

Abstract
The trapping of the elusive vinylogous position of a vinyl carbene with an aliphatic C(sp3)−H bond has been achieved for the first time during a silver‐catalyzed carbene/alkyne metathesis (CAM) process. A Tpx‐containing silver complex first promotes the generation of a donor‐acceptor silver carbene which triggers CAM, generating a subsequent donor‐donor vinyl silver carbene species, which then undergoes a selective vinylogous C(sp3)−H bond insertion, leading to the synthesis of a new family of benzoazepines. Density functional theory (DFT) calculations unveil the reaction mechanism, which allows proposing that the C−H bond insertion reaction takes place in a stepwise manner, with the hydrogen shift being the rate determining step.
Keywords: 1-Benzoazepine, C−H Bond Insertion, Carbene-Alkyne Metathesis, Silver Catalysis, Vinylogous Reactivity
The vinylogous position of a donor‐donor silver vinyl carbene is trapped with an aliphatic C(sp3)−H bond for the first time to afford 1‐benzoazepine scaffolds. The overall transformation entails a carbene alkyne metathesis of a donor‐acceptor silver carbene to furnish the key donor‐donor silver vinyl carbene whose vinylogous position is selectively intercepted in a stepwise C−H bond insertion.

The functionalization of C−H bonds by transition metal‐catalyzed reactions [1] represents an excellent alternative to classical methods for the construction of C−C and C‐heteroatom bonds. The fact that no pre‐functionalization of the substrate is required represents a clear advantage, although the ubiquity of C−H bonds in typical organic molecules poses a major challenge in the control of the selectivity. This is especially relevant in metal carbene complex C−H insertion reactions (Scheme 1a), [2] that are not assisted by a directing group [3] in the substrate. The site‐ and stereoselectivity in these processes, apart from the strength of the specific C−H bond, is mostly dictated by the metal‐catalyst and its surrounding ligands.
Scheme 1.
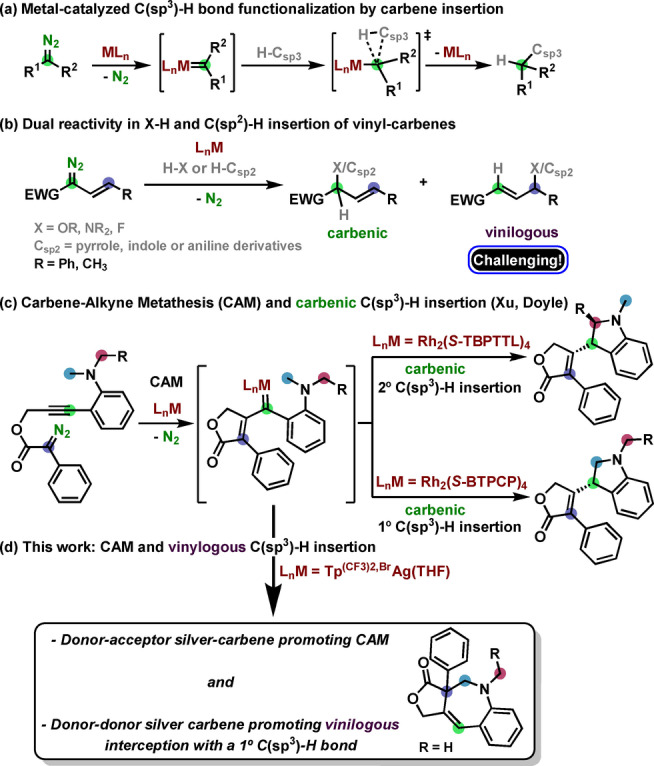
The metal‐catalyzed C−H bond functionalization by carbene insertion: carbenic vs vinylogous reactivity.
Vinylcarbene metal complexes, a fascinating subclass of metal carbene complexes, are extremely powerful reagents in cycloaddition reactions. [4] Furthermore, they have the particularity of displaying electrophilic reactivity at the vinylogous (or remote) position in addition to the typical carbene position (Scheme 1b). Due to the intrinsically higher reactivity of the carbene site, selective reaction onto the vinylogous one is synthetically very challenging. Pioneering work in this area was disclosed by Davies at the beginning of the 90s.[ 5a , 5b ] Since then, only a very limited number of examples have achieved insertion on the vinylogous site either with activated C(sp2)−H bonds under rhodium catalysis,[ 5c , 5d , 5e , 5f ] or with X−H bonds (X=O, N, F) using various metals. [6] Of note, works by Doyle [7a] and Harada and Nemoto [7b] have shown that silver carbenes are superior to rhodium ones in promoting O−H and N−H vinylogous insertion, respectively. However, the interception of the vinylogous position with a C(sp3)−H bond remains yet unknown.
The above examples for the generation of vinylcarbene metal complexes are based in the transition metal‐catalyzed decomposition of vinyldiazoacetates (or related arylsulfonylhydrazones). [8] An alternative route is the catalytic carbene/alkyne metathesis (CAM, Scheme 1c), [9] first reported by Padwa [10] and Hoye, [11] where a metal carbene complex, generated from a diazo functionality, reacts with an alkyne by transferring the carbene character to one of the alkyne carbon atoms thus generating a vinylcarbene metal complex. Construction of polycyclic frameworks can be efficiently achieved through CAM cascades terminated by various carbene reactions.[ 12 , 13 ] In this line, Hashmi [14a] and Zhang [14b] studied the reactivity of α‐oxo gold carbene complexes with alkynes followed by an intramolecular C(sp3)−H insertion on the carbenic position. On the other hand, Doyle and Xu [15] reported in 2018 the synthesis of chiral dihydroindole derivatives through a CAM cascade on propargyl diazoacetates terminated by a site‐selective intramolecular C(sp3)−H bond insertion on the carbenic site. By modifying the ligands on the dirhodium complex, either a primary C−H bond or a secondary benzylic C−H bond could be selectively inserted (Scheme 1c).
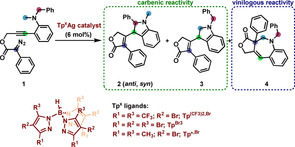
Highly active silver catalysts containing trispyrazolylborate ligands have been described for the catalytic functionalization of low reactive carbon‐hydrogen bonds, even for those of methane, using diazo compounds as the carbene source.[ 2d , 2e ] In view of the lack of reports on the use of silver for CAM‐cascade transformations, together with the precedents on the use of gold [16] for analogous transformations, we decided to investigate the potential of those complexes as catalysts for the transformation reported by Doyle and Xu with rhodium (Scheme 1c). Herein we describe the results showing that not only silver has been found to promote such transformation but also that the silver donor‐donor carbene intermediate induces the trapping of the vinylogous position with a methyl C−H bond, in the first example of such remote interception.
We first run a series of experiments to test the catalytic capabilities of three different silver complexes (Table 1), employing the model substrate propargyl diazoacetate 1. [15] Mixtures of up to four products were obtained, showing that silver catalyzes the carbene/alkyne metathesis reaction, which further undergoes C−H carbenic reactivity. The use of Tp(CF3)2,BrAg(THF) (entry 1) resulted in the formation of a mixture of the anti and syn diastereoisomers of the benzylic C(sp3)−H insertion product 2, and a minor subproduct as well that could not be fully identified due to the low yield. Repeating the reaction at lower concentration of 1 (3.8 mM, entry 2) increased the conversion and allowed the new product to be characterized. To our delight, 1‐benzoazepine scaffold 4 was identified, after isolation and full nuclear magnetic resonance (NMR) characterization (see Supporting Information), eventually resulting from an unprecedented primary C(sp3)−H bond insertion to the vinylogous position of a vinyl silver‐carbene intermediate. The other two silver complexes tested showed activity but decreased selectivity (entries 3 and 4). Moreover, they also induced the formation of the product resulting from the insertion of the carbene into the primary C(sp3)−H bond. Finally, the use of simple silver salts such as AgBF4 and AgSbF6 promoted complete consumption of substrate 1 in a very poor selective reaction that did not provide compound 4, assessing the relevance of the Tp(CF3)2,Br ligand in this catalytic reaction.
Table 1.
Initial screening of silver‐based catalysts for CAM‐cascade reactions.[a]
|
| ||
|---|---|---|
|
Entry |
Catalyst |
Yield [%] (2anti /2syn /3/4) |
|
1[b] |
Tp(CF3)2,BrAg(THF) |
45[d] (23/17/0/5) |
|
2 |
Tp(CF3)2,BrAg(THF) |
82[d] (39/22/0/21) |
|
3 |
[TpBr3Ag]2 |
95 (35/21/18/21) |
|
4[c] |
Tp*,BrAg(THF) |
39 (14/8/17/0) |
[a] Unless otherwise noted, reactions were carried out with 0.06 mmol of 1 ([1]=3.8 mM), at room temperature in 16 mL of DCM for 1.5 h. The yield was determined by NMR using 4‐chlorobenzaldehyde as internal standard. Product ratios determined by NMR. [b] Reaction carried out at [1]=20 mM for 1 h. [c] Reaction run for 18 h. [d] Isolated yield.
The novelty of this transformation does not only stand on how it proceeds through vinylogous interception by an aliphatic bond but also provides an alternative route to 1‐benzoazepine‐containing molecules. Therefore, we decided to optimize the synthesis of scaffolds analogous to 4 through the CAM cascade that terminated with this uncommon vinylogous C(sp3)−H addition reaction. Towards that end, propargyl diazoacetate 5 a was prepared and employed for optimization of the reaction conditions, which were set at using Tp(CF3)2,BrAg(THF) as catalyst, and running the reaction at 35 °C in chloroform in the presence of powdered molecular sieves (Scheme 2, see Supporting Information for optimization studies). In this manner, the desired 1‐benzoazepine product 7 a was synthesized in 71 % yield with a 1 : 7.1 (6 a : 7 a) selectivity. The structure of 7 a was confirmed by X‐ray diffraction analysis [17] (Scheme 2), assessing the structure assigned from NMR data (see Supporting Information).
Scheme 2.
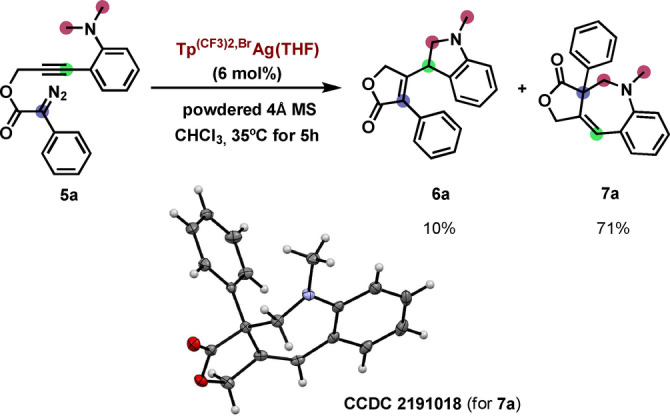
Isolation and structural characterization of 1‐benzoazepine 7 a.
The substrate scope for this reaction under the optimized conditions is shown in Scheme 3. Introduction of electron‐donating substituents in the phenyldiazo moiety led to moderate yields of 7 b–d. The presence of electron‐withdrawing halogen substituents in that ring not only decreased the overall conversion but also switched the selectivity towards the carbenic insertion product (vinylogous:carbenic ratio of 0.9 : 1 for 7 e and 0.6 : 1 for 7 f). On the contrary, a very good efficiency and selectivity was observed for the substrates with substituents in the dimethylaminophenyl ring. The yields remain in the 58–69 % interval, largely favoring the vinylogous reactivity for substrates bearing electron‐withdrawing or electron‐donating substituents either in meta‐ (7 g, 7 h) or in para‐ (7 i, 7 j) relative position to the dimethylamino moiety. Moreover, the reaction can be scaled up to a 1 mmol scale with only a minimal decrease of yield (see Supporting Information).
Scheme 3.
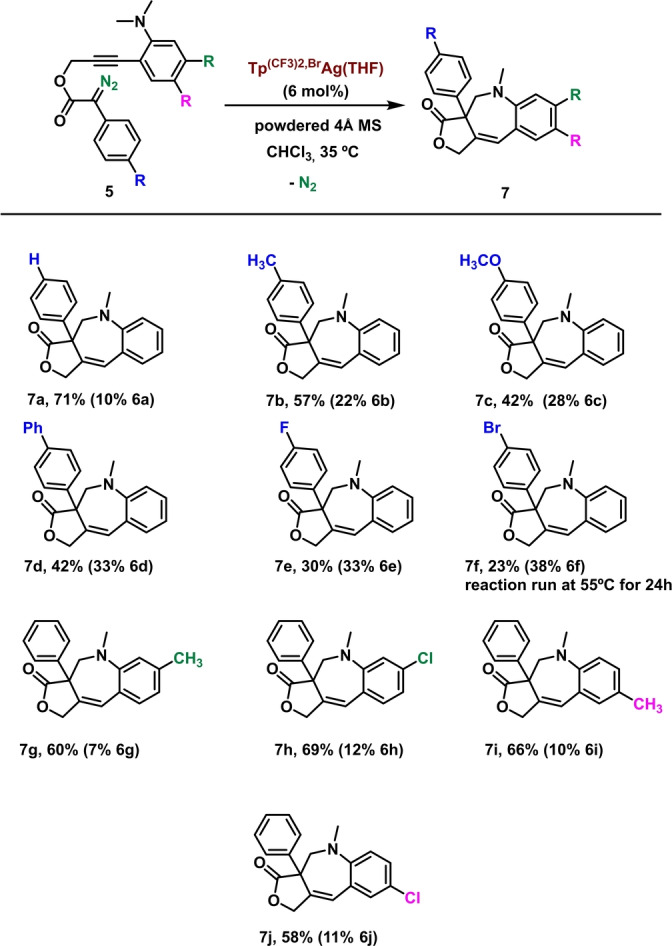
Reaction scope.
Since the formation of compounds 7 implies the migration of a hydrogen from the methyl groups of the dimethylamino fragment, we have prepared the corresponding N(CD3)2 derivative 5 a‐d6 to assess the destiny of the initial H(D). As shown in eqn 1, products 7a‐d6 and 6 a‐d6 were obtained in a 6.7 : 1 ratio, similarly to that for the protio derivatives (7.1 : 1, Scheme 2). The deuterium originated from the C−D cleavage was localized exclusively in one position, in both compounds, with no scrambling into other positions, a fact that must be considered for mechanistic proposal (see below).
Scheme 4 shows the application of the different pathways proposed in the literature for CAM processes catalyzed for different metals to our Ag‐based system. May [18] described the isolation and mechanistic relevance of ring‐fused cyclopropenes with rhodium catalysts (path I). Some of us [12a] postulated the formation of a rhodium(I) η3‐vinylcarbene intermediate through a [π2s+π2a] addition that subsequently evolves to the rhodium(I) η1‐vinylcarbene, a proposal also shared by Saá [19] in their intermolecular Ru‐catalyzed CAM cascade processes (path II, Scheme 4). Yu and Xu [20] proposed a 5‐exo‐dig cyclization leading to a very reactive zwitterionic vinyl cationic species that upon a Rh‐1,3‐shift converts to the rhodium(II) η1‐vinylcarbene (path III). An analogous mechanism was also postulated by Xue [21] under Rh2(OAc)4 catalysis. Under gold catalysis, Xu and Hashmi [22] and Hu [23] independently reported cascade reactions involving a CAM step postulated to occur through nucleophilic addition of the diazo compound onto the gold‐activated alkyne followed by the expulsion of dinitrogen (path IV).
Scheme 4.
Plausible mechanistic pathways for the silver‐catalyzed CAM.
On the basis of the plausible operation of several pathways, a series of density functional theory (DFT) calculations at the B3LYP‐D3/Def2TZVP‐SDD‐SMD(CHCl3)//BP86‐D3/Def2SVP‐SDD level of theory using the Gaussian16 software package were performed to unveil the reaction mechanism for our silver‐catalyzed CAM. Figure 1 shows the pathway leading to the silver carbene species F resulting from the CAM process. Coordination of the silver complex to the carbenic carbon of the diazo species is a mildly endergonic step (3.7 kcal mol−1) that is followed by extrusion of nitrogen through transition state TS‐BC with an overall energy barrier of 11.9 kcal mol−1 with respect to A leading to the silver carbene C (−16.2 kcal mol−1), which would be a common intermediate for the proposed paths I, II and III (Scheme 4). The computed single‐step path IV barrier was calculated to be 15.9 kcal mol−1, therefore it was discarded (see section S10 of the Supporting Information for a complete discussion on discarded paths). From carbene species C, the nucleophilic attack of the alkyne onto the carbenic carbon on a 5‐exo‐dig cyclization leads to the zwitterionic vinyl cationic species D in a barrierless exergonic process. [24] At this stage, the formation of the ring‐fused cyclopropene (path I) was computed requiring a kinetic effort of 31.5 kcal mol−1. In addition, the possibility of a direct hydride transfer to the vinyl cation [25] was considered showing an energy barrier of 12.6 kcal mol−1. Those paths were disregarded given the barrierless nucleophilic attack of the negatively charged silver atom to the carbocation, leading to silver η3‐vinylcarbene E, that rearranges to the almost isoenergetic silver η1‐vinylcarbene F. Overall, the formation of silver η1‐vinylcarbene F is very exergonic (−65.7 kcal mol−1) and does proceed through a new pathway that combines previously postulated paths II and III.
Figure 1.
Gibbs energy profile (in kcal mol−1) of the silver‐catalyzed CAM reaction ([Ag]=Tp(CF3)2,BrAg).
Next, the silver carbene insertion into the C−H bond was computed. This insertion generally occurs in a concerted although sometimes asynchronous manner (formation of the new C−H takes place earlier than formation of the C−C bond).[ 26 , 27 , 28 ] All attempts to locate the concerted C−H insertion from intermediate F in our system failed. Alternatively, a stepwise C−H insertion mechanism involving a zwitterionic intermediate was found (Figure 2), analogous to the mechanism reported by Shaw for the C−H insertion of donor/donor dirhodium carbenes. [29] Indeed, from silver vinyl carbene F a hydride shift surpassing an energy barrier of 18.0 kcal mol−1 leads to zwitterionic intermediate G in a slightly exergonic process (ΔG=−4.0 kcal mol−1), in the rate determining step of the overall process. An SE2 [30] C−C bond‐closing step takes place from G through TS‐GI with an energy barrier of 10.9 kcal mol−1 leading, upon silver decoordination, to I, the carbenic C−H insertion product 6. On the other hand, intermediate G may also undergo electrophilic substitution reaction at the γ carbon atom (in an SE2′ reaction or bimolecular electrophilic substitution reaction with rearrangement) (Figure 2). This reaction has a lower energy barrier of 10.5 kcal mol−1, although leads to the less thermodynamically stable product H, corresponding to the vinylogous C−H insertion product 7, which thus corresponds with the kinetic product of the reaction. Both pathways are in agreement with the observations from the deuterium‐labelling experiment (eq 1).
Figure 2.
Gibbs energy profile (in kcal mol−1) of the silver‐catalyzed C−H insertion step, showing the carbenic and vinylogous competing reactivity ([Ag]=Tp(CF3)2,BrAg).
To get more insight into the experimental ratio of products that inserted the C(sp3)−H in the vinylogous (7) and carbenic (6) positions (Scheme 3), some additional DFT calculations were performed. Intermediate G and transition states TS‐GI and TS‐GH were computed for compounds featuring Me, OMe, Ph, F and Br in para position in the phenyldiazo ring and compared to H. Computational results were in perfect agreement with experimental data, as they predicted a preference for the formation of the vinylogous product for substrates bearing H, Me, OMe, and Ph (ΔΔG≠ values of 0.4, 0.9, 1.4, and 1.3 kcal mol−1 were obtained, respectively). The F‐containing substrate that experimentally led to an almost equimolecular mixture of 7 e and 6 e led to a value of 0.0 kcal mol−1, while for Br that experimentally favored the carbenic 6 f over the vinylogous 7 f insertion, a kinetic barrier difference of −1.2 kcal mol−1 was computed. The kinetic preference for the vinylogous addition is explained by the fact that the vinylogous carbon is sterically less hindered than the carbenic one both in intermediate G and in transition states TS‐GH and TS‐GI. This is demonstrated by an 8.0 % lower buried volume (%VBur) [31] of the vinylogous carbon compared to the carbenic one in the case of intermediate G and a further 3.9 % in the subsequent TSs (see Supporting Information for details).
In conclusion, we have developed a silver‐based catalytic system for the carbene‐alkyne metathesis, which provides the unique unprecedented feature of the interception of the vinylogous position with a C(sp3)−H bond. The work shows that development of CAM cascades is a powerful method to access intermediate vinylcarbene metal complexes with otherwise inaccessible substitution patterns and unparalleled reactivity. Furthermore, the combination of experimental and computational techniques was key to reveal the mechanism and especially that the C(sp3)−H bond interception occurs through a stepwise, hydrogen shift pathway, at variance with previously described silver‐catalyzed C−H functionalization by carbene insertion reactions. The discovery of silver as catalyst for the new vinylogous reactivity observed toward alkylic C−H bonds, along with the venue of a stepwise mechanism, opens a new perspective in CAM‐cascade‐C−H activation reactions.
Supporting Information: all procedures and characterization data, computational data, and Cartesian coordinates of the optimized structures (PDF).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Acknowledgments
We are grateful for financial support from the Ministerio de Ciencia e Innovación (PGC2018‐097722‐B‐I00, PID2020‐113797RB‐C21 and PID2020‐113711GB‐I00 MCIN/AEI/10.13039/501100011033 and FPU grants to A.D.‐J. and R.M.‐C.) and the Generalitat de Catalunya (Project 2017‐SGR‐39). We also thank Junta de Andalucía (P18‐1536) and Universidad de Huelva (P.O.Feder UHU202024). A.P.Q. and A.P. are Serra Húnter Fellows and A.P. thanks ICREA Academia Prize 2019. M.A. thanks Junta de Andalucia for a postdoctoral fellowship. E.B. thanks Cátedra Cepsa‐Universidad de Huelva for financial support.
Díaz-Jiménez À., Monreal-Corona R., Poater A., Álvarez M., Borrego E., Pérez P. J., Caballero A., Roglans A., Pla-Quintana A., Angew. Chem. Int. Ed. 2023, 62, e202215163; Angew. Chem. 2023, 135, e202215163.
Contributor Information
Dr. Albert Poater, Email: albert.poater@udg.edu.
Prof. Dr. Pedro J. Pérez, Email: perez@dqcm.uhu.es.
Dr. Ana Caballero, Email: ana.caballero@dqcm.uhu.es.
Prof. Dr. Anna Roglans, Email: anna.roglans@udg.edu.
Dr. Anna Pla‐Quintana, Email: anna.plaq@udg.edu.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.For reviews, see:
- 1a. Gutekunst W. R., Baran P. S., Chem. Soc. Rev. 2011, 40, 1976–1991; [DOI] [PubMed] [Google Scholar]
- 1b. McMurray L., O'Hara F., Gaunt M. J., Chem. Soc. Rev. 2011, 40, 1885–1898; [DOI] [PubMed] [Google Scholar]
- 1c. Hartwig J. F., Larsen M. A., ACS Cent. Sci. 2016, 2, 281–292; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Rogge T., Kaplaneris N., Chatani N., Kim J., Chang S., Punji B., Schafer L. L., Musaev D. G., Wencel-Delord J., Roberts C. A., Sarpong R., Wilson Z. E., Brimble M. A., Johansson M. J., Ackermann L., Nat. Rev. Methods Primers 2021, 1, 43. [Google Scholar]
- 2.
- 2a. Doyle M. P., Duffy R., Ratnikov M., Zhou L., Chem. Rev. 2010, 110, 704–724; [DOI] [PubMed] [Google Scholar]
- 2b. Davies H. M. L., Morton D., Chem. Soc. Rev. 2011, 40, 1857–1869; [DOI] [PubMed] [Google Scholar]
- 2c. Bergstrom B. D., Nickerson L. A., Shaw J. T., Souza L. W., Angew. Chem. Int. Ed. 2021, 60, 6864–6878; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 6940–6954; [Google Scholar]
- 2d. Caballero A., Díaz-Requejo M. M., Fructos M. R., Olmos A., Urbano J., Pérez P. J., Dalton Trans. 2015, 44, 20295–20307; [DOI] [PubMed] [Google Scholar]
- 2e. He Y., Huang Z., Wu K., Ma J., Zhou Y.-G., Yu Z., Chem. Soc. Rev. 2022, 51, 2759–2852. [DOI] [PubMed] [Google Scholar]
- 3. Sambiagio C., Schönbauer D., Blieck R., Dao-Huy T., Pototschnig G., Schaaf P., Wiesinger T., Zia M. F., Wencel-Delord J., Besset T., Maes B. U. W., Schnürch M. A., Chem. Soc. Rev. 2018, 47, 6603–6743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Trost B. M., Hashmi A. S. K., J. Am. Chem. Soc. 1994, 116, 2183–2184; [Google Scholar]
- 4b. Trost B. M., Hashmi A. S. K., Angew. Chem. Int. Ed. Engl. 1993, 32, 1085–1087; [Google Scholar]; Angew. Chem. 1993, 105, 1130–1132; [Google Scholar]
- 4c. Briones J. F., Davies H. M. L., J. Am. Chem. Soc. 2013, 135, 13314–13317; [DOI] [PubMed] [Google Scholar]
- 4d. Xu X., Zavalij P. Y., Doyle M. P., Angew. Chem. Int. Ed. 2013, 52, 12664–12668; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12896–12900; [Google Scholar]
- 4e. Qian Y., Zavalij P. J., Hu W., Doyle M. P., Org. Lett. 2013, 15, 1564–1567; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4f. Cheng Q. Q., Yedoyan J., Arman H., Doyle M. P., J. Am. Chem. Soc. 2016, 138, 44–47; [DOI] [PubMed] [Google Scholar]
- 4g. López E., López L. A., Angew. Chem. Int. Ed. 2017, 56, 5121–5124; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5203–5206; [Google Scholar]
- 4h. Dong K., Humeidi A., Griffith W., Arman H., Xu X., Doyle M. P., Angew. Chem. Int. Ed. 2021, 60, 13394–13400; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 13506–13512. [Google Scholar]
- 5.
- 5a. Davies H. M. L., Saikali E., Clark T. J., Chee E. H., Tetrahedron Lett. 1990, 31, 6299–6302; [Google Scholar]
- 5b. Davies H. M. L., Hu B. H., Saikali E., Bruzinski P. R., J. Org. Chem. 1994, 59, 4535–4541; [Google Scholar]
- 5c. Lian Y., Davies H. M. L., Org. Lett. 2010, 12, 924–927; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Lian Y., Davies H. M. L., Org. Lett. 2012, 14, 1934–1937; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. Zhu D.-X., Xia H., Liu J.-G., Chung L. W., Xu M.-H., J. Am. Chem. Soc. 2021, 143, 2608–2619; [DOI] [PubMed] [Google Scholar]
- 5f. Wang T.-Y., Chen X.-X., Zhu D.-X., Chung L. W., Xu M.-H., Angew. Chem. Int. Ed. 2022, 61, e202207008; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2022, 134, e202207008. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Davies H. M. L., Yokota Y., Tetrahedron Lett. 2000, 41, 4851–4854; [Google Scholar]
- 6b. Qin C., Davies H. M. L., Org. Lett. 2013, 15, 6152–6154; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Xu G., Liu K., Dai Z., Sun J., Org. Biomol. Chem. 2017, 15, 2345–2348; [DOI] [PubMed] [Google Scholar]
- 6d. Zhang C., Li H., Pei C., Qiu L., Hu W., Bao X., Xu X., ACS Catal. 2019, 9, 2440–2447; [Google Scholar]
- 6e. Yang W., Pu M., Lin X., Chen M., Song Y., Liu X., Wu Y.-D., Feng X., J. Am. Chem. Soc. 2021, 143, 9648–9656. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Yue Y., Wang Y., Hu W., Tetrahedron Lett. 2007, 48, 3975–3977; [Google Scholar]
- 7b. Hansen J. H., Davies H. M. L., Chem. Sci. 2011, 2, 457–461; [Google Scholar]
- 7c. Ueda J., Harada S., Nakayama H., Nemoto T., Org. Biomol. Chem. 2018, 16, 4675–4682. [DOI] [PubMed] [Google Scholar]
- 8. Yang Y., Liu Z., Song Q., Sivaguru P., Zanoni G., Wang K., Bi Q., Bi X., Chem Catalysis 2022, 2, 563–577. [Google Scholar]
- 9.
- 9a. Pei C., Zhang C., Qian Y., Xu X., Org. Biomol. Chem. 2018, 16, 8677–8685; [DOI] [PubMed] [Google Scholar]
- 9b. Torres Ò., Pla-Quintana A., Tetrahedron Lett. 2016, 57, 3881–3891. [Google Scholar]
- 10.
- 10a. Padwa A., Blacklock T. J., Loza R., J. Am. Chem. Soc. 1981, 103, 2404–2405; [Google Scholar]
- 10b. Padwa A., Xu S. L., J. Am. Chem. Soc. 1992, 114, 5881–5882. [Google Scholar]
- 11. Hoye T. R., Dinsmore C. J., J. Am. Chem. Soc. 1991, 113, 4343–4345. [Google Scholar]
- 12.For examples from our group, see:
- 12a. Torres Ò., Parella T., Solà M., Roglans A., Pla-Quintana A., Chem. Eur. J. 2015, 21, 16240–16245; [DOI] [PubMed] [Google Scholar]
- 12b. Torres Ò., Roglans A., Pla-Quintana A., Adv. Synth. Catal. 2016, 358, 3512–3516; [Google Scholar]
- 12c. Torres Ò., Solà M., Roglans A., Pla-Quintana A., Chem. Commun. 2017, 53, 9922–9925. [DOI] [PubMed] [Google Scholar]
- 13.For selected examples, see:
- 13a. Marichev K. O., Qiu H., Offield A. C., Arman H., Doyle M. P., J. Org. Chem. 2016, 81, 9235–9246; [DOI] [PubMed] [Google Scholar]
- 13b. Zheng Y., Bao M., Yao R., Qiu L., Xu X., Chem. Commun. 2018, 54, 350–353; [DOI] [PubMed] [Google Scholar]
- 13c. Jansone-Popova S., May J. A., J. Am. Chem. Soc. 2012, 134, 17877–17880. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Nösel P., Nunes dos Santos Comprido L., Lauterbach T., Rudolph M., Rominger F., Hashmi A. S. K., J. Am. Chem. Soc. 2013, 135, 15662–15666; [DOI] [PubMed] [Google Scholar]
- 14b. Zheng Z., Zhang L., Org. Chem. Front. 2015, 2, 1556–1560. [Google Scholar]
- 15. Dong K., Pei C., Zeng Q., Wei H., Doyle M. P., Xu X., ACS Catal. 2018, 8, 9543–9549. [Google Scholar]
- 16.For a comparison on the reactivity of gold and silver, see: Silver in Organic Chemistry (Ed.: Harmata M.), John Wiley and Sons, Inc., Hoboken, 2010, Chapter 12. A Critical Comparison: Copper, Silver and Gold, pp. 357–379. [Google Scholar]
- 17.Deposition number 2191018 (for 7 a) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 18. Le P. Q., May J. A., J. Am. Chem. Soc. 2015, 137, 12219–12222. [DOI] [PubMed] [Google Scholar]
- 19. Padín D., Varela J. A., Saá C., Chem. Eur. J. 2020, 26, 7470–7478. [DOI] [PubMed] [Google Scholar]
- 20. Dong K., Fan X., Pei C., Zheng Y., Chang S., Cai J., Qiu L., Yu Z.-X., Xu X., Nat. Commun. 2020, 11, 2363–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y., Yang Y., Xue Y., Catal. Sci. Technol. 2020, 10, 5513–5524. [Google Scholar]
- 22. Zhang C., Hong K., Pei C., Zhou S., Hu W., Hashmi A. S. K., Xu X., Nat. Commun. 2021, 12, 1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang C., Hong K., Dong S., Pei C., Zhang X., He C., Hu W., Xu X., iScience 2019, 21, 499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hashmi A. S. K., Schuster A. M., Gaillard S., Cavallo L., Poater A., Nolan S. P., Organometallics 2011, 30, 6328–6337. [Google Scholar]
- 25. Cleary S. E., Li X., Yang L.-C., Houk K. N., Hong X., Brewer M., J. Am. Chem. Soc. 2019, 141, 3558–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 26a. Nakamura E., Yoshikai N., Yamanaka M., J. Am. Chem. Soc. 2002, 124, 7181–7192; [DOI] [PubMed] [Google Scholar]
- 26b. Laconsay C. J., Pla-Quintana A., Tantillo D. J., Organometallics 2021, 40, 4120–4132. [Google Scholar]
- 27. Flores J. A., Komine N., Pal K., Pinter B., Pink M., Chen C.-H., Caulton K. G., Mindiola D. J., ACS Catal. 2012, 2, 2066–2078. [Google Scholar]
- 28.
- 28a. Braga A. A. C., Maseras F., Urbano J., Caballero A., Díaz-Requejo M. M., Pérez P. J., Organometallics 2006, 25, 5292–5300; [Google Scholar]
- 28b. Olmos A., Gava R., Noverges B., Belleza D., Jacob K., Besora B., Sameera W. M. C., Etienne M., Maseras F., Asensio G., Caballero A., Pérez P. J., Angew. Chem. Int. Ed. 2018, 57, 13848–13852; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14044–14048. [Google Scholar]
- 29.
- 29a. Lamb K. N., Squitieri R. A., Chintala S. R., Kwong A. J., Balmond E. I., Soldi C., Dmitrenko O., Castiñeira Reis M., Chung R., Addison J. B., Fettinger J. C., Hein J. E., Tantillo D. J., Fox J. M., Shaw J. T., Chem. Eur. J. 2017, 23, 11843–11855; [DOI] [PubMed] [Google Scholar]
- 29b. Dishman S. N., Laconsay C. J., Fettinger J. C., Tantillo D. J., Shaw J. T., Chem. Sci. 2022, 13, 1030–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fukuto J. M., Jensen F. R., Acc. Chem. Res. 1983, 16, 177–184. [Google Scholar]
- 31.
- 31a. Poater A., Cosenza B., Correa A., Giudice S., Ragone F., Scarano V., Cavallo L., Eur. J. Inorg. Chem. 2009, 1759–1766; [Google Scholar]
- 31b. Falivene L., Cao Z., Petta A., Serra L., Poater A., Oliva R., Scarano V., Cavallo L., Nat. Chem. 2019, 11, 872–879. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.