

Key Points
-
•
Previously thought to be restricted to neural tissues, FBXW7β is the predominant FBXW7 isoform in normal and malignant human pro-B cells.
-
•
FBXW7β promotes transcriptional activation of the proinflammatory gene cluster that contains tumor necrosis factor superfamily members.
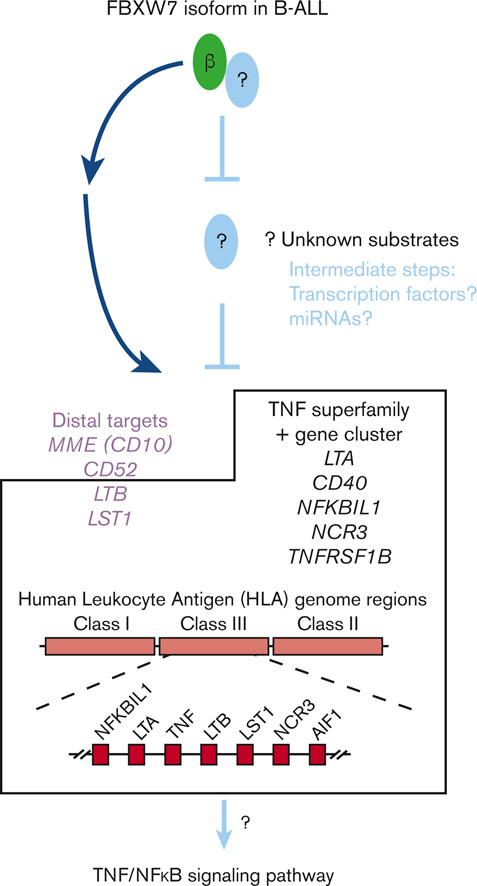
Visual Abstract
Abstract
Noncanonical exon usage plays many important roles in cellular phenotypes, but its contribution to human B-cell development remains sketchily understood. To fill this gap, we collected various B-cell fractions from bone marrow (BM) and tonsil donors, performed RNA sequencing, and examined transcript variants. We identified 150 genes that harbor local splicing variations in all pairwise comparisons. One of them encodes FBXW7, an E3 ubiquitin ligase implicated as a driver in several blood cancers. Surprisingly, we discovered that in normal human pro-B cells, the predominant transcript used an alternative first exon to produce the poorly characterized FBXW7β isoform, previously thought to be restricted to neural tissues. The FBXW7β transcript was also abundant in cell lines and primary samples of pediatric B-cell acute lymphoblastic leukemia (B-ALL), which originates in the BM. When overexpressed in a heterologous cell system, this transcript yielded the expected protein product, as judged by anti-FLAG immunoblotting and mass spectrometry. Furthermore, in REH B-ALL cells, FBXW7β mRNA was the only FBXW7 isoform enriched in the polyribosome fraction. To shed light on possible functions of FBXW7β, we used gain- and loss-of-function approaches and identified an FBXW7-dependent inflammatory gene signature, apparent in a subset of B-ALL with high FBXW7β expression. This signature contained several members of the tumor necrosis factor superfamily, including those comprising the HLA Class III cluster (LTB, LST1, NCR3, LTA, and NFKBIL1). Our findings suggest that FBXW7β expression drives proinflammatory responses, which could contribute to normal B-cell development, leukemogenesis, and responses to anticancer therapies.
Introduction
During normal B-cell development and neoplastic transformation, cellular transcriptomes undergo dramatic changes. Although these changes can be regulated by transcriptional activation and repression, transcript variation is another major mechanism that allows versatile gene regulation in response to cellular and environmental cues. Through tissue- or developmental stage–specific expression of distinct RNA isoforms from a given gene, transcript variation increases proteome diversity.
Transcript variation occurs via multiple contributing mechanisms, which could be divided into 3 major categories: alternative splicing (AS), alternative polyadenylation (APA), and alternative first exon usage (AFE). AS relies on the spliceosome for exon inclusion or exclusion. In contrast, APA and AFE are not directly mediated by the spliceosome and are therefore considered non-AS events.1 While APA involves different poly(A) site selection for pre-mRNA cleavage and the addition of a poly(A) tail, AFE isoforms are driven by distinct promoters. Nevertheless, regulation of these 3 types of transcript variation appears interrelated.1
There is a growing appreciation of the importance of transcript variation in immune system development.2,3 For example, the 2 splicing variants of the E2A transcription factor regulate early B-cell differentiation and class switching.4 However, the extent to which transcript variation contributes to B-cell development and malignancies is not clear, especially in humans. To this end, we previously examined splicing events in B-cell acute lymphoblastic leukemia (B-ALL) and found widespread aberrant splicing.5 Most of the top 20 mutated genes in B-ALL also exhibit transcript variation. Interestingly, among B-ALL samples, the prevalence of transcript variation far exceeds that of somatic mutations in these genes.5,6 One salient example is FBXW7, which is included in the Catalog of Somatic Mutations in Cancer (COSMIC) as a curated tier 1 census gene and cancer hallmark gene.7
FBXW7 has been suggested to play different roles in cancer. It can act as a tumor suppressor or an oncogene in a context-dependent manner.8, 9, 10, 11 To add to the complexity, AFEs of FBXW7 give rise to 3 coding isoforms: α, γ, and β. The protein isoforms localize to distinct cellular compartments and exhibit different tissue distribution.12,13 The α isoform is ubiquitously expressed and extensively studied. In contrast, the expression and function of the β isoform is not fully characterized. Although Fbxw7β is expressed in mouse brains,14 to our knowledge, there is no evidence of Fbxw7β expression in hematopoietic tissues in mice. Expression of the β isoform has been reported in normal human brain, breast, testis, and assorted cancer tissues.15, 16, 17, 18, 19 However, owing to technical constraints, such studies were often limited to RNA instead of protein analysis. In addition, FBXW7β mRNA structure was inferred from individual exon junctions, lacking evidence for the corresponding full-length transcripts. Finally, although experiments with epitope-tagged FBXW7β protein offered interesting insights into its subcellular localization,20 research on its endogenous counterpart was curbed by the lack of isoform-specific antibodies. As a result, relative contributions of the various FBXW7 isoforms to normal development and malignant transformation, especially in the hematological system, remain poorly understood.
To elucidate the role of FBXW7 isoforms in leukemia and normal human B-cell development, we used deep short-read RNA-Seq to discern isoform expression in normal progenitor subsets and primary B-ALL patient samples. We found that FBXW7β is the predominant transcript isoform in normal human pro-B cells, whereas α is the predominant isoform at other stages of development. We detected full-length FBXW7β transcript expression in B-ALL cells using long-read RNA-Seq and demonstrated its translatability using mass spectrometry (MS) and polysome profiling. With isoform-specific knockdown (KD), knockout (KO), and reconstitution systems, we showed that FBXW7β regulates the tumor necrosis factor (TNF) superfamily signaling pathways in B-ALL cells. Such regulation underpins the potential role of FBXW7β and its downstream effectors in leukemogenesis and antitumor cytotoxicity.
Methods
Additional information is contained in the supplemental Methods.
Primary sample acquisition from Children’s Hospital of Philadelphia (CHOP)
According to 45 CFR 102(f), research on deidentified, discarded, or excess specimens does not constitute human subject research. Institutional review board oversight was therefore not required for analyzing the normal pediatric bone marrow (BM) and tonsil samples. Eighteen primary pediatric B-ALL samples were obtained from the CHOP Center for Childhood Cancer Research leukemia biorepository, as previously described.5
BM fractionation
Normal human mononuclear cells and whole BM aspirates underwent Ficoll gradient and fluorescence-activated cell sorting (FACS) selection, as previously described.5 Live cells were sorted into early progenitor (CD34+CD19-IgM-), pro-B (CD34+CD19+IgM-), pre-B (CD34-CD19+IgM-), and immature B-cell (CD34-CD19+IgM+) fractions. Samples from 4 unrelated donors were analyzed.
Naïve mature B-cell isolation
Normal human tonsils from CHOP were manually homogenized with Precision Large Tissue Grinder (Covidien 3500SA) and passed through a 70-μm strainer. Cells were washed with phosphate-buffered saline (PBS), blocked with Human TruStain (Biolegend), and stained with anti-CD19 APC, anti-IgM FITC, and anti-IgD PE. Cells were pelleted by centrifugation at 250 × g for 5 minutes at 4°C and washed twice in PBS. Cells were resuspended in 2% fetal bovine serum (FBS) and 0.1 μg/mL DAPI in PBS. CD19+IgM+IgD+ or CD19+IgM+ cells were sorted on the MoFlo ASTRIOS into 10% FBS RPMI-1640 media with 25-mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES). Samples from 4 unrelated donors were analyzed. CD19+IgM+IgD+ cells were sorted from 3 donors, whereas CD19+IgM+ cells were sorted from 1 donor.
RNA-Seq analysis
RNA was isolated using Qiagen RNeasy Kit (Micro or Mini) or TRIzol. RNA integrity and concentration were determined using the Eukaryote Total RNA Nano assay on BioAnalyzer. RNA-Seq was performed on 10 ng to 1μg of total RNA according to the Genewiz Illumina HiSeq protocol for poly(A)-selected samples (2 × 100 or 150 bp, ∼350 M raw paired-end reads per lane). Paired-end reads were aligned using the RNA-sSq aligner STAR (version 2.7.9a) with flags “--alignSJoverhangMin 8” and “--quantMode GeneCounts.” For the genome reference and the subsequent gene quantification, the GENCODE gene annotation for hg38 (version GRCh38.p13) was provided to STAR. Differential gene expression was calculated using the limma-voom (version 3.52.2) bioconductor package.
Modeling alternative junction inclusion quantification (MAJIQ) splice junction analysis
The MAJIQ algorithm (v1.0.3a) was run with de novo junction detection, 10 reads minimum per junction, each sample being quantified individually against the Ensembl hg38 GFF3 annotation (v94), as described previously.21
Oxford Nanopore Technologies (ONT) long-read sequencing
Total RNA was extracted using the Qiagen RNeasy Kit following manufacturer’s instructions. The mRNA was isolated from 100 μg of total RNA using Dynabeads mRNA DIRECT kit (Invitrogen) and subjected to direct RNA (SQK-RNA002, ONT) library preparation, as described previously.22 Each library was loaded into a Spot-ON Flow Cell R9 version (FLO-MIN106D, ONT) and sequenced in a MinION Mk1B device (ONT) for 48 hours. Raw FAST5 files were converted to FASTQ with Guppy, followed by alignment to the GENCODE version of hg38 (v30) using minimap2; the resulting BAM file was visualized using the Integrative Genomics Viewer.
Polysome profiling
Whole-cell lysate was prepared by incubating 2 × 107 REH cells in lysis buffer supplemented with 100 μg/mL cycloheximide. It was layered onto a 10% to 50% sucrose density gradient, subjected to centrifugation at 35 000 rpm for 3 hours, and fractionated into 23 0.5-mL fractions. UV absorbance was monitored at 260 nm to record the polysome profile and RNA content. After quantitative reverse transcription polymerase chain reaction (qRT-PCR), the relative distribution of specific transcripts across the gradient was calculated using the formula 2Ct (fraction 1-X) × 100/sum, as described previously.23
Immunoprecipitation (IP) with FLAG antibody-agarose conjugate
Anti-FLAG M2 Affinity Gel (Sigma A2220) beads were washed twice with lysis buffer containing 1 × Halt protease and phosphatase inhibitors (Thermo Fisher 78446, 100×). FLAG beads were then applied to cell lysates and incubated at 4°C under rotary agitation for 2 hours. The mixtures were centrifuged at 1300 × g for 3 minutes at 4°C and washed with lysis buffer 3 times. To elute, samples were incubated in 0.1-M Glycine HCl buffer (pH 2-3) for 10 minutes at 4°C under agitation. Eluted supernatant was collected after centrifugation and neutralized by addition of 1-M Tris-HCl (pH 8) equivalent to 40% final volume.
SDS-PAGE and immunoblotting
Samples were mixed with reducing sample buffer (Thermo Fisher 39000, 5× containing DTT) and 12.5% final concentration of 2-Mercaptoethanol. To avoid aggregation of transmembrane proteins, samples were not boiled before running on 7.5% TGX gels (Biorad 4561023) at 130 V. Samples were transferred to Immobilon-P Membrane (Millipore Sigma IPVH00010) at 100 V for 90 minutes on ice. Membrane was blocked in 5% milk in tris-buffered saline containing 0.1% Tween-20 (TBST) at room temperature for 30 minutes before being incubated with primary antibody at 4°C for 16 hours, secondary antibody at room temperature for 30 minutes, and Immobilon Western Chemiluminescent Horseradish Peroxidase Substrate (Millipore WBKLS0500) for 5 minutes. Blots were visualized on Syngene G:Box Chemi XX6 imaging systems.
Data-dependent acquisition (DDA) MS
Samples were processed, cleaned up, and Trypsin/LysC-digested before reverse phase HPLC and MS/MS. MS data were acquired on Thermo Fisher Q Exactive HF Orbitrap at the CHOP Proteomics Core.
Morpholino (MO) transfection
Random control oligo and custom FBXW7 MOs (Gene Tools) were electroporated into REH cells via the Neon transfection system (1325 V, 10 ms, 3 pulses) at 5 μM in 2mL culture media. After 48 hours, cells were used for downstream assays.
Mixed primers RT-PCR
Total RNA was reverse transcribed using random primers and Multiscribe (Invitrogen). For PCR, all 3 FBXW7 first coding exon forward primers were mixed with the reverse primer in coding exon 2. Platinum Taq (Invitrogen) was used for 30 cycles of amplification. PCR products were visualized following gel electrophoresis. Individual bands were gel purified and Sanger sequenced.
Taqman real-time qRT-PCR
qRT-PCR was performed using Taqman Master Mix (Applied Biosystems 4369016). Reactions were performed on Applied Biosystems QuantStudio 7 Flex and analyzed with the embedded software. Comparative CT was calculated against GAPDH expression within the sample. Relative quantity of all samples was then normalized to the control sample within the experiment.
Cell cycle analysis
Cells were processed with FITC or APC BrdU Flow Kit (BD 559619 or 552598) for BrdU and 7-AAD staining following manufacturer’s instructions. Data were collected with FL1 or FL4 and FL3 on Accuri C6 or Accuri C6 Plus.
Cell death analysis
Cells were processed with FITC Annexin V Apoptosis Detection Kit (BD 556547) or APC Annexin V (BD 550474) for Annexin V and propidium iodide staining following manufacturer’s instructions. Data were collected with FL1 or FL4 and FL2 on Accuri C6 or Accuri C6 Plus. A dead-cell positive staining control was prepared by MO control transfection, 16-hour serum-free RPMI incubation, and flow cytometric staining.
Results
Normal human pro-B cells express FBXW7β as the predominant transcript isoform
To capture the extent of transcript variation in normal human B-cell development, we isolated hematopoietic progenitors and B-cell subsets from the BM and tonsils for RNA-Seq (Figure 1A). Cell subsets from 4 BM donors and 4 tonsil donors were analyzed. Using the MAJIQ algorithm,24 we compared splice junctions and exon usage in the early progenitors, pro-B, pre-B, immature, and naïve mature B cells. In these comparisons, thousands of genes were found to harbor at least 1 local splicing variation. Among them, 150 genes exhibit differential exon usage throughout the developmental process (Figure 1B and supplemental Table 5). Gene ontology (GO) analysis revealed that these 150 genes are overrepresented in biological pathways such as cell cycle process and positive regulation of proteolysis (Figure 1C). One of the genes, FBXW7, is involved in 4 of the top 10 biological pathways identified in the GO analysis. FBXW7 is known as the substrate adaptor of the Skp1-Cullin-F-box containing E3 ubiquitin ligase complex, and its 3 coding isoforms (α, γ, and β) are generated through distinct promoters and AFE usage (Figure 1D left).12 While the α isoform is ubiquitously expressed, FBXW7β expression has not previously been reported in human B cells.
Figure 1.

RNA-Seq of primary normal human BM and tonsil fractions identifies FBXW7 transcript variants. (A) FACS analysis of normal human progenitors and B-cell subsets from the BM and tonsils. Flow plots represent 1 of 4 BM donors and 1 of 4 tonsil donors. BM and tonsil donors were distinct. (B) Venn diagram of genes exhibiting differential exon usage throughout human B-cell development. Ensembl gene annotation was used. Cell subsets from 4 BM donors and 4 tonsil donors were analyzed. (C) GO analysis of the 150 genes highlighted in (B). (D) Genomic configuration of the FBXW7 gene (left) and its AFE usage in normal BM subsets (right). Each stacked bar represents means of frequencies of α, γ, and β exon usage across 4 donors.
To our surprise, FBXW7β isoform appears as the predominant isoform in pro-B cells, and the α isoform predominates in other stages of B-cell development (Figure 1D right). The transition from early progenitors to pro-B is accompanied by the α-to-β isoform switch, which is reversed later. We then investigated whether the Fbxw7 isoform switch occurs in the mouse. Upon analyzing existing transcriptional start site profiles (FANTOM5 Cap Analysis of Gene Expression) and RNA-Seq data (NCBI GEO: GSE72018), we found no evidence of Fbxw7β expression in murine hematopoietic tissues (supplemental Figure 1).25,26 Thus, the FBXW7 α-to-β isoform switch may be unique to human B-cell development.
Varying FBXW7 α-to-β transcript ratios in leukemic cell lines and primary B-ALL patient samples
After discovering FBXW7β expression in normal pro-B cells, we next sought to determine its expression status in hematologic malignancies. We visualized FBXW7 coding isoform expression in a panel of cell lines of lymphoid origin from the Cancer Cell Line Encyclopedia (CCLE) (Figure 2A). We observed expression of FBXW7β mostly (but not exclusively) in B-ALL cell lines arising from developing B cells, but generally not in lymphomas and plasma cell cancers originating in later stages of differentiation. Even within the B-ALL subset, the expression patterns of FBXW7 isoforms vary. For example, Nalm6 expresses FBXW7α as the predominant isoform (highlighted in red in Figure 2A), whereas REH predominantly expresses FBXW7β (highlighted in green in Figure 2A). We also examined primary B-ALL patient samples collected at CHOP and St. Jude Children’s Hospital (Figure 2B,C). Patients with B-ALL exhibited differential α-to-β ratios compared with normal counterparts (Figure 2B). We then stratified patient samples based on disease subtypes and profiled their gene expression (supplemental Figure 2). The Not Otherwise Specified (NOS), E2A, ETV6-RUNX1, and Philadelphia chromosome+ subtypes of B-ALL generally exhibited high β-to-α transcript ratios (Figure 2C). However, FBXW7α predominated in the ERG, hypodiploid, infantile, and MLL subtypes (Figure 2C). Intriguingly, usage of FBXW7β first exon is positively correlated with overall FBXW7 transcript expression in hundreds of patients with B-ALL (Figure 2D), possibly reflecting the strength of its promoter.
Figure 2.

RNA-Seq of B-lymphoid cancer cell lines and primary B-ALL patient samples revealed heterogeneous FBXW7 α and β isoform ratios. (A) FBXW7 AFE usage in B-lymphoid cancer cell lines from the CCLE RNA-Seq repository. Junctions per million (JPMs) were obtained by dividing each junction by the total sum of junctions multiplied by 1 million. Junctions are only counted if at least 5 unique spanning reads are observed. The y-axis shows the JPM fractions for each cell line, which approximates percent-spliced-in (PSI). Nalm6 and REH B-ALL cell lines are highlighted. BCL, B-cell lymphoma unspecified; Burkitt’s, Burkitt’s lymphoma; CLL, chronic lymphocytic leukemia-small lymphocytic lymphoma; DLBCL, diffuse large B-cell lymphoma; HL, Hodgkin lymphoma; LCAL, large cell anaplastic lymphoma; MCL, mantle cell lymphoma; MM, multiple myeloma (plasma cell myeloma). (B) FBXW7 AFE usage and transcript expression in patients with B-ALL at the CHOP (n = 18) and normal BM donors (n = 4). Each bar represents 1 sample. Plotted on the y-axis here and in the next panel are PSI values. (C) FBXW7 AFE usage and transcript expression in various B-ALL patient subtypes at St. Jude Children’s Hospital (n = 247). (D) Spearman correlation of β exon usage and overall FBXW7 transcript expression in patients with B-ALL at St. Jude Children’s Hospital.
To correlate FBXW7β expression with cells of origin of pediatric B-ALLs, we performed principal component analysis on transcriptomes of normal BM cell subsets and the primary B-ALL patient samples (supplemental Figure 2A). We also quantified transcript expression of B-cell markers. Most of the B-ALL subtypes in the datasets (except E2A and MLL subtypes) highly expressed CD34 and CD19 transcripts (supplemental Figure 2B), suggesting their pro-B origin. Interestingly, despite that, the FBXW7 isoform expression profiles of the ERG and infantile B-ALL subtypes were decoupled from that of normal pro-B cells; normal pro-B cells predominantly expressed FBXW7β, whereas the ERG and infantile B-ALL samples predominantly expressed FBXW7α (Figure 2C).
Full-length, translatable FBXW7β transcript is expressed in the REH B-ALL cell line and is enriched in the heavy polysome fraction
To further study the biology of FBXW7 isoforms, we performed our own deep short-read RNA-Seq on the REH cell line. Consistent with our previous analysis of the CCLE data, FBXW7β is the predominant isoform in REH cells (Figure 3A). However, short-read technology only provides local information near the reference exon. To study co-occurring junctions in the same transcript, we used ONT long-read RNA sequencing. Single-molecule, full-length FBXW7β transcript was successfully detected in the REH cell line (Figure 3B). Due to the limitations of the long-read technology, the minor RNA isoforms in REH, FBXW7α and γ, were below the detection limit.
Figure 3.

FBXW7β transcript is protein-coding and is enriched in the heavy polysome fraction. (A) Analysis of FBXW7 AFE usage based on short-read RNA-Seq in REH. (B) ONT long-read RNA sequencing demonstrates full-length FBXW7β transcript expression in REH. (C) Detection of FBXW7 protein isoforms following robust transient overexpression in 293T cells using anti-FLAG IP and immunoblotting. (D) DDA MS allowing detection of overexpressed FBXW7 isoform-specific peptides in 293T cells, in addition to peptide sequences common to all isoforms. (E) Polysome profiling followed by qPCR. Specific mRNA distribution was expressed as a percentage of the total signal.
Detection of the FBXW7β protein remained challenging due to lack of suitable antibodies and short protein half-life.27 In our hands, all commercial antibodies failed to detect endogenous FBXW7β by immunoblotting (supplemental Figure 3). Our subsequent attempts to generate mouse antibodies against FBXW7β-specific peptides did not lead to successful detection of FBXW7β (supplemental Figure 4). To circumvent these technical limitations, we overexpressed FLAG-tagged FBXW7β in the 293T cell line for IP and detected the FBXW7β protein by immunoblotting (Figure 3C). Furthermore, we identified 2 independent β-specific peptides in the whole proteome by DDA MS (Figure 3D). Besides isoform-specific peptides, peptides mapped to the regions common to all 3 coding isoforms were also found. To search for endogenous FBXW7 protein in the REH B-ALL cells, we next used the Data-Independent Acquisition (DIA) MS. However, the DIA method failed to detect any FBXW7 peptides in the REH B-ALL cells. We thus investigated the translational status of the FBXW7 isoforms in these cells.
To analyze mRNA distribution in relation to ribosome association, we performed polysome fractionation followed by qPCR. FBXW7α and γ transcripts were found in the monosome fraction, whereas FBXW7β transcript was predominantly found in the heavy polysome fraction in the REH cells (Figure 3E). This implied that FBXW7β transcript in REH cells is translated into a protein. To conclusively assign a function to that protein, we set out to perform both gain- and loss-of-function experiments in this cell line.
Gain- and loss-of-function of the FBXW7β isoform are well tolerated in the REH B-ALL cell model
To discern the relative contribution of each FBXW7 coding isoform to cellular phenotypes, we generated a pan-FBXW7 KO single-cell clone and stably reconstituted individual isoforms (Figure 4A and supplemental Figure 5). To this end, REH cells were first transduced with a Cas9 construct, yielding the REHCas9 cells. The REHCas9 cells were then transiently transfected with 2 CRISPR (clustered regularly interspaced short palindromic repeats) guide constructs simultaneously to remove the coding exon 2 of FBXW7, leading to a pan-FBXW7 KO single-cell clone. Finally, the pan-FBXW7 KO was stably transduced with individual FBXW7 isoform constructs that carried enhanced green fluorescent protein for FACS selection. We encountered no difficulty expanding the transduced cultures, attesting to REH cells’ high tolerance for modest (6- to 40-fold) FBXW7 overexpression (Figure 4B). Furthermore, in REH cells, FBXW7 isoform reconstitution did not affect cell cycle progression or viability (Figure 4C-F). We also used a loss-of-function approach via MO splice site blocking (Figure 5A). The MO targets respective FBXW7 first coding exon and its downstream intron, leading to transient isoform-specific KD (Figure 5B,C). This treatment does not affect cell cycle progression or viability (Figure 5D-G). The 2 complementary cell models, stable reconstitution and transient KD, facilitated our efforts to examine functions of FBXW7β.
Figure 4.

Generation of viable gain-of-function REH B-ALL cell models in which FBXW7 isoforms were knocked out and reconstituted. (A) Experimental approach for stable reconstitution of FBXW7 isoforms in a pan-FBXW7 KO REHCas9 single-cell clone. (B) Taqman qPCR to examine RNA expression in reconstituted REHCas9 FBXW7 KO cells. Relative expression was normalized to REH parental cells. (C-D) Lack of effects on cell cycle progression of FBXW7 isoform reconstitution in KO cells. (E-F) Lack of effects on cell viability of FBXW7 isoform reconstitution in KO cells. Data representative of 2 independent experiments are shown in panels C and E. Data represent means ± SEM of 2 independent experiments in panels D and F. One-way ANOVA was performed, and no significant difference was found between these groups.
Figure 5.

Generation of viable loss-of-function REH B-ALL cell models in which FBXW7 isoforms were transiently knocked down. (A) MO designed to target junctions of FBXW7 alternative first coding exons and corresponding downstream introns for transient isoform KD. (B) Reduction of FBXW7 transcripts by MO in an isoform-specific manner. Mixed primer RT-PCR was performed following FBXW7 KD in REH cells. FBXW7α and β transcripts were detected while γ was below detection limit in the 30-cycle PCR. (C) Taqman qPCR performed on REH cells following FBXW7 isoform KD. Error bars represent means ± SE of 2 independent experiments. One-way ANOVA of all groups was performed followed by Dunnett comparisons between the MO ctrl and other groups. ∗Adjusted P < .05, ∗∗adjusted P < .01. (D-E) Lack of effects on cell cycle progression of transient FBXW7 isoform KD in REH B-ALL cells. (F-G) Lack of effects on cell viability of transient FBXW7 isoform KD in REH B-ALL cells. An independent MO-ctrl–treated sample was serum-starved overnight, acting as a dead-cell positive control. Data representative of 2 independent experiments are shown in panels B, D, and F. Data represent means ± SEM of 2 independent experiments in panels E and G. One-way ANOVA was performed, and no significant difference was found between these groups.
Transcriptome profiling identifies FBXW7β distal targets in reciprocal REH-based B-ALL cell models
As a member of the Skp1-Cullin-F-box containing E3 ubiquitin ligase complex, FBXW7 interacts with a protein substrate, leading to polyubiquitination of the substrate. The fate of the substrate depends on the type of ubiquitin linkage, such as the K48 and K63 polyubiquitin chains. The polyubiquitinated substrate may engage in proteasome-independent signal transduction or, more often, be degraded by the proteasome.28 This makes identification of direct FBXW7 targets challenging. To uncover distal effects of FBXW7α and β isoforms, we performed RNA-Seq and profiled transcriptomes of our reciprocal REH-based B-ALL cell models (Figure 6A).
Figure 6.

Transcriptome profiling of REH B-ALL cell models after FBXW7 isoform KD or reconstitution reveals putative downstream targets. (A) Overlap of selected comparisons (α group vs β group) in RNA-Seq data from the 2 REH cell models. Blue: KD model gene list, differentially expressed genes in MO α vs MO β. Red: Reconstitution model gene list, differentially expressed genes in α reconstitution (KO+a) vs β reconstitution (KO+b). Each group includes replicates from 2 independent experiments. (B) List of overlapping downstream targets that exhibit opposite directionality of transcript changes in the 2 models. (C) Taqman qPCR of the putative downstream target genes in the KD (top) and reconstitution (bottom) models. Arrows indicate directionality of transcript changes comparing the α and β groups. Data represent means ± SEM of 2 independent experiments. One-way ANOVAs of all groups were performed followed by Tukey comparisons between the α and β groups. ∗Adjusted P < .05, ∗∗adjusted P < .01, ∗∗∗adjusted P < .001.
We first analyzed differential transcript expression between REH cells treated with MO FBXW7α or MO FBXW7β. We also compared transcript expression between pan-FBXW7 KO REHCas9 cells reconstituted with either FBXW7α or FBXW7β. Approximately 200 differentially expressed genes were revealed in each comparison (supplemental Tables 6 and 7), and a subset of 8 genes was found to overlap between the 2 lists (Figure 6A). In gain- and loss-of-function genetic perturbation models, distal targets are expected to be regulated in the opposite directions. Therefore, we further filtered the subset of genes based on directionality. Six genes fulfilled these criteria: Protocadherin 10 (PCDH10), Lymphotoxin beta (LTB), Leukocyte specific transcript 1 (LST1), CD52, Membrane metalloendopeptidase (MME, also known as CD10), and FSHD region gene 2 family member C (FRG2C) (Figure 6B). To corroborate the findings from the transcriptome analysis, we performed Taqman qPCR. Results demonstrated downregulation of LTB, LST1, CD52, MME (CD10), and FRG2C and upregulation of PCDH10 in response to FBXW7β KD (Figure 6C top). In contrast, the opposite trend was observed in the reconstitution model, where effects of FBXW7β re-expression were compared with those of FBXW7α re-expression (Figure 6C bottom). These results suggest FBXW7β indirectly promotes LTB, LST1, CD52, MME, and FRG2C transcript expression.
FBXW7β exon usage is positively correlated with the expression of the TNF superfamily and gene cluster in patients with B-ALL
To determine whether these distal targets are associated with FBXW7β expression in leukemia samples, we next examined the large B-ALL patient cohort at St. Jude Children’s Hospital (Figure 7A). Among the putative targets, the involvement of LTB, LST1, CD52, and MME (CD10) in inflammation has been reported. We found transcript expression of these 4 genes positively correlated with the frequency of FBXW7β exon usage (Figure 7A). As expected, there is no correlation between transcript expression of housekeeping genes (EMC7, ACTB, and GAPDH) and FBXW7β exon usage. On the other hand, PCDH10 and FRG2C transcripts are barely expressed in the primary B-ALL patient samples.
Figure 7.

Transcript expression of LTB, LST1, CD52, MME (CD10), and several members in the TNF superfamily pathway is positively correlated with FBXW7β exon usage in patients with B-ALL. (A) FBXW7β exon usage correlated with LTB, LST1, CD52, and MME transcript expression in primary B-ALL patient samples from St. Jude Children’s Hospital. (B) Top: STRING association network indicating functional association of LTB with members in the TNF superfamily signaling pathway. Bottom: STRING association network illustrating co-expression (black lines) of LST1 with other genes. (C) Schematic of HLA genomic loci. Within the HLA Class III region, a gene cluster containing TNF is involved in inflammation. (D) Transcript expression of several members in the TNF superfamily signaling pathway correlated with FBXW7β exon usage in patients with B-ALL from St. Jude Children’s Hospital. Among those that exhibit positive correlation in (C) and (D), LTB, LST1, NCR3, LTA, and NFKBIL1 gene loci are clustered in the same TNF genomic region. (E) Correlations between transcript expression of ∼40 000 genes and the FBXW7β exon usage. Genes were ranked by the Spearman correlation coefficients (rho values). Positive rho values indicate positive correlations. Near-zero rho values indicate lack of correlation.
To explore additional players that may interact with the distal targets, we turned to the STRING association network (Figure 7B).29 The STRING database consolidates prior knowledge from structure-function experiments, co-expression, and automated PubMed text mining. STRING analysis showed functional association of LTB with members in the TNF superfamily signaling pathway (Figure 7B top), while LST1 was associated with a network of genes based on co-expression prediction, including LTB, LTA, NCR3, and NFKBIL1 (Figure 7B bottom). Interestingly, this network of genes is involved in inflammatory responses and housed in the same gene cluster containing TNF in the HLA Class III genomic region (Figure 7C).
Overall, our analysis showed that transcript expression of members in the TNF superfamily signaling pathway and gene cluster (CD40, LTA, LTB, TNFRSF1B, LST1, NCR3, and NFKBIL1) positively correlates with FBXW7β exon usage in patients with B-ALL (Figure 7A and D). Strength of correlations was also analyzed and ranked by the Spearman correlation coefficients (rho values) (Figure 7E and supplemental Table 8). The expression of distal target genes identified in our 2 cell models (LTB, LST1, CD52, and MME) presented strongest correlation with FBXW7β exon usage, whereas other members identified by the STRING functional association network presented more moderate correlation. Taken together, our findings suggest a potential role of FBXW7β and its distal targets in inflammatory responses.
Discussion
Most studies in human malignancies, including T-cell acute lymphoblastic leukemia and colorectal cancers, identify FBXW7 as a tumor suppressor.30 For example, deep deletions and recurrent missense mutations in FBXW7 affect ubiquitination and half-life of the MYC oncoprotein, yielding leukemia-initiating cells.31 Paradoxically, FBXW7 can also promote normal and malignant B-cell survival in various contexts.9, 10, 11,32 These reports highlight the pleiotropic functions of FBXW7 that may be reprogrammed in response to cell-intrinsic and environmental cues. Another intriguing possibility is that its context-dependent roles rely on expression of alternative protein isoforms. FBXW7 encodes 3 coding isoforms (α, γ, and β) that are expressed from different promoters. These 3 protein isoforms differ in their N-termini but share the common nuclear localization signal, dimerization, F-box, and WD40 repeat domains. Of note, the α isoform contains an additional nuclear localization signal, whereas the β isoform contains a unique transmembrane domain,12 and the γ isoform is thought to be nucleolar.33
FBXW7α is the dominant isoform in most tissues, whereas FBXW7β is thought to be expressed in brain, and FBXW7γ in muscle. Most functional discoveries pertaining to FBXW7 are attributable to the α isoform. In colorectal cancer cells, FBXW7α carries out the bulk of the function.27 The FBXW7α isoform also enhances cell survival in the contexts of multiple myeloma and diffuse large B-cell lymphoma.9,11 However, studies that characterize the FBXW7β isoform are limited. Unexpectedly, our data showed FBXW7β to be the dominant isoform in normal and malignant human pro-B cells. Our results provide evidence for full-length FBXW7β transcript expression and support its translational status in B-ALL cells, which is a necessary first step toward understanding the biological relevance of FBXW7β.
We found that usage of the FBXW7β exon is positively correlated with overall FBXW7 transcript expression in primary B-ALL patient samples. This led us to ask what accounts for this tight regulation of FBXW7 isoform expression. Given that the germ line promoter DNA sequences are identical across cell types that express distinct dominant FBXW7 isoforms, the difference in transcript expression may be attributed to transcription factor expression, epigenetic regulation, chromatin landscape, and/or post-transcriptional regulation mediated by microRNA. FBXW7β transcription is induced by p53.34, 35, 36 We did not find a correlation between TP53 and FBXW7β transcript expression in the primary B-ALL patient samples, but because posttranslational modifications of p53 regulate its function as a transcriptional factor,37 additional investigation is needed to clarify the involvement of p53 in the regulation of FBXW7 in B-ALL. In addition, promoter hypermethylation has been suggested to inactivate FBXW7β expression in breast cancer.19 It remains to be determined whether this and other epigenetic mechanisms are reversed in normal and malignant B-cell precursors to allow for unfettered FBXW7β expression.
Regardless of the underlying gene regulation mechanism(s), various FBXW7 isoforms, in particular α and β, may target distinct substrates for ubiquitination to achieve temporal regulation of B-cell development at homeostasis or may play a pathological role in neoplastic transformation. Although FBXW7β-specific substrates in B-ALL still await discovery and validation, we demonstrated functional consequences of the FBXW7β isoform at the transcriptome level and identified LTB, LST1, CD52, and MME (CD10) as distal targets. Among these recognized players in immunity, LTB and LST1 genes reside in the tightly linked inflammatory TNF gene cluster in the HLA Class III genomic region.38 Consistent with this genetic linkage, several members of the TNF superfamily gene cluster (CD40, LTA, LTB, TNFRSF1B, LST1, NCR3, and NFKBIL1) were also found to be associated with FBXW7β expression, at least in B-ALL. The TNF superfamily controls expression of a constellation of genes through NFκB signal transduction and is critical in lymphoid development and regulation of immune responses.39 Our findings therefore corroborate previous reports that implicate FBXW7 in NFκB signaling and broaden the understanding of mechanisms by which FBXW7 controls gene expression.11,32,40,41 Unfortunately, the low number of available BM samples precluded us from establishing similar correlations in normal B-cell progenitors.
From a prognostic and therapeutic perspective, further research is warranted to reveal the role of FBXW7β and its distal targets in leukemogenesis and cytotoxicity. Augmentation of lymphotoxin signaling is known to inhibit tumor growth in multiple xenograft models.42,43 Moreover, activation of the TNF superfamily may enhance antitumor efficacy of cell-based therapies. For instance, lymphotoxin alpha (encoded by LTA) has been shown to potentiate the T-cell response against melanoma and prolong survival in mice.44 Aiming to improve cell therapies, a recent study found that lymphotoxin beta receptor overexpression boosts T-cell functions through activation of the canonical NFκB pathway.45 However, addition of exogenous soluble LTα did not affect the functions of these engineered T cells.45
Conversely, upregulation of the NFκB signaling in leukemic cells may enhance cell survival and promote cancer progression.46 For example, the fusion oncoprotein BCR-ABL1 activates NFκB signaling and drives leukemogenesis.47 As a result, several groups have sought to induce cell death and overcome drug resistance via NFκB inhibition.47,48 However, the success of these approaches is likely dependent on determining the correct timing with respect to cancer stage and treatment phase.49
Independently of their effects on NFκB signaling, several members of the TNF superfamily and death receptor pathways have been shown to synergize with chemotherapeutic agents to kill cancer cells.43,50 Very recently, molecules involved in TNF superfamily signaling, such as FADD and BID in leukemic cells, were implicated in sensitivity to killing by chimeric antigen receptor T cells, and their loss conferred resistance to immunotherapy. Even though the TNF superfamily members under investigation in this study do not contain death domains, they belong to the same interconnected network as the proteins that do. Thus, our testable hypothesis is that leukemias with varying levels of FBXW7β might exhibit differential sensitivity to adoptive cell therapy, and that, in this setting, FBXW7β exon usage could serve as a valuable predictive biomarker.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Acknowledgments
The authors thank former and current ATT laboratory members, former CHOP Clinical Hematopathology technical analyst Nicholas O’Grady, and CHOP ENT surgeons Steven Sobol and Steven Handler for their assistance with sample acquisition; Wei Tong, Kristen W. Lynch, David Allman, Rizwan Saffie, Luca Busino, Steven H. Seeholzer, and Hua Ding for consultation on experimental design; and Junwei Shi, Krista Budinich, and Caroline R. Bartman for consultation and generously gifting us with LentiV_Cas9_puro (cc60) and Lenti_gRNA-GFP(LRG)_2.1T CRISPR plasmids.
This work was supported by grants from the National Institutes of Health (U01 CA232563 to Y.B. and A.T.-T., T32 CA115299 and TL1 TR001880 to S.Y.Y.), St. Baldrick’s Stand Up to Cancer (SU2C-AACR-DT-27-17 to A.T.-T.), the V Foundation for Cancer Research (T2018-014 to A.T.-T.), and by the Alex’s Lemonade Stand Foundation Innovation Award (to A.T.-T.)
Authorship
Contribution: S.Y.Y. designed and performed research, analyzed data, and wrote the paper; K.E.H. contributed vital new reagents or analytical tools and analyzed data; H.F. performed research and analyzed data; L.A.S., M.A., and K.L.B. performed research; A.S.N. analyzed data; V.P. contributed vital new reagents; Y.B. contributed vital analytical tools; K.S.J.E.-J. designed research; and A.T.-T. designed research, analyzed data, and wrote the paper.
Footnotes
Sequencing data can be found in the Gene Expression Omnibus (accession numbers GSE212312 and GSE115656). Original RNA-Seq data from primary normal human tonsil mature B cells, REH parental cell line, REH cells with FBXW7 isoform knockdown, and pan-FBXW7 knockout REH cells with stable FBXW7 isoform reconstitution were deposited in the NCBI GEO database under accession number GSE212312. RNA-Seq from normal early progenitors, pro-B, pre-B, immature B cells (4 normal donors in each normal subset), and primary CHOP B-ALL (18 samples with names beginning with PHL) were deposited in the NCBI GEO database under accession number GSE115656.
All renewable materials and protocol are available on email request to the corresponding author, Andrei Thomas-Tikhonenko (andreit@pennmedicine.upenn.edu).
The full-text version of this article contains a data supplement.
Supplementary Material
References
- 1.van den Hoogenhof MM, Pinto YM, Creemers EE. RNA splicing: regulation and dysregulation in the heart. Circ Res. 2016;118(3):454–468. doi: 10.1161/CIRCRESAHA.115.307872. [DOI] [PubMed] [Google Scholar]
- 2.Ergun A, Doran G, Costello JC, et al. Differential splicing across immune system lineages. Proc Natl Acad Sci U S A. 2013;110(35):14324–14329. doi: 10.1073/pnas.1311839110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaub A, Glasmacher E. Splicing in immune cells-mechanistic insights and emerging topics. Int Immunol. 2017;29(4):173–181. doi: 10.1093/intimm/dxx026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beck K, Peak MM, Ota T, Nemazee D, Murre C. Distinct roles for E12 and E47 in B cell specification and the sequential rearrangement of immunoglobulin light chain loci. J Exp Med. 2009;206(10):2271–2284. doi: 10.1084/jem.20090756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Black KL, Naqvi AS, Asnani M, et al. Aberrant splicing in B-cell acute lymphoblastic leukemia. Nucleic Acids Res. 2018;46(21):11357–11369. doi: 10.1093/nar/gky946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi PS, Thomas-Tikhonenko A. RNA-binding proteins of COSMIC importance in cancer. J Clin Invest. 2021;131(18):e151627. doi: 10.1172/JCI151627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805–D811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yeh CH, Bellon M, Nicot C. FBXW7: a critical tumor suppressor of human cancers. Mol Cancer. 2018;17(1):115. doi: 10.1186/s12943-018-0857-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saffie R, Zhou N, Rolland D, et al. FBXW7 triggers degradation of KMT2D to favor growth of diffuse large B-cell lymphoma cells. Cancer Res. 2020;80(12):2498–2511. doi: 10.1158/0008-5472.CAN-19-2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reavie L, Buckley SM, Loizou E, et al. Regulation of c-Myc ubiquitination controls chronic myelogenous leukemia initiation and progression. Cancer Cell. 2013;23(3):362–375. doi: 10.1016/j.ccr.2013.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Busino L, Millman SE, Scotto L, et al. Fbxw7alpha- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat Cell Biol. 2012;14(4):375–385. doi: 10.1038/ncb2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8(2):83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 13.Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell. 2014;26(4):455–464. doi: 10.1016/j.ccell.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ekholm-Reed S, Goldberg MS, Schlossmacher MG, Reed SI. Parkin-dependent degradation of the F-box protein Fbw7beta promotes neuronal survival in response to oxidative stress by stabilizing Mcl-1. Mol Cell Biol. 2013;33(18):3627–3643. doi: 10.1128/MCB.00535-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu Z, Inomata K, Ishizawa K, Horii A. The FBXW7 beta-form is suppressed in human glioma cells. Biochem Biophys Res Commun. 2007;354(4):992–998. doi: 10.1016/j.bbrc.2007.01.080. [DOI] [PubMed] [Google Scholar]
- 16.Zhou D, Wang X, Liu Z, et al. The expression characteristics of FBXW7 in human testis suggest its function is different from that in mice. Tissue Cell. 2020;62:101315. doi: 10.1016/j.tice.2019.101315. [DOI] [PubMed] [Google Scholar]
- 17.Zheng Y, Song A, Wang C, et al. Isoform specific FBXW7 mediates NOTCH1 Abruptex mutation C1133Y deregulation in oral squamous cell carcinoma. Cell Death Dis. 2020;11(8):615. doi: 10.1038/s41419-020-02873-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vazquez-Dominguez I, Gonzalez-Sanchez L, Lopez-Nieva P, et al. Downregulation of specific FBXW7 isoforms with differential effects in T-cell lymphoblastic lymphoma. Oncogene. 2019;38(23):4620–4636. doi: 10.1038/s41388-019-0746-1. [DOI] [PubMed] [Google Scholar]
- 19.Akhoondi S, Lindstrom L, Widschwendter M, et al. Inactivation of FBXW7/hCDC4-beta expression by promoter hypermethylation is associated with favorable prognosis in primary breast cancer. Breast Cancer Res. 2010;12(6):R105. doi: 10.1186/bcr2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsumoto A, Tateishi Y, Onoyama I, Okita Y, Nakayama K, Nakayama KI. Fbxw7beta resides in the endoplasmic reticulum membrane and protects cells from oxidative stress. Cancer Sci. 2011;102(4):749–755. doi: 10.1111/j.1349-7006.2011.01851.x. [DOI] [PubMed] [Google Scholar]
- 21.Zheng S, Gillespie E, Naqvi AS, et al. Modulation of CD22 protein expression in childhood leukemia by pervasive splicing aberrations: implications for CD22-directed immunotherapies. Blood Cancer Discov. 2022;3(2):103–115. doi: 10.1158/2643-3230.BCD-21-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schulz L, Torres-Diz M, Cortes-Lopez M, et al. Direct long-read RNA sequencing identifies a subset of questionable exitrons likely arising from reverse transcription artifacts. Genome Biol. 2021;22(1):190. doi: 10.1186/s13059-021-02411-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asnani M, Hayer KE, Naqvi AS, et al. Retention of CD19 intron 2 contributes to CART-19 resistance in leukemias with subclonal frameshift mutations in CD19. Leukemia. 2020;34(4):1202–1207. doi: 10.1038/s41375-019-0580-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vaquero-Garcia J, Barrera A, Gazzara MR, et al. A new view of transcriptome complexity and regulation through the lens of local splicing variations. ELife. 2016;5:e11752. doi: 10.7554/eLife.11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lizio M, Harshbarger J, Shimoji H, et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015;16(1):22. doi: 10.1186/s13059-014-0560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brazao TF, Johnson JS, Muller J, Heger A, Ponting CP, Tybulewicz VL. Long noncoding RNAs in B-cell development and activation. Blood. 2016;128(7):e10–e19. doi: 10.1182/blood-2015-11-680843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grim JE, Gustafson MP, Hirata RK, et al. Isoform- and cell cycle-dependent substrate degradation by the Fbw7 ubiquitin ligase. J Cell Biol. 2008;181(6):913–920. doi: 10.1083/jcb.200802076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol. 2016;18(6):579–586. doi: 10.1038/ncb3358. [DOI] [PubMed] [Google Scholar]
- 29.Szklarczyk D, Franceschini A, Kuhn M, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yumimoto K, Nakayama KI. Recent insight into the role of FBXW7 as a tumor suppressor. Semin Cancer Biol. 2020;67(Pt 2):1–15. doi: 10.1016/j.semcancer.2020.02.017. [DOI] [PubMed] [Google Scholar]
- 31.King B, Trimarchi T, Reavie L, et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell. 2013;153(7):1552–1566. doi: 10.1016/j.cell.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramezani-Rad P, Leung CR, Apgar JR, Rickert RC. E3 ubiquitin ligase Fbw7 regulates the survival of mature B cells. J Immunol. 2020;204(6):1535–1542. doi: 10.4049/jimmunol.1901156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Welcker M, Orian A, Grim JE, Eisenman RN, Clurman BE. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr Biol. 2004;14(20):1852–1857. doi: 10.1016/j.cub.2004.09.083. [DOI] [PubMed] [Google Scholar]
- 34.Kimura T, Gotoh M, Nakamura Y, Arakawa H. hCDC4b, a regulator of cyclin E, as a direct transcriptional target of p53. Cancer Sci. 2003;94(5):431–436. doi: 10.1111/j.1349-7006.2003.tb01460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mao J-H, Perez-losada J, Wu D, et al. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004;432(7018):775–779. doi: 10.1038/nature03155. [DOI] [PubMed] [Google Scholar]
- 36.Sionov RV, Netzer E, Shaulian E. Differential regulation of FBXW7 isoforms by various stress stimuli. Cell Cycle. 2013;12(22):3547–3554. doi: 10.4161/cc.26591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20(4):199–210. doi: 10.1038/s41580-019-0110-x. [DOI] [PubMed] [Google Scholar]
- 38.Deakin JE, Papenfuss AT, Belov K, et al. Evolution and comparative analysis of the MHC Class III inflammatory region. BMC Genomics. 2006;7(1):281. doi: 10.1186/1471-2164-7-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fernandes MT, Ghezzo MN, Silveira AB, et al. Lymphotoxin-beta receptor in microenvironmental cells promotes the development of T-cell acute lymphoblastic leukaemia with cortical/mature immunophenotype. Br J Haematol. 2015;171(5):736–751. doi: 10.1111/bjh.13760. [DOI] [PubMed] [Google Scholar]
- 40.Arabi A, Ullah K, Branca RM, et al. Proteomic screen reveals Fbw7 as a modulator of the NF-kappaB pathway. Nat Commun. 2012;3(1):976. doi: 10.1038/ncomms1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fukushima H, Matsumoto A, Inuzuka H, et al. SCF(Fbw7) modulates the NFkB signaling pathway by targeting NFkB2 for ubiquitination and destruction. Cell Rep. 2012;1(5):434–443. doi: 10.1016/j.celrep.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reisfeld RA, Gillies SD, Mendelsohn J, Varki NM, Becker JC. Involvement of B lymphocytes in the growth inhibition of human pulmonary melanoma metastases in athymic nu/nu mice by an antibody-lymphotoxin fusion protein. Cancer Res. 1996;56(8):1707–1712. [PubMed] [Google Scholar]
- 43.Lukashev M, LePage D, Wilson C, et al. Targeting the lymphotoxin-beta receptor with agonist antibodies as a potential cancer therapy. Cancer Res. 2006;66(19):9617–9624. doi: 10.1158/0008-5472.CAN-06-0217. [DOI] [PubMed] [Google Scholar]
- 44.Schrama D, thor Straten P, Fischer WH, et al. Targeting of lymphotoxin-alpha to the tumor elicits an efficient immune response associated with induction of peripheral lymphoid-like tissue. Immunity. 2001;14(2):111–121. doi: 10.1016/s1074-7613(01)00094-2. [DOI] [PubMed] [Google Scholar]
- 45.Legut M, Gajic Z, Guarino M, et al. A genome-scale screen for synthetic drivers of T cell proliferation. Nature. 2022;603(7902):728–735. doi: 10.1038/s41586-022-04494-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weston VJ, Austen B, Wei W, et al. Apoptotic resistance to ionizing radiation in pediatric B-precursor acute lymphoblastic leukemia frequently involves increased NF-kappaB survival pathway signaling. Blood. 2004;104(5):1465–1473. doi: 10.1182/blood-2003-11-4039. [DOI] [PubMed] [Google Scholar]
- 47.Hsieh MY, Van Etten RA. IKK-dependent activation of NF-kappaB contributes to myeloid and lymphoid leukemogenesis by BCR-ABL1. Blood. 2014;123(15):2401–2411. doi: 10.1182/blood-2014-01-547943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Godwin P, Baird AM, Heavey S, Barr MP, O'Byrne KJ, Gately K. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front Oncol. 2013;3:120. doi: 10.3389/fonc.2013.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. 2013;12(1):86. doi: 10.1186/1476-4598-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harrington CT, Sotillo E, Robert A, et al. Transient stabilization, rather than inhibition, of MYC amplifies extrinsic apoptosis and therapeutic responses in refractory B-cell lymphoma. Leukemia. 2019;33(10):2429–2441. doi: 10.1038/s41375-019-0454-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.