

SUMMARY
Transient transgenic expression accelerates pharming and facilitates protein studies in plants. One embodiment of the approach involves leaf infiltration of Agrobacterium strains whose T‐DNA is engineered with the gene(s) of interest. However, gene expression during ‘agro‐infiltration’ is intrinsically and universally impeded by the onset of post‐transcriptional gene silencing (PTGS). Nearly 20 years ago, a simple method was developed, whereby co‐expression of the tombusvirus‐encoded P19 protein suppresses PTGS and thus enhances transient gene expression. Yet, how PTGS is activated and suppressed by P19 during the process has remained unclear to date. Here, we address these intertwined questions in a manner also rationalizing how vastly increased protein yields are achieved using a minimal viral replicon as a transient gene expression vector. We also explore, in side‐by‐side analyses, why some proteins do not accumulate to the expected high levels in the assay, despite vastly increased mRNA levels. We validate that enhanced co‐expression of multiple constructs is achieved within the same transformed cells, and illustrate how the P19 system allows rapid protein purification for optimized downstream in vitro applications. Finally, we assess the suitability of the P19 system for subcellular localization studies – an originally unanticipated, yet increasingly popular application – and uncover shortcomings of this specific implement. In revisiting the P19 system using contemporary knowledge, this study sheds light onto its hitherto poorly understood mechanisms while further illustrating its versatility but also some of its limits.
Keywords: transient expression, agro‐infiltration, RNA silencing, P19 protein, siRNAs, technical advance
Significance Statement
Since its disclosure in 2003, agro‐infiltration used in combination with the P19 RNA silencing suppressor has been a popular method to achieve high levels of transient gene expression in Nicotiana benthamiana, though the underlying molecular mechanisms have remained unclear. By shedding light on these mechanisms, the present contemporary reassessment of the method allows improvements to protein yields and further illustrates its versatility, but also exposes and rationalizes some of its limits.
INTRODUCTION
By respectively promoting nucleotide sequence‐specific chromatin compaction and enhanced mRNA turnover, transcriptional and post‐transcriptional gene silencing (TGS, PTGS) are major impediments to stable transgene expression in plants. Studies of transgene silencing in the model plant Arabidopsis thaliana have revealed that both TGS and PTGS are triggered by long double‐stranded RNA (dsRNA) (Beclin et al., 2002; Mourrain et al., 2000; Vaucheret et al., 1998). This molecule is processed into populations of small interfering RNA (siRNA) duplexes by one of four Dicer‐like (DCL) proteins encoded in the Arabidopsis and many other plants' genomes (Bologna & Voinnet, 2014). DCL4 and DCL2 have signature 21‐nt and 22‐nt‐long siRNA products, both of which can promote PTGS upon their incorporation into ARGONAUTE1 (AGO1)‐clade silencing effector proteins. AGO1 uses siRNAs as molecular guides to retrieve sequence‐complementary mRNAs and execute their silencing mainly via endonucleolytic cleavage. DCL3 processes dsRNA into 24‐nt siRNAs, which, loaded into AGO4‐clade members, guide de novo RNA‐directed DNA methylation (RdDM) at the siRNA loci of origin (Bologna & Voinnet, 2014). While RdDM mediated by promoter‐derived dsRNA/siRNAs usually results in TGS (Mette et al., 2000), gene body methylation caused by ORF‐derived siRNAs – which often accompanies PTGS – is largely inconsequential to transgene expression (Taochy et al., 2019). At least two non‐mutually exclusive sources of dsRNA have been identified as triggers of transgene silencing in stable transformants, ultimately converging in dsRNA production and downstream processing into siRNAs. In sense‐PTGS (S‐PTGS), aberrant (i.e., uncapped or PolyA−) mRNAs ((ab)mRNAs) spawned from presumably highly transcribed transgenes evade RNA quality control (RQC) and are instead competitively de novo converted into dsRNA by endogenous RNA‐dependent RNA polymerases (RDRs) (reviewed in Liu & Chen, 2016). In inverted repeat (IR)‐PTGS, rearranged transgene arrays, which are common transformation artifacts, form IRs whose transcription creates RDR‐independent sources of dsRNA (Beclin et al., 2002; Henderson et al., 2006; Himber et al., 2003). IR‐PTGS may also be deliberately achieved for the purpose of experimental gene knockdown via RNA interference (RNAi), using tailored ‘stem‐loop’ transgenes (Himber et al., 2003; Mette et al., 2000).
In Arabidopsis and other plants, both S‐ and IR‐PTGS pathways have endogenous gene regulatory counterparts (Henderson et al., 2006; Vaucheret, 2005) operating in complement to, or in conjunction with, a third endogenous PTGS pathway based on microRNAs (miRNAs) (Bologna & Voinnet, 2014). DCL1‐dependent 21‐ to 24‐nt miRNAs are often evolutionary conserved and excised as discrete species from genome‐encoded, imperfect stem‐loop primary transcripts. Incorporated into mostly AGO1, they regulate the abundance/translation of co‐evolving miRNA‐complementary transcripts via PTGS (Voinnet, 2009). Last but not least, mechanisms resembling IR‐ and S‐PTGS form the basis of a universal plant antiviral defense system primarily involving DCL4, DCL2, and AGO1/2. The IR‐PTGS‐like mechanism uses virus replication‐derived dsRNA to produce 21‐/22‐nt virus‐derived siRNAs (vsiRNAs), while the S‐PTGS‐like mechanism enables their amplification from single‐stranded viral RNA via RDR activities, thereby presumably keeping pace with the mounting viral load (Pumplin & Voinnet, 2013). As a counter‐defensive strategy, most plant viruses have evolved viral suppressors of RNA silencing (VSRs) targeting various and sometimes multiple steps of the plant antiviral silencing response (Pumplin & Voinnet, 2013; Wu et al., 2010). For instance, the tombusviral P19 protein, one of the best biochemically characterized VSRs, forms homodimeric ‘calipers’ structurally poised to bind, with extremely high affinity, the 21‐/22‐nt siRNA products of antiviral DCL4/DCL2 (Silhavy et al., 2002; Vargason et al., 2003; Voinnet et al., 1999). Binding presumably prevents fruitful loading of siRNAs into AGO1/AGO2, and is accompanied by a characteristic 1–2‐nt trimming of siRNA 3′‐ends by an unknown nuclease, a process also manifested on endogenous miRNAs under authentic infections (Kontra et al., 2016) and transgenic P19 expression conditions (Chapman et al., 2004; Iki et al., 2018; Papp et al., 2003).
Although stably transformed plants can be used as bioreactors to yield theoretically high levels of protein production, optimal performances are rarely achieved due to various impediments manifested all along the (trans)gene expression pathway, TGS and PTGS notwithstanding (Kjemtrup et al., 2014; Obembe et al., 2011). These include, but are not restricted to, protein degradation, poor translation, intrinsic mRNA instability, and mediocre transcription due to genomic position effects. Additionally, some proteins of interests might be toxic when constitutively or even conditionally expressed at high levels in stable transformants. Thus, embryo or seedling lethality during transgenesis naturally incurs selection of low‐ to moderate‐expression lines, or simply compromises altogether the viability of harvested tissues. Transient, as opposed to stable, protein expression has emerged as an interesting alternative overcoming at least some of the aforementioned limitations. While many approaches are available (reviewed in Tyurin et al., 2020), probably none exhibits the simplicity, rapidity, and scalability of the procedure whereby suspensions of Agrobacterium tumefaciens strains are pressure‐infiltrated, usually with a syringe, into the leaf air space, a procedure known as ‘agro‐infiltration’. In this process, so called ‘disarmed’ T‐DNAs deprived of the tumor‐inducing genes that cause crown gall disease are engineered with the transgene(s) of interest. Upon T‐DNA transfer into plant cells, transgene expression usually peaks between 3 and 5 days post‐infiltration, upon which infiltrated patches can be harvested for molecular studies or protein extraction (Chen et al., 2013). While the method is, in principle, universally applicable, its efficacy is substantially reduced in many Brassicales, including Arabidopsis, owing to an innate immune response triggered by the pathogen‐associated molecular pattern EF‐Tu produced by Agrobacterium (Zipfel et al., 2006). Due to its inability, among other Solanales, to perceive EF‐Tu, the wild tobacco relative Nicotiana benthamiana has emerged as a plant of choice with which to carry out transient expression via agro‐infiltration. Other advantages of N. benthamiana include its non‐demanding and high‐density growth conditions, leaves that are well adapted to pressure‐infiltration, and a reduced cellular protease profile compared to many other plant species (Kjemtrup et al., 2014).
Despite its clear advantages over stable transformation for pharming and other applications, agro‐infiltration in N. benthamiana leaves is almost invariably accompanied by a PTGS response diagnosed by accumulation of transgene‐derived siRNAs in the infiltrated tissues, paralleling transgene expression (Hamilton et al., 2002; Voinnet et al., 2000). In a search for a pragmatic remedy to this problem, it was realized that co‐infiltration with a second Agrobacterium strain engineered to transiently express a VSR reduces transgene PTGS and, hence, stabilizes transient expression (Hamilton et al., 2002). The approach was found particularly effective with the tombusviral P19 protein evoked here, granting not only stabilized but also, in many – albeit not all – cases, substantially enhanced expression (Voinnet et al., 2003). Due to its simplicity and efficacy, the enhanced transient expression system has been and remains widely employed by the plant community as well as commercially, with the co‐infiltrated P19 Agrobacterium strain often used as a default, as opposed to optional, setting in the procedure (Sainsbury & Lomonossoff, 2014). Despite this success, many questions have remained unanswered, not least what mechanisms underpin siRNA accumulation in agro‐infiltrated tissues, that impede transgene expression and, as a corollary, how P19 suppresses them. Indeed, while a plethora of TGS/PTGS mutants are available in Arabidopsis (Bologna & Voinnet, 2014), studying silencing mechanisms is much harder in the poorly genetically amenable species N. benthamiana.
Here, we revisit the P19‐enhanced transient expression system using fundamental knowledge gained in Arabidopsis to decipher the likely mechanisms of PTGS induction in infiltrated tissues and the molecular bases of their inhibition by P19. A better understanding of these processes helps us rationalize how a virus‐based self‐replicating transgene, poorly efficacious when expressed alone, rapidly achieves, under P19 co‐expression conditions, protein yields far exceeding those already obtained with the conventional embodiment of the method. While the approach enables a high degree of simultaneous expression of multiple constructs within single cells, we show that PTGS‐unrelated and protein‐intrinsic properties can negatively impact yield even under conditions of vastly enhanced mRNA accumulation enabled by P19. We further describe how an immuno‐purified enzyme performs substantially better in cell extracts prepared from P19‐co‐infiltrated tissues, indicating that high protein levels achieved with the method in vivo translate into high functionality in vitro. Last but not least, we assess not only the advantages but also the limits of the P19‐enhanced transient expression system for subcellular localization studies, a widespread yet so far largely non‐scrutinized application of the system. With this contemporary reassessment of the P19 system, we formulate both recommendations and cautions for its optimal use and performances in a variety of settings and applications, and discuss possible further improvements.
RESULTS AND DISCUSSION
21‐nt and 24‐nt siRNAs do not accumulate proportionally and are distinctively affected by P19 over time under non‐saturating transient gene expression conditions
Previous studies have shown how Agrobacterium‐mediated transient expression of reporter genes such as the endoplasmic reticulum (ER)‐targeted GFP5 allele (Angell & Page, 2002; Haseloff et al., 1997) is accompanied by the production of 21‐nt and 24‐nt siRNAs (Hamilton et al., 2002; Himber et al., 2003), the cognate products of plant DCL4 and DCL3 (Henderson et al., 2006). Certain VSRs co‐expressed in the transient assay impeded accumulation of these species and some displayed siRNA‐size‐selective effects demonstrating that 21‐nt siRNAs are both necessary and sufficient to mediate PTGS as a major impediment to transient expression (Hamilton et al., 2002; Himber et al., 2003). Among these VSRs, the tombusviral P19 protein exhibits potent effects, in agreement with its ability to suppress transgene, viral, and endogenous PTGS by forming homodimers with high and selective affinity for 21–22‐nt small (s)RNAs, including viral/endogenous siRNAs (Kontra et al., 2016) and endogenous miRNAs (Chapman et al., 2004; Iki et al., 2018; Vargason et al., 2003). Beside the efficacy of the P19 co‐expression approach, how siRNA accumulation and PTGS are triggered during Agrobacterium‐mediated transient expression and how, mechanistically, co‐expressed P19 impairs these processes have remained largely elusive.
To better dissect the molecular underpinnings of PTGS onset during transient expression and its suppression by P19, we revisited the GFP5‐P19 co‐expression assay in a time‐course analysis involving diluted (OD = 0.3 for each construct for a final OD = 0.6) as opposed to saturated Agrobacterium suspensions used previously (Hamilton et al., 2002, Himber et al., 2003). Nicotiana benthamiana leaves were infiltrated with an Agrobacterium strain carrying a disarmed T‐DNA engineered to express, under the control of the strong and ubiquitous p35S promoter, the open reading frame (ORF) of GFP5 (p35S::GFP5; Figure 1; Hamilton et al., 2002, Himber et al., 2003). Use of GFP5 enables measurements of not only mRNA/protein production, but also protein activity monitored visually and non‐invasively in planta with a handheld UV lamp and quantified with a fluorescence reader. The diluted p35S::GFP5 Agrobacterium strain was mixed 1:1 with a second strain engineered to express either an epitope‐tagged and intron‐containing GUS ORF or the P19 ORF under the control of the 35S promoter (p35S::FHA:GUS‐Intron or p35S::P19, respectively; Figure 1). Co‐expressing p35S::GFP5 with p35S::FHA:GUS‐Intron yielded a green fluorescence signal that progressively decreased in intensity from 4 days post‐infiltration (dpi) onward; it was lower at 7 dpi and nearly below visual detection at 10 dpi when only red fluorescence from chlorophyl remained clearly visible (Figure 2a). Correlating with this decrease in green fluorescence was a steady, approximately one‐order‐of‐magnitude decline in GFP5 mRNA levels from 4 to 10 dpi, as measured in patches individually collected over multiple independent experiments (Figure 2b). Total low‐molecular‐weight RNA analysis from the same tissues revealed accumulation of discrete 21‐nt and 24‐nt GFP5‐derived siRNAs reaching an accumulation peak at 7 dpi. At all three time points, however, 21‐nt‐long siRNAs accumulated at higher levels than the 24‐nt‐long species under the non‐saturating transient gene expression conditions used (Figure 2c). These results imply that large amounts of GFP5‐derived long dsRNA accumulate within agro‐infiltrated patches because dsRNA is the substrate used by Dicer proteins to produce siRNAs. They also suggest that the dsRNA molecules accounting for accumulation of more abundant 21‐nt siRNAs might be distinct from those underlying the production of 24‐nt siRNAs, reflecting, perhaps, the involvement of distinct dsRNA biosynthetic pathways.
Figure 1.
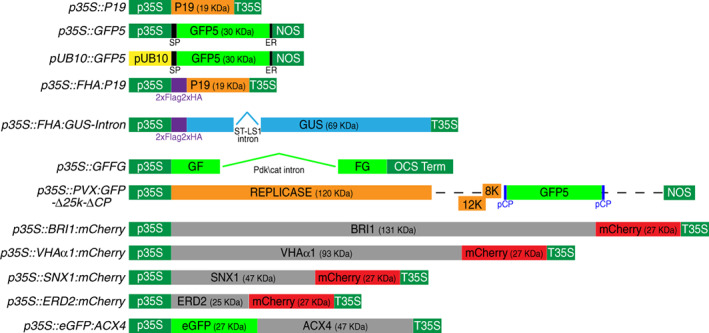
Schematics of the various constructs used in this study.
p35S: 35S promoter from Cauliflower mosaic virus (CaMV); pUB10: Arabidopsis UBIQUITIN10 promoter; T35S: 35S terminator from CaMV; OCS Term: octopine synthase terminator; NOS: nopaline synthase terminator; pCP: coat protein promoter; SP: basic chitinase B signal peptide; ER: HDEL endoplasmic reticulum retention signal.
Figure 2.
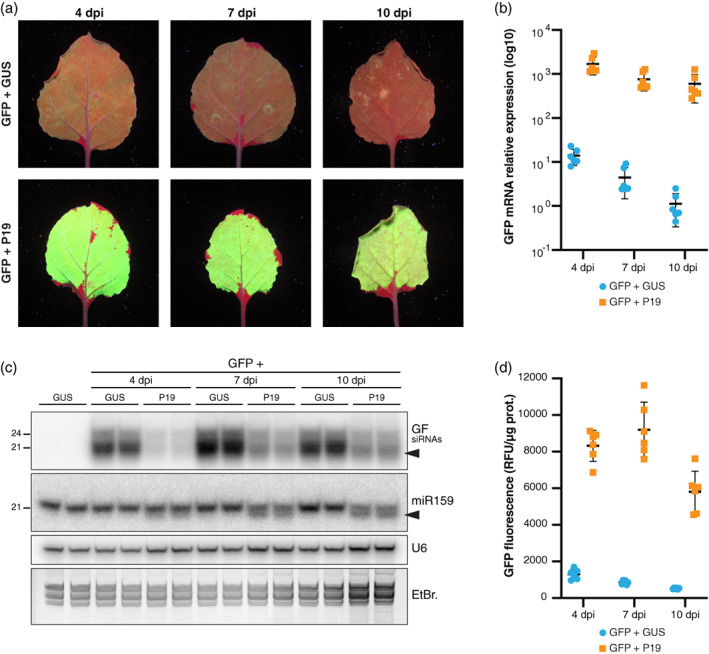
Transient P19 co‐expression dramatically enhances GFP5 mRNA accumulation and reduces 21‐nt siRNA production in infiltrated patches. (a) Images of Nicotiana benthamiana leaves, representative of six independent experiments, taken under UV illumination at 4, 7, and 10 days post‐infiltration (dpi) with the constructs p35S::GFP5 (GFP), p35S::FHA:GUS‐Intron (GUS), or p35S::P19 (P19). Note that chlorophyll fluoresces red under UV.
(b) Log10‐transformed relative GFP mRNA levels in samples depicted in (a) as quantified by RT‐qPCR. Black bars: mean. Error bars: standard deviations. n = 6.
(c) GF siRNA levels in biological duplicates of samples depicted in (a) as analyzed by Northern blot using a probe corresponding to the 5′ part of the GFP ORF (‘GF’). The probed miR159 and U6 small RNA were used as an endogenous P19 cargo and RNA loading control, respectively. EtBr.: Ethidium bromide staining provides an additional RNA loading control. Black arrows indicate 3′‐end‐trimmed sRNA species. The experiment was independently repeated three times with similar results.
(d) Relative GFP fluorescence units (RFU) measured from the samples depicted in (a). Black bars: mean. Error bars: standard deviations. n = 6.
In experiments conducted side‐by‐side, co‐expression of p35S::GFP5 with p35S::P19 vastly enhanced green fluorescence compared to co‐expression of p35S::GFP5 with p35S::FHA:GUS‐Intron (Figure 2a). This enhancement, moreover, persisted along the entire 4–10‐day observation time frame, in sharp contrast to the decline in fluorescence observed upon co‐expression of p35S::GFP5 with p35S::FHA:GUS‐Intron. Accordingly, measurements made at 4, 7, and 10 dpi in leaf patches individually collected over multiple independent experiments indicated that, at each time point, the GFP5 fluorescence levels were approximately one order of magnitude higher under p35S::P19 compared to p35S::FHA:GUS‐Intron co‐expression conditions (Figure 2d, confirmed independently by leaf fluorescence scanning in Figure S1). The GFP5 mRNA levels under p35S::P19 co‐expression conditions were up to three orders of magnitude (i.e., approximately 1000‐fold) higher than those under p35S::FHA:GUS‐Intron co‐expression conditions. Moreover, these mRNA levels remained extremely high over the 4–10‐day observation/sampling period (Figure 2b). Under p35S::P19 co‐expression conditions, the accumulation of GFP5‐derived siRNAs was strongly reduced at 4, 7, and 10 dpi and disproportionally affected the abundant 21‐nt siRNA species. Moreover, a portion of the 21‐nt GFP5 siRNAs remaining at 7 and 10 dpi showed increasing signs of trimming diagnosed by their enhanced electrophoretic mobility (Figure 2c); 3′‐end 1–2‐nt trimming has been ascribed to an as‐yet unidentified cellular exonuclease acting on the P19‐bound fraction of sRNAs (Iki et al., 2018; Kontra et al., 2016; Papp et al., 2003). An increasing proportion of the 21‐nt miR159, used as a representative of endogenous miRNAs, also displayed trimming over time, suggesting that transiently expressed P19 also binds these molecules as reported in stable transformants. Unlike those of the GFP5 siRNAs, however, the miR159 steady‐state levels were not overtly decreased. Similar observations on GFP5 siRNAs and miR159 were made when p35S from p35S::GFP5 was swapped for the Arabidopsis endogenous UBIQUITIN10 promoter (pUB10), which is also known to display strong and constitutive activity (Norris et al., 1993). When co‐agro‐infiltrated with p35S::FHA:GUS‐Intron, pUB10::GFP5 expression yielded a weak green fluorescence signal at 7 dpi similar in intensity to that yielded by co‐infiltrating p35S::GFP5 with p35S::FHA:GUS‐Intron (Figure S2a,b). Moreover, 21‐ and 24‐nt GFP5 siRNAs also accumulated under pUB10::GFP5 co‐expression (Figure S2c), suggesting that RNA silencing is a general response to transient transgene expression, regardless of p35S usage. Co‐expressing P19 with pUB10::GFP5 resulted in vastly enhanced green fluorescence (Figure S2a,b), correlating with a strong decrease in GFP5 siRNA accumulation and evidence of their trimming alongside that of endogenous miR159 (Figure S2c).
That P19 causes a net decrease in transgene‐derived siRNA but not endogenous miRNA levels in both systems suggests either that the protein acts differently on the former versus latter pool of sRNAs in agro‐infiltrated tissues, or that distinct silencing mechanisms involved in the biogenesis of these species influence the net output of P19 binding to sRNAs during transient gene expression.
The 21‐nt species are mainly secondary siRNAs produced by RDR6 via sense‐PTGS
No overt feature predisposes the transiently expressed p35S::GFP5 construct to strong siRNA production. This led us to consider that silencing accompanying agro‐infiltration might be akin, at least partly, to S‐PTGS elicited by sense‐transgenes upon stable, as opposed to transient, transformation. As evoked in the present study's introduction, a key feature of S‐PTGS is its strong dependency upon RDR6 activity, where the dsRNA products are processed by mainly DCL4 into 21‐nt siRNAs (Mourrain et al., 2000; Taochy et al., 2019). To test this idea, we used an established transgenic N. benthamiana line in which NbRDR6's activity is constitutively dampened by RNAi (rdr6 RNAi line; Schwach et al., 2005). In multiple independent experiments, co‐expressing p35S::GFP5 with p35S::FHA:GUS‐Intron resulted in consistently more intense GFP5 fluorescence at 4 dpi (quantified average of approximately 1.9‐fold gain) in leaves with the rdr6 RNAi compared with the wild‐type (WT) background (Figure 3a,b,d). This enhanced fluorescence was paralleled by a strong and selective reduction, albeit not elimination, of the abundant 21‐nt siRNA species accumulating in the assay, suggesting that silencing triggered during transient gene expression is indeed mostly a manifestation of RDR6‐dependent S‐PTGS (Figure 3c).
Figure 3.
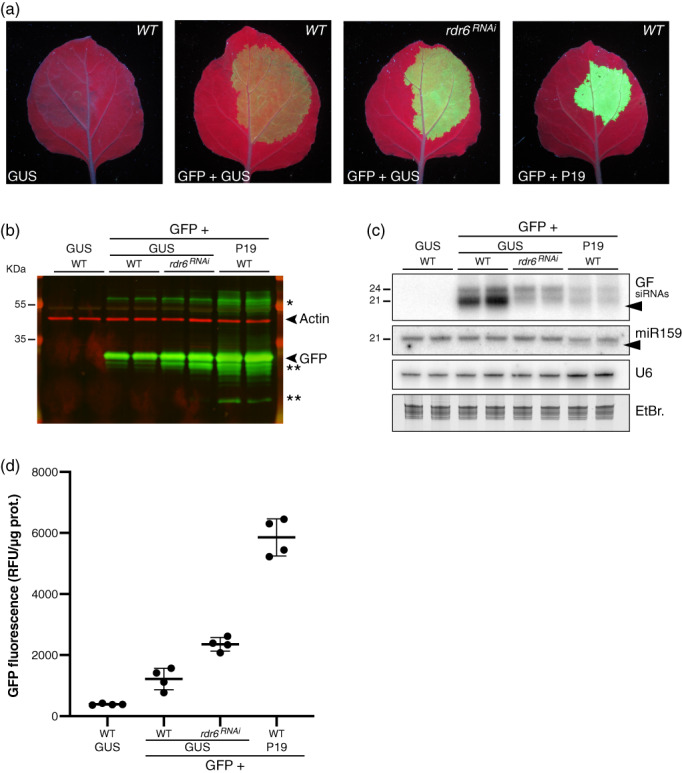
Compared effects of P19 transient co‐expression versus RNAi‐mediated knockdown of NbRDR6 on GFP5 expression and siRNA accumulation in infiltrated patches. (a) Images of Nicotiana benthamiana leaves in the WT or the rdr6 RNAi background, representative of six independent experiments, at 4 dpi with the constructs p35S::GFP5 (GFP), p35S::FHA:GUS‐Intron (GUS), and p35S::P19 (P19), as indicated, under UV illumination.
(b) GFP protein accumulation in biological duplicates of the samples depicted in (a) analyzed by Western blot using an anti‐GFP antibody (green signal). An anti‐actin antibody was used as a control for equal protein loading (red signal). *Indicates higher‐molecular‐weight forms of the GFP protein or potential concatemers thereof. **Indicates truncated byproducts of the GFP protein. The experiment was independently repeated two times with similar results.
(c) GF siRNA levels in biological duplicates of samples depicted in (a) analyzed by Northern blot. The probed miR159 and U6 small RNA were used as an endogenous P19 cargo and RNA loading control, respectively. EtBr.: Ethidium bromide staining provides an additional RNA loading control. Black arrows indicate 3′‐end‐trimmed sRNA species. The experiment was independently repeated two times with similar results.
(d) Relative GFP Fluorescence Units (RFU) measured from samples depicted in (a). Black bars: mean. Error bars: standard deviations. n = 4.
We anticipate that the two main and non‐exclusive pathways underpinning S‐PTGS activation upon stable plant transformation are also at work during transient expression via agro‐infiltration in a manner likely explaining the observed siRNA patterns. Firstly, (i) given the sheer levels of transgene expression potentially achievable by this method (as revealed under P19 co‐expression conditions; Figure 2b), elevated amounts of (ab)mRNAs are likely produced. By overwhelming RQC in infiltrated tissues (Gazzani et al., 2004; Moreno et al., 2013; Parent et al., 2015; reviewed in Liu & Chen, 2016), this excess of (ab)mRNA might spontaneously undergo dsRNA conversion by RDRs followed by 21‐nt siRNA production by RDR6‐coupled DCL4 in the cytosol, as previously reported (Mourrain et al., 2000, Taochy et al., 2019). Their 24‐nt counterpart could result from RDR2‐coupled DCL3 activity in the nucleus (Jauvion et al., 2012). Secondly, (ii) rearranged T‐DNA arrays are likely formed during transient expression, such that overlapping sense‐antisense and/or IR transcription would constitute RDR‐independent sources of dsRNA processed into 21‐nt and 24‐nt primary siRNAs, as reported during IR‐PTGS (Beclin et al., 2002; Henderson et al., 2006; Himber et al., 2003). In either scenario, the 21‐nt fraction of neo‐processed primary siRNAs would initiate PTGS of the main bulk of normal, sense‐transgene transcripts, with their breakdown products serving, upon their conversion into long dsRNAs by cytosolic RDR6, as further major sources of DCL4‐dependent 21‐nt secondary siRNAs. This second and prominent RDR6‐amplified phase likely underpins the disproportionate accumulation of 21‐nt versus 24‐nt siRNAs during S‐PTGS in stable transgenic lines (Taochy et al., 2019), as indeed also observed here during transient expression of p35S::GFP5 (Figures 2c and 3c).
The remaining 21‐nt GFP5 siRNAs observed upon p35S::GFP5 expression in rdr6 RNAi leaves are therefore likely RDR‐independent primary siRNAs formed according to scenario (ii) evoked above, although incomplete RNAi‐mediated knockdown or redundant RDR activities may also be involved. Regardless of their possible origin, these residual 21‐nt siRNAs likely display potent PTGS activity. Indeed, in side‐by‐side analyses, enhanced fluorescence was substantially less pronounced upon co‐expression of p35S::GFP5 with p35S::FHA:GUS‐Intron in rdr6 RNAi leaves than it was upon its co‐expression with p35S::P19 in WT leaves (quantified average of approximately 1.9‐fold versus 4.8‐fold gain) despite similar levels of GFP5 siRNAs remaining under each condition (Figure 3a–d). The key difference in silencing suppression efficacy probably reflects that the 21‐nt GFP5 siRNAs remaining in rdr6 RNAi leaves are PTGS‐proficient, whereas those remaining under p35S::P19 co‐expression are bound and inactivated by P19 as suggested by their enhanced electrophoretic mobility reflecting their likely 1–2‐nt trimming, a feature absent from the GFP5 siRNAs remaining in rdr6 RNAi tissues (Figure 3c). If the bulk of GFP5 siRNAs is mostly composed of RDR6‐dependent secondary molecules, as expected from S‐PTGS, P19‐mediated binding and inactivation of the primary siRNAs required for secondary siRNA amplification would explain the drastic net reduction in GFP5 siRNA levels observed in infiltrated tissues (Figures 2c and 3c). This would also explain why, in contrast, miR159 levels were not overtly altered by its binding to P19 because miRNA biogenesis does not involve RDR6 or sRNA amplification (Figures 2c and 3c; Voinnet, 2009).
P19 binding to primary siRNAs likely inhibits amplified S‐PTGS initiated by these molecules
The model in Figure 4 predicts that, unlike secondary siRNAs, primary siRNAs should not display reduced steady‐state accumulation, being predictably merely sequestered and thereby inactivated by P19. To test this hypothesis, we used an Agrobacterium strain expressing, under the control of the p35S promoter, an IR transgene encompassing the ‘GF’ portion of the GFP5 sequence (p35S::GFFG; Figure 1). As shown previously, the GF‐FG IR is genetically conditioned to produce dsRNA and, as such, spawns primary siRNAs in an RDR6‐independent manner (Himber et al., 2003). To test the binding of P19 to GF primary siRNAs in the absence of an immunoprecipitation (IP)‐graded antibody, we used an N‐terminal FLAG‐HA‐epitope‐tagged allele of the protein expressed under the control of the p35S promoter (p35S::FHA:P19; Figure 1). We verified that p35S::FHA:P19 is as efficient as p35S::P19 in suppressing GFP5 silencing in the co‐infiltration assay and that FHA:P19 can be robustly immunoprecipitated via anti‐HA IP used to assay binding (Figure 5a–d; Figure S3a). As previously reported in stably transformed WT and rdr6 Arabidopsis (Himber et al., 2003), transient expression of p35S::GFFG spawned, at 4 dpi, near‐equal amounts of DCL4‐dependent 21‐nt and DCL3‐dependent 24‐nt GF siRNAs, the cognate pattern of RDR6‐independent primary siRNA production from IRs (Beclin et al., 2002, Himber et al., 2003, Henderson et al., 2006; Figure 5e; Figure S3b). This pattern contrasted with the disproportionate levels of 21‐nt species accumulating during transient expression of p35S::GFP5 presumably as a consequence of their RDR6‐mediated amplification during S‐PTGS (Figures 2c and 3c). Co‐expressing p35S::GFFG with p35S::FHA:P19 did not cause any overt decrease in either GF siRNA species' levels compared to p35S::GFFG alone or in combination with p35S::FHA:GUS‐Intron, a further contrast to the strong reduction in siRNA levels incurred by co‐expressing p35S::P19 with p35S::GFP5 (Figures 2c, 3c, and 5e; Figure S3b). The only distinctive feature of the p35S::FHA:P19 + p35S::GFFG co‐expression conditions was the enhanced electrophoretic mobility of a fraction of the 21‐nt‐, but not 24‐nt‐, GF siRNAs likely reflecting their preferential binding by P19 and the linked 1–2‐nt trimming process (Figure 5e; Figure S3b). Directly supporting this idea, only the 21‐nt GF siRNAs were detected in FHA:P19 immune complexes isolated via HA IP, of which a fraction showed signs of trimming as did a fraction of the co‐immunoprecipitated endogenous miR159 (Figure 5e; Figure S3b). Thus, while their binding to P19 causes their trimming (and, presumably, inactivation), it does not reduce the steady‐state levels of primary 21‐nt siRNAs, supporting the proposed model of P19 action (Figure 4).
Figure 4.
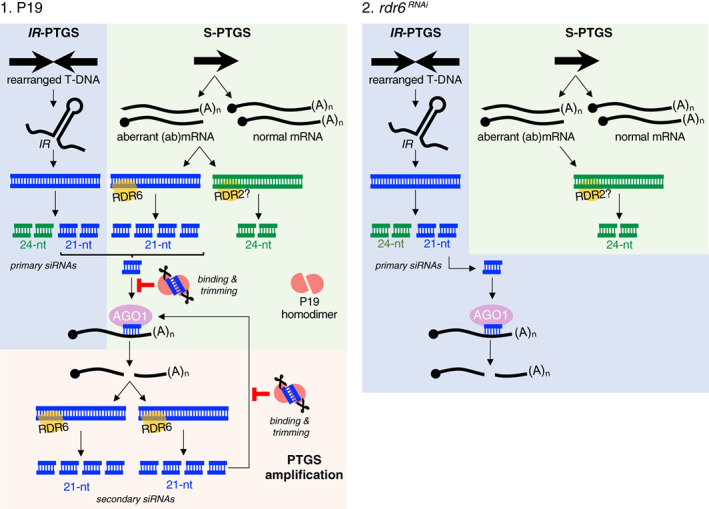
Proposed model for the multilayered action of P19 (scenario 1) during transient gene expression in Nicotiana benthamiana leaves as compared to the effect of RDR6 knockdown (scenario 2) using the rdr6 RNAi background.
In scenario 1, IR‐PTGS involves transgene arrays organized as inverted repeats (IRs). Upon transcription, these form RDR‐independent sources of dsRNA, which is processed into 21‐nt and 24‐nt siRNA species. An additional source of dsRNA is provided through S‐PTGS via RDR6 (leading to 21‐nt siRNA production) and possibly RDR2 (leading to 24‐nt siRNA production), using sense polyA− and uncapped (collectively referred to as to ‘aberrant’) RNAs as templates. 21‐nt siRNAs produced by both IR‐ and S‐PTGS are then loaded into AGO1 to guide endocleavage of complementary sense transgene mRNAs. The ensuing polyA− and uncapped RNA breakdown products being aberrant, they serve, in turn, as novel sources of RDR6‐dependent dsRNA and secondary 21‐nt siRNAs. This RDR6‐amplified phase would contribute the main siRNA bulk in infiltrated tissues, explaining the disproportionally high representation of 21‐nt RNAs. Co‐expressed P19 binds to and causes trimming of both primary and secondary 21‐nt siRNAs. Sequestration of the former, in particular, impedes PTGS amplification by RDR6, resulting in a substantial decrease of the dominant 21 nt siRNA fraction. The 24‐nt siRNA levels remain largely unchanged because they are not bound by P19 and, hence, not trimmed. In scenario 2, reduced RDR6 activity in the rdr6 RNAi background impedes the onset of amplified PTGS, leading to mostly primary (i.e., non‐amplified) 21‐nt siRNA accumulation. The levels of RDR2‐dependent 24‐nt siRNAs presumably accompanying S‐PTGS remain unchanged. While considerably reduced in levels as in scenario 1, the 21‐siRNAs would not undergo trimming.
Figure 5.
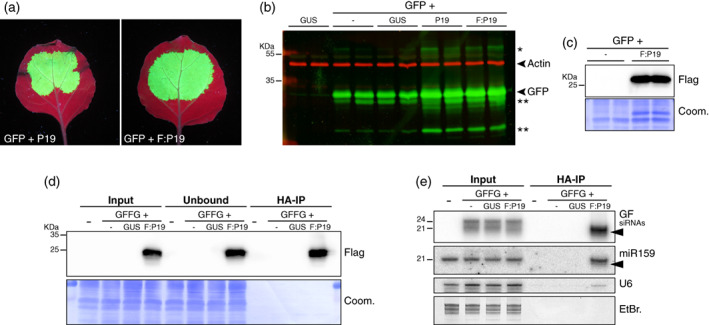
P19 selectively binds the 21‐nt siRNAs produced in infiltrated patches during IR‐PTGS. (a) Images of Nicotiana benthamiana leaves, representative of six independent experiments, at 4 dpi with the constructs p35S::GFP5 (GFP), p35S::P19 (P19), and p35S::FHA:P19 (F:P19), as indicated, under UV illumination.
(b) GFP protein accumulation (green signal) in biological duplicates of the samples depicted in (a) and their controls (p35S::FHA:GUS‐Intron: GUS), analyzed by Western blot. An anti‐actin antibody was used to provide a control for equal protein loading (red signal). *Indicates higher‐molecular‐weight forms of the GFP protein or potential concatemers thereof. **Indicates truncated byproducts of the GFP protein. The experiment was independently repeated three times with similar results.
(c) FHA:P19 protein levels in biological duplicates of one of the samples depicted in (a) and its controls analyzed by Western blot. Coomassie staining of total proteins (Coom.) provides a control for equal loading. The experiment was independently repeated three times with similar results.
(d) FHA:P19 protein levels in the input, unbound fraction, and IP fraction of N. benthamiana leaves at 4 dpi with infiltration medium (−), p35S::FHA:GUS‐Intron (GUS), p35S::FHA:P19 (F:P19), and p35S::GFFG (GFFG), as indicated. Coom.: as in (c). The experiment was independently repeated four times with similar results.
(e) GF siRNAs levels in samples depicted in (d) analyzed by Northern blot. miR159 and U6 small RNA were used as an endogenous P19 cargo and RNA loading control, respectively. EtBr.: Ethidium bromide staining provides an additional RNA loading control. Black arrows indicate 3′‐end‐trimmed sRNA species. The experiment was independently repeated four times with similar results.
Collectively, these results suggest that RDR6‐dependent S‐PTGS underpins the bulk of abundant 21‐nt siRNA levels accompanying transient transgene expression and that P19 strongly inhibits this amplified process by binding to and inactivating the 21‐nt primary siRNAs required for its initiation. The 21‐nt GFP5 siRNAs remaining in tissues co‐expressing p35S::GFP5 with p35S::P19 are, therefore, likely mostly composed of P19‐bound and hence, inactive, primary siRNAs.
Combining P19 expression with that of a recombinant Potato virus X replicon yields further increased GFP5 levels and activity
The sole known natural function of P19 is to protect the tombusviral RNA against RNA silencing triggered by vsiRNAs spawned during virus replication, which is a source of dsRNA (Vargason et al., 2003). Enhanced replication of recombinant viral vectors via P19 expression has been reported in N. benthamiana cell suspension‐ and hairy root‐based settings (Larsen & Curtis, 2012). We thus tested if replication of Potato virus X (PVX) would be enhanced by P19 co‐expression in the rapid and easy‐to‐implement N. benthamiana leaf agro‐infiltration setting used here. To that effect, we employed p35S::PVX‐Δ25ΔCP:GFP5 (Voinnet et al., 2000), which is devoid of the 25k and CP ORFs required for viral movement, encapsidation and, hence, disease symptom development (Figure 1). Being innocuous, non‐infectious, and non‐mobile, this near‐minimal PVX replicon is in principle suited to high‐level protein production via Agrobacterium‐mediated transient expression without biocontainment issues. The results in Figures 2, 3, and 5 suggested, however, that p35S::PVX‐Δ25ΔCP:GFP5 would trigger RNA silencing at least at three distinct, yet cumulative, levels. The first level would be akin to RDR6‐dependent S‐PTGS targeting the primary transgene RNA transcribed from the p35S promoter, as observed with p35S::GFP5 (Figures 2a and 3a). The second anticipated level of RNA silencing was that triggered by autonomous replication (and, hence, dsRNA production) of said primary transcripts independently of transgene expression. RNA silencing activated at this level would be conceptually similar to IR‐PTGS triggered by p35S‐GFFG. A third potential layer of RNA silencing would entail RDR‐dependent dsRNA amplification from replicated single‐stranded viral transcripts or breakdown products thereof, given the established sensitivity of PVX infection to at least RDR6 (Schwach et al., 2005). p35S::P19 co‐transient expression was predicted to suppress all three RNA silencing layers potentially restricting optimal GFP5 expression and activity from p35S::PVX‐Δ25ΔCP:GFP5. We thus tested if co‐expression of p35S::P19 with p35S::PVX‐Δ25ΔCP:GFP5 would yield higher GFP5 levels than co‐expression of p35S::P19 with the non‐replicative p35S::GFP5 transgene or co‐expression of the latter with p35S::FHA:GUS‐Intron (Figure 6a).
Figure 6.
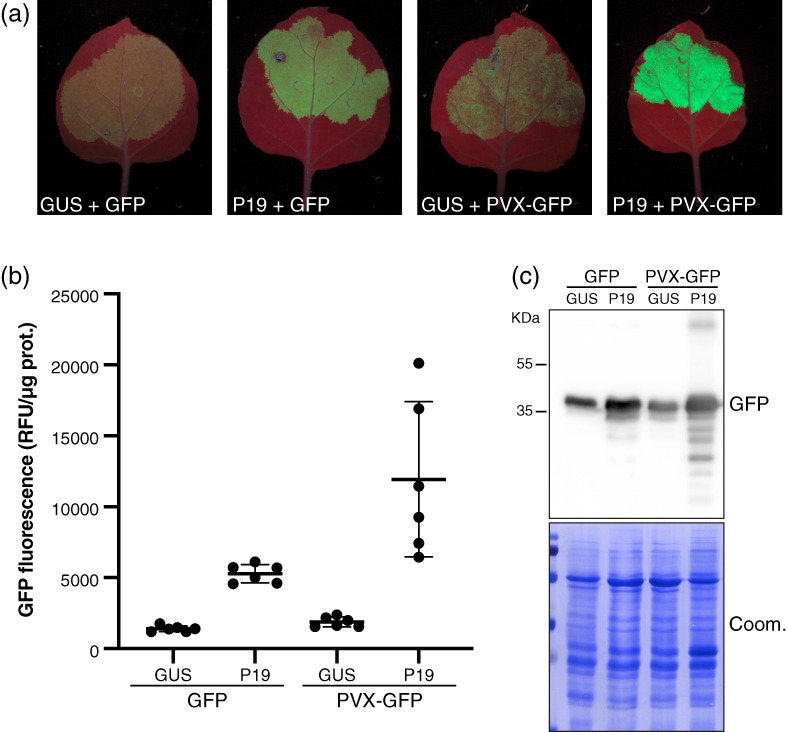
A PVX‐based minimal replicon boosts further GFP5 production in P19 co‐infiltrated patches. (a) Images of Nicotiana benthamiana leaves, representative of six independent experiments, at 4 dpi with the constructs p35S::FHA:GUS‐Intron (GUS), p35S::GFP5 (GFP), p35S::P19 (P19), and p35S::PVX:Δ25KΔCP:GFP5 (PVX‐GFP), as indicated, under UV illumination. To avoid saturation effects, the OD value of each individual Agrobacterium inoculate was 0.1, compared to 0.3 in Figures 2a, 3a, and 5a.
(b) Relative GFP fluorescence units (RFU) measured from samples depicted in (a). Black bars: mean. Error bars: standard deviations. n = 6.
(c) GFP protein accumulation in the samples depicted in (a) analyzed by Western blot. Coomassie staining of total proteins (Coom.) is used as a control for equal loading. The experiment was independently repeated two times with similar results.
To better quantify these anticipated differences, all Agrobacterium strains used in the following comparative experiments were diluted to an OD of 0.1, compared to 0.3 used in Figures 2, 3, and 5. The same UV light intensity and illumination time were used for imaging, however, as were the settings for fluorescence quantification conducted in patches individually collected over multiple independent experiments. In quantitative assessment of GFP5 fluorescence at 4 dpi, the effects of co‐expressing p35S::FHA:GUS‐Intron with p35S::GFP5 or p35S::PVX‐Δ25ΔCP:GFP5 were similar (Figure 6b). Thus, in the absence of silencing suppression, little to no significant gain was afforded by the replicative nature of the latter owing, presumably, to the full action of the aforementioned, intertwined RNA silencing layers. Co‐expressing p35S::GFP5 with p35S::P19 yielded higher GFP5 fluorescence under UV illumination, quantified as an average 3.8‐fold gain, which is lower than that reported in Figures 2, 3, and 5 as expected from the lower bacterial OD used (Figure 6b). However, and despite this suboptimal OD, the gain in GFP5 fluorescence was further increased to 8.5‐fold, on average, by co‐expressing p35S::PVX‐Δ25ΔCP:GFP5 with p35S::P19, resulting in the most intensely bright signal observed among all our GFP5‐based experiments (Figure 6a–c). Thus, the combined transient expression of P19 with a minimal PVX replicon yields expression levels substantially higher than those already achieved by expressing a non‐replicative transgene, either alone or with P19. The results also show that engineering the viral vector itself to co‐express P19 and the gene of interest (Mardanova et al., 2017) is not necessary for this approach to be successful.
Gains in protein accumulation granted by the P19 system may vary extensively, but are upheld upon expression of multiple, co‐delivered constructs
On the one hand, S‐PTGS is a likely general impediment to Agrobacterium‐mediated transient expression, while, on the other, co‐expressed P19 likely broadly prevents this caveat by protecting the mRNAs of interest from degradation. The levels of protein produced from such P19‐protected mRNAs may vary extensively, however. This likely reflects protein‐intrinsic translation efficacies, half‐lives, and, when applicable, varying turnover rates influenced by post‐translational degradation pathways. Distinct biochemical properties of the tested proteins, which ultimately influence the extraction and analytical methods used for their detection, may also artificially contribute to such variations independently of the abovementioned factors. To address this issue comprehensively, we engineered p35S‐driven mCherry‐translational fusions to the ORFs of three cellular membranous compartments' markers with dissimilar molecular weights. Hence, the same protocol of insoluble protein extraction and the same generic anti‐RFP antibody could be used in henceforth directly comparable Western blot analyses. Using a bacterial OD of 0.15 for each construct (for a total OD of 0.6, all combined together with p35S::P19 or p35S::FHA:GUS‐Intron), the cis‐Golgi marker ERD2 (25 kDa; Jaillais et al., 2008) was detected at 4 dpi without p35S::P19 co‐expression, while both the endosomal marker SNX1 (47 kDa; Jaillais et al., 2008) and the trans‐Golgi network marker VHAα1 (93 kDa; Jaillais et al., 2008) were below detection levels of Western blot analysis (Figures 1 and 7a). Under p35S::P19 co‐expression conditions, the initial ERD2 levels were substantially increased, whereas SNX1 and VHAα1 became detectable (Figure 7a). These results therefore indicate that the P19 system yields variable gains in otherwise identically extracted and detected proteins exhibiting similar biochemical properties, most likely due to protein‐intrinsic properties upon which P19 is expected to exert little or no effect. This notion is further illustrated by the discrepancy between the extremely high gains in GFP5 mRNA accumulation granted by P19 co‐expression (as presented in Figure 2b) and those observed at the GFP5 activity (Figure 2d) and protein steady‐state levels (Figures 3b and 5b). Accumulation of low‐molecular‐weight GFP5‐derived fragments revealed by Western blot analysis strongly suggest proteolysis as one possible source of this discrepancy (Figures 3b and 5b). Bands migrating at a higher than expected molecular weight, also detected with the anti‐GFP5 antibody, further suggest the involvement of ubiquitination and 26S proteasome activation (Figures 3b and 5b). Nonetheless, the results with SNX1 and VHAα1 also indicate that P19 can empower detection of transiently expressed proteins that are otherwise too low in abundance to enable their study, let alone purification. Using the rapid Agrobacterium leaf infiltration procedure granted, in these two cases, infinite gains in protein production. The results, finally, confirm that the P19 co‐expression effect is indiscriminative of transgene products, presumably because S‐PTGS is a universal impediment intrinsically inherent to the transient expression procedure itself.
Figure 7.
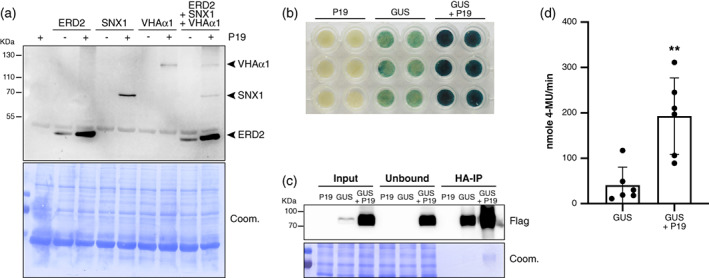
Simultaneous protein co‐expression and enzyme purification using the P19 system. (a) ERD2, SNX1, and VHAα1 protein accumulation in Nicotiana benthamiana leaves at 4 dpi with p35S::P19 (P19), p35S::ERD2:mCherry (ERD2), p35S::SNX1:mCherry (SNX1), or p35S::VHAα1:mCherry (VHAα1), as indicated, analyzed by Western blot using a generic RFP‐specific antibody. Coomassie staining of total proteins (Coom.) was used as a control for equal loading. The experiment was independently repeated two times with similar results.
(b) GUS staining of N. benthamiana leaf discs at 4 dpi with p35S::P19 (P19) and p35S::FHA:GUS‐Intron (GUS), as indicated. The experiment was independently repeated two times with similar results.
(c) FHA:GUS protein accumulation in the input, unbound fraction, and immunoprecipitated (IP) fraction of the samples depicted in (b). Coom.: as in (a). The experiment was independently repeated six times with similar results.
(d) GUS activity of the FHA:GUS IP fraction from samples depicted in (b) and (c). Black bars: mean. Error bars: standard deviations. n = 6. **P = 0.0087 (Mann–Whitney test).
We then tested if the above gains in protein production observed in separate agro‐infiltration experiments could be recapitulated if the three membrane markers' Agrobacterium strains were co‐infiltrated simultaneously in conjunction with the p35S::P19 strain. As shown in Figure 7a, this was indeed the case, with the protein gains detected by Western blot analysis being on par with those yielded by separate co‐expression, save slightly lower gains for SNX1. These results are most easily explained if a majority of cells in the co‐infiltrated patch had effectively undergone simultaneous transient transformation by the four T‐DNAs involved. This is consistent with the uniformity of GFP5 expression in infiltrated patches (Figures 2a, 3a, and 5a), the results of Figure 6a, and the previously reported spatial coincidence of reporter genes' expression upon co‐delivery from separate Agrobacterium strains (Hamilton et al., 2002; Himber et al., 2003; Voinnet et al., 2000). The ability provided by the P19 system to simultaneously enhance production of multiple proteins within the same cells allows interesting applications in which, for instance, a desirable compound is produced by multiple key enzymes as part of a given biosynthetic pathway.
The P19 system is suited for high‐level production of recombinant proteins for downstream in vitro applications
Increased GFP5 levels are accompanied by increased fluorescence in p35S::P19 co‐expressing cells, demonstrating that at least part of the over‐accumulated protein is biologically active in vivo (Figures 3a,b and 5a,b). Thus, another biotechnological application of the P19 system is to optimize production of recombinant proteins subsequently used for downstream applications upon their purification. For this purpose, the over‐accumulated protein should display high activity levels not only in vivo but also in vitro after purification. To quantitatively illustrate this additional application of the P19 system, we employed the p35S::FHA:GUS‐Intron Agrobacterium strain used so far as a mere negative control in co‐infiltration experiments. The 2×FLAG‐2×HA (FHA)‐tag in the construct enables immuno‐purification of the soluble bacterial β‐glucuronidase (GUS) enzyme, while the presence of an intron prevents its spurious production from Agrobacterium cells (Himber et al., 2003). This ensures that FHA:GUS activity measurements exclusively report that of the protein expressed in planta. Direct histochemical blue staining of leaf discs isolated from individually collected patches showed, visually, that substantially more FHA:GUS products accumulate upon co‐expression of p35S::FHA:GUS‐Intron and p35S::P19 (Figure 7b). This enhancement correlated with strongly increased FHA:GUS levels accumulating in the co‐infiltrated patches compared with those expressing p35S::FHA:GUS‐Intron alone (Figure 7c, Input lanes). Accordingly, more FHA:GUS was HA‐immuno‐purified from leaf patches co‐infiltrated with p35S::P19 (Figure 7c, HA IP lanes). To test if the enhanced FHA:GUS total enzyme activity observed in vivo persisted in vitro after purification, immunoprecipitates obtained under either condition were subjected to quantitative in vitro MUG assays involving six independent replicates each. These assays measure the hydrolysis rate of 4‐methylumbelliferyl β‐d‐glucuronide (4‐MUG) into 4‐methylumbelliferone (4‐MU), a fluorochrome emitting at 460 nm with negligible substrate background fluorescence (Blazquez, 2007). A 5‐min time‐course analysis revealed that the steady increase in GUS total enzyme activity yielded by FHA:GUS immuno‐purified from patches co‐infiltrated with p35S::P19 was substantially more pronounced than that yielded by FHA:GUS immuno‐purified from singly infiltrated patches, presumably reflecting the increased yield in FHA:GUS production under P19 co‐expression. Linear regression and use of a standard reference curve showed that an average yield of 193 nmol 4‐MU/min was achieved with the former compared to 40 nmol 4‐MU/min with the latter (Figure 7d; Figure S4). Although the assay displayed some variability, the gain in 4‐MUG production under P19 co‐expression conditions was up to 10‐ or even 20‐fold in some replicates (Figure 7d). These results therefore reinforce the proof of principle that the P19 system can be used to produce higher amounts of active recombinant protein for downstream in vitro applications.
Advantages and limits of the P19 transient expression system for cell biology studies
Possibly the most universal academic use of the Agrobacterium‐mediated transient expression system in N. benthamiana leaves is as a rapid method for the preliminary assessment of the functionality of transgene constructs. These notably include transcriptional and translational reporter gene fusions destined to in planta expression and subcellular localization studies in stable transformants. As shown in Figure 7a with the mCherry fusions to SNX1 and VHAα1, use of P19 as a suppressor of S‐PTGS is imperative, and thereby transformative, in enabling the mere detection of certain proteins of interest. Yet it simultaneously grants visualization of their cellular distribution within infiltrated tissues, including in the jigsaw puzzle‐shaped cells of the leaf epidermis. We present below three case studies conducted with well‐characterized endomembrane markers under the confocal microscope. These were chosen to document distinct outcomes of using P19 in preliminary subcellular localization studies conducted in agro‐infiltrated patches, two of which illustrate some limits of the method for this specific application. In a first experiment, the p35S::GFP5 Agrobacterium strain, which produces the ER‐targeted GFP5, was co‐infiltrated with a second strain engineered to express, under the control of the p35S promoter, a C‐terminal mCherry translational fusion to the plasma membrane‐associated brassinosteroid receptor BRI1 (Russinova et al., 2004) (p35S::BRI1:mCherry; Figure 1). Without co‐infiltrating the p35S::P19 strain, the green signal yielded by GFP5 was detected in epidermis cells, forming a reticulate network typical of the ER. BRI1::mCherry levels, by contrast, were below the detection limit (Figure 8a, left panels). Co‐infiltrating the two above strains with the p35S::P19 strain yielded enhanced and readily detectable signals for both p35S::GFP5 and p35S::BRI1:mCherry constructs, with the latter detected, as expected, on the plasma membrane (Figure 8a, right panels). Noteworthily, the enhanced reticulate green and plasma membrane mCherry signals were preponderantly simultaneously detected in epidermal cells. Since P19 is cell autonomous (Brosnan et al., 2019; Devers et al., 2020), as are the membrane‐bound GFP5 and BRI1:mCherry alleles, these observations demonstrate a high incidence of co‐delivery, within single cells, of the three T‐DNAs producing P19, GFP5, and BRI1:mCherry, thus directly supporting the notion already evoked in relation to Figure 7a.
Figure 8.
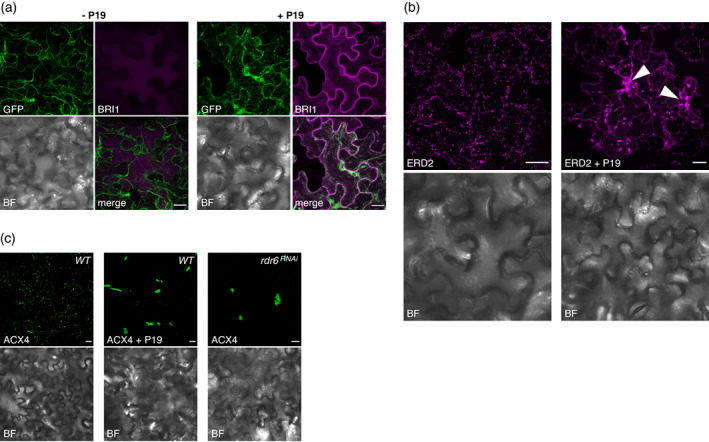
Advantages and limitations in using the transient P19 co‐expression system for subcellular localization studies. (a) Confocal images of Nicotiana benthamiana leaves at 4 dpi with p35S::GFP5 (GFP), p35S::BRI1:mCherry (BRI1), and p35S::P19 (P19), as indicated.
(b) Confocal images of N. benthamiana leaves at 4 dpi with p35S::ERD2:mCherry (ERD2) and p35S::P19 (P19), as indicated.
(c) Confocal images of N. benthamiana leaves at 4 dpi with p35S::eGFP:ACX4 (ACX4) and p35S::P19 (P19), in the WT or the rdr6 RNAi background, as indicated.
BF in (a–c): Bright field. Scale bars in (a–c): 20 μm. Images are representative of at least three pictures acquired in three (a) or two (b, c) independent experiments.
In a separate series of analyses, the mCherry fusion to the cis‐Golgi marker ERD2 used in the experiments of Figure 7a was imaged under a confocal microscope. Agreeing with ERD2:mCherry being detectable by Western blot analysis without the need for P19 co‐expression (Figure 7a), a signal highlighting a multitude of Golgi‐derived vesicles was detected when the p35S::ERD2:mCherry Agrobacterium strain was delivered alone into N. benthamiana leaves (Figure 8b, left panels). A similar, albeit enhanced signal was detected if the p35S::ERD2:mCherry strain was co‐delivered with the 35S::P19 strain (Figure 8b, right panels). Unlike in the singly infiltrated tissues, however, several vesicles' aggregates of various sizes also accumulated under the co‐infiltration condition (Figure 8b; arrows). These were likely caused by too high levels of p35S::ERD2:mCherry expression as a possible source of cellular stress and/or toxicity; accordingly, the co‐infiltrated leaves showed signs of necrosis observed with neither strain alone (Figure S5). In this case, therefore, use of P19 was not only unnecessary but it yielded, on top of a cognate signal, an undesirable and artifactual signal potentially confounding interpretation of ERD2 subcellular localization.
A third analysis involved an N‐terminal eGFP fusion to the peroxisomal protein ACX4 (Hayashi et al., 1999) expressed under the control of the p 35S promoter (p35S::eGFP:ACX4; Figure 1). As seen for p35S::ERD2:mCherry, expression of p35S::eGFP:ACX4 alone was sufficient to yield a cellular signal delineating punctate organelles as expected from the labeling of cognate peroxisomes (Figure 8c, left panels). This pattern was dramatically modified, however, if p35S::eGFP:ACX4 was co‐expressed with p35S::P19, a condition under which the green fluorescence signal delineated large structures presumably reflecting artifactual peroxisomal aggregates (Figure 8c, middle panels). Since peroxisomes have been suggested as subcellular sites of siRNA accumulation (Incarbone et al., 2017), we considered that the high affinity of P19 for these molecules might have caused these aggregates, independently of P19‐mediated PTGS suppression underpinning the visibly enhanced fluorescence signal in co‐infiltrated, compared to singly infiltrated, patches. This was not the case, however, because similar aggregates were observed in singly infiltrated N. benthamiana leaves with the rdr6 RNAi genetic background, suggesting that such aberrant structures result from mere silencing suppression and, hence, eGFP::ACX4 over‐accumulation (Figure 8c, right panels). In this particular example, P19 co‐expression was thus not only unnecessary, but it incurred an entirely artificial subcellular localization for ACX4. The latter two examples indicate that P19 co‐expression should not be used as a default setting during transient expression experiments in preliminary cell biology studies, and that singly infiltrated patches – assuming they yield sufficient signal – ought to be systematically inspected in parallel during such applications of the method.
CONCLUDING REMARKS
This contemporary re‐evaluation of the P19 system, a widely used method developed nearly 20 years ago, now sheds light on the thus far mysterious RNA silencing processes that intrinsically limit Agrobacterium‐mediated transient expression in tobacco leaves. Owing to the sheer amount of T‐DNA molecules likely involved in the assay, initial transcription of the transgenes of interest likely generates substantial amounts of (ab)mRNAs that, by overwhelming RQC, stimulate their RDR‐dependent conversion into dsRNA as a source of both 21‐nt (RDR6) and 24‐nt (RDR2) siRNAs. In parallel, T‐DNA rearrangements also likely trigger RDR‐independent IR‐PTGS further fueling production of both siRNA species. A priori, neither process needs to be activated in every T‐DNA‐transformed cell, since siRNAs are mobile within and outside infiltrated patches (Himber et al., 2003) in a manner that would create RDR substrates upon target mRNA cleavage in virtually all siRNA‐recipient cells (Figure 4). Perhaps counter‐intuitively, therefore, use of Agrobacterium suspensions at high ODs –while possibly enhancing initial mRNA transcription – will come at the cost of increased siRNA production as exemplified in previous studies employing saturated bacterial suspensions (Hamilton et al., 2002; Himber et al., 2003; Voinnet et al., 2000). We show here that ODs of approximately 0.1–0.3 will generally constitute a good compromise but, ultimately, optimal protein production (assuming this is the desired outcome) will require empirical adjustments made on a case‐by‐case basis. A transgene encoding the near‐minimal PVX replicon failed, on its own, to boost GFP5 production, presumably because the primary S‐PTGS targeting the non‐replicating p35S::GFP5 transgene was seconded, in this setting, by the strong and replication‐intrinsic IR‐PTGS‐like antiviral response naturally triggered by PVX. By presumably sequestering siRNAs emanating from both sources, P19 enabled protein gains that far exceeded those already obtained with co‐expression of p35S::GFP5 and p35S::P19. Given the universality of the plant RNA silencing‐based antiviral defense system (Lecellier & Voinnet, 2004; Pumplin & Voinnet, 2013), this likely explains why other bio‐contained viral vectors have been successfully used with P19 in the leaf agro‐infiltration procedure (Norkunas et al., 2018), including, for example, in antigen and antibody production (Liu et al., 2005). At least two factors could explain the potency of P19 in circumventing the multi‐layered RNA silencing triggered in agro‐infiltrated tissues. Firstly, being itself expressed from a T‐DNA‐encoded transgene, P19 likely stabilizes its own mRNA in addition to those of co‐delivered constructs. Secondly, the 1–2‐nt trimming of its si/miRNA cargoes likely promotes their release from P19 homodimers, as expected from their crystal structure (Kontra et al., 2016; Vargason et al., 2003). This would allow effective recycling of the VSR, consistent with a multiple‐turnover mode of action previously inferred from in vitro experiments and modeling (Rawlings et al., 2011).
Although P19 will broadly stabilize mRNAs of interest, it will remain ineffective in suboptimal downstream steps of the gene expression pathway including poor protein translatability or high turnover rates. This likely explains why great discrepancies might be observed between the gains in mRNA versus protein levels, as exemplified by GFP5 yet to a much lesser extent by GUS. This suggests that the P19 system might be further optimized by engineering mRNAs of interest via codon optimization (Sugio et al., 2010) and/or translation enhancer signals of viral (Carrington & Freed, 1990; Gallie & Walbot, 1992; Jobling & Gehrke, 1987), plant (Agarwal et al., 2014), or artificial (Kanoria & Burma, 2012) origins known to increase transgene expression not only in stable transformants but also during transient expression (Sainsbury & Lomonossoff, 2008). Concomitant inhibition of post‐translational degradation processes such as 26S proteasome‐ or autophagy‐mediated proteolysis using for example drugs (Derrien et al., 2012) or the transient CRISPR/Cas approach (Nekrasov et al., 2013) may further increase protein yield. Efforts to ameliorate the liquid infiltration medium with surfactants and antioxidants or to provide a more conducive ground to gene expression under the cellular stress incurred by the procedure by, for example, modifying the cell cycle or heat shocking samples (Norkunas et al., 2018) also hold great potential. Nonetheless, we also highlighted clear limits of the P19 system in preliminary cell biological studies by showing how artifactual agglomerates – due, presumably, to protein overaccumulation – may confound interpretations depending on the factor under study. We thus recommend that such observations be ultimately validated in stable transgenic lines.
METHODS
Plant material and growth conditions
Transgenic N. benthamiana rdr6 RNAi was previously described in Schwach et al. (2005). Plants were grown on soil at 21°C in 16 h light/8 h dark conditions (light intensity: 120 μE m−2 sec−1). All agro‐infiltrations were performed on the fourth and fifth leaves of approximately 4 week‐old plants.
Plasmids and cloning procedures
The p35S::P19, p35S::GFP5, and p35S::PVX:GFP‐∆25k‐∆CP expression vectors were described previously (Angell & Page, 2002; Hamilton et al., 2002; Voinnet et al., 2000). Briefly, removal of a cryptic intron by modifying codon usage in the GFP ORF (Aequorea victoria) led to mGFP4, also known as GFP4; mGFP4 was then further modified with an N‐terminal Chitin signal peptide and the ER‐targeting HDEL amino acid sequence in the C‐terminal region, leading to mGFP4‐ER, also known as GFP5 (Angell & Page, 2002; Haseloff et al., 1997). The complete sequence of the p35S::GFP5 expression cassette within the pBI121 binary vector backbone (Haseloff et al., 1997) is shown in Figure S6. Gateway cloning technology (Thermo Fisher Scientific, www.thermofisher.com) was used to assemble the other expression constructs presented in Figure 1. DNA cloning was conducted using Phusion High‐Fidelity DNA Polymerase (Thermo Scientific, www.thermofisher.com) and primers given in Table S1. The FHA:P19‐expressing construct was generated by introducing attB1, attB2, and FHA‐tag sequences into the P19 coding sequence by PCR. The attB‐flanked DNA fragment was BP‐recombined into pDONR221 and the resulting entry vector was LR‐recombined in the destination vector pB7m24GW (Karimi et al., 2007) together with an attL4/attR1 entry vector harboring the p35S promoter or the pUB10 promoter. The FHA:GUS‐Intron expression vector was generated by LR‐recombining entry vectors containing an attL4/attR1‐flanked 35S promoter sequence, an attL1/attL2‐flanked FHA‐tag sequence, and an attR2/attL3‐flanked ST‐LS1‐intron‐containing GUS sequence into the destination vector pB7m34GW (Karimi et al., 2007). The GFFG expression vector was generated by recombining an attL1/attL2 vector containing the first 400 nucleotides of the coding DNA sequence of mGFP6 in the destination vector pBm42GWIWG8 (Karimi et al., 2007). BRI1, VHAα1, SNX1, and ERD2 expression constructs were produced by LR‐recombining the relevant attL1/attL2 entry vectors together with an attL4/attR1 entry vector harboring a 35S promoter and an attR2/attL3 entry vector containing the mCherry coding sequence into the destination vector pK7m34GW (Karimi et al., 2007).
Transient expression in N. benthamiana leaves
Agro‐infiltrations were conducted by infiltrating leaves with equal volumes of agrobacterial suspensions in 10 mm MgCl2, 10 mm MES pH 5.6, and 200 μm acetosyringone. Final OD600 values were adjusted to 0.6, unless specifically indicated. mGFP5 fluorescence in agro‐infiltrated leaves was imaged either with a high‐intensity handheld UV lamp or with a Typhoon FLA 9000 system (GE Healthcare, www.gehealthcare.com) equipped with a 473 nm excitation laser and a 510–550 nm bandpass filter. Pictures obtained from the Typhoon system were quantified using ImageQuant TL software (GE Healthcare). Confocal imaging was carried out at 4 dpi.
IP experiments
Nicotiana benthamiana leaves (4 dpi) were ground in liquid nitrogen and 0.2 g of tissue powder was resuspended in 1 ml IP buffer (50 mm Tris–HCl pH 7.5, 150 mm NaCl, 10% glycerol, and 0.1% NP40) containing 2 μm MG‐132 and one tablet of cOmplete® protease inhibitor cocktail (Merck Roche, www.sigmaaldrich.com) per 10 ml. Lysates were incubated for 30 min on a rotating wheel and cleared from cell debris twice by centrifugation at 16 000 g for 10 min. For FHA:P19 IP experiments, cleared lysates were incubated for 30 min with 30 μl of Anti‐HA Magnetic Beads (Pierce, www.thermofisher.com) equilibrated in IP buffer. Bead conjugates were washed three times with IP buffer for 10 min. Immunoprecipitated proteins were retrieved from the beads from 10% of the last wash and resuspended in 1× Western blot loading buffer (10% glycerol, 4% SDS, 62.5 mm Tris–HCl pH 6.8, and 5% 2‐mercaptoethanol). The remaining beads were resuspended in 500 μl of TRI Reagent (Merck, www.sigmaaldrich.com) for RNA extraction. Immunoprecipitated RNA was precipitated from the aqueous phase with the addition of 20 μg of glycogen. RNA from input samples was extracted by adding one volume of Roti‐Phenol/Chloroform/Isoamyl Alcohol (Carl Roth, www.carlroth.com), precipitated from the aqueous phase with one volume of isopropanol in the presence of 0.3 m sodium acetate pH 5.2, and washed with 80% ethanol. For FHA‐GUS IP experiments, cleared lysates were incubated for 30 min on a rotating wheel with 50 μl of Anti‐HA Magnetic Beads (Pierce) equilibrated in IP buffer. Bead conjugates were washed four times with IP buffer for 10 min. Immunoprecipitated proteins were retrieved from 50% of the beads resuspended in 1× Western blot loading buffer (10% glycerol, 4% SDS, 62.5 mm Tris–HCl pH 6.8, and 5% 2‐mercaptoethanol). The remaining beads were resuspended in IP buffer containing 1 mm 4‐MUG (Merck Sigma, www.sigmaaldrich.com) and incubated at 37°C under gentle agitation (600 rpm) for 0, 1, 2, 3, and 5 min. Supernatants were collected and the reactions were stopped by adding 0.2 m Na2CO3 (Merck Sigma). Technical triplicates of 200 μl were transferred to black flat‐bottom 96‐well plates (Greiner Bio‐One, www.gbo.com) and 4‐MU production was measured with a plate reader (Polarstar Omega, www.bmglabtech.com; excitation: 355 nm, emission: 460–510 nm). A 0.1–10 μm 4‐MU standard curve was used to convert relative fluorescence units (RFU) to nmol 4‐MU. GUS activities (nmol 4‐MU/min) were obtained based on the average of the triplicates after correction using a blank sample and determination of the time‐course trendlines.
RNA extraction and Northern blot analysis
RNA was extracted from frozen tissues ground in liquid nitrogen using TRI Reagent (Merck) according to the manufacturer's instructions. Equal amounts of RNA (2 μg) or immunoprecipitated RNA fractions were resuspended in 50% formamide and 1× RNA loading buffer (12.5% glycerol, 12.5 mm Tris pH 7.7, 1.25 mM EDTA, and 0.008% bromophenol blue), resolved by electrophoresis on a denaturating polyacrylamide gel (0.5× TBE, 17.5% polyacrylamide/bisacrylamide 19:1, and 8 m urea), transferred to a Hybond‐NX Nylon membrane (Merck Sigma) in 0.5× TBE, and cross‐linked using 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide according to Pall and Hamilton (2008) for 2 h at 60°C. RNA blots were pre‐hybridized in PerfectHyb™ Plus Hybridization Buffer (Sigma‐Aldrich, www.sigmaaldrich.com) at 42°C for 30 min before adding probes of interest. Random‐radiolabeled probe GFP (Table S1) was generated by incubating PCR fragments, obtained using the p35S::GFFG plasmid as PCR template, with the Prime‐a‐Gene® labeling system (Promega, www.promega.com) in the presence of [α‐32P]‐dCTP (Hartmann Analytic, www.hartmann‐analytic.de). Oligonucleotide probes (Table S1) were end‐labeled by incubation with T4 PNK (Thermo Fisher Scientific) in the presence of [γ‐32P]‐dATP (Hartmann Analytic). Membranes were probed overnight at 42°C and then washed three times with 2× SSC, 2% SDS at 50°C for 15 min and exposed to a storage phosphor screen, followed by imaging on a Typhoon FLA9000 (GE Healthcare). Band quantification was conducted with Image Lab software (Bio‐Rad, www.bio‐rad.com) using auto‐contrasted images. For sequential hybridizations of several probes, membranes were stripped with boiling in 0.1% SDS three times for 15 min before re‐probing.
RT‐qPCR
Five hundred nanograms of RNA extracted with TRI Reagent was treated with 1 unit of DNAse I (Thermo Fisher Scientific) for 40 min at 37°C and reverse‐transcribed with the Maxima First Strand cDNA Synthesis kit (Thermo Fisher Scientific) according to the manufacturer's instructions. cDNA was diluted 1:3 and 1 μl was used in 10 μl PCR reactions with KAPA SYBR FAST qPCR 2× master mix (Merck Sigma) and gene‐specific primers (0.2 μm each) listed in Table S1. qPCR reactions were performed in triplicate in 384‐well plates using a LightCycler 480 System (Roche, www.diagnostics.roche.com) following the PCR program recommended with the KAPA SYBR FAST qPCR mix. In addition, a melting curve was drawn to verify the specificity of each PCR amplification. Cp values (cycle values of the maximum second derivative of the amplification curves) were calculated for each PCR reaction with LightCycler 480 software. Relative expression values were obtained by calculating 2−∆Cp, where ∆Cp represents the difference between the Cp value of the analyzed RNA and the average of the Cp values of ACTIN (ACT) and PHYTOENE DESATURASE (PDS), which were used as control mRNAs. Primers for ACT and PDS were designed based on the sequences from GenBank accession numbers JQ256516.1 and EU165355.1, respectively.
Western blot
Total proteins were extracted either following the protocol of Hurkman and Tanaka (1986) (Figures 3b, 5b, and 7a) or by adding 4 m urea/100 mm DTT to ground tissues prior to boiling for 5 min (Figure 6c). Proteins were separated by SDS‐PAGE before subsequent transfer onto Western blot membranes. For chemiluminescent detection, proteins were transferred onto Immobilon‐P PVDF membranes (Merck Millipore). Membranes were blocked for 30 min in 1× TBS supplemented with 0.5% non‐fat milk. For FLAG Western blot analysis, membranes were incubated for 1 h at room temperature with monoclonal anti‐FLAG M2‐Peroxidase (HRP) antibody (Sigma, www.sigmaaldrich.com; Table S2). For RFP and GFP Western blot analyses, membranes were incubated with primary antibodies overnight at 4°C (Table S2) and subsequently washed three times with 1× TBS‐T. Membranes were further incubated for 1 h at room temperature with 1:5000 dilutions of HRP‐conjugated goat anti‐rat secondary antibody (Table S2). Protein detection was carried out with Lumi‐LightPLUS Western Blotting ECL Substrate (Roche, www.sigmaaldrich.com) after three washes with 1× TBS‐T and protein bands were imaged with a ChemiDoc Touch Imaging System (Bio‐Rad). Membranes were stained with Coomassie blue to reveal total proteins. For fluorescence detection, proteins were transferred onto Immobilon‐L PVDF membranes (Merck Millipore, www.sigmaaldrich.com). Membranes were blocked for 30 min in 1× TBS supplemented with 0.5% non‐fat milk. Membranes were incubated with a mix of anti‐actin and anti‐GFP primary antibodies overnight at 4°C (Table S2) and subsequently washed three times with 1× TBS‐T. Membranes were further incubated for 1 h at room temperature with dilutions of fluorescent anti‐mouse and anti‐rat secondary antibodies (Table S2) in the dark. Membranes were washed three times with 1× TBS‐T and two times with 1× TBS. Protein detection was carried out on dry membranes by using an Odyssey CLx imaging system (Li‐Cor, www.licor.com) with automatic scanning settings. Protein band signal intensities were quantified using Image Studio Lite software (Li‐Cor).
Confocal microscopy
Confocal pictures were acquired using a Zeiss LSM 780 microscope (www.zeiss.com/microscopy) controlled by Zeiss Zen software. GFP and mCherry fusion proteins were imaged using a 488 nm excitation laser with detection at 495–550 nm and a 561 nm excitation laser with detection at 575–630 nm, respectively, in sequential acquisition mode. Images were further processed using ImageJ (www.imagej.net).
Measurements of RFU
Nicotiana benthamiana leaves were ground in liquid nitrogen, and 0.5 ml of tissue powder was resuspended in 1 ml of IP buffer (50 mm Tris–HCl pH 7.5, 150 mm NaCl, 10% glycerol, and 0.1% NP40) containing 2 μm MG‐132 and one tablet of cOmplete® protease inhibitor cocktail (Merck Roche) per 10 ml. Lysates were incubated for 30 min on a rotating wheel and then cleared from cell debris by centrifugation at 16 000 g for 30 min. Protein concentration was determined using the DC Protein Assay (Bio‐Rad), and lysates were adjusted to 250 μg ml−1. Technical triplicates of 100 μl were transferred to black flat‐bottom 96‐well plates (Greiner) and fluorescence was measured with a plate reader (Polarstar Omega; excitation: 485 nm, emission: 520 nm). RFU values were obtained as the average of the triplicates after correction using a blank sample and plotted as RFU per μg of protein.
AUTHOR CONTRIBUTIONS
FJ and FB performed the experiments. FJ, FB, and OV planned and designed the research, analyzed the data, and wrote the manuscript.
CONFLICT OF INTEREST
A technology employing the P19 protein as an enhancer of transient expression is licensed by Plant Bioscience Limited, Norwich, UK. The corresponding patents list Olivier Voinnet as a co‐inventor.
Supporting information
Figure S1. (a) GFP fluorescence signal at 473 nm of N. benthamiana leaves, representative of six independent experiments, 4 days post‐infiltration with the constructs p35S::FHA:GUS‐Intron (GUS) and p35S::GFP5 (GFP) on the left‐hand side, or p35S::P19 (P19) and p35S::GFP5 (GFP) on the right‐hand side. (b) Quantification of the GFP fluorescence signal obtained in (a). Black bars: mean. Error bars: standard deviations. n = 6.
Figure S2. (a) Images of N. benthamiana leaves, representative of four independent experiments, taken under UV illumination at 7 days post‐infiltration (dpi) with the constructs p35S::GFP5 or pUB10::GFP5, together with p35S::FHA:GUS‐Intron (GUS) or p35S::P19 (P19). (b) Relative GFP fluorescence units (RFU) measured from the samples depicted in (a). Black bars: mean. Error bars: standard deviations. n = 4. (c) GF siRNA accumulation in biological duplicates of samples depicted in (a) analyzed by Northern blot using a probe corresponding to the 5′ part of the GFP ORF (‘GF’). The probed miR159 and U6 small RNA were used as an endogenous P19 cargo and RNA loading control, respectively. EtBr.: Ethidium bromide staining provides an additional RNA loading control. Black arrows indicate 3′‐end‐trimmed sRNA species. The experiment was independently repeated two times with similar results.
Figure S3. (a) Independent biological replicate of experiments presented in Figure 5d. FHA:P19 protein levels in the input, unbound fraction, and IP fraction of N. benthamiana leaves at 4 dpi with infiltration medium (−), p35S::FHA:GUS‐Intron (GUS), p35S::FHA:P19 (F:P19), and p35S::GFFG (GFFG), as indicated. Coomassie staining of total proteins (Coom.) provides a control for equal loading. (b) Independent biological replicate of experiments presented in Figure 5e. GF siRNA levels in samples depicted in (a) analyzed by Northern blot. miR159 and U6 small RNA were used as an endogenous P19 cargo and RNA loading control, respectively. EtBr.: Ethidium bromide staining provides an additional RNA loading control. Black arrows indicate 3′‐end‐trimmed sRNA species.
Figure S4. GUS activity in relative MU fluorescence units (RFU) from the FHA:GUS IP fraction of samples depicted in Figure 7c at 0, 1, 2, 3, and 5 min. Plotted: mean. Error bars: standard deviations. n = 6.
Figure S5. Images of Nicotiana benthamiana leaves 4 days post‐infiltration with the constructs p35S::P19 (P19) and p35S::ERD2:mCherry (ERD2), as indicated, under normal light.
Figure S6. p35S::GFP5 expression cassette within the pBI121 binary vector backbone described in Angell and Page (2002) and used in this study.
Table S1. Sequences of oligonucleotide primers used in this study.
Table S2. List of antibodies used in this study.
ACKNOWLEDGMENTS
We thank the members of the Voinnet laboratory for critical reading of the manuscript. We thank André Imboden for assistance in plant growth. This research was funded by core funding to Olivier Voinnet for his professorship by ETH‐Zürich. We acknowledge support of the Scientific Center for Optical and Electron Microscopy (ScopeM) of ETH‐Zürich.
DATA AVAILABILITY STATEMENT
This study includes no data deposited in external repositories. All relevant data can be found within the manuscript and its supporting materials.
REFERENCES
- Agarwal, P. , Garg, V. , Gautam, T. , Pillai, B. , Kanoria, S. & Burma, P.K. (2014) A study on the influence of different promoter and 5′UTR (URM) cassettes from Arabidopsis thaliana on the expression level of the reporter gene beta glucuronidase in tobacco and cotton. Transgenic Research, 23, 351–363. [DOI] [PubMed] [Google Scholar]
- Angell, S. & Page, A. (2002) Transient expression of reporter proteins can alter plant gene expression. Plant Science, 163, 431–437. [Google Scholar]
- Beclin, C. , Boutet, S. , Waterhouse, P. & Vaucheret, H. (2002) A branched pathway for transgene‐induced RNA silencing in plants. Current Biology, 12, 684–688. [DOI] [PubMed] [Google Scholar]
- Blazquez, M. (2007) Quantitative GUS activity assay of plant extracts. CSH Protocols, 2007, prot4690. [DOI] [PubMed] [Google Scholar]
- Bologna, N.G. & Voinnet, O. (2014) The diversity, biogenesis, and activities of endogenous silencing small RNAs in Arabidopsis. Annual Review of Plant Biology, 65, 473–503. [DOI] [PubMed] [Google Scholar]
- Brosnan, C.A. , Sarazin, A. , Lim, P. , Bologna, N.G. , Hirsch‐Hoffmann, M. & Voinnet, O. (2019) Genome‐scale, single‐cell‐type resolution of microRNA activities within a whole plant organ. The EMBO Journal, 38, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington, J.C. & Freed, D.D. (1990) Cap‐independent enhancement of translation by a plant potyvirus 5′ nontranslated region. Journal of Virology, 64, 1590–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, E.J. , Prokhnevsky, A.I. , Gopinath, K. , Dolja, V.V. & Carrington, J.C. (2004) Viral RNA silencing suppressors inhibit the microRNA pathway at an intermediate step. Genes & Development, 18, 1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Q. , Lai, H. , Hurtado, J. , Stahnke, J. , Leuzinger, K. & Dent, M. (2013) Agroinfiltration as an effective and scalable strategy of gene delivery for production of pharmaceutical proteins. Advanced Techniques in Biology & Medicine, 1, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien, B. , Baumberger, N. , Schepetilnikov, M. , Viotti, C. , De Cillia, J. , Ziegler‐Graff, V. et al. (2012) Degradation of the antiviral component ARGONAUTE1 by the autophagy pathway. Proceedings of the National Academy of Sciences of the United States of America, 109, 15942–15946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devers, E.A. , Brosnan, C.A. , Sarazin, A. , Albertini, D. , Amsler, A.C. , Brioudes, F. et al. (2020) Movement and differential consumption of short interfering RNA duplexes underlie mobile RNA interference. Nature Plants, 6, 789–799. [DOI] [PubMed] [Google Scholar]
- Gallie, D.R. & Walbot, V. (1992) Identification of the motifs within the tobacco mosaic virus 5′‐leader responsible for enhancing translation. Nucleic Acids Research, 20, 4631–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazzani, S. , Lawrenson, T. , Woodward, C. , Headon, D. & Sablowski, R. (2004) A link between mRNA turnover and RNA interference in Arabidopsis. Science, 306, 1046–1048. [DOI] [PubMed] [Google Scholar]
- Hamilton, A. , Voinnet, O. , Chappell, L. & Baulcombe, D. (2002) Two classes of short interfering RNA in RNA silencing. The EMBO Journal, 21, 4671–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseloff, J. , Siemering, K.R. , Prasher, D.C. & Hodge, S. (1997) Removal of a cryptic intron and subcellular localization of green fluorescent protein are required to mark transgenic Arabidopsis plants brightly. Proceedings of the National Academy of Sciences of the United States of America, 94, 2122–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, H. , De Bellis, L. , Ciurli, A. , Kondo, M. , Hayashi, M. & Nishimura, M. (1999) A novel acyl‐CoA oxidase that can oxidize short‐chain acyl‐CoA in plant peroxisomes. The Journal of Biological Chemistry, 274, 12715–12721. [DOI] [PubMed] [Google Scholar]
- Henderson, I.R. , Zhang, X. , Lu, C. , Johnson, L. , Meyers, B.C. , Green, P.J. et al. (2006) Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nature Genetics, 38, 721–725. [DOI] [PubMed] [Google Scholar]
- Himber, C. , Dunoyer, P. , Moissiard, G. , Ritzenthaler, C. & Voinnet, O. (2003) Transitivity‐dependent and ‐independent cell‐to‐cell movement of RNA silencing. The EMBO Journal, 22, 4523–4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurkman, W.J. & Tanaka, C.K. (1986) Solubilization of plant membrane proteins for analysis by two‐dimensional gel electrophoresis. Plant Physiology, 81, 802–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iki, T. , Clery, A. , Bologna, N.G. , Sarazin, A. , Brosnan, C.A. , Pumplin, N. et al. (2018) Structural flexibility enables alternative maturation, ARGONAUTE sorting and activities of miR168, a global gene silencing regulator in plants. Molecular Plant, 11, 1008–1023. [DOI] [PubMed] [Google Scholar]
- Incarbone, M. , Zimmermann, A. , Hammann, P. , Erhardt, M. , Michel, F. & Dunoyer, P. (2017) Neutralization of mobile antiviral small RNA through peroxisomal import. Nature Plants, 3, 17094. [DOI] [PubMed] [Google Scholar]
- Jaillais, Y. , Fobis‐Loisy, I. , Miege, C. & Gaude, T. (2008) Evidence for a sorting endosome in Arabidopsis root cells. The Plant Journal, 53, 237–247. [DOI] [PubMed] [Google Scholar]
- Jauvion, V. , Rivard, M. , Bouteiller, N. , Elmayan, T. & Vaucheret, H. (2012) RDR2 partially antagonizes the production of RDR6‐dependent siRNA in sense transgene‐mediated PTGS. PLoS One, 7, e29785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobling, S.A. & Gehrke, L. (1987) Enhanced translation of chimaeric messenger RNAs containing a plant viral untranslated leader sequence. Nature, 325, 622–625. [DOI] [PubMed] [Google Scholar]
- Kanoria, S. & Burma, P.K. (2012) A 28 nt long synthetic 5′UTR (synJ) as an enhancer of transgene expression in dicotyledonous plants. BMC Biotechnology, 12, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi, M. , Depicker, A. & Hilson, P. (2007) Recombinational cloning with plant gateway vectors. Plant Physiology, 145, 1144–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjemtrup, S. , Talarico, T.L. & Ursin, V. (2014) Biotechnology: pharming. In: Van Alfen, N. K. (Ed.) Encyclopedia of agriculture and food systems. Amsterdam, the Netherlands: Elsevier, pp. 117–133. [Google Scholar]
- Kontra, L. , Csorba, T. , Tavazza, M. , Lucioli, A. , Tavazza, R. , Moxon, S. et al. (2016) Distinct effects of p19 RNA silencing suppressor on small RNA mediated pathways in plants. PLoS Pathogens, 12, e1005935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, J.S. & Curtis, W.R. (2012) RNA viral vectors for improved agrobacterium‐mediated transient expression of heterologous proteins in Nicotiana benthamiana cell suspensions and hairy roots. BMC Biotechnology, 12, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecellier, C.H. & Voinnet, O. (2004) RNA silencing: no mercy for viruses? Immunological Reviews, 198, 285–303. [DOI] [PubMed] [Google Scholar]
- Liu, L. , Canizares, M.C. , Monger, W. , Perrin, Y. , Tsakiris, E. , Porta, C. et al. (2005) Cowpea mosaic virus‐based systems for the production of antigens and antibodies in plants. Vaccine, 23, 1788–1792. [DOI] [PubMed] [Google Scholar]
- Liu, L. & Chen, X. (2016) RNA quality control as a key to suppressing RNA silencing of endogenous genes in plants. Molecular Plant, 9, 826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardanova, E.S. , Blokhina, E.A. , Tsybalova, L.M. , Peyret, H. , Lomonossoff, G.P. & Ravin, N.V. (2017) Efficient transient expression of recombinant proteins in plants by the novel pEff vector based on the genome of potato virus X. Frontiers in Plant Science, 8, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mette, M.F. , Aufsatz, W. , van der Winden, J. , Matzke, M.A. & Matzke, A.J. (2000) Transcriptional silencing and promoter methylation triggered by double‐stranded RNA. The EMBO Journal, 19, 5194–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno, A.B. , Martinez de Alba, A.E. , Bardou, F. , Crespi, M.D. , Vaucheret, H. , Maizel, A. et al. (2013) Cytoplasmic and nuclear quality control and turnover of single‐stranded RNA modulate post‐transcriptional gene silencing in plants. Nucleic Acids Research, 41, 4699–4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourrain, P. , Beclin, C. , Elmayan, T. , Feuerbach, F. , Godon, C. , Morel, J.B. et al. (2000) Arabidopsis SGS2 and SGS3 genes are required for posttranscriptional gene silencing and natural virus resistance. Cell, 101, 533–542. [DOI] [PubMed] [Google Scholar]
- Nekrasov, V. , Staskawicz, B. , Weigel, D. , Jones, J.D. & Kamoun, S. (2013) Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA‐guided endonuclease. Nature Biotechnology, 31, 691–693. [DOI] [PubMed] [Google Scholar]
- Norkunas, K. , Harding, R. , Dale, J. & Dugdale, B. (2018) Improving agroinfiltration‐based transient gene expression in Nicotiana benthamiana . Plant Methods, 14, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris, S.R. , Meyer, S.E. & Callis, J. (1993) The intron of Arabidopsis thaliana polyubiquitin genes is conserved in location and is a quantitative determinant of chimeric gene expression. Plant Molecular Biology, 21, 895–906. [DOI] [PubMed] [Google Scholar]
- Obembe, O.O. , Popoola, J.O. , Leelavathi, S. & Reddy, S.V. (2011) Advances in plant molecular farming. Biotechnology Advances, 29, 210–222. [DOI] [PubMed] [Google Scholar]
- Pall, G.S. & Hamilton, A.J. (2008) Improved northern blot method for enhanced detection of small RNA. Nature Protocols, 3, 1077–1084. [DOI] [PubMed] [Google Scholar]
- Papp, I. , Mette, M.F. , Aufsatz, W. , Daxinger, L. , Schauer, S.E. , Ray, A. et al. (2003) Evidence for nuclear processing of plant micro RNA and short interfering RNA precursors. Plant Physiology, 132, 1382–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent, J.S. , Jauvion, V. , Bouche, N. , Beclin, C. , Hachet, M. , Zytnicki, M. et al. (2015) Post‐transcriptional gene silencing triggered by sense transgenes involves uncapped antisense RNA and differs from silencing intentionally triggered by antisense transgenes. Nucleic Acids Research, 43, 8464–8475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pumplin, N. & Voinnet, O. (2013) RNA silencing suppression by plant pathogens: defence, counter‐defence and counter‐counter‐defence. Nature Reviews. Microbiology, 11, 745–760. [DOI] [PubMed] [Google Scholar]
- Rawlings, R.A. , Krishnan, V. & Walter, N.G. (2011) Viral RNAi suppressor reversibly binds siRNA to outcompete Dicer and RISC via multiple turnover. Journal of Molecular Biology, 408, 262–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russinova, E. , Borst, J.W. , Kwaaitaal, M. , Cano‐Delgado, A. , Yin, Y. , Chory, J. et al. (2004) Heterodimerization and endocytosis of Arabidopsis brassinosteroid receptors BRI1 and AtSERK3 (BAK1). Plant Cell, 16, 3216–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainsbury, F. & Lomonossoff, G.P. (2008) Extremely high‐level and rapid transient protein production in plants without the use of viral replication. Plant Physiology, 148, 1212–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainsbury, F. & Lomonossoff, G.P. (2014) Transient expressions of synthetic biology in plants. Current Opinion in Plant Biology, 19, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwach, F. , Vaistij, F.E. , Jones, L. & Baulcombe, D.C. (2005) An RNA‐dependent RNA polymerase prevents meristem invasion by potato virus X and is required for the activity but not the production of a systemic silencing signal. Plant Physiology, 138, 1842–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silhavy, D. , Molnar, A. , Lucioli, A. , Szittya, G. , Hornyik, C. , Tavazza, M. et al. (2002) A viral protein suppresses RNA silencing and binds silencing‐generated, 21‐ to 25‐nucleotide double‐stranded RNAs. The EMBO Journal, 21, 3070–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugio, T. , Matsuura, H. , Matsui, T. , Matsunaga, M. , Nosho, T. , Kanaya, S. et al. (2010) Effect of the sequence context of the AUG initiation codon on the rate of translation in dicotyledonous and monocotyledonous plant cells. Journal of Bioscience and Bioengineering, 109, 170–173. [DOI] [PubMed] [Google Scholar]
- Taochy, C. , Yu, A. , Bouche, N. , Bouteiller, N. , Elmayan, T. , Dressel, U. et al. (2019) Post‐transcriptional gene silencing triggers dispensable DNA methylation in gene body in Arabidopsis. Nucleic Acids Research, 47, 9104–9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyurin, A.A. , Suhorukova, A.V. , Kabardaeva, K.V. & Goldenkova‐Pavlova, I.V. (2020) Transient gene expression is an effective experimental tool for the research into the fine mechanisms of plant gene function: advantages, limitations, and solutions. Plants (Basel), 9, 1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargason, J.M. , Szittya, G. , Burgyan, J. & Hall, T.M. (2003) Size selective recognition of siRNA by an RNA silencing suppressor. Cell, 115, 799–811. [DOI] [PubMed] [Google Scholar]
- Vaucheret, H. (2005) MicroRNA‐dependent trans‐acting siRNA production. Science's STKE, 2005, pe43. [DOI] [PubMed] [Google Scholar]
- Vaucheret, H. , Beclin, C. , Elmayan, T. , Feuerbach, F. , Godon, C. , Morel, J.B. et al. (1998) Transgene‐induced gene silencing in plants. The Plant Journal, 16, 651–659. [DOI] [PubMed] [Google Scholar]
- Voinnet, O. (2009) Origin, biogenesis, and activity of plant microRNAs. Cell, 136, 669–687. [DOI] [PubMed] [Google Scholar]
- Voinnet, O. , Lederer, C. & Baulcombe, D.C. (2000) A viral movement protein prevents spread of the gene silencing signal in Nicotiana benthamiana . Cell, 103, 157–167. [DOI] [PubMed] [Google Scholar]
- Voinnet, O. , Pinto, Y.M. & Baulcombe, D.C. (1999) Suppression of gene silencing: a general strategy used by diverse DNA and RNA viruses of plants. Proceedings of the National Academy of Sciences of the United States of America, 96, 14147–14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voinnet, O. , Rivas, S. , Mestre, P. & Baulcombe, D. (2003) [Retracted] An enhanced transient expression system in plants based on suppression of gene silencing by the p19 protein of tomato bushy stunt virus. The Plant Journal, 33, 949–956. [DOI] [PubMed] [Google Scholar]
- Wu, Q. , Wang, X. & Ding, S.W. (2010) Viral suppressors of RNA‐based viral immunity: host targets. Cell Host & Microbe, 8, 12–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipfel, C. , Kunze, G. , Chinchilla, D. , Caniard, A. , Jones, J.D. , Boller, T. et al. (2006) Perception of the bacterial PAMP EF‐Tu by the receptor EFR restricts Agrobacterium‐mediated transformation. Cell, 125, 749–760. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (a) GFP fluorescence signal at 473 nm of N. benthamiana leaves, representative of six independent experiments, 4 days post‐infiltration with the constructs p35S::FHA:GUS‐Intron (GUS) and p35S::GFP5 (GFP) on the left‐hand side, or p35S::P19 (P19) and p35S::GFP5 (GFP) on the right‐hand side. (b) Quantification of the GFP fluorescence signal obtained in (a). Black bars: mean. Error bars: standard deviations. n = 6.
Figure S2. (a) Images of N. benthamiana leaves, representative of four independent experiments, taken under UV illumination at 7 days post‐infiltration (dpi) with the constructs p35S::GFP5 or pUB10::GFP5, together with p35S::FHA:GUS‐Intron (GUS) or p35S::P19 (P19). (b) Relative GFP fluorescence units (RFU) measured from the samples depicted in (a). Black bars: mean. Error bars: standard deviations. n = 4. (c) GF siRNA accumulation in biological duplicates of samples depicted in (a) analyzed by Northern blot using a probe corresponding to the 5′ part of the GFP ORF (‘GF’). The probed miR159 and U6 small RNA were used as an endogenous P19 cargo and RNA loading control, respectively. EtBr.: Ethidium bromide staining provides an additional RNA loading control. Black arrows indicate 3′‐end‐trimmed sRNA species. The experiment was independently repeated two times with similar results.
Figure S3. (a) Independent biological replicate of experiments presented in Figure 5d. FHA:P19 protein levels in the input, unbound fraction, and IP fraction of N. benthamiana leaves at 4 dpi with infiltration medium (−), p35S::FHA:GUS‐Intron (GUS), p35S::FHA:P19 (F:P19), and p35S::GFFG (GFFG), as indicated. Coomassie staining of total proteins (Coom.) provides a control for equal loading. (b) Independent biological replicate of experiments presented in Figure 5e. GF siRNA levels in samples depicted in (a) analyzed by Northern blot. miR159 and U6 small RNA were used as an endogenous P19 cargo and RNA loading control, respectively. EtBr.: Ethidium bromide staining provides an additional RNA loading control. Black arrows indicate 3′‐end‐trimmed sRNA species.
Figure S4. GUS activity in relative MU fluorescence units (RFU) from the FHA:GUS IP fraction of samples depicted in Figure 7c at 0, 1, 2, 3, and 5 min. Plotted: mean. Error bars: standard deviations. n = 6.
Figure S5. Images of Nicotiana benthamiana leaves 4 days post‐infiltration with the constructs p35S::P19 (P19) and p35S::ERD2:mCherry (ERD2), as indicated, under normal light.
Figure S6. p35S::GFP5 expression cassette within the pBI121 binary vector backbone described in Angell and Page (2002) and used in this study.
Table S1. Sequences of oligonucleotide primers used in this study.
Table S2. List of antibodies used in this study.
Data Availability Statement
This study includes no data deposited in external repositories. All relevant data can be found within the manuscript and its supporting materials.