

SUMMARY
Sorghum is an important food and feed crop globally; its production is hampered by anthracnose disease caused by the fungal pathogen Colletotrichum sublineola (Cs). Here, we report identification and characterization of ANTHRACNOSE RESISTANCE GENE 2 (ARG2) encoding a nucleotide‐binding leucine‐rich repeat (NLR) protein that confers race‐specific resistance to Cs strains. ARG2 is one of a cluster of several NLR genes initially identified in the sorghum differential line SC328C that is resistant to some Cs strains. This cluster shows structural and copy number variations in different sorghum genotypes. Different sorghum lines carrying independent ARG2 alleles provided the genetic validation for the identity of the ARG2 gene. ARG2 expression is induced by Cs, and chitin induces ARG2 expression in resistant but not in susceptible lines. ARG2‐mediated resistance is accompanied by higher expression of defense and secondary metabolite genes at early stages of infection, and anthocyanin and zeatin metabolisms are upregulated in resistant plants. Interestingly, ARG2 localizes to the plasma membrane when transiently expressed in Nicotiana benthamiana. Importantly, ARG2 plants produced higher shoot dry matter than near‐isogenic lines carrying the susceptible allele suggesting an absence of an ARG2 associated growth trade‐off. Furthermore, ARG2‐mediated resistance is stable at a wide range of temperatures. Our observations open avenues for resistance breeding and for dissecting mechanisms of resistance.
Keywords: sorghum [Sorghum bicolor (L.) Moench], anthracnose, NLRs, Colletotrichum sublineola, fungi, immunity
Significance Statement
Sorghum is a crop of global importance that is used for food, feed and biofuel. The crop feeds millions of people, especially small holder framers around the world. The productivity of sorghum is constrained by anthracnose diseases caused by the fungus Colletotrichum sublineola. Genetic resistance provides the most sustainable disease control method. We identified and characterized a sorghum anthracnose resistance gene encoding a nucleotide‐binding leucine‐rich repeat protein by exploring natural variation in the crop. Our findings will have significant implications for improvement of disease resistance in sorghum, as well as for future studies in the molecular genetics of pathogen resistance.
INTRODUCTION
Sorghum [Sorghum bicolor (L.) Moench] is a crop of global importance that is used for food, feed and biofuel. The productivity of sorghum is constrained by anthracnose diseases caused by the fungus Colletotrichum sublineola (Cs), which results in significant crop losses (Little & Perumal, 2019; Thakur, 2007). Although sorghum germplasm for disease resistance is widely available, the molecular genetics of anthracnose resistance has not been well studied. Generally, plant immune pathways are placed into two categories based primarily on pathogen‐derived molecules that are perceived and the kinetics of immune responses (Dangl et al., 2000). Recognition of pathogen derived signature molecules, pathogen associated molecular patterns (PAMPs), at the cell surface is mediated by pattern recognition receptors, which initiates pattern triggered immunity (PTI). PTI is associated with varying degrees of quantitative resistance (Godfrey & Rathjen, 2001). Accumulation of phytoalexin 3‐deoxyanthocyanidin at the site of infection has been linked to quantitative resistance to Cs (Cui et al., 2015; Lo, De Verdier, & Nicholson, 1999; Tenkouano et al., 1993). The second‐teir immunity pathway is mediated through the intracellular recognition of pathogen effectors by nucleotide‐binding leucine‐rich repeat (NLR) proteins, which initiate effector trigger immunity (ETI), an effective and often highly specific disease resistance (Rose et al., 2004). ETI is heightened immunity accompanied by hypersensitive response (HR) (Boyes et al., 1998). The highly polymorphic nature of NLRs and the strong selection pressure on pathogens drives an arms race between plants and pathogens. Resistance to anthracnose involves both qualitative traits controlled by major genes conferring strain‐specific resistance, as well as quantitative traits that involve many genes (Nelson et al., 2018; Patil et al., 2017). At the molecular level, ETI and PTI mechanisms are known to be mutually interdependent (Pruitt et al., 2021).
NLR genes are widely used in resistance breeding of crop plants and have been studied in different systems (Lagudah & Periyannan, 2018; Wang et al., 2017). The high penetrance of NLR regulated resistance makes resistance breeding easier, although it is prone to overcome by virulent pathogens. The sorghum genome encodes hundreds of putative NLRs, an estimated 97% of which occur in clusters (Cheng et al., 2010). By contrast, quantitative resistance is known to be durable and broad‐spectrum but confers only partial resistance, resulting in a better yield in infected fields rather than complete prevention of disease (St. Clair, 2010). Sorghum lines with race‐specific and quantitative anthracnose resistance have been reported (Burrell et al., 2015; Cuevas et al., 2014; Felderhoff et al., 2016; Patil et al., 2017). However, studies focused on the overall genetic architecture of resistance rather than the identity of individual resistance genes (Nelson et al., 2018).
The maize and Colletotrichum graminicola interaction is very similar to sorghum and Cs interactions that cause similar leaf blight and stalk rot diseases. The maize Rcg1 and Rcg1b genes encode a NLR protein that confers resistance to anthracnose leaf blight and stem rot caused by C. graminicola (Ma et al., 2022). Similarly, the interaction between the rice blast fungus (Magnaporthe oryzae) and rice (Oryza sativa) appears to closely mirror sorghum interaction with Cs. In both cases, the infection starts with a biotrophic phase followed by a necrotrophic phase. The genetic control and molecular mechanisms of resistance to rice blast disease have been well studied. More than 30 rice blast resistance genes and 12 M. oryzae effectors have been identified (Wang et al., 2017). Most of these rice genes encode NLRs that confer race‐specific resistance. There have been few studies on molecular mechanisms and specific genes that regulate sorghum resistance to this pathogen. Recently, the ARG1 gene encoding a sorghum NLR was shown to confer broad‐spectrum resistance to anthracnose (Lee et al., 2022). In addition, the global mRNA and microRNA expression dynamics in response to anthracnose define components of sorghum responses to the pathogen (Fu et al., 2020).
The present study was initiated to identify sorghum anthracnose resistance genes and determine their function. We report the identification of the ANTHRACNOSE RESISTANCE GENE 2 (ARG2) gene, encoding a canonical NLR protein that confers complete resistance to some Cs strains. The identification of ARG2 is supported by multiple and independent dominant and recessive ARG2 alleles in various sorghum lines that show resistance or susceptible responses, respectively, to Cs strains. The genomic and molecular characterization of ARG2 locus was performed, and the impact of ARG2 on defense gene expression, accumulation of secondary metabolites, pathogen resistance at elevated temperatures and defense growth tradeoff was determined.
RESULTS
Identification of resistant sorghum lines
Twenty‐five sorghum lines were screened using multiple strains of Cs to identify resistant genotypes (Table S1). One of these lines, SC328C, displayed a clear‐cut resistance to Cs strains Csgl1 and Csgrg and susceptibility to strains Csgl2, Cs27 and Cs29. SC328C is part of a collection of sorghum differential lines used for strain identification, and its resistance to other strains of Cs was reported in previous studies (Moore et al., 2008; Prom et al., 2012). As a result of the strong resistance phenotype in SC328C, we studied the genetic control of this resistance in detail.
The responses of the resistant line SC328C were compared with the susceptible line TAM428 after inoculation with the Csgl1 strain. In the detached leaf disease assay, the site of inoculated SC328C leaf tissue turns brown and remains intact and asymptomatic, whereas, TAM428 plants show severe disease symptoms in 7 days (Figure 1a). Staining of the inoculated tissue with WGA‐AF488, which stains the fungal hyphae (Redkar et al., 2018), revealed proliferous acervuli on TAM428 but not on SC328C (Figure 1b). Similarly, trypan blue staining of infected leaf tissue reveals a dense mass of mycelia on TAM428, whereas no pathogen growth occurred on SC328C (Figure S1). After spray inoculation, the susceptible line TAM428 shows widespread disease lesions and enhanced pathogen growth that often leads to complete collapse of infected leaves, whereas SC328C shows HR responses (Figure 1c). Similar to TAM428, the sorghum line BTx623 is susceptible to Csgl1, whereas Rio shows resistance response similar to SC328C (Figure 1c). In spray inoculated susceptible lines, pronounced disease symptom appears on the leaf sheath, and this leaf tissue often tears apart along the leaf venation (Figure 1d). At a temperature as high as 38°C, SC328C and the widely known resistant line SC748 show resistance (Figure S1), whereas the susceptible line BTx623 showed disease symptom (Figure S1). Detached TAM428 leaves did not survive the high temperature.
Figure 1.

Disease symptoms and fungal growth in sorghum lines.
(a) Disease symptom and resistance responses at 7 days after drop‐inoculation (dai) of Cs strain Csgl1 conidial suspension (20 μl, 1 × 106 conidia ml−1) on detached leaves.
(b) Fungal growth in drop inoculated leaves. Inoculated tissues with Cs Csgl1 were stained WGA‐AF488 and propidium iodide at 6 dai and examined using a confocal microscope. WGA‐AF488 (green) stains fungal hyphae and propidium iodide (red) stains dead host cells. Pathogen acervuli are visible on TAM428 but not on SC328C, which shows no pathogen growth.
(c) Disease symptoms on whole plants at 14 days after spray inoculation. The conidial suspension (Cs Csgl1; 1 × 106 conidia ml−1) was uniformly spray inoculated on sorghum plants.
(d) Disease symptom and resistance response on leaf sheath at 15 days after spray inoculation of the whole plant.
A single dominant locus in SC328C regulates resistance
To determine the inheritance of anthracnose resistance in SC328C to Cs, SC328C was crossed with two susceptible lines, TAM428 and BTx623 (Table S1). The F1 progenies of both crosses were resistant, as were the F1 progenies of the reciprocal SC328C × TAM428 cross (Figure 2a). The F2 progenies derived from self‐fertilization of the BTx623 × SC328C F1 plants resulted in 298 resistant and 91 susceptible progenies, which fitted to a 3:1 (χ2 = 0.464, P > 0.05) phenotypic ratio. Similarly, evaluation of 1131 F2 progenies of the TAM428 × SC328C cross resulted in 853 resistant and 278 susceptible plants that also fitted well to a 3:1 (χ2 = 0.704, P > 0.05) phenotypic ratio. The disease symptoms on BTx623 appeared later and were less severe compared to that of TAM428 (Figure 1c). Therefore, we used the TAM428 × SC328C population for gene identification because of the enhanced penetrance of the disease response phenotypes in this genetic background. The parental lines and progenies showed consistent disease responses to the two Cs strains Csgl1 and Csgrg, suggesting that the resistance to these two Cs fungal strains is regulated by the same locus. All the above genetic analyses strongly suggested that a single dominant locus in SC328C confers resistance to both strains, hereafter, designated as ANTHRACNOSE RESISTANCE GENE2 (ARG2).
Figure 2.
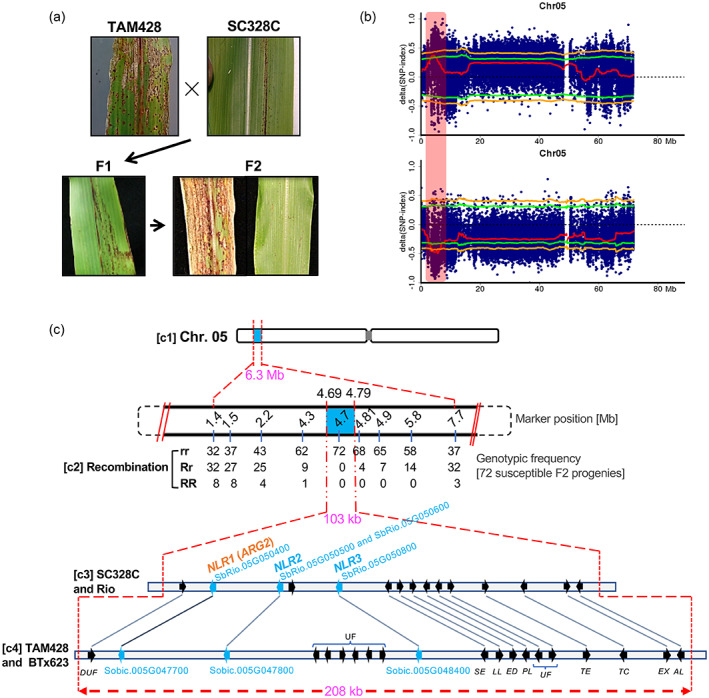
A diagrammatic summary of ARG2 genetic mapping.
(a) Disease symptom and resistance response on parents, F1 progenies and F2 progenies of the ARG2 mapping population.
(b) BSA‐Seq result showing ARG2 locus with respect to the resistant (upper) and the susceptible (lower) parental lines. The x‐axis is genomic coordinate and the y‐axis is ∆SNP‐index estimate. Each blue dot (highly overlapped) represents ∆SNP‐index estimate in the 2‐Mb sliding window and the red line is the ∆SNP‐index threshold. The green line shows a significance threshold (P = 0.05) of the ∆SNP‐index, with the orange line at P = 0.01. The vertical rectangular bar (light‐red shaded) marks the significant ARG2 mapping region.
(c) ARG2 fine mapping. (c1) ARG2 mapping region (6.3 Mb) as identified using BSA‐Seq. (c2) Genetic linkage analysis. The row of numbers (upper panel) are positions of indel markers that were used to generate the genotypic frequencies (c2, lower panel). (c3) The 103‐kb ARG2 mapping interval defined by the recombination analysis that carries 15 putative genes in the Rio reference genome. (c4) The corresponding genomic interval in the BTx623 reference genome (208 Mb) carries 20 putative genes. The short arrows in these two ARG2 genomic intervals show the relative position and orientation of the genes with the homologs linked using lines. The gene annotations are DUF, domain of unknown function; UF, unknown function; NLR, NBS‐LRR; SE, serine esterase; LL, limkain B Lkap; ED, Nad dependent epimerase/dehydratase; PL, TRNA‐nucleotidyl transferase/poly A polymerase family member; TE, TRANSCRIPTION ELONGATION FACTOR B POLYPEPTIDE 3; TC, CCR4‐NOT transcription complex subunit; EX, exocyst subunit EXO70 family protein C1‐related. ARG2 is NLR1 as identified later.
To identify the genomic region that carries ARG2, composite interval mapping was conducted using the bulked segregant analysis by sequencing (BSA‐Seq) (Takagi et al., 2013). Using F2 samples that are pooled based on responses to the pathogen, BSA‐Seq identifies the target locus that is expected to carry the causal gene as opposed to the random allelic frequency in the rest of the genome. Consistent with the above genetic data, BSA‐Seq analysis in F2 identified a single genomic region around the proximal end of chromosome 5 that was strongly associated with ARG2 resistance (Figure 2b,c). In BSA‐Seq, the statistical measure used to determine the causal genomic region of the disease phenotype is a ∆SNP‐index (Table S2), which is expressed as the difference in the average single nucleotide polymorphism (SNP) frequencies between the resistant and the susceptible pooled‐progeny genomic samples. ∆SNP‐indices were estimated using a 2‐Mb window that slides 50 kb at a time. The significant ∆SNP‐index estimate at 95% confidence interval (CI) ranged from 0.36 to 0.50; with the highest threshold estimate of 0.35 at 95% CI and 0.41 at 99% CI. The significant genomic interval was > 3.6 Mb in the 95% CI and approximately 2.8 Mb in the 99% CI. Within the 95% CI, the smallest depth of aligned raw‐reads ranged from 24.5 to 28.6 (mean = 26.2) and the SNP count was in the range 1067–2872 (mean = 1999) (Figure S2). This mapping interval carries more than 100 putative genes.
Fine mapping of the ARG2 gene
To narrow the ARG2 mapping interval that is generated using BSA‐Seq, genetic linkage analyses were conducted using indel markers for polymerase chain reaction (PCR) (Figure 2c). Linkage analysis was conducted using 72 susceptible F2 progenies, which carry the homozygous recessive allele of ARG2. This analysis resulted in a convergent gradient of allelic frequencies around the ARG2 locus that narrowed the ARG2 mapping interval to approximately 0.51 Mb, which is between the coordinates 4.3 Mb and 4.81 Mb in the Rio reference genome (Figure 2c). Additional genotyping of 336 F2 progenies within this 0.51 Mb interval showed complete co‐segregation between ARG2 phenotypic and genotypic data, although additional recombinant progeny was not found. With further recombinant analysis, two more recombinants were identified, one at 4.79 Mb in a homozygous resistant progeny and the other at 4.69 Mb in the ARG2‐near‐isogenic lines (NILs). In sum, linkage analyses resulted in a 103 kb ARG2 mapping interval in the Rio reference genome that is flanked by markers at 4686 kb and 4789 kb and the putative genes SbRio.05G050200 (4682 kb) and SbRio.05G051800 (4792 kb). In the BTx623 reference genome, this mapping interval is wider, 208 kb, which is between 4492 kb and 4700 kb, and the corresponding flanking genes are Sobic.005G047300 (at 4487 kb) and Sobic.005G049350 (at 4702 kb). The difference in the ARG2 mapping interval between Rio and BTx623 is similar between SC328C and TAM428 as well (Figure 2c).
Identification of a candidate ARG2 gene
Throughout the ARG2 mapping interval, the integrated genome view of the parental genome alignment map [binary alignment map (BAM) files] showed both striking sequence similarities and disparities among the four lines of sorghum: BTx623, TAM428, SC328C and Rio (Figures S3 and S4). Rio is a sweet sorghum line that produces high vegetative biomass (Cooper et al., 2019; Davila‐Gomez et al., 2011), whereas SC328C has a short stature and produces a large panicle. Despite these differences, both SC328C and Rio are resistant and carry nearly identical genomic sequences in the 103 kb ARG2 mapping interval, and TAM428 and BTx623 are susceptible and carry highly similar genomic sequences in their 208 kb ARG2 mapping interval. Thus, whole genome sequences of the Rio and BTx623 reference genomes were leveraged for the ARG2 fine mapping.
In these two ARG2 mapping intervals described above, the entire genomic sequence and putative genes were examined for sequence differences between the resistant and susceptible lines and previously known gene functions. The 103 kb ARG2 mapping interval carries 14 putative genes (Figure 2c), of which four genes (SbRio.05G050400, SbRio.05G050500, SbRio.05G050600 and SbRio.05G050800) encode putative nucleotide‐binding site leucine‐rich repeat (NB‐LRR, NLR) proteins with a N‐terminal coiled‐coil (CC) domain, CC‐NB‐LRR. These four NLR genes show high sequence similarity to each other, which caused ambiguous alignments to the reference genomes; parental DNA raw‐reads map to more than one of these gene regions (Figures S3 and S4). The other ten genes are identical or almost identical between the parental genomes and do not show any high impact SNP (Table S3).
ARG2 is inherited as a dominant allele, suggesting that resistance is likely conferred by a functional allele. Accordingly, we first searched for candidate genes that are intact in the resistant parent and are disrupted or altered in the susceptible parent. NLR3 (SbRio.05G050800) carries a > 9 kb transposon insertion that separates part of the CC domain and the promoter region from the rest of the gene and thus it is not likely to be functional (Figure 3). NLR2, which is predicted as two genes, SbRio.05G050600 and SbRio.05G050500, was found to encode a single NLR protein. In the resistant lines SC328C and Rio, NLR2 carries a 442 bp duplication in the middle of the gene, which disrupts the open reading frame (ORF) and results in mis‐annotation of the single gene as two separate genes. We amplified a single transcript that spans these two putative genes as verified by sequencing. In the susceptible lines BTx623 and TAM428, there is no duplicate segment in the middle of NLR2 and the prediction is a single gene in the BTx623 reference genome. We amplified a single NLR2 transcript from both susceptible lines as well, as verified by sequencing. Thus, similar to NLR3, NLR2 is also not likely to be functional in the resistant lines. Similarly, comparative genomic (CoGe) analysis of these three highly similar NLR genes indicated that two of the three NLR genes, NIR1 and NLR2, are disrupted in SC328C and Rio, leaving NLR1 (SbRio.05G050400) as the only viable candidate ARG2 gene (Figure 3a).
Figure 3.

Comparative genomics (CoGe) of ARG2 duplicate genes and sequence variation of ARG2 in parental lines.
(a) CoGe view of the three duplicate NLR genes in the ARG2 mapping interval using the Rio reference genome. The three panels show the three duplicate genes, and the colored connectors between the panels show homologous segments. NLR2 (in the middle panel) was predicted as two genes (SbRio.05G050500 and SbRio.05G050600), although these two putative genes are parts of a single gene and code for a single transcript. The dark green overlapping connector between NLR1 and NLR2 (marked with asterisk) shows a 442‐bp segment of NLR1 that is a duplicate in NLR2. The bottom panel shows a big transposon insertion (TE in NLR3) that disrupted the upstream gene region of NLR3.
(b) The integrated genomic view (igv) of NLR1 transcript raw‐reads of the parental lines of ARG2 mapping population that are aligned to the Rio reference genome. These sequences were obtained using WideSeq. The numbers on top are genomic coordinates of ARG2. The vertical distance of the gray area shows the depth of aligned reads, which ranges from 700 to > 7000. The uniformly gray area shows that aligned reads of SC328C are identical to the reference genome, and the SNPs in TAM428 are shown by red, orange, blue and purple vertical lines in the gray background. The gene regions in the TAM428 panel that does not show aligned reads (no gray area) are (iv) a 62 bp indel and (iii and v) exon segments with dense SNPs that prohibited alignment. The remaining no‐gray regions (i, ii and vi) are intronic segments and (vii) part of the 3′ UTR that was not included in the sequencing. The gene structure (below the panels) is the putative NLR1 gene in the reference genome.
(c) Aligned coding sequences that carry the premature stop codon in the susceptible parent.
(d) A snapshot of the top ARG2 homologs in rice, barley and wheat that were identified using CoGe. The different color connectors between the panels show the homologous segment between genes in the different species. (i) O. sativa has a cluster of three highly homologous genes, one of them has gene ID LOC_Os06g17880 (Apoptic ATPase, LRR annotations similar to ARG2). (iii) H. vulgare (barley) homolog has the gene ID HORVU7Hr1G006400. (iv) The homology with top homolog in T. aestivum covers 99% peptide segment, gene ID Traes_7DS_9881A234E [RPM1, Apoptic ATPase, NB‐ARC, LRR annotations].
Transcript analysis of these three NLR genes in the resistant parent supported the above structural genomic analyses (Figure 3b,c). Transcripts of NLR1 and NLR2 were amplified using gene‐specific primers in the untranslated regions (UTRs) and the coding sequences were cloned. However, in the resistant line, there is no transcript from NLR3, and the transcript from NLR2 carries a premature stop codon near the predicted translation start site, and downstream of the 442 bp duplication. Thus, NLR2 and NLR3 are disrupted in the resistant parent, and NLR1 is the only likely ARG2 candidate gene. NLR1 has a gene ID SbRio.05G050400 in the Rio v2.1 and Sobic.005G047700 in the BTx623 v3.1.1 reference genomes in the phytozome database. Because NLR1 is intact in the resistant parent, we refer to this as a wild‐type allele, whereas the gene in the susceptible parent carries a premature stop codon and hence is referred to as a mutant non‐functional allele. CoGe shows genes with strong homology to ARG2 in grasses, including a cluster of three NLR genes in rice (Figure 3d).
ARG2 has undergone numerous genomic rearrangements
The ARG2 locus contains a cluster of three duplicate NLR genes that have undergone many structural rearrangements, including high impact indels (Figures 2c and 3a). In the susceptible lines TAM428 and BTx623, these three duplicate genes are interspersed with several other putative genes (Figure 2c) within the 208 kb ARG2 mapping interval. By contrast to the resistant lines, the susceptible lines do not have the 442 bp duplicate segment in NLR2 and the large insertion in NLR3 (Figure 3a). The resistant ARG2 allele harbors a 480 bp intron in the 5' UTR region, and its transcript carries a 71codon upstream ORF (Figure 4a). One of the splice sites of this intron has an SNP, CT/CC in the wild‐type and CA/CC in TAM428 and BTx623, that eliminates the canonical splice site present in the resistant allele. Nevertheless, this intron carries many SNPs and three small indel sites, and the transcripts in TAM428 and BTx623 have another common splice site in that 480 bp intron, which is TGAT in the wild‐type, and TAGGAT in TAM428 and BTx623 (Figure 4a).
Figure 4.
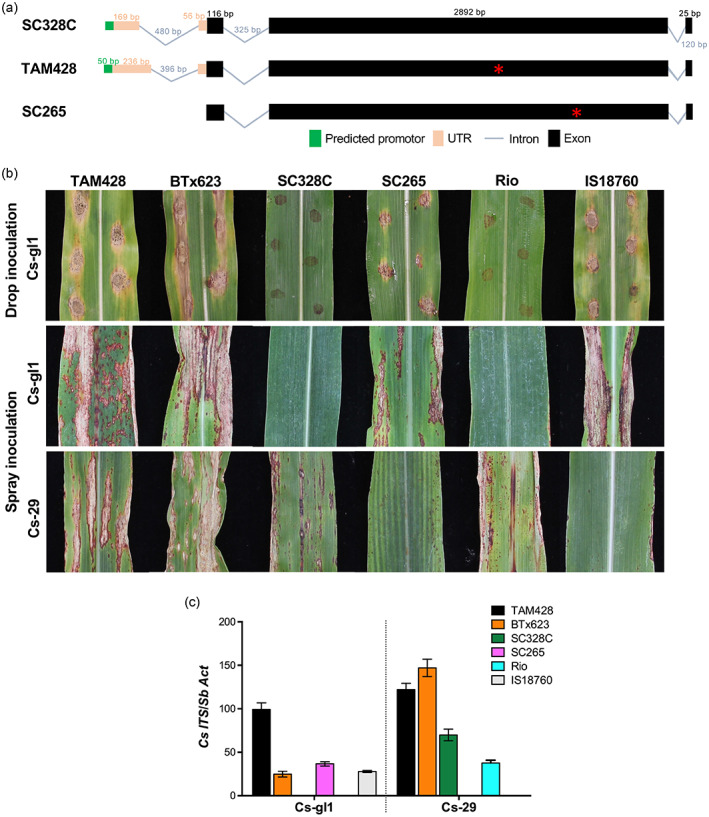
Genomic structure of ARG2 lines, disease symptoms and pathogen growth.
(a) ARG2 domain structure and variations in the upstream cis‐regulatory elements of the resistant and susceptible ARG2 alleles. ARG2 harbors three exons with a complete ORF in SC328C, whereas the allele in TAM428 and SC265 carry different premature stop codons. ARG2 shows structural rearrangement in the 5′ UTR between the parental lines of ARG2 mapping population.
(b) Sorghum lines that show differential responses to Cs strains Csgl1 and Cs29. TAM428 and BTx623 are susceptible to both strains of Cs. All images were taken 7 days after inoculation. (c) Growth of Cs strain on sorghum lines carrying different ARG2 alleles.
Validation of ARG2 with independent loss of function alleles
To validate the single ARG2 candidate gene, NLR1, we assessed sorghum lines with an emphasis on susceptible ones for identification of independent ARG2 loss of function alleles. In addition to phenotypic evaluations, extensive assessment of NLR1 was conducted using BAM files of publicly available genomic data. Subsequently, NLR1 transcripts were amplified from 11 representative sorghum lines that are susceptible to Csgl1 (SC265, TAM428, BTx642, SC23, IS9830, PQ‐434, IS18760, ICSV745, IS8525, Malisor84‐7 and BTx623) and four resistant sorghum lines (SC328C, Keller, SC237 and Rio) and the complete coding sequences of these NLR1 alleles were sequenced using WideSeq. There was no amplification of the NLR1 transcript and genomic sequence in the susceptible sorghum lines Greenleaf and IS3121 (Table S4; Data S1). The coding sequences in all the 15 sorghum lines were clustered into four similarity groups based on sequence identity, the presence of a complete ORF and the position of the premature stop codon (Data S2). The four resistant sorghum lines carry identical ARG2 coding sequences showing no sequence variation. By contrast, the coding sequences in the susceptible lines were clustered into three types: six lines carry the TAM428 allele, four lines carry the BTx623 allele and SC265 carries a unique allele. Figure 4b shows the disease phenotype of selected sorghum lines carrying the representative four different alleles after inoculation with Csgl1 and Cs29. SC328C and Rio show contrasting strain‐specific responses to Csgl1 and Cs29 as compared to SC265 and IS18760. TAM428 and BTx623 show an increased disease lesion size and disease symptoms to both Csgl1 and Cs29 (Figure 4b).
The IS18760 and SC265 sorghum lines are susceptible to Csgl1 and resistance to Cs29. Conversely, SC328C and Rio were free of disease symptoms caused by Csgl1 after drop and spray inoculations. Quantitative PCR (qPCR) using primers on the fungal‐specific nuclear ribosomal DNA internal transcribed spacer showed no fungal accumulation in SC328C and Rio plants inoculated with Csgl1 (Figure 4c). However, both resistant lines SC328C and Rio showed increased disease symptoms and fungal growth when inoculated with Cs29 (Figure 4b,c). These data confirmed the robust but strain‐specific resistance in SC328C and Rio to Csgl1.
NLR1 in TAM428 and SC265 carries premature stop codons at different positions, and sequence comparisons support independent divergence of these two alleles (Figure S5; Data S1 and S2). Compared with the wild‐type ARG2, the allele in SC265 carries < 8% SNPs whereas the allele in TAM428 carries > 10% SNPs. The amino acid sequence differences were also compared based on the shortest ORF, which is 1848 bp in the TAM428 allele (Data S3). Compared with the wild‐type ARG2, in this shortest ORF, the SC265 ARG2 allele carries 4% non‐synonymous codons, whereas the allele in TAM428 carries 14% non‐synonymous codons. This ORF in SC265 carries about 15% non‐synonymous codon compared to the one in TAM428. Thus, the sequence divergence between the susceptible alleles in SC265 and TAM428 is greater than their divergence from the resistance allele. These data show that the mutant allele in the susceptible lines TAM428 and SC265 are distinct because the mutation that inactivated the gene in SC265 must have occurred after the divergence of TAM428 from the resistance alleles in SC328C and Rio. In sum, the NLR1 allele in SC265 is an independent mutant allele that validates NLR1 as the functional ARG2 gene that was defined genetically.
The susceptible BTx623 carries a recessive allele of ARG2 but retains a complete ORF similar to the resistance allele in SC328C. The segregation of ARG2 alleles in the BTx623 × SC328C population also suggested the single locus resistance that is inherited as a dominant allele. To understand the mutant ARG2 allele in BTx623 that carries the complete ORF, nucleotide sequence analysis was carried out (Data S2). Consistent with loss of function in BTx623 and TAM428, their ARG2 sequences are more closely related to each other than either is to the resistance allele. In the LRR domain that is crucial for pathogen recognition, compared with the resistance allele, the allele in BTx623 shows approximately 19% non‐synonymous amino acid substitutions (Data S4). This sequence difference in the LRR domain includes two indels (2 and 4 bp) that flank a 45 bp coding sequence resulting in a reading frame shift of 15 amino acids. Regardless of the complete ORF, our sequence analyses supports the loss of ARG2 resistance in BTx623 to the Cs strains Csgl1 and Csgrg to which the allele in SC328C and Rio confers resistance.
The amino acid sequences of ARG2 proteins in SC328C and BTx623 were compared with the HOPZ‐ACTIVATED RESISTANCE 1 (ZAR1; template c6j5tC) due to amino acid sequence similarity to ARG2 and the availability of solved structure. This comparison revealed that the tryptophan at the 156th amino acid in SC328C is substituted with Serine in BTx623 (Data S2). A mutation from a conserved tryptophan to alanine at the 150th amino acid position of ZAR1 reduces NLR protein oligomerization and consequent cell death, resulting in the increased susceptibility of Arabidopsis to Xanthomonas campestris pv. campestris (Wang et al., 2019). To uncover more potential amino acids important for ARG2 function, we aligned sorghum ARG2 from SC283C and BTx623 with the top 100 monocot and dicot homologs. The amino acids in ARG2 that are highly conserved in the monocot and dicot ARG2 homologs also show a striking difference between the SC328C and BTx623 alleles (Table S5). We found that the conserved amino acids of ARG2 in SC328C are changed in BTx623 at positions 48, 103, 225, 242, 480 and 866 amino acids (Table S5). These changes in conserved amino acids imply impaired ARG2 function in BTx623. Particularly, valine (Val) at the 225th amino acid that is highly conserved both in monocots and dicots is retained in the resistant line SC328C but substituted by leucine (Leu) in the susceptible BTx623 (Table S5). Previously, NLR protein autoactivation was observed when Val at this site is substituted with alanine (van Ooijen et al., 2008).
To further understand differences in ARG2 protein between SC328C and BTx623, their predicted tertiary structures were compared using protein homology/analogy recognition engine v 2.0 (phyre2) (Kelley et al., 2015). These structures were compared with the disease resistance RPP13‐like protein 4 (ZAR1; template c6j5tC), which suggested that the ARG2 protein in SC328C has a unique structure. First, the beta‐sheet structure in BTx623 was replaced by an alpha‐helix structure at the nucleotide‐binding site (NBS) region in SC328C (Navy blue color, Figure S6a). These changes occurred between the subdomain RNBS‐A and Walk B (Data S2) and the substitution of the highly conserved Val by Leu occurred in this RNBS‐A motif. Second, an extra alpha‐helix is present between the conserved ARC1 and ARC2 in SC328C (green, Figure S6b; Data S2). The NBS‐ARC1‐ARC2 structure is important for NLR protein activation through ATP/ADP binding (Tameling et al., 2002). Third, more alpha‐helix was observed around the MHD motif (green, Figure S6c; Data S2). We also predicted the ligand binding site of ARG2 using cofactor and COACH of i‐tasser (Roy et al., 2010). This analysis revealed that there is no difference in ATP binding site residues in the deduced proteins from these two alleles. However, multiple ADP binding site residues between SC328C and BTx623 were altered (Figure S6d). The current model suggests that ARG2 protein bound with ADP is in an inactive form in BTx623 (El Kasmi, 2021). Although the impacts of changes in the tertiary structure have not been reported, apparently, there are clear differences between these two deduced proteins.
Similar to TAM428, BTx623 show increased disease lesion size and disease symptoms to Csgl1 (Figure 4b). qPCR using Cs internal transcribed spacer primers also showed fungal accumulation in BTx623 but not in the SC328C and Rio plants inoculated with Csgl1 (Figure 4c). However, similar to BTx623, the resistant lines SC328C and Rio showed increased disease symptoms and fungal growth when inoculated with Cs29 (Figure 4b,c). These data confirmed the susceptibility of BTx623 to Csgl1, whereas SC328C and Rio show resistance revealing strain‐specific resistance.
The ARG2 resistance allele localizes on the plasma membrane in Nicotiana benthamiana
To gain further insight into how ARG2 recognizes Cs, we investigated the subcellular localization of the resistance allele of ARG2 from SC328C in N. benthamiana (Figure 5). We fused enhanced GFP (mGFP) to the C‐terminus of full‐length ARG2 (ARG2:mGFP) and transiently expressed in N. benthamiana. The subcellular localization was evaluated at 48 h following agroinfiltration. Laser scanning confocal microscopy with ARG2:mGFP revealed an even distribution of mGFP fluorescence predominantly on the cell periphery with no observable localization to cytoplasmic strands, suggesting plasma membrane localization (Figure 5). To determine whether ARG2:mGFP localizes on the plasma membrane, we co‐expressed ARG2:mGFP with an Arabidopsis protein known to localize to the plasma membrane, AtFLS2:mCherry (Helm et al., 2019). The ARG2:mGFP fluorescence signal co‐localized with the AtFLS2‐mCherry fluorescence signal (Figure 5), confirming that ARG2:mGFP is indeed localized on the plasma membrane.
Figure 5.
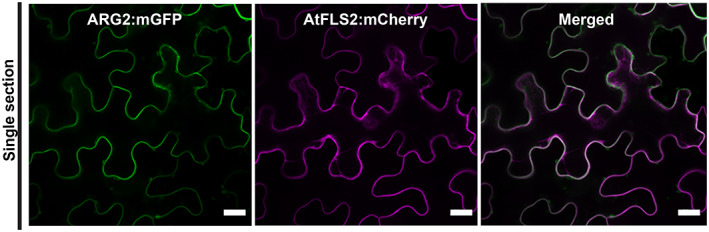
The ARG2 resistance protein, predominately localizes on the plasma membrane.
The indicated constructs were transiently co‐expressed in N. benthamiana and imaged using laser scanning confocal microscopy 48 h post agroinfiltration. mCherry‐tagged Arabidopsis FLS2 was included as a reference for plasma membrane localization (Helm et al., 2019). The mGFP and mCherry fluorophores were excited at 488 nm and 561 nm, respectively. mGFP (green) and mCherry (magenta) fluorescence signals were collected between 525 and 550, and at 610 nm, respectively. Images shown are single optical sections. Scale bar = 20 μm.
ARG2 homologs are widely conserved in monocot and dicot plant lineages
Comparative genome analyses revealed that ARG2 has homologs across monocot and dicot species. To examine the relationship between ARG2 in SC328C and BTx623, and other orthologs in monocots and dicots, phylogenetic analysis was conducted using amino acid sequences (Figure S7). ARG2 protein in SC328C and BTx623 cluster with the other sorghum NLR proteins encoded by the duplicate NLR genes in the ARG2 locus. Despite the widespread homology, except the duplicate genes in the ARG2 locus, the sequence identity of all ARG2 homologs is < 60%. No ARG2 homolog was identified in the maize genome despite their relatedness, and, from other species, Eragrostis curvula harbors the closest ARG2 homolog. Our analysis shows ARG2 is the RPM1 orthologue of sorghum.
Next, we performed a phylogenetic analysis using amino acid sequences of the NB‐ARC domain of ARG2 and functionally characterized NLRs (Kourelis et al., 2021) from diverse plant species. ARG2 clustered in the same clade as the rice resistance genes that confer resistance to the rice blast diseases (Wang et al., 2017) (Figure S8). The phylogenetic analysis displayed a clear separation between ARG1, a recently described sorghum NLR (Lee et al., 2022), and ARG2 (Figure S8).
ARG2 resistance is associated with early activation of defense response genes
To characterize the function of ARG2 with minimal interference from differences in the parental genomes, sorghum NILs were generated by crossing TAM428 and SC328C followed by backcrosses (Figure 6a). The resistant and susceptible NILs inherited the clear‐cut difference in disease symptom and pathogen growth of the parental lines (Figure 6b). Expression of eleven defense response genes, selected based on previous studies (Li et al., 2013; Lo, Hipskind, & Nicholson, 1999), was examined in the NILs using quantitative real‐time PCR (qRT‐PCR). Higher expression of flavon O‐methyltransferase (FOMT), chalcone synthase 08 (CHS8) and pathogenesis related gene 10 (PR10) was associated with resistance at early time points after inoculation (Figure 6c). At later time points, the susceptible NILs showed higher levels of expression, which is associated with elevated pathogen growth. These results suggest, in the presence of ARG2, increased expression of a subset of previously reported sorghum anthracnose immune response genes which may contribute to differences in resistance of susceptibility.
Figure 6.
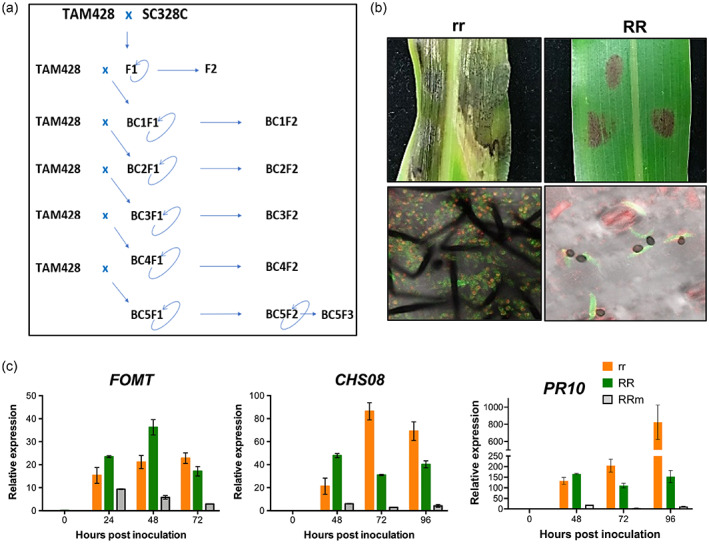
Defense gene expression in near‐isogenic lines of sorghum that differ in the ARG2 locus.
(a) Schematics showing the steps used to generate the NILs. The NILs carry the recessive (susceptible) arg2 allele in TAM428 (arg2/arg2, rr) and dominant (resistance) ARG2 allele (ARG2/ARG2, RR).
(b) Disease symptom and fungal growth in resistant NILs (RR) and susceptible NILs (rr).
(c) Expression of three defense response genes in the resistant NILs (RR) and the susceptible NILs (rr) after pathogen or mock inoculation. FOMT, flavon‐O methyltransferase; CHS8, chalcone synthase 08; PR10, pathogenesis related gene 10; hpi, hours post inoculation. Fungal growth on the inoculated tissue was examined using a confocal microscope at 6 days post inoculation (dpi). WGA‐AF488 stains fungal hyphae green and propidium iodide stains dead host cell red. The image was taken at 10 dpi in the detached‐leaf disease assay.
Expression of the ARG2 alleles in resistant and susceptible lines
To shed light on ARG2 function, we examined expression of ARG2 in response to pathogen inoculation and chitin treatment, as well as in different plant tissues and growth stages, using qRT‐PCR (Figure 7). In the resistant line SC328C, the flag leaf showed the highest ARG2 expression. In the leaf sheath and leaf blade, ARG2 showed relatively low expression at the two earlier sorghum growth stages, and ARG2 expression was hardly detectable in the stalk and the panicle tissues (Figure 7a). SC328C is resistant to Csgl1 and susceptible to Cs29, whereas BTx623 is susceptible to both strains. ARG2 expression was significantly increased at 24 h post inoculation (hpi) during both resistant and susceptible interactions but, at 48 hpi, its expression was strongly induced by Csgl1 in SC328C, but was unchanged in response to Cs29 (Figure 7b). In SC328C, Csgl1 induced expression of ARG2 was significantly higher than that in response to Cs29. BTx623 showed slightly induced ARG2 expression in response to both Csgl1 and Cs29 at 48 hpi, but was lower than in SC328C. Our results indicate that ARG2 in SC328C is induced significantly more by Csgl1 to which it confers resistance. The expression of ARG2 in SC328C was significantly induced in response to chitin treatment compared to the mock treated samples at 48 hpi (Figure 7c). Chitin is a fungal‐derived PAMP that is known to elicit basal resistance and ARG2 is a typical regulator of ETI. The induction of the resistance gene by fungal PAMP is consistent with the mutual interdependency between PTI and ETI (Pruitt et al., 2021) and potentiation of ETI by a general fungal elicitor. Consistent with this, we did not observe chitin induced expression of the ARG2 allele in the chitin‐treated BTx623 plants that are susceptible to the different Cs strains.
Figure 7.
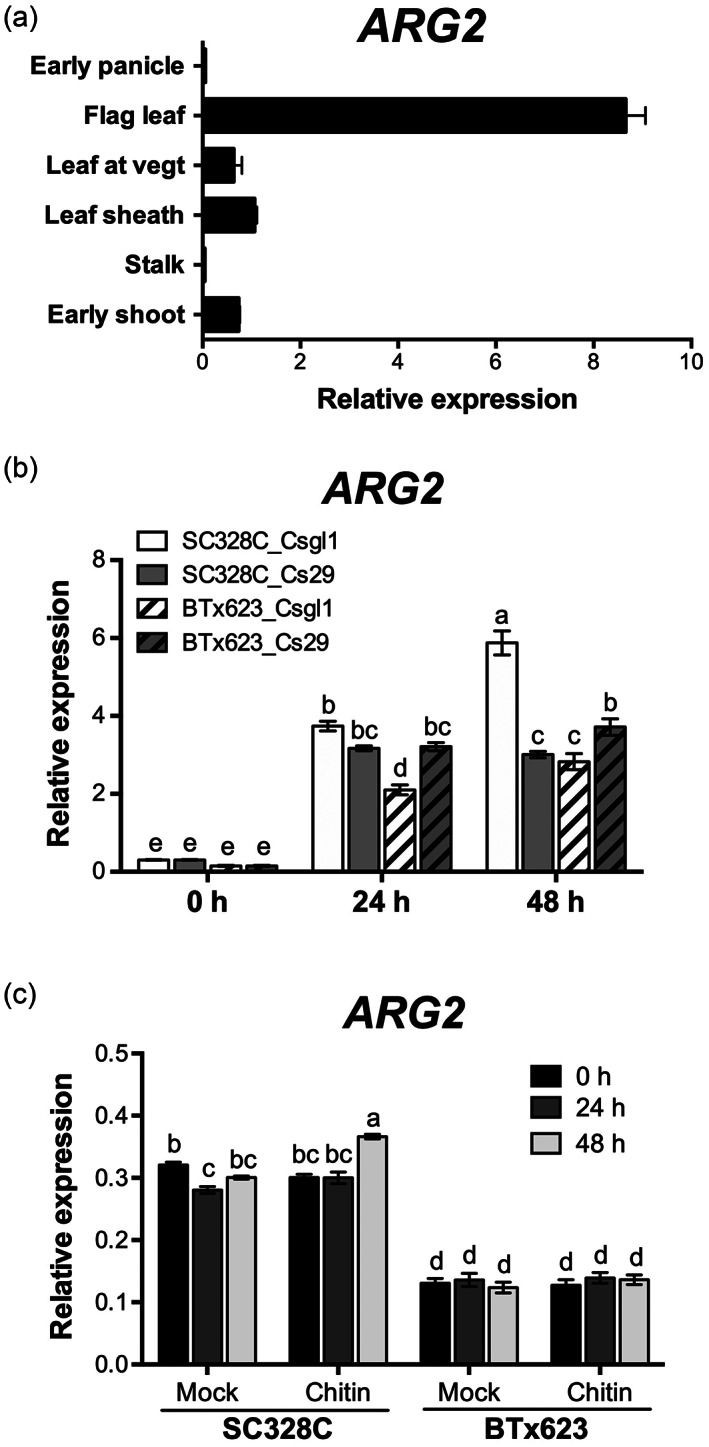
Regulation of ARG2 gene expression. (a) ARG2 expression in different tissues and growth stages in SC328C.
The tissue samples are the entire shoot at 10 days after planting (dap); the stalk, the leaf sheath and the leaf (leaf at vegt) all at 30 dap; the flag leaf at early flowering; and the panicle at early panicle development. Data are the mean ± SD from triplicate tissues. Expression level was analyzed using qRT‐PCR, and data were normalized by the comparative cycle threshold method with actin as the internal control. (b) ARG2 expression in response to two different strains of Cs. SC328C and BTx623 plants were inoculated with Cs strains Csgl1 or Cs29, and RNA was extracted at the indicated time points. (c) ARG2 expression in response to chitin. The leaf tissues of SC328C and BTx623 were treated with mock (water) or 2 nm chitin (β‐1,4 linked N‐acetylglucosamine) and RNA was extracted at the indicated time points. In (b,c), expression levels were analyzed using qRT‐PCR, and data were normalized by the comparative cycle threshold method with actin as the internal control. Data show the mean ± SE from three independent biological replicates (P < 0.05, Tukey–Kramer honestly significant difference).
Metabolite profiling data support ARG2 resistance function
To gain insight into pathogen induced accumulation of secondary metabolites that may contribute to ARG2‐mediated resistance, untargeted metabolite profiling was carried out using tissue samples from pathogen and mock inoculated ARG2 NILs. In total, 2916 preprocessed metabolite mass‐features were obtained: 1356 in the negative ionization mode and 1560 in the positive ionization mode. Regardless of genotypic difference, both in the resistant and susceptible NILs, many mass‐features were detected only after pathogen inoculation. However, none of these mass‐features were detected exclusively in Cs inoculated resistant NILs. In total, 474 mass‐features showed significant (P < 0.05) difference between the resistant and the susceptible NILs after Cs inoculation (Table S6a). Only 57 of these mass‐features showed higher accumulation in the resistant NILs, and the remaining 417 mass‐features showed higher accumulation in the susceptible NILs (Table S6a).
MS ‘peak to pathways’ analysis using the MetPA pipeline showed a total of 86 metabolite hits in 43 pathways (Table S6b). Most of these metabolite hits show the mass‐features carry abducts similar to H2O or a carbonate salt in addition to ionization feature (Table S6c). Six relatively highly enriched pathways are shown in Figure S9a. Zeatin and anthocyanin metabolisms showed the highest score upregulation in the Cs inoculated resistant NILs, followed by mock inoculated resistant NILs. By contrast, Cs inoculated resistant NILs showed downregulation of metabolites that are involved in lysine, tryptophan, arginine and proline metabolisms. Porphyrin and chlorophyll metabolism were downregulated only in Cs inoculated resistant NILs.
ARG2 resistance is not associated with growth tradeoff
The common genomic background in the ARG2 NILs was leveraged to examine the impact of the ARG2 locus on plant growth under controlled growth conditions in the absence of the pathogen (Figure 8; Figure S9b–f). Lower leaves of the resistant parent SC328C turn brownish at the vegetative growth stage and part of the panicle remains covered by the leaf sheath (Figure S10a,b), although none of these undesirable agronomic features are seen in the NILs. Significantly higher panicle and total dry matter weight were obtained in the true breeding resistant ARG2 NILs compared to the NILs that carry the recessive homozygote and heterozygote genotypes (Figure 8a). Total leaf area, photochemical reflectance index and nitrogen reflectance index at 97 and 119 days after planting revealed significant differences between the NILs (Figure 8b–d). In the true breeding resistant NILs, the shoot growth indicator morphometric and color related traits (Vyska et al., 2016; Zhu et al., 2021) and top‐plant‐surface and side‐average‐convex‐hull were also higher in most plant growth stages (Figure 8e,f). Furthermore, anthocyanin reflectance index and plant senescence reflectance index differed between the NILs (Figure S9c,d). Some color (hue) related traits showed relatively higher indices in the true breeding resistant NILs in the vegetative stage that is reversed in the reproductive stage (Figure S9e), suggesting that traits relatedness may vary with growth stage and a specific growth stage may not sufficiently explain a major aspect of the differences among the resistance and the susceptible NILs. Nitrogen reflectance index showed a negative correlation with most of these plant growth, anthocyanin and senescence traits (Figure S9f).
Figure 8.
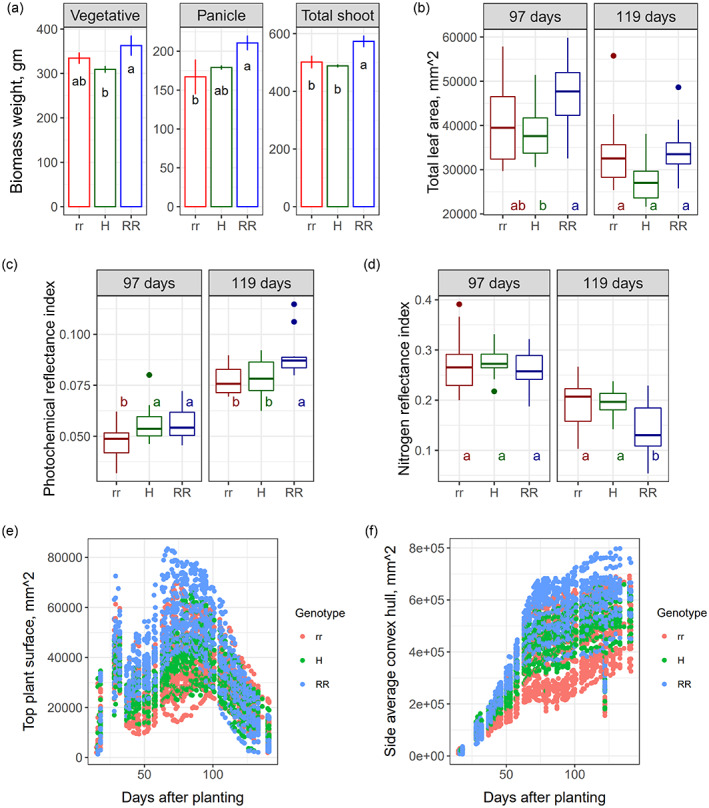
Effect of ARG2 on plant growth in the absence of disease in a controlled environment phenotyping facility.
(a) Shoot dry matter weight (gm) among ARG2 genotypes. rr, homozygote recessive; H, heterozygote; RR, homozygote dominant. Shoot dry matter is higher (P < 0.002) in RR; panicle dry matter is lower in rr (P < 0.02); and vegetative shoot dry matter is lower (P < 0.015) in the heterozygote, H.
(b) Total leaf area and (c) photochemical reflectance and (d) nitrogen reflectance indices at flowering time [97 days after planting (dap)] and grain filling stage (119 dap), based on hyperspectral image. Estimates of (e) top plant surface and (f) side average convex hull throughout the growth period. rr, homozygote recessive; H, heterozygote; RR, homozygote dominant genotypes. (b–f) Estimate of physiological and morphometric traits at flowering time (97 dap) and grain filling stage (119 dap) based on red–green–blue (RGB) and hyperspectral image data throughout the growth period. anova and Tukey honestly significant difference functions were used to test statistical significance of differences among genotypes.
DISCUSSION
Here, we report the identification and characterization of sorghum ARG2, encoding a NLR protein. ARG2 was identified based on a biparental F2 mapping population developed by crossing the Cs resistant sorghum line SC328C and the susceptible line TAM428. The resistance parent showed complete resistance to the Cs strains Csgl1 and Csgrg but susceptibility to other strains. SC328C is one of the sorghum differential cultivars used for pathogen‐strain identification (Moore et al., 2008; Prom et al., 2012). However, until now, a resistance locus in SC328C against any Cs strain had not been identified. In the present study, a single ARG2 candidate was identified through genetic mapping, as well as sequence comparisons between multiple and distinct ARG2 alleles and the corresponding plant responses. For example, the ARG2 locus is identical in the sweet sorghum Rio and the dwarf grain sorghum SC328C, consistent with their mutual resistance to Csgl1 and Csgrg, but despite major differences in the genetic backgrounds of these genotypes. In addition, multiple loss of functional alleles, including those in the susceptible TAM428 and SC265 lines, that do not encode the predicted ORF support the conclusion that loss of ARG2 is responsible for a loss of resistance. Previously, a minor effect anthracnose resistance QTL was mapped in the genomic region containing the ARG2 locus based on the susceptible BTx623 and resistant SC748‐5 biparental mapping population (Burrell et al., 2015). Resistance in that case was determined using fungal strains that are different from those in the present study. BTx623 and SC748‐5 both carry the same susceptible arg2 allele, and hence the QTL in SC748‐5 likely defined a different locus. Interestingly, ARG2 mediated resistance maintains effectiveness up to 38°C. Surprisingly, despite the reported growth and defense trade‐off associated with resistance genes (Vyska et al., 2016), with or without disease, the NILs that carry the resistance ARG2 allele grew better than the NILs that do not carry the resistance allele. Each of these observations suggests that the trade‐off between growth and disease resistance is ameliorated in this case, making this gene a particularly useful one for introgression.
ARG2 is found within a complex locus that contains NLR genes that have both copy number and structural variations. The ARG2 locus carries > 4 putative NLR genes in the genome of the sorghum line RTx430, and three copies in BTx623 and Rio genomes. Comparative studies revealed > 17 000 copy number variants in sorghum (Zheng et al., 2011) and hundreds of genome‐specific loci in rice (Schatz et al., 2014). Despite the complex nature of the ARG2 locus, the premature stop codon in NLR2 and the absence of a transcript from NLR3 reduced the genomic complexity and allowed us to determine ARG2. This is in addition to multiple independent arg2 recessive alleles that demonstrate loss of resistance.
Support for the role of ARG2 in disease resistance is provided by independent ARG2 alleles in distinct sorghum lines. Furthermore, ARG2 expression shows differences between the resistant SC328C and the susceptible BTx623 lines. Thus, multiple lines of evidence suggest functional differences between ARG2 from SC328C and BTx623. Given that, the ARG2 allele in BTx623 may be useful for a broader understanding of the mechanism of NLR action and evolution of NLR resistance and evolution of NLR specificity. Interestingly, although the ARG2 allele in BTx623 has an intact ORF, it does not confer resistance to the Cs strains Csgl1 and Csgrg. There is, however, enormous sequence divergence relative to the resistant ARG2 allele in SC328C. Multiple differences in conserved and critical amino acid residues and in predicted structures of ARG2 proteins from SC328C and BTx623 may account for functional differences between the two alleles. The LRR‐domain of the BTx623 ARG2 allele shows 19% amino acid sequence divergence relative to the SC328C ARG2 allele, which may have caused loss of Cs‐effector recognition from strains avirulent on SC328C. In addition, multiple ADP binding site residues show differences between SC328C and BTx623 and such differences are known to affect function of NLRs (Takken et al., 2006; Williams et al., 2011). The most critical differences include changes that disrupt NLR oligomerization and thus do not trigger cell death, resulting in disease susceptibility (Wang et al., 2020). In sorghum, a high proportion of large effect SNPs are in the LRR domain (Zheng et al., 2011). To our knowledge, there is no Cs strain to which BTx623 confers resistance. Although BTx623 is susceptible to all five Cs strains tested in the present study and to many more strains in earlier studies (Klein et al., 2017; Prom et al., 2012), the presence of a complete ORF suggests that ARG2 in BTx623 might still confer resistance to other strains (Panchy et al., 2016).
Sequence comparison using CoGe (Figure 3d) and blastp identified monocot and dicot homologs of ARG2. However, the sequence similarities of all the homologs, except that of the NLR genes in the ARG2 locus, are less than 60%. This is not unusual among NLR genes that are subject to frequent positive selection (Van de Weyer et al., 2019). A phylogenetic analysis of functionally characterized NLRs including ARG2 reveals that ARG2 clusters (Van de Weyer et al., 2019) with rice Pti2, Pti9, PigmR/PigmS and Piz‐t resistance genes, which confer resistance to rice blast (Wang et al., 2017). As opposed to the closely related genomes of sorghum and maize, comparative genomic studies in grasses show maize carries by far the smallest number of NLR genes (Yang & Wang, 2016). The absence of maize ARG2‐homolog is not surprising considering the relatively low number of maize NLR genes, and the high rate that NLR genes can be dispensed off the genome (Van de Weyer et al., 2019) along with genome instability. Finally, NLR‐mediated resistance is known to be modulated by temperature (Bao et al., 2014). The stability of ARG2 at high temperature and absence of resistance‐growth tradeoff suggests that ARG2 confers a particularly useful form of resistance. ARG2 confers race‐specific resistance; thus, identification of the Cs effector recognized by ARG2 will advance molecular dissection of resistance to Cs.
EXPERIMENTAL PROCEDURES
Plant materials and fungal culture
To identify resistant lines, a collection of sorghum lines (Table S1) was screened for resistance to Cs strains Csgl1, Csgl2, Csgrg, Cs27 and Cs29. Csgl1 and Csgl2 were collected in Indiana (USA) (Jamil & Nicholson, 1987). Most of the sorghum lines are differential lines defined as resistant or susceptible based on their reaction to a collection of prevalent Cs strains in the USA (Moore et al., 2008; Prom et al., 2012). Csgrg was a recent collection from Tiffon, Georgia (USA), and Cs29 and Cs27 were from Western Ethiopia. The fungal strains were cultured on potato dextrose agar (BD Difco, Franklin Lakes, NJ, USA). Fungal culture, spore collection and disease assays were conducted as described recently (Lee et al., 2022).
DNA extraction and sequencing
From the F2 mapping population of TAM428 × SC328C cross, 50 resistant and 50 susceptible plants were used to extract and sequence genomic DNA for mapping the genomic region of ARG2. DNA extraction was carried out using the DNeasy plant mini kit (Qiagen, Hilden, Germany). The quality and concentration of the DNA samples were determined using gel electrophoresis and Nanodrop (Thermo Fisher Scientific, Waltham, MA, USA). An equal amount of DNA of individual F2 plants was used to constitute two pooled DNA samples: one for the susceptible group and the other for the resistant group. The pooled samples were sequenced at 25× coverage using a HiSeq 2500 system (Illumina Inc., San Diego, CA, USA).
BSA‐Seq and fine‐mapping
Bulk‐segregant analysis of the genomic DNA raw‐reads (BSA‐Seq) was carried out using the QTL‐seq pipeline (Takagi et al., 2013) to identify the genomic region that confers resistance in SC328C to Csgl1. This pipeline uses the pooled‐genomic raw‐reads to locate the genomic regions that are significantly associated with the resistance. All of the default statistical parameters as specified in the pipeline were used. The BTx623 and the sweet sorghum Rio reference genomes were used to align the corresponding raw‐reads of the pooled samples. The basis of this analysis is the difference between the average frequency of SNPs in the pooled samples (∆SNP‐index) that are inherited from the two parental lines. In the genomic regions that are associated with the disease phenotype, the allelic frequency in the F2 pooled‐progeny samples is expected to be significantly different from random. Thus, ∆SNP‐indices that result from the random assortment of alleles approximate zero, whereas ∆SNP‐indices around the genomic regions that are significantly associated with the disease phenotype are different from zero and are observed as peaks in the BSA‐Seq result.
The detailed statistics of BSA‐Seq provide a better illustration of the potential genomic interval of the candidate locus around the peaks. The SNP‐index of the resistant and the susceptible pooled samples is estimated as the average frequency of all identified SNPs in the 2‐Mb and the 4‐Mb sliding windows. The windows slid 50 kb between successive SNP‐index estimates, and the majority of the genomic region overlaps between successive sliding windows. Thus, depending on the density and distribution of the SNPs, differences between adjacent ∆SNP‐indices arise from both the dropped 50‐kb sequence of the preceding window and the subsequently added 50‐kb sequence, and adjacent ∆SNP‐indices are estimated from highly overlapping SNPs. This nature of the BSA‐Seq results in a large genomic region that harbors the candidate resistance genes.
Indel markers were designed in the BSA‐Seq peak genomic region to narrow the wider genomic interval of the ARG2 locus. The BAM files of the parental and the pooled‐progeny genomes were examined using integrated genome viewer (igv) software (https://igv.org) to identify insertion–deletion (indel) sites to design PCR primers (Table S7). Accordingly, many polymorphic DNA markers between the parental genomes were designed. The frequency of mismatch between the disease phenotypes and the parental DNA marker‐type is expected to increase with distance from the ARG2 locus. Thus, to generate a strong gradient of allelic frequencies, markers were designed beyond the peak genomic region at which the ∆SNP‐indices dropped off to random recombination.
To design primers, once a potential‐indel site was identified via igv, the preferred primer annealing sites were picked manually to avoid SNPs between the parental genomes that may create a difference in the PCR efficiency as a result of the parental origins of templates. This manual method enabled the design of optimal indel‐to‐amplicon size proportion as well, which eased the gel electrophoresis separation of bands. The Rio reference genome was not integrated into the NCBI database. Therefore, the specificity of primer pairs was examined using the blast‐tool in the phytozome database (https://phytozome‐next.jgi.doe.gov). The sequences from the desired annealing sites were copied from igv and optimized as primers using the NCBI primer‐designing tool ( (https://www.ncbi.nlm.nih.gov/tools/primer‐blast). Finally, the specificity of primers was verified using amplicons from the parental lines prior to use for recombination analysis. The marker size determined the parental origin of amplified genomic segments in the progenies based on DNA size differences in gel electrophoresis.
The F3 families of recombinant F2 plants were evaluated to ensure the true‐to‐type disease phenotype. Genetic linkage analysis was considered complete when the marker‐type in a recombinant F2 plant was reconciled to the disease phenotype in the respective F3 progenies. This cycle of linkage analysis was carried out several times with an iterative primer design for polymorphic markers until the final 14 genes ARG2 mapping interval was obtained.
Once the number of candidate genes was reduced, we used comparative and functional analytical tools for further fine mapping. In addition to the sequence information of the parental lines, the similarity of ARG2 mapping interval and response to Cs strains of the resistance parent to the Rio reference genome and the susceptible parent to the BTx623 reference genome was instrumental in this analysis. The CoGe (Lyons et al., 2008) was used to examine the aligned BAM files of many sorghum lines. Functional domains were examined in the reference genomes in phytozome and using NCBI, SMART protein (http://smart.embl.de) and CoGe databases. WideSeq, comprising a targeted sequencing at Bindley Bioscience Center of Purdue University, of genomic DNA and cDNA was used to further examine SNPs, ORFs, functional domains and upstream 2‐kb regions of the likely candidate genes in the ARG2 mapping interval between the parental lines.
Generation of ARG2 near‐isogenic lines
NILs that differ in the ARG2 locus were developed through repeated backcrosses to the BC5F3 generation using the susceptible parental line as a recurrent mother plant. The controlled pollination in the backcrossing scheme was performed using the plastic‐bagging method (Lambright, 2019; Laxman, 1997). Because the pollen source is a heterozygote, the subsequent backcross progenies are expected to carry the heterozygote or the recessive homozygote ARG2. There was no molecular marker to track the ARG2 locus when the NILs were developed, and the F1 progenies of the backcross generations were identified primarily using their disease phenotype. This BCF1 screening method necessitated the evaluation of BCF2 progenies of each backcross generation to avoid disease escape rather than a resistant BCF1. The BCF1 progenies of all the backcross generations that carry the heterozygote genotype at the ARG2 locus are expected to show the 3:1 phenotypic ratio at BCF2.
Characterization of plant growth
ARG2 NILs were grown in Purdue University Ag Alumni Seed Phenotyping Facility (AAPF) growth chamber (120 μmol m−2 sec−1 and 65% relative humidity) to determine the effect of ARG2 on growth and physiological status of the plants in the absence of disease. Twenty plants each form the true‐to‐type recessive, or dominant genotypes and sixteen heterozygote NILs were used to examine differences throughout the growth period. The plants were supplied with minimal fertilizer to avoid compensatory growth, which may arise as a result of a higher amount of fertilizer and shadow any difference among NILs. RGB (red–green–blue) and hyperspectral images were captured once a day (sometimes once in a few to several days); a total of 66 times from the day 16 after planting to physiological maturity. Shoot dry matter was examined among a subset of well‐balanced group of genotypes for analysis of dry matter a analysis of variance (anova). The dry matter analysis was carried out in three replications, and each replication was represented by three plants, a single plant from each of three BC5F3 families. Plants in each of these families were raised from a single ARG2 BC5F2 heterozygote plant.
Transient expression assays and laser scanning confocal microscopy
To determine the subcellular localization of ARG2, full‐length ARG2 coding sequence amplified from SC328C and cloned by Gateway (Invitrogen, Waltham, MA, USA) recombination reactions into the pEarleyGate103 vector, upstream of the mGFP5 under the control of the CaMV 35 S promoter. Transient protein expression assays were performed as previously described (Helm et al., 2019). Bacterial suspensions were mixed in equal ratios (1:1) and infiltrated into the 3‐week‐old Nb leaves. Confocal laser scanning microscopy was performed at 48 h following agroinfiltration using a LSM880 (Carl Zeiss, Jena, Germany) upright confocal microscope. Leaf sections were examined on abaxial surface, and at least two different leaves from two different plants were imaged. mGFP protein fusions were excited using a 488‐nm argon laser and detected between 525 nm and 550 nm. mCherry fusions were excited using a 561 nm helium‐neon laser and detected at 610 nm. Confocal images were collected and processed using zen blue lite (Carl Zeiss).
Metabolite analysis
Ground tissue samples from the susceptible and the resistant ARG2 NILs at 48 h after inoculation, which were collected for the gene expression analysis, were used for metabolite analysis. Equal weights of the ground tissue from the three biological replicates were aliquoted to Eppendorf tubes for metabolite extraction using 50% methanol and resuspended by vortex. The samples were then incubated at 65°C for 2 h by vortexing every 30 min, followed by centrifugation at 17 900 g. The supernatant was transferred to a fresh tube leaving the residue. Each extract was dried for 24 h using a SpeedVac. Each sample was re‐dissolved in 50% methanol and sonicated for 15 min and centrifuged at 17 900 g for 10 min. The supernatant was transferred to a new tube. The metabolite samples were kept at −20°C until run on HPLC‐MS.
Untargeted metabolite data was generated using the HPLC‐MS platform in the negative and positive ionization modes. A thorough manual peak‐integration of the metabolite mass‐features was carried out using mass profiler professional (Agilent, Santa Clara, CA, USA) and the conversion of peaks to mass‐features was performed on the same platform. The MetPA analytical platform was used to visualize the summary statistics, to identify metabolites that showed significantly different concentrations among groups and to generate the input datasets for pathway analysis. The MetPA platform (https://www.metaboanalyst.ca) was also used for pathway analysis using several metabolite databases including the rice and the Arabidopsis Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa & Goto, 2000).
phyre2 analysis
phyre2 analysis was conducted essentially as described previously (Kelley et al., 2015). Briefly, we submitted two versions of ARG2 amino acids to phyre2 (http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index) using the default modeling mode. Then, we used pymol 2.5 (https://pymol.org) to observe the 3D protein structure presented in a classified pretty mode. The red circle or pink dots were labeled based on the position of the specific amino acids.
AUTHOR CONTRIBUTIONS
DBM conducted most of the experiments including the development of experimental population, genetic studies, ARG2 mapping and gene identification, characterization of the ARG2 locus, gene expression and metabolite analyses, and image‐based plant growth studies. SL conducted identification of ARG2 and cloning of candidate NLRs, generating vector constructs, disease assays and fungal growth analysis, gene expression, and phylogenetic analyses. AA conducted bioinformatic and genetic analyses. C‐JL conducted the tertiary structure predictions. MH conducted the localization experiment. DBM, SL, C‐JL, AA, MH, DL and TM designed the research, analyzed the data, and wrote the paper. TM oversaw the project.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Data S1. Alignment of the ARG2 nucleotide sequences from 15 sorghum lines. The ARG2 coding sequences in four resistant (SC328C, Keller, SC237 and Rio) and 11 susceptible lines were aligned. The alignment shows four different ARG2 alleles. ARG2 allele in SC328C is similar to that in Keller, SC237 and Rio. The susceptibility arg2 allele from SC265 is a unique allele. TAM428, BTx642, SC23, IS9830, PQ434 and IS18760 show the same allele. ICSV745, IS8525, Malisor84‐7 carry the susceptibility allele similar to BTx623. The premature stop codons are shaded in red. The premature stop codons are shaded in red. These amino acid sequences were aligned using ‘multiple sequence alignment’ tools: kalign (http://www.ebi.ac.uk/Tools/msa/kalign) and mview (http://www.ebi.ac.uk/Tools/msa/mview).
Data S2. Alignment of the ARG2 amino acid sequences in 15 sorghum lines. The ARG2 amino acid sequences in four resistant (SC328C, Keller, SC237 and Rio) and 11 susceptible lines were aligned. The alignment shows four different ARG2 alleles. ARG2 allele in SC328C is similar to that in Keller, SC237 and Rio. The susceptible ARG2 allele from SC265 is a unique allele. TAM428, BTx642, SC23, IS9830, PQ434 and IS18760 show the same allele. ICSV745, IS8525, Malisor84‐7 carry the susceptible allele similar to BTx623. These amino acid sequences were aligned using ‘multiple sequence alignment’ tools: kalign (www.ebi.ac.uk/Tools/msa/kalign) and mview (www.ebi.ac.uk/Tools/msa/mview).
Data S3. Comparison of the arg2 allele in SC265 and TAM428 (15.5% non‐synonymous codon).
Data S4. Comparison of the amino acid sequences of the LRR domain of ARG2 from SC328C and BTx623.
Figure S1. Fungal growth and anthracnose disease symptom at high temperature. (a) Fungal growth in resistant (SC328) and the susceptible lines (BTx623) differing in the ARG2 gene as revealed by trypan blue staining of spray inoculated tissues. The dark stain shows fungal hyphae. (b) Disease symptoms in SC328C at high temperature (38°C) and (c) Disease symptoms on the resistant and the susceptible ARG2 near‐isogenic lines at 38°C. The susceptible TAM428 genotype was killed by the high temperature. The resistance in SC328C to Csgl1 was compared with the widely known resistant line SC748. The appearance of disease symptoms at 38°C was delayed relative to the disease assays in laboratory (22–23°C) and greenhouse (30–32°C). The image was taken at 15 dpi.
Figure S2. BSA‐Seq analyses showing the mapping of the ARG2 locus. Pooled DNA from resistant and susceptible F3 plants was sequenced and used for the BSA‐Seq analyses with respect to (a) resistant parent, and (b) susceptible parent. The x‐axis is a genomic coordinate and the y‐axis is the ∆SNP‐index estimate. Each blue dot, highly overlapped, represents the ∆SNP‐index estimate in the 2 Mb sliding window, and the red line is the ∆SNP‐index threshold. The green line shows a significance threshold (P = 0.05) of the ∆SNP‐index and the orange line at P = 0.01.
Figure S3. Aligned BAM files as examined in igv. SC328C, TAM428 and BTx623 genome raw‐reads binary alignment map, BAM, files aligned to the Rio reference genome. The lower panel of each sorghum line shows aligned reads and the coverage, and the upper panel shows the frequency of aligned raw‐reads. The upper image shows the alignment at NLR1 (ARG2), and the lower image shows alignment at NLR2 and NLR3. During mapping, reads that found perfect match align without ambiguity and even highly similar reads from duplicate genes align without ambiguity, and hence the BAM coverage in IGV appears uniformly gray, as in SC328C. These uniformly gray shaded areas in the coverage panels show no SNP. ARG2 sequence in SC328C is identical to Rio whereas enormous variation exists with TAM428 and BTx623 that resulted in ambiguous alignment of reads as shown by red, blue, green and brown vertical lines (genomic coordinates) and some positions with no aligned reads. Each of these colors represents one of the four nucleotides as SNPs. Multiple colors in a single coordinate (vertical line) of a coverage panel (e.g. blue and red) show reads that belong to multiple duplicate genes.
Figure S4. General mapping pattern of sorghum lines in the ARG2 genomic region covering 80 kb. General mapping pattern of 16 representative sorghum lines in the ARG2 locus that covers the 22‐kb genomic region. The 16 panels show mapping statistics of the 16 variants that are mapped to the Rio reference genome. The top panel belongs to SC328C (the resistant parent), and all the rest belong to mutant arg2 variants. NLR1 (ARG2) and the non‐functional NLRs are shown.
Figure S5. The evolutionary relationship of ARG2 among 15 sorghum lines. Circles represent resistant and diamonds represent susceptible lines to Csgl1. Initial tree(s) for the heuristic search were obtained automatically by applying neighbor‐joining and BioNJ algorithms to a matrix of pairwise distances estimated using the JTT model, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. In total, there are 1011 positions in the final dataset. Evolutionary analysis was conducted in MEGA X. Bootstrap values are given at the node as a percentage of 1000 replicates.
Figure S6. Prediction of 3D structure of resistant and susceptible ARG2 alleles. (a–c) The tertiary structure of ARG2 predicted based on the template c6j5tC (RPP13‐like protein 4). The ARG2 structure of SC328C and BTx623 was predicted by phyre2 (Kelley et al., 2015). The specific regions of ARG2 were labeled with red circles or pink dots to represent the structural differences. (d) The ADP binding sites in ARG2 from BTx623 and SC328C are different. The amino acids required for ADP binding were predicted based on the 3D structure of 3pfiB using COFACTOR and COACH by I‐TASSER program and labeled as dots. The ARG2 tertiary structure in BTx623 was matched to the disease resistance RPP13‐like protein 4 (ZAR1; template c6j5tC).
Figure S7. Phylogenetic analyses of ARG2 and related proteins in monocot plant species. Phylogenetic relationship between ARG2 and (a) 36 related proteins from sorghum and other monocot species and (b) 17 closely related homologs from other monocot species. Phylogenetic trees constructed by MEGA X using the maximum likelihood method based on the JTT model with 1000 bootstrap replicates of amino acid sequences of ARG2 and selected species. Bootstrap percentages > 75% are shown at the nodes. Sb, Sorghum bicolor; Sv, Setaria viridis; Ttu,Triticum turgidum; Ome, Oryza meyeriana; Osi, Oryza sativa Indica Group; At, Aegilops tauschii; Tti, Triticum timopheevii; Osj, Oryza sativa Japonica Group; Oo, Oryza officinalis; Op, Oryza punctate; Ec, Eragrostis curvula; Si, Setaria italica; Hv, Hordeum vulgare; Omi, Oryza minuta; Bd, Brachypodium distachyon; Ob, Oryza brachyantha; Tu, Triticum urartu; Ath, Arabidopsis thaliana. Arabidopsis, dicot, is included as an outlier control.
Figure S8. Phylogenetic relationship between ARG2 and functionally characterized resistant proteins from different plant species. Intact NB‐ARC domains of the ARG2 and functionally characterized NLR genes from different plant species were used in the construction. Phylogenetic tree constructed by MEGA X using the maximum likelihood method based on the JTT model with 500 bootstrap replicates. Bootstrap percentages > 75% are shown at the nodes.
Figure S9. Metabolite analysis of ARG2‐mediated resistance and analysis of growth and physiological traits among ARG2 NILs in the absence of pathogen under a controlled environment. (a) MetPA ‘MS peak to pathway’ analysis of metabolites in ARG2 NILs in response to Csgl1 at 48 h post inoculation (hpi). Top hit metabolic pathways based on ‘mummichoh pathway enrichment’ algorithm and gene set enrichment analysis (GSEA) using the KEGG Oryza metabolite database. Zeatin biosynthesis, porphyrin and chlorophyll metabolism, tryptophan metabolism, anthocyanin biosynthesis, arginine and proline metabolism, glucosinolate biosynthesis, and lysine degradation. (b) NILs in the growth chamber without pathogen inoculation (left) and on the navigation belt for imaging (right). (c) Anthocyanin reflectance, and (d) plant senescence reflectance indices at flowering time (97 days after planting, dap) and grain filling stage (119 dap), based on hyperspectral image. Estimates of (e) top‐average‐hue throughout the growth period. rr, homozygote recessive; H, heterozygote; RR, homozygote dominant genotypes. (f) Correlation between physiological and growth traits estimates between the flowering time and the grain filling stage. ARI, anthocyanin reflectance index; NRI, nitrogen‐reflectance‐index; PRI, phytochemical‐reflectance‐index; PSRI, plant‐senescence‐reflectance‐index; SACH, side‐average‐convex‐hull; TLA, total‐leaf‐area; TPS, top‐plant‐surface. The suffices ‘1’ in the x‐axis and ‘2’ in the y‐axis labels show the flowering time (97 days after planting) and the grain filling stage (119 days after planting), respectively. anova and Tukey honestly significant difference functions were used to test statistical significance of differences among genotypes.
Figure S10. Panicle exertion and leaf color under greenhouse growth conditions in the parental lines, and the near‐isogenic lines that differ in ARG2 locus. (a) Leaf at maturity. (b) Panicle exertion.
Table S1. Response of sorghum differential lines and the sweet sorghum Rio to five strains of Colletotrichum sublineola.
Table S2. BSA‐Seq analysis. BSA‐Seq result showing the SNP index from the resistant and the susceptible bulks of the ARG2 mapping population.
Table S3. The 14 putative genes in the ARG2 mapping interval with summary of sequence differences between alleles in the resistant (R) and susceptible (S) alleles.
Table S4. Additional sorghum lines that were evaluated for resistance to Csgl1 strain. PCR‐based genotyping was carried out using an indel marker in the ARG2 mapping interval.
Table S5. Conserved NLR amino acids that vary between ARG2 proteins from SC328C and BTx623.
Table S6. Additional metabolite analysis results. All pathway hits in the Oryza KEGG based on ‘MS peak to pathway’ function in MetPA analytical pipeline. All pathway hits with their corresponding matched compound features. Only the proton was chosen in ‘Edit currency metabolites’ function as a result of the ionization mode that was used to generate the data. All other MetPA statistical parameters were under the default mode.
Table S7. List of primers used in the present study.
ACKNOWLEDGEMENTS
This study was made possible through funding by the Feed the Future Innovation Lab for Collaborative Research on Sorghum and Millet through grants from American People provided to the United States Agency for International Development (USAID) under cooperative agreement No. AID‐OAA‐A‐13‐00047. The contents are the sole responsibility of the authors and do not necessarily reflect the views of USAID or the United States Government. We also acknowledge grant from NSF (IOS‐1916893) to TM. We also thank the Purdue University Imaging Facility for access to the Zeiss LSM880 upright confocal microscope. We also thank Rachel Hiles (Purdue University) for technical assistance. This research was funded, in part, by the United States Department of Agriculture, Agriculture Research Service (USDA‐ARS) research project 5020‐21220‐019‐00D. The funding body had no role in designing the experiments, collecting the data, or writing the manuscript. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsements by the United States Department of Agriculture. USDA is an equal opportunity provider and employer.
DATA AVAILABILITY STATEMENT
All data supporting the findings of this work are available within the paper and its supporting materials.
REFERENCES
- Bao, F. , Huang, X. , Zhu, C. , Zhang, X. , Li, X. & Yang, S. (2014) Arabidopsis HSP90 protein modulates RPP4‐mediated temperature‐dependent cell death and defense responses. New Phytologist, 202, 1320–1334. [DOI] [PubMed] [Google Scholar]
- Boyes, D.C. , Nam, J. & Dangl, J.L. (1998) The Arabidopsis thaliana RPM1 disease resistance gene product is a peripheral plasma membrane protein that is degraded coincident with the hypersensitive response. Proceedings of the National Academy of Sciences, 95, 15849–15854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell, A.M. , Sharma, A. , Patil, N.Y. , Collins, S.D. , Anderson, W.F. , Rooney, W.L. et al. (2015) Sequencing of an anthracnose‐resistant sorghum genotype and mapping of a major QTL reveal strong candidate genes for anthracnose resistance. Crop Science, 55, 790–799. [Google Scholar]
- Cheng, X. , Jiang, H. , Zhao, Y. , Qian, Y. , Zhu, S. & Cheng, B. (2010) A genomic analysis of disease‐resistance genes encoding nucleotide binding sites in Sorghum bicolor. Genetics and Molecular Biology, 33, 292–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, E.A. , Brenton, Z.W. , Flinn, B.S. , Jenkins, J. , Shu, S. , Flowers, D. et al. (2019) A new reference genome for Sorghum bicolor reveals high levels of sequence similarity between sweet and grain genotypes: implications for the genetics of sugar metabolism. BMC Genomics, 20, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas, H.E. , Prom, L.K. & Erpelding, J.E. (2014) Inheritance and molecular mapping of anthracnose resistance genes present in sorghum line SC112‐14. Molecular Breeding, 34, 1943–1953. [Google Scholar]
- Cui, H. , Tsuda, K. & Parker, J.E. (2015) Effector‐triggered immunity: from pathogen perception to robust defense. Annual Review of Plant Biology, 66, 487–511. [DOI] [PubMed] [Google Scholar]
- Dangl, J. , Dietrich, R. & Thomas, H. (2000) Senescence and programmed cell death. Rockville, MD: American Society of Plant Physiologists Press. [Google Scholar]
- Davila‐Gomez, F. , Chuck‐Hernandez, C. , Perez‐Carrillo, E. , Rooney, W. & Serna‐Saldivar, S. (2011) Evaluation of bioethanol production from five different varieties of sweet and forage sorghums (Sorghum bicolor (L) Moench). Industrial Crops and Products, 33, 611–616. [Google Scholar]
- El Kasmi, F. (2021) How activated NLRs induce anti‐microbial defenses in plants. Biochemical Society Transactions, 49, 2177–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felderhoff, T.J. , McIntyre, L.M. , Saballos, A. & Vermerris, W. (2016) Using genotyping by sequencing to map two novel anthracnose resistance loci in Sorghum bicolor. G3‐Genes, Genomes, Genetics, 6, 1935–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, F. , Girma, G. & Mengiste, T. (2020) Global mRNA and microRNA expression dynamics in response to anthracnose infection in sorghum. BMC Genomics, 21, 760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey, D. & Rathjen, J.P. (2012) Recognition and response in plant PAMP‐triggered immunity. In: eLS. New York: John Wiley & Sons. 10.1002/9780470015902.a0023725 [DOI] [Google Scholar]
- Helm, M. , Qi, M. , Sarkar, S. , Yu, H. , Whitham, S.A. & Innes, R.W. (2019) Engineering a decoy substrate in soybean to enable recognition of the soybean mosaic virus NIa protease. Molecular Plant‐Microbe Interactions, 32, 760–769. [DOI] [PubMed] [Google Scholar]
- Jamil, F. & Nicholson, R. (1987) Susceptibility of corn to isolates of Colletotrichum graminicola pathogenic to other grasses. Plant Disease, 71, 809–810. [Google Scholar]
- Kanehisa, M. & Goto, S. (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Research, 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley, L.A. , Mezulis, S. , Yates, C.M. , Wass, M.N. & Sternberg, M.J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nature Protocols, 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, P. , Rooney, W.L. , Burrell, A.M. & Collins, D. (2017) Methods and compositions for producing sorghum plants with anthracnose resistance: Google patents.
- Kourelis, J. , Sakai, T. , Adachi, H. & Kamoun, S. (2021) RefPlantNLR is a comprehensive collection of experimentally validated plant disease resistance proteins from the NLR family. PLoS Biology, 19, e3001124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagudah, E. & Periyannan, S.K. (2018) Wheat stem rust resistance gene: Google patents.
- Lambright, L. (2019) Hybrid Sorghum Product Development and Production. In: Zhao, Z.Y. & Dahlberg, J. (Eds.) Sorghum. Methods in Molecular Biology, vol. 1931. New York: Springer. [DOI] [PubMed] [Google Scholar]
- Laxman, S. (1997) Plastic bag emasculation technique in Sorghum (Sorghum bicolor L. (Moench)). Journal of Research ANGRAU, 25(2), 38–39. [Google Scholar]
- Lee, S. , Fu, F.Y. , Liao, C.J. , Mewa, D.B. , Adeyanju, A. , Ejeta, G. et al. (2022) Broad‐spectrum fungal resistance in sorghum is conferred through the complex regulation of an immune receptor gene embedded in a natural antisense transcript. The Plant Cell, 34, 1641–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Zhu, F. , Liu, H. , Chu, A. & Lo, C. (2013) Isolation and expression analysis of defense‐related genes in sorghum–Colletotrichum sublineolum interaction. Physiological and Molecular Plant Pathology, 84, 123–130. [Google Scholar]
- Little, C.R. & Perumal, R. (2019) The biology and control of sorghum diseases. Sorghum: A State of the Art and Future Perspetives, 58, 297–346. [Google Scholar]
- Lo, S.‐C.C. , De Verdier, K. & Nicholson, R.L. (1999) Accumulation of 3‐deoxyanthocyanidin phytoalexins and resistance to Colletotrichum sublineolum in sorghum. Physiological and Molecular Plant Pathology, 55, 263–273. [Google Scholar]
- Lo, S.‐C.C. , Hipskind, J.D. & Nicholson, R.L. (1999) cDNA cloning of a sorghum pathogenesis‐related protein (PR‐10) and differential expression of defense‐related genes following inoculation with Cochliobolus heterostrophus or Colletotrichum sublineolum. Molecular Plant‐Microbe Interactions, 12, 479–489. [DOI] [PubMed] [Google Scholar]
- Lyons, E. , Pedersen, B. , Kane, J. , Alam, M. , Ming, R. , Tang, H. et al. (2008) Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar, and grape: CoGe with Rosids. Plant Physiology, 148, 1772–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, W. , Gao, X. , Han, T. , Mohammed, M.T. , Yang, J. , Ding, J. et al. (2022) Molecular genetics of anthracnose resistance in maize. Journal of Fungi, 8(5), 540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, J. , Ditmore, M. & TeBeest, D. (2008) Pathotypes of Colletotrichum sublineolum in Arkansas. Plant Disease, 92, 1415–1420. [DOI] [PubMed] [Google Scholar]
- Nelson, R. , Wiesner‐Hanks, T. , Wisser, R. & Balint‐Kurti, P. (2018) Navigating complexity to breed disease‐resistant crops. Nature Reviews Genetics, 19, 21–33. [DOI] [PubMed] [Google Scholar]
- Panchy, N. , Lehti‐Shiu, M. & Shiu, S.‐H. (2016) Evolution of gene duplication in plants. Plant Physiology, 171, 2294–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil, N.Y. , Klein, R.R. , Williams, C.L. , Collins, S.D. , Knoll, J.E. , Burrell, A.M. et al. (2017) Quantitative trait loci associated with anthracnose resistance in sorghum. Crop Science, 57, 877–890. [Google Scholar]
- Prom, L. , Perumal, R. , Erattaimuthu, S. , Little, C. , No, E. , Erpelding, J. et al. (2012) Genetic diversity and pathotype determination of Colletotrichum sublineolum isolates causing anthracnose in sorghum. European Journal of Plant Pathology, 133, 671–685. [Google Scholar]
- Pruitt, R.N. , Gust, A.A. & Nürnberger, T. (2021) Plant immunity unified. Nature Plants, 7, 382–383. [DOI] [PubMed] [Google Scholar]
- Redkar, A. , Jaeger, E. & Doehlemann, G. (2018) Visualization of growth and morphology of fungal hyphae in planta using WGA‐AF488 and propidium iodide co‐staining.
- Rose, L.E. , Bittner‐Eddy, P.D. , Langley, C.H. , Holub, E.B. , Michelmore, R.W. & Beynon, J.L. (2004) The maintenance of extreme amino acid diversity at the disease resistance gene, RPP13, in Arabidopsis thaliana. Genetics, 166, 1517–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, A. , Kucukural, A. & Zhang, Y. (2010) I‐TASSER: a unified platform for automated protein structure and function prediction. Nature Protocols, 5, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz, M.C. , Maron, L.G. , Stein, J.C. , Wences, A.H. , Gurtowski, J. , Biggers, E. et al. (2014) New whole genome de novo assemblies of three divergent strains of rice (O. sativa) documents novel gene space of aus and indica. Genome Biology, 15, 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St. Clair, D.A. (2010) Quantitative disease resistance and quantitative resistance loci in breeding. Annual Review of Phytopathology, 48, 247–268. [DOI] [PubMed] [Google Scholar]
- Takagi, H. , Abe, A. , Yoshida, K. , Kosugi, S. , Natsume, S. , Mitsuoka, C. et al. (2013) QTL‐seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. The Plant Journal, 74, 174–183. [DOI] [PubMed] [Google Scholar]
- Takken, F.L.W. , Albrecht, M. & Tameling, W.I.L. (2006) Resistance proteins: molecular switches of plant defence. Current Opinion in Plant Biology, 9, 383–390. [DOI] [PubMed] [Google Scholar]
- Tameling, W.I. , Elzinga, S.D. , Darmin, P.S. , Vossen, J.H. , Takken, F.L. , Haring, M.A. et al. (2002) The tomato R gene products I‐2 and MI‐1 are functional ATP binding proteins with ATPase activity. The Plant Cell, 14, 2929–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenkouano, A. , Miller, F.R. , Hart, G.E. , Frederiksen, R.A. & Nicholson, R.L. (1993) Phytoalexin assay in juvenile sorghum: an aid to breeding for anthracnose resistance. Crop Science, 33, 243–248. [Google Scholar]
- Thakur, R. (2007) Genetic resistance to foliar anthracnose in sorghum and pathogenic variability in Colletotrichum graminicola. Indian Phytopathology, 60, 13–23. [Google Scholar]
- Van de Weyer, A.L. , Monteiro, F. , Furzer, O.J. , Nishimura, M.T. , Cevik, V. , Witek, K. et al. (2019) A species‐wide inventory of NLR genes and alleles in Arabidopsis thaliana. Cell, 178(5), 1260–1272.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ooijen, G. , Mayr, G. , Kasiem, M.M. , Albrecht, M. , Cornelissen, B.J. & Takken, F.L. (2008) Structure‐function analysis of the NB‐ARC domain of plant disease resistance proteins. Journal of Experimental Botany, 59, 1383–1397. [DOI] [PubMed] [Google Scholar]
- Vyska, M. , Cunniffe, N. & Gilligan, C. (2016) Trade‐off between disease resistance and crop yield: a landscape‐scale mathematical modelling perspective. Journal of the Royal Society Interface, 13, 20160451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, B.‐H. , Ebbole, D.J. & Wang, Z.‐H. (2017) The arms race between Magnaporthe oryzae and rice: diversity and interaction of Avr and R genes. Journal of Integrative Agriculture, 16, 2746–2760. [Google Scholar]
- Wang, J. , Chen, T. , Han, M. , Qian, L. , Li, J. , Wu, M. et al. (2020) Plant NLR immune receptor Tm‐22 activation requires NB‐ARC domain‐mediated self‐association of CC domain. PLoS Pathogens, 16, e1008475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. , Hu, M. , Wang, J. , Qi, J. , Han, Z. , Wang, G. et al. (2019) Reconstitution and structure of a plant NLR resistosome conferring immunity. Science, 364, eaav5870. [DOI] [PubMed] [Google Scholar]
- Williams, S.J. , Sornaraj, P. , de Courcy‐Ireland, E. , Menz, R.I. , Kobe, B. , Ellis, J.G. et al. (2011) An autoactive mutant of the M flax rust resistance protein has a preference for binding ATP, whereas wild‐type M protein binds ADP. Molecular Plant‐Microbe Interactions, 24, 897–906. [DOI] [PubMed] [Google Scholar]
- Yang, X. & Wang, J. (2016) Genome‐wide analysis of NBS‐LRR genes in sorghum genome revealed several events contributing to NBS‐LRR gene evolution in grass species. Evolutionary Bioinformatics, 12, EBO.S36433–EBO.S36421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, L.‐Y. , Guo, X.‐S. , He, B. , Sun, L.‐J. , Peng, Y. , Dong, S.‐S. et al. (2011) Genome‐wide patterns of genetic variation in sweet and grain sorghum (Sorghum bicolor). Genome Biology, 12, R114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, F. , Saluja, M. , Dharni, J.S. , Paul, P. , Sattler, S.E. , Staswick, P. et al. (2021) PhenoImage: an open‐source graphical user interface for plant image analysis. The Plant Phenome Journal, 4, e20015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Alignment of the ARG2 nucleotide sequences from 15 sorghum lines. The ARG2 coding sequences in four resistant (SC328C, Keller, SC237 and Rio) and 11 susceptible lines were aligned. The alignment shows four different ARG2 alleles. ARG2 allele in SC328C is similar to that in Keller, SC237 and Rio. The susceptibility arg2 allele from SC265 is a unique allele. TAM428, BTx642, SC23, IS9830, PQ434 and IS18760 show the same allele. ICSV745, IS8525, Malisor84‐7 carry the susceptibility allele similar to BTx623. The premature stop codons are shaded in red. The premature stop codons are shaded in red. These amino acid sequences were aligned using ‘multiple sequence alignment’ tools: kalign (http://www.ebi.ac.uk/Tools/msa/kalign) and mview (http://www.ebi.ac.uk/Tools/msa/mview).
Data S2. Alignment of the ARG2 amino acid sequences in 15 sorghum lines. The ARG2 amino acid sequences in four resistant (SC328C, Keller, SC237 and Rio) and 11 susceptible lines were aligned. The alignment shows four different ARG2 alleles. ARG2 allele in SC328C is similar to that in Keller, SC237 and Rio. The susceptible ARG2 allele from SC265 is a unique allele. TAM428, BTx642, SC23, IS9830, PQ434 and IS18760 show the same allele. ICSV745, IS8525, Malisor84‐7 carry the susceptible allele similar to BTx623. These amino acid sequences were aligned using ‘multiple sequence alignment’ tools: kalign (www.ebi.ac.uk/Tools/msa/kalign) and mview (www.ebi.ac.uk/Tools/msa/mview).
Data S3. Comparison of the arg2 allele in SC265 and TAM428 (15.5% non‐synonymous codon).
Data S4. Comparison of the amino acid sequences of the LRR domain of ARG2 from SC328C and BTx623.
Figure S1. Fungal growth and anthracnose disease symptom at high temperature. (a) Fungal growth in resistant (SC328) and the susceptible lines (BTx623) differing in the ARG2 gene as revealed by trypan blue staining of spray inoculated tissues. The dark stain shows fungal hyphae. (b) Disease symptoms in SC328C at high temperature (38°C) and (c) Disease symptoms on the resistant and the susceptible ARG2 near‐isogenic lines at 38°C. The susceptible TAM428 genotype was killed by the high temperature. The resistance in SC328C to Csgl1 was compared with the widely known resistant line SC748. The appearance of disease symptoms at 38°C was delayed relative to the disease assays in laboratory (22–23°C) and greenhouse (30–32°C). The image was taken at 15 dpi.
Figure S2. BSA‐Seq analyses showing the mapping of the ARG2 locus. Pooled DNA from resistant and susceptible F3 plants was sequenced and used for the BSA‐Seq analyses with respect to (a) resistant parent, and (b) susceptible parent. The x‐axis is a genomic coordinate and the y‐axis is the ∆SNP‐index estimate. Each blue dot, highly overlapped, represents the ∆SNP‐index estimate in the 2 Mb sliding window, and the red line is the ∆SNP‐index threshold. The green line shows a significance threshold (P = 0.05) of the ∆SNP‐index and the orange line at P = 0.01.
Figure S3. Aligned BAM files as examined in igv. SC328C, TAM428 and BTx623 genome raw‐reads binary alignment map, BAM, files aligned to the Rio reference genome. The lower panel of each sorghum line shows aligned reads and the coverage, and the upper panel shows the frequency of aligned raw‐reads. The upper image shows the alignment at NLR1 (ARG2), and the lower image shows alignment at NLR2 and NLR3. During mapping, reads that found perfect match align without ambiguity and even highly similar reads from duplicate genes align without ambiguity, and hence the BAM coverage in IGV appears uniformly gray, as in SC328C. These uniformly gray shaded areas in the coverage panels show no SNP. ARG2 sequence in SC328C is identical to Rio whereas enormous variation exists with TAM428 and BTx623 that resulted in ambiguous alignment of reads as shown by red, blue, green and brown vertical lines (genomic coordinates) and some positions with no aligned reads. Each of these colors represents one of the four nucleotides as SNPs. Multiple colors in a single coordinate (vertical line) of a coverage panel (e.g. blue and red) show reads that belong to multiple duplicate genes.
Figure S4. General mapping pattern of sorghum lines in the ARG2 genomic region covering 80 kb. General mapping pattern of 16 representative sorghum lines in the ARG2 locus that covers the 22‐kb genomic region. The 16 panels show mapping statistics of the 16 variants that are mapped to the Rio reference genome. The top panel belongs to SC328C (the resistant parent), and all the rest belong to mutant arg2 variants. NLR1 (ARG2) and the non‐functional NLRs are shown.
Figure S5. The evolutionary relationship of ARG2 among 15 sorghum lines. Circles represent resistant and diamonds represent susceptible lines to Csgl1. Initial tree(s) for the heuristic search were obtained automatically by applying neighbor‐joining and BioNJ algorithms to a matrix of pairwise distances estimated using the JTT model, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. In total, there are 1011 positions in the final dataset. Evolutionary analysis was conducted in MEGA X. Bootstrap values are given at the node as a percentage of 1000 replicates.
Figure S6. Prediction of 3D structure of resistant and susceptible ARG2 alleles. (a–c) The tertiary structure of ARG2 predicted based on the template c6j5tC (RPP13‐like protein 4). The ARG2 structure of SC328C and BTx623 was predicted by phyre2 (Kelley et al., 2015). The specific regions of ARG2 were labeled with red circles or pink dots to represent the structural differences. (d) The ADP binding sites in ARG2 from BTx623 and SC328C are different. The amino acids required for ADP binding were predicted based on the 3D structure of 3pfiB using COFACTOR and COACH by I‐TASSER program and labeled as dots. The ARG2 tertiary structure in BTx623 was matched to the disease resistance RPP13‐like protein 4 (ZAR1; template c6j5tC).
Figure S7. Phylogenetic analyses of ARG2 and related proteins in monocot plant species. Phylogenetic relationship between ARG2 and (a) 36 related proteins from sorghum and other monocot species and (b) 17 closely related homologs from other monocot species. Phylogenetic trees constructed by MEGA X using the maximum likelihood method based on the JTT model with 1000 bootstrap replicates of amino acid sequences of ARG2 and selected species. Bootstrap percentages > 75% are shown at the nodes. Sb, Sorghum bicolor; Sv, Setaria viridis; Ttu,Triticum turgidum; Ome, Oryza meyeriana; Osi, Oryza sativa Indica Group; At, Aegilops tauschii; Tti, Triticum timopheevii; Osj, Oryza sativa Japonica Group; Oo, Oryza officinalis; Op, Oryza punctate; Ec, Eragrostis curvula; Si, Setaria italica; Hv, Hordeum vulgare; Omi, Oryza minuta; Bd, Brachypodium distachyon; Ob, Oryza brachyantha; Tu, Triticum urartu; Ath, Arabidopsis thaliana. Arabidopsis, dicot, is included as an outlier control.
Figure S8. Phylogenetic relationship between ARG2 and functionally characterized resistant proteins from different plant species. Intact NB‐ARC domains of the ARG2 and functionally characterized NLR genes from different plant species were used in the construction. Phylogenetic tree constructed by MEGA X using the maximum likelihood method based on the JTT model with 500 bootstrap replicates. Bootstrap percentages > 75% are shown at the nodes.
Figure S9. Metabolite analysis of ARG2‐mediated resistance and analysis of growth and physiological traits among ARG2 NILs in the absence of pathogen under a controlled environment. (a) MetPA ‘MS peak to pathway’ analysis of metabolites in ARG2 NILs in response to Csgl1 at 48 h post inoculation (hpi). Top hit metabolic pathways based on ‘mummichoh pathway enrichment’ algorithm and gene set enrichment analysis (GSEA) using the KEGG Oryza metabolite database. Zeatin biosynthesis, porphyrin and chlorophyll metabolism, tryptophan metabolism, anthocyanin biosynthesis, arginine and proline metabolism, glucosinolate biosynthesis, and lysine degradation. (b) NILs in the growth chamber without pathogen inoculation (left) and on the navigation belt for imaging (right). (c) Anthocyanin reflectance, and (d) plant senescence reflectance indices at flowering time (97 days after planting, dap) and grain filling stage (119 dap), based on hyperspectral image. Estimates of (e) top‐average‐hue throughout the growth period. rr, homozygote recessive; H, heterozygote; RR, homozygote dominant genotypes. (f) Correlation between physiological and growth traits estimates between the flowering time and the grain filling stage. ARI, anthocyanin reflectance index; NRI, nitrogen‐reflectance‐index; PRI, phytochemical‐reflectance‐index; PSRI, plant‐senescence‐reflectance‐index; SACH, side‐average‐convex‐hull; TLA, total‐leaf‐area; TPS, top‐plant‐surface. The suffices ‘1’ in the x‐axis and ‘2’ in the y‐axis labels show the flowering time (97 days after planting) and the grain filling stage (119 days after planting), respectively. anova and Tukey honestly significant difference functions were used to test statistical significance of differences among genotypes.
Figure S10. Panicle exertion and leaf color under greenhouse growth conditions in the parental lines, and the near‐isogenic lines that differ in ARG2 locus. (a) Leaf at maturity. (b) Panicle exertion.
Table S1. Response of sorghum differential lines and the sweet sorghum Rio to five strains of Colletotrichum sublineola.
Table S2. BSA‐Seq analysis. BSA‐Seq result showing the SNP index from the resistant and the susceptible bulks of the ARG2 mapping population.
Table S3. The 14 putative genes in the ARG2 mapping interval with summary of sequence differences between alleles in the resistant (R) and susceptible (S) alleles.
Table S4. Additional sorghum lines that were evaluated for resistance to Csgl1 strain. PCR‐based genotyping was carried out using an indel marker in the ARG2 mapping interval.
Table S5. Conserved NLR amino acids that vary between ARG2 proteins from SC328C and BTx623.
Table S6. Additional metabolite analysis results. All pathway hits in the Oryza KEGG based on ‘MS peak to pathway’ function in MetPA analytical pipeline. All pathway hits with their corresponding matched compound features. Only the proton was chosen in ‘Edit currency metabolites’ function as a result of the ionization mode that was used to generate the data. All other MetPA statistical parameters were under the default mode.
Table S7. List of primers used in the present study.
Data Availability Statement
All data supporting the findings of this work are available within the paper and its supporting materials.