

Abstract
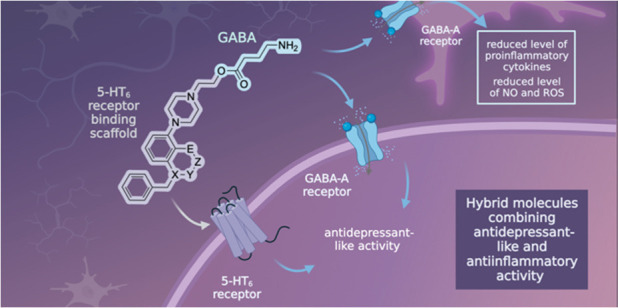
While monoaminergic deficits are evident in all depressed patients, nonresponders are characterized by impaired GABA-ergic signaling and the simultaneous presence of the inflammatory component. Pharmacological agents able to curb pathological immune responses and modulate ineffective GABA-ergic neurotransmission are thought to improve therapeutic outcomes in the treatment-resistant subgroup of depressed patients. Here, we report on a set of dually acting molecules designed to simultaneously modulate GABA-A and 5-HT6 receptor activity. The serotonin 5-HT6 receptor was chosen as a complementary molecular target, due to its promising antidepressant-like activities reported in animal studies. Within the study we identified that lead molecule 16 showed a desirable receptor profile and physicochemical properties. In pharmacological studies, 16 was able to reduce the secretion of proinflammatory cytokines and decrease oxidative stress markers. In animal studies, 16 exerted antidepressant-like activity deriving from a synergic interplay between 5-HT6 and GABA-A receptors. Altogether, the presented findings point to hybrid 16 as an interesting tool that interacts with pharmacologically relevant targets, matching the pathological dysfunction of depression associated with neuroinflammation.
Keywords: GABA-A receptor, hybrid molecules, 5-HT6 receptor, anti-inflammatory activity, depression associated with inflammation, neuroinflammation
1. Introduction
For decades, the primary objective of antidepressant therapy has been to increase the levels of monoamine neurotransmitters in the synaptic cleft.1,2 However, a selective focus on monoaminergic transmission is not effective for all patients. Despite the accessibility of the whole palette of antidepressants in the clinics, approximately 30% of patients do not respond to the marketed drugs.3 These data suggest that various mechanisms could play a role in the pathophysiology of depression, beyond monoamines. In this regard, several hypotheses were brought forward after clinical evidence confirmed changes in many neurotransmitter systems and various neurobiological variations.4,5
Significant progress has been made in understanding the specific neurochemical changes that play a role in the pathophysiology of treatment-resistant depression through brain imaging studies. For instance, magnetic resonance spectroscopy (MRS) revealed a significant reduction in GABA levels in plasma, cerebrospinal fluid, and cortical regions of depressed subjects.6 Neurochemical changes are consistent with changes in the levels and activity of GABA-A receptors.7 These findings, along with other neurochemical evidence, gave rise to the “GABA-ergic hypothesis of depression”.5 Highly noteworthy, GABA deficits are particularly evident in the subgroup of treatment-resistant patients. 1H MRS brain scans revealed substantially lower levels of GABA in the frontal cortex in the treatment resistant subgroup compared to patients without a history of treatment resistance.8 These findings suggest that compounds modulating the GABA-ergic signaling may help to optimize the patient’s response by interacting with the relevant target for the disease.
A plethora of evidence has confirmed that neuroinflammation is a widely recognized component of the pathophysiology of depression.4,9,10 Clinical studies showed that depressed patients suffer from increased serum levels of proinflammatory cytokines, particularly interleukin IL-6, IL-1β, and tumor necrosis α factor (TNF-α).9,11 Several clinical studies observed a robust association between a raised level of IL-6 and anhedonic states.12 Further studies indicated that persistent neuroinflammation may sabotage a patient’s response to the marketed antidepressants.13,14 The TNF-α was found to enhance the activity of neuronal SERT via stimulation of p38 MAPK.15 Therefore, the coexisting inflammatory factors circumvent the primary mechanism of serotonergic medications and can contribute to treatment resistance. Consequently, this scenario may be present in nearly 30% of all depressed subjects.3
Given the disappointing state related to the current pharmacotherapy for depression, the development of small molecule therapeutics with a novel mechanism of action has been intensively pursued to optimize the patient’s response to treatment. Interestingly it is possible to simultaneously target inflammatory responses and impaired GABA-ergic signaling with molecules modulating the activity of GABA-A receptors.16 Both microglia and neurons express GABA-A receptors, and modulation of microglia activity via GABA-A receptors decreases secreting proinflammatory cytokines IL-6 and TNF-α.16−18 At the same time, modulation of neuronal GABA-A receptor activity might regulate the impaired GABA-signaling.7
Inspired by the pallet of functions that GABA-A receptors can offer to mitigate depression, we designed a set of dually acting compounds that harness the GABA molecule, which is presumed to exert anti-inflammatory activity and antidepressant properties (Figure 1). The GABA molecule was assembled with a chemical scaffold that interacts with a complementary biological target, involved in the regulation of mood deficits, namely, the serotonin 5-HT6 receptor. Considering that regions of the brain involved in regulating emotions and memory processes (cortex and hippocampus) express the 5-HT6 receptor, we reasoned that it can be beneficial for therapeutic purposes.19 Both 5-HT6 agonists and 5-HT6 antagonists have demonstrated antidepressant-like activity in animal models. However, the current landscape of small molecules acting as 5-HT6 antagonists appears to be more developed.20 In fact, selective 5-HT6 antagonists hold great promise as small molecule therapeutics in neuropsychiatric diseases, due to their promising antidepressant-like efficacy in animal models.21 Continuing our previous research in this area,22,23 in the present work, we explored the novel 5-HT6 binding chemotypes bearing various heterocycles, which could be easily connected with the function of GABA to construct a series of bifunctional molecules 16–20. Within the in vitro profiling cascade, we identified hybrid molecule 16 characterized by the most desirable receptor profile and drug-like properties. Compound 16 was then assayed in BV-2 microglia cells to explore its ability to attenuate neuroinflammation. To verify the therapeutic potential of this novel chemotype, 16 was subsequently characterized in in vivo studies.
Figure 1.

General concept of hybrid molecules targeting GABA-A and 5-HT6 receptors, employing novel 5-HT6 binding chemotypes (5-HT6 antagonism): X = N, O; Y = CO, CH2, none; Z = CH2, none; E = N, O, CH3.
2. Results and Discussion
2.1. Design and Synthesis
Previously we have found that anchoring the GABA molecule via ethyl ester linker with a 5-HT6 receptor blocking scaffold yields dually active molecules, characterized by favorable chemical stability and optimal brain penetration.22 This strategy was confirmed for a series of indole derivatives acting as a 5-HT6 receptor binding scaffolds. The compounds containing the sulfonamide group showed superior activity, apparently due to hydrogen bonding with the Asn6.55 side chain. In the pursuit of novel biologically active chemotypes, in the present work we replaced the indole rings with the 5-HT6 receptor binding scaffolds containing hydrogen bond acceptors that would secure both the favorable interactions and physicochemical properties: 3-(benzyloxy)-2-methylphenyl (1),24 1,3-dihydro-2H-benzo[d]imidazol-2-one (2),25 and 2H-benzo[b][1,4]oxazin-3(4H)-one (4).26 Based on the molecular modeling studies described in detail in the next section, we presumed that the novel scaffolds would be beneficial for the interactions with the 5-HT6 receptor, due to the carbonyl/ether group that would allow for more efficient interaction with Asn6.55 which in turn stabilizes the aromatic interaction with Phe5.38. Alongside, we decided to investigate novel 5-HT6 binding fragments: 3-benzyl-7-(piperazin-1-yl)benzo[d]oxazol-2(3H)-one (3) and 4-benzyl-8-(piperazin-1-yl)-3,4-dihydro-2H-benzo[b][1,4]oxazine (5), which represent bioisosters of 2 and 4. Therefore, all the above-mentioned 5-HT6 receptor antagonism fragments were incorporated with GABA ethoxy moiety to compose a set of GABA/5-HT6 receptor hybrids (Scheme 1).
Scheme 1. Design Concept and Chemical Structure of a Series of Bifunctional Molecules.

The shaded portion of the molecule illustrates the key variations between the 5-HT6 binding scaffolds used.
The key 5-HT6 antagonists (1,242,25 and 4(26)) were synthesized according to previous protocols, while the 3 and 5 building blocks were prepared in our laboratory according to Scheme 2. Starting from the synthesis of 5-HT6 receptor binding scaffold 3, Boc protection of 7-(piperazin-1-yl)benzo[d]oxazol-2(3H)-one (a) delivered intermediate b, followed by functionalization with benzyl bromide to give c precursor. The key building block 3 was delivered by gentle deprotection using acetyl chloride in EtOH. The 5-HT6 scaffold 5 was prepared in three-step synthesis, starting from installation of benzyl moiety to deliver ii derivative. The latter was reacted with Boc-piperazine ring to deliver iii intermediate. Removal of Boc protecting moiety using 2 M HCl/Et2O liberated the final building block 5. With both building blocks 3 and 5 in hand, we proceeded with the synthesis of hybrid molecules 16–20 (Scheme 3). The key building blocks 1–5 were functionalized with bromoethanol to deliver 6–10 alcohols. Next, the alcohols 6–10 were reacted with Boc-GABA to deliver ester intermediates 11–15. Ultimate deprotection of 11–15 esters with HCl/Et2O liberated final molecules 16–20 in the form of dichloride salts (1H NMR spectra available in the Supporting Information).
Scheme 2. Preparation of Key Building Blocks 3 and 5.

Scheme 3. Synthesis of Final Hybrid Molecules 16–20.

X = N, O; Y = CO, CH2, none; Z = CH2, none; E = N, O, CH3.
2.2. Structure–Activity Relationships
In vitro radioligand binding studies were conducted to evaluate the affinity of all hybrid molecules for the muscimol site of the GABA-A receptor and the serotonin 5-HT6 receptor. Our starting point focused on determination of the affinity of 1–5 scaffolds for the 5-HT6 receptor. Consistent with previous reports, 1, 2, and 4 exerted nanomolar affinity for the 5-HT6 receptor (Ki = 0.2–2.7 nM). The novel fragments 3 and 5 also showed a high affinity with Ki values of 4.0 ± 0.2 nM and 60.0 ± 7.5 nM, respectively (Table 1). Next, we focused our attention on assessing the impact of the incorporation of the alkyl linker into 1–5 scaffolds. We observed that alcohols 6, 9, and 10 performed slightly better and maintained a high affinity for the 5-HT6 receptor (Table 1). The installation of the ethoxy chain in 6, 9, and 10 induced a minimal alteration in the affinity for the receptor of interest. However, the introduction of the ethoxy chain to the 3-benzyl-7-(piperazin-1-yl)benzo[d]oxazol-2(3H)-one scaffold (compound 8) and to the 1-benzyl-4-(piperazin-1-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one ring (7) led to a slight decrease in affinity for the 5-HT6 receptor (7, Ki = 40.0 ± 3.0 nM; 8, Ki = 150.0 ± 7.6 nM). The installation of the GABA function in the final hybrid molecules did not change the affinity for the 5-HT6 receptor. This conclusion is true for 16, 17, 18, and 19 hybrids. Hybrid molecules bearing 1-(3-(benzyloxy)-2-methylphenyl)piperazine (16) and 4-benzyl-8-(piperazin-1-yl)-2H-benzo[b][1,4]oxazin-3(4H)-one ring (19) showed the highest potency (16, Ki = 16.0 ± 0.4 nM; 19, Ki = 25.0 ± 1.0 nM) among all the series. Solely the 4-benzyl-8-(piperazin-1-yl)-3,4-dihydro-2H-benzo[b][1,4]oxazine analogue (20) did not show desired affinity (Ki = 1260.0 ± 82.0 nM). Then, the compounds 16–20 were subjected to functional studies, which showed that the compounds exhibited notable antagonist effectiveness at the 5-HT6 receptor, with KB values ranging from 11.9 ± 0.1 to 1670 ± 1.5 nM. The most effective antagonistic responses were observed for compounds 19 and 16 with KB values of 7 ± 0.1 and 11.9 ± 0.1.
Table 1. Radioligand Binding Affinity of 1–5, 6–10, and 16–20 for 5-HT6 Receptor and the Muscimol Site of the GABA-A Receptor: Antagonistic Activity at 5-HT6R (KB) and Electrophysiological Studies of the Selected Compounds.

The values of affinity are expressed as Ki and ± SEM and were determined from minimum three experiments conducted in duplicate. Competitive replacement of [3H]-LSD was used to measure the affinities for the 5-HT6 receptor.
The outcomes were standardized as a percentage of the maximum reaction observed in the absence of the antagonist.
Competitive replacement of [3H]muscimol was used to establish the affinities for the GABA-A receptor.
The electrophysiological studies examined the agonist mode of tested compounds at a concentration of 10 μM. Results are represented by the GABA current amplitude normalized to 10 μM GABA-induced response (10 μM). n.a.: “not active”. Blank spaces: not tested.
We next proceeded with the investigation of the affinity for the GABA-A receptor (Table 1). Considering that the GABA neurotransmitter binds to the GABA-A receptor at the muscimol site, we chose this site in the radioligand binding assay as it closely mimics the physiological activity of the receptor.27 Hybrids 16 and 20 displayed a binding affinity (Ki = 147.0 ± 12.7 nM, Ki = 184.0 ± 4.0 nM) close to the natural GABA neurotransmitter (Ki = 98 nM). The measured affinities for the remaining GABA derivatives (18 and 19) were Ki = 229.0 ± 28.0 nM for 18 and Ki = 247.0 ± 24.7 nM for 19. In the case of 1-benzyl-4-(piperazin-1-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one derivative 17 the affinity for GABA-A receptor dropped to 592.0 ± 52.5 nM. To confirm that the compounds interact with the receptors as hybrids and not hydrolysis products, we tested the stability of a representative hybrid 16 under radioligand binding assay conditions and found 98.21% of the compound remaining, after 1 h of incubation with brain tissue.
Based on the overall results of the radioligand binding assays, for electrophysiological studies we selected the most promising hybrids (16, 18, and 19), considering the complexity of the studies. The selection criteria focused on molecules that had an affinity for both the GABA-A and 5-HT6 receptors, with a Ki value of less than 250 nM. Therefore, molecules 16, 18, and 19 satisfied these criteria and were further evaluated in functional studies. The effects of selected hybrids molecules were evaluated at 10 μM concentration using a HEK293 cell line stably expressing human α1β2γ2-GABA-A receptors and were compared with effects provoked by GABA alone (at 10 μM concentration). According to the protocol, the intensity of the electrical current evoked by GABA was measured and established as the baseline value of 100%. The amplitudes of the compounds tested were then measured and expressed as a percentage of the amplitude produced by the natural GABA agonist (Table 1). Considering that GABA acts as an agonist at the GABA-A receptor, we hypothesized that the hybrid molecules containing a GABA ester moiety would also act similarly.28 We observed that the tested compounds induced an increase in the ion current, displaying weak agonistic properties. The increase in ion current for 16, 18, and 19 was 24.7% ± 1.5, 14.0% ± 2.6, and 9.5% ± 3.5 of the GABA responses (Table 1).
2.3. Molecular Modeling Studies
Data from radioligand binding studies were supported with in silico studies to survey the contribution of molecular interactions between both molecular targets: GABA-A and 5-HT6 receptors and representative hybrid molecule 16. Based on the overall receptor profile, we selected compound 16 as the main structure for further investigation, which showed a balanced receptor profile (Ki < 150 nM for both targets).
The predicted binding mode of a representative hybrid molecule 16 was similar to that observed for the selective 5-HT6 antagonist (1), and both molecules were anchored at the primary binding site, inside the 5-HT6 receptor (Figure 2). We observed that 1-(3-(benzyloxy)-2-methylphenyl)piperazine scaffold formed a salt bridge between Asp3.32 and the protonated nitrogen atom of 16, stabilized by π–π stackings with Phe6.51/6.52, the characteristic interactions observed for GPCRs. The benzyl moiety of hybrid 16 occupied a hydrophobic pocket between Val3.33 and Ala5.42 residues, interacting with Phe5.38. The analysis of the interactions of the compound series resulting from the scaffold hopping suggested the importance of a hydrogen bond acceptor exposed in the region of the polar interactions with Asn6.55. The superior affinity of compound 19 may be explained by the privileged formation of an H-bond with the latter residue (see Supporting Information), and its importance was confirmed by the drop in the affinity of compound 20. The ethyl linker and GABA fragment of 16 were anchored in an external pocket between TMH1, TMH2, and TMH7, and the protonated amine group of GABA formed ionic interaction with Asp7.36. This GABA fragment extended beyond the orthosteric binding site and did not cause a steric hindrance.
Figure 2.

Potential binding mode of 16 at the orthostatic sites of both targets: left, the 5-HT6 receptor; right, GABA-A receptor (canary, 16; cyan, 5-HT6 receptor ligand 1; green, GABA). 1-[3-(Benzyloxy)-2-methylphenyl]piperazine moiety of 16 established equal interactions to selective 5-HT6 antagonist 1, inside the 5-HT6 receptor. In the GABA-A receptor, the acyl fragment of 16 exhibited a binding pose akin to that of the natural agonist GABA. The remaining hybrid elements did not induce steric hindrance in the active sites of 5-HT6 and GABA-A receptors.
Further studies suggested that the 16 interacts with an orthosteric binding site of the α1β3γ2 GABA-A receptor, which is situated within the α1 and β3 subunits (Figure 2). Analogous to the natural agonist GABA, the GABA-fragment of 16 adopted an identical position in the cryo-EM structure and produced equivalent interactions. We could observe hydrogen bonds with both α1Arg-67 and β3Ser-156, a cation−π aromatic interaction with β3Tyr-205, and an ionic bond formed with β3Glu-155. The binding pose was also stabilized by the cation−π interactions formed between β3Phe-200 and protonated piperazine moiety, as well as β3Arg-207 and the benzyl ring, respectively. The remaining derivatives were characterized by an analogous binding mode, with the difference that in the case of hybrids 17 and 18 there was a lack of cation−π interaction with β3Arg-207 (corresponding figures in the Supporting Information).
2.4. Physicochemical and Preliminary ADMET Profiling
In addition to potency, key features that govern successful therapeutic-like activity are delineated by physicochemical profile. Selected hybrid 16 was subjected to further in vitro physicochemical profiling to determine its chemical stability in pH = 7.4, metabolic stability, solubility, and passive permeability (Table 2). In the thermodynamic solubility assay (PBS pH = 7.4), compound 16 was characterized by high aqueous solubility of >2 mg/mL. The parallel artificial membrane permeability assay (PAMPA) revealed that 16 exerts satisfying permeability (6.24 × 10–6 cm/s), suggesting good penetration through biological barriers. The metabolic stability evaluated using RLMs (rat liver microsomes) indicated a high metabolic stability of 16 (Table 2). Hepatotoxicity studies using HepG2 cells viability assay and highly sensitive probe ToxiLight29 indicated a not significant reduction of viability in concentrations up to 50 μM (see the Supporting Information). In addition, we tested selected compound 16 in various stability assays, including chemical stability in PBS, rat plasma stability, and brain tissue stability. Compound 16 displayed a desirable stability level ranging from 79.65 to 92.46% after 60 min under different conditions (Table 3). Overall, based on the results presented, we expected that 16 may produce the desired level of in vivo efficacy.
Table 2. Selected in Vitro Assays for 16 and the Reference Molecules.
| compound | solubility (pH = 7.4, mg/mL)a | PAMPA permeability (10–6 cm/s)b | rat liver microsomes t0,5 [min]c | rat liver microsomes Clint ((μL/min)/mg)c |
|---|---|---|---|---|
| 16 | >2 | 6.24 | >60 | <115.5 |
| perphenazine | 0.0306 | |||
| sulpiride | 0.016 | |||
| propranolol | >60 | <115.5 |
Thermodynamic solubility measured at rt.
Compounds tested at 10–5 M using the PAMPA Plate System Gentest.
Molecule assayed at 10–7 M. Empty cells: not examined.
Table 3. Stability Assays for 16 (Chemical Stability in PBS, Plasma, and Brain Homogenate).
| time point [min] | % compound remaining, rta | % compound remaining (rat plasma, 37 °C) | % compound remaining (brain homogenate)b |
|---|---|---|---|
| 0 | 100 ± 0.2 | 100 ± 0.1 | 100 ± 0.3 |
| 15 | 98.43 ± 1.8 | 97.21 ± 0.8 | 97.81 ± 2.1 |
| 30 | 94.23 ± 1.8 | 90.35 ± 1.4 | 92.56 ± 1.9 |
| 60 | 92.46 ± 2.2 | 79.65 ± 2.5 | 87.12 ± 3.2 |
| 120 | 86.63 ± 2.3 | 45.37 ± 2.2 | 49.73 ± 2.4 |
| 240 | 62.21 ± 1.7 | 21.46 ± 1.2 | 25.34 ± 1.6 |
Assay performed in PBS, rt.
Compound 16 was incubated with brain homogenate suspended in Tris-HCl buffer.
2.5. In Vitro Evaluation of Anti-Inflammatory Properties of 16
Activated microglia may disrupt neurotransmitter balance via enhanced production of proinflammatory cytokines and secretion of oxidative stress markers.11,10 Considering that modulation of the microglia’s activity by small molecules can be harnessed for therapeutic purposes, we sought to evaluate the potential of 16 to control inflammatory reactions. The relevance of this approach has been fueled by the fact that inflammatory signaling from microglia might be modulated by GABA-A receptor ligands.30,31 To stimulate an inflammatory response and oxidative stress, the microglia BV-2 cells were subjected to treatment with lipopolysaccharide (LPS) (Figure 3). The LPS stimulation assay reflects the clinical settings in which patients exposed to immune activation by bacterial LPS were characterized by depression symptoms.32 In first-line experiments, we assessed the morphology of microglial cells exposed to LPS and pretreated with selected ligands using fluorescence microscopy. Upon stimulation with LPS, we observed that mitochondrial metabolism and respiration in BV-2 cells changed, as there was a clear change in mitochondrial membrane potential (Figure 3B), detected with a fluorescent dye MitoTracker.33 The observed red fluorescence denoted elevated activation of mitochondria after stimulation with LPS (Figure 3B). In contrast, pretreatment with 16 (10 μM), protected the microglia from LPS insult, given that the red fluorescence was negligible and the fluorescence we observed was predominantly blue and green, characteristic for the cytoplasm and nucleus. When cells were pretreated with alcohol 6 (5-HT6 antagonist part deprived of the GABA part), we observed that the mitochondrial membrane potential was changed, suggesting that the alcohol 6 was deprived anti-inflammatory activity and the observed anti-inflammatory effects of 16 was arising from the modulation of GABA-A receptor activity.
Figure 3.
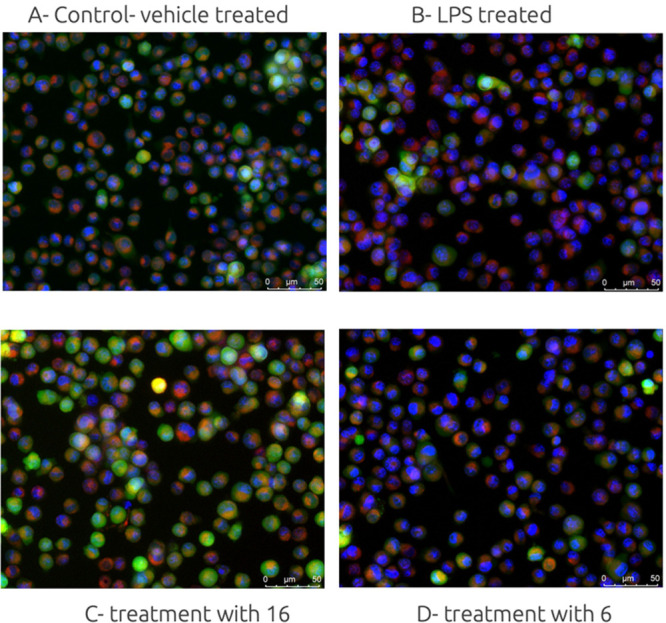
Representative images of microglia BV-2 cells: (A) control, vehicle treated cells, 0.1% DMSO; (B) cells treated with LPS (18 h); (C) cell pretreated with hybrid molecule 16 (10 μM, 1h); (D) cell pretreatment with alcohol 6 (10 μM, 1 h). BV-2 cells were stained with Calcein AM to highlight the outer membranes (green), Hoechst 33342 to detect the nucleus (blue), and MitoTracker to stain active mitochondria (red).
In the second line of experiments we aimed to establish if the hybrid molecule 16 can abolish oxidative stress and decrease the level of inflammatory markers. In response to LPS stimulation, microglia produced a substantial amount of toxic insults such as NO and ROS and proinflammatory cytokines IL-6 and TNF-α compared to the control group (Figure 4). In the group where BV-2 cells were pretreated with alcohol 6, the levels of ROS, NO, and proinflammatory mediators (IL-6 and TNF-α) did not change. On the contrary, pretreatment with 16 significantly decreased LPS-induced ROS, NO, IL-6, and TNF-α production, indicating anti-inflammatory activity of 16 (Figure 4). These results are in line with our previous studies in which we reported the anti-inflammatory efficacy of the GABA-A/5-HT6 hybrid molecules.22
Figure 4.

Determination of anti-inflammatory activity of 16 in comparison to the activity of 6. Pretreatment with 16 or 6 (10 μM), DMSO (0.1%, v/v) for 1 h followed by incubation with LPS for 18 h and measurement of nitric oxide (A), reactive oxygen species (B), IL-6 (C), and TNF-α (D). The results are reported as a percentage of the maximum cellular response to the LPS stimulation. The reported values are represented as the mean ± SD. To determine the differences between the treatment groups and the LPS-treated group, a one-way ANOVA was used, followed by post hoc analysis (Dunnett’s multiple comparison tests). Statistical significance was established if p < 0.05 (****p < 0.0001). The statistical analysis was conducted utilizing the GraphPad software. The studies were conducted using a plate reader (POLARstar Omega, BMG).
2.6. In Vivo Behavioral Studies
To test the in vivo antidepressant-like efficacy of selected lead molecule 16, we employed a forced swim test (FST, Porsolt test), due to its high predictive validity for selecting a broad range of antidepressant agents.34 The behavioral studies were carried out using female rats, given that the prevalence of depression in female subjects is estimated to double compared to male. Moreover, female subjects are particularly prone to fluctuations in GABA-ergic system.35 In the given experimental model, the activity of hybrid 16 was compared to the efficacy of parent alcohol 6 (5-HT6 antagonist part deprived of GABA function). GABA was not tested, as it does not produce antidepressant effects, as we reported previously.22
In the rat FST, selected molecule 16 administrated at 3 mg/kg (but not 1 or 8 mg/kg), in comparison with control group, significantly reduced immobility time by 31.9% (F(5,36) = 2.801, p < 0.05) and increased climbing behaviors by 336.5% (F(5,36) = 3.049, p < 0.05), without affecting the duration of swimming (F(5,36) = 1.680, p = 0.165) (Figure 5A). We observed that the alcohol 6, carrying solely 5-HT6 antagonistic activity, did not influence any of the above behaviors (Figure 5). Alongside, we did not observe any influence of 16 on the animal’s spontaneous locomotor when administered at a dose of 3 mg/kg, indicating that the antidepressant-like activity was specific (Table 3). Vortioxetine, a positive control antidepressant drug, at the dose 8 mg/kg significantly reduced immobility time by 24.7% (F(2,20) = 3.538, p < 0.05) and prolonged the swimming time by 88.1% (F(2,20) = 3.729, p < 0.05) but had no impact on the climbing (F(2,20) = 0.165, p = 0.849) (Figure 5B).
Figure 5.

Antidepressant-like properties of 16 in the modified FST in rats. Tested hybrid molecule and vortioxetine were administered ip 30 min prior the test. The 10% cyclodextrin was administered to the vehicle-treated groups. The one-way ANOVA followed by Newman–Keuls post hoc or Student t test was used in statistical analysis: *p < 0.05 vs vehicle-treated group, n = 7–8 per group.
The antidepressant-like activity of 16 was also confirmed after subchronic treatment, where 16 was administrated for 14 days at a dose of 3 mg/kg. Subchronic administration of compound 16 at the dose 3 mg/kg and vortioxetine at the dose 8 mg/kg compared with the vehicle-treated group significantly decreased immobility time in rats by 22.9% and 19.5% (F(2,27) = 5.330, p < 0.05), respectively (Figure 6). Notably, the tested dose of 3 mg/kg of substance 16 did not result in any significant changes in the spontaneous locomotor activity. This indicates that any observed antidepressant-like effect was specific and not due to a general increase in activity levels (Table 4). These findings correspond with our previous reports22 on antidepressant-like properties of dually acting GABA-A agonist and 5-HT6 antagonist hybrid molecules and, within the present study, have been confirmed for the novel chemotype.
Figure 6.

Antidepressant-like effect of 16 after chronic administration in the rat forced swim test. Both vortioxetine and 16 were administered intraperitoneally for 14 consecutive days. Sixty minutes after the last injection the test was performed. Vehicle-treated groups received 10% cyclodextrin for 14 days. Statistical analysis: one-way ANOVA followed by Newman-Keuls post hoc or Student t test: *p < 0.05 vs vehicle-treated group; n = 10 per group.
Table 4. Open Field Test in Rats Showing the Influence of Acute and Subchronic 16 and Vortioxetine Administration on the Locomotor Activity.
| treatment | dose | total distance traveled | rearing | X ambulation | Y ambulation | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acute Treatmenta | |||||||||||||
| vehicle | 485.3 | ± | 43.0 | 29.0 | ± | 2.0 | 187.5 | ± | 22.0 | 190.2 | ± | 18.7 | |
| 16 | 3 | 506.3 | ± | 36.7 | 34.5 | ± | 5.4 | 221.3 | ± | 17.9 | 213.7 | ± | 18.5 |
| Chronic Treatmentb | |||||||||||||
| vehicle | 2467 | ± | 142 | 53.8 | ± | 3.4 | 246.4 | ± | 14.7 | 243.1 | ± | 18.8 | |
| 16 | 3 | 2575 | ± | 92 | 52.2 | ± | 2.9 | 260.4 | ± | 15.0 | 255.3 | ± | 12.3 |
| vortioxetine | 8 | 2753 | ± | 120 | 50.6 | ± | 5.4 | 260.1 | ± | 10.4 | 265.9 | ± | 10.4 |
The compounds were administered ip 30 min prior the experiments. Vehicle-treated groups: 10% cyclodextrin (ip). Statistical analysis: one-way ANOVA followed by Newman–Keuls post hoc or Student t test. n = 6 per group.
Both vortioxetine and 16 were administered intraperitoneally for 14 consecutive days. Sixty minutes after the last injection, the test was performed. Vehicle-treated groups received 10% cyclodextrin for 14 days. Statistical analysis: one-way ANOVA followed by Newman–Keuls post hoc or Student t test. n = 10 per group.
3. Conclusions
Neuroinflammation and impaired GABA-ergic signaling often accompany the pathophysiology of depression, leading to poor clinical outcomes.11,13 Interest in this critical issue inspired us to design a set of hybrid compounds that include the GABA molecule and a 5-HT6 template that blocks the activity of this receptor. In the present study, we extended our previous research on the GABA-A/5-HT6 hybrid molecules by employing the novel “5-HT6 antagonists” scaffolds. Among novel chemotypes that were investigated in radioligand binding studies, we chose compound 16, characterized by high affinity for both targets (5-HT6Ki = 16.0 ± 0.4 nM, GABA-A Ki = 147.0 ± 12.7). In the physicochemical and ADMET assays 16 displayed favorable drug-like properties and promising chemical stability in rat plasma, brain tissue, and PBS buffer. We also investigated the anti-inflammatory properties of 16 in BV-2 microglia cells, which significantly reduced LPS-stimulated NO, ROS, IL-6, and TNF-α levels. In animal studies, we observed antidepressant-like activities of 16 in the forced swim test after acute administration of 16 at a dose of 3 mg/kg. This activity was maintained following subchronic administration of 16 during 14 consecutive days. Collectively, the present study suggests that combining the dual 5-HT6 antagonism and GABA-A receptor agonism could be useful in delivering compounds with anti-inflammatory and antidepressant-like activity. These activities were confirmed for another “5-HT6 antagonist” binding chemotype, suggesting the general applicability of this dual strategy. The present findings point to compound 16 as a novel lead for broader studies in the area of depression and coexisting neuroinflammation.
4. Methods
4.1. Molecular Modeling
The in silico studies were performed using SwissADME tool and Small-Molecule Drug Discovery Suite (Schrödinger, Inc.). The first one was used to test the structures of final molecules to detect notorious classes of reactive assay interference compounds (PAINS) and potential toxicophores (Brenk alert). The SMILES strings describing molecules 16–20 were verified by the server (http://www.swissadme.ch), showing no alerts. The Small-Molecule Drug Discovery Suite (Schrödinger, Inc.) was used for the docking procedures. The assignment of amino acid residues was performed according to their position in the protein sequence of the GABA-A receptor and Ballesteros–Weinstein nomenclature (5-HT6 receptor).36 Ligand structures were optimized using the LigPrep tool and the OPLS4 force field. The major ionization forms were calculated at pH 7 ± 2. Docking studies for the 5-HT6 receptor were carried out utilizing the 5-HT6 receptor model, established on the 7XTB experimental structure, preparation and validation of which were conducted previously.22,37,38 The Glide SP flexible docking procedure was applied with the centroid of a grid box and H-bond constraints set on Asp3.32. Docking studies for the GABA-A receptor were performed using the previously developed complex GABA-A receptor with dual GABA-A/5-HT6 agonist–compound 3B, which was prepared based on the previously reported cryo-EM structure of α1β3γ2L GABA-A receptor forming a complex GABA and picrotoxin (PDB code 6HUJ).22 The molecules were docked using Glide SP docking, in which the grid was set on the ligand and docking was controlled to its core structure (SMARTS: CCCC[N+]) and Glu-155. The docking scores were surveyed according to the presence of proper molecular interactions, considering values of Glide gscore scoring function.
4.2. Synthetic Procedures
4.2.1. General Chemistry Information
The chemical reagents were acquired as reagent grade and employed straightforwardly to the synthesis. Purification of the reaction mixtures was performed using automatic CombiFlash RF (Teledyne Isco) and RediSep Rf flash columns (silica gel 60, particle size 40–63 μm) or RediSep Gold (silica gel 60, particle size 20–40 μm). The progress of the reaction was monitored by TLC plates acquired from Merck containing an aluminum foil covered with silica gel 60 F254 and visualized with UV light (254 nm). The UPLC–MS purity of all molecules investigated in pharmacological studies was measured to be >95.2%. The UPLC–MS analyses were performed on a Waters ACQUITY UPLC (Waters Corporation, Milford, MA, USA) combined with a Waters tandem quadrupole mass spectrometer (TQD) (electrospray ionization (ESI) mode with TQD). The analytical measurements were conducted on C18 column ACQUITY UPLC BEH with the particle size of 2.1 mm × 100 mm and 1.7 μm particle size. The mobile phase conditions and gradient were the following: 10 min, flow rate = 0.3 mL/min, temperature 40 °C, gradient consisting of 95–0% eluent A that is a solution containing 0.1% formic acid in water (v/v) and eluent B that is 0.1% solution of formic acid in acetonitrile (v/v). 10 μL of each sample was injected, and chromatograms were recorded using a Waters eλ photodiode array detector and analyzed in the range of 200–700 nm with a resolution of 1.2 nm and a sampling rate of 20 points per second. The LCMS spectra were analyzed using Waters MassLynx 4.0 software. The 1H NMR, 13C NMR, and 19F NMR spectra were acquired on the Varian Mercury spectrometer (Varian Inc., Palo Alto, CA, USA) and the FT-NMR 500 MHz JEOL spectrometer (JEOL Ltd., Tokyo, Japan) JNM-ECZR500 RS1, version ECZR, operating at 300 or 500 MHz (1H NMR), 75 or 126 MHz (13C NMR), and 282 or 471 MHz (19F NMR), respectively. Chemical shifts δ are reported in ppm and analyzed adjusting the ppm value, first according to the appropriate reference solvents: chloroform-d, methanol-d4, or DMSO-d6. The signal multiplicities are described using the commonly found abbreviations: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br s (broad singlet), bd (broad doublet), etc. All the acquired spectra were analyzed using the ACD/Spectrus Procesor 2017.
4.2.2. Preparation of Key Building Blocks 3 and 5
tert-Butyl 4-(2-Oxo-2,3-dihydrobenzo[d]oxazol-7-yl)piperazine-1-carboxylate (b)
To a solution of 7-(piperazin-1-yl)benzo[d]oxazol-2(3H)-one (7.05 mmol, 1.8 g, 1 equiv) in DCM (35 mL), Boc anhydride (7,76 mmol, 11.7 g, 1.1 equiv) and triethylamine (12 mmol, 1.7 mL, 1.7 equiv) were added, and the reaction was stirred at room temperature for 12 h. Next, DCM was evaporated and the crude mixture was purified by column chromatography using Hex:EtOAc 6:4 as eluent. The final molecule was afforded as crystallizing oil, yield 54%. 1H NMR (chloroform-d, 500 MHz) δ 9.71 (s, 1H), 7.0–7.1 (m, 1H), 6.7–6.7 (m, 1H), 6.6–6.6 (m, 1H), 3.6–3.6 (m, 4H), 3.2–3.3 (m, 4H), 1.4–1.5 (m, 9H). 13C NMR (chloroform-d, 126 MHz) δ 155.4, 154.8, 135.8, 134.3, 130.4, 124.8, 110.7, 103.1, 80.1, 49.2, 28.4 (carbons overlap). UPLC–MS (ESI) calcd for C16H21N3O4 320.15 (M + H+), found 320.20 (M + H+).
tert-Butyl 4-(3-Benzyl-2-oxo-2,3-dihydrobenzo[d]oxazol-7-yl)piperazine-1-carboxylate (c)
To a solution of b (3.85 mmol, 1.23 g, 1 equiv) in dry THF at 0 °C the 1 M solution of t-BuOK (4.62 mmol, 4.62 mL, 1.2 equiv) was added, and the resulting slurry was stirred for 40 min at 0 °C. Next, benzyl bromide (4.62 mmol, 0.503 mL, 1.2 equiv) was added and the reaction mixture was stirred at room temperature for 12 h. After that time THF was removed under reduced pressure and the crude mixture was purified using Hex:EtOAc 6:4 as eluent affording the final compound as crystallizaing oil, yield 46%. 1H NMR (chloroform-d, 300 MHz) δ 7.3–7.4 (m, 5H), 7.00 (t, 1H, J = 1.0 Hz), 6.58 (d, 1H, J = 9.4 Hz), 6.4–6.5 (m, 1H), 4.98 (d, 2H, J = 1.0 Hz), 3.6–3.6 (m, 4H), 3.2–3.3 (m, 4H), 1.5–1.5 (m, 9H). 13C NMR (chloroform-d, 75 MHz) δ 154.7, 154.4, 135.7, 134.8, 132.9, 131.8, 128.9, 128.2, 127.6, 124.5, 110.8, 102.0 (aromatic carbons overlap), 80.0, 49.2, 46.1, 28.4. UPLC–MS (ESI) calcd for C23H27N3O4 410.15 (M + H+), found 410.20 (M + H+).
3-Benzyl-7-(piperazin-1-yl)benzo[d]oxazol-2(3H)-one (3)
To a solution of 2 (1.767 mmol, 0.721g, 1 equiv) in EtOH at 0 °C, acetyl chloride (10.21 mmol, 0.8 mL, 5.7 equiv) was added. The resulting reaction mixture was stirred at 55 °C for 2 h. After that time a white solid was precipitated, washed with hexane (15 mL), dried, and used in the next step without further purification (0.280 g, 52%). 1H NMR (DMSO-d6, 300 MHz) δ 9.43 (s, 1H), 7.2–7.4 (m, 5H), 7.0–7.1 (m, 1H), 6.8–6.8 (m, 1H), 6.72 (d, 1H, J = 8.8 Hz), 4.99 (s, 2H), 3.4–3.5 (m, 4H), 3.2–3.2 (m, 4H). 13C NMR (75 MHz; DMSO-d6): δ 153.9, 136.0, 134.6, 132.5, 132.1, 129.2, 128.3, 127.9, 125.0, 111.4, 103.3, 46.2, 45.5, 42.9. UPLC–MS (ESI) calcd for C18H19N3O2 310.15 (M + H+), found 310.19 (M + H+).
4-Benzyl-8-bromo-3,4-dihydro-2H-benzo[b][1,4]oxazine (ii)
A solution of 8-bromo-3,4-dihydro-2H-benzo[b][1,4]oxazine (1.63 mmol, 0.350g, 1 equiv) in DMF (10 mL) at 0 °C NaH was added portionwise, and the resulting mixture was stirred for 30 min following by addition of benzyl bromide (1.95 mmol, 0.212 g, 1.2 equiv). Resulting orange slurry was stirred at room temperature for 5 h. After that time, the reaction mixture was poured into water at 0 °C (20 mL), EtOAc (20 mL) was added, and the organic layer was separated. Purification over column chromatography using hexane/EtOAc 9:1 afforded ii as crystallizing oil (0.395g, 80%). 1H NMR (chloroform-d, 300 MHz) δ 7.2–7.5 (m, 3H), 6.91 (dd, 1H, J = 2.1, 7.3 Hz), 6.6–6.7 (m, 2H), 4.48 (s, 2H), 4.3–4.4 (m, 3H), 3.4–3.5 (m, 3H). 13C NMR (chloroform-d, 75 MHz) δ 140.6, 137.6, 136.7, 129.1, 128.8, 128.3, 127.4, 127.0, 122.1, 121.3, 111.6, 110.7, 65.1, 55.0, 47.2. UPLC–MS (ESI) calcd for C15H14BrNO 304.03 (M + H+), found 304.19 (M + H+).
tert-Butyl 4-(4-Benzyl-3,4-dihydro-2H-benzo[b][1,4]oxazin-8-yl)piperazine-1-carboxylate (iii)
A mixture of 4-benzyl-8-bromo-3,4-dihydro-2H-benzo[b][1,4]oxazine (1.72 mmol, 0.520 g, 1 equiv), Boc-piperazine (3.43 mmol, 0.638 g, 2 equiv), rec-BINAP (0.063 mmol, 0.042g, 0.04 equiv), Pd2(dba)3 (0.034 mmol 0.031g, 0.02 equiv), t-BuONa (2.41 mmol, 0.230g, 1.4 equiv) in toluene (10 mL) in sealted tube was heated at 100 °C for 12 h. After that time the reaction mixture was filtered over Celite, toluene was evaporated, and the crude mixture was filtered over silica gel and the compound was used directly in the next step.
4-(4-Benzyl-3,4-dihydro-2H-benzo[b][1,4]oxazin-8-yl)piperazin-1-ium Chloride (5)
A mixture of tert-butyl 4-(4-benzyl-3,4-dihydro-2H-benzo[b][1,4]oxazin-8-yl)piperazine-1-carboxylate (iii) (0.370g) and 1 M HCl/EtOAc (35 mL) was stirred for 6 h. The precipitated solid was washed with Et2O (15 mL) affording the final compound as a white solid (0.280 g, 75%). 1H NMR (DMSO-d6, 300 MHz) δ 9.6–9.7 (m, 2H), 7.2–7.4 (m, 5H), 6.6–6.7 (m, 1H), 6.5–6.6 (m, 2H), 4.47 (s, 2H), 4.25 (s, 2H), 3.39 (s, 6H), 3.29 (s, 4H). 13C NMR (DMSO-d6, 75 MHz) δ 138.5, 136.5, 136.5, 129.0, 127.5, 127.4, 121.5, 110.1, 108.3 (aromatic carbons overlap), 64.4, 54.7, 48.1, 47.3, 42.6. UPLC–MS (ESI) calcd for C19H23N3O 310.18 (M + H+), found 310.21 (M + H+).
4.2.3. General Procedure for the Preparation of Alcohols 6–10
A mixture of bromoethanol (1.2 equiv, 0.73 mmol, 0.060 mL), appropriate building block a–e (1 equiv, 0.61 mmol), and K2CO3 (1.2 equiv, 0.73 mmol, 0.105 g) in acetonitrile (15 mL) or dioxane (15 mL) was stirred at reflux overnight, then cooled to room temperature, and K2CO3 was filtered off. The solvent was removed, and the reaction mixture was purified using column chromatography with an eluent: ethyl acetate/dichloromethane/methanol, 2:7:1 (v/v/v).
2-(4-(3-(Benzyloxy)-2-methylphenyl)piperazin-1-yl)ethan-1-ol (6)
Crystallizing oil, yield 64%. 1H NMR (500 MHz, chloroform-d) δ 7.47–7.45 (m, 2H), 7.42–7.38 (m, 2H), 7.34 (d, J = 7.2 Hz, 1H), 7.13 (t, J = 8.2 Hz, 1H), 6.76–6.68 (m, 2H), 5.09 (s, 2H), 3.69–3.67 (m, 2H), 2.97 (br t, J = 4.7 Hz, 4H), 2.79–2.68 (m, 4H), 2.67–2.63 (m, 3H), 2.25 (s, 3H). 13C NMR (126 MHz, chloroform-d) δ 157.68, 152.50, 137.57, 128.47, 127.68, 127.08, 126.30, 121.18, 111.77, 107.01 (aromatic carbons overlap), 70.05, 59.28, 57.66, 53.32, 51.93, 11.03. UPLC–MS (ESI) calcd for C20H26N2O2 327.24 (M + H+), found 327.32 (M + H+).
1-Benzyl-4-(4-(2-hydroxyethyl)piperazin-1-yl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (7)
Crystallizing oil, yield 90%. 1H NMR (500 MHz, methanol-d4) δ 7.33–7.20 (m, 5H), 6.96–6.90 (m, 1H), 6.76–6.71 (m, 1H), 6.70–6.65 (m, 1H), 5.05–5.01 (m, 2H), 3.76–3.69 (m, 2H), 3.09–3.00 (m, 4H), 2.82–2.73 (m, 4H), 2.68–2.60 (m, 2H) (NH and OH protons not detected). 13C NMR (126 MHz, methanol-d4) δ 155.53, 136.72, 136.63, 130.81, 128.44, 128.41, 127.32, 127.03, 126.96, 121.78, 120.95, 111.03, 103.80 (aromatic carbons overlap), 59.97, 58.41, 53.30, 50.19, 43.82. UPLC–MS (ESI) calcd for C20H24N4O2 353.19 (M + H+), found 353.24 (M + H+).
3-Benzyl-7-(4-(2-hydroxyethyl)piperazin-1-yl)benzo[d]oxazol-2(3H)-one (8)
Crystallizing oil, yield 64%. 1H NMR (chloroform-d, 500 MHz) δ 7.3–7.3 (m, 5H), 6.9–7.0 (m, 1H), 6.6–6.6 (m, 1H), 6.4–6.5 (m, 1H), 4.96 (s, 2H), 3.6–3.7 (m, 2H), 3.3–3.3 (m, 5H), 2.7–2.7 (m, 4H), 2.6–2.6 (m, 2H). 13C NMR (75 MHz, chloroform-d) δ 155.36, 154.31, 135.63, 134.70, 132.55, 131.57, 128.76, 128.04, 127.40, 124.41, 110.49, 101.56 (aromatic carbons overlap), 64.48, 62.54, 59.34, 57.69, 52.71, 49.00, 45.92. UPLC–MS (ESI) calcd for C20H23N3O3 354.17 (M + H+), found 354.19 (M + H+).
4-Benzyl-8-(4-(2-hydroxyethyl)piperazin-1-yl)-2H-benzo[b][1,4]oxazin-3(4H)-one (9)
Crystallizing oil, yield 62%. 1H NMR (300 MHz, DMSO-d6) δ 7.35–7.18 (m, 5H), 6.87–6.79 (m, 1H), 6.66–6.60 (m, 2H), 5.14–5.09 (m, 2H), 4.76–4.71 (m, 2H), 3.52–3.47 (m, 4H), 3.39 (s, 1H), 3.02–2.92 (m, 4H), 2.56–2.53 (m, 2H), 2.46–2.39 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 165.09, 141.97, 137.97, 136.96, 129.82, 128.97, 127.45, 126.86, 122.82, 113.63, 109.95 (aromatic carbons overlap), 67.31, 63.05, 60.59, 58.76, 53.67, 50.49, 48.9, 44.22. UPLC–MS (ESI) calcd for C21H25N3O3 368.19 (M + H+), found 368.23 (M + H+).
2-(4-(4-Benzyl-3,4-dihydro-2H-benzo[b][1,4]oxazin-8-yl)piperazin-1-yl)ethan-1-ol (10)
Crystallizing oil, yield 29%. 1H NMR (chloroform-d, 300 MHz) δ 7.2–7.4 (m, 5H), 6.6–6.8 (m, 1H), 6.3–6.5 (m, 2H), 4.44 (s, 2H), 4.3–4.4 (m, 2H), 3.6–3.7 (m, 2H), 3.3–3.4 (m, 2H), 3.09 (m, 4H), 2.72 (br t, 4H, J = 4.4 Hz), 2.62 (t, 2H, J = 5.3 Hz), 2.35 (br s, 1H). 13C NMR (chloroform-d, 75 MHz) δ 154.8, 140.9, 138.2, 136.5, 136.2, 128.6, 127.1, 127.0, 121.0, 108.2, 107.9, 79.6, 64.4, 55.5, 50.9, 47.1 (aliphatic carbons overlap), 28.5. UPLC–MS (ESI) calcd for C21H27N3O2 354.21 (M + H+), found 354.27 (M + H+).
4.2.4. General Procedure for the Preparation of Boc-Intermediates 11–15 and Final Compounds 16–20
To a solution of appropriate alcohol 6–10 (1 equiv, 0.6 mmol) in dry DCM (10 mL), Boc-GABA (1.1 equiv, 0.72 mmol, 0.147 g), DCC (1 equiv, 0.72 mmol, 0.148 g), DMAP (0.1 equiv, 0.06 mmol, 0.007 g) were added, and the resulting reaction mixture was stirred for 12 h. Next, the solid was separated from the reaction mixture and the filtrate was evaporated. Boc intermediates 11–15 were used straightforwardly in the subsequent step without purification. Thus, a mixture of Boc intermediate (0.5 mmol) and 1 M solution of HCl in EtOAc (30 mL) was stirred for 12 h at room temperature. The resulting solid was filtered off, washed with EtOAc (20 mL), and dried.
1-(2-((4-Ammoniobutanoyl)oxy)ethyl)-4-(3-(benzyloxy)-2-methylphenyl)piperazin-1-ium Dichloride (16)
White solid, yield 52%. 1H NMR (300 MHz, DMSO-d6) δ 11.60–11.43 (br s, 1H), 8.10–7.97 (br s, 3H), 7.54–7.26 (m, 5H), 7.22–7.10 (m, 1H), 6.91–6.78 (m, 1H), 6.73–6.62 (m, 1H), 5.16–5.03 (m, 2H), 4.59–4.39 (m, 2H), 3.63–3.38 (m, 4H), 3.18–3.12 (m, 2H), 2.81–2.70 (m, 2H), 2.59–2.52 (m, 2H), 2.41 (t, J = 7.2 Hz, 2H), 2.15 (s, 3H), 1.94–1.62 (m, 4H). 13C NMR (75 MHz, DMSO-d6) δ 172.34, 171.52, 157.55, 151.29, 137.88, 128.88, 128.13, 127.74, 127.06, 120.53, 112.00, 108.35, 69.82, 52.31, 48.72, 38.41, 37.02, 35.28, 30.73, 29.79, 23.02, 22.58, 11.24. UPLC–MS (ESI) calcd for C24H33N3O3 412.25 (M + H+), found 412.35 (M + H+).
1-(2-((4-Ammoniobutanoyl)oxy)ethyl)-4-(1-benzyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-4-yl)piperazin-1-ium Dichloride (17)
White solid, yield 26%. 1H NMR (300 MHz, DMSO-d6) δ 10.92 (br s, 3H), 7.35–7.21 (m, 4H), 6.92–6.76 (m, 3H), 6.69 (d, J = 7.9 Hz, 1H), 6.60 (d, J = 8.2 Hz, 1H), 4.97 (s, 2H), 4.15 (br t, J = 5.8 Hz, 2H), 3.02–2.90 (m, 8H), 2.69–2.57 (m, 4H), 2.27 (td, J = 7.4, 12.3 Hz, 2H), 1.70–1.51 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 173.05, 156.03, 154.84, 137.77, 136.62, 130.98, 128.99, 127.75, 127.69, 121.54, 120.74, 110.59, 103.33 (aromatic carbons overlaps), 61.67, 56.58, 53.34, 50.51, 43.69, 35.22, 31.39, 25.37. UPLC–MS (ESI) calcd for C24H31N5O3 437.24 (M + H+), found 437.31 (M + H+).
1-(2-((4-Ammoniobutanoyl)oxy)ethyl)-4-(3-benzyl-2-oxo-2,3-dihydrobenzo[d]oxazol-7-yl)piperazin-1-ium Dichloride (18)
White solid, yield 43%. 1H NMR (500 MHz, methanol-d4) δ 7.27–7.18 (m, 5H), 7.05–6.96 (m, 1H), 6.73–6.60 (m, 2H), 4.92 (s, 2H), 4.52–4.38 (m, 2H), 3.89–3.76 (m, 2H), 3.73–3.60 (m, 2H), 3.55–3.44 (m, 2H), 3.38–3.28 (m, 4H), 3.00–2.86 (m, 2H), 2.63–2.50 (m, 2H), 1.98–1.78 (m, 2H). 13C NMR (126 MHz, methanol-d4) δ 172.23, 154.49, 135.33, 133.77, 133.06, 131.94, 128.66, 127.93, 127.46, 124.81, 111.56, 103.49 (aromatic carbons overlap), 58.51, 55.98, 52.83, 46.62, 45.64, 39.34, 30.81, 22.43. UPLC–MS (ESI) calcd for C24H30N4O4 439.23 (M + H+), found 439.42 (M + H+).
1-(2-((4-Ammoniobutanoyl)oxy)ethyl)-4-(4-benzyl-3-oxo-3,4-dihydro-2H-benzo[b][1,4]oxazin-8-yl)piperazin-1-ium Dichloride (19)
White solid, yield 68%. 1H NMR (500 MHz, methanol-d4) δ 7.34–7.09 (m, 5H), 6.91–6.63 (m, 3H), 5.14 (s, 2H), 4.72 (s, 2H), 3.98–3.84 (m, 2H), 3.79–3.45 (m, 4), 3.06–2.90 (6H), 2.41–2.33 (m, 2H), 2.00–1.81 (m, 4H). 13C NMR (126 MHz, methanol-d4) δ 173.15, 164.36, 139.31, 138.24, 136.18, 128.46, 127.07, 126.37, 122.65, 114.83, 114.10, 111.45 (aromatic carbons overlap), 67.42, 58.80, 58.63, 55.27, 52.41, 44.45, 39.30, 36.62, 34.63, 30.01, 22.52. UPLC–MS (ESI) calcd for C25H32N4O4 453.24 (M + H+), found 453.31 (M + H+).
1-(2-((4-Ammoniobutanoyl)oxy)ethyl)-4-(4-benzyl-3,4-dihydro-2H-benzo[b][1,4]oxazin-8-yl)piperazin-1-ium Dichloride (20)
White solid, yield 41%. 1H NMR (300 MHz, DMSO-d6) δ 11.41 (br s, 1H), 8.36–8.28 (s, 1H), 8.15–8.03 (br s, 2H), 7.38–7.17 (m, 5H), 6.69–6.57 (m, 1H), 6.46–6.36 (m, 1H), 6.31–6.17 (m, 1H), 4.60–4.37 (m, 4H), 4.31–4.13 (m, 2H), 3.32–2.90 (m, 10H), 2.91–2.70 (m, 4H), 2.59–2.51 (m, 2H), 1.93–1.76 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 172.38, 138.95, 138.95, 136.34, 136.23, 136.23, 128.92, 127.49, 127.29, 121.17, 107.46 (aromatic carbons overlap), 79.70, 64.30, 54.73, 52.39, 47.50, 47.41, 38.42, 33.78, 30.76, 22.62. UPLC–MS (ESI) calcd for C25H34N4O3 439.26 (M + H+), found 439.45 (M + H+).
4.3. Radioligand Binding Studies and Functional Assays
4.3.1. Determination of Affinity for GABA-A Receptor
The study was acquired according to the earlier protocols.39 The rats’ brains were homogenized and prepared accurately according to the previous protocol.39 On the day of the study, brain homogenates were thawed at room temperature and “diluted” in 20 volumes of ice-cold 50 mM Tris-HCl buffer (pH 7.4) and centrifuged (20.000g, 30 min, 0–4 °C). The assay was performed directly on 96-well microplates, which contained 50 mM Tris-HCl buffer (pH 7.4) in a total volume of 300 μL. Reaction mix included 240 μL of the brain tissue suspension, 30 μL of [3H]-muscimol, and 30 μL solution of tested compounds (administrated at various concentrations: 10–10–10–5 M). In order to determine potential nonspecific binding, 100 μM GABA was added. The 96-well microplates containing the reaction mix were incubated for 10 min at 0 °C, followed by rapid filtration over glass fiber filters FilterMate B (PerkinElmer, USA) using the Harvester-96 MACH III FM (Tomtec, USA). The filter mats were dried using a microwave and then placed inside a plastic bag (PerkinElmer, USA). After that, they were soaked in 10 mL of Ultima Gold MV liquid scintillation cocktail (PerkinElmer, USA). Subsequently, the radioactivity present on the filter was quantified using a MicroBeta TriLux 1450 scintillation counter (PerkinElmer, USA). Ki values were estimated according to the Cheng and Prusoff equation. The study was performed in duplicates. The statistical analysis was performed using (GraphPad Prism, version 4.0, San Diego, CA, USA).
4.3.2. Radioligand Binding Assays for 5-HT6 Receptors
To determine the affinity for 5-HT6, the cryopreserved membranes from HEK-293 cells stably transfected with the human recombinant 5-HT6 receptor were used.40 Preparation of the tested compounds included preparation of stock solutions in DMSO (1 mM) and subsequent serial dilutions (10–10–10–5 M) of tested compounds in the appropriate buffer (50 mM Tris buffer, pH 7.4, 10 mM MgCl2, 0.5 mM ethylenediaminetetraacetic acid). The serial dilutions were acquired directly on a 96-well microplate. The reaction mix included 50 μL of appropriate concentration of tested molecule, 50 μL of [3H]LSD 2.5/2.0 nM, and 150 μL of diluted membranes. The microplate containing the reaction mix was protected with a sealing tape and incubated for 60 min at 37 °C. Quenching of the reaction mixtire included: rapid filtration through UniFilter 96 GF/B filter microplate and rapid washes using 200 μL 50 mM assay buffer and vacuum manifold and 96-well pipettor. The collected microplates were dried overnight at 37 °C. The UniFilter bottoms were sealed, and then 30 μL of Betaplate Scint liquid scintillator (PerkinElmer) was added to each well. The radioactivity was quantified using a MicroBeta TriLux 1450 scintillation counter (PerkinElmer) with an estimated efficiency of 30%. The obtained data were fit to a one-site curve-fitting equation using Prism 5 software (GraphPad Software), and the Ki values were determined using the Cheng–Prusoff equation. Radioligand binding was performed in duplicates.
4.3.3. Functional Assays for 5-HT6 Receptor
The studies were acquired according to previous protocols.41 All the examined compounds (novel and the reference) were dissolved in DMSO (1 mM) and diluted accordingly using the assay buffer, directly in 96-well microplate. The recombinant CHO-K1 cells expressing human GPCR, mitochondrially targeted aequorin, and the promiscuous G protein α16 specific for 5-HT6 were used for the functional assay. The cells were thawed and suspended in the appropriate buffer containing DMEM/HAM’s F12 with 0.1% protease free BSA and were then centrifuged. After adding coelenterazine h to a final concentration of 5 μM, the cell pellet was resuspended in the buffer. The cell suspension was incubated at 16 °C for 16 h while being protected from light, gently shaken, and diluted with assay buffer to a concentration of 5000 cells/mL. Following that, 50 μL of the cell suspension was introduced to preloaded white opaque 96-well microplates containing the tested molecules. This was done using the automatic injectors integrated into the radiometric and luminescence plate counter MicroBeta2 LumiJET (PerkinElmer, USA). The light emission resulting from calcium mobilization was monitored for a duration of 60 s. For examination of the antagonistic properties of tested molecule, following 30 min of incubation, the reference agonist was administrated and the light emission was detected.
4.3.4. Electrophysiological Studies
Electrophysiological studies were acquired using the QPatch16X automatic patch clamp platform (Sophion Biosciences) as described before.42,43 The HEK293 cells stably expressing the human α1β3γ2 GABA-A receptor were used. Before the assay, cells were detached from the culture flask by treating them with TrypLE Express solution (Life Technologies) and subsequently suspended in serum-free media. The cells were loaded into a 1.5 mL microtube on the automated electrophysiology instrument, which subsequently performed a spin-down using its built-in centrifuge. Following this, the cells were washed with Ringer’s extracellular solution. The cells were moved to the pipetting wells of a 16-channel planar patch chip plate (QPlate 16X) designed for single-use. A combined suction/voltage protocol was then applied to establish gigaseals. Subsequent suction resulted in the establishment of whole-cell configuration. The chloride currents flowing through the GABA-A receptor were quantified for a duration of 7 s following the administration of the test molecule. During the entire whole-cell recording, the holding potential was maintained at −90 mV. The extracellular solution consisted of 2 mM 4KCl, 145 mM NaCl, 10 mM HEPES, CaCl2, 1 mM MgCl2, 10 mM glucose (pH 7.4, 300 mOsm). The intracellular solution contained 140 mM CsF, 1 mM EGTA, 5 mM CsOH, 10 mM HEPES, 20 mMNaCl (pH 7.2, 320 mOsm). The assay was configured within the instrument software in a sequence that involved the following steps: administration of 10 μM GABA (reference agonist AG1); administration of 1 μM test molecule (T1); coadministration of 1 μM test molecule and 10 μM GABA (T2); a second application of 10 μM GABA (AG2); and introduction of 10 μM bicuculline (reference antagonist) together with 10 μM GABA (ATG). The acquired data were examined with QPatch assay software (v5.0, Sophion Biosciences)44 and are represented as the mean of three separated measurements performed on distinct cells. The efficacy of the tested molecules was determined by calculating the baseline-corrected ratio of the maximum current amplitudes elicited by the tested compounds and the reference agonist (T1-ATG/AG1-ATG or T2-ATG/AG1-ATG). The raw current recordings were standardized and presented as a percentage of the current amplitude evoked by the reference agonist, using the QPatch assay software (v5.0, Sophion Biosciences).
4.4. Physicochemical and ADMET Assays
4.4.1. Thermodynamic Solubility in PBS and Stability Assays
The quantitative HPLC analyses were acquired using Waters Alliance e2695 separations module (Waters, Milford, CT, USA) containing 2998 photodiode array (PDA), a detector (Waters, Milford, CT, USA), and the SpeedROD RP-18e 50–4.6 mm column (Merck, KGaA, Darmstadt, Germany). The temperature of the column was preserved at 30 °C. The experiment was conducted under the following conditions: a flow rate of 5 mL/min, eluent A (water/0.1% HCOOH), eluent B (MeCN/0.1% HCOOH), a gradient of starting from 0% of B to 100% of B over a duration of 3 min. Each sample was injected at a volume of 10 μL in triplicate. Thermodynamic solubility in PBS measurement: The chromatograms were examined at 255 nm (perphenazine) and 212 nm (16). Stock solutions of analyzed compounds (16 and reference) were dissolved in methanol to achieve the concentration of 1 mg/mL. The stock solutions were mixed with methanol and diluted, resulting in various solutions with concentrations ranging from 1.0 to 0.125 μg/mL. These solutions were then utilized to generate calibration curves by plotting AUC versus concentration in μg/mL. The examined molecules (2 mg) were dissolved in 1 mL of Dulbecco’s phosphate buffered saline (DPBS). The mixture was then continuously agitated at 22 °C for 24 h using a thermoshaker. Following this period, the mixtures were filtered through a cellulose acetate syringe filter (with a pore size of 0.22 μm), moved to a chromatographic vial, and analyzed. To quantify the investigated compounds, the areas beneath their respective peaks on DAD chromatograms were employed. Solubility was calculated using the calibration curves. Chemical stability assay: Stock solution of 16 in DMSO (10 mg/mL) was used. Next, 25 μL of stock solution was mixed with 975 μL of PBS (Dulbecco’s phosphate-buffered saline, Sigma, Poland). The mix was gently stirred at 22 °C. HPLC analyses were used to quantify the percentage of molecules that remained at each time point relative to the 0 min time point. Brain tissue stability assay: Brain homogenate was prepared according to a previous protocol.22 Compound 16 was dissolved in DMSO to achieve the concentration of 5 mg/mL. Then, 50 μL of the stock solution was combined with 50 μL of ice-cold 50 mM Tris-HCl buffer (pH 7.4) and 150 μL of brain homogenate (suspended in 20 volumes of 50 mM Tris-HCl buffer, ice cold, pH 7.4). The mix was then incubated at 37 °C and quenched at several time points. To terminate the reaction mixture, 250 μL of the appropriate reaction mixture was mixed with 1000 μL of MeCM in the Eppendorf Tube. Following 10 min of shaking (1500 rpm), samples were centrifuged (10 000 rpm, 10 min, 4 °C) and the acquired supernatant was collected. Before conducting HPLC analysis, the supernatant was filtered via a cellulose acetate syringe filter with a pore size of 0.45 μm and then transferred to a chromatographic vial. HPLC analysis was used to determine the percentage of the tested molecule remaining at each time point, relative to the 0 min time point. Plasma stability assay was performed as described previously.45 Frozen rat plasma (Wistar) was thawed. Next, 4 μL of stock solution of tested compound 16 in DMSO (20 mg/mL) was mixed with 396 μL of rat plasma and incubated at 37 °C. The samples were collected at relative time points. The reaction was terminated by adding 1200 of μL acetonitrile/methanol mixture (50:50, v/v). The samples were centrifuge at 25 000 rpm for 10 min. HPLC analysis was used to determine the percentage of the tested molecule remaining at each time point relative to the 0 min time point. All the experiments were performed in duplicate.
4.4.2. Metabolic Stability
The assay was performed by Eurofins Discovery, a contract research organization, according to standard methodology, described previously.46
4.4.3. Permeability
To estimate the passive transport through cell membranes, we used specialized PAMPA Plate System Gentest acquired from Corning (Tewksbury, MA, USA). The assay was performed as described in the manufacturer’s protocol. The quantitative measurements of tested molecules in apical and basolateral wells were conducted using LC/MS (Waters ACQUITY TQD system with the TQ detector, Milford, USA) and a specific internal standard. The Pe permeability coefficient was calculated rendering the formulas described previously47,47 and compared to the reference compound sulpiride.
4.4.4. Hepatotoxicity Assay
Hepatotoxiticy assay was conducted according to our previously described procedures.42Cell culture and treatment: The human hepatocellular carcinoma (HepG2) cell line was acquired from ATCC, Manassas, USA (HB-8065), and maintained according to the manufacturer’s ATCC protocol. Cell culture conditions included a medium consisting of Dulbecco’s modified Eagle’s medium (DMEM, Merck), 10% fetal bovine serum, 100 mg/mL streptomycin, 100 IU/mL penicillin (acquired from ThermoFisher), humidified atmosphere containing 5% of carbon dioxide, temperature of 37 °C. The stock solution of the tested compound was prepared in DMSO (10 mM) and next diluted with buffered phosphate saline (PBS, Merck). Cell membrane damage was examined with the use of a ToxiLight bioassay (Lonza), rendering the manufacturer’s protocol described previously.42 A culture medium containing 10% Triton X-100 (Merck) was used as the positive control of cell damage, while the negative control contained solely vehicle. After incubating for 5 min, the luminescence was examined with the plate reader POLARstar Omega (BMG Labtech). The results were expressed as a percentage of the positive control, which was defined as the percentage of dead cells relative to the control sample. Cell viability assay: The viability of cells was examined with the use of the PrestoBlue reagent (ThermoFisher) and protocol provided by the manufacturer. After incubating for 24 h with the examined compounds, one-tenth of the remaining medium volume was mixed with the reagent (PrestoBlue) in a microplate well. Following incubation at 37 °C during 15 min, the fluorescence (EX 530; EM 580 nm) was measured using the plate reader POLARstar Omega, (BMG Labtech). The viability values obtained are presented as a percentage of live cells relative to the DMSO (control sample). The GraphPad program was used to perform the statistical analysis. The mean values with their corresponding coefficient of variation (CV) were reported for all data. The differences between the groups were analyzed using one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison tests as post hoc analysis. The statistical significance: p < 0.05.
4.5. In Vitro Anti-Inflammatory Assessment
4.5.1. Preparation of Cells
Mouse microglial (BV-2) cell line was a generous gift from Prof. Bożena Kamińska-Kaczmarek of the Nencki Institute of Experimental Biology, Polish Academy of Sciences, Warsaw, Poland. The cell culture medium: Dulbecco’s modified with high glucose (DMEM, Glutamx ThermoFisher) and supplemented with 10% inactivated fetal bovine serum heat (ThermoFisher), 100 IU/mL penicillin (Merck) and 100 μg/mL streptomycin (Merck). Cell culture conditions: culture flasks (area 175 cm2, Nunc), temperature of 37 °C, 5% CO2. For the assessment of the effect of tested compounds on the quantities of reactive oxygen species, nitric oxide, and inflammatory markers, the BV-2 cells were positioned in a 96-well culture plate (5 × 104 cells per well, Falcon). In the case of cell membrane damage assay, a 96-well culture plate (2 × 104 cells per well, Falcon) was used. The cells were cultured in the incubator (37 °C, 5% CO2) for 24 h prior to the assay. The tested compounds were dissolved in DMSO to achieve the concentration of 10–2 M. Serial dilutions of the compounds were prepared in PBS and added directly to the medium containing adherent cells. The resulting mixtures were examined for any evidence of precipitation or opalescence before the assay. BV-2 cells were incubated with selected molecules (6, 16 at a concentration of 10 μM) during 1 h. Subsequently, lipopolysaccharide (100 ng/mL) was added and the resulting mixture was incubated for 18 h. Next, the culture supernatant was collected to measure the levels of nitric oxide (NO), reactive oxygen species (ROS), IL-6, and TNF-α according to the procedures described in detail below. All experiments were performed in duplicates, in three independent experiments.
4.5.2. NO Measurement
The quantities of NO produced by the cells were determined using DAN reagent (2,3-diaminonaphthalene), rendering the method protocol described by Nussler et al.48 After incubating at room temperature for 15 min, the fluorescence intensity (EX 360; EM 440 nm) was measured using a microplate reader POLARstar Omega from BMG Labtech. The amounts of NO were then calculated as a percentage of the control (which represents the maximal response of LPS). ROS measurement. The fluorescent dye 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA)49 was utilized to determine the quantity of ROS released by BV-2 cells. After a 30 min incubation period at 37 °C with 10 μM DCFH-DA, the fluorescence intensity (EX 490; EM 520 nm) was examined using a microplate reader POLARstar Omega from BMG Labtech. The amounts of ROS were then expressed as a percentage of the control (which represents the maximal response of LPS). The IL-6 and TNF-α levels detected in the culture supernatants were quantified using LANCE Ultra TR-FRET detection kit (PerkinElmer), rendering the manufacturer’s procedure. The detection of IL-6 and TNF-α was performed independently in a 384-well plate according to the instructions provided. To each well of the plate, 15 μL of sample was added followed by the addition of 5 μL of premixed antibody solution. After incubating IL-6 for 1 h and TNF-α for 3 h in the absence of light, at 22 °C, the plates were transferred to an EnVision plate reader (PerkinElmer). Measurements were conducted at 320 nm wavelength for excitation, at 615 nm for donor emission, and at 660 nm for acceptor emission. The final data were determined as the ratio of the 660 nm signal to the 615 nm signal. The results represent the levels of each cytokine determined as a percentage of control (considered as maximal cell response to LPS). The GraphPad program was used to conduct statistical analysis, with all outcomes presented as mean values with standard deviation (SD). Differences between groups were assessed using one-way ANOVA, followed by post hoc analysis (Dunnett’s multiple comparison tests). Results were considered statistically significant if the p-value was less than 0.05. Evaluation of microglia morphology: BV-2 cell line was pretreated with the tested compounds (10 μM) and incubated for 1 h, followed by addition of LPS (100 ng/mL). The mixture was incubated during 18 h period of time. Next, the following fluorescent dyes were used: Calcein AM (ThermoFisher), Hoechst 33342 (ThermoFisher), and MitoTracker (ThermoFisher) as indicated previously.22 The images were taken with Leica DMI8 microscopy.
4.6. Behavioral Evaluation
4.6.1. Animals
Naïve female Wistar rats (weighing 180–200 g) were used in the study. The animals were housed in groups of three to four in standard Makrolon cages (37 cm × 21 cm × 15 cm) under strictly controlled laboratory conditions (ambient temperature 21–24 °C, relative humidity 45–65%) with a 12/12 h light–dark regime (light on at 6:00 a.m. and off at 6:00 p.m.). Animals had unrestricted access to tap water and food (typical pellets). Prior the experiment the animals were randomly appointed to the group. The behavioral studies were accomplished between 8:00 a.m. and 5:00 p.m. with the researcher blind to the study. The experiments were performed according to protocols approved by the Local Ethical Committee in Krakow (Approval Number 147/2018), Poland. Housing and experimental procedures were conducted under the European Union Directive of September 22, 2010 (2010/63/EU) and Polish legislation regarding animal studies. All efforts were made to minimize animal suffering and the number of animals used in the study.
4.6.2. Drugs
Vortioxetine (Sigma, Germany) and the tested compounds (16 and 6) were dissolved in 10% cyclodextrin and administered ip 30 min before the test or during 14 consecutive days, with the last injection 60 min before the test. 10% cyclodextrin was administrated to control group.
4.6.3. Forced Swim Test in Rats
A modified forced swim test was carried out according to Detke et al.50,51 First, the animals were individually placed inside Plexiglas cylinders, which were 40 cm high and 18 cm in diameter, filled with water at a temperature of 23–25 °C for 15 min. Afterward, the rats were transferred to a Plexiglas box and kept under a 60 W bulb for 30 min to dry. The day after, exactly 24 h later, the rats were once again placed in the cylinder, and their duration of immobility, swimming, and climbing was observed and recorded during a 5 min. The swimming behavior involved energetic movements such as horizontal swimming around the cylinder, while climbing activity referred to upward movements of the forepaws along the swim chamber’s side. Immobility was recorded when the rat only made necessary movements to keep its head above the water. For each test, a fresh water was provided. Open field test in rats: The test was conducted as previously reported by our research group.52 The test was performed in a darkened room employing the Motor Monitor System (Campden Instruments, Ltd., U.K.). The system was equipped with two SmartFrame Open Field stations, each measuring 40 cm × 40 cm × 38 cm and consisting of 16 × 16 beams. These stations were placed inside sound-attenuating chambers and connected to a PC software using Motor Monitor System (Campden Instruments, Ltd., U.K.), equipped with two SmartFrame Open Field stations (40 cm × 40 cm × 38 cm) with 16 × 16 beams, placed in sound-attenuating chambers, which was linked to a PC software. The animals, belonging to either the vehicle-injected or drug-injected group, were individually positioned at the center of the station. The Motor Monitor System, which was automated, recorded the animal’s ambulation in both the X and Y axes, as well as the number of rearing and peeping episodes. Additionally, the total distance traveled by each animal during the 5 min test period was also recorded. Statistical analysis: Results are presented as the mean ± SEM values. They were estimated using one-way analysis of variance (ANOVA), followed by Newman–Keuls post hoc or unpaired two-tailed Student t test. Statistical significance was considered when p < 0.05 for differences between groups.
Acknowledgments
The authors thank Professor Bożena Kamińska-Kaczmarek from Nencki Institute of Experimental Biology, Polish Academy of Sciences, Warsaw, Poland, for the BV-2 cell line. The biorender.com server was used to prepare the graphical abstract and Figure 1.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.3c00033.
1H NMR, 13C NMR, COSY, and LCMS/MS spectra of selected final compounds, predicted binding modes of selected molecules, and hepatotoxicity assay (PDF)
Author Contributions
M.M.: study design and conception, synthesis of compounds, data collection and analysis, preparation of the original draft of the manuscript. B.M.: anti-inflammatory studies, data analysis. A.S.: radioligand binding and electrophysiological studies, data analysis. M.G.-L.: functional assays. T.K.: electrophysiological studies. A.G., M.S., A.B.: in silico studies, preparation of the manuscript. K.S.: resynthesis of compound 16, experimental part preparation. B.P.: confocal images of the BV-2 cell line. K.P., M.Kot., K.M.: in vivo studies, data analysis, manuscript preparation. M.Kol.: discussion of the result, manuscript review. All authors have given their approval for the final version of the manuscript.
The studies were supported by the Funds for Statutory Activity of Jagiellonian University, Medical College, Krakow, Poland, Projects K/ZDS/005536 and N42/DBS/000307. This research was funded by National Science Centre, Poland, Grant UMO-2018/31/D/NZ7/01433 (synthesis of 5-HT6 receptor binding scaffolds and radioligand binding studies for 5-HT6 receptor). The publication was created with the use of equipment cofinanced by the qLIFE Priority Research Area under the program “Excellence Initiative—Research University” (Grant 06/IDUB/2019/94) at Jagiellonian University.
The authors declare the following competing financial interest(s): M. Kolaczkowski is an employee of Adamed Pharma.
Special Issue
Published as part of the ACS Chemical Neuroscience special issue “Monitoring Molecules in Neuroscience 2023”.
Supplementary Material
References
- Hirschfeld R. M. History and Evolution of the Monoamine Hypothesis of Depression. J. Clin. Psychiatry 2000, 61 (Suppl. 6), 4–6. [PubMed] [Google Scholar]
- Cosci F.; Chouinard G.. The Monoamine Hypothesis of Depression Revisited: Could It Mechanistically Novel Antidepressant Strategies? In Neurobiology of Depression; Elsevier, 2019; pp 63–73, 10.1016/B978-0-12-813333-0.00007-X. [DOI] [Google Scholar]
- Voineskos D.; Daskalakis Z. J.; Blumberger D. M. Management of Treatment-Resistant Depression: Challenges and Strategies. NDT 2020, 16, 221–234. 10.2147/NDT.S198774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roohi E.; Jaafari N.; Hashemian F. On Inflammatory Hypothesis of Depression: What Is the Role of IL-6 in the Middle of the Chaos?. J. Neuroinflammation 2021, 18 (1), 45. 10.1186/s12974-021-02100-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher B.; Shen Q.; Sahir N. The GABAergic Deficit Hypothesis of Major Depressive Disorder. Mol. Psychiatry 2011, 16 (4), 383–406. 10.1038/mp.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G.; Mason G. F.; Rothman D. L.; Behar K. L.; Hyder F.; Petroff O. A. C.; Berman R. M.; Charney D. S.; Krystal J. H. Reduced Cortical γ-Aminobutyric Acid Levels in Depressed Patients Determined by Proton Magnetic Resonance Spectroscopy. Arch. Gen. Psychiatry 1999, 56 (11), 1043 10.1001/archpsyc.56.11.1043. [DOI] [PubMed] [Google Scholar]
- Fogaça M. V.; Duman R. S. Cortical GABAergic Dysfunction in Stress and Depression: New Insights for Therapeutic Interventions. Front. Cell. Neurosci. 2019, 13, 87. 10.3389/fncel.2019.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price R. B.; Shungu D. C.; Mao X.; Nestadt P.; Kelly C.; Collins K. A.; Murrough J. W.; Charney D. S.; Mathew S. J. Amino Acid Neurotransmitters Assessed by Proton Magnetic Resonance Spectroscopy: Relationship to Treatment Resistance in Major Depressive Disorder. Biol. Psychiatry 2009, 65 (9), 792–800. 10.1016/j.biopsych.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes M.; Bosmans E.; De Jongh R.; Kenis G.; Vandoolaeghe E.; Neels H. Increased serum il-6 and il-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine 1997, 9 (11), 853–858. 10.1006/cyto.1997.0238. [DOI] [PubMed] [Google Scholar]
- Sowa-Kućma M.; Styczeń K.; Siwek M.; Misztak P.; Nowak R. J.; Dudek D.; Rybakowski J. K.; Nowak G.; Maes M. Lipid Peroxidation and Immune Biomarkers Are Associated with Major Depression and Its Phenotypes, Including Treatment-Resistant Depression and Melancholia. Neurotox. Res. 2018, 33 (2), 448–460. 10.1007/s12640-017-9835-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes M.; Galecki P.; Chang Y. S.; Berk M. A Review on the Oxidative and Nitrosative Stress (O&NS) Pathways in Major Depression and Their Possible Contribution to the (Neuro)Degenerative Processes in That Illness. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35 (3), 676–692. 10.1016/j.pnpbp.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Wang D.; Salvadore G.; Hsu B.; Curran M.; Casper C.; Vermeulen J.; Kent J. M.; Singh J.; Drevets W. C.; Wittenberg G. M.; Chen G. The Effects of Interleukin-6 Neutralizing Antibodies on Symptoms of Depressed Mood and Anhedonia in Patients with Rheumatoid Arthritis and Multicentric Castleman’s Disease. Brain Behav Immun. 2017, 66, 156–164. 10.1016/j.bbi.2017.06.014. [DOI] [PubMed] [Google Scholar]
- Haroon E.; Daguanno A. W.; Woolwine B. J.; Goldsmith D. R.; Baer W. M.; Wommack E. C.; Felger J. C.; Miller A. H. Antidepressant Treatment Resistance Is Associated with Increased Inflammatory Markers in Patients with Major Depressive Disorder. Psychoneuroendocrinology 2018, 95, 43–49. 10.1016/j.psyneuen.2018.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C.-B.; Blakely R. D.; Hewlett W. A. The Proinflammatory Cytokines Interleukin-1beta and Tumor Necrosis Factor-Alpha Activate Serotonin Transporters. Neuropsychopharmacol. 2006, 31 (10), 2121–2131. 10.1038/sj.npp.1301029. [DOI] [PubMed] [Google Scholar]
- Samuvel D. J.; et al. A Role for P38 Mitogen-Activated Protein Kinase in the Regulation of the Serotonin Transporter: Evidence for Distinct Cellular Mechanisms Involved in Transporter Surface Expression. J. Neurosci. 2005, 25 (1), 29–41. 10.1523/JNEUROSCI.3754-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.; Schwab C.; Mcgeer P. Astrocytes Are GABAergic Cells That Modulate Microglial Activity. Glia 2011, 59, 152. 10.1002/glia.21087. [DOI] [PubMed] [Google Scholar]
- Nieman A. N.; Li G.; Zahn N. M.; Mian M. Y.; Mikulsky B. N.; Hoffman D. A.; Wilcox T. M.; Kehoe A. S.; Luecke I. W.; Poe M. M.; Alvarez-Carbonell D.; Cook J. M.; Stafford D. C.; Arnold L. A. Targeting Nitric Oxide Production in Microglia with Novel Imidazodiazepines for Nonsedative Pain Treatment. ACS Chem. Neurosci. 2020, 11 (13), 2019–2030. 10.1021/acschemneuro.0c00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y. The Immunological Function of GABAergic System. Front. Biosci. 2017, 22 (7), 1162–1172. 10.2741/4539. [DOI] [PubMed] [Google Scholar]
- Hirano K.; Searle K. L.; Nasir S.; Aw C.-C.; Browne E. R.; Rutter A. R. In Vivo 5-HT6 Receptor Occupancy by Antipsychotic Drugs in the Rat Brain. Neurosci. Lett. 2011, 503 (3), 240–243. 10.1016/j.neulet.2011.08.044. [DOI] [PubMed] [Google Scholar]
- Kucwaj-Brysz K.; Baltrukevich H.; Czarnota K.; Handzlik J. Chemical Update on the Potential for Serotonin 5-HT6 and 5-HT7 Receptor Agents in the Treatment of Alzheimer’s Disease. Bioorg. Med. Chem. Lett. 2021, 49, 128275 10.1016/j.bmcl.2021.128275. [DOI] [PubMed] [Google Scholar]
- Wesołowska A. Potential Role of the 5-HT6 Receptor in Depression and Anxiety: An Overview of Preclinical Data. Pharmacol Rep. 2010, 62 (4), 564–577. 10.1016/S1734-1140(10)70315-7. [DOI] [PubMed] [Google Scholar]
- Marcinkowska M.; Mordyl B.; Fajkis-Zajaczkowska N.; Siwek A.; Karcz T.; Gawalska A.; Bucki A.; Żmudzki P.; Partyka A.; Jastrzębska-Więsek M.; Pomierny B.; Walczak M.; Smolik M.; Pytka K.; Mika K.; Kotańska M.; Kolaczkowski M. Hybrid Molecules Combining GABA-A and Serotonin 5-HT6 Receptors Activity Designed to Tackle Neuroinflammation Associated with Depression. Eur. J. Med. Chem. 2023, 247, 115071 10.1016/j.ejmech.2022.115071. [DOI] [PubMed] [Google Scholar]
- Kołaczkowski M.; Marcinkowska M.; Bucki A.; Śniecikowska J.; Pawłowski M.; Kazek G.; Siwek A.; Jastrzębska-Więsek M.; Partyka A.; Wasik A.; Wesołowska A.; Mierzejewski P.; Bienkowski P. Novel 5-HT6 Receptor Antagonists/D2 Receptor Partial Agonists Targeting Behavioral and Psychological Symptoms of Dementia. Eur. J. Med. Chem. 2015, 92, 221–235. 10.1016/j.ejmech.2014.12.045. [DOI] [PubMed] [Google Scholar]
- Anand N.; et al. Kinase Modulators and Methods of Use. WO/2005/117909, Dec 15, 2005.
- Owens T.; et al. Benzimidazolone and Dihydroindolone Derivatives and Uses Thereof. US2007015744A1, Jan 18, 2007.
- Maag H.; et al. Substituted Benzoxazinones and Uses Thereof. US2004/92512, Nov 6, 2003.
- Chandra D.; Halonen L. M.; Linden A.-M.; Procaccini C.; Hellsten K.; Homanics G. E.; Korpi E. R. Prototypic GABAA Receptor Agonist Muscimol Acts Preferentially Through Forebrain High-Affinity Binding Sites. Neuropsychopharmacol. 2010, 35 (4), 999–1007. 10.1038/npp.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallard E.; Letourneur D.; Legendre P. Electrophysiology of Ionotropic GABA Receptors. Cell. Mol. Life Sci. 2021, 78 (13), 5341–5370. 10.1007/s00018-021-03846-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miret S.; De Groene E. M.; Klaffke W. Comparison of In Vitro Assays of Cellular Toxicity in the Human Hepatic Cell Line HepG2. SLAS Discovery 2006, 11 (2), 184–193. 10.1177/1087057105283787. [DOI] [PubMed] [Google Scholar]
- Cheung G.; Kann O.; Kohsaka S.; Făerber K.; Kettenmann H. GABAergic Activities Enhance Macrophage Inflammatory Protein-1α Release from Microglia (Brain Macrophages) in Postnatal Mouse Brain: Invading Microglia and GABAergic Activities. J. Physiol. 2009, 587 (4), 753–768. 10.1113/jphysiol.2008.163923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.; Schwab C.; Mcgeer P. L. Astrocytes Are GABAergic Cells That Modulate Microglial Activity. Glia 2011, 59 (1), 152–165. 10.1002/glia.21087. [DOI] [PubMed] [Google Scholar]
- Yang S.; Yang Y.; Chen C.; Wang H.; Ai Q.; Lin M.; Zeng Q.; Zhang Y.; Gao Y.; Li X.; Chen N. The Anti-Neuroinflammatory Effect of Fuzi and Ganjiang Extraction on LPS-Induced BV2 Microglia and Its Intervention Function on Depression-Like Behavior of Cancer-Related Fatigue Model Mice. Front. Pharmacol. 2021, 12, 670586 10.3389/fphar.2021.670586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro L. de B.; Davanzo G. G.; de Aguiar C. F.; Moraes-Vieira P. M. M. Using Flow Cytometry for Mitochondrial Assays. MethodsX 2020, 7, 100938 10.1016/j.mex.2020.100938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankelevitch-Yahav R.; Franko M.; Huly A.; Doron R. The Forced Swim Test as a Model of Depressive-like Behavior. JoVE 2015, (97), 52587 10.3791/52587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslau J.; Gilman S. E.; Stein B. D.; Ruder T.; Gmelin T.; Miller E. Sex Differences in Recent First-Onset Depression in an Epidemiological Sample of Adolescents. Transl. Psychiatry 2017, 7 (5), e1139–e1139. 10.1038/tp.2017.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J. A.; Weinstein H. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G Protein-Coupled Receptors. Methods in Neurosciences 1995, 25, 366–428. 10.1016/S1043-9471(05)80049-7. [DOI] [Google Scholar]
- Huang S.; Xu P.; Shen D.-D.; Simon I. A.; Mao C.; Tan Y.; Zhang H.; Harpso̷e K.; Li H.; Zhang Y.; You C.; Yu X.; Jiang Y.; Zhang Y.; Gloriam D. E.; Xu H. E. GPCRs Steer Gi and Gs Selectivity via TM5-TM6 Switches as Revealed by Structures of Serotonin Receptors. Mol. Cell 2022, 82 (14), 2681–2695. 10.1016/j.molcel.2022.05.031. [DOI] [PubMed] [Google Scholar]
- Kołaczkowski M.; Bucki A.; Feder M.; Pawłowski M. Ligand-Optimized Homology Models of D 1 and D 2 Dopamine Receptors: Application for Virtual Screening. J. Chem. Inf. Model. 2013, 53 (3), 638–648. 10.1021/ci300413h. [DOI] [PubMed] [Google Scholar]
- Sari S.; Barut B.; Marcinkowska M.; Sabuncuoğlu S.; Avci A.; Koçak Aslan E.; Özel A.; Siwek A. Potential of Nafimidone Derivatives against Co-morbidities of Epilepsy: In Vitro, in Vivo, and in Silico Investigations. Drug Dev Res. 2022, 83 (1), 184–193. 10.1002/ddr.21858. [DOI] [PubMed] [Google Scholar]
- Marcinkowska M.; Bucki A.; Sniecikowska J.; Zagórska A.; Fajkis-Zajączkowska N.; Siwek A.; Gluch-Lutwin M.; Żmudzki P.; Jastrzebska-Wiesek M.; Partyka A.; Wesołowska A.; Abram M.; Przejczowska-Pomierny K.; Cios A.; Wyska E.; Mika K.; Kotańska M.; Mierzejewski P.; Kolaczkowski M. Multifunctional Arylsulfone and Arylsulfonamide-Based Ligands with Prominent Mood-Modulating Activity and Benign Safety Profile, Targeting Neuropsychiatric Symptoms of Dementia. J. Med. Chem. 2021, 64 (17), 12603–12629. 10.1021/acs.jmedchem.1c00497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinkowska M.; Bucki A.; Panek D.; Siwek A.; Fajkis N.; Bednarski M.; Zygmunt M.; Godyń J.; Del Rio Valdivieso A.; Kotańska M.; Kołaczkowski M.; Więckowska A. Anti-Alzheimer’s Multitarget-directed Ligands with Serotonin 5-HT 6 Antagonist, Butyrylcholinesterase Inhibitory, and Antioxidant Activity. Arch. Pharm. Chem. Life Sci. 2019, 352 (7), 1900041 10.1002/ardp.201900041. [DOI] [PubMed] [Google Scholar]
- Marcinkowska M.; Kołaczkowski M.; Kamiński K.; Bucki A.; Pawłowski M.; Siwek A.; Karcz T.; Mordyl B.; Starowicz G.; Kubowicz P.; Pękala E.; Wesołowska A.; Samochowiec J.; Mierzejewski P.; Bienkowski P. Design, Synthesis, and Biological Evaluation of Fluorinated Imidazo[1,2- a ]Pyridine Derivatives with Potential Antipsychotic Activity. Eur. J. Med. Chem. 2016, 124, 456–467. 10.1016/j.ejmech.2016.08.059. [DOI] [PubMed] [Google Scholar]
- Marcinkowska M.; Kołaczkowski M.; Kamiński K.; Bucki A.; Pawłowski M.; Siwek A.; Karcz T.; Starowicz G.; Słoczyńska K.; Pękala E.; Wesołowska A.; Samochowiec J.; Mierzejewski P.; Bienkowski P. 3-Aminomethyl Derivatives of 2-Phenylimidazo[1,2- a ]-Pyridine as Positive Allosteric Modulators of GABA A Receptor with Potential Antipsychotic Activity. ACS Chem. Neurosci. 2017, 8 (6), 1291–1298. 10.1021/acschemneuro.6b00432. [DOI] [PubMed] [Google Scholar]
- Schupp M.; Park S. H.; Qian B.; Yu W. Electrophysiological Studies of GABA A Receptors Using QPatch II, the Next Generation of Automated Patch-Clamp Instruments. Curr. Protoc. Pharmacol. 2020, 89 (1), e75. 10.1002/cpph.75. [DOI] [PubMed] [Google Scholar]
- Hartman D. A. Determination of the Stability of Drugs in Plasma. Curr. Protoc. Pharmacol. 2002, 19 (1), 7.6. 10.1002/0471141755.ph0706s19. [DOI] [PubMed] [Google Scholar]
- Di L.; Kerns E. H.; Hong Y.; Chen H. Development and Application of High Throughput Plasma Stability Assay for Drug Discovery. Int. J. Pharm. 2005, 297 (1–2), 110–119. 10.1016/j.ijpharm.2005.03.022. [DOI] [PubMed] [Google Scholar]
- Chen X.; Murawski A.; Patel K.; Crespi C. L.; Balimane P. V. A Novel Design of Artificial Membrane for Improving the PAMPA Model. Pharm. Res. 2008, 25 (7), 1511–1520. 10.1007/s11095-007-9517-8. [DOI] [PubMed] [Google Scholar]
- Nussler A. K.; Glanemann M.; Schirmeier A.; Liu L.; Nüssler N. C. Fluorometric Measurement of Nitrite/Nitrate by 2,3-Diaminonaphthalene. Nat. Protoc. 2006, 1 (5), 2223–2226. 10.1038/nprot.2006.341. [DOI] [PubMed] [Google Scholar]
- Sauer H.; Klimm B.; Hescheler J.; Wartenberg M. Activation of P90RSK and Growth Stimulation of Multicellular Tumor Spheroids Are Dependent on Reactive Oxygen Species Generated after Purinergic Receptor Stimulation by ATP. FASEB j. 2001, 15 (13), 2539–2541. 10.1096/fj.01-0360fje. [DOI] [PubMed] [Google Scholar]
- Detke M. J.; Rickels M.; Lucki I. Active Behaviors in the Rat Forced Swimming Test Differentially Produced by Serotonergic and Noradrenergic Antidepressants. Psychopharmacology 1995, 121 (1), 66–72. 10.1007/BF02245592. [DOI] [PubMed] [Google Scholar]
- Porsolt R. D.; Anton G.; Blavet N.; Jalfre M. Behavioural Despair in Rats: A New Model Sensitive to Antidepressant Treatments. Eur. J. Pharmacol. 1978, 47 (4), 379–391. 10.1016/0014-2999(78)90118-8. [DOI] [PubMed] [Google Scholar]
- Jastrzębska-Więsek M.; Siwek A.; Partyka A.; Antkiewicz-Michaluk L.; Michaluk J.; Romańska I.; Kołaczkowski M.; Wesołowska A. Study of a Mechanism Responsible for Potential Antidepressant Activity of EMD 386088, a 5-HT6 Partial Agonist in Rats. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2016, 389 (8), 839–849. 10.1007/s00210-016-1245-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.