

In this study, Song et al. sought to understand the molecular mechanism of polyglutamine-expanded mutant huntingtin mHTT-mediated actions, which causes Huntington's disease (HD), and showed that expression of the N-terminal fragment containing the expanded polyglutamine (HTTQ94) of mHTT is able to promote both ACSL4-dependent and ACSL4-independent ferroptosis. They also identified ALOX5 as a major factor required for the ACSL4-independent ferroptosis induced by HTTQ94, and their findings provide new insights into mHTT-mediated ferroptosis.
Keywords: HTT, ROS, ALOX5, GPX4, ACSL4, ferroptosis, oxidative stress
Abstract
Although it is well established that Huntington's disease (HD) is mainly caused by polyglutamine-expanded mutant huntingtin (mHTT), the molecular mechanism of mHTT-mediated actions is not fully understood. Here, we showed that expression of the N-terminal fragment containing the expanded polyglutamine (HTTQ94) of mHTT is able to promote both the ACSL4-dependent and the ACSL4-independent ferroptosis. Surprisingly, inactivation of the ACSL4-dependent ferroptosis fails to show any effect on the life span of Huntington's disease mice. Moreover, by using RNAi-mediated screening, we identified ALOX5 as a major factor required for the ACSL4-independent ferroptosis induced by HTTQ94. Although ALOX5 is not required for the ferroptotic responses triggered by common ferroptosis inducers such as erastin, loss of ALOX5 expression abolishes HTTQ94-mediated ferroptosis upon reactive oxygen species (ROS)-induced stress. Interestingly, ALOX5 is also required for HTTQ94-mediated ferroptosis in neuronal cells upon high levels of glutamate. Mechanistically, HTTQ94 activates ALOX5-mediated ferroptosis by stabilizing FLAP, an essential cofactor of ALOX5-mediated lipoxygenase activity. Notably, inactivation of the Alox5 gene abrogates the ferroptosis activity in the striatal neurons from the HD mice; more importantly, loss of ALOX5 significantly ameliorates the pathological phenotypes and extends the life spans of these HD mice. Taken together, these results demonstrate that ALOX5 is critical for mHTT-mediated ferroptosis and suggest that ALOX5 is a potential new target for Huntington's disease.
Huntington's disease (HD) is an autosomal dominant hereditary neurodegenerative disease characterized by progressive cognitive, behavioral, and motor dysfunctions and short life spans (Bates et al. 2015). HD is caused by a CAG trinucleotide repeat expansion in exon 1 of the HTT gene, which results in an expanded polyglutamine (polyQ) tract in the encoded huntingtin protein, referred to as mutant huntingtin (mHTT). The mHTT exhibits toxic gain of functions, causing neuronal dysfunction and cell death (MacDonald et al. 1993; Bates 2003). The proteolytic cleavage of mHTT is a key event in the molecular pathogenesis of HD, and it is believed that the N-terminal fragment containing the expanded polyglutamine of mHTT plays an important role in the pathogenesis of HD (Martindale et al. 1998; Lunkes et al. 2002; Graham et al. 2006). Although the genetic cause of HD is well established, the cellular and molecular mechanisms involved in mHTT-mediated early neuronal dysfunction and late neurodegeneration are not fully understood.
Redox signaling is essential for normal brain function, being involved in memory consolidation, neuronal differentiation, and plasticity. Ferroptosis is a regulated form of nonapoptotic cell death driven by excess accumulation of lipid peroxidates critically regulated by the redox signaling (Stockwell et al. 2020). Accumulating evidence indicates that ferroptosis may be involved in both animal models and human patients with Huntington's disease. For example, in transgenic HD mouse models and patients, most neuronal deaths did not exhibit the classic apoptotic features (Turmaine et al. 2000; Hickey and Chesselet 2003). Moreover, high levels of lipid peroxidation are observed as a principal characteristic in HD patients (Klepac et al. 2007). It was reported that increased levels of lipid peroxidation were detected in corticostriatal brain slices (Skouta et al. 2014) and were colocalized with mHTT inclusions in the striatal neurons (Lee et al. 2011). Inhibition of lipid peroxidation with ferrostatin-1 (Ferr-1) significantly rescued the cell death in cellular models of Huntington's disease (HD) (Skouta et al. 2014). On the other hand, low GSH levels are another characteristic in HD patients (Klepac et al. 2007). Indeed, decreased GSH and GSH-S transferase were detected in the striatum, cortex, and hippocampus in 3-nitropropionic acid (3-NP)-induced HD mice (Kumar et al. 2010). mHTT can directly interact with mitochondrial proteins, such as translocase of the inner membrane 23 (TIM23), disrupting mitochondrial proteostasis and favoring ROS production and HD progression (Yablonska et al. 2019). Thus, it is very important to examine whether mHTT is directly involved in regulating ferroptosis, and, more importantly, the molecular factors that mediate mHTT-dependent ferroptosis need to be delineated.
Ferroptosis, an iron-dependent form of nonapoptotic cell death driven by lipid-based reactive oxygen species (ROS), is tightly linked with human diseases including neurodegenerative diseases (Stockwell et al. 2020). Lipid peroxides can be eliminated by glutathione peroxidase 4 (GPX4) and its cofactor, glutathione (GSH) (Stockwell et al. 2017). Thus, ferroptosis is commonly induced by pharmacological agents that directly or indirectly lead to GPX4 inhibition and disruption of this lipid repair system (Stockwell et al. 2017). Notably, by using genome-wide haploid and CRISPR–Cas9-based screening, acyl-CoA synthetase long chain family member 4 (ACSL4) was identified as an essential factor for ferroptosis induced by GPX4 inhibitors (Doll et al. 2017; Kagan et al. 2017). The critical role of ACSL4 in ferroptosis relies on its ability to incorporate arachidonic acid and adrenic acid into phosphatidylethanolamines and thereby provides the main substrates for peroxidation (Kagan et al. 2017). In addition to GPX4-mediated neutralization of lipid peroxidation, the levels of cellular lipid peroxides can be induced enzymatically by the lipoxygenases. Indeed, our recent studies showed that the ferroptotic response induced by the ALOX12 lipoxygenase plays an important role in catalyzing lipid peroxidation and p53-mediated effect on Myc-induced lymphomagenesis (Chu et al. 2019). Together, these studies indicate that two types of ferroptosis are critical for oxidative stress responses: ACSL4-dependent ferroptosis is induced by these common ferroptosis inducers mainly including GPX4 inhibitors, whereas ACSL4-independent ferroptosis is induced by high levels of reactive oxygen species (ROS).
Here, we showed that expression of the mHTT fragment with the expanded 94 glutamine residues (HTTQ94) directly promotes ferroptosis in both neuronal cells and neuroblastoma cells upon erastin treatment. As expected, inhibition of ACSL4 in those cells completely abrogated the ferroptotic response induced by HTTQ94; surprisingly, however, inactivation of the Acsl4 gene failed to show any significant effect in the Huntington's disease transgenic mouse model (HD-N171-82Q). Moreover, we identified ALOX5 as a major mediator for HTTQ94-driven ferroptosis upon ROS-induced stress. Interestingly, neither ACSL4 nor GPX4 is required for ALOX5-dependent ferroptosis induced by HTTQ94. Notably, knockout of the Alox5 gene abrogated ferroptosis in the striatal neurons from the HD mice and significantly improved the pathological phenotypes of Huntington's disease mice. Thus, our study reveals a novel ferroptosis pathway critically involved in the pathological mechanism of Huntington's disease.
Results
HTTQ94 expression sensitizes neuronal cells and other cell types to ferroptosis
To elucidate the potential role of ferroptosis in Huntington's disease, we first examined whether mHTT is involved in regulating ferroptotic responses. To this end, we used the mouse hippocampal neuronal cell line HT-22 to establish HTTQ94 tet-on-inducible cell lines in which expression of the N-terminal fragment of mutant huntingtin protein containing the expanded 94 glutamine residues (HTTQ94) can be induced by doxycycline. As shown in Figure 1A, high levels of HTTQ94 were indeed induced upon the treatment of doxycycline in those cells. Notably, although induction of HTTQ94 alone did not significantly induce any cell death (Fig. 1B,C), the combination of HTTQ94 expression and the erastin treatment produced higher levels of cell death than either treatment alone, and the cell death was completely inhibited by ferrostatin-1, a specific ferroptosis inhibitor (Fig. 1B,C). To corroborate these findings, we performed similar assays by establishing a human neuroblastoma SK-N-BE(2)C HTTQ94-inducible cell line (Supplemental Fig. S1A). Notably, although native SK-N-BE(2)C cells are highly resistant to the erastin treatment, high levels of ferroptosis were induced upon HTTQ94 expression in those cells (Supplemental Fig. S1B,C). Moreover, we also established a control tet-on-inducible cell line in which the N-terminal fragment of wild-type huntingtin protein containing the normal 19 glutamine residues is expressed (HTTQ19) (Fig. 1D). In contrast to the effects induced by HTTQ94 expression, expression of HTTQ19 failed to sensitize the cells to ferroptosis (Fig. 1E). Taken together, these data demonstrate that HTTQ94 is involved in promoting ferroptosis in neuronal cells as well as other cell types.
Figure 1.

HTTQ94 expression sensitizes neuronal cells and other cell types to ferroptosis. (A) Western blot analysis for HTTQ94 from HTTQ94 tet-on HT-22 cells treated with 0.5 μg/mL doxycycline for 2, 4, 8, and 16 h. (B) HTTQ94 tet-on HT-22 cells were preincubated with 0.5 μg/mL doxycycline for 4 h and then treated with 1 μM erastin for 12 h with/without 2 μM Ferr-1. (C) Representative phase-contrast images of cell death, related to B. (D) Western blot analysis for mutant HTT fragment (HTTQ94) and normal HTT fragment (HTTQ19) expression in the tet-on H1299 cells treated with 0.5 μg/mL doxycycline (tet) for 16 h. (E) Cell death assay. Control, HTTQ94, and HTTQ19 fragment tet-on H1299 cells preincubated with 0.5 μg/mL doxycycline for 16 h were treated with 30 μM erastin for 48 h with/without 2 μM Ferr-1. Cell deaths were calculated from three replicates. Data shown in B and E are the means ± SD. P-values were derived from two-tailed unpaired t-test. (***) P ≤ 0.001, (n.s.) P > 0.05.
Inactivation of ACSL4 has no obvious effect on the pathological phenotypes and the life span of Huntington's disease mice
Recent studies indicate that acyl-CoA synthetase long chain family member 4 (ACSL4) is essential for the ferroptotic responses induced by either erastin or GPX4 inhibitors (Doll et al. 2017; Kagan et al. 2017; Stockwell et al. 2017). To dissect the molecular mechanism of mHTT-mediated ferroptosis, we first examined the effect of the ACSL4 inhibitors on HTTQ94-mediated ferroptosis. Both the ACSL4 inhibitors rosiglitazone (ROSI) and troglitazone (TRO) prevented HTTQ94-mediated ferroptosis in both HT-22 and SK-N-BE(2)C cells (Fig. 2A,B). To further validate the role of ACSL4 in HTTQ94-mediated ferroptosis, we generated Acsl4 knockout cells from the HTTQ94 tet-on SK-N-BE(2)C cell line using the CRISPR/cas9 method. Western blot analysis revealed that ACSL4 protein was undetectable in Acsl4-Crispr cells, whereas the same levels of HTTQ94 were induced in both control and Acsl4-Crispr cells (Fig. 2C). Indeed, the ferroptosis response induced by HTTQ94 and erastin was completely abrogated in Acsl4-null cells (Fig. 2D). The results were further confirmed by four independent Acsl4 knockout cell lines under the same conditions (Supplemental Fig. S1D,E). These results demonstrate that HTTQ94 plays an important role in promoting the ferroptotic response upon GPX4 inhibition and that this activity can be completely abolished upon loss of ACSL4.
Figure 2.
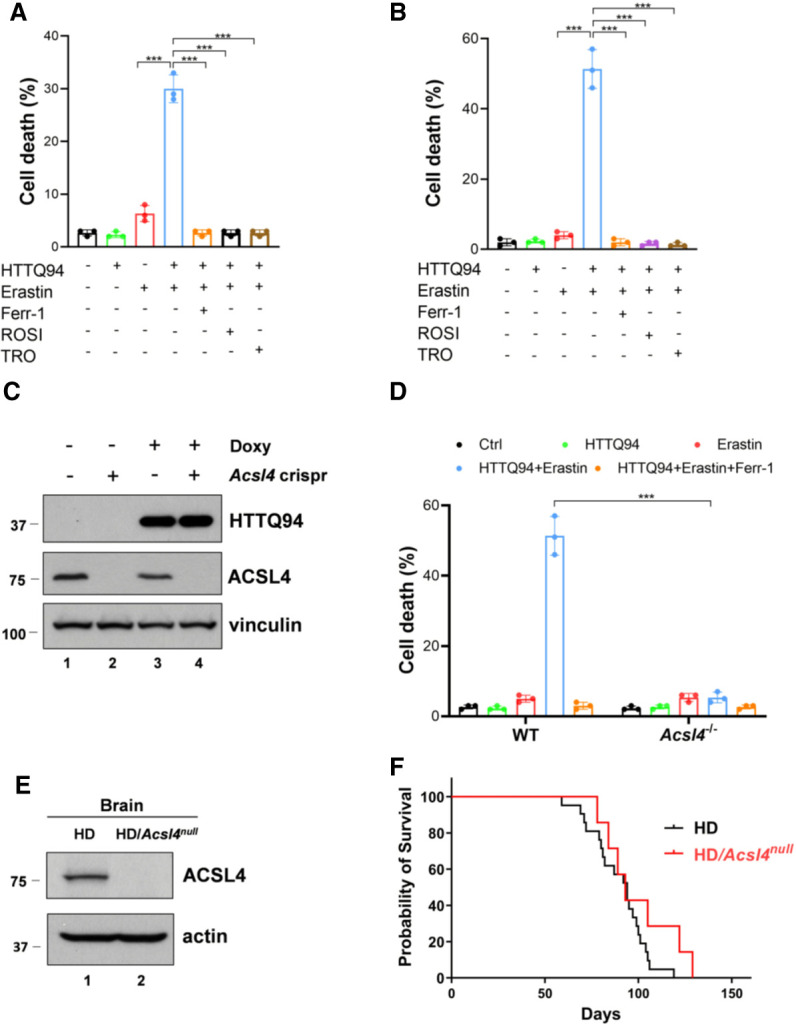
The role of ACSL4 in mHTT-induced ferroptosis and the life span of the HD mice. (A) Cell death assay. HTTQ94 tet-on HT-22 cells preincubated with 0.5 μg/mL doxycycline for 4 h were treated with 1 μM erastin for 12 h in the presence or absence of 2 μM ferrostatin-1 (Ferr-1) or ACSL4 inhibitors (10 μM rosiglitazone [ROSI] and 10 μM troglitazone [TRO]). (B) Cell death assay. HTTQ94 tet-on SK-N-BE(2)C cells preincubated with 0.5 μg/mL doxycycline for 16 h were treated with 40 μM erastin for 32 h in the presence or absence of 2 μM ferrostatin-1 (Ferr-1), 10 μM rosiglitazone (ROSI), and 10 μM troglitazone(TRO). (C) Western blot analysis for ACSL4 and HTTQ94 in HTTQ94 tet-on SK-N-BE(2)C control and Acsl4-Crispr cells treated with 0.5 μg/mL doxycycline for 16 h. (D) Cell death assay. HTTQ94 tet-on SK-N-BE(2)C control Crispr and Acsl4-Crispr cells were preincubated with 0.5 μg/mL doxycycline for 16 h and then treated with 40 μM erastin for 32 h with/without 2 μM Ferr-1. (E) Western blot analysis for ACSL4 from the cerebral cortex tissues of HD and HD/Acsl4-null mice. (F) Kaplan–Meier survival curves of HD (n = 21 independent mice) and HD/Acsl4-null (n = 7 independent mice) mice. Cell deaths were calculated from three replicates. Data shown in A, B, and D are the means ± SD. P-values were derived from two-tailed unpaired t-test. (***) P ≤ 0.001. In F, P-value (HD vs. HD/Acsl4-null) was calculated using log-rank Mantel–Cox test. P = 0.1356.
Using Acsl4 knockout mice, we previously showed that the GPX4-dependent ferroptosis can also be inactivated upon loss of ACSL4 expression in vivo (Chu et al. 2019). To further elucidate the role of mHTT-mediated ferroptosis in the pathogenesis of Huntington's disease, we examined whether inactivation of the GPX4-dependent ferroptosis has any effect on the phenotypes observed in Huntington's disease mouse models. To this end, we used a well-established HD transgenic mouse model (HD-N171-82Q) in which a HTTQ82 protein with the N-terminal fragment (171 amino acids) and an 82-residue glutamine repeat was constitutively expressed (Schilling et al. 1999). These mice developed behavioral abnormalities, including loss of coordination, tremors, hypokinesis, and abnormal gait and died prematurely within ∼120 d after birth (Schilling et al. 1999). After crossing these transgenic HD mice with Acsl4 knockout mice, we were able to generate HD/Acsl4-null mice. As expected, the ACSL4 protein was undetectable in the brains of the HD/Acsl4-null mice (Fig. 2E). More importantly, there was no statistically significant difference of the median survival comparing the HD/Acsl4-null mice with the control HD mice (Fig. 2F), suggesting loss of Acsl4 does not affect the life span of HD mice. Future studies are required to further characterize the pathological phenotypes of the HD/Acsl4-null mice. Nevertheless, although ACSL4 is essential for HTTQ94-mediated ferroptosis induced by GPX4 inhibition, we failed to show that this pathway has major effects on the life span of Huntington's disease mice.
HTTQ94 is able to induce the ACSL4-independent ferroptosis upon ROS-induced stress
In our previous study, we found that the ferroptosis can also be induced upon ROS stress independent of the ACSL4 status (Chu et al. 2019). Since numerous studies indicate the importance of ROS stress in Huntington's disease pathogenesis (Paul and Snyder 2019), we first examined whether HTTQ94 promotes ROS-induced ferroptosis in the established HT-22 HTTQ94-inducible cell line upon ROS-induced stress, generated by tert-butyl hydroperoxide (TBH) treatment. As shown in Figure 3, A and B, although HTTQ94 expression or TBH treatment alone failed to induce any cell death, high levels of cell death were induced upon the combination of HTTQ94 expression and ROS stress. Moreover, these HTTQ94-mediated responses were specifically blocked by several well-known ferroptosis inhibitors (e.g., Ferr-1, Lipro-1, and DFO) (Fig. 3A,B) but not by the inhibitors of other types of cell death, such as apoptosis, autophagy, or necroptosis (Fig. 3A,B). Similar results were also obtained in the SK-N-BE(2)C HTTQ94-inducible cell line upon ROS-induced stress (Supplemental Fig. S2A,B). Moreover, in contrast to the effects induced by HTTQ94 expression, no obvious ferroptotic cell death was induced upon expression of the N-terminal fragment wild-type huntingtin protein containing the normal 19 glutamine residues (HTTQ19) under the same treatment (Supplemental Fig. S2C).
Figure 3.

HTTQ94 is able to induce the ACSL4-independent ferroptosis upon ROS stress. (A) Representative phase-contrast images of cell death from the HTTQ94 tet-on HT-22 cells. HTTQ94 tet-on HT-22 cells were preincubated with 0.5 μg/mL doxycycline for 4 h and then treated with 10 μM TBH for 8 h in the presence or absence of the ferroptosis inhibitors (2 μM ferrostatin-1 [Ferr1], 2 μM liproxstatin-1 [Lipor1], and 100 μM DFO), apoptosis inhibitor (10 μM Z-VADFMK [zVAD]), autophagy inhibitor (2 mM 3-methylademine [3MA]), or necroptosis inhibitor (10 μM necrostatin-1 [Nec1]). (B) Quantification of cell death, related to A. Three replicates were used for each group. (C) HTTQ94 tet-on SK-N-BE(2)C control Crispr and Acsl4Crispr cells were preincubated with 0.5 μg/mL doxycycline for 16 h and then treated with 350 μM TBH for 24 h with/without 2 μM Ferr-1. (D) Western blot analysis of GPX4, ACSL4, and HTTQ94 in HTTQ94 tet-on SK-N-BE(2)C control Crispr and GPX4/Acsl4 double-Crispr cells treated with 0.5 μg/mL doxycycline for 16 h. (E) HTTQ94 tet-on SK-N-BE(2)C control Crispr and GPX4/Acsl4 double-Crispr cells were preincubated with 0.5 μg/mL doxycycline for 16 h and then treated with 350 μM TBH for 24 h with/without 2 μM Ferr-1. Cell deaths were calculated from three replicates; Data shown in B, C, and E are means ± SD. P-values were derived from two-tailed unpaired t-test. (***) P ≤ 0.001, (n.s.) P > 0.05.
Next, we examined whether this type of ferroptosis is dependent on ACSL4 by using the established Acsl4 knockout cell lines. Surprisingly, HTTQ94-mediated ferroptosis remained unaffected in Acsl4-null cells under the same conditions (Fig. 3C). Similar results were observed in four independent Acsl4 knockout cell lines (Supplemental Fig. S3A). Since ACSL4 is essential for ferroptosis induced by GPX4 inhibition, it is likely that HTTQ94-mediated ferroptosis upon ROS-induced stress is independent of GPX4 function. However, this notion cannot be directly tested, since GPX4-null cells do not survive under normal conditions unless Acsl4 is codeleted. To address whether HTTQ94-mediated ferroptosis can be induced independent of GPX4 function, we generated Acsl4/GPX4 double-knockout derivatives of the SK-N-BE(2)C HTTQ94-inducible cell line. As shown in Figure 3D, neither ACSL4 nor GPX4 was detectable in the Acsl4/GPX4-null cells with inducible HTTQ94 expression. Nevertheless, the ferroptotic cell death was readily induced upon HTTQ94 expression in Acsl4/GPX4-null cells under ROS stress conditions (Fig. 3E). The result was again confirmed by using several independent Acsl4/GPX4 knockout cell lines under the same conditions (Supplemental Fig. S3B,C). Taken together, these data demonstrate that HTTQ94 is able to induce ferroptosis under ROS stress conditions independent of either ACSL4 or GPX4.
Finally, in addition to GPX4, several important cellular factors have been identified as critically involved in regulating ferroptotic responses through completely different mechanisms such as FSP1, GCH1, and DHODH (Bersuker et al. 2019; Doll et al. 2019; Kraft et al. 2020; Mao et al. 2021). To examine whether these ferroptosis regulators play a role in HTTQ94-mediated ferroptosis upon ROS stress, we performed RNAi-mediated depletion in the tet-on HTTQ94-inducible SK-N-BE(2)C cells (Supplemental Fig. S4A) and overexpression in the tet-on HTTQ94-inducible H1299 cells (Supplemental Fig. S4B) to test whether HTTQ94-mediated ferroptosis is affected by depletion or overexpression of FSP1, GCH1, or DHODH. As shown in Supplemental Figure S4, C and D, HTTQ94-mediated ferroptotic responses upon ROS stress remained intact upon either depletion or overexpression of FSP1, GCH1, and DHODH. Together, these data suggest that HTTQ94-mediated cell death upon high levels of ROS may represent a new ferroptosis pathway that needs to be further elucidated.
ALOX5 is required for HTTQ94-mediated ferroptosis upon ROS-induced stress
Notably, our recent studies showed that ferroptosis can be induced by activation of the lipoxygenases (Chu et al. 2019). The mammalian lipoxygenase family consists of six isoforms (ALOXE3, ALOX5, ALOX12, ALOX12B, ALOX15, and ALOX15B) with differing substrate specificities (Mashima and Okuyama 2015). To examine whether any of these lipoxygenases is required for mHTT-mediated ferroptosis upon ROS stress, we performed an RNAi-mediated loss-of-function screen to test whether depletion of individual isoforms affects HTTQ94-dependent ferroptosis. To this end, the tet-on HTTQ94-inducible SK-N-BE(2)C cells were transfected with siRNAs (Dharmacon SMART-pools) specific for each of the six lipoxygenases and treated with TBH to induce ferroptosis. Significantly, mHTTQ94-mediated ferroptosis upon ROS stress was markedly blocked by RNAi-mediated depletion of ALOX5 but not by the depletion of the other five lipoxygenases (Fig. 4A). Quantitative polymerase chain reaction (qPCR) analyses confirmed that the expression of each of the six lipoxygenase isoforms was individually abrogated by RNAi-mediated depletion (Fig. 4B). To corroborate this finding, we performed RNAi-mediated depletion of ALOX5 in the HTTQ94 tet-on HT-22 cells (Fig. 4C). Indeed, HTTQ94-mediated ferroptosis upon ROS stress was abolished upon loss of ALOX5 expression (Fig. 4C).
Figure 4.

ALOX5 is required for HTTQ94-mediated ferroptosis induced by ROS stress and glutamate. (A) Cell death assay in HTTQ94 tet-on SK-N-BE(2)C cells with different ALOX knockdowns. Cells were transfected with control siRNA (ctrl) or ALOX family-specific siRNAs, followed by preincubation with 0.5 µg/mL doxycycline for 16 h and then treated with 350 μM TBH for 24 h. (B) qPCR analysis of the knockdown efficiency of ALOX family members in HTTQ94 tet-on SK-N-BE(2)C cells transfected with control siRNA or ALOX family-specific siRNAs. (C) Cell death assay in HTTQ94 tet-on HT-22 cells with ALOX5 knockdown. Cells were transfected with control siRNA (ctrl) or ALOX5-specific siRNA and then preincubated with 0.5 µg/mL doxycycline for 4 h, followed by 10 μM TBH treatment for 8 h. (Left panel) ALOX5 knockdown efficiency. (Right panel) Cell death assay. (D) Western blot analysis of ALOX5 and HTTQ94 in HTTQ94 tet-on SK-N-BE(2)C control Crispr and Alox5-Crispr cells treated with 0.5 μg/mL doxycycline for 16 h. (E) Cell death assay for mHTT tet-on SK-N-BE(2)C control Crispr and Alox5-Crispr cells. Cells were preincubated with 0.5 μg/mL doxycycline for 16 h, followed by incubation with 350 μM TBH for 24 h with/without 2 μM Ferr-1. (F) FACS analysis of lipid ROS production in HTTQ94 tet-on SK-N-BE(2)C control Crispr and Alox5-Crispr cells. Cells were preincubated with 0.5 μg/mL doxycycline for 16 h and then treated with 350 μM TBH for 6 h. Lipid ROS was stained with C11-BODIPY. (G) Cell death assay in HTTQ94 tet-on HT-22 cells. Cells were transfected with control siRNA (ctrl) or ALOX5 siRNA, followed by incubation with 0.5 μg/mL doxycycline and 10 mM glutamate for 16 h in the presence or absence of 2 μM Ferr-1. Cell deaths were calculated from three replicates. Data shown in A, C, E, and G are means ± SD. P-values were derived from two-tailed unpaired t-test. (***) P ≤ 0.001.
To further validate the role of ALOX5 in HTTQ94-mediated ferroptosis, we generated Alox5 knockout cells by CRISPR/cas9 technology using the HTTQ94 tet-on SK-N-BE(2)C cells (Supplemental Fig. S5A). As shown in Figure 4D, the ALOX5 protein was undetectable in Alox5-null cells, and loss of ALOX5 expression dramatically abolished the HTTQ94-mediated ferroptosis upon ROS stress (Fig. 4E). The results were further confirmed by four independent Alox5 knockout cell lines under the same conditions (Supplemental Fig. S5B). In contrast, the ferroptosis response induced by HTTQ94 remained intact in Alox5-null cells treated with erastin, an inhibitor of the cystine–glutamate antiporter system X−c (Supplemental Fig. S5C). Moreover, by using flow cytometry analysis with C11-BODIPY staining, the levels of endogenous membrane lipid peroxidation, a key marker of ferroptosis, were significantly induced upon HTTQ94 expression, but these effects were largely abrogated upon loss of Alox5 (Fig. 4F; Supplemental Fig. S5D). Taken together, these data demonstrate that ALOX5 is essential for HTTQ94-mediated ferroptosis upon ROS stress and increased lipid peroxidation levels.
Glutamate, the most abundant endogenous excitatory neurotransmitter in the brain, plays a crucial role in neuronal tissue damage. Recently, several studies showed that ferroptosis is regulated by glutamate (Liu et al. 2015; Maher et al. 2020; Xie et al. 2022). We treated the HTTQ94-inducible HT-22 cell line with glutamate to investigate whether HTTQ94 also promotes glutamate-induced ferroptosis. Indeed, high levels of cell death were induced upon HTTQ94 expression in the presence of glutamate (Supplemental Fig. S5E), which was inhibited by ferrostatin-1, indicating that glutamate-induced ferroptosis is significantly enhanced by HTTQ94 expression. More importantly, HTTQ94-mediated ferroptosis upon glutamate induction could be completely blocked by zileuton, a well-known inhibitor of ALOX5 (Supplemental Fig. S5E; Liu et al. 2015). Next, we performed RNAi-mediated knockdown of ALOX5 (Supplemental Fig. S5F) to further examine the role of ALOX5 in regulating glutamate-mediated ferroptosis. As shown in Figure 4G, HTTQ94-mediated ferroptosis was indeed abrogated in ALOX5 knockdown cells. Thus, these data indicate that ALOX5 is also critical for glutamate-mediated ferroptosis upon HTTQ94 expression.
Mechanistic insight into ALOX5 activation induced by HTTQ94 expression
To dissect the mechanism by which HTTQ94 promotes ALOX5-dependent ferroptosis, we first tested whether the protein levels of ALOX5 are regulated by HTTQ94 expression. As shown in Supplemental Figure S6A, Western blot analysis revealed that HTTQ94 overexpression had no obvious effect on the levels of ALOX5 protein, suggesting that ALOX5 protein stability is not affected in HTTQ94-overexpressing cells. Like ALOX12, ALOX5 is a member of the lipoxygenase family containing intrinsic lipoxygenase activity (Chu et al. 2019). However, unlike other lipoxygenases, ALOX5 requires a specific cofactor called FLAP (the 5-lipoxygenase-activating protein) for catalyzing the lipid oxygenation reaction (Peters-Golden and Brock 2003; Mashima and Okuyama 2015). Next, we examined whether the protein stability of FLAP can be regulated by HTTQ94. Notably, the steady-state levels of FLAP were markedly increased upon HTTQ94 coexpression (Fig. 5A) but were not affected by coexpression of the N-terminal fragment wild-type huntingtin protein containing the normal 19 glutamine residues (HTTQ19) (Fig. 5A). Moreover, the levels of endogenous FLAP, but not ALOX5, were increased in a dosage-dependent manner upon HTTQ94 induction (Fig. 5B); the half-life of endogenous FLAP was significantly extended upon HTTQ94 expression from 6 to >12 h (Fig. 5C; Supplemental Fig. S6B). These data indicate that HTTQ94, but not the wild-type counterpart (HTTQ19), is able to increase the protein stability of FLAP.
Figure 5.

Mechanistic insight into HTTQ94-induced ALOX5 activation. (A) Western blot analysis of FLAP and HTTQ94 or HTTQ19 in HEK293 cells transfected with an HA-FLAP-expressing plasmid and either an empty vector, HTTQ94-GFP-expressing vector, or HTTQ19-GFP-expressing vector. (B) Western blot analysis of endogenous FLAP and ALOX5 in HTTQ94 tet-on SK-N-BE(2)C cells incubated with 0.5 μg/mL doxycycline for 4, 8, and 16 h. (C) Densitometry quantification of FLAP protein levels calculated using ImageJ software and plotted for half-life determination corresponding to Supplemental Figure S5B. (D) Co-IP of GFP-tagged HTTQ94/HTTQ19 and FLAG-tagged FLAP in HEK293 cells. Cell lysates were immunoprecipitated with anti-FLAG-coupled beads (M2), followed by Western analysis of HTTQ94, HTTQ19, and FLAP. (E) Ubiquitination analysis of FLAP in the presence of HTTQ94 or HTTQ19. HEK293 cells were cotransfected with FLAG-FLAP and HA-ubiquitin (Ub) in the presence of HTTQ94 or HTTQ19. Cell lysates were immunoprecipitated with anti-FLAG-coupled beads (M2), followed by Western blot analysis of HA, FLAP, and HTT proteins. (F) Cell death assay for HTTQ94 tet-on SK-N-BE(2)C cells with FLAP knockdown. Cells were transfected with control siRNA (ctrl) or FLAP-specific siRNA and then preincubated with 0.5 µg/mL doxycycline for 16 h, followed by 350 μM TBH treatment for 24 h. (G) Cell death assay. HTTQ94 tet-on HT-22 cells preincubated with 0.5 μg/mL doxycycline for 4 h were treated with 10 μM TBH for 8 h in the presence or absence of 2 μM Ferr-1, 10 μM zileuton, or 10 μM MK886. Cell deaths were calculated from three replicates. Data shown in F and G are the means ± SD. P-values were derived from two-tailed unpaired t-test. (***) P ≤ 0.001.
Next, we examined the interaction between HTTQ94 (or HTTQ19) and FLAP in human cells. To this end, we transfected cells with a HTTQ94 or HTTQ19 expression vector in the presence or absence of a vector encoding FLAG-FLAP in 293 cells. As shown in Figure 5D, HTTQ94 was readily detected in the immunoprecipitated complexes of FLAG-FLAP; however, the wild-type counterpart (HTTQ19) failed to be detected in the immunoprecipitated complexes of FLAG-FLAP under the same conditions (Fig. 5D), suggesting that HTTQ94 specifically interacts with FLAP. To further dissect the mechanism by which HTTQ94 promotes the stabilization of FLAP, we tested the effect of the expression of HTTQ94 on the levels of FLAP ubiquitination. Indeed, the levels of ubiquitinated FLAP were dramatically reduced upon the expression of HTTQ94 but not HTTQ19 (Fig. 5E), suggesting that HTTQ94 stabilizes FLAP by suppressing FLAP ubiquitination. Finally, to evaluate the importance of FLAP in HTTQ94-mediated effects, we tested whether HTTQ94-mediated ferroptosis also requires FLAP. To this end, we first knocked down the endogenous FLAP by RNAi in the HTTQ94-inducible cells and then tested the effects of depletion of FLAP on HTTQ94-mediated ferroptosis. Indeed, upon RNAi-mediated depletion of FLAP (Supplemental Fig. S6C), HTTQ94-mediated ferroptosis upon ROS stress was largely abrogated (Fig. 5F). Moreover, we investigated whether pharmacological inhibition of FLAP or ALOX5 with specific inhibitors of FLAP or ALOX5 (Pergola et al. 2014; Liu et al. 2015) has any effect on HTTQ94-mediated ferroptosis. Indeed, HTTQ94-mediated ferroptosis was largely abrogated by specific inhibitors of either FLAP or ALOX5 in both HT-22 (Fig. 5G) and SK-N-BE(2)C (Supplemental Fig. S6D) cells. Taken together, these data demonstrate that HTTQ94 promotes ALOX5-mediated ferroptosis by interacting with FLAP and up-regulating its stability.
Loss of ALOX5 ameliorates the pathological phenotypes and significantly extends the life spans of these HD mice
To examine the role of HTTQ94-mediated, ALOX5-dependent ferroptosis in contributing to the pathophysiological phenotypes in the HD mice, we crossed these HD mice (HD-N171-82Q) with Alox5-null mice to generate compound HD/Alox5-null mutant mice. As shown in Figure 6A, ALOX5 protein levels were undetectable in the brains of HD/Alox5-null mutant mice. As expected, the HD-N171-82Q mice displayed a progressive neurological phenotype at 9 wk old. For example, the clasping reflex, an abnormal posturing of the hind limb during the tail suspension, is a pathological characteristic of HD mice that reflects the progression of brain damage (Schilling et al. 1999; Paul and Snyder 2019). We examined the phenotypes of the HD/Alox5-null mice in comparison with the age-matched HD littermates. Strikingly, we found that the levels of the HD mice showing clasping were significantly reduced in the HD/Alox5-null mice (Fig. 6B; Supplemental Fig. S7A). Moreover, the travelled distance was significantly improved in the HD/Alox5-null mice (Fig. 6C). More importantly, the HD/Alox5-null mutant mice lived much longer than the HD control mice, with the median survival increased to 134 d compared with 94 d for the HD mice (Fig. 6D).
Figure 6.

Loss of ALOX5 ameliorates the phenotypes and significantly extends the life spans of HD mice. (A) Western blot analysis for ALOX5 expression in HD and HD/Alox5-null mouse brains. (B) Representative images of limb clasping from the HD and HD/Alox5-null mice. (C) Open field test. Thirteen-week-old mice were subjected to open field test, and the distance moved was calculated. Six mice were used for each group (HD vs. HD/Alox5-null; P < 0.01). (D) Kaplan–Meier survival curves of HD (n = 21 independent mice) and HD/Alox5-null (n = 25 independent mice) mice. P-value was calculated using log-rank Mantel–Cox test (HD vs. HD/Alox5-null; P < 0.0001). (E,F) TfR1 staining on mouse brain slides. WT, HD-N171-82Q, and HD-N171-82Q/Alox5-null mouse brain paraffin slides were dewaxed, and antigens were retrieved by PH6.0 citric acid solution followed by incubation with TfR1 antibody. (E) Representative images of TfR1 staining (four mice for each group). Nuclei were stained with DAPI. (F) Quantification of TfR1-positive striatal neurons. Data are represented as mean ± SEM. (G) Representative images of TfR1 and DARPP-32 double staining on mouse brain paraffin slides. DARPP-32 was used as a marker for striatal medium spiny neurons (Naia and Rego 2018). (H) Quantification of TfR1-positive striatal neurons, related to G. Data shown in C, F, and H are the means ± SEM. n = 6. P-values were derived from two-tailed unpaired t-test. (***) P ≤ 0.001, (**) P ≤ 0.01.
To evaluate the role of ferroptosis in contributing to the pathological phenotypes of these mice, we examined whether mHTT-mediated ferroptosis is indeed elevated in the HD mice and whether loss of ALOX5 is effective to reduce the ferroptotic response in vivo. Ferroptosis is a new type of programmed cell death driven by the iron-dependent accumulation of lipid ROS. To examine the levels of ferroptosis in the HD mice, we established a reliable way to specifically detect ferroptotic cells in tissue sections to gauge the extent of ferroptosis in animal models. Notably, by screening ∼4750 monoclonal antibodies generated for their ability to selectively detect cells undergoing ferroptosis, one antibody—3F3 ferroptotic membrane antibody (3F3-FMA), which recognizes the human transferrin receptor 1 protein (TfR1)—was discovered as a specific ferroptosis-staining reagent (Feng et al. 2020).
To validate whether this ferroptosis marker is effective for staining ferroptotic cells in the HD mice, we first examined whether the anti-TfR1 antibody staining is able to specifically recognize ferroptotic cells induced by HTTQ94 expression in mouse neuronal cells. To this end, we used the tet-on-HTTQ94 mouse hippocampal HT-22 neuronal-inducible cell line. As shown in Supplemental Figure S7B, upon the induction of HTTQ94 expression in the presence of tert-butyl hydroperoxide (TBH), the levels of the anti-TfR1 antibody-stained cells were dramatically increased. As expected, the anti-TfR1 antibody stained with greater intensity at the cell boundaries of those ferroptotic cells, and intracellular puncta also became brighter. In addition, the levels of the staining were abolished in the presence of the ferroptosis inhibitor ferrostatin-1 (Ferr-1) (Supplemental Fig. S7B). Moreover, the levels of the anti-TfR1 antibody staining were well correlated with the levels of ferroptosis cell death (Supplemental Fig. S7C,D). These data indicate that the anti-TfR1 antibody is able to selectively stain ferroptotic cells induced by HTTQ94 expression. Next, we performed the same assay to examine the role of ALOX5 in modulating the ferroptosis levels in vivo. As shown in Figure 6E, the levels of the bright staining with great intensity by anti-TfR1 antibody were very low in the wild-type mouse striatum; in contrast, the anti-TfR1-positive-stained neurons were significantly increased in HD-N171-82Q mouse striatum. More importantly, Alox5 deficiency completely abolished the levels of these anti-TfR1-positive-stained neurons (Fig. 6E,F). Thus, by using the anti-TfR1 antibody staining, we confirmed that ALOX5 is crucial for ferroptotic cell death in the HD mice. Since the medium spiny projection neuron cells constitute ∼95% of striatal neurons, it is very likely that the ferroptotic cells detected in the HD mice (HD-N171-82Q) are the medium spiny projection neurons. To this end, we used dopamine- and cAMP-regulated phosphoprotein 32 kDa (DARPP-32) as a specific maker for striatal neurons (Naia and Rego 2018). Indeed, all the striatal neuronal cells were stained by the anti-DARPP-32 antibody (Fig. 6G, green). Simultaneously, the ferroptotic neuronal cells from the HD brains were also brightly stained with the anti-TfR1 antibody (Fig. 6G, red). Notably, all the anti-TfR1-positive cells (Fig. 6G, red) were also stained with the anti-DARPP-32 antibody (Fig. 6G, green). Moreover, the levels of ferroptotic striatal neurons with TfR1-positive staining were completely abolished in HD/Alox5-null mice (Fig. 6G,H). Taken together, these data demonstrate that the levels of ferroptosis are up-regulated in the striatal neurons of the HD mice and that deletion of the Alox5 gene effectively abrogates the ferroptosis activity in the HD mice.
Discussion
Cell death induced by mHTT toxicity is a pathological hallmark in HD, characterized by significant neuronal loss in the striatum and cerebral cortex, followed by widespread neuronal loss in other brain regions (Thu et al. 2010; Nana et al. 2014; Bates et al. 2015). Previous studies have indicated that apoptosis is apparently not the major factor for the neuronal cell death in HD (Turmaine et al. 2000; Hickey and Chesselet 2003). Ferroptosis is a newly identified form of cell death that is morphologically, biochemically, and genetically distinct from other known forms of cell death (Dixon et al. 2012; Stockwell et al. 2017). Indeed, we found that expression of the mHTT fragment with the expanded 94 glutamine residues (HTTQ94), but not the wild-type counterpart (HTTQ19), sensitizes neuronal cells to ferroptosis. Although numerous studies implicate ACSL4 as a central factor in modulating ferroptotic responses induced by GPX4 inhibition, we failed to show that inactivation of the ACSL4-dependent ferroptosis has any significant effect on pathophysiological phenotypes and the life spans of the HD mice. Notably, we demonstrated that expression of HTTQ94 induces ferroptosis upon ROS-induced stress through a distinct pathway. Our results further demonstrated that HTTQ94-mediated ferroptosis upon ROS stress is mediated by ALOX5. Moreover, we showed that HTTQ94 promotes the function of ALOX5 through stabilizing its cofactor, FLAP.
Signs and symptoms of Huntington's disease typically begin at age 30–50 yr and progress over the next 10–20 yr (Bates et al. 2015), suggesting that besides mHTT toxic function, aging also plays an important role in neuronal death. The production of ROS is progressively increased in aging, which is one of the key factors in cellular damage (Floyd and Hensley 2002; Tower 2015). Although the precise mechanism of mHTT-mediated ferroptosis in vivo needs further elucidation, based on our data, it is very likely that mHTT-mediated ferroptotic responses are tightly regulated by the levels of ROS production in HD patients. Moreover, glutamate, the most common endogenous excitatory neurotransmitter in the brain, plays a crucial role in neuronal tissue damage. Under normal physiological condition, glutamate regulates memory, learning, cognitive, emotional, endocrine, and other visceral functions (Liu et al. 2015; Maher et al. 2020; Xie et al. 2022). Interestingly, our data indicate that glutamate-induced ferroptosis is significantly enhanced by HTTQ94 expression. More importantly, we further showed that ALOX5 is essential for these effects. Numerous studies indicate that glutamate-mediated cell death is involved in the pathogenesis of almost all neurological diseases, including Huntington's diseases (Coyle and Puttfarcken 1993; Caudle and Zhang 2009; Lewerenz and Maher 2015; McGrath et al. 2022). These data reveal a potential physiological pathway in modulating mHTT-induced ferroptosis during the pathological process of Huntington's diseases. Ferroptosis driven by lethal amounts of lipid peroxide is critical for ROS stress responses (Yang and Stockwell 2016). Lipid peroxidation has been observed in the brains of the HD mice (Lee et al. 2011; Skouta et al. 2014), indicating the involvement of ferroptosis in the progression of HD. Our data further showed that loss of ALOX5 expression abrogates ferroptosis in the striatal neurons of the HD mice. More importantly, the HD/Alox5-null mutant mice survived significantly longer than HD mice with much improved pathological phenotypes. Further studies are clearly warranted to further characterize the phenotypes of the HD/Alox5-null mutant mice and, more importantly, to validate these findings in the HD mouse model expressing full-length mHTT and other HD mouse models. Nevertheless, our findings indicate that the ALOX5-dependent ferroptosis pathway is crucial in mHTT-induced pathophysiological phenotypes of the HD mice and suggest that inhibition of the ALOX5 activity may be beneficial for treatment of Huntington's disease.
Limitations of the study
Although the role of the ACSL4-mediated ferroptosis in contributing to several types of neurodegenerative diseases has been well recognized, our study indicates that a new ferroptosis pathway controlled by ALOX5 is critical for mHTT-mediated effects in HD mice. There are several additional mouse models of HD, including the knock-in and BAC transgenic mice that express full-length mutant HTT. HdhQ111 knock-in mice express 111-glutamine mutant huntingtin from the mouse HTT endogenous locus and exhibit early dominant abnormalities selective for medium spiny striatal neurons, including nuclear retention of full-length mutant huntingtin (Wheeler et al. 2000). A bacterial artificial chromosome (BAC)-mediated transgenic mouse model (BACHD) was developed expressing full-length mHTT with 97 glutamine repeats under the control of endogenous HTT regulatory machinery on the BAC (Gray et al. 1998). Both HD mice exhibit progressive motor deficits, neuronal synaptic dysfunction, and selective neuropathology, which includes significant cortical and striatal atrophy and striatal dark neuron degeneration. Since limited pathological phenotypes of HD mice were analyzed in this study, future studies are clearly warranted to examine whether loss of ALOX5 expression is able to effectively ameliorate the pathological phenotypes and significantly extend the life spans of those HD mice. Moreover, mHTT-mediated ferroptosis is effectively blocked by zileuton, a specific inhibitor of ALOX5. Interestingly, zileuton is an FDA-approved drug that has been used for the treatment of asthma (Liu et al. 2015). It will be very interesting to test the effect of this compound in the pathological phenotypes and the life spans of HD mice for repurposing this drug to potential therapeutic application in Huntington's disease.
Materials and methods
Mammalian cell culture
The SK-N-BE(2)C neuroblastoma, H1299, and HEK293 cell lines were obtained from American Type Culture Collection (ATCC). The HT-22 mouse hippocampal neuronal cell line was obtained from Sigma-Aldrich in 2018. All cells have been tested to be negative for mycoplasma contamination. No cell lines used in this work were listed in the International Cell Line Authentication Committee database. All cells were cultured in a 37°C incubator with 5% CO2. All media were supplemented in DMEM with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin (all from Gibco). Stable cell lines derived from these cell lines, and experimental treatments are described in detail in the Materials and Methods.
Plasmids
pTreTight-HTTQ94-GFP was a gift from Nico Dantuma (Addgene plasmid 23966, http://n2t.net/addgene:23966, RRID: Addgene_23966). HTTQ94-GFP was subcloned into pcDNA3.1/v5-His-Topo vector (Invitrogen). The HTTQ94 sequence was amplified from pTreTight-HTTQ94-GFP. The HTTQ19 (nHTT) sequence was amplified from the DNA isolated from human cells. Both the HTTQ94 and HTTQ19 sequences were subcloned into pDONR221 vector and then subcloned into pInducer vector (Gateway LR kit, Thermo) to generate pInducer-HTTQ19 and pInducer-HTTQ94, respectively. V5-ALOX5 was previously described (Chu et al. 2019). pDONR221-FLAP was obtained from Harvard Medical School PlasmID (HsCD00043730). To generate HA-FLAP and FLAG-FLAP, full-length FLAP was amplified using forward primer with the HA sequence and the FLAG sequence, respectively, and then subcloned into pcDNA3.1/v5-His-Topo vector (Invitrogen). cDNA of AIFM2 (FSP1), GCH1, and DHODH was amplified using forward primer with the FLAG sequence and cloned into pcDNA3.1/v5-His-Topo vector (Invitrogen).
Cell lines
To generate the HTTQ94 and HTTQ19 fragment tet-on stable cell lines, pInducer-HTTQ94 and pInducer-HTTQ19 were transfected into H1299 cells. To generate the HTTQ94 tet-on stable neuronal cell lines, pInducer-HTTQ94 was transfected into HT-22 and SK-N-BE(2)C cells. The transfected cells were selected and maintained with 500 μg/mL G418 (Sigma) in DMEM medium containing 10% FBS. Single clones were selected and screened by Western blot. Acsl4 and Alox5 CRISPR–cas9 knockout cells were generated by transfecting ACSL4 and ALOX5 double-nickase plasmid (ACSL4 : Santa Cruz Biotechnology sc-401649-NIC, and ALOX5: Santa Cruz Biotechnology sc-401239-NIC) into the HTTQ94 tet-on SK-N-BE(2)C cells. Acsl4/GPX4 double-knockout cells were generated by transfecting ACSL4 and GPX4 double-nickase plasmid (GPX4; Santa Cruz Biotechnology sc-401558-NIC) simultaneously into the HTTQ94 tet-on SK-N-BE(2)C cells. Forty-eight hours later, CRISPR efficiency was determined by Western blot analysis, and then pool cells were seeded in 10-cm dishes to grow clones at a density of 100–200 cells/dish. One week or 2 wk later, moloclones were picked and seeded into 12-well plates, followed by identification by Western blot analysis.
Western blotting and antibodies
Protein extracts were analyzed by Western blotting according to standard protocols using primary antibodies specific for ALOX5 (1:500 dilution; Cell Signaling 3289S), ACSL4 antibody (A5; 1:1000 dilution; Santa Cruz Biotechnology sc-271800), HA (1:1000 dilution; Sigma 11867423001), FLAG (1:1000 dilution; Sigma F-3040), FLAP (1:1000 dilution; Abcam ab85227), β-actin (1:2000 dilution; Abcam ab8227), and vinculin (1:5000 dilution; Sigma-Aldrich V9264). HRP-conjugated antimouse (SouthernBiotech 1031-05) and antirabbit secondary antibody (SouthernBiotech 4050-05) and antirat secondary antibody (Southern Biotech 3051-05) were used.
RNA interference
Cells were plated at 20%–30% density 1 d prior to siRNA transfection. Knockdown of ALOX family proteins and FLAP was performed by transfection of HTTQ94 tet-on SK-N-BE(2)C cells with 60 μM siRNA duplex oligo set (ON-TARGET plus SMARTpool: ALOXE3: L-009022-00-0005, ALOX12B: L-009025-00-0005, ALOX12: L-004558-00-0005, ALOX5: L-004530-00-0005, ALOX15B: L-009026-00-0005, ALOX15: L-003808-00-0005, and FLAP: L-010166-01-0005; Horizon Discovery) with Lipofectamine 3000 (Invitrogen) according to the manufacturer's protocol. HTTQ94 tet-on HT-22 cells were transfected with 60 μM mouse ALOX5 siRNA duplex oligo set (ON-TARGET plus SMARTpool, L-065695-01-0005; Horizon Discovery). HTTQ94 tet-on SK-N-BE(2)C cells were transfected with 40 μM AIFM2/FSP1, GCH1, and DHODH siRNA pools (Invitrogen). Cells were collected at 48–72 h after transfection and subjected to functional assays. The siRNA target sequences were follows: AIFM2/FSP1 (siRNA-1, 5′-CAACAUCGUCAACUCUGUGAA-3′ and siRNA-2, 5′-GAUUCUCUGCACCGGCAUCAA-3′), GCH1 (siRNA-1, 5′-GCGAGGAUUGUAGAAAUCUAU-3′ and siRNA-2, 5′-GCAACACACAUGUGUAUGGUA-3′), and DHODH (siRNA-1, 5′- GUGAGAGUUCUGGGCCAUAAA-3′ and siRNA-2, 5′-CGAUGGGCUGAUUGUUACGAA-3′).
RNA extraction and qRT-PCR
Total RNA was extracted using TRIzol (Thermo Fisher Scientific 15596018) according to the manufacturer's protocol. cDNA was generated using SuperScript IV VILO master mix (Thermo Fisher Scientific 11756500). Quantitative PCR was done using a 7500 Fast real-time PCR system (Applied Biosystems) with standard protocol. Reactions were done in triplicate. The following primers were used: human ALOX family and mouse CHAC1 described previously (Chu et al. 2019), human FLAP (forward 5′-CTTGCCTTTGAGCGGGTCTA-3′ and reverse 5′-CATCAGTCCAGCAAACGCAG-3′), mouse ALOX5 (forward 5′-ATCGAGTTCCCATGTTACCGC-3′ and reverse 5′-AATTTGGTCATCTCGGGCCA-3′), human AIFM2/FSP1 (forward 5′-AGACAGGGTTCGCCAAAAAGA-3′ and reverse 5′-CAGGTCTATCCCCACTACTAGC-3′), human GCH1 (forward 5′-ACGAGCTGAACCTCCCTAAC-3′ and reverse 5′-GAACCAAGTGATGCTCACACA-3′), and human DHODH (forward 5′-GTTCTGGGCCATAAATTCCGA-3′ and reverse 5′-TCTGGGTCTAGGGTTTCCTTC-3′).
Drugs and cell death inhibitors
All drugs (except for those listed below) were ordered from Sigma-Aldrich. Ferrostatin-1 was from Xcess Biosciences, and MK886 was from MedChemExpress (MCE). For ROS generation, tert-butyl hydroxide solution (TBH) was used at different doses depending on the experiment; see the respective figure legends. Erastin (ferroptosis inducer), rosiglitazone (ACSL4 inhibitor), troglitazone (ACSL4 inhibitor), zileuton (ALOX5 inhibitor), MK886 (FLAP inhibitor), ferrostatin-1 (ferroptosis inhibitor), DFO (ferroptosis inhibitor), liproxstatin-1(ferroptosis inhibitor), 3-methylademine (autophagy inhibitor), Z-VADFMK (apoptosis inhibitor), and necrostatin-1(necroptosis inhibitor) were used at different doses depending on the experiment; see the related figure legends.
Cell death assay
Cells were treated with ferroptosis inducer TBH or with erastin or glutamate combined with other drugs or conditions. At the indicated time points, cells were trypsinized and stained with trypan blue followed by counting with a hemocytometer using the cell number counter (Life Technologies Countess II). Living cells and dead cells were all counted, and cells stained by trypan blue were considered dead cells. Quantification of cell death was further confirmed using ToxiLight nondestructive cytotoxicity bioassay kit (Lonza LT07-117). Data were collected using a GloMax Explorer multimode microplate reader (Promega).
Lipid ROS assay using flow cytometer
Cells were incubated with DMEM containing 5 μM BODIPY 581/591 C11 (Thermo Fisher Scientific D3861) for 25–30 min at 37°C in serum-free medium. Cells were then harvested and washed twice with PBS followed by resuspension in 500 μL of PBS. Lipid ROS levels were analyzed using a Becton Dickinson FACSCalibur machine through the FL1 channel, and the data were analyzed using CellQuest software. Ten-thousand cells were collected and analyzed for each sample.
Mice
The Alox5 knockout mice and Huntington transgenic mice (HD-N171-82Q) were purchased from The Jackson Laboratory (stock nos. 004155 and 003627, respectively). The Acsl4 knockout mice were described previously (Chu et al. 2019). All experimental protocols using mice were approved by the Institutional Animal Care and Use Committee (IACUC) of Columbia University.
Immunofluorescence staining
For cell lines, HT-22 HTTQ94-inducible cells were fixed with 2% paraformaldehyde for 20 min at room temperature and then incubated with blocking buffer (10% donkey serum, 0.3% Triton X-100 in PBS), followed by incubation with TfR1 antibody (1: 250; Thermo Fisher Scientific 13-6800) for 1 h, and then incubated with Alexa 568-conjugated secondary antibody (1:250; Thermo Fisher Scientific A11004) for an additional 1 h.
For mouse brain slides, brain paraffin slides were dewaxed by xylene, and antigens were retrieved with PH6.0 citrate buffer using a pressure cooker. Slides were blocked with 5% horse serum for 30 min and then incubated with primary antibody (TfR1: 1:1000 [Thermo Fisher Scientific 13-6800] and DARPP-32: 1:200 [Cell Signaling 2306]) for 1–1.5 h, followed by incubation with biotinylated secondary antibody (1: 200; Vector Laboratories BA-2000 and BA-1000) for 30 min, and then incubated with streptavidin-conjugated Alexa 594/488 dye (1:1000; Thermo Fisher Scientific S32356 and S32354) for another 30 min. In the double-staining experiment, Alexa 488-conjugated secondary antibody (Thermo Fisher Scientific A11008) was also used for detecting DARPP-32.
Behavioral tests
Both female and male mice were included for behavioral tests. There was no significant effect on gender for the behavioral tests. Motor activity was measured at 13 wk of age in an open field. Mice were allowed to habituate to the experimental environment for at least 1 h before assessment. Mice were placed in the center for 10 min, during which the distance travelled was recorded for the last 5 m by a digital camera coupled to the Smart Junior system. The distance travelled within each group was averaged separately. As for the clasping test, each mouse was suspended upside down by its tail for 15 sec. They were administered three trials per week at 9, 11, and 13 wk of age. The weekly percentage of limb clasping within each group was averaged separately. The weight of each mouse was recorded weekly.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism 9 software. Results are presented as the mean ± SD or ± SEM. Statistical significance of the mouse Kaplan–Meier survival curves was determined by log-rank Mantel–Cox test.
Supplementary Material
Acknowledgments
We thank Dr. Ai Yamamoto and Dr. Mu Yang for helpful discussions. W.G. is supported by the National Cancer Institute (NCI) of the National Institutes of Health (NIH) under awards R35CA253059, RO1CA258390, and RO1CA254970, and also acknowledges support from the Herbert Irving Comprehensive Cancer Center (HICCC; P30 CA13696) and the Molecular Pathology, Genomics, and Proteomics of Shared Resources of HICCC. X.J. is supported by NIH R01CA204232 and R01CA258622, as well as NCI Cancer Center Core Grant P30 CA008748 to Memorial Sloan-Kettering Cancer Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authors’ contributions. S.S. and W.G. conceived the study and designed the experiments. S.S., Z.S., N.K., H.L., and B.C. performed the methodology and acquired the data. S.S., Z.S., N.K., and W.G. analyzed and interpreted the data. S.S., Z.S., N.K., X.J., B.R.S. and W.G. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.350211.122.
Competing interest statement
B.R.S. is an inventor on patents and patent applications involving ferroptosis; cofounded and serves as a consultant to ProJenX, Inc., and Exarta Therapeutics; holds equity in Sonata Therapeutics; serves as a consultant to Weatherwax Biotechnologies Corporation and Akin Gump Strauss Hauer and Feld LLP; and receives sponsored research support from Sumitomo Dainippon Pharma Oncology. X.J. is an inventor on patents related to autophagy and cell death, and holds equity in and consults for Exarta Therapeutics and Lime Therapeutics. The other authors declare no competing interests.
References
- Bates G. 2003. Huntingtin aggregation and toxicity in Huntington's disease. The Lancet 361: 1642–1644. 10.1016/S0140-6736(03)13304-1 [DOI] [PubMed] [Google Scholar]
- Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, Nance M, Ross CA, Scahill RI, Wetzel R, et al. 2015. Huntington disease. Nat Rev Dis Primers 1: 15005. 10.1038/nrdp.2015.5 [DOI] [PubMed] [Google Scholar]
- Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. 2019. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575: 688–692. 10.1038/s41586-019-1705-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudle WM, Zhang J. 2009. Glutamate, excitotoxicity, and programmed cell death in Parkinson disease. Exp Neurol 220: 230–233. 10.1016/j.expneurol.2009.09.027 [DOI] [PubMed] [Google Scholar]
- Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, Song S, Tavana O, Gu W. 2019. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Boil 21: 579–591. 10.1038/s41556-019-0305-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P. 1993. Oxidative stress, glutamate, and neurodegenerative disorders. Science 262: 689–695. 10.1126/science.7901908 [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. 2012. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149: 1060–1072. 10.1016/j.cell.2012.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. 2017. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13: 91–98. 10.1038/nchembio.2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius E, Scheel CH, et al. 2019. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575: 693–698. 10.1038/s41586-019-1707-0 [DOI] [PubMed] [Google Scholar]
- Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM, Rajbhandari P, Stokes ME, Bender HG, Csuka JM, et al. 2020. Transferrin receptor is a specific ferroptosis marker. Cell Rep 30: 3411–3423.e7. 10.1016/j.celrep.2020.02.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd RA, Hensley K. 2002. Oxidative stress in brain aging: implications for therapeutics of neurodegenerative diseases. Neurobiol Aging 23: 795–807. 10.1016/S0197-4580(02)00019-2 [DOI] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, et al. 2006. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 125: 1179–1191. 10.1016/j.cell.2006.04.026 [DOI] [PubMed] [Google Scholar]
- Gray M, Shirasaki DI, Cepeda C, André VM, Wilburn B, Lu XH, Tao J, Yamazaki I, Li SH, Sun YE, et al. 1998. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci 28: 6182–6195. 10.1523/JNEUROSCI.0857-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey MA, Chesselet MF. 2003. Apoptosis in Huntington's disease. Prog Neuropsychopharmacol Biol Psychiatry 27: 255–265. 10.1016/S0278-5846(03)00021-6 [DOI] [PubMed] [Google Scholar]
- Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. 2017. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13: 81–90. 10.1038/nchembio.2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klepac N, Relja M, Klepac R, Hećimović S, Babić T, Trkulja VJ. 2007. Oxidative stress parameters in plasma of Huntington's disease patients, asymptomatic Huntington's disease gene carriers and healthy subjects: a cross-sectional study. J Neurol 254: 1676–1683. 10.1007/s00415-007-0611-y [DOI] [PubMed] [Google Scholar]
- Kraft V, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, Merl-Pham J, Bao X, Anastasov N, Kössl J, et al. 2020. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci 6: 41–53. 10.1021/acscentsci.9b01063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Kalonia H, Kumar A. 2010. Nitric oxide mechanism in the protective effect of antidepressants against 3-nitropropionic acid-induced cognitive deficit, glutathione and mitochondrial alterations in animal model of Huntington's disease. Behav Pharmacol 21: 217–230. 10.1097/FBP.0b013e32833a5bf4 [DOI] [PubMed] [Google Scholar]
- Lee J, Kosaras B, Del Signore SJ, Cormier K, McKee A, Ratan RR, Kowall NW, Ryu H. 2011. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington's disease mice. Acta Neuropathol 121: 487–498. 10.1007/s00401-010-0788-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Maher P. 2015. Chronic glutamate toxicity in neurodegenerative diseases-what is the evidence? Front Neurosci 9: 469. 10.3389/fnins.2015.00469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang W, Li Y, Xiao Y, Cheng J, Jia J. 2015. The 5-lipoxygenase inhibitor zileuton confers neuroprotection against glutamate oxidative damage by inhibiting ferroptosis. Biol Pharm Bull 38: 1234–1239. 10.1248/bpb.b15-00048 [DOI] [PubMed] [Google Scholar]
- Lunkes A, Lindenberg KS, Ben-Haı¨em L, Weber C, Devys D, Landwehrmeyer GB, Mandel JL, Trottier Y. 2002. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell 10: 259–269. 10.1016/S1097-2765(02)00602-0 [DOI] [PubMed] [Google Scholar]
- MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, Groot N, et al. 1993. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72: 971–983. 10.1016/0092-8674(93)90585-E [DOI] [PubMed] [Google Scholar]
- Maher P, Currais A, Schubert D. 2020. Using the oxytosis/ferroptosis pathway to understand and treat Age-associated neurodegenerative diseases. Cell Chem Biol 27: 1456–1471. 10.1016/j.chembiol.2020.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, Koppula P, Wu S, Zhuang L, Fang B, et al. 2021. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593: 586–590. 10.1038/s41586-021-03539-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim SU, et al. 1998. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet 18: 150–154. 10.1038/ng0298-150 [DOI] [PubMed] [Google Scholar]
- Mashima R, Okuyama T. 2015. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol 6: 297–310. 10.1016/j.redox.2015.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath T, Baskerville R, Rogero M, Castell L. 2022. Emerging evidence for the widespread role of glutamatergic dysfunction in neuropsychiatric diseases. Nutrients 14: 917. 10.3390/nu14050917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naia L, Rego AC. 2018. Isolation and maintenance of murine embryonic striatal neurons. Bio Protoc 8: e2823. 10.21769/BioProtoc.2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nana AL, Kim EH, Thu DC, Oorschot DE, Tippett LJ, Hogg VM, Synek BJ, Roxburgh R, Waldvogel HJ, Faull RL. 2014. Widespread heterogeneous neuronal loss across the cerebral cortex in Huntington's disease. J Huntingtons Dis 3: 45–64. 10.3233/JHD-140092 [DOI] [PubMed] [Google Scholar]
- Paul BD, Snyder SH. 2019. Impaired redox signaling in Huntington's disease: therapeutic implications. Front Mol Neurosci 12: 68. 10.3389/fnmol.2019.00068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pergola C, Gerstmeier J, Mönch B, Çalışkan B, Luderer S, Weinigel C, Barz D, Maczewsky J, Pace S, Rossi A, et al. 2014. The novel benzimidazole derivative BRP-7 inhibits leukotriene biosynthesis in vitro and in vivo by targeting 5-lipoxygenase-activating protein (FLAP). Br J Pharmacol 171: 3051–3064. 10.1111/bph.12625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters-Golden M, Brock TG. 2003. 5-lipoxygenase and FLAP. Prostaglandins Leukot Essent Fatty Acids 69: 99–109. 10.1016/S0952-3278(03)00070-X [DOI] [PubMed] [Google Scholar]
- Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, et al. 1999. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet 8: 397–407. 10.1093/hmg/8.3.397 [DOI] [PubMed] [Google Scholar]
- Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, et al. 2014. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 136: 4551–4556. 10.1021/ja411006a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR, Angeli JPF, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK, Kagan VE, et al. 2017. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171: 273–285. 10.1016/j.cell.2017.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR, Jiang X, Gu W. 2020. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol 30: 478–490. 10.1016/j.tcb.2020.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thu DC, Oorschot DE, Tippett LJ, Nana AL, Hogg VM, Synek BJ, Luthi-Carter R, Waldvogel HJ, Faull RL. 2010. Cell loss in the motor and cingulate cortex correlates with symptomatology in Huntington's disease. Brain 133: 1094–1110. 10.1093/brain/awq047 [DOI] [PubMed] [Google Scholar]
- Tower J. 2015. Programmed cell death in aging. Ageing Res Rev 23: 90–100. 10.1016/j.arr.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. 2000. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci 97: 8093–8097. 10.1073/pnas.110078997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler VC, White JK, Gutekunst CA, Vrbanac V, Weaver M, Li XJ, Li SH, Yi H, Vonsattel JP, Gusella JF, et al. 2000. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet 9: 503–513. 10.1093/hmg/9.4.503 [DOI] [PubMed] [Google Scholar]
- Xie Z, Xu M, Xie J, Liu T, Xu X, Gao W, Li Z, Bai X, Liu X. 2022. Inhibition of ferroptosis attenuates glutamate excitotoxicity and nuclear autophagy in a CLP septic mouse model. Shock 57: 694–702. 10.1097/SHK.0000000000001893 [DOI] [PubMed] [Google Scholar]
- Yablonska S, Ganesan V, Ferrando LM, Kim J, Pyzel A, Baranova OV, Khattar NK, Larkin TM, Baranov SV, Chen N, et al. 2019. Mutant huntingtin disrupts mitochondrial proteostasis by interacting with TIM23. Proc Natl Acad Sci 116: 16593–16602. 10.1073/pnas.1904101116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Stockwell BR. 2016. Ferroptosis: death by lipid peroxidation. Trends Cell Biol 26: 165–176. 10.1016/j.tcb.2015.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.