

Abstract
Increased activity of the lysine methyltransferase NSD2 driven by translocation and activating mutations is associated with multiple myeloma and acute lymphoblastic leukemia, but no NSD2-targeting chemical probe has been reported to date. Here, we present the first antagonists that block the protein–protein interaction between the N-terminal PWWP domain of NSD2 and H3K36me2. Using virtual screening and experimental validation, we identified the small-molecule antagonist 3f, which binds to the NSD2-PWWP1 domain with a Kd of 3.4 μM and abrogates histone H3K36me2 binding to the PWWP1 domain in cells. This study establishes an alternative approach to targeting NSD2 and provides a small-molecule antagonist that can be further optimized into a chemical probe to better understand the cellular function of this protein.
Graphical Abstract
INTRODUCTION
NSD2 (nuclear receptor-binding SET domain-containing 2, also known as WHSC1 and MMSET) is a protein lysine methyltransferase that belongs to the NSD family, which also includes NSD1 and NSD3, and that predominantly mono- and dimethylates lysine 36 of histone 3 (H3K36).1 NSD2 is an oncoprotein that is aberrantly expressed, amplified, or somatically mutated in multiple types of cancer.2 Notably, the t(4;14) NSD2 translocation in multiple myeloma and the hyperactivating NSD2 mutation E1099K in a subset of pediatric acute lymphoblastic leukemia result in altered chromatin methylation that drives oncogenesis.3–5
While NSD2 is an attractive therapeutic target, efforts to target the catalytic SET domain with small-molecule inhibitors have so far met with little success,6–9 and only recently was the first selective inhibitor of an NSD family protein reported, a compound that binds covalently to the catalytic site of NSD1.10 Aside from the catalytic domain, NSD2 has multiple protein–protein interaction domains that may be clinically relevant, including plant homeodomain and PWWP (proline–tryptophan–tryptophan–proline) domains (Figure 1).11,12 The N-terminal PWWP domain of NSD2 (NSD2-PWWP1) binds H3K36me2, presumably through a conserved aromatic cage composed of three orthogonally positioned aromatic side chains (Y233, W236, F266) that can engage in cation–π and hydrophobic interactions with the ammonium group of the methylated lysine;13 the F266A mutation at the aromatic cage destabilizes the chromatin occupancy of full-length NSD2 and inhibits cancer cell proliferation, but without significantly affecting H3K36 dimethylation.12 Small molecules selectively targeting the aromatic cage of NSD2-PWWP1 would be valuable chemical tools to probe the therapeutic relevance of this domain in NSD2-driven tumors or to design NSD2-targeting PROTACs.
Figure 1.
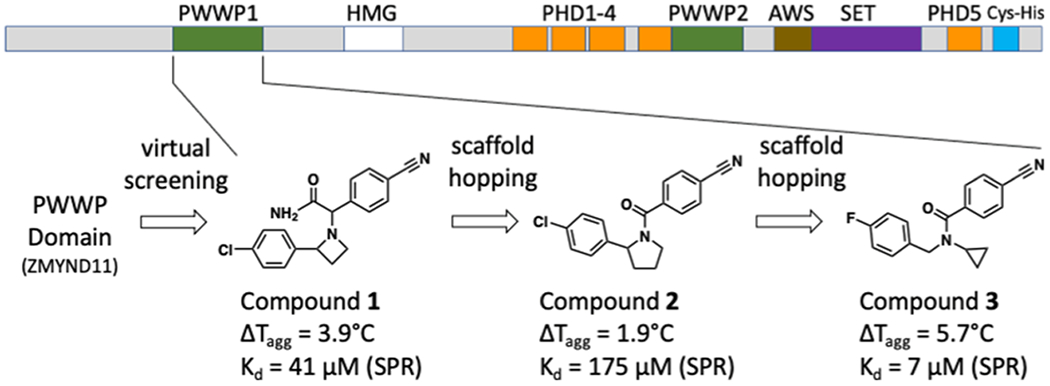
Domain architecture of NSD2 and chemical evolution of NSD2-PWWP1 antagonists. Receptor-based virtual screening followed by target and scaffold hopping led to the development of 3.
In this study, we report the first chemical antagonists targeting the NSD2-PWWP1:H3K36me2 interaction in biophysical and cellular assays. X-ray crystallography established the structural determinants for ligand binding and provided crucial information for future ligand optimization. To our knowledge, the present study is the first successful example of using a virtual screening approach to discover antagonists that directly and specifically target a PWWP domain.
RESULTS AND DISCUSSION
Structure-Based Discovery of NSD2-PWWP1 Ligands.
At the onset of this project, no structure of NSD2 PWWP domains was available. Of the PWWP domain structures available from the Protein Data Bank (PDB), that of the chromatin factor ZMYND11 had a structurally well-defined methyl-lysine binding pocket and was identified as a good candidate for a PWWP-focused virtual screening campaign. A library of ~2 million commercial compounds was therefore screened virtually against the PWWP domain of ZMYND11.14 A total of 39 compounds were purchased and experimentally screened using differential static light scattering (DSLS—Table S1). Since no hit was confirmed for ZMYND11, we took a target class approach and screened the purchased compound set against other members of the PWWP family. Compound 1 (Figure 1) displayed a stabilization effect on the N-terminal PWWP domain of NSD2, increasing its melting temperature by 4 °C at 400 μM. The interaction of 1 with NSD2-PWWP1 was further confirmed by surface plasmon resonance (SPR), which yielded a dissociation constant (Kd) of 41 ± 8 μM (Figure S1a). Given the poor solubility of 1, a reverse isothermal titration calorimetry (ITC) titration was carried out by titrating a concentrated NSD2-PWWP1 solution (1 mM) into the sample cell containing a solution of compound 1 at 40 μM, which produced a Kd of 8.9 μM (Figure S1b). Compound 1 did not bind six other PWWP-containing proteins when tested at 400 μM using DSLS (Table S2), indicating that this compound is selective for NSD2-PWWP1. Compound 1 contains two chiral centers but was found to be a single diastereoisomer where both enantiomers bound the target equipotently (Figures S2–S4).
Ligand-Guided Optimization Leads to Improved Compounds.
The encouraging potency and selectivity of 1 prompted us to further explore this scaffold. However, no analogues were commercially available, and only a limited number of building blocks could be purchased for analogue synthesis. We therefore sought to identify a new series of antagonists that would be more tractable for follow-up medicinal chemistry efforts. Since no structure of the NSD2-PWWP1 domain had been reported, we used ligand-based scaffold hopping approaches.
First, a low-energy 3D conformation of compound 1 was generated with the computational chemistry suite ICM15 and used as a reference to screen a virtual library of ~8 million commercially available compounds with the shape-matching tool ROCS (OpenEye).16 Second, we used FTrees (Bio-SolveIT)17 to perform a 2D similarity search against the same library. This approach uses a fuzzy similarity searching, ignoring the three-dimensional structure and chirality of the reference. This feature was helpful in this case, as at the time, we knew neither the bioactive conformation of 1 nor the correct stereochemistry of the two chiral centers. In the third approach, we performed a substructure search using Filter (OpenEye).18 Finally, based on the structure of 1, a series of scaffold-hopping candidates were manually designed and the closest commercial compounds selected. The atomic property field19 alignment method implemented in ICM was used to flexibly align the top hits of each approach to the lowest energy conformation of 1 used in the ROCS screen. Visual analysis and clustering with LibMCS (ChemAxon)20 led to the selection of 24 compounds (Figure 1 and Table S1).
Of the 24 compounds tested, only one hit (compound 2) showed a modest stabilization ΔTagg = 1.9 °C) of NSD2-PWWP1 (Figure 1). The binding of 2 was further confirmed by SPR with a K of 175 μM. Although 2 was weaker than the initial hit, this new scaffold allowed us to perform a structure–activity relationship (SAR) by catalog, as several analogues were commercially available. From a substructure search, 34 analogues of 2 were purchased and tested (Table S1). The initial SAR showed that removing the chlorine group improved potency (2a), while replacing it with a methoxy group had no significant effect (2b, Table 1). The chlorophenyl group could be replaced with other heterocycles without a significant effect on potency (2c–2e) but removing the nitrile or replacing it with groups lacking a hydrogen bond acceptor abolished activity (Table 1: 2h vs 2a, 2i vs 2), suggesting that the nitrile group was forming a specific interaction with the protein.
Table 1.
SARs of the Analogues of Compound 2 and Close Analogues
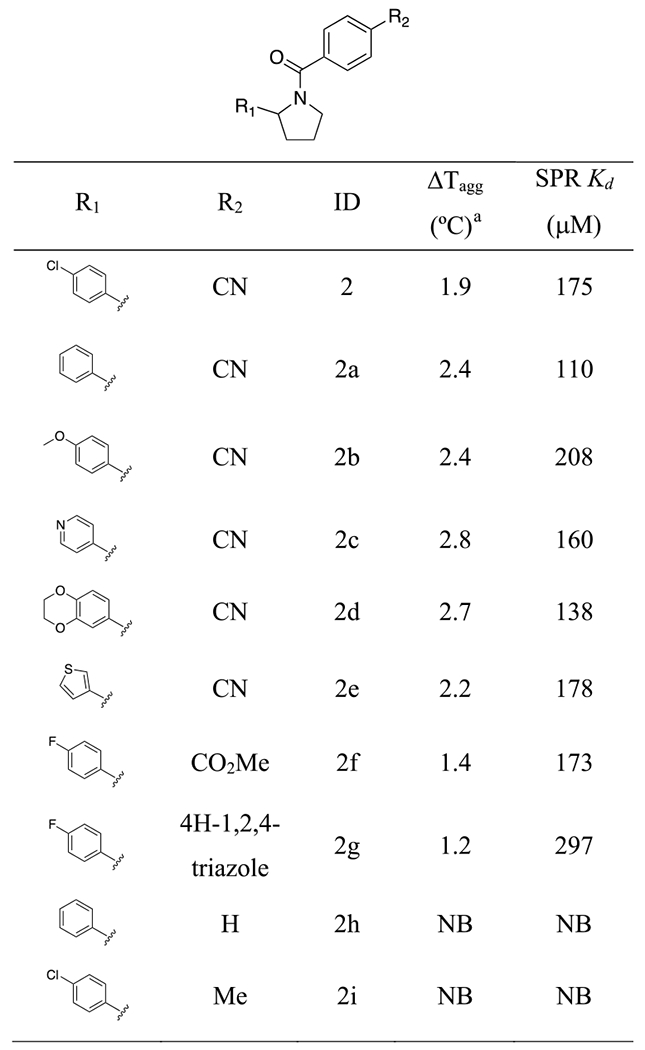 |
Compound concentration was 500 μM.
On the basis of the preliminary SAR provided by 2 and its close derivatives, we set out to identify novel chemical templates that would preserve the two phenyl groups and the nitrile of 2 using FTrees. The seven compounds selected from this exercise (Table S1) included the breakthrough antagonist 3, which binds NSD2-PWWP1 with a Kd of 7 μM by SPR and shows strong protein stabilization ΔTagg = 5.7 °C) by DSLS (Figure 1). Compound 3 is 6 and 25-times more potent than 1 and 2, respectively, is chemically more accessible than the previous scaffolds as it has no chiral center, and can easily be derivatized.
To further understand the molecular basis for the increased potency of 3, we tested 40 analogues that were either purchased or synthesized (Tables 2, S1). The initial SAR demonstrated that the cyclopropyl group is an essential structural element, since compounds where it is absent (3b) or is replaced with smaller (3a) or larger (3c—3e) groups are much weaker or completely inactive (Table 2). Moreover, replacing the fluorophenyl group with thiophene (3f) or tetrahydrofuran (THF) (3g) resulted in similar potency (Table 2). When tested against 10 PWWP domains by DSLS, compound 3f only bound NSD2-PWWP1 and was found to be about 6 times more potent than the initial compound 1 (SPR Kd = 7 ± 3 μM) (Figure 2). 3f did not bind to NSD2-PWWP1 mutants where aromatic cage residues Y233 or F266 were mutated to alanine, indicating binding at the methyl-lysine binding site (Figure S5). It was previously shown that F266A antagonizes association of NSD2 to chromatin, indicating that 3f binds at a site that contributes to chromatin engagement.12 In agreement with a recent observation that high dimethyl sulfoxide (DMSO) concentrations can limit the potency of PWWP ligands,21 the Kd value of 3f was measured as 3.4 ± 0.4 μM in an SPR experiment with 0.5% DMSO (Figure S6).
Table 2.
SAR of the Analogues of Compound 3
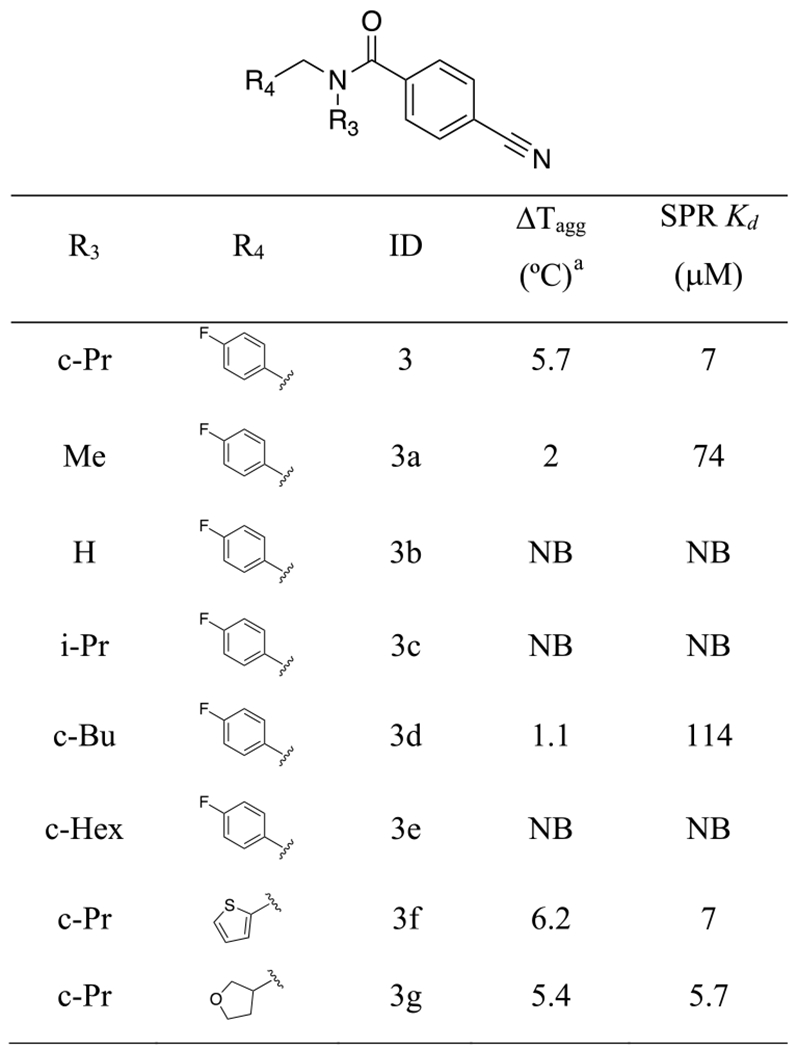 |
Compound concentration was 500 μM.
Figure 2.
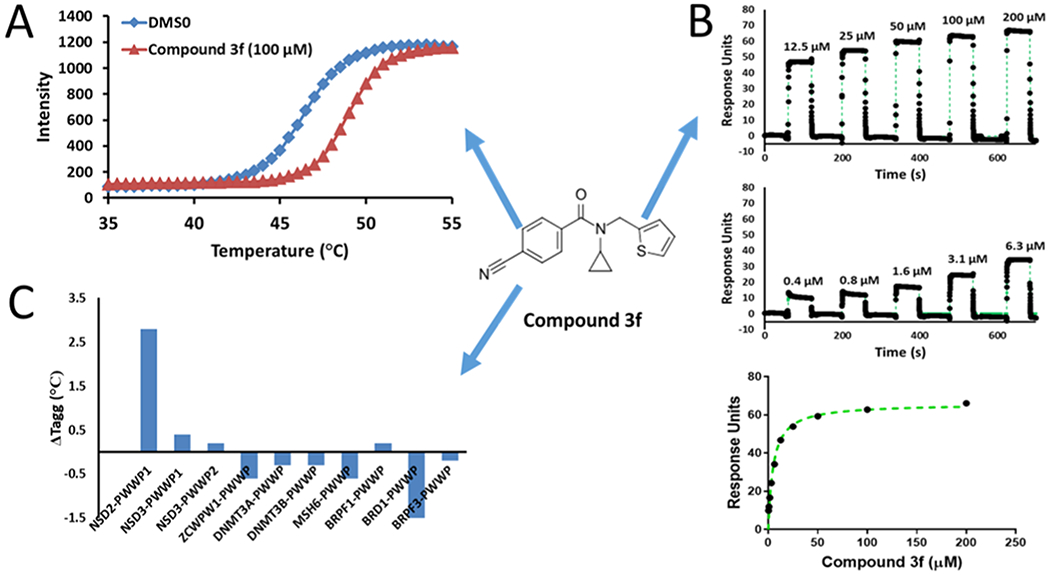
Compound 3f (structure shown in the center) selectively binds NSD2 PWWP1. (A) Treatment with 3f increases the melting temperature of NSD2-PWWP1 in a DSLS assay. (B) 3f binds NSD2-PWWP1 in an SPR assay. (C) 3f only affects the melting temperature of NSD2-PWWP1 in a panel of 10 PWWP domains at 100 μM.
Crystal Structure Confirms Binding at the PWWP Aromatic Cage.
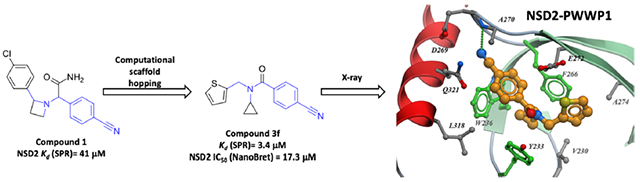
To support follow-up chemistry focused on this chemical series, we solved the crystal structure of NSD2-PWWP1 in complex with compound 3f at a resolution of 2.4 Å (Figure 3, Table S3, and Figure S7). Examination of the complex structure revealed the molecular basis for the high affinity of the antagonist and confirmed that it was binding in the Kme reader aromatic cage. The cyclopropyl ring is bound in a narrow and deep lipophilic pocket formed by the aromatic cage residues (Y233, W236, and F266) and V230, while a tightly coordinated water molecule is found at the bottom of the pocket. This rationalizes previous SAR showing that compounds lacking the cyclopropyl ring are inactive, as they do not fill the hydrophobic pocket. Conversely, compounds featuring larger rings such as cyclohexane (3e, Table 2) or even an iso-propyl group (3c, Table 2) probably clash with the amino acids of the aromatic cage (Figure S8). The cyanophenyl group is enclosed in a pocket formed by W236, G268, D269, A270, E272, L318, and Q321. The nitrile group is completely shielded from the solvent and the nitrogen accepts a hydrogen bond from the amide nitrogen of A270. This supports our previous SAR showing that compounds without the nitrile group are weak or inactive. In addition, the carbonyl oxygen of 3f forms a hydrogen bond with the side chain of Y233 (Figure 3). Finally, the thiophene ring binds within a hydrophobic pocket defined by V230 and A274, and partially exposed to the solvent, which is in agreement with the fact that chemical modifications are tolerated at this end of the molecule.
Figure 3.
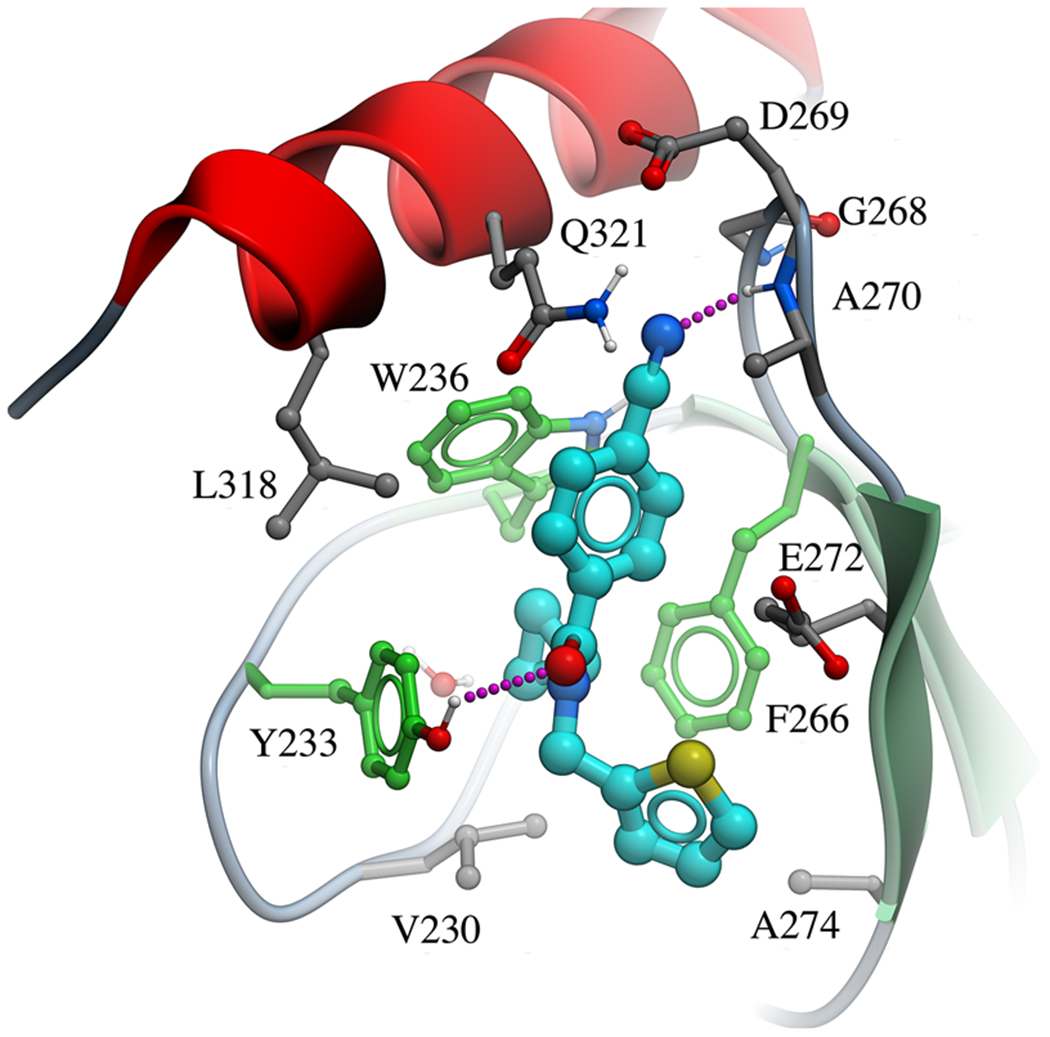
Crystal structure of the NSD2-PWWP1 domain in complex with 3f [PDB code 6UE6]. Binding pocket residues (gray) and the antagonist (cyan) are displayed as sticks and hydrogen bonds as magenta dashed lines. Aromatic cage residues are colored in green.
Compared to the recently published structure of NSD2-PWWP1 in complex with DNA (PBD ID: 5VC8), the loop residues G268, D269, A270, and P271 connecting the β3 and β4 strands undergo significant conformational changes when the ligand binds (Figure S9). Also, 3f induces a conformational change of Y233 and E272, opening-up the aromatic cage which was occluded in the apo structure.
Compound 3f Engages NSD2-PWWP1 in Cells.
Cellular target engagement and displacement of NSD2-PWWP1 from histone H3.3 were tested using a NanoBRET assay measuring the displacement of the NanoLuc-tagged NSD2-PWWP1 domain from Halo-tagged histone H3.3. Encouragingly, we found that compound 3f decreased the interaction of NSD2-PWWP1 but not NSD3-PWWP1 with histone 3 in cells in a dose-dependent manner with an IC50 of 17.3 μM (Figure 4).
Figure 4.
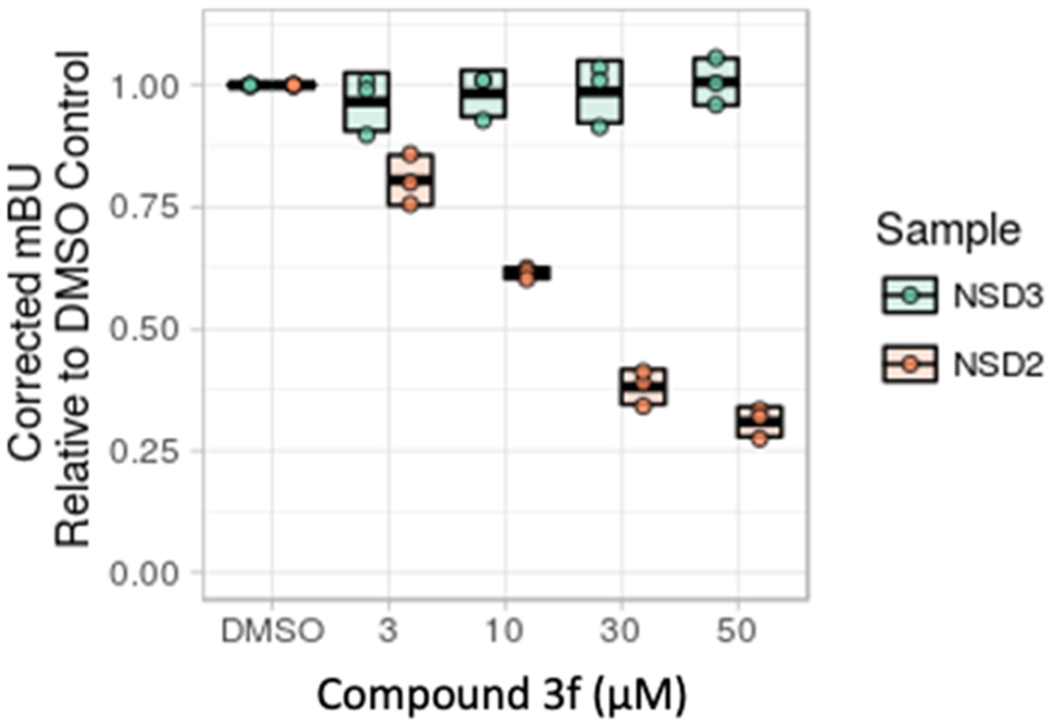
Compound 3f disengages NSD2 PWWP1 from histone H3 in cells. Mean-corrected NanoBRET ratios (mBU) are shown for the interaction between NSD2 (orange) and NSD3 (green) with increasing doses of compound 3f.
The first potent antagonist for a PWWP domain was recently reported against NSD3-PWWP1.21 Our results further support the idea that PWWP domains represent a chemically tractable target class. PWWP domains are often found in multi-modular proteins involved in transcriptional regulation or DNA repair,13 and pharmacological targeting of PWWP domains could provide an avenue to deregulate the function of these proteins. PWWP ligands could also serve as chemical handles toward the development of PROTACs that catalyze the proteasomal degradation of PWWP-containing proteins.
CONCLUSIONS
In summary, using a combination of receptor-based virtual screening and ligand-based scaffold hopping, we identified chemically tractable small-molecule antagonists targeting the NSD2-PWWP1 domain with low micromolar affinity. A crystal structure showed that compound 3f occupies the Kme binding aromatic cage of NSD2-PWWP1 and 3f inhibits histone H3K36me2 binding in cells. This study establishes an alternative approach to targeting NSD2 and reveals a class of compounds that we hope to further optimize into a high-quality chemical probe.
EXPERIMENTAL SECTION
Computational Methods.
Docking.
The X-ray structure of the PWWP domain of ZMYND11 (PDB ID: 4N4I)14 was prepared with PrepWizard (Schrodinger, New York) using the standard protocol, including the addition of hydrogens, the assignment of bond order, assessment of the correct protonation states, and a restrained minimization using the OPLS-AA 2005 force field. Receptor grids were calculated at the centroid of the trimethyllysine (M3L) with the option to dock ligands of similar size.
A lead-like library of ~2 million compounds was prepared with LigPrep (Schrodinger, New York). The resulting library was then docked using the virtual screening workflow using Glide HTVS in the first stage, followed by Glide SP in the second stage (Schrodinger, New York). After removing the compounds with a docking score worse than −7 kcal/mol and that do not contain a tertiary amine, the remaining 18k compounds were docked with Glide XP. Next, compounds with a docking score better than −8 kcal/mol or with a score better than −6 kcal/mol and two hydrogen bonds with nearby residues (R317, F311, H313, N316, W319, H314, S340) were selected for visual inspection. This step selected 431 compounds, which were rescored with the GBSA method (AMBER 12, UCSF). The compounds were clustered and the ones with GBSA > −15 kcal/mol were removed. Finally, after a visual inspection, 40 compounds were selected to be purchased.
ROCS Search.
A library of ~8 million compounds was processed with the Omega software (OpenEye Scientific Software, Santa Fe) to generate 200 lowest energy conformations for each compound. Then, ROCS (OpenEye Scientific Software, Santa Fe) was used to perform a 3D shape-based search against the resulting library using a low-energy 3D conformation of 1 generated by ICM (Molsoft, San Diego) as the query. The compounds were ranked by the TanimotoCombo score, which is a combination of the shape and color similarities, and the top 7500 were saved for analysis.
FTrees Search.
The 2D structure of 1 was used as the reference to search the virtual library described before using the Ftrees (Feature Trees) software (BioSolveIT GmbH). Feature trees are descriptors that represent the molecule as a reduced graph. The graph encodes functional groups and rings to single nodes. The similarity between two molecules is calculated by comparing their associated feature trees by superposing (matching) similar subtrees onto each other. The top 1980 compounds with the highest similarity with compound 1 were selected for further analysis.
Substructure Search.
The software Filter (OpenEye Scientific Software, Sant Fe) was used to perform a substructure search against the virtual library using a substructure of 1 as the query. The substructure used in the search encoded in SMARTS was NC-(=[OX1])C([ar5,ar6])[Ni;!$(N=C);!$(N=N);!$(N-[!#6;!#1]);!$(N–C=[O,N,S])][CX4][ar5,ar6]. This searched resulted in 1041 compounds.
Isothermal Titration Calorimetry.
Purified NSD2-PWWP1 was dialyzed in ITC buffer (20 mM Tris, pH 7.5, and 100 mM NaCl). Protein at 1 mM was injected into the sample cell containing about 300 μL of 40 μM 1 with 1% DMSO. ITC titrations were performed on a Nano ITC from TA Instruments (New Castle, DE) at 25 °C by using 4 μL injections with a total of 12 injections. The concentration of DMSO was adjusted to 1% for NSD2-PWWP1 solution. Data were fitted with a one-binding-site model using Nano Analyze software.
Differential Static Light Scattering.
Experiments were carried out by determining the effect of compounds on the thermal stability by DSLS using StarGazer (from Harbinger). All PWWP domains for selectivity experiments were used at a final concentration of 0.2 mg/mL in a buffer consisting of 0.1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.5) and 150 mM NaCl. As described previously,22 this method assesses ligand binding through ligand-induced stability where protein aggregation/denaturation is measured while heating the sample from 25 to 85 °C at 1 °C/min in a 50 μL volume (covered with 50 μL of mineral oil to prevent evaporation) in a clear-bottom 384-well plate (from Nunc). Aggregation was monitored by the increase of scattered light using CCD camera detection. Pixel intensities were integrated using image analysis software, plotted against temperature and data was then fitted to a Boltzmann sigmoid function to obtain the aggregation temperature (Tagg) from the midpoint of the transition.
Surface Plasmon Resonance.
SPR studies were performed using a Biacore T200 (GE Health Sciences Inc.) at 20 °C. Biotinylated NSD2-PWWP1 was captured onto one flow cell of a streptavidin-conjugated SA chip at approximately 6000 response units (according to the manufacturer’s protocol), while another flow cell was left empty for reference subtraction. Compound 3f was tested at 200 μM as the highest concentration and dilution factor of 0.5 in HBS-EP buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 3 mM ethylenediaminetetraacetic acid, 0.05% Tween-20) was used to yield 10 concentrations. Experiments were performed using the same buffer with 5% DMSO in single cycle kinetics with 60 s contact time and a dissociation time of 120 s at a flow rate of 75 μL/min. Kinetic curve fittings and Kd value calculations were done with a 1:1 binding model using the Biacore T200 Evaluation software (GE Health Sciences Inc.). All compounds that were evaluated in biochemical and biophysical assays had >95% purity as determined by 1H NMR and liquid chromatography/mass spectrometry (LC–MS).
Crystal Structure.
The NSD2 fragment (aa 211-350) containing the first human PWWP domain of NSD2 was cloned into a pET28a-MHL vector with a His-tag in its N-terminus for affinity purification. The recombinant plasmid was transformed in the Escherichia coli BL21 (DE3), and the target protein overexpression was induced by 0.2 mM IPTG at 18 °C overnight. The cells were harvested and lysed by sonication. The cell lysis was first purified by Ni-NTA, and the eluted protein was treated by tobacco etch virus to remove the His-tag by dialysis against dialysis buffer containing 20 mM Tris pH 7.5, 150 mM NaCl, 1 mM dithiothreitol (DTT). The sample was then purified by gel filtration chromatography using a Superdex 75 column in a buffer containing 20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM DTT. The peak fractions from the gel filtration column were pooled and concentrated to 20 mg/mL for crystallization. The purified protein was mixed with the ligand at a molar ratio of 1:3 and cocrystallized using the sitting drop vapor diffusion method at 18 °C. The crystals were obtained in a buffer containing 20% PEG 3350, 0.2 M KSCN. Crystals were soaked in a cryoprotectant consisting of 100% reservoir solution and 15% glycerol for data collection. X-ray diffraction data for NSD2 + 3f was collected at 100k at beamline 08ID-1 of Canadian Light Source (CLS). The data set was processed using the XDS23 suite. The structures of NSD2 + 3f were solved by molecular replacement using PHASER24 with PDB entry 5VC8 as the search template. Graphics program COOT25 was used for model building and visualization. Geometry restraints for the compound refinement were prepared with GRADE (Global Phasing Ltd.) developed at Global Phasing Ltd. Restrained refinement and validation using BUSTER (Global Phasing Ltd.) and MOLPROBITY,26 respectively.
NanoBRET Assay.
U2OS cells were plated in 12-well plates (1 × 105/well) in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL), and streptomycin (100 μ/mL). 4 h after plating, cells were transfected with 1 μg of histone H3.3-HaloTagFusion Vector DNA (Promega) + 0.1 μg of NSD2 PWWP1—NanoLuc Fusion Vector DNA (C-terminal) (Promega) using X-tremeGENE HP DNA Transfection Reagent (Sigma) following manufacturer’s instructions. Next day, cells were trypsinized, spun down, and resuspended in DMEM/F12 (no phenol red) supplemented with 4% FBS, penicillin (100 U/mL), and streptomycin (100 μg/mL) at a density of 1.1 × 105/mL. Cells were divided into two pools. To the first pool, 1 μL/mL DMEM/F12 (no phenol red) of HaloTag NanoBRET 618 Ligand (Promega) was added and to the second pool DMSO. Cells were plated (90 μL/well) in 96-well plates (white, 655083, Greiner Bio One). 10× concentrated compound and DMSO control were prepared in DMEM/F12 and added to cells (10 μL/well). Next day, 25 μL of NanoBRET Nano-GloR Substrate (Promega) solution in DMEM/F12 (10 μL/mL) was added to each well. Cells were shaken for 30 s, and donor emission at 450 nm (filter: 450 nm/BP 80 nm) and acceptor emission at 618 nm (filter: 610 nm/LP) were measured within 10 min of substrate addition using a CLARIOstar microplate reader (Mandel). Mean-corrected NanoBRET ratios (mBU) were determined by subtracting the mean of the 618/460 signal from cells without NanoBRET 618 Ligand × 1000 from the mean of the 618/460 signal from cells with NanoBRET 618 Ligand × 1000.
Synthesis and Separation of All Four Isomers of Compound
1. All reagents were purchased from commercial vendors and used without further purification. Volatiles were removed under reduced pressure by rotary evaporation or by using the V-10 solvent evaporator system by Biotage. Very high boiling point (6000 rpm, 0 mbar, 56 °c), mixed volatile (7000 rpm, 30 mbar, 36 °C), and volatile (6000 rpm, 30 mbar, 36 °C) methods were used to evaporate solvents. The yields given refer to chromatographically purified and spectroscopically pure compounds. Compounds were purified using a Biotage Isolera One system by normal phase chromatography using Biotage SNAP KP-Sil or Sfar Silica D columns (part no: FSKO-1107/FSRD-0445) or by reverse-phase chromatography using Biotage SNAP KP-C18-HS or Sfar C18 D columns (Part no.: FSLO-1118/FSUD-040). If additional purification was required, compounds were purified by solid-phase extraction (SPE) using Biotage Isolute Flash SCX-2 cation exchange cartridges (part no: 532-0050-C and 456-0200-D). Products were washed with two cartridge volumes of MeOH and eluted with a solution of MeOH and NH4OH (9:1 v/v). Preparative chromatography was carried out using a Waters 2767 injector with the collector attached to PDA UV/Vis and SQD mass detectors. An XSelect CSH Prep C18 5 μm OBD 19 mm × 100 mm (part no: 186005421) or XSelect CSH Prep C18 5 μm 10 mm × 100 mm (part no: 186005415) column was used for purification. Final compounds were dried using the Labconco Benchtop FreeZone Freeze-Dry System (4.5 L model). 1H and proton-decoupled 19F NMRs were recorded on a Bruker AVANCE-III 500 MHz spectrometer at ambient temperature. Residual protons of CDCl3, DMSO-d6, and CD3OD solvents were used as internal references. Spectral data are reported as follows: chemical shift (δ in ppm), multiplicity (br = broad, s = singlet, d = doublet, dd = doublet of doublets, m = multiplet), coupling constants (J in Hz), and proton integration. Compound purity was determined by UV absorbance at 254 nm during tandem LC–MS using a Waters Acquity separations module. All final compounds had a purity of ≥95% as determined using this method. Low-resolution mass spectrometry was conducted in positive ion mode using a Waters Acquity SQD mass spectrometer [electrospray ionization (ESI) source] fitted with a PDA detector. Mobile phase A consisted of 0.1% formic acid in water, while mobile phase B consisted of 0.1% formic acid in acetonitrile. Column 1 (method 1): Acquity UPLC CSH C18 (2.1 × 50 mm, 130 Å, 1.7 μm. Part no. 186005296). The gradient went from 90% to 5% mobile phase A over 1.8 min, maintained at 5% for 0.5 min, and then increased to 90% over 0.2 min for a total run time of 3 min. The column was used with the temperature maintained at 25 °C.
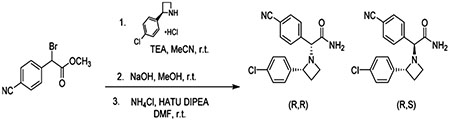
A solution of (R)-2-(4-chlorophenyl)azetidine, hydrochloride (67.5 mg, 0.331 mmol), and N,N-diisopropylethylamine (121 μL, 0.694 mmol) in acetonitrile (0.5 mL) was added dropwise to methyl 2-bromo-2-(4-cyanophenyl)acetate (84 mg, 0.331 mmol) in acetonitrile (1.18 mL) at 0 °C. The mixture was allowed to warm to room temperature and stirred for 5 h. All volatiles were then removed under reduced pressure, and the residue was partitioned between EtOAc and brine. The combined organic layers were then dried over Na2SO4, concentrated under reduced pressure, and column chromatographed (silica gel, hexanes/EtOAc, 10:0 to 7:3 v/v) to afford methyl 2-((R)-2-(4-chlorophenyl)azetidin-1-yl)-2-(4-cyanophenyl)acetate (72 mg, 64% yield) as a yellow oil. LC–MS method 1, RT = 1.84 and 2.04 min, MS (ESI): m/z = 341.3 [M + 1]+, purity (UV254) = 99%
To the above ester (72 mg, 0.211 mmol) in wet MeOH (10 mL) was added potassium hydroxide (59 mg, 1.06 mmol), and the reaction was stirred at room temperature overnight. All volatiles were evaporated, and the residue was column chromatographed (RP-C18, H2O (0.1% v/v FA)/MeCN, 98:2 to 10:90 v/v) to afford 2-2R-((4-chlorophenyl)azetidin-1-yl)-2-(4-cyanophenyl)acetic acid formic acid salt (39 mg, 50% yield) as a white solid. LC–MS method 1, RT = 1.36 and 1.39 min, MS (ESI): m/z = 327.4 [M + 1]+, purity (UV254) = 99%
To the above acid salt in dimethylformamide (5 mL) were added N,N-diisopropylethylamine (121 μL, 0.694 mmol) and 1-[bis-(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (69.6 mg, 0.183 mmol) at room temperature. To this mixture was added, with vigorous stirring, ammonium chloride (26.1 mg, 0.488 mmol) in one portion, and the reaction was stirred for 1 h. Water (20 mL) was added to quench the reaction, and the aqueous layer was extracted with EtOAc (3 × 10 mL) to give the desired compound as a mixture of diastereomers. LC–MS method 1, RT = 1.52 min, MS (ESI): m/z = 326.4 [M + 1]+, purity (UV254) = 99%.
This synthetic mixture was separated by preparative chiral SFC using an AD-H column (2 × 25 cm) using 35% MeOH/CO2 as the eluant at a flow rate of 70 mLּmin−1 and 100 bar pressure. Detection by UV at 220 nm. Injection volume was 1.0 mL of 5 mgּmL−1 in MeOH/dichloromethane. Analytical chiral SFC (Figure 1) used AD-H column (0.46 × 25 cm) with 300% MeOH/CO2 as the eluant at a flow rate of 3 mLּmin−1 and 120 bar pressure. Detection by UV at 220 nm.
2R-(2-(4-Chlorophenyl)azetidin-1-yl)-2R-(4-cyanophenyl)-acetamide (29 mg).
Chiral SFC retention time 2.6 min. 1H NMR (500 MHz, MeOD): δ 7.38 (d, J = 8.3 Hz, 2H), 7.33 (d, J = 8.4 Hz, 2H), 7.12 (d, J =8.6 Hz, 2H), 7.08 (d, J =8.6 Hz, 2H), 4.11 (t, J =8.2 Hz, 1H), 4.07 (s, 1H), 3.73–3.69 (m, 1H), 3.13 (dt, J = 9.3, 8.0 Hz, 1H), 2.36 (dtd, J = 10.3, 8.0, 2.3 Hz, 1H), 2.24–2.11 (m, 1H).
2R-(2-(4-Chlorophenyl)azetidin-1-yl)-2S-(4-cyanophenyl)-acetamide (13 mg).
Chiral SFC retention time 4.1 min. 1H NMR (500 MHz, MeOD): δ 7.72 (d, J = 8.3 Hz, 2H), 7.63 (d, J = 8.3 Hz, 2H), 7.54 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 8.4 Hz, 2H), 4.27 (t, J = 8.3 Hz, 1H), 4.11 (s, 1H), 3.27–3.20 (m, 1H), 2.89–2.80 (m, 1H), 2.32 (dtd, J = 10.4, 7.9, 2.4 Hz, 1H), 2.25–2.15 (m, 1H).
Similarly Prepared Were the Diastereoisomers from (S)-2-(4-Chlorophenyl)azetidine.
2S-(2-(4-Chlorophenyl)azetidin-1-yl)-2S-(4-cyanophenyl)acetamide (29 mg).
Chiral SFC retention time 3.1 min. 1H NMR (500 MHz, MeOD): δ 7.36 (d, J = 8.3 Hz, 2H), 7.31 (d, J = 8.4 Hz, 2H), 7.10 (d, J = 8.6 Hz, 2H), 7.07 (d, J = 8.6 Hz, 2H), 4.09 (t, J = 8.2 Hz, 1H), 4.05 (s, 1H), 3.72–3.66 (m, 1H), 3.11 (dt, J = 9.4, 8.0 Hz, 1H), 2.34 (dtd, J = 10.3, 8.0, 2.3 Hz, 1H), 2.202–2.10 (m, 1H).
2S-(2-(4-Chlorophenyl)azetidin-1-yl)-2R-(4-cyanophenyl)-acetamide (7 mg).
Chiral SFC retention time 4.9 min. 1H NMR (500 MHz, MeOD): δ 7.72 (d, J = 8.4 Hz, 2H), 7.63 (d, J = 8.3 Hz, 2H), 7.54 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 8.4 Hz, 2H), 4.27 (t, J = 8.3 Hz, 1H), 4.11 (s, 1H), 3.28–3.20 (m, 1H), 2.89–2.80 (m, 1H), 2.32 (dtd, J = 10.4, 8.0, 2.4 Hz, 1H), 2.24–2.15 (m, 1H).
Chemical Synthesis of Compounds 3c and 3d.
All starting materials were commercially procured and were used without further purification unless specified. Analytical LC–MS data for all compounds were acquired using an Agilent 6110 Series system with the UV detector set to 220 nm. Samples were injected (<10 μL) onto an Agilent Eclipse Plus 4.6 × 50 mm, 1.8 μm, C18 column at room temperature. Mobile phases A (H2O + 0.1% acetic acid) and B (MeOH + 0.1% acetic acid) were used with a linear gradient from 10 to 100% B in 5.0 min, followed by a flush at 100% B for another 2 min with a flow rate of 1.0 mL/min. Mass spectroscopy (MS) data were acquired in positive ion mode using an Agilent 6110 single quadrupole mass spectrometer with an ESI source. Nuclear magnetic resonance (NMR) spectra were recorded on a Varian Mercury spectrometer at 400 MHz for proton (1H NMR); chemical shifts are reported in ppm (δ) relative to residual protons in deuterated solvent peaks. Normal phase column chromatography was performed with a Teledyne Isco CombiFlashRf using silica RediSepRf columns with the UV detector set to 220 nm and 254 nm. The mobile phases used are indicated for each compound. Reverse phase column chromatography was performed with a Teledyne Isco CombiFlashRf 200 using C18 RediSepRf Gold columns with the UV detector set to 220 nm and 254 nm. Mobile phases of A (H2O + 0.1% TFA) and B (MeCN) were used with default column gradients. Preparative HPLC was performed using an Agilent Prep 1200 series with the UV detector set to 220 nm and 254 nm. Samples were injected onto a Phenomenex Luna 250 × 30 mm, 5 μm, C18 column at room temperature. Mobile phases of A (H2O + 0.1% TFA) and B (MeOH or MeCN) were used with a flow rate of 40 mL/min. A general gradient of 0–15 min increasing from 10 to 100% B followed by a 100% B flush for another 5 min was used. Small variations in this purification method were made as needed to achieve ideal separation for each compound. Analytical LC–MS (at 254 nm) and NMR were used to establish the purity of targeted compounds. All compounds that were evaluated in biochemical and biophysical assays had >95% purity as determined by 1H NMR and LC–MS.
Synthesis of Compound 3c.
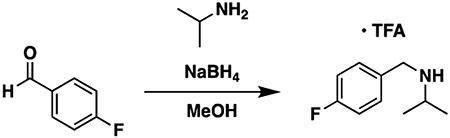
To a flask equipped with a stir bar were added 4-fluorobenzaldehyde (0.25 g, 0.21 mL, 1 equiv, 2.0 mmol) and methanol (10 mL). The flask was sealed with a septum, and isopropylamine (0.59 g, 0.86 mL, 5 equiv, 10 mmol) was introduced via a syringe. The mixture was stirred at room temperature for 3 h to allow for imine formation. Next, the flask was cooled in an ice bath, and NaBH4 (0.15 g, 2 equiv, 4.0 mmol) was added in one portion. The reaction was allowed to come to room temperature with stirring overnight. The next day, the reaction was diluted with water and extracted three times with ethyl acetate. The combined organic layers were washed three times with water and once with brine, then dried over sodium sulfate, and concentrated to a clear oil. The oil was taken up in ether (10 mL) and cooled in an ice bath, and TFA (0.34 g, 0.23 mL, 1.5 equiv, 3.0 mmol) was added with vigorous stirring. The voluminous white precipitate formed was filtered off, rinsed with additional ether, and air-dried to provide N-(4-fluorobenzyl)propan-2-amine 2,2,2-trifluoroacetate (111.5 mg, 396.4 μmol, 20%) as a fluffy white solid.
1H NMR (400 MHz, methanol-d4): δ 7.54 (dd, J = 8.6, 5.4 Hz, 2H), 7.20 (t, J = 8.7 Hz, 2H), 4.20 (s, 2H), 3.44 (hept, J = 6.5 Hz, 1H), 1.39 (d, J = 6.6 Hz, 6H). LC–MS (ESI, +ve mode) expected m/z for C10H15FN [M + H] + 168.12, found 168.20.
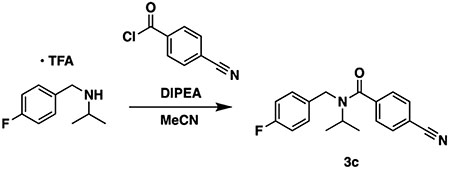
To a vial containing N-(4-fluorobenzyl)propan-2-amine 2,2,2-trifluoroacetate (75 mg, 1.1 equiv, 0.27 mmol) were added acetonitrile (1 mL) and triethylamine (74 mg, 0.10 mL, 3 equiv, 0.73 mmol). The vial was cooled in an ice bath, and 4-cyanobenzoyl chloride (40 mg, 1 equiv, 0.24 mmol) was added in one portion. The reaction was allowed to come to room temperature with stirring overnight. The next day, the reaction was diluted with water and extracted three times with ethyl acetate. The combined organic layers were washed once with 0.5 M citric acid, once with water, once with saturated sodium bicarbonate, and once with brine, dried over sodium sulfate, and concentrated to a white solid. Normal phase chromatography over silica gel (0-50% ethyl acetate in hexanes) afforded compound 3c (47 mg, 0.16 mmol, 65%) as a clear residue that solidified on standing to a white solid.
1H NMR (400 MHz, chloroform-d) complicated by rotamers. δ 7.79–7.65 (br s, 2H), 7.65–7.38 (br s, 2H), 7.37–7.27 (br s, 2H), 7.05–6.93 (m, 2H), 4.62–4.26 (br m, 2H), 4.05–3.81 (br s, 1H), 1.33–1.01 (br s, 6H). 13C NMR (101 MHz, chloroform-d): δ 170.3, 162.0 (d, 1JCF = 245.1 Hz), 141.6, 134.6, 132.7, 129.0, 127.0, 118.2, 115.5 (d, 2JCF = 21.7 Hz), 113.3, 51.1, 43.2, 21.6. 19F NMR (376 MHz, chloroform-d): δ −114.6 (minor rotamer), −115.7 (major rotamer). LC–MS (ESI, +ve mode) expected m/z for C18H18FN2O [M + H] + 297.14, found 297.10.
Synthesis of Compound 3d.
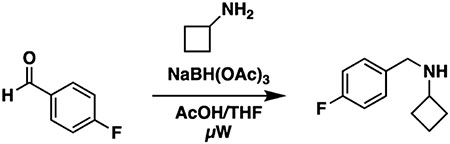
To a flame-dried microwave vial equipped with a stir bar were added 4-fluorobenzaldehyde (100 mg, 86.4 μL, 0.806 mmol) and cyclobutanamine (57.3 mg, 68.8 μL, 0.806 mmol), followed by acetic acid and THF (ratio 1:4; 4mL). The sealed vial was irradiated in the microwave at 60 °C (250 W) for 10 min. Upon cooling, SiliaBond cyanoborohydride was added (50.6 mg, 0.806 mmol) and stirred at room temperature for 10 min. The mixture was irradiated in the microwave at 120 °C (250 W) for 10 min. The crude mixture was filtered, concentrated under vacuum, and used in the next step without further purification.
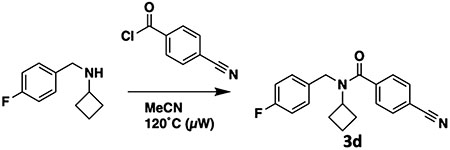
To a flame-dried microwave vial equipped with a stir bar were added the N-(4-fluorobenzyl)cyclobutanamine synthesized previously, 4-cyanobenzoyl chloride (100 mg, 0.604 mmol), and acetonitrile (2 mL). The sealed vial was irradiated in the microwave at 120 °C for 60 min (250 W). Once the reaction was complete, the volatiles were evaporated. The crude mixture was first purified by normal phase gradient column chromatography (0–100% ethyl acetate in hexanes). The desired fractions were concentrated, redissolved in methanol (1 mL), and then purified further by preparative-HPLC [(H2O + 0.1% TFA)/MeCN] to afford compound 3d (56.9 mg, 184 μmol, 30% over two steps) as a colorless gum.
1H NMR (400 MHz, chloroform-d) complicated by rotamers. δ 7.81–7.59 (m, 2H), 7.57–7.38 (m, 2H), 7.23 (br s, 2H), 7.07–6.95 (m, 2H), 4.90–4.00 (multiple br signals, 3H), 2.18–1.33 (multiple br signals, 6H). 13C NMR (101 MHz, chloroform-d): δ 162.08 (d, 1JCF = 245.7 Hz), 141.27, 134.02, 132.49, 128.32, 127.44, 118.21, 115.73 (d, 2JCF = 21.5 Hz), 113.60, 77.48, 77.16, 76.84, 53.71, 44.71, 29.43, 14.60. NB—carbonyl carbon is too weak to be detected. 19F NMR (376 MHz, chloroform-d): δ −115.21 (minor rotamer), −116.12 (major rotamer). NB—trace TFA in spectrum. LC–MS (ESI, +ve mode) expected m/z for C19H18FN2O [M + H] + 309.14, found 309.20
Supplementary Material
ACKNOWLEDGMENTS
The SGC is a registered charity (no. 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada through Ontario Genomics Institute, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and the Wellcome Trust. We received additional funding from the Collaborative Research and Training Experience grant (Aled Edwards 432008-2013) from the Natural Sciences and Engineering Research Council of Canada. We thank BioSolveIT GmbH for supplying an evaluation copy of the Ftrees. We are grateful to OpenEye Scientific Software, Inc. for providing us with an academic license for their software. M.S. gratefully acknowledges funding from NSERC (grants RGPIN-2019-04416 and ALLRP 555329-20). This work was further supported by the National Cancer Institute, NIH (grant R01CA242305 to L.I.J.).
ABBREVIATIONS
- Kd
dissociation constant
- H3K36
lysine 36 of histone 3
- DSLS
differential static light scattering
- SPR
surface plasmon resonance
- ΔTagg
difference in aggregation temperature
- ITC
isothermal titration calorimetry
- PDB
Protein Data Bank
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01768.
Experimental details, 1H NMR spectra of synthesized compounds, 3D docked conformations of virtual screening hits, and compound purity: analytical spectra (PDF)
Molecular string formula and activity of all compounds (PDB)
X-ray crystallographic data and refinement statistics (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c01768
The authors declare no competing financial interest.
Contributor Information
Renato Ferreira de Freitas, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
Yanli Liu, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
Magdalena M. Szewczyk, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada
Naimee Mehta, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Fengling Li, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
David McLeod, Drug Discovery Program, Ontario Institute for Cancer Research, Toronto, Ontario M5G 0A3, Canada.
Carlos Zepeda-Velázquez, Drug Discovery Program, Ontario Institute for Cancer Research, Toronto, Ontario M5G 0A3, Canada.
David Dilworth, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
Ronan P. Hanley, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Elisa Gibson, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
Peter J. Brown, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada
Rima Al-Awar, Drug Discovery Program, Ontario Institute for Cancer Research, Toronto, Ontario M5G 0A3, Canada.
Lindsey I. James, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Cheryl H. Arrowsmith, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada; Princess Margaret Cancer Centre and Department of Medical Biophysics, University of Toronto, Toronto, Ontario M5G 2M9, Canada.
Dalia Barsyte-Lovejoy, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada; Department of Pharmacology & Toxicology, University of Toronto, Toronto, Ontario M5S 1A8, Canada.
Jinrong Min, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada; Department of Physiology, University of Toronto, Toronto, Ontario M5S 1A8, Canada.
Masoud Vedadi, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada; Department of Pharmacology & Toxicology, University of Toronto, Toronto, Ontario M5S 1A8, Canada.
Matthieu Schapira, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada; Department of Pharmacology & Toxicology, University of Toronto, Toronto, Ontario M5S 1A8, Canada.
Abdellah Allali-Hassani, Structural Genomics Consortium, University of Toronto, Toronto, Ontario M5G 1L7, Canada.
REFERENCES
- (1).Kuo AJ; Cheung P; Chen K; Zee BM; Kioi M; Lauring J; Xi Y; Park BH; Shi X; Garcia BA; Li W; Gozani O NSD2 Links Dimethylation of Histone H3 at Lysine 36 to Oncogenic Programming. Mol. Cell 2011, 44, 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Vougiouklakis T; Hamamoto R; Nakamura Y; Saloura V The NSD Family of Protein Methyltransferases in Human Cancer. Epigenomics 2015, 7, 863–874. [DOI] [PubMed] [Google Scholar]
- (3).Keats JJ; Reiman T; Maxwell CA; Taylor BJ; Larratt LM; Mant MJ; Belch AR; Pilarski LM In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood 2003, 101, 1520–1529. [DOI] [PubMed] [Google Scholar]
- (4).Jaffe JD; Wang Y; Chan HM; Zhang J; Huether R; Kryukov GV; Bhang H.-e. C.; Taylor JE; Hu M; Englund NP; Yan F; Wang Z; Robert McDonald E; Wei L; Ma J; Easton J; Yu Z; deBeaumount R; Gibaja V; Venkatesan K; Schlegel R; Sellers WR; Keen N; Liu J; Caponigro G; Barretina J; Cooke VG; Mullighan C; Carr SA; Downing JR; Garraway LA; Stegmeier F Global Chromatin Profiling Reveals NSD2 Mutations in Pediatric Acute Lymphoblastic Leukemia. Nat. Genet. 2013, 45, 1386–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Oyer JA; Huang X; Zheng Y; Shim J; Ezponda T; Carpenter Z; Allegretta M; Okot-Kotber CI; Patel JP; Melnick A; Levine RL; Ferrando A; Mackerell AD; Kelleher NL; Licht JD; Popovic R Point Mutation E1099K in MMSET/NSD2 Enhances Its Methyltranferase Activity and Leads to Altered Global Chromatin Methylation in Lymphoid Malignancies. Leukemia 2014, 28, 198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chinnaiyan AM; Lnu S; Cao Q; Asangani I Compositions and Methods for Inhibiting MMSET. U.S. Patent 20,110,207,198 A1, 2011.
- (7).Morishita M; Mevius DEHF; Shen Y; Zhao S; di Luccio E BIX-01294 Inhibits Oncoproteins NSD1, NSD2 and NSD3. Med. Chem. Res. 2017, 26, 2038–2047. [Google Scholar]
- (8).Tisi D; Chiarparin E; Tamanini E; Pathuri P; Coyle JE; Hold A; Holding FP; Amin N; Martin ACL; Rich SJ; Berdini V; Yon J; Acklam P; Burke R; Drouin L; Harmer JE; Jeganathan F; van Montfort RLM; Newbatt Y; Tortorici M; Westlake M; Wood A; Hoelder S; Heightman TD Structure of the Epigenetic Oncogene MMSET and Inhibition byN-Alkyl Sinefungin Derivatives. ACS Chem. Biol. 2016, 11, 3093–3105. [DOI] [PubMed] [Google Scholar]
- (9).Coussens NP; Kales SC; Henderson MJ; Lee OW; Horiuchi KY; Wang Y; Chen Q; Kuznetsova E; Wu J; Chakka S; Cheff DM; Cheng KC-C; Shinn P; Brimacombe KR; Shen M; Simeonov A; Lal-Nag M; Ma H; Jadhav A; Hall MD High-throughput screening with nucleosome substrate identifies small-molecule inhibitors of the human histone lysine methyltransferase NSD2. J. Biol. Chem. 2018, 293, 13750–13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Huang H; Howard CA; Zari S; Cho HJ; Shukla S; Li H; Ndoj J; González-Alonso P; Nikolaidis C; Abbott J; Rogawski DS; Potopnyk MA; Kempinska K; Miao H; Purohit T; Henderson A; Mapp A; Sulis ML; Ferrando A; Grembecka J; Cierpicki T Covalent Inhibition of NSD1 Histone Methyltransferase. Nat. Chem. Biol. 2020, 16, 1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Huang Z; Wu H; Chuai S; Xu F; Yan F; Englund N; Wang Z; Zhang H; Fang M; Wang Y; Gu J; Zhang M; Yang T; Zhao K; Yu Y; Dai J; Yi W; Zhou S; Li Q; Wu J; Liu J; Wu X; Chan H; Lu C; Atadja P; Li E; Wang Y; Hu M NSD2 Is Recruited through Its PHD Domain to Oncogenic Gene Loci to Drive Multiple Myeloma. Cancer Res. 2013, 73, 6277–6288. [DOI] [PubMed] [Google Scholar]
- (12).Sankaran SM; Wilkinson AW; Elias JE; Gozani O A PWWP Domain of Histone-Lysine N-Methyltransferase NSD2 Binds to Dimethylated Lys-36 of Histone H3 and Regulates NSD2 Function at Chromatin. J. Biol. Chem. 2016, 291, 8465–8474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Qin S; Min J Structure and Function of the Nucleosome-Binding PWWP Domain. Trends Biochem. Sci 2014, 39, 536–547. [DOI] [PubMed] [Google Scholar]
- (14).Wen H; Li Y; Xi Y; Jiang S; Stratton S; Peng D; Tanaka K; Ren Y; Xia Z; Wu J; Li B; Barton MC; Li W; Li H; Shi X ZMYND11 Links Histone H3.3K36me3 to Transcription Elongation and Tumour Suppression. Nature 2014, 508, 263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Abagyan R; Totrov M; Kuznetsov D ICM?A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar]
- (16).ROCS 3.2.2.2; OpenEye Scientific Software: Santa Fe, New Mexico. [Google Scholar]
- (17).FTree, 3.4; BioSolveIT GmbH: Sankt Augustin, Germany. [Google Scholar]
- (18).Filter, 2.4; OpenEye Scientific Software: Santa Fe, New Mexico. [Google Scholar]
- (19).Totrov M Atomic Property Fields: Generalized 3D Pharmacophoric Potential for Automated Ligand Superposition, Pharmacophore Elucidation and 3D QSAR. Chem. Biol. Drug Des. 2008, 71, 15–27. [DOI] [PubMed] [Google Scholar]
- (20).LibMCS, 16.6.13.0; ChemAxon: Budapest, Hungary. [Google Scholar]
- (21).Böttcher J; Dilworth D; Reiser U; Neumüller RA; Schleicher M; Petronczki M; Zeeb M; Mischerikow N; Allali-Hassani A; Szewczyk MM; Li F; Kennedy S; Vedadi M; Barsyte-Lovejoy D; Brown PJ; Huber KVM; Rogers CM; Wells CI; Fedorov O; Rumpel K; Zoephel A; Mayer M; Wunberg T; Böse D; Zahn S; Arnhof H; Berger H; Reiser C; Hörmann A; Krammer T; Corcokovic M; Sharps B; Winkler S; Häring D; Cockcroft X-L; Fuchs JE; Müllauer B; Weiss-Puxbaum A; Gerstberger T; Boehmelt G; Vakoc CR; Arrowsmith CH; Pearson M; McConnell DB Fragment-Based Discovery of a Chemical Probe for the PWWP1 Domain of NSD3. Nat. Chem. Biol. 2019, 15, 822–829. [DOI] [PubMed] [Google Scholar]
- (22).Senisterra GA; Markin E; Yamazaki K; Hui R; Vedadi M; Awrey DE Screening for Ligands Using a Generic and High-Throughput Light-Scattering-Based Assay. J. Biomol. Screening 2006, 11, 940–948. [DOI] [PubMed] [Google Scholar]
- (23).Kabsch W XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phasercrystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Emsley P; Cowtan K Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (26).Davis IW; Murray LW; Richardson JS; Richardson DC MOLPROBITY: Structure Validation and All-Atom Contact Analysis for Nucleic Acids and Their Complexes. Nucleic Acids Res. 2004, 32, W615–W619. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.