


Abstract
Homochirality of the cellular proteome is attributed to the L-chiral bias of the translation apparatus. The chiral specificity of enzymes was elegantly explained using the ‘four-location’ model by Koshland two decades ago. In accordance with the model, it was envisaged and noted that some aminoacyl-tRNA synthetases (aaRS) that charge larger amino acids are porous to D-amino acids. However, a recent study showed that alanyl-tRNA synthetase (AlaRS) can mischarge D-alanine and that its editing domain, but not the universally present D-aminoacyl-tRNA deacylase (DTD), is responsible for correcting the chirality-based error. Here, using in vitro and in vivo data coupled with structural analysis, we show that AlaRS catalytic site is a strict D-chiral rejection system and therefore does not activate D-alanine. It obviates the need for AlaRS editing domain to be active against D-Ala-tRNAAla and we show that it is indeed the case as it only corrects L-serine and glycine mischarging. We further provide direct biochemical evidence showing activity of DTD on smaller D-aa-tRNAs that corroborates with the L-chiral rejection mode of action proposed earlier. Overall, while removing anomalies in the fundamental recognition mechanisms, the current study further substantiates how chiral fidelity is perpetuated during protein biosynthesis.
INTRODUCTION
Biological macromolecules are homochiral, which is essentially due to the use of homochiral building blocks (1). The chiral specificity of enzymes towards their substrate was explained by using the ‘four-location’ model proposed by Koshland. According to this model, which is a modification of the ‘three-point attachment (TPA)’ model, three points of attachment and the direction of entry of the ligand are required for selecting chiral entities based on insights from high resolution isocitrate dehydrogenase structures (2). In the context of translation apparatus, it is well known that there is an absolute bias against the usage of D-amino acids to perpetuate homochirality during synthesis of proteins, universally across the tree of life (3). Aminoacyl-tRNA synthetases (aaRS) are a specialized class of enzymes that dictate the genetic code by preferentially pairing specific L-amino acids onto their corresponding tRNAs and thereby contributing significantly to proteome homochirality (3). However, aaRSs are known to commit errors occasionally by charging a wrong amino acid on a cognate tRNA (amino acid misselection), and rarely, charging a correct amino acid on the non-cognate tRNA (tRNA misselection) (4–10). Amino acid misselection is of two types: charging (i) a non-cognate L-amino acid or (ii) a D-amino acid (11). Ten out of the 20 aaRSs are equipped with proofreading domains to avoid the substitution of non-cognate L-amino acids (except AlaRS editing domain, which can act on achiral glycine also) (10). In case of non-cognate D-amino acids, in vivo studies on Escherichia coli and Saccharomyces cerevisiae have shown that chiral errors are committed by only a few aaRSs such as aspartyl-tRNA synthetase (AspRS), phenylalanyl-tRNA synthetase (PheRS), tyrosyl-tRNA synthetase (TyrRS) and tryptophanyl-tRNA synthetase (TrpRS) as envisaged by Koshland's ‘four-location’ model (2,12). Unlike the L-amino acid misselection, these erroneously formed D-aminoacyl-tRNAs are not proofread by their respective editing domains but by a specialized chiral proofreader called D-aminoacyl-tRNA deacylase (DTD) (13,14).
Extensive genetic and biochemical studies have shown that DTD is selective towards mischarged D-amino acids but not L-amino acids (12,15,16). The structural basis of such absolute chiral specificity of DTD was deciphered by solving the crystal structure of DTD in complex with D-aminoacyl-tRNA analogue. DTD achieves such specificity by employing an invariant cross-subunit Gly-cisPro motif, which captures the chiral centre of the incoming D-amino acid by interacting with the amino group and the β-carbon (15). The side chain of the D-amino acid protrudes out from the active site pocket, which provides the basis of how a single enzyme, DTD, can act on multiple D-amino acids with varying side chains attached to tRNAs. Interestingly, this mode of operation allows DTD to act on achiral glycine, which is advantageous for avoiding the mistranslation of Ala codons (17). Such a discrimination profile can be explained only by the L-chiral rejection mechanism of DTD (16). However, the cognate Gly-tRNAGly escapes DTD action due to the presence of an anti-determinant in the acceptor arm of tRNAGly, whereas the non-cognate Gly-tRNAAla possesses a positive determinant at the same position (N73) that enable its proofreading. Thus, DTD employs a discriminator base based sorting of cognate and non-cognate species of Gly-tRNAs for proofreading (18). However, the activity of DTD was not tested on the tRNAs charged with smaller side-chain D-amino acids such as alanine, which is necessary to establish the L-chiral rejection mode of substrate discrimination across the entire spectrum of amino acid sizes.
Alanyl-tRNA synthetase (AlaRS) charges alanine on tRNAAla and presents an interesting paradigm of mischarging smaller glycine and larger serine compared to its cognate amino acid (19). The mistranslation of alanine to glycine and serine is effectively prevented by employing both cis- and trans-editing domains including DTD that decouples erroneously attached Gly on tRNAAla (17,20). Very recently, it was shown that AlaRS also mischarges D-alanine on tRNAAla apart from glycine and L-serine, which is proofread by the AlaRS cis-editing domain and surprisingly not by DTD (21). These results deviate from previous works on multiple counts viz. the ‘four-location’ model of Koshland, in vivo studies in yeast and bacteria showing lack of growth defects in D-alanine enriched media, and the mechanistic model of DTD’s function proposed earlier (2,12,15,16). Considering the disagreement between the results of Rybak et al., NAR, 2019 with multiple earlier works, it is imperative that the results are thoroughly evaluated to aid in making any conclusion on the fundamental mechanisms involved in protein biosynthesis and chiral proofreading.
Here, by using in vitro and in vivo experiments, our study unequivocally demonstrates that the L-chiral rejection mechanism of DTD holds true even on the smallest side chain proteogenic amino acid substrate, i.e. D/L-Ala-tRNAAla as well. Based on the analysis of the available AlaRS structures in the Protein Data Bank (PDB) (22), we show that D-alanine is sterically excluded from the active site and hence cannot be charged by AlaRS. Our studies further show that the cis-editing domain of AlaRS is inactive on D-Ala-tRNAAla thus disproving all the major claims made in the previous work (21) and clears the serious anomalies ensued on the most fundamental reaction mechanisms that ensure chiral fidelity of the cellular aminoacyl-tRNA pool. Overall, the work experimentally substantiates the design principles governed by Koshland's ‘four-location’ model to explain the size-based disparity in activation of D-amino acids by aaRSs and the universality of L-chiral rejection based chiral proofreading by DTD.
MATERIALS AND METHODS
Cloning, expression and purification
DTD genes with C-terminal 6x-His tag from E. coli (Ec), S. cerevisiae (Sc), Danio rerio (Dr) and Mus musculus (Mm) were cloned in pET28b (Novagen) expression vector. Similarly, N-terminal 6x- His tagged EcAlaRS (wildtype), EcAlaRS C666A mutant and TtAlaRS genes were also cloned in pET28b vector. C-terminal 6x His tag T. thermophilus (Tt) DTD is cloned in pTRC99 vector. The vectors carrying genes were first confirmed by Sanger sequencing (Eurofins Genomics, India) and then transformed in E. coli BL-21(DE3) expression cells for IPTG induction-based protein overexpression (17). TtDTD was expressed in Terrific Broth (TB) under IPTG induction. All the His-tagged proteins were purified from cell lysate of respective overexpression strain using immobilised metal affinity chromatography (Ni-NTA) in buffer containing 50 mM Tris (pH 8), 150 mM sodium chloride, 5% Glycerol and 5 mM β-mercaptoethanol. Proteins were eluted by gradient elution between 10 mM to 250 mM imidazole. Obtained fractions were further purified using size exclusion chromatography in buffer containing 150 mM NaCl, 50 mM Tris (pH 7.5). The purified and concentrated proteins were stored in -20°C in buffer containing 150 mM NaCl, 100 mM Tris (pH 7.5) and 50% glycerol for biochemical assays. All the steps after expression were carried out on ice or 4°C.
Biochemical assays
EctRNAAla was generated by in vitro-transcription using MEGAshortscript T7 Transcription Kit (Thermo Fisher Scientific, USA). Transcribed tRNA was 3′ end radiolabelled with [α-32P]-ATP (BRIT-Jonaki, India) using E. coli CCA adding enzyme. L-alanine was charged onto 3′ radiolabelled EctRNAAla (10 μM) in a reaction mix containing ATP (2 mM), β-mercaptoethanol (5 mM), L-alanine (100 mM) and EcAlaRS-WT (500 nM) (23). Alternatively, D-Ala-tRNAAla was generated by two strategies, 1) by EcAlaRS-WT with the same aminoacylation procedure used for L-Ala-tRNAAla and 2) Flexizyme-based charging method described in the following section (24). All the deacylation assays were carried out as reported earlier (15,16). In brief, all the aminoacylated substrates (2 μM) were incubated with 20 mM Tris pH 7.2, 5 mM MgCl2, 5 mM DTT and 0.2 mg/ml BSA along with 10 pM to 1μM DTD or 100 nM of EcAlaRS-WT/C666A mutant or 500 nM of TtAlaRS. Time course studies were performed by aliquoting 0.8 μl of the deacylation mix for each time point (2, 5, 10 and 15 minutes) and followed by S1 nuclease digestion. The incubated samples were then fractionated in Thin Layer Chromatography (TLC) and the proportion of aminoacylation in the samples were estimated after phosphor imaging the TLC exposed storage phosphor screen (Typhoon FLA 9000 biomolecular imager, GE Healthcare). All the aminoacylation and deacylation reactions were carried out in 37°C. EctRNAAla was used throughout the study.
Aminoacylation titration assays with decreasing concentration of amino acid were performed using serially diluted amino acid substrates (L/D-alanine) from 100 mM to 100 nM in the reaction containing 500 nM of EcAlaRS WT or editing defective C666A enzyme.
Flexizyme based aminoacylation
Flexizymes were generated through in vitro-transcription as reported earlier (25). All the amino acid substrates were activated by either CME (Cyanomethyl ester) or DBE (Dinitro benzyl ester). These activated amino acid substrates were charged using enhanced Flexizyme (eFx) (for CME activated substrate) and di-nitro flexizyme (dFx) (for DBE activated substrate) based on the previous reports (25–28). Briefly, flexizyme is mixed step wise with 3′ radiolabelled tRNA, MgCl2 and respective amino acid substrate and then incubated for 12–14 h on ice. After incubation, all the aminoacylated-tRNAAlas were then subjected to ethanol precipitation and the resultant pellets were washed twice with 70% ethanol and 0.1% sodium acetate. Air-dried pellets were resuspended in 5 mM sodium acetate (pH 4.5) and used for further deacylations.
Viability assays
Viability assays were performed with both the MG1655 single mutant ΔalaS (contains editing defective AlaRS) and as well as double mutant ΔalaSΔdtd (dtd gene deleted strain containing editing defective AlaRS) in M9 minimal agar as per the protocol described in Pawar et al. (17). Briefly, ΔalaS, Δdtd, ΔalaSΔdtd mutant strains of E. coli MG1655 were inoculated in the minimal media containing 0.002% arabinose and 0.2% maltose, till the cultures reach 0.6 OD600. The cultures were serially diluted from 10−1 to 10−5 and 5 μl of each dilution was spotted on the agar plates containing respective amino acids (L-serine (3 mM), L-alanine (10, 50, 100 mM), D-alanine (10, 50, 100 mM), L-tyrosine (6 mM) and D-tyrosine (6 mM)) along with the above-mentioned concentration of arabinose and maltose.
Growth curve experiments were done with the same strains which we utilised in the spot dilution assay, in M9 minimal liquid media containing 0.0002% of arabinose and 0.2% of maltose and supplemented with the amino acid of interests (L-serine (3 mM), L-alanine (100 mM), D-alanine (100 mM)) in the specified concentrations. Since D-tyrosine is known to get turbid at higher concentration (>5 mM) in the liquid culture (29), we used a mixture of D-amino acids containing 3mm of D-tyrosine, 10 mM of D-aspartate and 1.5 mM of D-tryptophan for the growth curve assay. The secondary cultures were initiated with 1% of overnight grown culture and allowed to grow till 0.6 OD600. Grown secondary cultures were utilised to initiate the tertiary cultures with the initial OD600 of 0.06Cell density was measured at an interval of 2 h from the time of inoculation. All the viability assays were done in triplicates
Mass spectrometry
Both L-Ala-tRNAAla and D-Ala-tRNAAla were generated using EcAlaRS WT as above mentioned. The resultant products were ethanol precipitated and digested using aqueous ammonia solution (25% of v/v NH4OH) at 70°C for 12 h. These samples were then dried and resuspended in 10% methanol and 1% acetic acid. ESI-based mass spectrometry analysis of the selected precursor ion was carried out exactly as previously reported (30). Briefly, the samples were subjected to Heated Electrospray Ionization (HESI) and the spectra of the selected ions were analysed using Xcalibur™ 4.0 software (Thermo Fisher Scientific, USA).
Structural analysis
Highest resolution structures of both HsAlaRS (PDB id: 4XEM, resolution-1.28Å) and EcAlaRS (PDB id: 3HXU, resolution-2.1Å) bound with L-AlaSA (5'-O-(N-(L-alanyl)-sulfamoyl adenosine) were selected from Protein Data Bank (PDB) and used to model D-AlaSA in the active site. Pymol plugin ‘ProteinInteractionViewer’ was used with default parameters to visualize and represent the small and bad overlaps in both of the AlaRS structures with L-AlaSA and modelled D-AlaSA in the active site. MolProbity server (https://molprobity.biochem.duke.edu/) was used to evaluate the small, bad, and worse clashes/overlaps based on van der Waals radii of the individual atom in the model (31). It was also employed to confirm and quantify clashes in AlaRS structures with D-AlaSA and L-AlaSA. From the resultant clash score tables, values that were common in both the structures were eliminated to obtain scores for unique clashes ensued by modelling D-AlaSA in the active site pocket.
Detection of chiral compounds
To probe the chiral impurity in the amino acid supply, we utilised Marfey's reagent (Nα-(2,4-dinitro-5-fluorophenyl)-L-valinamide) to derivatise the D-alanine and L-alanine reagents to separate and quantitate the D- and L-enantiomeric levels in each of them by following the method as previously mentioned (32). Briefly, amino acid dissolved in autoclaved milliQ water was added with acetone, 1% of Marfey's solution prepared in acetone and aqueous sodium bicarbonate. After incubating the mixture at 37°C for 90 min, the reaction was quenched with formic acid. The resultant mixture was further diluted 10-fold with 1% acetone and subjected to UPLC (Acuity H-Class UPLC connected to Xevo TQ-S micro-MS system) with Agilent ZORBRAX Eclipse Plus C18 (2.1 mm × 100 mm) reverse phase column. Gradient elution was done using a mobile phase consisting of solution A (ammonium acetate, pH 4.6) and solution B (Acetone). A linear gradient from 20% to 80% of solution B was developed in 10 min.
RESULTS
AlaRS editing defective strain of bacteria is not sensitive to D-alanine
Recently it has been shown in vitro that AlaRS charges D-alanine on tRNAAla, and is proofread by the cis-editing domain of AlaRS (21). In order to test these observations physiologically, we hypothesized that bacterial stain with AlaRS editing defective (AlaRSED) gene would be sensitive to excess D-alanine supplementation in growth media, while the wild type bacterial strain would be insensitive to the same. To test this, we have used E. coli MG1655 strain with AlaRS gene deleted at the genomic level carrying a shelter plasmid bearing editing defective EcAlaRS (T567F, S587W, C666F-AlaRSED) gene under arabinose inducible promoter (MG1655 ΔalaS Para::AlaRSED) (17). As expected, the MG1655 ΔalaS Para::AlaRSED strain was sensitive to glycine and 6 mM L-serine supplementation, while the growth of E. coli strain containing wild type AlaRS was unperturbed (Figure 1A). Similarly, both the strains did not show any growth defect on L and D-tyrosine supplementation (Supplementation Figure S1A). Surprisingly, MG1655 ΔalaS Para::AlaRSED had no growth defect upon supplementation of excess D-alanine, ranging from of 10 to 100 mM, similar to that of the wild type strain (Figure 1B). The spot dilution assay results were further confirmed by growth curve assays with the same strains in the liquid media supplemented with respective concentration of amino acids (Supplementary figure S1B and C). These results were perplexing as to how AlaRS, which was earlier shown to misacylate and edit both L-serine and D-alanine under in vitro conditions (21), shows toxicity towards L-serine but not to D-alanine in viability assays. These observations raise the possibility of two scenarios: that there might be a redundant proofreader for D-Ala-tRNAAla or D-alanine charging by AlaRS in vitro may not have any physiological relevance.
Figure 1.

D-alanine supplementation is not toxic to AlaRS editing defective bacterial strain. Spot dilution assay of E. coli wild type MG1655 strain and E. coli MG1655 ΔalaS/para :: alaS (TM)-T567F, S587W, C666F supplemented with (A) No amino acid, 20 mM glycine and 6 mM L-serine, (B) 10, 50 and 100 mM of L-alanine and D-alanine. Increasing concentration of D-alanine even to 100 mM in the growth media does not cause any toxicity in both MG1655 wild type and MG1655 ΔalaS/para :: alaS TM strains.
DTD’s L-chiral rejection-based mechanism is amino acid size independent
Lack of toxicity in MG1655 ΔalaS Para::AlaRSED strain to D-alanine in our viability assays raised a possibility for presence of a redundant proofreader for D-Ala-tRNAAla in the cells other than AlaRS editing domain. Based on earlier studies on D-aminoacyl-tRNAs proofreading modules, the only possible redundant factor for editing D-Ala-tRNAAla could be D-aminoacyl-tRNA deacylase (DTD) (13,15). Insights from multiple ligand-bound crystal structures of DTD and its biochemical activity, led to the proposition that DTD is a strict L-chiral rejection module (15–17). Previous NMR and modelling studies clearly showed that D-Ala-tRNAAla could be accommodated and acted upon by DTD (16,33). D-alanine fits snugly in the active site pocket of DTD when modelled based on the D-Tyr-3AA that was captured in the co-crystal structure (PDB id: 4NBI). Like any other D-amino acid, R group (methyl group)/ Cβ of D-alanine makes a C-H…O hydrogen bond with the carbonyl oxygen of Pro150 from the cross-subunit Gly-cisPro motif. This further reinforces the fact that D-aa-tRNAs are positioned in such a way that the side chain beyond Cβ atom is protruding outside the active site of DTD (Figure 2A). However, the biochemical activity of DTD was shown only on tRNAs charged with larger D-amino acids like tyrosine, phenylalanine, aspartate and tryptophan (11,15). Therefore, the activity of DTD on tRNAs charged with smaller D-amino acids was only a logical extrapolation. We checked whether DTD’s L-chiral rejection based enantioselectivity is effective on smaller amino acid charged tRNAs or not, experimentally, by performing in vitro deacylation assays. We generated L-Ala-tRNAAla, L-Phe-tRNAAla, D-Ala-tRNAAla, D-Phe-tRNAAla, Gly-tRNAAla and D-Ser-tRNAAla using flexizyme-based aminoacylation system. The substrates were incubated with E. coli DTD (EcDTD) and analysed for its ability to deacylate the aminoacyl-tRNAs. As expected, DTD was inactive on both L-Ala-tRNAAla (Figure 2B) and L-Phe-tRNAAla (Figure 2C) but efficiently deacylated all tRNAs charged with D-amino acids irrespective of the side chain size (Figure 2D, E and G) and glycine (Figure 2F). To show the conservation of this activity across species, we also tested the D-Ala-tRNAAla deacylation activity of DTD from various organisms such as P. aeruginosa (PaDTD), D. melanogaster (DmDTD), M. musculus (MmDTD) (Supplementary figure S2). In line with the EcDTD, all DTDs tested were able to deacylate D-Ala-tRNAAla at 100 pM concentrations. Thus, the above biochemical results experimentally substantiate the side chain independent L-chiral amino acid rejection mechanism of DTD. This observation contradicts the previous report that DTD cannot act on D-Ala-tRNAAla (21), thereby implying its possible role as a redundant proofreader for D-Ala-tRNAAla in the cells other than AlaRS editing domain.
Figure 2.
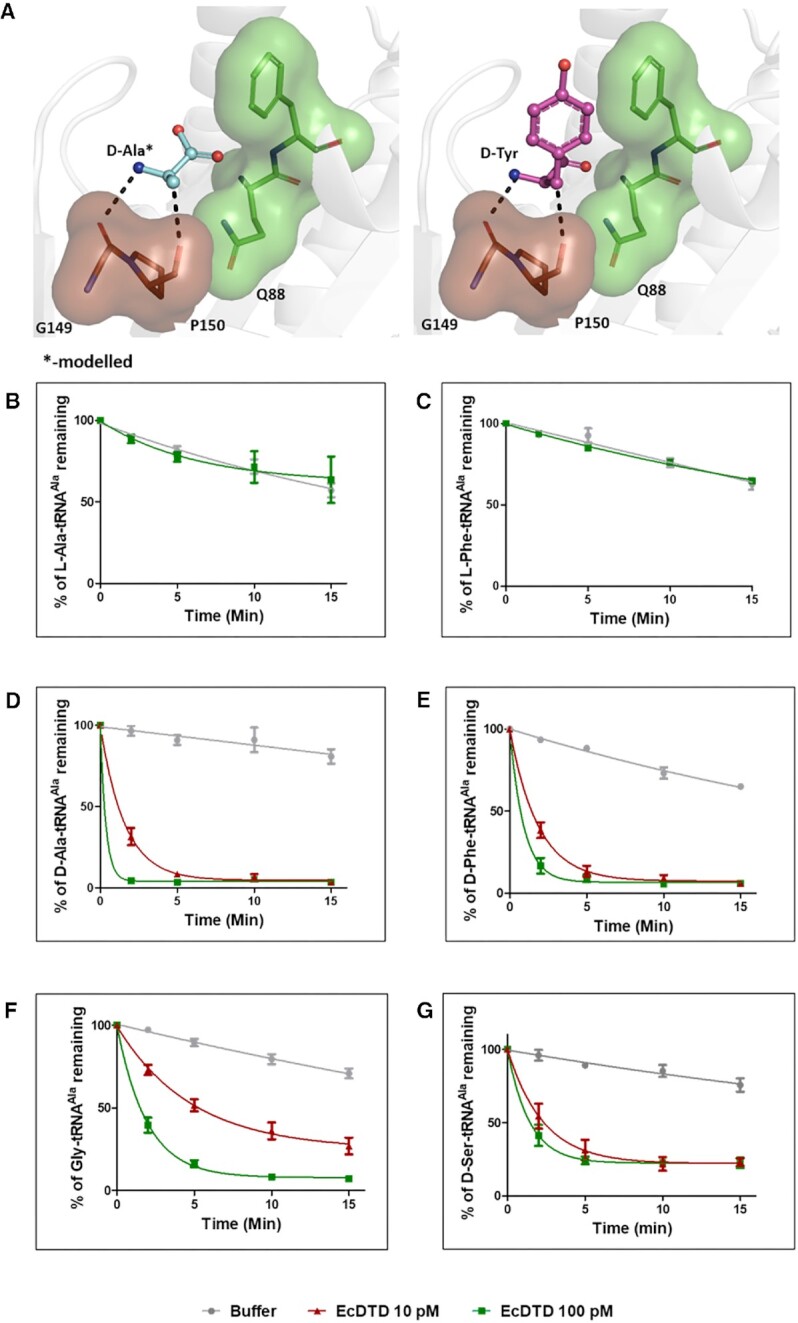
DTD is a strict L-Chiral rejection module. (A) D-alanine modelled in the active site of DTD, showing D-alanine can fit in the active site like any other larger side chain D-amino acids (D-tyrosine). Deacylation assay at 10 pM and 100 pM concentrations of EcDTD on (B) L-Ala-tRNAAla, (C) L-Phe-tRNAAla, (D) D-Ala-tRNAAla, (E) D-Phe-tRNAAla, (F) Gly-tRNAAla and (G) D-Ser-tRNAAla (all the substrates from B to F were generated using flexizyme) (*:modelled).
E. coli lacking both DTD and AlaRS editing activity is not sensitive to D-alanine
Many trans-editing factors act in redundancy with the aaRS borne cis-editing domains towards correction of mischarged aa-tRNAs, which can obscure the physiological impact of abrogation of the aaRS cis-editing domain activity (6,17,34,35). Our viability assay with MG1655 ΔalaS Para::EcAlaRSED upon supplementation of excess D-alanine in growth medium did not lead to any growth defect (Figure 1). Moreover, our biochemical data confirms that DTD can effectively deacylate D-Ala-tRNAAla generated with the help of flexizyme (Figure 2D; Supplementary figure S2). To probe the possibility of DTD being a redundant proofreader of D-alanine charged by AlaRS in vivo, we employed MG1655 bacterial strains lacking dtd gene (MG1655 Δdtd), AlaRS editing defective (MG1655 ΔalaS Δdtd Para::EcAlaRSED – Double knockout editing defective (DKO-ED)) and AlaRS-wild type complemented (MG1655 ΔalaS Δdtd Para::EcAlaRSWT – DKO-WT) background. As expected, all the three strains were sensitive to D-tyrosine and the DKO-ED showed sensitivity towards glycine and L-serine supplementation (Figure 3A). In addition, the growth of the all the strain remains unperturbed even after supplementing with 100 mM D-alanine in the media. However, high concentration of L-alanine (50 mM and 100 mM) showed severe growth perturbation as it is known to result in metabolic imbalance or growth defect (Figure 3B) (36). Similarly, growth curve assays with DKO-ED strain in the presence of 100 mM D-alanine, did not exhibit any cellular toxicity or reduced growth (Supplementary figure S3A) in comparison with ΔDTD and DKO complemented with wildtype EcAlaRS (Supplementary figure S3B and C). The absence of any toxicity towards D-alanine in ΔDTD and DKO-ED strain raise the question as to whether D-alanine charging by AlaRS is at all physiologically relevant or is it just an in vitro artefact (21).
Figure 3.

DTD knockout and AlaRS editing defective strain is insensitive to D-alanine. Toxicity assay of E. coli MG1655 Δdtd compared with that of E. coli MG1655 Δdtd::ΔalaS/para:: alaS-(TM)T567F, S587W, C666F and E. coli MG1655 Δdtd::ΔalaS/para::alaS in the presence of (A) no amino acid, 3 mM of L- and D-tyrosine, 20 mM glycine 3 mM and 6 mM L-serine, (B) Supplementation of 10, 50 and 100 mM of L-alanine and D-alanine, wherein even at 100 mM concentration of D-alanine all the three strains does not show any toxicity.
Aminoacylation domain of AlaRS operates by strict D-chiral rejection mode
The Protein Data Bank has multiple entries of high-resolution structures of AlaRS aminoacylation domain with substrate analogue bound to the catalytic pocket. These substrate analogue bound structures helped glean mechanistic understanding at atomic level resolution of the alanylation reaction carried out by AlaRS (10). We sought to understand the structural basis of the in vitro D-alanine charging by AlaRS reported in previous study (21). In order to figure out how D-alanine fits in the aminoacylation site of AlaRS, we choose the highest resolution (1.28 Å) structure of AlaRS catalytic domain complexed with L-AlaSA (‘5’-O-(N-(L-alanyl)-sulfamoyl adenosine) (PDB ID: 4XEM- HsAlaRS) (Figure 4A) from PDB and modelled D-AlaSA in the active site. The model showed that D-alanine could not fit inside the active site pocket of AlaRS aminoacylation site as Cβ of D-alanine had a serious clash with Cγ2 atom of V218 and Cδ2 of Trp176 (Figure 4B). Furthermore, we confirmed the clashes using MolProbity server (https://molprobity.biochem.duke.edu/). With the bad clash default overlap cut-off of 0.4Å, the modelled D-AlaSA exhibited severe clash with Val218 and Trp176 residues with the highest clash score (Figure 4C). To test the universality of the clashes, we repeated similar analysis with the high-resolution structure of EcAlaRS (2.1Å) with L-AlaSA in the active site (PDB: 3HXU). Expectedly, same clashes were observed when D-AlaSA was modelled in the active site but not for L-AlaSA (Supplementary figure S4A and B). MolProbity scores from D-AlaSA modelled EcAlaRS also gave similar scores as that of HsAlaRS (PDB ID: 4XEM) (Supplementary figure S4C). Moreover, the residues (valine and tryptophan) that are clashing with modelled D-alanine are conserved across the species (Figure 4D). Interestingly, Thermus thermophilus (Tt) AlaRS, which is shown to charge D-alanine by Rybak et al., NAR, 2019 is also possess the invariant valine and tryptophan in the active site pocket (Figure 4D). Structural modelling and alignment clearly suggest that AlaRS catalytic pocket has conserved features responsible for rejecting D-alanine that supports the ‘four-location’ model proposed by Koshland. In the case of AlaRS bound with L-AlaSA, the amino group of L-alanine is fixed by the side chain of residue Asp236, carbonyl group is held by guanidinium group of Arg70 (10,19). With this mode of selection of the amino acid, the residues Val218 and Trp176 sterically excludes Cβ of any D-amino acid, thus serving as the third attachment point and the fourth constraint is imposed by the orientation of amino acid to form amino acyl-AMP intermediate and aminoacyl-tRNA. Therefore, based on the structural insights from multiple substrate analogue (data not shown) bound crystal structures of AlaRS aminoacylation domain, D-alanine cannot bind to AlaRS aminoacylation domain and hence cannot be aminoacylated.
Figure 4.

AlaRS active site operates as a strict D-chiral rejection module. (A) L-AlaSA in the active site of the HsAlaRS catalytic domain (PDB id:4XEM). (B) D-AlaSA modelled in the active site of the HsAlaRS catalytic domain shows serious clashes with Val-218 and Trp-176. (C) Table showing clash distances calculated using MolProbity server between modelled D-AlaSA and surrounding residues in AlaRS aminoacylation domain. This suggests that D-alanine cannot fit into AlaRS catalytic pocket (with clash distance in red). (D) Structure based sequence alignment showing the invariant valine and tryptophan (marked by black arrows) in the catalytic site of AlaRS in all the domains of life.
The species charged by AlaRS in D-alanylation reaction is a contaminant
There exists a contradiction between the proposed AlaRS aminoacylation of D-alanine in earlier work (21) and the observed in vivo D-alanine toxicity data as well as structural insights from the substrate analogue bound AlaRS aminoacylation domain. To resolve this, we probed the reported aminoacylation of tRNAAla with D-alanine by editing defective C666A mutant of EcAlaRS. In our aminoacylation reactions, along with L-alanine, L-serine, and glycine, EcAlaRS C666A also charged D-alanine (albeit at a very high concentration of 100 mM of amino acid) on tRNAAla. We then tried deacylating the L-Ala/D-Ala aminoacylated tRNAs by DTD and AlaRS. Interestingly, both L-Ala-tRNAAla and D-Ala-tRNAAla were not deacylated by DTD and AlaRS (Figure 5A and B, respectively). Next, we used L-Ala-tRNAAla and D-Ala-tRNAAla generated by flexizyme as substrates for deacylation by AlaRS and DTD. Unlike aaRSs, flexizyme do not possess features for substrate specificity, therefore, this method allows aminoacylation irrespective of the side chain chemistry or Cα chirality (28). Surprisingly, AlaRS could not deacylate either L- or D-Ala-tRNAAla and D-Ser-tRNAAla substrates generated using flexizyme. However, flexizyme generated D-Ala-tRNAAla and D-Ser-tRNAAla were deacylated by DTD at 100 pM but not L-Ala-tRNAAla (Figure 5C and D; Supplementary figure S5A). The above results clearly indicate the possibility of some unknown contaminant in D-alanine reagent being charged in aminoacylation reaction with AlaRS. Yet another noteworthy observation is that AlaRS was not able to edit the presumed AlaRS generated D-Ala-tRNAAla substrate as effectively as L-Ser/Gly-tRNAAla (Figure 5B and supplementary figure S5B and C, respectively). It is important to note that the earlier paper (21) authors have used Thermus thermophilus DTD (Tt) and TtAlaRS. Though the residues responsible for the D-alanine rejection is present in the active site of TtAlaRS, we wanted to check the ability of the same and TtDTD to deacylate the D-Ala-tRNAAla generated using flexizyme. Similar to EcDTD, TtDTD was inactive against L-Ala-tRNAAla but readily deacylated the flexizyme generated D-Ala-tRNAAla (Supplementary figure S6A and C, respectively). Tt AlaRS efficiently deacylated L-Ser-tRNAAla but was inert against D-Ala-tRNAAla (Supplementary figure S6B and C, respectively). Taking the structural, biochemical and in vivo experiments into account, it is evident that AlaRS cannot charge or deacylate D-alanine. Hence, it is most likely that the observed D-alanine aminoacylation might be a contaminant, which is present in trace amount in the D-alanine reagent.
Figure 5.

Chiral selectivity of AlaRS aminoacylation site. (A) Deacylation of L-Ala-tRNAAla by both EcDTD and EcAlaRS-WT. (B) Deacylation of D-Ala-tRNAAla (AlaRS generated aminoacylated substrate) by EcAlaRS-WT and EcDTD showing that D-Ala-tRNAAla is not cleaved by either of them. (C) Deacylation of L-Ala-tRNAAla charged by flexizyme by EcAlaRS-WT and EcDTD showing similar results as AlaRS charged substrate. (D) Deacylation of flexizyme charged D-Ala-tRNAAla by EcAlaRS-WT and EcDTD shows that EcDTD can effectively cleave D-Ala-tRNAAla but not by EcAlaRS-WT. (E) Aminoacylation of L-Ala and D-Ala on tRNAAla by EcAlaRS C666A with reducing concentration of the amino acid substrate in the reaction (100 mM, 10 mM, 1 mM, 100 μM, 10 μM, 1 μM and 100 nM) showing L-alanine getting charged at 10 μM amino acid concentration, which is 1000-fold less than the minimum concentration at which D-alanine charging is seen, i.e. 10 mM.
To investigate the possibility that the aminoacylated specimen in D-alanine aminoacylation reaction could be a contaminant, we titrated a range of amino acid (D-alanine) concentrations lesser than the set concentration in the standard alanylation protocol (from 100 to 100 nM). Considering the contaminant in the D-alanine reagent is expected to be in trace amount, only a high amount of D-alanine in the reaction will have optimal amount of the contaminant for charging to happen. Indeed, upon reducing the concentration of D-alanine in the reaction with EcAlaRS C666A, there was a clear reduction in formation of aminoacyl-tRNAAla, however reducing L-alanine in aminoacylation reaction did not lead to reduction in formation of aminoacyl-tRNAAla as abruptly like in case of D-Ala (Figure 5E). Compared to L-Ala, D-Ala could be charged on tRNAAla only at 1000-fold excess amino acid concentration in the reaction. To probe the effect of AlaRS editing domain, we repeated the amino acid titration in aminoacylation reaction for both L-and D-alanine with EcAlaRS WT. Remarkably, EcAlaRS WT enzyme could aminoacylate D-alanine at around 1000-fold more concentration than L-Ala (Supplementary Figure S7). The ∼1000-fold excess concentration of D-Ala required to get comparable aminoacylation to that of L-Ala by AlaRS raises the possibility of enantiomeric contaminant L-alanine being the species charged on tRNAAla in D-alanine aminoacylation reactions.
AlaRS charges enantiomeric contaminant L-alanine in D-alanylation reaction
Based on the previous studies, AlaRS can charge L-serine, glycine and non-proteogenic amino acids such as azetidine-2-carboxylic acid (AZE) and βN-methylamino-L-alanine (BMAA) apart from L-alanine (31,33,34). Among these, it is known that AlaRS-editing domain can deacylate all the above-mentioned amino acids but not its cognate L-alanine and BMAA. To find out the nature of the contaminant present in the D-alanine reagent which can be charged by AlaRS catalytic domain, we performed mass spectrometry-based analysis of D-alanine and L-alanine for the presence of above-mentioned substrates. Interestingly, we could see a single prominent peak corresponding to alanine in both the samples whereas contaminant peaks were negligible (Supplementary Figure S8A and B). To probe the identity of the charged contaminant, we charged both L-alanine and D-alanine with EcAlaRS-WT on EctRNAAla and subjected the charged product to mass spectrometry. We observed similar mass spectrum for both the samples with a single predominant peak with m/z corresponding to alanine (Figure 6A). The mass spectra from both the sample suggests identical species alanine being charged by AlaRS, but the chirality of the charged alanine remain undetermined.
Figure 6.

AlaRS charges L-Ala contaminant in D-alanylation reaction. (A) ESI-MS profile of resultant product after aminoacylated by EcAlaRS using D- and L-alanine. (B) Chromatograms showing that area of the peak corresponding to L-alanine in D-alanine sample is increasing upon titration of increasing concentration of D-alanine in the reaction.
In order to identify the enantiomeric status of the alanine charged onto tRNAAla, we utilised Marfey's reagent which helps in achieving enantio-separation and quantitation owing to the formation of diastereomers with different polarity with D- and L-enantiomers (32,37). When the derivatised D-alanine was subjected to reverse phase chromatography, the chromatogram showed a prominent D-alanine peak along with a comparatively smaller but observable peak. The retention time of the smaller peak correspond to that of L-alanine (Supplementary Figure S9A). To further validate this observation, we increased the concentration of D-alanine by 1:1, 2:1, 5:1, 10:1 and 20:1 with Marfey's reagent in the reaction. Concomitantly, the smaller peak gradually increased with the increasing D-alanine concentration in the reaction (Figure 6B). Notably, when the same set of reactions were repeated using L-alanine, conversely a smaller peak corresponding the retention time of D-alanine was observed (Supplementary Figure 9B). These observations clearly indicate that the D-alanine supply contains trace amount of enantiomeric impurity and vice versa. Relative quantification of the peaks suggests that the enantiomeric impurity contributes to around 0.1% of total derived amino acids. This result is also in line with the reported enantiomeric excess (ee) of D-alanine supply of 99% (Sigma-Aldrich). Ee of 99% denotes that ≤0.5% of L-alanine contamination is always possible in the commercially available D-alanine, which is in agreement with the aminoacylation titrations, where ∼1000-fold excess of D-alanine was required to achieve comparable aminoacylation with L-alanine (Figure 5E). This also highlights the strict chiral selective nature of the AlaRS aminoacylation site that even a 0.1% L-alanine in the enantiomeric mixture can be selected and charged onto tRNAAla leading to inadvertent synthesis of cognate L-Ala-tRNAAla that does not get deacylated by both AlaRS editing domain and DTD (Figure 5B). Moreover, using chirality non-discriminatory aminoacylation system Flexizyme, we generated D-ala-tRNAAla that is effectively deacylated by DTDs but not AlaRS (Figure 5D).
Taken together, it is evident that AlaRS cannot charge or deacylate D-alanine and DTD can proofread small side chain D-aa-tRNAs although some of them may not be physiologically generated (Figure 7). Combining mass spectroscopy analysis of the charged species and derivatization of the D-alanine followed by HPLC analysis concludes that the observed aminoacylation is due to a trace amount of enantiomeric L-alanine contaminant in D-alanine reagent. Hence, DTD and AlaRS editing domain were not able to deacylate it. Our results conclusively show that it is indeed the aminoacylation site and not the editing site of AlaRS that enforces enantiomeric fidelity.
Figure 7.

Overall model showing the modus operandi of AlaRS and DTD. AlaRS sterically excludes D-alanine from the aminoacylation site but can charge L-serine and glycine along with L-alanine and its editing domain can deacylate mischarged L-Ser-tRNAAla and Gly-tRNAAla. DTD acts on D-aminoacyl-tRNAs independent of side chain size and deacylates the misacylated product into free amino acids and tRNAs.
DISCUSSION
Proofreading by aaRSs have been studied majorly from the point of view of non-cognate L-amino acids charging by aminoacyl-tRNA synthetases. Several cis- as well as trans-editing domains have been characterized for their mechanistic and functional basis of operation (34,38,39). It is now well known that nearly ten aaRSs charge non-cognate L-amino acids and proofreading functions are invoked to clear the errors generated using both double- and triple-sieve models (39–43). However, in the context of opposite chiral D-amino acids, only the larger side chains have been shown to be activated biochemically by tRNA synthetases and this phenomenon could be explained by Koshland's ‘four-location’ model (2,12,14). Therefore, it was puzzling to note from a recent work that AlaRS mis-aminoacylates D-alanine (21). Our modelling and biochemical work identifies the residues responsible for the L-chiral selection mechanisms and shows that AlaRS does not charge D-alanine as previously reported (Supplementary Figure S10A). AlaRS being one of the early synthetases evolved an efficient mechanism which allows it to discriminate and exclude D-alanine from the active site. The high enantio-specificity towards L-alanine permits it to effectively select and activate even ∼0.1% of L-alanine from the D-alanine mixture.
The casualty of misinterpreting observation arising due to the interference by trace amount of contaminant present in reagent used in reaction is not new in the field. Classic examples of the presence of L-enantiomeric contamination in the D-amino acids and its effect on experimental outcome have been well documented in the case of D-aspartate and D-lysine (12). Similar instances of contaminants were reported and rectified in other studies (44–46). In this regard, the present study emphasises that even a ∼0.1% contamination in the starting material, would lead to misinterpretation and affects well defined mechanistic principles.
While showing D-alanine charging, the Rybak et al. study also presented the first evidence of a cis-editing domain of an aaRS to clear such a chiral error (21). It would imply that the editing domain of AlaRS could remove opposite chiral D-alanine in addition to non-cognate L-serine and achiral glycine. Our experimental work unambiguously demonstrated that the AlaRS editing module, while retaining the long-established proofreading activity against L-serine and glycine (10,47–50), does not possess any detectable deacylase activity against D-alanine. The fact that AlaRS editing domain being inactive against flexizyme generated D-Ala-tRNAAla is in line with the literature that an aminoacyl-tRNA synthetase that charges D-amino acid cannot deacylate it using its cis-editing domain (Supplementary Figure S10B). As observed in the case of PheRS, which mischarges D-phenylalanine and L-tyrosine, the editing domain can only deacylate L-tyrosine but not D-phenylalanine (14). This is in accordance with the proposition that editing domains would have evolved to effectively deacylate the mischarged, chemically similar, non-cognate amino acids by aaRSs. It would be startling to identify a cis-editing domain with an ability to clear both mischarged non-cognate amino acid and D-form of amino acid, if at all it exists, as this would have important implications for the evolution of chiral fidelity in protein biosynthesis.
Earlier, we proposed based on the mechanistic mode of action, DTD can carry out chiral proofreading of smaller amino acids (16,33). Now with the direct experimental proof of it acting on both smaller and larger D-amino acids, it is highly likely that DTD will act on the entire range of D-amino acids charged on tRNAs. The early translation machinery, which includes aaRSs, EF-Tu and the ribosome, in a pre-LUCA era may not have been as specific for L-chiral forms as the evolved ones that are present today. It is therefore tempting to propose that a DTD with its activity on the entire spectrum of D-aa-tRNA would have been beneficial in clearing the chiral mistakes thus enforcing homochirality during evolution of the protein biosynthetic machinery. Also, as we begin to understand how the metabolite fluxes are changing under different environmental conditions in organisms, retaining activity towards the total range of D-amino acids mischarged on tRNA may be beneficial and this aspect of DTDs physiological role, particularly in eukaryotes, needs to be explored further.
DATA AVAILABILITY
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Rumit Maini and Prof Hiroaki Suga from the University of Tokyo for sharing the protocol and materials for the flexizyme standardisation. We also thank Dr Srivari Chandrasekhar, CSIR-IICT for his encouragement and support during the synthesis of compounds required for flexizyme assays.
Author contribution: K.S., V.L.V., G.A., K.J.A., J.G., S.P.K. designed and performed the experiments. G.K.G., K.N. P.S.M. synthesised the activated flexizyme substrates. R.S. conceived and supervised the study. All the authors analysed the data. K.S., J.G. and R.S. wrote the manuscript with the help from S.K.K., and all the authors reviewed it.
Contributor Information
Koushick Sivakumar, CSIR-Centre for Cellular and Molecular Biology (CSIR-CCMB), Hyderabad, Telangana 500007, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Vinitha Lakshmi Venkadasamy, CSIR-Centre for Cellular and Molecular Biology (CSIR-CCMB), Hyderabad, Telangana 500007, India.
Gurumoorthy Amudhan, CSIR-Centre for Cellular and Molecular Biology (CSIR-CCMB), Hyderabad, Telangana 500007, India.
Kezia J Ann, CSIR-Centre for Cellular and Molecular Biology (CSIR-CCMB), Hyderabad, Telangana 500007, India.
Gadela Karteek Goud, Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India; Department of Organic Synthesis & Process Chemistry, CSIR-Indian Institute of Chemical Technology (CSIR-IICT), Hyderabad, Telangana 500007, India.
Kiranmai Nayani, Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India; Department of Organic Synthesis & Process Chemistry, CSIR-Indian Institute of Chemical Technology (CSIR-IICT), Hyderabad, Telangana 500007, India.
Jotin Gogoi, CSIR-Centre for Cellular and Molecular Biology (CSIR-CCMB), Hyderabad, Telangana 500007, India.
Santosh Kumar Kuncha, CSIR-Centre for Cellular and Molecular Biology (CSIR-CCMB), Hyderabad, Telangana 500007, India.
Prathama S Mainkar, Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India; Department of Organic Synthesis & Process Chemistry, CSIR-Indian Institute of Chemical Technology (CSIR-IICT), Hyderabad, Telangana 500007, India.
Shobha P Kruparani, CSIR-Centre for Cellular and Molecular Biology (CSIR-CCMB), Hyderabad, Telangana 500007, India.
Rajan Sankaranarayanan, CSIR-Centre for Cellular and Molecular Biology (CSIR-CCMB), Hyderabad, Telangana 500007, India; Academy of Scientific and Innovative Research (AcSIR), CSIR–CCMB campus, Uppal Road, Hyderabad, Telangana 500007, India.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
K.S. thank Council of Scientific and Industrial Research for the research fellowship; R.S. acknowledges health care theme projects - Fundamental and Innovative Research in Science of Tomorrow (FIRST) [MLP-0162]; Niche Creation Project (NCP) [MLP-0138] of Council of Scientific and Industrial Research, India; J.C. Bose Fellowship of SERB, India, Centre of Excellence Project of Department of Biotechnology, India. Funding for open access charge: Fundamental and Innovative Research in Science of Tomorrow (FIRST) [MLP-0162].
Conflict of interest statement. None declared.
REFERENCES
- 1. Blackmond D.G. The origin of biological homochirality. Cold Spring Harb. Perspect. Biol. 2010; 2:a002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mesecar A.D., Koshland D.E.. Sites of binding and orientation in a four-location model for protein stereospecificity. IUBMB Life. 2000; 49:457–466. [DOI] [PubMed] [Google Scholar]
- 3. Kuncha S.K., Kruparani S.P., Sankaranarayanan R.. Chiral checkpoints during protein biosynthesis. J. Biol. Chem. 2019; 294:16535–16548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kuncha S.K., Mazeed M., Singh R., Kattula B., Routh S.B., Sankaranarayanan R.. A chiral selectivity relaxed paralog of DTD for proofreading tRNA mischarging in Animalia. Nat. Commun. 2018; 9:511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wiltrout E., Goodenbour J.M., Fréchin M., Pan T.. Misacylation of tRNA with methionine in Saccharomyces cerevisiae. Nucleic Acids Res. 2012; 40:10494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuncha S.K., Venkadasamy V.L., Amudhan G., Dahate P., Kola S.R., Pottabathini S., Kruparani S.P., Shekar P.C., Sankaranarayanan R.. Genomic innovation of atd alleviates mistranslation associated with multicellularity in animalia. Elife. 2020; 9:e58118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jakubowski H., Goldman E.. Editing of errors in selection of amino acids for protein synthesis. Microbiol. Rev. 1992; 56:412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fukai S., Nureki O., Shun-Ichi Sekine S.,A., Tao J., Vassylyev D.G., Yokoyama S.. Structural basis for double-sieve discrimination of L-valine from L-isoleucine and L-threonine by the complex of tRNA val and valyl-tRNA synthetase. Cell. 2000; 103:793–803. [DOI] [PubMed] [Google Scholar]
- 9. Kamtekar S., Kennedy W.D., Wang J., Stathopoulos C., Sö Ll D., Steitz T.A.. The structural basis of cysteine aminoacylation of tRNA pro by prolyl-tRNA synthetases. Proc. Natl. Acad. Sci. U.S.A. 2002; 100:1673–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guo M., Chong Y.E., Shapiro R., Beebe K., Yang X.-L., Schimmel P.. Paradox of mistranslation of serine for alanine caused by AlaRS recognition dilemma. Nature. 2009; 462:800–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Calendar R., Berg P.. d-tyrosyl RNA: formation, hydrolysis and utilization for protein synthesis. J. Mol. Biol. 1967; 26:39–54. [DOI] [PubMed] [Google Scholar]
- 12. Soutourina J., Plateau P., Blanquet S.. Metabolism of D-aminoacyl-tRNAs in Escherichia coli and Saccharomyces cerevisiae cells. J. Biol. Chem. 2000; 275:32535–32542. [DOI] [PubMed] [Google Scholar]
- 13. Calendar R., Berg P.. Purification and physical characterization of tyrosyl ribonucleic acid synthetases from Escherichia coli and Bacillus subtilis. Biochemistry. 1966; 5:1681–1690. [DOI] [PubMed] [Google Scholar]
- 14. Ling J., Yadavalli S.S., Ibba M.. Phenylalanyl-tRNA synthetase editing defects result in efficient mistranslation of phenylalanine codons as tyrosine. RNA. 2007; 13:1881–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ahmad S., Routh S.B., Kamarthapu V., Chalissery J., Muthukumar S., Hussain T., Kruparani S.P., Deshmukh M.v., Sankaranarayanan R.. Mechanism of chiral proofreading during translation of the genetic code. Elife. 2013; 2:e01519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Routh S.B., Pawar K.I., Ahmad S., Singh S., Suma K., Kumar M., Kuncha S.K., Yadav K., Kruparani S.P., Sankaranarayanan R.. Elongation factor tu prevents misediting of gly-tRNA(Gly) caused by the design behind the chiral proofreading site of D-aminoacyl-tRNA deacylase. PLoS Biol. 2016; 14:e1002465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pawar K.I., Suma K., Seenivasan A., Kuncha S.K., Routh S.B., Kruparani S.P., Sankaranarayanan R.. Role of D-aminoacyl-tRNA deacylase beyond chiral proofreading as a cellular defense against glycine mischarging by AlaRS. Elife. 2017; 6:e24001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuncha S.K., Suma K., Pawar K.I., Gogoi J., Routh S.B., Pottabathini S., Kruparani S.P., Sankaranarayanan R.. A discriminator code–based DTD surveillance ensures faithful glycine delivery for protein biosynthesis in bacteria. Elife. 2018; 7:e38232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Swairjo M.A., Schimmel P.R.. Breaking sieve for steric exclusion of a noncognate amino acid from active site of a tRNA synthetase. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beuning P.J., Musier-Forsyth K.. Hydrolytic editing by a class II aminoacyl-tRNA synthetase. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:8916–8920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rybak M.Y., Rayevsky A.v., Gudzera O.I., Tukalo M.A.. Stereospecificity control in aminoacyl-tRNA-synthetases: new evidence of D-amino acids activation and editing. Nucleic Acids Res. 2019; 47:9777–9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Berman H., Henrick K., Nakamura H.. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003; 10:980. [DOI] [PubMed] [Google Scholar]
- 23. Pasman Z., Robey-Bond S., Mirando A.C., Smith G.J., Lague A., Francklyn C.S.. Substrate specificity and catalysis by the editing active site of alanyl-tRNA synthetase from Escherichia coli. Biochemistry. 2011; 50:1474–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goto Y., Katoh T., Suga H.. Flexizymes for genetic code reprogramming. Nat. Protoc. 2011; 6:779–790. [DOI] [PubMed] [Google Scholar]
- 25. Goto Y., Goto Y., Katoh T., Suga H.. Preparation of materials for flexizyme reactions and genetic code reprogramming. Protoc. Exch. 2011; 1–11. [Google Scholar]
- 26. Karmakar A., Basha M., Venkatesh Babu G.T., Botlagunta M., Malik N.A., Rampulla R., Mathur A., Gupta A.K.. Tertiary-butoxycarbonyl (Boc) – A strategic group for N-protection/deprotection in the synthesis of various natural/unnatural N-unprotected aminoacid cyanomethyl esters. Tetrahedron Lett. 2018; 59:4267–4271. [Google Scholar]
- 27. Wagner A.M., Fegley M.W., Warner J.B., Grindley C.L.J., Marotta N.P., Petersson E.J.. N-terminal protein modification using simple aminoacyl transferase substrates. J. Am. Chem. Soc. 2011; 133:15139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murakami H., Ohta A., Ashigai H., Suga H.. A highly flexible tRNA acylation method for non-natural polypeptide synthesis. Nat. Methods. 2006; 3:357–359. [DOI] [PubMed] [Google Scholar]
- 29. Shinitzky M., Nudelman F., Barda Y., Haimovitz R., Chen E., Deamer D.W.. Unexpected differences between D- and L-tyrosine lead to chiral enhancement in racemic mixtures. Orig. Life Evol. Biosphys. 2002; 32:285–297. [DOI] [PubMed] [Google Scholar]
- 30. Mazeed M., Singh R., Kumar P., Roy A., Raman B., Kruparani S.P., Sankaranarayanan R.. Recruitment of archaeal DTD is a key event toward the emergence of land plants. Sci. Adv. 2021; 7:eabe8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B.et al.. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 2018; 27:293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Atila M., Luo Y.. Profiling and tandem mass spectrometry analysis of aminoacylated phospholipids in Bacillus subtilis. F1000Res. 2016; 5:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Routh S.B., Sankaranarayanan R.. Enzyme action at RNA-protein interface in DTD-like fold. Curr. Opin. Struct. Biol. 2018; 53:107–114. [DOI] [PubMed] [Google Scholar]
- 34. Kuzmishin Nagy A.B., Bakhtina M., Musier-Forsyth K.. Trans-editing by aminoacyl-tRNA synthetase-like editing domains. Enzymes. 2020; 48:69–115. [DOI] [PubMed] [Google Scholar]
- 35. Vargas-Rodriguez O., Bakhtina M., McGowan D., Abid J., Goto Y., Suga H., Musier-Forsyth K.. Human trans-editing enzyme displays tRNA acceptor-stem specificity and relaxed amino acid selectivity. J. Biol. Chem. 2020; 295:16180–16190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim S., Ihara K., Katsube S., Hori H., Ando T., Isogai E., Yoneyama H.. Characterization of the l-alanine exporter AlaE of Escherichia coli and its potential role in protecting cells from a toxic-level accumulation of l-alanine and its derivatives. Microbiologyopen. 2015; 4:632–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marfey P. Determination of D-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res. Commun. 1984; 49:591–596. [Google Scholar]
- 38. Dock-Bregeon A.C., Sankaranarayanan R., Romby P., Caillet J., Springer M., Rees B., Francklyn C.S., Ehresmann C., Moras D.. Transfer RNA–mediated editing in threonyl-tRNA synthetase: the class II solution to the double discrimination problem. Cell. 2000; 103:877–884. [DOI] [PubMed] [Google Scholar]
- 39. Angel M., Gomez R., Ibba M.. Aminoacyl-tRNA synthetases. RNA. 2020; 26:910–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fersht A.R., Dingwall1 C.. Evidence for the double-sieve editing mechanism in protein synthesis. Steric exclusion of isoleucine by valyl-tRNA synthetases. Biochemistry. 1979; 18:2627–2631. [DOI] [PubMed] [Google Scholar]
- 41. Roy H., Ling J., Irnov M., Ibba M.. Post-transfer editing in vitro and in vivo by the b subunit of phenylalanyl-tRNA synthetase. EMBO J. 2004; 23:4639–4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen L., Tanimoto A., So B.R., Bakhtina M., Magliery T.J., Wysocki V.H., Musier-Forsyth K.. Stoichiometry of triple-sieve tRNA editing complex ensures fidelity of aminoacyl-tRNA formation. Nucleic Acids Res. 2019; 47:929–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ibba M., Söll D.. Aminoacyl-tRNA synthesis. Annual Reviews. 2000; 69:671–650. [DOI] [PubMed] [Google Scholar]
- 44. Erb T.J., Kiefer P., Hattendorf B., Günther D., Vorholt J.A.. GFAJ-1 is an arsenate-resistant, phosphate-dependent organism. Science. 2012; 337:467–470. [DOI] [PubMed] [Google Scholar]
- 45. Reaves M.L., Sinha S., Rabinowitz J.D., Kruglyak L., Redfield R.J.. Absence of detectable arsenate in DNA from arsenate-grown GFAJ-1 cells. Science. 2012; 337:470–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wolfe-Simon F., Blum J.S., Kulp T.R., Gordon G.W., Hoeft S.E., Pett-Ridge J., Stolz J.F., Webb S.M., Weber P.K., Davies P.C.W.et al.. A bacterium that can grow by using arsenic instead of phosphorus. Science. 2010; 332:1163–1166. [DOI] [PubMed] [Google Scholar]
- 47. Sokabe M., Okada A., Yao M., Nakashima T., Tanaka I.. Molecular basis of alanine discrimination in editing site. Proc. Natl Acad. Sci. U.S.A. 2005; 102:11669–11674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Beebe K., Merriman E., Schimmel P.. Structure-specific tRNA determinants for editing a mischarged amino acid. J. Biol. Chem. 2003; 278:45056–45061. [DOI] [PubMed] [Google Scholar]
- 49. Beebe K., Mock M., Merriman E., Schimmel P.. Distinct domains of tRNA synthetase recognize the same base pair. Nature. 2007; 451:90–93. [DOI] [PubMed] [Google Scholar]
- 50. Schimmel P. Mistranslation and its control by tRNA synthetases. Philos. Trans. Roy. Soc. B: Biol. Sci.s. 2011; 366:2965–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.