

SUMMARY
The histone-like protein HU plays a diverse role in bacterial physiology from the maintenance of chromosome structure to the regulation of gene transcription. HU binds DNA in a sequence-non-specific manner via two distinct binding modes: (i) random binding to any DNA through ionic bonds between surface-exposed lysine residues (K3, K18, and K83) and phosphate backbone (non-specific); (ii) preferential binding to contorted DNA of given structures containing a pair of kinks (structure-specific) through conserved proline residues (P63) that induce and/or stabilize the kinks. First, we show here that the P63-mediated structure-specific binding also requires the three lysine residues, which are needed for non-specific binding. Second, we demonstrate that substituting P63 to alanine in HU had no impact on non-specific binding but caused differential transcription of diverse genes previously shown to be regulated by HU, such as those associated with organonitrogen compound biosynthetic process, galactose metabolism, ribosome biogenesis, and cell adhesion. The structure-specific binding also helps create DNA supercoiling, which, in turn, may influence directly or indirectly the transcription of other genes. Our previous and current studies show that non-specific and structure-specific HU binding appear to have separate functions- nucleoid architecture and transcription regulation- which may be true in other DNA-binding proteins.
Keywords: HU, structure-specific binding, non-specific binding, transcription regulation, nucleoid architecture
Graphical Abstract
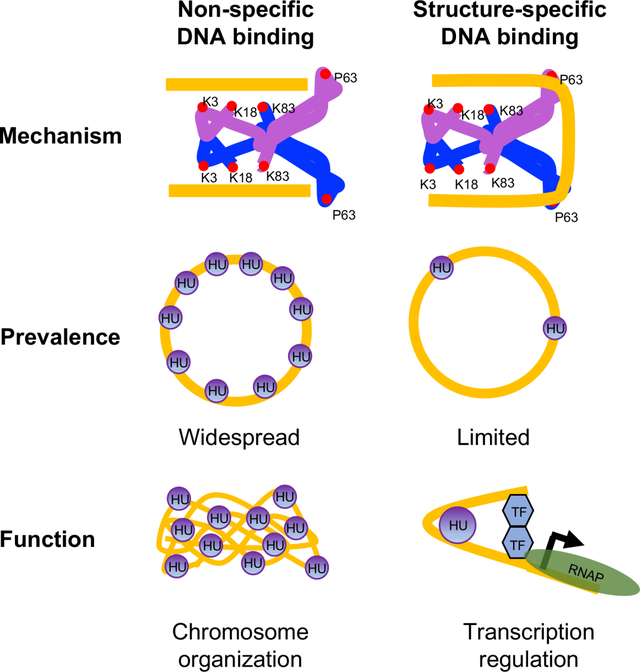
HU, the only evolutionarily conserved histone-like protein in bacteria, exhibits a non-specific or random binding mode that allows it to bind any duplex DNA without inducing bends and a DNA-structure-specific binding mode that stabilizes or indues bends in DNA. This study shows that non-specific and specific binding modes have separate functions-chromosome organization and transcription regulation, which may be true in other architectural DNA-binding proteins.
INTRODUCTION
The Escherichia coli chromosome of 1.5-millimeter contour length is condensed and organized into a helical ellipsoid called nucleoid (Fisher et al., 2013; Verma et al., 2019). Many biochemical factors (RNA and proteins) contribute to the formation of the nucleoid (Lioy et al., 2018; Lioy et al., 2021; Verma et al., 2019). Among them, the histone-like protein HU is a crucial player in maintaining a dynamic chromosome by promoting long-range DNA-DNA contacts, including the clustering of RNA-encoding loci (Lioy et al., 2018; Walker et al., 2020). HU is also associated with many biological processes, including gene transcription (Aki and Adhya, 1997) and DNA metabolic reactions e.g. replication, repair, and site-specific recombination (Chodavarapu et al., 2008; Kamashev and Rouviere-Yaniv, 2000; Montano et al., 2012). E. coli lacking HU exhibits a highly pleiotropic phenotype and an altered transcription profile with differential expression of about 8% of the genes (Oberto et al., 2009; Prieto et al., 2012). The HU regulon comprises highly conserved genes involved in essential biological processes, such as translation and ribosome biogenesis, and adaptive response to anaerobiosis, acid stress, and high osmolarity (Oberto et al., 2009; Prieto et al., 2012). Thus, HU plays a multitude of roles in bacterial physiology. Not surprisingly, HU is omnipresent in the bacterial kingdom and even essential in some bacterial species including pathogens (Bhowmick et al., 2014; Fernandez et al., 1997). HU in E. coli is composed of two homologous subunits α and β, encoded by hupA and hupB genes respectively, and exists in three dimeric forms: HUα2, HUβ2, and HUαβ. The levels of α and β subunits varies during the growth cycle, resulting in different composition of HU at different stages. HUα2 is predominant in the exponential phase and HUαβ in the stationary phase while HUβ2 is nearly undetectable in any growth phase (Claret and Rouviere-Yaniv, 1997).
The E. coli HU binds DNA in a sequence-non-specific manner with binding affinities ranging from 1 nM to up to 66,000 nM depending on DNA conformation, the form of HU, and ionic strength (Pinson et al., 1999). The extensive in vitro characterization of HU DNA binding using many different methodologies suggested several DNA binding modes (Hammel et al., 2016; Koh et al., 2011; Swinger et al., 2003; van Noort et al., 2004), but all of them fall into basically into two distinct types at the molecular level. We refer to them as (i) non-specific and (ii) structure-specific binding modes. The non-specific binding mode describes random binding of HU to any duplex DNA through ionic bonds between positively charged amino acid residues and phosphate backbone without inducing significant structural changes in the DNA. The structure-specific binding mode describes strong and preferential binding of HU that stabilizes or induces major DNA structural changes. The latter includes binding to contorted DNA (non-B DNA) with kinks, nicks, gaps, or cruciform structures, bent DNA within loops e.g. in the Mu transpososome and the gal repressosome (Bonnefoy et al., 1994; Geanacopoulos et al., 2001; Kamashev and Rouviere-Yaniv, 2000; Montano et al., 2012; Pinson et al., 1999; Pontiggia et al., 1993; Vitoc and Mukerji, 2011). Crystal structures of E. coli HUα2 or HUαβ bound to a 20-base pair DNA of random sequence showed that three highly conserved surface-exposed lysine residues, K3, K18, and K83, form electrostatic bonds with the DNA phosphate backbone (phosphate locks) (Fig. 1.). They are exposed on both sides of the HU dimer “body”, allowing a single HU dimer to interact simultaneously with either two different segments of the same DNA molecule or two different DNA molecules (Hammel et al., 2016; Remesh et al., 2020). Crystal structures of Anabaena HU in a complex with a kinked DNA showed that structure-specific binding, on the other hand, involves β-strands or “arms” instead of the dimer body (Swinger et al., 2003). While the positively charged surfaces of the two arms reach around the opposite faces of the DNA and wrap the minor grooves, a nearly universally conserved proline residue P63 at the tip of each arm intercalates into bases at the kink sites in the corresponding minor grooves (Swinger et al., 2003) (Fig. 1). Since the kinks coincide with the intercalation of P63 residues, the intercalation must induce and/or stabilize the kinks. The binding does not involve specific amino acid-nucleobase contacts and instead solely depends on the readout of the DNA structure containing bent DNA helices, consistent with preferential binding of HU to other forms of contorted DNA such as cruciform or nicked DNA and the binding within DNA loops e.g. in the Mu transpososome, the Hin invertasome, or the gal repressosome (Balandina et al., 2002; Geanacopoulos et al., 2001; Kamashev et al., 1999; Montano et al., 2012).
Fig. 1.
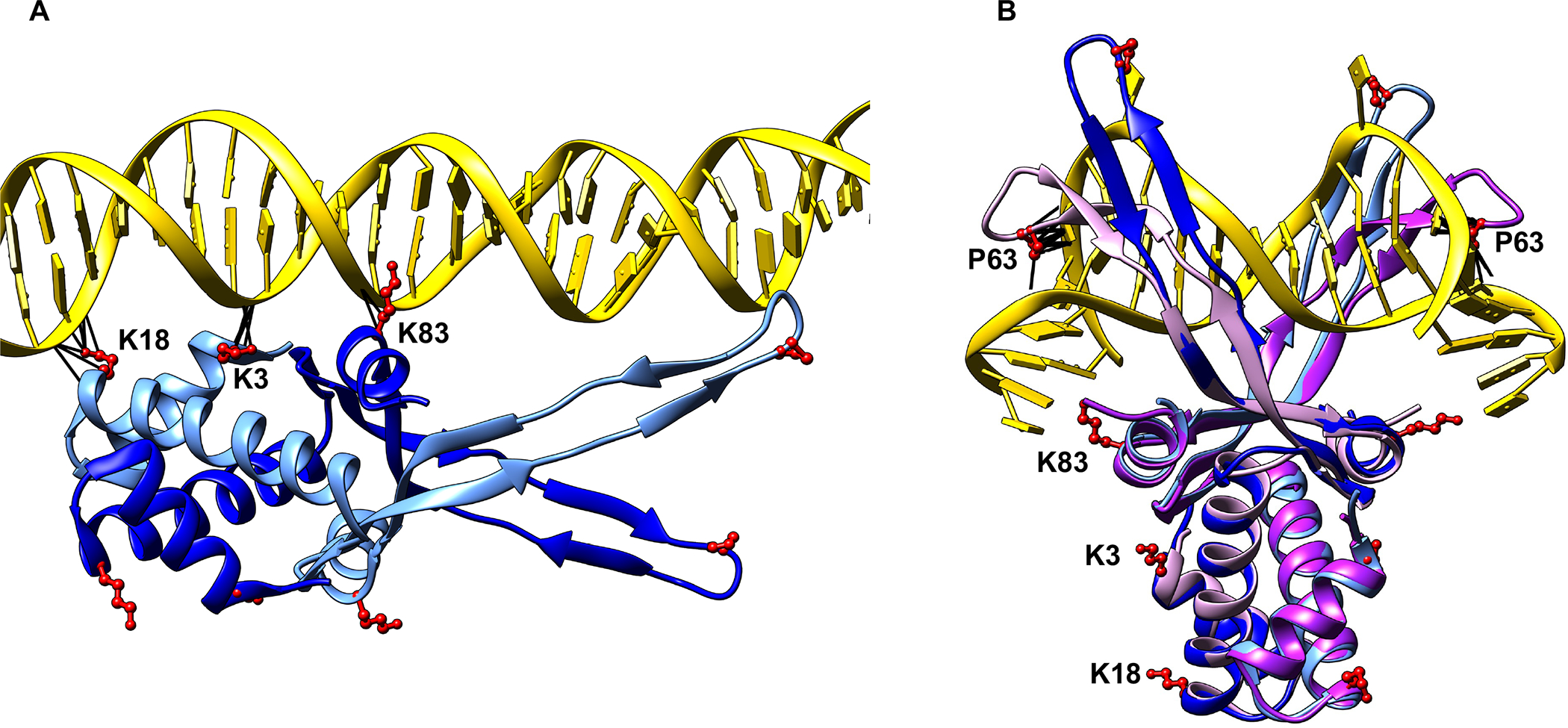
Crystal structures of HU-DNA complexes.
(A) Crystal structure of E. coli HUα2 (α subunits in blue and cornflower blue) in a complex with linear DNA (PDB 6O8Q).
(B) Crystal structure of Anabaena HU (two subunits in plum and purple) in a complex with a distorted DNA (PDB 1P51), with E. coli HUα2 (shown in A) superimposed on it. The lysine residues, K3, K18, and K83, and the proline residue P63 of both HU subunits are shown in red. Any contacts between these residues and DNA are shown as black lines.
Mounting evidence suggests that HU associates with the nucleoid mostly through non-specific binding. Genomic analysis of DNA binding of the E. coli HU by chromatin immunoprecipitation followed by sequencing (ChIP-Seq) revealed a uniformly distributed binding profile of HU across the genome with a strong resemblance to that of mock-IP, indicating mostly weak, non-specific binding of HU to the chromosome (Prieto et al., 2012). Recent studies probing the binding of HU to chromosomal DNA by single-molecule tracking demonstrated that HU exhibits fast diffusion within the nucleoid with rapid binding and unbinding to the chromosomal DNA, indicative of a weak, non-specific, and transitory binding (rapid association/dissociation kinetics) (Bettridge et al., 2020; Floc’h et al., 2019; Kamagata et al., 2021). Substituting the three lysine residues to alanine in the α subunit caused almost complete loss of HU binding to the chromosome (Bettridge et al., 2020), consistent with the non-specific binding mode involving the lysine residues as shown by crystallographic studies (Hammel et al., 2016; Remesh et al., 2020). Substituting P63 to alanine in the α subunit had a modest effect on overall HU binding to the chromosome in the presence of the β subunit and had very little effect in the absence of the β subunit (Bettridge et al., 2020), suggesting that P63-mediated structure-specific binding is not as widespread as non-specific binding and occurs at presumably a limited number of sites in the chromosome. The only demonstrated example of a structure-specific binding of HU in the E. coli chromosome is at the gal operon for regulation of the gal promoters. HU binds specifically between GalR binding sites (Majumdar and Adhya, 1984) and promotes the formation of a DNA loop by DNA-bound GalR-GalR interactions, blocking the transcription of the gal promoters located within the looped DNA (Lyubchenko et al., 1997). Furthermore, while non-specific binding is likely involved in the architectural role of HU in chromosome structure and chromosome remodeling (Hammel et al., 2016; Lioy et al., 2021; Remesh et al., 2020), the role of structure-specific binding in HU-dependent physiological processes other than the gal gene regulation is unknown, leaving the role of structure-specific binding in vivo for further exploration. In this study, by substituting them to alanine individually or in combination, we biochemically determined the specific contribution of the lysine and P63 residues of HUα2 in structure-specific and non-specific DNA binding. We then examined the effect of the substitutions on three HU-dependent physiological processes, plasmid DNA supercoiling, gal gene expression, and global gene expression. We found that non-specific DNA binding uses only the lysine residues, corroborating the structural studies, but structure-specific binding of HUα2 to an artificially designed cruciform DNA in vitro and a potential cruciform structure at the gal operon in vivo requires both P63 and lysine residues. Furthermore, the ability of HUα2 to induce supercoiling in a relaxed plasmid was dependent on both P63 and lysine residues, suggesting an involvement of DNA structure-specific binding in HU-induced supercoiling. More strikingly, while HUα2 was sufficient for HU-mediated control of gene expression, the P63A amino acid change in HUα2 resulted in a gene expression profile that resembled that of an HU null strain, suggesting a major role of structure-specific binding in global gene regulation by HU. We further demonstrated the hitherto unknown role of HU in regulating fimbriae formation via transcription control of type I fimbriae genes through structure-specific binding. Our findings together with those of recent studies (Bettridge et al., 2020; Floc’h et al., 2019; Kamagata et al., 2021) suggest that two modes of DNA binding by HU appear to have distinct physiological roles. While the widespread non-specific binding is responsible for the dynamic organization of the chromosome, likely by enabling DNA-DNA contacts, the limited structure-specific binding is responsible for gene expression control by HU, most likely by promoting the formation of local higher-order DNA structures at or near promoters.
RESULTS
Amino acid residues of HU involved in structure-specific and non-specific DNA binding
The contributions, if any, of the lysine residues in structure-specific binding, or that of the P63 residue in non-specific binding are so far unresolved. The electron density of the β arms was either not visible in the crystal structures of HU with the random dsDNA or the β arms made no contact with the DNA (Fig. 1), implying that the P63 residue was dispensable for non-specific binding (Hammel et al., 2016; Remesh et al., 2020). Similarly, the contorted DNA in the crystal structure of a single HU-DNA complex was not long enough to enable contact with the lysine residues (Fig. 1.) (Swinger et al., 2003). We note, however, that the contorted DNA formed a pseudo-continuous helix in the crystal packing and HU dimer contacted the phosphate backbone of the neighboring DNA (Swinger et al., 2003), indicating that HU probably contacts DNA beyond the kink sites in structure-specific binding, possibly via electrostatic interactions using lysine residues and or other positively charged residues. To test whether the lysine residues contribute to structure-specific binding, or vice versa, whether the P63 residue contributes to non-specific binding, we purified HUα2 wild-type and HUα2 variants with P63A single, K3A-K83A double, or K3A-K18A-K83A triple amino acid substitutions and investigated their two modes of DNA binding. Since HUα2 homodimer exhibits DNA binding affinities similar to HUαβ (Pinson et al., 1999) and fulfills most of the roles of HUαβ heterodimer (Wada et al., 1988), we used HUα2 and its variants for technical ease for this study. We used a 40-bp dsDNA of random sequence (hereafter linear DNA) to measure non-specific binding and an artificially designed cruciform DNA (Vitoc and Mukerji, 2011) to measure structure-specific binding. Binding was measured and quantified in solution by fluorescence polarization, but electrophoretic mobility shift assays (EMSA) were also carried out to monitor the formation of discrete DNA-protein complexes. Binding was measured at low salt (15 mM or 30 mM NaCl) and high salt (200 mM NaCl), as reported previously (Pinson et al., 1999), to distinguish weak non-specific binding mediated by electrostatic interactions from structure-specific binding. We note that the low salt concentrations used here are below the physiological ionic strength of E. coli, and 200 mM is within the physiological range. Wild-type HUα2 exhibited binding to linear DNA at low salt but no binding at high salt (Fig. 1), consistent with the previous study (Pinson et al., 1999). On the other hand, wild-type HUα2 showed binding to cruciform DNA at both low and high salt (Fig. 1 and S1). The cruciform DNA-HU complexes migrated as four discrete bands in EMSA under low salt conditions and as two discrete bands under high salt conditions (Fig. S2), consistent with previous findings (Bonnefoy et al., 1994; Pinson et al., 1999). We estimated the apparent dissociation constants (Kd) from binding curves by nonlinear curve-fitting to a Hill equation. The apparent Kd for linear DNA at 15 mM salt was 51 nM ± 4.0. Our estimate of the apparent Kd for linear DNA is much smaller than the Kd reported previously under these buffer conditions (Pinson et al., 1999). The difference could be due to the much higher sensitivity of fluorescence polarization assays in detecting weak interactions directly in solution without the need to separate free and bound DNA. Also, estimates of Kd for HU binding to a linear DNA have been reported to range from 200 nM to 2500 nM, depending on the ionic strength and DNA binding assays. HU has been reported to bind a 1000 bp linear DNA non-specifically at <100 nM concentration in atomic force microscopy experiments (van Noort et al., 2004). The estimated apparent Kd for cruciform DNA at high salt was 178 nM ± 22, which is about 2-fold higher than that at low salt as reported previously (Fig. 1 and S1) (Pinson et al., 1999). We next measured the binding of HUα2 variants to linear and cruciform DNA to determine specific contributions of the lysine and P63 residues in structure-specific and non-specific DNA binding. The HUα2P63A protein showed moderately reduced binding to cruciform DNA at low salt (Fig. S1) but almost a two-fold lower binding affinity at high salt compared to wild-type HUα2 (Fig. 2 and Fig. S1) (Kd at 200 mM salt 352 nM), validating the importance of the P63 residue in structure-specific binding to a distorted DNA as suggested by crystal structures. However, we observed no difference in the binding of wild-type HUα2 and HUα2P63A proteins to linear DNA at low salt (Fig. 2). In contrast, HUα2K3A-K83A and HUα2K3A-K18A-K83A proteins showed reduced binding not only to linear DNA, as expected, but also to cruciform DNA at both low and high salt (Fig. S1). From these results, we conclude that i) the P63 residue is dispensable for non-specific binding and ii) besides the P63 residue, the lysine residues are also critical for structure-specific binding of HU to cruciform DNA.
Fig. 2.
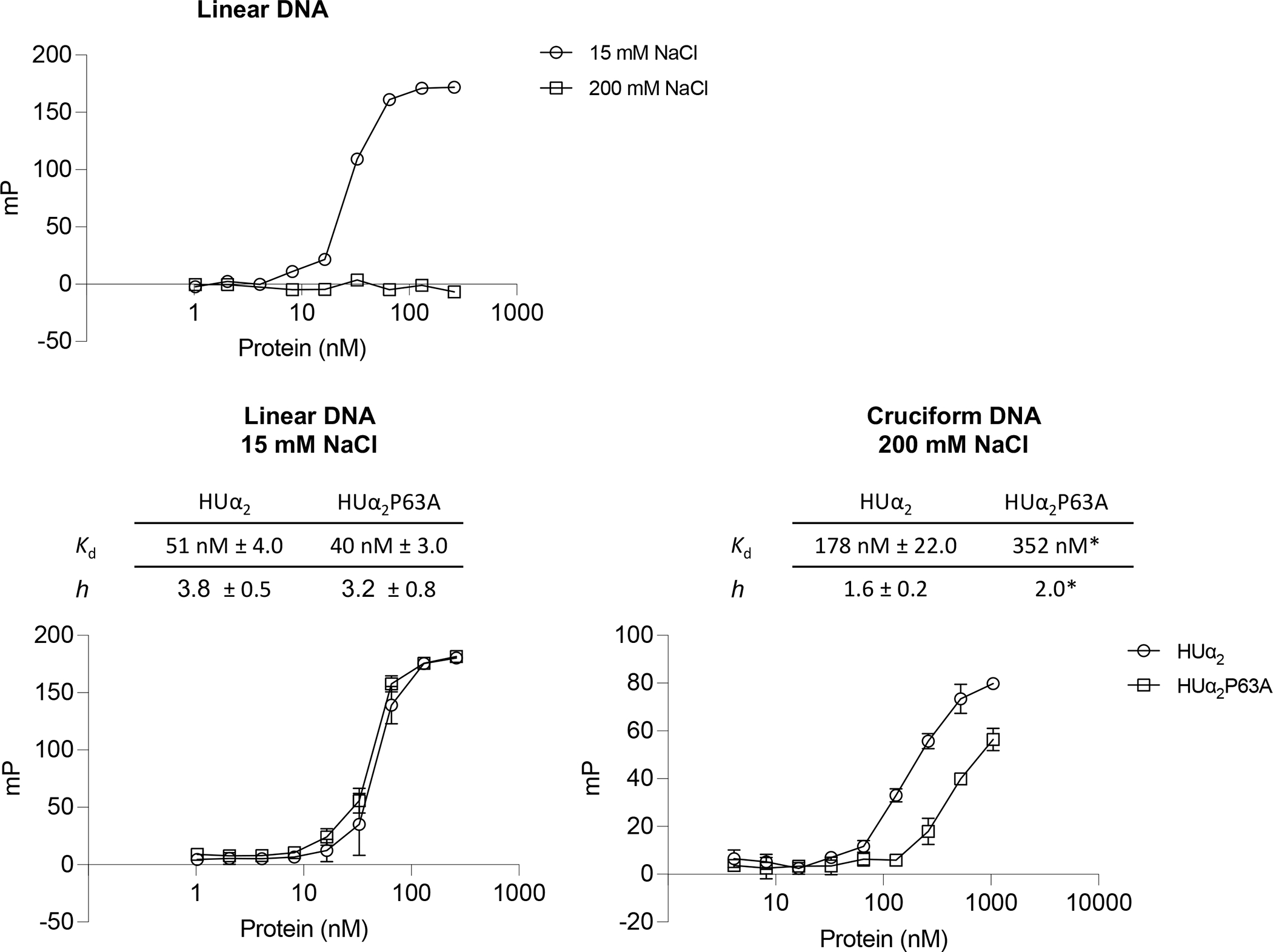
Effect of substituting lysine and P63 residues of HUα2 on DNA binding.
Binding of purified wild-type HUα2 or its variants harboring indicated amino acid substitutions to a 6-carboxy fluorescein labeled linear DNA or cruciform DNA. Each data point on Y-axis represents a change in milipolarization (mP) units at indicated protein concentrations in fluorescence polarizations assays. Kd (nM) estimated by fitting binding curves to a Hill equation are given in the table. ND = not determined.
Involvement of structure-specific binding of HU in DNA supercoil formation
HU induces negative supercoiling in a relaxed DNA in the presence of topoisomerase I (Guo and Adhya, 2007; Tanaka et al., 1995). How HU induces negative supercoiling is not fully understood. The crystal structures of HU with a contorted (artificially kinked) DNA suggest that the P63 residue may play a critical role in the formation of negative supercoils by inducing/stabilizing flexible bends with bend angles of opposite angular orientations (Swinger et al., 2003). Therefore, we tested the importance of the P63 residue, and the lysine residues, in the supercoil inducing ability of HU by incubating the wild-type and variant HUα2 proteins with a relaxed plasmid DNA made by introducing a nick in one of the strands. After incubation, the nicks were sealed with DNA ligase, the proteins were removed, and the resulting DNA topoisomers were resolved on gels by EMSA. Wild type HUα2 induced supercoils at 400 nM with saturation at 1600 nM protein concentrations (Fig. 3). The HUα2P63A protein was significantly impaired in the ability to form supercoils even at the saturating levels of the protein, demonstrating a critical role of the P63 residue in forming DNA supercoils. We also found the lysine residues played a significant role in the process because the HUα2 K3A-K83A and HUα2 K3A-K18A-K83A harboring double and triple lysine substitutions respectively were also impaired in promoting supercoil formation (Fig. 3). Although it is unclear whether HU binds to any specific structures in the relaxed plasmid DNA, these results show that the P63-dependent highly bent binding mode of HU is critical for inducing supercoiling. Additionally, they confirm that this mode requires the lysine residues besides the P63 residue.
Fig. 3.
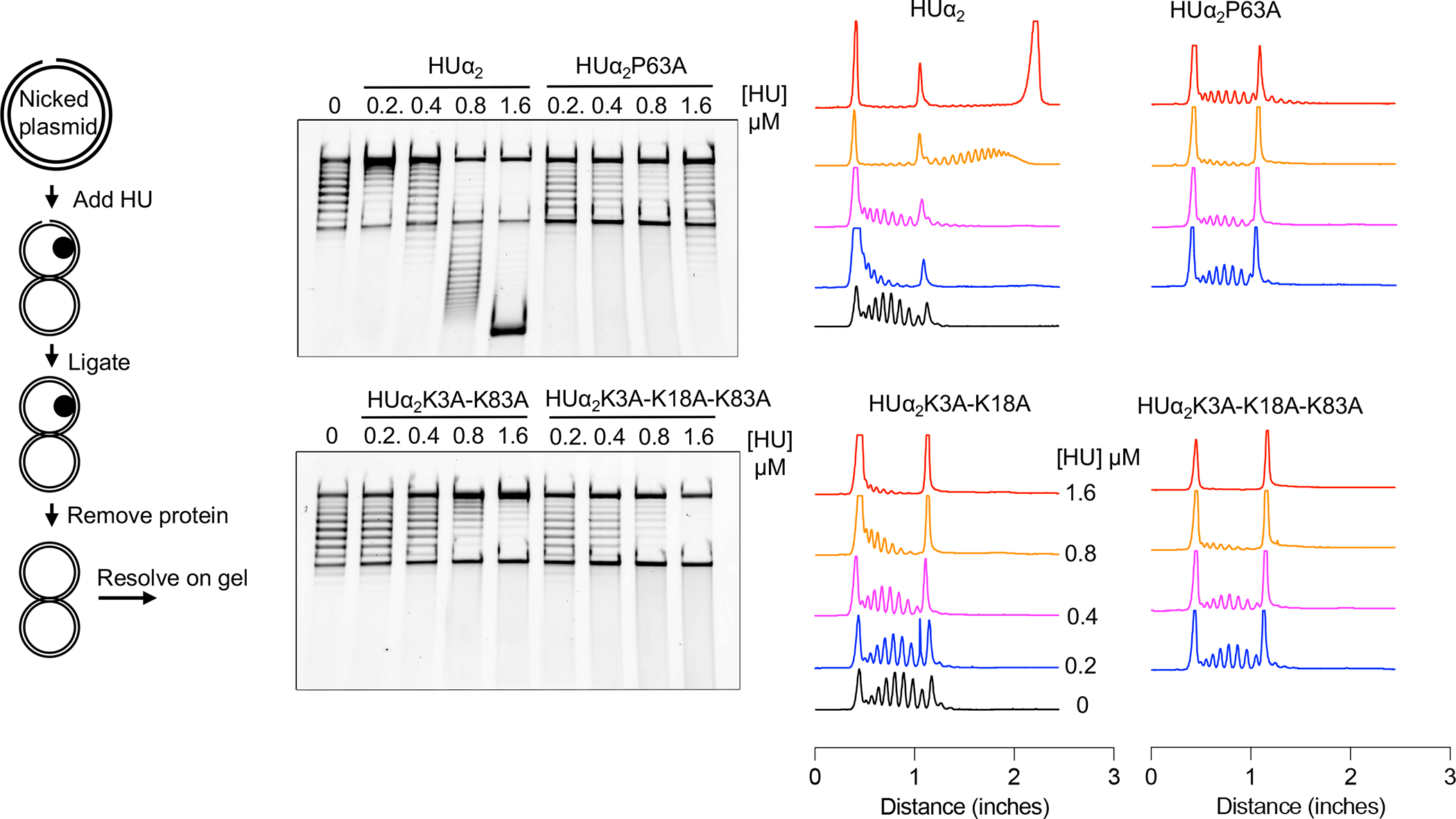
Effect of substituting lysine and P63 residues of HUα2 on supercoiling inducing ability.
The left panel outlines the assay carried out to determine the ability of HUα2 to induce negative supercoils in a nicked plasmid DNA. The middle panel shows the electrophoresis of the DNA products from the assay in the presence of the indicated concentrations of wild-type HUα2 protein or its variants. The right panel shows densitometric lane traces of the gel shown in the middle panel.
Structure-specific HU binding involved in gal transcription
HU binds to a specific (kinked) DNA structure in the E. coli chromosome at the gal locus as part of a higher-order DNA-multiprotein complex, called repressosome, consisting of a DNA loop, HU, and GalR, that represses transcription from the gal promoters (Geanacopoulos et al., 2001). The HU binding site (hbs) in the gal operon contains a palindrome sequence that can potentially form a cruciform structure (Fig. S3). The fact that negative supercoiling promotes the formation of a cruciform structure in a palindrome sequence and that HU repression of the gal operon in vitro requires a negatively supercoiled DNA template suggests that the cruciform structure may determine the binding specificity of HU in the gal system. Therefore, to validate the importance of P63 and the lysine residues in structure-specific binding of HU in a physiologically relevant system, we examined the ability of purified wild-type and variant HUα2 proteins to repress in vitro transcription from the DNA looping-sensitive P2 promoter in a supercoiled DNA template (Lewis et al.,1999). We observed that, compared to wild-type HUα2 protein, the HUα2P63A protein showed a significantly reduced ability to repress gal transcription (Fig. 4A and Fig. S4), consistent with DNA binding data. Similarly, the HUα2K3A-K83A and HUα2K3A-K18A-K83A proteins also showed reduced ability to repress the transcription (Fig. 4A and Fig. S4), confirming that lysine residues also contribute to the binding.
Fig. 4.

Effect of substituting lysine and P63 residues of HUα2 on gal transcription
(A) Amount of galP2 transcript originating from the galP2 promoter of pSA850 plasmid in the presence of 200 nM GalR and the indicated concentrations of HUα2 protein or its variants. The amount of galP2 transcript was normalized with that of RNAI and RNAII transcripts originating from the same plasmid. Each data point represents the galP2/(RNAI + RNAII) transcript ratio relative to that in the presence of GalR alone which was set to 100. The gel image used to quantify transcript amount is shown in Fig. S3).
(B) Levels of β-galactosidase activity of the chromosomal galE-lacZ transcriptional reporter in the E. coli strains of indicated genotypes: Graphical and error bars represent averages and standard deviation of triplicate biological replicates, respectively. Experiment was performed twice, with similar results. Statistically significant differences were determined by 1-way ANOVA with Tukey’s multiple comparisons test. ns not significant; ** adjusted p-value 0.001; *** adjusted p-value <0.001.
We also investigated the involvement of the P63 and the lysine residues in the HUα2-mediated gal repression in vivo. To do this, we replaced the promoter of the lac operon in the chromosome with the gal promoter segment to create a gal-lacZ transcription reporter fusion and carried out β-galactosidase assays in cells growing exponentially in M63 minimal media. The levels of β-galactosidase activity were low in the WT strain, which carries wild-type hupA and hupB genes and thus produces a mixture of HUα2 homodimer and HUαβ heterodimer, in the ΔhupB strain, which produces only HUα2 homodimer, and in the ΔhupA strain, which produces only HUβ2 homodimer (Fig. 4B), suggesting that all three forms of HU can repress the gal promoter equally well. However, deleting both hupA and hupB genes caused more than a 4-fold increase in the levels of β-galactosidase activity compared to the WT strain, consistent with de-repression of the gal promoters due to no DNA loop formation in the absence of HU (Lewis et al., 1999). The P63A substitution in HUα2 also de-repressed the gal promoter, causing a 2.5-fold increase in the levels of β-galactosidase compared to the ΔhupB parent strain expressing wild-type HUα2 (Fig. 4B). As suggested by crystal structures of the HU-DNA complexes, the P63 residues probably facilitate a DNA loop formation by inducing/stabilizing kinks in the gal DNA. We reasoned that glutamic acid due to its negative charge will be even more unfavorable than alanine for the intercalation into DNA bases and thus for inducing kinks in DNA. Indeed, the P63E substitution caused even more de-repression of the gal promoter, resulting in a 3.5-fold increase in the levels of β-galactosidase (Fig. 4B). These results demonstrate that P63 is a critical amino acid residue in HUα2 for binding to DNA and to promote the formation of the DNA loop at the gal operon. We could not ascertain the impact of substituting lysine residues because substituting lysine residues dramatically reduced the steady-state levels of HUα2 protein (Fig. S5). The K3A substitution alone reduced the steady-state levels to almost half of that without any change. The triple K3A-K18A-K83A substitution resulted in almost undetectable levels of the protein (Fig. S5). The P63A substitution, however, did not reduce the steady-state levels of the protein (Fig. S5). On the contrary, the P63A substitution caused a two-fold increase in the steady-state levels of the protein. This increase in the amount of the HUα2 protein caused by the P63A substitution is consistent with previously reported autorepression of the transcription of the hupA gene by HU, which, as this data suggests, appears to be mediated by structure-specific binding of HUα2 to the hupA promoter.
Involvement of structure-specific binding in the global gene transcription
After demonstrating the role of structure-specific binding in the gal gene regulation, we next examined the impact of substituting the P63 residue in HUα2 on global gene transcription to test whether the disruption of structure-specific binding would result in the differential expression of many other HU-regulated genes (Oberto et al., 2009; Prieto et al., 2012). We introduced the P63A substitution in HUα2 fused to mVenus to test its impact on both gene expression and the association of HUα2 with the nucleoid. HUα2-mVenus showed repression of the transcription from gal-lacZ reporter to the same extent as HUα2, and the P63A substitution in HUα2-mVenus showed effects like those in HUα2 (Fig. S6), demonstrating that the mVenus fusion does not interfere with the functionality of the HUα2 protein. The HUα2P63A-mVenus protein co-localized with the chromosomal DNA just as well as the HUα2-mVenus protein, with a distribution pattern resembling the shape of the chromosome (Fig. S7). This result indicates that the P63 residue does not significantly contribute to the association or the confinement of HU to the nucleoid, which is largely mediated by non-specific binding through the lysine residues (Bettridge et al., 2020).
We then analyzed the transcriptome of a strain expressing either HUα2-mVenus (hupA-mVenus ΔhupB) or HUα2P63A-mVenus (hupAP63A-mVenus ΔhupB) by next-generation total RNA sequencing. In addition, we analyzed the transcriptome of a wild-type strain with functional hupA and hupB genes (WT) and an HU null strain (ΔhupA ΔhupB) to identify all HU-regulated genes. Consistent with previous reports that the absence of HU causes differential expression of many genes in E. coli (Oberto et al., 2009; Prieto et al., 2012), 438 genes showed differential expression in HU null strain compared to the WT strain (Fig. 5 and Table S1). Among those genes, 203 genes showed up-regulation and 235 genes showed down-regulation. We manually assigned the differentially expressed genes (DEGs) to biological processes based on their established functions (Table 1 and Table S2) and performed a gene ontology over-representation test of DEGs. We found the enrichment of many genes involved in the organonitrogen compound biosynthetic process (amino acid, nucleotide, and biotin synthesis), translation, ribosome biogenesis, protein folding, defense response, and cell adhesion (Table S2 and Fig. S8) as reported previously (Oberto et al., 2009; Prieto et al., 2012). Only the ppiD gene, which encodes the periplasmic chaperone PpiD, showed lower expression in the ΔhupB strain compared to the WT strain (Fig. 5), suggesting that (i) the ppiD gene is regulated by HUβ2, and (ii) HUα2 is sufficient for HU-mediated control of global gene expression. The same result was observed in the strain expressing HUα2-mVenus when compared to the WT strain, except the hupA gene showed about 3-fold lower expression besides the ppiD gene (Fig. 5), suggesting that the mVenus fusion caused the lower expression of the hupA gene. The hupA gene was indeed detected as the only differentially expressed genes in the strain expressing HUα2-mVenus when compared to the strain expressing HUα2 without the fusion (Fig. 5), confirming that the mVenus fusion caused the lower expression of the hupA gene. Nonetheless, since we did not observe differential expression of any other genes despite the 3-fold lower expression of the hupA gene that would presumably significantly lower the amount of the HUα2-mVenus protein, we conclude that HUα2-mVenus is as good as HUα2 in HU-mediated control of global gene expression. However, the strain expressing HUα2P63A-mVenus showed differential expression of 261 genes compared to the strain expressing HUα2-mVenus (Fig. 5 and Table S3). Among those, 201 genes including the gal operon genes also showed differential expression in the HU null strain, and the differential expression was in the same direction, up or down-regulation, in both strains (Table S2). Moreover, the enrichment analysis showed an overrepresentation of genes associated with the same biological processes as those in the HU null strain, such as the organonitrogen compound biosynthetic process, ribosome biogenesis, and cellular response to hydrogen peroxide (Table 1 and Fig. S8).
Fig. 5.
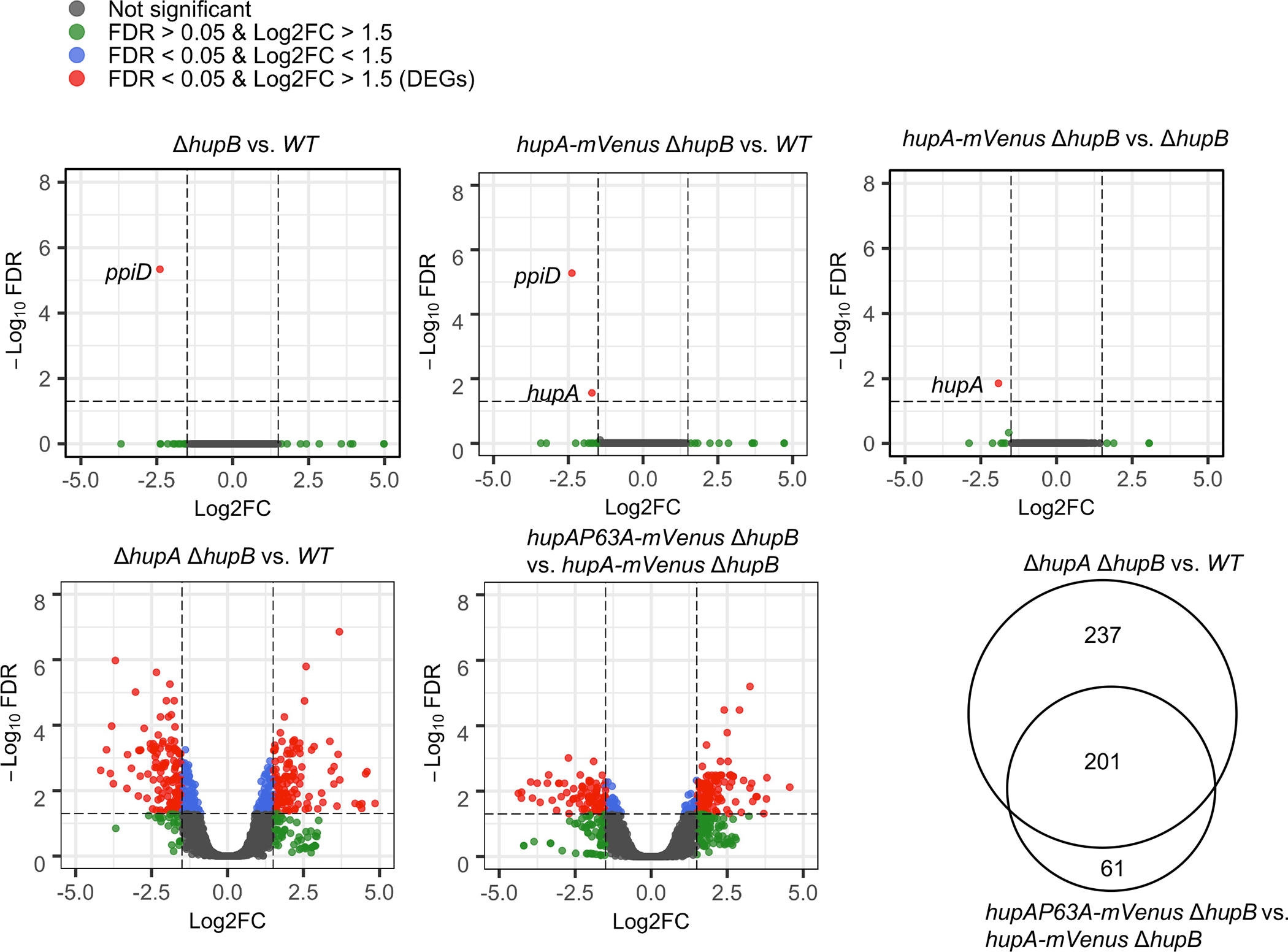
Effect of substituting the P63 residue of HUα2 on global gene expression.
Volcano plots showing differential expression of genes between E. coli strains of the indicated genotypes. X-axis represents log2 of the fold-change (Log2FC) in RNA levels and y-axis represents the −log10 of false discovery rate (FDR) of each gene. Vertical dotted lines are positioned at a log2 fold-change of 0.5 or −0.5 and horizontal dotted lines are positioned at the −log10 of 0.05 FDR. Genes in red are identified as differentially expressed genes (DEGs), determined using glmTreat function at FDR<0.05 and log2 fold-change >0.5 (plus or minus). Venn diagram shows DEGs common between two the comparison groups.
Table 1.
Number of genes associated with various biological processes that were differentially expressed in a E. coli strain lacking HU (relative to WT strain harboring hupA and hupB genes) or a E. coli strain producing HUɑ2P63A (relative to a strain producing HUɑ2) or both.
| Biological process | ΔhupA ΔhupB vs. WT | hupAmP63A ΔhupB vs. hupAm ΔhupB | Common | Total | Unique |
|---|---|---|---|---|---|
| Others | 111 | 58 | 45 | 169 | 124 |
| Amino acid metabolism | 42 | 32 | 28 | 74 | 46 |
| Transport | 32 | 11 | 9 | 43 | 34 |
| Transcription/Cell signaling | 31 | 8 | 6 | 39 | 33 |
| Translation | 29 | 28 | 18 | 57 | 39 |
| Carbohydrate metabolism | 28 | 14 | 10 | 42 | 32 |
| Stress response | 25 | 17 | 13 | 42 | 29 |
| Adhesion | 24 | 12 | 9 | 36 | 27 |
| DNA metabolism | 18 | 13 | 9 | 31 | 22 |
| Nucleotide biosynthesis | 16 | 12 | 9 | 28 | 19 |
| Protein homeostasis | 14 | 9 | 7 | 23 | 16 |
| Prophage | 11 | 7 | 4 | 18 | 14 |
| Nitrate assimilation | 8 | 2 | 1 | 10 | 9 |
| ATP synthesis | 7 | 8 | 7 | 15 | 8 |
| Putrescine metabolism | 7 | 7 | 7 | 14 | 7 |
| Cell cycle | 7 | 4 | 4 | 11 | 7 |
| Biotin synthesis | 6 | 6 | 6 | 12 | 6 |
| Pseudogenes | 6 | 3 | 2 | 9 | 7 |
| Methylglyoxal catabolism | 4 | 2 | 2 | 6 | 4 |
| Fatty acid oxidation | 4 | 2 | 2 | 6 | 4 |
| Defense | 4 | 0 | 0 | 4 | 4 |
| TCA cycle | 2 | 5 | 2 | 7 | 5 |
| ncRNAs | 2 | 1 | 1 | 3 | 2 |
| 438 | 261 | 201 | 699 | 498 |
The P63A substitution did not disrupt structure-specific binding completely in our DNA binding assays. Therefore, we predicted that the remaining genes may not have enough differential expression to be detected as DEGs by our criteria, but their differential expression pattern would resemble that of the HU null strain. To test this prediction, we constructed the heatmap of normalized read counts (counts per million) of 438 DEGs identified in the HU null strain. We found that the overall expression pattern of these genes in the HU null strain and the strain expressing HUα2P63A-mVenus was strikingly similar (Fig. S9), supporting the prediction that the complete disruption of the binding may account for even more DEGs observed in the HU null host. These results show that the structure-specific HU binding regulates many genes besides the gal operon. HU binds not only DNA but also RNA, with a preference for RNA containing secondary structures, such as mRNA of the rpoS gene, encoding the stress sigma factor of RNA polymerase (Balandina et al., 2001; Balandina et al., 2002). HU binding to RNA may prevent the degradation of certain mRNA species. RNA-seq measures differential expression based on the relative RNA abundance. Therefore, it is possible that lower expression of some genes caused by the loss of structure-specific binding, or the absence of HU, is due to the degradation of mRNAs of those genes in the absence of HU binding. But it does not appear to be the case because we did not observe lower expression of any of the mRNAs and the non-coding RNAs known to associate with HU (Macvanin et al., 2012).
HU regulation of type I fimbriae
To confirm that structure-specific binding of HU controls the expression of genes other than the gal operon, we analyzed in detail the HU regulation of genes encoding type I fimbriae using a chromosomal fimA-gfpmut2 transcription reporter fusion. The fimA gene encodes the major subunit of the type 1 fimbriae and is the first gene of the fimAICDFGH operon. The expression of the fim genes in E. coli is phase-variable due to the inversion of a 314 bp DNA element, called the fimS switch, located before the fimA gene (Abraham et al., 1985). The inversion of the switch between OFF and ON orientations by FimB and FimE recombinases regulates the fim gene expression (Klemm, 1986). GFP intensity of most wild-type cells harboring the fimA-gfpmut2 transcription reporter resembled that of the control cells that did not harbor fimA-gfpmut2 (Fig. 6A). However, 7% of the cells showed fluorescence intensity higher than the control cells (Fig 6A and 6B), consistent with phase variable expression of fimbriae genes. We defined these cells as fim+ cells. The strain expressing either HUα2 or HUβ2 produced about the same number of fim+ cells as the wild-type strain (Fig. 6B). However, the HU null strain produced three-fold more fim+ cells than the wild-type strain (Fig. 6B), suggesting that HU favors the OFF orientation of the fimS switch in E. coli. To confirm the involvement of structure-specific binding of HU, we measured the number of fim+ cells in the strain expressing the HUα2P63A variant protein. We observed a significant increase in the number of fim+ cells in this strain compared to the strain expressing HUα2 (Fig. 6B). By observing cells under scanning electron microscopy, we saw one or two long appendages protruding from wild-type cells that were most likely flagella but found many smaller appendages on the surface of cells lacking HU (Fig. 7). These appendages disappeared upon the deletion of the fim operon, showing that these structures were type I fimbriae (Fig. 7). We directly determined the orientation of the fimS switch by polymerase chain reaction (PCR) amplification of the fimS chromosomal region followed by the Hinfl restriction digestion of the PCR products (Fig. 8A). While we detected the fimS switch in the ON orientation in ~11% of wild-type cells or cells expressing HUα2 or HUβ2, ~34% of cells lacking HU had the switch in the ON orientation (Fig. 8B), consistent with the results of the fimA-gfpmut2 reporter. We detected the switch in the ON orientation in ~18% of cells expressing HUα2P63A, about a 1.5-fold increase compared to cells expressing HUα2 (Fig. 8B). We conclude from these results that structure-specific binding is, directly or indirectly, involved in maintaining the OFF position of the fimS switch and thereby controlling the number of cells expressing type I fimbriae genes in the bacterial population.
Fig. 6.
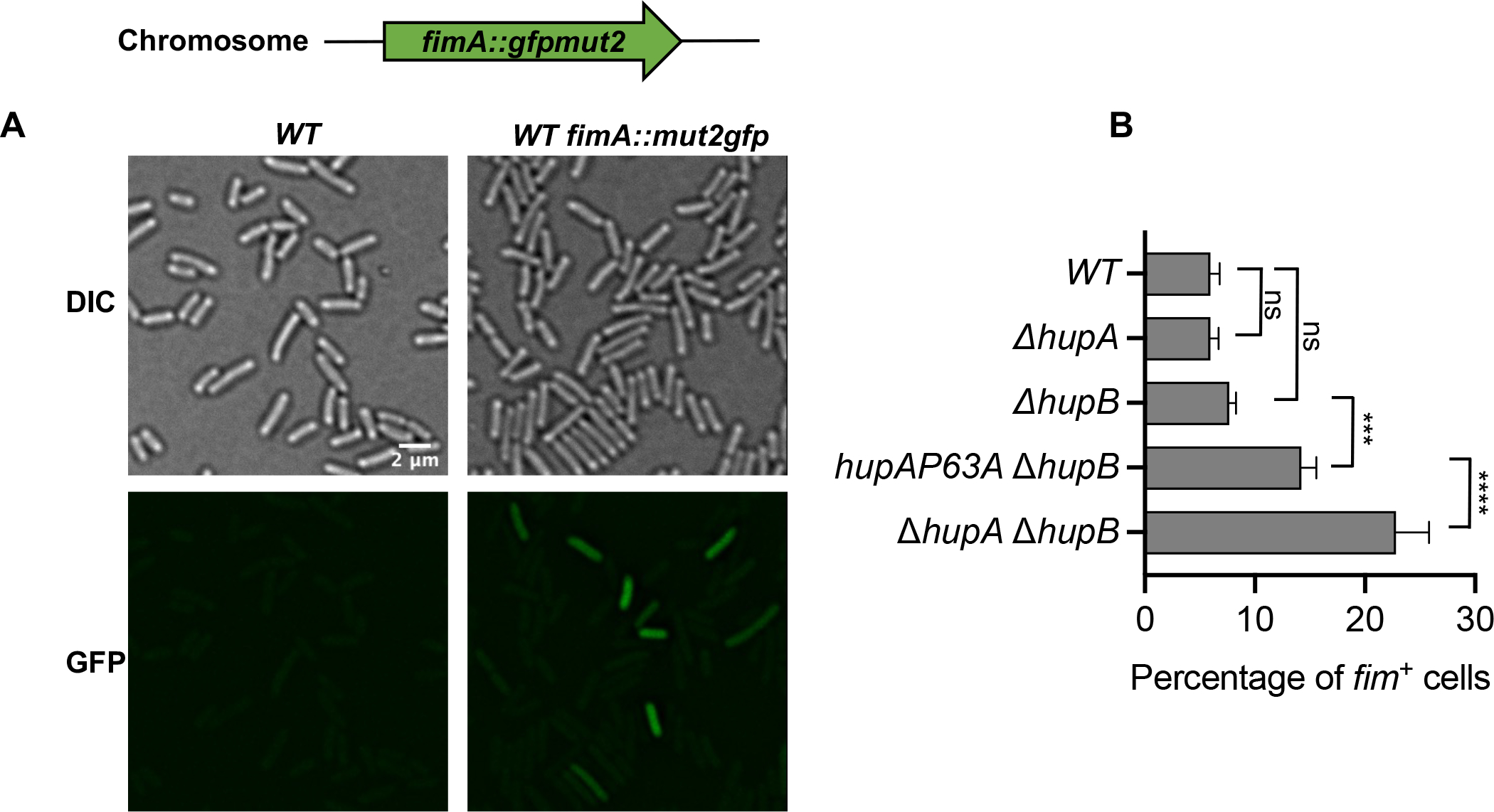
Role of HU in transcription regulation of type 1 fimbriae genes
(A) Representative fluorescence and differential interference contrast images of WT cells, or WT cells harboring fimA::gfpmut2 transcriptional reporter.
(B) Percentage of cells expressing GFP fluorescence above background in E. coli strains harboring the indicated deletion mutations in hupA and hupB genes and mutations in the hupA gene to introduce P63A amino acid substitution in the HUα subunit. Graphical and error bars represent averages and standard deviation of at least four fluorescence images each containing more than 200 cells. Experiment was performed twice, with similar results. Statistically significant differences as determined by 1-way ANOVA with Dunnett’s multiple comparisons test. ns not significant; *** adjusted p-value 0.0001; **** adjusted p-value <0.0001.
Fig. 7.
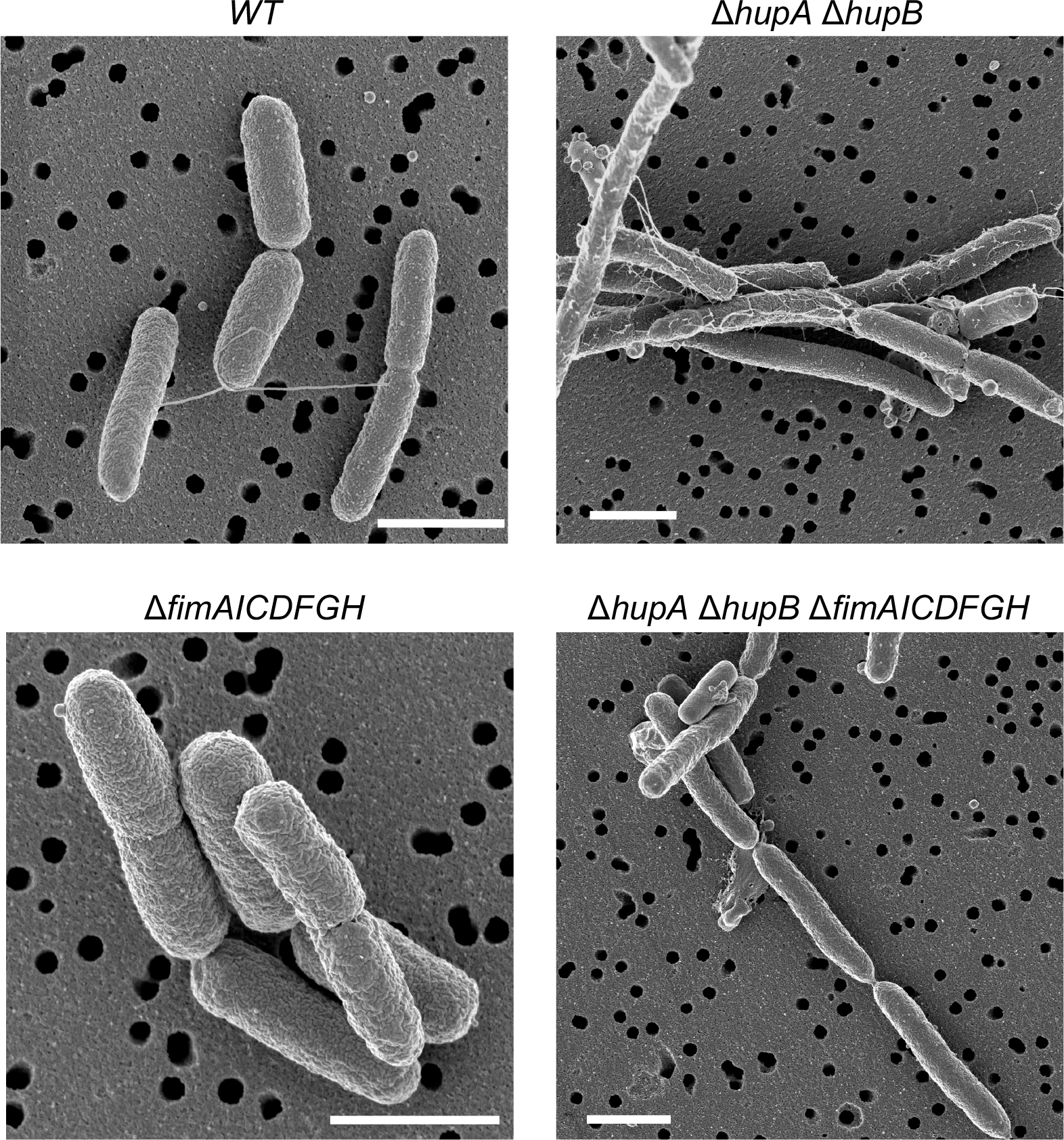
Role of HU in the formation of type I fimbriae on the surface of E. coli cells
Scanning electron micrographs of representative cells of E. coli strains harboring the indicated deletion mutations in hupA, hupB, and type I fimbriae genes. Scale bar 1 μm.
Fig. 8.
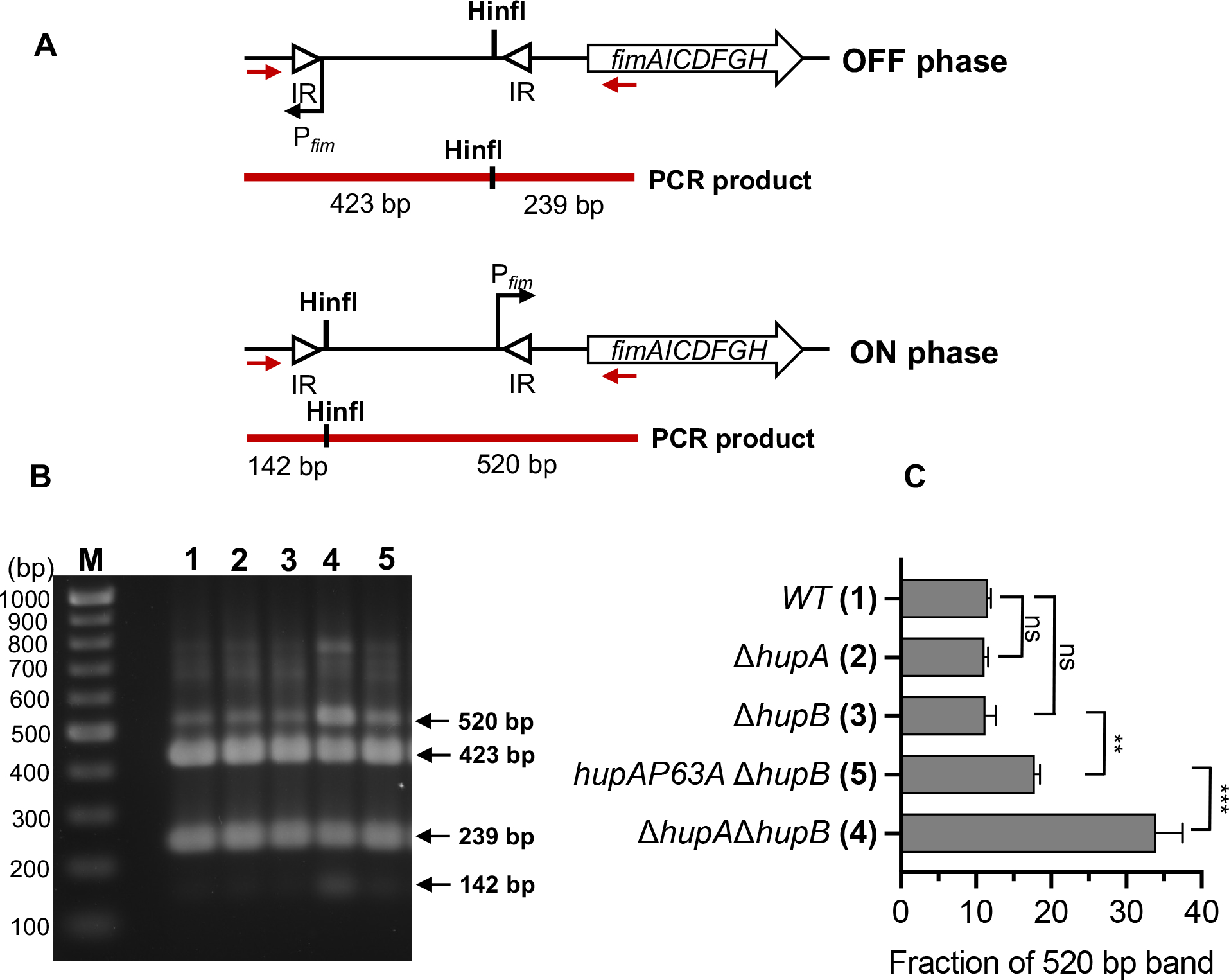
Role of HU in regulating the orientation of the fimS switch
(A) The position of Hinfl restriction site when fimS switch is in ON or OFF phase. Red arrows represent the binding sites of the primers fimE-u1 and fimA-l1 used for polymerase chain reaction of fimS region. Solid red lines represent the PCR products and sizes of Hinfl restriction fragments in ON or OFF phase. IR inverted repeats
(B) Agarose geel after electrophoresis of Hinfl digestion reactions of the PCR products of chromosomal DNA of E. coli strains harboring the indicated deletion mutations in hupA and hupB genes and mutations in the hupA gene to introduce P63A amino acid substitution in the HUα subunit. Numbers correspond to genotypes in panel B. M DNA marker.
(C) Percentage amount of 520 bp fragment from total amount of 520 bp and 423 bp fragments. Graphical and error bars represent averages and standard deviation of three biological replicates respectively. Experiment was performed twice, with similar results. Statistically significant differences as determined by 1-way ANOVA with Dunnett’s multiple comparisons test. ns not significant; ** adjusted p-value 0.003; *** adjusted p-value <0.001.
DISCUSSION
The role of two modes of HU binding to DNA, non-specific and structure-specific, in a multitude of physiological processes that depend on HU such as chromosome structure and gene expression is poorly understood. Here, we attempted to elucidate the molecular mechanism of the two binding modes and understand their role in HU-dependent physiological processes. We provide new insight into the mechanism of structure-specific binding and propose that the non-specific and the structure-specific HU binding have distinct functions.
Non-specific binding:
In this binding, three highly conserved lysine residues in HU electrostatically interact with DNA phosphates without inducing bends in DNA (Hammel et al., 2016; Remesh et al., 2020). Here, we biochemically confirm the role of the lysine residues in non-specific DNA binding of HUα2 and show that the proline residue P63, which is required for structure-specific binding, is dispensable for non-specific binding. This is consistent with crystal structures of HU bound to a random dsDNA in which only the lysine residues made ionic contacts with the DNA. The HU “arms” in which the P63 residues are located were either not visible or made no contact with the DNA (Fig. 1) (Hammel et al., 2016; Remesh et al., 2020). We note that HU may also use other binding modes for non-specific binding that do not require the lysine residues, for example, using the basic saddle (Koh et al., 2011). HU appears to interact with the chromosome mostly through non-specific binding as evidenced by single-molecule tracking studies (Bettridge et al., 2020; Kamagata et al., 2021). It diffuses along the DNA making weak, random, and transient interactions (with rapid association/dissociation kinetics). Most of the HU in which the three lysine residues, K3, K18, and K83, were substituted to alanine lost these interactions, suggesting that HU uses the binding mode involving the lysine residues. But because the lysine residues are also needed for structure-specific binding the triple lysine substitution must destroy both non-specific and structure-specific binding of HU to the chromosome. Function-wise, the widespread non-specific binding is most likely responsible for the architectural role of HU in chromosome organization demonstrated by recent studies (Lioy et al., 2018; Lioy et al., 2021; Walker et al., 2020). The rapid kinetics of non-specific binding can allow the newly replicated genome to rapidly reorganize as the genome progressively segregates during DNA replication (Nielsen et al., 2006).
Structure-specific binding:
Structural studies previously identified the P63 amino acid residue as a critical residue for HU binding to contorted DNA (Swinger et al., 2003). We have further demonstrated here that structure-specific binding of HU to a cruciform DNA in vitro and to a potential cruciform structure at the gal operon in vivo requires not only the P63 residue but also the lysine residues that are needed for non-specific binding. The involvement of the lysine residues is consistent with a binding mode that resembles the DNA binding mode of integration host factor (IHF) and IHF family proteins (PDB IDs: 2NP2 and 1IHF) (Mouw and Rice, 2007; Rice et al., 1996), which are almost structurally identical to HU. In this mode, the DNA is not just bound between the β “arms”, but it is also bound further along to the lysine residues on both sides of HU protein (Fig. 9). The length of the bound DNA in this mode is more than the 17-bp length of the DNA in the Anabaena HU-DNA complex (Fig. 1), which is supported by a 34 bp highly bent binding mode observed by isothermal titration calorimetry and fluorescence energy transfer studies (Koh et al., 2008b; Koh et al., 2011). Additionally, the footprinting of chemically converted HU-nuclease revealed that HU binds to a ~40 bp long segment of the gal DNA (Aki and Adhya, 1997). Although HU and IHF appear to use a similar binding mode, there are striking differences between the two proteins in binding specificity and DNA bending. IHF and IHF family proteins bind to a specific DNA sequence and bend DNA by 180° (Mouw and Rice, 2007; Rice et al., 1996), whereas HU binds DNA independent of the sequence and induces flexible bends (bend angles vary between 10–180°) (Koh et al., 2008a; Koh et al., 2011; van Noort et al., 2004). Since β-arms containing P63 residues bind and bend DNA almost in an identical manner in both HU and IHF (Swinger and Rice, 2004), the differences appear to lie in how the ɑ-helical “body” binds with the DNA. It appears that while residues such as arginine at position 46 in the ɑ-helical body of IHF contact specific nucleobases in the cognate sequence and sharply bend the DNA (Swinger and Rice, 2004), the lysine residues in the ɑ-helical “body” of HU make non-specific ionic bonds with the phosphate backbone, perhaps allowing HU to bind to contorted DNA regardless of the constituent base sequence and to induce flexible bends. Interestingly, the lysine residues K18 and K83 are conserved in both α and β subunits of HU, but they are present in either α or β subunit of IHF. Since IHF exists only as a heterodimer, this indicates that non-specific contacts may not be as important in IHF as in HU and may have been even replaced by specific amino acid-nucleobase contacts, making IHF a sequence-specific DNA binding protein despite it sharing the same structure with HU.
Figure 9.
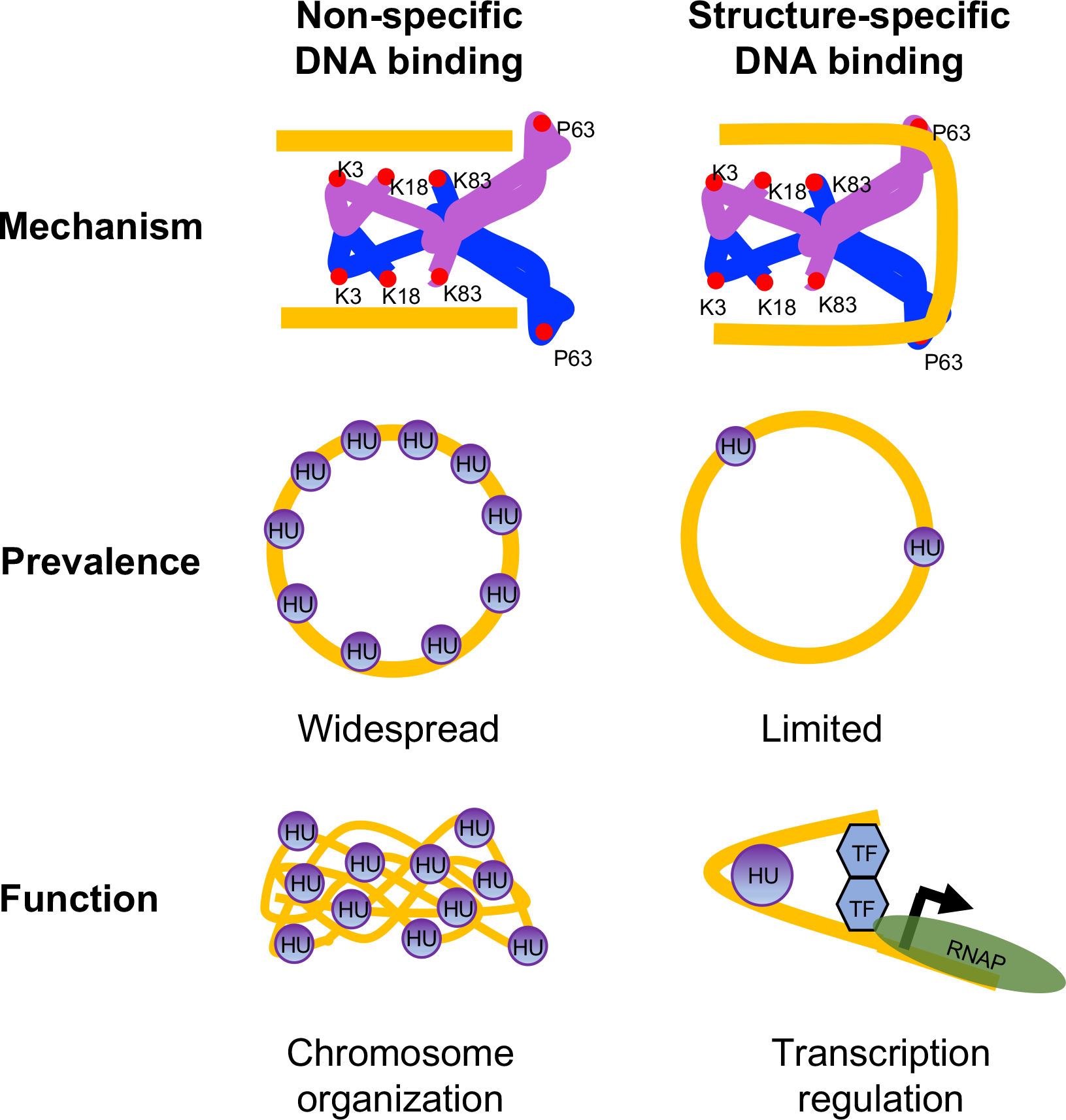
A model for mechanism, prevalence, and function of non-specific and structure-specific DNA binding modes of HU in E. coli
Two subunits of HU are depicted as blue and purple with DNA binding amino acid residues shown as red circles. DNA is depicted in gold. The bound DNA is in the straight conformation in non-specific mode and sharpy bent in structure-specific mode. While non-specific binding mode is widespread in the chromosome and primarily involved in chromosome organization, structure-specific binding mode is limited and involved in transcription regulation. TF Transcription factor; RNAP RNA polymerase.
We recently showed that substituting P63 to alanine in HUα2 only slightly altered the diffusive behavior of the protein in the nucleoid. The bound fraction of the variant HU determined by modeling the distribution of diffusion coefficients of individual HU molecules using Hidden Markov model was 30%, compared to 38% of the wild-type (Bettridge et al., 2020), suggesting that P63-mediated structure-specific binding is a small fraction of the global HU binding to the chromosome, occurring presumably at limited sites in the chromosome. Yet, we found that while HUα2 was sufficient for the HU-mediated control of gene expression, the P63A change alone in HUα2 resulted in an overall gene expression profile that resembled that of the HU null strain with an overlap of 201 DEGs, demonstrating that the ability of HU to induce or stabilize DNA bends is critical for the role of HU in transcription regulation. We propose that a multitude of cellular processes that HU carries out are mostly due to the regulation of gene transcription through structure-specific binding to DNA at some step of the processes, which provides the molecular basis for the highly pleiotropic phenotype of HU null cells observed in previous studies. Although identifying direct targets of the binding and elucidating the molecular mechanism involved therein need further investigation, the binding may directly regulate gene expression via the following mechanisms: (i) by promoting the formation of repressosome or enhanceosome structures, which help bring a DNA-bound repressor or an activator located at a distal site to the proximity of the RNA polymerase bound at the promoter by forming a DNA loop (e.g., gal operon) (Lyubchenko et al., 1997). Interestingly, out of 48 operons of E. coli predicted to be regulated by a DNA loop, 35 operons show differential expression upon deletion of HU encoding genes either in this study or previous two studies that analyzed global gene expression control by HU (Table S4) (Cournac and Plumbridge, 2013; Oberto et al., 2009; Prieto et al., 2012); (ii) by facilitating site-specific recombinational flipping of invertible promoter orientation (fim operon) as DNA inversion frequently needs HU structure-specific binding for the formation of invertasome structure (Paull et al., 1994; Wada et al., 1989); (iii) by inducing or restraining negative supercoils of the promoter regions (Berger et al., 2016). Many promoters including the gal promoters are heavily dependent upon a given amount of DNA superhelicity for optimal transcription (Dorman and Dorman, 2016). Alternatively, DNA superhelicity may influence a promoter indirectly by promoting the formation of repressosome, enhanceosome, or invertasome-like structures. We note that some of the differential expression caused by the disruption of structure-specific binding could reflect secondary changes in cell physiology caused by the direct effects of the binding on gene regulation. We found that the P63A change recapitulated the previously reported anucleate phenotype of HU null strain (Fig. S10).
In summary, we propose that HU intervenes in numerous physiological events in E. coli by two different binding modes in distinctly different mechanisms. While non-specific binding is widespread throughout the chromosome and dictates chromosome structure, structure-specific binding occurs at limited sites in the chromosome and regulates gene expression (Fig. 9). Other architectural DNA binding proteins such as the yeast chromatin protein Nhp6A, the factor for inversion stimulation (FIS), and IHF, and even well-studied specific DNA binding proteins such as CRP display both non-specific and specific binding to the chromosome (Kamagata et al., 2021; Visweswariah and Busby, 2015). Therefore, it will not be surprising if the two kinds of binding in other proteins also have distinct biological functions.
EXPERIMENTAL PROCEDURES
Media and growth conditions.
Cells were grown in M63 minimal media supplemented with 0.1% glycerol (v/v), 10 μg ml−1 thiamine, and 0.5 mg ml−1 casamino acids. Cells were grown overnight, diluted 1000-fold, and grown to A600 of 0.4–0.6 for all experiments.
Strains.
All E. coli strains are listed in Table S5 and were derived from MG1655 (Blattner et al., 1997). Primers used in this study are listed in Table S6. Strains were constructed by Lambda Red recombineering (Yu et al., 2000) using the plasmid pSIM6 (Datta et al., 2006). To introduce amino acid substitutions in the α subunit of HU, the pBAD-ccdB-kan genetic element that was PCR amplified using homology primers hupA-kan-ccdB-u2 and hupA-kan-ccdB-l2 was inserted after the stop codon of the hupA open reading frame. The pBAD-ccdB-kan expresses CcdB toxin under the arabinose inducible promoter pBAD and confers kanamycin resistance. The recombinants were selected on LB agar containing 1% glucose and 30 μg ml−1 kanamycin and verified by PCR using the primers hupA-u1 and hupA-l1 that bind outside the hupA gene and by the inability of the recombinants to grow on LB agar supplemented with 0.02% arabinose. In the second step, the pBAD-ccdB-kan element was replaced with a synthetic double-stranded DNA (synthesized by Integrated DNA Technologies) encoding the hupA gene with mutations introducing desired amino acid substitutions in the protein. Similarly, a synthetic DNA containing the hupA gene genetically fused to yellow fluorescent protein mVenus via the glycine-serine-isoleucine (GSI) linker was used to construct the strain with hupA-mVenus fusion. The recombinants were selected on LB agar with 0.02% arabinose verified by Sanger sequencing. The plasmid pSIM6 was removed by repeatedly growing the strain at 37 °C and verifying for ampicillin sensitivity. The ΔhupB in the genotypes mentioned in the figures refers to the hupB11 allele (Wada et al., 1988) in which the part of the hupB gene between EcoRV and AatI sites is replaced with a 1.4 kb HaeII DNA fragment containing the chloramphenicol-resistance (CmR) gene of the pACYC184 plasmid. The hupB11 allele was transferred using P1 phage transduction. The gal-lacZ reporter was constructed using Lambda Red recombineering by replacing the chromosomal region of MG1655 starting from the lacI up to the start codon of the lacZ with a DNA fragment containing ampicillin resistance gene bla, the gal promoter, and the galE gene. The DNA fragment was amplified from the plasmid pSA813 using the primers lacI-bla-u1 and lacZ-galE-l1. The gal-lacZ reporter was transferred to other strains using P1 phage transduction.
Protein purification.
HU proteins used for supercoiling assays were purified by GenScript Biotech Corporation. A 6x-histidine tag containing the Enterokinase cleavage site was placed at the N-terminus. Proteins were expressed in E. coli strain BL21(DE3) and purified with >95% purity using Ni-NTA affinity chromatography followed by Q-Sepharose and size-exclusion chromatography. The proteins were stored in the buffer containing 50 mM Tris HCl pH 8.0, 150 mM NaCl, and 10% Glycerol. The histidine tag was cleaved by incubating 1 ml of protein (concentration in the range of 1 to 2 mg/ml) in the storage buffer additionally containing 2 mM CaCl2 with 100 units of histidine-tagged Bovine Enterokinase (GenScript Biotech Corporation) at 22°C for one hour. The tag and the enzyme were removed by incubating the reaction mix for one hour at 4°C with high-affinity Ni-NTA resin (GenScript Biotech Corporation) that was prewashed and equilibrated with the protein storage buffer. The proteins were eluted by centrifugation at 1000 × g for 1 min at 4°C and analyzed by SDS PAGE.
DNA binding assays
The 6-carboxy fluorescein (6-FAM) labeled duplex DNA substrates made by annealing the following oligonucleotides (synthesized and annealed by Integrated DNA Technologies) were used for DNA binding.
linear DNA of random sequence:
CCGACTAAGTACATGTGAGAATTTTGCTGCCTTCGAACCT and /56-FAM/AGGTTCGAAGGCAGCAAAATTCTCACATGTACTTAGTCGG
Cruciform DNA:
CCTAGCAAGGGGCTGCTACCTTTGGTAGCAGCCTGAGCGGTGG and /56-FAM/CCACCGCTCAACTCAACTGCTTTGCAGTTGAGTCCTTGCTAGG
The binding assays were carried out in the 100 μl reaction volume containing 10 mM Tris-HCl pH 8.0, 15 % (v/v) glycerol, 0.1 mM EDTA, 15 mM, 30 mM, or 200 mM NaCl, 1 nM DNA, and varying concentrations of the protein. The reactions were incubated in 6 mm × 50 mm glass test tubes at room temperature for 2 min and then fluorescence polarization measurements were taken in the Beacon™ 2000 instrument at 25°C. Subsequently, 20 μl reaction volume was loaded onto 6% DNA retardation gels (Invitrogen), and electrophoresis was carried out at room temperature in 0.5x Tris borate buffer, pH 8.0. The gels were imaged using Bio-Rad ChemiDoc MP Imaging System. The milipolarization (mP) units were plotted in GraphPad Prism v9. The data points were fitted using the Hill slope equation wherein is the maximum binding in the mP units, is the protein concentration, is the hill slope, and is the protein concentration needed to achieve the half-maximum binding at equilibrium.
In vitro transcription assay.
In vitro transcription reactions were carried out as described earlier (Lewis, 2003). Supercoiled plasmid pSA850 (40 nM) was preincubated at 37°C for 5 min with 20 nM RNA polymerase; 200 nM GalR and/or varying concentrations of wild-type HUα2 or its mutant variants in a total reaction volume of 50 μl containing transcription buffer (20 mM Tris-acetate/10 mM Mg acetate/50 mM NaCl) supplemented with 1 mM DTT, 1 mM ATP, and 0.8 units recombinant ribonuclease inhibitor. Transcription reactions were initiated by adding nucleotides to a final concentration of 0.1 mM GTP and CTP, 0.01 mM UTP, and 5 μCi [α−32P] UTP (1 Ci = 37 GBq). The reactions were incubated for an additional 10 min before they were terminated by the addition of an equal volume (50 μl) of loading dye (90% formamide/10 mM EDTA/0.1% xylene cyanol/0.1% bromophenol blue). Samples were heated to 90°C for 2–3 min, chilled, then loaded on an 8% sequencing gel and electrophoresed at a constant power of 60 W in TBE (90 mM Tris/64.6 mM boric acid/2.5 mM EDTA, pH 8.3). The RNAI transcripts (106 and 108 nts) were used as an internal control to normalize the relative amount of transcript from P2 promoter.
Supercoiling assay.
The singly nicked pCG09 plasmid was prepared by incubating CsCl purified negatively supercoiled pCG09 (60 μg) in 600 μl of 1X NEB Smart Cut buffer with the nicking endonuclease Nb.BbVCI (NEB, 8.5 units) at 37 °C for 30 min. The plasmid was purified by phenol: chloroform: isoamyl alcohol extraction, recovered by standard ethanol precipitation, and dissolved in 1X buffer containing 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, and 0.05 % (v/v) IGEPAL 630 (Sigma).
HUα2 protein and its variants were incubated with singly nicked pCG09 plasmid for 5 min in a buffer containing 50 mM HEPES-KOH (pH 7.5), 20 mM KCl, 30 mM NaCl (from protein stocks containing 150 mM NaCl), 10 mM DTT, 5 mM MgOAc, 7.5 % glycerol, 2 mM ATP-Mg and 5 ng/μL tRNA. T4 DNA ligase was added to seal the nick. The reactions were stopped by the addition of NaCl and EDTA followed by deproteination with SDS and proteinase K for 30 min at 37°C. Topoisomers were resolved on a 0.8 % agarose gel at room temperature and 23 volts for 18 hours. Gels stained with SYBR Gold and imaged by Typhoon scanner.
β-galactosidase assay.
The β-galactosidase assay was carried out using the standard protocol. Briefly, cells were diluted in Z-buffer and permeabilized by adding 100 μl chloroform and 50 μl 0.1% sodium dodecyl sulfate. The reaction was initiated by adding 0.2 ml of 4 mg/ml o-nitrophenyl-β-D-galactoside in 0.1M phosphate buffer (pH 7.0), incubated at 28°C, and stopped after sufficient yellow color developed by adding 0.5 ml 1M Na2CO3. 1 ml of the reaction was transferred to a centrifuge tube, spun at maximum speed to remove debris and chloroform, and the absorbance at 420 nm was recorded for each tube. The following equation was used to calculate units of enzyme activity: 1000 × (A420) / (A600 × T × V) where T is the time of reaction in minutes and V is the volume of cells used in the reaction in milliliters.
RNA sequencing.
RNA isolation and sequencing were carried out as described previously (Remesh et al., 2020). For total RNA isolation, a frozen pellet of cells was resuspended and homogenized in 1 ml TRIzol reagent (Life Technologies) and incubated at room temperature for 5 min. To the resuspension, 0.2 ml chloroform was added and mixed by inverting the tube for 15 seconds. The mixture was incubated at room temperature for 10 min and then centrifuged at 20,000 × g for 10 min at 4 °C. After centrifugation, ~0.6 ml of the upper phase was transferred to a new centrifuge tube containing 0.5 ml isopropanol. The mixture was incubated at room temperature for 10 min and then centrifuged at 20,000 × g for 15 min at 4 °C. After centrifugation, the supernatant was discarded and the pellet was washed twice with 1 ml 75% ethanol in Diethyl pyrocarbonate (DEPC)-treated water by centrifugation at 13,000 × g, for 5 min at 4 °C. After the second wash, the tube was left open for 10–15 min at room temperature to dry the pellet. To the pellet, 50 μl DEPC treated water was added, and the tube was left at 37 °C for 10–15 min and then the pellet was fully resuspended using a pipette. DNA was removed using TURBO DNA-free Kit (Invitrogen). The quality of total RNA was determined by electrophoresis on the TapeStation system (Agilent). Paired-end sequencing libraries were prepared with 2.5 μg of total RNA using Illumina TruSeq Stranded Total RNA library prep workflow with Ribo-Zero. Samples were pooled and sequenced on HiSeq4000 with a read length of 150. Samples were barcode demultiplexed allowing one mismatch using Bcl2fastq v2.17. The reads were trimmed for adapters and low-quality bases using Cutadapt software. Alignment of the reads to the annotated transcriptome of E. coli K12 MG1655 was done using STAR. Transcript abundances were calculated by RSEM, and differential expression analysis was done using the glmTreat function of edgeR. We identified differential expression based on a false discovery rate (FDR) cut-off of 0.05 and log2 (log with base 2) fold change of 1.5. The gene ontology over-representation test was performed using the clusterProfiler package implementing enrichGO fucntion. The RNA-Seq data have been deposited to the Gene Expression Omnibus (GEO) database and can be accessed with the GEO accession number GSE175465.
Fluorescence microscopy.
1 ml bacterial culture was stained with 10 μg ml−1 Hoechst 33342 for 10 min and centrifuged at 1000 × g for 3 min at room temperature. All supernatant was removed except the volume in microliters equal to A600 × 200. The resuspended cells were stained with 15 μg ml−1 FM464 for 10 min. Cells were then spotted onto 35 mm Poly-D-Lysine coated glass petri dish (MatTek Life Sciences) and covered by M63 glycerol 1% (w/v) agarose pad. Z- sections were collected using DeltaVision imaging system (GE Healthcare) equipped with CoolSNAP_HQ2 camera. The pixel size of each image was 0.064 μm × 0.064 μm × 0.2 μm. mVenus or GFP fluorescence was detected using a fluorescein isothiocyanate (FITC) filter (excitation: 475/28; emission: 525/48) with 100% light transmission. Hoechst 33342 was detected using a 4′,6-diamidino-2-phenylindole (DAPI) filter (excitation: 390/18; emission: 435/48) with 50% light transmission. FM464 was detected using a tetramethylrhodamine filter (excitation: 542/27; emission: 597/45) with 100% light transmission. Exposure time for Hoechst 33342 and FM464 was 0.5 s, and for mVenus was 0.2 s. The images were deconvoluted using the recommended method in the SoftWoRx software. Image quantitation was performed in ImageJ using the middle section of the z-stack. Cell length was manually measured using FM464 fluorescence. fim+ cells were defined as cells containing GFP fluorescence after subtracting the average fluorescence intensity of cells containing no fimA-gfp fusion.
Scanning electron microscopy.
An aliquot of bacterial culture growing statically was carefully pipetted and gently placed onto a Sterlitech PETE membrane filter and incubated for 2 hours without disturbance or vacuum, allowing the bacteria to settle on the substrate and maintain the structural integrity of the fine fimbriae. The samples were covered to prevent contamination and drying out. The samples were then fixed in a cocktail of 4% formaldehyde and 2% glutaraldehyde in 0.1M cacodylate buffer and subsequently post-fixed using a 1% osmium tetroxide solution. They were then dehydrated in a series of graded alcohols ranging from 35% to 100% with the final dehydration completed using a Tousimis (Rockville, MD) critical point dryer. The dried samples were then coated with a thin layer of iridium using an EMITECH K575X high-resolution sputter coater and imaged with the Zeiss 450 FE-SEM (Oberkochen, DE) at 1.50kV using the InLens SE detector.
Phase switch orientation assay.
The orientation of the fimS switch was determined as described previously (Stentebjerg-Olesen et al., 2000). Briefly, the chromosomal region containing the fimS switch was amplified by PCR using the primers fimE-u1 and fimA-l1. The PCR product was purified and 2 ug DNA was cut with Hinfl. The digestion fragments were separated on 2% Tris-acetate-EDTA agarose gel. The intensities of 520 bp in the ON state and 423 bp in the OFF state were quantified using the ImageJ gel analysis function.
Western Blot.
HU levels were determined by Western blotting. Cells were pelleted by centrifugation and lysed by incubating at 95°C for 10 min. Proteins were separated on 4–12% Bis-tris gels (Invitrogen) and transferred to polyvinylidene difluoride (PVDF) membrane using the iBlot 2 System (Life Science Technologies). The membrane was incubated in phosphate-buffered saline with 0.1% Tween 20 (PBST) containing mouse anti-EFTU antibody (Hycult Biotech) and either rabbit anti-HU antibody (custom made by GenScript Biotech Corporation) or rabbit anti-GFP antibody (ab290; Abcam). Subsequently, the membrane was incubated with secondary fluorescent antibodies, StarBright Blue 700 Goat Anti-Rabbit IgG (Bio-Rad) and DyLight 800 Goat Anti-Mouse IgG (Bio-Rad). After antibody incubation steps, the membrane was washed twice with PBST, and imaged using the ChemiDoc imaging system (Bio-Rad). Fluorescence intensities of EFTU and HU bands were quantified using gel analysis function of ImageJ.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research to S. A. We thank Rupesh Kumar at Memorial Sloan Kettering Cancer Center for help in supercoiling assays. We thank our laboratory members for regular discussions on the subject covered here and Dale Lewis for his additional help in in vitro transcription assays.
Footnotes
Conflict of Interest Statement: The authors declare no conflict of interest.
REFERENCES
- Abraham JM, Freitag CS, Clements JR, and Eisenstein BI (1985). An invertible element of DNA controls phase variation of type 1 fimbriae of Escherichia coli. Proc Natl Acad Sci U S A 82, 5724–5727. 10.1073/pnas.82.17.5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aki T, and Adhya S (1997). Repressor induced site-specific binding of HU for transcriptional regulation. The EMBO journal 16, 3666–3674. 10.1093/emboj/16.12.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balandina A, Claret L, Hengge-Aronis R, and Rouviere-Yaniv J (2001). The Escherichia coli histone-like protein HU regulates rpoS translation. Molecular microbiology 39, 1069–1079. [DOI] [PubMed] [Google Scholar]
- Balandina A, Kamashev D, and Rouviere-Yaniv J (2002). The bacterial histone-like protein HU specifically recognizes similar structures in all nucleic acids. DNA, RNA, and their hybrids. The Journal of biological chemistry 277, 27622–27628. 10.1074/jbc.M201978200. [DOI] [PubMed] [Google Scholar]
- Bettridge K, Verma S, Weng X, Adhya S, and Xiao J (2020). Single-molecule tracking reveals that the nucleoid-associated protein HU plays a dual role in maintaining proper nucleoid volume through differential interactions with chromosomal DNA. Molecular microbiology. 10.1111/mmi.14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick T, Ghosh S, Dixit K, Ganesan V, Ramagopal UA, Dey D, Sarma SP, Ramakumar S, and Nagaraja V (2014). Targeting Mycobacterium tuberculosis nucleoid-associated protein HU with structure-based inhibitors. Nature communications 5, 4124. 10.1038/ncomms5124. [DOI] [PubMed] [Google Scholar]
- Blattner FR, Plunkett G 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. (1997). The complete genome sequence of Escherichia coli K-12. Science (New York, N.Y.) 277, 1453–1462. 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Bonnefoy E, Takahashi M, and Yaniv JR (1994). DNA-binding parameters of the HU protein of Escherichia coli to cruciform DNA. Journal of molecular biology 242, 116–129. 10.1006/jmbi.1994.1563. [DOI] [PubMed] [Google Scholar]
- Chodavarapu S, Felczak MM, Yaniv JR, and Kaguni JM (2008). Escherichia coli DnaA interacts with HU in initiation at the E. coli replication origin. Molecular microbiology 67, 781–792. 10.1111/j.1365-2958.2007.06094.x. [DOI] [PubMed] [Google Scholar]
- Claret L, and Rouviere-Yaniv J (1997). Variation in HU composition during growth of Escherichia coli: the heterodimer is required for long term survival. Journal of molecular biology 273, 93–104. 10.1006/jmbi.1997.1310. [DOI] [PubMed] [Google Scholar]
- Cournac A, and Plumbridge J (2013). DNA looping in prokaryotes: experimental and theoretical approaches. Journal of bacteriology 195, 1109–1119. 10.1128/JB.02038-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S, Costantino N, and Court DL (2006). A set of recombineering plasmids for gram-negative bacteria. Gene 379, 109–115. 10.1016/j.gene.2006.04.018. [DOI] [PubMed] [Google Scholar]
- Fernandez S, Rojo F, and Alonso JC (1997). The Bacillus subtilis chromatin-associated protein Hbsu is involved in DNA repair and recombination. Molecular microbiology 23, 1169–1179. [DOI] [PubMed] [Google Scholar]
- Fisher JK, Bourniquel A, Witz G, Weiner B, Prentiss M, and Kleckner N (2013). Four-dimensional imaging of E. coli nucleoid organization and dynamics in living cells. Cell 153, 882–895. 10.1016/j.cell.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floc’h K, Lacroix F, Servant P, Wong YS, Kleman JP, Bourgeois D, and Timmins J (2019). Cell morphology and nucleoid dynamics in dividing Deinococcus radiodurans. Nature communications 10, 3815. 10.1038/s41467-019-11725-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geanacopoulos M, Vasmatzis G, Zhurkin VB, and Adhya S (2001). Gal repressosome contains an antiparallel DNA loop. Nat Struct Biol 8, 432–436. 10.1038/87595. [DOI] [PubMed] [Google Scholar]
- Guo F, and Adhya S (2007). Spiral structure of Escherichia coli HUalphabeta provides foundation for DNA supercoiling. Proceedings of the National Academy of Sciences of the United States of America 104, 4309–4314. 10.1073/pnas.0611686104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel M, Amlanjyoti D, Reyes FE, Chen JH, Parpana R, Tang HY, Larabell CA, Tainer JA, and Adhya S (2016). HU multimerization shift controls nucleoid compaction. Sci Adv 2, e1600650. 10.1126/sciadv.1600650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamagata K, Itoh Y, Tan C, Mano E, Wu Y, Mandali S, Takada S, and Johnson RC (2021). Testing mechanisms of DNA sliding by architectural DNA-binding proteins: dynamics of single wild-type and mutant protein molecules in vitro and in vivo. Nucleic acids research 49, 8642–8664. 10.1093/nar/gkab658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamashev D, Balandina A, and Rouviere-Yaniv J (1999). The binding motif recognized by HU on both nicked and cruciform DNA. The EMBO journal 18, 5434–5444. 10.1093/emboj/18.19.5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamashev D, and Rouviere-Yaniv J (2000). The histone-like protein HU binds specifically to DNA recombination and repair intermediates. The EMBO journal 19, 6527–6535. 10.1093/emboj/19.23.6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm P (1986). Two regulatory fim genes, fimB and fimE, control the phase variation of type 1 fimbriae in Escherichia coli. EMBO J 5, 1389–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh J, Saecker RM, and Record MT (2008a). DNA Binding Mode Transitions of Escherichia coli HU alpha beta: Evidence for Formation of a Bent DNA - Protein Complex on Intact, Linear Duplex DNA. Journal of molecular biology 383, 324–346. 10.1016/j.jmb.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh J, Saecker RM, and Record MT Jr. (2008b). DNA binding mode transitions of Escherichia coli HU(alphabeta): evidence for formation of a bent DNA--protein complex on intact, linear duplex DNA. Journal of molecular biology 383, 324–346. 10.1016/j.jmb.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh J, Shkel I, Saecker RM, and Record MT Jr. (2011). Nonspecific DNA binding and bending by HUalphabeta: interfaces of the three binding modes characterized by salt-dependent thermodynamics. Journal of molecular biology 410, 241–267. 10.1016/j.jmb.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DE, Geanacopoulos M, and Adhya S (1999). Role of HU and DNA supercoiling in transcription repression: specialized nucleoprotein repression complex at gal promoters in Escherichia coli. Molecular microbiology 31, 451–461. 10.1046/j.1365-2958.1999.01186.x. [DOI] [PubMed] [Google Scholar]
- Lewis DEA (2003). Identification of promoters of Esherichia coli and phage in transcription section plasmid pSA850. Method Enzymol 370, 618–645. [DOI] [PubMed] [Google Scholar]
- Lioy VS, Cournac A, Marbouty M, Duigou S, Mozziconacci J, Espeli O, Boccard F, and Koszul R (2018). Multiscale Structuring of the E. coli Chromosome by Nucleoid-Associated and Condensin Proteins. Cell 172, 771–783 e718. 10.1016/j.cell.2017.12.027. [DOI] [PubMed] [Google Scholar]
- Lioy VS, Junier I, and Boccard F (2021). Multiscale Dynamic Structuring of Bacterial Chromosomes. Annu Rev Microbiol 75, 541–561. 10.1146/annurev-micro-033021-113232. [DOI] [PubMed] [Google Scholar]
- Lyubchenko YL, Shlyakhtenko LS, Aki T, and Adhya S (1997). Atomic force microscopic demonstration of DNA looping by GalR and HU. Nucleic acids research 25, 873–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macvanin M, Edgar R, Cui F, Trostel A, Zhurkin V, and Adhya S (2012). Noncoding RNAs binding to the nucleoid protein HU in Escherichia coli. Journal of bacteriology 194, 6046–6055. 10.1128/JB.00961-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A, and Adhya S (1984). Demonstration of two operator elements in gal: in vitro repressor binding studies. Proceedings of the National Academy of Sciences of the United States of America 81, 6100–6104. 10.1073/pnas.81.19.6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montano SP, Pigli YZ, and Rice PA (2012). The mu transpososome structure sheds light on DDE recombinase evolution. Nature 491, 413–417. 10.1038/nature11602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouw KW, and Rice PA (2007). Shaping the Borrelia burgdorferi genome: crystal structure and binding properties of the DNA-bending protein Hbb. Molecular microbiology 63, 1319–1330. 10.1111/j.1365-2958.2007.05586.x. [DOI] [PubMed] [Google Scholar]
- Nielsen HJ, Li Y, Youngren B, Hansen FG, and Austin S (2006). Progressive segregation of the Escherichia coli chromosome. Molecular microbiology 61, 383–393. 10.1111/j.1365-2958.2006.05245.x. [DOI] [PubMed] [Google Scholar]
- Oberto J, Nabti S, Jooste V, Mignot H, and Rouviere-Yaniv J (2009). The HU regulon is composed of genes responding to anaerobiosis, acid stress, high osmolarity and SOS induction. PloS one 4, e4367. 10.1371/journal.pone.0004367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Haykinson MJ, and Johnson RC (1994). HU and functional analogs in eukaryotes promote Hin invertasome assembly. Biochimie 76, 992–1004. 10.1016/0300-9084(94)90024-8. [DOI] [PubMed] [Google Scholar]
- Pinson V, Takahashi M, and Rouviere-Yaniv J (1999). Differential binding of the Escherichia coli HU, homodimeric forms and heterodimeric form to linear, gapped and cruciform DNA. Journal of molecular biology 287, 485–497. 10.1006/jmbi.1999.2631. [DOI] [PubMed] [Google Scholar]
- Pontiggia A, Negri A, Beltrame M, and Bianchi ME (1993). Protein HU binds specifically to kinked DNA. Molecular microbiology 7, 343–350. [DOI] [PubMed] [Google Scholar]
- Prieto AI, Kahramanoglou C, Ali RM, Fraser GM, Seshasayee AS, and Luscombe NM (2012). Genomic analysis of DNA binding and gene regulation by homologous nucleoid-associated proteins IHF and HU in Escherichia coli K12. Nucleic acids research 40, 3524–3537. 10.1093/nar/gkr1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remesh SG, Verma SC, Chen JH, Ekman AA, Larabell CA, Adhya S, and Hammel M (2020). Nucleoid remodeling during environmental adaptation is regulated by HU-dependent DNA bundling. Nature communications 11, 2905. 10.1038/s41467-020-16724-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice PA, Yang S, Mizuuchi K, and Nash HA (1996). Crystal structure of an IHF-DNA complex: a protein-induced DNA U-turn. Cell 87, 1295–1306. [DOI] [PubMed] [Google Scholar]
- Stentebjerg-Olesen B, Chakraborty T, and Klemm P (2000). FimE-catalyzed off-to-on inversion of the type 1 fimbrial phase switch and insertion sequence recruitment in an Escherichia coli K-12 fimB strain. FEMS microbiology letters 182, 319–325. 10.1111/j.1574-6968.2000.tb08915.x. [DOI] [PubMed] [Google Scholar]
- Swinger KK, Lemberg KM, Zhang Y, and Rice PA (2003). Flexible DNA bending in HU-DNA cocrystal structures. The EMBO journal 22, 3749–3760. 10.1093/emboj/cdg351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinger KK, and Rice PA (2004). IHF and HU: flexible architects of bent DNA. Curr Opin Struct Biol 14, 28–35. 10.1016/j.sbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Yasuzawa K, Kohno K, Goshima N, Kano Y, Saiki T, and Imamoto F (1995). Role of HU proteins in forming and constraining supercoils of chromosomal DNA in Escherichia coli. Molecular & general genetics : MGG 248, 518–526. 10.1007/BF02423446. [DOI] [PubMed] [Google Scholar]
- van Noort J, Verbrugge S, Goosen N, Dekker C, and Dame RT (2004). Dual architectural roles of HU: formation of flexible hinges and rigid filaments. Proceedings of the National Academy of Sciences of the United States of America 101, 6969–6974. 10.1073/pnas.0308230101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma SC, Qian Z, and Adhya SL (2019). Architecture of the Escherichia coli nucleoid. PLoS genetics 15, e1008456. 10.1371/journal.pgen.1008456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visweswariah SS, and Busby SJ (2015). Evolution of bacterial transcription factors: how proteins take on new tasks, but do not always stop doing the old ones. Trends in microbiology 23, 463–467. 10.1016/j.tim.2015.04.009. [DOI] [PubMed] [Google Scholar]
- Vitoc CI, and Mukerji I (2011). HU binding to a DNA four-way junction probed by Forster resonance energy transfer. Biochemistry 50, 1432–1441. 10.1021/bi1007589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada M, Kano Y, Ogawa T, Okazaki T, and Imamoto F (1988). Construction and characterization of the deletion mutant of hupA and hupB genes in Escherichia coli. Journal of molecular biology 204, 581–591. [DOI] [PubMed] [Google Scholar]
- Wada M, Kutsukake K, Komano T, Imamoto F, and Kano Y (1989). Participation of the hup gene product in site-specific DNA inversion in Escherichia coli. Gene 76, 345–352. 10.1016/0378-1119(89)90174-1. [DOI] [PubMed] [Google Scholar]
- Walker DM, Freddolino PL, and Harshey RM (2020). A Well-Mixed E. coli Genome: Widespread Contacts Revealed by Tracking Mu Transposition. Cell 180, 703–716 e718. 10.1016/j.cell.2020.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, and Court DL (2000). An efficient recombination system for chromosome engineering in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America 97, 5978–5983. 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.