

Key Points
-
•
CH is common at the time of stem cell mobilization in poor mobilizers and matched controls.
-
•
Mutations in PPM1D and TP53 correspond with poor stem cell mobilization.
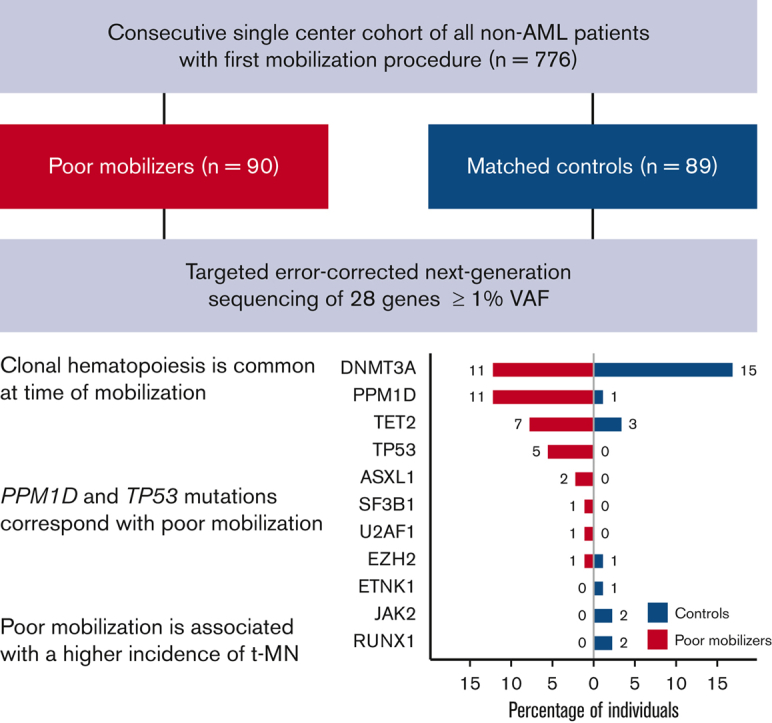
Visual Abstract
Abstract
Inadequate mobilization of peripheral blood progenitor cells (PBPCs) is a limiting factor to proceeding with autologous hematopoietic cell transplantation (auto-HCT). To assess the impact of clonal hematopoiesis (CH) on mobilization failure of PBPC for auto-HCT, we investigated the characteristics of poor mobilizers (with a total PBPC collection <2 × 106 CD34+ cells per kg) in a consecutive single-center cohort of 776 patients. Targeted error-corrected next-generation sequencing of 28 genes was performed in a nested case-control cohort of 90 poor mobilizers and 89 matched controls. CH was detected in 48 out of 179 patients (27%), with most patients carrying a single mutation. The presence of CH (detected at variant allele frequency [VAF] ≥ 1%) did not associate with poor mobilization potential (31% vs 22% in controls, odds ratio, 1.55; 95% confidence interval, 0.76-3.23; P = .238). PPM1D mutations were detected more often in poor mobilizers (P = .005). In addition, TP53 mutations in this cohort were detected exclusively in patients with poor mobilization potential (P = .06). The incidence of therapy-related myeloid neoplasms (t-MN) was higher among patients with mobilization failure (P = .014). Although poor mobilizers experienced worse overall survival (P = .019), this was not affected by the presence of CH. We conclude that CH at low VAF (1%-10%) is common at the time of stem cell mobilization. TP53 mutations and PPM1D mutations are associated with poor mobilization potential and their role in subsequent development of t-MN in these individuals should be established.
Introduction
High-dose chemotherapy followed by autologous hematopoietic progenitor cell reinfusion is widely used in the treatment of multiple myeloma (MM) and malignant lymphoma (ML). A prerequisite for this procedure is the collection of adequate numbers of CD34+ stem and progenitor cells, which is achieved by the administration of granulocyte colony-stimulating factor (G-CSF), often combined with chemotherapy. In approximately 15% of patients, the peripheral blood progenitor cells (PBPCs) collection is unsuccessful with G-CSF, irrespective of the administration of chemotherapy. This failure percentage can be reduced to 5% by the addition of the CXCR4 antagonist plerixafor.1,2 Different risk factors have been identified that contribute to mobilization failure, including advanced age, low bone marrow cellularity, low platelet count, and previous type and amount of cytostatic treatment.3,4 Poor mobilization is usually defined as the collection of less than 2 × 106 CD34+ cells per kg or a peak concentration of <20 CD34+ cells /μL in peripheral blood upon stimulation with G-CSF.3,5
Previous reports have shown that poor mobilization is associated with worse overall survival (OS)6 and a higher incidence of therapy-related myeloid neoplasms (t-MN),7 suggesting that in some cases, stem and progenitor cells can be affected by mutations at the time of mobilization predisposing to subsequent transformation to t-MN.
Recently, recurrent somatic mutations have been identified in blood and bone marrow of otherwise healthy individuals, so-called clonal hematopoiesis (CH). CH is defined by detectable clonal somatic mutations associated with hematological malignancies without morphological evidence of a hematological neoplasm.8 CH has a rate of progression to hematological neoplasm of ∼0.5% to 1% per year.9,10 Depending on the sensitivity of the sequencing technique, CH is detected at a variant allele frequency (VAF) of ≥1% to 2% in 10% to 40% of individuals >60 years. The incidence increases with age, with a prevalence of up to 62% reported in people over 80 years.11, 12, 13, 14, 15
The prevalence of CH in patients who were treated for cancer (including lymphoma and various solid tumors) is higher (∼19% to 33% of patients, VAF ≥ 1% or VAF ≥ 2%) compared with patients without cancer.16, 17, 18, 19 In patients with ML, CH was detected in 25% to 30% of them at the time of stem cell mobilization.20,21 For patients with MM, the prevalence was reported to be 21%.6 It has also been demonstrated that patients with CH after previous cancer treatment and a VAF ≥ 2% have an increased risk of developing t-MN.17,22, 23, 24 Subsequent development of t-MN corresponded with a significantly lower CD34+ yield at the time of stem cell collection.22
To determine whether CH or specific gene mutations are related to poor mobilization, we investigated the mutational spectrum of CH in 90 poor mobilizers and 89 matched controls nested within a consecutive single-center cohort of 776 patients undergoing mobilization for autologous hematopoietic cell transplantation (auto-HCT).
Methods
Cohort selection
We included all consecutive patients without myeloid malignancy who underwent a first PBPC mobilization cycle at the University Medical Center Groningen between 01 January 2007 and 31 December 2018. Clinical data were retrospectively collected from the medical records. Patients were included when treated with G-CSF with or without preceding chemotherapy. Patients with myeloid neoplasms or nonhematological diseases were excluded, along with patients with failure or discontinuation of mobilization owing to problems involving the central venous line or comorbid conditions. All patients gave written informed consent for the use of their clinical data for research purposes. The study was conducted in accordance with the Declaration of Helsinki and institutional guidelines and regulations as approved by the local institutional board.
Procedure of mobilization
Collection of peripheral blood stem cells was preceded by stimulation with G-CSF at a dose of 5 or 10 μg/kg per day. G-CSF was dosed at 10 μg/kg per day when preceded by cyclophosphamide mobilization chemotherapy in the case of MM, or when preceded by high-dose cytarabine and thiotepa for CNS relapse of lymphoma as previously described.25 For all other chemotherapy regimens, G-CSF was dosed at 5 μg/kg per day. G-CSF was started between days 4 and 8 after chemotherapy, according to local protocol. CD34+ counts were assessed from day 8 after the start of G-CSF onward, with expected apheresis from day 9 to 11 after the start of G-CSF.
Plerixafor has been available at our center since August 2009. The indication for plerixafor is expected mobilization failure, defined at our center as (1) CD34+ counts below collection threshold on 2 consecutive days at day 9 and 10 after the start of G-CSF, (2) rapidly declining CD34+ count or apheresis yield, or (3) second mobilization attempt after initial mobilization failure. Plerixafor was added to the mobilization regimen, dosed at 0.24 mg/kg and administered the evening before collection. Leukapheresis was started at the discretion of the treating physician, generally when CD34+ apheresis yield was expected to exceed 0.5 × 106 cells per L. Apheresis was not started if the expected yield was lower than 0.5 × 106 cells per L.
Definition of mobilization failure
Mobilization failure was defined as the inability to collect ≥2.0 × 106 CD34+ cells after mobilization with G-CSF.3 Failure of mobilization was divided into 3 categories: (1) group 1: failure to collect ≥2 × 106 CD34+ cells per kg after treatment with G-CSF; (2) group 2: failure to collect ≥2 × 106 CD34+ cells per kg after treatment with G-CSF and plerixafor; and (3) group 3: need for plerixafor and subsequent successful collection of ≥2 × 106 CD34+ cells per kg.
Sample collection and preparation
We used biobanked genomic DNA collected for high resolution HLA-typing (standard procedure in our center) for a period of maximum 6 months around the mobilization procedure (supplemental Figure 2). Before storage at −20°C, genomic DNA was purified from fresh whole blood specimens using either the QIAamp DNA mini kit (Qiagen) or the QIAcube automated instrument (Qiagen) according to the manufacturer’s instructions.
Error-corrected targeted next-generation sequencing (NGS) to detect clonal gene mutations
For determination of CH, target regions in 28 driver genes were covered with single-molecule–tagged molecular inversion probes (supplemental Table 1). Detailed procedures have been previously described.13,15 For PPM1D, a separate single-molecule–tagged molecular inversion probe pool was designed, equipped with 2 × 5N single-molecule tags. Paired-end sequencing of MIP libraries was performed on the NovaSeq 6000 platform (Illumina, San Diego, CA) or MiniSeq system (Illumina, San Diego, CA) for PPM1D. Mean sequencing depth was 5619 consensus reads for all samples and regions, and the consensus read depth was >500× for 96.8%. Somatic variants were called if they met the following criteria: ≥1% VAF and ≥10 unique variant reads. Subsequently, variants were curated by manual inspection and recurrent artifacts and polymorphisms were excluded.
Statistical analyses
Statistical comparison of parametric variables was performed using Student’s t test. Wilcoxon rank-sum tests were used to test the association between poor mobilization and nonparametric variables, including the VAF and number of mutations. Between-group comparisons of mutation frequencies were carried out using Fisher exact test. OS and time to t-MN were defined as the time from the first (planned) apheresis day until the death from any cause or diagnosis of t-MN, and were censored at the previous follow-up. Visualization of OS was done using the Kaplan-Meier method. Multivariable analyses for OS were performed using Cox regression and corrected for age, sex, and major histological subtype. For the development of t-MNs (in this cohort, t-MDS or t-AML), cumulative incidence curves were constructed using the Aalen-Johansen estimator, with death as the competing risk, and P values from Gray’s test were reported. Hazard ratios (HRs) and odds ratios (ORs) are reported with their 95% confidence intervals (CI). Statistical analyses were performed using R statistical computing software version 3.6.1 (supplemental Methods), and a P value of <.05 was considered statistically significant.
Results
Cohort characteristics
After excluding those with a malignant myeloid disorder, we collected data on all patients ≥18 years of age undergoing a first stem cell mobilization between 01 January 2007 and 31 December 2018 at our institution (Figure 1). Individuals who failed mobilization (n = 13) because of problems involving central venous access, intercurrent infection, and other intercurrent comorbidities interrupting their oncological treatment were excluded from this study. Among 776 included patients, poor mobilization occurred in 106 (13.7%). We identified 21 patients failing to collect ≥2 × 106 CD34+ cells per kg after mobilization with G-CSF (group 1). A cohort of 85 patients received additional plerixafor for mobilization after the failure of G-CSF mobilization. In this cohort, 14 patients did not collect ≥2 × 106 CD34+ cells per kg (group 2), and 71 patients underwent successful stem cell mobilization after the addition of plerixafor (group 3).
Figure 1.

Flowchartdepicting the nestedcase-control study cohort and availability of samples.
Patient characteristics are listed in Table 1. Patients with poor mobilization potential (groups 1, 2, and 3) did not significantly differ from those with successful mobilization in the entire cohort of 776 patients, regarding sex (P = .11) and age (median 59 years in both groups, P = .45). Among poor mobilizers, 47% of patients were diagnosed with MM vs 57% of successful mobilizers (P = .059).
Table 1.
Baseline characteristics of poor mobilizers andcontrols
| Total cohort |
Total cohort |
NGS cohort |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Successful mobilizers |
Poor mobilizers |
P value | n | Matched controls |
Poor mobilizers |
P value | n | ||
| n = 776 | n = 670 | n = 106 | n = 89 | n = 90 | |||||
| Male sex - n (%) | 499 (64.3%) | 423 (63.1%) | 76 (71.7%) | .109 | 776 | 64 (71.9%) | 65 (72.2%) | 1.000 | 179 |
| Age at apheresis (y) – median (IQR) | 59.0 (52.0-63.0) | 59.0 (52.0-63.0) | 59.0 (52.0-64.0) | .453 | 776 | 59.0 (52.0-63.0) | 59.0 (51.0-64.0) | .948 | 179 |
| Major histological subtype - n (%) | .036 | 776 | .845 | 179 | |||||
| Aggressive B-cell lymphoma | 127 (16.4%) | 104 (15.5%) | 23 (21.7%) | 21 (23.6%) | 19 (21.1%) | ||||
| Follicular lymphoma | 22 (2.84%) | 18 (2.69%) | 4 (3.77%) | 4 (4.49%) | 4 (4.44%) | ||||
| Hodgkin lymphoma | 51 (6.57%) | 43 (6.42%) | 8 (7.55%) | 8 (8.99%) | 8 (8.89%) | ||||
| Mantle cell lymphoma | 78 (10.1%) | 67 (10.0%) | 11 (10.4%) | 11 (12.4%) | 11 (12.2%) | ||||
| Other non-Hodgkin lymphoma | 11 (1.42%) | 6 (0.90%) | 5 (4.72%) | 0 (0.00%) | 3 (3.33%) | ||||
| Plasma cell dyscrasia | 433 (55.8%) | 383 (57.2%) | 50 (47.2%) | 42 (47.2%) | 42 (46.7%) | ||||
| T-cell lymphoma | 54 (6.96%) | 49 (7.31%) | 5 (4.72%) | 3 (3.37%) | 3 (3.33%) | ||||
| Remission status at the time of mobilization - n (%) | 179 | ||||||||
| CR | 35 (39%) | 35 (39%) | |||||||
| PR or VGPR | 50 (56%) | 46 (51%) | |||||||
| Stable disease or PD | 4 (4%) | 9 (10%) | |||||||
| Bone marrow infiltration - n∗ | 2 | 5 | 43 | ||||||
| Number of lines of therapy – median (IQR) | 2.00 [1.00-2.00] | 2.00 [1.00-2.00] | .076 | 179 | |||||
| Chemomobilization regimen - n (%) | .269 | 179 | |||||||
| BV-DHAC/DHAP | 0 (0.00%) | 3 (3.33%) | |||||||
| CAD | 7 (7.87%) | 3 (3.33%) | |||||||
| CHO(E)P | 3 (3.37%) | 2 (2.22%) | |||||||
| Cyclophosphamide | 35 (39.3%) | 44 (48.9%) | |||||||
| (R)-DHAP or (R)-VIM | 29 (32.6%) | 20 (22.2%) | |||||||
| HD ARA-C | 10 (11.2%) | 11 (12.2%) | |||||||
| Other | 5 (5.62%) | 7 (7.77%) | |||||||
| CD34 yield (106/kg) - median (IQR) | 9.78 (6.70-13.6) | 10.3 (7.37-14.1) | 5.42 (3.68-7.880) | <.001 | 751 | 10.4 (7.76-12.90) | 5.58 (3.54-7.81) | <.001 | 157 |
| Number of apheresis days - median (IQR) | 1.00 (1.00-2.00) | 1.00 (1.00-2.00) | 2.00 (1.00-3.00) | <.001 | 776 | 1.00 (1.00-2.00) | 2.00 (1.00-3.00) | .007 | 179 |
| Allogeneic transplantation - n (%) | 101 (13.0%) | 86 (12.8%) | 15 (14.2%) | .827 | 776 | 11 (12.4%) | 12 (13.3%) | 1.000 | 179 |
| Peripheral blood counts† - mean (SD) | |||||||||
| Hemoglobin level (g/dL) | 12.0 (1.54) | 11.5 (1.75) | .046 | 179 | |||||
| Platelet count (×109/L) | 273 (94.3) | 239 (113) | .032 | 179 | |||||
| WBC (×109/L) | 7.05 (3.36) | 5.83 (3.27) | .015 | 179 | |||||
| ANC (×109/L) | 4.37 (2.44) | 3.30 (1.84) | .009 | 112 | |||||
Data are presented as mean (SD), median (IQR) or number (%), as appropriate.
ANC, absolute neutrophil count; BV, brentuximab vedotin; CAD, cyclophosphamide, doxorubicin; CHO(E)P, cyclophosphamide, doxorubicin, vincristine, (etoposide), prednisolone; CR, complete remission; DHAC, dexamethasone, cytarabine, carboplatin; HD ARA-C, high-dose cytarabine; IQR, interquartile range; PD, progressive disease; PR, partial remission; (R-)DHAP, (rituximab) dexamethasone, cytarabine, cisplatin; (R-)VIM, (rituximab) etoposide, iphosphamide, methotrexate; SD, standard deviation; VGPR, very good partial response; WBC, white blood cell count. Other chemomobilization regimens include: CYVE (cytarabine, etoposide), mini-BEAM (carmustine, etoposide, cytarabine, melphalan), AraC/TT (high dose cytarabine and thiotepa).
For lymphoma patients with stable disease or PD or PR.
Peripheral blood levels were recorded before start of the chemomobilization regimen.
For 90 out of 106 poor mobilizers, peripheral blood-derived DNA samples were available. A cohort of controls with a successful collection of CD34+ cells was matched for age, sex, and major histological subtype. All blood samples were collected around the time of mobilization, most of the samples within 1 month before (median 0.5 months, IQR 0.3-0.8 months, supplemental Figure 2). The cohort of controls was successfully matched for age (P = 1.00) and sex (P = .95). There was no significant difference in the number of preceding therapy regimens (P = .08). Compared with matched controls, poor mobilizers had significantly lower platelet counts at the start of mobilization chemotherapy (P = .032). In addition, mean hemoglobin levels (P = .046) and white blood cell counts (P = .015) before the start of mobilization chemotherapy were also significantly lower for poor mobilizers than the controls. Absolute neutrophil counts were decreased in poor mobilizers (P = .009) at the onset of mobilization chemotherapy, although the neutrophil counts were only available for a proportion of individuals (n = 112).
Landscape of CH associated with poor mobilization potential
Sequencing was successfully performed for all available samples from poor mobilizers (n = 90) and matched controls (n = 89). Somatic mutations were called so when they are present at a VAF ≥ 1%, corresponding to ≥2% of peripheral blood cells when heterozygosity is assumed. In the sequenced cohort of 179 patients, 79 mutations were detected in 48 individuals. We identified CH in 28 patients from the group of poor mobilizers (31%) and in 20 patients from the group of matched controls (22%) (OR, 1.55; 95% CI, 0.76-3.23; P = .238) (Figure 2A). Furthermore, we corrected the association between poor mobilization and CH for the time difference between sample collection and (planned) apheresis (OR, 1.56; 95% CI, 0.80-3.08; P = .192). VAFs of detected mutations ranged between 1% and 45% (median 2.6%). The median VAF in the cohort of poor mobilizers was 2.8% (1.1%-45%) as compared with 3.5% (1.1%-30%) in the matched control group (P = .391) (Figure 2B). A total of 51 variants in 34 individuals were retained when restricted to the proposed CHIP definition (VAF ≥ 2%).8 Using CHIP definition, again there was no association between poor mobilization potential and CH (OR, 1.17; 95% CI, 0.52-2.67; P = .707, supplemental Figure 3). Most patients had a single mutation, whereas 13 poor mobilizers and 7 controls had 2 or more mutations (with no significant difference in the number of mutations between cases and controls, P = .319) (Figure 2C).
Figure 2.

Spectrum of CH detected in poor mobilizers and controls. (A) Prevalence of CH in 90 poor mobilizers and 89 matched controls. (B) Distribution in the highest VAF for poor mobilizers (red) and matched controls (blue) carrying CH. Boxplots indicate median, first, and third quartiles, with whiskers extending to 1.5× IQR. (C) Violin plot displaying the distribution in number of detected mutations in poor mobilizers (red) and controls (blue) carrying CH. (D) Prevalence of CH according to age (n = 179). Red, poor mobilizers (n = 90); blue, matched controls (n = 89). (E) Proportion of poor mobilizers and matched controls carrying specific gene mutations. The absolute number of individuals with the respective gene mutation is given. (F) Prevalence of CH in failure subgroups 1 (n = 17), 2 (n = 13) and 3 (n = 60), as compared with their respective matched controls.
As expected, the prevalence of CH increased with advancing age, reaching a prevalence of 30% for those ≥60 years of age (Figure 2D). The mean age for patients with CH at the time of mobilization was 59 years, as compared with 56 years for those without somatic mutations (P = .020). Limited by small numbers, no significant differences in the prevalence of CH were observed for mobilization failure subgroups as compared with their respective controls (Figure 2F).
TP53 and PPM1D mutations are associated with poor mobilization potential
Consistent with age-related CH, DNMT3A was the most prevalent mutated gene. PPM1D, previously identified as wild-type p53-induced phosphatase 1 (WIP1),26 was also detected at high frequency (Figures 2E and 3; supplemental Table 2). Most PPM1D mutations were truncating mutations, except for 1 missense mutation (supplemental Table 3). Most interestingly, PPM1D mutations were detected in 11 poor mobilizers vs 1 of the controls (P = .005). Among these, 3 patients were treated for MM, and 8 had received previous treatment with topoisomerase II inhibitors and/or cisplatin for ML. TP53 mutations were exclusively detected in poor mobilizers (P = .06). Notably, 2 patients were identified who carried 2 TP53 mutations. Most individuals with mutated TP53 or PPM1D were diagnosed with ML (69% vs 52% of individuals with other mutational spectra, supplemental Table 4). Furthermore, a significantly lower CD34+ yield was confirmed for individuals carrying mutations in TP53 or PPM1D (4.26 × 106 CD34+ per kg vs 8.20 × 106 CD34+ per kg in individuals without CH, P = .007). Mutations in ASXL1, SF3B1, U2AF1, and EZH2, previously identified to be typical for secondary AML,27 were detected in higher frequencies in poor mobilizers though not significantly (in n = 4 poor mobilizers vs in n = 1 of controls, P = .368).
Figure 3.

Landscape of concurrent gene mutations in poor mobilizers and controls. Each row indicates a specific gene mutation, with columns representing individual patients. The darker shade indicates multiple mutations within the same gene. Subgroups of poor mobilizers are indicated. Individuals developing t-MNs are indicated by an asterisk.
Poor mobilization is associated with t-MN development
After a median follow-up of 43.6 months for the entire cohort, 12 out of 776 patients developed a t-MN after a median period of 43.5 months following stem cell collection. t-MN occurred in 4 out of 106 poor mobilizers (3.8%) and 8 out of 670 successful mobilizers (1.2%) (Figure 4A and supplemental Figure 6, P = .014 from Gray’s test). Patients developing t-MN had relatively lower CD34+ yields (P = .064) (supplemental Figure 5). NGS data at the time of mobilization was available from 3 patients that mobilized poorly and developed t-MN. All carried CH at the time of mobilization: 2 patients harbored 2 TP53 mutations, 1 of them also carrying a PPM1D mutation and 2 DNMT3A mutations, and the third harbored mutations in ASXL1, DNMT3A, TET2, and U2AF1 (Figure 3).
Figure 4.

Outcomes for poor mobilizers and controls. (A) Cumulative incidence of t-MNs among poor mobilizers (n = 106) and successful mobilizers (n = 670). (B) OS for poor mobilizers and controls. HRs for OS are shown for poor mobilization potential in the entire cohort (n = 776) in the upper graph. Within the case-control cohort with NGS data, HRs are shown for the presence of CH among cases (n = 90) and controls (n = 89) in the lower part of the graph. Forest plots display HR and 95% CI from Cox proportional hazard regression, corrected for age, sex, and major histological subtype.
Adverse prognosis associated with poor mobilization is not modified by the presence of CH
We subsequently examined whether the presence of CH affected outcomes for patients with and without successful PBPC mobilization. After a median follow-up of 43.6 months, 294 out of all 776 patients died. Poor mobilizers (groups 1, 2, and 3) experienced worse OS when compared with successful mobilizers (HR, 1.48; 95% CI, 1.07-2.04) (Figure 4B and supplemental Figure 7). The presence of CH was not associated with inferior OS in poor mobilizers (HR, 1.13; 95% CI, 0.51-2.50) and controls (HR, 0.79; 95% CI, 0.31-2.05) (Figure 4B). The cause of death could be evaluated in 89% of deceased patients (for n = 57 out of n = 64) in the sequenced cohort (n = 179). Most often, death was owing to disease progression (67%). None of the patients experienced death due to cardiovascular disease.
Discussion
In this single-center cohort study of 776 consecutive patients undergoing auto-HCT for ML or multiple myeloma, we characterized 90 patients with poor mobilization and evaluated the presence of CH at the time of PBPC mobilization in a nested case-control study. CH at low VAF ≥ 1% was common at the time of PBPC mobilization, in poor mobilizers as well as in matched controls and in general was not associated with poor mobilization potential. However, the distribution of specific mutations was different between both groups: TP53 mutations and PPM1D mutations were mainly detected in patients with poor mobilization potential. The incidence of t-MN was significantly higher in poor mobilizers.
The prevalence of CH at the time of stem cell mobilization in our cohort was similar to findings reported by others.6,20,21 Depending on cutoff values of VAF, CH after cytotoxic therapy is observed in approximately 19% to 33% of patients and increases with age.16, 17, 18, 19 The prevalence of CH after chemotherapy as well as the number of mutations and the VAF are higher compared with healthy controls.16,18,19 Mutations in our cohort were most frequently detected in DNMT3A, PPM1D, TET2, and TP53. This resembles the mutational spectrum that was previously detected in the context of CH after chemotherapy.6,16,18,19,21,24,28, 29, 30, 31
A recently reported study conducted in a similar patient group suggested an association between the presence of CH and poor stem cell mobilization, based on a relatively high percentage of CH in patients with mobilization failure, defined as a collection of <10 × 106 CD34+ PBPC following G-CSF stimulation.30 In this study, only 12 patients with CH were identified in a cohort of 96 patients. Because 7 of these 12 patients were poor mobilizers, the authors conclude that CH may predispose to mobilization failure. In contrast, we observed no significant difference in the overall prevalence of CH, number of mutations, or VAFs in poor mobilizers compared with controls. Both studies differ in the composition of the study cohort in size, patient characteristics, and sensitivity of the sequencing technique to detect CH, which may explain the different findings. In addition, we screened for a panel of 28 genes, including PPM1D, as opposed to 6 genes (ASXL1, DNMT3A, JAK2, SF3B1, TET2, and TP53) in the report by Gifford et al. The most important difference, and strength of our study, is reflected in the nested case-control setup of our study within a consecutive cohort, which allowed for an unbiased comparison of CH in a large number of poor mobilizers and matched controls. In healthy (allogeneic) donors, the presence of CHIP does not seem to affect the ability to mobilize PBPC.32 However, in this healthy population, the mutational spectrum resembled age-related CH, with mutations predominantly detected in DNMT3A, TET2, and ASXL1, which is in contrast to the mutational spectrum detected in poor mobilizers in our study, possibly reflecting clonal selection due to previous therapy.
Previous studies have found a significantly higher incidence of t-MN in patients with cancer with CH than in patients without CH (30% compared with 7% at 5 years),23 with a higher incidence of t-MN with increased VAFs.16,22,24 In addition, patients who developed t-MN had significantly lower CD34+ yields during the mobilization procedure.22 Studies of biobanked patient samples preceding the diagnosis of t-MN have shown that premalignant clones can be present more than 7 years before t-MN diagnosis33 and that mutations present in CH often remain present at the time of t-MN.34 This was also observed for poor mobilizers with the subsequent development of t-MN. Our results confirm the reported relationship between reduced mobilization potential and t-MN development, with a higher risk for those who fail to mobilize sufficient numbers of CD34+ cells.
In our nested case-control study, we identified mutations in TP53 and PPM1D as being associated with poor mobilization potential. Patients with CH after previous cancer treatment who develop t-MN frequently carry TP53 mutations (40%) or PPM1D mutations (20%), often already detectable before the development of t-MN.16,28,35 Mouse models have shown that TP53 mutations are not induced by cytotoxic stress, but rather that TP53-mutated cells expand preferentially through chemoresistance, resulting in a selective advantage.36 Thus, the early presence of TP53 may predict t-MN development. Another recent report has shown that lenalidomide promotes the development of TP53-mutated cells.31 The prognostic impact of TP53 mutations in AML and MDS is dependent on co-occurring genetic aberrations, and specifically higher risks were observed for biallelic mutations and the concurrent presence of adverse cytogenetics.37, 38, 39 Two patients with double-mutant TP53 in this study developed t-MN. Additional studies will establish whether the presence of TP53 mutant clones is useful to predict the risk of t-MN development and guide treatment decisions in patients eligible for auto-HCT, especially those with poor mobilization potential.
PPM1D is upregulated by TP53 and is a negative-feedback regulator of TP53 and other DNA damage response proteins.26,40,41 PPM1D mutations are detected frequently after chemotherapy treatment41 and are associated with prior treatment with platinum and topoisomerase II inhibitors.16, 17, 18,28 Indeed, most patients with PPM1D mutant clones in our study had previously been treated with these agents. PPM1D mutations are more common in t-MN than in primary MDS and are not associated with complex karyotypes or adverse prognosis, in contrast to TP53 mutations.35 Other reports have shown that mutated PPM1D is not sufficient as a driver of myeloid malignancies.17,28 PPM1D mutations seem to affect hematopoietic reserve, as shown by in vitro studies that demonstrated reduced engraftment potential of PPM1D mutated cells.28 This may also be the case in vivo because patients with nonhematological cancers with CH and PPM1D mutations were more likely to require growth factor therapy during treatment.17 This suggests that the high prevalence of PPM1D mutations in poor mobilizers might be explained in the context of a general impairment of hematopoiesis, possibly by an increased fitness advantage in such conditions. The mechanisms underlying the fitness of PPM1D clones and their possible role as a potential driver in t-MN development in these individuals need to be established.
Collectively, these data show that CH at low VAF is common at the time of stem cell mobilization, even in patients <60 years of age. Although the incidence of CH and the risk of poor mobilization increases with age,1,42,43 this is not caused by the presence of CH per se, but the presence of specific mutations (TP53 and PPM1D) associated with mobilization failure.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Acknowledgment
This work was supported by a grant from the Tekke Huizinga Fund (STHF-171) (I.A.v.Z.). The funder had no role in study design, collection, analysis, and interpretation of data, and writing or approval of the manuscript.
Authorship
Contribution: I.A.v.Z., A.O.d.G, J.H.J., E.V., and G.A.H. contributed to the study design; I.A.v.Z. performed the statistical analyses; L.B. provided DNA samples; A.O.d.G., M.G.J.M.v.B., and J.H.J. performed sequencing and variant curation; C.L.E.H., R.M., L.B., A.O.d.G., M.G.J.M.v.B., J.H.J., A.B.M., G.C., M.R.d.G., E.V., G.H., and I.A.v.Z. contributed to the interpretation of data and analysis; C.L.E.H. and I.A.v.Z. wrote the first version of the manuscript; and all authors read and approved the final version of the manuscript for submission.
Footnotes
Data are available on request from the corresponding authors, Isabelle A. van Zeventer (i.a.van.zeventer@umcg.nl) or Gerwin Huls (g.huls@umcg.nl).
The full-text version of this article contains a data supplement.
Contributor Information
Gerwin Huls, Email: g.huls@umcg.nl.
Isabelle A. van Zeventer, Email: i.a.van.zeventer@umcg.nl.
Supplementary Material
References
- 1.Perseghin P, Terruzzi E, Dassi M, et al. Management of poor peripheral blood stem cell mobilization: incidence, predictive factors, alternative strategies and outcome. A retrospective analysis on 2177 patients from three major Italian institutions. Transfus Apher Sci. 2009;41(1):33–37. doi: 10.1016/j.transci.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 2.DiPersio JF, Stadtmauer EA, Nademanee A, et al. Plerixafor and G-CSF versus placebo and G-CSF to mobilize hematopoietic stem cells for autologous stem cell transplantation in patients with multiple myeloma. Blood. 2009;113(23):5720–5726. doi: 10.1182/blood-2008-08-174946. [DOI] [PubMed] [Google Scholar]
- 3.Wuchter P, Ran D, Bruckner T, et al. Poor mobilization of hematopoietic stem cells-definitions, incidence, risk factors, and impact on outcome of autologous transplantation. Biol Blood Marrow Transplant. 2010;16(4):490–499. doi: 10.1016/j.bbmt.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 4.Bensinger W, Appelbaum F, Rowley S, et al. Factors that influence collection and engraftment of autologous peripheral-blood stem cells. J Clin Oncol. 1995;13(10):2547–2555. doi: 10.1200/JCO.1995.13.10.2547. [DOI] [PubMed] [Google Scholar]
- 5.Olivieri A, Marchetti M, Lemoli R, et al. Proposed definition of “poor mobilizer” in lymphoma and multiple myeloma: an analytic hierarchy process by ad hoc working group Gruppo ItalianoTrapianto di Midollo Osseo. Bone Marrow Transplant. 2012;47(3):342–351. doi: 10.1038/bmt.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mouhieddine TH, Sperling AS, Redd R, et al. Clonal hematopoiesis is associated with adverse outcomes in multiple myeloma patients undergoing transplant. Nat Commun. 2020;11(1):2996. doi: 10.1038/s41467-020-16805-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barlogie B, Tricot G, Haessler J, et al. Cytogenetically defined myelodysplasia after melphalan-based autotransplantation for multiple myeloma linked to poor hematopoietic stem-cell mobilization: the Arkansas experience in more than 3000 patients treated since 1989. Blood. 2008;111(1):94–100. doi: 10.1182/blood-2007-06-097444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9–16. doi: 10.1182/blood-2015-03-631747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zink F, Stacey SN, Norddahl GL, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017;130(6):742–752. doi: 10.1182/blood-2017-02-769869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van den Akker EB, Makrodimitris S, Hulsman M, et al. Dynamic clonal hematopoiesis and functional T-cell immunity in a supercentenarian. Leukemia. 2021;35(7):2125–2129. doi: 10.1038/s41375-020-01086-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Zeventer IA, Salzbrunn JB, de Graaf AO, et al. Prevalence, predictors, and outcomes of clonal hematopoiesis in individuals aged ≥80 years. Blood Adv. 2021;5(8):2115–2122. doi: 10.1182/bloodadvances.2020004062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossi M, Meggendorfer M, Zampini M, et al. Clinical relevance of clonal hematopoiesis in the oldest-old population. Blood. 2021;138(21):2093–2105. doi: 10.1182/blood.2021011320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Zeventer IA, de Graaf AO, Wouters HJCM, et al. Mutational spectrum and dynamics of clonal hematopoiesis in anemia of older individuals. Blood. 2020;135(14):1161–1170. doi: 10.1182/blood.2019004362. [DOI] [PubMed] [Google Scholar]
- 16.Bolton KL, Ptashkin RN, Gao T, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet. 2020;52(11):1219–1226. doi: 10.1038/s41588-020-00710-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coombs CC, Zehir A, Devlin SM, et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell. 2017;21(3):374–382.e4. doi: 10.1016/j.stem.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong TN, Miller CA, Jotte MRM, et al. Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nat Commun. 2018;9(1):455. doi: 10.1038/s41467-018-02858-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olszewski AJ, Chorzalska AD, Kim AS, et al. Clonal haematopoiesis of indeterminate potential among cancer survivors exposed to myelotoxic chemotherapy. Br J Haematol. 2019;186(3):e31–e35. doi: 10.1111/bjh.15861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibson CJ, Lindsley RC, Tchekmedyian V, et al. Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J Clin Oncol. 2017;35(14):1598–1605. doi: 10.1200/JCO.2016.71.6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Husby S, Favero F, Nielsen C, et al. Clinical impact of clonal hematopoiesis in patients with lymphoma undergoing ASCT: a national population-based cohort study. Leukemia. 2020;34(12):3256–3268. doi: 10.1038/s41375-020-0795-z. [DOI] [PubMed] [Google Scholar]
- 22.Soerensen JF, Aggerholm A, Kerndrup GB, et al. Clonal hematopoiesis predicts development of therapy-related myeloid neoplasms post-autologous stem cell transplantation. Blood Adv. 2020;4(5):885–892. doi: 10.1182/bloodadvances.2019001157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi K, Wang F, Kantarjian H, et al. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: a case-control study. Lancet Oncol. 2017;18(1):100–111. doi: 10.1016/S1470-2045(16)30626-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gillis NK, Ball M, Zhang Q, et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol. 2017;18(1):112–121. doi: 10.1016/S1470-2045(16)30627-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korfel A, Elter T, Thiel E, et al. Phase II study of central nervous system (CNS)-directed chemotherapy including high-dose chemotherapy with autologous stem cell transplantation for CNS relapse of aggressive lymphomas. Haematologica. 2013;98(3):364–370. doi: 10.3324/haematol.2012.077917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fiscella M, Zhang H, Fan S, et al. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci U S A. 1997;94(12):6048–6053. doi: 10.1073/pnas.94.12.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367–1376. doi: 10.1182/blood-2014-11-610543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsu JI, Dayaram T, Tovy A, et al. PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell Stem Cell. 2018;23(5):700–713.e6. doi: 10.1016/j.stem.2018.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jongen-Lavrencic M, Grob T, Hanekamp D, et al. Molecular minimal residual disease in acute myeloid leukemia. N Engl J Med. 2018;378(13):1189–1199. doi: 10.1056/NEJMoa1716863. [DOI] [PubMed] [Google Scholar]
- 30.Gifford G, Hesson L, Wong JWH, et al. Poor mobilization of autologous CD34+ peripheral blood stem cells in haematology patients undergoing autologous stem cell transplantation is associated with the presence of variants in genes implicated in clonal haematopoiesis of indeterminant potential. Br J Haematol. 2021;193(4):841–844. doi: 10.1111/bjh.17316. [DOI] [PubMed] [Google Scholar]
- 31.Sperling AS, Guerra VA, Kennedy JA, et al. Lenalidomide promotes the development of TP53-mutated therapy-related myeloid neoplasms. Blood. 2022;140(16):1753–1763. doi: 10.1182/blood.2021014956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frick M, Chan W, Arends CM, et al. Role of donor clonal hematopoiesis in allogeneic hematopoietic stem-cell transplantation. J Clin Oncol. 2019;37(5):375–385. doi: 10.1200/JCO.2018.79.2184. [DOI] [PubMed] [Google Scholar]
- 33.Berger G, Kroeze LI, Koorenhof-Scheele TN, et al. Early detection and evolution of preleukemic clones in therapy-related myeloid neoplasms following autologous SCT. Blood. 2018;131(16):1846–1857. doi: 10.1182/blood-2017-09-805879. [DOI] [PubMed] [Google Scholar]
- 34.Katagiri S, Makishima H, Azuma K, et al. Predisposed genomic instability in pre-treatment bone marrow evolves to therapy-related myeloid neoplasms in malignant lymphoma. Haematologica. 2020;105(7):E337–E339. doi: 10.3324/haematol.2019.229856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindsley RC, Saber W, Mar BG, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med. 2017;376(6):536–547. doi: 10.1056/NEJMoa1611604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong TN, Ramsingh G, Young AL, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518(7540):552–555. doi: 10.1038/nature13968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Short NJ, Montalban-Bravo G, Hwang H, et al. Prognostic and therapeutic impacts of mutant TP53 variant allelic frequency in newly diagnosed acute myeloid leukemia. Blood Adv. 2020;4(22):5681–5689. doi: 10.1182/bloodadvances.2020003120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sasaki K, Kanagal-Shamanna R, Montalban-Bravo G, et al. Impact of the variant allele frequency of ASXL1, DNMT3A, JAK2, TET2, TP53, and NPM1 on the outcomes of patients with newly diagnosed acute myeloid leukemia. Cancer. 2020;126(4):765–774. doi: 10.1002/cncr.32566. [DOI] [PubMed] [Google Scholar]
- 39.Bernard E, Nannya Y, Hasserjian RP, et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. 2020;26(10):1549–1556. doi: 10.1038/s41591-020-1008-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takekawa M, Adachi M, Nakahata A, et al. p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 2000;19(23):6517–6526. doi: 10.1093/emboj/19.23.6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kahn JD, Miller PG, Silver AJ, et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood. 2018;132(11):1095–1105. doi: 10.1182/blood-2018-05-850339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morris CL, Siegel E, Barlogie B, et al. Mobilization of CD34+ cells in elderly patients (≥ 70 years) with multiple myeloma: Influence of age, prior therapy, platelet count and mobilization regimen. Br J Haematol. 2003;120(3):413–423. doi: 10.1046/j.1365-2141.2003.04107.x. [DOI] [PubMed] [Google Scholar]
- 43.Hosing C, Saliba RM, Ahlawat S, et al. Poor hematopoietic stem cell mobilizers: a single institution study of incidence and risk factors in patients with recurrent or relapsed lymphoma. Am J Haematol. 2009;84(6):335–337. doi: 10.1002/ajh.21400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.