

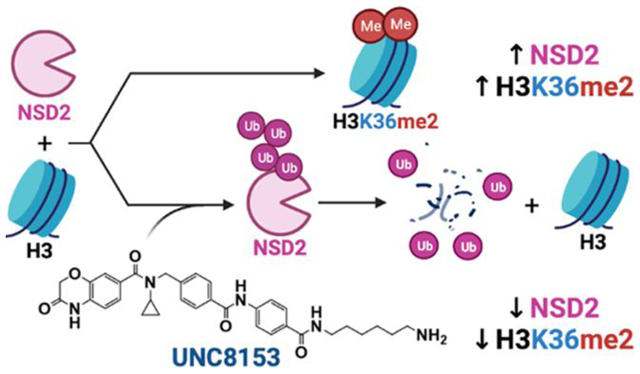
Abstract
Nuclear receptor-binding SET domain-containing 2 (NSD2) plays important roles in gene regulation, largely through its ability to dimethylate lysine 36 of histone 3 (H3K36me2). Despite aberrant activity of NSD2 reported in numerous cancers, efforts to selectively inhibit the catalytic activity of this protein with small molecules have been unsuccessful to date. Here, we report the development of UNC8153, a novel NSD2-targeted degrader that potently and selectively reduces the cellular levels of both NSD2 protein and the H3K36me2 chromatin mark. UNC8153 contains a simple warhead that confers proteasome-dependent degradation of NSD2 through a novel mechanism. Importantly, UNC8153-mediated reduction of H3K36me2 through the degradation of NSD2 results in the downregulation of pathological phenotypes in multiple myeloma cells including mild antiproliferative effects in MM1.S cells containing an activating point mutation and antiadhesive effects in KMS11 cells harboring the t(4;14) translocation that upregulates NSD2 expression.
Graphical Abstract
INTRODUCTION
Epigenetics is a growing area of interest among biomedical and drug discovery researchers. Over the last decade, advances in epigenomics technologies and chemical biology have demonstrated the central role that epigenetics plays in health and disease.1,2 A major class of epigenetic regulatory proteins is the protein lysine methyltransferases (PKMTs).3 These enzymes mono-, di- and/or trimethylate lysine residues (Kme1, Kme2, Kme3) on multiple target proteins, one class of which is histones. Histone lysine methylation plays an important role in gene regulation, and aberrant histone methylation states are associated with most cancers.4,5 Accordingly, research in recent years has led to a number of PKMT inhibitors entering clinical trials in oncology, and the recent approval of Tazemetostat, an EZH2 inhibitor, for the treatment of epithelioid sarcoma and follicular lymphoma further strengthens the therapeutic relevance and druggability of this target class.6
Nuclear receptor-binding SET domain-containing 2 (NSD2, also known as WHSC1 and MMSET) is the primary methyltransferase that installs the dimethyl mark on lysine 36 of histone 3 (H3K36me2), which is associated with active gene transcription.7 NSD2 is associated with several types of cancers through aberrant expression or somatic mutation of the protein.7,8 A t(4;14) translocation, which leads to overexpression of NSD2, is present in 15−20% of multiple myeloma (MM) patients.7 Additionally, an NSD2 E1099K gain-of-function point mutation is strongly enriched in a subset of pediatric acute lymphoblastic leukemia (ALL) cases and has been detected in individuals with lung adenocarcinoma.9,10 Both mutations lead to an upregulation of H3K36me2 and are associated with malignant tumor progression.11 Importantly, genetic knockdown of NSD2 reduces H3K36me2 levels and tumor growth in vivo.7,12,13 Despite the demonstrated significance of NSD2 in cancer, there has been little success in potently and selectively targeting the catalytic activity of this protein or removing the NSD2-deposited H3K36me2 mark from chromatin with small molecules.14,15
Full-length NSD2 contains both a catalytic SET [Su(var)3–9, Enhancer-of-zeste, and Trithorax] domain that “writes” the H3K36me2 mark, and multiple chromatin “reader” domains (two PWWP domains and five PHD domains) that bind to chromatin in a multivalent fashion (Figure 1A). Specifically, the N-terminal PWWP domain (NSD2-PWWP1) binds H3K36me2-marked nucleosomes through a conserved aromatic cage, helping to stabilize NSD2 on chromatin.16,17 Additional shorter isoforms that contain only a subset of these domains also exist.
Figure 1.
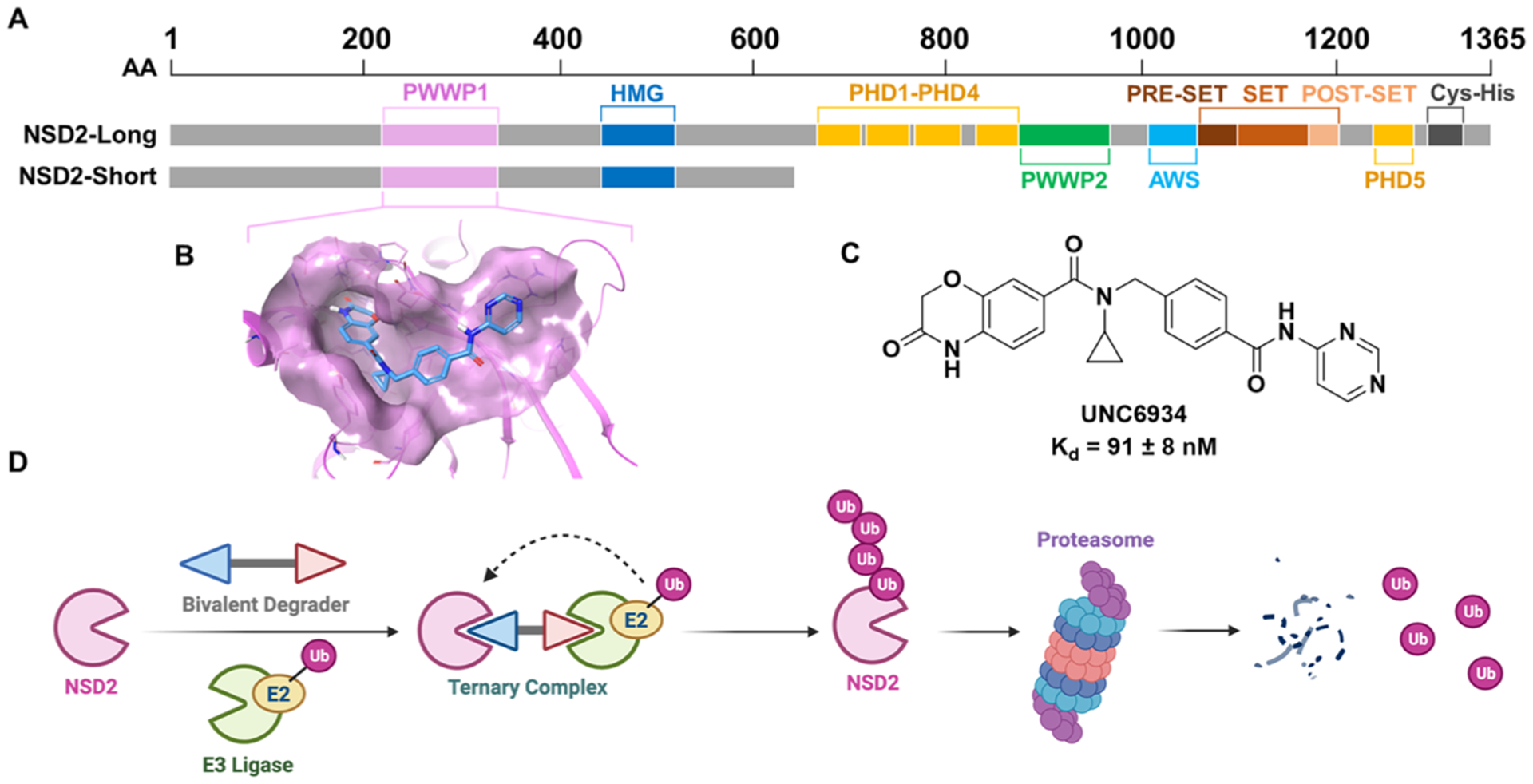
UNC6934 enables the development of NSD2-targeted degraders. (A) Domain architecture of the two main isoforms, NSD2-short (MMSET I) and NSD2-long (MMSET II). (B) UNC6934 (cyan) bound to NSD2-PWWP1 (magenta) (PDB: 6XCG). (C) Chemical structure of the NSD2-PWWP1 chemical probe, UNC6934. (D) Schematic of induced degradation of NSD2 via a heterobifunctional degrader. Created with BioRender.com.
We recently reported the first antagonists to target the PWWP1 domain of NSD2.16,17 UNC6934 potently and selectively binds to the aromatic cage of NSD2-PWWP1 (Kd = 91 ± 8 nM) (Figure 1B,C, PDB: 6XCG) and disrupts the binding of PWWP1 to H3K36me2-marked nucleosomes (IC50 = 104 ± 13 nM). We further demonstrated that UNC6934 increased the subcellular localization of NSD2 within the nucleolus; however, UNC6934 does not affect the catalytic activity of NSD2, and as a result, global H3K36me2 levels remain unaltered in UNC6934-treated KMS11 t(4;14) multiple myeloma cells. In contrast, genetic knockdown of NSD2 in KMS11 cells leads to a substantial decrease in H3K36me2.7 Furthermore, it has been reported that NSD2 translocation knockout KMS11 cells show reduced cancer phenotypes,7,18 whereas KMS11 cells treated with UNC6934 do not. Thus, alternative strategies are needed to target NSD2 in t(4;14) multiple myeloma and other NSD2-linked cancers.
Targeted protein degradation (TPD) is a rapidly growing strategy in drug discovery. Excitement around this approach stems from the fact that TPD can often overcome difficulties encountered with direct chemical inhibition of a target of interest.19 Generally, TPD employs heterobifunctional molecules that contain two important functional moieties—a ligand for a protein of interest and a ligand for an E3 ligase—separated by a linker, which can vary in length and composition (Figure 1D). Through simultaneous binding to the target protein and an E3 ubiquitin ligase, these molecules induce the formation of a ternary complex and enable proximity-induced polyubiquitination of the target protein, marking it for degradation by the proteasome (Figure 1D). Here, we leveraged the potency and specificity of UNC6934 for NSD2-PWWP1 to design and characterize highly selective NSD2-targeted degraders. Our NSD2 degraders induce proteasome-dependent degradation of both major isoforms of NSD2 in cells and reduce global cellular levels of H3K36me2 while also reducing cellular adhesion in KMS11 cells and resulting in mild antiproliferative effects in MM1.S cells. Excitingly, these molecules facilitate NSD2 degradation through a mechanism that is not well-established in the TPD field and may provide new opportunities for TPD more broadly.
RESULTS
Design and Evaluation of NSD2-Targeted Degraders.
We hypothesized that we could achieve NSD2 degradation by derivatizing UNC6934 with chemical moieties intended to mimic a specific set of N-terminal residues known as N-degrons.20 These N-degrons serve as signals that are commonly recognized by a class of E3 ligases, known as “N-recognins,” to facilitate protein degradation via the N-end rule pathway.21,22 This E3 family is comprised of at least four proteins—UBR1, UBR2, UBR4, and UBR5—named for their conserved ubiquitin-recognin box (UBR-box) domain that recognizes specific N-terminal protein sequences. Type 1 N-degrons are composed of basic residues such as arginine, lysine, and histidine. Inspired by a singular application of this approach in the literature,23 we therefore sought to synthesize heterobifunctional molecules containing UNC6934 and one of these basic amino acids to recruit a UBR E3 ligase into proximity with NSD2.
The crystal structure of UNC6934 in complex with NSD2-PWWP1 revealed that the terminal pyrimidine ring is fairly solvent-exposed and may be amenable to derivatization without disruption of binding to the PWWP1 domain (Figure 1C).16,17 Furthermore, we previously showed that an analogue of UNC6934 biotinylated at this position, UNC7096, maintains binding to NSD2, confirming that this position could serve as a suitable exit vector from which we could begin modifying UNC6934. Our initial design strategy involved the derivatization of the terminal aromatic ring of our NSD2 ligand with alkyl amine linkers of varying lengths to enable subsequent tethering to the carboxylic acid of an arginine or histidine residue. This afforded the eight arginine- and histidine-containing compounds shown in Figures 2A and S1A, respectively. Using surface plasmon resonance (SPR), we confirmed that the bifunctional compounds maintain potent binding to NSD2-PWWP1 with dissociation constants ranging from 10 to 60 nM (Figures 2A,B and S1A,B). This clearly demonstrates that the addition of the linker and basic amino acid does not compromise binding and the compounds are sufficiently potent to potentially facilitate NSD2 degradation.
Figure 2.
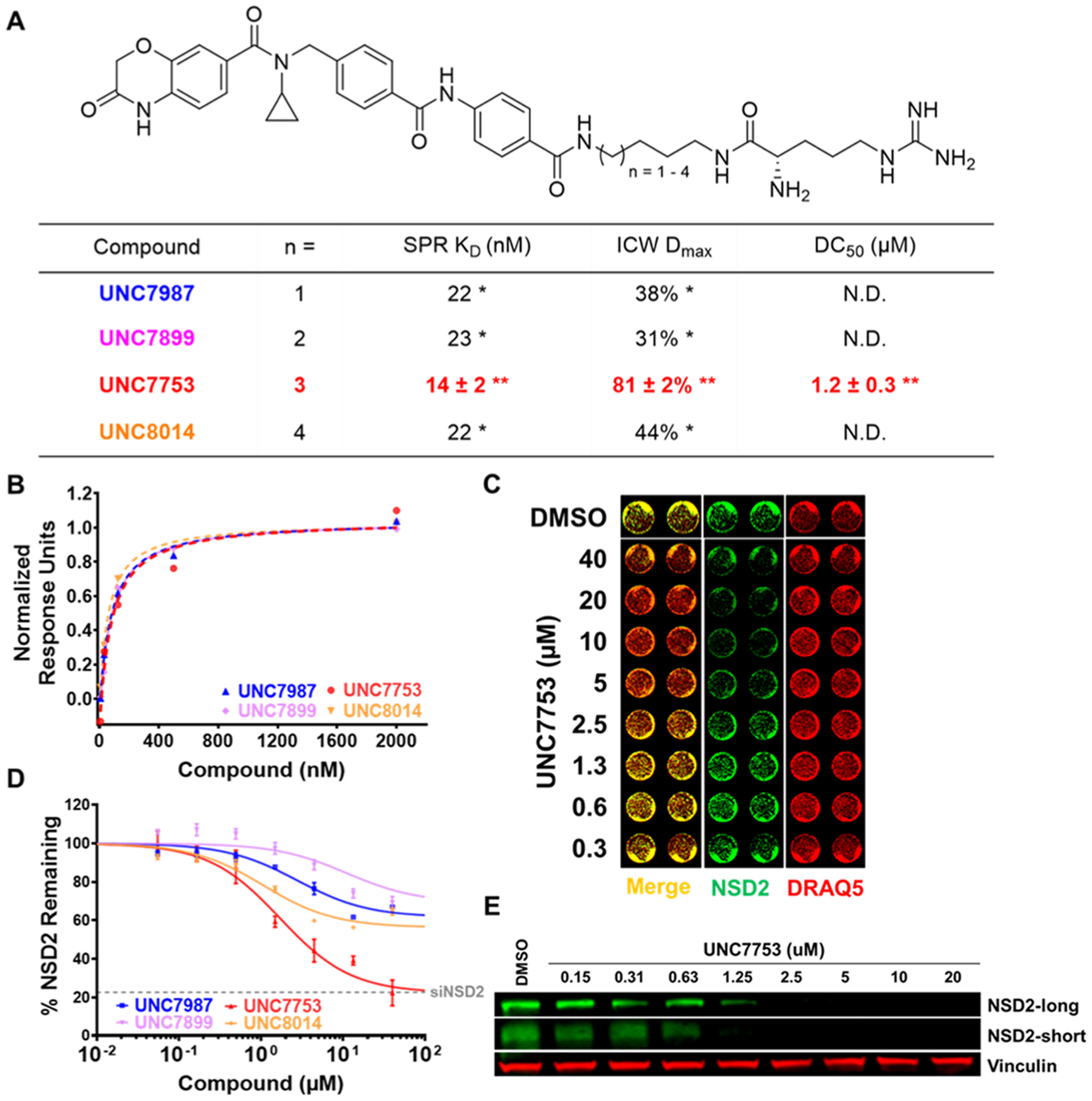
Analysis of NSD2 binding and degradation with arginine-containing degraders. (A) Chemical structures of arginine-containing degraders. Compound binding affinities (Kd values) for recombinant NSD2-PWWP1 were determined by SPR. NSD2 degradation was evaluated by an ICW assay in U2OS cells dosed with compound for 24 h. Maximal observed degradation values (Dmax) and half-maximal degradation concentrations (DC50) were determined based on total NSD2 including both isoforms. *Determined from a single experiment. **Mean of three independent experiments ± SEM. N.D: DC50 values not determined for compounds with Dmax values < 50%. (B) Representative NSD2-PWWP1 SPR traces of compounds listed in panel (A). SPR response units were normalized with the highest response unit = 1.00 for each set of curves. (C) Representative image of ICW data upon UNC7753 treatment for 24 h, showing a dose-dependent reduction of NSD2 levels. Image contains NSD2 intensities (green), DRAQ5 loading control intensities (red), and merged signals of NSD2 and DRAQ5. (D) ICW dose−response curves for compounds listed in panel (A). Four technical replicates were performed for each condition with mean ± SEM plotted. A representative curve from one experiment is shown for UNC7753. Gray dashed line denotes NSD2 levels measured by ICW upon siRNA knockdown of NSD2 in U2OS cells. (E) Confirmation of reduction of NSD2-long and NSD2-short by western blot upon UNC7753 treatment of U2OS cells for 24 h. Vinculinis a loading control.
Next, to better enable the evaluation of many potential NSD2 degraders, we developed a robust in-cell western (ICW) assay to readily monitor and quantify NSD2 protein levels in U2OS cells. As the antibody used for this assay recognizes the N-terminus of NSD2, we were able to detect both major isoforms, NSD2-long (MMSET II) and NSD2-short (MMSET I). Using this ICW assay, we were very pleased to see that the arginine-containing compounds resulted in at least a partial decrease in the levels of NSD2 at 24 h (Figure 2C,D), while the histidine-containing compounds were slightly less effective overall (Figure S1A,C). Of the four arginine-containing compounds tested, which differ only by the number of methylene groups in the linker, UNC7753 was the most effective degrader of NSD2, resulting in a maximal observed degradation (Dmax) value of 81% and a half-maximal degradation concentration (DC50) of 1.2 ± 0.3 μM as determined by ICW. These results suggest that a six-carbon linker is optimal in this context, and a similar trend was observed with the analogous histidine-containing compounds. Encouragingly, UNC7753 reduces NSD2 protein levels to a similar extent as that of NSD2 knockdown by siRNA (Figure 2D, siRNA validation shown in Figure S1D). To further investigate the degradation potential of UNC7753 and confirm the ICW results, we analyzed NSD2 degradation by traditional Western Blot. Upon treatment of U2OS cells for 24 h, we observed that UNC7753 enabled degradation of both the long and short isoforms of NSD2 in a dose-dependent manner, giving DC50 values that are consistent with our ICW results (IB: NSD2-long DC50 = 1.18 [95% CI = 0.98−1.41] μM; Figure 2E).
SAR Studies Reveal That NSD2 Degradation is Not Dependent on UBR E3 Ubiquitin Ligases.
Encouraged by our preliminary findings, and with the aim of improving compound potency and permeability and gaining a better understanding of the structural requirements for degradation, we next performed structure−activity relationship (SAR) studies focused on the E3 recruiting moiety of UNC7753. This effort was initially guided by available structural information of UBR E3 ligases and the known structural requirements for N-degron recognition.21,24 Crystal strucures of the UBR-box domains in complex with known N-degrons reveal a large, negative groove on the surface of the UBR-box binding site that engages the side chains of basic amino acids (Figure S2A,B). In addition to this negative groove, key hydrogen bonding interactions enable binding of N-degrons to the UBR-box domain. As a result, we synthesized a series of UNC7753 analogues modified at the arginine residue and evaluated their ability to reduce NSD2 protein levels in our in-cell western assay (Figure 3A,B).
Figure 3.
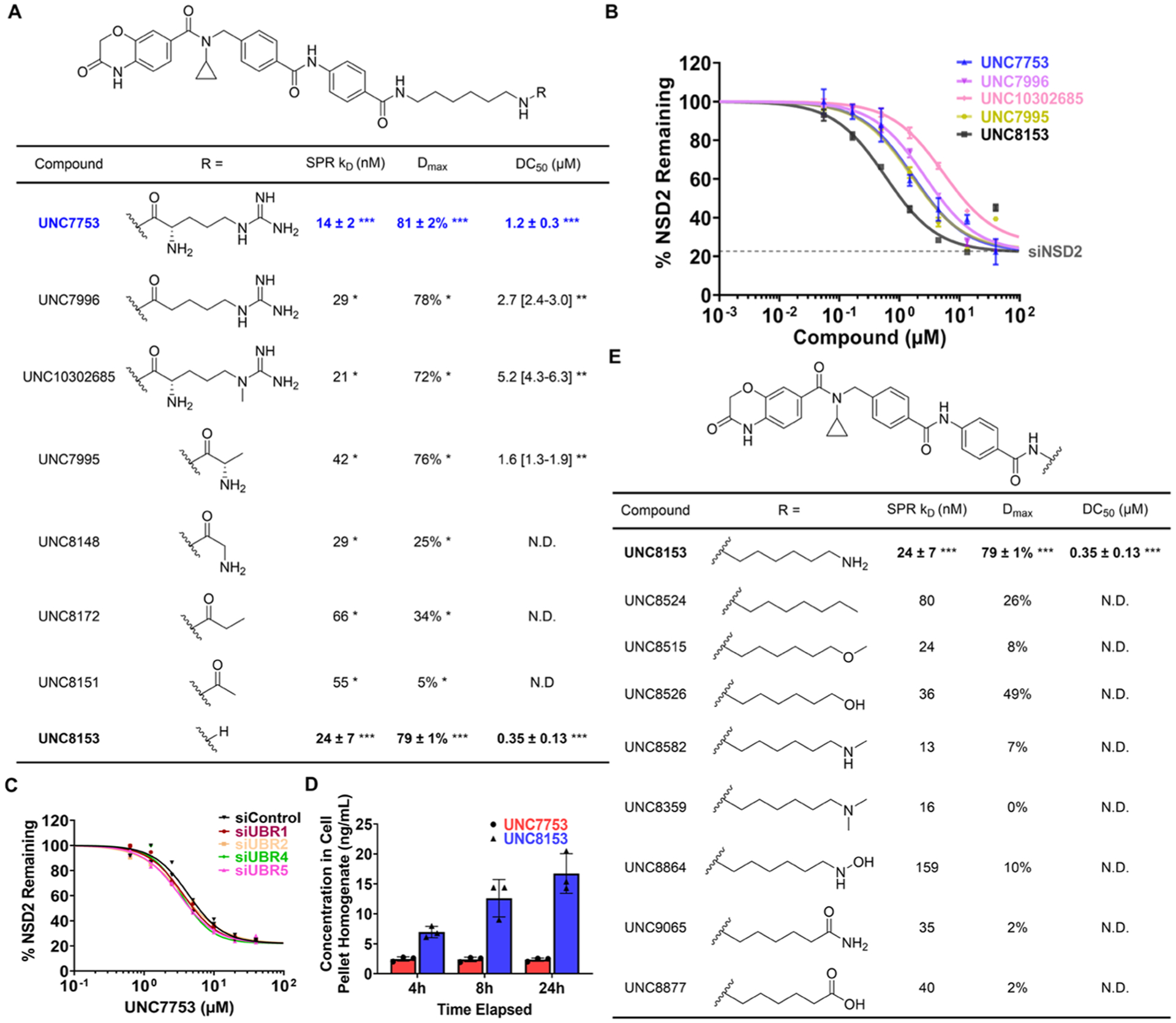
Conversion of UNC7753 to UNC8153 promotes NSD2 degradation via a UBR-independent mechanism. (A) Analysis of NSD2 binding and degradation with UNC7753 analogues modified at the arginine moiety. SPR KD values, ICW Dmax, and DC50 values were determined as described in Figure 2A. *Determined from a single experiment. **Determined from curve fitting of four technical replicates and [95% CI]. ***Mean of three independent experiments each containing 2−4 technical repeats ± SEM. N.D: DC 50 values not determined for compounds with Dmax values < 50%. (B) ICW dose−response curves for the compounds with DC50 values listed in panel (A). Four technical replicates were performed for each condition with mean ± SEM plotted. A representative curve from one experiment is shown for UNC7753 and UNC8153. Gray dashed line denotes NSD2 levels measured by ICW upon siRNA knockdown of NSD2 in U2OS cells. (C) ICW dose−response curves upon treatment with UNC7753 for 24 h after siRNA knockdown of UBR-1, −2, −4, or −5. Two technical replicates were performed for each condition. (D) Concentrations of UNC7753 and UNC8153 in the cell pellet homogenate after treating with 10 μM UNC7753 for the indicated amount of time. Three independent replicates were performed for each condition. Bar height represents mean ± SD. (E) Analysis of NSD2 binding and degradation with UNC8153 analogues modified at the free amine. SPR KD and ICW Dmax values were determined from a single experiment. ***Mean of three independent experiments each containing 2−4 technical repeats ± SEM. N.D: DC50 values not determined for compounds with Dmax values < 50%.
According to previous work, a free N-terminal α-amino group is required for recognition of an N-degron by the UBR-box domain.21 Therefore, we were surprised to see that UNC7996, in which the α-amino group is absent, was able to effectively degrade NSD2 with similar potency to UNC7753 (Figure 3A,B). Additionally, in the UBR-box binding pocket, a water molecule mediates a key hydrogen bond between the arginine ε-nitrogen of the N-degron and Asp118 of the UBR-box domain (Figure S2B).21 UNC10302685, in which the ε-N-methyl would be expected to hinder this stabilizing hydrogen bond, is similarly potent to UNC7753. Together, these results called into question the mechanism of NSD2 degradation and suggested that degradation may not be mediated by a UBR E3 ubiquitin ligase as we previously expected.
Further truncation of the arginine side chain of UNC7753 continued to reveal similar inconsistencies with a degradation mechanism involving UBR recruitment. Although alanine is incapable of serving as an N-degron for UBR-box domains,21 UNC7995, in which arginine is substituted with alanine, potently degrades NSD2. We next sought to determine the minimum pharmacophore required for degradation by synthesizing four additional truncated analogues. Remarkably, UNC8153, which lacks the amino acid moiety entirely, was over three times more potent than UNC7753 with a DC50 of 0.35 μM and a Dmax of 79%. While the glycinamide and propionamide analogues (UNC8148 and UNC8172, respectively) were still able to degrade NSD2, albeit with much lower Dmax values, the acetamide analogue (UNC8151) did not demonstrate measurable NSD2 degradation despite potent binding to NSD2-PWWP1. Similarly, our NSD2-PWWP1 chemical probe, UNC6934, was also unable to degrade NSD2 (Figure S1E).17
To further confirm that NSD2 degradation is not dependent on the recruitment of a UBR E3 ligase, we evaluated compound-mediated degradation upon siRNA knockdown of UBR1, −2, −3, or −4 by ICW. In each case, siRNA knockdown of either UBR1, −2, −3, or −4 had no effect on the reduction of NSD2 by UNC7753 (Figure 3C) or UNC8153 (Figure S2C− G). Collectively, these efforts identified UNC8153 as a more potent degrader of NSD2 and provided mechanistic insight, suggesting that NSD2 degradation is not mediated by a UBR E3 ubiquitin ligase.
UNC7753 Functions as a Prodrug.
Considering the efficacy of the truncated compound UNC8153, we hypothesized that our amino acid-containing NSD2 degraders may be acting as prodrugs, whereby they are cleaved by a cellular protease(s) to yield an active degrader. The observation that the degraders in Figure 3A have differing degrees of degradation efficiency may be caused by a combination of parameters such as differences in the rate of proteolysis of each amino acid derivative and/or variable levels of cell permeability. To test this hypothesis, we treated U2OS cells with 10 μM UNC7753 and looked for the presence of UNC8153 in the cell lysates by mass spectrometry. Indeed, upon treatment of cells with 10 μM UNC7753, UNC8153 was readily detected in the cell lysate, increasing in concentration over a period of 24 h (Figure 3D). Overall, this data overwhelmingly suggests that UNC8153 is generated from the proteolysis of UNC7753 and enables NSD2 degradation via a non-UBR-dependent mechanism. This also helps to explain why UNC8153 is a more effective degrader than the other compounds tested in Figure 3A, all of which must first undergo proteolysis.
A primary Amine Confers NSD2 Degradation.
Intrigued by these results, we continued to explore the structural requirements for NSD2 degradation. We synthesized and screened eight compounds where the primary amine was either substituted or replaced with a different functional group while keeping the NSD2 ligand and six-carbon linker constant (Figure 3E). This revealed that compounds containing a terminal alkyl group (UNC8524), ether (UNC8515), secondary amine (UNC8582), or tertiary amine (UNC8359) are not effective degraders, resulting in minimal observed loss of NSD2, if any. Interestingly, the hydroxyl analogue of UNC8153 (UNC8526) appears to give some degree of degradation, though it is much less effective than UNC8153. As a primary amine might be a labile functional group under cellular conditions, we also synthesized several potential metabolites of UNC8153 to rule out their involvement in the degradation process. We replaced the amine of UNC8153 with a hydroxylamine (UNC8864), amide (UNC9065), or carboxylic acid (UNC8877), none of which gave any appreciable amount of degradation. Unfortunately, attempts to synthesize and purify the corresponding aldehyde were unsuccessful; therefore, oxidation of UNC8153 to the corresponding aldehyde and its possible involvement in the degradation mechanism cannot be ruled out. Overall, this data reveals a preference for the primary amine of UNC8153 and suggests that, unlike the other functional groups tested, the primary amine can promote NSD2 degradation.
UNC8153 is a Bivalent Degrader.
Our ICW results reveal that a hook effect occurs at high concentrations of UNC8153 in U2OS cells, as the extent of NSD2 reduction begins to decrease (Figure 3B). This result was further confirmed by immunoblotting (Figure S3A). This suggests that UNC8153 acts like a classic bivalent degrader. To further investigate the mode of degradation and determine which regions of UNC8153 are involved in ternary complex formation, we performed several competition experiments—one between UNC8153 and the parent NSD2-PWWP1 ligand, UNC6934, and another between UNC8153 and (6-aminohexyl)benzamide (UNC8603). Treatment of U2OS cells with 2 μM UNC8153 in combination with increasing concentrations of UNC6934 significantly decreased NSD2 degradation in a dose−response fashion (Figure 4A). This is expected, as UNC6934 should compete with UNC8153 for binding to NSD2-PWWP1 at high concentrations. Importantly, treatment with UNC8603 also caused a reduction in the degradation efficiency of UNC8153 in a dose-dependent manner, which suggests that UNC8603 is in competition with UNC8153. This was somewhat surprising due to the small size and simplicity of UNC8603. The abilities of UNC6934 and UNC8603 to separately outcompete UNC8153 for the recruitment of NSD2 and a putative E3 ubiquitin ligase, respectively, and consequently block NSD2 degradation demonstrate that both moieties facilitate ternary complex formation during the degradation mechanism. These results also demonstrate that the two moieties are independent of each other in targeting their respective proteins, further suggesting that UNC8153 behaves as a bivalent degrader.
Figure 4.
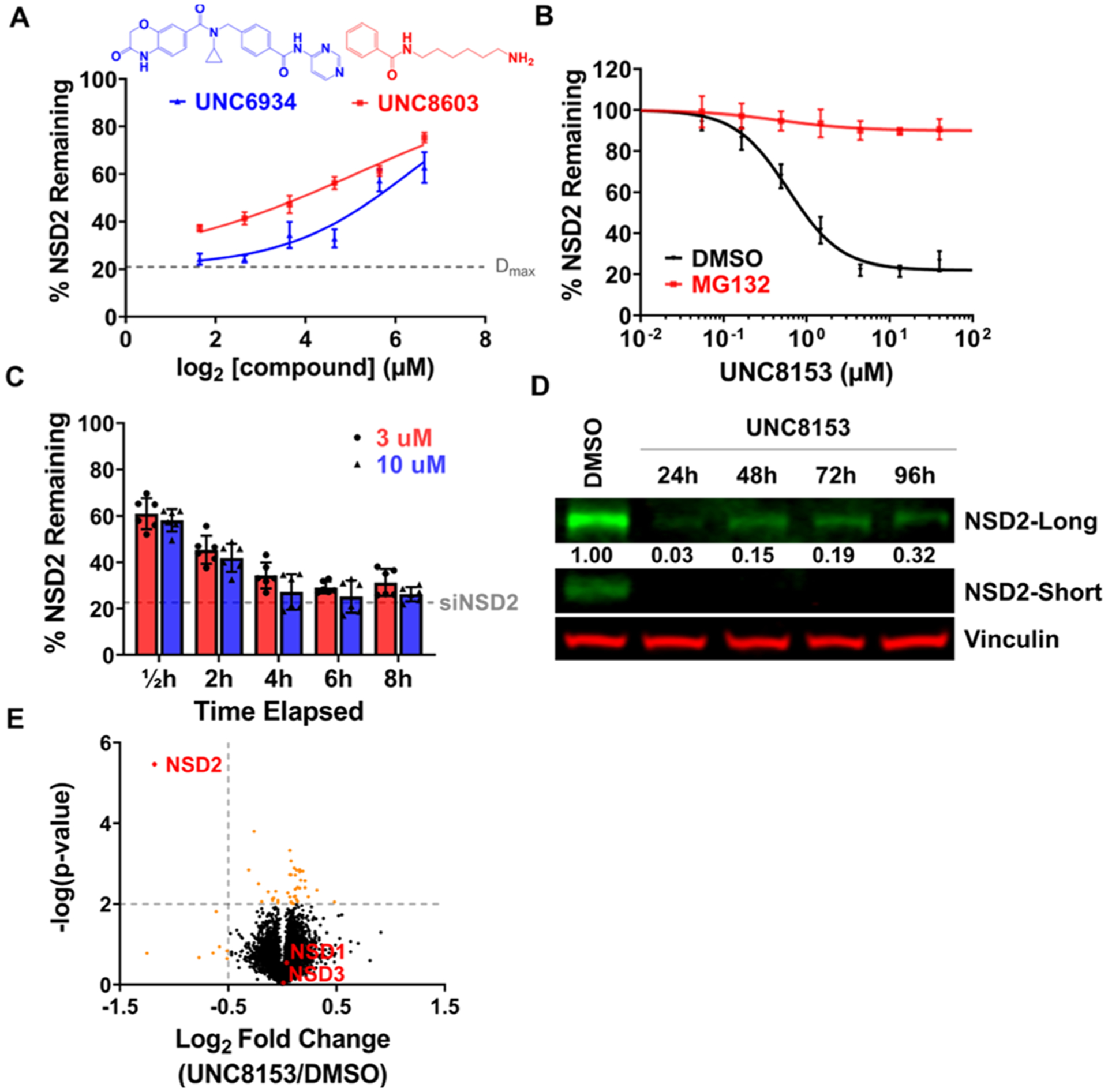
UNC8153 is a fast-acting bivalent degrader targeting NSD2 for proteasomal-dependent degradation with high selectivity. (A) Quantified NSD2 levels by ICW following treatment of U2OS cells with 2 μM UNC8153 and varying concentrations of UNC8603 (red) or UNC6934 (blue) for 6 h. Error bars represent ± SEM from n = 3 technical replicates. (B) ICW dose−response curves upon UNC8153 treatment for 6 h in the presence (red) or absence (black) of the MG132 proteasome inhibitor in U2OS cells. Error bars represent ± SEM from n = 3 technical replicates. (C) Quantified NSD2 levels by ICW after UNC8153 treatment at 3 and 10 μM from 0.5 to 8 h in U2OS cells. The dotted gray line indicates NSD2 levels with siRNA knockdown. Six technical replicates from two independent experiments were performed for each condition. Bar height represents mean ± SD. (D) Immunoblot of NSD2-long and NSD2-short from U2OS cells treated with 10 μM UNC8153 for 24−96 h. Vinculin is a loading control. Numbers represent relative intensities of NSD2-long bands normalized to vinculin as a proportion of DMSO treatment (set as 1.00). (E) Unbiased global proteomics experiment using tandem mass tag quantification comparing U2OS cells treated with DMSO vs 5 μM UNC8153 for 6 h.
UNC8153-Mediated Degradation of NSD2 is Protea-some- and Neddylation-Dependent.
To further understand the mechanism of action of UNC8153-mediated NSD2 degradation, we monitored the degradation efficiency of UNC8153 in the presence of a proteasome inhibitor, MG132,25 in our in-cell western assay. Treatment with MG132 completely ablates NSD2 degradation at all concentrations of UNC8153 tested (Figure 4B). Next, we performed a NanoBRET ubiquitination assay in cells expressing Nano-Luciferase fused to NSD2-PWWP1 at the N- or C-terminus and HaloTag-Ubiquitin. We observed increased BRET signals between HaloTag-ubiquitin and NanoLuc-NSD2-PWWP1 upon UNC8153 treatment compared to UNC6934 or the vehicle, suggesting that UNC8153 promotes NSD2 ubiquitylation (Figure S3B). Furthermore, treatment with UNC8153 in either the presence of TAK243,26 an inhibitor of the E1-ubiquitin-activating enzyme, or MLN4924, an inhibitor of the Nedd8 activating enzyme that is essential for the activity of Cullin-RING family E3 ubiquitin ligases, also prevented NSD2 degradation (Figure S3C).27 Together, these data strongly support an NSD2 degradation mechanism involving a Cullin-RING family E3 ubiquitin ligase complex and the ubiquitin-proteasome system.
UNC8153 is Fast-Acting and Delivers Persistent Degradation Effects.
Having identified UNC8153 as our most effective degrader, we next evaluated its ability to degrade NSD2 in a time-dependent fashion. First, U2OS cells were treated with 3 and 10 μM UNC8153 for various time points up to 8 h and evaluated by ICW (Figure 4C). An appreciable degree of degradation (~40%) was observed as early as 30 min after compound treatment, and this level gradually increased to reach maximal degradation in 4−6 h, demonstrating that UNC8153 is fast-acting in its ability to facilitate degradation of NSD2. At these time points, NSD2 levels were similar to those achieved upon siRNA knockdown of NSD2. The persistence of UNC8153-mediated NSD2 degradation was subsequently evaluated by traditional western blot in U2OS cells over a 24−96 h time course (Figure 4D). It appears that UNC8153-mediated NSD2 degradation is relatively long-lasting, as 96 h after a single treatment of UNC8153 (without replenishing the compound), the extent of degradation of NSD2-long is still above 60%.
UNC8153 Degrades NSD2 with High Selectivity.
To assess the effects of UNC8153 treatment on cellular protein levels more broadly, we performed global proteomics experiments using tandem mass tag quantification comparing U2OS cells treated with 5 μM UNC7753, 5 μM UNC8153, or DMSO for 6 h. Whole proteome analysis resulted in the identification of about 9000 proteins and, of these, 7863 were quantifiable. Significant degradation was defined by a −log p-value of >2 and a log 2 fold change ratio (compound-treated/DMSO-treated) of less than −0.5. Excitingly, these data revealed that NSD2 was selectively degraded by UNC8153 within the measurable proteome (Figure 4E). Treatment with UNC7753 also resulted in similar findings (Figure S3D). The related NSD family members NSD1 and NSD3 were not degraded by either compound, and this result was further confirmed by immunoblotting (Figure S3E). Additionally, this revealed that this novel mechanism of degradation can enable selective degradation of a protein of interest without off-target degradation effects, unlike other more well-established mechanisms of degradation. Overall, these data reveal that UNC8153 enables degradation of NSD2 in a highly selective fashion, establishing UNC8153 as an excellent tool to link cellular phenotypes observed upon compound treatment to loss of NSD2 protein.
UNC8153 Reduces H3K36me2 Levels in Multiple Cell Lines.
Next, we were interested in evaluating the functional consequences of UNC8153-mediated NSD2 degradation. Before doing so, we synthesized UNC8587 as a negative control compound that contains an isopropyl moiety in place of the cyclopropyl in UNC8153 (Figure 5A). We have previously shown that this subtle modification is sufficient to completely abrogate binding to NSD2.17
Figure 5.
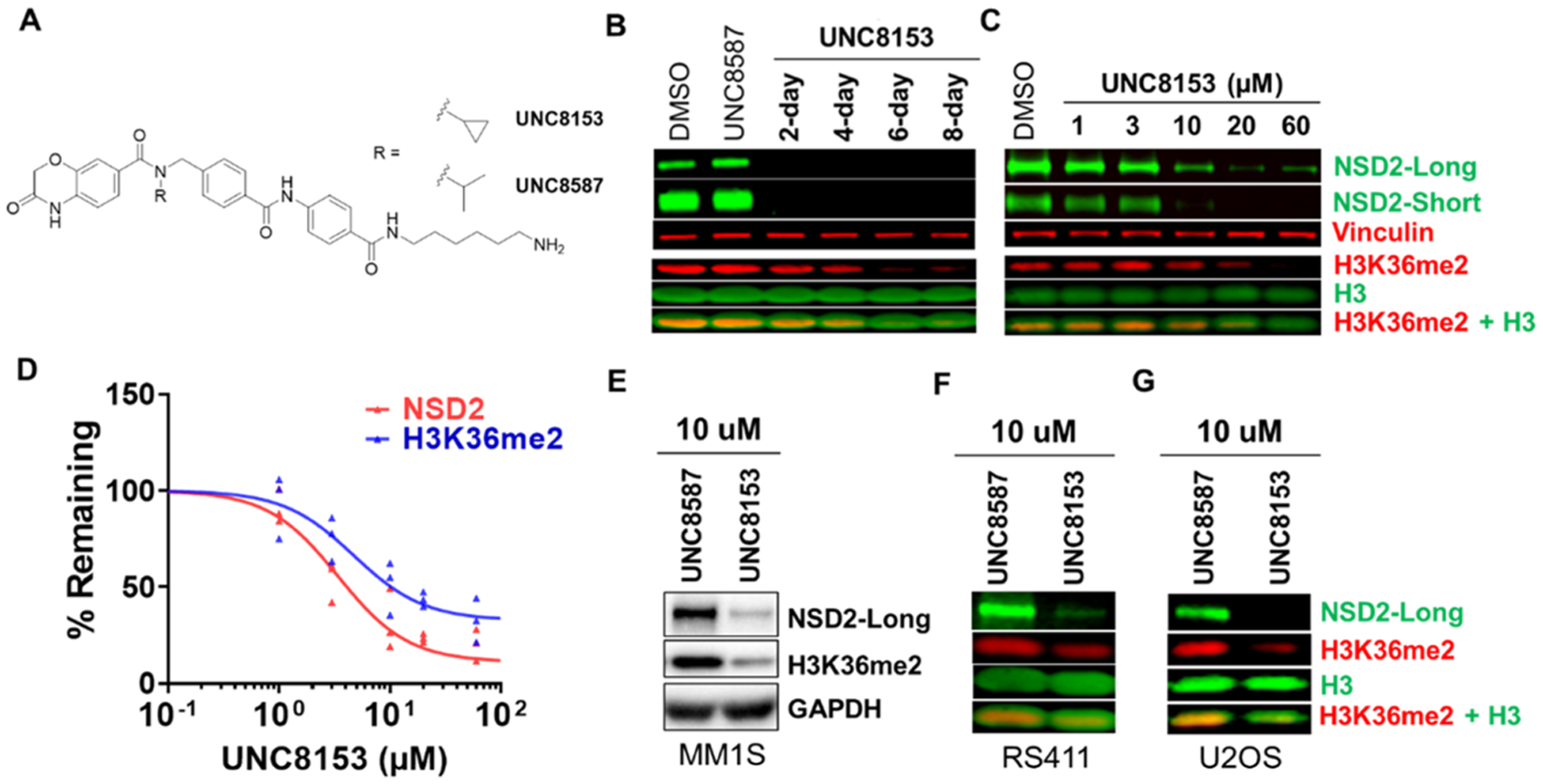
UNC8153 degrades NSD2 and modulates H3K36me2 levels. (A) Structure of UNC8153 (active compound) compared to UNC8587 (negative control). (B) Representative NSD2 and H3K36me2 immunoblots of KMS11 cell pellets from a 2−8 day time-course treatment with 20 μM UNC8153 and an 8-day treatment with UNC8587 and DMSO. Vinculin and H3 are loading controls. H3K36me2 blot was performed three times with similar results. NSD2 blot was performed once. (C) Representative NSD2 and H3K36me2 immunoblot of KMS11 cells treated with UNC8153 at 1−60 μM for 6 days. Vinculin and H3 are loading controls. The experiment was repeated three times with similar results. (D) Quantified NSD2-long and H3K36me2 levels from immunoblotting (as shown in panel (C)) normalized to loading control and as a percentage of control treatment from three independent experiments. (E) NSD2-long and H3K36me2 immunoblot after a 6-day treatment of MM1S cells with 10 μM UNC8153 or UNC8587. GAPDH is a loading control. (F) NSD2-long and H3K36me2 immunoblot after a 6-day treatment of RS411 cells with 10 μM UNC8153 or UNC8587. H3 is a loading control. (G) NSD2-long and H3K36me2 immunoblot after a 6-day treatment of U2OS cells with 10 μM UNC8153 or UNC8587. H3 is a loading control.
As NSD2 is the primary methyltransferase that installs the H3K36me2 mark, we evaluated H3K36me2 levels in UNC8153-treated (20 μM) KMS11 cells over 2−8 days by immunoblotting. KMS11 cells were chosen as they are a model t(4;14)+ multiple myeloma cell line in which the IgH enhancer drives overexpression of NSD2.7 Significant loss of the H3K36me2 mark was observed after 6 days, while cells treated with the inactive control compound, UNC8587, or DMSO showed no change (Figure 5B). As it is thought that loss of methylation occurs via dilution through cell division, it is not surprising that full loss of H3K36me2 occurs days after NSD2 is effectively degraded. Next, we investigated the effect of UNC8153 on NSD2 degradation and H3K36me2 levels in KMS11 cells in a dose-dependent manner (1−60 μM) after 6 days. Significant dose-dependent reduction in both the NSD2-short and long isoforms (IB: NSD2-long DC50 = 3.41 [95% CI: 2.22–5.55] μM), as well as dose-dependent reduction of the H3K36me2 mark, was observed (Figure 5C,D). Similar results were obtained with UNC7753 and the corresponding negative control compound UNC8592 (Figure S4A) in both time-course and dose−response experiments (Figure S4B,C). Additionally, upon treatment with UNC8153, we observed an increase in H3K36 monomethylation (H3K36me1), potentially due to its accumulation in the absence of NSD2-mediated dimethylation of H3K36me1 (Figure S4D). In contrast, H3K36 trimethylation (H3K36me3) decreased slightly, likely due to the fact that H3K36me2 serves as a substrate for the generation of H3K36me3. This provides further reinforcement that dimethylation of H3K36 is the primary activity of NSD2 in KMS11 cells and is consistent with the reported effects of NSD2 genetic knockdown.7 In addition to t(4;14)-translocated KMS11 cells, we also evaluated the effects of NSD2 degradation in MM1S (multiple myeloma) and RS411 (acute lymphocytic leukemia) cells, which carry the activating NSD2 E1099K mutation. Encouragingly, both cell lines were similarly responsive to the NSD2 and H3K36me2 modulating effects of UNC8153 (Figure 5E,F). UNC8153 treatment also led to a reduction in H3K36me2 in U2OS cells that have unaltered, wildtype NSD2 (Figure 5G).
Phenotypic Consequences of UNC8153 Treatment Vary with NSD2 Genomic Status.
Because NSD2 has been implicated as a molecular target in MM, we investigated the phenotypic effects of NSD2 degradation in several MM cell lines. Treatment of t(4;14)-translocated KMS11 cells with up to 30 μM UNC8153 did not result in a significant change in proliferation despite effective NSD2 degradation (Figure 6A). In contrast, a mild antiproliferative effect was observed in MM1S cells harboring the activating NSD2 E1099K mutation upon treatment with increasing concentrations of UNC8153 for 8 days, whereas the negative control compound (UNC8587) had no effect (Figure 6B). This is consistent with the antiproliferative effects observed upon CRISPR/cas9-mediated gene depletion of NSD2 in MM1S cells (Figure S5A). Additionally, like in U2OS cells, UNC8153 selectively degrades NSD2 over other NSD family members in MM1S cells, further supporting that the antiproliferative effect observed is on target (Figure S5B). Cell lines without mutation or disruption of the NSD2 gene, including MDA-MB-231 (breast), U2OS (bone), or HEK293 (kidney), also showed no significant changes in proliferative potential upon UNC8153 treatment over 5 days, despite significantly reduced levels of NSD2 (Figures 6C and S5C−H). These data suggest that the E1099K activating mutation may confer dependence on NSD2 catalytic activity for the growth or survival of MM1S cells, consistent with prior studies suggesting that the E1099K mutation is a driver of oncogenesis.28
Figure 6.
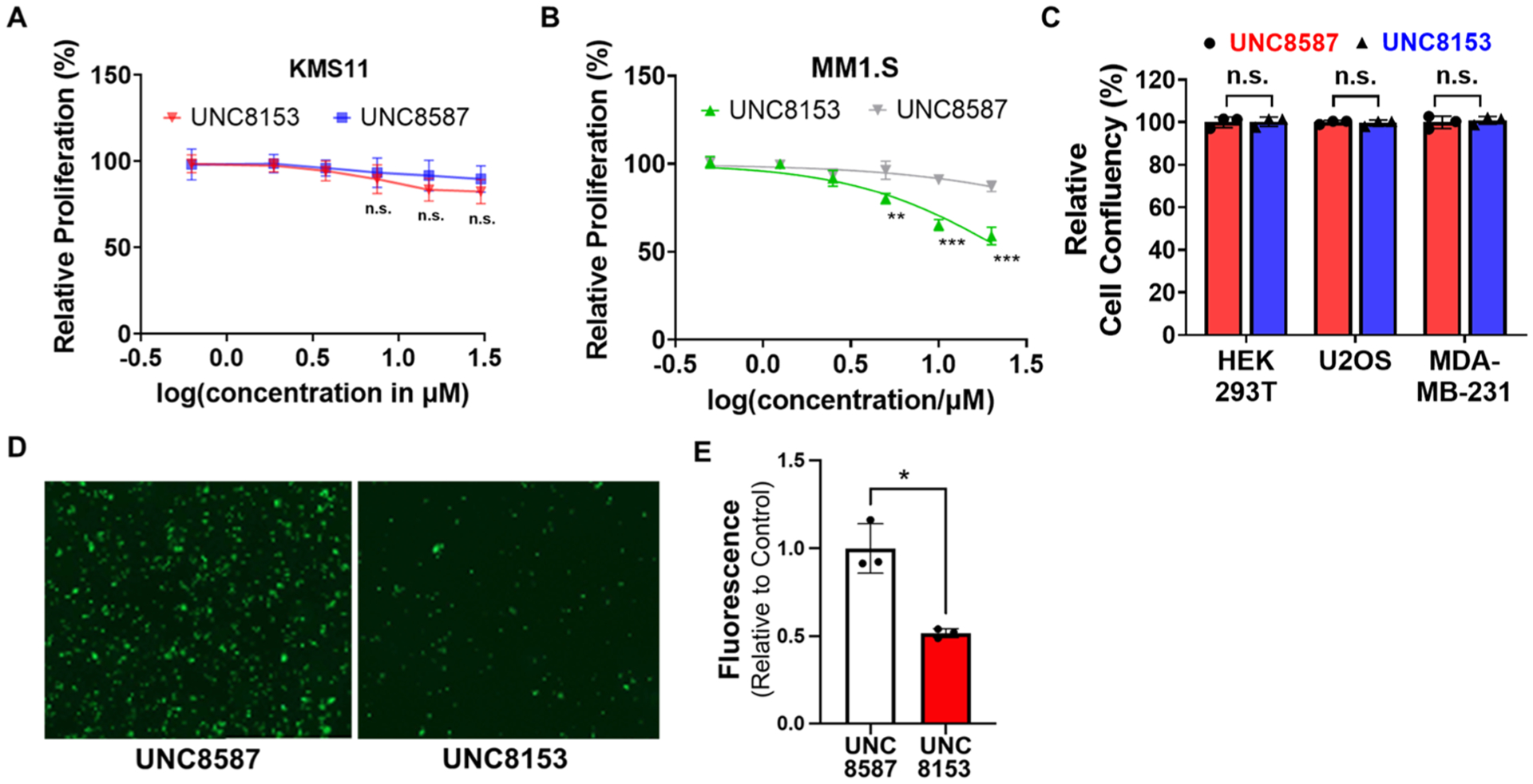
Degradation of NSD2 results in antitumor phenotypes. (A) KMS11 cells were treated for 8 days with UNC8153 or UNC8587 (control) at varying concentrations and quantified for cell viability by CellTiter-Glo. Data are reported as the mean of four independent experiments ± SD. Not significant: n.s. (B) MM1S cells were treated for 8 days with UNC8153 or UNC8587 (control) at varying concentrations and counted for cell viability after trypan blue staining. Data are reported as the mean of three replicates ± SD. **p < 0.01, ***p < 0.001 (one-tailed t-test). (C) Relative cell confluency of cells treated with 20 μM UNC8153 or UNC8587 (control) for 5 days. Data are represented as the endpoint mean confluency relative to control from three independent experiments ± SD. Not significant: n.s. (D) Representative microscopic images from adhesion assays with green fluorescent KMS11 cells treated with 25 μM UNC8153 or UNC8587 for 14 days. Cells were plated in Matrigel-coated plates for 12 h, and after washing the plates with PBS, the images were acquired at 10× magnification. (E) Fluorescence intensity of adherent cells treated with 25 μM UNC8153 or UNC8587. Data are represented as mean fluorescence intensities from three independent experiments relative to control ± SD. *p < 0.05 (one-tailed t-test).
Given numerous reports suggesting an important role of NSD2 in regulating MM cell adhesion,18,29 we performed an adhesion assay in KMS11 cells in the presence of UNC8153 or UNC8587 for 14 days. Significant antiadhesive effects were observed for UNC8153-treated cells compared to the control (UNC8587)-treated cells as shown by reduced cell attachment to Matrigel (Figure 6D,E). Similarly, this result was observed in UNC7753-treated cells, albeit with a slightly less pronounced effect (Figure S5I,J). Thus, UNC8153-mediated degradation of NSD2 phenocopies the reduced adhesion effects that have been observed upon NSD2 knockdown in MM cells.18
DISCUSSION
NSD2 has been a long sought-after drug target due to the fact that it is aberrantly expressed or mutated in numerous cancers; however, efforts to manipulate NSD2 activity and H3K36me2 levels via chemical approaches have been largely unsuccessful. Meng et al. recently reported the degradation of NSD2 via the recruitment of Cereblon, a well-established protein degradation mechanism; however, the resultant reduction of H3K36me2 was not demonstrated.30 Herein, we report the discovery of a novel NSD2-targeted degrader. UNC8153 potently binds to the PWWP1 domain of NSD2 in vitro and degrades both NSD2-short and NSD2-long with high potency and remarkable selectivity in a concentration- and time-dependent manner. Additionally, UNC8153 degrades NSD2 via a novel mechanism that is both proteasome- and neddylation-dependent. Our SAR efforts revealed that primary amine-containing compounds can promote potent compound-mediated degradation. Intrigued by the minimalistic nature of the primary amine pharmacophore, we demonstrated via a competition assay that UNC8153 appears to engage its binding partners in a bivalent fashion. We still have more to learn about the mechanistic details through which the primary amine handle recruits an E3 ligase, which will be critical to gauge the potential value of this approach. If broadly applicable, this small, minimalistic E3-recruitment handle has the potential to improve the properties of degrader molecules.
Epigenetic mechanisms are often context- and cell-type-dependent, and the phenotypic effects that we observe upon compound-mediated NSD2 degradation are consistent with this. While UNC8153 dramatically reduced the levels of NSD2 in both t(4;14)-translocated MM and NSD2 E1099K gain-of-function mutant cancer cell lines, the phenotypic consequences were different. Specifically, loss of NSD2 and subsequent H3K36me2 reduction is associated with mild antiproliferative effects in MM1.S cells but not KMS11 cells. These results are supported by data from Depmap (https://depmap.org/portal/) that show that NSD2 is not essential for the cell fitness of MM t(4;14) cell lines. Interestingly, Meng et al. reported strong antiproliferative effects upon treatment of KMS11 cells with their Cereblon-based degrader.30 This apparent discrepancy may be explained by the fact that their Cereblon-based NSD2 degrader also degrades IKZF1 and IKZF3, two transcription factors whose degradation is known to cause antiproliferative activity in MM.31
Multiple myeloma cells rely on interactions within the bone marrow microenvironment through a combination of cytokine and adhesion molecules that form a complex signaling network. These physical interactions can activate critical signaling responses, which regulate the survival, proliferation, migration, and sometimes drug resistance of MM cells. Thus, targeting the adhesion system in MM is an attractive therapeutic approach. Our data suggest that targeted degradation of NSD2 and subsequent global reduction of H3K36me2 can reduce the adherent properties of KMS11 cells. Thus, we anticipate that the chemical degraders described here will be useful tools for the biomedical community to investigate NSD2-mediated pathways in MM and their associated therapeutic potential. Overall, we present NSD2 degradation as a tractable and attractive strategy for future drug discovery efforts to enable the treatment of NSD2-dependent cancers.
METHODS
Surface Plasmon Resonance (SPR).
The binding affinity of compounds was assessed by surface plasmon resonance (SPR, Biacore T200, GE Health Sciences Inc.) as previously described.17
Cell Culture.
Cell lines were cultured in 5% CO2 at 37 °C in accordance with standard mammalian tissue culture protocols and tested for mycoplasma contamination via the MycoAlert Mycoplasma Detection kit (Lonza). U2OS, HEK293T, and MDA-MB-231 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) (Wisent) and 100 U/mL penicillin and 100 μg/mL streptomycin (Wisent). KMS11, RS411, and MM1S were cultured in RPMI supplemented with 10% fetal bovine serum (FBS) (Wisent) and 100 U/mL penicillin and 100 μg/mL streptomycin (Wisent).
In-Cell Western (ICW) Assay.
Cell Seeding and Treatment.
U2OS cells were seeded to achieve 70−80% confluency in Nunc MicroWell 96-Well Flat Clear Bottom Black Microplate (Thermo Fisher 165305) or Corning 384-well Flat Clear Bottom Black plates (Corning 3764). Upon cell attachment to the plate, the cells were treated by compounds diluted in medium to achieve the correct final concentration, for the prescribed amount of time (up to 24 h).
Fixation, Permeabilization, and Staining.
Upon completion of treatment, the medium was removed from all wells and was rinsed once gently with PBS. The cells were fixed in each well with 50 μL of 2% formaldehyde (Sigma F8775 diluted in 1× PBS) for 10 min at room temperature. The wells were washed three times with 100 μL of PBS in each well to remove the residual formaldehyde and media from the well. The cells were then permeabilize with 100 μL of 0.25% Triton X-100 in 1× PBS for 15 min at room temperature and was subsequently washed three times with 100 μL of PBS. The cells were then blocked by incubating all wells with 100 μL of 5% bovine serum albumin (BSA) in PBS-T for 1 h at room temperature, followed by NSD2 antibody staining by 50 μL of primary antibody (Abcam ab75359 diluted 1:1000 in 1% BSA in PBS-T) for 1 h at room temperature with agitation. At least two wells were intentionally not stained by the primary antibody to act as controls for secondary antibody background staining. Following primary antibody staining, the cells were washed three times again, then stained by secondary antibody IRDye 800CW (anti-mouse green) diluted 1:1000 and DRAQ5 (Cell Signaling #4084) diluted 1:2000 in 5% BSA diluted in PBS-T for 1 h at room temperature with agitation. The cells were lastly washed again three times by PBS and imaged on a LiCOR near-IR scanner, and the intensities of the 800CW and 700CW channels were quantified.
Data Analysis.
For each well, the NSD2 800CW intensity was normalized to DRAQ5 700CW intensity, and the no primary antibody control normalized intensity was subtracted from each normalized intensity. For each compound condition, the background-subtracted normalized intensity will be expressed as a percentage of the average background-subtracted normalized intensity of DMSO control-treated wells; this would be the “% NSD2 remaining.” For each set of dose−response, the % NSD2 remaining will be plotted against the log of concentration. The curves are fitted by [inhibitor] vs response nonlinear fit model in GraphPad PRISM 8 with the “Top” constrained = 100, “Bottom” constrained = lowest % NSD2 Remaining for the set of dilution series, and Hill slope constrained = −1. The DC50 was extracted from the “IC50” value calculated for the curve fitting, and the Dmax was extracted from lowest % NSD2 Remaining for the set of dilution series subtracted from 100%. For UNC7753 and UNC8153, three independent experiments, each containing 2−4 technical repeats, were analyzed and each individually curve fitted. For all other compounds, four technical repeats for each set of dose−response concentrations were analyzed from one experiment.
Cell Culture Compound Treatment Conditions for Immunoblotting NSD1–3.
5 × 105 cells were seeded in 1 mL of media per well in a 24-well plate and the compound was added to specific wells immediately (for MM1S cells) or upon cell attachment to the plate (for U2OS cells) to achieve specified concentrations. The cells were collected the next day and were subsequently lysed for immunoblotting.
Immunoblotting.
Cell pellets were lysed for 3 min at room temperature with lysis buffer containing 20 mM Tris-HCl pH 8, 150 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 0.5% Triton X-100, protease inhibitor (Roche 11873580001), and benzonase (Millipore Sigma E1014). After 3 min, SDS was added until a final concentration of 1%. Next, the mixtures were spun on a tabletop centrifuge at maximum RPM for 10 min, and the supernatants (cell lysates) were transferred to a new tube. The concentration of protein in each lysate was measured by a Thermo Scientific Pierce BCA Protein Assay Kit (Cat. # 23225) per supplier protocol. 10−30 μg of protein per loading was prepared and diluted by 6× sample loading buffer to a minimum 1× sample loading buffer concentration. Samples were loaded into Invitrogen NuPAGE Bis-Tris Gels. The gels were run in 1× NuPAGE MOPS buffer (Invitrogen NP0001) for 90 min at 120 V. The gel was subsequently transferred to a PVDF membrane in transfer solution containing 3 g/L Tris, 14.4 g/L glycine, and 10% methanol diluted by ddH2O. Gels were transferred by Invitrogen XCell II Blot Module (EI9051) at 40 V/500 mA, for 2 h, or by Bio-Rad Mini Trans-Blot Cell, at 70 V/500 mA, for 1.5 h, per manufacturer protocol. Membranes were blocked in 5% BSA for 1 h at room temperature, then blotted overnight by primary antibodies at a working concentration diluted by 5% BSA. Upon primary antibody blotting, the membrane was washed for 5 min by PBS-T three times and subsequently blotted by secondary antibodies for 1 h at room temperature and washed for 5 min by PBS-T three times. See Table S1 for the antibodies used.
siRNA Knockdown.
1 × 104 cells were seeded per 90 μL of media per well in a NUNC MicroWell 96-Well Flat Clear Bottom Black Microplate (Thermo Fisher 165305), for ICW, or 1 × 105 per mL media per well in a 24-well tissue culture plate. The cells were transfected at seeding by Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher 13778150), per manufacturer protocol. For every 8 wells in 96-well plates, or 2 wells in 24-well plates (proportionally scaled up or down accordingly): 50 μL of Opti-MEM Medium (Gibco 31985070) was mixed with 3 μL of RNAiMAX reagent and 50 μL of Opti-MEM was mixed with 1 μL of 10 μM siRNA, separately. The two mixtures were then mixed to form a 100 μL mixture and incubated at room temperature for 5 min to allow the formation of siRNA−lipid complexes. The siRNA−lipid complex mixture was added to the wells at 10 μL/well for 96-well plates, or 50 μL per well for 24-well plates. For ICW experiments, the cells were incubated for at least 24 h post-transfection before any compound treatment. For UBR siRNA knockdown ICW, cells in 96-well plates were treated in dose−response by 25 μL of 5× concentrated UNC7753 diluted in media 24 h post-transfection, for a treatment duration of 24 h (end at 48 h post-transfection). For UBR immunoblotting samples, cells in 24-well plates were treated 24 h post-transfection by either 1 μL of DMSO or 20 mM UNC8153 diluted in DMSO to achieve 20 μM final UNC8153 concentration. For qRT-PCR validation of UBR-1 knockdown, cells in 24-well plates were collected 48 h post-transfection without treatment. See Table S1 for siRNAs used.
qRT-PCR for UBR-1 Knockdown Validation.
Total RNA was isolated using QIAGEN RNeasy Mini RNA Extraction Kit (QIAGEN 74106). 1 μg of RNA was used to synthesize cDNA using the iScript gDNA Clear cDNA Synthesis Kit (Bio-Rad) in 20 μL volume of the reaction mixture. Upon completion, 180 μL of ddH2O was added (10× dilution). RT-qPCR was performed using the PowerUp SYBR Green Mastermix (Applied Biosystems) on 2 μL of the synthesized cDNA per reaction. The relative abundance of transcripts was normalized to α-tubulin and assessed via the 2−ΔΔCT method. Two biological replicates were collected and analyzed for control siRNA and UBR-1 siRNA. See Table S1 for the primers used.
Cellular Metabolism.
Cell Seeding and Treatment.
For each replicate, 2 mL of U2OS cells were seeded into 6-well plates at a density of 3.0 × 105 cells/mL and incubated at 37 °C for 24 h. The media was aspirated from each well and replaced with 2 mL of fresh media containing 10 μM UNC7753 and 0.1% DMSO. The plates were then incubated at 37 °C for a predetermined incubation time at which point media and pellet were collected. Biological triplicates were done for each time point.
Pellet Collection.
After the prescribed incubation time, for each replicate, media was removed and the wells were rinsed with an additional 0.5 mL of media. The wells were subsequently washed with 1 mL of cold PBS, which was then aspirated and discarded. To each well, 0.5 mL of versene was added, and the plate was incubated at 37 °C for 5 min. The versene was quenched with 1 mL of media. Cells were scraped into a corner and the suspension transferred to a microfuge tube and centrifuged at 1000 rpm for 5 min. The supernatant was aspirated, and the pellet was resuspended in 1 mL of cold PBS by pipetting up and down 30 times. The suspension was again centrifuged at 1000 rpm for 5 min and the supernatant was again aspirated. The cell pellets were then snap frozen in liquid nitrogen.
Sample Preparation.
After the addition of 60 μL of methanol/water 1:1 (v/v) in each cell pellet, cell pellet samples were homogenized. 20 μL of the cell pellet homogenate was pipetted out for analysis. After the addition of 10 μL of DMSO/acetonitrile 1:1 (v/v), 20 μL of 200 ng/mL propranolol in methanol/water 1:1 (v/v) as internal standard (IS), and 200 μL of chilled acetonitrile, cell pellet samples were vortexed and centrifuged at 3500 rpm for 30 min. 0.5 μL of the supernatant was injected onto an AB Sciex Triple Quad 5500 LC-MS/MS system coupled with a Shimadzu Prominence HPLC for analysis.
LC-MS/MS Analysis.
LC separation was performed on a Agilent Zorbax Extended-C18 column (5 μm, 2.1 × 50 mm2) with 0.1% acetic acid 1 mM ammonium acetate in acetonitrile/water 1:9 (v/v) as mobile phase A and 50 mM acetic acid in acetonitrile as mobile phase B. A gradient elution at 0.75 mL/min started with 0% B. The B component was increased linearly to 95% in 1 min. After holding at 95% B for 0.5 min, the column was reequilibrated with 0% B for 0.5 min. Mass spectrometric detection was performed with TurboSpray ionization in positive ion mode. The multiple reaction monitoring transitions for UNC7753, UNC8153, and the Internal Standard (propranolol) are 740.3 → 176, 584.2 → 176.2, and 260.15 → 116.1, respectively. The calibration ranges for the cell pellet and medium sample analysis are 0.5−1500 ng/mL cell pellet homogenate and 0.2−600 ng/mL medium, respectively.
UNC6934 and UNC8603 Competition Assay.
1 × 104 U2OS cells per 100 μL per well were seeded in Nunc MicroWell 96-Well Flat Clear Bottom Black Microplate (Thermo Fisher 165305). A dose−response series of 5× concentrated UNC6934 or UNC8603 was prepared in a medium containing 10 μM UNC8153. 25 μL of the 5× compound-containing mixture was added to each well, reaching 1× concentration and 2 μM UNC8153. The plate was incubated for 6 h at 37 °C before being fixed for ICW (see the ICW protocol above).
NanoBRET Ubiquitination Assay.
100 μL of resuspended U2OS cells at 4 × 105 cells/mL were seeded per well in 96-well white opaque bottom plates to allow for cell attachment (2−4 h). A mastermix of NanoLuc and HT plasmids was prepared by mixing 0.01 ng of NSD2-PWWP1 NanoLuc plasmid (as previously used in Dilworth et al.), 0.3 ng of HaloTag-Ubiquitin plasmid (Promega N272A), and 0.69 ng of empty vector pcDNA to each 100 μL of opti-MEM (scaled up accordingly). XtremeGene HP (Millipore Sigma 6366236001) was subsequently added to the mastermix at 1 μL per 50 μL of mastermix. The mixture was incubated at room temperature for 15 min before being added to the wells at 10 μL per well and allowed to incubate overnight in a cell culture incubator. The next day, the medium was removed from each well and 20 μL of 1:1000 (diluted in phenyl red free DMEM with 4% FBS) 618 ligand was added to each well. This was followed by the addition of 20 μL of 2× concentration (diluted in phenyl red free DMEM with 4% FBS) of compound (or DMSO control) treatment to give 1× treatment at 40 μL total mixture in the wells. At least two wells were reserved for no ligand control (40 μL of phenyl red free DMEM only). The cells were placed back in the incubator and incubated for the prescribed time (0.5 or 2 h). At the endpoint, 10 μL of 1:100 (diluted in phenyl red free DMEM with 4% FBS) of NanoBRET Nano-Glo substrate (Promega N1572) was added in each well and immediately scanned (with orbital mixing) on a Clariostar plate reader (BMGlabtech). The data collection and subsequent background subtraction follows the Promega technical manual (Literature # TM616). The background-subtracted (adjusted) BRET units were normalized to DMSO treatment in the same experiment run in parallel (set = 1.00).
MG132, MLN4924, and TAK243 Inhibitor Assay.
1 × 104 U2OS cells per 100 μL per well were seeded in a Nunc MicroWell 96-Well Flat Clear Bottom Black Microplate (Thermo Fisher 165305). A dose−response series of 5× concentrated UNC8153 was prepared in a medium containing 50 μM MG132 (Abcam ab141003), 5 μM MLN4924 (Cayman Chemical 15217), or 5 μM TAK243 (Active Biochem A-1384). 25 μL of the 5× compound-containing mixture was added to each well, reaching a final concentration of 1× UNC8153 and 10 μM MG132, 1 μM MLN4924, or 1 μM TAK243. The plate was incubated for 6 h at 37 °C before being fixed for ICW (see the ICW protocol above).
Global Proteomics.
Sample Preparation for Proteomics Analysis.
For each replicate, 10 mL of U2OS cells were seeded into 15 cm plates in triplicate at a density of 2.5 × 105 cells/mL and incubated at 37 °C for 24 h. To each plate was then added 2.5 mL of media containing 25 μM test compound and 1.25% DMSO, to a final concentration of 5 μM test compound and 0.25% DMSO. The plates were then incubated at 37 °C for 6 h. Each plate was washed three times with 5 mL of ice-cold DPBS (Gibco), then 1 mL of ice-cold DPBS was added and the cells were scraped into a prechilled microfuge tube and centrifuged at 14 000 rpm for 5 min at 4 °C. The supernatant was aspirated, and the pellet was resuspended in 200 μL of the lysis buffer (0.05M Tris pH 8, 8 M urea, 1× Roche PhosSTOP phosphatase inhibitor cocktail, 1× ActiveMotif protease inhibitor) by pipetting up and down 30 times. Each lysate was vortexed for 30 s, incubated on ice for 30 min, vortexed again for 30 s, and incubated a further 30 min on ice before centrifugation at 14 000 rpm for 15 min at 4 °C. The supernatants were transferred into clean, prechilled microfuge tubes and the protein concentration determined by the Bradford assay. The samples were then snap-frozen in liquid nitrogen. Each set of replicates was processed in parallel and staggered by 30 min from the next set, to minimize variability due to cell cycle differences. Protein lysates (n = 3) were reduced with 5 mM DTT at 56 °C for 30 min, then alkylated with 15 mM iodoacetamide at room temperature in the dark for 45 min. The samples were acetone precipitated, then reconstituted in 1 M urea, and subjected to digestion with LysC (Wako) for 2 h and trypsin (Promega) overnight at 37 °C at a 1:50 enzyme/protein ratio. The resulting peptide samples were acidified, desalted using Thermo desalting spin columns, and then the eluates were dried via vacuum centrifugation. The peptide concentration was determined using Pierce Quantitative Colorimetric Peptide Assay. 40 μg of each sample was reconstituted with 50 mM HEPES pH 8.5, then individually labeled with 150 μg TMT 10plex reagent (Thermo Fisher) for 1 h at room temperature. Prior to quenching, the labeling efficiency was evaluated by LC-MS/MS analysis of a pooled sample consisting of 1 μL of each sample. After confirming >99% efficiency, the samples were quenched with 50% hydroxylamine to a final concentration of 0.4%. The labeled peptide samples were combined, desalted using Thermo desalting spin column, and dried via vacuum centrifugation. The dried TMT-labeled samples were fractionated using high-pH reverse-phase HPLC.32 Briefly, the samples were offline fractionated over a 96 min run, into 96 fractions by high-pH reverse-phase HPLC (Agilent 1260) using an Agilent Zorbax 300 Extend-C18 column (3.5 μm, 4.6 × 250 mm2) with mobile phase A containing 4.5 mM ammonium formate (pH 10) in 2% (vol/vol) LC-MS-grade acetonitrile, and the mobile phase B containing 4.5 mM ammonium formate (pH 10) in 90% (vol/vol) LC-MS-grade acetonitrile. The 96 resulting fractions were then concatenated in a noncontinuous manner into 24 fractions and then dried via vacuum centrifugation.
LC/MS/MS Analysis.
The 24 fractions were analyzed by LC/MS/MS using an Ultimate 3000 nLC coupled to an Orbitrap Exploris480 mass spectrometer (Thermo Scientific) using a turboTMT method.33 Samples were injected onto an Ion Opticks Aurora C18 column (75 μm id × 15 cm, 1.6 μm particle size) and separated over a 70 min method. The gradient for separation consisted of 5−42% mobile phase B at a 250 nl/min flow rate, where mobile phase A was 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in 80% ACN. The Exploris480 was operated in turboTMT mode with a cycle time of 3 s. Resolution for the precursor scan (m/z 375−1400) was set to 60 000 with a AGC target set to standard and a maximum injection time set to auto. MS2 scans (30 000 resolution) consisted of higher collision dissociate (HCD) set to 38, AGC target set to 300%, maximum injection time set to auto, isolation window of 0.7 Da, and fixed first mass of 110 m/z.
Data Analysis.
Raw data files were processed using Proteome Discoverer version 2.5, set to “reporter ion MS2” with “10plex TMT.” Peak lists were searched against a reviewed Uniprot human database (downloaded Feb 2020 containing 20 350 sequences), appended with a common contaminants database, using Sequest HT within Proteome Discoverer. Data were searched with up to two missed trypsin cleavage sites; fixed modifications: TMT6plex peptide N-terminus and Lys, carbamidomethylation Cys, dynamic modification: N-terminal protein acetyl, oxidation Met. Precursor mass tolerance was 10 ppm and fragment mass tolerance was 0.02 Da. Peptide false discovery rate was set to 1%. Reporter abundance based on intensity, coisolation threshold set to 50, and proteins identified by >1 razor and unique peptides were used for quantitation. Data were further analyzed in Argonaut for statistical analysis and visualization.34 GraphPad was used to generate the volcano plots. Proteins with −log p-value > 2 and log 2 fold change < −0.5 were considered significant.
KMS11 Time-Course Treatments for Immunoblotting.
1.2 × 105 cells were seeded in 1 mL of media per well in a 24-well plate and compound treatments were done to specific wells every 2 days, until day 6, allowing for a time-course treatment with 2-day intervals. The cells were collected on day 8 and were subsequently lysed for immunoblotting.
KMS11 Dose−Response Treatments for Immunoblotting.
1.5 × 105 cells were seeded in 1 mL of media per well in a 24-well plate and compound treatments were done to specific wells immediately achieving specified concentrations. Cells were collected on day 6 and were subsequently lysed for immunoblotting.
KMS11 CellTiter-Glo Luminescence Cell Viability Assay.
1.5 × 103 KMS11 cells were seeded per well in 40 μL of media in a 384-well plate. Cells were immediately treated with 20 μL of 3× compound solutions diluted in media, making 1× compound 60 μL culturing conditions, and then incubated for 8 days. At the endpoint, 25 μL of Promega CellTiter-Glo reagent (Promega G7572) was added per well. The plates were mixed 2 min on an orbital shaker gently, then incubated for 20 min at room temperature while protected from light. The luminescence signal intensity of each well was subsequently scanned on a CLARIOstar plate reader (BMGlabtech).
MM1S Dose−Response Treatment and Proliferation Assay.
3.0 × 105 cells per well were seeded in triplicate into 24-well plates, subjected to treatment with various final concentrations of the compound. Fresh medium containing the compound was changed every 2 days. All flow-growing cells were periodically diluted to keep the cell density less than 1 million per mL. Cell numbers were counted by an automated TC10 cell counter (Bio-Rad).
CRISPR/cas9-Mediated Gene Depletion.
The sgRNAs targeting NSD2 were cloned into a pLenti LRG-2.1 Neo vector (Addgene 125593). A lentiviral plasmid that allows the doxycycline-inducible expression of SpCas9 was obtained from Dr. David Sabatini. The sgRNA sequences for targeting NSD2 are sgRNA #3 (5′-GCGGGCGAGTTCTGCCAGAA-3′) and sgRNA #5 (5′-CGGGCGAGTTCTGCCAGAAC-3′). All plasmid sequences were verified by sequencing.
U2OS/HEK293T/MDA-MB-231 Proliferation Assays and Immunoblotting.
1.0 × 105 of U2OS or MDA-MB-231 cells or 0.5 × 105 of HEK293T cells were seeded in triplicates in 2 mL of media per well in a 12-well plate (for 5-day proliferation and immunoblotting studies) and were subject to treatment with various final concentrations of compounds upon cell attachment to the plate (at minimum 6 h later). Plates from one independent experiment were immediately placed in IncuCyte ZOOM for periodic confluency measurements every 12 h. Plates from two independent experiments were imaged on IncuCyte ZOOM for cell confluency at the endpoint. Cells were collected at the endpoint and were lysed for immunoblotting.
Adhesion Assay.
Cells were transduced with Incucyte Nuclight Green Lentivirus (Sartorius) and selected with puromycin to generate green fluorescent stable cell lines. Cells were treated with control or NSD2 degrader compounds (25 μM) for 14 days and plated in Matrigel-coated 12-well plates (250 000 cells/well) for 12 h after which media and suspended cells were aspirated. The wells were washed with PBS and images of fluorescent adherent cells were taken at 10× magnification on an EVOS imaging system (Thermo Fisher). To quantify fluorescence in each well, plates were scanned on a Clariostar Plus plate reader (BMGlabtech).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the members of the James laboratory, as well as Stephen Frye and Jon Collins for helpful discussions and input throughout the project. The authors thank Dmitri Kireev and Tristan Kenney for assistance with figure generation. This work is supported by grants from the NIH to L.I.J. (R01CA242305), the Canadian Institutes of Health Research (FDN154328) and the Princess Margaret Cancer Foundation to C.H.A., the Leukemia and Lymphoma Society Specialized Center of Research (NIH R01) to J.D.L., a Leukemia and Lymphoma Society Special Fellow Award to A.S., and the Structural Genomics Consortium, a registered charity (No. 1097737) that receives funds from Bayer AG, Boehringer Ingelheim, Bristol Myers Squibb, Genentech, Genome Canada through Ontario Genomics Institute [OGI-196], EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking [EUbOPEN Grant 875510], Janssen, Merck KGaA (aka EMD in Canada and US), Pfizer and Takeda. D.Y.N. is supported by a Doctoral Training Scholarship from Fonds de recherche du Québec—Santé (320128). D.B-L. is supported by CRS grant 25418. This research is based in part upon work conducted using the UNC Proteomics Core Facility, which is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. This material is based in part upon work supported by the National Science Foundation under Grant No. CHE-1726291. The authors thank the University of North Carolina’s Department of Chemistry Mass Spectrometry Core Laboratory for their assistance with mass spectrometry analysis. G.G.W. is an American Cancer Society (ACS) Research Scholar, a Leukemia and Lymphoma Society (LLS) Scholar.
R.P.H., J.R.T., and L.I.J. report patents from the University of North Carolina. The James lab receives funding from Pinnacle Hill and the Licht lab receives funding from Epizyme. J.D.L. is a consultant for AstraZeneca.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c01421.
Raw SPR data and curve fitting, ICW dose−response curves, ICW plate scans, full western blot images, general chemistry procedures, synthesis schemes and methods, NMR spectra, and LC-MS spectra (Figures S1−S5 and Tables S1 and S2) (PDF)
Raw proteomics data (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.3c01421
The authors declare no competing financial interest.
Contributor Information
Ronan P. Hanley, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
David Y. Nie, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada; Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario M5G 1L7, Canada; Department of Medical Biophysics, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
John R. Tabor, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Fengling Li, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
Amin Sobh, University of Florida Health Cancer Center, Gainesville, Florida 32610, United States.
Chenxi Xu, Lineberger Comprehensive Cancer Center and Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, North Carolina 27599, United States.
Natalie K. Barker, UNC Proteomics Core Facility, Department of Pharmacology, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
David Dilworth, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
Taraneh Hajian, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
Elisa Gibson, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
Magdalena M. Szewczyk, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada
Peter J. Brown, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada
Dalia Barsyte-Lovejoy, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada; Department of Pharmacology and Toxicology, University of Toronto, Toronto, Ontario M5S 1A8, Canada.
Laura E. Herring, UNC Proteomics Core Facility, Department of Pharmacology, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Gang Greg Wang, Lineberger Comprehensive Cancer Center, Department of Biochemistry and Biophysics, and Department of Pharmacology, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, North Carolina 27599, United States.
Jonathan D. Licht, University of Florida Health Cancer Center, Gainesville, Florida 32610, United States
Masoud Vedadi, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada; Department of Pharmacology and Toxicology, University of Toronto, Toronto, Ontario M5S 1A8, Canada.
Cheryl H. Arrowsmith, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada; Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario M5G 1L7, Canada; Department of Medical Biophysics, University of Toronto, Toronto, Ontario M5G 1L7, Canada
Lindsey I. James, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States; Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, North Carolina 27599, United States
REFERENCES
- (1).Tost J 10 Years of Epigenomics: A Journey with the Epigenetic Community through Exciting Times. Epigenomics 2020, 12, 81–85. [DOI] [PubMed] [Google Scholar]
- (2).Bates SE Epigenetic Therapies for Cancer. N. Engl. J. Med 2020, 383, 650–663. [DOI] [PubMed] [Google Scholar]
- (3).Hyun K; Jeon J; Park K; Kim J Writing, Erasing and Reading Histone Lysine Methylations. Exp. Mol. Med 2017, 49, No. e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Husmann D; Gozani O Histone Lysine Methyltransferases in Biology and Disease. Nat. Struct. Mol. Biol 2019, 26, 880–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Vougiouklakis T; Bernard BJ; Nigam N; Burkitt K; Nakamura Y; Saloura V Clinicopathologic Significance of Protein Lysine Methyltransferases in Cancer. Clin. Epigenet 2020, 12, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bhat KP;Ümit Kaniskan H; Jin J; Gozani O Epigenetics and beyond: Targeting Writers of Protein Lysine Methylation to Treat Disease. Nat. Rev. Drug Discovery 2021, 20, 265–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kuo AJ; Cheung P; Chen K; Zee BM; Kioi M; Lauring J; Xi Y; Park BH; Shi X; Garcia BA; Li W; Gozani O NSD2 Links Dimethylation of Histone H3 at Lysine 36 to Oncogenic Programming. Mol. Cell 2011, 44, 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bennett RL; Swaroop A; Troche C; Licht JD The Role of Nuclear Receptor−Binding SET Domain Family Histone Lysine Methyltransferases in Cancer. Cold Spring Harb. Perspect. Med 2017, 7, No. a026708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jaffe JD; Wang Y; Chan HM; Zhang J; Huether R; Kryukov Gv.; Bhang HC; Taylor JE; Hu M; Englund NP; Yan F; Wang Z; Robert McDonald E; Wei L; Ma J; Easton J; Yu Z; deBeaumount R; Gibaja V; Venkatesan K; Schlegel R; Sellers WR; Keen N; Liu J; Caponigro G; Barretina J; Cooke VG; Mullighan C; Carr SA; Downing JR; Garraway LA; Stegmeier F Global Chromatin Profiling Reveals NSD2 Mutations in Pediatric Acute Lymphoblastic Leukemia. Nat. Genet 2013, 45, 1386–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sengupta D; Zeng L; Li Y; Hausmann S; Ghosh D; Yuan G; Nguyen TN; Lyu R; Caporicci M; Morales Benitez A; Coles GL; Kharchenko V; Czaban I; Azhibek D; Fischle W; Jaremko M; Wistuba II; Sage J; Jaremko Ł; Li W; Mazur PK; Gozani O NSD2 Dimethylation at H3K36 Promotes Lung Adenocarcinoma Pathogenesis. Mol. Cell 2021, 81, 4481–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yuan S; Natesan R; Sanchez-Rivera FJ; Li J; Bhanu Nv; Yamazoe T; Lin JH; Merrell AJ; Sela Y; Thomas SK; Jiang Y; Plesset JB; Miller EM; Shi J; Garcia BA; Lowe SW; Asangani IA; Stanger BZ Global Regulation of the Histone Mark H3K36me2 Underlies Epithelial Plasticity and Metastatic Progression. Cancer Discovery 2020, 10, 854–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Popovic R; Martinez-Garcia E; Giannopoulou EG; Zhang Q; Zhang Q; Ezponda T; Shah MY; Zheng Y; Will CM; Small EC; Hua Y; Bulic M; Jiang Y; Carrara M; Calogero RA; Kath WL; Kelleher NL; Wang J-P; Elemento O; Licht JD Histone Methyltransferase MMSET/NSD2 Alters EZH2 Binding and Reprograms the Myeloma Epigenome through Global and Focal Changes in H3K36 and H3K27 Methylation. PLoS Genet 2014, 10, No. e1004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Huang Z; Wu H; Chuai S; Xu F; Yan F; Englund N; Wang Z; Zhang H; Fang M; Wang Y; Gu J; Zhang M; Yang T; Zhao K; Yu Y; Dai J; Yi W; Zhou S; Li Q; Wu J; Liu J; Wu X; Chan H; Lu C; Atadja P; Li E; Wang Y; Hu M NSD2 Is Recruited through Its PHD Domain to Oncogenic Gene Loci to Drive Multiple Myeloma. Cancer Res 2013, 73, 6277–6288. [DOI] [PubMed] [Google Scholar]
- (14).Coussens NP; Kales SC; Henderson MJ; Lee OW; Horiuchi KY; Wang Y; Chen Q; Kuznetsova E; Wu J; Chakka S; Cheff DM; Cheng KC-C; Shinn P; Brimacombe KR; Shen M; Simeonov A; Lal-Nag M; Ma H; Jadhav A; Hall MD High-Throughput Screening with Nucleosome Substrate Identifies Small-Molecule Inhibitors of the Human Histone Lysine Methyltransferase NSD2. J. Biol. Chem 2018, 293, 13750–13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Shen Y; Morishita M; Lee D; Kim S; Lee T; Mevius DEHF; Roh Y; di Luccio E Identification of LEM-14 Inhibitor of the Oncoprotein NSD2. Biochem. Biophys. Res. Commun 2019, 508, 102–108. [DOI] [PubMed] [Google Scholar]
- (16).De Freitas RF; Liu Y; Szewczyk MM; Mehta N; Li F; McLeod D; Zepeda-Velázquez C; Dilworth D; Hanley RP; Gibson E; Brown PJ; Al-Awar R; James LI; Arrowsmith CH; Barsyte-Lovejoy D; Min J; Vedadi M; Schapira M; Allali-Hassani A Discovery of Small-Molecule Antagonists of the PWWP Domain of NSD2. J. Med. Chem 2021, 64, 1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Dilworth D; Hanley RP; Ferreira de Freitas R; Allali-Hassani A; Zhou M; Mehta N; Marunde MR; Ackloo S; Carvalho Machado RA; Khalili Yazdi A; Owens DDG; Vu V; Nie DY; Alqazzaz M; Marcon E; Li F; Chau I; Bolotokova A; Qin S; Lei M; Liu Y; Szewczyk MM; Dong A; Kazemzadeh S; Abramyan T; Popova IK; Hall NW; Meiners MJ; Cheek MA; Gibson E; Kireev D; Greenblatt JF; Keogh M-C; Min J; Brown PJ; Vedadi M; Arrowsmith CH; Barsyte-Lovejoy D; James LI; Schapira M A Chemical Probe Targeting the PWWP Domain Alters NSD2 Nucleolar Localization. Nat. Chem. Biol 2022, 18, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lauring J; Abukhdeir AM; Konishi H; Garay JP; Gustin JP; Wang Q; Arceci RJ; Matsui W; Park BH The Multiple Myeloma−Associated MMSET Gene Contributes to Cellular Adhesion, Clonogenic Growth, and Tumorigenicity. Blood 2008, 111, 856–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Békés M; Langley DR; Crews CM PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discovery 2022, 21, 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ryan A; Liu J; Deiters A Targeted Protein Degradation through Fast Optogenetic Activation and Its Application to the Control of Cell Signaling. J. Am. Chem. Soc 2021, 143, 9222–9229. [DOI] [PubMed] [Google Scholar]
- (21).Matta-Camacho E; Kozlov G; Li FF; Gehring K Structural Basis of Substrate Recognition and Specificity in the N-End Rule Pathway. Nat. Struct. Mol. Biol 2010, 17, 1182–1187. [DOI] [PubMed] [Google Scholar]
- (22).Bachmair A; Finley D; Varshavsky A In Vivo Half-Life of a Protein Is a Function of Its Amino-Terminal Residue. Science 1986, 234, 179–186. [DOI] [PubMed] [Google Scholar]
- (23).Shanmugasundaram K; Shao P; Chen H; Campos B; McHardy SF; Luo T; Rao H A Modular PROTAC Design for Target Destruction Using a Degradation Signal Based on a Single Amino Acid. J. Biol. Chem 2019, 294, 15172–15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Varshavsky A Discovery of Cellular Regulation by Protein Degradation. J. Biol. Chem 2008, 283, 34469–34489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kisselev AF; Goldberg AL Proteasome Inhibitors: From Research Tools to Drug Candidates. Chem. Biol 2001, 8, 739–758. [DOI] [PubMed] [Google Scholar]
- (26).Hyer ML; Milhollen MA; Ciavarri J; Fleming P; Traore T; Sappal D; Huck J; Shi J; Gavin J; Brownell J; Yang Y; Stringer B; Griffin R; Bruzzese F; Soucy T; Duffy J; Rabino C; Riceberg J; Hoar K; Lublinsky A; Menon S; Sintchak M; Bump N; Pulukuri SM; Langston S; Tirrell S; Kuranda M; Veiby P; Newcomb J; Li P; Wu JT; Powe J; Dick LR; Greenspan P; Galvin K; Manfredi M; Claiborne C; Amidon BS; Bence NF A Small-Molecule Inhibitor of the Ubiquitin Activating Enzyme for Cancer Treatment. Nat. Med 2018, 24, 186–193. [DOI] [PubMed] [Google Scholar]
- (27).Soucy TA; Smith PG; Milhollen MA; Berger AJ; Gavin JM; Adhikari S; Brownell JE; Burke KE; Cardin DP; Critchley S; Cullis CA; Doucette A; Garnsey JJ; Gaulin JL; Gershman RE; Lublinsky AR; McDonald A; Mizutani H; Narayanan U; Olhava EJ; Peluso S; Rezaei M; Sintchak MD; Talreja T; Thomas MP; Traore T; Vyskocil S; Weatherhead GS; Yu J; Zhang J; Dick LR; Claiborne CF; Rolfe M; Bolen JB; Langston SP An Inhibitor of NEDD8-Activating Enzyme as a New Approach to Treat Cancer. Nature 2009, 458, 732–736. [DOI] [PubMed] [Google Scholar]
- (28).Oyer JA; Huang X; Zheng Y; Shim J; Ezponda T; Carpenter Z; Allegretta M; Okot-Kotber CI; Patel JP; Melnick A; Levine RL; Ferrando A; MacKerell AD; Kelleher NL; Licht JD; Popovic R Point Mutation E1099K in MMSET/NSD2 Enhances Its Methyltranferase Activity and Leads to Altered Global Chromatin Methylation in Lymphoid Malignancies. Leukemia 2014, 28, 198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chen J; Li N; Yin Y; Zheng N; Min M; Lin B; Zhang L; Long X; Zhang Y; Cai Z; Zhai S; Qin J; Wang X Methyltransferase Nsd2 Ensures Germinal Center Selection by Promoting Adhesive Interactions between B Cells and Follicular Dendritic Cells. Cell Rep 2018, 25, 3393–3404. [DOI] [PubMed] [Google Scholar]
- (30).Meng F; Xu C; Park K-S; Kaniskan HÜ; Wang GG; Jin J Discovery of a First-in-Class Degrader for Nuclear Receptor Binding SET Domain Protein 2 (NSD2) and Ikaros/Aiolos. J. Med. Chem 2022, 65, 10611–10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Cippitelli M; Stabile H; Kosta A; Petillo S; Gismondi A; Santoni A; Fionda C Role of Aiolos and Ikaros in the Antitumor and Immunomodulatory Activity of IMiDs in Multiple Myeloma: Better to Lose Than to Find Them. Int. J. Mol. Sci 2021, 22, No. 1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Mertins P; Tang LC; Krug K; Clark DJ; Gritsenko MA; Chen L; Clauser KR; Clauss TR; Shah P; Gillette MA; Petyuk VA; Thomas SN; Mani DR; Mundt F; Moore RJ; Hu Y; Zhao R; Schnaubelt M; Keshishian H; Monroe ME; Zhang Z; Udeshi ND; Mani D; Davies SR; Townsend RR; Chan DW; Smith RD; Zhang H; Liu T; Carr SA Reproducible Workflow for Multiplexed Deep-Scale Proteome and Phosphoproteome Analysis of Tumor Tissues by Liquid Chromatography−Mass Spectrometry. Nat. Protoc 2018, 13, 1632–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).McAlister GC; Nusinow DP; Jedrychowski MP; Wühr M; Huttlin EL; Erickson BK; Rad R; Haas W; Gygi SP MultiNotch MS3 Enables Accurate, Sensitive, and Multiplexed Detection of Differential Expression across Cancer Cell Line Proteomes. Anal. Chem 2014, 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Brademan DR; Miller IJ; Kwiecien NW; Pagliarini DJ; Westphall MS; Coon JJ; Shishkova E Argonaut: A Web Platform for Collaborative Multi-Omic Data Visualization and Exploration. Patterns 2020, 1, No. 100122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.