J, Cho JE, Carvajal-Garcia P, Feng W, Wood RD, Sekelsky J, Gupta GP, Roberts SA, Ramsden DA, Mechanistic basis for microhomology identification and genome scarring by polymerase theta, Proc. Natl. Acad. Sci. USA 117 (2020) 8476–8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Allen F, Crepaldi L, Alsinet C, Strong AJ, Kleshchevnikov V, De Angeli P, Palenikova P, Khodak A, Kiselev V, Kosicki M, Bassett AR, Harding H, Galanty Y, Munoz-Martinez F, Metzakopian E, Jackson SP, Parts L, Predicting the mutations generated by repair of Cas9-induced double-strand breaks, Nat. Biotechnol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].van Overbeek M, Capurso D, Carter MM, Thompson MS, Frias E, Russ C, Reece-Hoyes JS, Nye C, Gradia S, Vidal B, Zheng J, Hoffman GR, Fuller CK, May AP, DNA repair profiling reveals nonrandom outcomes at Cas9-Mediated breaks, Mol. Cell 63 (2016) 633–646. [DOI] [PubMed] [Google Scholar]
- [42].Taheri-Ghahfarokhi A, Taylor BJM, Nitsch R, Lundin A, Cavallo AL, Madeyski-Bengtson K, Karlsson F, Clausen M, Hicks R, Mayr LM, Bohlooly YM, Maresca M, Decoding non-random mutational signatures at Cas9 targeted sites, Nucleic Acids Res. 46 (2018) 8417–8434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Behjati S, Gundem G, Wedge DC, Roberts ND, Tarpey PS, Cooke SL, Van Loo P, Alexandrov LB, Ramakrishna M, Davies H, Nik-Zainal S, Hardy C, Latimer C, Raine KM, Stebbings L, Menzies A, Jones D, Shepherd R, Butler AP, Teague JW, Jorgensen M, Khatri B, Pillay N, Shlien A, Futreal PA, Badie C, McDermott U, Bova GS, Richardson AL, Flanagan AM, Stratton MR, Campbell PJ, Mutational signatures of ionizing radiation in second malignancies, Nat. Commun 7 (2016) 12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Rose Y, Li Halliwill KD, Adams CJ, Iyer V, Riva L, Mamunur R, Jen KY, Del Rosario R, Fredlund E, Hirst G, Alexandrov LB, Adams D, Balmain A, Mutational signatures in tumours induced by high and low energy radiation in Trp53 deficient mice, Nat. Commun 11 (2020) 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kocakavuk E, Anderson KJ, Varn FS, Johnson KC, Amin SB, Sulman EP, Lolkema MP, Barthel FP, Verhaak RGW, Radiotherapy is associated with a deletion signature that contributes to poor outcomes in patients with cancer, Nat. Genet 53 (2021) 1088–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Morton LM, Karyadi DM, Stewart C, Bogdanova TI, Dawson ET, Steinberg MK, Dai J, Hartley SW, Schonfeld SJ, Sampson JN, Maruvka YE, Kapoor V, Ramsden DA, Carvajal-Garcia J, Perou CM, Parker JS, Krznaric M, Yeager M, Boland JF, Hutchinson A, Hicks BD, Dagnall CL, Gastier-Foster JM, Bowen J, Lee O, Machiela MJ, Cahoon EK, Brenner AV, Mabuchi K, Drozdovitch V, Masiuk S, Chepurny M, Zurnadzhy LY, Hatch M, Berrington de Gonzalez A, Thomas GA, Tronko MD, Getz G, Chanock SJ, Radiation-related genomic profile of papillary thyroid carcinoma after the Chernobyl accident, Science 372 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Waters CA, Strande NT, Pryor JM, Strom CN, Mieczkowski P, Burkhalter MD, Oh S, Qaqish BF, Moore DT, Hendrickson EA, Ramsden DA, The fidelity of the ligation step determines how ends are resolved during nonhomologous end joining, Nat. Commun 5 (2014) 4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Perrino FW, Harvey S, McMillin S, Hollis T, The human TREX2 3’ -> 5’-exonuclease structure suggests a mechanism for efficient nonprocessive DNA catalysis, J. Biol. Chem 280 (2005) 15212–15218. [DOI] [PubMed] [Google Scholar]
- [49].Bennardo N, Gunn A, Cheng A, Hasty P, Stark JM, Limiting the persistence of a chromosome break diminishes its mutagenic potential, PLoS Genet. 5 (2009), e1000683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gunn A, Bennardo N, Cheng A, Stark JM, Correct end use during end joining of multiple chromosomal double strand breaks is influenced by repair protein RAD50, DNA-dependent protein kinase DNA-PKcs, and transcription context, J. Biol. Chem 286 (2011) 42470–42482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bennardo N, Stark JM, ATM limits incorrect end utilization during non-homologous end joining of multiple chromosome breaks, PLoS Genet. 6 (2010), e1001194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Certo MT, Gwiazda KS, Kuhar R, Sather B, Curinga G, Mandt T, Brault M, Lambert AR, Baxter SK, Jacoby K, Ryu BY, Kiem HP, Gouble A, Paques F, Rawlings DJ, Scharenberg AM, Coupling endonucleases with DNA end-processing enzymes to drive gene disruption, Nat. Methods 9 (2012) 973–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Munoz MC, Yanez DA, Stark JM, An RNF168 fragment defective for focal accumulation at DNA damage is proficient for inhibition of homologous recombination in BRCA1 deficient cells, Nucleic Acids Res. 42 (2014) 7720–7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yin J, Lu R, Xin C, Wang Y, Ling X, Li D, Zhang W, Liu M, Xie W, Kong L, Si W, Wei P, Xiao B, Lee HY, Liu T, Hu J, Cas9 exo-endonuclease eliminates chromosomal translocations during genome editing, Nat. Commun 13 (2022) 1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Chari R, Mali P, Moosburner M, Church GM, Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach, Nat. Methods 12 (2015) 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bothmer A, Phadke T, Barrera LA, Margulies CM, Lee CS, Buquicchio F, Moss S, Abdulkerim HS, Selleck W, Jayaram H, Myer VE, Cotta-Ramusino C, Characterization of the interplay between DNA repair and CRISPR/Cas9-induced DNA lesions at an endogenous locus, Nat. Commun 8 (2017) 13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Cejka P, Symington LS, DNA end resection: mechanism and control, Annu. Rev. Genet 55 (2021) 285–307. [DOI] [PubMed] [Google Scholar]
- [58].Bennardo N, Cheng A, Huang N, Stark JM, Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair, PLoS Genet. 4 (2008), e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Grabarz A, Guirouilh-Barbat J, Barascu A, Pennarun G, Genet D, Rass E, Germann SM, Bertrand P, Hickson ID, Lopez BS, A role for BLM in double-strand break repair pathway choice: prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining, Cell Rep. 5 (2013) 21–28. [DOI] [PubMed] [Google Scholar]
- [60].Howard SM, Ceppi I, Anand R, Geiger R, Cejka P, The internal region of CtIP negatively regulates DNA end resection, Nucleic Acids Res. 48 (2020) 5485–5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Biehs R, Steinlage M, Barton O, Juhasz S, Kunzel J, Spies J, Shibata A, Jeggo PA, Lobrich M, DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination, Mol. Cell 65 (2017), 671–684 e675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Shamanna RA, Lu H, de Freitas JK, Tian J, Croteau DL, Bohr VA, WRN regulates pathway choice between classical and alternative non-homologous end joining, Nat. Commun 7 (2016) 13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Nickoloff JA, Sharma N, Taylor L, Allen SJ, Lee SH, Hromas R, Metnase and EEPD1: DNA repair functions and potential targets in cancer therapy, Front. Oncol 12 (2022), 808757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Howard SM, Yanez DA, Stark JM, DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining, PLoS Genet. 11 (2015), e1004943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Mengwasser KE, Adeyemi RO, Leng Y, Choi MY, Clairmont C, D’Andrea AD, Elledge SJ, Genetic screens reveal FEN1 and APEX2 as BRCA2 synthetic lethal targets, Mol. Cell 73 (2019), 885–899 e886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chang HH, Lieber MR, Structure-specific nuclease activities of Artemis and the Artemis: DNA-PKcs complex, Nucleic Acids Res. 44 (2016) 4991–4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ma Y, Schwarz K, Lieber MR, The Artemis:DNA-PKcs endonuclease cleaves DNA loops, flaps, and gaps, DNA Repair 4 (2005) 845–851. [DOI] [PubMed] [Google Scholar]
- [68].Woodbine L, Brunton H, Goodarzi AA, Shibata A, Jeggo PA, Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and Artemis for their repair, Nucleic Acids Res. 39 (2011) 6986–6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, Parker AR, Jackson SP, Gennery A, Jeggo PA, Lobrich M, A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci, Mol. Cell 16 (2004) 715–724. [DOI] [PubMed] [Google Scholar]
- [70].Waters CA, Strande NT, Wyatt DW, Pryor JM, Ramsden DA, Nonhomologous end joining: a good solution for bad ends, DNA Repair 17 (2014) 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Pryor JM, Conlin MP, Carvajal-Garcia J, Luedeman ME, Luthman AJ, Small GW, Ramsden DA, Ribonucleotide incorporation enables repair of chromosome breaks by nonhomologous end joining, Science 361 (2018) 1126–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Nick McElhinny SA, Havener JM, Garcia-Diaz M, Juarez R, Bebenek K, Kee BL, Blanco L, Kunkel TA, Ramsden DA, A gradient of template dependence defines distinct biological roles for family X polymerases in nonhomologous end joining, Mol. Cell 19 (2005) 357–366. [DOI] [PubMed] [Google Scholar]
- [73].Liang S, Thomas SE, Chaplin AK, Hardwick SW, Chirgadze DY, Blundell TL, Structural insights into inhibitor regulation of the DNA repair protein DNA-PKcs, Nature 601 (2022) 643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Durant ST, Zheng L, Wang Y, Chen K, Zhang L, Zhang T, Yang Z, Riches L, Trinidad AG, Fok JHL, Hunt T, Pike KG, Wilson J, Smith A, Colclough N, Reddy VP, Sykes A, Janefeldt A, Johnstrom P, Varnas K, Takano A, Ling S, Orme J, Stott J, Roberts C, Barrett I, Jones G, Roudier M, Pierce A, Allen J, Kahn J, Sule A, Karlin J, Cronin A, Chapman M, Valerie K, Illingworth R, Pass M, The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models, Sci. Adv 4 (2018) eaat1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Chen S, Lees-Miller JP, He Y, Lees-Miller SP, Structural insights into the role of DNA-PK as a master regulator in NHEJ, Genome Instab. Dis 2 (2021) 195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Graham TG, Walter JC, Loparo JJ, Two-stage synapsis of DNA ends during non-homologous end joining, Mol. Cell 61 (2016) 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Liu L, Chen X, Li J, Wang H, Buehl CJ, Goff NJ, Meek K, Yang W, Gellert M, Autophosphorylation transforms DNA-PK from protecting to processing DNA ends, Mol. Cell 82 (2022), 177–189 e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Stinson BM, Moreno AT, Walter JC, Loparo JJ, A mechanism to minimize errors during non-homologous end joining, Mol. Cell 77 (2020), 1080–1091 e1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Buehl CJ, Goff NJ, Hardwick SW, Gellert M, Blundell TL, Yang W, Chaplin AK, Meek K, Evidence for Separate Functions for Two Distinct Non-Homologous End Joining Long-Range Synaptic Complexes, Available at SSRN, 2022. [DOI] [PMC free article] [PubMed]
- [80].Deshpande RA, Myler LR, Soniat MM, Makharashvili N, Lee L, Lees-Miller SP, Finkelstein IJ, Paull TT, DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP, Sci. Adv 6 (2020), eaay0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Shiloh Y, Ziv Y, The ATM protein kinase: regulating the cellular response to genotoxic stress, and more, Nat. Rev. Mol. Cell Biol 14 (2013) 197–210. [PubMed] [Google Scholar]
- [82].Bermudez-Cabrera HC, Culbertson S, Barkal S, Holmes B, Shen MW, Zhang S, Gifford DK, Sherwood RI, Small molecule inhibition of ATM kinase increases CRISPR-Cas9 1-bp insertion frequency, Nat. Commun 12 (2021) 5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Balmus G, Pilger D, Coates J, Demir M, Sczaniecka-Clift M, Barros AC, Woods M, Fu B, Yang F, Chen E, Ostermaier M, Stankovic T, Ponstingl H, Herzog M, Yusa K, Martinez FM, Durant ST, Galanty Y, Beli P, Adams DJ, Bradley A, Metzakopian E, Forment JV, Jackson SP, ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks, Nat. Commun 10 (2019) 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Aditi S.M. Downing, Schreiner PA, Kwak YD, Li Y, Shaw TI, Russell HR, McKinnon PJ, Genome instability independent of type I interferon signaling drives neuropathology caused by impaired ribonucleotide excision repair, Neuron 109 (2021), 3962–3979 e3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Rass E, Chandramouly G, Zha S, Alt FW, Xie A, Ataxia telangiectasia mutated (ATM) is dispensable for endonuclease I-SceI-induced homologous recombination in mouse embryonic stem cells, J. Biol. Chem 288 (2013) 7086–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Kass EM, Helgadottir HR, Chen CC, Barbera M, Wang R, Westermark UK, Ludwig T, Moynahan ME, Jasin M, Double-strand break repair by homologous recombination in primary mouse somatic cells requires BRCA1 but not the ATM kinase, Proc. Natl. Acad. Sci. USA 110 (2013) 5564–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Weigelt B, Bi R, Kumar R, Blecua P, Mandelker DL, Geyer FC, Pareja F, James PA, Couch FJ, Eccles DM, Blows F, Pharoah P, Li A, Selenica P, Lim RS, Jayakumaran G, Waddell N, Shen R, Norton L, Wen HY, Powell SN, Riaz N, Robson ME, Reis-Filho JS, Chenevix-Trench G, The landscape of somatic genetic alterations in breast cancers from ATM germline mutation carriers, J. Natl. Cancer Inst (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Lee JH, Paull TT, Cellular functions of the protein kinase ATM and their relevance to human disease, Nat. Rev. Mol. Cell Biol 22 (2021) 796–814. [DOI] [PubMed] [Google Scholar]
- [89].Rogakou EP, Boon C, Redon C, Bonner WM, Megabase chromatin domains involved in DNA double-strand breaks in vivo, J. Cell Biol 146 (1999) 905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Feng YL, Xiang JF, Liu SC, Guo T, Yan GF, Feng Y, Kong N, Li HD, Huang Y, Lin H, Cai XJ, Xie AY, H2AX facilitates classical non-homologous end joining at the expense of limited nucleotide loss at repair junctions, Nucleic Acids Res. 45 (2017) 10614–10633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Wang H, Shi LZ, Wong CC, Han X, Hwang PY, Truong LN, Zhu Q, Shao Z, Chen DJ, Berns MW, Yates JR 3rd, Chen L, Wu X, The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair, PLoS Genet. 9 (2013), e1003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Zhou Y, Lee JH, Jiang W, Crowe JL, Zha S, Paull TT, Regulation of the DNA damage response by DNA-PKcs inhibitory phosphorylation of ATM, Mol. Cell 65 (2017) 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Zimmermann M, de Lange T, 53BP1: pro choice in DNA repair, Trends Cell Biol. 24 (2014) 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Guirouilh-Barbat J, Gelot C, Xie A, Dardillac E, Scully R, Lopez BS, 53BP1 protects against CtIP-dependent capture of ectopic chromosomal sequences at the junction of distant double-strand breaks, PLoS Genet. 12 (2016), e1006230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Zhao F, Kim W, Gao H, Liu C, Zhang Y, Chen Y, Deng M, Zhou Q, Huang J, Hu Q, Chen SH, Nowsheen S, Kloeber JA, Qin B, Yin P, Tu X, Guo G, Qin S, Zhang C, Gao M, Luo K, Liu Y, Lou Z, Yuan J, ASTE1 promotes shieldin-complex-mediated DNA repair by attenuating end resection, Nat. Cell Biol 23 (2021) 894–904. [DOI] [PubMed] [Google Scholar]
- [96].Lottersberger F, Karssemeijer RA, Dimitrova N, de Lange T, The role of the LINC complex in the 53BP1-driven mobility and NHEJ of dysfunctional telomeres, Mol. Biol. Cell 25 (2014). [Google Scholar]
- [97].Lottersberger F, Karssemeijer RA, Dimitrova N, de Lange T, 53BP1 and the LINC complex promote microtubule-dependent DSB mobility and DNA repair, Cell 163 (2015) 880–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Becker A, Durante M, Taucher-Scholz G, Jakob B, ATM alters the otherwise robust chromatin mobility at sites of DNA double-strand breaks (DSBs) in human cells, PLoS One 9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Caron P, Choudjaye J, Clouaire T, Bugler B, Daburon V, Aguirrebengoa M, Mangeat T, Iacovoni JS, Alvarez-Quilon A, Cortes-Ledesma F, Legube G, Non-redundant functions of ATM and DNA-PKcs in response to DNA double-strand breaks, Cell Rep. 13 (2015) 1598–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Dimitrova N, Chen YCM, Spector DL, de Lange T, 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility, Nature 456 (2008) 524–U551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Barnes RP, Thosar SA, Fouquerel E, Opresko PL, Targeted formation of 8-oxoguanine in telomeres, Methods Mol. Biol 2444 (2022) 141–159. [DOI] [PubMed] [Google Scholar]
- [102].Clouaire T, Legube G, A snapshot on the Cis chromatin response to DNA double-strand breaks, Trends Genet. 35 (2019) 330–345. [DOI] [PubMed] [Google Scholar]