

Abstract
One major determinant of systemic immunity during homeostasis and in certain complex multifactorial diseases (e.g. cancer and autoimmune conditions), is the gut microbiota. These commensals can shape systemic immune responses via translocation of metabolites, microbial cell wall components, and viable microbes. In the last few years, bacterial translocation has revealed itself as playing a key, and potentially causal role in mediating immunomodulatory processes in nongastrointestinal diseases. Moreover, recent observations regarding the presence of complex microbial communities and viable bacteria within gut-distal tissues during homeostasis challenge the current paradigm that healthy mammals are entirely sterile at nonmucosal sites. This review discusses our current understanding of how the gut microbiota orchestrates systemic immunity during non-infectious extraintestinal diseases and homeostasis, focusing on the translocation of viable bacteria to gut-distal sites.
How Do Gut Bacteria Govern Gut-Distal Immunity?
All humans are inhabited by microorganisms; these form a metaorganism comprising a multicellular host and communities of associated microorganisms, which colonize the gastrointestinal tract and other mucosal sites (including the skin, lungs, and vagina). This inherently symbiotic coexistence is in part maintained by spatial separation; epithelial, vascular, and lymphatic barriers serve to avert uncontrolled microbial influx to extraintestinal (systemic) sites [1–3]. Here, the mesenteric lymph nodes (mLNs) and liver are considered ‘firewalls’ that shield internal organs from invading pathobionts (see Glossary) that breach the lymphatic or vascular barrier, respectively [4,5]. Although translocation of viable bacteria could potentially occur from other mucosal sites, this review focuses primarily on the translocation of gut bacteria. Host germline-encoded disease susceptibility risks and environmental factors, such as diet, age, and dysbiosis, can lead to loss of intestinal barrier integrity, allowing viable enteric commensal bacteria to escape to systemic sites, including the mLNs, liver, spleen, and the blood circulatory system. This process is referred to as bacterial translocation [6]. The mechanisms that trigger gut barrier dysfunction are beyond the scope of this review and are described in detail elsewhere [5,7].
Over a century ago, the late Nobel Laureate and visionary Elie Metchnikoff theorized, ‘many diseases which lead to chronic systemic inflammation occur as a result of increased gut bacterial translocation into peripheral organs’ [8]. Numerous studies have provided the important and descriptive foundation of a clear association between gut bacterial translocation and a multitude of gastrointestinal, as well as nongastrointestinal, diseases. In particular, studies that used complex mechanistic gnotobiotic approaches have highlighted potential causal implications of specific translocated gut microbes in mediating key immunomodulatory processes in gut-distal diseases and homeostasis (Table 1, Figure 1, Key Figure). Despite the importance of bacterial translocation in host physio-pathology, the majority of studies focused their efforts on defining the mechanisms of how the microbiota modulates systemic immunity via microbial cell wall components or their produced metabolites, including lipopolysaccharides (LPS) and short-chain fatty acids (SCFAs), respectively [9,10]. The detection of gut commensals at systemic sites is complex, and the most reliable strategy requires a combination of several independent approaches (see Figure I in Box 1). Enabled by technological advancements that have facilitated a substantial increase in the sensitivity and specificity of the detection and characterization of low abundant microbial consortia and viable microbes, accumulating evidence of the existence of a tissue microbiome at nonmucosal sites and the occurrence of viable bacterial translocation during homeostatic conditions is on the rise, challenging the paradigm that nonmucosal tissue is entirely sterile in the absence of overt pathology.
Table 1.
Correlative and Causal Immunomodulatory Consequences of Gut Bacterial Translocation during Extraintestinal Diseases and Homeostasis in Mice and Humans
| Disease model | Detection | Impact on systemic immunity | Causality | Refs |
|---|---|---|---|---|
| Disease: myeloid cancer - preleukemic myeloid proliferation Model: Tet2fl/fl VAVcre |
Culture, sequencing Organs: spleen, mLN, blood |
Spleen: splenomegaly, ↑ GMP cells, ↑CD11b+Gr1+ cells, ↑ IL-6 Blood: ↑GMP cells, ↑CD11b+Gr1+ cells, ↑IL-6 Bone marrow: ↑GMP |
Requirement: ABX in GF mice Sufficiency: DSS, Pam3CSK4 Specificity: TLR-2 signaling | [31] |
| Disease: PDAC Model: K-rasLSLG12D/+; Trp53R172H/+; Pdx-1-Cre (KPC) mice |
Culture, imaging, sequencing Organs/tissue: tumor |
Human LTS: Tumor: ↑CD3+ T cells, ↑CD8+ T cells, ↑Granzyme B FMT with LTS feces: Blood: −IL-2, ↑ (IFN)γ Tumor: ↑CD8+ T cells, ↓ MDSCs, ↓FoxP3+ CD4+ Treg cells FMT with STS feces: Tumor: ↑MDSCs, ↑Treg cells |
Requirement: ABX Sufficiency: FMT from human STS/LTS or control donors | [35] |
| Disease: PDAC Model: KPC mice; K-rasLSLG12D/+; Pdx-1-Cre (KC) mice |
Imaging, sequencing Organs: pancreas |
Pancreas: ↑Intratumoral fibrosis, ↑pancreatic dysplasia, ↑MDSCs, ↑immunosuppressive TAMs, ↓proinflammatory TAMs, ↓IFNγ + CD4+ T cells (Th1), ↓IFNγ + CD8+ T cells (Tc1), ↓CD3+ T cell infiltration, ↓CD8+:CD4+ T cell ratio | Requirement: ABX, GF mice Sufficiency: FMT KPC feces, Bifidobacterium pseudolongum monocolonization | [36] |
| Disease: Cancer Model: Nod2−/− mice; MCA205 sarcoma, MC38 colon carcinoma, TC-1 cervical cancer or RET melanoma |
Culture Organs/tissue: tumor, spleen, mLN |
Spleen: ↑IL-17A producing CD4+ T cells (Th17), ↑IFNγ+ Th17, ↑Thl, ↑Tcl Tumor: E. hirae-induced:↑ CD8+ T cell/Treg cell ratio, ↓Foxp3+Treg cells, ↓IL-17A+ γδT cells B. intestihominis induced: ↑IFNγ+ γδT cells, ↓IL-17A+ γδT cells |
Requirement: ABX Sufficiency: Oral administration of B. intestihominis or E. hirae in B6 or Nod2−/− mice treated with cyclophosphamide Specificity: Comparison to L. johnsonii and B. intestihominis |
[52] |
| Disease: Type 1 diabetes Model: Nod2−/− mice |
Culture, sequencing Organs: mLN, pancreatic LN |
Pancreas: ↑Th17, ↑Thl, insulitis Pancreatic LN: ↑ DCs, ↑macrophages, ↑IL-6, ↑IL-12, ↑Thl, ↑Th17 | Requirement: ABX | [72] |
| Disease: Model: (NZW × BXSB) F1 mice; autoimmune hepatitis (AIH) patients |
Culture, imaging, sequencing Organs: mesenteric veins, mLN, liver, spleen |
Systemic autoantibodies: ↑Anti-dsDNA IgG, ↑anti-RNA IgG, ↑anti-endogenous retrovirus glycoprotein-70, ↑anti-β2 glycoprotein mLN and spleen: ↑Th17, ↑T follicular helper cells (Tfh) Liver: ↑IFN-α1, ↑IFN-α2, ↑ TNF-α |
Requirement: ABX Sufficiency: E. gallinarum monocolonization of GF C57BL/6 (B6) mice Specificity: Comparison to E. faecalis, B. thetaiotaomicron and S. typhimurium |
[62] |
| Disease: SLE Model: TLR7.1 mice; IMQ-treated WT mice |
Culture, sequencing Organs: mesenteric veins, mLN, liver, spleen |
Systemic:↑Type I IFN, ↑plasmacytoid DCs Spleen: Splenomegaly Bone marrow: ↑GMP, ↑common myeloid progenitors, ↑ megakaryocyte-erythrocyte progenitors Kidney: ↑Immune cell infiltration, ↑extramedullary hematopoiesis, nephritis, proteinuria |
Requirement: ABX, GF Sufficiency: FMT of TLR7.1 mice cecal contents, L. reuteri oral administration Specificity: Comparison to L. johnsonii and B. thetaiotamicron |
[63] |
| Disease: SLE Model: Triple congenic (TC) B6. Sle1. Sle2. Sle3 |
Culture, sequencing Organs: mLN, liver | Systemic autoantibodies: ↑Anti-dsDNA IgG mLN: ↑Germinal center B cells, ↑Tfh cells Spleen: ↑IFNγ, ↑IL-17A and IL-10 producing CD4+T cells, ↑eftector memory CD4+ T cells, splenomegaly | Requirement: ABX Sufficiency: FMT from aged TC mouse feces into GF B6 mice | [65] |
| Disease: Primary sclerosing cholangitis (PSC) Model: DDC-fed WT mice with FMT from PSC patients |
Culture, sequencing Organs: mLN, liver, spleen | Liver: ↑Th17, ↑inflammatory genes (serum amyloid A (Saa) and //1b), ↑fibrosis (Colla, and Tmp1) Blood: ↑SAA, ↑bilirubin, ↑alkaline phosphatase |
Requirement: ABX, pore-forming function of K. pneumonia Sufficiency: GF mice inoculated with K. pneumoniae, P. mirabilis E. gallinarum |
[78] |
| Disease: None Model: JH −/−, TLR2−/− 4−/−, TLR4-/ mice |
Culture, sequencing Organs: mLN, liver, blood, spleen |
Mouse blood: tanticommensal IgG (IgG recognizing murein lipoprotein (MLP) antigen) Human blood: ↑Anti-gut commensal IgG, ↑anti-MLP IgG |
Requirement: GF mice; TLR4; bacterial MLP Specificity: Anti-Gram-negative commensal IgG |
[82] |
Abbreviations: ABX, antibiotic treatment; DC, dendritic cell; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; DSS, dextran sodiumsulfate; FMT, fecalmicrobiota transplant; GF, germ free; GMP, granulocyte–monocyte progenitor; IFN, interferon; Ig, immunoglobulin; IL, interleukin; IMQ, imiquimod; LTS, long-term survivor; MDSC, myeloid derived suppressor cells; mLNs,mesenteric lymph nodes; SLE, systemic lupus erythematosus; STS, short-termsurvivor; TAM, tumor-associated macrophage; Tfh, follicular T cell; TLR, Toll-Like receptor; TNF, tumor necrosis factor; Treg cell, T regulatory cell.
Figure 1. Key Figure. Tissue-Specific Immunoregulatory Effects of Translocated Gut Bacteria during Extra-intestinal Diseases and Homeostasis in Mice and Humans.
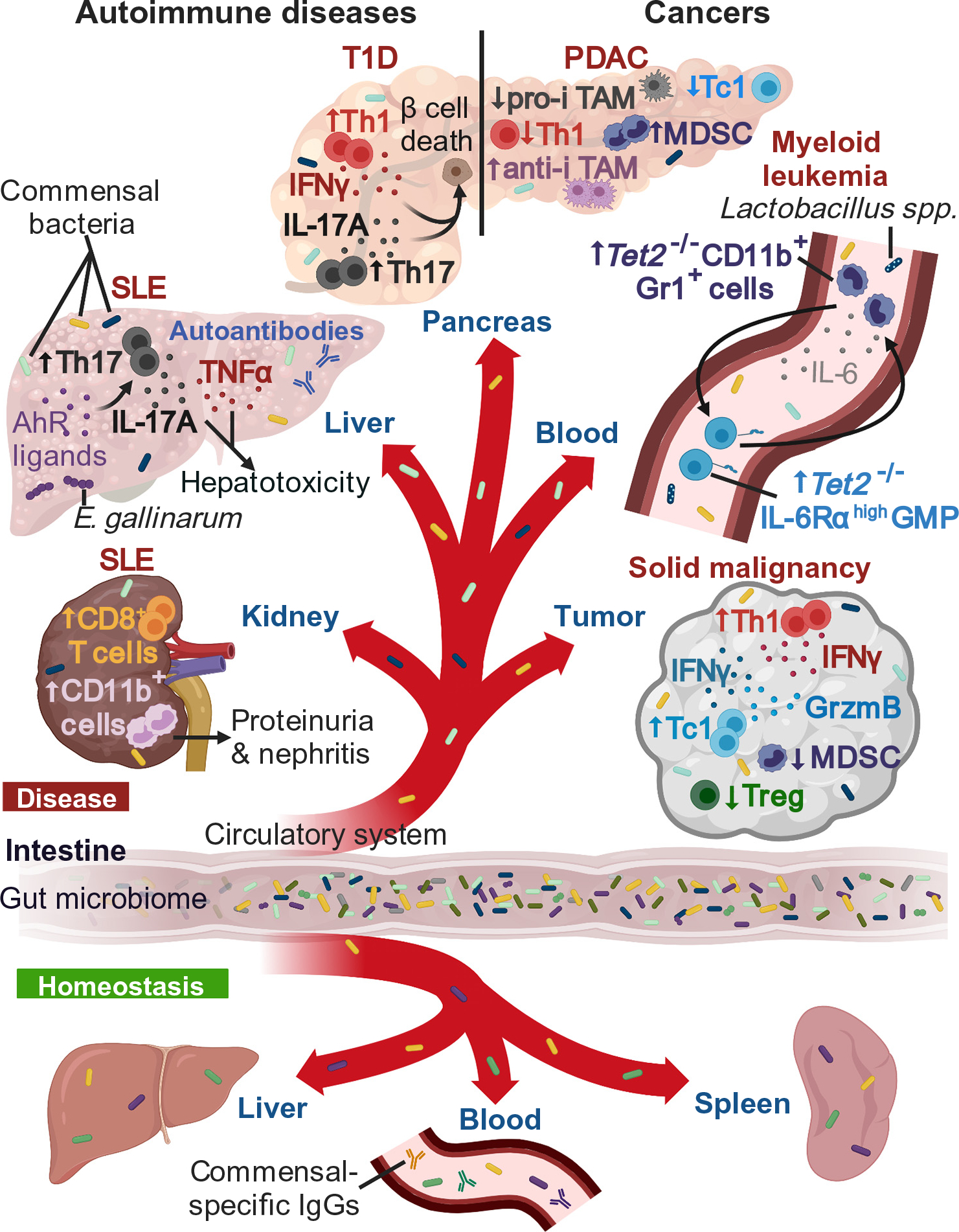
The diagram depicts example of homeostasis and diseases, including cancers and autoimmune diseases. In solid malignancies, bacterial translocation to extraintestinal tumors in mice is associated with an increase in interferon (IFN)γ+ CD4+ T cells (Th1 cells) and cytotoxic IFNγ+/Granzyme B+ (GrzmB) CD8+ T cells (Tc1 cells), with a reduction in immunosuppressive myeloid derived suppressor cells (MDSCs) and regulatory T (Treg) cells [35,52]. Within the pancreas of a pancreatic ductal adenocarcinoma (PDAC) mouse model, intratumoral bacteria are associated with a reduction of Th1 cells, Tc1 cells, and proinflammatory tumor-associated macrophages (pro-i TAMs) and a concomitant increase in MDSCs and anti-inflammatory (anti-i) TAMs [36]. In a myeloid leukemia-like Tet2-deficient mouse model, bacterial translocation (mainly Lactobacillus spp.) triggers high interleukin (IL)-6 production – sensed by IL-6 receptor (IL-6Rα)-overexpressing granulocyte-myeloid progenitor (GMP) cells, which subsequently differentiate into IL-6-producing CD11b+Gr1+ myeloid cells. This cycle results in the development of preleukemic myeloproliferation [31]. For certain autoimmune diseases, such as type 1 diabetes (T1D), bacterial translocation to the pancreas, in a T1D-like mouse model, is associated with an accumulation of Th1 cells and IL-17A+ CD4+ T cells (Th17 cells), resulting in insulin-producing β cell death [72]. Within a systemic lupus erythematosus (SLE)-like mouse model, Enterococcus gallinarum translocation to the liver causes an increase in tumor necrosis factor-α (TNF-α) and triggers an aryl hydrocarbon receptor (AhR)-dependent Th17 cell expansion, which increases autoantibodies and hepatotoxicity [62]. Within the kidney of an SLE-like mouse model, bacterial translocation is associated with an influx of CD8+ T cells and CD11b+ myeloid cells, leading to proteinuria and nephritis [63]. During homeostasis, viable bacteria [50,63,65,76] and anticommensal IgG [62,82,85] have been detected in human and mouse organs, including spleen, liver, and blood.
Figure I.
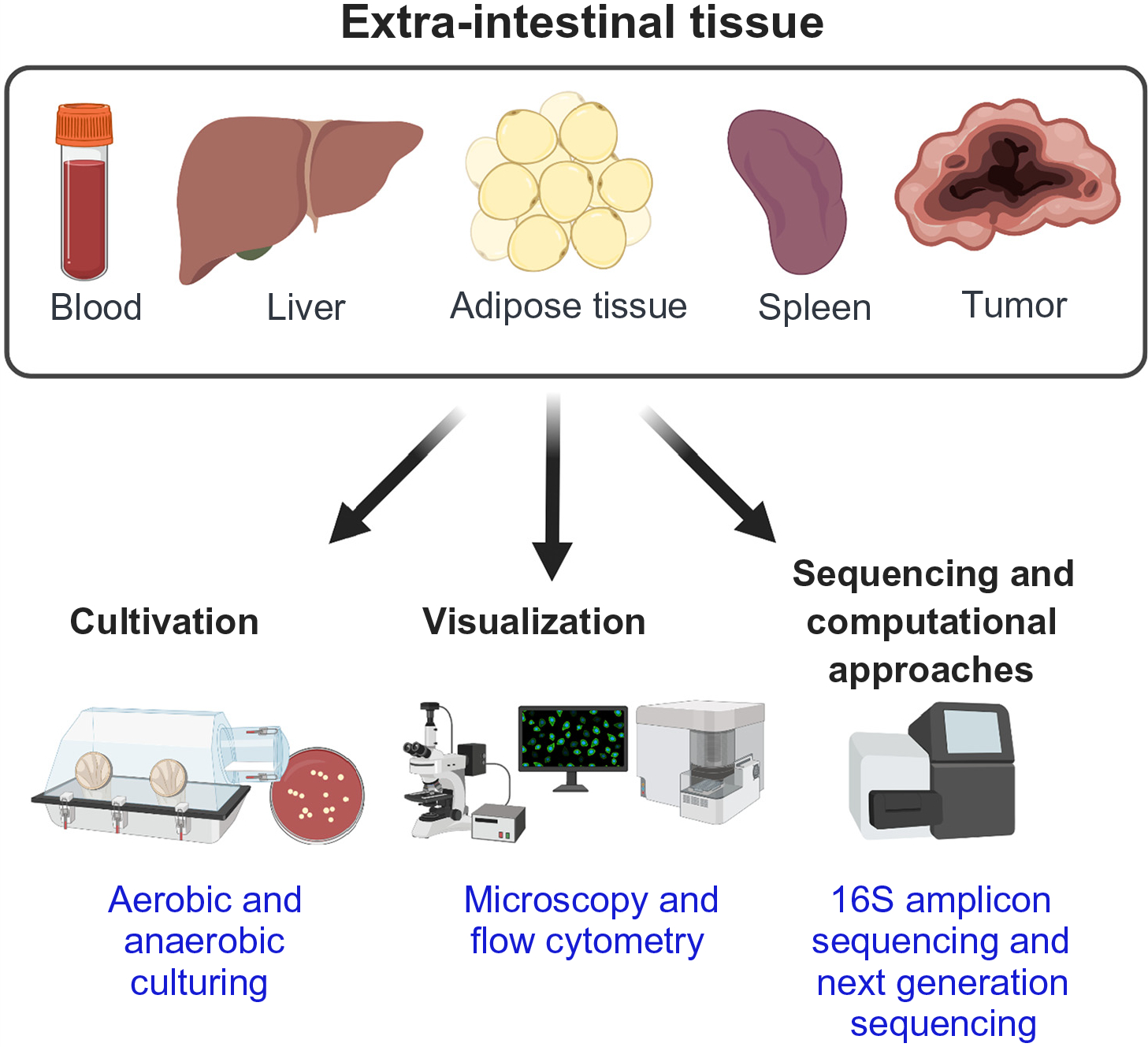
Methods to Detect and Characterize Bacterial Signals and Viable Bacteria in Extra-intestinal tissue.
Box 1. Methods to Detect and Characterize Bacterial Signals and Viable Bacteria at Gut-Distal Sites: Advantages, Disadvantages, and Future Potential (see Figure I).
Cultivation [4,31,35,37,62,63,75,82]
Key Purpose: quantitative assessment of viable bacteria.
Examples: incubation of homogenized tissue: agar plates and broth media.
Advantages: gold standard to demonstrate viability of bacteria in tissues; selective species detection based on culture conditions; broth-expansion promotes detection of low abundant bacteria; individual colonies can be taxonomically classified when combined with sequencing.
Disadvantages: many bacteria are difficult to cultivate; biased growth selection depending on culture conditions; quantification of colony-forming units a broth-expansion does not reflect the quantity of tissue resident viable bacteria in situ.
Future potential: Cultureomics: application of a wide variation of culture conditions to reduce the bias of selective expansions.
Visualization [34–37,62,87]
Key Purpose: quantitative assessment and special distribution of bacteria within tissues or cell types.
Examples (Microscopy): fluorescence in situ hybridization; Gram, lipopolysaccharide, or lipoteichoic acid staining; electron microscopy.
Advantages: enables quantification and cellular localization of bacteria within host tissue in situ.
Disadvantages: unable to differentiate viable from dead bacteria.
Examples (Flow Cytometry): detection of fluorescently stained bacteria within homogenized tissue: SYTO (live and dead bacteria stain) and propidium iodine (PI) (dead bacteria stain).
Advantages: possible to differentiate viable (SYTO+PI-) from dead (SYTO+PI+) bacteria and define the frequency of bacteria within a specific tissue; sorting of viable fraction with subsequent sequencing would enable characterization and quantification of viable bacteria within a tissue in the absence of selective culture-dependent expansion.
Disadvantages: specificity issues, given the low bacterial abundance; facultative anaerobes may be selected against during sample preparation.
Future Potential: single cell sorting of tissue-resident bacteria.
Sequencing and Computational Approaches [35,37–40,62,76,82,83]
Key Purpose: quantitative and qualitative assessment of complex microbial communities or single bacterial taxa.
Examples: 16S rRNA PCR, 16S rRNA quantitative PCR, 16S rRNA amplicon sequencing (full length or variable region); metagenomic or metatranscriptomic sequencing; machine learning approaches.
Advantages: quantitative assessment of bacterial load; detailed taxonomic classification of complex microbial communities or single bacterial taxa within a tissue; reveals functional/metabolic microbial pathways; high throughput; machine learning-approaches enable unbiased stratification of large-scale data sets.
Disadvantages: cannot differentiate between live or dead bacteria; contamination of DNA extraction kits; environment confounds low biomass sample data.
Future Potential: single cell sequencing of tissue-resident bacteria, machine learning approaches.
These described methods can be applied to extraintestinal tissues, independent of a healthy or diseased state. Altogether, the most powerful strategy to detect and define translocated commensal gut bacteria at systemic sites is a combination of all three techniques.
A void exists in the comprehension of the mechanisms and consequences of how translocated gut bacteria shape systemic immunity during health and disease. Thus, the goal of this Review is to provide a detailed overview of the evolving trend of host–gut microbiome interactions geared towards a more comprehensive understanding of the immunomodulatory role of translocated gut bacteria during homeostasis and in noninfectious extraintestinal diseases. Several recent discoveries have highlighted a key relationship between gut dysbiosis, systemic bacterial translocation, and non-autoimmune-mediated liver diseases, but are beyond the scope of this review and discussed elsewhere [11–26]. Our discussion specifically focuses on gut bacterial translocation in cancer and certain autoimmune diseases.
Cancer and Bacterial Translocation
Can Translocated Gut Commensals Drive Malignant Transformation?
The mammalian gut microbiome has emerged as one major factor that exerts a profound impact on the etiopathogenesis of cancer [27–29]. In accordance with epidemiological studies that report an association between myeloid malignancies with antecedent chronic inflammatory conditions of infectious origin [30], our laboratory group recently showed that gut bacterial translocation of Lactobacillus spp. (mainly), resulting from small-intestinal barrier dysfunction, drove the development of preleukemic myeloproliferation (PMP) in mice lacking Tet methylcytosine dioxygenase 2 (Tet2) specifically in hematopoietic cells [31]. PMP was reversed by antibiotic treatment and failed to develop in germ-free (GF) Tet2−/− mice; in addition, both dextran sulfate sodium (DSS)-mediated loss of intestinal barrier integrity or systemic challenge with the Toll-like receptor 2 (TLR2) ligand Pam3CSK4 sufficiently triggered persistent signs of PMP, including CD11b+Gr1+ myeloid cell expansion, in mice lacking Tet2 expression in hematopoietic cells, but not in littermate controls. This demonstrated the importance of microbial signals in the development of PMP in the context of Tet2 deficiency [31]. Mechanistically, microbial-induced interleukin (IL)-6 is sensed by IL-6Rα-overexpressing Tet2−/− myeloid progenitor cells that are, relative to wild-type (WT) myeloid progenitor cells, highly sensitive to exogenous IL-6 and proliferate, giving rise to more CD11b+Gr1+ myeloid cells. These findings are relevant as these myeloid cells in turn continue to produce IL-6, thus creating a vicious cycle that ultimately leads to the development of PMP in mice lacking Tet2 expression in hematopoietic cells relative to WT littermates (Figure 1). This indicates that IL-6 is required for the development of PMP, and intraperitoneal treatment of mice with neutralizing anti-IL-6 antibody reverses PMP in the context of Tet2 deficiency [31]. From a therapeutic perspective, these findings suggest that targeting microbial-induced inflammatory signals, such as IL-6, may be an important therapeutic approach in patients harboring TET2 loss of function mutations and PMP and thus, might potentially reduce the risk of progression to myeloid cancer, although this remains to be assessed. These findings provide evidence that gut barrier dysfunction and microbial-mediated systemic inflammation – in conjunction with unfavorable host genetics – can lead to malignant transformation, here, in the context of myeloid cancer.
Tumor Microbiome at Gut-Distal Sites
In the last decade, several groups have highlighted the crucial immunomodulatory role of the tumor microbiome at mucosal sites [32,33]. Evidence that microbial signatures are present within gut-distal tumors continues to grow. Recently, the tumors of patients with pancreatic ductal adenocarcinoma (PDAC) were shown to harbor a complex microbiome which included bacterial communities expressing the enzyme cytidine deaminase, enabling them to potentially metabolize and inactivate the chemotherapeutic drug gemcitabine, possibly leading to worsened clinical outcomes [34].Indeed, cultured bacteria from human pancreatic tumors mediated gemcitabine resistance in in vitro cell culture systems, suggesting certain microbial communities within pancreatic tumors might potentially abate the efficacy of anticancer chemotherapeutic treatments. Considering that the pancreatic tumor microbiome might be of substantial prognostic value, the tumor microbiome signature was used to stratify PDAC patients according to their length of survival post-diagnosis [35]. Accordingly, the pancreatic tumor microbiome landscape of long-term, in contrast to short-term survivors, was characterized by a substantially increased diversity and an enrichment of three taxa communities: Pseudoxanthomonas, Streptomyces, and Saccharopolyspora, combined with a concomitant increase of tumor-infiltrating, granzyme-B-producing, CD8+ T cells [35]. Within individual patients, taxonomic comparison of feces, PDAC tumor tissue, and normal (adjacent) tissue revealed that one quarter of the tumor microbiome composition matched corresponding gut microbiome samples, suggesting that gut microbes translocated to pancreatic tumor sites. Moreover, a small fraction of advanced pancreatic cancer patients’ fecal microbiome could be detected within implanted pancreatic tumors of K-rasLSL.G12D/+; Trp53R172H/+; Pdx-1-Cre (KPC) mice, post fecal microbiota transplant (FMT) [35]. Furthermore, FMT with long-term, but not short-term, survivor feces potently suppresses tumor growth, triggers increased infiltration of cytotoxic interferon γ (IFNγ)-producing CD8+ T cells (Tc1 cells), and reduces the accumulation of immunosuppressive Foxp3+ CD4+ regulatory T (Treg) cells and myeloid-derived suppressor cells (MDSCs) in tumors. These findings suggest that the gut microbiome could, to some degree, colonize gut-distal tumors and thereby shape systemic tumor immunity [35]. Moreover, fluorescently labeled Enterococcus faecalis fragments are detected after oral administration in healthy WT pancreata. This suggests that intestinal bacteria might directly influence the pancreatic microenvironment. An increased pancreatic bacterial load is detected in both KPC mice and PDAC patients when compared to healthy WT mice or healthy individuals, respectively, indicating that the local pancreatic microbiome could potentially play a pathogenic role in the context of pancreatic cancer [35]. In line with this hypothesis, antibiotic-induced microbial ablation in KPC mice leads to enhanced expansion of antitumorigenic intratumoral IFNγ-producing CD4+ (Th1 cells), Tc1 cells, and proinflammatory tumor-associated macrophages (TAMs), with concomitant suppression of protumorigenic MDSCs and anti-inflammatory TAMs, relative to tumor-bearing mice that do not receive antibiotic treatment [36]. FMT of antibiotic-treated KPC mice, using KPC-mouse derived feces, partially reverses antibiotic-mediated antitumor immune responses, as evidenced by the expansion of intratumoral MDSCs and compression of T cell frequencies. This suggests that a cancer-derived microbiome could aggravate tumor progression in a genetically predisposed host (Figure 1) [36].
A comprehensive characterization of human gut-distal tumor microbiomes (such as pancreas, bone, breast and melanoma tumors) on a species-level resolution revealed a cancer-type-specific tumor microbiome signature [37] (Figure 2). Intratumoral bacterial fragments reside within tumor cells, macrophages, and lymphocytes, and viable bacteria are detected in five breast tumors (predominantly Staphylococcus spp.). Several metabolic functions encoded by intratumoral bacteria have correlated with clinical features of certain tumor subtypes, implying that intratumoral microbes might be active participants in the tumor microenvironment. In the case of human bone cancer, which is associated with elevated concentrations of hydroxyproline – a degradation product of bone collagen – a microbial hydroxyproline-degradation pathway is enriched relative to non-bone tumors [37]. However, while an intratumoral presence of bacteria and bacterial fragments has been established in many models, a causal role of tumor-intrinsic bacteria at gut-distal sites remains to be determined. Aiming to establish a microbiome-based oncological diagnostic tool, one study has deployed complex computational approaches, including independently trained machine-learning (ML) models to characterize the composition of microbial communities in plasma, tumor tissue, and normal (adjacent) tissue samples of ~10 000 cancer patients (33 cancer types) from The Cancer Genome Atlas (TCGA) platform [38]. The established computational model successfully discriminates between cancer types and between tumor and normal tissue. Application of the developed ML tools on metagenomic reads from plasma-derived microbial DNA collected from individuals with prostate, lung, or skin cancer, as well as healthy controls, has shown that blood plasma microbial profiles could be used to achieve healthy-versus-cancer and cancer-versus-cancer discrimination, in line with similar findings by others [39]. The finding that the taxonomic compositions of gut bacteria found in nontumorigenic adjacent tissue shows clear demarcations from the tumor microbiome signatures [35–40] suggests that the tumor provides a niche for specific bacterial communities (Figure 2). This might, depending on the tumor microbiome composition, provide novel opportunities in the diagnosis and treatment of gut-distal cancer types.
Figure 2. Tumor Microbiome at Gutdistal Sites.
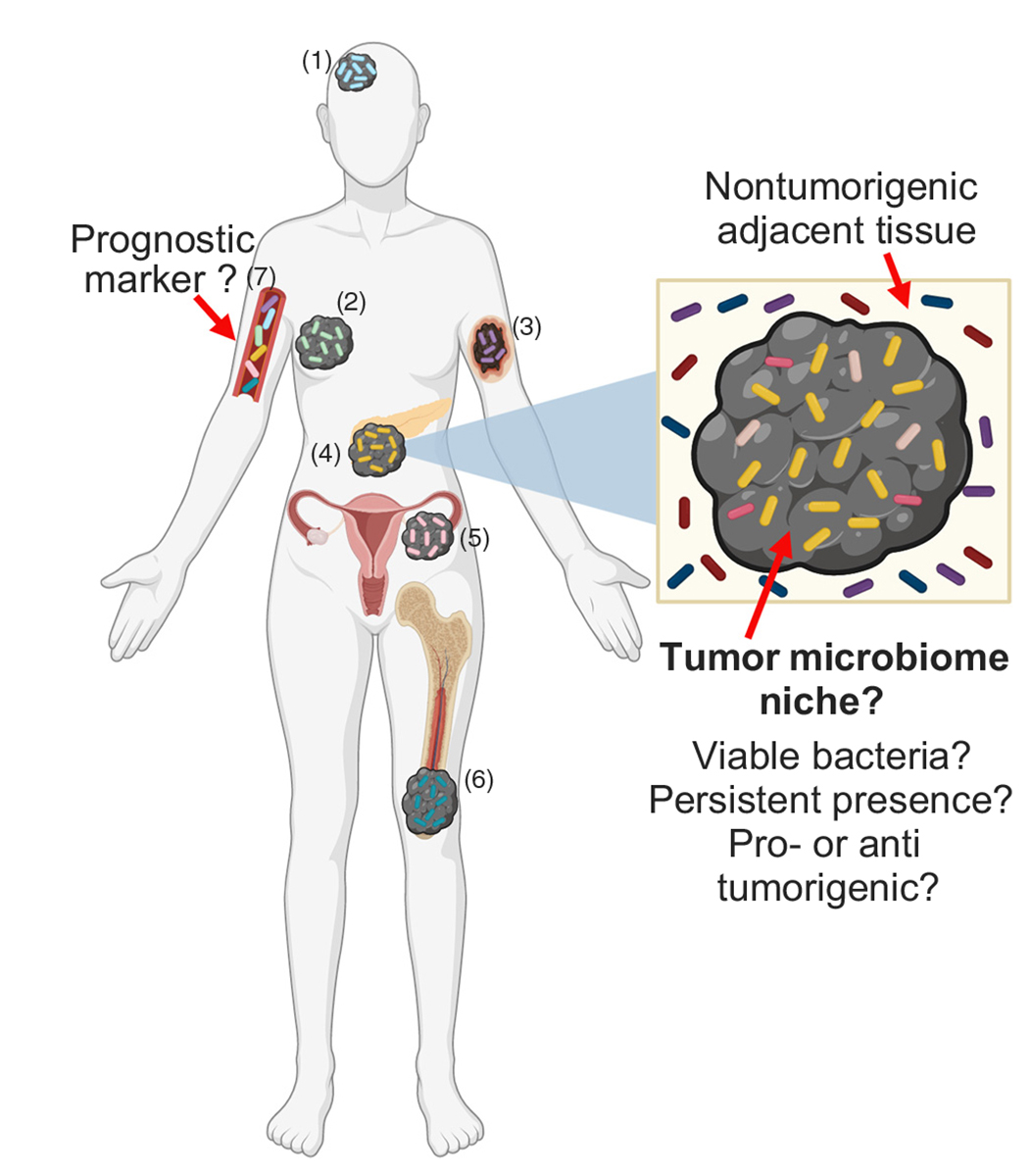
Gut-distal tumor types such as glioblastoma (1), breast (2), melanoma (3), pancreatic (4), ovarian (5), and bone (6) cancer provide a biological niche for specific bacterial growth [35–40]. These distinct human cancer-type-specific tumor microbiomes may provide a novel, powerful diagnostic marker for distinguishing between normal and cancerous tissue, as well as between specific cancer types via analysis of microbial signatures in the plasma (7) [38,39].
One postulated mechanism of how viable gut microbes might gain access to remote tumors is via loss of intestinal barrier integrity [41,42]. However, (i) the underlying mechanisms of how bacteria translocate from the gut to remote tumors and persist in cancerous tissue; (ii) the presence of viable bacteria in systemic tumors; and (iii) the mechanisms of how viable tumor-intrinsic microbes can mediate tumor-specific immune responses and modulate the effects of cancer therapies remain enigmatic [43]. Future studies are warranted that address these key questions when aiming to improve microbial-based cancer therapies. Although the tumor microbiome field is in its early stages, recent cumulative scientific discoveries point towards an active, rather than passive, role of tumor-intrinsic microbes in mediating or contributing to malignant transformation, oncogenesis, and cancer-driven immune responses. New advances on the pro- and antitumorigenic functions of intratumoral microbes have been reviewed elsewhere [41]. A better understanding of the gut-distal tumor microbiome might provide a basis for a new era of microbialbased cancer diagnostics, risk stratification, and novel personalized cancer therapies.
How Do Gut Microbes Shape Systemic Tumor Immunity during Cancer Therapy?
Several recent studies have highlighted the robust ability of the gut microbiome to orchestrate systemic antitumor immunity [32,44–49] in the context of chemotherapeutic regimens [50–52] and immune checkpoint inhibitor (ICI) therapy [44,48,49,53–57]; moreover, probiotic bacteria are gaining traction as a crucial component in successful ICI therapies in mice and humans [48,58–60].
Commensal-specific Th1 cells [49,50,52,53], IL-17A-producing CD4+ Th17 cells [50] and Tc1 cells [52,53] have been identified in the periphery of murine MCA205 sarcoma tumor bearers [49,50], as well as in lung, kidney, and ovarian cancer patients [49,52,53]. Adoptive transfer of ex vivo Bacteroides fragilis-stimulated CD4+ T cells into sarcoma-tumor-bearing mice partially restores ICI efficacy by delaying tumor outgrowth relative to tumor bearers that receive either unstimulated or Bacteroides distasonis-stimulated CD4+ T cells [49]. This attests to the functional relevance and potential commensal specificity of T cell responses in preclinical studies. Moreover, oral gavage of Enterococcus hirae promotes antitumor cognate Th1 cell effector function and CD8+ T cell activation in two distinct preclinical tumor models: the ovalbumin-expressing sarcoma model and the HPV16-E7-expressing TC-1 lung tumor model [52]. This suggests that gut commensals can potentially enhance tumor-antigen-specific immunity, although this warrants further investigation and is likely context dependent. To study systemic (peripheral blood) commensal T cell responses of advanced lung and ovarian cancer patients, one study performed ex vivo cocultures of monocyte and memory CD4+ T cells, stimulated with distinct commensals [52]. Stratifying cancer patients according to the median IFNγ produced by bacteria-specific memory CD4+ T cells has revealed that memory Th1 cells – recognizing selectively E. hirae and Barnesiella intestinihominis, but not other commensals used in this assay – are associated with prolonged progression-free survival of cancer patients [52]. This finding suggests that commensal-specific Th1 cells are of prognostic value in certain cases, and thus, merit further attention.
Another potential mechanism of how gut microbes might orchestrate systemic antitumor immunity is via antigen mimicry. A recent report demonstrates that antigen mimicry between the SVYRYYGL (SVY) epitope expressed on gut commensal Bifidobacterium breve and the B16 melanoma-antigen SIYRYYGL (SIY) promotes antitumor CD8+ T cell immunity in the mouse B16.SIY melanoma model [61]. The accelerated B16.SIY tumor growth in mice lacking B. breve is potently suppressed by either cohousing with B. breve-colonized mice or by adoptive transfer of ex vivo expanded SVY-specific CD8+ T cells [61].
Translocation of viable commensal bacteria might be a potential contributor to systemic antitumor immunity in the context of chemotherapeutic cancer treatments [50,52]. The chemotherapeutic drug, cyclophosphamide (CTX) induces small intestinal morphological abnormalities, such as villi shortening and mononuclear immune cell infiltration, leading to gut barrier dysfunction in naïve mice. This subsequently triggers increased translocation of commensal bacteria Lactobacillus johnsonii, Lactobacillus murinus, E. hirae, Lactobacillus intestinalis, and Lactobacillus reuteri to secondary immune organs relative to untreated mice [50]. The CTX-mediated splenic IL-17A- and IFNγ-producing Th17 cell expansion and tumor-growth-suppressing effect in MCA205 sarcoma and B16-F10 melanoma models are reduced in both antibiotic-treated specific pathogen free (SPF) and GF sarcoma-bearing mice, relative to untreated SPF or GF tumor bearers, respectively. Adoptive transfer of ex vivo expanded IL-17A+ and IFNγ+ Th17 cells into antibiotic-treated SPF sarcoma-bearers re-establishes the CTX-mediated tumor growth retardation [50]. E. hirae, but not L. johnsonii, monocolonization restores CTX-mediated tumor growth retardation in antibiotic-treated SPF sarcoma-bearing mice. This indicates that only specific commensal microbes found to translocate to secondary lymphoid organs upon CTX treatment [50] can mediate the CTX-induced tumoricidal activity [52]. These findings suggest possible mechanisms by which gut commensal bacteria are able to modulate/influence systemic antitumor immunity and perhaps potentiate cancer therapy efficacy, although these possibilities will require rigorous future testing.
Autoimmune Diseases and Bacterial Translocation
Systemic Lupus Erythematosus
Gut dysbiosis and gut barrier dysfunction are two key events that contribute to systemic lupus erythematosus (SLE) etiopathogenesis in mice and humans [5,62–64]. Compelling evidence from three independent studies using different SLE-prone models (see following text) suggest a causal relationship between gut commensal translocation and the development of SLE-like pathology (Figure 1, Table 1). Specifically, one study reported that intestinal barrier dysfunction and translocation of the Gram-positive gut commensal Enterococcus gallinarum in the (NZW×BXSB)F1-SLE-like mouse model play a causal role in SLE-like pathology [62]. Indeed, bacterial monocolonization studies using GF WT hosts have revealed that E. gallinarum and the close phylogenetic relative, E. faecalis, are equally sufficient to mediate gut barrier dysfunction and translocation to systemic sites, as demonstrated from the presence of similar amounts of viable E. gallinarum or E. faecalis in mLNs and liver when compared to GF control mice, which are devoid of viable commensals at systemic sites [62]. However, only E. gallinarum induces autoimmunity-promoting proteins: endogenous retrovirus glycoprotein 70 and β2-glycoprotein 1; a possible mechanism for induction of this aberrant autoimmune response is proposed to be via aryl hydrocarbon receptor (AhR)-mediated Th17 cell priming. Indeed, AhR antagonist treatment suppresses both systemic autoantibody responses and Th17 cell effector functions in antibiotic-treated (NZW×BXSB)F1 mice that are orally gavaged with E. gallinarum, compared to vehicle-treated mice. This observation suggests that microbial AhR ligands, produced by translocated E. gallinarum, which encodes the enzymatic machinery to convert the essential amino acid tryptophan into AhR ligands (as shown by whole-genome sequencing), could promote autoimmunity in this context [62]. Highlighting the translational relevance of these findings, E. gallinarum DNA is present in the livers of SLE patients, and E. gallinarum vaccination of (NZW×BXSB)F1 mice reduces SLE-associated symptoms, including serum titers of autoantibodies and systemic bacterial burden relative to E. faecalis- or B. thetaiotaomicron-vaccinated mice [62]. Additionally, the findings of this study [62] in combination with others [26,65] highlight a potential important role of microbial tryptophan catabolites in the etiopathogenesis of certain mouse models of autoimmune disease.
Intestinal bacterial overgrowth – associated with bacterial translocation – commonly occurs in SLE patients and is reviewed elsewhere [5]. One study has reported that, similar to a fraction of SLE patients, L. reuteri is overabundant in SLE-prone TLR7.1 Tg mouse feces relative to WT feces [63]. Fecal L. reuteri overgrowth is associated with increased intestinal permeability and a dominant translocation of L. reuteri and L. johnsonii in approximately 50% of TLR7.1 Tg mice compared to WT controls or antibiotic-treated TLR7.1 Tg mice, in which systemic translocation of viable bacteria is not detected [63]. Oral administration of L. reuteri, but not L. johnsonii, is sufficient to exacerbate imiquimod-induced SLE-like symptoms in WT mice, suggesting that only specific bacteria that are translocated aggravate pathology. Moreover, establishing a potential therapeutic rationale, dietary-starch-induced promotion of microbial SCFAs suppresses fecal L. reuteri outgrowth and reduces systemic signs of SLE-like pathology in TLR7.1 Tg mice relative to TLR7.1 Tg mice fed a standard diet – possibly via restoration of gut barrier integrity, although this remains to be tested [63].
The observation that a single enteric pathobiont can trigger SLE-like pathology in a healthy host [62] in combination with the fact that SLE patients display gut barrier dysfunction raises the provocative hypothesis that the aberrant immune response in SLE is not solely attributed to increased reactivity to self-antigens but could be, in some cases, synergistic with an increased reactivity to commensal gut microbes [66]. In line with this hypothesis, monocolonization of GF WT mice with the Ro60 ortho-autoantibody-containing commensal B. thetaiotaomicron – commonly found in the human gut microbiota – leads to a substantial crossreactive immune response in vivo, as demonstrated by an increased binding of anti-human-Ro60 IgG to commensal Ro60 mimotopes present in mouse sera ex vivo, when compared to sera derived from GF control mice [67]. Supporting T cell crossreactivity in humans, Ro60-autoantigen-specific CD4+ memory T cell clones from SLE patients have shown reactivity to and proliferate upon stimulation with Ro60-containing bacteria in ex vivo assays [67]. Additionally, crossreactivity between mimotopes expressed on human gut commensal Roseburia intestinalis and autoepitopes present on autoantigen β2-glycoprotein 1, which activated both self-recognizing B and T cells in ex vivo assays, has been reported in a fraction of patients with autoimmune antiphospholipid syndrome (APS), but not in healthy controls [68]. This suggests that commensal antigens can trigger the activation of self-recognizing adaptive immune cells, meriting further investigation to determine whether this aberrant immune response can be causally involved in worsening the outcome of certain autoimmune pathologies. A more in-depth discussion of microbial–host crossreactivity has been recently discussed elsewhere [1]. It appears that gut bacterial translocation may be a common pathophysiological feature in SLE animal models and SLE patients and, consequently, might not be solely restricted to a specific genetic-susceptibility gene.
Type 1 Diabetes Mellitus
A recent report demonstrated that hyperglycemia, which precedes the onset of type 1 diabetes (T1D) [69,70], was both required and sufficient to trigger gut barrier dysfunction in both environmental and genetic models of T1D [71]. In both a streptozotocin (STZ)-induced T1D mouse model and in hyperglycemic Akita mice compared to control mice, hyperglycemia-induced gut barrier dysfunction leads to increased systemic dissemination of Citrobacter rodentium upon enteric infection. Compared to controls, STZ-treated diabetic mice harboring gut epithelial– specific GLUT2 deletion (GLUT2ΔIEC), as well as STZ-treated WT mice with 2-deoxyglucose-mediated inhibition of glucose metabolism, show restored barrier function [71]. This suggests that glucose metabolism plays a protective role in enteric infections in the context of T1D.
In the context of bacterial translocation, one study used nucleotide-binding oligomerization domain-containing protein 2 (NOD2) deficient mice (NOD2−/−) to emphasize a correlative connection between gut bacterial translocation with a NOD2-dependent increase in IL-6- and IL-12-producing dendritic cells and macrophages. This innate inflammatory response drives an expansion of IL-17A- and IFNγ-producing CD4+ and CD8+ effector T cells in pancreatic lymph nodes (pLNs) and pancreatic tissues in STZ-treated mice, relative to controls [72]. This indicates that NOD2-dependent recognition of translocated bacterial products can contribute to STZ-induced T1D development and is associated with proinflammatory immune responses (Figure 1). Highlighting the importance of systemic commensal bacteria recognition in this model, antibiotic treatment confers resistance to STZ-induced proinflammatory immune responses and bacterial translocation; moreover, NOD2-ligand treatment sufficiently re-installs STZ-induced proinflammatory immune responses in antibiotic-treated mice [72]. These are examples of recent studies that underscore the association between gut barrier dysfunction and bacterial translocation in T1D-like mouse models – an area that certainly deserves future investigation.
Bacterial Translocation during Homeostasis
From our current understanding, gut bacterial translocation only occurs with ongoing intestinal or systemic pathology [5], and live commensal bacteria are not thought to reach internal organs under steady state conditions [2,4,73,74].
A study from 1979 investigated whether indigenous bacteria could translocate to systemic tissues during homeostasis [75]. Indeed, viable E. coli were recovered from systemic sites, such as the mLNs, spleen, and liver up to 4 months after monocolonization of healthy GF WT mice. A variety of viable gut commensals, such as Escherichia coli, Proteus mirabilis (both Proteobacteria) and Lactobacillus spp. (Firmicutes), were recovered in mLNs 1 week after colonizing GF mice with cecal contents derived from healthy SPF mice, indicating that gut bacterial translocation in a gnotobiotic setting is not taxonomically restricted [75]. In contrast to one study where E. coli was not detectable at systemic sites upon monocolonization in gnotobiotic mice [4], a recent report demonstrates that 53 distinctive human gut commensal isolates, including E. coli, translocate to mLNs and systemic lymphoid organs during homeostasis in monocolonized gnotobiotic mice [47]. Attesting that gut commensals can similarly translocate to systemic sites in the presence of a complex microbiota, both viable E. coli and Lactobacillus acidophilus are detected in systemic tissues upon oral administration to healthy SPF mice [75]. These findings indicate that bacterial translocation can indeed occur during homeostasis and are in line with recent studies reporting viable commensal bacteria at systemic sites within healthy SPF WT mice [50,63,65,76,77]. Suggesting a symbiotic relationship with the host, commensal bacteria, once translocated to systemic immune organs, can persist up to several months post colonization in mice [47,62,75,78]. Accordingly, several reports identified microbial signals, such as LPS and bacterial 16S rRNA gene fragments, or even a complex microbiome at systemic sites in the absence of overt pathology in mice [65,76,79–82] and humans [38,62,83]. Furthermore, commensal-specific serum IgG antibodies, which are considered a surrogate marker for systemic commensal penetration [4], have been detected in both mice and humans in the absence of overt pathology [76,82,84,85] and have been implicated in conferring protection to systemic infection [82] (Figure 1). Noteworthy, several potential confounding factors can impact the detection of viable gut bacteria in healthy SPF mice, including the genetic background [31,62,63,76], age [65], microbiome [62,75], and diet [65] of mice, in addition to microbial culture techniques [86].
Collectively, these data demonstrate (i) the presence of a complex tissue microbiome at extra-intestinal sites during homeostasis; and (ii) that live commensal bacteria can translocate to systemic tissues, both in the absence and presence of a complex gut microbiome at steady state. Thus, future studies are necessary to define the mechanisms of bacterial translocation during homeostasis, and the subsequent physiological consequences on systemic immunity as well as the evolutionary benefits to the host when conferring protection from noninfectious or infectious pathologies.
Concluding Remarks
Even though many of the detailed mechanisms governing the complex interplay between gut-distal microbiota and systemic immunity remain undeciphered, cumulative research efforts have unveiled the major impact of microbial signals and viable commensal bacteria in mediating systemic immune responses during homeostasis and nongastrointestinal diseases. The consequences of bacterial translocation, including its impact on systemic immune responses, are seemingly context-, tissue- and disease-dependent and possibly influenced by other environmental factors; therefore, they remain an exciting area of future research (see Outstanding Questions). A better understanding of gut bacterial translocation and its systemic immunomodulatory potential will presumably unlock a new wave of microbial-based therapies to treat certain extraintestinal diseases and potentially help us to maintain host-microbial mutualism.
Outstanding Questions.
What is the physiological relevance of bacterial translocation during homeostasis? Can viable commensal microbes use secondary immune organs as a niche, or is there constant low-grade translocation of commensal bacteria from the gut? How do commensal bacteria travel and persist within the host? Is there an evolutionary benefit of bacterial translocation for the host in protecting from noninfectious or infectious diseases? Can we modulate the systemic tissue microbiome in a tissue-specific manner?
Can we manipulate the tumormicrobiome to boost antitumor immunity and potentiate responses to cancer therapies? Are gut bacteria chemoattracted to tumors at gutdistal sites? Does systemic tumor formation per se trigger gut barrier dysfunction enabling gut bacteria to escape to the periphery? If yes, what are the mechanisms?
Can antigen mimicry between gut microbial antigens and tumor antigens boost systemic antitumor immunity by generating crossreactive antitumor T cells? What is the role of commensal specific T cells during cancer?
Is the aberrant immune response in certain autoimmune diseases a combination of increased reactivity to both self-antigens and gut microbial antigens?
Is bacterial translocation contributing to the systemic immunomodulatory effects mediated by FMT?
Besides bacteria, can other gut microbes, such as archaea, bacteriophages, fungi, or protists, translocate to systemic sites during homeostasis or extraintestinal diseases? If yes, how do they modulate systemic immunity during health and disease?
A better understanding of gut bacterial translocation during health and disease might unlock a new wave of microbial-based therapies to treat complex diseases such as cancer and autoimmunity, improve current therapy approaches, and maintain host– microbial mutualism.
Highlights.
A complex tissue microbiome, containing viable bacteria, is present at gut distal sites during homeostasis.
A crucial, and potentially causal role of gut bacterial translocation in modulating systemic immunity during extraintestinal diseases, such as autoimmunity and cancer, is emerging.
The systemic immunoregulatory consequences of gut bacterial translocation are contextual.
Gut-distal tumors and circulatory systems of cancer patients can harbor cancer-type-specific microbial communities of prognostic value.
The efficacy of cancer immunotherapy can be strongly associated with gut bacterial translocation.
Bacterial translocation can exacerbate the severity of certain autoimmune diseases, potentially through modulating self-reactivity.
The aberrant immune response in certain autoimmune diseases may not be attributable to self-antigen reactivity alone, but may be a synergistic effect, including a reaction to gut microbes.
Acknowledgments
The authors would like to apologize that due to length requirements, not all work in this growing field could be discussed and properly cited. The figures were created using BioRender (https://biorender.com/). This work was supported by the UPMC Hillman Cancer Center Pilot Award and the Liver Research Pilot and Feasibility Award (NIH/NIDDK P30DK120531). The authors would like to thank Reinhard Hinterleitner and Mohit Rana for critical reading of the manuscript and helpful discussions.
Glossary
- Antigen mimicry
the sharing of antigen epitopes between microbes and host
- Antiphospholipid syndrome
autoimmune disorder characterized by increased formation of blood clots often due to autoantigen β2-GP1 T cell responses
- Aryl hydrocarbon receptor
ligand-dependent transcription factor; binds several exogenous factors such as microbial tryptophan catabolites; plays important roles regulating metabolism and T cell differentiation
- Cytidine deaminase
enzyme catalyzing the hydrolytic deamination of cytidine and deoxycytidine to uridine and deoxyuridine, respectively
- Dextran sulfate sodium
sulfated polysaccharide, toxic to colonic epithelial cells causing human ulcerative-colitis-like diseases when used in mice
- Dysbiosis
any change to the composition of resident commensal communities relative to the community found in healthy individuals; often associated with disease
- Fecal microbiota transplant
transfer of donor stool, containing fecal microbes, to a recipient
- Gnotobiotic
environment/model organism in which all microorganisms present are defined
- HPV16-E7
human papilloma viral oncoprotein expressed in TC-1 lung tumor cells
- Hyperglycemia
abnormally high glucose concentrations in the blood
- Imiquimod
stimulates the innate immune system by activating TLR7
- Immune checkpoint inhibitor therapy
drugs that block immune checkpoint proteins (from binding to their partner ligands), enabling T cell activation and tumor-cell killing.
- Lipopolysaccharide
endotoxin expressed on the outer membrane of Gram-negative bacteria with potential immunostimulatory activity
- Melanoma
form of skin cancer developed from melanocytes
- Mimotope
peptide sequences which mimic epitopes of specific antigens and are therefore capable of eliciting an antigen-specific immune response
- Murein lipoprotein
outer membrane lipoprotein found in the cell wall of Gram-negative bacteria
- Nucleotide-binding oligomerization domain-containing protein 2
pathogen recognition receptor recognizing bacterial products which include muramyl dipeptide
- Ovalbumin
model antigen for immunology research
- Pathobiont
typically nonharmful symbiont which, when specific genetic or environmental conditions are altered, can become pathogenic to the host
- Preleukemic myeloproliferation
expansion of mature myeloid cells including myeloid progenitor cells in the peripheral blood, spleen, and bone marrow, accompanied by splenomegaly
- Ro60
human RNA-binding protein; ortholog present in bacteria
- Short-chain fatty acid
fatty acid containing less than six carbons derived from intestinal microbial fermentation
- Streptozotocin-induced type 1 diabetes
experimental model of T1D, induced by glucosamine–nitrosourea compound streptozotocin which displays preferential toxicity toward pancreatic β cells
- Systemic lupus erythematosus
chronic, debilitating, multiorgan autoimmune disease with unclear etiology
- Tc1 cells
cytotoxic CD8+ T cells characterized by the production of IFNγ and Granzyme B, triggering cell death of infected/damaged cells
- Tet2
gene encoding a methylcytosine dioxygenase protein; catalyzes the conversion of 5-methyl-cytosine to 5-hydroxymethyl-cytosine and promotes DNA demethylation
- Th1 cells
CD4+ T cells characterized by the production of IFNγ, IL-2, and TNF; promote cell-mediated immune responses
- Th17 cells
CD4+ T cells characterized by the production of IL-17A, IL-17F, IL-21, and IL-22; promote cell-mediated immune responses
- Toll-like receptor 2
pattern recognition receptor expressed on immune cells; recognizes lipid-containing pathogen associated molecular patterns
- Type 1 diabetes
autoimmune disease characterized by the destruction of the endocrine pancreas, specifically insulin-producing β-islet cells required for glucose homeostasis
References
- 1.Ruff WE et al. (2020) Host-microbiota interactions in immune-mediated diseases. Nat. Rev. Microbiol. 18, 521–538 [DOI] [PubMed] [Google Scholar]
- 2.Spadoni I et al. (2015) A gut-vascular barrier controls the systemic dissemination of bacteria. Science 350, 830–834 [DOI] [PubMed] [Google Scholar]
- 3.Spadoni I et al. (2017) Organ-specific protection mediated by cooperation between vascular and epithelial barriers. Nat. Rev. Immunol. 17, 761–773 [DOI] [PubMed] [Google Scholar]
- 4.Balmer ML et al. (2014) The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci. Transl. Med. 6, 237ra266. [DOI] [PubMed] [Google Scholar]
- 5.Fine RL et al. (2020) Mechanisms and consequences of gut commensal translocation in chronic diseases. Gut Microbes 11, 217–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolochow H et al. (1966) Translocation of microorganisms across the intestinal wall of the rat: effect of microbial size and concentration. J. Infect. Dis. 116, 523–528 [DOI] [PubMed] [Google Scholar]
- 7.Marchiando AM et al. (2010) Epithelial barriers in homeostasis and disease. Annu. Rev. Pathol. 5, 119–144 [DOI] [PubMed] [Google Scholar]
- 8.Metchnikoff E and Mitchell PC (1910) The Prolongation of Life. Optimistic Studies, G.P. Putnam [Google Scholar]
- 9.Dorrestein PC et al. (2014) Finding the missing links among metabolites, microbes, and the host. Immunity 40, 824–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rooks MG and Garrett WS (2016) Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 16, 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Llorente C and Schnabl B (2015) The gut microbiota and liver disease. Cell. Mol. Gastroenterol. Hepatol. 1, 275–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarin SK et al. (2019) Microbiome as a therapeutic target in alcohol-related liver disease. J. Hepatol. 70, 260–272 [DOI] [PubMed] [Google Scholar]
- 13.Sharpton SR et al. (2019) Gut microbial metabolism and non-alcoholic fatty liver disease. Hepatol. Commun. 3, 29–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chopyk DM and Grakoui A (2020) Contribution of the intestinal microbiome and gut barrier to hepatic disorders. Gastroenterology 159, 849–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma C et al. (2018) Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360, eaan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brenner DA et al. (2015) Role of gut microbiota in liver disease. J. Clin. Gastroenterol. 49, S25–S27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolodziejczyk AA et al. (2019) The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 11, e9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lelouvier B et al. (2016) Changes in blood microbiota profiles associated with liver fibrosis in obese patients: a pilot analysis. Hepatology 64, 2015–2027 [DOI] [PubMed] [Google Scholar]
- 19.Schierwagen R et al. (2019) Circulating microbiome in blood of different circulatory compartments. Gut 68, 578–580 [DOI] [PubMed] [Google Scholar]
- 20.Hartmann P et al. (2012) Toll-like receptor 2-mediated intestinal injury and enteric tumor necrosis factor receptor I contribute to liver fibrosis in mice. Gastroenterology 143, 1330–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fouts DE et al. (2012) Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J. Hepatol. 56, 1283–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bluemel S et al. (2020) Intestinal and hepatic microbiota changes associated with chronic ethanol administration in mice. Gut Microbes 11, 265–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang AM et al. (2017) Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Invest. 127, 2829–2841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Llorente C et al. (2017) Gastric acid suppression promotes alcoholic liver disease by inducing overgrowth of intestinal Enterococcus. Nat. Commun. 8, 837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duan Y et al. (2019) Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 575, 505–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sonner JK et al. (2019) Dietary tryptophan links encephalogenicity of autoreactive T cells with gut microbial ecology. Nat. Commun. 10, 4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zitvogel L et al. (2015) Cancer and the gut microbiota: an unexpected link. Sci. Transl. Med. 7, 271ps271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elinav E et al. (2019) The cancer microbiome. Nat. Rev. Cancer 19, 371–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gopalakrishnan V et al. (2018) The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 33, 570–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kristinsson SY et al. (2011) Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. J. Clin. Oncol. 29, 2897–2903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meisel M et al. (2018) Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 557, 580–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bullman S et al. (2017) Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 358, 1443–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin C et al. (2019) Commensal microbiota promote lung cancer development via gammadelta T cells. Cell 176, 998–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geller LT et al. (2017) Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 357, 1156–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riquelme E et al. (2019) Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell 178, 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pushalkar S et al. (2018) The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov. 8, 403–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nejman D et al. (2020) The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 368, 973–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poore GD et al. (2020) Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 579, 567–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson KM et al. (2017) Distinguishing potential bacteria-tumor associations from contamination in a secondary data analysis of public cancer genome sequence data. Microbiome 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith A et al. (2019) Distinct microbial communities that differ by race, stage, or breast-tumor subtype in breast tissues of non-Hispanic Black and non-Hispanic White women. Sci. Rep. 9, 11940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cogdill AP et al. (2018) The impact of intratumoral and gastrointestinal microbiota on systemic cancer therapy. Trends Immunol. 39, 900–920 [DOI] [PubMed] [Google Scholar]
- 42.Penn RL et al. (1985) Increased translocation of bacteria from the gastrointestinal tracts of tumor-bearing mice. Infect. Immun. 47, 793–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Atreya CE and Turnbaugh PJ (2020) Probing the tumor micro(b)environment. Science 368, 938–939 [DOI] [PubMed] [Google Scholar]
- 44.Matson V et al. (2018) The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ter Horst R et al. (2016) Host and environmental factors influencing individual human cytokine responses. Cell 167, 1111–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zitvogel L et al. (2017) Anticancer effects of the microbiome and its products. Nat. Rev. Microbiol. 15, 465–478 [DOI] [PubMed] [Google Scholar]
- 47.Geva-Zatorsky N et al. (2017) Mining the human gut microbiota for immunomodulatory organisms. Cell 168, 928–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sivan A et al. (2015) Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vetizou M et al. (2015) Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350, 1079–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Viaud S et al. (2013) The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 342, 971–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iida N et al. (2013) Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342, 967–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daillere R et al. (2016) Enterococcus hirae and Barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity 45, 931–943 [DOI] [PubMed] [Google Scholar]
- 53.Routy B et al. (2018) Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97 [DOI] [PubMed] [Google Scholar]
- 54.Gopalakrishnan V et al. (2018) Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frankel AE et al. (2017) Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia 19, 848–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanoue T et al. (2019) A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 565, 600–605 [DOI] [PubMed] [Google Scholar]
- 57.Mager LF et al. (2020) Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 369, 1481–1489 [DOI] [PubMed] [Google Scholar]
- 58.Poutahidis T et al. (2014) Gut microbiota and the paradox of cancer immunotherapy. Front. Immunol. 5, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.West NR and Powrie F (2015) Immunotherapy not working? Check your microbiota. Cancer Cell 28, 687–689 [DOI] [PubMed] [Google Scholar]
- 60.Wan MLY and El-Nezami H (2018) Targeting gut microbiota in hepatocellular carcinoma: probiotics as a novel therapy. Hepatobiliary Surg. Nutr. 7, 11–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bessell CA et al. (2020) Commensal bacteria stimulate antitumor responses via T cell cross-reactivity. JCI Insight 5, e135597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Manfredo Vieira S et al. (2018) Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science 359, 1156–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zegarra-Ruiz DF et al. (2019) A diet-sensitive commensal Lactobacillus strain mediates TLR7-dependent systemic autoimmunity. Cell Host Microbe 25, 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mu Q et al. (2017) Control of lupus nephritis by changes of gut microbiota. Microbiome 5, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Choi SC et al. (2020) Gut microbiota dysbiosis and altered tryptophan catabolism contribute to autoimmunity in lupus-susceptible mice. Sci. Transl. Med. 12, eaax2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Silverman GJ (2019) The microbiome in SLE pathogenesis. Nat. Rev. Rheumatol. 15, 72–74 [DOI] [PubMed] [Google Scholar]
- 67.Greiling TM et al. (2018) Commensal orthologs of the human autoantigen Ro60 as triggers of autoimmunity in lupus. Sci. Transl. Med. 10, eaan2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ruff WE et al. (2019) Pathogenic autoreactive T and B cells cross-react with mimotopes expressed by a common human gut commensal to trigger autoimmunity. Cell Host Microbe 26, 100–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Winer DA et al. (2016) The intestinal immune system in obesity and insulin resistance. Cell Metab. 23, 413–426 [DOI] [PubMed] [Google Scholar]
- 70.Tilg H et al. (2020) The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 20, 40–54 [DOI] [PubMed] [Google Scholar]
- 71.Thaiss CA et al. (2018) Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 359, 1376–1383 [DOI] [PubMed] [Google Scholar]
- 72.Costa FR et al. (2016) Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J. Exp. Med. 213, 1223–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Macpherson AJ and Uhr T (2004) Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 303, 1662–1665 [DOI] [PubMed] [Google Scholar]
- 74.Rescigno M et al. (2001) Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2, 361–367 [DOI] [PubMed] [Google Scholar]
- 75.Berg RD and Garlington AW (1979) Translocation of certain indigenous bacteria from the gastrointestinal tract to the mesenteric lymph nodes and other organs in a gnotobiotic mouse model. Infect. Immun. 23, 403–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martinez-Lopez M et al. (2019) Microbiota sensing by MincleSyk axis in dendritic cells regulates interleukin-17 and −22 production and promotes intestinal barrier integrity. Immunity 50, 446–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fung TC et al. (2016) Lymphoid-tissue-resident commensal bacteria promote members of the IL-10 cytokine family to establish mutualism. Immunity 44, 634–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nakamoto N et al. (2019) Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat. Microbiol. 4, 492–503 [DOI] [PubMed] [Google Scholar]
- 79.Berg RD and Savage DC (1975) Immune responses of specific pathogen-free and gnotobiotic mice to antigens of indigenous and nonindigenous microorganisms. Infect. Immun. 11, 320–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gordon LE et al. (1955) Studies on susceptibility to infection following ionizing radiation. IV. The pathogenesis of the endogenous bacteremias in mice. J. Exp. Med. 102, 413–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hale P and Hill A (1973) The recovery of Lactobacillus sp. from the livers of healthy mice. Lab. Anim. 7, 119–124 [DOI] [PubMed] [Google Scholar]
- 82.Zeng MY et al. (2016) Gut microbiota-induced immunoglobulin g controls systemic infection by symbiotic bacteria and pathogens. Immunity 44, 647–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Anhe FF et al. (2020) Type 2 diabetes influences bacterial tissue compartmentalisation in human obesity. Nat. Metab. 2, 233–242 [DOI] [PubMed] [Google Scholar]
- 84.Koch MA et al. (2016) Maternal IgG and IgA antibodies dampen mucosal T helper cell responses in early life. Cell 165, 827–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ansaldo E et al. (2019) Akkermansia muciniphila induces intestinal adaptive immune responses during homeostasis. Science 364, 1179–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bonnet M et al. (2020) Bacterial culture through selective and non-selective conditions: the evolution of culture media in clinical microbiology. New Microbes New Infect. 34, 100622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bunker JJ et al. (2015) Innate and adaptive humoral responses coat distinct commensal bacteria with immunoglobulin A. Immunity 43, 541–553 [DOI] [PMC free article] [PubMed] [Google Scholar]