

Abstract
Background:
Preeclampsia is a syndrome of high blood pressure (BP) with end organ damage in late pregnancy that is associated with high circulating soluble VEGF receptor (sFlt1). Women exposed to preeclampsia have a substantially increased risk of hypertension after pregnancy, but the mechanism remains unknown, leaving a missed interventional opportunity. After preeclampsia, women have enhanced sensitivity to hypertensive stress. Since smooth muscle cell mineralocorticoid receptors (SMC-MR) are activated by hypertensive stimuli, we hypothesized that high sFlt1 exposure in pregnancy induces a post-partum state of enhanced SMC-MR responsiveness.
Methods:
Post-partum BP response to high salt intake was studied in women with prior preeclampsia. MR transcriptional activity was assessed in vitro in sFlt1-treated SMC by reporter assays and PCR. preeclampsia was modeled by transient sFlt1 expression in pregnant mice. Two months post-partum, mice were exposed to high salt and then to AngII and BP and vasoconstriction were measured.
Results:
Women exposed to preeclampsia had significantly enhanced salt-sensitivity of BP verses those with a normotensive pregnancy. sFlt1 overexpression during pregnancy in mice induced elevated BP and glomerular endotheliosis, which resolved post-partum. The sFlt1 exposed post-partum mice had significantly increased BP response to 4% salt diet and to AngII infusion. In vitro, SMC-MR transcriptional activity in response to aldosterone or AngII was significantly increased after transient exposure to sFlt1 as was aldosterone-induced expression of AngII type 1 receptor (AT1R). Post-partum, SMC-MR-KO mice were protected from the enhanced response to hypertensive stimuli after PE. Mechanistically, preeclampsia mice exposed to post-partum hypertensive stimuli develop enhanced aortic stiffness, microvascular myogenic tone, AngII constriction, and AT1R expression, all of which were prevented in SMC-MR-KO littermates.
Conclusions:
These data support that sFlt1-induced vascular injury during PE produces a persistent state of enhanced sensitivity of SMC-MR to activation. This contributes to postpartum hypertension in response to common stresses and supports testing of MR antagonism to mitigate the increased cardiovascular risk in women after PE.
Keywords: preeclampsia, hypertension, smooth muscle cell, angiotensin II, mineralocorticoid receptor
Introduction
Preeclampsia occurs in 5% of pregnancies and is characterized by new onset of high blood pressure (BP) combined with signs of kidney or other target organ dysfunction during the third trimester.1 Women who survive preeclampsia are at two times the risk of premature cardiovascular disease (CVD), which becomes evident around 10 years post-partum.2–5 The most substantial increased risk after preeclampsia is the development of post-partum hypertension, for which the risk is increased almost 4 fold and may also contribute to other CVD risks such as heart attack and stroke.2, 6 Racial disparities also exist in preeclampsia and post- preeclampsia hypertension prevalence, both of which are higher in Black, American Indian and Alaskan Native populations.7, 8 As CVD and preeclampsia share common risk factors9, it is remains unclear if preeclampsia simply uncovers preexisting CVD risk or if preeclampsia exposure directly contributes to, and is sufficient to induce future CVD.
While the initiating mechanism has not been completely elucidated, preeclampsia is characterized by impaired placental vascular development resulting in placental ischemia and restricted fetal growth.10 The ischemic placenta releases anti-angiogenic factors, including a soluble form of the vascular endothelial growth factor receptor (sFlt1). Increased sFlt1 occurs in approximately 90% of preterm preeclampsia cases11, precedes the development of symptoms including hypertension,12, 13 and correlates with severity of preeclampsia in humans.14–16 High levels of sFlt1 sequester vascular endothelial growth factor (VEGF) and placental growth factor, which are necessary for normal vascular function. This contributes to impaired microvascular function resulting in hypertension and organ damage characteristic of preeclampsia.10 As such, overexpression of sFlt1 in rodents is sufficient to phenocopy signs of preeclampsia, including hypertension and renal glomerular damage, suggesting a role for high sFlt1 in the pathophysiology of the systemic syndrome that accompanies the placental defect in preeclampsia.14, 17, 18
Despite normalization of sFlt1 levels and resolution of preeclampsia signs after delivery of the placenta, clinical studies reveal persistent microvascular damage and enhanced post-partum sensitivity to hypertensive stimuli after preeclampsia.4, 19–21 Specifically, women with a prior hypertensive pregnancy have a greater BP response to angiotensin II (AngII) infusion19, 22 and increased microvascular constriction to AngII.21 In addition, in women who had severe preeclampsia (SBP > 160 mmHg during pregnancy), enhanced post-partum salt-sensitivity of BP has also been demonstrated.23 Women are routinely exposed to hypertensive stimuli since renin-angiotensin-aldosterone system (RAAS) activity increases with stress and aging24, 25 and most western societies are exposed to high salt diet. Thus, enhanced response to future hypertensive stimuli may contribute to post- preeclampsia CVD however, the molecular mechanisms remain unknown.
One of the terminal steps of the RAAS is activation of the mineralocorticoid receptor (MR).26 The MR is a hormone-activated transcription factor that traditionally regulates renal sodium retention in response to aldosterone (Aldo) to control BP. MR is also expressed in vascular SMC where it can be activated by its ligand Aldo or, in a ligand-independent manner, by AngII via PKC signaling.27, 28 Recent data shows a minimal role for SMC-MR in basal homeostasis, but rather, vascular MR is poised to respond to cardiovascular stress as its activation and subsequent target gene transcription promote hypertension, vascular remodeling, vasoconstriction and vascular stiffness in aging or injured male mice.29, 30 However, the role of SMC-MR has never been investigated in the context of post-partum vascular dysfunction or hypertension after preeclampsia. Therefore, we used the sFlt1-induced mouse preeclampsia model to test the hypothesis that exposure to high sFlt1 during pregnancy results in a persistently exacerbated response to hypertensive stimuli mediated by vasoconstriction due to enhanced activation of SMC-MR.
Methods
Data Availability.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
See Supplemental materials for detailed methods
Post- preeclampsia human BP study
The protocol was approved by the Partners Healthcare Human Subjects Committee and all participants signed written informed consent. Women who were within 10 years of a Prior Normotensive Pregnancy (n=13) were compared to those with prior preeclampsia diagnosis31(n=8) and baseline characteristics are compared in Table S1. Women with prior normotensive pregnancies were on average 4.3 ± 3.0 years post-partum and women with prior preeclamptic pregnancies were on average 3.2 ± 2.2 years post-partum (p=0.30). A crossover study design was used, exposing both groups of women to high salt (200 mEq/d) and low salt (10 mEq/d) for a week, each followed by systolic BP (SBP) measurement to calculate salt-sensitivity of BP (difference in BP on high versus low salt balance).
Animals
All animal studies were approved by the Tufts University institutional animal care and use committee and conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Pregnant C57Bl/6 (Jackson Labs), smooth muscle mineralocorticoid receptor knockout mice (SMC-MR-KO), and SMC-MR-WT littermates (bred and induced as previously validated32) were randomized in an alternating fashion as they became pregnant to control adenovirus (CMV-null vector= “control”) or sFlt1 adenovirus injected on gestation day (GD)9. BP was recorded using implanted telemetry during pregnancy or post-partum in response to hypertensive stimuli (high salt and subsequent AngII (600ng/kg/min)). Kidneys were removed from a subset of mice upon sacrifice at GD18 or 2 months post-partum and glomeruli blindly scored for severity of endotheliosis. Blood was collected to measure sFlt1 and Aldo. No significant difference in any phenotype was found between Cre− or Cre+ mice injected with control virus, therefore data from control-injected SMC-MR-KO and SMC-MR-WT were combined for analysis.
Vascular Function
Vasoconstriction of third order mesenteric arteries was measured using wire myograph (response to AngII) or pressure myograph (myogenic tone) as in 33, 34.
In Vitro Experiments
Pac1 rat SMC biological replicates were treated with sFlt1 for 24 hours and MR mRNA and protein were quantified. sFlt1 was then removed from the media and fresh media added for 24 hours after which MR transcriptional activity was quantified using luciferase-reporter assay or by QRT-PCR of MR target genes after MR activation with AngII or Aldo as indicated. Primers are listed in Table S2–S3.
Statistics
Statistical analysis was performed in Graph Pad Prism version 8. Two group comparison was done using unpaired student’s t-test. Comparison of three groups was done by one-way ANOVA followed by appropriate post hoc test. Four group comparison was done by 2-way ANOVA with appropriate post hoc and mixed effects model was used if there were missing values. Myograph studies were analyzed by 2-way repeated measures ANOVA and appropriate post hoc to determine differences at specific concentrations. Datasets with significantly different standard deviations were compared using unpaired t-test with Welch’s correction or Brown-Forsythe/Welch one way ANOVA. Normal distribution was tested using Shapiro-Wilk test and a nonparametric test was used if a group was not normally distributed (p<0.05). Statistical and post hoc tests used are indicated in each figure legend. Values are mean +/− standard error. Significance was set at p<0.05.
Results
Prior exposure to preeclampsia is associated with salt sensitivity of BP in women
Women exposed to a preeclampsia pregnancy have previously been shown to have an enhanced BP response to hypertensive stimuli such as RAAS activation induced by AngII infusion compared to woman after a normal pregnancy.19 Women with severe preeclampsia also have enhanced BP response to high salt intake post-partum.23 We expanded on this finding by measuring salt-sensitivity of BP in women who had a previous normotensive versus a prior PE pregnancy, without limiting to severe preeclampsia. Prior to salt challenge, there was no significant difference in age, time since pregnancy, body mass index, creatinine, or hemoglobin A1C between women with normotensive or prior preeclamptic pregnancy. Urine sodium was not different between groups in response to low or high sodium intake (Table S1). Women were exposed to low salt or high salt diet, each for 1 week, in a crossover fashion, and SBP was measured at the end of each diet. The difference between the BP on low versus high salt is a measure of the responsiveness of BP to high salt intake. Women with a prior normotensive pregnancy had no significant difference in SBP while on low versus high salt diet. However, women with prior preeclampsia had a significant increase in SBP on high compared to low salt diet (Figure 1A). This was evidenced by 9 ± 3 mmHg increase in SBP on high salt compared to low salt diet in the women with prior preeclampsia which was significantly greater than those with prior normotensive pregnancy (Figure 1B), consistent with salt-sensitivity of BP.
Figure 1. Prior preeclampsia is associated with salt sensitive blood pressure in women.
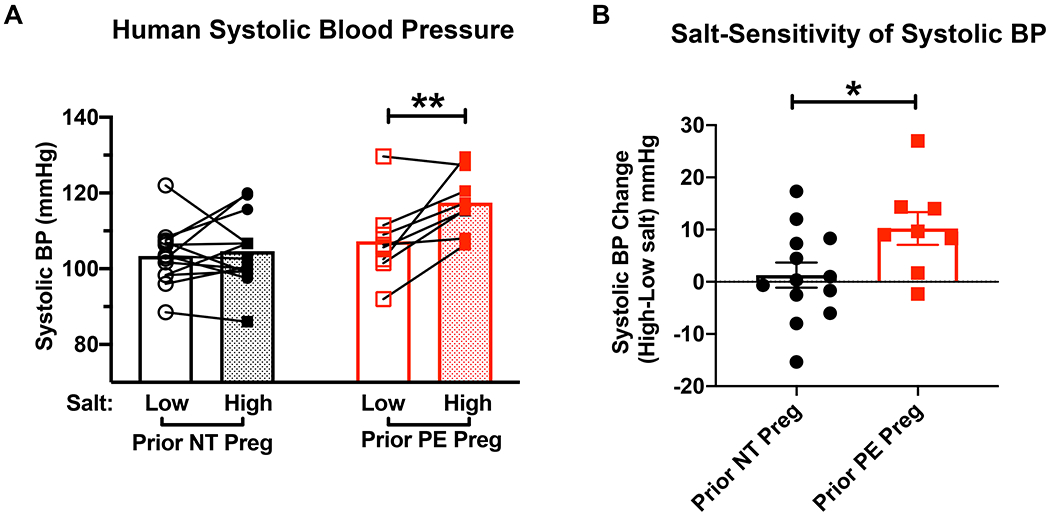
(A) Women with prior normotensive (NT, n=13) or preeclamptic (preeclampsia, n=8) pregnancies were subjected to one week of low salt diet and one week of high salt diet in a crossover design. Systolic blood pressure was measured at the end of each condition. **P=7.6x10−3 via repeated measures 2 way ANOVA with Sidak post hoc. (B)The change in systolic blood pressure from low to high salt measurements in mmHg. *P=3.5x10−2 via unpaired t-test
High sFlt1 exposure in mid-gestation in mice induces the features of human preeclampsia which resolve post-partum
As prior studies using the sFlt1-induced preeclampsia model utilized outbred CD1 mice, we first established the model in C57Bl/6 mice to enable use of KO mice on the C57Bl6 genetic background. Pregnant C57Bl6 mice were injected on GD9 with control adenovirus or sFlt1 adenovirus that transiently expresses sFlt1 during late gestation. sFlt1 virus injection into pregnant mice significantly increased SBP in late pregnancy (Figure 2A). To account for any variability of baseline BP, SBP for each mouse was also compared to their own baseline SBP and the area under the BP curve from GD8-18 was also found to be significantly greater for sFlt1-exposed mice (Figure 2B). Kidney damage in the form of glomerular endotheliosis, the hallmark histological lesion of human preeclampsia, was also significantly increased in pregnant sFlt1-injected females compared to control-injected females at the end of gestation. Two months post-partum, renal histology was normal in both groups (Figure 2C). As expected, sFlt1 virus injection significantly increases plasma sFlt1 level during pregnancy (Figure 1D). Two months post-partum, sFlt1 levels in both groups were down to non-pregnant levels and did not differ between groups (Figure 2E). Similarly, while SBP was significantly increased in the sFlt1 mice at the end of pregnancy, BP returned to normal 2 months post-partum with no statistically significant difference observed between control and sFlt1 exposed mice (Figure 2F). Thus, exposure to high sFlt1 during late gestation in C57Bl6 mice is sufficient to induce the preeclampsia phenotype of hypertension and glomerular endotheliosis during pregnancy, all of which resolve post-partum, similar to humans.
Figure 2. Exposure to elevated plasma sFlt1 in pregnant C57Bl/6 females is sufficient to phenocopy hypertension and renal damage seen in human preeclampsia.
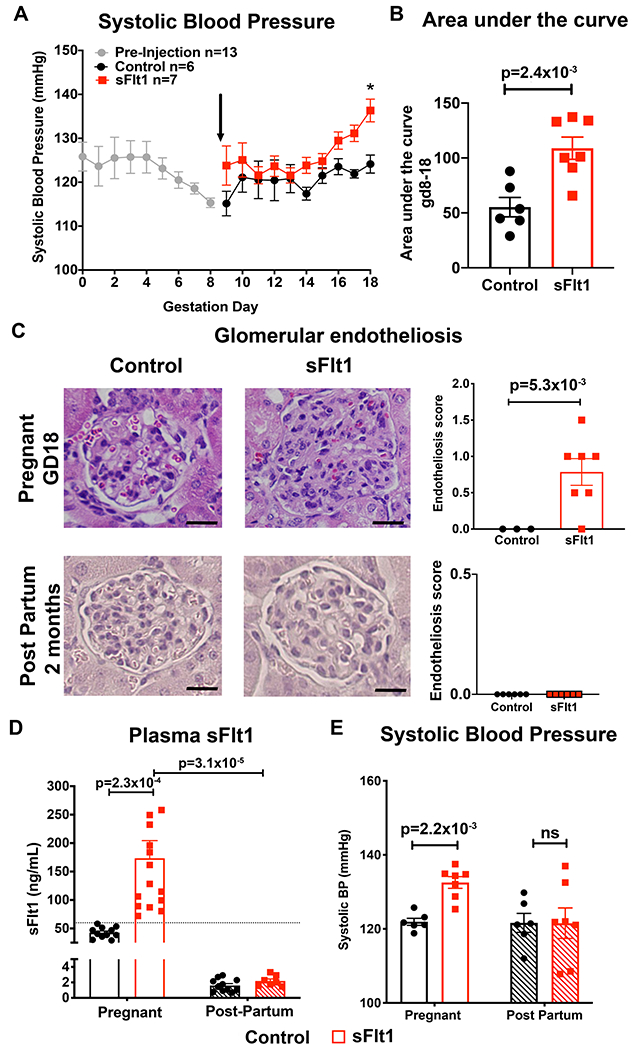
(A) Systolic blood pressure (SBP) measured during pregnancy with radiotelemetry. Black arrow denotes randomization to injection with either “Control” (CMV-Null transgene) or “sFlt1” adenovirus at gestation day (GD) 9. All mouse BP data were averaged together prior to randomization (GD0-8, n=13). Control n=6, sFlt1 n=7. *p=1.6x10−2 via two way repeated measures ANOVA with Sidak post hoc. (B) Area under the curve for SBP between GD8 before injection and the end of pregnancy (GD18) for each mouse. Control n=6, sFlt1 n=7. p value determined by unpaired student t test. (C) Representative images of glomeruli from kidneys at GD18 and 2 months post-partum, stained with hematoxylin and eosin. Average glomerular endotheliosis severity scored based on blinded analysis. Scale bar=20μm. Control n=3, sFlt1 n=7. p value determined by unpaired t-test with Welch correction. (D) Plasma was taken on GD17 and two months post-partum and sFlt1 measured using ELISA. Dashed line indicates exclusion value for sFlt1 levels of 60ng/mL. Control pregnant n=11, sFlt1 pregnant n=15, Control post-partum n=11 , sFlt1 post-partum n=7. p values determined via two way ANOVA with Sidak post hoc. (E) SBP at the end of pregnancy (GD17) and two months post-partum. Control pregnant n=6, sFlt1 pregnant n=7, Control post-partum n=6, sFlt1 post-partum n=7. p value determined by two way ANOVA with Sidak post hoc.
Prior exposure to preeclampsia is sufficient to induce sensitivity to future hypertensive stimuli in the mouse sFlt1-induced preeclampsia model
We next investigated whether the mouse sFlt1-induced preeclampsia model reproduces the human phenotype of enhanced BP responses to hypertensive stimuli using the paradigm in Figure 3A. Beginning 2 months post-partum, mice with implanted radiotelemetric BP monitors were subjected to normal sodium diet for 1 week (0.5% NaCl) followed by moderate (2% NaCl) salt intake for a week, then normal salt for 1 week followed by high salt intake (4% NaCl) for one week. Finally, mice were returned to normal salt intake for a week and then infused with AngII at a dose known to induce a moderate pressor response in female mice.35 Exposure to 2% salt diet resulted in no significant difference in the area under the curve (AUC) of the SBP response between groups (Figure 3B). However, previous exposure to sFlt1-induced preeclampsia during pregnancy resulted in a significantly greater AUC of SBP in response to high salt intake (4% NaCl), compared to mice exposed to a control pregnancy (Figure 3C). Similarly, mice exposed to prior sFlt1-induced preeclampsia had a significantly greater BP response to AngII (Figure 3D). AngII increases BP by two main mechanisms: inducing Aldo production by the adrenal to enhance renal sodium retention and by direct vasoconstriction of resistance vessels. The enhanced pressure response in sFlt1 mice was not associated with a difference in plasma Aldo level at the end of pregnancy, two months post-partum, or after AngII infusion (Figure 3E). Thus, to determine if this was a vascular-mediated effect, ex vivo mesenteric microvascular constriction was quantified after the hypertensive stimuli. Resistance vessel constriction to AngII was significantly greater in vessels taken from mice exposed to prior sFlt1-induced preeclampsia compared to those with prior control pregnancy (Figure 3F). These data together support that women with prior preeclampsia have enhanced BP response to high salt intake and that in mice, exposure to sFlt1 during pregnancy is sufficient to reproduce this finding of a persistently enhanced response to future hypertensive stimuli accompanied by enhanced vasoconstriction to AngII.19, 21
Figure 3. Prior preeclampsia results in sensitivity to post-partum hypertensive stimuli in mice.
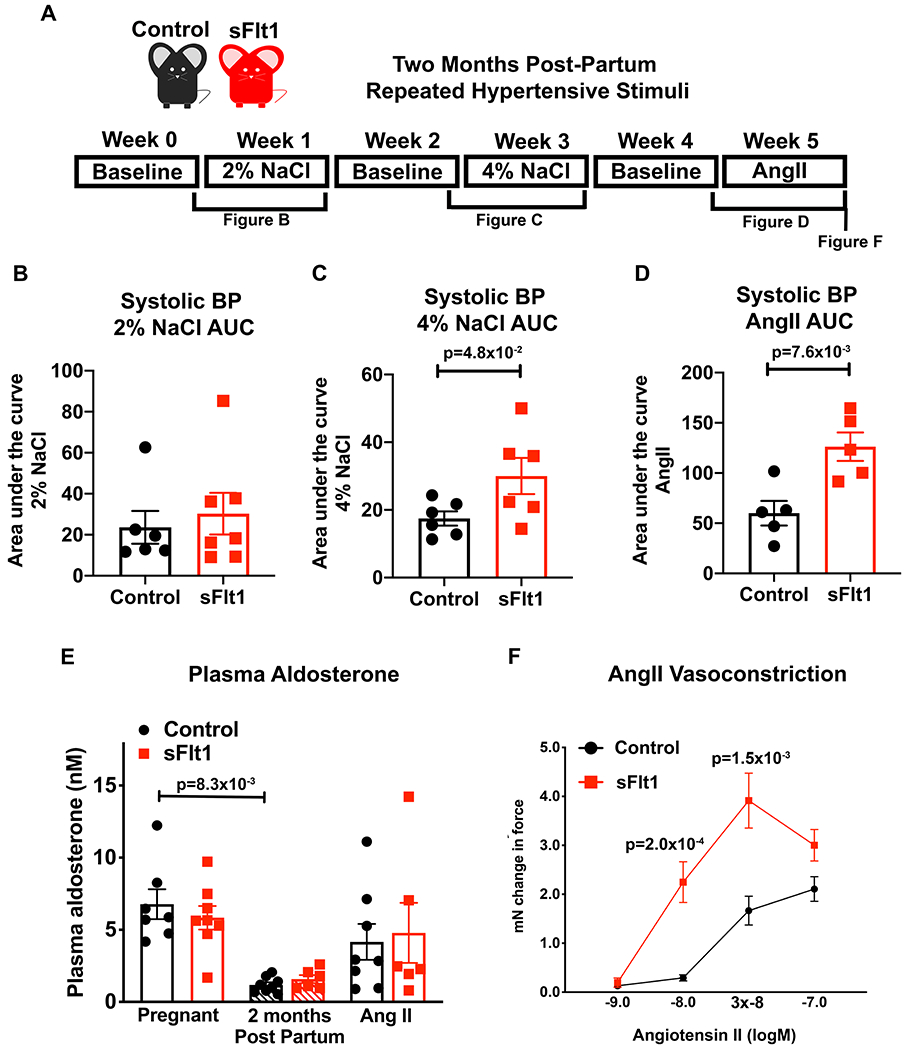
(A) Timeline of post-partum hypertensive stimuli protocol with baseline measurements on standard chow (0.5% NaCl) taken at two months post-partum. Mice were then subjected to 2%NaCl diet for one week, a week of washout, 4% NaCl for one week, a week of washout, and then one week of Angiotensin II (AngII) infusion (600ng/kg/day). The area under the SBP curve quantifies the change in BP response to 1 week exposure to: (B) 2% NaCl (Control n=6, sFlt1 n=7, p=9.5x10−1 via Mann-Whitney test), (C) 4% NaCl (Control n=6, sFlt1 n=6, p value determined by unpaired t-test), and (D) AngII infusion compared to each individual mouse’s baseline SBP (Control n=5, sFlt1 n=5, p value determined by unpaired student t test.) (E) Plasma Aldo at the end of pregnancy, two months post-partum and after repeated hypertensive stimuli. Control pregnant n=7, sFlt1 pregnant n=8, Control post-partum n=8, sFlt1 post-partum n=6, Control AngII n=8, sFlt1 AngII n=6, p value determined by Kruskal-Wallis with Dunn post hoc. (F) Ex vivo microvascular mesenteric artery vasoconstriction to AngII. Control n=7, sFlt n=9, p values determined by 2 way repeated measures mixed effects model, Sidak post hoc test.
SMC transiently exposed to sFlt1 exhibit increased mineralocorticoid receptor (MR) activity in response to subsequent agonist stimulation
We previously demonstrated that SMC-MR mediates the vasoconstrictive impact of AngII.28, 36 Since the AngII vasoconstriction response is enhanced after preeclampsia exposure in women and after sFlt1-induced preeclampsia and hypertensive stimuli in the mouse model (Figure 3F), we next investigated the impact of transient sFlt1 exposure on MR expression and activity in SMC (Figure 4). Pac1 cells, a transfectable SMC line, express endogenous MR as well as VEGFR1 and VEGFR2 (Figure S1) and VEGF-A is present in the serum-containing cell culture supernatant at a concentration of 112.7+/− 19.72 pg/mL (N=6). Pac1 SMCs were exposed to vehicle control or sFlt1 for 24 hours and then MR mRNA (Figure 4A) and protein (Figure 4B) expression were quantified. There was no statistically significant difference observed in SMC-MR expression after sFlt1 exposure. MR is a hormone-activated transcription factor so MR activity can be measured using a MR-responsive element (MRE)-driven luciferase reporter. Thus, SMC were treated with sFlt1 for 24 hours followed by sFlt1 washout for 24 hours. Then cells were transfected with the MRE-luciferase reporter and 6 hours later, MR was activated with either AngII (300 nM) or the traditional MR ligand, Aldo (10 nM). Prior sFlt1 exposure significantly enhanced MR responsiveness but the difference was only significant after stimulation with Aldo or AngII, not with vehicle alone (Figure 4C and 4D). This is consistent with no statistically significant difference observed in baseline BP in the women or mice with prior preeclampsia but a signficant change in the response to hypertensive stimuli.
Figure 4. Transient exposure of smooth muscle cells (SMC) to sFlt1 in vitro enhances mineralocorticoid receptor (MR) transcriptional activity and target gene expression upon receptor stimulation.
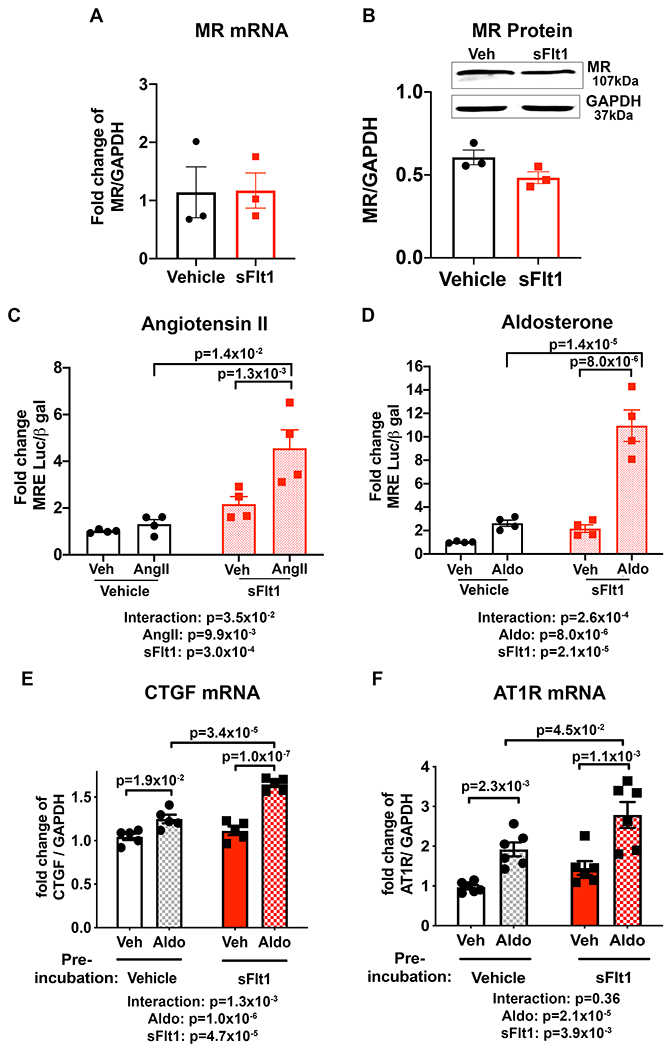
Pac1 SMC biological replicates were exposed to 50 ng/mL sFlt1 for 24 hours and expression of MR; (A) mRNA and (B) protein was measured. Control n=3, sFlt1 n=3. Representative western blots of MR and GAPDH in SMC and quantification are shown. 24 hours after sFlt1 was removed, MR transcriptional reporter activity was measured in response to MR stimulation with (C) angiotensin II (AngII, all groups n=4 independent experiments, p values determined by 2-way ANOVA with Sidak post hoc or (D) aldosterone (Aldo, all groups n=4 independent experiments, p values determined by 2-way ANOVA with Sidak post hoc compared to vehicle treated controls. Aldo-stimulated mRNA expression of MR target genes; (E) connective tissue growth factor (CTGF, all groups n=5 independent experiments, p values determined by 2-way ANOVA with Sidak post hoc and, (F) AngII type 1 receptor (AT1R), all groups n=6 independent experiment, p values determined by 2-way ANOVA with Sidak post hoc.
As these data support enhanced MR responsiveness on a heterologous promoter, we next tested whether this translates into changes in expression of a well characterized SMC-MR transcriptional target gene, connective tissue growth factor (CTGF). Consistent with published data37, Aldo increases CTGF mRNA expression in SMC (Figure 4E). Pretreatment of SMC with sFlt1 significantly enhanced the subsequent Aldo-induced increase in CTGF mRNA expression, further supporting that transient sFlt1 exposure enhances the future responsiveness of SMC-MR. Since SMC-MR has been shown to mediate AngII vasoconstriction at least in part by regulating angiotensin type 1 receptor (AT1R)36, the impact of sFlt1 pre-treatment on AT1R expression was also quantified. Once again, Aldo induction of AT1R expression in SMC was significantly exacerbated when SMC were previously exposed to sFlt1 (Figure 4F). As with the reporter assays in Fig 4C–D, enhanced induction of MR target genes after sFlt1 exposure of SMC was only significant after stimulation with Aldo. Expression of angiotensin type 2 receptor (AT2R), which opposes AT1R and mediates vasodilation, was decreased in vitro when SMC were pretreated with sFlt1 (Figure S2A). These data support that transient sFlt1 exposure enhances the future responsiveness of SMC-MR to hypertensive stimuli including Aldo and AngII.
Mice lacking SMC-MR are protected from enhanced responsiveness to hypertensive stimuli after exposure to sFlt1-induced preeclampsia
To test the role of SMC-MR activation in mediating the post- preeclampsia sensitivity to hypertension in vivo, the sFlt1-induced preeclampsia model followed by post-partum exposure to hypertensive stressors was repeated, comparing mice with the MR specifically deleted from SMC (SMC-MR-KO) versus SMC-MR-WT littermates. For all studies, there were no statistically significant differences observed in any measured outcomes in SMC-MR-WT and SMC-MR-KO after exposure to control pregnancy, thus, those data were combined for further analysis. The preeclampsia phenotype was compared between pregnant SMC-MR-WT and SMC-MR-KO mice. We confirmed that plasma sFlt1 levels at the end of pregnancy were significantly and similarly increased by sFlt1 injection in pregnant SMC-MR-WT and SMC-MR-KO mice (Figure 5A). There was also no significant difference in in plasma Aldo levels during pregnancy (Figure 5B) between the two genotypes. Telemetry devices were implanted into SMC-MR-KO mice and SMC-MR-WT littermates and BP was measured during pregnancy in response to sFlt1 expression by adenoviral injection. Systolic BP was not different between groups in the first half of pregnancy prior to sFlt1 injection. Upon injection at GD9, blood pressure increased significantly and equivalently between WT and KO (Figure S3). As such, the area under the curve for BP from GD8-17 was the same regardless of the presence of SMC-MR (Figure 5C). Mice exposed to elevated sFlt1 developed glomerular endotheliosis during pregnancy, consistent with end organ damage from the preeclampsia model which also did not differ by genotype (Figure 5D). These data demonstrate that lack of SMC MR during pregnancy does not impact the preeclampsia phenotype in response to high sFlt1.
Figure 5. SMC-MR-KO mice exposed to high sFlt1 in pregnancy are protected from exacerbated response to post-partum hypertensive stress.
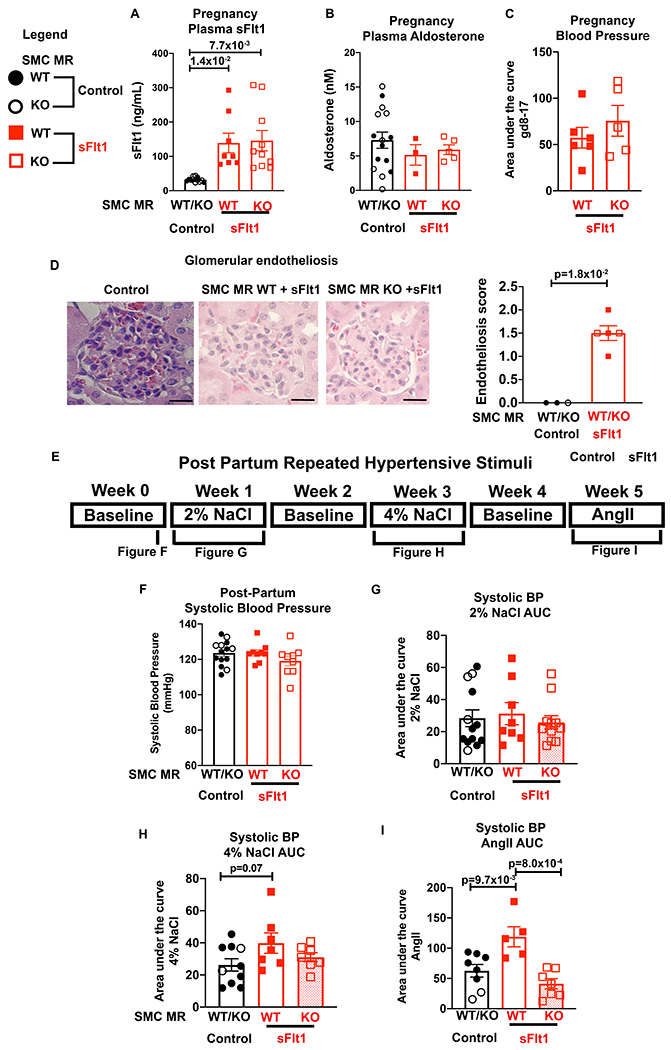
Pregnant SMC-MR-WT or SMC-MR-KO females were injected with Control or sFlt1 on GD9, allowed to deliver, and then exposed to hypertensive stimuli 2 months post-partum. Control injected females were combined for analyses as there were no statistically significant differences between WT and KO. (A) Plasma sFlt1 levels on GD17. Control n=12, WT sFlt n=8, KO sFlt n=10, p values determined via Welch’s one way ANOVA and Dunnett’s multiple comparisons. (B) Plasma aldosterone level on GD17. Control n=15, SMC-MR-WT sFlt n=3, SMC-MR-KO sFlt n=5. (C) Area under the curve for SBP between GD8 before injection and the end of pregnancy (GD17) for each mouse. SMC-MR-WT sFlt1 n=6, SMC-MR-KO sFlt1 n=5. (D) Representative images of glomeruli from mice on GD18, stained with hematoxylin and eosin. Scale bar=20μm. Average glomerular endotheliosis severity scored based on blinded analysis. Control n=3, WT sFlt n=3, KO sFlt n=2, p values determined by Mann-Whitney test. E) Timeline of post-partum hypertensive stimuli protocol. (F) SBP was measured via radiotelemetry two months post-partum. Control n=10, WT sFlt n=5, KO sFlt n=5. (G) The SBP area under the curve (AUC) for 2% NaCl. Control n=13, WT sFlt n=8, KO sFlt n=11. P=8.3x10−1 via Kruskal-Wallis test. (H) The SBP AUC during 4% NaCl. Control n=10, WT sFlt n=7, KO sFlt n=7, p values determined by one way ANOVA and Dunnett’s post hoc test. (I) The SBP AUC during AngII. Control n=8, WT sFlt n=5, KO sFlt n=7, p values determined by one way ANOVA and Dunnett’s post hoc test. All SBP was compared to each individual mouse’s baseline.
Two months post-partum, mice were implanted with radiotelemetry devices and subjected to repeated hypertensive stimuli (Figure 5E). Prior to initiation of hypertensive stress, SBP was normal and not significantly different between groups (Figure 5F). As in the WT C57Bl6 mice, the response to 2% NaCl diet was not impacted by exposure to sFlt1 during pregnancy (Figure 5G). When mice received excess dietary sodium at 4% NaCl, the MR-intact mice exposed to sFlt1 during pregnancy tended to have an increased SBP response (similar to Figure 3C) but this was not statistically significant (p= 0.07, Figure 5H). Once again, MR-intact mice exposed to sFlt1-induced preeclampsia had a significantly greater BP response to AngII infusion and this enhanced responsiveness to AngII was prevented in SMC-MR-KO mice exposed to sFlt1 (Figure 5I). These data support that SMC-MR is necessary for the enhanced hypertensive response to AngII after exposure to sFlt1-induced preeclampsia.
Mice lacking SMC-MR are protected from enhanced vascular stiffness, microvascular tone and AngII constriction after exposure to sFlt1-induced preeclampsia
SMC-MR has been shown to contribute to conduit vessel stiffness and rising BP in the aging vasculature by contributing to microvascular myogenic tone (the vasoconstrictive response to increasing BP) and vasoconstriction to AngII.29, 36, 38 Preeclampsia in women has also been linked with premature vascular aging.39 Thus, we examined the impact of sFlt1-induced preeclampsia on aortic stiffness and resistance vessel myogenic tone and constriction to AngII, comparing mice with MR intact to SMC-MR-KO littermates. Two months post-partum, there was no difference in aortic stiffness, as measured by pulse wave velocity (PWV), between SMC-MR-WT or SMC-MR-KO mice exposed to control pregnancy or sFlt1-induced preeclampsia (Figure S4). However, exposure to repeated hypertensive stimuli significantly increased vascular stiffness in MR intact mice exposed to prior preeclampsia and this was completely inhibited in SMC-MR-KO mice (Figure 6A). Next, resistance vessel myogenic tone after hypertensive stress was measured by pressure myography. sFlt1-induced preeclampsia significantly increased mesenteric myogenic tone in SMC-MR intact mice compared to WT mice exposed to control pregnancy. This enhanced vasoconstrictive response to increasing pressure after preeclampsia was prevented in SMC-MR-KO (Figure 6B). As in Figure 3F, ex vivo mesenteric microvascular constriction to AngII was significantly increased in vessels from SMC-MR-WT mice after prior sFlt1 exposure and subsequent hypertensive stress, compared to control pregnancy. This enhanced constriction to AngII was also prevented in SMC-MR-KO mice (Figure 6C). To examine the potential mechanism, we measured mesenteric vessel mRNA expression of the vasodilatory AT2R and the vasoconstrictive AngII type 1b receptor (AT1Rb is the murine AT1R expressed in mesenteric vessels). AT2R expression was not significantly altered in mesenteric vessels of sFlt1- injected SMC-MR-WT or SMC-MR-KO vessels after post-partum hypertensive stimuli, a pattern distinct from that found in SMC in vitro (Figure S2B). AT1Rb expression in mesenteric resistance vessels from SMC-MR-WT mice was significantly increased after exposure of to sFlt1-induced preeclampsia followed by hypertensive stimuli (Figure 6D). The increased expression of AT1Rb was prevented in SMC-MR-KO mice resulting in significantly less AT1Rb expression in SMC-MR-KO compared to MR-intact mice exposed to preeclampsia. These results are consistent with the in vitro data in Figure 4 in which pretreatment of SMC in vitro with sFlt1 exacerbates MR driven upregulation of AT1R expression in SMC and MR responsiveness to AngII.
Figure 6. SMC-MR is necessary for post- preeclampsia aortic stiffness, microvascular dysfunction and AT1R upregulation in response to hypertensive stimuli.
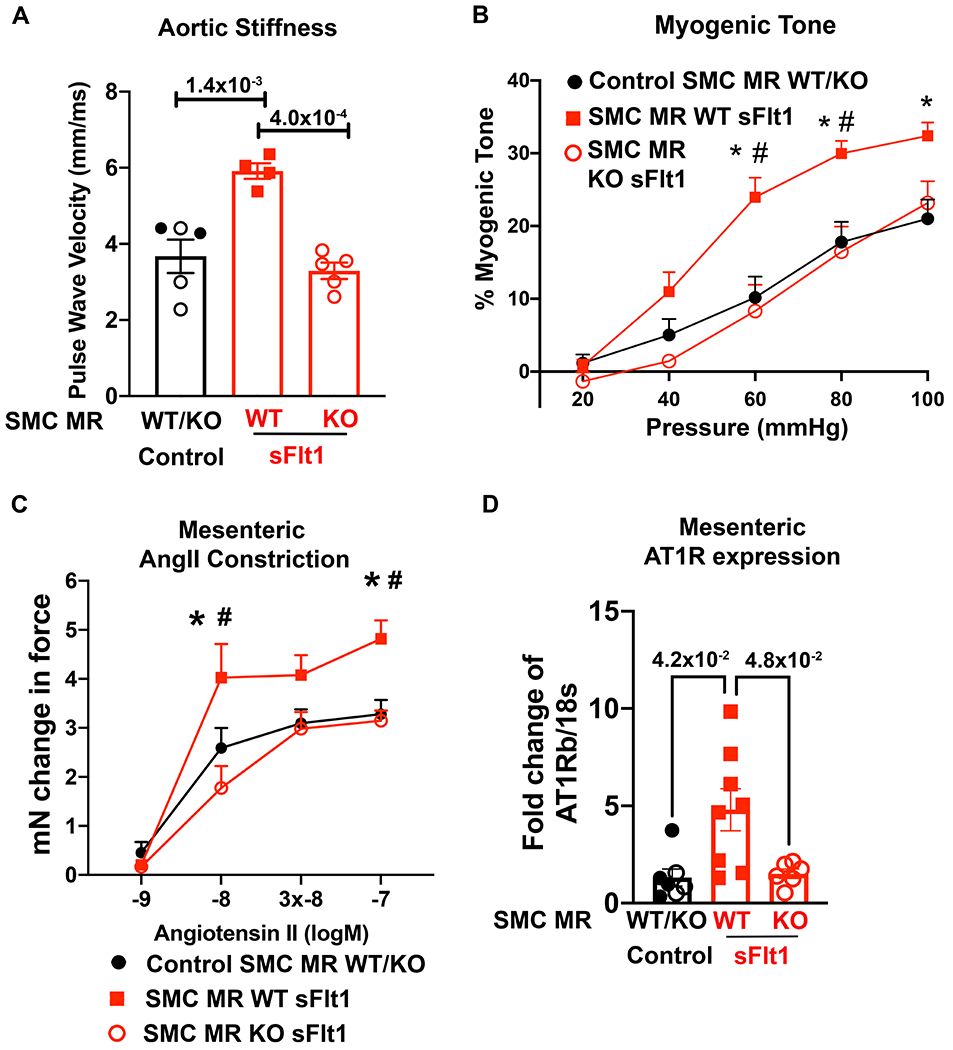
In vivo aortic stiffness and ex vivo mesenteric microvessels were compared between Control mice (SMC-MR-WT and SMC-MR-KO exposed to control pregnancy, SMC-MR-WT exposed to sFlt1-induced preeclampsia, and SMC-MR-KO exposed to sFlt1-induced preeclampsia, all after hypertensive stimuli. (A) Aortic stiffness was measured using pulse wave velocity of the abdominal aorta. Control n=5, WT sFlt n=4, KO sFlt n=5, p values determined by one way ANOVA with Tukey post hoc test. (B) Myogenic tone was measured in control mice (n=15), SMC-MR-WT sFlt (n=11) and SMC-MR-KO mice (n=9). At 60mmHg: *p=7.0x10−4 Control versus SMC-MR-WT sFlt, #p=7.0x10−4 SMC-MR-WT sFlt vs SMC-MR-KO sFlt. At 80mmHg: *p=4.7x10−3 Control versus SMC-MR-WT sFlt, #p=6.0x10−3 SMC-MR-WT sFlt vs SMC-MR-KO sFlt. At 100mmHg: *p=1.1x10−2 Control versus SMC-MR-WT sFlt. p values determined by 2 way repeated measures ANOVA with Sidak post hoc. (C) Ex vivo microvascular mesenteric constriction to AngII was measured in controls (n=14), SMC-MR-WT sFlt (n=5), and SMC-MR-KO mice (n=9). At 10−8 concentration: *p=3.0x10-2 Control vs SMC-MR-WT sFlt, #p=7.0x10−4 SMC-MR-WT sFlt vs SMC-MR-KO sFlt. At 10−7 concentration: *p=1.8x10−2 Control vs SMC-MR-WT sFlt, #p=1.6x10−2 SMC-MR-WT sFlt vs SMC-MR-KO sFlt via two-way repeated measures ANOVA with Sidak post hoc. (D) Mesenteric vessels were isolated and mRNA expression of AT1Rb was quantified. Control n=7, WT sFlt n=8, KO sFlt n=6, p values determined by Brown-Forsyth/Welch ANOVA and Dunnett’s post hoc test.
Discussion
In summary, this study demonstrates that short term sFlt1 exposure in pregnant mice is sufficient to induce a future state of enhanced sensitivity to hypertensive stress, as seen in women after preeclampsia. However, in mice, this can be interpreted to be independent of preexisting CVD risk factors. Further exploration of the mechanism reveals that vascular SMCs transiently exposed to sFlt1 develop a persistent hypersensitivity of the MR to hormone-dependent or hormone–independent activation that drives post-partum BP response via enhanced resistance vessel myogenic tone, AT1R expression and vasoconstriction. Specifically, this study: 1) validated adenoviral-mediated expression of sFlt1 in pregnant C57Bl6 mice as a model to explore the mechanism for post-partum CVD risk by confirming key features of preeclampsia during pregnancy, including elevated late term BP, high sFlt1 levels, and renal endotheliosis that all resolve post-partum; 2) Two months after exposure to sFlt1-induced preeclampsia, the BP response to high salt diet and AngII infusion was significantly increased in this mouse model. This is consistent with enhanced AngII responsiveness previously shown in post- preeclampsia women19 and our new data showing that a prior preeclampsia diagnosis in women associates with post-partum salt sensitivity of BP that is not seen in women after normotensive pregnancy; 3) Mechanistic studies show that transient exposure of SMC to sFlt1 in vitro results in persistently enhanced MR transcriptional responsiveness and target gene expression, including MR-induced expression of the AT1R; 4) As such, mice previously exposed to sFlt1-induced preeclampsia that are challenged with hypertensive stress from high salt and RAAS activation, develop increased; microvascular AT1bR expression, AngII vasoconstriction, myogenic tone, and blood pressure compared to mice with a prior control pregnancy; 5) Finally, SMC-MR-KO mice were protected from the detrimental response to hypertensive stimuli in this model, including protection from enhanced vascular stiffness, increased resistance vessel myogenic tone, upregulation of AT1bR and sensitivity to AngII-induced vasoconstriction and BP elevation. These data support the model in Figure 7 where transient sFlt1 exposure during pregnancy modifies MR responsiveness in vascular SMC. This persists after resolution of preeclampsia in a latent state until it manifests upon exposure to poor diet, stress or aging post-partum. Upon exposure to hypertensive stress, SMC-MR is hyperactivated, enhancing AT1R transcription, promoting microvascular constriction and myogenic tone resulting in increased BP response when exposed to hypertensive stress. In the mouse model, this occurs even in the absence of other pre-existing or predisposing risk factors for CVD such as obesity or family history which may further exacerbate risk in women.
Figure 7. Summary Figure.
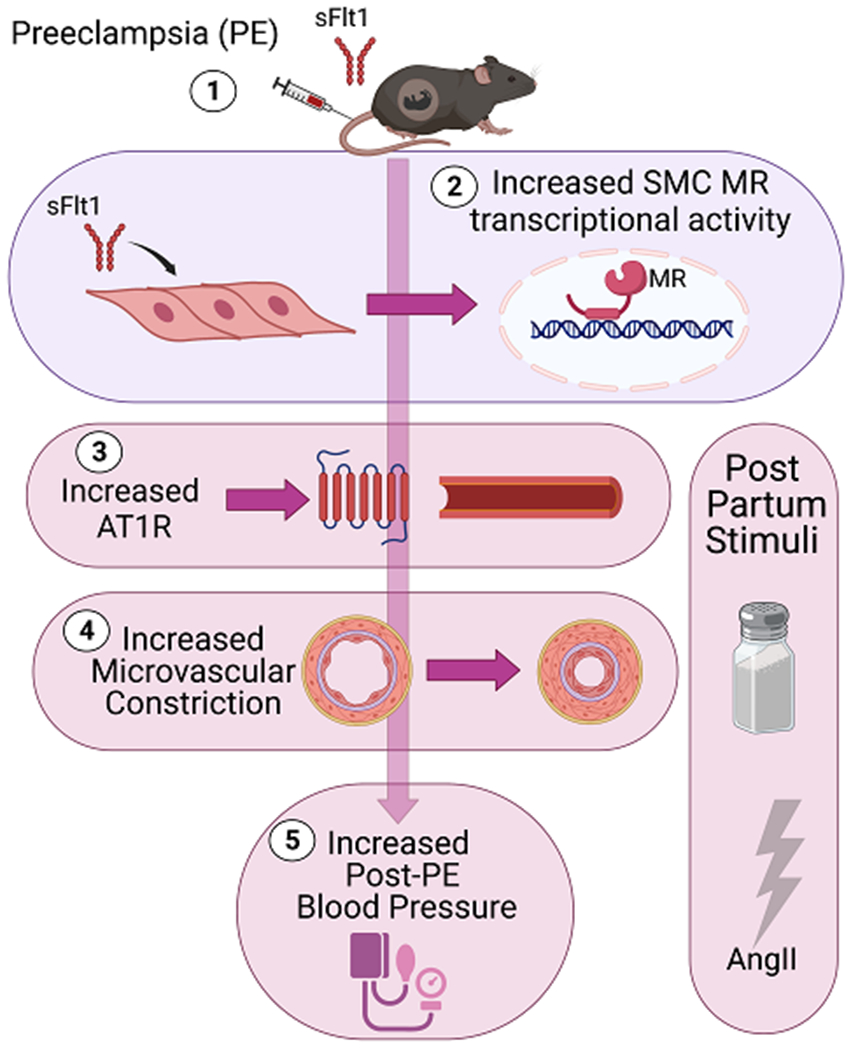
(1) Adenoviral sFlt1 injection during mid-gestation induces preeclampsia -like signs in mice that resolve post-partum. (2) sFlt1 exposure enhances SMC-MR transcriptional activity and target gene expression, including increased AT1R expression. When formerly preeclamptic mice are challenged post-partum with hypertensive stimuli (high dietary salt, AngII) they exhibit; (3) increased microvascular AT1R and (4) increased microvascular constriction that contributes to (5) exacerbated BP response to hypertensive stress. SMC-MR gene deletion in mice protects against the increased microvascular AT1R expression, enhanced myogenic tone and vasoconstriction and mitigates the exacerbated BP responsiveness after preeclampsia. Thus, the sFlt1-induced preeclampsia model in mice reproduces the post-partum hypertensive sensitivity seen in post- preeclampsia women in a manner that is independent of preexisting risk factors and depends on the presence of SMC-MR.
Females are typically protected from CVD prior to menopause, but P preeclampsia E negates this protection. Ample published data shows that MR expression rises with age in SMC and contributes to cardiovascular aging by promoting vascular fibrosis and stiffness38, 40, enhancing AT1R expression and vasoconstriction to AngII36, 41, and contributing to myogenic tone and rising BP with age.29 Thus, one unifying concept might be that sFlt1 exposure during preeclampsia leads to accelerated vascular aging by activating SMC-MR. A recent study showed that sFlt1 expression increases with age in mice and that the decline in VEGF signaling with age contributes to the multi-organ aging phenotype by unknown mechanisms.42 Our study suggests that the accelerated vascular aging phenotype in preeclampsia is mediated by sFlt1-induced SMC-MR activation and thus SMC-MR-KO is protective by preventing this aging phenotype. There are distinct mechanisms of vascular aging in females and these mechanisms are differentially impacted by SMC-MR. For example, SMC-MR expression is increased with age in both sexes but this happens sooner in males.36, 41, 43 A similar pattern is seen with microvascular AngII constriction and aortic stiffness as both increase with age, but this occurs sooner in males, consistent with the timing of changes in SMC-MR expression.41 Pregnancy or AngII infusion increases Aldo levels, and Aldo exposure leads to ubiquitination and degradation of MR.44, 45 However, in this study, the preeclampsia signs (blood pressure, kidney damage) were not changed by SMC-MR-KO nor did sFlt1 exposure alter circulating Aldo in vivo. In vitro, sFlt1 exposure did not modulate SMC-MR mRNA expression either. While we cannot measure MR protein in mice due to the lack of a murine-specific antibody, these data suggest that the effects are likely independent of changes in Aldo levels or MR expression. Rather, SMC-MR appears to be more sensitive to activation by Aldo or AngII after preeclampsia, thereby leading to increased AT1R expression.36 It has been previously shown that MR can also be activated in a ligand-independent manner by Rac1 signaling, which contributes to salt-sensitive hypertension.46, 47 Ligand-independent SMC-MR activation also occurs through AT1R signaling via PKC delta, resulting in post-translational MR modification that is synergistic with ligand to enhance MR transcriptional function.27, 28 Here we focused on MR in SMC since it is implicated in vasoconstriction and BP control. In a leptin infusion model of preeclampsia, EC-MR deletion protected against EC dysfunction and hypertension during pregnancy.48 The long term effects of EC MR on post- preeclampsia enhanced CVD risk remain to be determined. In our sFlt1 preeclampsia model, the presence of SMC-MR did not impact the preeclampsia phenotype during pregnancy but rather had beneficial effects in the post-partum ‘premature vascular aging’ phenotype. Further studies are needed to determine the mechanism by which sFlt1 modifies SMC-MR sensitivity in the setting of preeclampsia.
Clinical data also reveal exacerbated AngII responsiveness in post-partum women with prior preeclampsia. In response to AngII infusion, women exposed to preeclampsia have a significantly greater BP response22 and greater skin microvasculature constriction21 compared to women with prior normotensive pregnancies. In both of these published studies, skin biopsies showed increased expression of AT1R21 and a correlation between AT1R expression and the AngII BP response in women after preeclampsia.22 Preclinical evidence shows that resistance vessel AT1R also functions as a mechanosensor to mediate myogenic constriction in response to increased pressure.49, 50 Our data provide a potential novel mechanism underlying these observations in post- preeclampsia women. High sFlt1 during pregnancy, which is observed in up to 90% of preeclampsia patients51, may have enhanced SMC-MR sensitivity resulting in greater upregulation of AT1R expression in response to the hypertensive stresses experienced by most women, and leading to enhanced myogenic tone and microvascular constriction and increased BP response to hypertensive stress in post- preeclampsia women. We also examined the impact of sFlt1 on expression of the vasodilatory AT2R that opposes AT1R vasoconstriction. Here we found distinct effects in cultured SMC treated in vitro versus mesenteric vessels exposed to sFlt1 in vivo. As the vessels also include endothelial cells that express AT2R and gene expression in vivo may also be modulated in response to changes in BP, the differences between the in vitro and in vivo results could have multiple explanations. Overall, the in vitro and in vivo data consistently support a role for SMC-MR induction of AT1R in the post- preeclampsia enhanced responsiveness to hypertensive stress in mice and humans.
preeclampsia This is important clinically, as it suggests this phenotype may apply to the larger population of women exposed to preeclampsia regardless of severity. Both our study and the severe preeclampsia study showed that post-partum BP was within the normotensive range prior to salt challenge. Since the majority of people worldwide consume more salt than recommended54, most women exposed to preeclampsia during pregnancy are subsequently exposed to high salt and other hypertensive stresses (psychological stress, RAAS activation, aging).22, 24, 55 Whether enhanced salt-sensitive BP after preeclampsia is a direct consequence of preeclampsia or due to pre-existing salt sensitivity that predispose to both preeclampsia and early onset hypertension has been a critical question in the field. Indeed, in our small clinical study, a subset of women with a normotensive pregnancy also appear to be salt sensitive. This is consistent with recent data demonstrating that women are more likely than men to have salt-sensitive hypertension and that some normotensive premenopausal women have salt sensitive BP due to adrenal, renal, or genetic factors.56 Since our animal model uses WT C57Bl/6 mice without any genetic or environmental predisposition to hypertension, the data in this study support that exposure to sFlt1 during pregnancy is sufficient to enhance the future response to hypertensive stimuli, even in the absence of predisposition or preexisting to CVD. This does not rule out the potential for synergy between pre-existing risk factors and the damage caused by sFlt1 exposure during preeclampsia.
preeclampsia SMC-MR can contribute to vascular stiffness by increasing vessel fibrosis38, 40, 41, calcification59 or enhancing SMC adhesion to fibronectin in the extracellular matrix via regulation of integrin expression.40 Since vascular stiffness contributes to heart and kidney failure, exacerbates hypertension (and is induced by hypertension), and correlates with adverse CVD outcomes, the mechanism for increased vascular stiffness after preeclampsia warrants further investigation.
As preeclampsia and CVD share common risk factors, it has been proposed that preeclampsia is a manifestation of pre-existing cardiovascular risk due to pregnancy as a stress test on the cardiovascular system. That hypothesis has been difficult to test in humans, but mouse models provide the ability to study preeclampsia in the absence of any predisposition to CVD. Indeed, post-partum study of the sFlt1-induced preeclampsia mouse model indicates that preeclampsia can induce persistent cardiovascular damage in the absence of contributing CVD risk factors like hypertension or dyslipidemia.60, 61 A previously published study showed that mice exposed to prior preeclampsia increased vascular remodeling with SMC proliferation and fibrosis in response to post-partum carotid injury.62 Together with this study, these findings are consistent with the concept that sFlt1 exposure during pregnancy is “remembered” upon encounter with subsequent cardiovascular injury. Our data showing that prior preeclampsia results in enhanced mesenteric vasoconstriction post-partum is consistent with a study using a rat model of preeclampsia induced by reduced uteroplacental perfusion, showing enhanced mesenteric vasoconstriction to phenylephrine post-partum63 with no impairment in vasodilation.64 The complement component 1q (C1q) KO model of preeclampsia showed persistent aortic endothelial dysfunction post-partum, although hypertension never fully resolved in that model.65 Thus, multiple preclinical preeclampsia models support the concept that exposure to preeclampsia is sufficient to induce persistent post-partum vascular dysfunction with enhanced response to cardiovascular stress in the absence of preexisting risk factors.
Limitations of this study are that the sFlt1 model bypasses placental ischemia, posited to be an initiating factor of preeclampsia. We chose the model in part because it is reversible, unlike some models. However, this approach is likely modeling only a subset of the preeclampsia phenotype as there are surely other factors that also contribute to post- preeclampsia CVD risk. sFlt1 is produced by the placenta, increases in the majority of preeclampsia patients prior to diagnostic signs, and circulates in significantly higher concentrations with increasing severity of preeclampsia 12, 14, therefore ample clinical data support an important role of sFlt1 in preeclampsia pathophysiology. Since sFlt1 is sufficient to induce a post-partum phenotype in this mouse model, the findings provide more specific information about the mechanism of post- preeclampsia sensitivity to hypertensive stimuli that are specifically downstream of sFlt1. Although VEGF receptors are classically thought to be expressed in endothelial cells, VEGFR1 (Flt1) is also expressed in vascular smooth muscle cells.66 In models of vascular injury, vascular smooth muscle cells upregulate Flt1 expression and thus are particularly sensitive to endothelial growth factors.67 Additional studies are needed to elucidate how interruption of Flt1 signaling leads to enhanced SMC-MR activation. Preeclampsia also predisposes to cardiac dysfunction and cardiac fibrosis, which could be mediated by SMC-MR; we did not investigate this as it is outside the scope of the current study. We also did not explore the role of auto-antibodies to the AT1R that have been shown to increase with preeclampsia and persist in a subset of women after preeclampsia.68 However, it has been suggested that AT1R auto-antibodies are upstream of sFlt1 production69 and it is unclear how the auto-antibodies affect AT1R expression, so this mechanism is unlikely to contribute to the post- preeclampsia phenotype observed in this model. There are also some obvious differences between the in vitro and in vivo studies in our study. SMC were exposed only to sFlt1 in vitro while in the mouse model, in vivo SMC are exposed to sFlt1 and hypertension during pregnancy. In vitro, transient sFlt1 exposure is sufficient to enhance SMC-MR transcriptional activity. Whether transient hypertension alone is sufficient to induce future CVD by the same mechanism (without elevated sFlt1) cannot be determined from the studies reported here, thus future experiments are needed. Finally, while we have used the Shapiro-Wilk test to determine normality, using parametric tests for sample sizes smaller than 6 may reduce reproducibility or rigor of the findings
Despite these limitations, this mouse model phenocopies the human post preeclampsia condition quite well. The novel mechanism linking elevated sFlt1 to enhanced sensitivity of SMC-MR activation to mediate hypertension has clinical implications for women with a history of preeclampsia as well as other conditions like cancer therapy-associated vascular toxicity. VEGF receptor inhibitors, including small molecule kinase inhibitors, anti-VEGF antibodies, or VEGF traps have reduced cancer related mortality by inhibiting angiogenesis. However, VEGFR inhibitors have the undesired effect of raising BP and causing proteinuria, a syndrome that resembles preeclampsia and can lead to discontinuation or dose reduction of life-saving cancer treatment.70 sFlt1 acts as a VEGF trap in circulation to inhibit VEGF action during the high angiogenic state of pregnancy, similar to the way VEGF inhibitors blunt tumor angiogenesis. Patients who received RAAS inhibition prior to anti-VEGF therapy for cancer had blunted BP response to the vasculotoxic drugs71, supporting a similar mechanism to what we identified in preeclampsia and post- preeclampsia hypertension. Thus, further studies are warranted to determine if post-partum MR inhibition in women with a history of preeclampsia (or cancer patients treated with anti-angiogenic therapy) may be a viable strategy to mitigate future hypertension and CVD.
Supplementary Material
Novelty and Significance.
What is known?
Preeclampsia is a common hypertensive disease of pregnancy that is associated with a high risk of early cardiovascular disease (CVD) post-partum, with a particularly high risk of developing hypertension.
Women with prior preeclampsia have an increased blood pressure and microvascular vasoconstriction response to hypertensive stimuli like angiotensin II.
Since preeclampsia and CVD share common risk factors, it has been difficult to determine how much of the post-partum CVD risk is due to pre-existing risk versus direct damage caused by preeclampsia.
What new information does this article contribute?
In a mouse model of soluble fms-like tyrosine 1 (sFlt1)-induced preeclampsia in the absence of pre-existing risk factors, preeclampsia is sufficient to drive post-partum hyperresponsiveness to hypertensive stimuli.
Mice exposed to prior preeclampsia develop increased aortic stiffness and microvascular vasoconstriction in response to post-partum hypertensive stimuli.
Smooth muscle mineralocorticoid receptor is more transcriptionally active after sFlt1 exposure and its deletion in mice prevents the detrimental cardiovascular response to hypertensive stimuli after preeclampsia.
Preeclampsia occurs in 5-8 percent of pregnancies. Epidemiological and clinical studies demonstrate that women exposed to preeclampsia during pregnancy have a substantially increased risk of future hypertension. These women have increased salt-sensitive blood pressure and microvascular responsiveness to angiotensin II but whether this is caused by preeclampsia exposure or simply a correlation is unknown. Using a mouse model of preeclampsia that reproduces the human phenotype, this study demonstrates that exposure to sFlt1 during pregnancy is sufficient to increase future response to hypertensive stimuli, including high salt diet and angiotensin II infusion. This model also reveals that preeclampsia exposure increases post-partum microvascular vasoconstriction and large vessel stiffness. The mechanism involves enhanced activation of smooth muscle mineralocorticoid receptor in the vasculature after exposure to preeclampsia. This may provide a viable therapeutic target to prevent future hypertension in these high risk women.
Acknowledgements
Suzanne Burke, PhD for assistance in establishing the sFlt1 model in C57Bl/6 mice, Greg Martin and Mark Aronovitz at the Molecular Cardiology Research Institute Mouse Surgical Core for surgical expertise, Nathan Li at the Tufts Histology Core for processing and staining of kidney tissue, and Estelle Kelty at Brigham and Women’s Hospital for clinical research data analysis. Figure 7 was created using Biorender.com
Sources of Funding:
This work was supported by grants from the National Institutes of Health: T32HL007609 (LAB), K99HL161321 (LAB), NIH F30HL152505 (JJM), R25GM066567 (BVC), R56 HL118085 (ESW), K24HL096141 (ESW), R01HL119290 (IZJ) and the American Heart Association: 22POST903910 (JI) and 20AIREA35120469 (ZZ).
Disclosures:
Dr. Karumanchi is a co-inventor on numerous biomarker patents in preeclampsia that are held by Beth Israel Deaconess Medical Center and financial interest in Comanche Biopharma and Aggamin Therapeutics. In addition, Dr. Karumanchi has received grants from Thermofisher and Siemens and has served as a consultant to Roche diagnostics.
Abbreviations
- Aldo
Aldosterone
- AngII
angiotensin II
- AT1R
angiotensin II receptor type 1
- BP
blood pressure
- CTGF
connective tissue growth factor
- CVD
cardiovascular disease
- GD
gestation day
- MR
mineralocorticoid receptor
- RAAS
renin angiotensin Aldosterone system
- SBP
systolic blood pressure
- sFlt1
soluble Fms-like tyrosine kinase 1
- SMC
smooth muscle cell
- SMC-MR-WT
smooth muscle cell mineralocorticoid receptor intact mouse
- SMC-MR-KO
smooth muscle cell mineralocorticoid receptor knockout mouse
- VEGF
vascular endothelial growth factor
- WT
wild type
References:
- 1.Chappell LC, Cluver CA, Kingdom J,Tong S. Pre-eclampsia. Lancet. 2021;398:341–354. [DOI] [PubMed] [Google Scholar]
- 2.Bellamy L, Casas JP, Hingorani AD,Williams DJ. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: systematic review and meta-analysis. BMJ. 2007;335:974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ray JG, Vermeulen MJ, Schull MJ,Redelmeier DA. Cardiovascular health after maternal placental syndromes (CHAMPS): population-based retrospective cohort study. Lancet. 2005;366:1797–1803. [DOI] [PubMed] [Google Scholar]
- 4.Tooher J, Thornton C, Makris A, Ogle R, Korda A,Hennessy A. All Hypertensive Disorders of Pregnancy Increase the Risk of Future Cardiovascular Disease. Hypertension. 2017;70:798–803. [DOI] [PubMed] [Google Scholar]
- 5.Mongraw-Chaffin ML, Cirillo PM,Cohn BA. Preeclampsia and cardiovascular disease death: prospective evidence from the child health and development studies cohort. Hypertension. 2010;56:166–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garovic VD,August P. Preeclampsia and the future risk of hypertension: the pregnant evidence. Curr Hypertens Rep. 2013;15:114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson JD,Louis JM. Does race or ethnicity play a role in the origin, pathophysiology, and outcomes of preeclampsia? An expert review of the literature. Am J Obstet Gynecol. 2022;226:S876–S885. [DOI] [PubMed] [Google Scholar]
- 8.Hauspurg A, Lemon L, Cabrera C, Javaid A, Binstock A, Quinn B, Larkin J, Watson AR, Beigi RH,Simhan H. Racial Differences in Postpartum Blood Pressure Trajectories Among Women After a Hypertensive Disorder of Pregnancy. JAMA Netw Open. 2020;3:e2030815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Romundstad PR, Magnussen EB, Smith GD,Vatten LJ. Hypertension in pregnancy and later cardiovascular risk: common antecedents? Circulation. 2010;122:579–584. [DOI] [PubMed] [Google Scholar]
- 10.Rana S, Lemoine E, Granger JP,Karumanchi SA. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ Res. 2019;124:1094–1112. [DOI] [PubMed] [Google Scholar]
- 11.Noori M, Donald AE, Angelakopoulou A, Hingorani AD,Williams DJ. Prospective study of placental angiogenic factors and maternal vascular function before and after preeclampsia and gestational hypertension. Circulation. 2010;122:478–487. [DOI] [PubMed] [Google Scholar]
- 12.Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, Schisterman EF, Thadhani R, Sachs BP, Epstein FH, Sibai BM, Sukhatme VP,Karumanchi SA. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350:672–683. [DOI] [PubMed] [Google Scholar]
- 13.Zeisler H, Llurba E, Chantraine F, Vatish M, Staff AC, Sennstrom M, Olovsson M, Brennecke SP, Stepan H, Allegranza D, Dilba P, Schoedl M, Hund M,Verlohren S. Predictive Value of the sFlt-1:PlGF Ratio in Women with Suspected Preeclampsia. N Engl J Med. 2016;374:13–22. [DOI] [PubMed] [Google Scholar]
- 14.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP,Karumanchi SA. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huhn EA, Kreienbuhl A, Hoffmann I, Schoetzau A, Lange S, Martinez de Tejada B, Hund M, Hoesli I,Lapaire O. Diagnostic Accuracy of Different Soluble fms-Like Tyrosine Kinase 1 and Placental Growth Factor Cut-Off Values in the Assessment of Preterm and Term Preeclampsia: A Gestational Age Matched Case-Control Study. Front Med (Lausanne). 2018;5:325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rana S, Powe CE, Salahuddin S, Verlohren S, Perschel FH, Levine RJ, Lim KH, Wenger JB, Thadhani R,Karumanchi SA. Angiogenic factors and the risk of adverse outcomes in women with suspected preeclampsia. Circulation. 2012;125:911–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burke SD, Zsengeller ZK, Khankin EV, Lo AS, Rajakumar A, DuPont JJ, McCurley A, Moss ME, Zhang D, Clark CD, Wang A, Seely EW, Kang PM, Stillman IE, Jaffe IZ,Karumanchi SA. Soluble fms-like tyrosine kinase 1 promotes angiotensin II sensitivity in preeclampsia. J Clin Invest. 2016;126:2561–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu F, Longo M, Tamayo E, Maner W, Al-Hendy A, Anderson GD, Hankins GD,Saade GR. The effect of over-expression of sFlt-1 on blood pressure and the occurrence of other manifestations of preeclampsia in unrestrained conscious pregnant mice. Am J Obstet Gynecol. 2007;196:396 e391–397; discussion 396 e397. [DOI] [PubMed] [Google Scholar]
- 19.Saxena AR, Karumanchi SA, Brown NJ, Royle CM, McElrath TF,Seely EW. Increased sensitivity to angiotensin II is present postpartum in women with a history of hypertensive pregnancy. Hypertension. 2010;55:1239–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stanhewicz AE, Jandu S, Santhanam L,Alexander LM. Alterations in endothelin type B receptor contribute to microvascular dysfunction in women who have had preeclampsia. Clin Sci (Lond). 2017;131:2777–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stanhewicz AE, Jandu S, Santhanam L,Alexander LM. Increased Angiotensin II Sensitivity Contributes to Microvascular Dysfunction in Women Who Have Had Preeclampsia. Hypertension. 2017;70:382–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hladunewich MA, Kingdom J, Odutayo A, Burns K, Lai V, O’Brien T, Gandhi S, Zimpelmann J, Kiss A, Miller J,Cherney D. Postpartum assessment of the renin angiotensin system in women with previous severe, early-onset preeclampsia. J Clin Endocrinol Metab. 2011;96:3517–3524. [DOI] [PubMed] [Google Scholar]
- 23.Martillotti G, Ditisheim A, Burnier M, Wagner G, Boulvain M, Irion O,Pechere-Bertschi A. Increased salt sensitivity of ambulatory blood pressure in women with a history of severe preeclampsia. Hypertension. 2013;62:802–808. [DOI] [PubMed] [Google Scholar]
- 24.Lakatta EG. The reality of getting old. Nat Rev Cardiol. 2018;15:499–500. [DOI] [PubMed] [Google Scholar]
- 25.Alfaras I, Di Germanio C, Bernier M, Csiszar A, Ungvari Z, Lakatta EG,de Cabo R. Pharmacological Strategies to Retard Cardiovascular Aging. Circ Res. 2016;118:1626–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorini S, Kim SK, Infante M, Mammi C, La Vignera S, Fabbri A, Jaffe IZ,Caprio M. Role of Aldosterone and Mineralocorticoid Receptor in Cardiovascular Aging. Front Endocrinol (Lausanne). 2019;10:584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jaffe IZ,Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ Res. 2005;96:643–650. [DOI] [PubMed] [Google Scholar]
- 28.Lu Q, Davel AP, McGraw AP, Rao SP, Newfell BG,Jaffe IZ. PKCdelta Mediates Mineralocorticoid Receptor Activation by Angiotensin II to Modulate Smooth Muscle Cell Function. Endocrinology. 2019;160:2101–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCurley A,Jaffe IZ. Mineralocorticoid receptors in vascular function and disease. Mol Cell Endocrinol. 2012;350:256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biwer LA, Wallingford MC,Jaffe IZ. Vascular Mineralocorticoid Receptor: Evolutionary Mediator of Wound Healing Turned Harmful by Our Modern Lifestyle. Am J Hypertens. 2019;32:123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hypertension in Pregnancy: Executive Summary. 2013;122:1122–1131. [Google Scholar]
- 32.McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, Chambon P, Hill MA, Dorrance AM, Mendelsohn ME,Jaffe IZ. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med. 2012;18:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biwer LA, Carvajal BV, Lu Q, Man JJ,Jaffe IZ. Mineralocorticoid and Estrogen Receptors in Endothelial Cells Coordinately Regulate Microvascular Function in Obese Female Mice. Hypertension. 2021;77:2117–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Good ME, Eucker SA, Li J, Bacon HM, Lang SM, Butcher JT, Johnson TJ, Gaykema RP, Patel MK, Zuo Z,Isakson BE. Endothelial cell Pannexin1 modulates severity of ischemic stroke by regulating cerebral inflammation and myogenic tone. JCI Insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mirabito KM, Hilliard LM, Kett MM, Brown RD, Booth SC, Widdop RE, Moritz KM, Evans RG,Denton KM. Sex- and age-related differences in the chronic pressure-natriuresis relationship: role of the angiotensin type 2 receptor. Am J Physiol Renal Physiol. 2014;307:F901–907. [DOI] [PubMed] [Google Scholar]
- 36.DuPont JJ, McCurley A, Davel AP, McCarthy J, Bender SB, Hong K, Yang Y, Yoo JK, Aronovitz M, Baur WE, Christou DD, Hill MA,Jaffe IZ. Vascular mineralocorticoid receptor regulates microRNA-155 to promote vasoconstriction and rising blood pressure with aging. JCI Insight. 2016;1:e88942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newfell BG, Iyer LK, Mohammad NN, McGraw AP, Ehsan A, Rosano G, Huang PL, Mendelsohn ME,Jaffe IZ. Aldosterone regulates vascular gene transcription via oxidative stress-dependent and -independent pathways. Arterioscler Thromb Vasc Biol. 2011;31:1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim SK, McCurley AT, DuPont JJ, Aronovitz M, Moss ME, Stillman IE, Karumanchi SA, Christou DD,Jaffe IZ. Smooth Muscle Cell-Mineralocorticoid Receptor as a Mediator of Cardiovascular Stiffness With Aging. Hypertension. 2018;71:609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akhter T, Wikstrom AK, Larsson M, Larsson A, Wikstrom G,Naessen T. Association between angiogenic factors and signs of arterial aging in women with pre-eclampsia. Ultrasound Obstet Gynecol. 2017;50:93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ibarrola J, Kim SK, Lu Q, DuPont J, Creech A, Sun Z, Hill MA, Jaffe JD,Jaffe IZ. Smooth muscle mineralocorticoid receptor as an epigenetic regulator of vascular aging. Cardiovasc Res. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DuPont JJ, Kim SK, Kenney RM,Jaffe IZ. Sex differences in the time course and mechanisms of vascular and cardiac aging in mice: role of the smooth muscle cell mineralocorticoid receptor. Am J Physiol Heart Circ Physiol. 2021;320:H169–H180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grunewald M, Kumar S, Sharife H, Volinsky E, Gileles-Hillel A, Licht T, Permyakova A, Hinden L, Azar S, Friedmann Y, Kupetz P, Tzuberi R, Anisimov A, Alitalo K, Horwitz M, Leebhoff S, Khoma OZ, Hlushchuk R, Djonov V, Abramovitch R, Tam J,Keshet E. Counteracting age-related VEGF signaling insufficiency promotes healthy aging and extends life span. Science. 2021;373. [DOI] [PubMed] [Google Scholar]
- 43.Krug AW, Allenhofer L, Monticone R, Spinetti G, Gekle M, Wang M,Lakatta EG. Elevated mineralocorticoid receptor activity in aged rat vascular smooth muscle cells promotes a proinflammatory phenotype via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase and epidermal growth factor receptor-dependent pathways. Hypertension. 2010;55:1476–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Faresse N, Vitagliano JJ,Staub O. Differential ubiquitylation of the mineralocorticoid receptor is regulated by phosphorylation. FASEB J. 2012;26:4373–4382. [DOI] [PubMed] [Google Scholar]
- 45.Yokota K, Shibata H, Kobayashi S, Suda N, Murai A, Kurihara I, Saito I,Saruta T. Proteasome-mediated mineralocorticoid receptor degradation attenuates transcriptional response to aldosterone. Endocr Res. 2004;30:611–616. [DOI] [PubMed] [Google Scholar]
- 46.Nagase M,Fujita T. Role of Rac1-mineralocorticoid-receptor signalling in renal and cardiac disease. Nat Rev Nephrol. 2013;9:86–98. [DOI] [PubMed] [Google Scholar]
- 47.Kawarazaki W,Fujita T. Aberrant Rac1-mineralocorticoid receptor pathways in salt-sensitive hypertension. Clin Exp Pharmacol Physiol. 2013;40:929–936. [DOI] [PubMed] [Google Scholar]
- 48.Faulkner JL, Wright D, Antonova G, Jaffe IZ, Kennard S,Belin de Chantemele EJ. Midgestation Leptin Infusion Induces Characteristics of Clinical Preeclampsia in Mice, Which Is Ablated by Endothelial Mineralocorticoid Receptor Deletion. Hypertension. 2022;79:1536–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pires PW, Ko EA, Pritchard HAT, Rudokas M, Yamasaki E,Earley S. The angiotensin II receptor type 1b is the primary sensor of intraluminal pressure in cerebral artery smooth muscle cells. J Physiol. 2017;595:4735–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong K, Zhao G, Hong Z, Sun Z, Yang Y, Clifford PS, Davis MJ, Meininger GA,Hill MA. Mechanical activation of angiotensin II type 1 receptors causes actin remodelling and myogenic responsiveness in skeletal muscle arterioles. J Physiol. 2016;594:7027–7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rana S, Burke SD,Karumanchi SA. Imbalances in circulating angiogenic factors in the pathophysiology of preeclampsia and related disorders. Am J Obstet Gynecol. 2022;226:S1019–S1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schulman IH, Aranda P, Raij L, Veronesi M, Aranda FJ,Martin R. Surgical menopause increases salt sensitivity of blood pressure. Hypertension. 2006;47:1168–1174. [DOI] [PubMed] [Google Scholar]
- 53.Pechere-Bertschi A,Burnier M. Female sex hormones, salt, and blood pressure regulation. Am J Hypertens. 2004;17:994–1001. [DOI] [PubMed] [Google Scholar]
- 54.Powles J, Fahimi S, Micha R, Khatibzadeh S, Shi P, Ezzati M, Engell RE, Lim SS, Danaei G, Mozaffarian D, Global Burden of Diseases N,Chronic Diseases Expert G. Global, regional and national sodium intakes in 1990 and 2010: a systematic analysis of 24 h urinary sodium excretion and dietary surveys worldwide. BMJ Open. 2013;3:e003733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Obrochta CA, Chambers C,Bandoli G. Psychological distress in pregnancy and postpartum. Women Birth. 2020;33:583–591. [DOI] [PubMed] [Google Scholar]
- 56.Shukri MZ, Tan JW, Manosroi W, Pojoga LH, Rivera A, Williams JS, Seely EW, Adler GK, Jaffe IZ, Karas RH, Williams GH,Romero JR. Biological Sex Modulates the Adrenal and Blood Pressure Responses to Angiotensin II. Hypertension. 2018;71:1083–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Usselman CW, Adler TE, Coovadia Y, Leone C, Paidas MJ,Stachenfeld NS. A recent history of preeclampsia is associated with elevated central pulse wave velocity and muscle sympathetic outflow. Am J Physiol Heart Circ Physiol. 2020;318:H581–H589. [DOI] [PubMed] [Google Scholar]
- 58.Perry H, Gutierrez J, Binder J, Thilaganathan B,Khalil A. Maternal arterial stiffness in hypertensive pregnancies with and without small-for-gestational-age neonate. Ultrasound Obstet Gynecol. 2020;56:44–50. [DOI] [PubMed] [Google Scholar]
- 59.Jaffe IZ, Tintut Y, Newfell BG, Demer LL,Mendelsohn ME. Mineralocorticoid receptor activation promotes vascular cell calcification. Arterioscler Thromb Vasc Biol. 2007;27:799–805. [DOI] [PubMed] [Google Scholar]
- 60.Bytautiene E, Bulayeva N, Bhat G, Li L, Rosenblatt KP,Saade GR. Long-term alterations in maternal plasma proteome after sFlt1-induced preeclampsia in mice. Am J Obstet Gynecol. 2013;208:388 e381–388 e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bytautiene E, Lu F, Tamayo EH, Hankins GD, Longo M, Kublickiene K,Saade GR. Long-term maternal cardiovascular function in a mouse model of sFlt-1-induced preeclampsia. Am J Physiol Heart Circ Physiol. 2010;298:H189–193. [DOI] [PubMed] [Google Scholar]
- 62.Pruthi D, Khankin EV, Blanton RM, Aronovitz M, Burke SD, McCurley A, Karumanchi SA,Jaffe IZ. Exposure to experimental preeclampsia in mice enhances the vascular response to future injury. Hypertension. 2015;65:863–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brennan L, Morton JS, Quon A,Davidge ST. Postpartum Vascular Dysfunction in the Reduced Uteroplacental Perfusion Model of Preeclampsia. PLoS One. 2016;11:e0162487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Paauw ND, Joles JA, Spradley FT, Bakrania B, Zsengeller ZK, Franx A, Verhaar MC, Granger JP,Lely AT. Exposure to placental ischemia impairs postpartum maternal renal and cardiac function in rats. Am J Physiol Regul Integr Comp Physiol. 2017;312:R664–R670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garrett N, Pombo J, Umpierrez M, Clark JE, Simmons M,Girardi G. Pravastatin therapy during preeclampsia prevents long-term adverse health effects in mice. JCI Insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ishida A, Murray J, Saito Y, Kanthou C, Benzakour O, Shibuya M,Wijelath ES. Expression of vascular endothelial growth factor receptors in smooth muscle cells. J Cell Physiol. 2001;188:359–368. [DOI] [PubMed] [Google Scholar]
- 67.Parenti A, Brogelli L, Filippi S, Donnini S,Ledda F. Effect of hypoxia and endothelial loss on vascular smooth muscle cell responsiveness to VEGF-A: role of flt-1/VEGF-receptor-1. Cardiovasc Res. 2002;55:201–212. [DOI] [PubMed] [Google Scholar]
- 68.Hubel CA, Wallukat G, Wolf M, Herse F, Rajakumar A, Roberts JM, Markovic N, Thadhani R, Luft FC,Dechend R. Agonistic angiotensin II type 1 receptor autoantibodies in postpartum women with a history of preeclampsia. Hypertension. 2007;49:612–617. [DOI] [PubMed] [Google Scholar]
- 69.LaMarca B, Wallace K,Granger J. Role of angiotensin II type I receptor agonistic autoantibodies (AT1-AA) in preeclampsia. Curr Opin Pharmacol. 2011;11:175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Camarda N, Travers R, Yang VK, London C,Jaffe IZ. VEGF Receptor Inhibitor-Induced Hypertension: Emerging Mechanisms and Clinical Implications. Curr Oncol Rep. 2022;24:463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bottinor WJ, Shuey MM, Manouchehri A, Farber-Eger EH, Xu M, Nair D, Salem JE, Wang TJ,Brittain EL. Renin-Angiotensin-Aldosterone System Modulates Blood Pressure Response During Vascular Endothelial Growth Factor Receptor Inhibition. JACC CardioOncol. 2019;1:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.