

Abstract
Background
CARD9 deficiency is an autosomal recessive primary immunodeficiency underlying increased susceptibility to fungal infection primarily presenting as invasive CNS Candida and/or cutaneous/invasive dermatophyte infections. More recently, a rare heterozygous dominant negative CARD9 variant c.1434 + 1G > C was reported to be protective from inflammatory bowel disease.
Objective
We studied two siblings carrying homozygous CARD9 variants (c.1434 + 1G > C) and born to heterozygous asymptomatic parents. One sibling was asymptomatic and the other presented with candida esophagitis, upper respiratory infections, hypogammaglobulinemia, and low class-switched memory B cells.
Methods and Results
The CARD9 c.1434 + 1G > C variant generated two mutant transcripts confirmed by mRNA and protein expression: an out-of-frame c.1358–1434 deletion/ ~ 55 kDa protein (CARD9Δex.11) and an in-frame c.1417–1434 deletion/ ~ 61 kDa protein (CARD9Δ18 nt.). Neither transcript was able to form a complete/functional CBM complex, which includes TRIM62. Based on the index patient’s CVID-like phenotype, CARD9 expression was tested and detected in lymphocytes and monocytes from humans and mice. The functional impact of different CARD9 mutations and gene dosage conditions was evaluated in heterozygous and homozygous c.1434 + 1 G > C members of the index family, and in WT (two WT alleles), haploinsufficiency (one WT, one null allele), and null (two null alleles) individuals. CARD9 gene dosage impacted lymphocyte and monocyte functions including cytokine generation, MAPK activation, T-helper commitment, transcription, plasmablast differentiation, and immunoglobulin production in a differential manner.
Conclusions
CARD9 exon 11 integrity is critical to CBM complex function. CARD9 is expressed and affects particular T and B cell functions in a gene dosage-dependent manner, which in turn may contribute to the phenotype of CARD9 deficiency.
Keywords: T cells; B cells, Th17; Candida; fungal infection; inflammatory bowel disease; CARD9; BCL10; MALT1; TRIM62; CBM complex; MAPK signaling; human immunology; mouse immunology
Introduction
Caspase recruitment domain-containing protein 9 (CARD9) is an adaptor molecule reportedly expressed in myeloid cells consisting of 536 amino acids with an N-terminal CARD domain and a C-terminal coiled-coil domain. The C-type lectin receptors as Dectin 1, Dectin 2, and Mincle, bind to fungal components (e.g., β-glucan or α-mannan) and utilize CARD9 to form the CBM complex (CARD9/B cell lymphoma 10 [BCL10]/Mucosal-associated lymphoid tissue lymphoma translocation protein 1 [MALT1]), followed by downstream activation of the NF-κB pathway [1–6]. CARD9-dependent CBM complex-independent functions have also been reported in mouse and human PBMCs where CARD9 induces Erk phosphorylation through the Ras-GRF1/H-Ras axis. Erk activation supports antifungal immunity and secretion of pro-inflammatory cytokines including IL-6, TNF-α, and IL-1β, while p38/c-fos-dependent IL-1β production in microglia promotes CXCL1-dependent neutrophil migration to the fungal-infected central nervous system (CNS) [4, 7–9]. CARD9 deficiency was first described in an extended Iranian family with increased susceptibility to superficial and invasive fungal infections (i.e., Candida albicans meningoencephalitis) [10]. Later, superficial and invasive dermatophyte infections were also associated with this disease [11]. Currently, more than 50 cases have been documented to present with similar clinical phenotypes, with manifestations arising at any age and associated with biallelic loss-of-function CARD9 mutations [11–19]. More recently, genome-wide association studies (GWAS) have shown that particular monoallelic CARD9 variants could be associated either with detrimental or protective effects toward inflammatory bowel disease (IBD) [20–24]. Individuals carrying a monoallelic CARD9 p.S12N activating variant have shown increased production of inflammatory cytokines supporting IBD’s autoinflammatory pathophysiology and disease [25, 26]. Moreover, an IBD consortium reported that monoallelic CARD9 variant c.1434 + 1G > C, reportedly leading to CARD9 exon 11 deletion (Δ ex.11), was associated with an IBD-protective effect due to a dominant negative mechanism inhibiting inflammatory cytokine production [24, 25].
In this study, we report a family carrying CARD9 c.1434 + 1G > C in heterozygous or homozygous state. The index patient with the homozygous mutation had recurrent episodes of mucocutaneous candidiasis (i.e., oral thrush, Candida esophagitis), recurrent upper respiratory infections, and hypogammaglobinemia resembling a common variable immunodeficiency (CVID)-like phenotype. Family segregation analysis and other CARD9 gene dosage conditions allowed us to explore the role of CARD9 linked to fungal susceptibility and autoinflammatory pathways. Moreover, we found that the expression of CARD9 extends beyond myeloid cells involving T and B cells both in humans and mice, suggesting a previously unrecognized CARD9/Card9 role in lymphoid function.
Patients, Materials, and Methods
Patients
All research subjects or their guardians provided written informed consent in accordance with the Declaration of Helsinki under institutional review board-approved protocols of the National Institute of Allergy and Infectious Diseases, NIH. Blood from healthy donors was obtained under approved protocols.
Patient II.2 is the second of 3 children born to healthy, non-consanguineous white American parents. At the time of this report, II.2 was 9 years old with a history of recurrent and severe oropharyngeal candidiasis from the age of 2 to 18 months. At age 2 years, he was diagnosed with Candida esophagitis and had a 2nd episode at age 3. Since then, he presented with several episodes of upper respiratory infections and sinusitis that responded adequately to antibiotic treatment and then prophylaxis; he also developed a superficial dermatophyte infection (tinea pedis). His humoral immune evaluation (last at age 9 years) consistently showed low IgG 460 mg/dL (reference ranges for age 673–1734 mg/dL), IgA 17 mg/dL (41–368 mg/dL), and IgM 44 mg/dL (47–311 mg/dL); but protective titers to diphtheria toxoid, tetanus toxoid, Haemophilus influenzae B, as well as to 13/13 pneumococcal polysaccharide antigens. His lymphocyte immunophenotype showed low class switched memory B cells (CD20+/CD27+/IgM−) as well as plasmablasts (CD19+/CD38high/CD24−). His family history was remarkable for his healthy and asymptomatic father (I.1), mother (I.2), and 11-year-old older brother (II.1); his younger brother (II.3) was also healthy and asymptomatic at 5 years of age.
PBMCs Isolation and Cell Culture
Peripheral blood mononuclear cells (PBMCs) were isolated according to density centrifugation method. Due to COVID restrictions and intrinsic limitations to recruit pediatric normal controls (NCs) to NIH research protocols, our in vitro experiments were performed using young adult NIH blood bank donors/volunteers, age- and sex-matched to the young adults in our study; some of the in vitro comparisons with the pediatric patients might be influenced by age differences. HEK-293 T cell lines were cultured in DMEM media following standard techniques.
Whole-Exome Sequencing
A commercial clinical whole exome sequencing (WES) test and analysis was performed at Baylor Genetics Laboratory Inc, Houston TX.
Sanger Sequencing
Sanger sequencing was performed to confirm WES-detected variants and to screen family members.
RNA Isolation, cDNA Amplification and Real-Time PCR
Total RNA was isolated from PBMCs and reverse transcribed. cDNA was PCR-amplified using exon-specific CARD9 primers. For the quantitative real-time PCR study, CARD9 exon 11-specific TaqMan Probe was used.
Sorting of Mouse T Cells, B Cells and Monocytes
Spleens from four WT C57BL/6 J mice were digested using Liberase TL (Roche) and DNase. T cell, B cells, and monocytes were sorted using a BD FACS Aria™ Il Cell Sorter.
Western Blotting
Protein lysates were prepared, separated by electrophoresis, transferred to nitrocellulose membranes, and blotted with indicated antibodies in figures.
Immunophenotyping and Th17 Evaluation
Whole blood from NCs and patients were processed for detailed T, B, and NK-cell immunophenotyping and total PBMCs were used for Th17 commitment evaluation by flow cytometry.
Cytokine Evaluation
Cytokines production from PBMCs from NCs and patients after heat-killed Candida (HKC) or LPS stimulation was evaluated by Luminex.
Plasmid Preparation
Flag-CARD9 WT was purchased, and mutants (CARD9 Δ ex.11 and CARD9 Δ 18 nt) were generated for transfection and expression experiments.
CBM Complex Evaluation
TRIM62, MALT1, and BCL10 expression plasmids were co-transfected either with WT CARD9 or mutant CARD9 vectors (CARD9 Δ ex. 11 or CARD9 Δ18 nt) in HEK293T, and co-immunoprecipitation was performed to evaluate the CBM complex composition.
Plasmablast Differentiation and Immunoglobulin Production
PBMCs from NCs and patients were stimulated with CD40L plus IL-21 for 5 days. Plasmablast (CD19+CD27+CD38+) generation was evaluated by flow cytometry. Culture supernatants were collected from the stimulated cells and evaluated for IgG, IgM, and IgA production by ELISA.
Transcriptomic Analysis on T Cells
NCs and patients’ enriched CD3 T cells were co-cultured with monocytes from a healthy control in the presence of HKC. After 3 days, T cells were enriched again and total RNA was extracted for RNASeq analysis. The RNASeq data generated for this publication have been deposited in NCBI’s Gene Expression Omnibus [27] and are accessible through GEO Series accession number GSE181718 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE181718).
Statistical Analysis
GraphPad Prism software (Version 8.1.2, GraphPad Software, La Jolla, CA, USA) was used for the statistical analysis. P values less than 0.05 were considered significant.
Results
Clinical and Immunological Features
Laboratory immune evaluation in the 4 family members tested (parents I.1 and I.2; siblings II.2 and II.3) revealed that T, B, and NK-cell numbers were predominantly within reference ranges. Specific abnormalities included the percentage elevation of CD8+ central memory (CM) cells in all family members and the decrease of CD8+ TEMRA in both siblings and one parent. B cell subsets showed reduced percentages of class-switched memory in II.2 and the parents, and reduced plasmablast cells in all subjects. Immunoglobulin levels were within normal ranges in I.1, I.2, and II.3, while IgG and IgA were diminished in II.2. T cell proliferation in response to phytohemagglutinin (PHA) and heat-killed Candida (HKC), as well as B cell proliferation in response to CD40L plus IL-21, were normal in all subjects tested (I.1, I.2, II.2, and II.3; not shown). A more detailed description of the baseline immunological evaluation is shown in Table 1.
Table.1.
Immunological features in individuals carrying CARD9 c.143 + 1 G >C mutation
| Sample ID | I.1 | I.2 | II.2 | II.3 |
|---|---|---|---|---|
| Age/Gender | 48Y/M | 39Y/F | 9Y/M | 5/M |
| Zygosity | Heterozygous | Heterozygous | Heterozygous | Heterozygous |
|
| ||||
| T cells | ||||
| CD3 (cells/ul) | 2158 | 1821 | 1700 | 2714 |
| CD4 (cells/ul) | 1592 | 912 | 1162 | 1423 |
| CD8 (cells/ul) | 517 | 789 | 400 | 808 |
| CD3 (%) | 84.3 | 86.3 | 82.5 | 80.3 |
| CD4 (%) | 73.8 | 50 | 68.4 (H) | 52.4 |
| CD8 (%) | 24 | 43.3 (H) | 23.5 | 29.7 |
| CD4/CD8 ratio | 3.08 | 1.16 | 2.91 (H) | 1.76 |
| CD3 +/CD4−CD8−(DNT) (%) | 1.7 | 6 (H) | 7.7 (H) | 0.8 (L) |
| CD62L +CD45RA +/CD4 +(Naive) (%) | 33.2 | 41.9 | 72.1 | 70.5 |
| CD62L +CD45RA−/CD4 +(central memory) (%) | 55.2 | 46.3 | 24.5 | 25.9 |
| CD62L−CD45RA−/CD4 +(effector memory) (%) | 11.5 | 11.6 | 3.4 | 3.4 |
| CD62L−CD45RA +/CD4 +(TEMRA) (%) | 0.1 | 0.2 | 0.1 | 0.1 |
| CD62L +CD45RA +/CD8 +(Naive) (%) | 53.9 | 39.4 | 76.1 | 68.8 |
| CD62L +CD45RA−/CD8 +(central memory) (%) | 24.5 (H) | 21.7 (H) | 11.4 (H) | 20.3 (H) |
| CD62L−CD45RA−/CD8 +(effector memory) (%) | 18 | 24.2 | 8 | 8.2 |
| CD62L−CD45RA +/CD8 +(TEMRA) (%) | 3.7 (L) | 14.7 | 4.5 (L) | 2.7 (L) |
| B cells | ||||
| CD19 +(cells/ul) | 223 | 205 | 192 | 392 |
| CD19 +(%) | 8.7 | 9.7 | 9.3 | 11.6 |
| IgM +CD27 +/CD20 +(unswitched memory) (%) | 34.9 (H) | 10.7 | 4.1 | 9.5 |
| IgM-CD27 +/CD20 +(class-switched memory) (%) | 4.8 (L) | 3.5 (L) | 1.4 (L) | 4.1 |
| CD24-CD38 + +/CD19 +(Plasmablast) (%) | 0.4 (L) | 0.2 (L) | 0.4 (L) | 0.5 (L) |
| NK cells | ||||
| CD16 +CD56 +/CD3− (cells/ul) | 169 | 89 (L) | 177 | 281 |
| CD16 +CD56 +/CD3− (%) | 6.5 | 4.1 (L) | 8.3 | 8.1 |
| Serum immunoglobulin levels (mg/dL) | ||||
| IgG | 875 | 1030 | 460 (L) | 960 |
| IgM | 186 | 82 | 44 | 74 |
| IgA | 107 | 145 | 17 (L) | 89 |
Mutation Characterization
PBMCs-extracted gDNA from patient II.2 was tested by WES and a homozygous CARD9 variant c.1434 + 1G > C (NM_052813; dbSNP rs141992399); gnomAD allele frequency = 0.003325 with 924 heterozygous and 2 homozygous occurrences reported. The nucleotide substitution occurs at the highly conserved + 1 position of a canonical donor splice donor site (GERPN = 3.33, GERPS = 2.41). This variant is reported in ClinVar (Variation ID:535,816) with conflicting interpretations of pathogenicity (likely benign and uncertain significance). It is classified as pathogenic according to ACMG criteria PVS1 (null variant within canonical site in a gene where LOF is a known mechanism of disease), PM2 (extremely low frequency in recessive gene in gnomAD), and PP3 (multiple lines of computational evidence support a deleterious effect; pathogenic computational verdict based on 5 pathogenic predictions CADD [phred = 28.7], MutationTaster, SpliceAI [donor loss 0.88], dbscSNV-ada [0.99998], dbscSNV-rf [0.844]). This transversion maps to the exon 11’s canonical donor splice site. Targeted Sanger sequencing determined that both parents (I.1 and I.2) and the proband’s elder brother (II.1) carried the same variant in the heterozygous state, while his asymptomatic younger brother (II.3) was homozygous for the mutation (Fig. 1A).
Fig. 1.
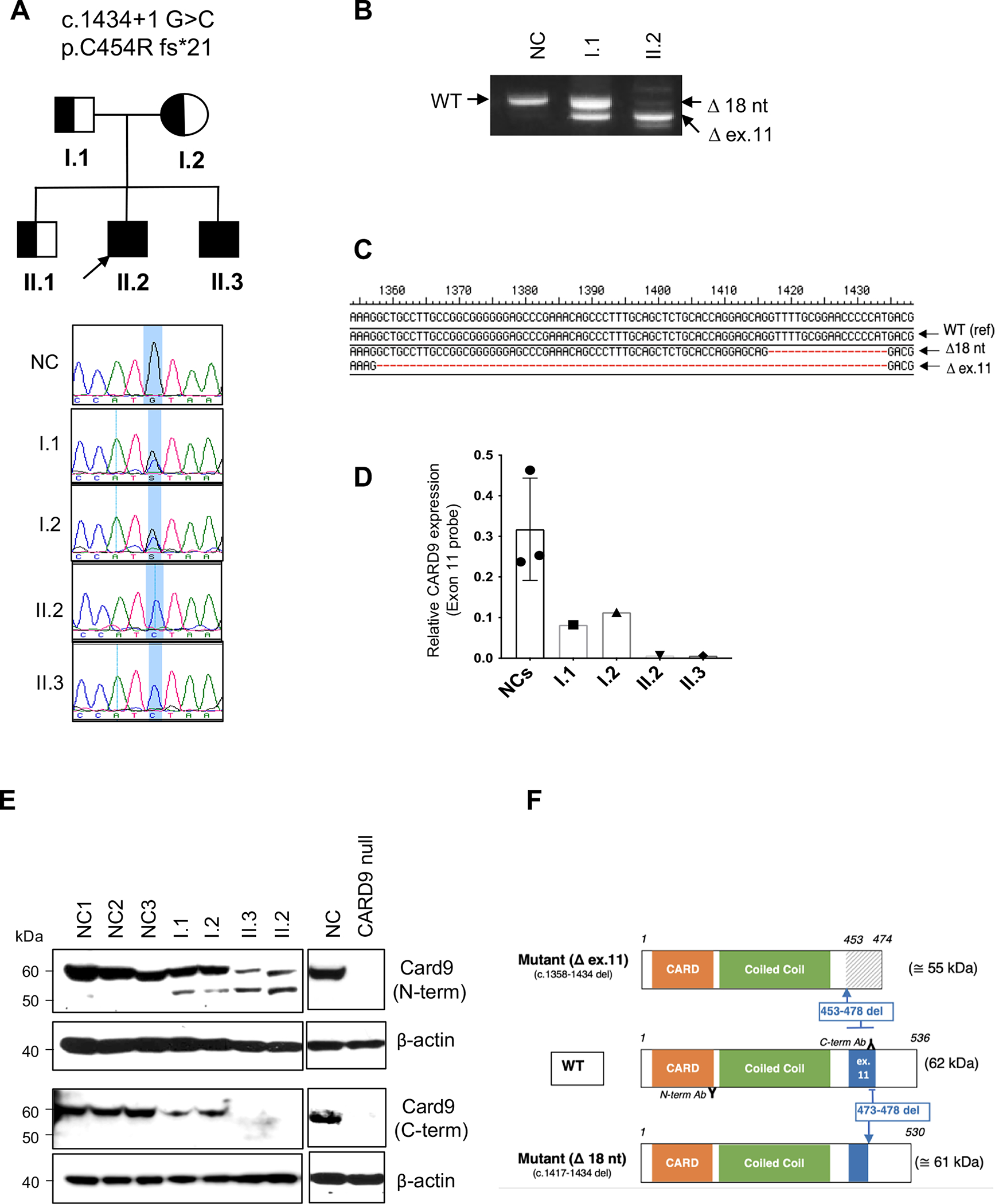
Genetic and molecular characterization of the CARD9 c.143 + 1 G > C mutation. A Pedigree of a family with the CARD9 mutation (upper panel). Half-filled symbols indicate the heterozygous CARD9 mutation and black filled symbols denote the homozygous mutation. The proband is indicated with an arrow; clinical expressivity and disease penetrance are discussed in the text. Chromatogram of CARD9 Sanger sequencing in a normal control (NC), patients (II.2 and II.3) with a homozygous change, and parents (I.1 and I.2) with a heterozygous change (lower panel). The blue color indicates the site of the mutation. B The agarose gel electrophoresis showing PCR products of WT and mutant alleles of CARD9 using cDNA from PBMCs. PCR was performed using exon-8-F and exon-13-R primers. C The PCR product from II.2 (in B) was cloned into a TA vector and sequenced 24 single colonies. Two transcript variants are detected: exon 11 deletion (c.1358–1434; Δ ex.11) and 18 nt deletion (c.1417–1434; Δ 18 nt). D Scatter plot showing quantitative real time PCR of CARD9 mRNA expression from PBMCs. The CARD9 TaqMan probe specific to exon 11 was used to discriminate the WT and mutant allele, and the expression was normalized to GAPDH as a housekeeping gene. Data represent the mean ± SD of three normal controls and the indicated individuals. E CARD9 protein expression from PBMCs from three different normal controls and indicated CARD9 mutation carriers. Immunoblotting with the CARD9 C-terminal antibody (Immunogen 467–480 amino acid) and N-terminal antibody (Immunogen 98–187 amino acid) are shown. CARD9 expression was not detected in a known CARD9 deficiency patient (CARD9 null) with compound heterozygous mutations (Q289*/D8Afs*10). Data are representative of three replicates. F Schematic diagram of WT and CARD9 mutant proteins (CARD9 Δ ex.11 and CARD9 Δ 18 nt); CARD9 domains and exon 11-coded regions are color identified. The protein resulting from the out-of-frame full exon 11 deletion (Mutant Δ ex.11) generates a missense C-terminal region, depicted by diagonal lines. The protein resulting from the in-frame exon 11 18 nucleotide deletion (Mutant Δ 18 nt) is identified by a smaller ex.11. Binding sites for CARD9 N- and C-terminal-directed antibodies used in (E) are also shown
CARD9 RNA was extracted, reverse transcribed to cDNA, PCR-amplified, and sequenced in II.2 (proband, biallelic mutation carrier) and I.1 (father, monoallelic carrier). PCR amplification of CARD9 cDNA using exonic primers spanning exons 8 to 13 showed three different patterns in a normal control (NC), I.1 and II.2, respectively. The NC showed a single sharp band, size corresponding to the WT transcript, including exon 11. The CARD9 c.1434 + 1G > C heterozygous father I.1 demonstrated an upper band grossly matching the expected WT transcript, and a lower band matching the expected mutant transcript with full deletion of exon 11 (Δ ex.11). The CARD9 c.1434 + 1G > C homozygous patient II.2 showed one primary band corresponding to the Δex.11 mutant transcript, along with other less prevalent bands; one of them had a slightly lower molecular weight than the WT band (Fig. 1B). Sanger sequencing of the single NC’s band confirmed it was WT, and II.1’s two bands corresponded to WT and Δex.11 sequence. TA-cloning of patient’s II.2 CARD9 cDNA using the same exon 8 to 13 primer set yielded no WT but 2 mutant transcripts: an outof-frame full exon 11 deletion (c.1358–1434, 77 nucleotides deletion, Δex.11) was found in 8-of-24 colonies sequenced, and an in-frame partial exon 11 deletion (c.1417–1434, 18 nucleotides deletion, Δ18nt) was found in the remaining 16 (Fig. 1C). A real-time PCR approach using an exon 11-specific CARD9 TaqMan probe showed no exon 11-containing CARD9 transcription in the homozygous patients; whereas intermediate WT CARD9 transcript levels were detected in the heterozygous parents when compared to NCs (Fig. 1D).
N-terminal and C-terminal CARD9-directed antibodies were used to characterize the expression of the CARD9 protein. While a single ~ 62/61 kDa band was identified in NCs with the N-terminal antibody (matching either a WT or an 18 nt. in-frame deleted CARD9 protein), two bands (at ~ 62/61 and ~ 55 kDa, the latter matching the Δ ex.11-mutant predicted molecular weight) were detected in the heterozygous and homozygous mutated family members, although at markedly different ratios. When the C-terminal antibody was tested, a single band at ~ 62/61 kDa was seen in NCs and the c.1434 + 1G > C heterozygous individuals, while no bands were detected in the c.1434 + 1G > C homozygous brothers. No CARD9 protein was detected with neither of the antibodies in the cell lysates of a CARD9 compound heterozygous Q289*/D8Afs*10 patient (CARD9 null) [19] (Fig. 1E).
Based on our results, CARD9 alleles carrying the c.1434 + 1G > C can generate 2 main alternative transcripts: (a) one carrying an exon 11 with an 18 nt in-frame-deletion and coding for a ~ 61 kDa protein that could only be detected by the N-terminal directed antibody; and (b) the other with a fully deleted out-of-frame exon 11 transcript coding for a ~ 55 kDa shortened protein with a missense C-terminal tail such that it can also only be detected by the N-terminal directed antibody. The C-terminal directed CARD9 antibody can only detect the 62 kDa WT protein (Fig. 1F).
CBM Complex Composition and CARD9 c.1434 + 1G > C Mutation Effect
Tripartite motif containing-62 (TRIM62) has recently been identified as a CARD9-binding partner through the C-terminal domain; however, its partnership with the CBM complex has not been formally established [25]. TRIM62, along with CARD9 WT or mutants, MALT1, and BCL10, were co-transfected in HEK293T cells and evaluated as CBM complex integral components (Fig. 2A). While WT CARD9 was able to bind and co-precipitate with MALT1, BCL10, and TRIM62, mutated CARD9Δ ex.11 and CARD9 Δ18 nt. proteins were unable to bind to MALT1 and BCL10. However, the two mutants differed in terms of their ability to bind TRIM62, as it was preserved with CARD9Δ18 nt. but abolished with CARD9Δex.11. This result suggests that a structurally preserved exon 11 is indeed required to bind BCL10, MALT1, and TRIM62 to form a full CBM complex, while a partially preserved exon 11 (i.e., Δ18 nt.) allows binding to TRIM62, but not BCL10 and MALT1 (Fig. 2A; right panel). Different attempts to pull down the CBM complex using antibodies other than CARD9 and TRIM62 (i.e., anti-BCL10 and anti-MALT1) proved to be technically challenging and not successful (not shown).
Fig. 2.
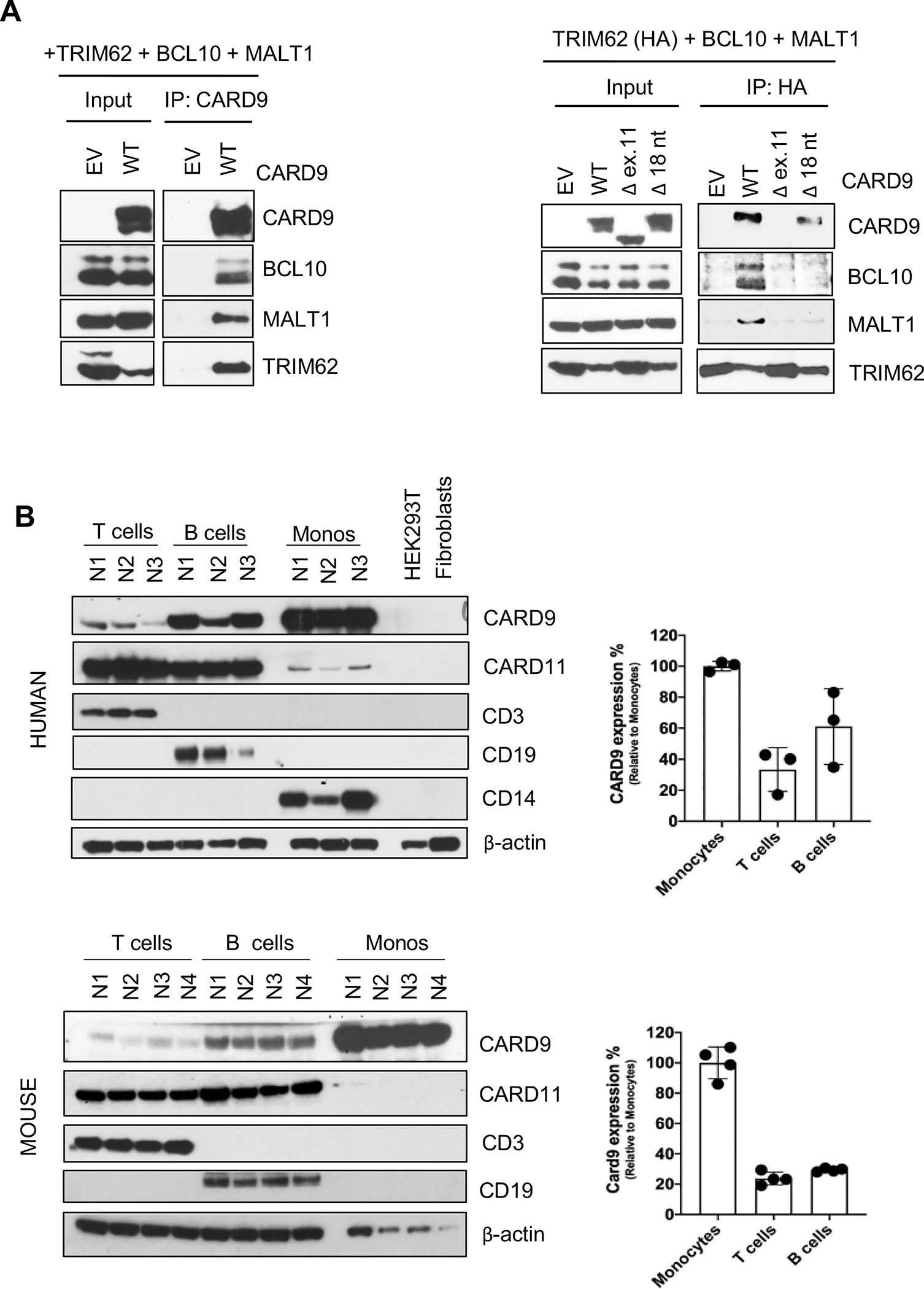
CBM complex formation and CARD9 protein expression. A HEK293T cells were co-transfected with plasmids expressing TRIM62, BCL10, and MALT1 together with either CARD9 WT or mutants (CARD9 Δ ex.11 or CARD9 Δ18 nt) plasmids. Immunoprecipitations were performed either with an anti-CARD9 antibody (left panel) or with an anti-HA (TRIM62) antibody (right panel) as indicated. Western blotting was performed for the detection of CARD9, BCL10, MALT1 (CBM complex), and TRIM62. Data are representative of three independent experiments. B Immunoblot showing CARD9 protein expression from T-, B-, and monocyte cells of three different healthy normal controls (upper panel) and four different wild type C57BL/6 J mice (lower panel). The CARD9/Card9 protein bands were quantified using densitometry, and the levels of protein were normalized to β-actin. The relative CARD9/Card9 protein levels in T cells, B cells compared with the levels in monocytes are shown. The values are expressed as the mean ± SD
CARD9 Protein Expression in Myeloid and Lymphoid Lineages in Humans and Mice
T and B cells, as well as monocytes isolated from human NC PBMC’s, and C57BL/6 WT mouse splenocytes were enriched and tested for species/lineage specific CARD9/Card9 expression. CARD9/Card9 proteins were readily expressed in human and mouse monocytes and lymphocytes. When compared to their species-specific CARD9/Card9 monocyte control (arbitrarily defined as 100%), human B cells expressed 34–83%, and T cells 17–42%, while mouse B cells expressed 28–30% and T cells 19–29% levels of the protein. Residual expression of CARD11 was also detected in human monocytes but not in mice (Fig. 2B). These results show that besides myeloid cells, CARD9 protein is efficiently expressed in T, and more prominently, in B lymphocytes. Moreover, CARD9 lymphoid expression is not restricted to humans, as mice showed a similar expression pattern in T and B cell lineages (Fig. 2B).
CARD9 Effects on Myeloid Cells
Generation of inflammatory cytokines IL-6, TNF-α, and IL-1β following LPS or HKC stimulation of PBMCs was tested in CARD9 c.1434 + 1G > C homozygous and heterozygous, and a CARD9 null patient. While all the research subjects responded similarly to NCs when stimulated with LPS (except the TNFα response in the CARD9 null patient), both c.1434 + 1 G > C homozygous and heterozygous showed markedly reduced cytokine productions upon HKC stimulation (Fig. 3A). This result suggests a c.1434 + 1G > C dominant negative effect on particular inflammatory cytokine generation upon HKC stimulation.
Fig. 3.
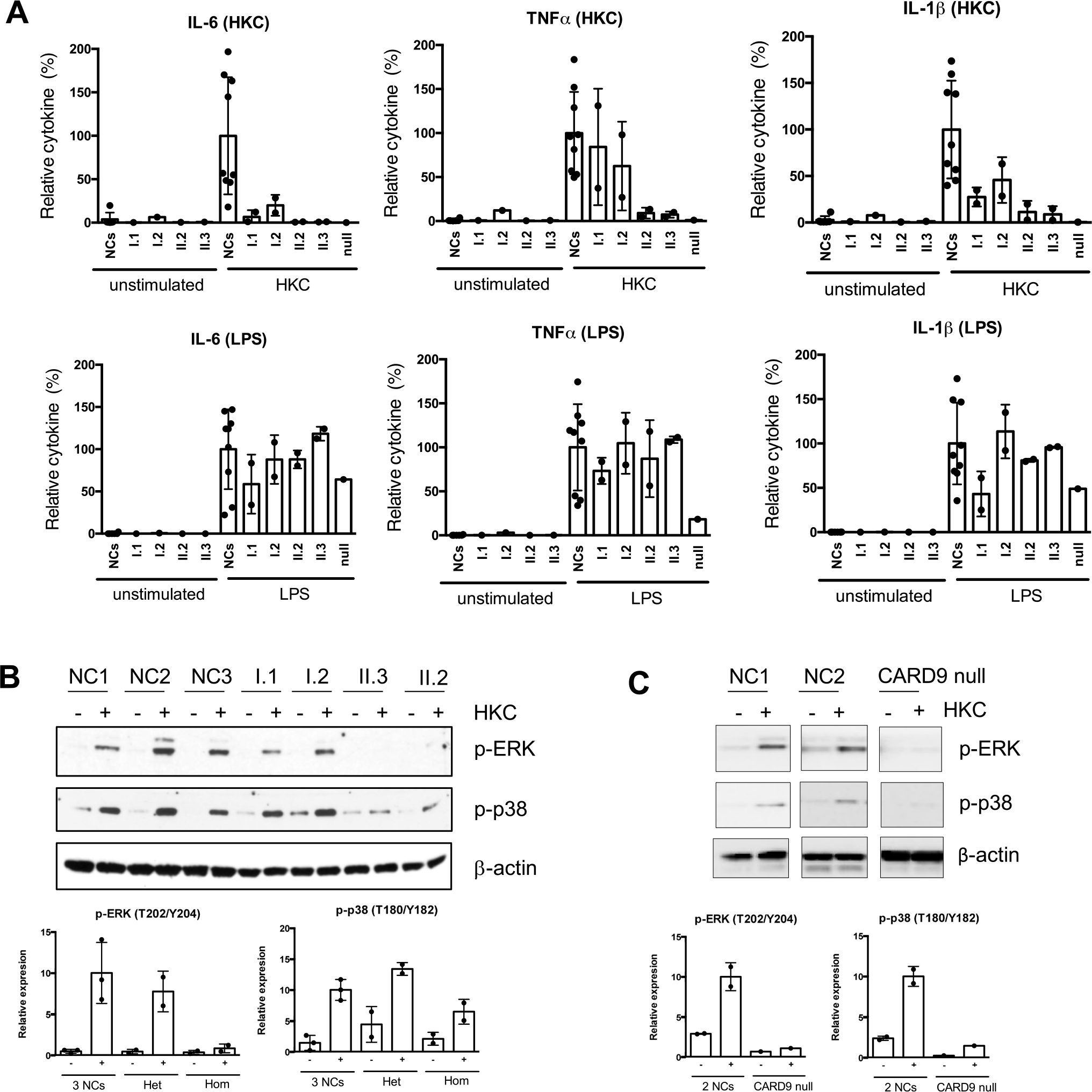
Cytokine generation and MAPK activation in patients with CARD9 mutations. A PBMCs from nine different normal controls and indicated patients were stimulated with HKC for 24 h. Supernatants were collected, and cytokines were tested by a multiplex Luminex kit. Data were pulled from two independent experiments and normalized each data set’s stimulated-normal controls to 100%. Data are plotted as mean ± SD. B, C Total PBMCs were stimulated with HKC for 60 min. Cell lysates were prepared and subjected to the western blot analysis with p-Erk and p-p38 antibodies. β-actin was used as a loading control. The p-ERK and p-p38 bands were quantified using densitometry, and the levels of protein were normalized to β-actin. The relative phosphorylation levels compared with the levels in normal controls are shown. The values are expressed as the mean ± SD from 2–3 normal controls and indicated groups (Het: heterozygous [I.1 and I.2], Hom: homozygous [II.2 and II.3]). A CARD9 null patient (p.Q289*/D8Afs*10) was included as a control to see the defect of CARD9-mediated signaling
The MAPK (ERK and p38) pathway, shown to regulate cytokine production, was tested in CARD9 c.1434 + 1G > C homozygous and heterozygous family members, as well as on the CARD9 null patient (Fig. 3B and C). While signaling following PMA/Ionomycin stimulation was similar to NCs in all research subjects tested (data not shown), the heterozygous parents displayed comparable levels of phosphorylated (p)-ERK and p-p38 to the NCs, and the homozygous individuals and the CARD9 null patient showed a marked decrease in p-ERK and p-p38 response. In contrast to cytokine generation, the c.1434 + 1G > C allele seems to behave in an autosomal recessive fashion in terms of p-ERK and p38 signaling upon HKC stimulation as only individuals with homozygous mutations presented with a HKC-specific abnormal response.
The Role of CARD9 Effects in T Cells
PBMCs from c.1434 + 1G > C heterozygous and homozygous family members, as well as the CARD9 null patient and her haploinsufficient brother were evaluated for in vitro Th17, Th1, and Th2 commitment/differentiation. Th17 commitment and differentiation was decreased in all subjects tested except the haploinsufficient individual (Fig. 4A). Besides, while the heterozygous parents and the haploinsufficient individual showed normal Th1 cells, the homozygous siblings and the CARD9 null patient showed decreased Th1 cells (Fig. 4A). Th2 cells, however, were comparable to NCs in all subjects tested except the haploinsufficient individual (who was later diagnosed with enteroparasitosis) (Fig. 4A). These results suggest that CARD9 c.1434 + 1 G > C heterozygosity behaves in a dominant negative manner for Th17 commitment/differentiation. In contrast, CARD9 c.1434 + 1G > C homozygosity seems necessary to negatively affect Th1 cells.
Fig. 4.
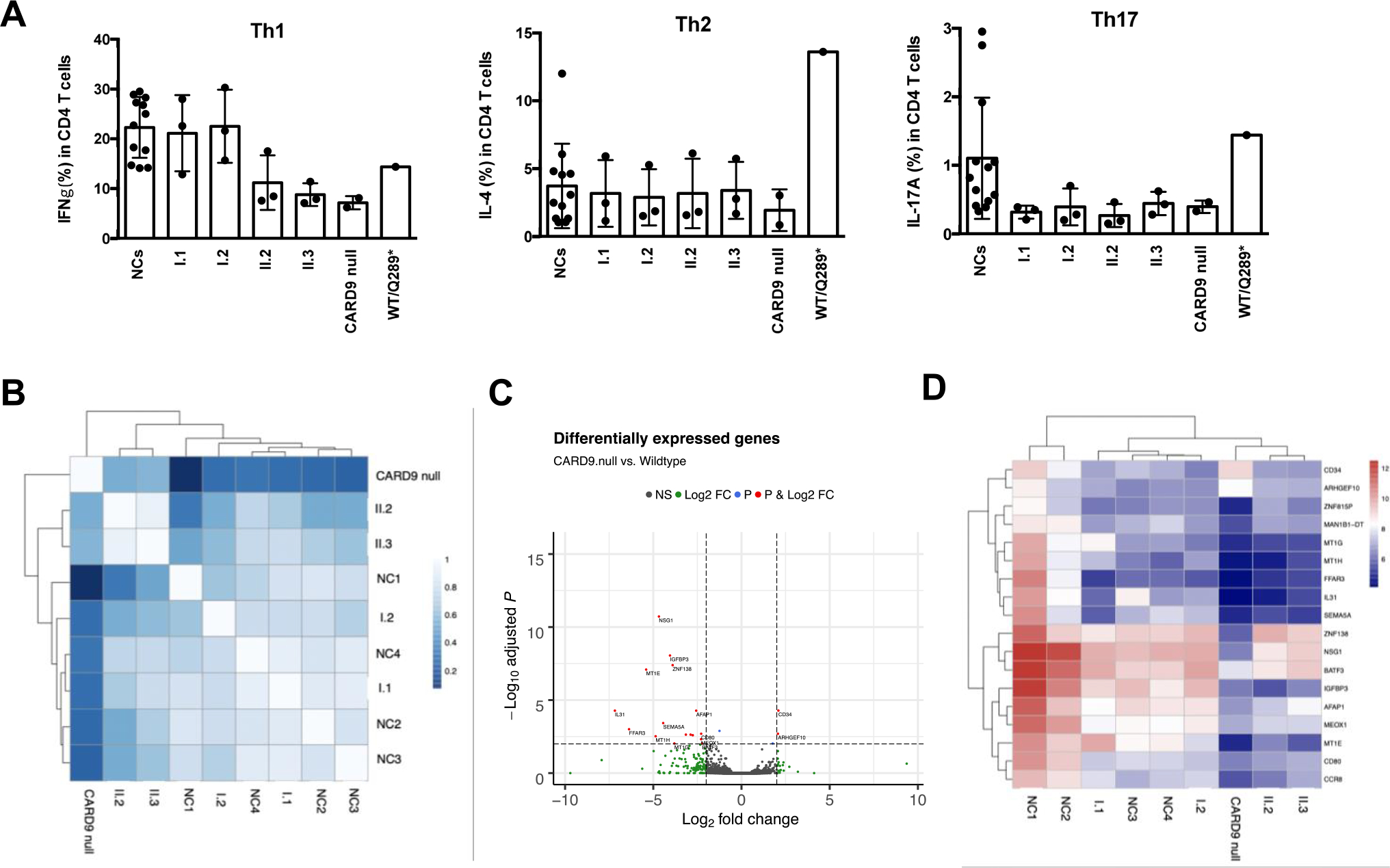
The effect of CARD9 on T cells. A Percentages of Th1, Th2, and Th17 cells from CD4+ T cells upon PMA/Ionomycin stimulation. Data are presented as mean ± SD from 12 healthy normal controls (NCs). Each dot in the research subjects indicates values from replicates. B–D RNAseq analyses of HKC-stimulated T cells from normal controls (NC) and indicated CARD9-mutated individuals. B Pearson correlation heatmap, C volcano plot, and D heatmap of the variance stabilized counts for all differentially expressed genes
T cell-oriented gene-level RNASeq analyses were evaluated in NCs (n = 4), the CARD9 c.1434 + 1G > C heterozygous, the CARD9 c.1434 + 1G > C homozygous, and the CARD9 null patient. RNASeq was performed using total RNA extracted from unstimulated enriched T cells, and from enriched T cells stimulated with HKC in the presence of WT CARD9 monocytes. This latter condition was designed to allow for HKC antigen presentation by WT CARD9 monocytes as a constant, leaving the CARD9 status in T cells as the only variable. The global gene expression signatures for the unstimulated enriched T cell were not unique among the study groups (i.e., the gene expression patterns did not cluster according to the study groups upon Pearson correlation and heatmap analyses with unsupervised clustering; Fig. 4B). However, the gene expression signatures for the HKC-stimulated enriched T cells were unique when based on the statistical contrast of data from the NCs vs. the CARD9 null patient, indicating a Candida-specific treatment effect. The CARD9 null patient served as a CARD9-dependent gene transcript normalization factor to focus the analyses on genes that are differentially expressed based on CARD9 expression. The differentially expressed genes for the HKC-stimulated T cells are shown in the volcano plot (Fig. 4C) and heatmap of the variance stabilized counts (Fig. 4D). Expression of MT1G, MT1H, MT1E, FFAR3, SEMA5A, IGFBP3, and MEOX1was found to be decreased in the CARD9 c.1434 + 1G > C homozygous and CARD9 null patients (unstimulated and HKC-treated T cells), while IL-31 expression was found to be significantly lower only in the HKC-stimulated T cells from the CARD9 null patient. IL-31, mainly produced by Th2 cells but also expressed by epithelial cells, is involved in the physical and antimicrobial barrier function of the skin [28].
The Role of CARD9 in B Cells
PBMCs from both CARD9 c.1434 + 1G > C heterozygous and homozygous family members, the CARD9 null patient, and her heterozygous brother were tested for ex-vivo plasmablast differentiation and immunoglobulin production upon CD40L and IL-21 stimulation. Both, the heterozygous and homozygous CARD9 c.1434 + 1G > C individuals, as well as the CARD9 null patient, showed markedly decreased in vitro CD19+CD27+CD38+ plasmablast differentiation when compared to NCs (Fig. 5A). The in vitro immunoglobulin production showed a decreased IgG/IgA secretion in all individuals tested when compared to the NCs, although the effect was more striking in CARD9 c.1434 + 1G > C homozygous and the CARD9 null patient. While IgM production was less consistently affected, it correlated with IgM plasma levels as the heterozygous father (I.1) and mother (I.2) had the highest, and the homozygous siblings II.2 and II.3 the lowest levels for both test (Fig. 5B and Table 1). As all individuals tested in these experiments had CD19+ B cells within normal limits for age, the initial absolute number of B cells seems unlikely to have influenced the final results. On the other hand, the starting B cell subset distribution, as well as their functional response to the stimuli applied and linked to their genetic background, are more likely causes to have determined the differences detected. Altogether, these results suggest that CARD9 c.1434 + 1G > C behaves in a likely autosomal recessive manner in relation to plasmablast differentiation and Ig generation from PBMCs.
Fig. 5.
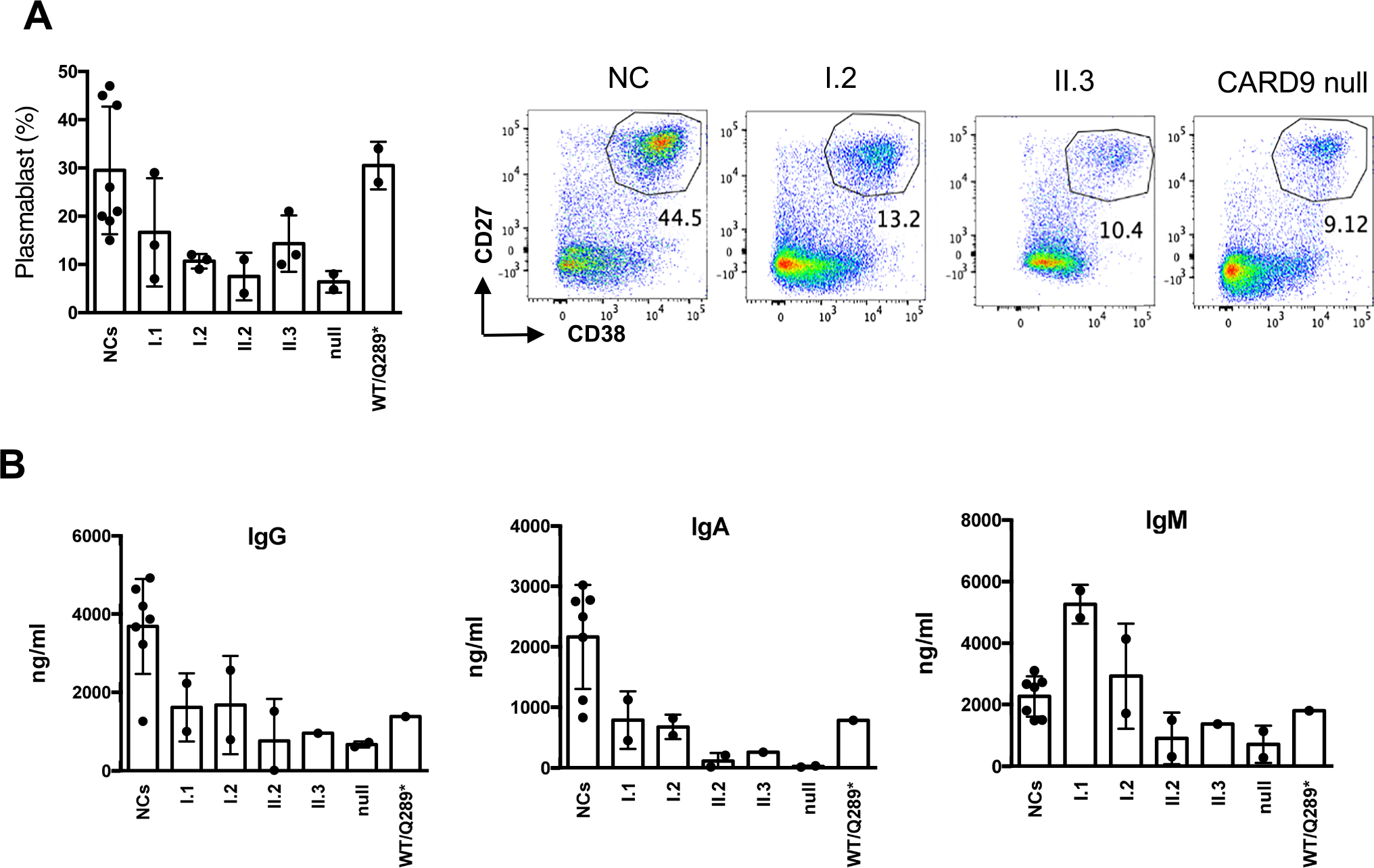
The effect of CARD9 on B cells. A PBMCs were stimulated with CD40L (100 ng/ml) together with IL-21(100 ng/ml) for 5 days, followed by cell surface staining with antibodies as indicated. A scatter plot of percentages of plasmablasts (% of CD27+CD38+ population from CD19+ B cells) and representative flowcytometric dot plots are shown. The values are expressed as the mean ± SD. B The culture supernatant was saved from the simulated samples from (A) and used for the immunoglobulin production assay. Scatter plot to show IgG, IgA, and IgM immunoglobulin (ng/ml) production, respectively. Data represent mean ± SD from 7 different healthy normal controls (NCs). Each dot in the research subjects indicates values from replicates
Discussion
CARD9 deficiency is an autosomal recessive primary immune deficiency caused by biallelic (homozygous or compound heterozygous) loss-of-function mutations [11–19, 29]. CARD9 deficiency can manifest at any age in life, as persistent, life-threatening, superficial, and/or invasive Candida or dermatophyte fungal infections [15, 29, 30]. Besides the abovementioned genotype/phenotype correlation, CARD9 variants c.1434 + 1G > C has been described to be associated with an IBD-protective effect [24]. Single patient case reports have also associated CARD9 c.1434 + 1G > C to specific infections with one heterozygous patient diagnosed with a nontuberculous mycobacterial pulmonary infection [31], whereas another patient with a homozygous CARD9 c.1434 + 1G > C variant plus a homozygous MYD88 in-frame deletion leading to no protein expression presenting with recurrent severe pyogenic bacterial infections and persistent EBV viremia [32]. Two reportedly healthy homozygotes have been detected among non-Finnish Europeans in gnomAD. However, in-depth molecular and functional characterization of CARD9 c.1434 + 1G > C in either heterozygous or homozygous state has not been reported. Herein, we evaluated two siblings with biallelic CARD9 c.1434 + 1G > C mutations (II.2 and II.3), one of them (II.2) showing increased mucosal Candida infection susceptibility along with hypogammaglobulinemia of 2 isotypes and low memory B cells consistent with a CVID-like phenotype, and the other one (II.3) asymptomatic by the age of 5 years. Of note, it has been well established that manifestations associated with CARD9 deficiency could have late onset and present beyond the 4th decade of life [10]; therefore, a close follow up, as well as antifungal prophylaxis, has been recommended for the symptomatic, as well as the asymptomatic sibling.
As previously reported, CARD9 deficiency has a limited impact on lymphocyte development and function [4, 29, 33]. The fact that the index case in our family presented with CMC, a CVID-like B cell phenotype, and carried a rare homozygous CARD9 variant, prompted us to study the biologic role of this mutation in myeloid and also lymphoid cells. We identified CARD9 protein expression in enriched T and B lymphocytes in both humans and mice, although protein accumulation was reduced compared to monocytes. Our study also provides the first lymphoid lineage-oriented CARD9 function evaluation in humans, although evidence suggestive of this fact could be inferred from previously published data. Ishikuza et al. [34] explored the role of Card9 in mice linked to Streptococcus pneumoniae pneumonia, a common pathogen in human humoral immunodeficiencies [35, 36]. The bronchoalveolar lavage (BAL) of Card9-deficient mice intratracheally-infected with S. pneumoniae demonstrated fewer neutrophils, and reduced TNF-a (greek), CXCL1 and MIP-2 production when compared to control and Dectin2 knockout mice. Moreover, production of TNF-α and CXCL1 by alveolar macrophages was also reduced, which indicates a role of Card9 in myeloid cell biology and neutrophil migration. Interestingly, production of anti-PPS3 IgG3, a S. pneumoniae infection neutralizing factor (that the authors linked to reduced IFNγ production) was decreased only in Card9 knockout mice. These results suggest that in addition to the myeloid effect, a B cell defect is also associated with Card9 deficiency. When Uematsu et al. [37] evaluated Card9 deficiency in a murine influenza pneumonia model, loss of Card9 (Card9−/−) was associated with a markedly attenuated clinical phenotype resulting from a reduced local (i.e., BAL) inflammatory cytokine and chemokine production while IFNγ was significantly increased contrasting with the S. pneumoniae model results [34]. However, when white blood cells (WBC) were evaluated in the BAL of infected mice, significantly decreased neutrophils and T cell numbers were detected in the Card9-deficiency. These results show that Card9 −/− animals produce a deficient/less robust immune response to particular viral infections resulting from myeloid and T cell defects associated with Card9 deficiency [34, 37]. More recently, Doron et al. found that gut-induced antifungal IgG generation, a humoral mechanism protective from systemic Candida albicans and Candida auris infection, was CARD9 dependent. The authors concluded that this mechanism was exclusively controlled by CARD9+CX3CR1+ macrophages, but no T or B cells, as they only detected CARD9 transcripts in myeloid cells based on single-cell RNAseq analysis of human biopsies [38]. Noteworthy, CARD9 expression has been detected in human NC T cells by Chiriaco et al., without specific comment by the authors [32].
Gene dosage refers to the variation, as well as the correlation, between a particular gene copy number (affected or not by mutations/allelic variants), the production of its encoded protein, its function, and the impact of these variations in cell biology [39–41]. CARD9 is transcribed and expressed as a biallelic trait and is involved in (a) a canonical CBM-dependent signaling that engages the NF-kB inflammatory/anti-infectious pathway [4, 5, 42], and (b) a non-canonical CBM-independent signaling through the CARD9/G-RAF1/H-RAS axis, that following dectin ligand/receptor interaction and p-Erk and p-p38 activation contributes to antifungal immunity [7, 8]. In this context, we focused our analysis of CARD9 gene dosage in relation to fungal infection immunity, inflammatory responses, and lymphocyte biology by analyzing these functions in NCs, patients with monoallelic/heterozygous and biallelic/homozygous c.1434 + 1 G > C splicing mutations, a patient with biallelic/compound heterozygous null (Q289*/D8Afs*10) variants (CARD9 null), and (when sample availability permitted), her monoallelic/heterozygous brother (WT/Q289*) (CARD9 haploinsufficiency). While NCs and the CARD9 null patient defined the definitive boundaries for these functions, the biallelic and monoallelic splice mutant carriers, as well as the CARD9 haploinsufficiency individual, would provide the “shades of gray” for each of the evaluated functions.
Interestingly, each of the CARD9-related mechanisms tested displayed a particular and independent type of functional outcome linked to the underlying gene dosage/mutation condition that could also be associated with its clinical impact:
Fungal, but no LPS-dependent cytokine production by PBMCs/myeloid cells was decreased in the CARD9 c.1434 + 1G > C heterozygous condition in similar to the homozygous and the CARD9 null patients, supporting a dominant negative effect for this variant in this particular function that could also explain the protective effect for IBD.
Th17 commitment was affected in all gene dosage conditions tested with the exception of the haploinsufficient individual (i.e., WT/Q289*) and correlated with increased fungal infection susceptibility in the proband. However, Th1 commitment seemed more affected in the CARD9 c.1434 + 1 G > C homozygous and CARD9 null patients, suggesting a Th1 additive role to fungal control. In terms of T cell gene regulation upon fungal stimulation, the CARD9 c.1434 + 1 G > C heterozygous cells were indistinguishable from the NCs, the CARD9 null patient showed a marked defect, and the CARD9 c.1434 + 1 G > C homozygous cells presented an intermediate defect. Taken together, these findings suggest that one WT allele was sufficient to support a relatively normal fungal response.
When CARD9-related B cell functions were evaluated ex-vivo, plasmablast differentiation was impaired in all gene dosage conditions tested with the exception of the haploinsufficient individual (i.e., WT/Q289*), suggesting that for this readout, one CARD9 WT allele was sufficient to sustain complete function. On the other hand, ex-vivo IgG and IgA production were more markedly affected in the CARD9 c.1434 + 1G > C homozygous and CARD9 null patients, which correlate with the serum immunoglobulin levels found in proband II.2 and the CARD9 null patient (the latter had a partial IgA deficiency).
While the T and B cell role, if any, in the clinical phenotype of patients with CARD9 deficiency is overshadowed by the major myeloid impairments, when analyzed in detail, particular lymphocyte defects have been detected among these patients. Corvilain et al. found that Candida spp.-mediated lymphocyte proliferation was defective in 3 out of 7 patients tested in the largest series of CARD9 deficient patients published (n = 58) [29]. Moreover, in addition to the CARD9 deficient patients presented herein (i.e., II.1, II.2, and CARD9 null), another four patients were evaluated at NIH. Among them, one patient had low IgG levels (486 mg/dL at age 10) and another had non-protective antibody titers to tetanus toxoid, mumps, and rubella despite an adequate vaccination schedule (unpublished data).
In summary, while exploring the biology associated with the CARD9 c.1434 + 1 G > C mutation identified in a patient with features of CMC and CVID, we were able to establish the central role of CARD9 exon 11 in the conformation of the CBM complex, which includes CARD9, BCL10, MALT1, as well as TRIM62. Moreover, our data demonstrates that CARD9/Card9 is indeed expressed in T and B cells from both humans and mice, and CARD9 defects can affect particular T and B cell functions (in addition to the well-established myeloid defects) in a gene dosage manner depending on the gene burden (e.g., haploinsufficiency) or genetic mutation effect (e.g., dominant negative). Altogether, this allows for the speculation that T and B cells might have a contributory role on the clinical phenotype of CARD9 deficiency.
Supplementary Material
Acknowledgements
We thank the patients and their families for their contributions to the study. These studies were supported by the Intramural Research Program, NIH Clinical Center, US National Institutes of Health (NIH); the French National Research Agency (ANR) under the “Investments for the Future” program (ANR-10-IAHU-01), the ANR-FNS LTh-MSMD-CMCD (ANR-18-CE93-0008-01), The Rockefeller University, and the National Institutes of Health (# R01AI127564). The content of this article does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
Funding
These studies were supported by the Intramural Research Program, NIH Clinical Center, US National Institutes of Health (NIH); the French National Research Agency (ANR) under the “Investments for the Future” program (ANR-10-IAHU-01), the ANR-FNS LTh-MSMD-CMCD (ANR-18-CE93-0008-01), The Rockefeller University, and the National Institutes of Health (# R01AI127564).
Abbreviations
- BAL
Bronchoalveolar lavage
- BCL10
B cell lymphoma 10
- CARD9
Caspase recruitment domain-containing protein 9
- CBM
CARD9/BCL10/MALT1
- CM
Central memory
- CNS
Central nervous system
- CVID
Common variable immunodeficiency
- GWAS
Genome-wide association studies
- HKC
Heat-killed Candida
- MALT1
Mucosal-associated lymphoid tissue lymphoma translocation protein 1
- IBD
Inflammatory bowel disease
- NCs
Normal controls
- nt
Nucleotides
- PBMCs
Peripheral blood mononuclear cells
- PHA
Phytohemagglutinin
- RNASeq
RNA sequencing assay
- TRIM62
Tripartite motif containing-62
- WBC
White blood cells
- WES
Whole exome sequencing
- WT
Wild type
Footnotes
Declarations
Conflict of Interest The authors declare no conflict of interest.
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s10875-021-01173-6.
References
- 1.Bertin J, Guo Y, Wang L, Srinivasula SM, Jacobson MD, Poyet J-L, et al. CARD9 is a novel caspase recruitment domain-containing protein that interacts with BCL10/CLAP and activates NF-κB. J Biol Chem. 2000;275(52):41082–6. [DOI] [PubMed] [Google Scholar]
- 2.Hsu Y-MS, Zhang Y, You Y, Wang D, Li H, Duramad O, et al. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat Immunol. 2007;8(2):198–205. [DOI] [PubMed] [Google Scholar]
- 3.Roth S, Ruland J. Caspase recruitment domain-containing protein 9 signaling in innate immunity and inflammation. Trends Immunol. 2013;34(6):243–50. [DOI] [PubMed] [Google Scholar]
- 4.Hara H, Ishihara C, Takeuchi A, Imanishi T, Xue L, Morris SW, et al. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat Immunol. 2007;8(6):619–29. [DOI] [PubMed] [Google Scholar]
- 5.Hara H, Saito T. CARD9 versus CARMA1 in innate and adaptive immunity. Trends Immunol. 2009;30(5):234–42. [DOI] [PubMed] [Google Scholar]
- 6.Zhong X, Chen B, Yang L, Yang Z. Molecular and physiological roles of the adaptor protein CARD9 in immunity. Cell Death Dis. 2018;9(2):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gavino C, Hamel N, Zeng JB, Legault C, Guiot M-C, Chankowsky J, et al. Impaired RASGRF1/ERK–mediated GM-CSF response characterizes CARD9 deficiency in French-Canadians. J Allergy Clin Immunol. 2016;137(4):1178–88.e7. [DOI] [PubMed] [Google Scholar]
- 8.Jia X-M, Tang B, Zhu L-L, Liu Y-H, Zhao X-Q, Gorjestani S, et al. CARD9 mediates Dectin-1–induced ERK activation by linking Ras-GRF1 to H-Ras for antifungal immunity. J Exp Med. 2014;211(11):2307–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drummond RA, Swamydas M, Oikonomou V, Zhai B, Dambuza IM, Schaefer BC, et al. CARD9(+) microglia promote antifungal immunity via IL-1beta- and CXCL1-mediated neutrophil recruitment. Nat Immunol. 2019;20(5):559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glocker E-O, Hennigs A, Nabavi M, Schäffer AA, Woellner C, Salzer U, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. 2009;361(18):1727–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lanternier F, Pathan S, Vincent QB, Liu L, Cypowyj S, Prando C, et al. Deep dermatophytosis and inherited CARD9 deficiency. N Engl J Med. 2013;369(18):1704–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drummond RA, Collar AL, Swamydas M, Rodriguez CA, Lim JK, Mendez LM, et al. CARD9-dependent neutrophil recruitment protects against fungal invasion of the central nervous system. PLoS Pathog. 2015;11(12):e1005293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drummond RA, Lionakis MS. Mechanistic insights into the role of C-type lectin receptor/CARD9 signaling in human antifungal immunity. Front Cell Infect Microbiol. 2016;6:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lanternier F, Barbati E, Meinzer U, Liu L, Pedergnana V, Migaud M, et al. Inherited CARD9 deficiency in 2 unrelated patients with invasive Exophiala infection. J Infect Dis. 2015;211(8):1241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lanternier F, Mahdaviani SA, Barbati E, Chaussade H, Koumar Y, Levy R, et al. Inherited CARD9 deficiency in otherwise healthy children and adults with Candida species–induced meningoencephalitis, colitis, or both. J Allergy Clin Immunol. 2015;135(6):1558–68.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gavino C, Cotter A, Lichtenstein D, Lejtenyi D, Fortin C, Legault C, et al. CARD9 deficiency and spontaneous central nervous system candidiasis: complete clinical remission with GM-CSF therapy. Clin Infect Dis. 2014;59(1):81–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grumach AS, de Queiroz-Telles F, Migaud M, Lanternier F, Rosario Filho N, Palma SM, et al. A homozygous CARD9 mutation in a Brazilian patient with deep dermatophytosis. J Clin Immunol. 2015;35(5):486–90. [DOI] [PubMed] [Google Scholar]
- 18.Drewniak A, Gazendam RP, Tool AT, van Houdt M, Jansen MH, van Hamme JL, et al. Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood. 2013;121(13):2385–92. [DOI] [PubMed] [Google Scholar]
- 19.Arango-Franco CA, Moncada-Vélez M, Beltrán CP, Berrío I, Mogollón C, Restrepo A, et al. Early-onset invasive infection due to Corynespora cassiicola associated with compound heterozygous CARD9 mutations in a Colombian patient. J Clin Immunol. 2018;38(7):794–803. [DOI] [PubMed] [Google Scholar]
- 20.Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449(7164):851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42(12):1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McGovern DP, Gardet A, Törkvist L, Goyette P, Essers J, Taylor KD, et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet. 2010;42(4):332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burghardt KM, Avinashi V, Kosar C, Xu W, Wales PW, Avitzur Y, et al. A CARD9 polymorphism is associated with decreased likelihood of persistent conjugated hyperbilirubinemia in intestinal failure. PLoS One. 2014;9(1):e85915. 10.1371/journal.pone.0085915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43(11):1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao Z, Conway KL, Heath RJ, Rush JS, Leshchiner ES, Ramirez-Ortiz ZG, et al. Ubiquitin ligase TRIM62 regulates CARD9-mediated anti-fungal immunity and intestinal inflammation. Immunity. 2015;43(4):715–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beaudoin M, Goyette P, Boucher G, Lo KS, Rivas MA, Stevens C, et al. Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet. 2013;9(9):e1003723. 10.1371/journal.pgen.1003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanel KH, Pfaff CM, Cornelissen C, Amann PM, Marquardt Y, Czaja K, et al. Control of the physical and antimicrobial skin barrier by an IL-31-IL-1 Signaling Network. J Immunol. 2016;196(8):3233–44. [DOI] [PubMed] [Google Scholar]
- 29.Corvilain E, Casanova J-L, Puel A. Inherited CARD9 deficiency: invasive disease caused by ascomycete fungi in previously healthy children and adults. J Clin Immunol. 2018;38(6):656–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Puel A Human inborn errors of immunity underlying superficial or invasive candidiasis. Hum Genet. 2020;139:1011–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szymanski EP, Leung JM, Fowler CJ, Haney C, Hsu AP, Chen F, et al. Pulmonary nontuberculous mycobacterial infection. A multisystem, multigenic disease. Am J Respir Crit Care Med. 2015;192(5):618–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiriaco M, Di Matteo G, Conti F, Petricone D, De Luca M, Di Cesare S, et al. First Case of Patient With Two Homozygous Mutations in MYD88 and CARD9 Genes Presenting With Pyogenic Bacterial Infections, Elevated IgE, and Persistent EBV Viremia. Front Immunol. 2019;10:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Diego RP, Sánchez-Ramón S, López-Collazo E, Martínez-Barricarte R, Cubillos-Zapata C, Cerdán AF, et al. Genetic errors of the human caspase recruitment domain–B-cell lymphoma 10–mucosa-associated lymphoid tissue lymphoma-translocation gene 1 (CBM) complex: Molecular, immunologic, and clinical heterogeneity. J Allergy Clin Immunol. 2015;136(5):1139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishizuka S, Yokoyama R, Sato K, Shiroma R, Nakahira A, Yamamoto H, et al. Effect of CARD9 deficiency on neutrophil-mediated host defense against pulmonary infection with Streptococcus pneumoniae. Infect Immun. 2020;89:e00305–20. 10.1128/IAI.00305-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuehn HS, Boisson B, Cunningham-Rundles C, Reichenbach J, Stray-Pedersen A, Gelfand EW, et al. Loss of B Cells in Patients with Heterozygous Mutations in IKAROS. N Engl J Med. 2016;374(11):1032–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saffran DC, Parolini O, Fitch-Hilgenberg ME, Rawlings DJ, Afar DE, Witte ON, et al. Brief report: a point mutation in the SH2 domain of Bruton’s tyrosine kinase in atypical X-linked agammaglobulinemia. N Engl J Med. 1994;330(21):1488–91. [DOI] [PubMed] [Google Scholar]
- 37.Uematsu T, Iizasa E, Kobayashi N, Yoshida H, Hara H. Loss of CARD9-mediated innate activation attenuates severe influenza pneumonia without compromising host viral immunity. Sci Rep. 2015;5:17577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doron I, Leonardi I, Li XV, Fiers WD, Semon A, Bialt-DeCelie M, et al. Human gut mycobiota tune immunity via CARD9-dependent induction of anti-fungal IgG antibodies. Cell. 2021;184(4):1017–31e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rice AM, McLysaght A. Dosage-sensitive genes in evolution and disease. BMC Biol. 2017;15(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Veitia RA, Bottani S, Birchler JA. Cellular reactions to gene dosage imbalance: genomic, transcriptomic and proteomic effects. Trends Genet. 2008;24(8):390–7. [DOI] [PubMed] [Google Scholar]
- 41.Fisher E, Scambler P. Human haploinsufficiency—one for sorrow, two for joy. Nat Genet. 1994;7(1):5. [DOI] [PubMed] [Google Scholar]
- 42.De Bruyne M, Hoste L, Bogaert DJ, Van den Bossche L, Tavernier SJ, Parthoens E, et al. A CARD9 founder mutation disrupts NF-κB signaling by inhibiting BCL10 and MALT1 recruitment and signalosome formation. Front Immunol. 2018;9:2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schatorje EJ, Gemen EF, Driessen GJ, Leuvenink J, van Hout RW, de Vries E. Paediatric reference values for the peripheral T cell compartment. Scand J Immunol. 2012;75(4):436–44. [DOI] [PubMed] [Google Scholar]
- 44.Morbach H, Eichhorn EM, Liese JG, Girschick HJ. Reference values for B cell subpopulations from infancy to adulthood. Clin Exp Immunol. 2010;162(2):271–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.