

Abstract
Rhamnogalacturonan I (RG-I) is a major plant cell wall pectic polysaccharide defined by its repeating disaccharide backbone structure of [4)-α-d-GalA-(1,2)-α-l-Rha-(1,]. A family of RG-I:Rhamnosyltransferases (RRT) has previously been identified, but synthesis of the RG-I backbone has not been demonstrated in vitro because the identity of Rhamnogalacturonan I:Galaturonosyltransferase (RG-I:GalAT) was unknown. Here a putative glycosyltransferase, At1g28240/MUCI70, is shown to be an RG-I:GalAT. The name RGGAT1 is proposed to reflect the catalytic activity of this enzyme. When incubated together with the rhamnosyltransferase RRT4, the combined activities of RGGAT1 and RRT4 result in elongation of RG-I acceptors in vitro into a polymeric product. RGGAT1 is a member of a new GT family categorized as GT116, which does not group into existing GT-A clades and is phylogenetically distinct from the GALACTURONOSYLTRANSFERASE (GAUT) family of GalA transferases that synthesize the backbone of the pectin homogalacturonan. RGGAT1 has a predicted GT-A fold structure but employs a metal-independent catalytic mechanism that is rare among glycosyltransferases with this fold type. The identification of RGGAT1 and the 8-member Arabidopsis GT116 family provides a new avenue for studying the mechanism of RG-I synthesis and the function of RG-I in plants.
Pectins are a galacturonic acid (GalA)-rich class of polysaccharides present in the cell wall of nearly every plant species and cell type. The traditionally recognized roles of pectic polysaccharides as structural components of plant cell walls have been studied extensively in model organisms, most notably Arabidopsis1, and also in biomass feedstock species including switchgrass and poplar2. Having well-established uses as a safe food additive and proposed roles in gut microbiome health, contemporary interest in pectin research extends to uncovering the positive health effects that are expected to result from human metabolic pathways affected by pectin consumption3,4. As a result of the chemical complexity and structural heterogeneity that exists within pectic polysaccharides, several challenges limit the current understanding of pectins as a family of functionally active macromolecules. These include difficulties in isolating homogeneous pectic domains for use in biological studies and characterizing the families of biosynthetic enzymes that synthesize the individual sugar linkages.
The simplest glycan domain of pectin, homogalacturonan (HG), is a linear polysaccharide of repeating α-d-1,4-linked GalA. More complex pectins, such as rhamnogalacturonan II and xylogalacturonan, have HG backbones substituted with additional sugar side chains1. Polymerization of the HG backbone is catalysed by GALACTURONOSYLTRANSFERASEs (GAUTs), which are members of the glycosyltransferase (GT) GT8 family in the Carbohydrate Active Enzymes (CAZy) database5–7. Rhamnogalacturonan I (RG-I) is a pectic domain with a backbone that contains rhamnose (Rha) in a repeating disaccharide [4)-α-d-GalA-(1,2)-α-l-Rha-(1,] structure. The GalA in the RG-I backbone is partially acetylated, and approximately 50% of the Rha residues in the backbone are branched at O-4 with side branches largely composed of arabinan, galactan and arabinogalactan1. GAUT family enzymes have not been shown to incorporate GalA into RG-I oligosaccharide acceptors5,8, which suggests that a distinct, unidentified GT family may function in polymerization of the RG-I backbone.
The in vivo functions of RG-I are poorly understood; however, the cell wall structural and compositional changes that occur during fruit ripening have provided some initial insight into RG-I function9,10. RG-I has been proposed to contribute to cell wall structural integrity through interactions with other polysaccharides and to cellular adhesion by interacting with HG within the primary wall and middle lamella2,9,11. The isolation of RG-I polysaccharides has previously required extensive sequential selective hydrolysis and extraction of pectin-rich tissues such as citrus peels, the major source of commercial pectins12. Seed mucilages, water-retentive polysaccharide fractions secreted by many species to maintain seed viability and hydration, have been identified as an ideal source of RG-I polysaccharide material for biosynthesis studies13,14. The polysaccharide components of seed mucilages vary across species, but Arabidopsis seed epidermal cells have been shown to secrete a non-adherent mucilage highly enriched in RG-I with minimal or no backbone substitution15. Arabidopsis seed mucilage RG-I is a polysaccharide with a molecular mass greater than 600 kDa15.
Several activities related to RG-I biosynthesis have been identified and added to the families of GTs categorized within the CAZy database. A family of RG-I:Rhamnosyltransferases, annotated as RRT, transfers Rha to RG-I acceptors, resulting in an α-4 linkage to GalA on the non-reducing end16. The discovery of the RRT activity resulted in the establishment of CAZy family GT106. The RRT clade in Arabidopsis has recently been expanded to 10 members, of which 5 have been shown to have RG-I:RRT activity17. Consistent with a role in RG-I mucilage synthesis, the founding member, RRT1, was discovered due to its high expression in the late stages of Arabidopsis seed development when mucilage production is elevated16. A GT family has also been identified that functions in the elongation of the RG-I-specific β-1,4-linked galactans. Each of the three members of the GALS family, a sub-clade of GT92 in Arabidopsis, has been confirmed to exhibit RG-I galactan synthase function18,19.
Synthesis of the RG-I backbone has not been demonstrated in vitro because Rhamnogalacturonan I:Galaturonosyltransferase (RG-I:GalAT), the enzyme that transfers GalA to Rha-containing RG-I acceptors, has not been identified. Here, At1g28240, a gene encoding a protein currently annotated as MUCILAGE-RELATED70 (MUCI70), was selected as a candidate RG-I:GalAT. Similar to RRT1, MUCI70 was originally discovered due to its high expression in the Arabidopsis seed coat during a developmental period consistent with upregulated RG-I biosynthesis20. The putative domain structure of MUCI70 has characteristics common with known GT families, including an N-terminal transmembrane domain and a predicted C-terminal putative GT domain currently annotated as DUF616 (PF04765)21. This putative GT domain has been predicted to be most closely related to GT8 family proteins22, but DUF616-domain proteins have not been identified as members of the GAUT1-related superfamily23. Mutants of MUCI70 have reduced staining of the mucilage that is released from seeds upon hydration and a reduced amount of both GalA and Rha in total mucilage extracts, phenotypes also observed in mutants of RRT116,20. Concurrent with the work described in this study, MUCI70 was also identified in a genome-wide study of phenotypes resulting from single nucleotide polymorphisms, with mutant alleles of muci70 resulting in reduced molecular weight of the mucilage polysaccharide24. The association of MUCI70 expression with the size and composition of RG-I polysaccharides recovered from Arabidopsis seed mucilage supported a proposed function in RG-I biosynthesis. Here we show that MUCI70 is a GalAT that functions with RRT to synthesize the RG-I backbone.
Results
Heterologous expression of a candidate glycosyltransferase
Polymerization of the RG-I backbone in vitro requires an enzymatic source of RG-I:GalAT and RhaT activities. On the basis of the structure of RG-I, the predicted RG-I:GalAT activity is illustrated in Fig. 1 as the transfer of GalA to RG-I oligosaccharide acceptors containing Rha on the non-reducing end. MUCI70 was selected as a putative RG-I:GalAT. Consistent with other families of Golgi-localized glycosyltransferases that synthesize cell wall matrix proteins, MUCI70 has a predicted transmembrane domain in the N-terminal region and a C-terminal putative GT domain (Pfam: DUF616/PF04765) (Fig. 2a). To purify MUCI70 as a putative enzymatic source of RG-I:GalAT activity, the transmembrane-truncated coding region of At1g28240 was cloned into a vector for recombinant expression as a secreted protein in human embryonic kidney 293 (HEK293) cells.
Fig. 1 |. Representative chemical structure of rhamnogalacturonan I (RG-I) backbone.
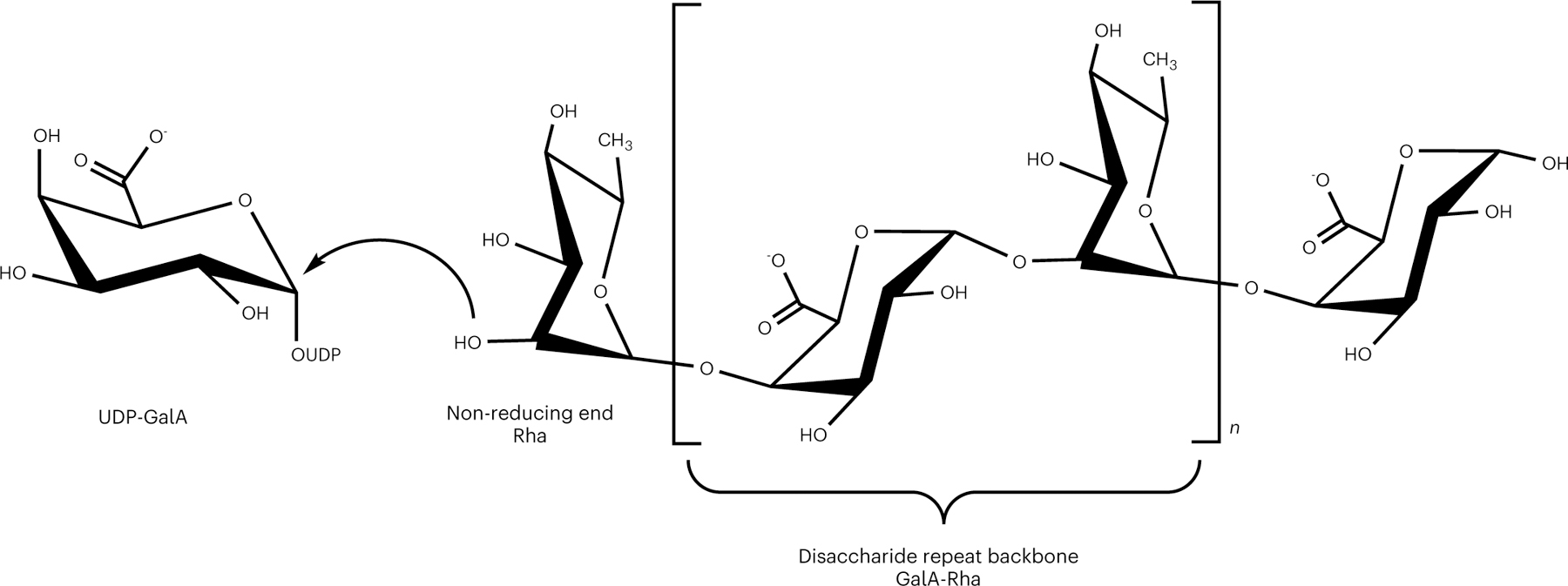
RG-I is a polysaccharide with a [4)-α-d-GalA-(1,2)-α-l-Rha-(1,] disaccharide repeat backbone. The curved arrow represents the nucleophilic attack of an acceptor oligosaccharide with a non-reducing end Rha to the anomeric carbon of UDP-GalA. This putative transfer of a GalA monosaccharide to elongate the RG-I acceptor with retention of stereochemistry is referred to here as RG-I:GalAT activity. Chemical structure was created with ChemDraw Professional 16.0.
Fig. 2 |. MUCI70 protein domain structure, expression construct and purified protein.
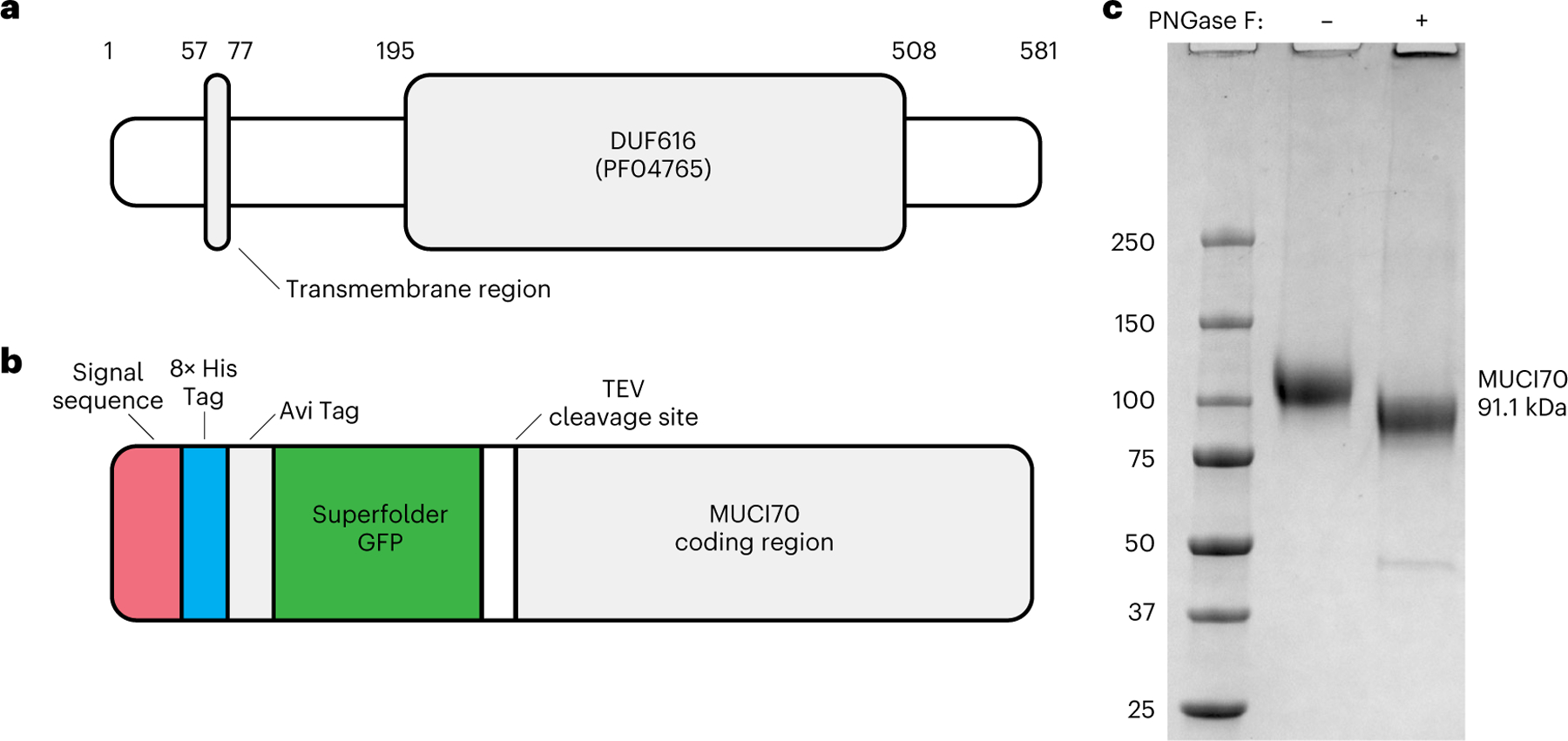
a, Representation of the conserved domains within the 581 amino acid coding region of At1g28240 (MUCI70). The region from amino acid 57 to 77 is a putative transmembrane domain that was truncated to create the expression construct. Residues 195–508 are the putative GT domain. b, Expression construct pGEn2 containing the truncated coding region (residues 78–581) of MUCI70. The fusion protein has N-terminal tags that include a signal sequence for secretion, 8× His Tag, Avi Tag, ‘superfolder’ GFP and a site for tag removal by Tobacco Etch Virus (TEV) protease. c, Coomassie blue-stained SDS–polyacrylamide gel of the MUCI70 fusion protein. Expression of the fusion protein in HEK293 cells resulted in secretion of the protein into the cell culture medium. The protein was purified by Ni2+-NTA affinity (HisTrap HP) and size-exclusion chromatography (Superdex 200), resulting in a protein that resolves near the expected molecular weight of the fusion protein (91.1 kDa) after removal of N-linked glycosylation by PNGase F. Similar results were obtained from two independent purifications of MUCI70. The gel shown is representative of two independent digest experiments.
Mammalian HEK293 cells have been extensively developed as a system for the recombinant expression of glycosyltransferases secreted in a soluble form that can be purified for use in in vitro activity assays25. In recent years, this expression system has been used to express plant cell wall GTs from a range of different GT families, including the GAUTs, XYLAN SYNTHASE-1 and Fucosyltransferases6,8,20,26–28. The transmembrane-truncated coding region of the target protein was inserted into the pGEn2 vector, resulting in expression of a fusion protein with N-terminal tags including a signal sequence to target the protein for secretion, 8× His Tag, Avi Tag and a ‘superfolder’ green fluorescent protein (GFP) domain followed by the predicted ectodomain of MUCI70 (residues 78–581, abbreviated as MUCI70Δ77, Fig. 2b). Expression in HEK293 cells and purification using Ni2+-NTA affinity followed by size-exclusion chromatography resulted in a fusion protein that was soluble and resolved as a highly purified monomer by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) (Fig. 2c). Expression of MUCI70Δ77 and secretion into the culture medium was high in both small-scale (20 ml) and larger-scale (250 ml) HEK293 cell cultures (Extended Data Fig. 1), with 11 mg of purified protein obtained from the latter culture. Treatment of the fusion protein with peptide:N-glycosidase F (PNGase F) resulted in a reduction of the molecular weight of the monomer, indicating that MUCI70 is N-glycosylated when expressed in HEK293 cells. Following PNGase F treatment, the fusion protein resolved near the expected size (91.1 kDa) in a non-reducing SDS–PAGE gel (Fig. 1c). The glycosylation state of MUCI70 may differ in planta. The fully glycosylated protein was used for all subsequent enzyme activity assays.
RGGAT1 (MUCI70) adds GalA to RG-I acceptors in vitro
The purified MUCI70 protein was tested for the ability to transfer GalA to different pectin acceptors. RG-I acceptor oligosaccharides were generated by digesting Arabidopsis seed mucilage with a rhamnogalacturonan endohydrolase from Aspergillus aculeatus, RGase A29,30 (Extended Data Fig. 2a). Following the method originally developed by Ishii et al.31, RG-I oligosaccharides of defined chain lengths were derivatized to include a 2-aminobenzamide (2AB) fluorescent tag at the reducing terminus and were purified from the mixture of digested mucilage (Extended Data Fig. 2b and Supplementary Fig. 1). Elongation of the oligosaccharide by transfer to the non-reducing end, as depicted in Fig. 3a, would be consistent with the elongation mechanism of the pectic biosynthetic GAUT6,32 and RRT16 enzyme families. The abbreviation RG-I (R) signifies RG-I oligosaccharides generated by digestion of RG-I with RGase A and resulting in non-reducing terminal rhamnose.
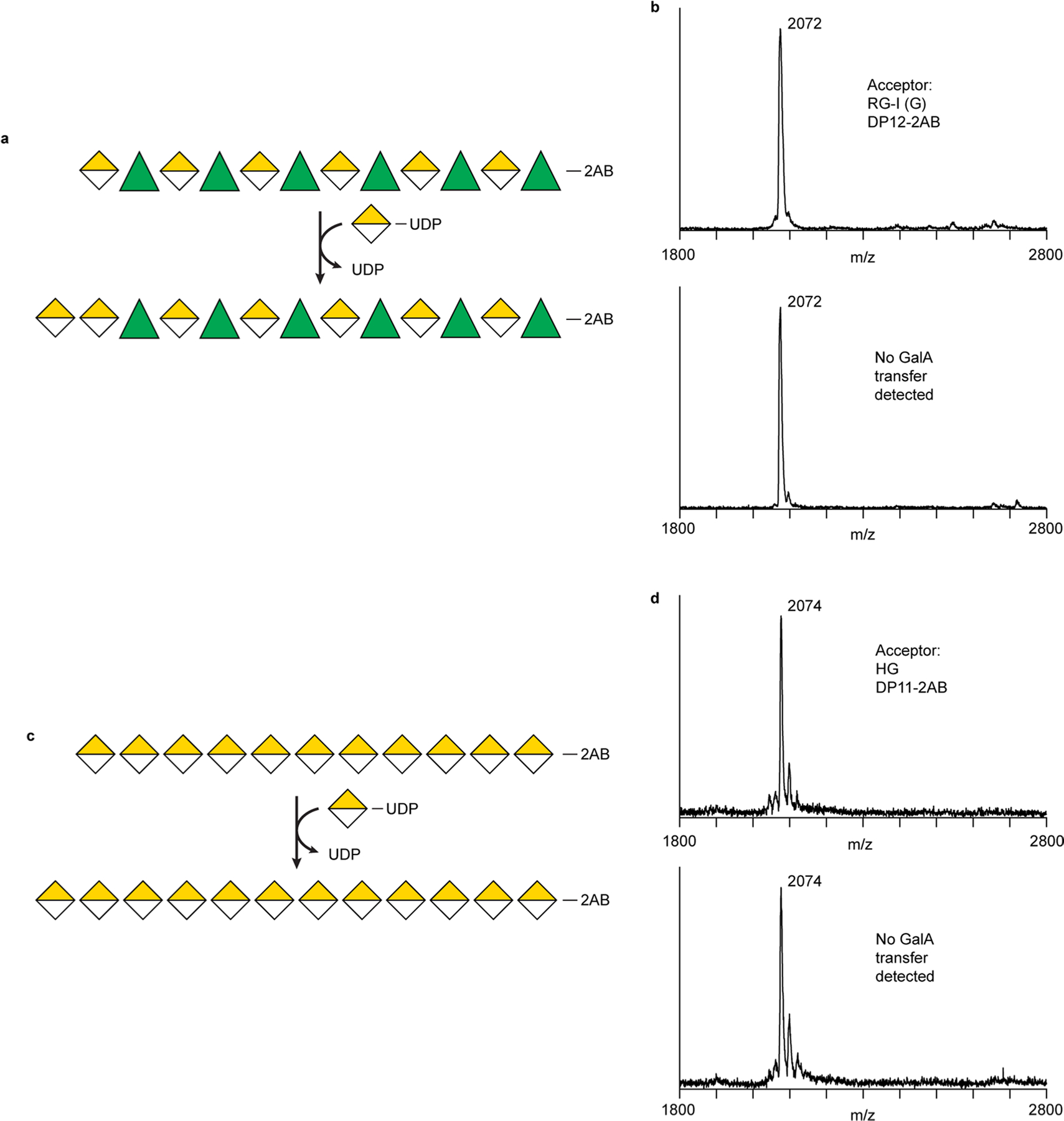
Fig. 3 |. Recombinant RGGAT1 is an RG-I:Galacturonosyltransferase.
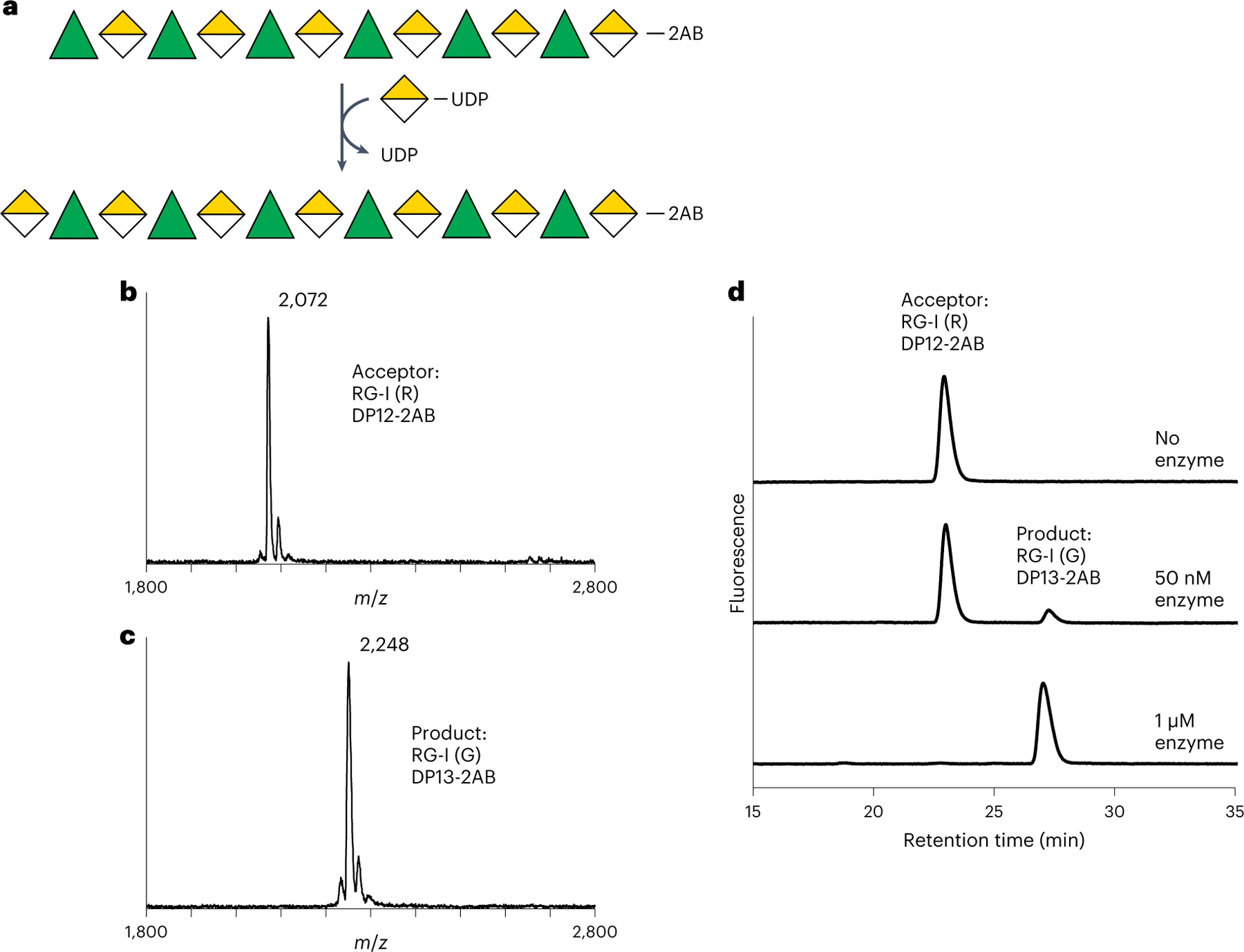
a, Reaction scheme representing RG-I:GalAT activity using the Symbol Nomenclature for Glycans63. Rha, green triangles. GalA, yellow divided diamond. The oligosaccharide acceptor contains 2AB at the reducing end. Transfer of GalA from UDP-GalA results in conversion of an RG-I (R) DP12-2AB acceptor to an RG-I (G) DP13-2AB product. The abbreviation RG-I (R) signifies RG-I oligosaccharides obtained from digestion of RG-I with RGase A resulting in a non-reducing terminal rhamnose, while RG-I (G) represents an RG-I oligosaccharide with a GalA at the non-reducing terminus, in this case resulting from the catalytic activity of RGGAT1 on RG-I (R). b, MALDI-TOF-MS spectrum of the DP12-2AB oligosaccharide acceptor of predicted mass 2,071.8 Da. c, MALDI-TOF-MS spectrum of the product after GalA transfer. The mass increase of 176 Da is consistent with the addition of a single GalA unit to the acceptor oligosaccharide shown in b. d, The product of RGGAT1 activity was detected by anion exchange chromatography with fluorescence detection. RGGAT1 enzyme (50 nM or 1 µM) was incubated with 1 mM UDP-GalA and 100 µM RG-I (R) DP12-2AB acceptor for 10 min. The reaction was boiled, and an aliquot representing 2.5 nmol of the starting acceptor was separated using a CarboPac PA-1 anion exchange column. The starting acceptor is represented by the peak with a retention time of 22 min. The top panel is a control reaction with no enzyme. In the middle panel, use of 50 nM enzyme resulted in approximately 10% of the acceptor converted into the product based on the peak areas. In the bottom panel, a reaction containing 1 µM enzyme resulted in 100% conversion of the acceptor into the product. The assay is representative of at least three independent replicates. Quantitation of the peak area of the reaction containing 50 nM enzyme is shown in Extended Data Fig. 4a.
RG-I:GalAT activity was assayed by incubating MUCI70 with an RG-I (R) oligosaccharide acceptor of a degree of polymerization (DP) of 12 total sugar units. The hypothetical reaction scheme (Fig. 3a) represents elongation of a DP12 (R) to a DP13 (G) oligosaccharide. On the basis of a mass shift of 176 Da corresponding to the addition of a GalA monomer, MUCI70 catalysed the transfer of GalA to the RG-I (R) acceptor (Fig. 3b,c). The activity of MUCI70 is limited to the addition of a single GalA to this acceptor and does not catalyse the transfer of GalA to RG-I acceptors containing a GalA on the non-reducing end or to HG acceptors (Extended Data Fig. 3). On the basis of this activity, we propose the name RG-I:GALACTURONOSYLTRANSFERASE1 (RGGAT1) for this enzyme.
The initial test of RGGAT1 activity, described in the previous paragraph, used a high enzyme concentration (1 µM) that resulted in the complete conversion of a DP12 (R) acceptor to a product elongated by a single GalA monosaccharide. A separate set of reaction conditions was established to measure the biochemical parameters of the enzyme activity. In these reactions, the activity was tested using a lower enzyme concentration (50 nM) to limit the reaction progress. A 10 min incubation with the DP12 (R) acceptor under these conditions resulted in a 9.7% conversion of the DP12 (R) acceptor to the DP13 (G) product (Fig. 3d and Extended Data Fig. 4).
Biochemical characterization of RGGAT1 activity
The kinetics of RG-I:GalA activity was determined using a commercial UDP-Glo assay that detects activity on the basis of the conversion of the UDP released during the glycosyltransfer reaction to a luminescent signal. The RGGAT1 reaction progress curve was monitored from 0 to 60 min using the DP12 (R) acceptor substrate and UDP-GalA as a donor, indicating a 6% conversion to products in a reaction containing 1 mM UDP-GalA, 100 µM acceptor and 50 nM enzyme when measured at 10 min (Extended Data Fig. 4b). Similar levels of activity were detected using anion exchange chromatography (Extended Data Fig. 4a) under equivalent reaction conditions, indicating that both methods are suitable for biochemical characterization of RG-I:GalAT activity.
The pH optimum of RGGAT1 was 6.5 (Fig. 4a). Comparison of RGGAT1 activity using a series of RG-I acceptors revealed that RGGAT1 can detectably transfer GalA to acceptors of at least DP6, with an approximately 4-fold increase in activity with acceptors of DP ≥ 10 (Fig. 4b). DP6 was the smallest size acceptor purified from the Arabidopsis mucilage digest.
Fig. 4 |. Biochemical characterization of RG-I:GalAT activity by RGGAT1.
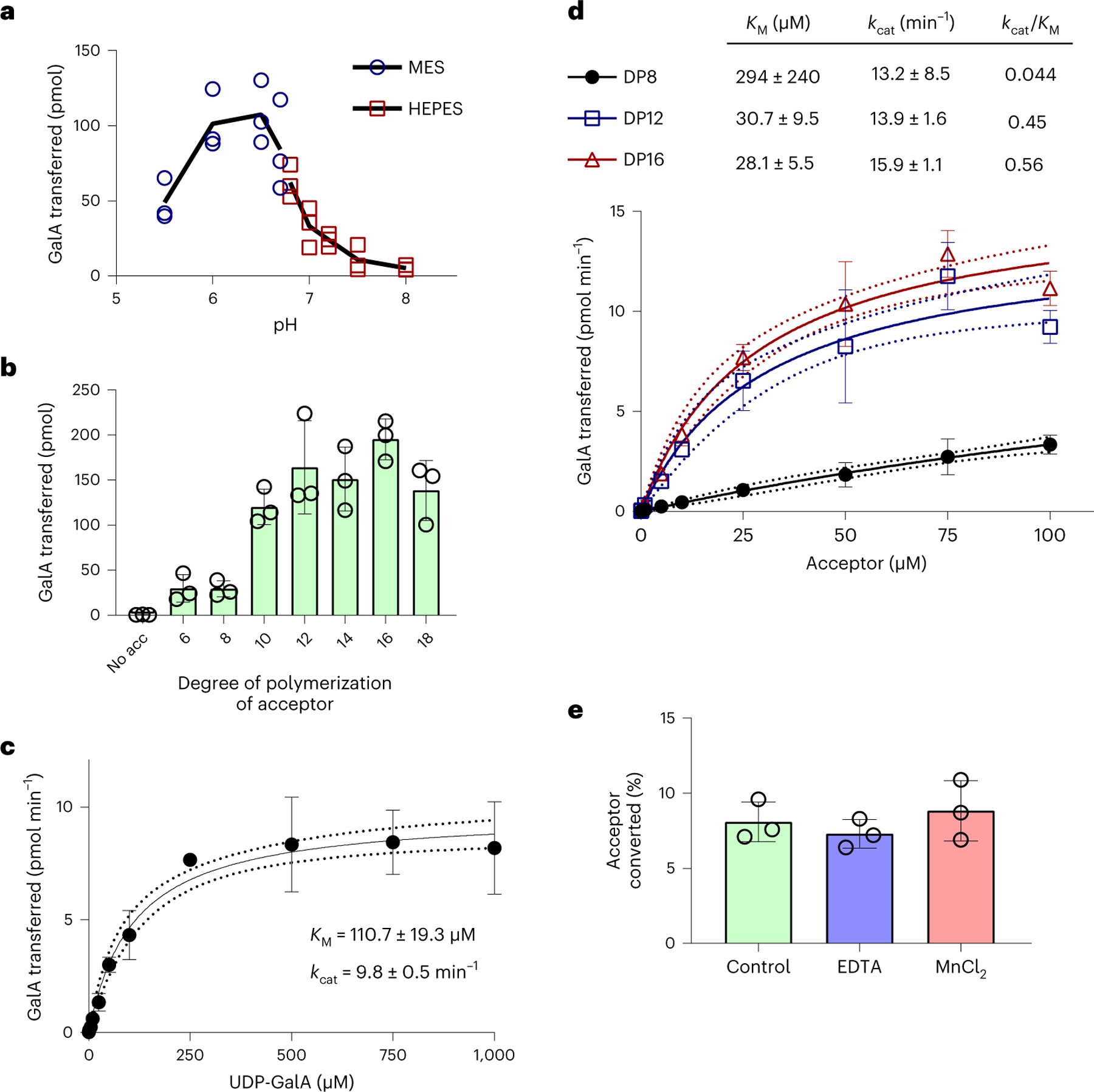
a, The pH optimum of RGGAT1 activity was measured using UDP-Glo in 10 min reactions containing 50 nM enzyme, 1 mM UDP-GalA and 100 µM of a DP12-2AB acceptor. Reactions were incubated with 50 mM of MES buffer (blue circles) of pH 5.5–6.7 or HEPES buffer (red squares) of pH 6.7–8.0. The buffer MES pH 6.5 was used for standard condition assays. The black line represents the average value of n = 3 independent assays. Individual data points from the three assays are shown. b, RGGAT1 activity was measured in 10 min reactions containing 50 nM enzyme, 1 mM UDP-GalA and 100 µM of acceptors with degrees of polymerization ranging from DP6 to DP18 or no acceptor (no acc) using UDP-Glo. Error bars represent standard deviations of n = 3 independent experiments. c, Michaelis-Menten kinetics for the UDP-GalA donor. RGGAT1 was incubated for 10 min with 100 µM DP16 acceptor and variable concentrations of UDP-GalA (0–1,000 µM). Kinetic constants were calculated by nonlinear regression using GraphPad Prism. Error bars represent standard deviations from n = 4 independent experiments. Dotted lines represent 95% confidence intervals. KM and kcat are reported as mean ± s.e.m. d, Michaelis-Menten kinetics for RG-I oligosaccharide acceptors of DP8, DP12 and DP16. RGGAT1 was incubated for 10 min with 1 mM UDP-GalA and variable concentrations of the indicated acceptors (0–100 µM). Kinetic constants were calculated by nonlinear regression using GraphPad Prism. Error bars represent standard deviations of n = 3 independent experiments. Dotted lines represent 95% confidence intervals. KM and kcat are reported as mean ± s.e.m. e, RGGAT1 was incubated in a 50 mM MES pH 6.5 buffer (control) or with buffer containing either 10 mM EDTA or 10 mM MnCl2 for 30 min before the assay. After a 30 min incubation period, the enzyme was diluted and assayed as described. The final concentration during the reaction was 10 mM for EDTA and 0.25 mM for MnCl2. No difference in activity compared to the control reaction was detected. Error bars represent standard deviations of n = 3 independent experiments.
Michaelis-Menten kinetics were measured for the UDP-GalA donor and for acceptor oligosaccharides of different chain lengths (Fig. 4c). RGGAT1 has a Michaelis constant (KM)for UDP-GalA of 110 µM. Using a range of acceptor concentrations from 0 to 100 µM, we were able to model Michaelis-Menten kinetics, with similar results for the DP12 and DP16 acceptors, yielding an estimated KM of 28–31 µM; however, the estimated KM of 294 µM for the DP8 acceptor was outside of the range of acceptor concentrations available for assay (Fig. 4d). The inability of the DP8 acceptor to saturate the active site under this range of concentrations resulted in a catalytic efficiency kcat/KM, where kcat is the catalytic constant, that was >10-fold higher for the longer-chain acceptors.
Most families of GT-A fold enzymes require divalent cations for activity since they coordinate interactions between the diphosphate of the sugar nucleotide donor and the enzyme active site DxD motif33. The most common divalent cation utilized by glycosyltransferases is Mn2+, which is also required for transferase activity by the HG biosynthetic complex GAUT1:GAUT76. Following Ni2+-NTA affinity purification, RGGAT1 was dialysed against Chelex-100, a resin used to remove any residual metal ions by chelation. In the assays presented above, activity was observed without the addition of exogenous sources of metal ions to the reaction mixture, suggesting that RGGAT1 might not require metal ions for catalysis. To verify that RGGAT1 is a metal-independent GT, the enzyme was incubated in a MES buffer containing no additives (control), 10 mM EDTA or 10 mM MnCl2. After a 30 min incubation period, the assay was performed. Identical enzyme activity was observed in control reactions as well as in those containing EDTA or MnCl2. The results indicate that divalent cations are not required for activity (Fig. 4e).
In assays containing 50 nM enzyme, the reactions were limited to approximately 20% conversion of the acceptor, as measured at 60 min (Extended Data Fig. 4c). Increased reaction times, including overnight incubation of samples, did not result in complete conversion of the acceptor at limiting enzyme concentrations. When a phosphatase (potato apyrase) was included in the reaction, at least a 2-fold increase in the conversion of the acceptor was detected at 60 min (Extended Data Fig. 4c), suggesting that RGGAT1 is inhibited by UDP released during the transferase reaction.
In vitro polymerization of RG-I by RGGAT1 and RRT4
The data presented established that RGGAT1 transfers a single GalA to RG-I acceptors. It has been previously shown that a family of RG-I:Rhamnosyltransferases (RRT) transfer Rha to RG-I acceptors16. If the linkages transferred by these two enzymes were consistent with the linkages of the GalA-Rha disaccharide repeat backbone of RG-I, then we predicted that the combined activities would result in polymerization of longer-chain RG-I polysaccharides through sequential addition of GalA and Rha to the non-reducing end of elongating acceptor oligosaccharides.
To purify a source of RG-I:RhaT activity, the coding sequences of the original four RRT enzymes with known activity16, truncated by their predicted N-terminal transmembrane domains, were cloned into the pGEn2 vector for expression in HEK293 cells. Compared to RGGAT1, all four members of the RRT family expressed relatively poorly in HEK293 cells, as indicated by the low fluorescence of secreted protein (Extended Data Fig. 5). Of the four proteins tested, RRT4Δ51 resulted in the highest yield of soluble protein. RRT4Δ51 was expressed in a 500 ml culture and purified using Ni2+-NTA affinity. The protein eluted from this purification was resolved on an SDS–PAGE gel under reducing (+DTT (dithiothreitol)) and non-reducing (−DTT) conditions (Fig. 5a). Under reducing conditions, the major band detected was consistent with the expected molecular weight of the RRT4Δ51 fusion protein, but the appearance of a higher-molecular-weight band under non-reducing conditions suggested that an aggregated form of the protein co-purified with the monomeric protein during Ni2+-NTA affinity chromatography. Size-exclusion chromatography was unable to separate the active monomer from the aggregates. Despite the lower apparent purity of this enzyme compared with RGGAT1, RRT4 was able to transfer Rha to RG-I acceptors containing a GalA residue on the non-reducing end. A product with an increased mass of 146 Da was detected when RRT4 was added to a reaction mixture containing UDP-Rha and a DP12-2AB (G) RG-I oligosaccharide acceptor (Extended Data Fig. 6), consistent with previously published data showing that RRT4 is an RG-I:RhaT16. Conversion of the RG-I acceptor oligosaccharide required higher enzyme concentrations (1–5 µM, Extended Data Fig. 6) than were necessary for the measurement of activity by RGGAT1, suggesting that the specific activity of RRT4 was low due to the relatively low expression and purity. To compensate for this low conversion efficiency, higher enzyme concentrations were used in reactions with RRT4 to maximize the observable polymerization of longer-chain polysaccharide products.
Fig. 5 |. The combined activities of RGGAT1 and RRT4 polymerize the RG-I backbone.
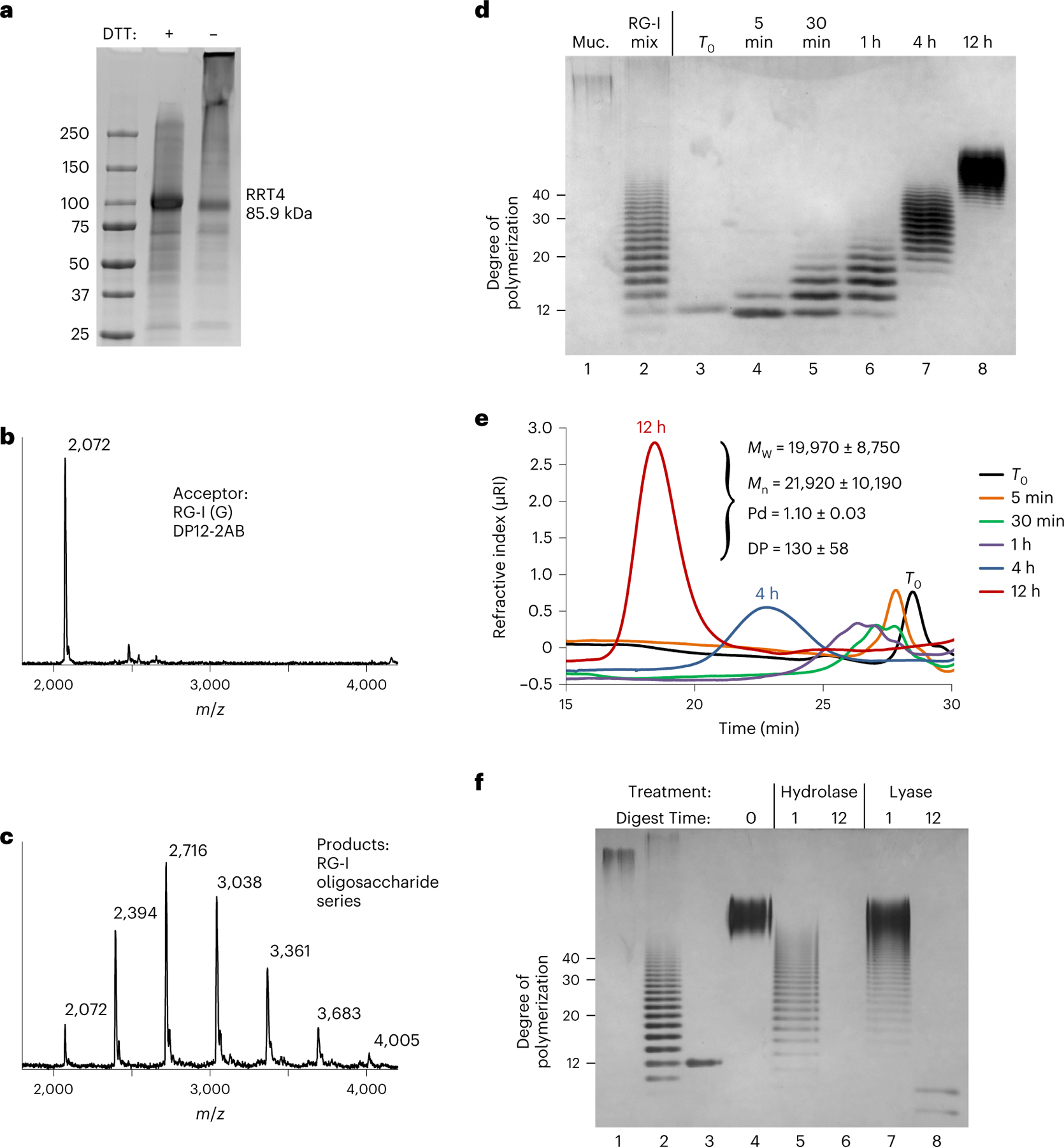
a, Coomassie blue-stained SDS–polyacrylamide gel of the RRT4 fusion protein. The location of the RRT4 monomer with a predicted mass of 85.9 kDa is indicated. Similar results were obtained from three independent purifications of RRT4. The gel shown is a single experiment from the purified protein used in all assays. b, MALDI-TOF-MS spectrum of a control reaction containing a DP12-2AB oligosaccharide acceptor of predicted mass 2,071.8 Da. The acceptor is labelled as RG-I (G) to identify that it contains GalA on the non-reducing end. c, MALDI-TOF-MS spectrum of a reaction containing 5 µM of both RGGAT1 and RRT4 enzymes, 1 mM UDP-GalA, 1 mM UDP-Rha and 100 µM of the RG-I (G) acceptor detected in b. After 1 h incubation, a series of peaks separated by 322 Da is consistent with the addition of GalA-Rha disaccharide units added by the combined activities of GalAT and RhaT. d, In vitro polymerization of RG-I detected by alcian blue-stained polyacrylamide gel electrophoresis. A reaction containing 5 µM of both RGGAT1 and RRT4 enzymes, 1 mM UDP-GalA, 1 mM UDP-Rha and 10 µM of RG-I (R) DP12-2AB acceptor was incubated for the indicated amounts of time. Aliquots equivalent to 300 ng of starting acceptor were removed from the reaction at each time point and were boiled. Control samples are undigested mucilage (Muc., lane 1) and an RG-I oligosaccharide mixture enriched for DP10-40 (lane 2). Reaction samples (lanes 3–8) represent an equal amount of starting reaction material. The degree of polymerization of RG-I oligosaccharides is indicated. The data are representative of duplicate experiments. e, In vitro polymerization of RG-I detected by size-exclusion chromatography with refractive index detection. Reactions were incubated for the amount of time as in d. Aliquots equivalent to 5 µg of starting acceptor were removed from the reaction at individual times, boiled and injected into the column. Selected time points are labelled on the chromatogram. Characteristics of the polysaccharide product synthesized after 12 h measured by SEC-MALS are shown (MW, weight-averaged molecular mass; Mn, number-averaged molecular mass; Pd, polydispersity). Measured values represent the mean ± s.d. from n = 2 experiments. f, Digest of the in vitro polymerized material by RG-I hydrolase and RG-I lyase from Aspergillus aculeatus. In vitro polymerization of RG-I from a DP12-2AB starting acceptor (lane 3) was performed for 12 h (lane 4). The polymerized material was digested with RG-I hydrolase (lanes 5 and 6) or RG-I lyase (lanes 7 and 8) for 1 or 12 h, as indicated. Control lanes contained undigested mucilage (lane 1) and RG-I oligosaccharide mixture (lane 2) as in d. All lanes represent a reaction aliquot equivalent to 300 ng of starting DP12-2AB acceptor.
The potential for the combined activities of RGGAT1 and RRT4 to elongate RG-I acceptors was tested by incubating both enzymes (5 µM) in a reaction mixture containing 1 mM UDP-GalA, 1 mM UDP-Rha and 100 µM DP12 RG-I (G) acceptor. The reaction resulted in a series of peaks separated by 322 Da, consistent with the size of an RG-I disaccharide containing both GalA (176 Da) and Rha (146 Da) residues (Fig. 5b,c). The absence of a detectable intermediate mass resulting from a single Rha addition indicates that the GalA transfer reaction proceeded at a significantly faster rate than the Rha addition under these reaction conditions. The activities of RGGAT1 and RRT4 were limited to a single GalA or Rha transfer when incubated as individual enzymes, and neither RGGAT1 nor RRT4 was able to polymerize RG-I in the absence of the other enzyme (Extended Data Fig. 7).
Having established that the RG-I oligosaccharide acceptor can be elongated by at least 6 disaccharide repeat units when incubated with both RGGAT1 and RRT4 enzymes (Fig. 5c), the enzyme pair was tested for the ability to polymerize longer-chain RG-I polysaccharides. The enzymes were incubated with 2.5 mM UDP-GalA, 2.5 mM UDP-Rha and 25 µM of the DP12-2AB acceptor to create a 100:1 molar ratio of each donor molecule to the acceptor. If the reaction was able to consume the respective sugar nucleotide donors, it would theoretically result in the synthesis of an RG-I polysaccharide of DP212 as a result of the addition of 100 disaccharide units to the initial acceptor. At the indicated time points ranging from 0 to 12 h, aliquots were removed and products were detected using high-percentage polyacrylamide gels stained with alcian blue (Fig. 5d) and size-exclusion chromatography with refractive index detection (Fig. 5e). Both of these methods have previously been used to detect the polymerization of HG by GAUT family enzymes6. The reaction resulted in the synthesis of RG-I polysaccharides that increased in size during the 12 h incubation to a final mixture of polysaccharides of at least DP40 compared to RG-I standards of known size based on alcian blue staining in polyacrylamide gels. The products separated by size-exclusion chromatography were also coupled to a multi-angle light scattering (MALS) detector, which estimated a product size of DP130 for the RG-I polysaccharides synthesized in a 12 h incubation.
The in vitro polymerized RG-I polysaccharides were digested by two enzymes specific to the two linkages in the RG-I backbone. RG-I hydrolase from Aspergillus aculeatus (RGase A) is an endohydrolase that cleaves the [4)-α-d-GalA-(1,2)-α-l-Rha-(1,] linkage, resulting in oligosaccharides containing Rha residues on the non-reducing end30. Alternatively, RG-I lyase (RGase B) is an endolyase that cleaves the [2)-α-l-Rha-(1, 4)-α-d-GalA-(1,] linkage, resulting in oligosaccharides containing 4,5-unsaturated GalA residues on the non-reducing end30. An RG-I polysaccharide was polymerized in vitro, as described above. After termination of the reaction by boiling, the polysaccharide was incubated with RG-I hydrolase or RG-I lyase for 1-12 h and the digested products were detected by alcian blue-stained PAGE (Fig. 5f). The ability of these two enzymes to degrade the in vitro polymerized RG-I polysaccharides confirmed that the linkages synthesized by RGGAT1 and RRT4 are the expected backbone linkages for an RG-I polysaccharide.
The sequential addition of GalA and Rha units to polymerize long-chain RG-I polysaccharides invites the hypothesis that RGGAT1 and RRT family enzymes interact and function as a biosynthetic complex. Co-expression in HEK293 cells of two HG biosynthetic enzymes, GAUT1 and GAUT7, resulted in the formation of a heterocomplex with enhanced expression compared with expression of the individual enzymes in the same system6. We tested whether co-expression of RGGAT1 with four RRT family members in HEK293 cells resulted in enhanced expression of RRT as a preliminary test of interactions between these two GT families. Only RGGAT1 protein was detected in all samples in sufficient amounts to observe monomer bands, suggesting that co-expression with RGGAT1 did not result in increased expression of any RRT family enzymes tested (Extended Data Fig. 8). Although no evidence currently exists for an RG-I biosynthetic heterocomplex, such a complex may require a specific permutation of RGGAT (GT116) and RRT (GT106) family members.
RGGAT1 is a GT116 family enzyme with a predicted GT-A fold
Before this study, RGGAT1 was not annotated as a member of any existing GT family in the CAZy database34. RGGAT1 has now been included as a member of the new family GT116 as a result of the GalA transferase activity presented here. At least 154 plant species and 143 bacterial species listed in the Pfam database have additional uncharacterized sequences containing a GT116 domain (previously DUF616)21. While some of the members of this family may function in pectin biosynthesis, a broad range of substrate utilization can exist within a single GT family. Rather than being grouped by substrate specificity, enzymes within a given GT family are predicted to share a similar overall structural fold34.
Despite the large number of GT families, glycosyltransferases have generally been found to belong to one of three different structural fold types33. The most common fold type, GT-A, includes the GT8 family that contains the GAUTs. We were interested in determining whether RGGAT1 is also predicted to share this fold as a basis for future studies on the structures and mechanisms of the pectin biosynthetic machinery. The GT-A fold shares elements of secondary structure that are highly conserved across many families, including a series of alternating α-helices and β-sheets that make up a Rossman-like domain and four landmark active site motifs (DxD, G-loop, xED and C-His)35. Because RGGAT1 shares limited sequence similarities with other GT sequences, generating an accurate primary sequence alignment for the comparison of these motifs is difficult. Thus, we first used a sequence alignment-independent deep-learning-based method that was recently developed to determine GT fold type on the basis of primary sequence information using a module trained on nearly 50,000 GT sequences36. In contrast with typical methods of sequence or structural alignment, this method recognizes patterns of conserved secondary structure shared within the GT fold classes and uses these common elements for GT fold prediction. By applying this method to 678 representative GT116/DUF616 sequences, the family of proteins was predicted to adopt a GT-A fold with high confidence (Extended Data Fig. 9).
On the basis of the prediction that RGGAT1 contains structural features representative of the broad GT-A fold, we used AlphaFold2 (v2.0.1)37 to model the structure of RGGAT1. The resulting protein structural model was generated with high confidence and conformed to the general structural features of a GT-A fold domain (Supplementary Fig. 2). A structural comparison with a well-characterized GT-A fold from a GT31 family protein38 validates the prediction that the GT116 family of enzymes conforms to a GT-A fold with a core alignment to the GT31 protein structure with a 3.2 Å root-mean-square deviation (RMSD) (Fig. 6a). The alignment has highest structural similarity in the secondary structural elements that are specific to this fold type, which include several α-helices and β-sheets of the Rossman domain. The aligned structural model predicts that three of the GT-A fold common core conserved motifs are positioned into a putative active site (Fig. 6b). The DxD motif (DGK in RGGAT1), xED motif (RDQ) and G-Loop (EGC) are regions with substrate-binding and catalytic functions, but variations occur across the many GT-A fold families and contribute to the mechanistic diversity observed in this enzyme superfamily35. Additional variations occur in the hyper-variable regions, which are regions of secondary structure that are specific to individual GT families and may contribute to the binding of acceptor substrates (Fig. 6b).
Fig. 6 |. Predicted GT-A fold of RGGAT1.
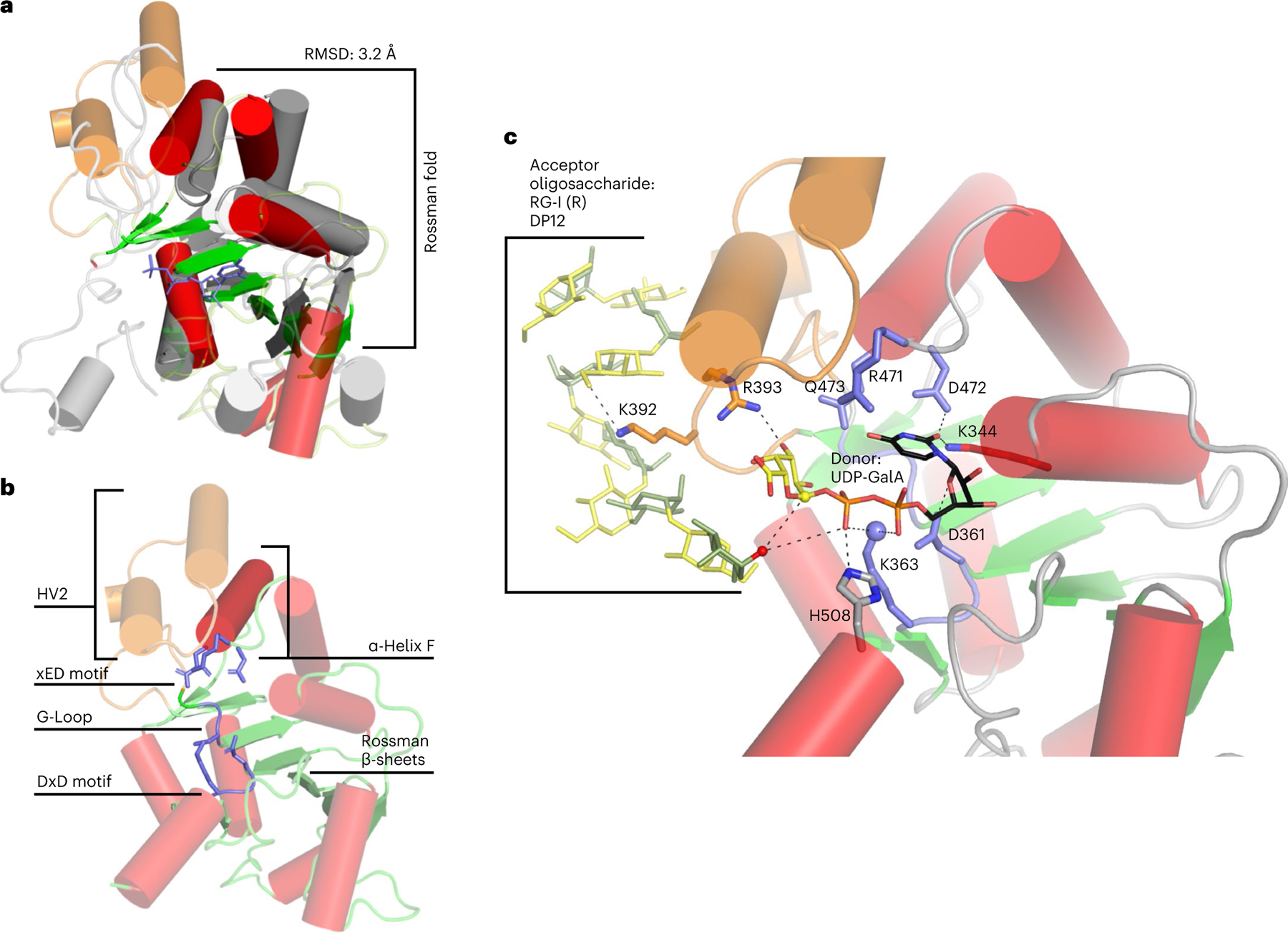
a, Structural alignment of RGGAT1 AlphaFold2 structure (red and green) to a GT31 structure (grey) (pdb: 6wmo) at an RMSD of 3.2 Å across 155 residues, validating that the 3D structure matches a GT-A fold topology. The secondary structure matching algorithm64 in the molecular graphics programme Coot 0.9.7 was used to produce an alignment that was restricted to the core Rossman α-helices (red) and β-sheets (green) shown as opaque structures. b, The AlphaFold2 structure of RGGAT1 has elements of the canonical GT-A fold structure that includes β-sheets of the Rossman fold (green), α-Helix F (dark red) and three conserved motifs of the GT-A fold core (xED, G-Loop and DxD motifs, blue). Hypervariable region 2 (HV2, orange) has helices that are poorly aligned to the template. c, Docked structure of RGGAT1 with the donor UDP-GalA and acceptor RG-I (R) DP12 oligosaccharide. Selected amino acids predicted to interact with the donor and acceptor substrates are shown in stick representation. Dashed lines represent putative hydrogen bonding interactions within 2.7–3.4 Å distance. Residues in blue indicate putative GT-A motifs. Residues in orange are residues present on the HV2 region in contact with the acceptor. Based on a retaining mechanism, the acceptor nucleophile (red sphere) is deprotonated by the β-phosphate oxygen of the UDP donor, allowing a nucleophilic attack on the anomeric carbon of the GalA (yellow sphere)33. The side-chain amine of K363 (blue sphere) may function in place of a divalent cation to interact with the nucleotide phosphate diester of UDP-GalA.
The classification of RGGAT1 as part of a new GT family suggested that GT116 is phylogenetically distinct from the existing GT families. We evaluated the phylogeny using a structure-based sequence alignment of RGGAT1 and related GT116 sequences to previously published GT-A profiles35. This analysis revealed that RGGAT1/GT116 does not group into existing GT-A clades. The observed metal-independent activity (Fig. 4c), which is uncommon among GT-A fold enzymes, is consistent with this family being divergent from other GT-A families with the same overall structural fold.
The AlphaFold2 structure of RGGAT1 was used to predict candidate residues with roles in binding to the donor and acceptor substrates. Molecular docking was performed with UDP-GalA and an RG-I (R) oligosaccharide, DP12 substrate (Fig. 6c). Notably, a lysine residue (K363) was found to be well-positioned to interact with the diphosphate group of UDP-GalA. K363 is part of the DGK motif which replaces the DxD motif that is normally highly conserved in GT-A fold enzymes35 with a crucial role in coordinating metal ions, typically Mn2+, that interact with the diphosphate common to nucleotide sugar donors33. Several additional interactions with UDP-GalA were also predicted from the docked structure, including D361 (also part of the DGK motif), K344, R393, D472 (part of the RDQ motif that replaces the canonical xED motif) and H508. One of the residues of the hypervariable region (K392) was predicted to interact with a carboxyl group of a GalA residue within the RG-I acceptor oligosaccharide.
The Arabidopsis genome includes 8 sequences with GT116/DUF616 domains (Extended Data Fig. 10). Using 9 plant species, including Arabidopsis, 4 ancestral lineages, and 4 angiosperms with applications as agricultural crops and biomass feedstocks, a phylogenetic tree was created from a total of 77 protein sequences containing GT116/DUF616 domains (Fig. 7a). This analysis expands on a similar previously created phylogenetic tree20, but here the 8 Arabidopsis sequences are grouped into 5 distinct clades with the inclusion of sequences from additional species. The GT116 family represents putative GTs that may be predicted to also have RG-I:GalAT activity, but proof of function in RG-I synthesis for other family members will require confirmation of enzyme activity.
Fig. 7 |. Phylogenetic tree and tissue expression of GT116 family proteins.
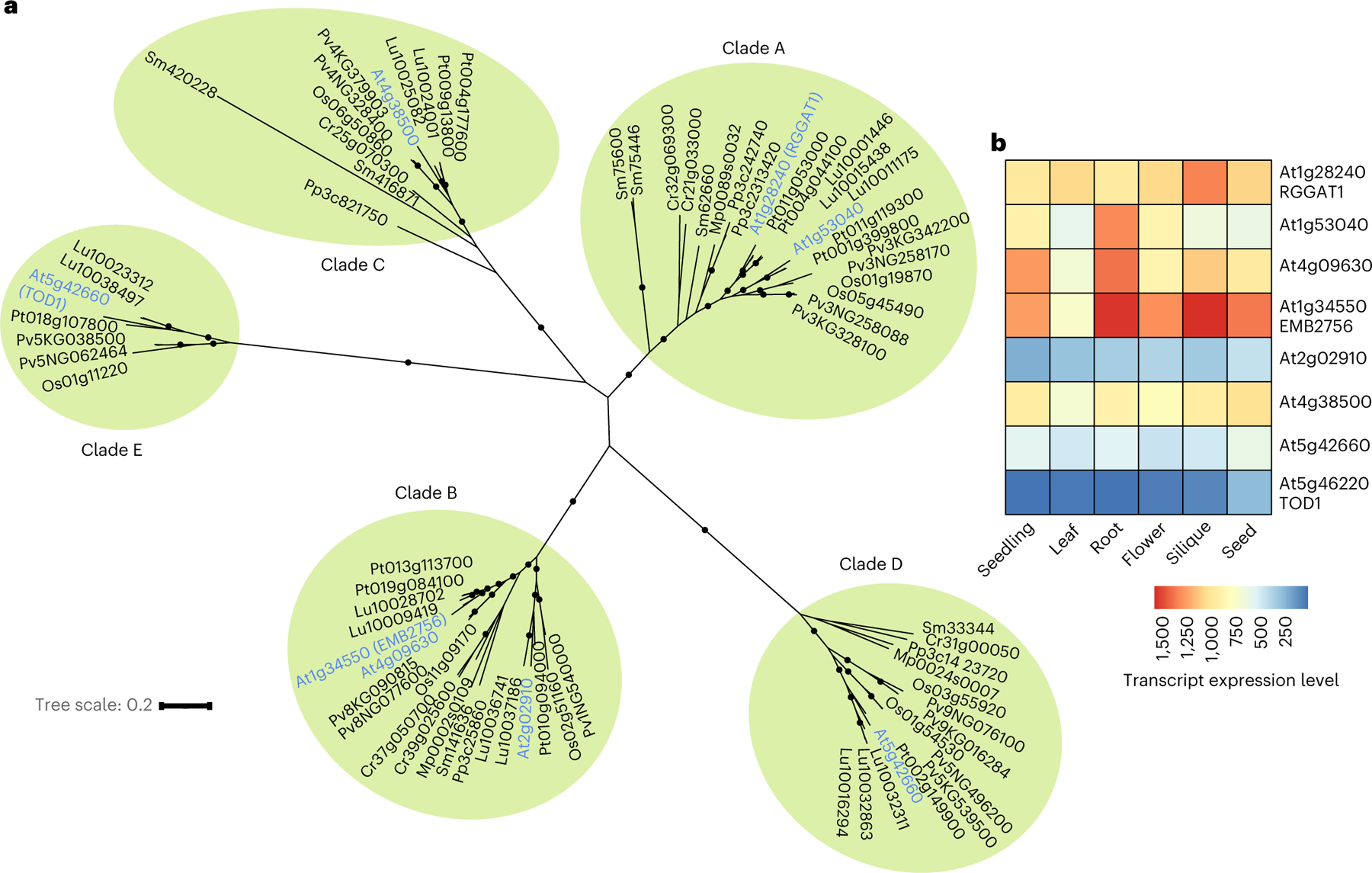
a, Phylogenetic tree containing 77 GT116 sequences from 9 plant species. Arabidopsis gene names are in blue and organized into 5 clades. The clades (A, B, C, D and E) were ranked according to the primary amino acid sequences with the highest similarity to RGGAT1. Black circles indicate support for nodes with bootstrap values of greater than 90% from 500 bootstrap trials. Branch lengths indicate genetic divergence, with the scale bar indicating the average number of substitutions per amino acid. At, Arabidopsis thaliana; Cr, Ceratopteris richardii (fern); Lu, Linum usitatissimum (flax); Mp, Marchantia polymorphia (liverwort); Os, Oryza sativa (rice); Pv, Panicum virgatum (switchgrass); Pt, Populus trichocarpa (poplar); Pp, Physcomitrium patens (bryophyte); Sm, Selaginella moellendorffi (lycophyte). b, Heat map of RNA-seq data retrieved from TravaDB39 for each GT116 family member from 6 Arabidopsis tissues. Expression values are absolute read counts from selected tissues. For mature tissues in which data from several developmental stages were available (flowers, siliques, seeds), younger tissues that had not reached senescence were selected.
One possible reason for the existence of an expanded GT family is that members with similar catalytic activities have a tissue-specific functional specialization. The availability of RNA-seq genome-wide Arabidopsis expression data has enabled facile analysis of differential gene expression39. The expression of the eight Arabidopsis GT116 family members was compared in six different tissues representing both early developmental and mature stages (Fig. 7b). RGGAT1 and several of the GT116 family members are expressed broadly in plant tissues. Combined with the observation that lower plant paralogues of RGGAT1 are also present in Clade A (Fig. 7a), RGGAT1 is likely to function beyond mucilage synthesis in other tissues. Two genes from Clade B, At4g09630 and At1g34550 (EMB2756), are highly expressed in seedlings and mature tissues (Fig. 7b). The enzymes coded for by these genes have relatively large amino acid chain lengths of 711 and 735 residues, respectively (Extended Data Fig. 10), suggesting that they have an expanded domain structure that could facilitate possible interactions with other RG-I biosynthetic enzymes or complex glycan acceptors. Due to the high expression profiles, these Clade B genes are putative targets for RG-I biosynthesis in other cell types and developmental stages.
Discussion
Pectins are a heterogeneous family of cell wall polysaccharides that have proven challenging to define as functional macromolecules. For most plant tissues, pectins are extracted as heteropolysaccharides composed of distinct domains that require enzymatic or chemical digestion for isolation40,41. One of the difficulties associated with the study of pectins is the existence of different backbone and side-chain structures. More than 60 individual transferase activities have been estimated to be necessary for synthesis of the full range of pectic glycan linkages1. Understanding the scope of plant cell wall biosynthetic machinery is further complicated by the existence of large families of GTs with sometimes redundant catalytic activities42.
All pectic polymers contain a homogalacturonan backbone (HG; repeating unit [-4-d-GalA-α-1-]) or rhamnogalacturonan backbone (RG-I; repeating unit [-4-α-d-GalA-1,2-α-l-Rha-1-]). Synthesis of the HG backbone is catalysed by at least six members of the GALACTURONOSYLTRANSFERASE (GAUT) family (GAUT1, 4, 10, 11, 13, 14 and the GAUT1:GAUT7 complex)2,5,8,23. The present work establishes that the α-1,2-GalA transferase that catalyses biosynthesis of the RG-I backbone is a novel GalAT and a founding member of family GT116. Annotated in previous studies as MUCILAGE-RELATED70, a new name for this enzyme has been proposed here—RHAMNOGALACTURONAN GALACTURONOSYLTRANSFERASE1 (RGGAT1)—to distinguish this activity from the HG biosynthetic activity of the GAUT family. Genes homologous to RGGAT1 were identified in ancestral plant lineages and modern crops of industrial and agricultural interest (Fig. 7a), providing opportunities to study RG-I synthesis in plant species beyond the model organism Arabidopsis.
Two previous publications describing muci70 gene mutants yielded results that are consistent with the revelation that RGGAT1/MUCI70 functions in RG-I biosynthesis. The levels of RGGAT1/MUCI70 transcript measured from silique tissues were reduced by at least 60% in two T-DNA insertion mutant lines (muci70-1 and muci70-2)20. These knockdown mutants resulted in a reduction of the surface area of the mucilage layer released on hydration of seeds and at least a 50% reduction of both GalA and Rha from total mucilage20. A study of the macromolecular properties of Arabidopsis seed mucilage identified several natural variants containing single nucleotide polymorphisms in RGGAT1/MUCI70 that resulted in reduced molar mass of the mucilage polysaccharide24. Water-extracted mucilage, which has been shown to be mostly composed of a >600 kDa polysaccharide of unbranched RG-I15, was reduced in molar mass by >70% in the muci70-1 and muci70-2 mutants24. These mutants are transcriptional knockdowns of an RG-I:GalAT, but the reduction of this activity does not appear to have been compensated by the presence of up to 7 other putative RG-I:GalATs. This lack of compensation suggests that RGGAT1/MUCI70 may be functionally specialized for the production of the high-molecular-weight RG-I polysaccharides specifically synthesized by seed epidermal cells, but the expression analysis suggests that RGGAT1 also functions to synthesize RG-I within other tissues (Fig. 7b).
The seed mucilage phenotypes of muci70 mutants were instrumental in the discovery that RGGAT1 functions in RG-I biosynthesis in seed mucilage, but RG-I also exists broadly in other plant tissues9. RNA-seq data obtained from the database Transcriptome Variation Analysis (TraVA) indicate that some GT116 family members are transcribed in all Arabidopsis tissues39. One member of the family, At1g34550, is included within a curated dataset for mutants that result in an ‘embryo-defective’ phenotype43. These mutants are classified by the production of defective seeds due to arrested embryonic development. The goal of establishing the EMB dataset, currently containing 510 EMBRYO-DEFECTIVE genes, was to identify the minimal set of genes necessary for plant growth and development43,44. The EMBRYO-DEFECTIVE gene EMB2756, corresponding to At1g34550, encodes a GT116-domain protein that is a putative RG-I:GalAT. At the time of the original study, EMB2756 was classified as a protein of unknown function44. If EMB2756 is an RG-I:GalAT, then the ‘embryo-defective’ phenotype of the emb2756 mutant suggests that RG-I synthesis is an essential cellular function necessary for the completion of embryonic development.
Because the other members of the GT116 family have not yet been shown to have RG-I:GalAT activity, we have not proposed changing the gene annotations of the other family members to RGGAT. The GT116 family is complicated by the existence of the family member At5g46220 (TOD1), which has previously been identified as having alkaline ceramidase activity45. In addition to the substrates being lipids rather than polysaccharides, this activity has notable differences from the activity of RGGAT1, such as being calcium-dependent and having an optimal pH of 9.545. Of the eight family members, TOD1 shares the least sequence identity with RGGAT1 (Extended Data Fig. 10), but it does have a characteristic DGK motif that was found to distinguish the GT116 family from other GT-A fold families. On identifying TOD1 as an alkaline ceramidase, Chen et al. noted that TOD1 has low sequence similarity to alkaline ceramidases from other organisms, including mammals and Saccharomyces cerevisiae45. Future studies will be needed to determine whether At5g46220 (TOD1) is a GT116 family member with RG-I:GalAT activity or whether it should be categorized as a separate family of alkaline ceramidases.
With the identification of RGGAT1, it is now possible to compare the catalytic properties of RG-I and HG backbone biosynthesis. Comparison of the in vitro synthesis rates reveals that for both backbones, the rate of transfer of GalA is dependent on the chain length of the acceptor. For RGGAT1, increases in activity for acceptors of lengths greater than DP8 appear to be due to an increased affinity of the enzyme for the longer-chain acceptors, as represented by an estimated ~10-fold lower KM value for DP ≥ 8 acceptors. In a study of HG biosynthesis by the GAUT1:GAUT7 complex, transfer to short-chain HG acceptors (DP ≤ 7) was also marked by reduced catalytic efficiency relative to acceptors of increased chain length (DP ≥ 11)6. Measurements of the reaction kinetics of pectin biosynthesis have been limited due to the resource-intensive requirement for purified acceptor substrates. However, the results provided here suggest that RG-I elongation has a mechanism consistent with the previously discovered mechanism for HG6 in which the transition to longer-chain oligosaccharides (approximately 10 sugar units) represents a considerable increase in the catalytic rate.
The metal-independent catalysis by RGGAT1 is unusual for GT-A fold enzymes33, and contrasts with the Mn2+-dependent catalysis by galacturonosyltransferases involved in HG biosynthesis6,8. The metal-independent activity is shared by the RRT-family enzymes16,17, allowing both GalAT and RhaT activities of RG-I backbone polymerization tooccurwithoutthe addition ofexogenous metalcations andsuggesting a common feature of enzymes involved in RG-I biosynthesis. The DxD motif (Asp-x-Asp), which is highly conserved in metal-dependent GT-A fold enzymes33, is changed to 361Asp-Gly-Lys363 in RGGAT1. Partial loss of the DxD motif was also found to have occurred in a GT from Bacteroides ovatus, BoGT6a, one of the GT-A fold families with a metal-independent mechanism of catalysis46. This study of RGGAT1 illustrates the power of using new deep-learning-based tools36 to investigate the relationships between anomalous mechanistic properties and predicted structures of newly discovered GTs. The initial structural model for RGGAT1 has provided a template for future studies of the unique catalytic properties of this enzyme and its divergence from related GT families.
Recent efforts have focused on discovering mechanisms by which pectin consumption contributes broadly to human health through proposed roles in metabolic pathways, including immune system function and cholesterol metabolism47,48. Because differences such as polymer size and sugar composition are likely to affect the bioactivity of pectins as components of dietary fibre, increasing recognition has been placed on the need to develop methods to purify pectic glycans with reduced biological variability41,49,50. The development of in vitro tools for the controlled synthesis of pectic glycans presents an avenue for production of pure substrates for use in biological studies of pectin function. Controlled chemoenzymatic methods have the potential for broader glycobiology applications, as similar methods have been explored for the synthesis of oligosaccharide domains for use in glycoconjugate vaccines51. Continued improvements to heterologous expression systems will allow for higher-yield purifications of GTs and the potential to expand the current capabilities of in vitro polysaccharide synthesis.
Methods
Extraction of Arabidopsis mucilage and purification of RG-I oligosaccharide acceptors
Arabidopsis mucilage used as the source of RG-I oligosaccharides was extracted using a scaled-up version of the protocol outlined previously15. Arabidopsis wild-type (Col-0) seeds (10 g total) placed in five 50 ml conical tubes each containing 2 g were mixed with deionized water to a total volume of 40 ml. Non-adherent mucilage was extracted by head-over-tail mixing for 3 h. The mixture of seeds and water containing extracted mucilage was centrifuged (2,500 × g, 5 min) and the water removed. The seeds were washed with water by mixing for 10 min and the water recovered after centrifugation. The mucilage extracted and water washes (600 ml total) were filtered using a polycarbonate filter of 3 µM pore size (Osmonics) and the filtrate lyophilised. Dry mucilage was resuspended in water at 10 mg ml−1.
Recombinant rhamnogalacturonan hydrolase (RGase A from Aspergillus aculeatus) was obtained as a gift from Novo Nordisk as previously described30. Resuspended mucilage was digested under a range of RG-I hydrolase concentrations (0.01–1.0 µg ml−1) at 40 °C in a sodium acetate buffer (20 mM, pH 5.0). The resulting oligosaccharide mixtures were visualized by high-percentage PAGE and stained with a combination of alcian blue and silver staining (described below). For the scaled-up preparation of RG-I oligosaccharides, 50 mg mucilage was digested in a reaction containing 0.2 µg ml−1 RG-I hydrolase for 21 h in 6.5 ml total volume. This mixture was boiled to terminate the hydrolase reaction, dialysed against water using a 3,500 Da cut-off membrane (SpectraPor) and lyophilised. The resulting oligosaccharides contained Rha at the non-reducing end and were designated RG-I (R).
Resuspended mucilage was also digested by acid hydrolysis using 0.1 M hydrochloric acid at 80 °C for up to 48 h. The resulting oligosaccharide mixture was visualized by high-percentage PAGE, as above. The digested oligosaccharides were neutralized by addition of 0.1 M sodium hydroxide, dialysed against water using a 3,500 Da cut-off membrane and lyophilised. The resulting oligosaccharides contained GalA at the non-reducing end and were designated RG-I (G).
The lyophilised mixture of digested RG-I oligosaccharides was fluorescently labelled on the reducing end by resuspending at 10 mg ml−1 in 10% acetic acid containing 0.2 M 2-aminobenzamide (2AB) and 1 M sodium cyanoborohydride31. The mixture was incubated at 45 °C for 16 h, dialysed against water using a 3,500 Da cut-off membrane and lyophilised. After resuspension in water, the concentration of 2AB-labelled oligosaccharides was determined using UV-visible spectroscopy (Nanodrop) with a molar absorptivity coefficient for 2AB at 330 nm of 2,500 M−1 cm−1. RG-I oligosaccharides were separated using a semi-preparative CarboPac PA-1 column (22 × 250 mm) connected to a Dionex system with fluorescence detection (excitation 330 nm, emission 420 nm). Peaks containing RG-I oligosaccharides ranging from DP6 to DP18 were separated using an ammonium formate gradient. Peaks enriched for the target oligosaccharides eluted as the ammonium formate concentration increased from 350 mM to 450 mM. Samples containing up to 10 µmol of RG-I oligosaccharides were injected into the system for semi-preparative scale purification. Individual peaks containing homogenous RG-I oligosaccharides were collected, dialysed against water using a 3,500 Da cut-off membrane and lyophilised. The purity of the collected fractions containing RG-I oligosaccharides was assessed by an analytical-scale injection of 5 nmol into a CarboPac PA-1 column (4 × 250 mm) and matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS).
High-percentage PAGE
RG-I oligosaccharides were separated over a 30% acrylamide resolving gel (38 mM Tris, pH 8.8). Samples ranging from 300 ng (homogeneously purified DP12-2AB oligosaccharide) to 10 µg (undigested mucilage) were mixed with loading buffer (100 mM Tris, pH 6.8, 0.01% phenol red and 10% glycerol) and loaded into a stacking gel (5% acrylamide, 64 mM Tris, pH 6.8). Current (25 mA) was applied for up to 90 min. The gel was soaked for 20 min in a fixative solution (40% methanol, 10% acetic acid) and stained for 2 h in a solution of 0.1% alcian blue in 40% ethanol. After staining, the gel was washed with at least three changes of water for a total of 12 h. Silver staining and developing was completed using a silver staining kit (Bio-Rad). Staining was terminated by addition of 5% acetic acid. Gel images were captured using Bio-Rad Image Lab 5.2.1.
MALDI-TOF mass spectrometry
Negative ion mode MALDI-TOF-MS spectra were acquired using an LT Bruker Microflex spectrometer. Nafion 117 solution (Sigma) was applied to a Bruker MSP 96 ground steel target. RG-I oligosaccharides labelled with 2AB were mixed 1:1 with a 20 mg ml−1 2,5-dihydroxybenzoic acid matrix solution in 50% methanol. Purified RG-I oligosaccharides were resuspended at a concentration of at least 20 µM for detection by MALDI-TOF-MS. Reaction samples containing 100 µM acceptor oligosaccharides were diluted 1:4 in water containing 100 mM ammonium hydroxide before mixing with the sample matrix. Ammonium hydroxide was added to hydrolyse any sugar lactone structures present in the purified RG-I oligosaccharides. Data were collected with Bruker Daltonik FlexControl 3.0 software.
Cloning and expression of recombinant glycosyltransferases in HEK293 cells
All glycosyltransferase constructs were cloned for heterologous expression in HEK293F cells as previously described6,25. The expression construct for MUCI70Δ77 was cloned for use in a previous study20. The sequences for five Arabidopsis proteins (MUCI70/RGGAT1, RRT1, RRT2, RRT3 and RRT4) were analysed using The Arabidopsis Plant Membrane Protein Database (Aramemnon) to identify putative N-terminal transmembrane domains on the basis of the consensus results from hydrophobicity prediction servers. Primers for PCR amplification of the protein coding sequences truncated by the N-terminal transmembrane domain were designed with overhanging universal sequences for attB sites to enable Gateway cloning. The template for PCR amplification was complementary DNA produced from RNA extracted from 7 d old Arabidopsis seedlings for MUCI70Δ7720 and RNA extracted from Arabidopsis leaf tissue for RRT1Δ61, RRT2Δ62, RRT3Δ54 and RRT4Δ51. Following the first round of PCR to amplify the truncated gene sequence, a second round of PCR was done using PCR products as templates to insert Gateway cloning-specific sequences using Universal Primers.
PCR products were inserted into the Gateway entry vector pDONR221 by reaction with BP clonase (Invitrogen). Sequences were verified after insertion into vector pDONR221 using M13F and M13R primers. The coding sequences were inserted into the mammalian destination vector pGEn2 by reaction with LR clonase (Invitrogen). All primer sequences used are listed in Supplementary Table 1.
Following LR cloning, the expression constructs containing each truncated coding region in the pGEn2 expression plasmids were purified using Purelink HiPure Plasmid Gigaprep kits (Invitrogen). Fusion proteins were expressed in HEK293F cells and cell culture medium containing secreted proteins was collected after an incubation of 6 d. Secreted proteins were purified from the medium using Ni2+-NTA affinity chromatography with a column (HisTrap HP, GE Healthcare) equilibrated in 50 mM HEPES buffer, pH 7.2, with 20 mM imidazole. The column was washed and protein was eluted in steps containing 40 mM, 100 mM and 300 mM imidazole. The protein in the fraction eluted with 300 mM imidazole was dialysed into a storage buffer containing 50 mM MES, pH 6.5, using the metal-chelating ion resin Chelex-100. Protein was dialysed against two changes of storage buffer for 4 h each. Recovered protein was concentrated in centrifugal concentrator units with a 30 kDa cut-off (Amicon Ultra-15, Millipore). The protein concentration was determined using UV-visible spectroscopy (Nanodrop).
Protein purity was assessed using SDS–PAGE. An aliquot containing 5–10 µg of the purified protein was mixed with loading buffer at a final concentration of 20 mM Tris-HCl, pH 6.8, 2% SDS, 5% glycerol and 0.01% bromophenol blue. For reducing SDS–PAGE, 25 mM DTT was included in the loading buffer. Samples were boiled for 5 min to denature proteins before loading into the gel (MINI-PROTEAN 4–15% gradient gel, Bio-Rad). Proteins were detected by Coomassie blue staining. Gels were destained by mixing with a solution of 40% methanol and 10% acetic acid, followed by repeated washes with water.
RG-I:GalA transferase reactions
Unless noted otherwise, all reactions were incubated at 30 °C in 50 mM MES buffer, pH 6.5, with 1 mM UDP-GalA and 100 µM RG-I (R), DP12-2AB oligosaccharide acceptor. RGGAT1/MUCI70 enzyme was added at concentrations ranging from 50 nM to 5 µM.
Enzyme activity measurements completed using the UDP-Glo glycosyltransferase assay (Promega) were carried out according to the manufacturer’s instructions. A standard curve of UDP concentration vs luminescence established the linear range of the assay to be 50 nM–20 µM. From each 20 µl reaction, 5 µl aliquots were removed and mixed 1:1 with the UDP detection reagent at the indicated times to stop the reactions. All activity measurements were in duplicate, and the data report the averages. Unless noted otherwise, all assays were replicated in three independent experiments. The luminescence reading was converted to µM UDP released on the basis of comparison to a UDP standard curve carried out in duplicate for each set of reaction samples. Data were acquired using BioTek Gen5 3.05.11 software and imported into Microsoft Office Excel 2007 for conversion calculations. The UDP-GalA donor substrate was incubated with calf intestinal alkaline phosphatase (CIAP, Promega) to remove residual UDP from the sample. CIAP was removed from the UDP-GalA preparation by centrifugation using a Microcon 10 kDa centrifugal filter unit (EMD Millipore). The filtrate was collected and concentrated to 10 mM as determined by UV-visible spectroscopy (Nanodrop 1000, Thermo Fisher, v3.7.1) with a molar extinction coefficient for UDP of 10,000 M−1 cm−1 at 260 nm. Nonlinear regression Michaelis-Menten kinetics analysis was performed using Graphpad Prism 9.0.2 for Windows (www.graphpad.com).
All RG-I synthesis activity measurements using anion exchange chromatography were done under similar conditions. All samples were boiled at the indicated time points tostop the reaction. From each 30 µl reaction, an aliquot of 25 µl containing the equivalent of 2.5 nmol total DP12-2AB acceptor was mixed with water and 100 mM ammonium hydroxide to a total volume of 1 ml. Ammonium hydroxide was included before injection tohydrolyse sugar lactone structures that resolve as peaks in the chromatogram in addition to the parent RG-I oligosaccharide structure. The sample was injected into a CarboPac PA-1 (4 × 250 mm) column and resolved using an ammonium formate gradient. From 5 to 45 min, the ammonium formate concentration was increased from 200 mM to 600 mM, resulting in a DP12-2AB acceptor with a retention time of 23 min and a DP13-2AB product with a retention time of 27.4 min. 2AB-labelled acceptors and reaction products were detected using an RF-2000 fluorescence detector set to high sensitivity. The peak areas of the DP12 acceptor and DP13 products weremeasured using Chromeleon 6.80. Percentage ofacceptor conversion was calculated on the basis of the proportion of product peak area to total combined peak area of acceptors and products.
For the test of metal dependence of RGGAT1, enzyme at a concentration of 4 µM was diluted into a mixture of MES buffer, pH 6.5, containing 10 mM of either EDTA, MnCl2 or no additive. This mixture was incubated at room temperature for 30 min. Enzyme from this mixture was diluted in MES buffer and added to reactions containing a total EDTA concentration of 10 mM, a total MnCl2 concentration of 0.25 mM or no additives and incubated under standard reaction conditions for 10 min. For assays containing alkaline phosphatase (Potato apyrase, Sigma A6535) to reduce inhibitory UDP formed during the reaction, a total of 0.2 U of the phosphatase was added to the reaction.
RG-I polymerization reactions
In vitro polymerization of RG-I was completed using conditions described for RG-I:GalAT activity with the following modifications. Two enzymes, RGGAT1 and RRT4, at concentrations of 5 µM were mixed with two nucleotide sugar donors, UDP-GalA and UDP-Rha, and with an RG-I oligosaccharide acceptor. For the detection of reaction products using MALDI-TOF-MS, UDP-GalA and UDP-Rha at a concentration of 1 mM and an RG-I (G), DP12-2AB acceptor at a concentration of 100 µM were incubated in a total volume of 20 µl. For the detection of reaction products by alcian blue-stained PAGE and size-exclusion chromatography, UDP-GalA and UDP-Rha at a concentration of 2.5 mM, an RG-I (R), DP12-2AB acceptor at a concentration of 25 µM, and 1 U potato apyrase (Sigma) were incubated in a total volume of 120 µl. Each reaction was incubated for the indicated time (0–12 h) and boiled. An aliquot of 6 µl of the full reaction, representing 300 ng of the starting DP12-2AB acceptor, was removed and mixed with loading buffer for detection by alcian blue-stained PAGE. An aliquot of 100 µl from the full reaction, representing 5 µg of the starting DP12-2AB acceptor, was removed and injected into a Superdex 75 10/300 GL column attached to an Agilent 1260 Infinity II high-pressure liquid chromatography system at a flow rate of 0.5 ml min−1 of 50 mM ammonium formate. RG-I products were detected by multi-angle light scattering coupled with size-exclusion chromatography (SEC-MALS) as described for the determination of RG-II molecular mass52. Detection was performed using an Optilab t-rEX differential refractometer (Wyatt Technology) connected in series with a Dawn Heleos 8 MALS detector. The molecular mass was calculated using a dn/dc value of 0.122 mg ml−1. Data were processed using ASTRA 7 software (Wyatt Technology).
Prediction of DUF616 structural fold
Representative DUF616 sequences were collected using PSI-BLAST with the A. thaliana RGGAT1 sequence as query and a stringent e-value cut-off; 679 representative sequences were selected. These sequences were then passed through the fold prediction pipeline previously described36. In brief, NetSurfP2.053 was used to predict secondary structures for the 679 sequences. The 3-state secondary structure prediction results were then evaluated using the deep-learning model to calculate the reconstruction errors and the fold assignment score. On the basis of these scores, the final fold prediction was made. The average reconstruction error was well below the 95% confidence interval limit of 0.107, indicating that GT116 adopted a known fold, and the fold assignment score was positive and highest for the GT-A fold, indicating that members of the GT116 family adopt a GT-A fold.
Generation of the RGGAT1 predicted model using AlphaFold2
A local version of AlphaFold2 (v2.0.1)37 was used to generate models for the RGGAT1 GT-A domain. After an additional relaxation using Rosetta (v3.9) minimization54, a structural comparison was performed in PyMOL using the ceAlign 2.5 algorithm with a well-studied GT31 domain (pdb: 6wmo)55 to validate that the RGGAT1 sequence indeed formed a GT-A fold enzyme.
Phylogenetic comparison of GT-A fold families
An expansive GT-A phylogenetic tree was previously published, providing evolutionary relationships between GT-A enzymes35. With this new enzyme family, we sought to update the tree. As this new GT-A is highly variant, it failed to map to previously published profiles of GT-A sequences. Thus, we opted to utilize the highest ranked Alpha-Fold2 predicted structure and performed a structure-based sequence alignment, comparing it with a GT31 domain. We additionally ran Blast using the RGGAT1 sequence to collect divergent RGGAT1 sequences and create a consensus RGGAT1 sequence. We then added the RGGAT1 consensus and the AlphaFold2 sequence to the profile, and manually aligned the sequences on the basis of the structural alignment. As the three-dimensional (3D) topologies aligned quite well, we were able to integrate the RGGAT1 consensus sequence into the existing GT-A profiles. To evaluate that the profiles were accurate, we ran the software MapGaps 2.156, which picks up sequences that match a constructed profile, and found that the profile matched to RGGAT1 sequences.
Sequence analysis of DUF616-domain enzymes
The amino acid sequence for At1g28240 was searched using Pfam (https://pfam.xfam.org/). The corresponding family, DUF616 (Pf04765), was sorted by species for A. thaliana. Redundant sequences were removed by manual curation. The amino acid position of the DUF616 domain for each of the eight unique Arabidopsis sequences was identified by searching individual sequences in Pfam. The amino acid sequence for At1g28240 was entered as a query sequence against A. thaliana (taxid:3702) using Protein BLAST (blast.ncbi.nlm.nih.gov). Each of the eight DUF616-domain-containing target sequences were identified. The residues aligned to the query, query coverage percentage, amino acid sequence identity percentage and sequence similarity percentage were identified in the BLAST results report.
Phylogenetic tree of DUF616-domain enzymes
The amino acid sequences for DUF616-domain proteins from 9 plant species were obtained from Phytozome v1357. The Biomart tool was used to extract protein sequences containing Pfam ID PF04765 from each selected species. In cases where more than one sequence was identified for each individual gene annotation, redundant sequences were removed by manual curation to create a list of 77 sequences from 9 species. MEGA11 software58 was used to create an alignment, compute the best substitution model and construct a maximum-likelihood tree. The phylogenetic analysis was completed following the guidelines for this software as previously published59. The MUSCLE method was used for primary sequence alignment, and the LG model (G+I) with 500 bootstrap replicates was used. All data were imported into the Interactive Tree of Life tool for visualization60.
RNA-seq expression analysis
Average expression values were obtained from TraVA39 (http://travadb.org/) as absolute read counts normalized using the median-of-ratio method. Expression values from selected tissues (germinating seeds 3, whole mature leaf, root without apex, flower 3, silique 8 and seeds 7) were used for comparison. A heat map corresponding to the mean expression values was plotted using the ‘pheatmap’ package in RStudio 2022.02.3.
Molecular docking
Molecular docking studies were conducted on the AlphaFold2 protein model. We generated the acceptor substrate with the GLYCAM carbohydrate builder tool (GLYCAM Web, http://legacy.glycam.org). The donor substrate, UDP-GalA, was acquired from a UDP-phosphorylase crystal structure (pdb: 3OH1). The grid and docking parameters were created using AutoDock Tools61. Molecular docking was performed using Autodock Vina with the Vina-Carb scoring function to treat carbohydrate molecules62 using an 80 Å3 grid placed at the centre of the active site. After docking each molecule, the top scoring conformations were analysed together.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Expression of MUCI70Δ77 in HEK293 cells.
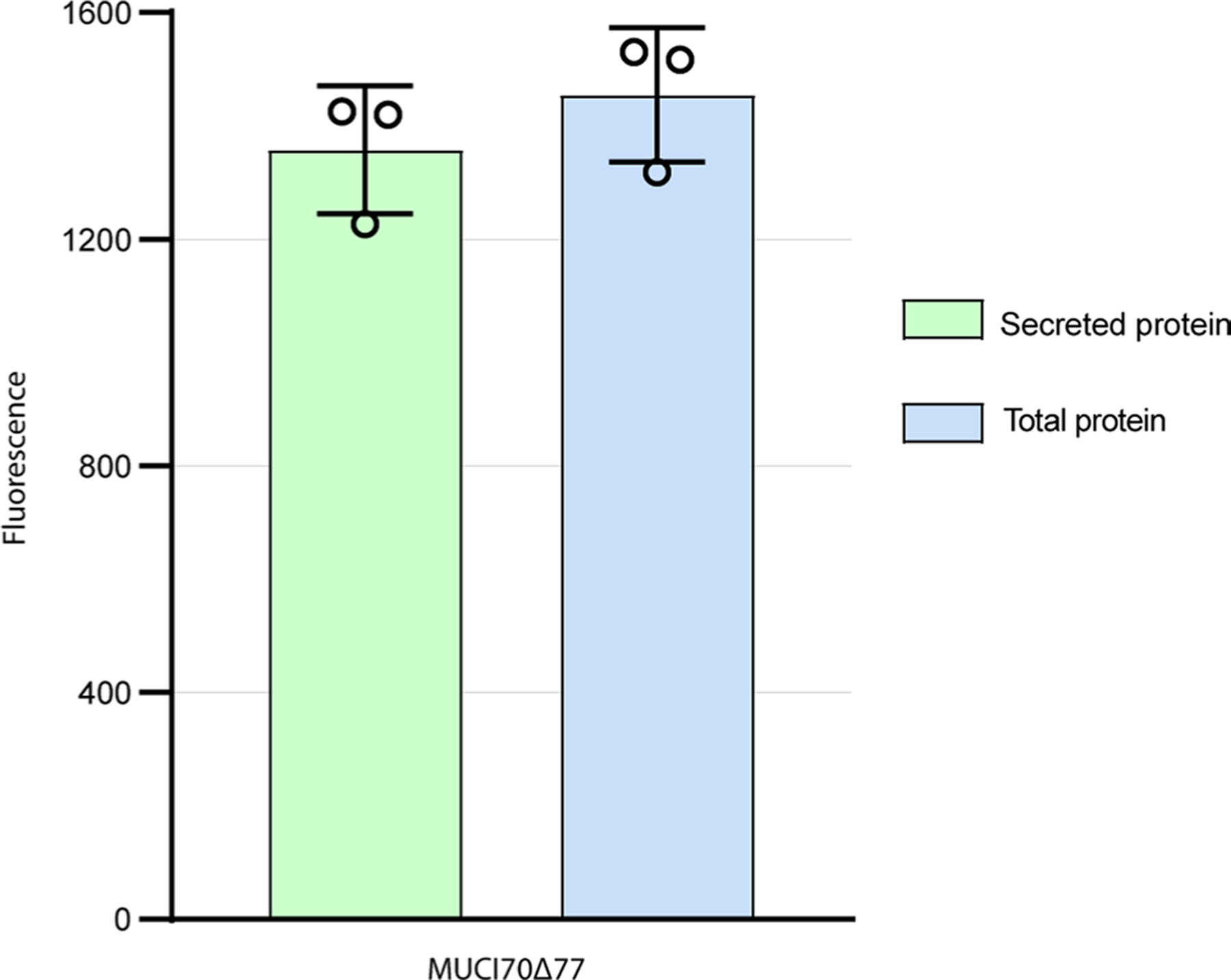
MUCI70Δ77 was expressed in a total of two small-scale (20 mL) and one large-scale (250 mL) cultures. Total protein is the measure of fluorescence of total GFP fluorescence from cells + culture medium. Secreted protein is the measure of fluorescence of cell-free medium. All samples were taken from a 100 µL aliquot from the cell culture after 6 days. MUCI70Δ77 was expressed with 93% secretion efficiency, defined as the proportion of secreted protein to the total protein fluorescence. Error bars represent the standard deviation from three biological replicates.
Extended Data Fig. 2 |. Digest of RG-I mucilage and purification of RG-I acceptor oligosaccharides.
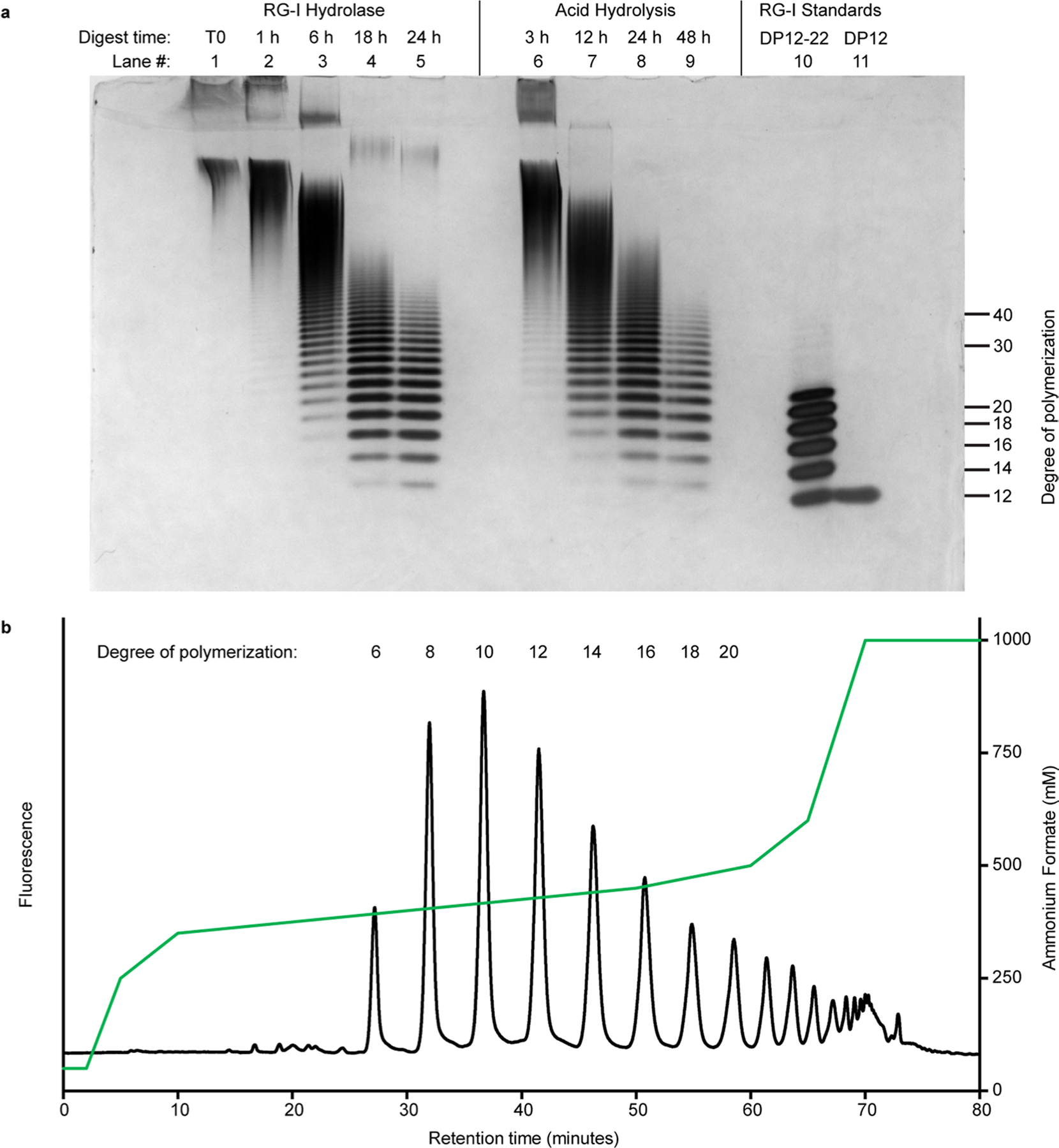
a, Arabidopsis mucilage was digested for the indicated times with RG-I hydrolase and by acid hydrolysis. Digests were carried out using 10 mg of mucilage and 0.1 µg RG-I hydrolase from Aspergillus aculeatus at 40 °C or 0.1 M HCl at 80 °C for the indicated times. b, RG-I oligosaccharides from digested mucilage were injected into a CarboPac PA-1 semi-preparative (22×250 mm) column following labeling with 2AB. Fractions were collected as individual peaks containing RG-I oligosaccharides of the indicated degree of polymerization (indicated above peak). Peaks were eluted in a gradient ranging from 50–1000 mM ammonium formate indicated by the green line.
Extended Data Fig. 3 |. RGGAT1 does not transfer GalA to RG-I acceptors containing GalA on the non-reducing end or to HG acceptors.
a. Hypothetical transfer of GalA to the non-reducing end GalA of an RG-I acceptor, resulting in RG-I oligosaccharides containing at least two contiguous GalA residues on the non-reducing end. Such an enzyme should exist in plants since HG:RG-I heteroglycans are known to be present in plant cell walls. The reaction depicted represents the elongation of homogalacturonan onto an RG-I acceptor. b. RGGAT1 does not catalyze the transfer of GalA to the RG-I (G) acceptor. RGGAT1 (1 mM) was incubated with UDP-GalA and an RG-I (G) acceptor for 1 hour. Longer incubation times did not result in any detectable activity. c. Hypothetical transfer of GalA to the non-reducing end of an HG acceptor, resulting in elongation of the HG backbone by at least one GalA monosaccharide. d. RGGAT1 does not catalyze the transfer of GalA to the HG acceptor. RGGAT1 (1 mM) was incubated with UDP-GalA and an HG acceptor for 1 hour. Longer incubation times did not result in any detectable activity.
Extended Data Fig. 4 |. Biochemical characterization of RGGAT1 activity.
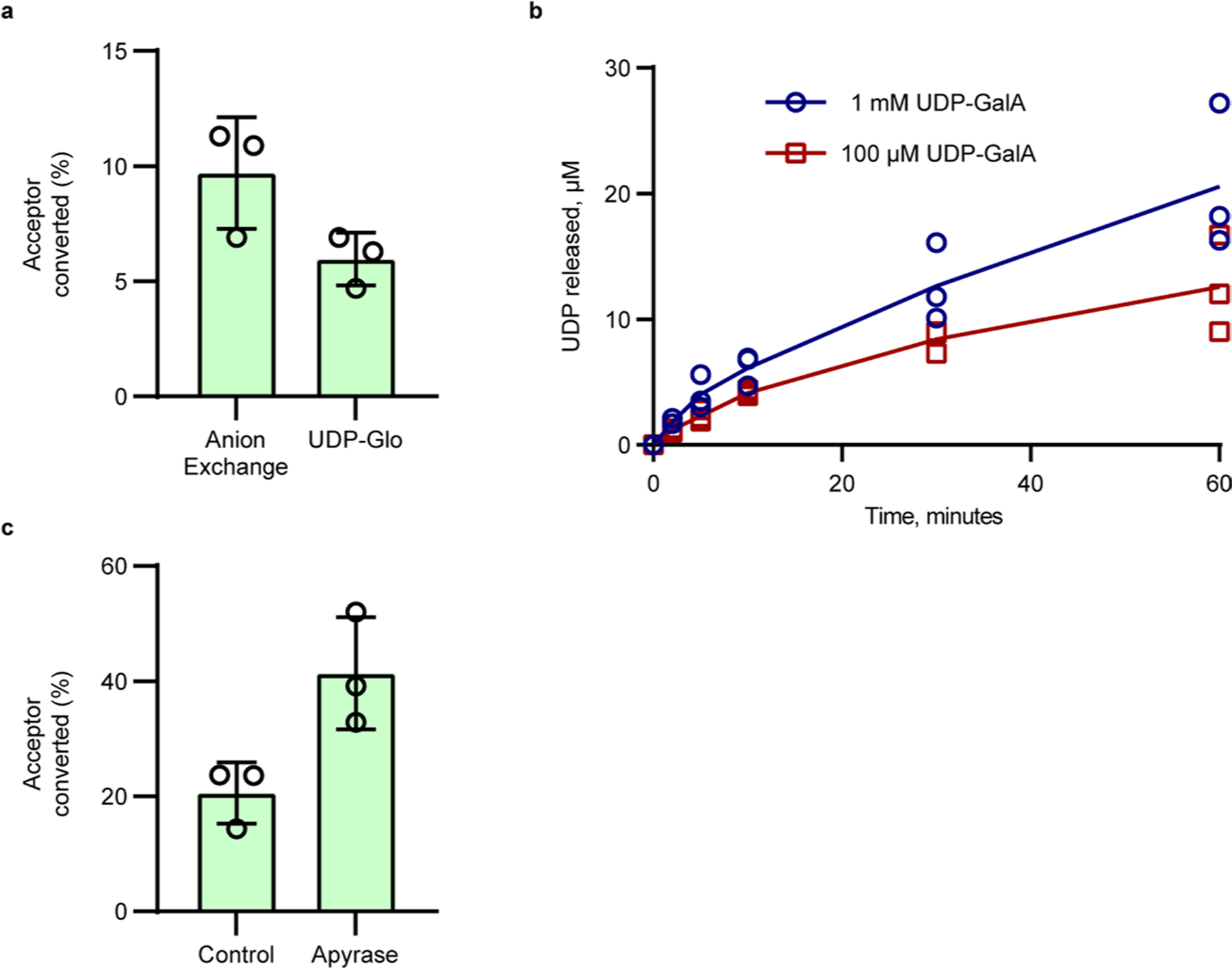
a, Comparison of RGGAT1 activity using two independent methods. For anion exchange, percentage of acceptor converted was calculated based on the relative proportion of the peaks for the DP12 (R) acceptor and DP13 (G) in the fluorescence chromatogram. For UDP-Glo, activity was measured as a function of UDP released in a 10 min assay containing 1 mM UDP-GalA and 100 µM acceptor. This activity value was presented as “percentage of acceptor converted” based on the conversion that 1 µM UDP released is equal to conversion of 1% of the starting DP12 (R) acceptor to a DP13 (G) product. Reactions contained 50 nM enzyme. Error bars represent the standard deviation from three independent experiments. b, Progress curve of activity using UDP-Glo. In all assays, each point represents the average of duplicate luminescence readings. The blue (assay with 1 mM UDP-GalA) and red (assay with 100 µM UDP-GalA) lines represent the average activity from three independent assays containing 50 nM enzyme. The results from independent assays are shown as individual points. c, Percentage of acceptor conversion was enhanced by addition of a phosphatase (potato apyrase, Sigma A6132) to the reaction. Percentage of acceptor converted was measured as the relative proportion of the peak area of the product to the remaining acceptor at 60 minutes in a reaction containing 50 nM enzyme, 1 mM UDP-GalA, and 100 µM DP12-2AB (R) acceptor. Error bars represent the standard deviation from three independent experiments.
Extended Data Fig. 5 |. Expression of RRT1, RRT2, RRT3, RRT4, and co-expression of RRT1:RRT2.
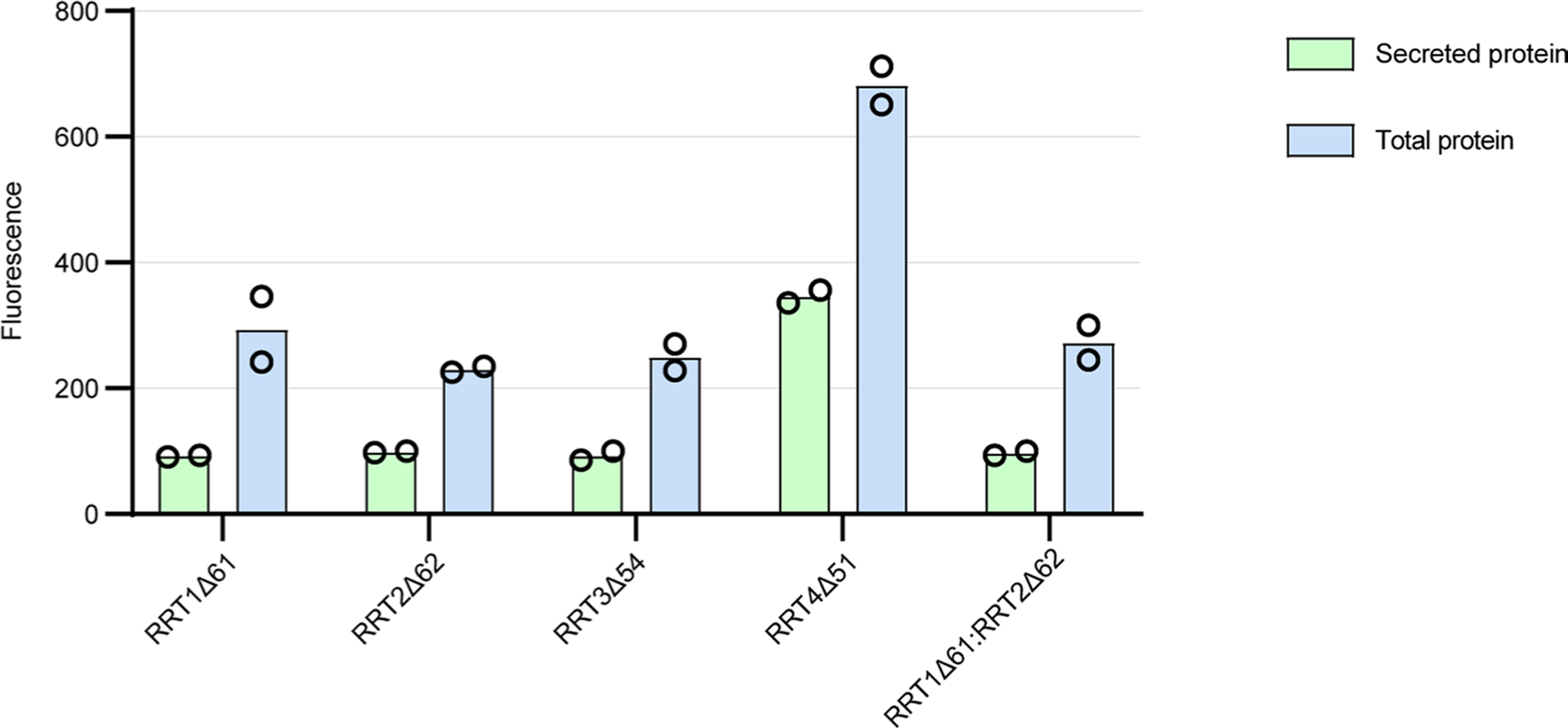
Four proteins from the RRT family were expressed in HEK293 cells. A co-expression experiment in which RRT1 and RRT2 were co-transfected into the cells was also performed. Total protein is the measure of fluorescence in the cells + culture medium. Secreted protein is the measure of fluorescence in cell-free medium. Of the four RRT-family proteins expressed in this system, RRT4Δ51 yielded the highest total protein. RRT4 protein expressed with 50% secretion efficiency. Error bars represent the standard deviation of two biological replicates. Co-expression of RRT1Δ61 with RRT2Δ62 did not result in increased expression, suggesting that these two proteins do not form a heterocomplex in vitro.
Extended Data Fig. 6 |. The purified RRT4 protein has RG-I:RhaT activity.
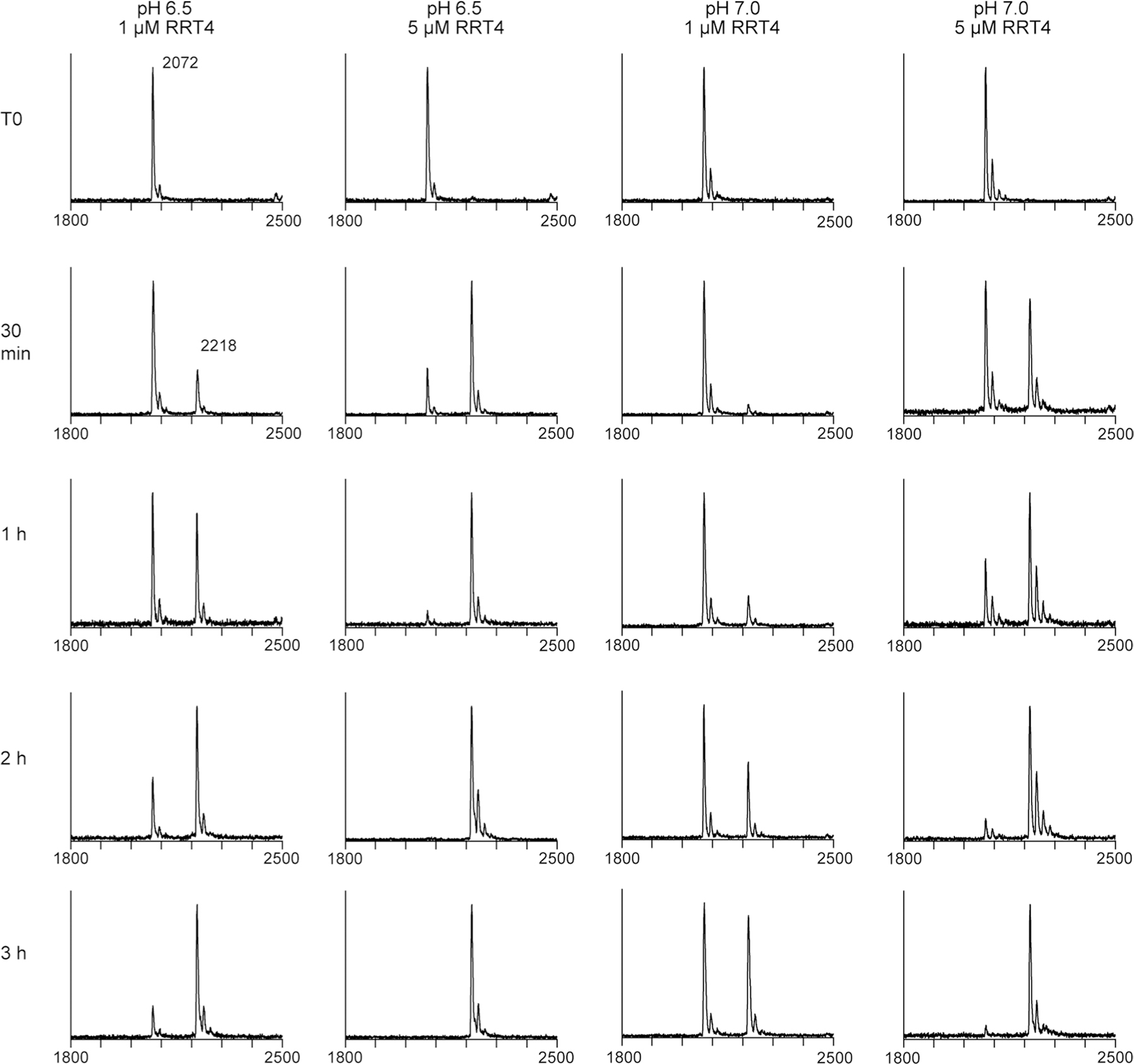
The purified RRT4 enzyme was incubated with 1 mM UDP-Rha and an RG-I (G) acceptor, DP12. Activity was tested at pH 6.5 and 7.0 with either 1 µM or 5 µM enzyme. The reaction progress was detected by MALDI-MS at the indicated time points. Activity at pH 6.5 was higher based on the relative conversion of the acceptor (2072 Da) to the RhaT product (2218 Da).
Extended Data Fig. 7 |. Individual RGGAT1 and RRT4 enzymes do not polymerize RG-I.
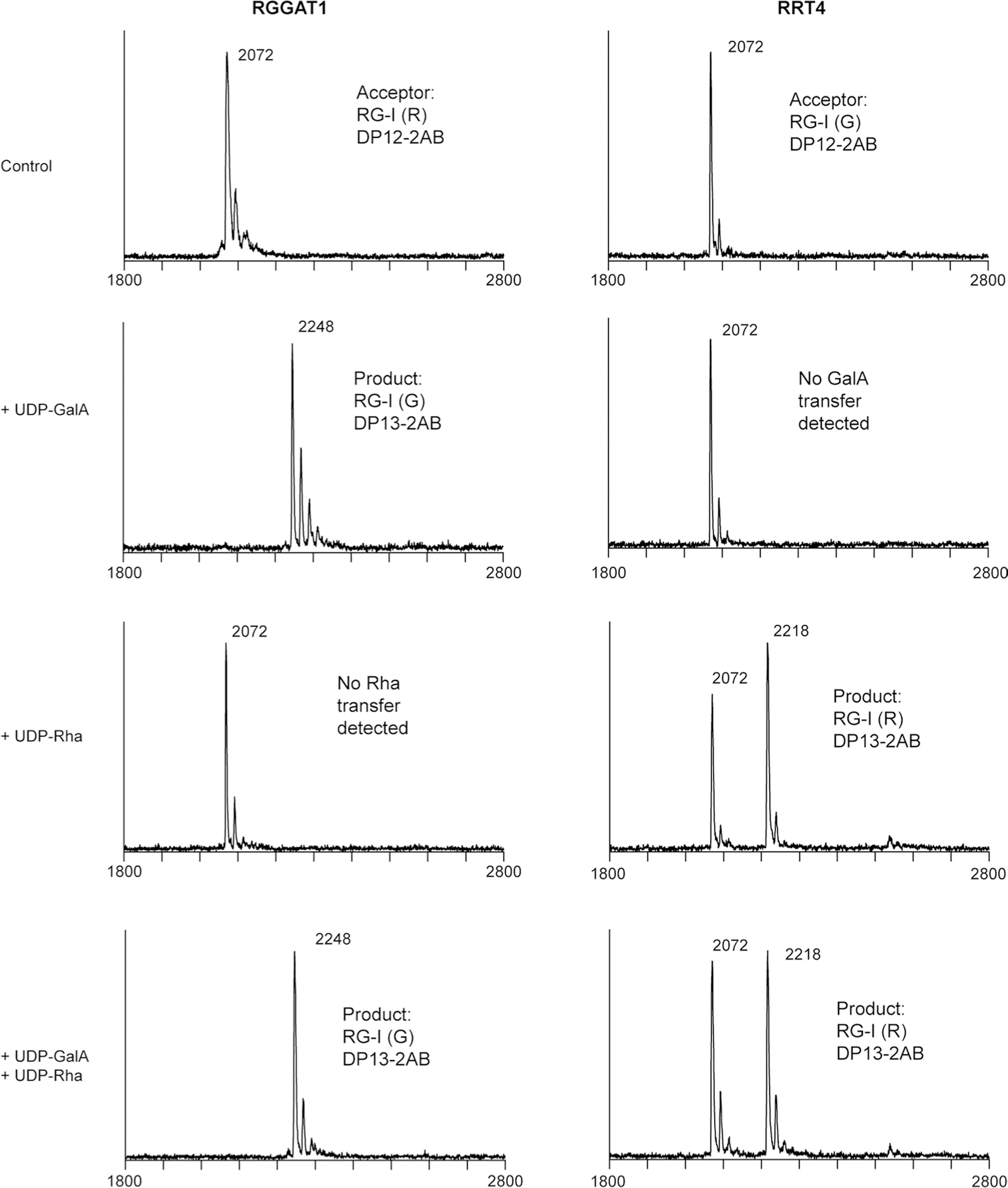
RGGAT1 enzyme (1 µM) was incubated with 100 µM RG-I (R) acceptor and 1 mM of UDP-GalA, UDP-Rha, or a combination of UDP-GalA and UDP-Rha. The activity was limited to addition of a single GalA residue with no additional products detected when UDP-Rha was included in the reaction. RRT4 enzyme (1 µM) was incubated with 100 µM RG-I (G) acceptor and 1 mM of UDP-GalA, UDP-Rha, or a combination of UDP-GalA and UDP-Rha. The activity was limited to addition of a single Rha residue with no additional products detected when UDP-GalA was included in the reaction.
Extended Data Fig. 8 |. Coexpression of RGGAT1 with RRT family members does not improve RRT expression.
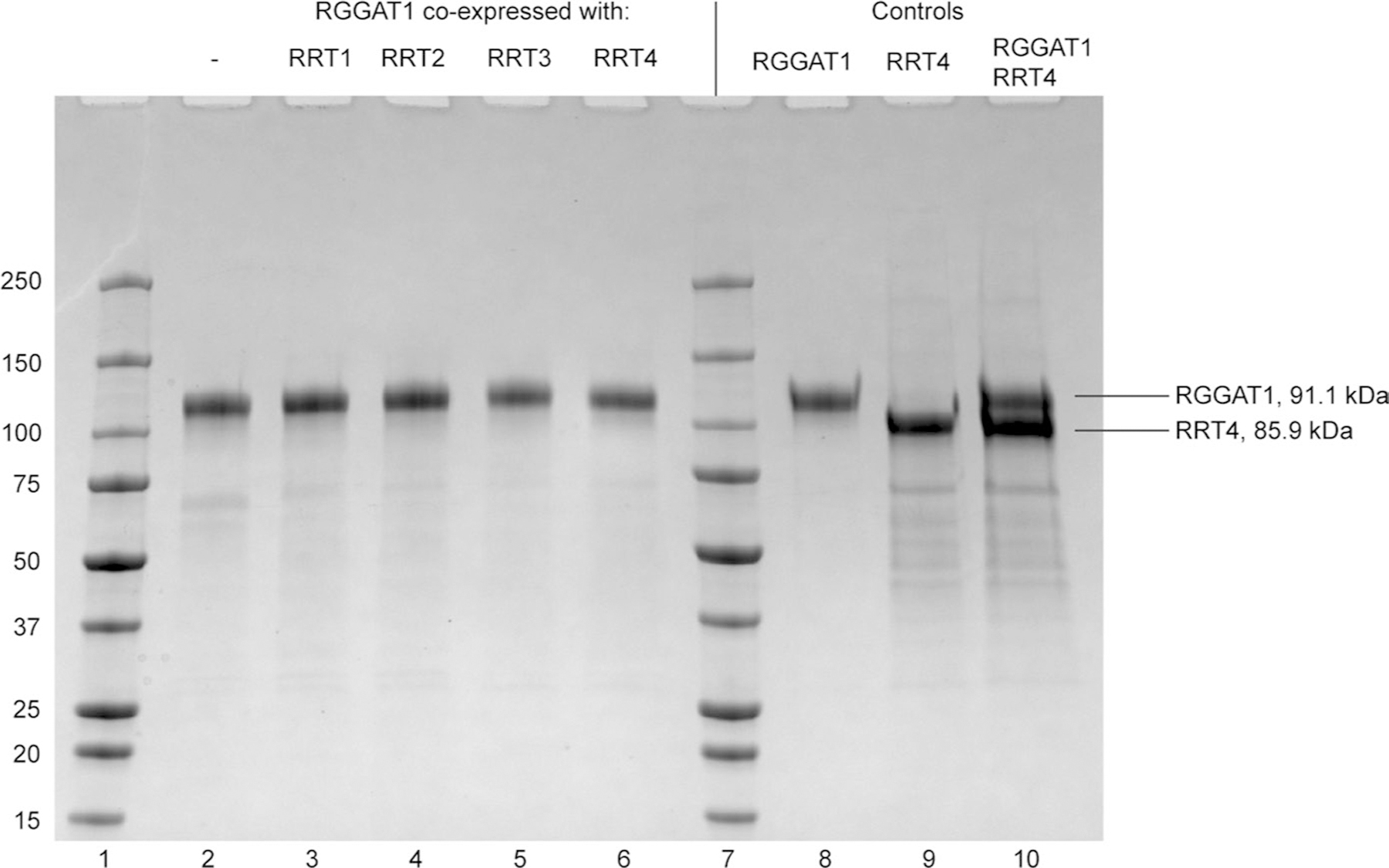
RGGAT1 (91.1 kDa) was expressed alone (lane 2) or coexpressed with RRT1 (81.3 kDa), RRT2 (86.4 kDa), RRT3 (86.7 kDa), or RRT4 (85.9 kDa) in HEK293 cells (lanes 3–6). The proteins were purified by Ni2+-NTA affinity from the cell culture medium. Protein concentration was measured by fluorescence. Proteins were loaded into an SDS-PAGE gel based on an equal amount of fluorescence corresponding to an estimated 1 µg total protein. All samples were separated under reducing conditions (+DTT) to observe the presence of monomers. Proteins were compared to previously-purified controls (Lanes 8–10). Lane 10, containing both RGGAT1 and RRT4 protein, was used as a control to demonstrate that the RGGAT1 and RRT4 monomers can be distinguished when an equal amount of both proteins was present. Although some RRT protein may be present in each co-expression lane, the results indicate that they were poorly expressed compared to RGGAT1. The gel represents a single experiment of the coexpression of RGGAT1 with RRT family members.
Extended Data Fig. 9 |. DUF616 family sequences are predicted to be a GT-A fold type.
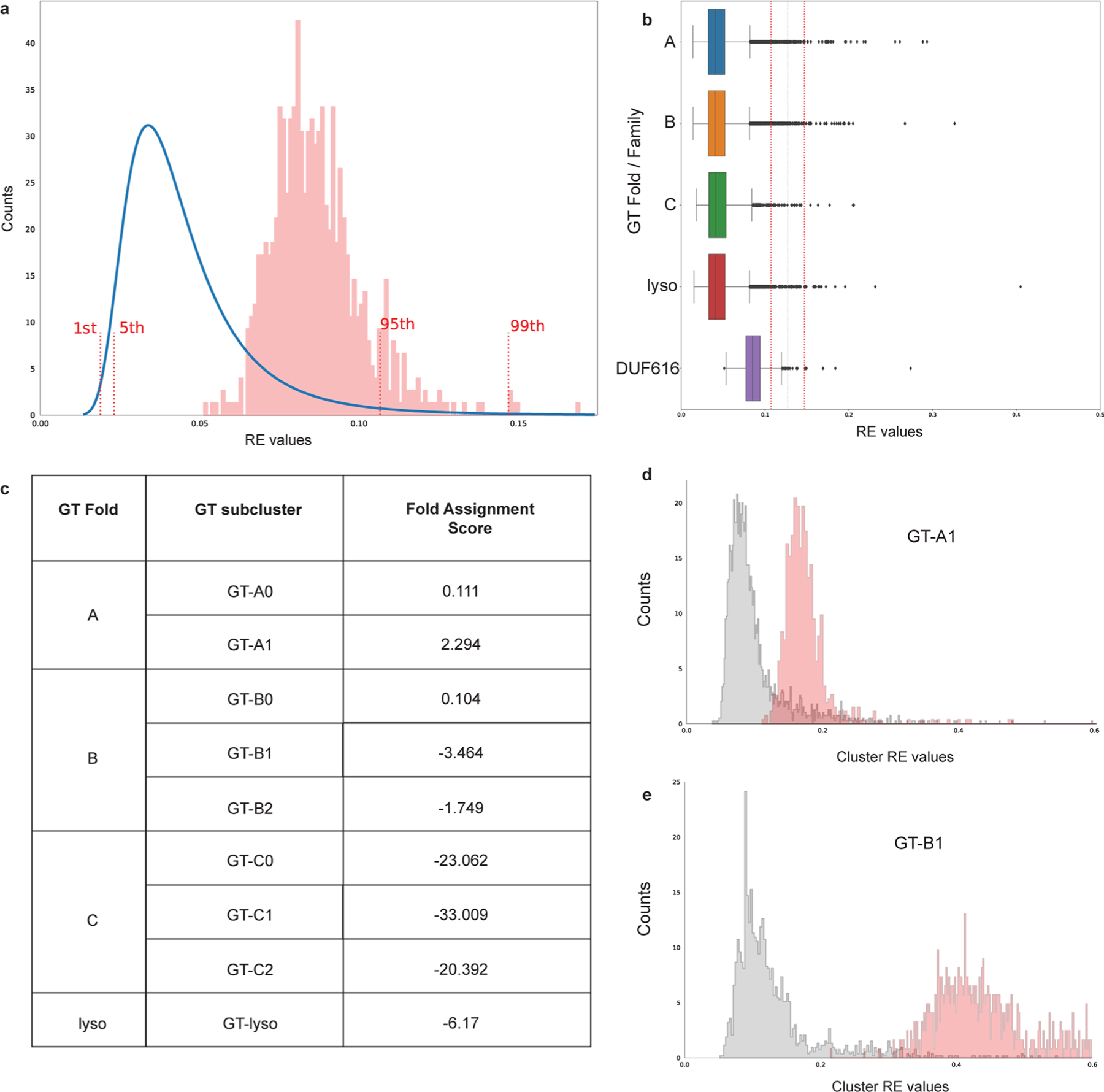
a, Reconstruction error (RE) values are calculated for DUF616 (n = 678) sequences and fall within 95% CI of the RE values for GT-A, B, C and lyso type folds suggesting that DUF616 belongs to one of the known folds. The reference RE values (blue line) were combined from the training set consisting of 39713 GT-A, GT-B, GT-C and GT-lyso sequences. b, RE values for the GT-A (n = 12,316), B (n = 20,397), C (n = 1,518), lyso (n = 5482) and DUF616 (n = 678) sequences are shown as boxplots. Dotted lines mark the 95th and the 99th percentile upper bounds. Boxes show the first and third quartiles. The line within the box indicates the median value. The whiskers mark 1.5 times the interquartile range, excluding the outliers shown as individual diamonds. c, Highest Fold Assignment Scores are found to be for the GT-A1 subcluster for the DUF616 sequences, suggesting that the sequences from this novel family adopt a GT-A type fold. d and e, The RE values against sub cluster GT-A1 and GT-B1 are plotted for DUF616 sequences. As seen, the RE values for GT-A1 are much closer to the true RE values, suggesting overall similarity in core structural fold.
Extended Data Fig. 10 |. The GT116 family contains eight putative members.
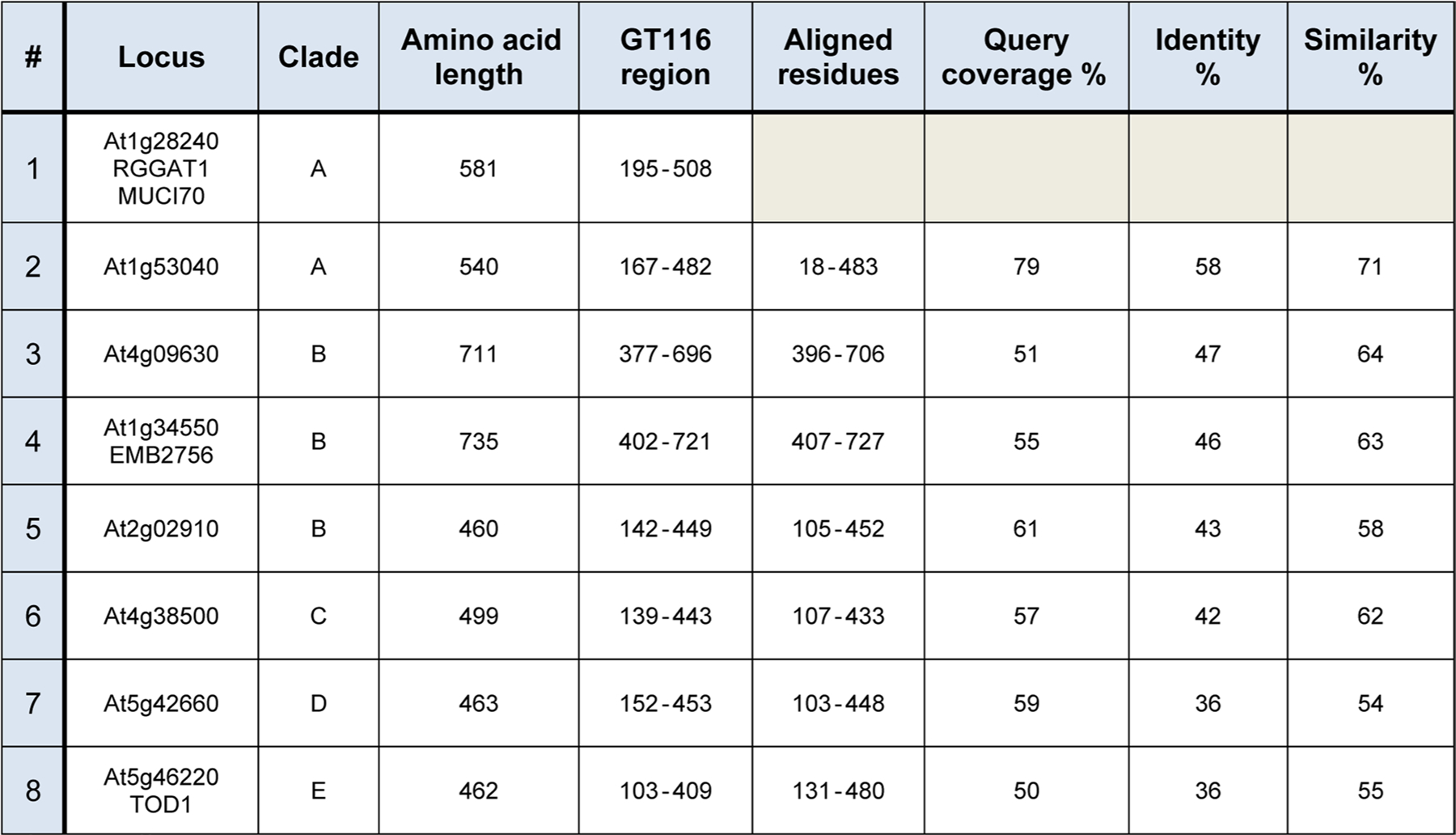
The GT116 domain was annotated as DUF616 (PF04765) in pfam. There are 12 Arabidopsis thaliana DUF616 sequences in pfam corresponding to 8 unique gene loci. The GT116 region is the envelope region containing the DUF616 domain predicted by pfam. The At1g28240/RGGAT1/MUCI70 sequence was entered as a query sequence in Protein BLAST. For the 7 additional GT116 sequences, aligned amino acid residues, query coverage, identity, and similarity are target sequence values obtained using At1g28240 as a query.
Supplementary Material
Acknowledgements
Funding was provided by the US Department of Energy, Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences and Biosciences Division, under award no. DE-SC0015662 (D.M.); the National Institutes of Health Grants P41GM103390 (K.W.M.), R01-GM130915 (K.W.M.) and R35 GM139656 (N.K.); and partially by The Center for Bioenergy Innovation, a US Department of Energy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science (DE-AC05-000R22725, D.M.). We thank M. O’Neill, M. Pena, B. Urbanowicz and P. Prabhakar for technical guidance; S. A. E. Garcia and A. Banks for laboratory support; P. J. Glatz for substrate production; and T. Ishimizu for identifying RRT.
Footnotes
Code availability
The code used to predict the glycosyltransferase fold structure was a deep-learning framework previously described in ref. 36, available at https://www.nature.com/articles/s41467-021-25975-9. The published version of the code with the manuscript is available at https://doi.org/10.5281/zenodo.5173136.
Competing interests
The authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41477-022-01270-3.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41477-022-01270-3.
Peer review information Nature Plants thanks Wei Zeng, Jesper Harholt and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
All data generated or analysed during this study are included in this published article (and its supplementary information files) or are available from the corresponding author upon request. UDP-GalA structure was accessed from Protein Data Bank: 3OH1 (https://www.rcsb.org/structure/3OH1). Plant genome sequences were accessed from Phytozome v1357 (https://phytozome-next.jgi.doe.gov/): A. thaliana TAIR10, C. richardii v2.1 ( JAIKUY010000000), L. usitatissimum v1.0, M. polymorpha v3.1 (PNPG01000000), O. sativa v7.0, P. virgatum v5.1 ( JABWAI010000000), P. trichocarpa v4.1, P. patens v3.3 and S. moellendorffii v1.0. RNA-seq data were accessed from Transcriptome Variation Analysis (http://travadb.org/). Source data are provided with this paper.
References
- 1.Atmodjo MA, Hao Z & Mohnen D Evolving views of pectin biosynthesis. Annu. Rev. Plant Biol 64, 747–779 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Biswal AK et al. Sugar release and growth of biofuel crops are improved by downregulation of pectin biosynthesis. Nat. Biotechnol 36, 249–257 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Wu D et al. Dietary pectic substances enhance gut health by its polycomponent: a review. Compr. Rev. Food Sci. Food Saf 20, 2015–2039 (2021). [DOI] [PubMed] [Google Scholar]
- 4.Bonnin E, Garnier C & Ralet MC Pectin-modifying enzymes and pectin-derived materials: applications and impacts. Appl. Microbiol. Biotechnol 98, 519–532 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Atmodjo MA et al. Galacturonosyltransferase (GAUT)1 and GAUT7 are the core of a plant cell wall pectin biosynthetic homog alacturonan:galacturonosyltransferase complex. Proc. Natl Acad. Sci. USA 108, 20225–20230 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amos RA et al. A two-phase model for the non-processive biosynthesis of homogalacturonan polysaccharides by the GAUT1:GAUT7 complex. J. Biol. Chem 293, 19047–19063 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM & Henrissat B The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42, D490–D495 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engle KA et al. Multiple Arabidopsis galacturonosyltransferases synthesize polymeric homogalacturonan by oligosaccharide acceptor-dependent or de novo synthesis. Plant J 10.1111/tpj.15640 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaczmarska A, Pieczywek PM, Cybulska J & Zdunek A Structure and functionality of rhamnogalacturonan I in the cell wall and in solution: a review. Carbohydr. Polym 278, 118909 (2022). [DOI] [PubMed] [Google Scholar]
- 10.Pena MJ & Carpita NC Loss of highly branched arabinans and debranching of rhamnogalacturonan I accompany loss of firm texture and cell separation during prolonged storage of apple. Plant Physiol 135, 1305–1313 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molina-Hidalgo FJ et al. The strawberry (Fragaria×ananassa) fruit-specific rhamnogalacturonate lyase 1 (FaRGLyase1) gene encodes an enzyme involved in the degradation of cell-wall middle lamellae. J. Exp. Bot 64, 1471–1483 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Yapo BM, Lerouge P, Thibault J-F & Ralet M-C Pectins from citrus peel cell walls contain homogalacturonans homogenous with respect to molar mass, rhamnogalacturonan I and rhamnogalacturonan II. Carbohydr. Polym 69, 426–435 (2007). [Google Scholar]
- 13.Arsovski AA, Haughn GW & Western TL Seed coat mucilage cells of Arabidopsis thaliana as a model for plant cell wall research. Plant Signal. Behav 5, 796–801 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haughn GW & Western TL Arabidopsis seed coat mucilage is a specialized cell wall that can be used as a model for genetic analysis of plant cell wall structure and function. Front. Plant Sci 3, 64 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macquet A, Ralet MC, Kronenberger J, Marion-Poll A & North HM In situ, chemical and macromolecular study of the composition of Arabidopsis thaliana seed coat mucilage. Plant Cell Physiol 48, 984–999 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Takenaka Y et al. Pectin RG-I rhamnosyltransferases represent a novel plant-specific glycosyltransferase family. Nat. Plants 4, 669–676 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Wachananawat B et al. Diversity of pectin rhamnogalacturonan I rhamnosyltransferases in glycosyltransferase family 106. Front. Plant Sci 11, 997 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liwanag AJ et al. Pectin biosynthesis: GALS1 in Arabidopsis thaliana is a β-1,4-galactan β-1,4-galactosyltransferase. Plant Cell 24, 5024–5036 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebert B et al. The three members of the Arabidopsis glycosyltransferase family 92 are functional β-1,4-galactan synthases. Plant Cell Physiol 59, 2624–2636 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Voiniciuc C et al. Identification of key enzymes for pectin synthesis in seed mucilage. Plant Physiol 178, 1045–1064 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mistry J et al. Pfam: the protein families database in 2021. Nucleic Acids Res 49, D412–D419 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nikolovski N et al. Putative glycosyltransferases and other plant Golgi apparatus proteins are revealed by LOPIT proteomics. Plant Physiol 160, 1037–1051 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sterling JD et al. Functional identification of an Arabidopsis pectin biosynthetic homogalacturonan galacturonosyltransferase. Proc. Natl Acad. Sci. USA 103, 5236–5241 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fabrissin I et al. Natural variation reveals a key role for rhamnogalacturonan I in seed outer mucilage and underlying genes. Plant Physiol 181, 1498–1518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moremen KW et al. Expression system for structural and functional studies of human glycosylation enzymes. Nat. Chem. Biol 14, 156–162 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Urbanowicz BR, Pena MJ, Moniz HA, Moremen KW & York WS Two Arabidopsis proteins synthesize acetylated xylan in vitro. Plant J 80, 197–206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Urbanowicz BR et al. Structural, mutagenic and in silico studies of xyloglucan fucosylation in Arabidopsis thaliana suggest a water-mediated mechanism. Plant J 91, 931–949 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soto MJ et al. AtFUT4 and AtFUT6 are arabinofuranose-specific fucosyltransferases. Front. Plant Sci 12, 589518 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kofod LV et al. Cloning and characterization of two structurally and functionally divergent rhamnogalacturonases from Aspergillus aculeatus. J. Biol. Chem 269, 29182–29189 (1994). [PubMed] [Google Scholar]
- 30.Azadi P, O’Neill MA, Bergmann C, Darvill AG & Albersheim P The backbone of the pectic polysaccharide rhamnogalacturonan I is cleaved by an endohydrolase and an endolyase. Glycobiology 5, 783–789 (1995). [DOI] [PubMed] [Google Scholar]
- 31.Ishii T, Ichita J, Matsue H, Ono H & Maeda I Fluorescent labeling of pectic oligosaccharides with 2-aminobenzamide and enzyme assay for pectin. Carbohydr. Res 337, 1023–1032 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Scheller HV, Doong RL, Ridley BL & Mohnen D Pectin biosynthesis: a solubilized α1,4-galacturonosyltransferase from tobacco catalyzes the transfer of galacturonic acid from UDP-galacturonic acid onto the non-reducing end of homogalacturonan. Planta 207, 512–517 (1999). [Google Scholar]
- 33.Moremen KW & Haltiwanger RS Emerging structural insights into glycosyltransferase-mediated synthesis of glycans. Nat. Chem. Biol 15, 853–864 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drula E et al. The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res 10.1093/nar/gkab1045 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taujale R et al. Deep evolutionary analysis reveals the design principles of fold A glycosyltransferases. eLife 10.7554/eLife.54532 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taujale R et al. Mapping the glycosyltransferase fold landscape using interpretable deep learning. Nat. Commun 12, 5656 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kadirvelraj R et al. Comparison of human poly-N-acetyl-lactosamine synthase structure with GT-A fold glycosyltransferases supports a modular assembly of catalytic subsites. J. Biol. Chem 296, 100110 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klepikova AV, Kasianov AS, Gerasimov ES, Logacheva MD & Penin AA A high resolution map of the Arabidopsis thaliana developmental transcriptome based on RNA-seq profiling. Plant J 88, 1058–1070 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Round AN, Rigby NM, MacDougall AJ & Morris VJ A new view of pectin structure revealed by acid hydrolysis and atomic force microscopy. Carbohydr. Res 345, 487–497 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Zdunek A, Pieczywek PM & Cybulska J The primary, secondary, and structures of higher levels of pectin polysaccharides. Compr. Rev. Food Sci. Food Saf 20, 1101–1117 (2021). [DOI] [PubMed] [Google Scholar]
- 42.Amos RA & Mohnen D Critical review of plant cell wall matrix polysaccharide glycosyltransferase activities verified by heterologous protein expression. Front. Plant Sci 10, 915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meinke DW Genome-wide identification of EMBRYO-DEFECTIVE (EMB) genes required for growth and development in Arabidopsis. New Phytol 226, 306–325 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Tzafrir I et al. Identification of genes required for embryo development in Arabidopsis. Plant Physiol 135, 1206–1220 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen LY et al. The Arabidopsis alkaline ceramidase TOD1 is a key turgor pressure regulator in plant cells. Nat. Commun 6, 6030 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pham TT et al. Structures of complexes of a metal-independent glycosyltransferase GT6 from Bacteroides ovatus with UDP-N-acetylgalactosamine (UDP-GalNAc) and its hydrolysis products. J. Biol. Chem 289, 8041–8050 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu D et al. Rethinking the impact of RG-I mainly from fruits and vegetables on dietary health. Crit. Rev. Food Sci. Nutr 60, 2938–2960 (2020). [DOI] [PubMed] [Google Scholar]
- 48.Naqash F, Masoodi FA, Rather SA, Wani SM & Gani A Emerging concepts in the nutraceutical and functional properties of pectin—a review. Carbohydr. Polym 168, 227–239 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Singh RP et al. Generation of structurally diverse pectin oligosaccharides having prebiotic attributes. Food Hydrocoll 108, 105988 (2020). [Google Scholar]
- 50.Cui J et al. Dietary fibers from fruits and vegetables and their health benefits via modulation of gut microbiota. Compr. Rev. Food Sci. Food Saf 18, 1514–1532 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Micoli F et al. Glycoconjugate vaccines: current approaches towards faster vaccine design. Expert Rev. Vaccines 18, 881–895 (2019). [DOI] [PubMed] [Google Scholar]
- 52.Barnes WJ et al. Protocols for isolating and characterizing polysaccharides from plant cell walls: a case study using rhamnogalacturonan-II. Biotechnol. Biofuels 14, 142 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klausen MS et al. NetSurfP-2.0: improved prediction of protein structural features by integrated deep learning. Proteins 87, 520–527 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Rohl CA, Strauss CE, Misura KM & Baker D Protein structure prediction using Rosetta. Methods Enzymol 383, 66–93 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Osawa T et al. Crystal structure of chondroitin polymerase from Escherichia coli K4. Biochem. Biophys. Res. Commun 378, 10–14 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Neuwald AF Rapid detection, classification and accurate alignment of up to a million or more related protein sequences. Bioinformatics 25, 1869–1875 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goodstein DM et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 40, D1178–D1186 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tamura K, Stecher G & Kumar S MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol 38, 3022–3027 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hall BG Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol 30, 1229–1235 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Letunic I & Bork P Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49, W293–W296 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morris GM et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem 30, 2785–2791 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nivedha AK, Thieker DF, Makeneni S, Hu H & Woods RJ Vina-Carb: improving glycosidic angles during carbohydrate docking. J. Chem. Theory Comput 12, 892–901 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Neelamegham S et al. Updates to the symbol nomenclature for glycans guidelines. Glycobiology 29, 620–624 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krissinel E & Henrick K Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D 60, 2256–2268 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article (and its supplementary information files) or are available from the corresponding author upon request. UDP-GalA structure was accessed from Protein Data Bank: 3OH1 (https://www.rcsb.org/structure/3OH1). Plant genome sequences were accessed from Phytozome v1357 (https://phytozome-next.jgi.doe.gov/): A. thaliana TAIR10, C. richardii v2.1 ( JAIKUY010000000), L. usitatissimum v1.0, M. polymorpha v3.1 (PNPG01000000), O. sativa v7.0, P. virgatum v5.1 ( JABWAI010000000), P. trichocarpa v4.1, P. patens v3.3 and S. moellendorffii v1.0. RNA-seq data were accessed from Transcriptome Variation Analysis (http://travadb.org/). Source data are provided with this paper.